
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integration of functional peptides into nucleic acid-based nanostructures
Jessica S.
Freitag
 a,
Christin
Möser
a,
Robel
Belay
a,
Basma
Altattan
a,
Nico
Grasse
a,
Bhanu Kiran
Pothineni
a,
Jörg
Schnauß
abc and
David M.
Smith
*abd
a,
Christin
Möser
a,
Robel
Belay
a,
Basma
Altattan
a,
Nico
Grasse
a,
Bhanu Kiran
Pothineni
a,
Jörg
Schnauß
abc and
David M.
Smith
*abd
aFraunhofer Institute for Cell Therapy and Immunology, 04103 Leipzig, Germany. E-mail: david.smith@izi.fraunhofer.de
bPeter Debye Institute for Soft Matter Physics, Leipzig University, 04103 Leipzig, Germany
cUnconventional Computing Lab, UWE, Bristol, BS16 1QY, UK
dInstitute of Clinical Immunology, University of Leipzig Medical Faculty, 04103 Leipzig, Germany
First published on 5th April 2023
Abstract
In many applications such as diagnostics and therapy development, small peptide fragments consisting of only a few amino acids are often attractive alternatives to bulky proteins. This is due to factors such as the ease of scalable chemical synthesis and numerous methods for their discovery. One drawback of using peptides is that their activity can often be negatively impacted by the lack of a rigid, 3D stabilizing structure provided by the rest of the protein. In many cases, this can be alleviated by different methods of rational templating onto nanomaterials, which provides additional possibilities to use concepts of multivalence or rational nano-engineering to enhance or even create new types of function or structure. In recent years, nanostructures made from the self-assembly of DNA strands have been used as scaffolds to create functional arrangements of peptides, often leading to greatly enhanced biological activity or new material properties. This review will give an overview of nano-templating approaches based on the combination of DNA nanotechnology and peptides. This will include both bioengineering strategies to control interactions with cells or other biological systems, as well as examples where the combination of DNA and peptides has been leveraged for the rational design of new functional materials.
 From left to right: Christin Möser, Jessica S. Freitag, David M Smith | Jessica S. Freitag, Christin Möser and David M Smith are members of the “DNA Nanodevices Group” at the Fraunhofer Institute for Cell Therapy and Immunology (IZI) in Leipzig, Germany. The group was established in 2013, and its research focuses on applying approaches from DNA nanotechnology and other forms of molecular design to diagnostics, virology, and biological/polymer physics. They have published numerous manuscripts and patents on the usage of DNA-peptide conjugate materials to stimulate cells, modulate biological soft materials, bind to specific biological targets, analyze binding properties to pathogens or inhibit the infectivity of respiratory viruses. In recent years this has included a strong focus in the implementation of the advantages of DNA-based nanofabrication into commonly used analytical and diagnostic methods such as flow cytometry, ELISA, surface plasmon resonance (SPR), immunohistochemistry (IHC) and many more. Their multidisciplinary range of research topics includes methods and background knowledge in biological physics, biochemistry, immunology, pathogen biology, biomechanics and oncology. Motivation for many of their approaches comes from their proximity to real-world, commercial applications, particularly in the biomedical sector, as well as frequent “creative coffees” to spark the creative half of their brains. |
1. Introduction
Since the invention of solid-phase peptide synthesis nearly 60 years ago by Robert Bruce Merrifield,1 these short polymers of amino acids have become integral components in the essential biomolecular toolbox. While Merrifield's 1984 Nobel Prize for the technique perhaps stands out as the most overtly notable measure of acclaim,2 synthetically produced peptides have become the core of many therapeutic approaches3 as well as an increasing number of innovative strategies for biomaterial design.4 In the most simplified view, oligo- or polypeptides can be thought of as tiny fragments of proteins, which lack much of the secondary, tertiary, and quaternary features that are necessary for their precisely defined 3D structure and complex functions. Nevertheless, even relatively short motifs of just a few amino acids can be used as powerful tools for applications ranging from regenerative medicine,5 to drug discovery and delivery6 and the engineering of smart biomaterials.7 Currently, many methods for the identification or discovery of biologically active peptide sequences are available, including peptide arrays,8in silico modelling to optimize active motifs interpolated from crystal structures or known protein sequences,9 peptide phage display,10 or combinations thereof. These can be used to pinpoint the minimal active sequences, i.e., the shortest sequence of amino acids, that are necessary to trigger or inhibit a specific biological function. By using short peptides, one can also bypass some main drawbacks of therapeutic antibodies, such as expensive manufacturing or poor tissue penetration and biodistribution due to their large size and the resulting steric hindrance.When they are being used to target biological reactions such as stimulating signalling receptors or blocking a viral fusion protein, small peptides can suffer a major disadvantage compared to large, structured proteins; namely that the lack of the rigidly defined 3D structure provided by the rest of the protein can lead to weaker effect.11 Recently implemented synthesis strategies have enhanced the toolbox, using methods for chemical stabilization of secondary motifs or even the use of non-naturally-occurring amino acids in order to enhance their desired function.12 Alternatively, the two- or three-dimensional arrangement of multiple peptides on structural scaffolds can be used as a functional enhancement strategy. Small-molecule scaffolds like adamantane provide specificity in terms of stoichiometry and spatial arrangement,13 however precise fine-tuning of these types of scaffolds to specific binding is practically unavailable to all but the most highly trained of medicinal chemists. Alternatively, the integration of ligands onto large polymeric14 or nanoparticle scaffolds15 does allow the leveraging of brute polyvalency to simultaneously present nearly all possible configurations of bioactive ligands, however these can be wasteful in terms of materials, and ignore the potential to optimize individual oligovalent interactions.
As a pragmatic middle ground, we present the templating of peptides by nanostructures constructed from DNA or other nucleotide materials (Fig. 1a). While providing structural programmability on the nanometer size-scale of most biological constructs such as receptor dimers or trimers, the simplicity of altering stoichiometric, e.g. density of peptides attached, and structural parameters, e.g. 3D orientation also opens the possibility for their use as important tools for the rapid-prototyping of peptide-based bioengineering. Nanostructures based on the assembly of oligonucleotides such as DNA are increasingly used as structural scaffolds for arranging functional molecules such as peptides, since they have certain advantages over other nanoparticle-based templating methods. Due to the well-defined structural properties of DNA coupled with the ability to rapidly synthesize hundreds or even thousands of high-quality oligonucleotides at low cost, the different methods for DNA-based nanofabrication have become an important rapid-prototyping tool for creating precise, yet complex molecular patterns16–18 (Fig. 1b and c).
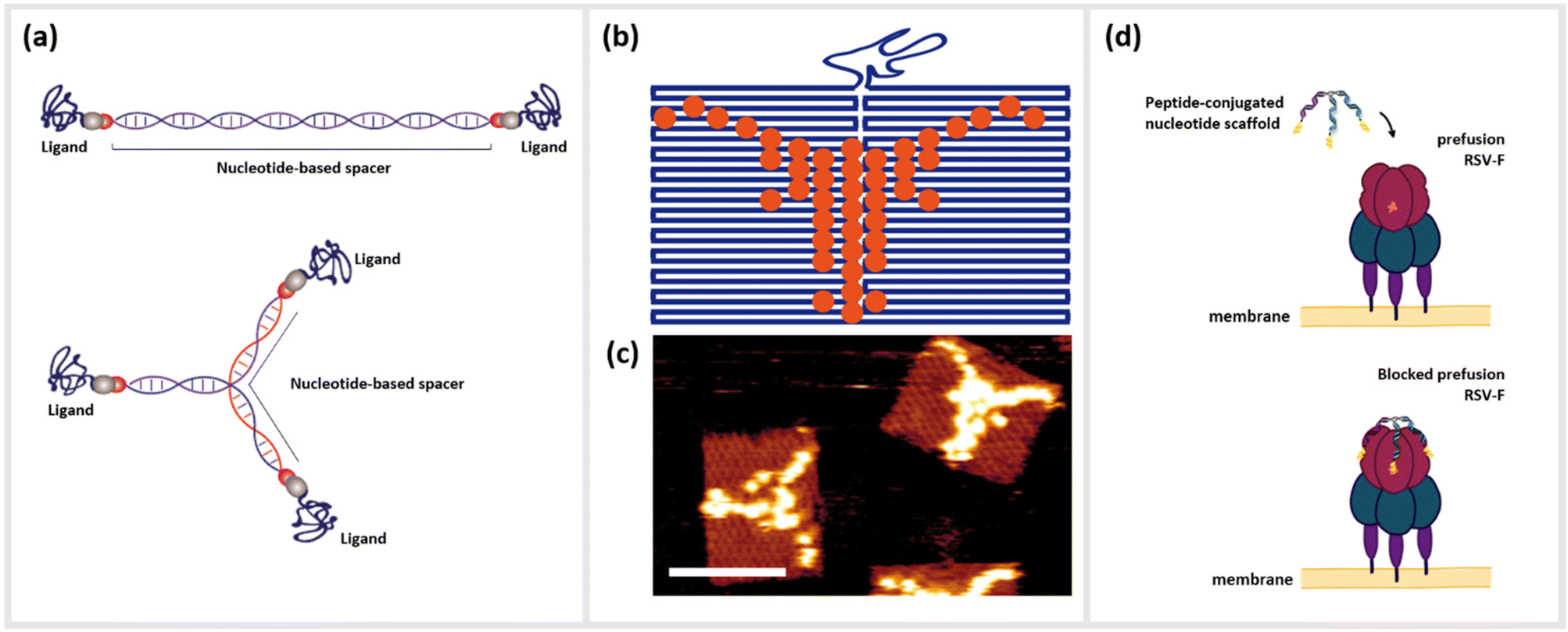 | ||
| Fig. 1 Nanometer-precise programming of DNA nanostructures with various ligands. (a) Rod-like and multimeric DNA nanostructures formed by (partially) complementary base-pairing of ssDNA strands, which can further be functionalized with various ligands in a nanometer-precise manner. (b) Scheme of “painting” on DNA origami in the pattern of the head of a Longhorn Steer, and (c) the corresponding atomic force microscopy image of Streptavidin-coupled DNA origami (scale bar: 100 nm). (d) Schematic drawing of the Respiratory syncytial virus (RSV) prefusion receptor F and its blocking through a trimeric, peptide-conjugated nucleotide scaffold. | ||
Techniques such as DNA origami,19 DNA bricks,20 DNA tiles21 or even the creation of simple branched structures from a handful of strands22,23 can create two- or three-dimensional structural templates, with the possibility to localize single or collections of molecules at single- or even sub-nanometer resolution. Due to this unprecedented level control, a large number of studies in recent years have examined how arrangements of optically-active molecules and nanoparticles can control the characteristics of visible light,24 or alternatively how biologically active molecules can be optimally arranged to interact with complex biomolecules such as antibodies,25 cells or living organisms.26–28 As an example, DNA origami functionalized with anti-freezing threonine peptides helped to protect the cell membranes of living cells during cryopreservation.29 Short peptides as described above represent an important class of biomolecules that have benefitted from nano-templating onto DNA or other nucleotide nanostructures. Here, we define peptides as small polymer chains comprised of up to approximately 50 amino acids, which can be distinguished from proteins by a lack of a tertiary and in most cases quaternary structure. These molecules can be associated with DNA strands and structures through direct chemical conjugation22,30–32 or by non-covalent association arising from hydrophobic or electrostatic interactions.33,34 The simplest variants consisting of one peptide conjugated to a single oligonucleotide already can lead to significantly enhanced or synergistic effects well beyond those of the individual components in isolation.35 These hybrid complexes are referred to as oligonucleotide peptide conjugates (OPCs), and have been well-studied for use in the therapy and diagnosis of various chronic diseases.36 For example, so-called cell-penetrating peptides (CPPs) have been exploited to directly shuttle regulatory nucleotides such as small interfering RNA (siRNA) or antisense oligonucleotides (ASO) across the membrane barriers with varying degrees of success.37–40
Alternatively, chimeras of peptides linked together with DNA or RNA aptamers, oligonucleotides that are selected to bind strongly and specifically to a particular target41,42 have shown drastic amplification of binding or inhibitory activity, often by several order of magnitude.43,44 Moving up the ladder of complexity, the combination of peptides with structured DNA templates can enhance the effect of the peptide in many ways, including the addition of targeting molecules for cells or bacteria,45 adding a protective element against harsh biological environments46,47 integrating synergistic functional nucleotide elements like aptamers,44 or amplifying function through cooperative oligo- or multivalence.22 This last concept of multivalent cooperativity is a particularly convenient advantage of DNA-based templating; enhancement of functions such as binding or receptor activation by the presentation of multiple ligands on a structural scaffold heavily depends upon precisely controlling both their number and nanoscale positioning relative to each other.48 Arranging these ligands in a geometrical complement to their targeted binding sites allows them to act cooperatively as a single unit. Structural information obtained from various imaging modalities such as Cryo-Electron Microscopy (CEM), X-ray crystallography, and nuclear magnetic resonance (NMR), which show the cognate receptor and its interaction with their native peptide ligands, can give information such as spatial orientation, distances between binding epitopes, conformational changes of the receptor during binding, or steric hindrances.49 The structural properties of DNA, specifically the roughly 3.4 Å length of single-base-pair for double-stranded DNA (dsDNA), mean that several small peptide ligands can be accurately arranged on a similar size scale to the distances between binding pockets on multimeric receptors on the surfaces of cells or viruses. Furthermore, the persistence length, or the approximate contour length of a polymer under which it will appear to be quite rigid, of dsDNA, is approximately 50 nm, while unpaired single-stranded DNA (ssDNA) segments appear completely flexible even on the length scale of a few nanometers. This means that even extremely simple DNA nanostructures can combine rigid and flexible features,50,51 thus enabling a final relaxation of the ligands into the optimal configuration of the target (Fig. 1d).
