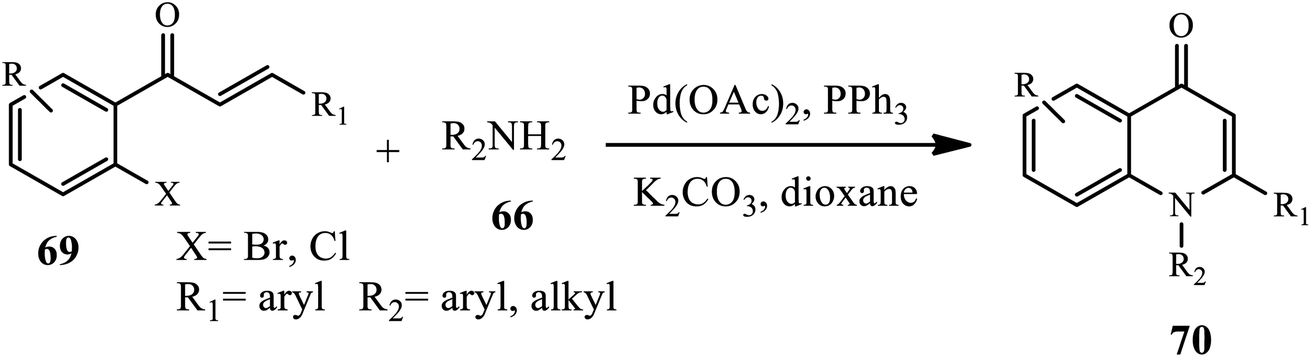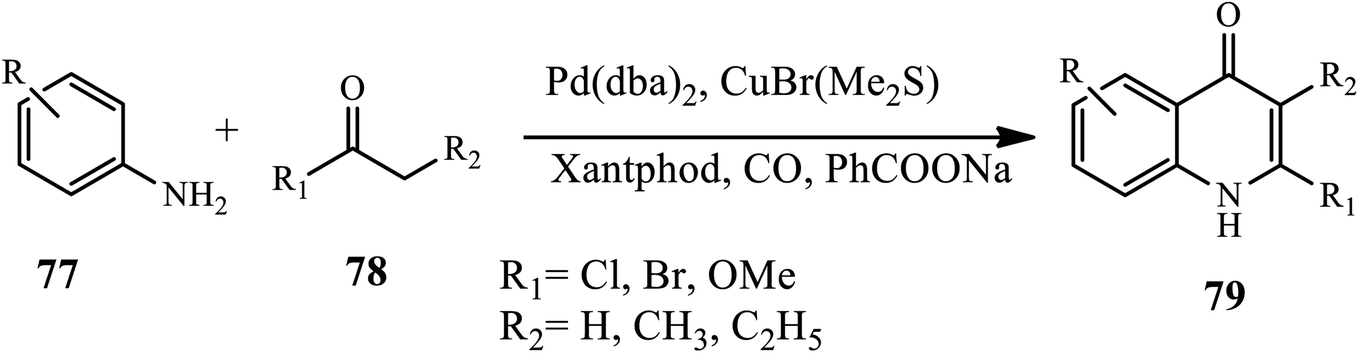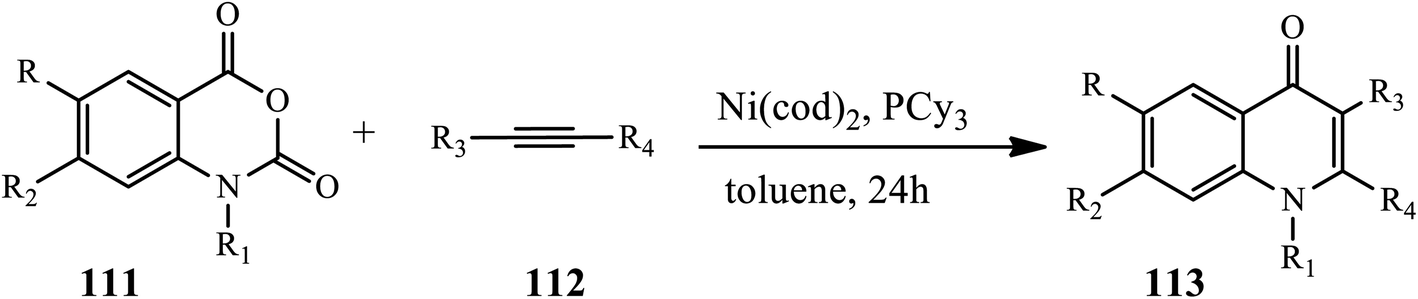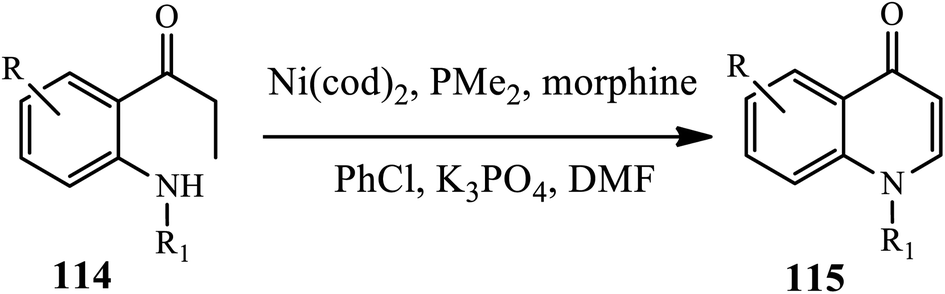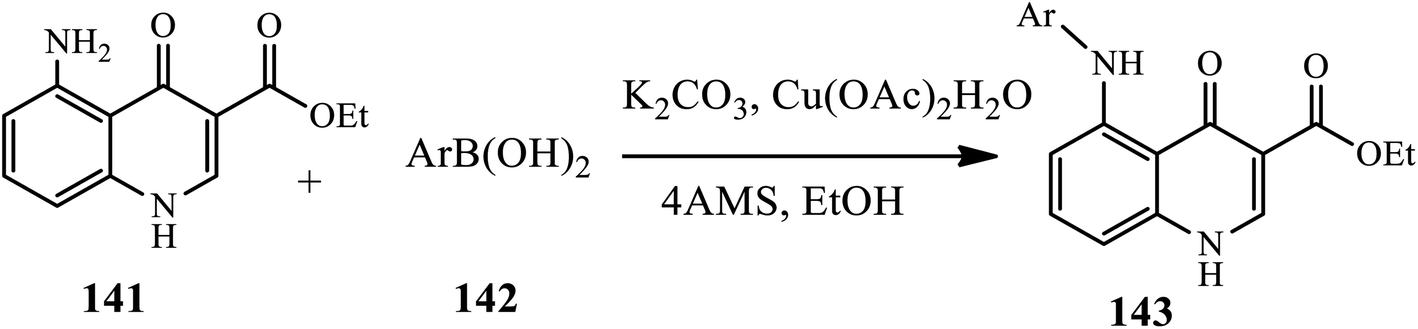DOI:
10.1039/D3RA00749A
(Review Article)
RSC Adv., 2023,
13, 8657-8682
Recent advances in the synthesis of pharmaceutically active 4-quinolone and its analogues: a review
Received
3rd February 2023
, Accepted 1st March 2023
First published on 15th March 2023
Abstract
4-Quinolone and its analogs are heterocyclic classes of organic compounds displaying biologically active and a broad spectrum of pharmaceutical drug scaffolds. 4-Quinolone is the first-line chemotherapeutic treatment for a wide spectrum of bacterial infections. Recently, 4-quinolone and its derivatives have been shown to have the potential to cure and regulate various acute and chronic diseases, including pain, ischemia, immunomodulation, inflammation, malarial, bacterial infection, fungal infection, HIV, and cancer, based on several reports. This review highlights and provides brief information to better understand the development of experimental progress made to date in the synthetic protocol towards 4-quinolone and its analogs. Thus, classical synthesis protocol, metal-free reaction protocol, and transition metal-catalyzed reaction procedures are briefly discussed along with the pharmaceutical activities of selected 4-quinolone derivatives.
1. Introduction
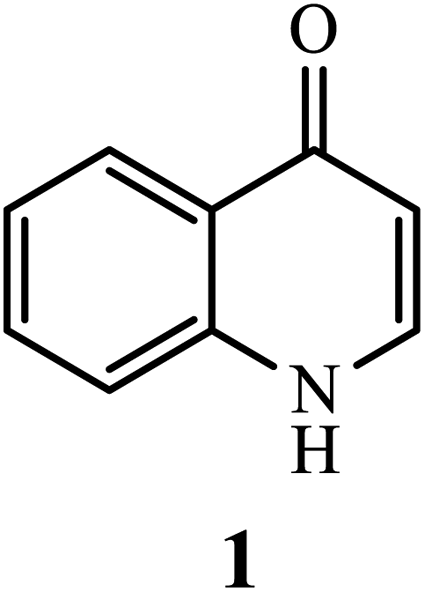
Currently, quinolones have captured the attention of numerous researchers from various scientific disciplines. In the last two decades, researchers have contributed significantly to the scientific advancements in the pharmaceutical arena that have occurred with title compounds. The investigations and synthesis modifications suggested by researchers promote future synthesis advancements in quinolone and its derivatives as a vital scaffold for pharmaceutical industries. Microbial infections are becoming a global issue and a major public health concern because they cause diseases and mortality, placing tremendous strain on healthcare systems.1 In the fight against microbial illnesses, antimicrobial agents are effective weapons.2 Following the discovery of penicillin, the first antibiotic agent used in human clinical trials in the 1940s, a plethora of antibiotics was developed.3 In the field of antibiotics, the quinolone scaffold has been widely explored, and it represents an interesting synthetic candidate for chemical synthesis.4,5 Among the family of quinolones, the most prominent structure is 4-quinolone, which possesses the feature of a crucial substructure that exists in various quinolone derivatives with excellent biological activities (Fig. 1). 4-Quinolone is one of the most widely used N-hetero-aromatic compounds, which possess a bicyclic system of pyridine moiety fused with aromatic or hetero-aromatic scaffold at two adjacent carbons and have a carbonyl group at C-4.6,7
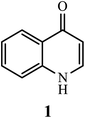 |
| | Fig. 1 Chemical structure of 4-quinolone (1). | |
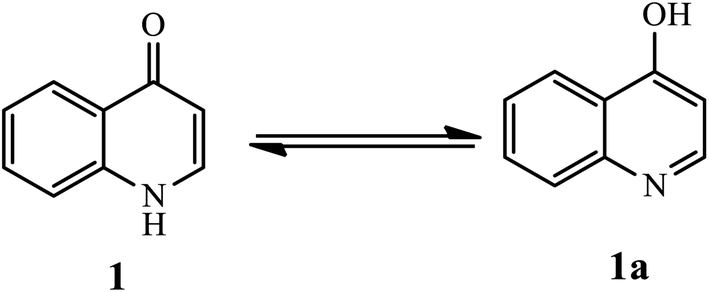
Quinolones are present in equilibrium through tautomerism, the minor tautomeric form of 4-quinolone (1) and its other version 4-hydroxyquinolone (1a), as depicted in Fig. 2.8
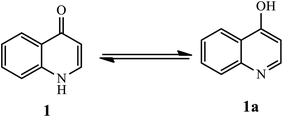 |
| | Fig. 2 Tautomeric form of 4-quinolone (1). | |
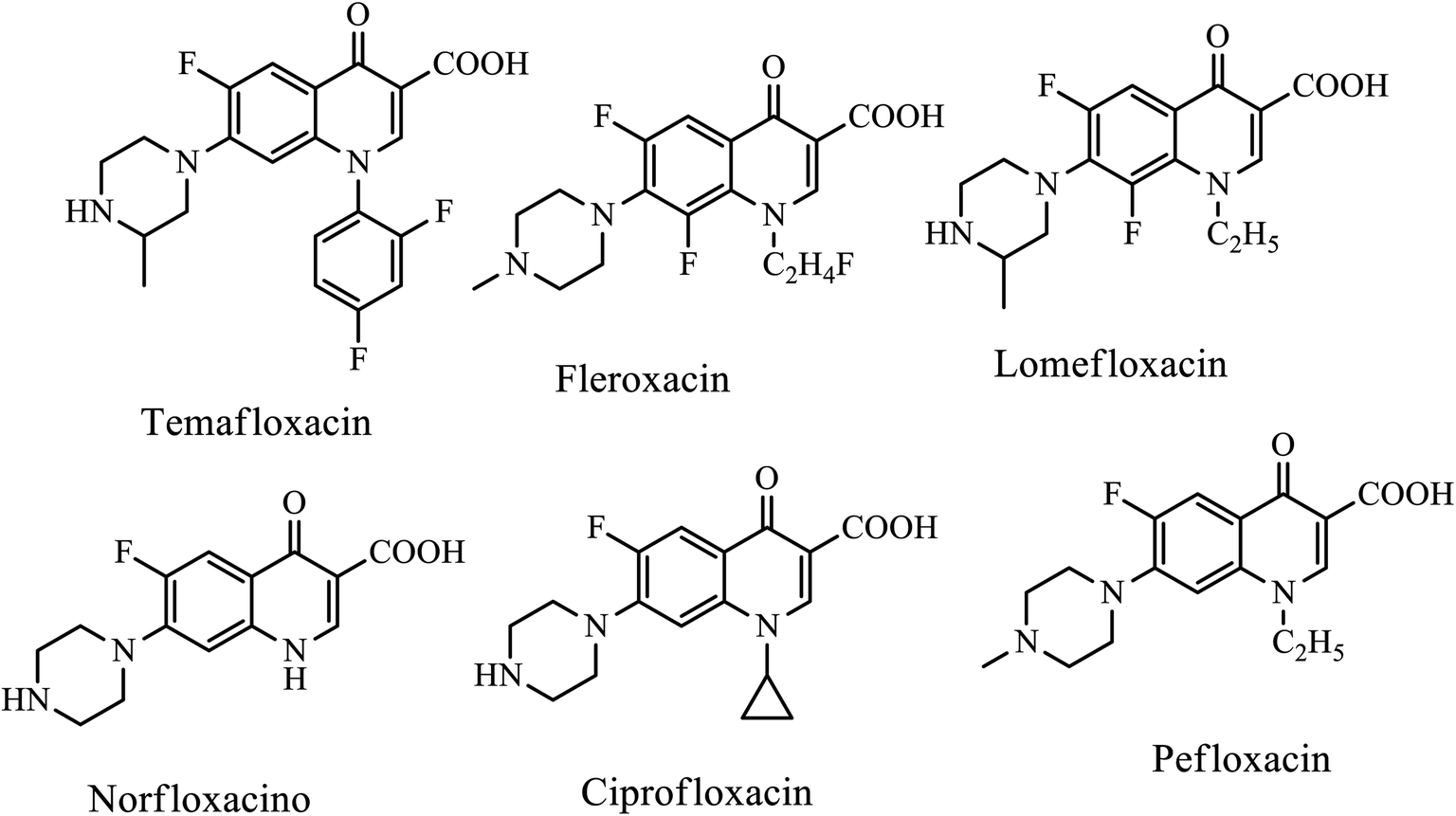
Chemical modification of the parent 4-quinolone scaffold is a potential resource for accessing biologically and pharmacologically important molecules. Quinolone derivatives are now being used as lead compounds in the drug industries or being developed as drug candidates to treat various human diseases, including infectious diseases, such as malaria, parasitic infections, bacterial infections, and fungal infections, and viral infections such as hepatitis, HIV, and herpes.5 Quinolone and its derivatives exhibit a potential antimicrobial agent that acts as therapy for many infections with broad-spectrum properties. The comment examples with the quinolone scaffold are as follows (Fig. 3).
 |
| | Fig. 3 Chemical structure of biologically active compounds containing 4-quinolone scaffold. | |
2. Structural and historical background of 4-quinolone
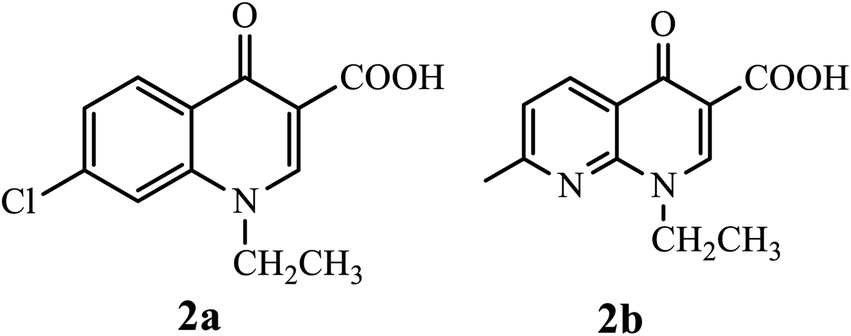
In 1962, Lesher and coworkers showed that 7-chloro-1,4-dihydro-1-ethyl-4-oxoquinolone-3-carboxylic acid 2a, obtained as an impurity during the synthesis of chloroquine, possessed antibacterial activity (Fig. 4).10
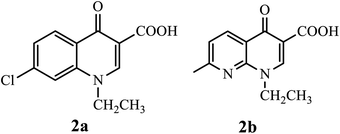 |
| | Fig. 4 Chemical structure of 7-chloro-1,4-dihydro-1-ethyl-4-oxoquinolone-3-carboxylic acid and nalidixic acid. | |
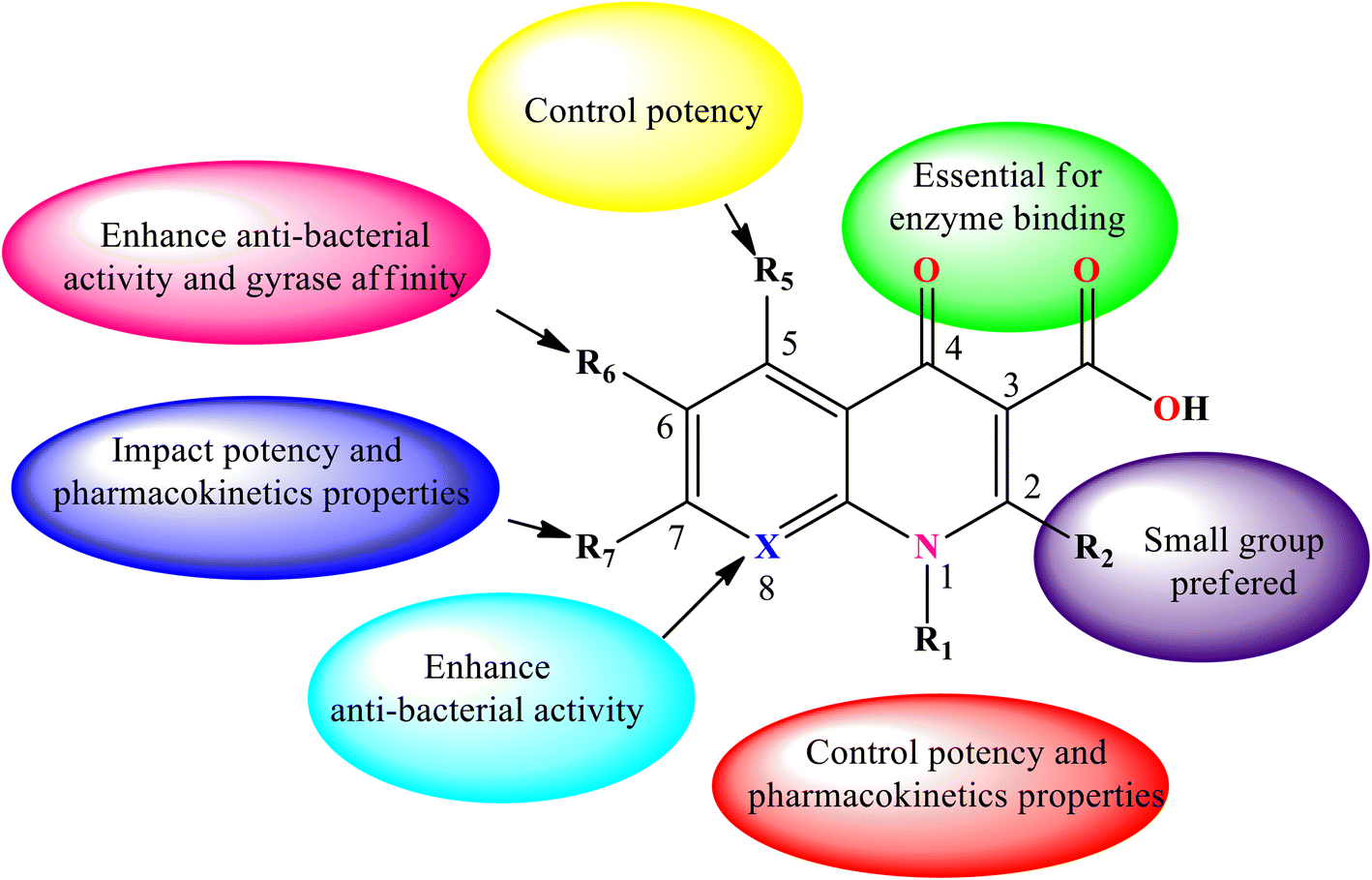
Consequently, various derivatives of the title compound were synthesized and evaluated owing to their antibacterial activities. For instance, nalidixic acid 2b is one of the oldest quinolone derivatives used as an antibiotic to treat urinary tract infections.4 Currently, 4-quinolone derivatives used as antibacterial agents are halogenated at the 6-position, and some are halogenated at position 8. Thus, by modifying the chemical structure of the 4-quinolone scaffold, the action or potency of the compound can be altered. The quinolone scaffold can be structurally modified using various substituents by introducing it at various sites of the skeleton. The chemical modifications on the quinolone scaffold at the N-1, C-2, C-3, C-5, C-6, C-7, and C-8 positions have resulted in molecules that boost the physical, chemical, pharmacokinetic, and pharmacological properties.9 The substituents and their positions improve the properties of the biological activities of 4-quinolone and its derivatives (Fig. 5).
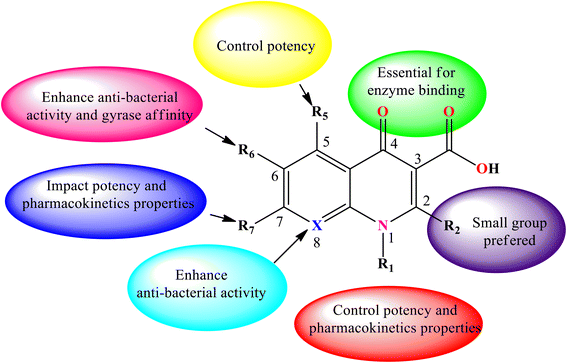 |
| | Fig. 5 Structural–activity relationship (SAR) of 4-quinolone derivatives. | |
In the past few years, efficient, various new, convenient with broad scope synthetic approaches towards the title compound and its derivatives have been designed and developed to prepare diverse quinolone-based scaffolds. In this review, we summarize recent advancements in the synthesis of 4-quinolones and their derivatives as well as the pharmacological potential properties of the drug with the scaffold of the title compound. The reactions discussed in this review were selected owing to their high yield, relative simplicity to conduct and highly selective catalytic processes under mild reaction conditions. This review is divided into two sections: general catalytic synthesis and post modification of the scaffold.
3. Development of the syntheses of 4-quinolone and its derivatives
Because of their broad spectrum of antibacterial, antimalarial, antiviral, antifungal, and anti-cancer activities, quinolones and their derivatives are an omnipotent class of drugs. An update in the syntheses of 4-quinolone and its derivatives via selectivity of functionalization at a specified position on the scaffold is an interesting field of research planned to enhance their bioactivities. Generally, there are various methods for synthesizing title scaffolds. Herein, metal-free or green synthesis, metal-catalyzed synthesis, and post-functionalization at various positions of the skeleton are discussed briefly.
3.1 Metal-free 4-quinolone synthesis
To date, various synthesis protocols have been reported in the literature for preparing 4-quinolone and its derivatives. Among them, the metal free synthesis method has attracted the interest of various researchers owing to its simplicity, low cost, and non-hazardous materials.