This review therefore summarizes recent advancements where the symbiotic combination of nucleotide-based nanostructures – in most cases DNA – and peptides has been used as a strategy to either enhance the activity of a peptide, or to create entirely new structures or function. We aim to elucidate on how nucleic acid-based nanotechnology can interface with biological systems and create more efficient compounds, in a more cost-effective manner, and resulting in more precise and stable interactions. While our main focus is on instances where these structural scaffolds are used to precisely template arrangements of multiple peptides or connect single peptides with other functional moieties, we do nevertheless highlight some studies that are based on non-specific self-organization of the two types of components.
2. Conjugation strategies
Currently, a large number of strategies exist for the coupling of biomolecules to DNA, many of which can be directly utilized for creating peptide-nucleotide chimeras. Peptide synthesis as well as oligonucleotide synthesis are both highly optimized and versatile methods for incorporating functional modifications that can act as points of attachment. In both molecules, modifications can be attached at nearly any position along the sequence. Due to the conflicting chemistries of both peptide and oligonucleotide synthesis, automated solid phase syntheses of the two molecules still have to be typically performed separately, prior to conjugation. Specific strategies for chemical functionalization of DNA strands relevant to DNA nanotechnology are discussed in more detail in a review by Madsen and Gothelf.52An enormous variety of methods exist for the conjugation of biomolecules, however not all of them are optimal for the conjugation of DNA to peptides due to various chemical and kinetic reasons. An overview of several conjugation strategies is given in Table 1. Known reactions described in literature on DNA and peptide conjugation are: native chemical ligation (NCL),53 thiol ether linkage,54,55 carbonyl ligation techniques,56–58 Staudinger Ligation59,60 and N-hydroxy-succinimide chemistry.61,62 Currently, the most commonly used techniques for high-yield ligation of peptides to nucleotides are based on the cycloadditions: Diels–Alder reaction,63–66 Inverse-Electron-Demand Diels–Alder cycloaddition (IED-DA),67,68 Copper catalyzed alkyne–azide cycloaddition (CuAAC)69,70 and strain promoted alkyne–azide-cycloaddition reaction (SPAAC).71,72 The IED-DA reaction between trans-cyclooctenes (TCO) and tetrazine derivatives is characterized by its high reactivity and fast kinetics, and is the fastest known biorthogonal conjugation strategy.67 However, the routine application of IED-DA for biological systems is limited due to high costs and short shelf life of the trans-cyclooctene derivatives caused by isomerization reactions to the nonreactive cis-conformation.66,73 Nevertheless, an advantage of most conjugation strategies based on cycloadditions is the commercial availability of conjugation reagents.
| Reaction | Mechanism | Advantage | Disadvantage | Ref. |
|---|---|---|---|---|
| NCL | Thioester-transesterification | High chemoselectivity; feasibility in aqueous media; no protective groups needed; biorthogonal; suitable for in vivo studies; direct conjugation of proteins after purification | Cys dependence; no coupling of proline possible | 53 and 78–80 |
| Thiol-ether-linkage | Michael addition | No additional protecting group; high selectivity; low number of side products; reaction in aqueous solution | Cys dependence | 54 and 81–83 |
| Staudinger | Addition–elimination-reaction | No unspecific reaction with endogenous molecules; remarkable selectivity and compatibility with cells, tissues, and even animals; stable amide bond using traceless staudinger ligation; biorthogonal; easy handling | Interfering triarylphosphaine oxide group using classical Staudinger ligation; slower than other reactions; needs more reagents | 59 and 73 |
| NHS ester | Addition–elimination-reaction | Commercial availability; half-life in the range of hours under physiological conditions; high yields; easy handling | Hydrophobic | 61, 62 and 84–88 |
| IED-DA | [4 + 2]-Cycloaddition | Fastest known biorthogonal transformation; suitable for live animal imaging; small amounts of reagents needed; high signal to noise ratios using fluorescent probes; commercially available | Isomerization of trans-cyclooctene; costly | 66 and 73 |
| Diels Alder | [4 + 2]-Cycloaddition | No naturally occurring compounds; high chemoselectivity; rapid reaction conversion; aqueous environment; incorporation via automated DNA synthesis | cis-Dienes more reactive than trans-dienes, but less conformationally stable | 64 and 73 |
| CuAAC | [3 + 2]-Cycloaddition | Takes place in aqueous environment and forms stable triazoles; fast kinetic; relative simplicity; most accessible reaction to date; commercially available | Not suitable for in vivo experiments due to cytotoxicity of copper(I)-catalyst | 72, 73 and 82 |
| SPAAC | [3 + 2]-Cycloaddition | No need of a catalyst; easy handling; fast kinetics; stable triazole product; non-toxic and hence suitable for in vivo experiments; 10 diverse cyclooctynes for customized application commercially available; applicable for extracellular labelling | Increased hydrophobicity of biomolecule; not applicable for intracellular labelling | 71, 73, 82 and 89 |
| Carbonyl-reactions | Addition–elimination-reaction | Easy chemical access; high stability in biological media; incorporation via automated DNA synthesis; no excess of peptide needed; operational simplicity; good yields; small size of aldehydes/ketones | Not biorthogonal; product hydrolysis in cellular environments; decreasing duplex stability of DNA | 57, 58, 73 and 82 |
The 1,3-dipolar cycloaddition allows the synthesis of a wide range of 5-membered heterocycles. Numerous, highly regioselective reactions as well as enantioselective reactions of prochiral reactants have been described in literature.74–76 In particular, the alkyne-acid cycloaddition for the synthesis of various triazole derivatives has proven to be a useful tool for the simple modification of biomolecules. The problem with these reactions was the uncontrolled formation of 1,4- and 1,5-difunctionalized triazoles. Sharpless et al. have been able to solve this problem via copper-catalyzed alkyne–azide cycloaddition (CuAAC), in which the 1,4-product is selectively formed.69,70 However, due to the cytotoxicity of copper, this method is unsuitable for bioorthogonal reactions. Only with the development of the strain-promoted alkyne–azide cycloaddition (SPAAC) by Bertozzi et al., could a bioorthogonal reaction be established.71 Using this strategy, the high ring tension of cyclooctynes, the only known stable cycloalkynes, was exploited (Fig. 2). As a result, this reaction does not require the addition of a catalyst. For the preparation of protein/peptide-oligonucleotide conjugates, the use of SPAAC coupled with NHS ester chemistry is convenient due to its simplicity, its feasibility in an aqueous environment, and high yields.22,77 Here, an NHS-ester is coupled to a DBCO-unit via a variable linker. By introducing an amino functional group at the 3′or 5′ end of the DNA nanostructure, it can be coupled to the DBCO-NHS-ester via nucleophilic substitution. In the next step, an azide-modified peptide reacts via a SPAAC reaction with the cyclooctyne derivative to form a stable triazole derivative.
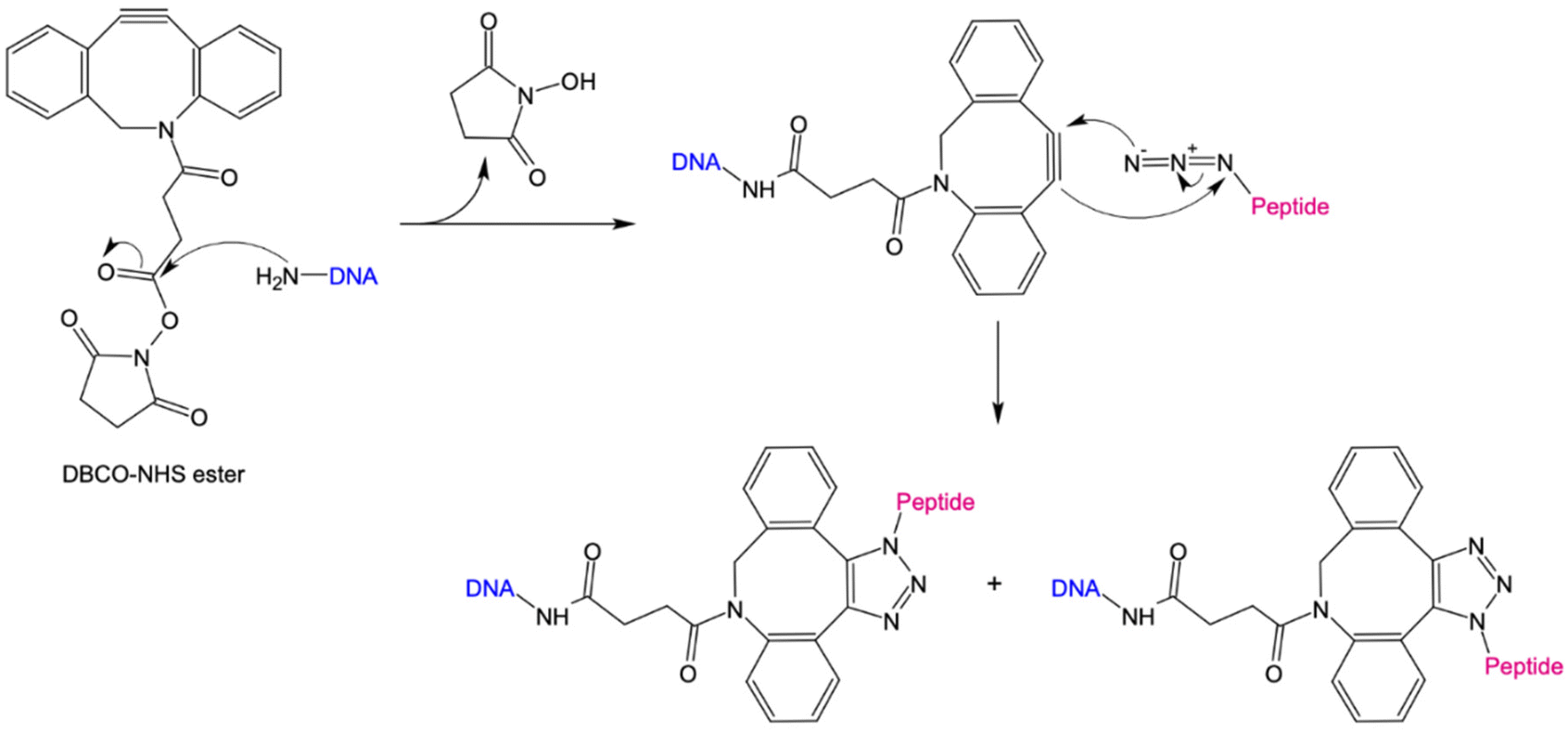 | ||
| Fig. 2 Schematic representation of covalent peptide-DNA conjugate (POC) formation through strain-promoted alkyne–azide cycloaddition (SPAAC). Amine-reactive Dibenzocyclooctyne-N-hydroxysuccinimidylesters (DBCO-NHS) react with primary amine (NH2) groups of DNA. NHS is released and DNA covalently coupled to the strained alkyne. DBCO further reacts with an azide-containing peptide in a [3 + 2] cycloaddition under the formation of 1,4- or 1,5-functionalized triazoles. | ||
3. Electrostatic templating of peptides onto DNA nanostructures
We first give brief attention to several approaches where the anionic, polyelectrolyte nature of DNA strands is exploited as a structural guide for the non-covalent attachment of cationic peptides. Analogous to the art of Papier-mâché, electrostatic attraction between the oppositely charged polyelectrolytes allows the small, and mostly flexible peptide chains to form a coating around the rigid nanostructure template. Studies using this strategy have primarily focused on two distinct classes of peptides: short antimicrobial peptides (AMPs) for disrupting bacteria membranes and long, cationic peptide chains used to provide stability in harsh, typically biological environments.Several studies have used strategies based on electrostatic coupling to attach antimicrobial peptides (AMPs) to different forms of nucleic acid structures, in order to enhance their antimicrobial effects, biological activity, and stability. In many cases, AMPs have an overall positive charge,90 thus facilitating their electrostatic adhesion to negatively-charged DNA nanostructures. As an example, the AMP GL13K has been conjugated to tetrahedral framework nucleic acids (tFNAs), a rigid, three-dimensional DNA structure formed by the hybridization of four oligonucleotides, each with an approximate size of 10 nm.34 Due to their simplicity to assemble and modify, these structures have become ubiquitous within the field of DNA nanotechnology, and will appear several times within this review. These DNA-peptide mixtures formed small polyplexes up to 150 nm in diameter, consisting of several DNA tetrahedron packaged together with the peptide (Fig. 3a). When used as an antimicrobial-delivery vehicle, these DNA-peptide complexes displayed an enhanced antimicrobial effect against Escherichia coli compared to the peptide alone, and furthermore protected the peptide from degradation by Porphyromonas gingivalis.91 Furthermore, using DNA nanostructures as encapsulation tools for AMPs can also facilitate their release in a controlled manner without losing their biological effect. Examples of that were shown in a pair of studies carried out by Obuobi et al., where L12 antimicrobial peptides were loaded into DNA hydrogels92 and cage-like DNA nanogels,93 that delivered superior anti-inflammatory activity against nuclease-releasing susceptible and methicillin-resistant Staphylococcus aureus wound infections.
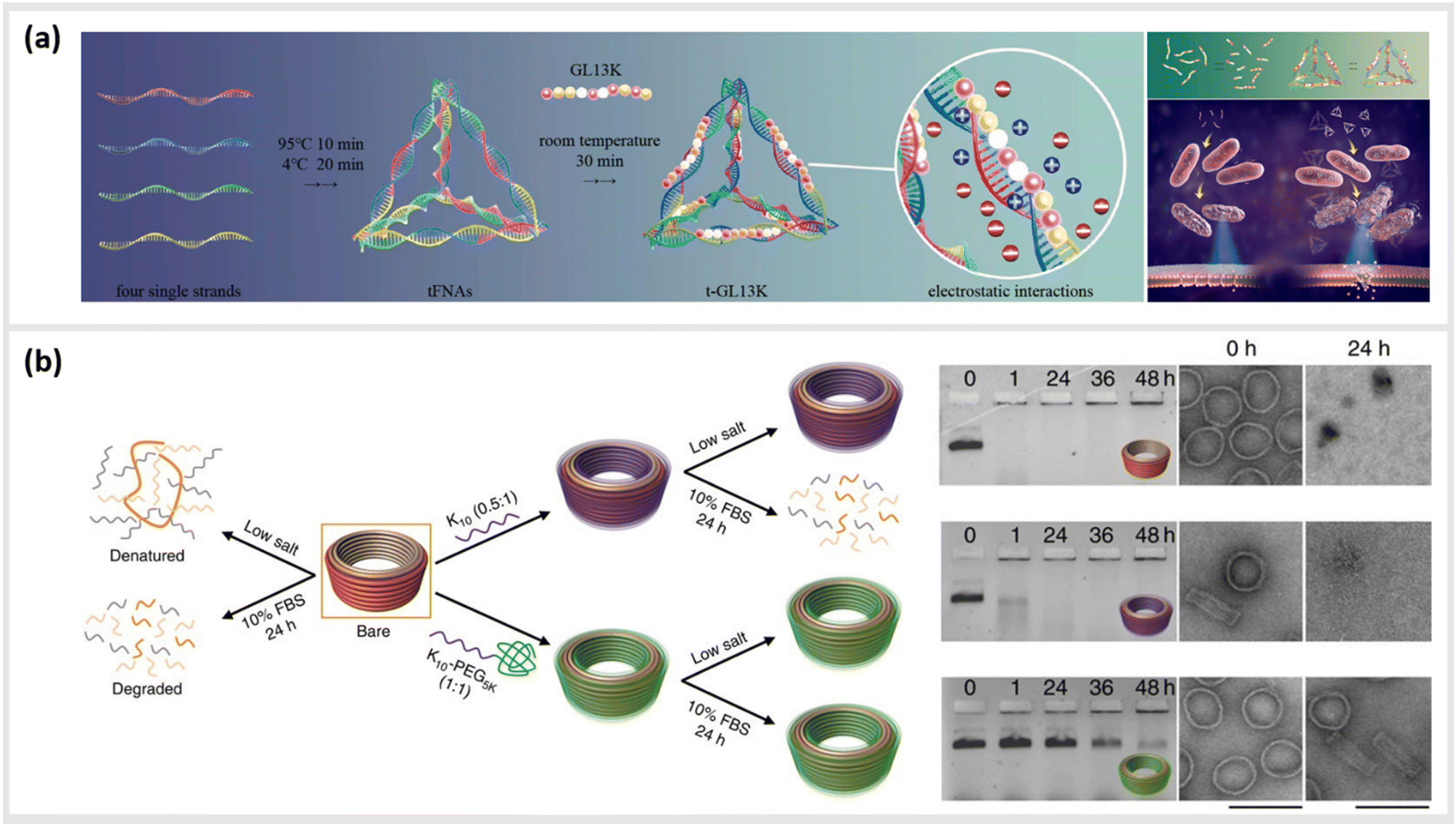 | ||
| Fig. 3 Electrostatic templating of peptides onto DNA nanostructures. (a) Illustration of the generation of tetrahedral framework nucleic acids (tFNAs) and antimicrobial peptide GL13K loaded tFNAs as well as their effect on Escherichia coli (sensitive to GL13K) and Porphyromonas gingivalis (capable of degrading GL13K). The tFNA vehicle increased bacterial uptake, promoted membrane destabilization and enhanced the effects of GL13K against P. gingivalis by protecting the peptide against degradation,91 reprinted with permission from,91 Copyright 2020 American Chemical Society. (b) Schematic representation of the fate of bare (orange) and coated (oligolysine (purple); polyethylene glycol (PEG)-oligolysine (green)) DNA nanostructures in physical buffers at 37 °C containing low salt and/or 10% (v/v) FBS. Coating with oligolysine only modestly protected DNs against nucleases whereas coating with oligolysine conjugated to PEG significantly slowed down nuclease degradation (agarose and TEM images (right)),47 reprinted from Springer Nature, under Creative Commons CC BY license, copyright 2017. | ||
This strategy for the noncovalent, electrostatic association of cationic polypeptides with DNA nanostructures has also been used as means to create a protective corona, in order to ensure stability in biological environments. In particular, coating DNA origami structures with long-chain polymers containing cationic polylysine46,47,94–96 or peptide-like peptoids97 has led to an increased protection against nucleases or low-salt environments.
In the first case, Agrawal et al. used a block co-polymer consisting of a polyethylene glycol (PEG) chain conjugated to a poly-L-lysine segment of either 10 or 18 units in length to create DNA origami polyplex micelles (DOPMs).46 The complexes proved to be resistant against DNAse digestion, stable in low-salt conditions, and retained their ability to be functionalized with additional accessory molecules either by DNA hybridization or protein–ligand interactions at the outer surface. Similarly, Ponnuswamy et al. employed several different cationic polyamines as protective coatings for an assortment of eight different DNA origami structures. They found the most effective protection against denaturation in cation-depleted conditions at 37 °C when oligolysines of different lengths were applied, with a 10-unit polypeptide leading to the best mix of stability and low aggregation (Fig. 3b).47 This stabilizing approach enabled systematic studies into the impact of the DNA origami shape and mass96 as well as different surface properties95 on their uptake into different types of cells. Furthermore, Anastassacos and colleagues were able to enhance the protective effect of the PEG-oligolysine coatings by chemically crosslinking the polymers with glutaraldehyde, increasing the survival time of the underlying DNA structures against enzymatic degradation by an additional factor of 250.94
More recently, Wang et al. took a similar approach using a class of peptidomimetic materials commonly referred to as peptoids,97 which have, amongst other advantages, resistance to degradation by proteases due to their modified backbone structure. The authors synthesized brush- and block-like peptoid structures, which both protected the underlying DNA origami structure from proteolytic hydrolysis by DNAse I. The authors showed that release rate of the anti-cancer drug doxorubicin in solution, which can be heavily impacted by the structure of the underlying DNA origami,98 could also be modulated by the protective peptoid layer.
However, it should be stated that numerous types of protection strategies for DNA nanostructures also go far beyond only peptide-based materials described here, and represent a rich topic unto itself that deserves broader coverage in its own review paper.
4. Templated interactions with biological systems
An increasing number of studies have utilized DNA or other nucleotide-based nanostructures to structurally “program” the direct interactions of peptides with different types of biological entities. Here, the underlying DNA scaffold offers two strategic advantages over alternative nanoparticle approaches. First, multiple peptide and non-peptide moieties for imaging, targeting, therapy or other functions can be associated onto a single structure with molecularly precise stoichiometry. Second, the number and arrangements of bioactive peptides that mediate binding or stimulation can be optimized to the specific target. As will be described in the following, these aspects can be used to localize molecules to specific cell compartments, improve binding kinetics to a specific target, or control complex biological signalling events and responses in cells and organisms.4.1. Cellular penetration and subcellular localization
Due to their inherent biocompatibility, peptides, DNA and their conjugates are ideal candidates for biological and medical applications that involve delivering a therapeutic payload into the interior of a cell, or guiding a molecule to a particular subcellular compartment for direct imaging. Liang et al. used a similar tetrahedral DNA nanostructure (TDN) as described earlier to study cellular uptake and transport pathways within cells.99 By using a click reaction, TDNs were functionalized with peptides that display nuclear localization signals (NLSs).100 Indeed, intact NLS-TDNs could be found in nuclei of HeLa cells. The addressability of DNA nanostructures also enables the direct and highly specific integration of CPPs known to shuttle materials through the membrane of cells into multifunctional nanoparticles. For example, Guo et al. demonstrated that a CPP-decorated DNA nanopore, formed from the hybridization of six oligonucleotides into a barrel-like structure, could both target the surface, and penetrate into the cytoplasm of tumor cells.101 While these structures did not carry any cytotoxic agent, and therefore, left the cells healthy and in-tact, a related study carried out by Xia et al. demonstrated the possibility for the direct delivery of compounds into the cell. In this case, doxorubicin hydrochloride was intercalated into the helix of a double-stranded TDN modified with a tetrahedron-tumor penetrating peptide (TPP) (Fig. 4a).102 This combination was developed to significantly improve the endocytosis of the drug into intracranial human primary glioblastoma cells. The TPP was conjugated to TDN via click chemistry and could be specifically internalized by the cancer cells.102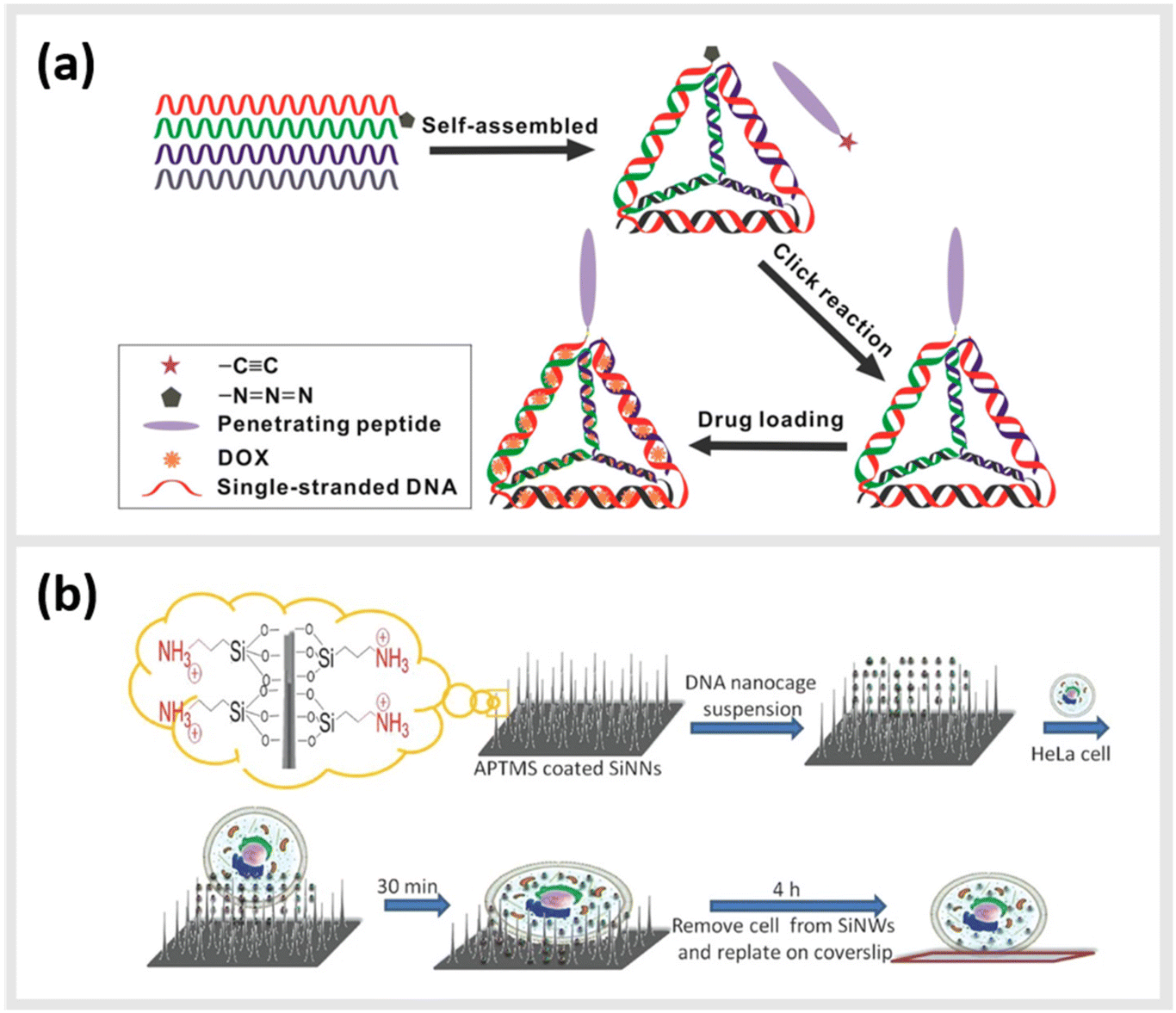 | ||
| Fig. 4 Peptide-conjugated DNA nanostructures for subcellular localization. (a) Schematic representation of self-assembly of tetrahedral DNA nanostructure (TDN) via partially complementary DNA sequences TDN-1 to TDN-4. The TDN was functionalized with the tumor-penetrating peptide (TPP) and subsequently doxorubicin hydrochloride (DOX), a well-known chemotherapy drug, was intercalated into the double-stranded DNA helix (DOX@p-TDN). These drug carriers were tested on glioblastoma U87MG cells regarding their intracellular uptake and their potency to inhibit cell growth,102 reprinted with permission from,102 Copyright 2016 American Chemical Society. (b) Illustration of a nanoneedle-assisted delivery platform which shows the intracellular delivery of DNA nanocages to HeLa cells via silicon nanoneedle array. These peptide-functionalized DNA nanostructures selectively targeted mitochondria and the nucleus by means of nanoneedle-assisted delivery. Additionally, this technology preserved the cell viability and structural integrity of nanostructures and assisted their endosomal escape,103 reprinted with permission from John Wiley and Sons, copyright 2013. | ||
Besides enhanced delivery of drugs to specific target sites, DNA-based constructs can be also used to lower cytotoxicity and increase the stability of functional constructs within cells. A 2013 study from Chan et al. described a method for the delivery of DNA nanostructures through vertical silicon nanowire arrays (Fig. 4b).103 This technique enables the delivery to subcellular organelles through peptides. They developed this technology in order to escape cellular endocytosis processes. The group synthetized DNA nanocages tagged with specific organelle localization peptides (mitochondrial localization signalling peptides or nuclear localization signalling peptides) and transferred them into cells through silicon nanowire arrays.103 In detail, 5′ amino modified DNA oligomers were synthesized on a controlled pore glass by using an automated DNA synthesizer and standard cyanoethylphosphoramidite chemistry. The oligomers were subsequently treated with 1,6-diisocyanatohexane. The peptides at their N-terminus were attached with β-alanine to increase the nucleophilicity of the coupling reactions. All ε-amino groups of lysine residues were protected with a trifluoroacetyl group. The modified DNA oligomers on controlled pore glass were further treated with partially protected peptides. The peptide-modified oligomers were cleaved from the solid support and lysine residues and were deprotected using ammonium hydroxide. These peptide-modified oligomers were added to the DNA nanocages and proved to have a low cytotoxicity and a high stability.