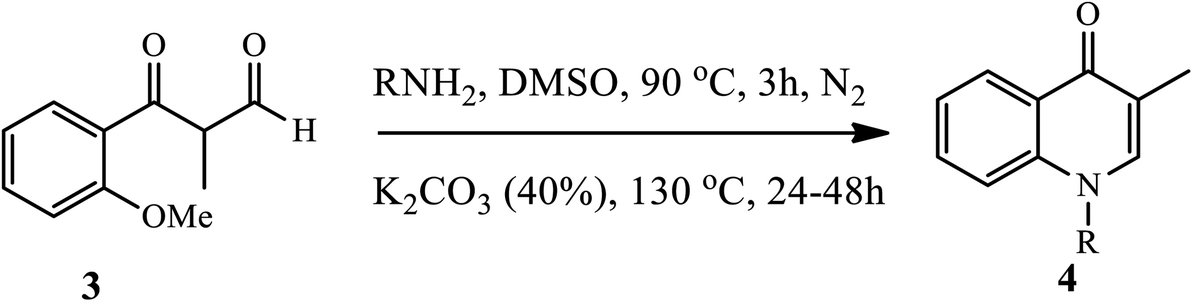
3.1.1 Synthesis of N-substituted 4-quinolone. Fu and coworkers reported a convenient and efficient method through the selective cleavage of aromatic carbon–oxygen (C–O) bonds under metal free and in the presence of a base to afford 4-quinolone derivatives in moderate yield (Scheme 1).11 However, the cleavage of aromatic C–O bonds is extremely difficult because of their energy requirements. The synthesis of N-substituted-4-quinolone 4 was accomplished via the cleavage of aromatic C-from the O-methoxy group, followed by condensation of aldehyde derivatives 3 with a primary amine in the presence of K2CO3 base and DMSO solvent under inert conditions to generate an imine intermediate, which is subjected to intramolecular aromatic nucleophilic substitution to afford the N-substituted-4-quinolone derivatives 4.11
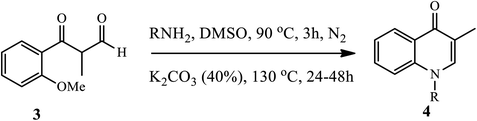 |
| | Scheme 1 Synthesis of N-substituted-4-quinolone derivatives via cleavage of aromatic C–O bonds. | |
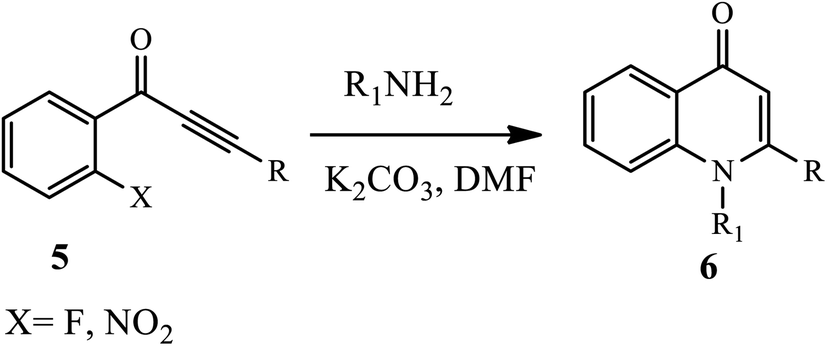
Through a tandem fashion amination followed by conjugated Michael addition reaction under a free catalyst, Zhao and coworkers reported functionalized N-substituted-4-quinolone derivatives 6 (Scheme 2) under high temperature and an inert atmosphere.12 Aliphatic amines afford the target product a good to excellent yield. The electron-withdrawing group substituents, such as –NO2 or –F, at the ynones 5 moiety participated in the reaction smoothly.12
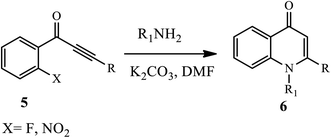 |
| | Scheme 2 Synthesis of functionalized N-substituted-4-quinolinone derivatives. | |
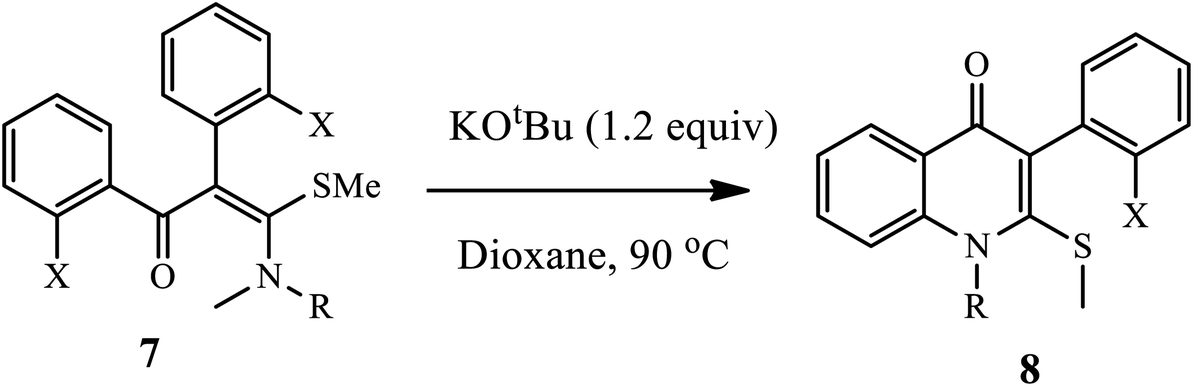
Janni and coworkers disclosed a new metal-free protocol to prepare fused heterocycle quinolone derivatives 8 from single S,N-acetal precursors 7 through double hetero-annulation under basic conditions (Scheme 3).13 Furthermore, the resulting quinolones 8 are transformed into fused heterocycles upon C–S and C–O coupling reactions under radical and metal-mediated conditions, respectively. It has been observed that the corresponding products from the substituted aniline nitrogen atom of S,N-acetals bear both electron-withdrawing and electron releasing groups synthesized in moderate to excellent yields. Unfortunately, aliphatic amino S,N-acetals exhibited poor yield.
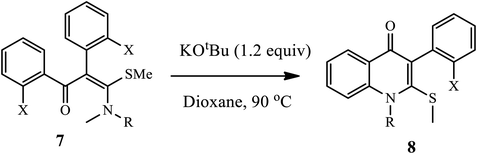 |
| | Scheme 3 Synthesis of 4-quinolone derivative 8 using KOtBu. | |
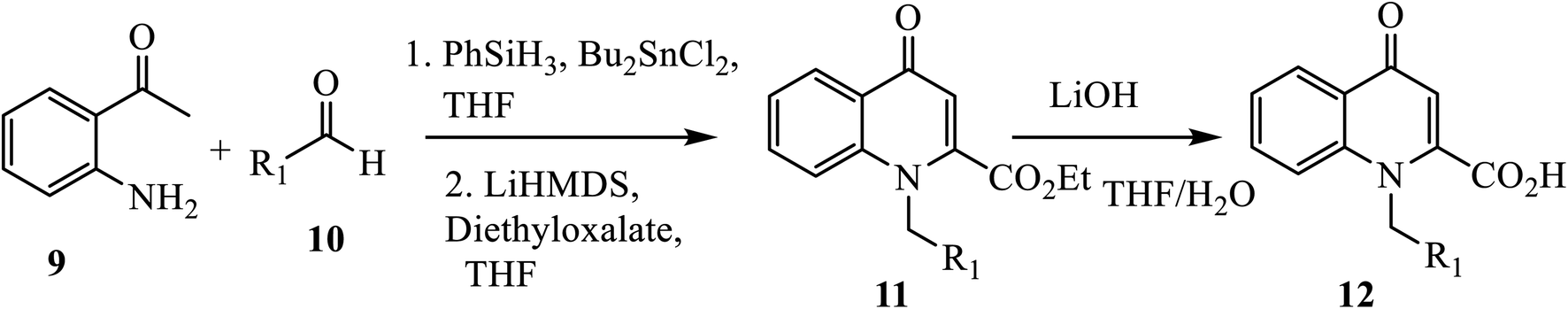
An efficient strategy based on direct reductive amination, followed by LiHMDS-induced in situ cyclo-condensation of aminoacetophenone 9 and aldehyde derivative 10, was reported for the synthesis of N-substituted 2-carboxy-4-quinolones 12 (Scheme 4).14 Treatment of 2′ aminoacetophenone 9 and aldehyde 10 with phenyl silane (PhSiH3)/dibutyltin dichloride (Bu2SnCl2), followed by in situ cyclo-condensation in the presence of LiHMDS, affords ester 11 in a very good yield under reflux. Further, the hydrolysis of ester 11 gave the corresponding acid 12 in moderate to good yield with a broad scope.
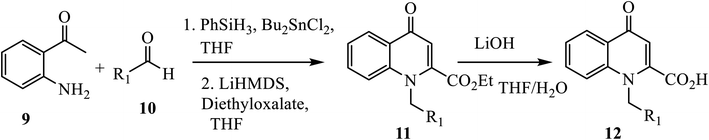 |
| | Scheme 4 Synthesis of 4-quinolone derivative 12 using LiHMDS-induced cyclo-condensation. | |
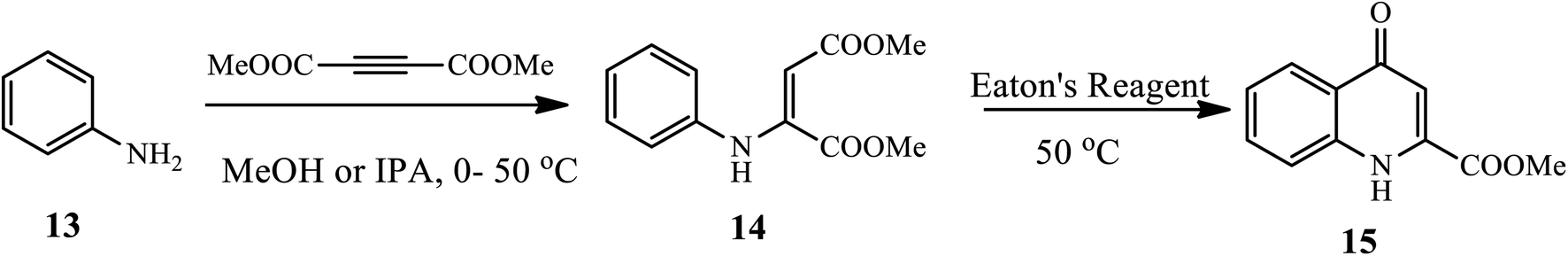
3.1.2 Synthesis of 2-substituted 4-quinolone. Zewge and coworkers reported a very mild, efficient and scalable method for the synthesis of 4-quinolone derivative 15 from aniline 13 using an inexpensive and commercially available Eaton's reagent under mild conditions (Scheme 5).15 Broad scopes of functionalized anilines participate and afford the target product in very good to excellent yields.
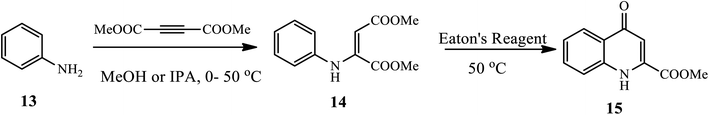 |
| | Scheme 5 Synthesis of 4-quinolone-2-carboxylic acid 15 using Eaton's reagent. | |
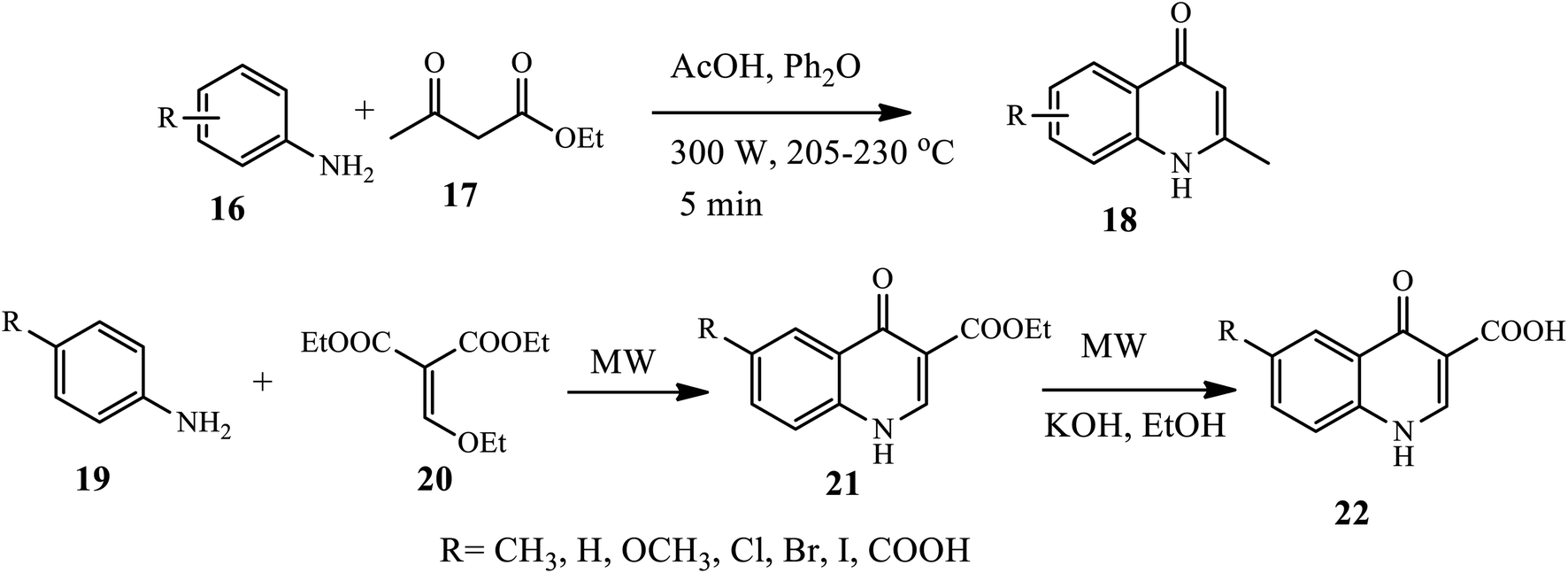
Duarte's group reported a microwave-assisted synthesis of various substituted 2-methyl-4-quinolone derivatives 18 in a one-step reaction procedure. Here, using electron-rich aniline 16 and ethyl acetoacetate 17 in the presence of diphenyl ether as a solvent under reflux at very high temperatures afforded the target compound in good yields. Anilines substituted with electron-donating groups afforded the required 4-quinolones 18 in a good yield. However, using the same procedure with electron withdrawing substituents N,N′-diarylureas was afforded in moderate to good yields.16 Similarly, an efficient microwave-assisted procedure for the synthesis of ethyl-quinolone-4-one-3-carboxylates 21 from p-substituted aniline 19 and diethyl-ethoxymethylenemalonate 20 in the presence of diphenyl ether at 80 °C under MW irradiation was reported by Malvacio and coworkers (Scheme 6).17 Further, the quinolone obtained after hydrolysis under MW irradiation furnished the corresponding quinolone-4-one-3-carboxylic acids 22 in very good to excellent yields. The advantage of this procedure is that it works for a wide substrate scope, and this route offers the benefits of a very short reaction time and relatively good yield.
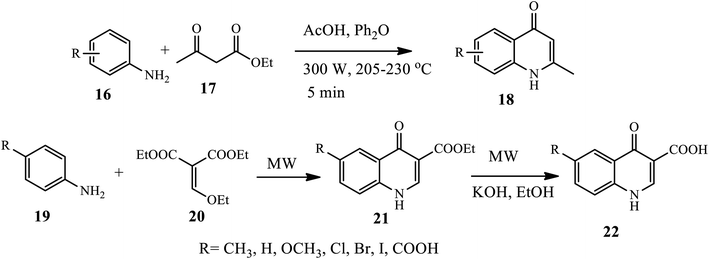 |
| | Scheme 6 Microwave-assisted synthesis of 4-quinolone derivatives. | |
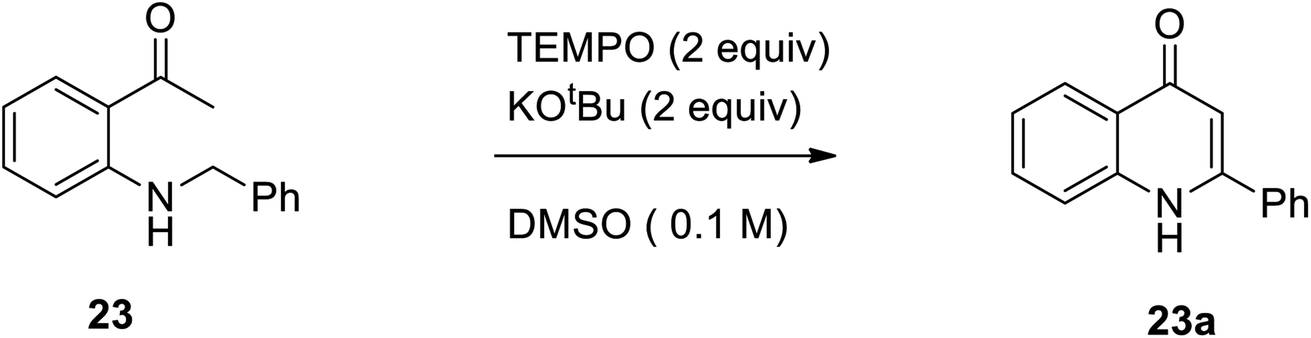
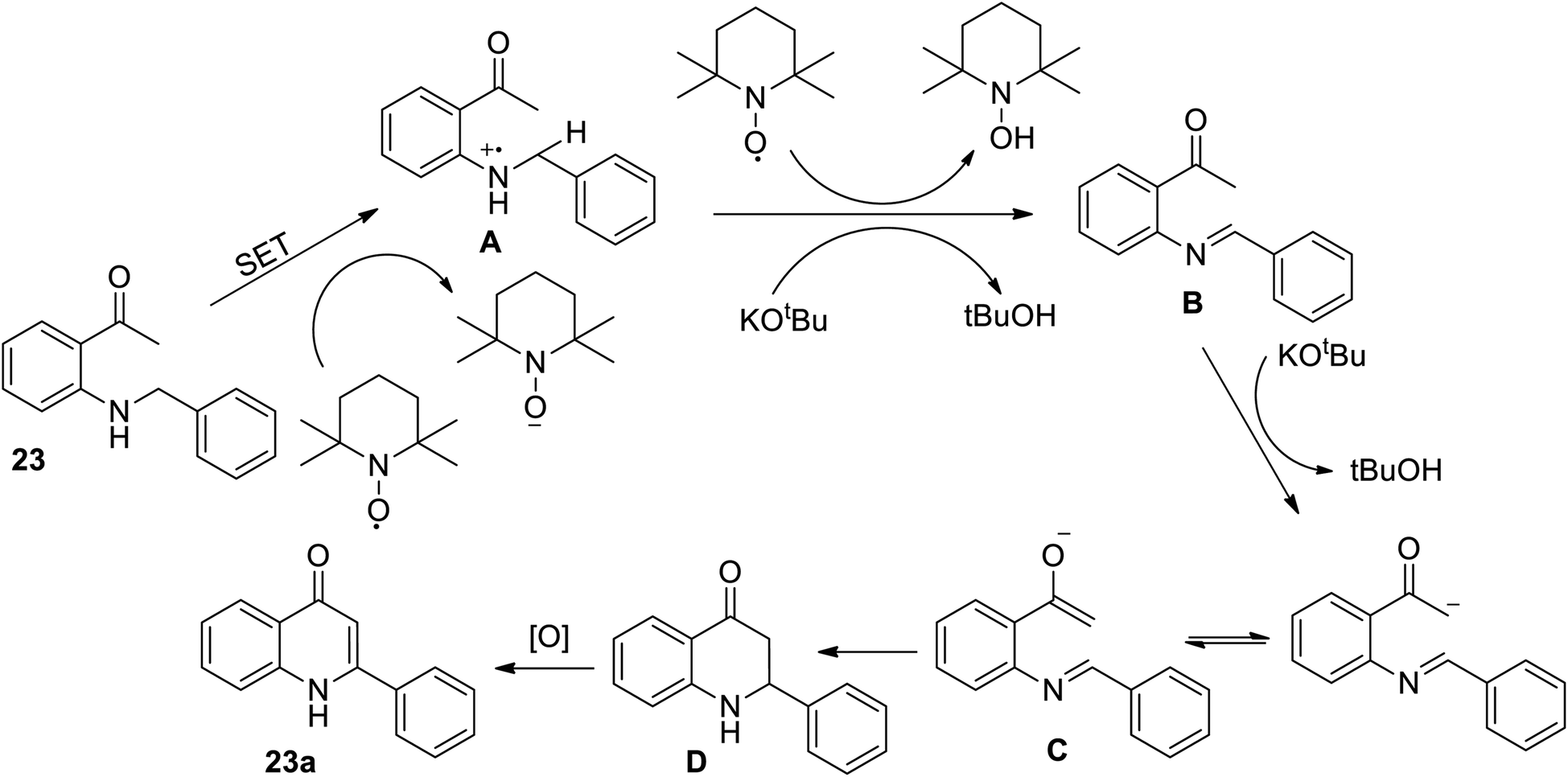
In 2015, Hu et al. designed a straight forward method for the synthesis of numerous 2-aryl-4-quinolone derivatives 23 from N-aryl methyl-2-aminophenyl ketones 23a through a metal-free oxidative intramolecular oxidative Mannich reaction, followed by C(sp3)–H/C(sp3)–H coupling and aromatization using TEMPO as an oxidant and KOtBu as the base (Scheme 7).18
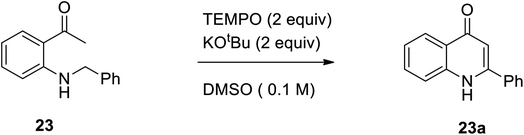 |
| | Scheme 7 TEMPO catalyzed intramolecular tandem oxidative C(sp3)–H/C(sp3)–H coupling. | |
The above reaction between secondary amine and ketone derivatives was developed simply and directly to afford the title compound and its derivatives with a broad scope. This reaction is further described using a plausible mechanism (Scheme 8).
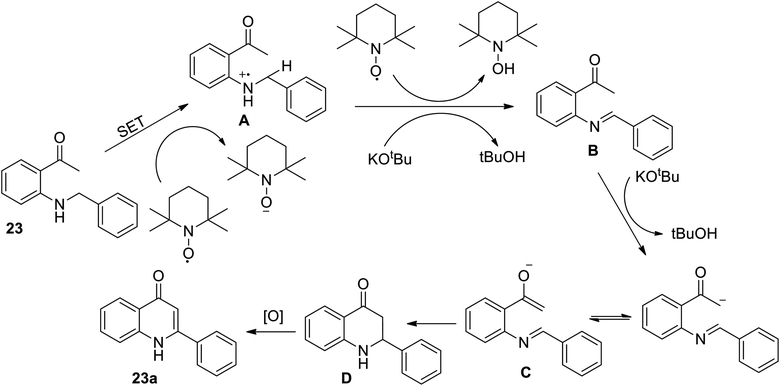 |
| | Scheme 8 Proposed mechanism for metal free promoted oxidative annulation. | |
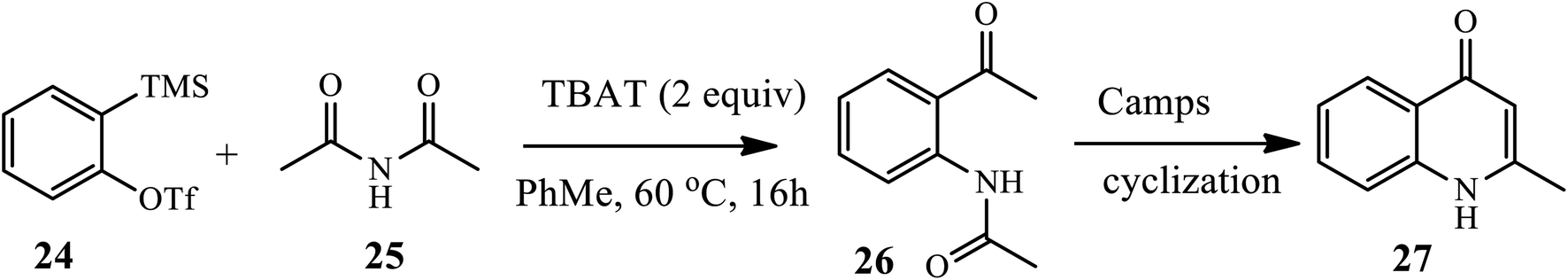
In 2016, Wright et al. established the insertion of arynes 24 into acyclic imides and anhydrides 25 to afford aryl ketoamides 26 under mild conditions in the presence of TBAT, which was then cyclized through base-initiated camp cyclization to provide quinolones 27 in a moderate to good yield (Scheme 9).19 However, electron withdrawing substituents, such as F, failed to afford aryl ketoamide.
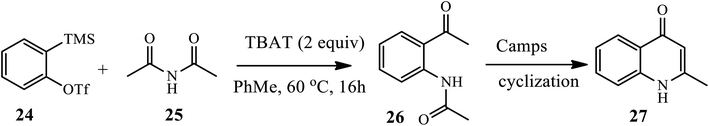 |
| | Scheme 9 Camp cyclization of ketoamide insertion products to provide quinolones. | |
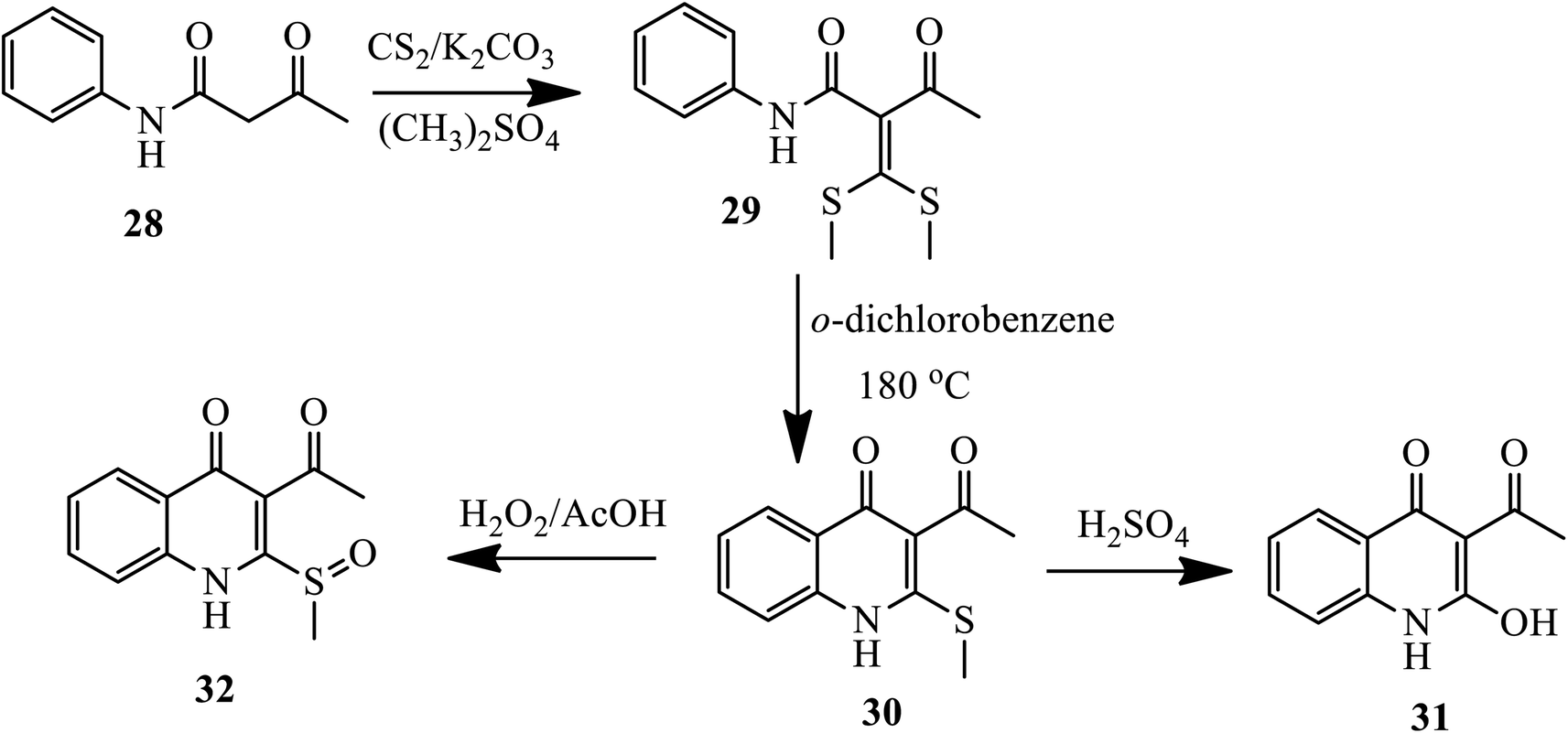
Later on, in 2018, Xu and coworkers presented the synthesis of 2-methyl-thioquinolin-4(1H)-ones as an antibacterial agent via free radical reaction. The nucleophilic reaction of aceto-acetanilide 28 with carbon bisulfide in the presence of K2CO3 following methylation with dimethyl sulfate forms the intermediate (bis(methylthio)methylene)-3-oxo-N-phenylbutanamide 29, which was then cyclized thermally to yield compound 30 in the presence of o-dichlorobenzene. Further, upon hydrolysis in 50% sulfuric acid and oxidation using hydrogen peroxide in AcOH, the 2-methylthio of quinolone 30 was converted to compounds 31 and 32 (Scheme 10).20
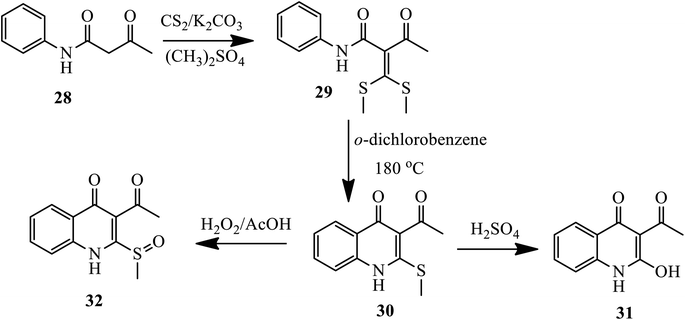 |
| | Scheme 10 Synthesis of sulfur-containing 4-quinolone by free-radical reaction. | |
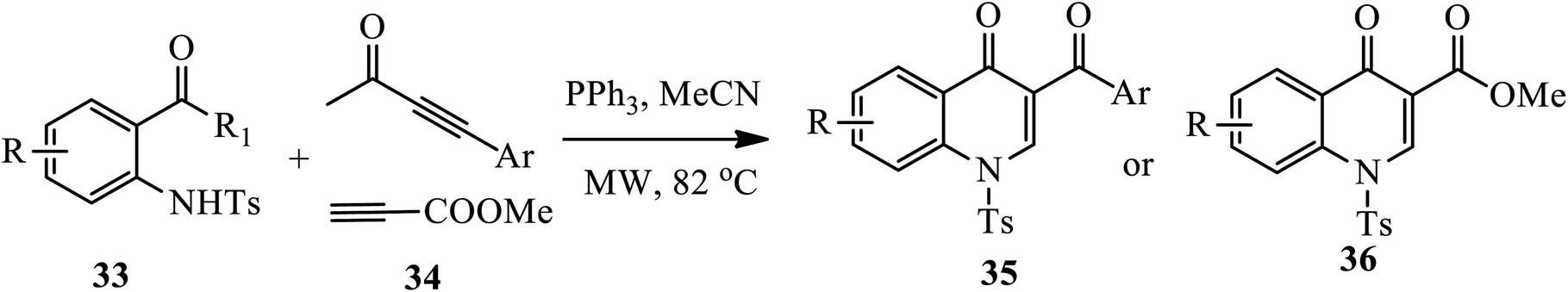
3.1.3 Synthesis of 3-substituted 4-quinolone. A phosphino-catalysis approach toward the synthesis of several 3-aroyl-4-quinolones 35 and methyl-4-quinolone-3-carboxylate esters 36 from S-phenyl-2-(N-tosylamido) benzothioates 33 and activated alkynes 34 under microwave irradiation at 82 °C for 2 h was reported by Khong and Kwon (Scheme 11).21 The method provides moderate to very good yields for both electron rich and electron withdrawing substituents containing starting materials. However, the electron deficient aromatic ring resulted in a lower yield compared to the electron-rich aromatic rings.