4.2. Optimization of binding kinetics to protein targets
As already described in earlier sections, the ability to precisely arrange ligands such as peptides onto DNA-based structural templates is ideal for exploiting cooperative, multivalent binding to complex targets. In several studies, this has been demonstrated using a variety of different DNA templates ranging in complexity from short dsDNA segments to complex two-dimensional (2D) tiled arrays. In 2009, Williams et al. published a study exploring options for introducing multivalent interaction of small peptide fragments on a synthetic tether or polymer with affinity to yeast regulatory protein Gal80. They were able to show that this yielded a significantly higher (approximately a 1000-fold increase) affinity compared to the individual peptides. The group used a novel DNA linking strategy called CELL (combinatorial examination of ligands and linkers) to determine the optimal distance and strand orientation required to transform two weak affinity ligands to high affinity reagents.104 A similar study carried out in 2011 by Liu et al. demonstrated the method of “ligand interactions by nucleotide conjugates” (LINC) as an attractive strategy for creating so-called DNA “synbodies” (Fig. 5a and b).105 In this study, they chose growth factor receptor-bound protein 2 (Grb2) to create an anti-Grb2 DNA synbody and two peptides that bind to the Grb2 at its SH2 and SH3 domains. They attached one peptide at the 5′-end of the sense strand and the other peptide to a downstream position on the antisense strands. By conducting a series of experiments involving SPR, western blot analysis and ELISA, it could be proved that DNA synbodies bound 5–10-fold stronger than commercially available antibodies.105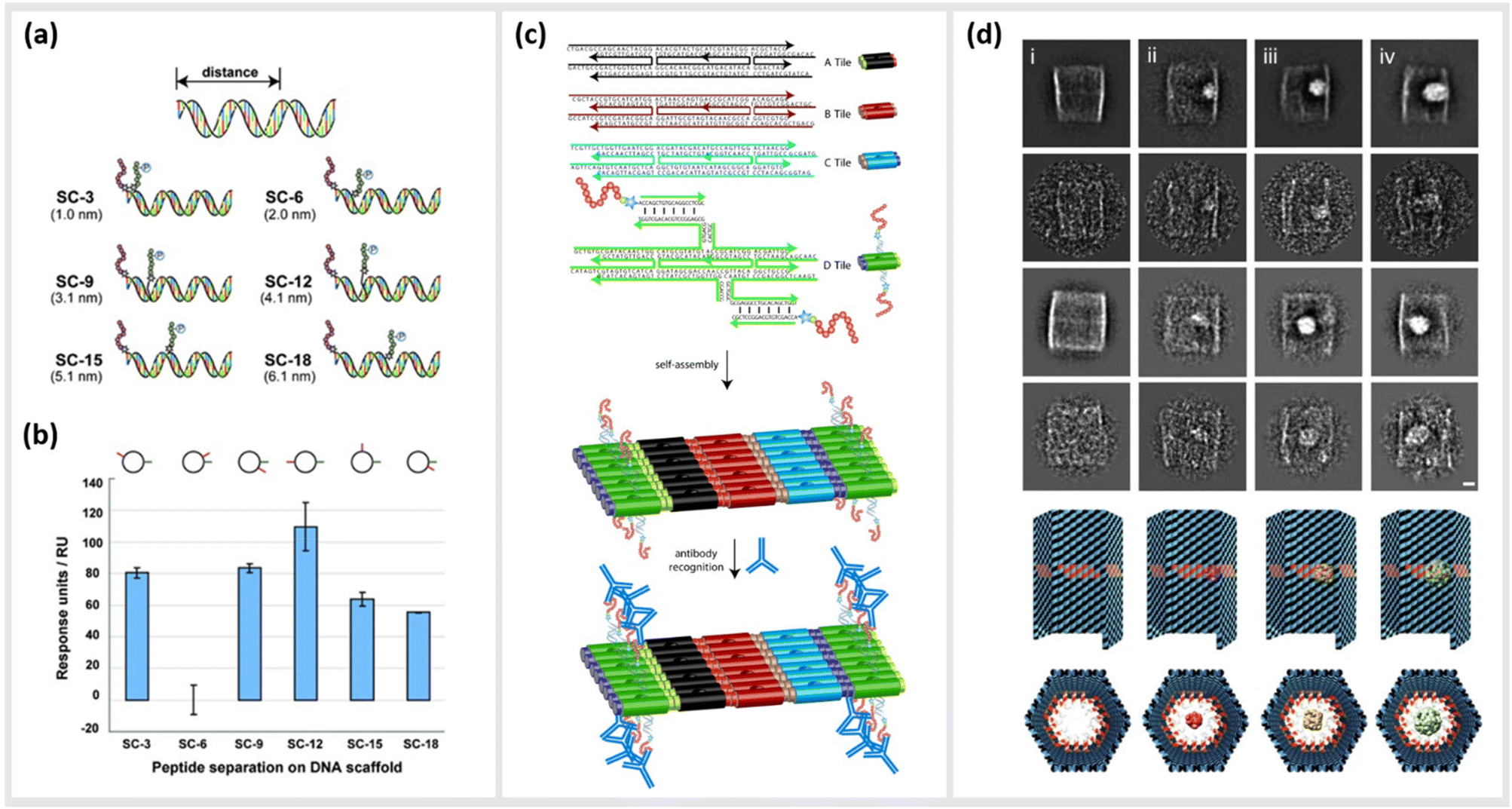 | ||
| Fig. 5 Utilization of DNA nanostructures for the optimization of binding kinetics to protein targets. (a) Cartoon representation illustrating the design of six “synbody” constructs (SCs). SCs were designed to spatially separate two human growth factor receptor-bound protein 2 (Grb2)-binding peptides by 3, 6, 9, 12, 15, and 18 bp (nanometer distance given in parenthesis). (b) The affinity of these synthetic antibodies was further analysed via surface plasmon resonance (SPR),105 reprinted with permission from John Wiley and Sons, copyright 2011. (c) Scheme of self-assembled, high-density peptide arrays that are capable of displaying many different amino acid sequences at well-defined and addressable locations on the same DNA nanostructure. In a first step, DX tiles A–D are preassembled from a set of 22 DNA strands. These tiles further assemble into 2D arrays with the D tile displaying a unique capture strand for hybridization to a complement DNA-peptide conjugate. After hybridization to the DNA nanoarray the conjugated peptide retains its conformation allowing for antibody recognition,106 reprinted with permission from John Wiley and Sons, copyright 2007. (d) DNA nanocontainers were rationally designed to trap single guest molecules in their native form, mimicking natural strategies of molecular recognition and anticipating a new method of protein caging. Two-dimensional negative stain electron microscopy analysis of empty (i), DegP6 (ii), DegP12 (iii) and DegP24 (iv) loaded DNA origami cages bearing 18 convergent protruding arms (Pas) in their cavity. DegP is a serine protease accessible in many interchangeable oligomerization states of known crystal structure, ranging from the 6-mer (DegP6; ca. 250 kDa) to the 12-mer (DegP12; 500 kDa) till the highest 24-mer (DegP24; 1 MDa),107 reprinted (adapted) from Springer Nature, under Creative Commons CC BY license, copyright 2017. | ||
In addition to the precise positioning of bioactive constructs on DNA scaffolds to achieve higher binding affinities or to control multivalent binding, DNA structures can be used to arrange and study target structures with nanometer precision. Williams et al., for instance, reported the use of self-assembling nanoarrays composed of DNA-peptide nanostructures that were used to investigate interactions between proteins. In a method they referred to as “nanodisplay”, they used a 2D layer of DNA with capture strands to serve as a nanometer-scale patterning substrate, which was a major improvement in spatial precision compared to the traditional, micrometer-scale glass microarray (Fig. 5c). This had the additional advantage that multiple molecules, in this case proteins, could be studied in solution and simultaneously could be addressed to a specific location (at near single-molecule level) on the 2D array due to the complementary base pairing of the capture strands. To demonstrate the potential of their platform, they coupled DNA to a myc–epitope peptide and localized it onto DNA tiles, which they subsequently used to capture an antibody against this peptide.106
Another study by Sprengel et al. presented DNA cages and DNA origami nanochannels of various sizes and mechanical rigidities, to allow for ligand- and size-selective encapsulation of proteins within the DNA structure (Fig. 5d). The encapsulation occurred by taking both the chemical affinity and the geometric compatibility of the interacting species into consideration for efficient binding.107 Through modifying the inner cavity of the DNA cage, multiple proteins could be arranged inside to form protein scaffolds, thus modulating local concentration effects and multivalent short-range interactions.
4.3. Templated stimulation of in vitro and in vivo pathways
Moving to more complex biological systems, DNA-based templating of peptides is also a powerful strategy for directly interacting with living cells or other microorganisms, thus influencing their behaviour. The surfaces of cells, bacteria and viruses are covered with glycoprotein receptors, which are responsible for interacting with the surrounding environment, and particularly in the case of cells, serving as a functional gateway for the generation of biochemical signals and responses. Many of these are activated by homo-/heterodimerization,108–111 sub-membrane crosslinking112 or the triggering of a conformational change upon simultaneous binding of ligands in a rigid arrangement of binding pockets.113–115 This makes DNA-based construction, where parameters such as distance, orientational flexibility and ligand number can be precisely controlled, an ideal basis for approaches to “program” the behaviours of cells and organisms with bioactive peptide ligands.An example demonstrating the usage of DNA-peptide constructs in a biological system using a DNA templated approach is found in the work of Möser et al., where up to three peptides were conjugated to a three-armed DNA nanostructure, in order to target and activate the Erythropoietin-producing hepatocellular carcinoma (EphA2) receptor (Fig. 6a and b).22 These receptors were found to be overexpressed in many cancerous cells compared to healthy cells, however can also act as tumor suppressors once their downstream pathway is activated via canonical signalling. This is initiated when an EphA2 homodimer is bivalently stimulated by a natural agonist such as the ephrin protein or, as shown here, synthetic agonist ligand.116 In particular, the peptide SWL is a 12mer agonistic-mimicking peptide that binds specifically to the EphA2 receptor and activates its downstream signalling pathway.117 While having a relatively low efficacy when applied monovalently, a chemically-dimerized version of the peptide was found to exhibit stronger binding to recombinant EphA2 in ELISA assay,118 albeit with low stability in biological conditions. Therefore, a modular, DNA-templated approach in delivering the SWL peptide to PC-3 prostate cancer cells was pursued in order to provide an oligovalent, simultaneous binding of the peptides to the EphA2 receptors,119 which would provide stronger collective interaction than a single peptide interaction. This stronger interaction is due to the trimeric templating allowing for a tripod-like stabilization of the peptides to the EphA2 receptor, thus inducing a minimal cluster. Binding and activation of DNA-peptide constructs showed a remarkable enhancement of signaling activity compared to the peptide alone. It was reported that EphA2 phosphorylation was significantly increased by DNA trimers carrying three SWL peptides compared to monovalent SWL.