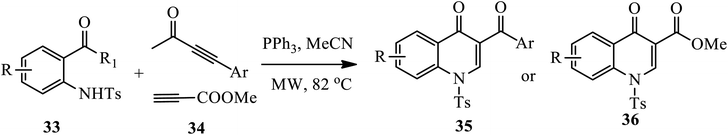 |
| | Scheme 11 Triphenylphospine-catalyzed synthesis of 4-quinolone derivatives. | |
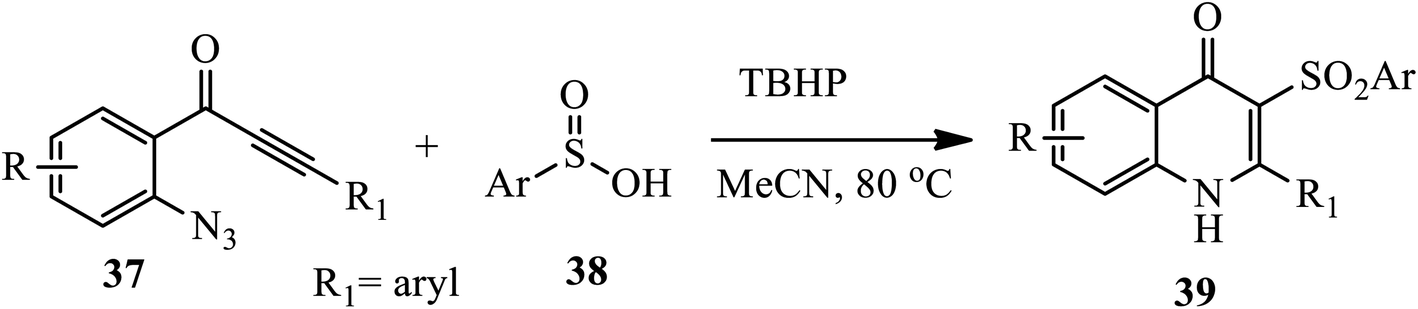
In 2016, Zhou and coworkers reported a novel one-pot metal free protocol for the synthesis of sulfone-containing 4-quinolones upon radical oxidative cyclization of o-azidoaryl acetylenic ketones 37 and sulfinic acids 38 in the presence of radical initiators (TBHP) at 80 °C under Ar for 0.5 h affording phenyl-3-(phenylsulfonyl) quinolin-4(1H)-one 39 (Scheme 12).22 Both electron-donating and electron-withdrawing groups on the aromatic ring produced the corresponding phenyl-3-(phenylsulfonyl) quinolin-4(1H)-one 39 in good to excellent yields. However, the reaction was influenced by the steric effect, and aliphatic sulfinic acid, such as methanesulfinic acid, failed to produce the desired 4-quinlone.
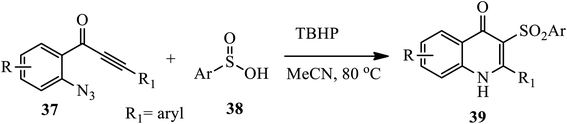 |
| | Scheme 12 Metal-free synthesis of sulfone containing 4-quinolones. | |
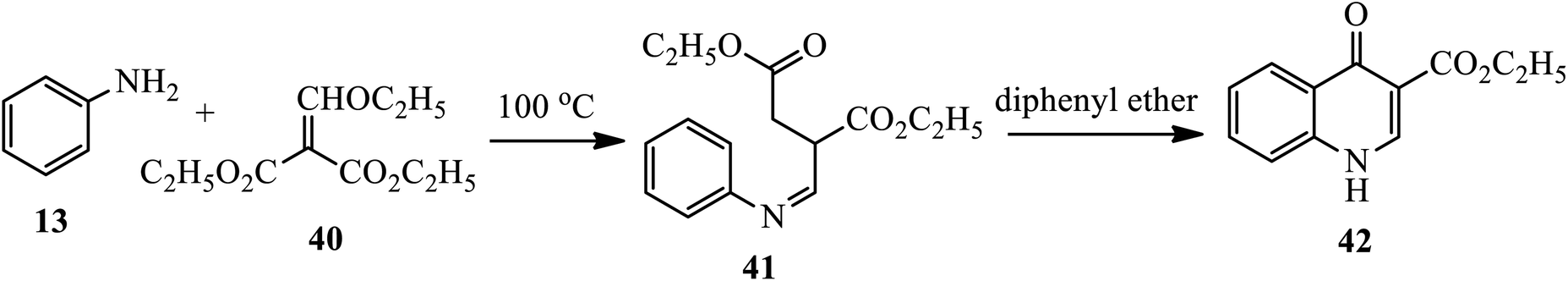
Novel water-soluble antiproliferative agents, 4-quinolones 42, were synthesized by Zhang's group (Scheme 13).23 Heating aniline 13 with ethoxy methylene malonic ester 40 at 100 °C afforded Schiff bases 41, which was then cyclized to ethyl 4(1H)-quinolone-3-carboxylates 42 using diphenyl ether under reflux conditions.
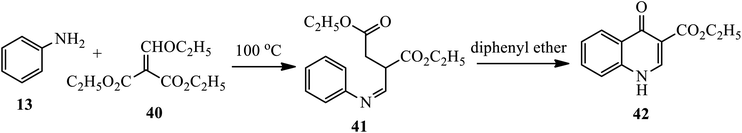 |
| | Scheme 13 Synthesis of 4-quinolone using diphenyl ether. | |
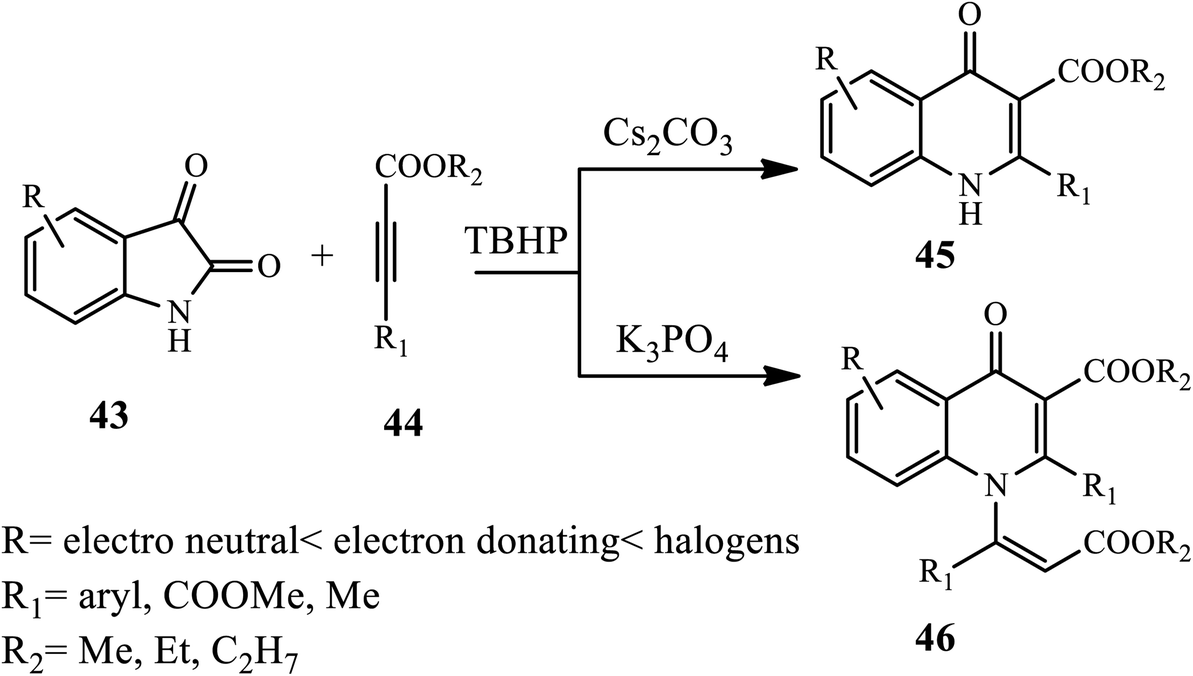
In 2018, Jiang and coworkers developed an efficient transition-metal-free oxidative cyclization reaction for the synthesis of 3-carboxylate-4-quinolones 45 and 1-vinyl-3-carboxylate-4-quinolone 46 using isatins 43 and alkynes 44 in the presence of DMSO solvent, Cs2CO3 base, and TBHP as an oxidant, while in the case of 1-vinyl-3 carboxylate-4-quinolones, K3PO4 was used as a base. The authors examined various substrate effects, including electron-neutral, electron-donating and electron withdrawing substituted isatins, which provided excellent yields of the desired product (Scheme 14).24
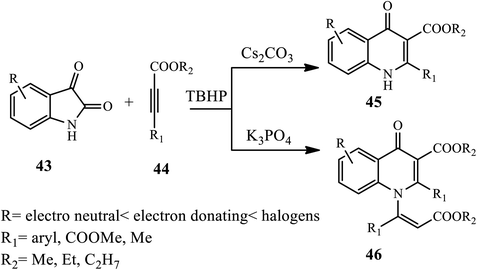 |
| | Scheme 14 Synthesis of 4-quinolones through transition-metal-free oxidative cyclization. | |
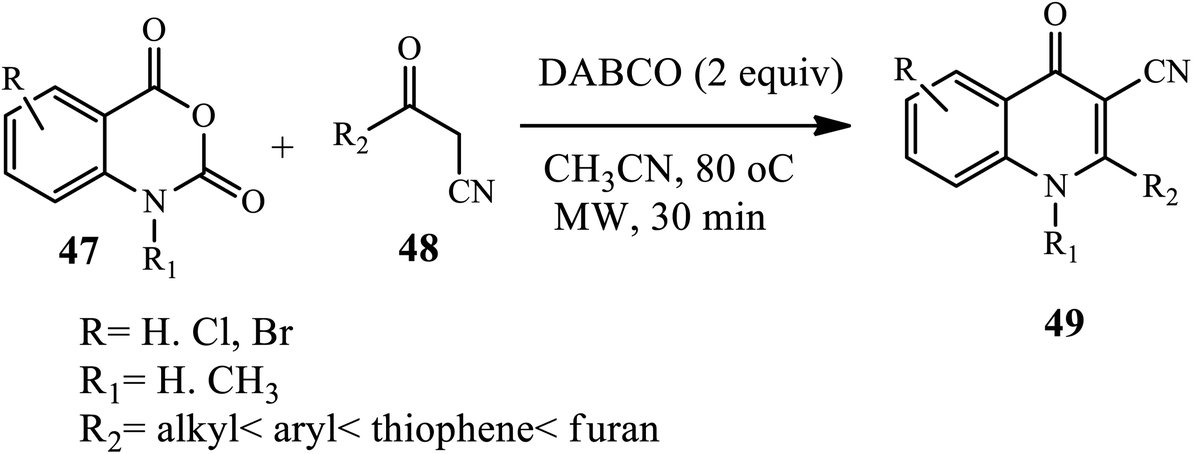
Recently, 3-cyano 4-quinolone derivatives 49 were synthesized in good to excellent yields upon DABCO-mediated decarboxylative cyclization of isatoic anhydrides 47 with active methylene groups 48, such as aroyl/heteroaryl/alkoylacetonitriles, in CH3CN under microwave irradiation at 80 °C for 30 min. Further, this method provides mild reaction conditions and a short reaction time compatible with a wide substrate scope (Scheme 15).25
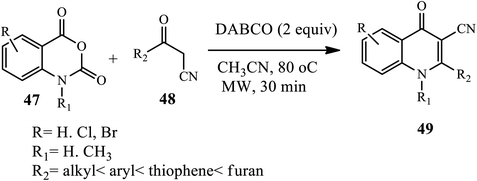 |
| | Scheme 15 Synthesis of 4-quinolone via one-pot DABCO-mediated decarboxylative cyclization. | |
3.1.4 Synthesis of di-substituted 4-quinolone. In 2017, Chen's group successfully synthesized 4-quinolone derivatives 51 in one pot using a tandem procedure in good to excellent yields (Scheme 16).26 An intermolecular nucleophilic addition of various amines to (Z)-β-chlorovinyl ketones 50 followed by elimination of chlorine anion gives Z-enamine intermediates, which can be transformed to 4-quinolones 51 through intramolecular SNAr reaction in the presence of a base (Cs2CO3) in DMSO at 140 °C. Various substitution patterns and functionalities (such as Me, t-Bu, MeO, F, and Cl) are applicable and tolerated in this protocol. Interestingly, the dihalo-substituted ketone substrates converted to the corresponding quinolones in very good yields.
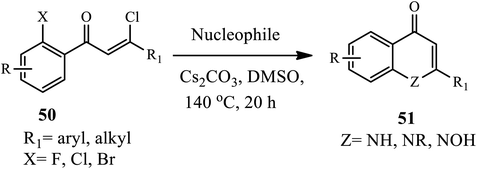 |
| | Scheme 16 Synthesis of 4-quinolone through nucleophilic addition–elimination-SNA reaction. | |
In 2018, Bandatmakuru and Arava reported a direct procedure for the synthesis of N-functionalized 4-quinolones 54 and 1,2-substituted 4-quinolones 53 from 2-aminoacetophenones 52. Various 2-aminoacetophenone derivatives 52 were transformed into the corresponding 4-quinolones 54 and 1,2-substituted 4-quinolones 53 upon refluxing with benzoyl chloride in TEA and with PTSA in DMFDMA, respectively. The authors examined various substrate effects, including electron-donating/electron-withdrawing groups on the phenyl ring, ortho-, meta- and para-substituted aminoacetophenones, along with N-alkyl and N-aryl aminoacetophenones that can be successfully transformed into the corresponding 4-quinolones in good yields (Scheme 17).27
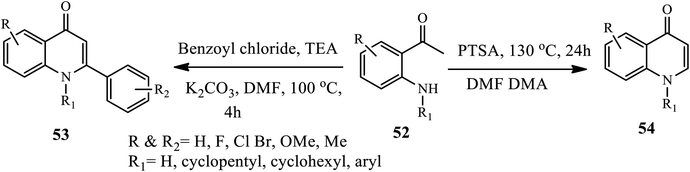 |
| | Scheme 17 Synthesis of N-functionalized 4-quinolone and 1,2-substituted 4-quinolones. | |
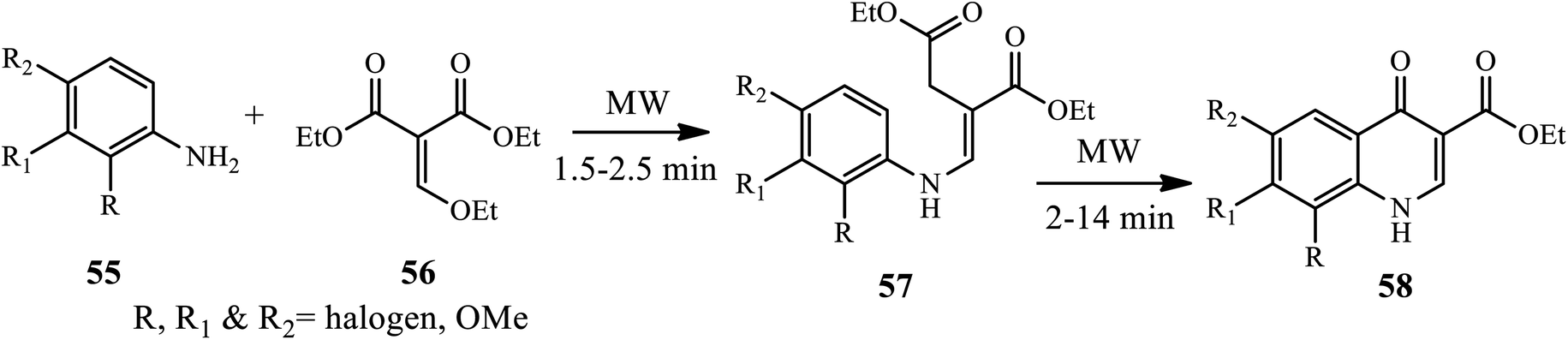
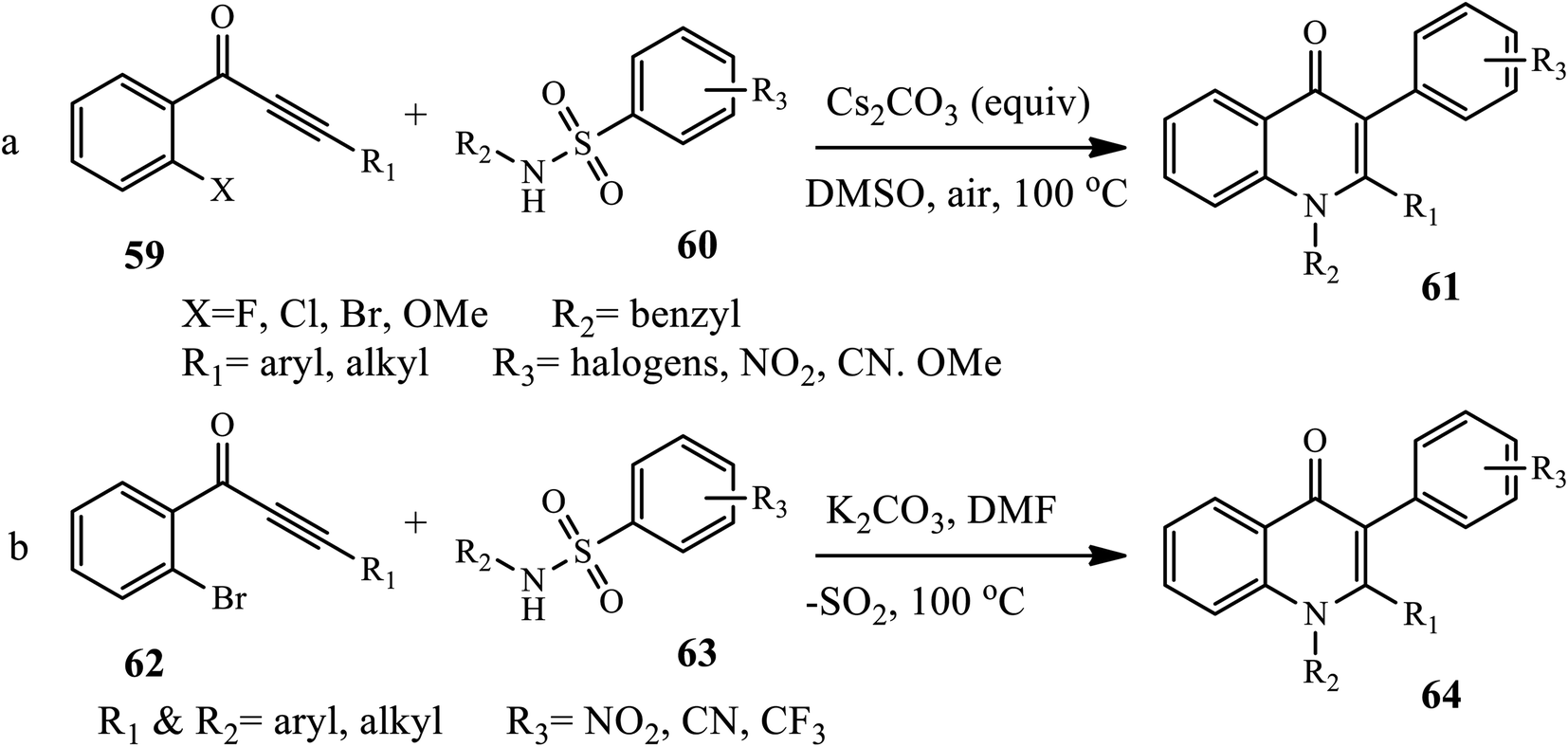
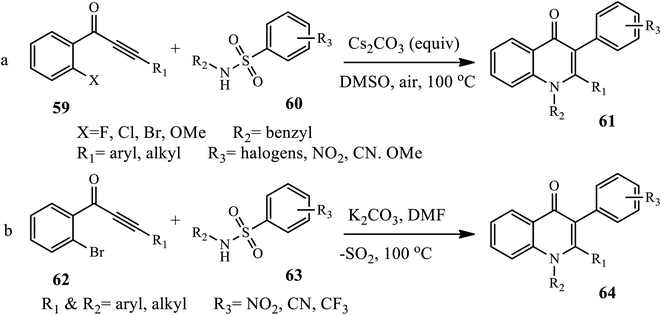
3.1.5 Synthesis of tri-substituted 4-quinolone. Dave and Joshipura reported a microwave-assisted protocol for the synthesis of 4-quinolone by the condensation of aromatic amines 55 with diethyl ethoxy methylene malonates 56, giving an intermediate 57 cyclized to 4-quinolones 58 at 2–14 min in good to excellent yield (Scheme 18).28 Recently, Liu and co-worker developed a one-step tandem Michael addition/Smiles rearrangement/N-arylation reaction for the synthesis of 1,2,3-trisubstituted 4-quinolones 61 between ynone 59 and sulfonamide 60 under transition-metal free conditions.29 The author examined various substrate scopes and functional groups tolerated to give good to excellent yields.
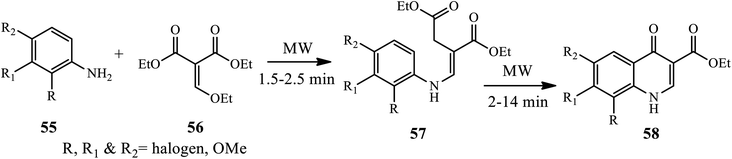 |
| | Scheme 18 Synthesis of tri-substituted 4-quinolone under microwave assistance. | |
Similarly, Xie et al. recently synthesized tri-substituted 4-quinolone derivatives 64 via a cascade reaction of Michael addition and Truce–Smiles rearrangement between benzene-sulfonamide derivatives 62 and 1-(2-bromophenyl)-3-phenyl prop-2-yn-1-ones 63 in the presence of K2CO3 in DMF at 100 °C in excellent yield (Scheme 19).30
 |
| | Scheme 19 Synthesis of 1,2,3-trisubstituted 4-quinolone derivatives. | |
3.2 Transition metal-catalyzed synthesis of 4-quinolone
Over the last decade, the metal-catalyzed synthesis of 4-quinolones has become increasingly relevant in organic processes. As catalysts, various transition-metal complexes, including those of Pd, Cu, Co, Au, Ni, and Rh, have been utilized. Transition-metal-based catalysts are essential for achieving a wide range of beneficial chemical transformations that would otherwise be difficult or impossible to achieve.31
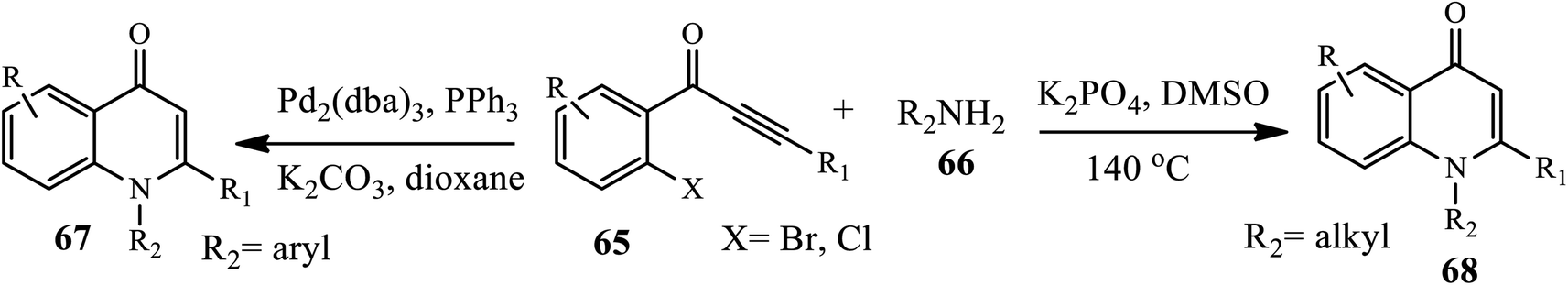
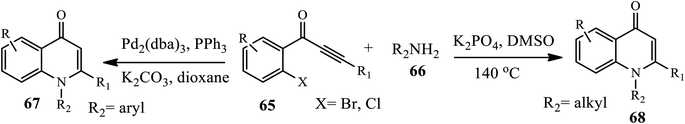
3.2.1 Palladium-catalyzed synthesis. In 2010, Zhao and coworkers reported the palladium-catalyzed synthesis of N-aryl-4-quinolones 67 from o-haloaryl acetylenic ketones 65 and primary amines 66 in PPh3 as a ligand, K2CO3 as a base and dioxane as a solvent. Numerous electronically and structurally varied aromatic amines participated well under the optimized conditions to provide the desired product in moderate to good yields. Similarly, Shao's group developed a palladium-catalyzed synthesis of N-alkyl-substituted 4-quinolones 68 using o-chloroaryl acetylenic ketones 65 and various alkyl amines 66 in the presence of K3PO4 in DMSO at 140 °C in good to excellent yields (Scheme 20).12a,b
 |
| | Scheme 20 Synthesis of N-functionalized 4-quinolones via palladium-catalyzation. | |
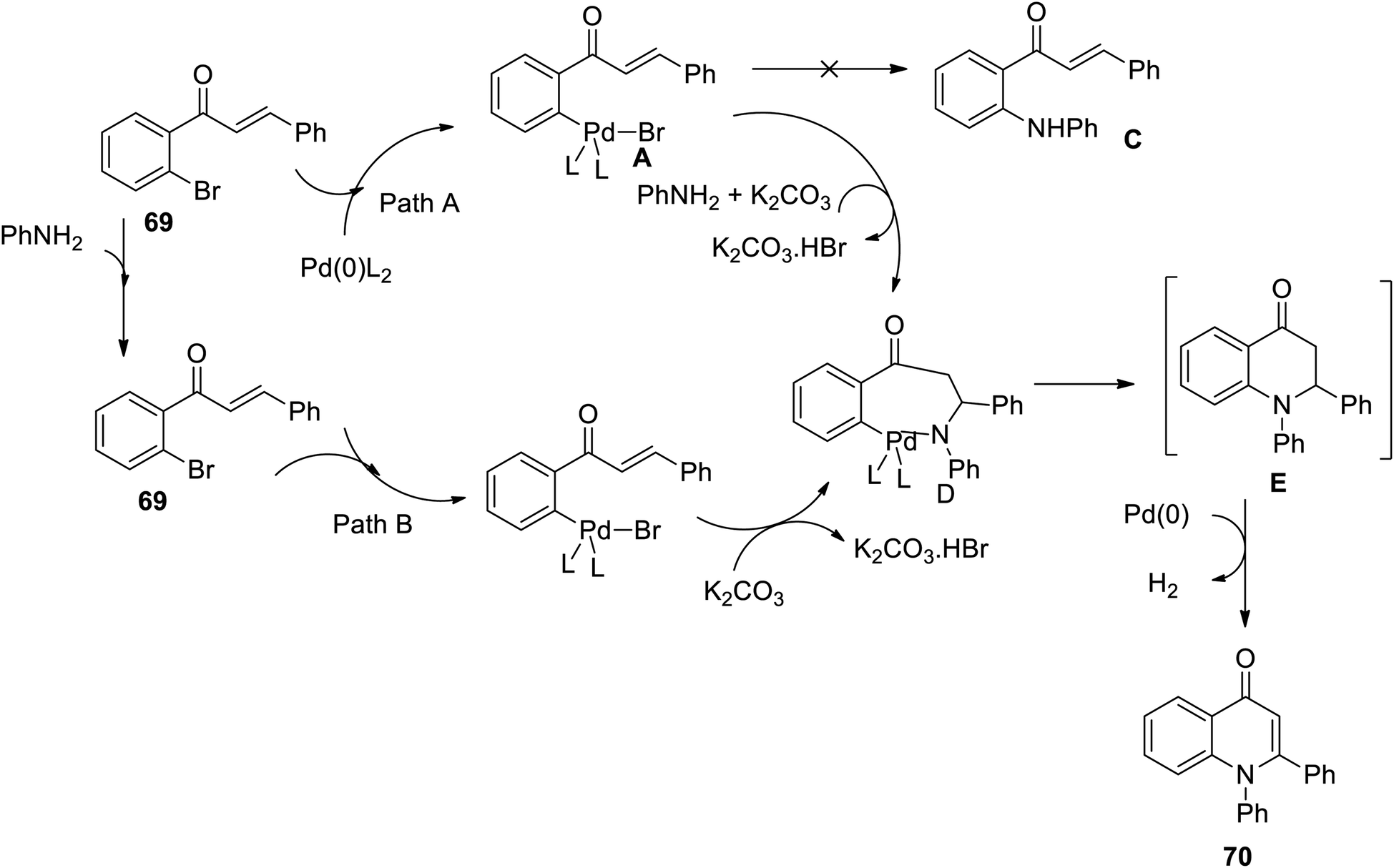
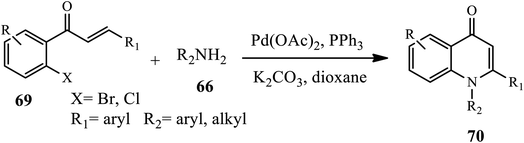
An efficient palladium-catalyzed one-pot synthesis of 1,2-disubstituted 4-quinolones 70 was developed by Fei and coworkers through Buchwald–Hartwig coupling, followed by Michael addition reaction of chalcone 69 and primary amines 66 under PPh3 as a ligand in anhydrous dioxane with K2CO3 as the base. Both aromatic and aliphatic amines gave rise to 4-quinolones in moderate to high yields except for a chloro-substituted substrate that gives low yields of product because of the poor reactivity of the C–Cl bond during the oxidative addition step. Further, arylamines containing electron-donating groups gave higher yields than those with electron-withdrawing groups (Scheme 21).32
 |
| | Scheme 21 Synthesis of 1,2-disubstituted 4-quinolones through Buchwald–Hartwig Coupling. | |
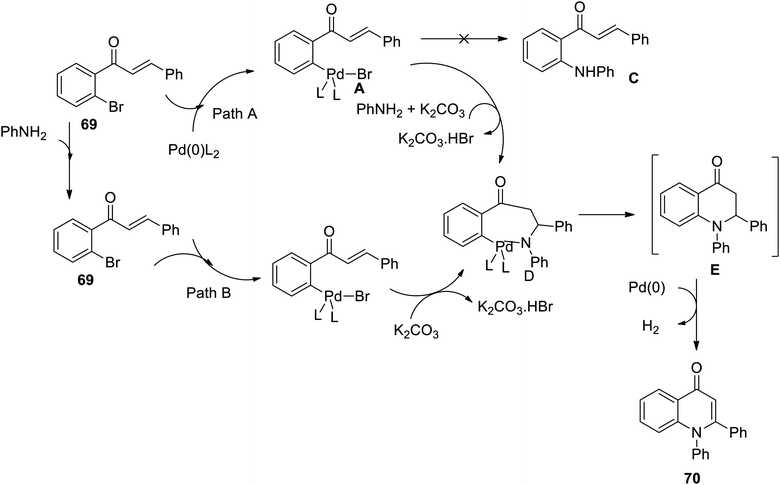
The Buchwald–Hartwig coupling followed by the Michael addition reaction has been successfully applied and reported for the synthesis of functionalized 1,2-disubstituted 4-quinolone derivatives using Pd(OAc)2 as a catalyst and PPh3 as a ligand. Under these conditions, the intermediate products first formed from chalcones and primary amines underwent catalytic dehydrogenation to afford the 1,2-disubstituted 4-quinolone derivative.
A plausible reaction mechanism for the one-pot synthesis of 1,2-disubstituted 4-quinolones is outlined in Scheme 22 (Paths A and B).32 In Path A, the oxidative addition of 69a to the palladium zero catalyst leads to palladium complex A. The carbon–carbon double bond in A can be activated through coordination to Pd(II) and attacked by aniline to form intermediate D. In the course of the reaction, intermediate C was not formed from complex A under these reaction conditions. Path B involves the Michael addition of aniline to 69a and oxidative addition, and then intermediate D is formed by eliminating HBr.32
 |
| | Scheme 22 Plausible mechanism for the synthesis of 4-quinolone derivative 70. | |
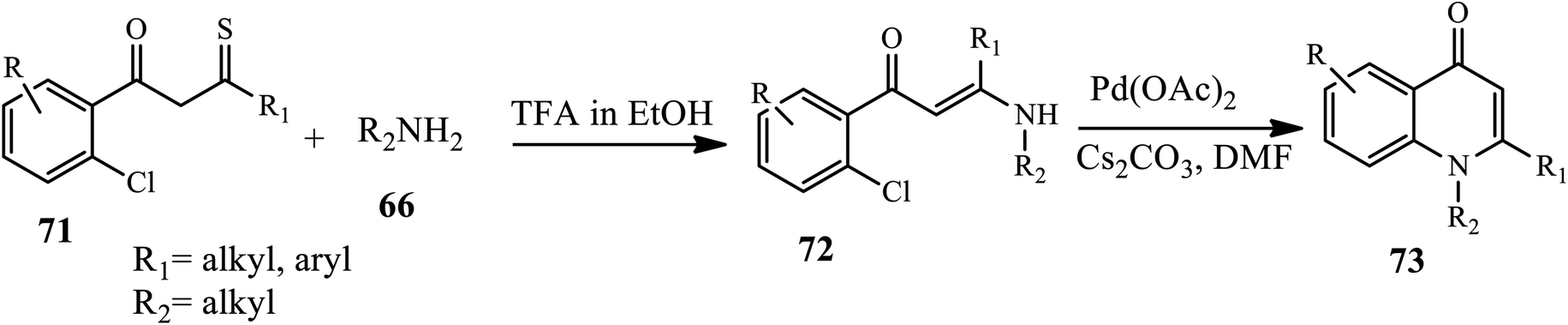
Vinayaka groups synthesized 1,2-disubstituted 4-quinolone derivative 73 starting from monothiodiketone substrate 71 and arylamine 66 in the presence of TFA in ethanol through the formation of an intermediate 72, which is further subjected to Pd(OAc)2 catalyst in the presence of Cs2CO3 as a base cyclized to afford 73 (Scheme 23).33
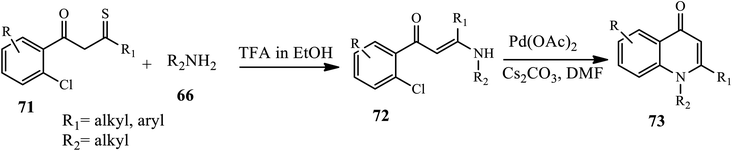 |
| | Scheme 23 Synthesis of 1,2-disubstituted 4-quinolones via Pd-catalyzed. | |
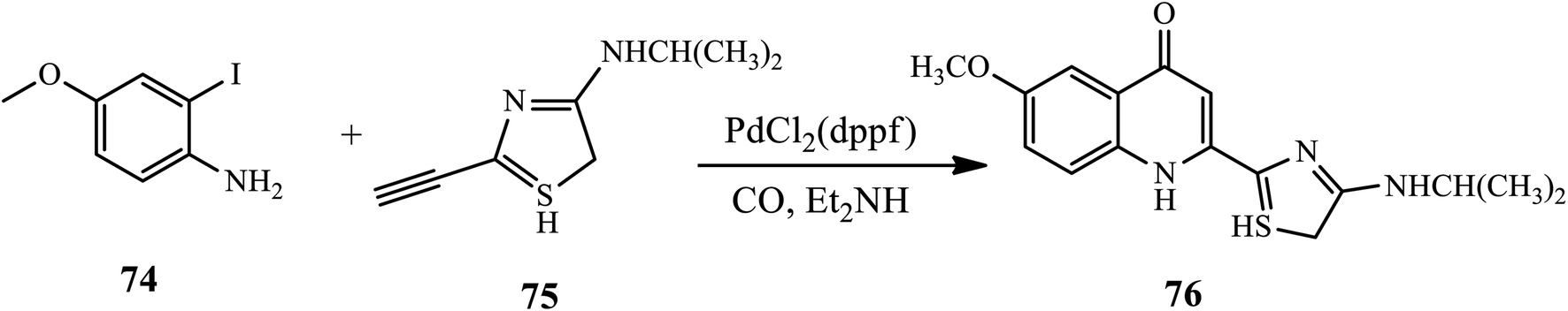
3.2.2 Sonogashira coupling. 4-Quinolone derivative 76, which is the key substructure of protease inhibitor BILN 2061, was synthesized and reported through carbonylative Sonogashira coupling, followed by cyclization of 2-iodo-5-methoxyaniline derivative 74 with thiazolylacetylene 75 in the presence of PdCl2 (dppf) as the catalyst in a good yield (Scheme 24).34
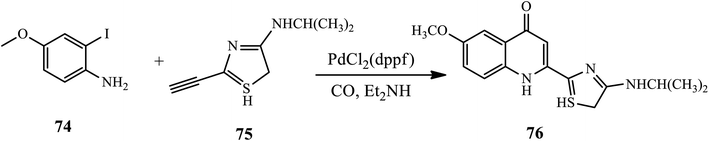 |
| | Scheme 24 Synthesis of 4-quinolone via carbonylative Sonogashira coupling/cyclization. | |
In 2017, 1,2 di-substituted 4-quinolone derivatives 79 were synthesized via palladium-catalyzed oxidative carbonylation by Wu and coworkers. In this reaction, various amines 77 and ketones 78 were reacted using carbon monoxide as a carbonyl source at atmospheric pressure in the presence of Pd(dba)2 and CuBr(Me2S). Substituted ketones with electron-rich groups afford the corresponding 4-quinolone derivative in good yields. Interestingly, halogen substituents participated smoothly, affording the desired products in moderate to good yields (Scheme 25).35
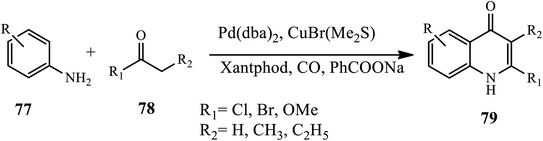 |
| | Scheme 25 Synthesis of 1,2 disubstituted 4-quinolone derivative through palladium-catalyzed. | |
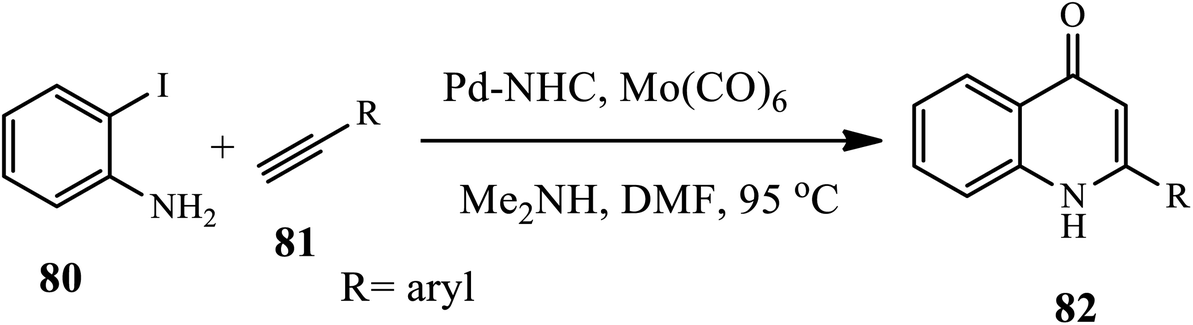
A CO gas free synthesis of 4-quinolone 82 via carbonylative Sonogashira annulation sequence in the presence of a Pd–NHC catalyst was reported by Ghosh and coworkers (Scheme 26).36 The reaction occurred between substituted 2-iodoanilines 80 and various acetylenes 81 using Pd–NHC as a catalyst and Mo(CO)6 as a source of CO at 95 °C under N2. The authors observed a slight decrease in the yield of the final products when an electron-withdrawing group is present in the phenylacetylene moiety, whereas, in the presence of electron-donating groups, the products were obtained in good to excellent yields. The absence of toxic CO gas, lower catalyst loading and use of inexpensive salt are the advantages of this approach over others.
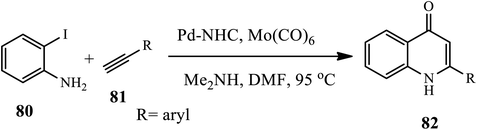 |
| | Scheme 26 Synthesis of 4-quinolone by palladium-catalyzed carbonylative Sonogashira. | |
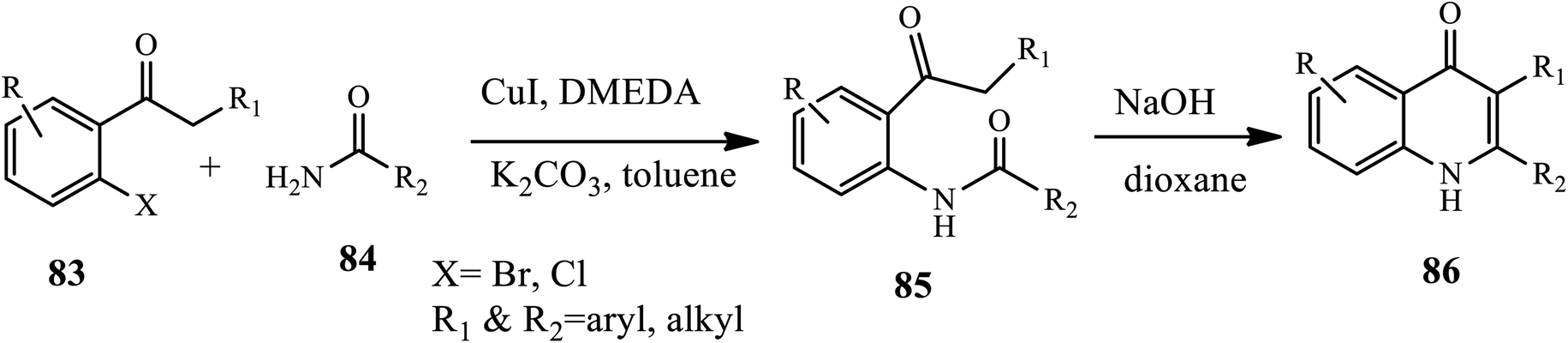
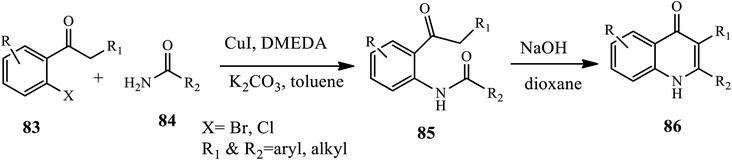
3.2.3 Copper-catalyzed synthesis. Jones and coworkers established a two-step method, Cu-catalyzed amidation followed by base-promoted camp cyclization for the synthesis of 2,3-substituted 4-quinolones 86 (Scheme 27).37 Cu-catalyzed amidation of 2-halophenones 83 with amide derivatives 84 afforded N-(2-ketoaryl) amides intermediate 85, which then underwent base-catalyzed Camps cyclization at 110 °C to form the desired product in good to excellent yields. The 2-halophenones bearing both electron-withdrawing and electron-donating groups, both aryl and alkyl vinyl amides, were well tolerated in good yields. Unfortunately, acyclic secondary amides were ineffective coupling partners.
 |
| | Scheme 27 Synthesis of 2,3-substituted 4-quinolone through Cu-catalyzed reaction. | |
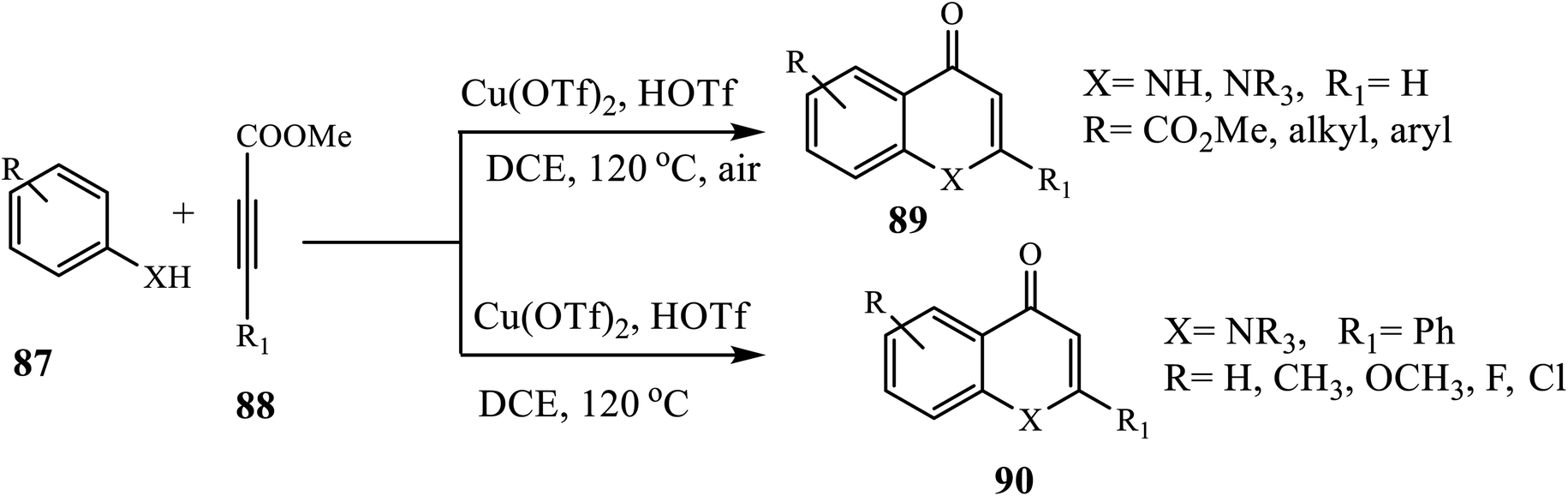
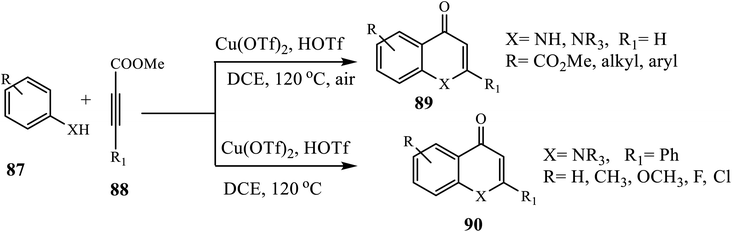
The Cu-catalyzed intermolecular cyclization protocol for the synthesis of 4-quinolones 91 was investigated by Zhang and Xu (Scheme 28).38 Heating secondary anilines 89 with alkynes 90 using CuI as a catalyst, HOTf and Tf2O as additives and DCE as solvent at 120 °C for 12 h formed the target compound in good yields. Similarly, under the same conditions, this group reported the construction of 4-quinolone 89 from primary anilines 87 and alkynoate 88. The authors observed that N-alkyl and N-aryl substituted anilines carrying electron-withdrawing and electron-donating functional groups provided a corresponding 4-quinolone 90 in good to excellent yield.
 |
| | Scheme 28 Synthesis of 4-quinolones through Cu-mediated cyclization. | |
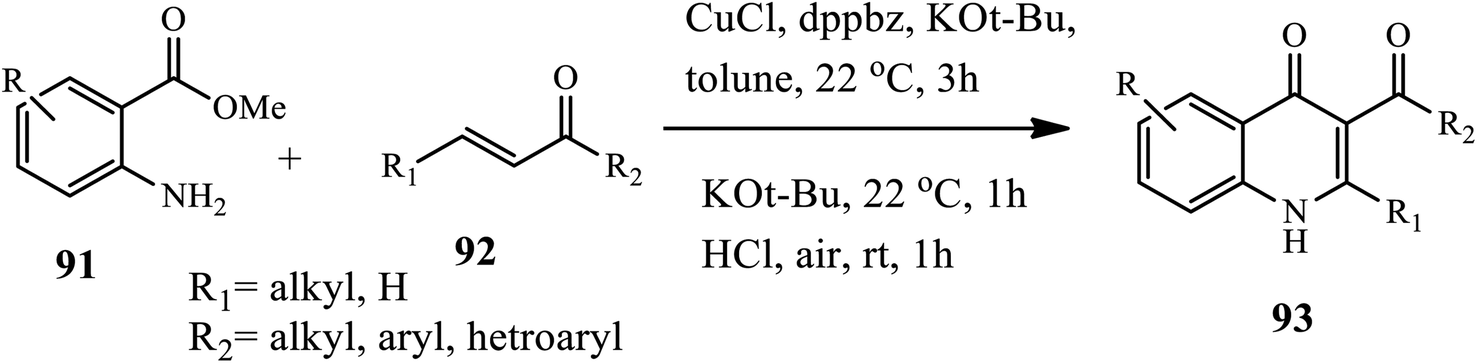
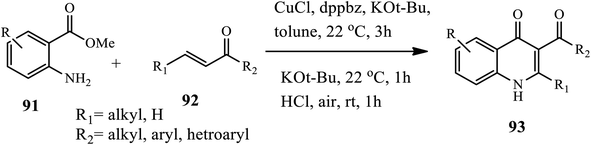
In 2018, Kang's group reported the synthesis of 3-acyl-2-substituted 4-quinolone 93 via the Cu-catalyzed aza-Michael addition of 2-aminobenzoates 91 to β-substituted α,β-unsaturated ketones 92, followed by base-mediated cyclization and oxidation (Scheme 29).39 In this method, the treatment of 2-aminobenzoates with β-substituted α,β-unsaturated ketones in the presence of CuCl as a catalyst, phosphine (dppbz) as a ligand, and KOt-Bu as a base afforded β-aminoketone intermediate, which was then cyclized to form 3-carbonyl-2,3-dihydroquinolin-4(1H)-one intermediate upon dehydrogenation with 6 N HCl in toluene under open-air at room temperature to give the desired product. The procedure showed good tolerance to various functional groups and substrates, with good to excellent yields. However, the sterically demanding aryl substituent on the β-position showed lower yields.