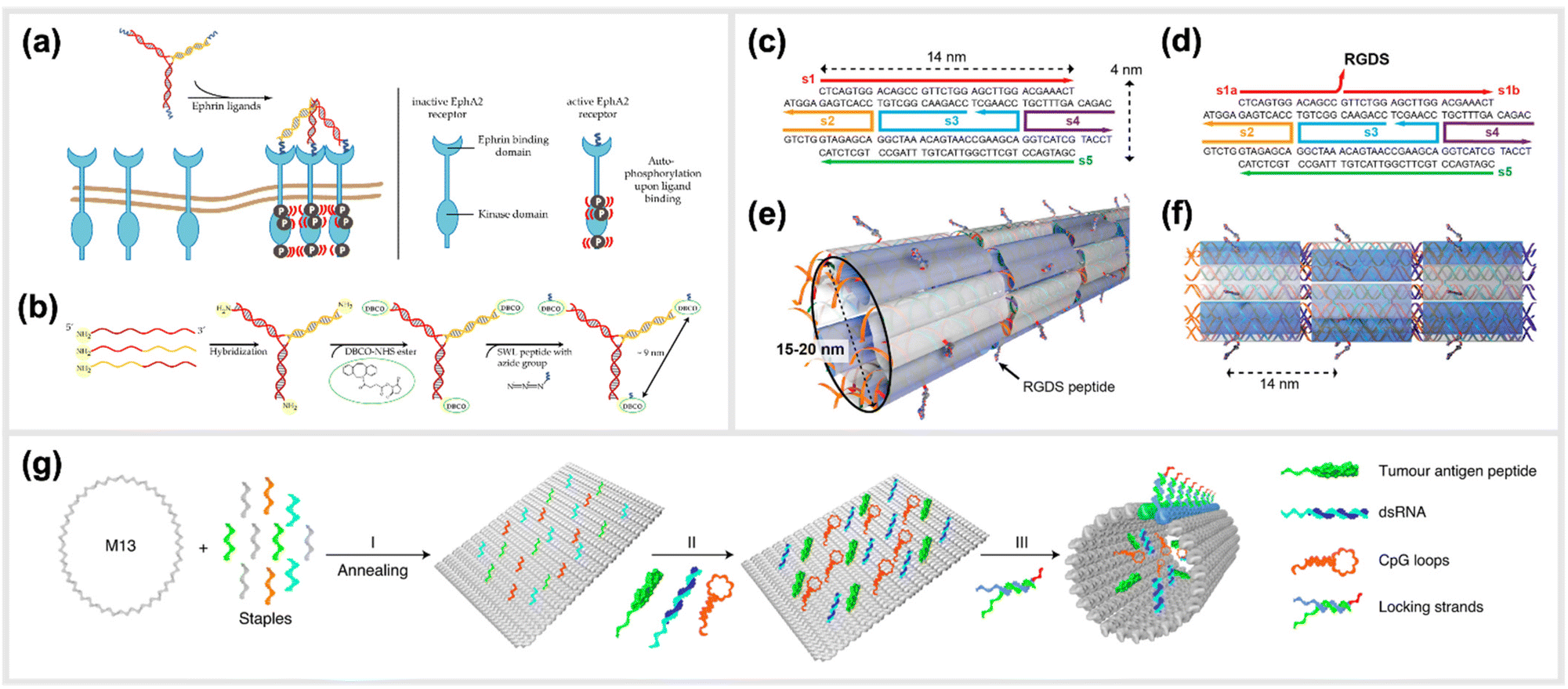 | ||
| Fig. 6 Templated stimulation of in vitro and in vivo pathways. (a) Inactive Ephrin A2 (EphA2) receptors are loosely distributed on cell membranes and only become ordered when activated. Due to the presence of a DNA-trimer coupled to three SWL peptides, EphA2 receptors form clusters and subsequently undergo autophosphorylation which activates downstream signalling. (b) Schematic synthesis of SWL-conjugated DNA trimers. Three partially complementary strands are hybridized to form a DNA trimer. Each primary amine group on the 5′end reacts with DBCO-NHS esters. Azide-containing SWL-peptides further react with the DBCO group under the formation of stable triazoles,22 reprinted from MDPI, Basel, Switzerland, images taken from an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license, copyright 2018. (c–f) Illustration of the design of cell adhesion peptide RGDS containing DNA nanotubes as a bioactive substrate for neural stem cell differentiation,121 reprinted with permission from,121 Copyright 2014 American Chemical Society (https://pubs.acs.org/doi/full/10.1021/nl504079q), further permission related to the material excerpted should be directed to the ACS. (g) Schematic representation of the formation of a structurally well-defined DNA nanodevice vaccine. It is generated by precisely assembling two types of molecular adjuvants and an antigen peptide within the inner cavity of a tubular DNA nanostructure that can be activated in the subcellular environment to trigger T-cell activation and cancer cytotoxicity,28 reprinted from Springer Nature, under Creative Commons CC BY license, copyright 2020. | ||
Using a similar concept, Wang and colleagues later demonstrated that cluster formation of death receptors often overexpressed on the surface of cancer cells could also be induced due to binding of peptide-decorated DNA origami structures. As a demonstration, they generated precise hexagonal arrangements of up to 6 apoptosis-inducing peptides on the surface of a plate-like DNA origami structure, and showed induction of apoptosis in human breast cancer cells,120 with an optimal spacing of approximately 5 nm between peptides needed to attain maximum efficacy.
Another example of an in vivo demonstration of DNA-based peptide presentation, was reported by Stephanopoulos et al. The fibronectin-derived RGDS peptide was conjugated periodically along DNA nanotube structures with a 14 nm spacing, which helped to enhance neural stem cells adhesion to the target site, and subsequent differentiation into neurons (indirectly enhanced neurogenesis) (Fig. 6c–f).121 An RGDS peptide was also used in a publication from Sumida et al.122 Together with the peptide sequence REDV, it was coupled to DNA to function as well-positioned cell-recognition ligand. These peptide-DNA conjugates were specifically attached to the polymer poly(L-lactide) (PLLA), and the cell-specific attachment of different target cells was demonstrated. Additionally, DNA-scaffolded bivalent RGD ligands, displaying two RGD sequences in different spacings, were engineered to explore spatial distribution of integrin α5β1 on the cell surfaces of various cell lines.123
A study carried out by Liu et al. reported the generation and in vivo application of a DNA-structured cancer vaccine. The underlying nanostructures were composed of a DNA nanotube, which contained two types of toll-like receptor agonists (double-stranded RNA (dsRNA) and CpG DNA) along with a tumor antigen peptide (Fig. 6g).28 This DNA tubular structure was locked by pH sensitive DNA strands, which unlock in acidic environments of the endosomes in antigen-presenting cells. Subsequently, the constructs can activate tumor-specific T-cell responses that induced a promising cancer cytotoxicity in mice.28
Several recent studies applied the concept of DNA-templated oligovalent presentation of peptides to bind the surface glycoproteins of viruses, and in one case, the inhibition of virus fusion and entry into host cells. This concept exploited the well-defined multimeric structure of the viral surface proteins responsible for attachment and fusion to targeted structures on the outer membrane of host cells. The strategy used simple, branched Holliday junctions, typically consisting of three DNA oligonucleotides, as structural scaffolds to display virus-binding peptides in a geometrical complement to the arrangement of binding pockets of the targeted virus protein. The peptides were coupled to the DNA strands via NHS ester chemistry and copper-free click chemistry. Two recent publications by Kruse et al. utilized fluorescence proximity sensing, a technology called switchSENSE124 to investigate the binding of peptide-decorated DNA trimeric structures to influenza A virus (IAV)125 and severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2).126 For IAV, the peptide PeB was used, which has been designed to interfere with the function of IAV's Hemagglutinin (HA) protein. It was observed that three PeB peptides linked to DNA structures showed oligovalent binding to different strains of IAV.11 Since the multivalent binding of IAV to PeB-DNA structures on the electrode substrate was very strong, IAV could be immobilized and rate constants of binding interactions to IAV-HA could be calculated.125 For binding SARS-CoV-2, peptides derived from human angiotensin converting enzyme 2 (hACE2) receptor were used.127 Those peptides, named SBP1, were reported to bind to the receptor binding domain (RBD) of the spike protein of SARS-CoV-2. Compared to the conjugation of only one SBP1-peptide per DNA nanostructure, the affinity to recombinantly produced, full-length spike protein homotrimers was significantly enhanced for three SBP1-peptides per nanostructure.126 More recently, Issmail et al. demonstrated that the above-mentioned concept is also applicable for respiratory syncytial viruses (RSV).128 Notably, while the previously described studies focused on binding of the peptide-DNA structures to viral proteins or whole viruses, this study showed that the structures are also capable of acting as an effective RSV entry inhibitor in vitro. On the basis of the RSV-F binding antibody D25, peptides were designed and optimized.129 The most effective peptide candidate was Pep30, a linear sequence containing non-canonical amino acids to increase biostability and efficacy. DNA nanostructures displaying three Pep30 were able to block RSV infection on cells in a concentration-dependent manner with a 214-fold higher potency than the monomeric peptides.
In addition, preliminary data presented in two patent filings substantiated that the in vitro antiviral efficacy of peptides, typically effective in the mid- to high-micromolar range, could be significantly enhanced when they were presented in an oligovalent fashion. Examples of this strategy were shown for blocking the entry of IAV, Dengue virus (DENV)130 and RSV131 using peptides against hemagglutinin (IAV-HA), envelope protein ectodomain III (DENV-ED3)9 and fusion protein (RSV-F), respectively.
5. Generation of bio-hybrid nanomaterials
Besides the apparent applications in biological and medical contexts due to the general biocompatibility of the components, DNA-peptide hybrids can be also used to tune properties of biologically derived or bio-inspired materials. These approaches may involve methods which can be related to biological structures of physiological importance, but also for purely artificial applications. Due to the extraordinary diversity of functions that are possible with even relatively small peptides, the following will cover a broad scope of applications, ranging from approaches to generate bio-hybrid networks and hydrogels, to stimuli-responsive nano-photonic systems. The common, underlying connecting point of these studies is that the defined structure of the DNA-based templates, and some function of the peptides synergistically combine to create entirely new, emergent properties beyond what is possible with either component on its own.5.1. Mechanical modulation of bio-hybrid networks
Two recent studies exploited a similar approach of using simple, linear, dsDNA segments conjugated to peptides as a way to modulate the mechanical properties of hydrogels. One of the approaches is reported by Lorenz et al., which utilized simple DNA-peptide nanostructures as biomimetic constructs to non-covalently crosslink reconstituted cytoskeletal actin networks. Within this study, the construction and impact of synthetic DNA-based actin crosslinkers were described, which revealed different alterations in reconstituted actin networks depending on the peptide used for the DNA-peptide hybrids (Fig. 7).30 Within the study, NHS ester and SPAAC chemistry was used to covalently attach the actin-binding peptides, LifeAct or phalloidin, to both sides of a 20 nm long DNA double-stranded segment. This dsDNA segment acted as a spacer for the binding peptides and effectively connected pairs of actin filaments as a crosslinking complex.30 The binding affinities of the peptides to actin are comparable to the natural crosslinkers α-actinin and fascin, respectively. While LifeAct and α-actinin bind weakly to actin filaments, phalloidin and fascin are strong actin binders. Shear rheology measurements revealed that the artificial crosslinkers indeed induced similar (non-trivial) mechanical fingerprints in actin networks as their natural counterparts did. Furthermore, the DNA spacer between the peptides could be easily cleaved by an EcoRV-HF restriction enzyme implementing an effective switch in the system. Upon cutting the DNA spacer, the crosslinkers were no longer able to physically connect the actin filaments, and thus their crosslinking effects were fully reversible.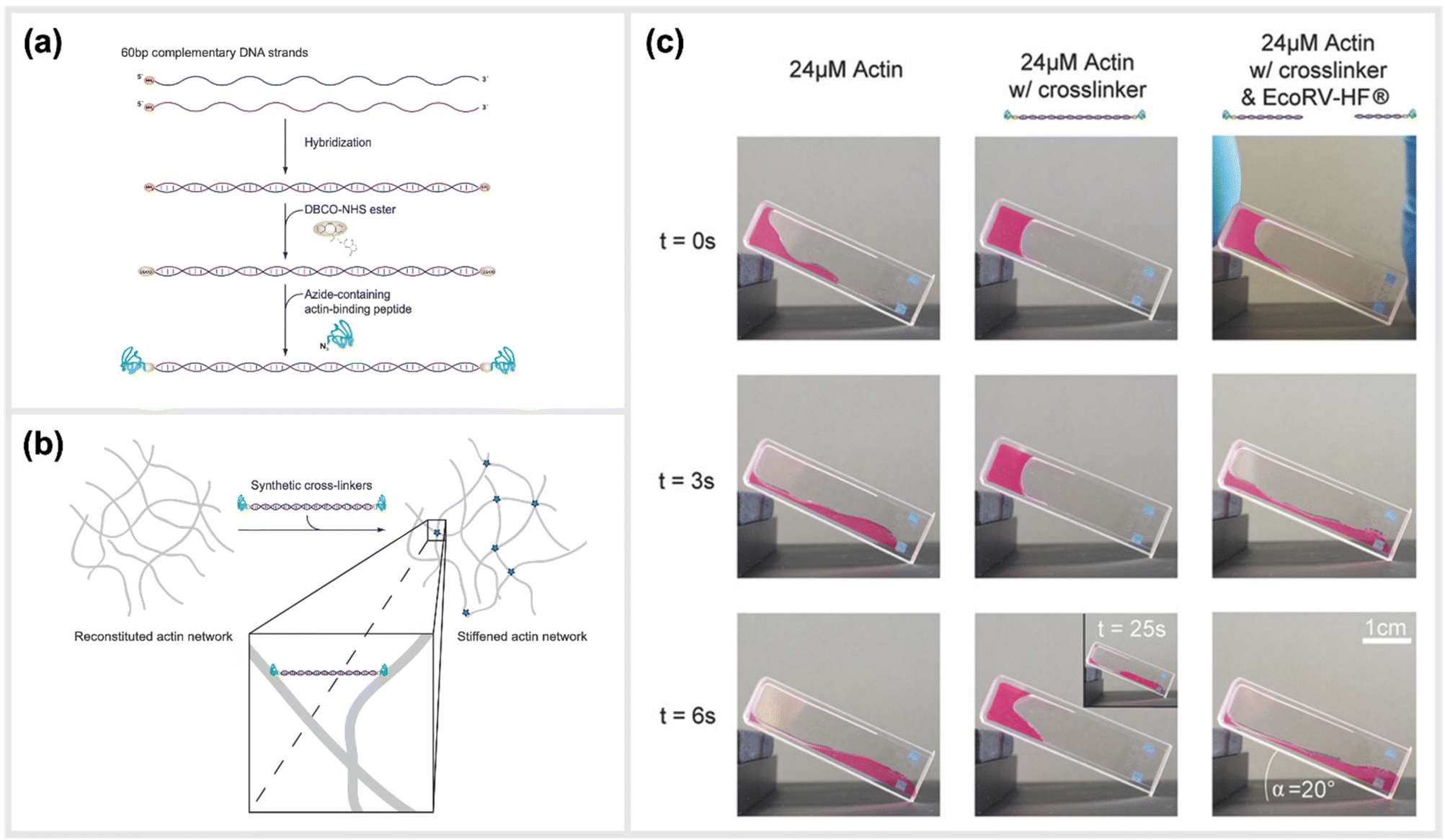 | ||
| Fig. 7 Schematic construction of synthetic actin crosslinkers and their effect on reconstituted in vitro actin networks. (a) Two actin-binding peptides were covalently attached to dsDNA via a copper-free click chemistry approach (SPAAC). In a first step, DBCO-NHS ester covalently reacted with primary amine groups on each end of the double-stranded DNA spacer that was previously hybridized. The strained alkyne subsequently reacted with an azide-containing actin binding peptide in a covalent manner. (b) Schematic representation of the crosslinking of actin filaments within the reconstituted system via the artificial crosslinkers. (c) Macroscopic behaviour of crosslinked actin networks as well as their reversibility. Reconstituted actin was polymerized at 24 μM (left: pure; middle: with artificial crosslinkers; right: with EcoRV-HF cleaved artificial crosslinkers) and observed over time in an inclined cuvette. It was shown that a cleavage of the crosslinkers reversed the stiffening effect and illustrated the possibility to switch between different mechanical states,30 reprinted with permission from John Wiley and Sons, image taken from an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license, copyright 2018. | ||
A related approach of generating stimuli-responsive, crosslinked hydrogels by Deshpande et al. was based on conjugates of DNA with a form of long-chain peptide polymer that is known to coil into so-called β-helices.132 Rather than using a natural biological polymer such as filamentous actin as their underlying support structure, they integrated complementary DNA oligonucleotides into an entirely synthetic network consisting of bundled polyisocyanopeptide (PIP) polymers, which are known to form stable, elastic networks.133 Here, the DNA oligonucleotides were covalently attached to the PIP networks, therefore the crosslinking strength was dictated by interactions (e.g. stable hybridization for the formation of noncanonical structures) between the conjugated strands. By chemically conjugating different ssDNA motifs, such as i-motifs or aptamers, along the PIP backbone with an average spacing of 6 nm between oligonucleotides, they were able to create networks that were responsive to temperature, pH or a particular ligand.