 |
| | Scheme 29 Cu-catalyzed synthesis of 3-acyl-2-substituted 4-quinolone derivatives. | |
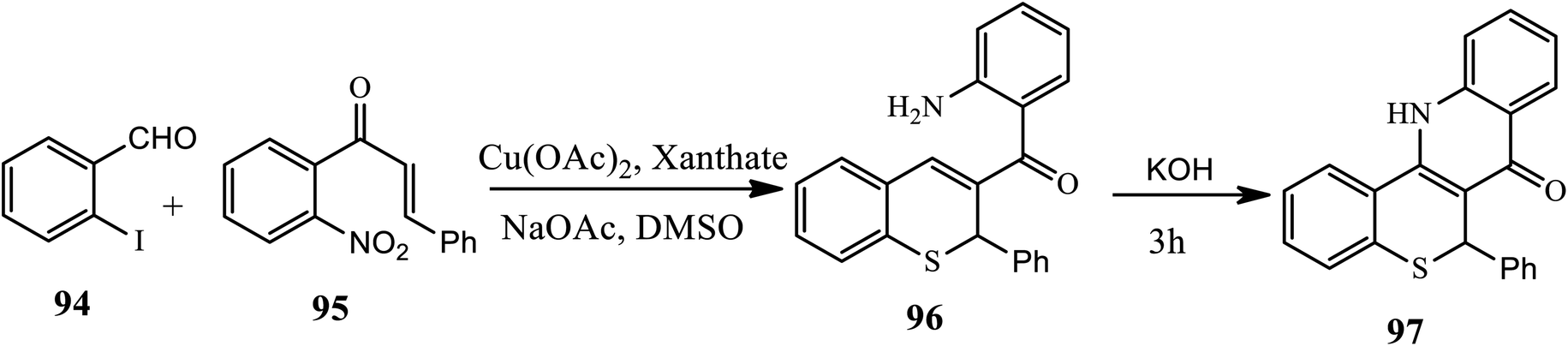
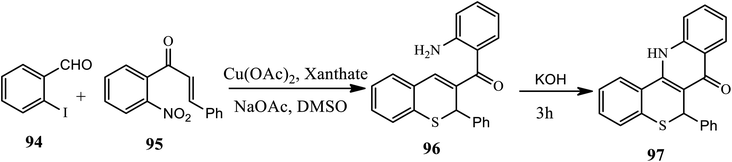
Recently, Sundaravelu and coworker developed an efficient Cu-catalyzed one-pot strategy for the synthesis of novel tetracyclic fused π-conjugated thiochromenoquinolinone (97) through regioselective double hetero Michael addition adduct-generated aza-nucleophiles (Scheme 30).40 The treatment of 2-iodobenzaldehyde 94 with 2-nitrochalcone 95 in the presence of Cu(OAc)2, xanthate and NaOAc in DMSO affords an isolable intermediate thiochromene 96 after 2 h through an intermolecular Michael addition reaction and aldol condensation, and further oxidation leads to the formation of the desired product in very good yields. In this procedure, xanthate played a dual role as an odorless sulfur source and a chemoselective reducing agent. Various substituted 2-halobenzaldehydes and chalcones with both electron donating and electron withdrawing groups at various positions effectively participated to afford the products in moderate to good yields.
 |
| | Scheme 30 Synthesis of thiochromenoquinolinone through Cu-catalyzed. | |
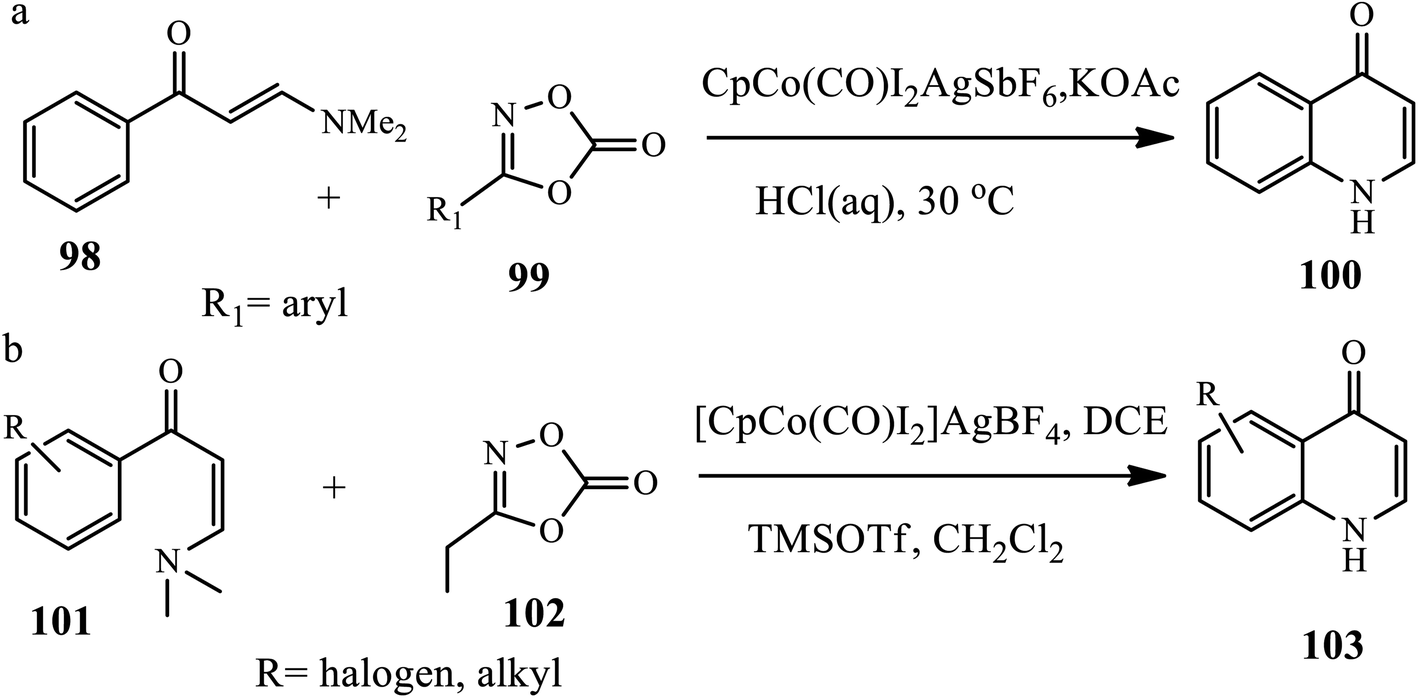
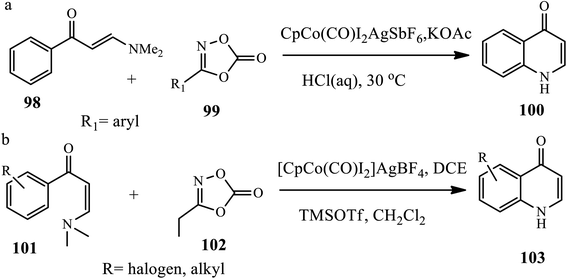
3.2.4 Cobalt-catalyzed synthesis. Wang and coworkers reported the synthesis of N-acyl 4-quinolones 100 through co-catalyzed C–N amidation of enaminone 98 and dioxazolone 99 (30a). The authors used Co(III)-catalyzed C–H bond functionalization of enaminones as a coordinating directing group to promote the C–H amidation of a series of arenes in the presence of CpCo(CO)I2, KOAc and AgSbF6 in 1,4-dioxane solvent (Scheme 31).41 This procedure was found to be suitable for enaminones bearing electron donating, withdrawing or halogen groups generally coupled under cobalt-catalyzed conditions in excellent yields.
 |
| | Scheme 31 Cobalt-catalyzed C–H amidation for the synthesis of 4-quinolone derivatives. | |
Similarly, Shi et al. synthesized 7-substituted 4-quinolones 103 from (E)-3-dimethylamino-1-phenyl prop-2-en-1-one 100 and 3-ethyl-1,4,2-oxazole-5-one 102 using [CpCo(CO)2] as a catalyst, AgBF4 as an additive, and DCE as a reaction medium, which was then deacetylated by refluxing in HCl or by treating with trimethylsilyl trifluoromethane sulfonate (TMSOTf), thereby generating a free amino group, which in turn undergoes nucleophilic attack at the alkene moiety of enaminone to afford a diverse range of quinolone derivatives (30b). The authors claim that substituent effects, such as para-, meta-, and ortho-substituted enaminone carrying both electron donating and electron withdrawing groups at different positions of the phenyl ring, also reacted smoothly under the procedural reaction conditions.42
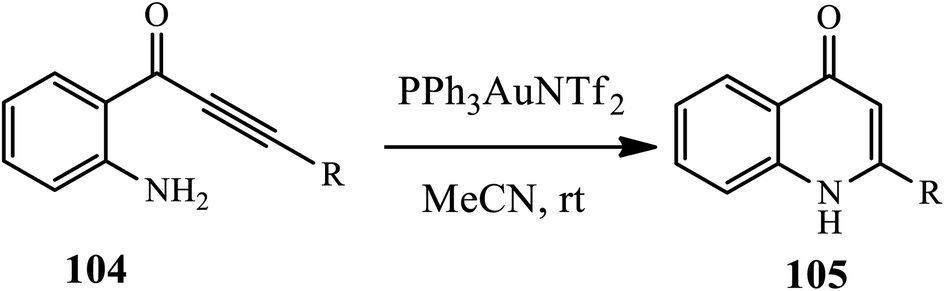
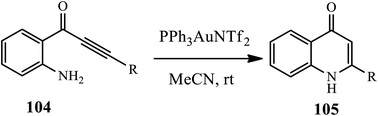
3.2.5 Gold catalyst. Gold-catalyzed cyclization of alkyl- or aryl-substituted 1-(aminophenyl)-2-propyn-1-ones 104 towards the synthesis of the corresponding 2-substituted 4-quinolones 105 was reported by Helaja and coworkers (Scheme 32).43 Various substrates, including both electron-withdrawing and donating substituents and straight chain alkyl substrates, were cyclized in good to excellent yields. However, TMS-protected substrates failed to be cyclized.
 |
| | Scheme 32 Gold-catalyzed synthesis of 4-quinolone 105. | |
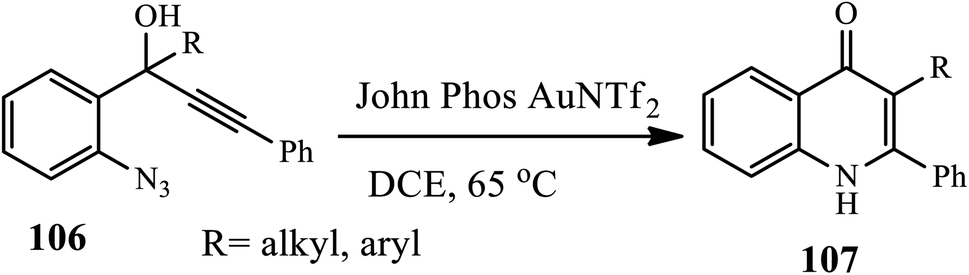
Wu et al. reported the synthesis of 1,2-disubstituted-4-quinolone 107 via gold-catalyzed cyclization of 1-(2′-azidoaryl) propynols 106 in the presence of John Phos AuNTf2 and DCE at 65 °C under N2 (Scheme 33).44 The authors found that the yield of the product increased when different aryl or alkyl substituents were adjacent to the carbon–carbon triple bond. Additionally, the presence of electron-donating or electron-withdrawing groups on the azido-substituted phenyl ring led to the generation of the desired products in good yields except for benzo1,3 dioxolyl-substituted propynol, which gave a relatively lower yield.
 |
| | Scheme 33 Gold-catalyzed synthesis of 1,2 disubstituted 4-quinolone derivative 107. | |
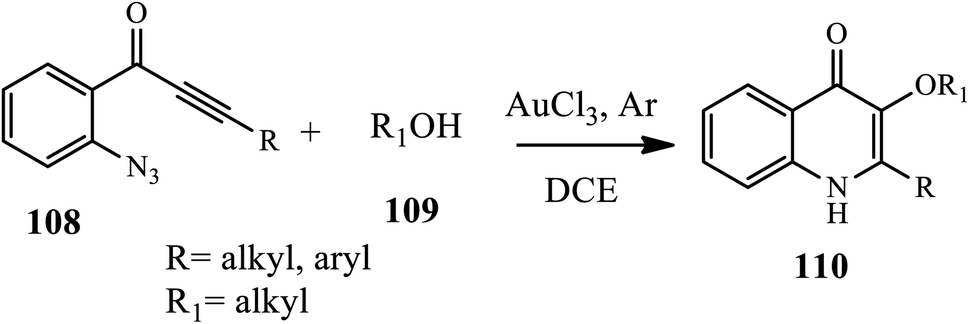
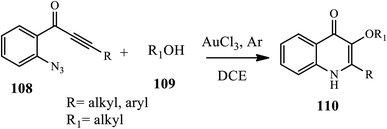
A straightforward method for the synthesis of 3-alkoxy-4-quinolone 110 derivatives via a gold-catalyzed 6-endo-dig azide-yne cyclization, followed by an intermolecular O–H insertion cascade reaction of o-azidoacetylenic ketones 108 with alcohols 109 at room temperature under an argon atmosphere, was described by Huang's group (Scheme 34).45 In this method, both electron-donating and electroneutral halides were cyclized in good yields, whereas 8-methyl substituted substrate and alkyne tethered with electron-deficient aryl group gave the cyclized product in lower yields.
 |
| | Scheme 34 Gold(III)-catalyzed synthesis of 3-alkoxy-4-quinolone. | |
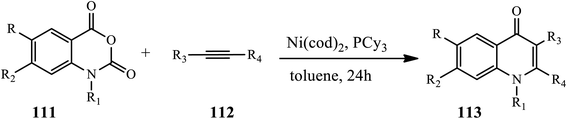
3.2.6 Nickel catalyst. Yoshino and coworkers disclosed a Ni-catalyzed protocol for the synthesis of polysubstituted 4-quinolones 113 (Scheme 35).46 The reaction occurred between alkynes 111 and isotonic anhydrides 112 in the presence of Ni (cod)2, PCy3, and toluene at 80 °C for 24 h in excellent yields. Decarboxylative carboamination was proposed to be initiated by the oxidative addition of an anhydride O–CO bond to Ni. Then, an Ni(II) intermediate was formed via subsequent decarboxylation and coordination by the alkyne. After insertion by an alkyne into the acyl Ni bond, a nickel cycle species was obtained. The nickel cycle intermediate then underwent reductive elimination to form the desired product along with the formation of Ni(0). Various alkyl- and aryl-substituted alkynes are well tolerated in this protocol.
 |
| | Scheme 35 Nickel-catalyzed 4-quinolone synthesis. | |
Another (Ni)-catalyzed synthesis of 4-quinolones 115 through the intramolecular amination of o-(N-alkylamino) propiophenones 114 was developed by Ueno and coworkers (Scheme 36).47 The Ni(0) catalyst reacts with propiophenones to form a C-bound Ni enolate, yielding a β-unsaturated ketone through β-hydride elimination. This is followed by a 1,4-addition of the morpholine catalyst to give a β-aminoketone intermediate. Then, the β-aminoketone is transformed into β-enaminone through the formation of Ni enolate and ensuing elimination. Subsequently, the addition of an intramolecular conjugate followed by β-elimination of morpholine reaction yields the 4-quinolone derivatives in good yields.
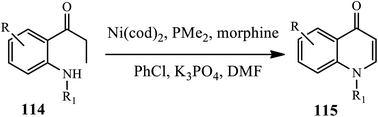 |
| | Scheme 36 Nickel(0) catalyzed synthesis of 4-quinolones from o-(N-alkylamino) propiophenones. | |
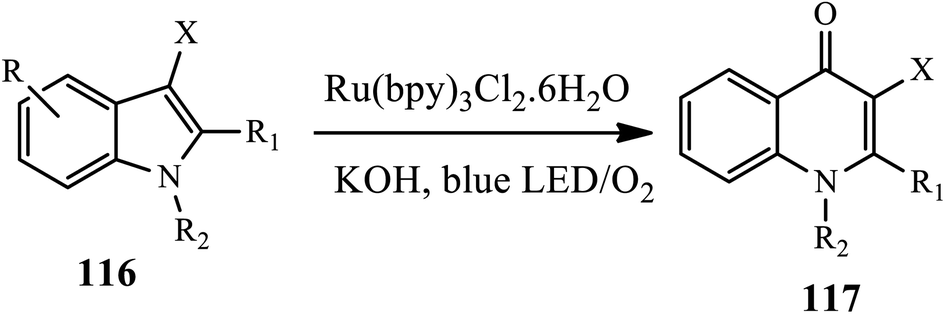
3.2.7 Other metal-catalyzed synthesis of 4-quinolone. A one-pot method for the synthesis of 4-quinolones 117 from indoles (N-methylindole) 116 in the presence of a Ru catalyst was developed by Ji group in 2018 (Scheme 37).48 This method involves the photocatalytically aerobic oxidation of indoles with a blue LED light and subsequent base-promoted Camps cyclization in the presence of Ru(bpy)3Cl2·6H2O catalyst at room temperature, affording the desired product in moderate to high yield. In this reaction, various N-substituted-3-methylindoles bearing N-ethyl, N-benzyl, and N-allyl provide corresponding 4-quinolone products in moderate to good yields. Unfortunately, N–H indole and N-Ts indole were found to be ineffective substrates for this reaction.
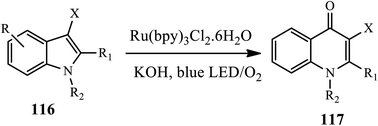 |
| | Scheme 37 Synthesis of 4-quinolones via photocatalysis using ruthenium catalysis. | |
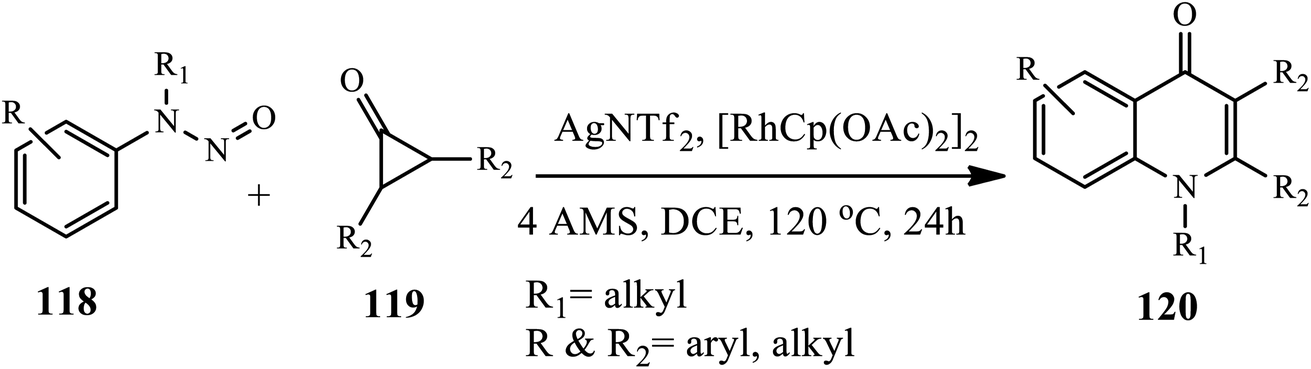
In 2020, Shi and coworkers reported a condition-controlled divergent synthesis strategy for constructing 1,2,3 tri-substituted 4-quinolones 120 (Scheme 38).49 A C–H activation of N-nitrosoaniline 118 and diphenylcyclopropenone 119 in the presence of AgNTf2 and [RhCp(OAc)2]2 catalysts in 4 AMS at 120 °C afforded the corresponding quinolones after 24 h in good yields. Broad substrate scope, mild reaction conditions, high functional group tolerance and amenability to gram-scale synthesis are the characteristics of this protocol.
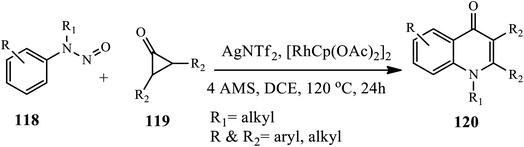 |
| | Scheme 38 Synthesis of 1,2,3 tri-substituted 4-quinolones by C–H activation. | |
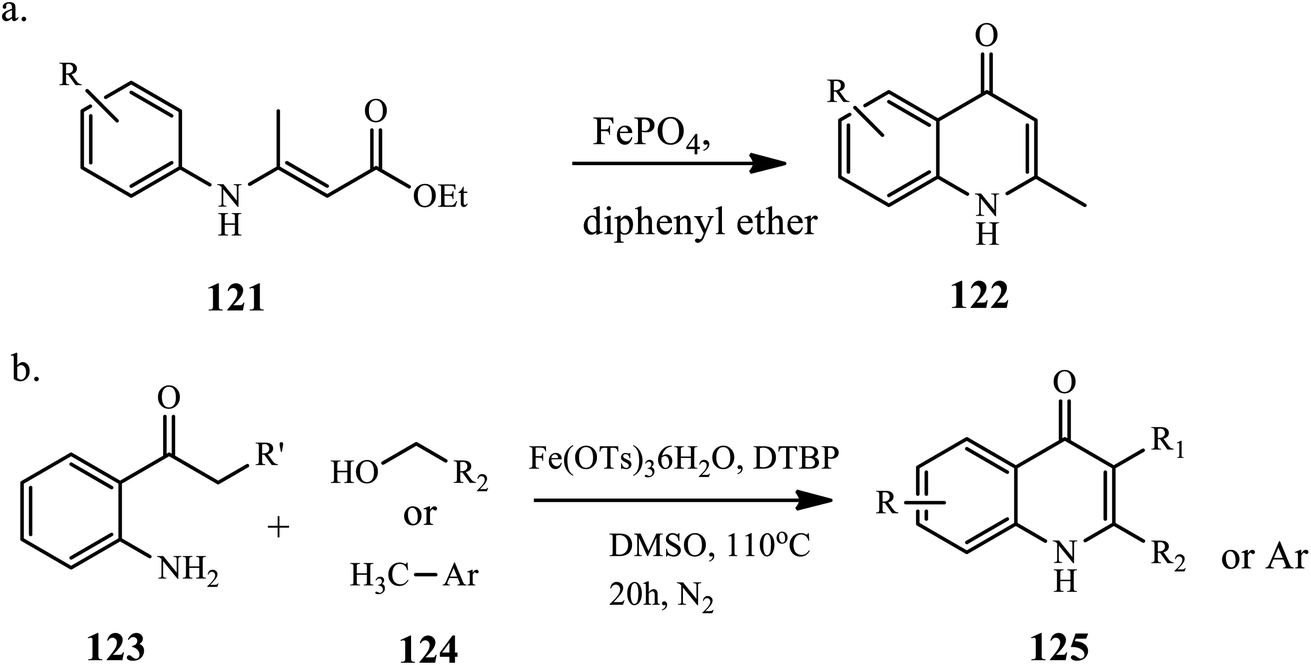
The synthesis of 4-quinolone derivative 122 from arylaminocrotonate 121 through a Conrad–Limpach reaction in the presence of a catalytic amount of iron(III) phosphate under refluxing conditions was reported by Samadi et al. (Scheme 39a).79 The reaction was carried out in toluene, ethanol, chloroform and acetonitrile solvents; with the standard reaction protocol, no product was observed. However, when the reaction was carried out in diphenyl ether solvent at room and high temperature, a high yield of product was observed. The reaction affords a good-to-excellent yield.
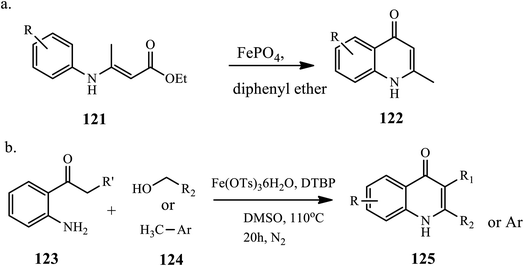 |
| | Scheme 39 Synthesis of 4-quinolone derivatives using iron(III) catalyzed reactions. | |
Similarly, Lee and coworkers demonstrated the synthesis of 4-quinolones via the iron(III)-catalyzed oxidative coupling of alcohol/methyl arene 124 with 2-amino phenyl ketone 123. Initially, alcohols and methyl arenes are oxidized to aldehyde in the presence of an iron catalyst and di-tert-butyl peroxide, followed by a tandem process condensed with amine/Mannich-type cyclization/oxidation, to complete the 4-quinolone ring 125 (Scheme 39b).80 This method tolerates various functional groups and provides a direct approach for synthesizing 4-quinolones from less functionalized substrates.
In 2016, Joussot and coworkers described a one-pot synthesis of 3-substituted-4-quinolones 127 via a silver catalyst. In this transformation, the desired 3-substituted-4-quinolones were prepared by reacting enaminones 126 and electrophiles in the presence of silver (Ag(I)) catalyst (Scheme 40).50 The authors suggested a mechanism for this reaction in which they expected the reaction to follow two different routes. Initially, in path 1, the alkylation of enaminones 126 led to the formation of iminium salt, which was then cyclized to a new ammonium salt. In pathway 2, cyclization precedes alkylation, resulting in identical iminium salt. Finally, with the removal of dimethylamine, 3-substituted quinolone 129 was produced in low to moderate yields.
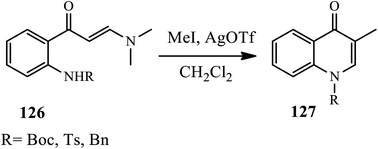 |
| | Scheme 40 Silver(I) mediated cyclization of enaminone to 4-quinolone derivative. | |
3.3 Functionalization of 4-quinolone and its derivatives
The addition of new functional groups and the fusing of a polycyclic ring to a quinolone core could result in a novel class of chemicals with biological and material science uses. Various ways to functionalize the 4-quinolone scaffold have been reported in the literature, such as C–C cross-coupling, C-hetero cross-coupling, C–H bond activation, decarboxylative cross-coupling, and regioselective electrophilic insertion.
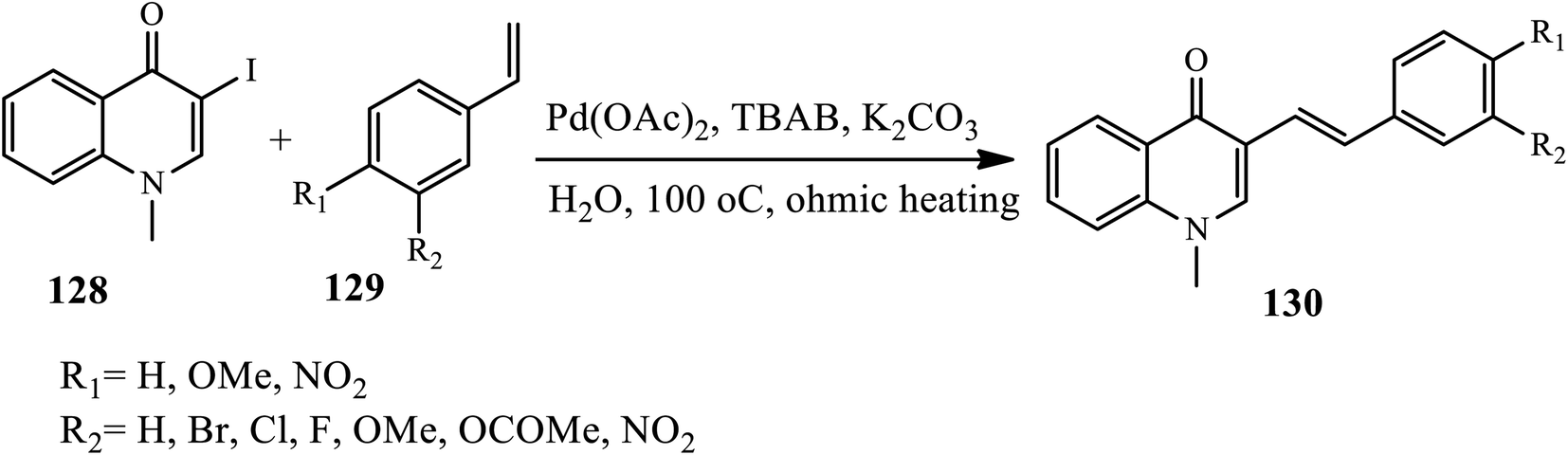
3.3.1 C–C cross-coupling reactions. Silva et al. reported the synthesis of (E)-3-styrylquinolin-4(1H)-ones 130 via the Heck coupling reaction of 3-iodo substituted 4-quinolones 128 and styrene derivatives 129 under heating in the presence of Pd(OAc)2 catalyst in TBAB as a phase transfer catalyst in aqueous media (Scheme 41).51 Using water as a solvent instead of other toxic solvents, avoiding toxic phosphine ligands, and using ohmic heating instead of conventional and microwave heating are the main advantages of this method. Styrenes that contain electron-withdrawing and electron-donating substituents participate to give the desired product in moderate to good yields.
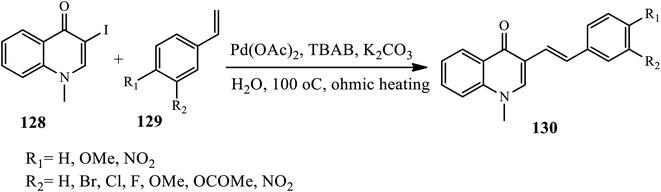 |
| | Scheme 41 Heck cross-coupling reaction through ohmic heating induced. | |
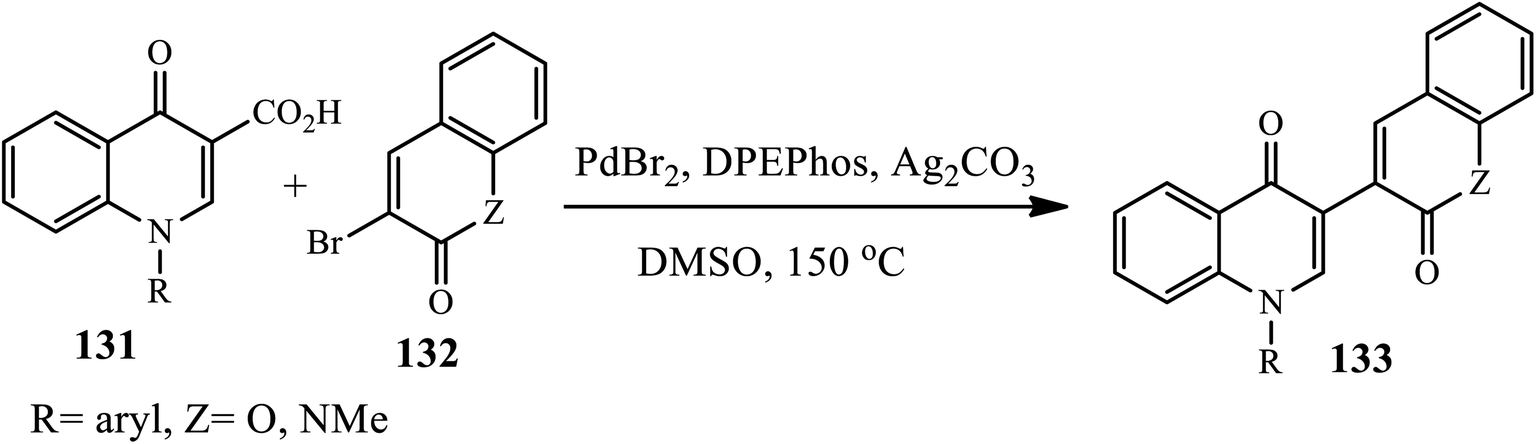
Reddy's group reported a novel palladium-catalyzed synthesis of a series of bis-heterocycle derivatives 133 through the direct decarboxylative coupling of quinolin-4-ones 3-carboxylic acids 131 with 3-bromo-coumarins or quinolin-2(1H)-one 132 in the presence of PdBr2, DPEphos, Ag2CO3 and DMSO in a sealed tube at 150 °C for 10 min (Scheme 42).52 The authors examined the effect of substrates as all the couplings proceeded cleanly and selectively in good to excellent yields regardless of the nature of the substituents on the aromatic ring of the quinolin-4-one 3-carboxylic acid or coumarin/quinolin-2-one moieties.
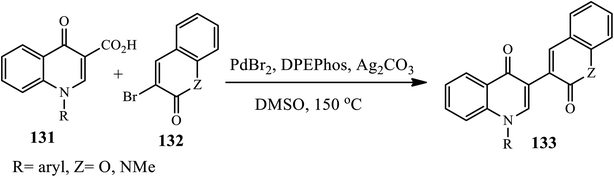 |
| | Scheme 42 Pd-catalyzed decarboxylative cross-coupling of 4-quinolone with heterocyclic halides. | |
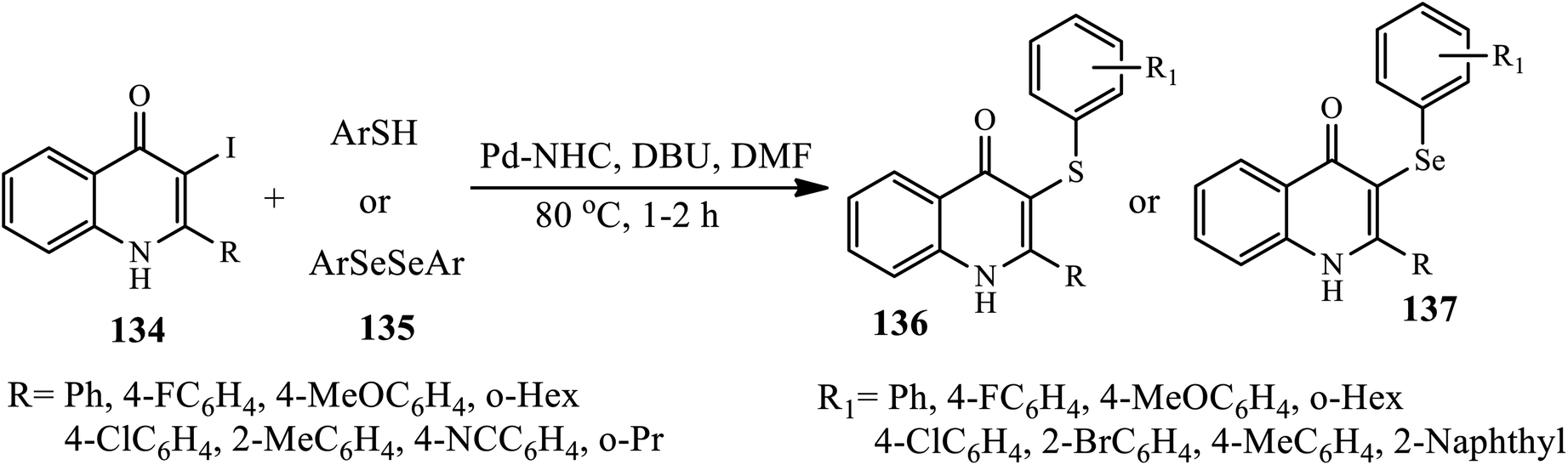
Recently, a novel, simple and efficient protocol for Pd–NHC-catalyzed the etherification and selenylation of 3-iodo-4-quinolones 134 under aerobic conditions was developed by Ghosh and Das. The target products 136 and 137 were synthesized via Pd–NHC catalyzed cross-coupling reactions of 3-iodo-2-phenyl-4-quinolone 136 and thiophenol/diphenyl diselenide 135 in the presence of DBU as a base in DMF at 80 °C (Scheme 43).53 A broad range of thiophenols was coupled with various 3-iodo-2-phenylquinolin-4-(1H)ones, affording the corresponding 3-aryl sulfide-4-quinolones in good to excellent yields. Both thiophenols possessing electron-donating and electron-withdrawing groups performed well in this transformation. Interestingly, the electron-withdrawing groups at C-2 increased the yield, whereas the steric bulk groups lowered the yield. The highest yield was obtained when 2-(4-fluorophenyl)-3-iodo-4-quinolone was coupled with 4-fluorothiophenol; however, 2-(2-methylphenyl)-3-iodo-4-quinolone did not afford the corresponding product. In the case of 134, 4-quinolones possessing 2-cyclohexyl and 2-cyclopropyl substituents gave the highest yields, whereas 2-aryl substituents possessing electron-withdrawing groups, such as 4-chloro and 4-fluoro, resulted in low yields.
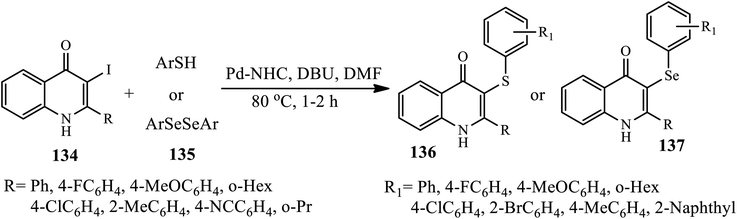 |
| | Scheme 43 Synthesis of ipso-C–S and C–Se-substituted 4-quinolone derivatives through Pd–NHC-catalyzed cross-coupling. | |
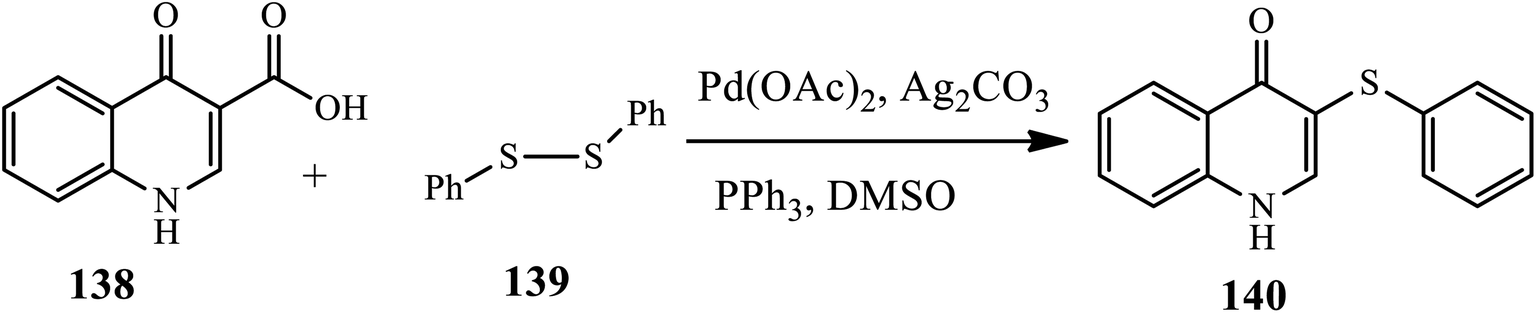
3.3.2 C-hetero cross-coupling. Xia and coworkers established a highly efficient and practical procedure for palladium-catalyzed direct the thioetherification of quinolinone-3-carboxylic acids 138 with diaryl disulfides 139 through decarboxylative C–S coupling under an air atmosphere in the presence of Pd(OAc)2 and Ag2CO3 in DMSO at 130 °C to prepare derivative 140. The authors claim that the effect of substrates such as diaryl disulfides bearing an electron-donating group, such as diphenyl disulfide, p-tolyldisulfide, and bis(4-methoxyphenyl)disulfide, increase the yield, whereas bis(4-chlorophenyl)disulfide substituted with an electron-withdrawing group coupled decrease the yield (Scheme 44).54
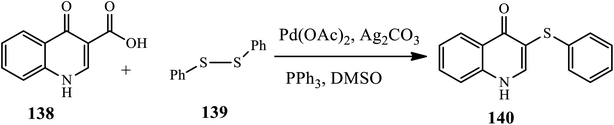 |
| | Scheme 44 Palladium-catalyzed thioetherification of quinolones through C–S cross coupling. | |
A ligand free copper(II) mediated selective C–NH2 arylation of both unprotected and protected 4-quinolones under ambient conditions was reported by Ghosh and Das in 2018 (Scheme 45).55 The coupling of substituted amino 4-quinolone 141 obtained through the reduction of the corresponding nitro 4-quinolone with phenylboronic acid 142 in the presence of Cu(OAc)2·H2O and K2CO3 in EtOH at room temperature afforded the corresponding N-arylated derivatives 143 in good to excellent yields. Functional groups, such as –Br, –Cl and –NO2, were well compatible in this reaction, whereas thiopheneboronic acid did not participate in the reaction at all. Additionally, sterically hindered 2-bromophenylboronic acid gave the highest yield product, but 2-napthylboronic acid resulted in a slight decrease in yield.
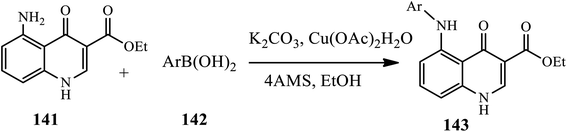 |
| | Scheme 45 Copper(II) catalyzed selective C–NH2 arylation of 4-quinolones. | |
3.3.3 C–H bond activation. Li et al. demonstrated a cascade two-step one-pot synthesis of quinolinone-fused isoquinolines 146. In this method, the desired N-fused polyheterocycles were synthesized by regio- and stereo-selective nucleophilic addition of 4-quinolinones 144 to alkynyl bromides 145, which provided (Z)-N-alkenylated bromides, followed by ligand-free Pd-catalyzed intramolecular C–H alkenylation in the presence of PdCl2 and K3PO4/K2CO3. The authors examined the substrate effects, including both electron withdrawing and electron releasing groups bearing 4-quinolone at the C-2 position, which gave the desired product in moderate to excellent yields. Additionally, para- and meta-substituted arylbromo acetylenes participated smoothly in this reaction. However, ortho substituents decrease the yield of the desired product (Scheme 46).56
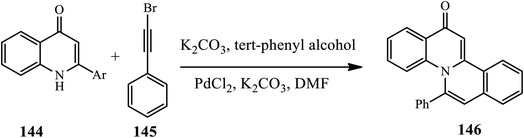 |
| | Scheme 46 Synthesis of quinolone-fused isoquinolines via intramolecular C–H alkenylation. | |
3.3.4 Nitration. In 2013, Azad and coworkers developed a decarboxylative nitration method for the conversion of 3-carboxy-4-quinolones 147 to 3-nitro-4-quinolones 148 via ipso nitration using polysaccharide-supported Cu nano-particles in the presence of Chit-Cu-NP and NO2BF4 in DMF at 100 °C. The bi-functional groups, such as hydroxyl and amine, in chitosan were found to be responsible for providing the excellent stability of the metal nanoparticles (Scheme 47).57
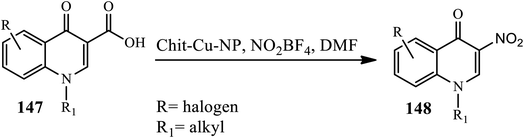 |
| | Scheme 47 Chitosan Cu-NP catalyzed ipso-nitration of 4-quinolons. | |
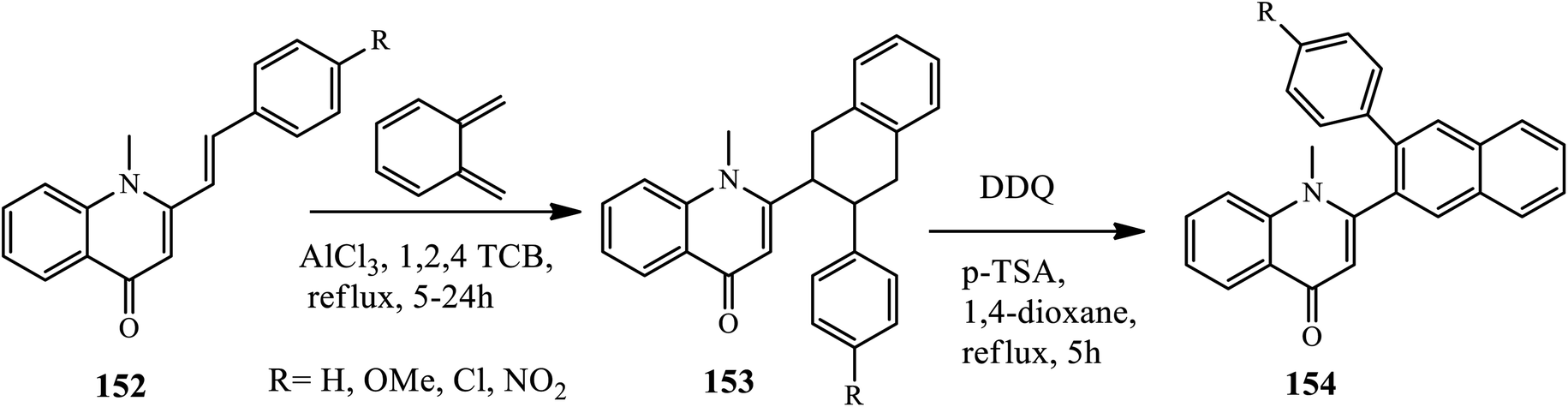
3.3.5 Regioselective electrophilic insertion. A Lewis acid-catalyzed Diels–Alder reaction for the synthesis of new 2-(3-arylnaphthalen-2-yl)-1-methylquinolin-4(1H)-ones 151 was disclosed by Silva's group in 2016. Cycloaddition reaction of (E)-1-methyl-2-styrylquinolin-4(1H)-ones 149 with ortho-benzoquinodimethane afforded trans-2-(3-aryl-1,2,3,4-tetrahydronaphthalen-2-yl)-1-methylquinolin-4(1H)-ones 150 in the presence of Lewis acid (AlCl3) by refluxing 1,2,4-TCB, which further converted to a 150 dehydrogenation reaction using DDQ and the catalytic amount of p-toluenesulfonic acid (p-TSA) in dry 1,4-dioxane. The authors claimed that the effect of substitutes as an electron donating group (–OMe) decreases the reactivity towards cycloaddition reaction, whereas the electron withdrawing group showed higher reactivity to afford an excellent yield of the desired product (Scheme 48).58
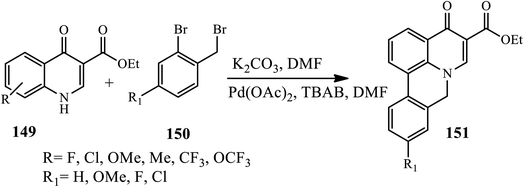 |
| | Scheme 48 Synthesis of phenanthridone fused 4-quinolone derivative. | |
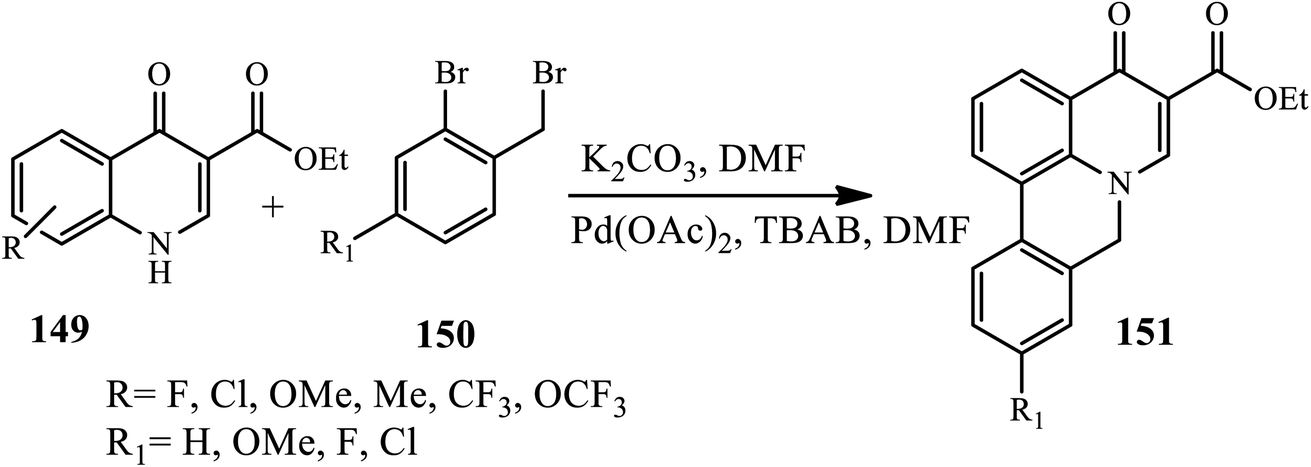
Similarly, Arasakumar and coworkers reported a direct protocol towards the formation of phenanthridone fused heterocycles 151 by the intramolecular C–H arylation of N-benzylquinolone 149 with 2-bromobenzyl bromide 155 mediated by Pd(OAc)2 in the presence of K2CO3 as a base and tetrabutylammonium bromide (TBAB) as an additive in DMF at 80 °C for 3 h under an argon atmosphere (Scheme 48). Electron-withdrawing groups (F, Cl, or CF3) or electron-donating groups (Me, OMe, OEt, or OCF3) on the phenyl ring of 4-quinolones gave moderate to excellent yields (Scheme 49).59
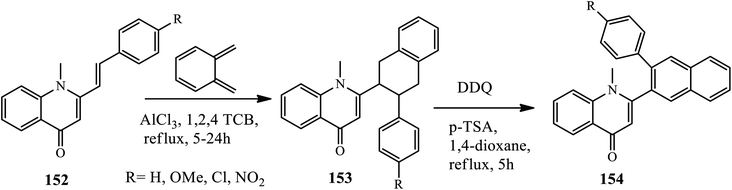 |
| | Scheme 49 Synthesis of 4-quinolone via acid-catalyzed Diels–Alder Reactions. | |
4. Bioactivities of 4-quinolones scaffolds and their derivatives
4-Quinolones possess diverse pharmacological properties, such as anticancer, anti-HIV, antibacterial, antifungal, antimalarial, anti-tubercular, anti-Alzheimer's disease activities, playing an intriguing role in the development of new drugs. Some of them are discussed below.