5.2. Peptide-mediated mechanical actuation of DNA-based devices
In some cases, the primary function of a peptide comes not from its interaction with some external target, but rather through how its underlying amino acid sequence leads to specific binding with other identical peptides, or even with itself. This interaction can often be triggered or controlled by conditions such as pH, local ionic strength or temperature, and thus can be exploited as a stimuli-responsive “switch” to control some sort of assembly state, or serve as a kinetic actuator. A pair of recent studies used so-called elastin-like peptides (ELPs), which undergo a conformational change above a certain sequence-specific critical temperature leading hydrophobic-driven aggregation and self-association,134 to control the configuration and optical properties of different DNA origami-based particles.In an initial study conducted by Vogele et al., the authors introduced an active plasmonic waveguide through a solid-phase-based assembly of a single-layered rectangular DNA origami structure used as a molecular circuit board. These DNA structures were assembled with gold nanoparticles (AuNPs) to facilitate the spectroscopic excitations and visualization of transferred energy through two different fluorescent dyes. By including elastin-like peptides that can be thermodynamically regulated by the DNA structures, it was possible to create a temperature-driven nanomechanical transmittance switch for the plasmonic waveguide.135
In line with implementing kinetic switches in systems to enforce changes in configuration, Goetzfried et al. later developed a responsive macromolecular assembly of ELP-functionalized DNA origami rectangles, which can be reconfigured on demand through salt concentration and temperature stimuli, as a way to produce machine-like behaviour in DNA origami structures. The rectangular DNA in this structure serves as the backbone and the ELPs are the side chains. This conformational change of the DNA rectangle was possible through the fully reversible phase transition of elastin-like polypeptides, as ELPs can change from hydrophilic to hydrophobic states depending on the change in their transition temperature (Fig. 8a and b).136 Conclusively, these modified DNA devices can serve as a mechanical framework that could be reconfigured on demand through environmental and external stimuli.
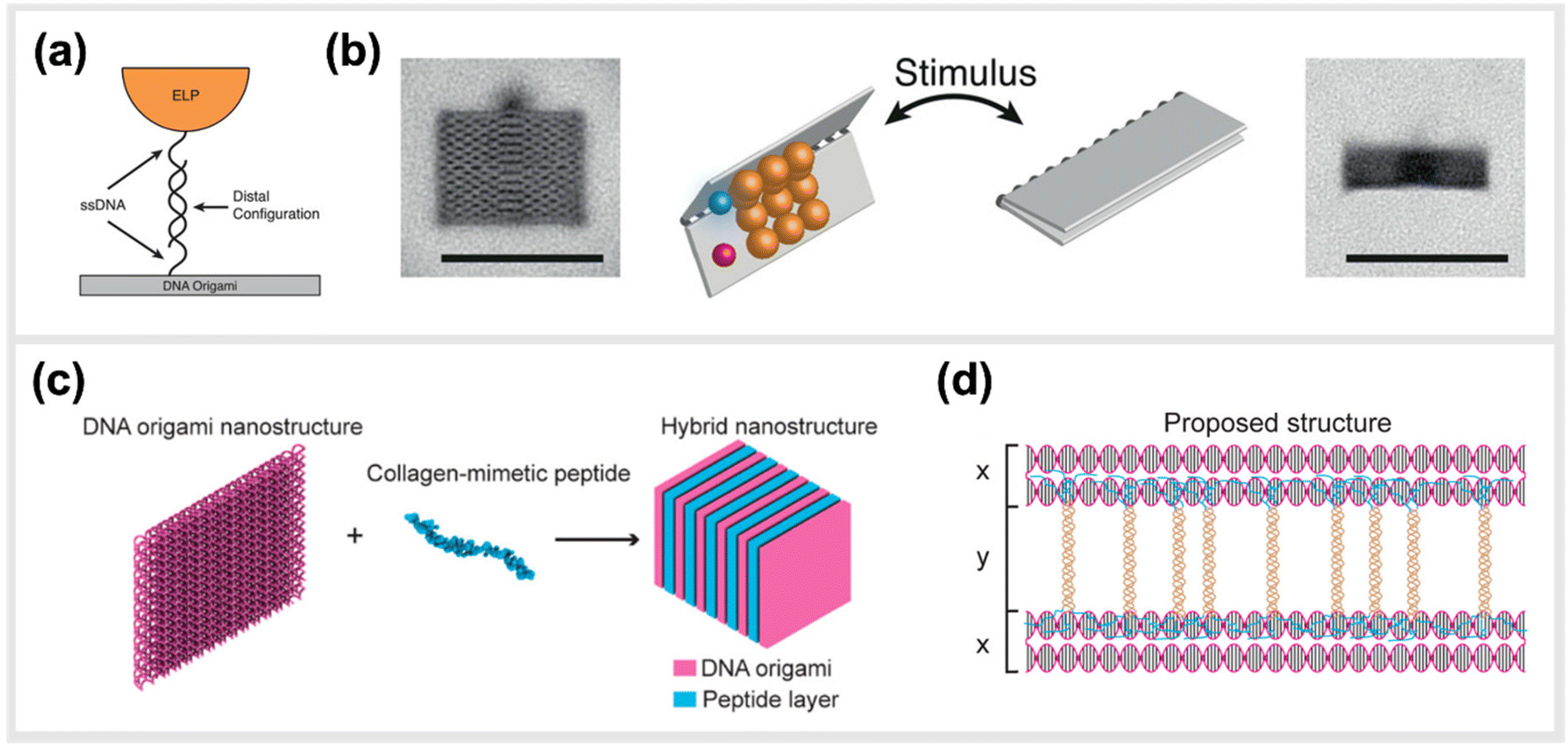 | ||
| Fig. 8 Hybrid material DNA Nanodevices. Hybrid devices utilizing other components than DNA have the potential to considerably expand the library of functionalities. (a) Schematic design of elastin-like peptide functionalized DNA origami sheet. (b) Class-averaged transmission electron microscopy (TEM) images of an open rectangle (left) and a closed rectangle (right) (scale bar: 100 nm). By applying an external stimulus, such as a change in salt concentration, the hydrophilic–hydrophobic phase transition of these peptides actuate the folding of the DNA origami structure,136 reprinted with permission from John Wiley and Sons, image taken from an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license, copyright 2019. (c) Hybrid nanostructure assembled from DNA origami and collagen-like peptides. The co-assembly of two peptides and DNA origami two-layer (TL) nanosheets afforded the formation of one-dimensional nanowires with repeating periodicity of ∼10 nm. (d) Illustration of the proposed co-assembly structure,33 reprinted with permission from ref. 33, Copyright 2017 American Chemical Society. | ||
5.3. Hybrid self-assembly of DNA-peptide suprastructures
Finally, we address several studies focused on the organized self-assembly of large, DNA-peptide suprastructures. Here, the well-defined, rigid, structural support of even simple DNA motifs like short dsDNA segments can be mixed with peptides that have natural self-assembly tendencies, to lead to entirely new phases. In several cases, the relative amounts of the two ingredients, or even the morphological structure of the DNA (e.g., dsDNA plasmids vs. rigid DNA origami of differing shapes) can be used to effectively “tune” the types of assemblies.Chotera et al. investigated the self-assembling properties of DNA-peptide conjugates when mixed with peptides alone.137 They were using this as a proxy to study supramolecular assemblies that often underpin and drive many biological functions. They wanted to understand the environmental as well as stoichiometric effects of gradually mixing these different components, and were able to obtain a variety of structures that varied from long narrow fibrils to complex multi-lamellar spherical structures, depending on the mixing speed and stoichiometric ratios.137 Subsequently, Kye & Lim reported on a multimodal, self-assembling, DNA-peptide system that was intended to mimic a nucleoprotein complex. The reported work demonstrated the creation of a uniform, supramolecular nanostructure that could be formed reliably either through the formation of a β-sheet between the peptides or through the normal base-pairing properties of DNA. They then went on to carry out in vitro testing on HeLa cells, using this nanostructure as a delivery mechanism for antisense oligonucleotides (ASOs), and measured the levels of GFP expression. They found it to have a notable effect, revealing that the hybrid structure could act a reliable gene regulator.138
A study by Buchberger et al. investigated the self-assembly of DNA-peptide nanofibers. These micrometer-long nanofibers consisted of DNA origami structures, which carry DNA handles that are connected to two peptides. Due to the peptides’ tendency to form coiled-coil heterodimers, DNA origami structures could be linked to form supramolecular polymers.139
Another study done by Jiang et al. presented a hybrid molecular co-assembly between collagen-mimetic peptides and DNA origami nanosheets. Through the intrinsic physical and chemical properties of these biomolecules, the assembly between the peptides and the DNA origami produced nanowire formations of DNA nanosheets that stacked every 10 nm, with the peptide aligned on their surfaces (Fig. 8c and d).33
Similarly, a more recent study from Hanke et al. explored the co-self-assembly of DNA origami structures and the human islet amyloid polypeptide (hIAPP) involved in type 2 diabetes mellitus.140 Here, the authors focused on how the formation of typical hIAPP fibrillar aggregates was impacted by the addition of two different architectures of DNA origami structures (a rodlike 6-helix bundle and a thin, triangular sheet) or alternatively polydisperse dsDNA segments. Since the 37-amino-acid peptide is highly positively charged, it has a strong electrostatic interaction with the negatively charged DNA molecules. The co-assembly kinetics, as measured by the evolution of turbidity in the mixtures, and the final structural architecture, imaged by atomic force microscopy (AFM) were found to be dependent upon both the presence of DNA and the shape of the applied DNA nanostructure. Most notably, both DNA origami as well as dsDNA hindered the assembly of large agglomerates seen in hIAPP alone. While the specific shape of the DNA structures did not seem to be a factor in the overall retardation of hIAPP aggregation, differences were seen in the final morphology of the co-assembled structures. For example, the rod-like, 6-helix bundle structures seemed to favour the formation of longer fibril structures compared to the other DNA structures or hIAPP alone. Interestingly, these architectures resembled those previously seen to result from the co-mixture of rod-like DNA nanotubes with small molecules that induce an entropically-based attractive force.141
6. Summary and perspectives
Short peptide fragments, and indeed other minimally-sized, biomolecular ligands, represent a class of molecules that is of great use in basic biological research, development of therapeutic and diagnostic approaches, and for the construction of innovative bio-hybrid materials. When compared to full-sized proteins, these minimalized ligands often suffer some drawbacks in terms of their activity due to the lack of a supporting structural framework that comes from the rest of a protein's typically rigid 3D conformation. This major disadvantage can be alleviated by associating peptides together with some sort of synthetic, nano-sized structural scaffold, thus enhancing their function through cooperative multivalence or the synergetic linking to other functional moieties. Nanoparticles based on the self-assembly of a few or even up to several hundred DNA strands have proven to be ideal scaffolds for presenting precise arrangements small peptides, due to the ability to control both their nanoscale arrangement and absolute number. In this review, we have aimed to present an exhaustive account of promising applications that merge peptide science and DNA nanotechnology, with a heavy focus on strategies for interacting with, and/or controlling the behaviours of biological entities such as protein complexes, cells or viruses.In order for DNA-peptide nanostructures to unleash their great potential as tool in diagnostics and therapeutic applications, researchers have to find out whether these interesting proof-of-concept studies can be transformed into or inspire real-world implementations of these hybrid materials. The translational value of peptides, particularly in medicine, is already established, so the determining factor will be whether the added value resulting from hybrid nanostructures can offset the inevitable increases in production complexity and cost on a commercial scale. Over the last years, DNA-based nanofabrication techniques have begun to make inroads into industrial development, with the notable commercial products on the market being high-precision calibration tools for microscopes, materials and services for other researchers looking to implement complex DNA-based techniques,142 and a platform using simple, linear dsDNA “nanolevers” to analyse binding to conjugated ligands based on effects of either hydrodynamic friction or proximity-quenching of dye molecules.124,143,144 Recent, in-depth analysis has indicated that even complex DNA nanostructures, such as those formed by DNA origami, are likely financially viable for commercial application to cancer therapy.144 This broadly implies that the cost of the DNA scaffolds might not be the limiting factor for applications of DNA-peptide structures, already clearing one of the most significant hurdles for broader entry into relevant diagnostic or therapeutic markets.