4.1 Anti-cancer activity
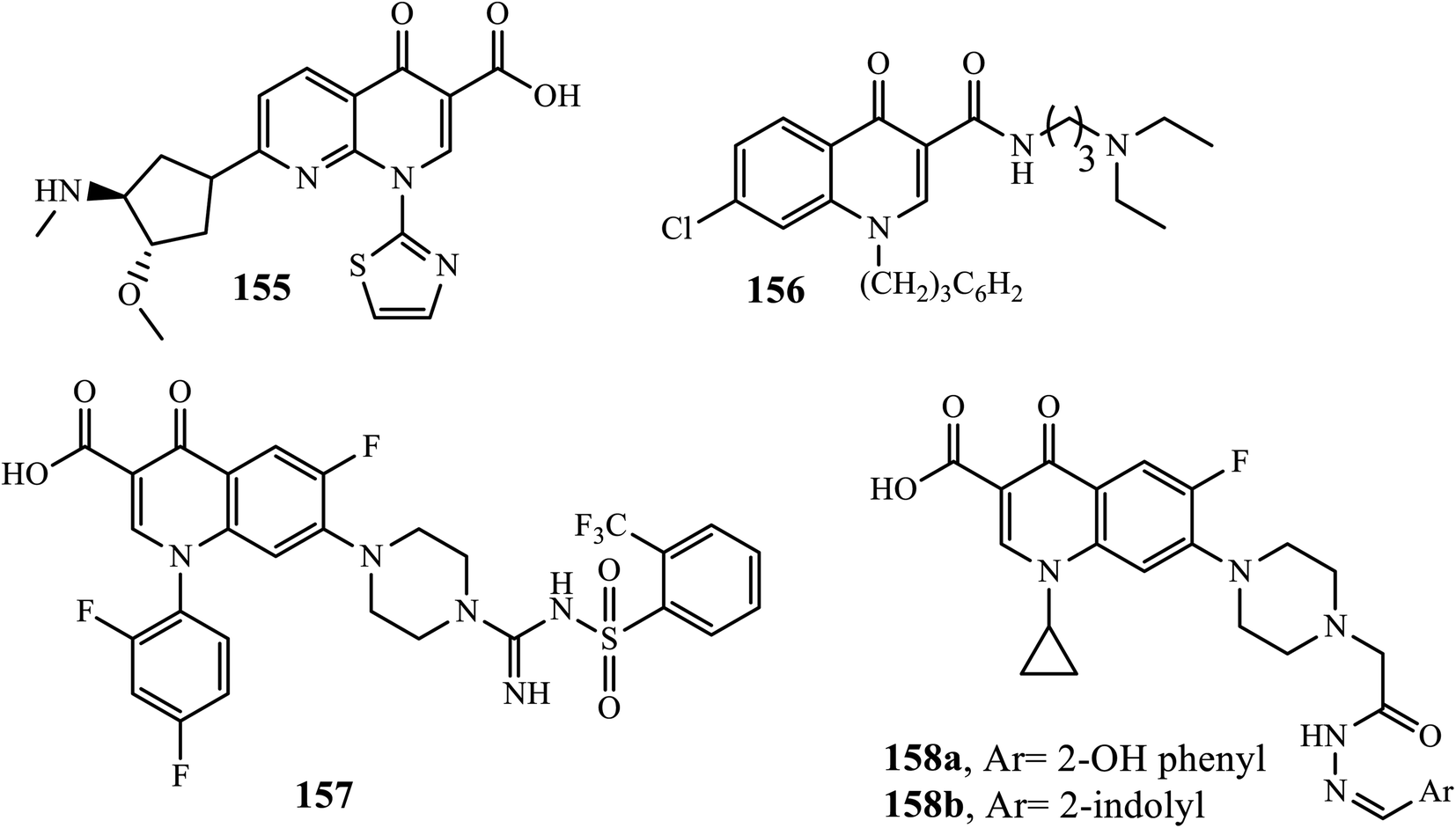
In an organism, cancer is defined as aberrant cell division, proliferation, and accumulation. It is one of the leading causes of death in the world. It might affect just one organ and spread to other parts of the body.60 DNA topoisomerases play a key role in cancer because they regulate cell division through the modulation of the DNA supercoiling process during replication. Quinolone can inhibit DNA topoisomerase, leading to its potential for the treatment and control of solid tumors.61 Voreloxin 155 was the first quinolone derivative approved by the FDA as a therapy for acute myeloid leukemia.6 In 2017, Zhang and coworkers reported the synthesis of a series of novel water-soluble 4-quinolone-3-carboxamide derivatives 156 for anticancer activity against seven human tumor cell lines. The authors revealed that compound 156 showed reasonable anticancer activities against tested tumor cell lines (laryngeal carcinoma (Hep-2), breast carcinoma (MCF-7), gastric carcinoma (BGC-823), liver carcinoma (HepG2), cervical carcinoma (HeLa), prostate carcinoma (PC-3) and colorectal carcinoma (HCT-8, HCT-116 and RKO)) with IC50 values lower than 10.0 μM.62
Li and coworkers also demonstrated the synthesis of anticancer active compounds. Among synthesized compounds, compound 157 showed excellent topoisomerase inhibitory activity against human cancer cell lines (A549, HL-60 and HeLa cell line). The outers revealed that the compound was found to be 5 times more potent in cell-killing activity for cell lines A549, HL-60, and HeLa than the positive control irinotecan or cisplatin, with IC50 of 0.009, 0.008 and 0.010 μM, respectively. Docking studies of compound 157 showed that it was actively bound to the active site of the Top I-DNA complex via a hydrogen bond, cation–π interaction, and π–π interaction.63
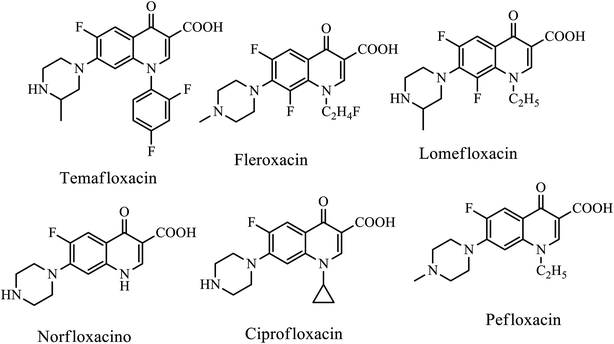
Ciprofloxacin has been reported to be a Topo-II inhibitor and antitumor active against various human cancer cell lines, such as colorectal, leukemia, and osteosarcoma cell lines. Thus, ciprofloxacin derivatives also showed potent activities. Hydrazone derivatives attached to ciprofloxacin 158a and 158b showed significant antitumor activities against the UO-31 cancer cell line with IC50 values of 0.75 μM and 0.72 μM, respectively. Additionally, compound 158b displayed cytotoxicity against NCI-H226 and IGROV1 cancer cell lines with IC50 of 1.02 and 0.75 μM, respectively. Moreover, they revealed a potent pro-apoptotic effect through the induction of apoptosis and an increase in the level of active caspase-3 compared to control and cell cycle arrest at the G2/M phase.6 Generally, SAR studies revealed that Azoles connecting to the C-8 and N-1 positions, small ring at the C-2 position, amide, ester, and heterocycles, such as azoles at the C-3 position, –H, –F, –Me, and –OMe substitutes at the C-5 position, and N, C–H, C–F substituted at the C-8 position, can improve anticancer activity (Fig. 6).2
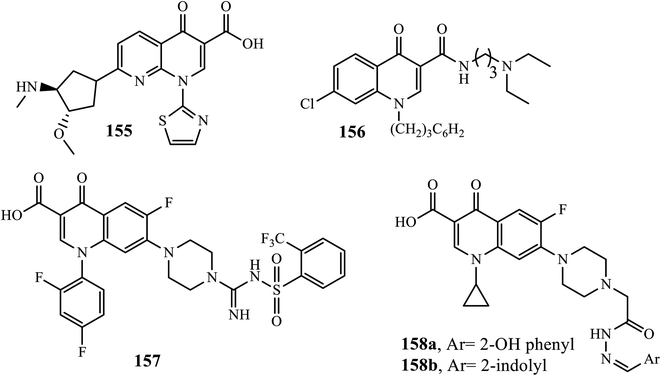 |
| | Fig. 6 Chemical structures of some anticancer containing 4-quinolone derivatives. | |
4.2 Antiviral activity
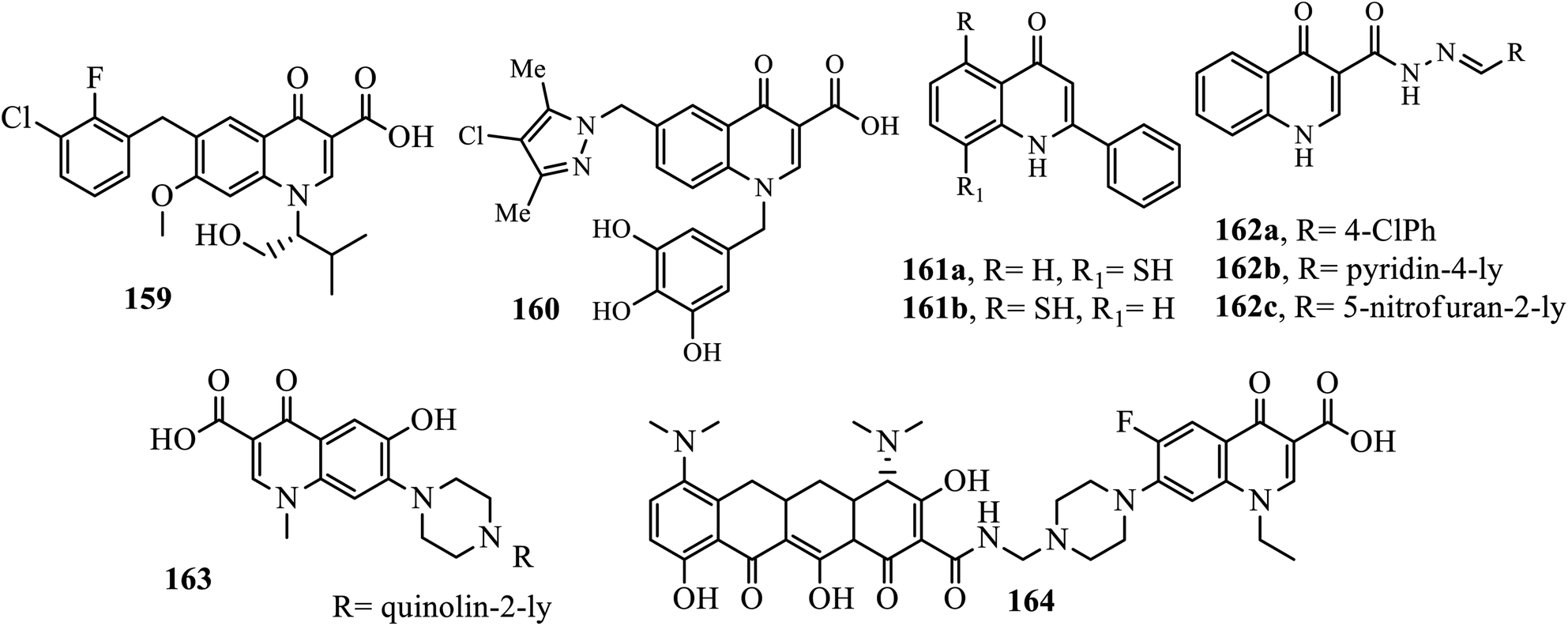
Acquired immune deficiency syndrome (AIDS) is caused by the human immunodeficiency virus (HIV), which is one of the world's deadliest and most deadly public health pandemics. In the recent decade, several quinolone compounds have been studied for anti-HIV activity, and some of them have shown promising results. Elvitegravir 159, the first quinolone integrase inhibitor, has already been approved by the U.S. Food and Drug Administration for the treatment of HIV infection, demonstrating that quinolone derivatives might have potential for anti-HIV activity.64
Xu and coworkers reported the synthesis of numerous quinolone acid derivatives as potential HIV-1 integrase inhibitors. Among the synthesized quinolone acid derivatives, compound 160 with 3,4,5-trihydroxylated aromatic substituents on the N-1 position of the quinolone ring exhibited the most significant inhibitory activity to the strand transfer process of HIV-1 integrase with IC50 of 2.6 μM using S-1360 as the control compound (IC50 of 0.50 μM). It was revealed that the presence of an appropriate substituent at the N-1 position is crucial to enhance the structure–activity relationships for quinolone derivatives.65 Among the synthesized quinolone derivatives by Wang and coworkers, compounds 161a and 162b displayed selective antiviral activity (ED50 0.65–9.5 μg mL−1) against HIV in acutely infected H9 lymphocytes with therapeutic indexes in the range of 0.6–5.7. Compound 161b, bearing acetoxy groups on both the 6- and 7-positions, was the most active (ED50 value, 0.65 μg mL−1) with a therapeutic index of about 5.66
Three quinolone-acyl hydrazones 162a–c were synthesized by Yonedas' group and evaluated for their antiviral activities against HIV-1 (PBMC cells) and herpes simplex virus (HSV). All hybrids showed potential anti-HIV activity with EC50 of 3.4, 100, and 56 μM. It was revealed that oxoquinolone-acylhydrazone 162a is the best candidate for an anti HSV-1 agent.67 Compound 163 synthesized and reported by Massari et al. is highly active against HIV-1IIIB and HIV-2ROD (MT-4 cells) with the EC50 values of 1.85 ± 0.76 μg mL−1 and 1.31 ± 0.89 μg mL−1, respectively. It is also better when compared to nevirapine (EC50: 23 and >4000 nM). Sriram and coworkers synthesized a series of prodrugs for the treatment of HIV/AIDS. Hybrids compound 164 was identified as the most promising compound against HIV-1 replication with EC50 of 5.2 μM and was nontoxic towards the CEM cells (CC50: 200 μM), and MIC of 0.2 μg mL−1 against MTB H37Rv, with the moderate inhibition of both 3′-P and ST steps of HIV-1 IN (IC50 0: 20 and 18 μM) 204 and 188 μM).68,69
Generally, the SAR study relieved that liner, branched alkyl, hydoxyalkyl, and benzyl substituted at the N-1 position, small alkyl at the C-2 position, carbocyclic acid at the C-3 position, ring formation of C-5 at the C-6 position, NH2, 2-F-ClBn at the C-7 position, OMe, hydroxyl-containing ether, and nitrogen-containing heterocycles at the C-7 position. Finally, N, CH, C–OMe, and COCHF2 substitution at the C-8 position increases anti-HIV activities (Fig. 7).64
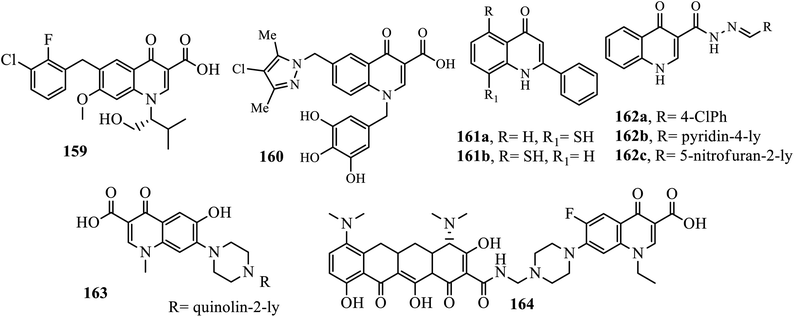 |
| | Fig. 7 Chemical structures of some anti-HIV containing 4-quinolone derivatives. | |
4.3 Anti-bacterial activity
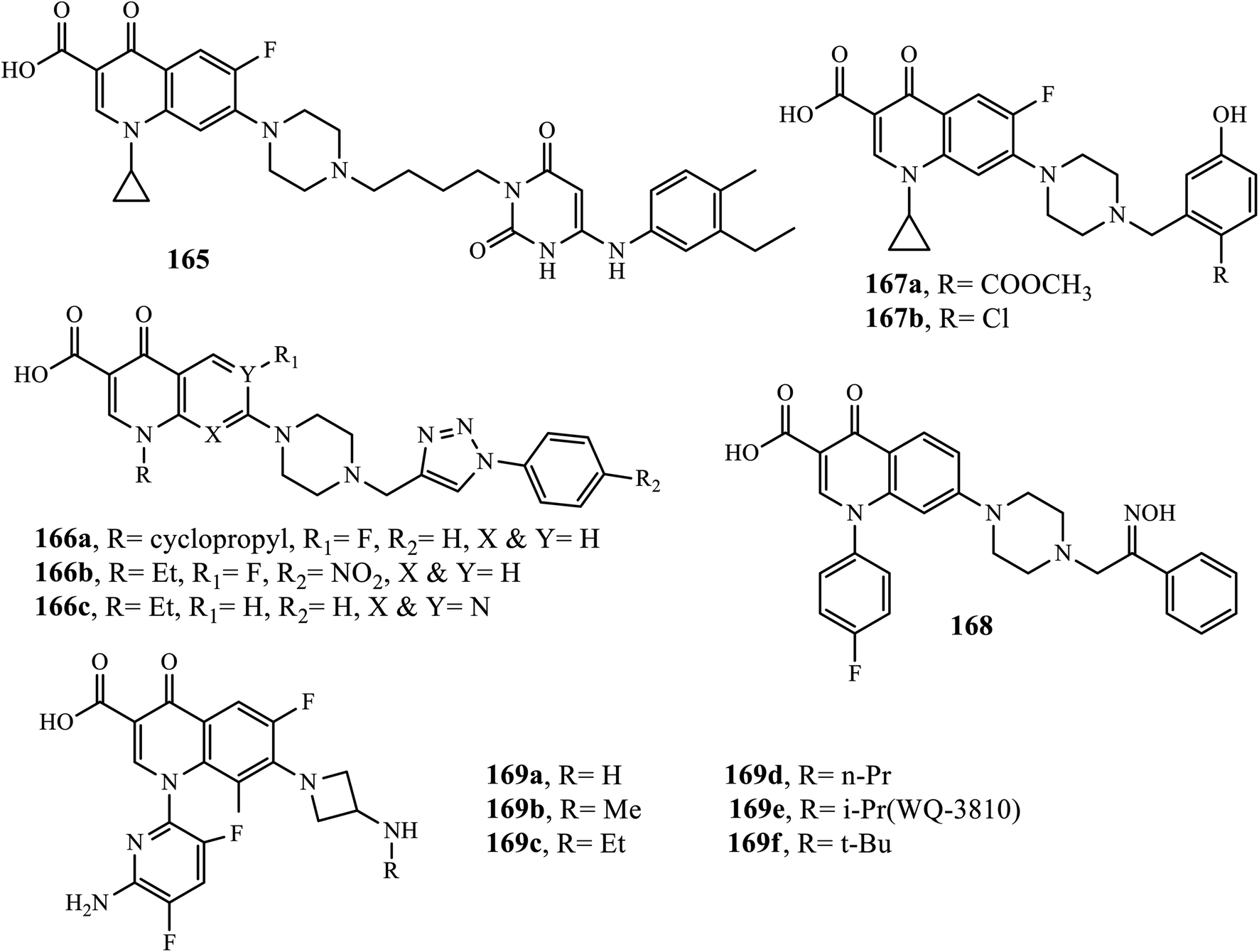
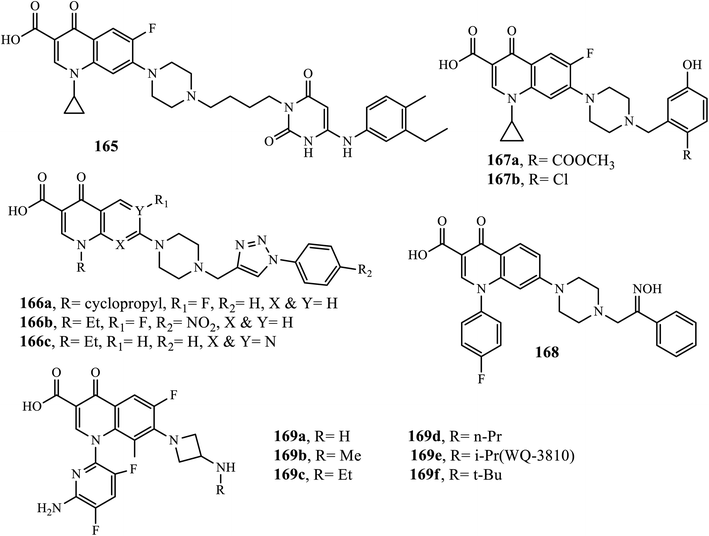
Quinolones are known for their antibacterial activity because the discovery of nalidixic acid and fluoroquinolones is one of the most important antibacterials that are widely used for the treatment of various infectious diseases. Recently, the hybridization of quinolone with other compounds has attracted much attention because it has led to the development of new drugs with the potential to overcome cross-resistance and improve affinity and efficacy. Compound 165 (a hybrid of fluoroquinolone) showed inhibition activities against DNA polymerase IIIC in addition to topoisomerase enzymes. The results indicated that this hybrid exhibited potent anti-bacterial activities against B. subtilis and DNA polymerase IIIC (Ki: 0.024 and 0.019 μM) in addition to drug-resistant bacteria with a MIC of 0.313–2.5 μg mL−1.70
The quinolone-triazole hybrid substituted compounds 166a, 166b, and 166c synthesized by Faidallah's group via microwave-assisted click chemistry exhibited good antibacterial activities against Gram-positive bacteria (S. aureus and St. Pyogenes) and Gram-negative bacteria (Salmonella typhi and E. coli) higher than parent drugs, ciprofloxacin, norfloxacin, and pipemidic acid with a MIC range of 0.2–0.5 μg mL−1. A series of the novel Mannich bases of ciprofloxacin was synthesized and reported by Abdel-Rahman and coworkers. Among the synthesized compounds, compound 166a exhibited high antibacterial activities against E. coli P. aeruginosa species with MIC values of 0.036 and 0.043 μg mL−1, respectively. Compound 167b showed potent activities against S. aureus and MRSA with MIC values of 0.061 and 0.066 μg mL−1, respectively, which is higher than the reference drug, ciprofloxacin, with MIC values of 1.6 and 1.5 μg mL−1, respectively.71,72
Asadipour and coworkers reported the synthesis and in vitro antibacterial evaluations of N-substituted piperazinyl sarafloxacin derivatives. Among them, compound 168 exhibited comparable activity as sarafloxacin (positive control) against S. aureus, S. epidermidis and B. subtilis (MIC ≤ 0.125 μg mL−1) and Gram-negative bacteria E. Coli (MIC = 0.2–2 μg mL−1) compared to the standard drug sarafloxacin (MIC ≤ 0.125 μg mL−1). It was revealed that the antibacterial activities against both Gram-negative and Gram-positive bacteria were reduced upon the introduction of bulky substituents on the piperazine ring at the C-7 position of fluoroquinolones.73
Novel 7-(3-alkylaminoazetidin-1-yl)fluoroquinolones were designed, synthesized and evaluated owing to their antibacterial activities by Itoh's group. Interestingly, all compounds 169a–f exhibited strong inhibition of E. coli DNA gyrase with IC50 values ranging from 0.078 to 0.33 mg L−1. Furthermore, the compounds showed better antibacterial activity against E. coli (MIC of 0.004 to 0.012 mg L−1 for 169a–e and 0.038 mg L−1 for 169f) and Gram-negative bacteria (11 species, 15 strains, 0.061 to 0.21 mg L−1 for 169a–e and 0.57 mg L−1 for 169f). Additionally, the oral absorption rates (in rats) of various analogs (169a–e) were determined, and the results indicated that oral absorption rates (%) increased as the chain length increased, which in turn may be due to the increased lipophilicities of the compounds, thereby resulting in enhancing membrane permeability. From the series of fluoroquinolone synthesized, WQ-3810 (169e) was identified as an orally active antibacterial agent with potent in vitro activity along with better oral bioavailability. Thus, compound 169e (WQ-3810) is considered a promising lead molecule for further exploration (Fig. 8).74
 |
| | Fig. 8 Chemical structure of some antibacterial containing 4-quinolone derivatives. | |
4.4 Anti-malarial activity
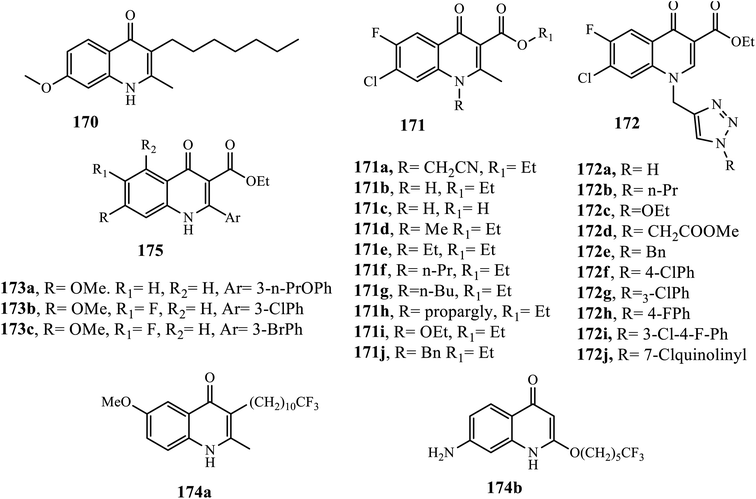
Quinolones have been known as potential anti-malarial agents starting from endochin 170 since the 1940s. In vitro, quinolones showed potential anti-malarial activities against the erythrocytic and hepatic stages of CQ-sensitive (CQS) and CQ-resistant (CQR) P. falciparum as well as exhibited excellent activities in vivo potency.75
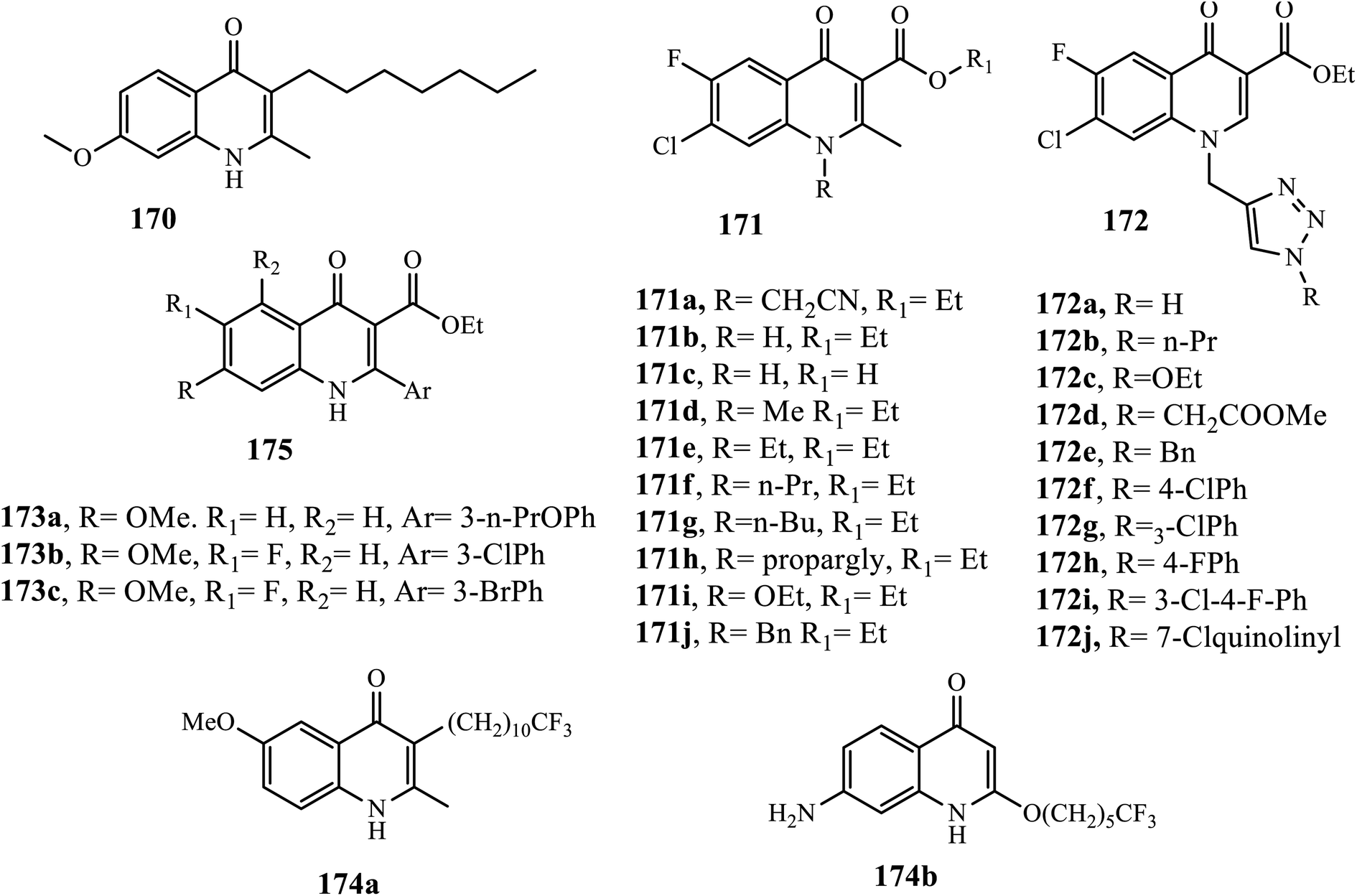
Awasthi et al. synthesized several fluoroquinolone analogs 171 and 172. All derivatives (IC50: 1.33–15.20 μM) showed promising malarial activities and negligible or very low toxicity profiles, and the majority of them were more potent than ciprofloxacin (IC50: 8.82 μM).76 Among quinolone synthesized by Dixit et al., 173a was the most potent with ED50 ranging from <1 to 20 nM against CQS D10, MDR K1, and TM90-C2B strains, and it was more active than atovaquone. Additionally, derivatives 173b, c exhibited excellent anti-plasmodia activities in vitro (ED50: 80–200 nM), and showed the highest hepatic metabolic stability in the in vitro mouse liver microsome assay, demonstrating 38 and 31 μM maximum concentrations in plasma (Cmax at a 50 mg kg−1 dose, while Cmax was 66 and 39 μM, respectively, at 200 mg kg−1). Both of them also displayed significant AUC values at both dosages (>125 μM h for 173b and >70 μM h for 173c), suggesting significant systemic exposure for those two derivatives relative to their potency. In vivo antimalarial potency was evaluated in a P. berghei infected mouse model; the results indicated that both compounds 173b, c displayed in vivo activity. Ester 173b exhibited suppression activity comparable to that of amodiaquine but was not curative, while 173c showed high in vivo toxicity with no survival after day 14. In general, compound 173b exhibited the best balance between potency, physicochemical properties, in vitro mouse liver microsome stability, in vivo pharmacokinetic profile, and the best efficacy, which was equivalent to that of amodiaquine, so 173b is promising for further development as antimalarials.77
Winter et al. synthesized endochin-like quinolones 174, and half of the derivatives exhibited promising antiplasmodial activities with IC50 values ranging from 0.02 to around 30 nM against CQS D6, MDR Dd2, and TM90-C2B P. falciparum strains. Compound 174a (IC50: 0.02 nM) was found to be most active against CQSD6 and MDRDd2, which was 15–320 fold more potent than the references, chloroquine, artemisinin, and atovaquone (IC50: 6.4, 0.81 and 0.3 nM, respectively) against CQS 3D7, and 25–5650 times more active than the three references (IC50: 113, 0.6 and 0.5 nM, respectively) against MDR Dd2. Compound 174b (IC50: 5 nM) was the most potent against TM90-C2B, which was comparable to artemisinin (IC50: 2.1 nM), 28- and 1083-fold more potent than chloroquine and atovaquone (IC50: 134 and 5090 nM, respectively). Moreover, both derivatives displayed non-cytotoxicity in murine splenic lymphocytes even at a concentration of >25 μM. Combined with excellent antiplasmodial activity with low cytotoxicity, these novel quinolones suggest a favorable therapeutic index in vivo and emphasize the developmental potential of this structural class for clinical use.78 Generally, the SAR study showed that the electro-donating group at the C-2 position, ester at the C-3 position, hydrophobic, electronegative, and large volume atom at the C-7 position, a small hydrophobic electron donating group at the C-6 position is preferred, whereas dimethoxy, dihalogen substituted at the C-5, C-7, and C-6 positions is disfavored (Fig. 9).75
 |
| | Fig. 9 Chemical structures of some anti-malaria containing 4-quinolone derivatives. | |
5. Conclusion
To date, various studies have been carried out based on the synthesis procedure and pharmaceutical potential for various diseases of 4-quinolone and its derivatives. Regarding synthetic methods, researchers have focused on using classical routes to obtain simpler, cheaper, cleaner, and faster methods with the highest yield. Recently, researchers have synthesized hybrid 4-quinolone and its derivatives scaffolds with compounds containing other heterocyclic compounds using various synthetic procedures, such as metal-free or green synthesis, metal-catalyzed synthesis, microwave irradiation or multicomponent one-pot synthesis methodologies and further post modification techniques. 4-Quinolone and its derivatives are found in a broad spectrum range of physiologically and therapeutically active compounds. They play an important role in drug development and discovery. Researchers pay attention not only to the synthesis of 4-quinolone and its derivatives but also to the development and design of eco-friendly reaction processes towards the title compound. Reactions catalyzed by typical metal-free, solvent-free, aqueous media, and ionic liquids are environmentally friendly protocols and are discussed well in this review. Furthermore, quinolone and its derivatives exhibit a broad spectrum of bioactivities and have been applied in the treatment of various types of human infections.
Conflicts of interest
There are no conflicts to declare.
References
- Y. Hu, S. Zhang, Z. Xu, Z. Lv and M. Liu, 4-Quinolone hybrids and their antibacterial activities, Eur. J. Med. Chem., 2017, 335–345 CrossRef CAS PubMed.
- F. Gao, J. Xiao and G. Huang, Current scenario of tetrazole hybrids for antibacterial activity, Eur. J. Med. Chem., 2019, 184, 111744–111785 CrossRef CAS PubMed.
- P. Lucia, Quinolone: Synthesis and Antibacterial Activity, National Institute for Chemical-Pharmaceutical Research and Development, Bucharest, 2012, pp. 255–272 Search PubMed.
- C. Shen, A. Wang, J. Xu, Z. An, K. Y. Loh, P. Zhang and X. Liu, Recent Advances in the Catalytic Synthesis of 4-Quinolones, Chem, 2019, 1059–1107 CAS.
- V. L. M. Silva and A. M. S. Silva, Palladium-Catalysed Synthesis and Transformation of quinolones, MDPI, Molecules, 2019, 1–74 Search PubMed.
- M. Samir, M. Ramadan, M. H. Abdelrahman, M. Abdel-Aziz and G. E.-D. A. Abuo-Rahma, Recent Strategies in Design of Antitumor and Antibacterial Fluoroquinolones, J. Adv. Biomed. Pharm. Sci., 2021, 134–151 CrossRef.
- R. I. Al-Bayati, M. R. Ahamad and L. S. Ahamed, Synthesis and Biological Activity Investigation of Some Quinoline-2-One Derivatives, Am. J. Org. Chem., 2015, 5(4), 125–135 CAS.
- P. Dhiman, N. Arora, P. V. Thanikachalam and V. Monga, Recent advances in the synthetic and medicinal perspective of quinolones: A review, Bioorg. Chem., 2019, 92, 1–45 CrossRef PubMed.
- P. Dhiman, N. Arora, P. V. Thanikachalam and V. Monga, Recent advances in the synthetic and medicinal perspective of quinolones: A review, Bioorg. Chem., 2019, 92, 1–45 CrossRef PubMed.
- G. Y. Lesher, E. J. Froelich, M. D. Gurett, J. H. Baily and R. P. Brundage, 1,8-Naphthyridine derivatives. A new class of chemotherapeutic agents, J. Med. Pharm. Chem., 1962, 91, 1063–1065 CrossRef CAS PubMed.
- J. Zhao, Y. Zhao and H. Fu, K2CO3-Catalyzed Synthesis of Chromones and 4-Quinolones through the Cleavage of Aromatic C-O Bonds, Org. Lett., 2012, 14, 2710–2713 CrossRef CAS PubMed.
-
(a) T. Zhao and B. Xu, Palladium-Catalyzed Tandem Amination Reaction for the Synthesis of 4-Quinolones, Org. Lett., 2010, 12, 212–215 CrossRef CAS PubMed;
(b) J. Shao, X. Huang, X. Hong, B. Liu and B. Xua, Synthesis of N-Alkyl-Substituted 4-Quinolones via Tandem Alkenyl and Aryl C–N Bond Formation, Synthesis, 2012, 44, 1798–1808 CrossRef CAS.
- M. Janni, S. Aroraa and S. Peruncheralathan, Double heteroannulation of S,N-acetals: a facile access to quinolone derivatives, Org. Biomol. Chem., 2016, 14, 8781–8788 RSC.
- P. Hasan, B. Aneja, M. Masood, B. Ahmad, U. Yadava, C. Daniliuc and M. Abid, Efficient synthesis of novel N-substituted 2-carboxy-4-quinolones via lithium bis(trimethylsilyl)amide (LiHMDS)-induced in situ cyclocondensation reaction, RSC Adv., 2017, 7, 11367–11372 RSC.
- D. Zewge, C.-y. Chen, C. Deer, P. G. Dormer and D. L. Hughes, A Mild and Efficient Synthesis of 4-Quinolones and Quinolone Heterocycles, J. Org. Chem., 2007, 72, 4276–4279 CrossRef CAS PubMed.
- P. D. Duarte, M. W. Paixão and A. G. Corr êa, Microwave assisted synthesis of 4-quinolones and N,N′ -diarylureas, Green Process. Synth., 2013, 2, 19–24 CAS.
- I. Malvacio, D. Vera and E. Moyano, Microwave assisted synthesis of ethyl-quinolon-4- one-3-carboxylates and hydrolysis to quinolon-4-one-3-carboxylic acids, Curr. Microwave Chem., 2014, 1, 52–58 CrossRef CAS.
- W. Hu, J.-P. Lin, L.-R. Song and Y.-Q. Lon, Direct Synthesis of 2-Aryl-4-quinolones via Transition-Metal-Free Intramolecular Oxidative C(sp3)–H/C(sp3)–H Coupling, Org. Lett., 2015, 5(17), 1268 CrossRef PubMed.
- A. C. Wright, C. K. Haley, G. Lapointe and B. M. Stoltz, Synthesis of Aryl Ketoamides via Aryne Insertion into Imides, Org. Lett., 2016, 1–3 Search PubMed.
- K. Xu, S. He, S. Chen, G. Qiu, J. Shi, X. Liu, X. Wu, J. Zhang and W. Tang, Free radical rearrangement synthesis and microbiological evaluation of novel 2-sulfoether-4-quinolone scaffolds as potential antibacterial agents, Eur. J. Med. Chem., 2018, 154, 144–154 CrossRef CAS PubMed.
- S. Khong and O. Kwon, Phosphine-Initiated General-Base-Catalyzed Quinolone Synthesis, Asian J. Org. Chem., 2014, 3, 453–457 CrossRef CAS PubMed.
- N. Zhou, Z. Yan, H. Zhang, Z. Wu and C. Zhu, Metal-free radical Oxidative Cyclization of o-Azidoaryl Acetylenic Ketones with Sulfinic acids to Access Sulfone-Containing 4-Quinolones, J. Org. Chem., 2016, 1–19 Search PubMed.
- Z. Zhang, X. Xiao, T. Sun, J. Wub, J. Ren, J. Zhu, X. Zhanga, R. Cao and R. Du, Synthesis, structure-activity relationships and preliminary mechanism of action of novel water-soluble 4-quinolone-3-carboxamides as antiproliferative agents, Eur. J. Med. Chem., 2017, 1–41 Search PubMed.
- S.-F. Jiang, C. Xu, Z.-W. Zhou, Q. Zhang, X.-H. Wen, F.-C. Jia and A.-X. Wu, Switchable Access to 3 - Carboxylate-4-quinolones and 1 - Vinyl-3-carboxylate-4-quinolones via Oxidative Cyclization of Isatins and Alkynes, Org. Lett., 2018, 1–4 Search PubMed.
- M. S. Rao and S. Hussain, DABCO-mediated decarboxylative cyclization of isatoic anhydride with aroyl/heteroaroyl/alkoylacetonitriles under microwave conditions: Strategy for the synthesis of substituted 4-quinolones, Tetrahedron Lett., 2021, 1–8 Search PubMed.
- D. Wang, P. Sun, P. Jia, J. Peng, Y. Yue and C. Chen, Transition-Metal-Free One-Pot Tandem Synthesis of 4-Quinolone and 4 H-Thiochromen-4-one Derivatives Through Sequential Nucleophilic Addition–Elimination–SNAr Reaction, Synthesis, 2017, 49, 1–11 CrossRef.
- S. Bandatmakuru and V. Arava, Novel Synthesis of 1,2-Substituted 4-Quinolones, SynOpen, 2018, 2, 285–292 CrossRef CAS.
- C. G. Dave and H. M. Joshipura, Microwave assisted Gould-Jacob reaction: Synthesis of 4-quinolones under solvent free conditions, Indian J. Chem., 2002, 650–652 CAS.
- J. Liu, D. Ba, W. Lv, Y. Chen, Z. Zhao and G. Cheng, Base-Promoted Michael Addition/Smiles Rearrangement/N Arylation Cascade: One-Step Synthesis of 1,2,3-Trisubstituted 4-Quinolones from Ynones and Sulfonamides, Adv. Synth. Catal., 2019, 1–13 Search PubMed.
- C. Xie, D. Yang, X. Wang and C. Ma, A Cascade Reaction of Michael Addition and Truce-Smiles Rearrangement to Synthesize Trisubstituted 4 - Quinolone Derivatives, J. Org. Chem., 2020, 1–8 Search PubMed.
- B.-H. Zhu, Y.-Q. Zhang, H.-J. Xu, L. Li, G.-C. Deng, P.-C. Qian, C. Deng and L.-W. Ye, Regio- and Stereoselective Synthesis of Diverse 3,4-Dihydro-2-quinolones through Catalytic Hydrative Cyclization of Imine- and Carbonyl-Ynamides with Water, ACS Catal., 2021, 11, 1706–1713 CrossRef CAS.
- X.-D. Fei, Z. Zhou, W. Li, Y.-M. Zhu and J.-K. Shen, Buchwald–Hartwig Coupling/Michael Addition Reactions: One-Pot Synthesis of 1,2-Disubstituted 4-Quinolones from Chalcones and Primary Amines, Eur. J. Org. Chem., 2012, 3001–3008 CrossRef CAS.
- C. Shen, A. Wang, J. Xu, Z. An, K. Y. Loh, P. Zhang and X. Liu, Recent Advances in the Catalytic Synthesis of 4-Quinolones, Chem, 2019, 5, 1059–1107 CAS.
- N. Haddad, J. Tan and V. Farina, Convergent synthesis of the quinolone substructure of BILN 2061 via carbonylative sonogashira coupling/cyclization, J. Org. Chem., 2006, 71, 5031–5034 CrossRef CAS PubMed.
- J. Wu, Y. Zhou, T. Wu, Y. Zhou and A. Lei, From ketones, amines, and carbon monoxide to 4-quinolones: palladium-catalyzed oxidative carbonylation, Org. Lett., 2017, 19, 6432–6435 CrossRef CAS PubMed.
- P. Ghosh, A. K. Nandi and S. Das, Carbonylative Sonogashira annulation sequence: One-pot synthesis of 4-quinolone and 4H-chromen-4-one derivatives, Tetrahedron Lett., 2018, 1–4 Search PubMed.
- C. Jones, K. Anderson and S. L. Buchwald, Sequential Cu-catalyzed amidation-base-mediated camps cyclization: a two-step synthesis of 2-aryl-4-quinolones from o- halophenones, J. Org. Chem., 2007, 72, 7968–7973 CrossRef CAS PubMed.
- X. Xu and X. Zhang, Direct synthesis of 4-quinolones via copper-catalyzed anilines and alkynes, Org. Lett., 2017, 19, 4984–4987 CrossRef CAS PubMed.
- S. Kang, S. Park, K.-s. Kim, C. Song and Y. Lee, Copper-Catalyzed Aza-Michael Addition of 2-Aminobenzoate to β-Substituted α,β-Unsaturated Ketones: One-Pot Synthesis of 3-Carbonyl-2-Substituted Quinolin-4(1H)-ones, J. Org. Chem., 2018, 83(5), 2694–2705 CrossRef CAS PubMed.
- N. Sundaravelu and G. Sekar, Cu-Catalyzed one-pot synthesis of thiochromeno quinolinone and thiochromeno-thioflavone via oxidative double hetero Michael addition using in situ generated nucleophiles, Chem. Commun., 2020, 56, 8826–8829 RSC.
- F. Wang, L. Jin, L. Kong and X. Li, Cobalt(III)- and rhodium(III)-catalyzed C-H amidation and synthesis of 4-quinolones: C-H activation assisted by weakly coordinating and functionalizable enaminone, Org. Lett., 2017, 19, 1812–1815 CrossRef CAS PubMed.
- P. Shi, L. Wang, K. Chen, J. Wang and J. Zhu, Co(III)-catalyzed enaminone-directed C–H amidation for quinolone synthesis, Org. Lett., 2017, 19, 2418–2421 CrossRef CAS PubMed.
- O. Seppänen, M. Muuronen and J. Helaja, Gold-Catalyzed Conversion of Aryl- and Alkyl-Substituted 1-(o-Aminophenyl)-2-propyn-1-ones to the Corresponding 2-Substituted 4-Quinolones, Eur. J. Org. Chem., 2014, 4044–4052 CrossRef.
- X. Wu, L.-L. Zheng, L.-P. Zhao, C.-F. Zhu and Y.-G. Li, Gold-catalyzed cyclization of 1-(2 0 -azidoaryl) propynols: synthesis of polysubstituted 4-quinolones, Chem. Commun., 2019, 55, 14769–14772 RSC.
- J. Huang, H. Su, M. Bao, L. Qiu, Y. Zhang and X. Xu, Gold(III)-catalyzed azide-yne cyclization/O-H insertion cascade reaction for the expeditious construction of 3-alkoxy-4-quinolinone frameworks, Org. Biomol. Chem., 2020, 18, 3888–3892 RSC.
- Y. Yoshino, T. Kurahashi and S. Matsubara, Nickel-catalyzed decarboxylative carboamination of alkynes with isatoic anhydrides, J. Am. Chem. Soc., 2009, 131, 7494–7495 CrossRef CAS PubMed.
- S. Ueno, R. Shimizu, R. Maeda and R. Kuwano, Synthesis of 4-quinolones through nickel-catalyzed intramolecular amination on the b-carbon of o-(N-alkylamino) propiophenones, Synlett, 2012, 23, 1639–1642 CrossRef CAS.
- X. Ji, D. Li, Z. Wang, M. Tan, H. Huang and G.-J. Deng, visible light-induced aerobic oxidation of indoles: one-pot formation of 4-quinolones at room temperature, Asian J. Org. Chem., 2018, 7, 711–714 CrossRef CAS.
- Y. Shi, H. Xing, T. Huang, X. Liu, J. Chen, X. Guo, G.-B. Li and W. Yong, Divergent C-H Activation Synthesis of Chalcones, Quinolones and Indoles, ChemComm, 2020, 1–4 Search PubMed.
- J. Joussot, A. Schoenfelder, L. Larquetoux, M. Nicolas, J. Suffert and G. Blond, Synthesis of 3-Substituted Chromones and Quinolones from Enaminones, Synthesis, 2016, 48, 3364–3372 CrossRef CAS.
- J. Pinto, V. Silva, L. Santos and A. Silva, Synthesis of (E)-3-styrylquinolin 4(1H)-ones in water by ohmic heating: A comparison with other methodologies, Eur. J. Org. Chem., 2016, 2888–2896 CrossRef CAS.
- K. H. V. Reddy, J.-D. Brion, S. Messaoudi and M. Alami, Synthesis of Biheterocycles based on Quinolinone, Chromone and Coumarin Scaffolds by Palladium-Catalyzed Decarboxylative Couplings, J. Org. Chem., 2016, 81, 424–432 CrossRef PubMed.
- P. Ghosh and S. Das, Creation of 4-Quinolone Thioether and Selenoether Derivatives via Pd-NHC Catalysed Cross-Coupling Reaction, SynOpen, 2020, 4, 33–37 CrossRef CAS.
- C. Xia, Z. Wei, Y. Yang, W. Yu, H. Liao, C. Shen and P. Zhang, Palladium-Catalyzed Thioetherification of Quinolone Derivatives via Decarboxylative C-S Cross-Couplings, Chem.–Asian J., 2016, 11, 360–366 CrossRef CAS PubMed.
- P. Ghosh and S. Das, Ligand Free Approach for the Copper(II)-Mediated C-NH2 Arylation of 4-Quinolone Derivatives Under Ambient condition, ChemistrySelect, 2018, 3, 8624–8627 CrossRef CAS.
- X. Li, Y. Bian, X. Chen, H. Zhang, W. Wang, S. Ren, X. Yang, C. Lu, C. Chen and J. Peng, Tunable synthesis of quinolinone-fused isoquinolines through sequential one-pot nucleophilic addition and palladium-catalyzed intramolecular C– H alkenylation, Org. Biomol. Chem., 2019, 17, 321–332 RSC.
- C. S. Azad and K. A. Narula, An operational transformation of 3-carboxy-4-quinolones into 3-nitro-4-quinolones via ipso-nitration using polysaccharide supported copper nanoparticles: synthesis of 3-tetrazolyl bioisosteres of 3-carboxy-4-quinolones as antibacterial agents, RSC Adv., 2016, 6, 19052–19059 RSC.
- G. D. Silva, L. M. V. Silva and M. S. A. Silva, Lewis Acid Catalyzed Diels–Alder Reactions of ( E)-1-Methyl-2-styrylquinolin-4(1 H)-ones with ortho-Benzoquinodimethane: Synthesis of New 2-(3-Arylnaphthalen-2-yl)-1-methylquinolin-4(1 H)-ones, Synthesis, 2016, 48, 4519–4524 CrossRef.
- S. Arasakumar, S. Shyamsivappan, S. Gopalan, A. Ata and P. S. Mohan, One-Pot Approach to Pyrido-4-phenanthridinones by PalladiumCatalyzed Annulation of 4 Quinolones with 2-Bromobenzyl Bromides, Synlett, 2019, 30, 63–68 CrossRef.
- E. Akkol, Y. Genc, B. Karpuz, E. S. Sanchez and R. Capasso, Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer, Cancers, 2020, 12, 1959–1984 CrossRef CAS PubMed.
- P. Horta, A. Secrieru, A. Coninckx and M. L. S. Cristiano, Quinolones for Applications in Medicinal Chemistry: Synthesis and Structure, Med. Chem., 2019, 261–297 Search PubMed.
- Z. Zhang, X. Xiao, T. Su, J. Wu, J. Ren and J. Zhu, Synthesis, structure-activity relationships and preliminary mechanism of action of novel water-soluble 4-quinolone-3-carboxamides as antiproliferative agents, Eur. J. Med. Chem., 2017, 140, 239–251 CrossRef CAS PubMed.
- J. Li, T. Zheng, Y. Jin, J. Xu, J. Yu and Y. Lv, synthesis, molecular docking and biological evaluation of quinolone derivatives as novel anticancer agents, chemical and pharmaceutical bulletin, Chem. Pharm. Bull., 2018, 66, 55–60 CrossRef CAS PubMed.
- R. Wang, K. Xu and W. Shi, Quinolone derivatives: Potential anti-HIV agent development and application, Arch. Pharm., 2019, 1–17 Search PubMed.
- X. Xu, Z. Luo, K. He and M. Zhan, Synthesis and Biological Evaluation of Quinolone Acid Derivatives Having Polyhydroxylated Aromatics as HIV-1 Integrase Inhibitions, Adv. Mater. Res., 2013, 634–638, 1116–1119 Search PubMed.
- H.-K. Wang, K. F. Bastow, L. M. Cosent and K.-H. Lee, Antitumor Agents. 166. Synthesis and Biological Evaluation of 5,6,7,8-Substituted-2-phenylthiochromen-4-ones, J. Med. Chem., 1996, 39, 1975–1980 CrossRef CAS PubMed.
- J. D. Yoneda, M. G. Albuquerque, K. Z. Leal, F. d. CostaSantos, P. N. Batalah, L. Brozeguinia, S. R. Peter, R. B. Alencastro, A. C. Cunhaa, M. C. de Souza, V. F. Ferreira, V. A. Giongo, C. C. Sanos and g. C. P. Paixãs, Docking of anti-HIV-1 oxoquinoline-acylhydrazone derivatives as potential HSV-1 DNA polymerase inhibitors, J. Mol. Struct., 2014, 1074, 263–270 CrossRef CAS.
- S. Massari, D. Daelemans, G. Manfroni, t. Sabatini, O. Tabarrini, C. Pannecouque and V. Cecchetti, Studies on anti-HIV quinolones: New insights on the C-6 position, Bioorg. Med. Chem., 2009, 17(2), 667–674 CrossRef CAS PubMed.
- D. Sriram, P. Yogeeswari, N. Srichakravarthy and T. R. Bal, Synthesis of stavudine amino acid ester prodrugs with broad-spectrum chemotherapeutic properties for the effective treatment of HIV/AIDS, Bioorg. Med. Chem. Lett., 2005, 14(5), 1085–1087 CrossRef PubMed.
- A. Chrzanowska, P. Roszkowski, A. Bielenica, W. Olejarz, K. Stępień and M. Struga, Anticancer and antimicrobial effects of novel ciprofloxacin fatty acids conjugates, Eur. J. Med. Chem., 2020, 185, 111810 CrossRef CAS PubMed.
- H. M. Faidallah, A. S. Girgis, A. D. Tiwari, H. H. Honkanadavar, S. J. Thomas, A. Samir, A. Kalmouch, K. A. Alamry, K. A. Khan and T. S. Ibrahim, Synthesis, antibacterial properties and 2D-QSAR studies of quinolone-triazole conjugates, Eur. J. Med. Chem., 2018, 143, 1524–1534 CrossRef CAS PubMed.
- I. M. Abdel-Rahman, M. Mustafa, S. A. Mohamed, R. Yahia, M. Abdel-Aziz, G. E.-D. A. Abuo-Rahma and A. M. Hayallah, Novel Mannich bases of ciprofloxacin with improved physicochemical properties, antibacterial, anticancer activities and caspase-3 mediated apoptosis, Bioorg. Chem., 2021, 107, 104629 CrossRef CAS PubMed.
- A. Asadipour, M. H. Moshaf, L. Khosravani, S. Moghimi, E. Amou, L. Firoozpour, G. Ilbeigi, K. Beiki, E. Soleimani and A. Foroumadi, N-substituted piperazinyl saraflxacin derivatives: synthesis and in vitro antibacterial evaluation, DARU J. Pharm. Sci., 2018, 26, 199–207 CrossRef CAS PubMed.
- K. Itoh, Y. Kuramoto, H. Amano, D. Kazamori and A. Yazaki, Discovery of WQ-3810: design, synthesis, and evaluation of 7-(3-alkylaminoazetidin-1-yl)floro-quinolones as orally active antibacterial agents), Eur. J. Med. Chem., 2015, 103, 354–360 CrossRef CAS PubMed.
- Y.-L. Fan, X.-W. Cheng, J.-B. Wub, M. Liu, F.-Z. Zhanga, Z. Xuc and L.-S. Feng, Antiplasmodial and antimalarial activities of quinolone derivatives: An overview, Eur. J. Med. Chem., 2018, 1–48 CrossRef PubMed.
- S. K. Dixit, N. Mishra, M. Sharma, S. Singh, A. Agarwal, S. K. Awasthi and V. Bhasin, Synthesis and in vitro antiplasmodial activities of fluoroquinolone analogs, Eur. J. Med. Chem., 2012, 51, 52–59 CrossRef CAS PubMed.
- Y. Zhang, W. A. Guiguemde, M. Sigal, F. Zhu, M. C. Connelly, S. Nwaka and R. K. Guy, Synthesis and structure–activity relationships of antimalarial 4-oxo-3-carboxyl quinolones, Bioorg. Med. Chem., 2010, 18, 2756–2766 CrossRef CAS PubMed.
- W. Rolf, X. K. Jane, J. S. Martin, H. David, R. K. Dennis and K. R. Michael, Optimization of endochin-like quinolones for antimalarial activity, Exp. Parasitol., 2011, 127, 545–551 CrossRef PubMed.
- S. Samadi and F. K. Behbahani, Iron (III) Phosphate Catalyzed the Synthesis of 4-quinolones, Chem. Methodol., 2018, 2, 181–185 CAS.
- S. B. Lee, Y. Jang, J. Ahn, S. Chun, D.-C. Oh and S. Hong, One-Pot Synthesis of 4-Quinolone via Iron-Catalyzed Oxidative Coupling of Alcohol and Methyl Arene, Org. Lett., 2020, 22, 8382–8386 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yadessa Melaku* and
Melis Belete
*,
Yadessa Melaku* and
Melis Belete