Instead, scaling up of production and navigating the relevant regulatory frameworks likely present the most significant challenges for potentially promising technologies to make the jump from robust demonstration to actual product development and implementation. In regard to the latter, the necessary regulatory certifications and resulting process to achieve approval will be defined by the specific legal classification of the prospective product. I.E., a hypothetical therapy based on a complex DNA-peptide nanostructure will face a drastically different landscape and associated challenges than similar molecular components implemented in a diagnostic test or material application.
Rather, the former set of issues, related to scaling up of production and manufacturing, more likely represents a shared challenge to all prospective large-scale, real-world implementations. Currently, and in all of the studies described throughout this review, peptides and oligonucleotides are synthesized separately, then chemically conjugated or otherwise associated with each other through additional synthesis steps. This chemical synthesis is even a separate process from the actual self-assembly of the DNA structural template, which is typically mediated by a thermal denaturation and gradual re-hybridization process that can range from seconds to days, depending on the structural complexity. While this is by no means out of line for small-scale production carried out by a researcher in a typical academic research lab, implementing such processes in robust, error-free, large-scale operations that are developed according to established International Organization for Standardization (ISO) standards and follow Good Manufacturing Practice (GMP) is a substantial challenge. Nevertheless, this points to a range of opportunities for researchers to develop new synthesis, conjugation and assembly strategies, and amalgamations thereof, which would help to meet the inherent challenges associated with upscaling. Therefore, we suggest that an important, if not disruptive leap in the field of broadly applying DNA-peptide nanostructures to different real-world problems will come from the painstaking development of new methods for generating these types of hybrid constructs in continuous, perhaps one-pot reactions.
Author contributions
David M Smith conceptualized, wrote and edited the manuscript. Jessica S. Freitag, Christin Möser and Robel Belay wrote and edited the manuscript, as well as prepared figures. Basma Altattan, Nico Grasse, Bhanu Kiran Pothineni and Jörg Schnauß wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr. Jessica S. Freitag and Dr. Christin Möser contributed equally to this work. This research was funded by the Bundesministerium für Bildung und Forschung (BMBF) (Grant No. 161B0986) and Fraunhofer Gesellschaft (Project No. 600962, 601748 and 600025). The authors would also like to acknowledge the researchers who carried out the work cited in this manuscript.References
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- A. R. Mitchell, Biopolymers, 2008, 90, 175–184 CrossRef CAS PubMed.
- A. K. Sato, M. Viswanathan, R. B. Kent and C. R. Wood, Curr. Opin. Biotechnol., 2006, 17, 638–642 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- S. L. Bellis, Biomaterials, 2011, 32, 4205–4210 CrossRef CAS PubMed.
- Y. Miura, T. Takenaka, K. Toh, S. Wu, H. Nishihara, M. R. Kano, Y. Ino, T. Nomoto, Y. Matsumoto, H. Koyama, H. Cabral, N. Nishiyama and K. Kataoka, ACS Nano, 2013, 7, 8583–8592 CrossRef CAS PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- R. A. Houghten, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5131–5135 CrossRef CAS PubMed.
- M. A. Alhoot, A. K. Rathinam, S. M. Wang, R. Manikam and S. D. Sekaran, Int. J. Med. Sci., 2013, 10, 719–729 CrossRef CAS PubMed.
- G. P. Smith, Science, 1985, 228, 1315–1317 CrossRef CAS PubMed.
- H. Memczak, D. Lauster, P. Kar, S. Di Lella, R. Volkmer, V. Knecht, A. Herrmann, E. Ehrentreich-Förster, F. F. Bier and W. F. M. Stöcklein, PLoS One, 2016, 11(7), e0159074 CrossRef PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- M. Grillaud and A. Bianco, J. Pept. Sci., 2015, 21, 330–345 CrossRef CAS PubMed.
- D. Lauster, M. Glanz, M. Bardua, K. Ludwig, M. Hellmund, U. Hoffmann, A. Hamann, C. Böttcher, R. Haag, C. P. R. Hackenberger and A. Herrmann, Angew. Chem., Int. Ed., 2017, 56, 5931–5936 CrossRef CAS PubMed.
- C. Roma-Rodrigues, A. Heuer-Jungemann, A. Fernandes, A. Kanaras and P. Baptista, Int. J. Nanomed., 2016, 2633 CAS.
- S. Dey, C. Fan, K. V. Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Methods Primers, 2021, 1, 13 CrossRef CAS.
- N. Stephanopoulos, Chem, 2020, 6, 364–405 CAS.
- A. Keller and V. Linko, Angew. Chem., Int. Ed., 2020, 59, 15818–15833 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS PubMed.
- B. Wei, M. Dai and P. Yin, Nature, 2012, 485, 623–626 CrossRef CAS PubMed.
- C. Möser, J. S. Lorenz, M. Sajfutdinow and D. M. Smith, Int. J. Mol. Sci., 2018, 19, 3482 CrossRef PubMed.
- P. S. Kwon, S. Ren, S.-J. Kwon, M. E. Kizer, L. Kuo, M. Xie, D. Zhu, F. Zhou, F. Zhang, D. Kim, K. Fraser, L. D. Kramer, N. C. Seeman, J. S. Dordick, R. J. Linhardt, J. Chao and X. Wang, Nat. Chem., 2020, 12, 26–35 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- A. Shaw, I. T. Hoffecker, I. Smyrlaki, J. Rosa, A. Grevys, D. Bratlie, I. Sandlie, T. E. Michaelsen, J. T. Andersen and B. Högberg, Nat. Nanotechnol., 2019, 14, 184–190 CrossRef CAS PubMed.
- A. Shaw, V. Lundin, E. Petrova, F. Fördős, E. Benson, A. Al-Amin, A. Herland, A. Blokzijl, B. Högberg and A. I. Teixeira, Nat. Methods, 2014, 11, 841–846 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- S. Liu, Q. Jiang, X. Zhao, R. Zhao, Y. Wang, J. Liu, Y. Shang, S. Zhao, T. Wu, Y. Zhang, G. Nie and B. Ding, Nat. Mater., 2021, 20, 421–430 CrossRef CAS PubMed.
- C. Lee, Y. Lee, W. H. Jung, T.-Y. Kim, T. Kim, D.-N. Kim and D. J. Ahn, Sci. Adv., 2022, 8(43), eadd0185 CrossRef CAS PubMed.
- J. S. Lorenz, J. Schnauß, M. Glaser, M. Sajfutdinow, C. Schuldt, J. A. Käs and D. M. Smith, Adv. Mater., 2018, 30, 1706092 CrossRef PubMed.
- A. A. Fuaad, F. Azmi, M. Skwarczynski and I. Toth, Molecules, 2013, 18, 13148–13174 CrossRef PubMed.
- C.-H. Tung and S. Stein, Bioconjugate Chem., 2000, 11, 605–618 CrossRef CAS PubMed.
- T. Jiang, T. A. Meyer, C. Modlin, X. Zuo, V. P. Conticello and Y. Ke, J. Am. Chem. Soc., 2017, 139, 14025–14028 CrossRef CAS PubMed.
- R. P. Goodman, I. A. T. Schaap, C. F. Tardin, C. M. Erben, R. M. Berry, C. F. Schmidt and A. J. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS PubMed.
- T. MacCulloch, A. Buchberger and N. Stephanopoulos, Org. Biomol. Chem., 2019, 17, 1668–1682 RSC.
- J. Winkler, Ther. Delivery, 2013, 4, 791–809 CrossRef CAS PubMed.
- F. Madani, S. Lindberg, Ü. Langel, S. Futaki and A. Gräslund, J. Biophys., 2011, 2011, 1–10 CrossRef PubMed.
- M. Lindgren, M. Hällbrink, A. Prochiantz and Ü. Langel, Trends Pharmacol. Sci., 2000, 21, 99–103 CrossRef CAS PubMed.
- E. L. Snyder and S. F. Dowdy, Pharm. Res., 2004, 21, 389–393 CrossRef CAS PubMed.
- E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- Y. Lin, A. Padmapriya, K. M. Morden and S. D. Jayasena, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 11044–11048 CrossRef CAS PubMed.
- A. Aviñó, A. F. Jorge, C. S. Huertas, T. F. G. G. Cova, A. Pais, L. M. Lechuga, R. Eritja and C. Fabrega, Biochim. Biophys. Acta, Gen. Subj., 2019, 1863, 1619–1630 CrossRef PubMed.
- I. Mela, P. P. Vallejo-Ramirez, S. Makarchuk, G. Christie, D. Bailey, R. M. Henderson, H. Sugiyama, M. Endo and C. F. Kaminski, Angew. Chem., 2020, 132, 12798–12802 CrossRef.
- N. P. Agarwal, M. Matthies, F. N. Gür, K. Osada and T. L. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 5460–5464 CrossRef CAS PubMed.
- N. Ponnuswamy, M. M. C. Bastings, B. Nathwani, J. H. Ryu, L. Y. T. Chou, M. Vinther, W. A. Li, F. M. Anastassacos, D. J. Mooney and W. M. Shih, Nat. Commun., 2017, 8, 15654 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- C. Vénien-Bryan, Z. Li, L. Vuillard and J. A. Boutin, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2017, 73, 174–183 CrossRef PubMed.
- J. Nangreave, H. Yan and Y. Liu, Biophys. J., 2009, 97, 563–571 CrossRef CAS PubMed.
- J. Nangreave, H. Yan and Y. Liu, J. Am. Chem. Soc., 2011, 133, 4490–4497 CrossRef CAS PubMed.
- M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384–6458 CrossRef CAS PubMed.
- F. Diezmann, H. Eberhard and O. Seitz, Biopolymers, 2010, 94, 397–404 CrossRef CAS PubMed.
- N. J. Ede, G. W. Tregear and J. Haralambidis, Bioconjugate Chem., 1994, 5, 373–378 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- T. S. Zatsepin, D. A. Stetsenko, A. A. Arzumanov, E. A. Romanova, M. J. Gait and T. S. Oretskaya, Bioconjugate Chem., 2002, 13, 822–830 CrossRef CAS PubMed.
- D. Forget, O. Renaudet, D. Boturyn, E. Defrancq and P. Dumy, Tetrahedron Lett., 2001, 42, 9171–9174 CrossRef CAS.
- M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825–839 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- S. Mädler, C. Bich, D. Touboul and R. Zenobi, J. Mass Spectrom., 2009, 44, 694–706 CrossRef PubMed.
- E. M. Zubin, E. A. Romanova, E. M. Volkov, V. N. Tashlitsky, G. A. Korshunova, Z. A. Shabarova and T. S. Oretskaya, FEBS Lett., 1999, 456, 59–62 CrossRef CAS PubMed.
- S. Ameta, J. Becker and A. Jäschke, Org. Biomol. Chem., 2014, 12, 4701–4707 RSC.
- K. W. Hill, J. Taunton-Rigby, J. D. Carter, E. Kropp, K. Vagle, W. Pieken, D. P. C. McGee, G. M. Husar, M. Leuck, D. J. Anziano and D. P. Sebesta, J. Org. Chem., 2001, 66, 5352–5358 CrossRef CAS PubMed.
- V. Steven and D. Graham, Org. Biomol. Chem., 2008, 6, 3781 RSC.
- J. Schoch, M. Wiessler and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8846–8847 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- J. Šečkutė, J. Yang and N. K. Devaraj, Nucleic Acids Res., 2013, 41, e148–e148 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC.
- K. Krell, D. Harijan, D. Ganz, L. Doll and H.-A. Wagenknecht, Bioconjugate Chem., 2020, 31, 990–1011 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- F. Zhou, C. Tan, J. Tang, Y.-Y. Zhang, W.-M. Gao, H.-H. Wu, Y.-H. Yu and J. Zhou, J. Am. Chem. Soc., 2013, 135, 10994–10997 CrossRef CAS PubMed.
- L. S. Campbell-Verduyn, W. Szymański, C. P. Postema, R. A. Dierckx, P. H. Elsinga, D. B. Janssen and B. L. Feringa, Chem. Commun., 2010, 46, 898 RSC.
- W. D. G. Brittain, B. R. Buckley and J. S. Fossey, ACS Catal., 2016, 6, 3629–3636 CrossRef CAS.
- H. Gong, I. Holcomb, A. Ooi, X. Wang, D. Majonis, M. A. Unger and R. Ramakrishnan, Bioconjugate Chem., 2016, 27, 217–225 CrossRef CAS PubMed.
- D. A. Stetsenko and M. J. Gait, J. Org. Chem., 2000, 65, 4900–4908 CrossRef CAS PubMed.
- S. Thorand and N. Krause, J. Org. Chem., 1998, 63, 8551–8553 CrossRef CAS.
- S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- S. Soukchareun, J. Haralambidis and G. Tregear, Bioconjugate Chem., 1998, 9, 466–475 CrossRef CAS PubMed.
- K. Lu, Q.-P. Duan, L. Ma and D.-X. Zhao, Bioconjugate Chem., 2010, 21, 187–202 CrossRef CAS PubMed.
- F. T. Liu, M. Zinnecker, T. Hamaoka and D. H. Katz, Biochemistry, 1979, 18, 690–693 CrossRef CAS PubMed.
- G. T. Hermanson, in Bioconjugate Techniques, Elsevier, 2013, pp. 229–258 Search PubMed.
- P. Cuatrecasas and I. Parikh, Biochemistry, 1972, 11, 2291–2299 CrossRef CAS PubMed.
- S. Kalkhof and A. Sinz, Anal. Bioanal. Chem., 2008, 392, 305–312 CrossRef CAS PubMed.
- A. J. Lomant and G. Fairbanks, J. Mol. Biol., 1976, 104, 243–261 CrossRef CAS PubMed.
- J. V. Staros, Acc. Chem. Res., 1988, 21, 435–441 CrossRef CAS.
- P. M. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442–3444 CrossRef CAS PubMed.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- Y. Liu, Y. Sun, S. Li, M. Liu, X. Qin, X. Chen and Y. Lin, Nano Lett., 2020, 20, 3602–3610 CrossRef CAS PubMed.
- S. Obuobi, H. K.-L. Tay, N. D. T. Tram, V. Selvarajan, J. S. Khara, Y. Wang and P. L. R. Ee, J. Controlled Release, 2019, 313, 120–130 CrossRef CAS PubMed.
- S. Obuobi, V. Mayandi, N. A. M. Nor, B. J. Lee, R. Lakshminarayanan and P. L. R. Ee, Nanoscale, 2020, 12, 17411–17425 RSC.
- F. M. Anastassacos, Z. Zhao, Y. Zeng and W. M. Shih, J. Am. Chem. Soc., 2020, 142, 3311–3315 CrossRef CAS PubMed.
- M. M. Koga, A. Comberlato, H. J. Rodríguez-Franco and M. M. Bastings, Biomacromolecules, 2022, 23, 2586–2594 CrossRef CAS PubMed.
- M. M. Bastings, F. M. Anastassacos, N. Ponnuswamy, F. G. Leifer, G. Cuneo, C. Lin, D. E. Ingber, J. H. Ryu and W. M. Shih, Nano Lett., 2018, 18, 3557–3564 CrossRef CAS PubMed.
- S.-T. Wang, M. A. Gray, S. Xuan, Y. Lin, J. Byrnes, A. I. Nguyen, N. Todorova, M. M. Stevens, C. R. Bertozzi, R. N. Zuckermann and O. Gang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6339–6348 CrossRef CAS PubMed.
- H. Ijäs, B. Shen, A. Heuer-Jungemann, A. Keller, M. A. Kostiainen, T. Liedl, J. A. Ihalainen and V. Linko, Nucleic Acids Res., 2021, 49, 3048–3062 CrossRef PubMed.
- L. Liang, J. Li, Q. Li, Q. Huang, J. Shi, H. Yan and C. Fan, Angew. Chem., Int. Ed., 2014, 53, 7745–7750 CrossRef CAS PubMed.
- M. A. Zanta, P. Belguise-Valladier and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 91–96 CrossRef CAS PubMed.
- X.-L. Guo, D.-D. Yuan, T. Song and X.-M. Li, Anal. Bioanal. Chem., 2017, 409, 3789–3797 CrossRef CAS PubMed.
- Z. Xia, P. Wang, X. Liu, T. Liu, Y. Yan, J. Yan, J. Zhong, G. Sun and D. He, Biochemistry, 2016, 55, 1326–1331 CrossRef CAS PubMed.
- M. S. Chan and P. K. Lo, Small, 2014, 10, 1255–1260 CrossRef CAS PubMed.
- B. A. R. Williams, C. W. Diehnelt, P. Belcher, M. Greving, N. W. Woodbury, S. A. Johnston and J. C. Chaput, J. Am. Chem. Soc., 2009, 131, 17233–17241 CrossRef CAS PubMed.
- R. Liu, B. Jiang, H. Yu and J. C. Chaput, ChemBioChem, 2011, 12, 1813–1817 CrossRef CAS PubMed.
- B. A. R. Williams, K. Lund, Y. Liu, H. Yan and J. C. Chaput, Angew. Chem., Int. Ed., 2007, 46, 3051–3054 CrossRef CAS PubMed.
- A. Sprengel, P. Lill, P. Stegemann, K. Bravo-Rodriguez, E.-C. Schöneweiß, M. Merdanovic, D. Gudnason, M. Aznauryan, L. Gamrad, S. Barcikowski, E. Sanchez-Garcia, V. Birkedal, C. Gatsogiannis, M. Ehrmann and B. Saccà, Nat. Commun., 2017, 8, 14472 CrossRef CAS PubMed.
- M. M. Davis and P. J. Bjorkman, Nature, 1988, 334, 395–402 CrossRef CAS PubMed.
- J. Schlessinger, Cell, 2002, 110, 669–672 CrossRef CAS PubMed.
- A. Weiss and J. Schlessinger, Cell, 1998, 94, 277–280 CrossRef CAS PubMed.
- A. N. Plotnikov, J. Schlessinger, S. R. Hubbard and M. Mohammadi, Cell, 1999, 98, 641–650 CrossRef CAS PubMed.
- M. R. Gold, L. Matsuuchi, R. B. Kelly and A. L. DeFranco, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3436–3440 CrossRef CAS PubMed.
- A. Ashkenazi, Nat. Rev. Cancer, 2002, 2, 420–430 CrossRef CAS PubMed.
- M. Kielian, Annu. Rev. Virol., 2014, 1, 171–189 CrossRef PubMed.
- M. Kielian and F. A. Rey, Nat. Rev. Microbiol., 2006, 4, 67–76 CrossRef CAS PubMed.
- D. Jackson, J. Gooya, S. Mao, K. Kinneer, L. Xu, M. Camara, C. Fazenbaker, R. Fleming, S. Swamynathan, D. Meyer, P. D. Senter, C. Gao, H. Wu, M. Kinch, S. Coats, P. A. Kiener and D. A. Tice, Cancer Res., 2008, 68, 9367–9374 CrossRef CAS PubMed.
- M. Koolpe, M. Dail and E. B. Pasquale, J. Biol. Chem., 2002, 277, 46974–46979 CrossRef CAS PubMed.
- S. Duggineni, S. Mitra, I. Lamberto, X. Han, Y. Xu, J. An, E. B. Pasquale and Z. Huang, ACS Med. Chem. Lett., 2013, 4, 344–348 CrossRef CAS PubMed.
- J. W. Astin, J. Batson, S. Kadir, J. Charlet, R. A. Persad, D. Gillatt, J. D. Oxley and C. D. Nobes, Nat. Cell Biol., 2010, 12, 1194–1204 CrossRef CAS PubMed.
- Y. Wang, I. Baars, F. Fördös and B. Högberg, ACS Nano, 2021, 15, 9614–9626 CrossRef CAS PubMed.
- N. Stephanopoulos, R. Freeman, H. A. North, S. Sur, S. J. Jeong, F. Tantakitti, J. A. Kessler and S. I. Stupp, Nano Lett., 2015, 15, 603–609 CrossRef CAS PubMed.
- H. Sumida, Y. Yoshizaki, A. Kuzuya and Y. Ohya, Biomacromolecules, 2020, 21, 3713–3723 CrossRef CAS PubMed.
- E. E. Kurisinkal, V. Caroprese, M. M. Koga, D. Morzy and M. M. C. Bastings, Molecules, 2022, 27(15), 4968 CrossRef CAS PubMed.
- U. Rant, K. Arinaga, S. Scherer, E. Pringsheim, S. Fujita, N. Yokoyama, M. Tornow and G. Abstreiter, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17364–17369 CrossRef CAS PubMed.
- M. Kruse, C. Möser, D. M. Smith, H. Müller-Landau, U. Rant, R. Hölzel and F. F. Bier, Adv. Mater. Technol., 2022, 7, 2101141 CrossRef CAS.
- M. Kruse, B. Altattan, E.-M. Laux, N. Grasse, L. Heinig, C. Möser, D. M. Smith and R. Hölzel, Sci. Rep., 2022, 12, 12828 CrossRef CAS PubMed.
- G. Zhang, S. Pomplun, A. R. Loftis, X. Tan, A. Loas and B. L. Pentelute, Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD, 2020 Search PubMed.
- L. Issmail, C. Möser, C. Jäger, B. Altattan, D. Ramsbeck, M. Kleinschmidt, M. Buchholz, D. Smith and T. Grunwald, Front. Virol., 2022, 2, 90 Search PubMed.
- J. S. McLellan, M. Chen, S. Leung, K. W. Graepel, X. Du, Y. Yang, T. Zhou, U. Baxa, E. Yasuda, T. Beaumont, A. Kumar, K. Modjarrad, Z. Zheng, M. Zhao, N. Xia, P. D. Kwong and B. S. Graham, Science, 2013, 340, 1113–1117 CrossRef CAS PubMed.
- D. M. Smith, J. S. Lorenz, C. Möser, J. Fertey, W. Stöcklein, A. Herrmann and D. Lauster, WO2018215660A1, 2018.
- D. M. Smith, C. Möser, T. Grunwald, L. Issmail, C. Jäger, M. Kleinschmidt, D. Ramsbeck and M. Buchholz, WO2020212576A1, 2020.
- S. R. Deshpande, R. Hammink, F. H. T. Nelissen, A. E. Rowan and H. A. Heus, Biomacromolecules, 2017, 18, 3310–3317 CrossRef CAS PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- A. Valiaev, D. W. Lim, S. Schmidler, R. L. Clark, A. Chilkoti and S. Zauscher, J. Am. Chem. Soc., 2008, 130, 10939–10946 CrossRef CAS PubMed.
- K. Vogele, J. List, G. Pardatscher, N. B. Holland, F. C. Simmel and T. Pirzer, ACS Nano, 2016, 10, 11377–11384 CrossRef CAS PubMed.
- M. A. Goetzfried, K. Vogele, A. Mückl, M. Kaiser, N. B. Holland, F. C. Simmel and T. Pirzer, Small, 2019, 15, 1903541 CrossRef CAS PubMed.
- A. Chotera, H. Sadihov, R. Cohen-Luria, P.-A. Monnard and G. Ashkenasy, Chem. – Eur. J., 2018, 24, 10128–10135 CrossRef CAS PubMed.
- M. Kye and Y.-B. Lim, Angew. Chem., Int. Ed., 2016, 55, 12003–12007 CrossRef CAS PubMed.
- A. Buchberger, C. R. Simmons, N. E. Fahmi, R. Freeman and N. Stephanopoulos, J. Am. Chem. Soc., 2020, 142, 1406–1416 CrossRef CAS PubMed.
- M. Hanke, A. G. Orive, G. Grundmeier and A. Keller, Nanomaterials, 2020, 10, 2200 CrossRef CAS PubMed.
- M. Glaser, J. Schnauß, T. Tschirner, B. U. S. Schmidt, M. Moebius-Winkler, J. A. Käs and D. M. Smith, New J. Phys., 2016, 18, 55001 CrossRef.
- K. E. Dunn, Molecules, 2020, 25, 377 CrossRef CAS PubMed.
- J. Knezevic, A. Langer, P. A. Hampel, W. Kaiser, R. Strasser and U. Rant, J. Am. Chem. Soc., 2012, 134, 15225–15228 CrossRef CAS PubMed.
- E. L. Coleridge and K. E. Dunn, Biomed. Phys. Eng. Express, 2020, 6, 65030 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |