
An interrupted Heyns rearrangement approach for the regioselective synthesis of acylindoles†
Minakshi
Altia
and
Pazhamalai
Anbarasan
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai–600036, India. E-mail: anbarasansp@iitm.ac.in; Web: https://home.iitm.ac.in/anbarasansp
First published on 24th October 2023
Abstract
An efficient and general method for the synthesis of 2- and 3-acylindoles has been achieved with high regioselectivity from o-acylanilines and α-hydroxycarbonyl or its equivalent. The strategy involves the intramolecular trapping of an in situ generated aminoenol intermediate and an interrupted Heyns rearrangement pathway, followed by aromatization or rearrangement/aromatization. Important features include excellent regiocontrol, good functional group tolerance, operational simplicity and application to gram-scale synthesis and the synthesis of an anti-tumor agent.
Indoles are some of the frequently encountered N-heterocycles in natural products, pharmaceuticals, and agrochemicals, as well as in materials science.1 Among them, 2-acyl indoles and 3-acyl indoles are of particular importance because of their presence as core moieties in many drug molecules with diverse pharmacological effects (Fig. 1).2 Due to their high importance, the synthesis of functionalized indoles has been of long-lasting interest in organic synthesis3 and a number of metal catalyzed synthetic methods for indoles4 have been developed in contemporary organic synthesis research. However, methods known for the construction of 2- or 3-acylindole are scarce.
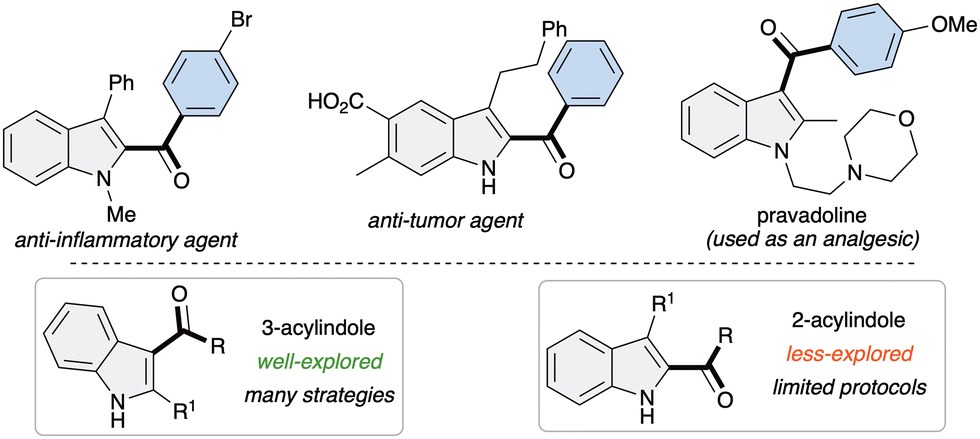 | ||
| Fig. 1 Representative examples of bioactive 2 and 3-acylindoles. | ||
The synthesis of 3-acylindole derivatives5 is relatively well studied compared to the synthesis of 2-acylindoles,6 possibly due to the inherent reactivity of the indole moiety. However, most of the known methods suffer from poor regioselectivity, harsh conditions, side reactions and the need for precious metal catalysts as well as the use of pre-functionalized indoles as precursors, which limit their synthetic applicability. Therefore, the development of a unified, regioselective, metal-free and complementary strategy to access both 2- and 3-acylindoles remains highly desirable.
The Heyns rearrangement is an important and renowned reaction in carbohydrate chemistry and the key reaction in the biosynthesis of D-glucosamine-6-phosphate from fructose and ammonia.7 This rearrangement involves the initial reaction of α-hydroxycarbonyls with amines to form aminoenol intermediates, which upon tautomerism afford α-aminocarbonyls, called Heyns adducts (Scheme 1). Although the Heyns rearrangement is well known in carbohydrate chemistry, its application in organic synthesis is rather limited.
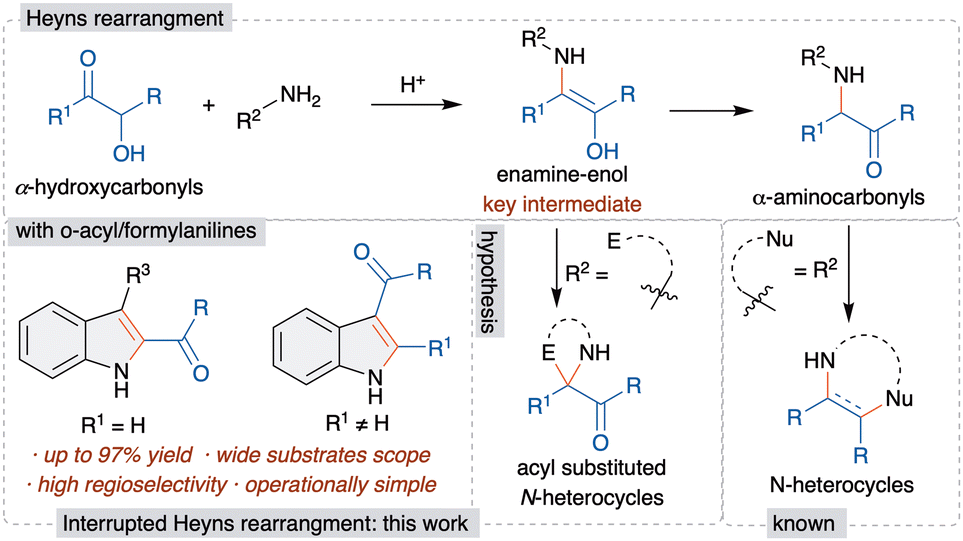 | ||
| Scheme 1 Heyns and interrupted Heyns rearrangements. | ||
The known transformations involve the one-pot functionalization of the formed Heyns adduct, derived from a nucleophile tethered amine derivative, for the synthesis of various nitrogen based heterocycles (Scheme 1).8 However, these methods are limited to the functionalization of the Heyns adduct and offer poor regioselectivity for the unsymmetrically substituted α-hydroxycarbonyls. Importantly, the in situ trapping of the aminoenol intermediate with a suitable electrophile, by way of interrupted Heyns rearrangement, is scarcely studied. The only known method has been demonstrated most recently by Ghorai et al. utilizing an enone as an electrophile and highly reactive α-hydroxycyclobutanone for the synthesis of azaspirocyclobutanones.9 Thus, the development of a general and efficient interrupted Heyns rearrangement is highly warranted. The successful development of this method would offer novel interrupted Heyns rearrangement and will open a new avenue for the functionalization of the Heyns intermediate. Thus, we herein disclose the regioselective interrupted Heyns rearrangement for the unified synthesis of various substituted 2- and 3-acylindole derivatives (Scheme 1).
To test our hypothesis, α-hydroxydimethylacetal 1a, an equivalent of α-hydroxyaldehyde, and o-aminoacetophenone 2a were chosen as model substrates. Initially, 1a and 2a mixed at an equimolar ratio were treated with 1 N HCl in ethanol at room temperature. As postulated, the formation of 2-benzoylindole derivative 3a was observed in 34% yield (Table 1, entry 1). The formation of 3a proves the potential intramolecular trapping of aminoenol with the carbonyl motif, a successful interrupted Heyns rearrangement. Having identified the formation of 3a, conditions were optimized to improve the yield of 3a. Reaction in the absence of acid did not afford 3a, which demonstrates the importance of acid for this reaction (Table 1, entry 2).
|
|
||||
|---|---|---|---|---|
| Entry | Acid | Temp (°C) | Solvent | Yielda (%) |
| Reaction conditions: 1a (0.23 mmol, 1 equiv.), 2a (0.23 mmol, 1 equiv.), acid (equiv.), solvent (3 mL for 0.23 mmol), temp, 18 h.a All are isolated yields.b 1a and 2a were recovered.c 1a was decomposed and 2a was recovered.d 80 °C.e 120 °C. | ||||
| 1 | 1N HCl (2) | rt | EtOH | 34 |
| 2 | — | rt | EtOH | —b |
| 3 | 0.5N H2SO4 (2) | rt | EtOH | —c |
| 4 | AcOH (1) | rt | EtOH | —c |
| 5 | PTSA (1) | rt | EtOH | —c (60)d |
| 6 | 1N HCl (2) | 60 | EtOH | 50 |
| 7 | 1N HCl (2) | 90 | EtOH | 84 (83)d |
| 8 | 1N HCl (2) | 90 | Dioxane | 90 |
| 9 | 1N HCl (2) | 90 | CH3CN | 87 |
| 10 | 1N HCl (2) | 90 | H2O | 66 |
| 11 | 1N HCl (2) | 100 | Dioxane | 94 (93)e |
Subsequently, various acids were examined to understand their influence in the reaction. Most of the Brønsted acids examined showed inferior results (Table 1, entries 3–5). Next, using 1 N HCl as the promoter, critical parameters were varied to improve the yield of 3a. Increasing the temperature to 60 °C improved the yield of 3a to 50% (Table 1, entry 6). Further increasing the temperature to 80 and 90 °C furnished 3a in ∼83% yield (Table 1, entry 7). The use of dioxane and acetonitrile resulted in a slight improvement in the yield, but the reaction in water furnished 3a in 66% yield (Table 1, entries 9 and 10). Finally, the use of dioxane at 100 °C gave 3a in 94% yield (Table 1, entry 11). Thus, 1a (1 equiv.), 2a (1 equiv.), 1 N HCl, dioxane, and 100 °C for 18 h were chosen as the best optimized conditions for further studies.
Having the suitable optimized conditions in hand, we next moved on to study the scope and limitations with respect to o-acylaniline derivatives. Gratifyingly, all of the examined o-acylaniline derivatives led to the formation of 2-acylindole derivatives in good to excellent yield (Scheme 2). For instance, the reaction of alkylketone derived o-acylaniline furnished products 3a, 3b and 3d in >85% yield. The synthesis of 3c was achieved in 72% yield from o-aminobenzaldehyde.
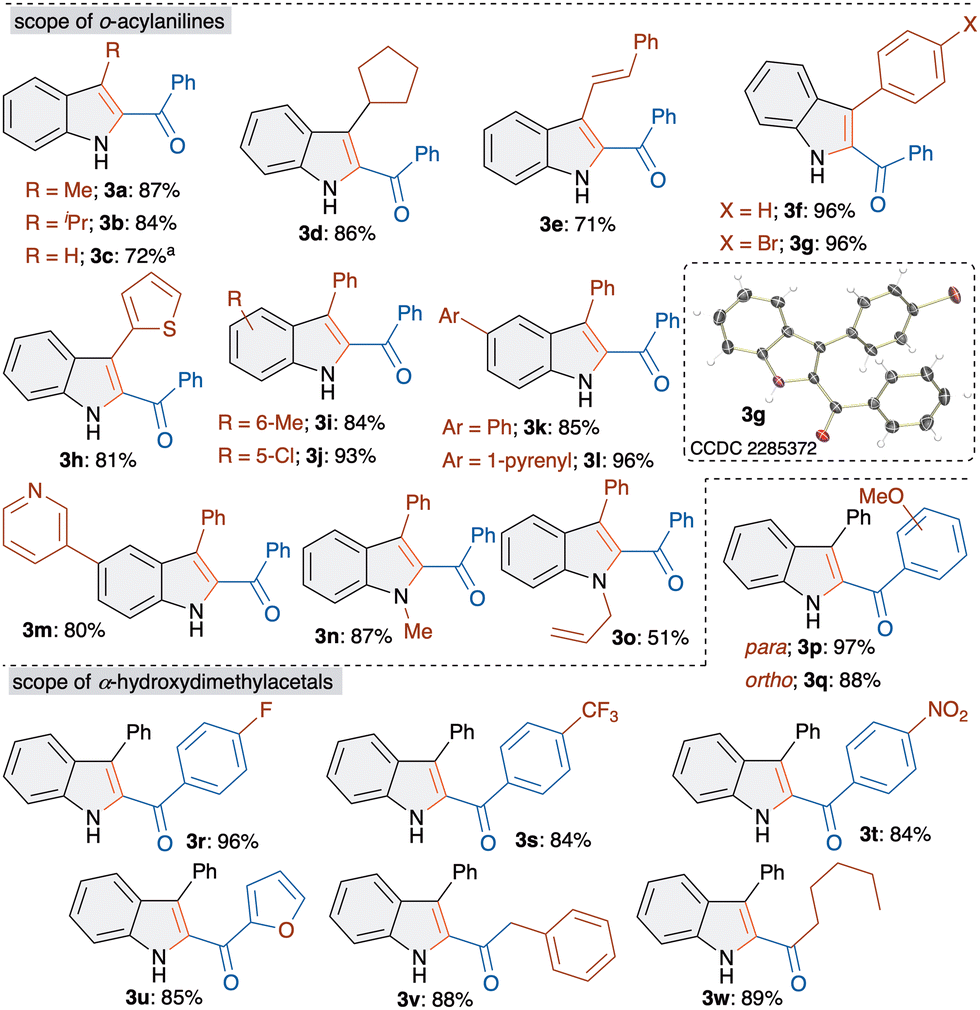 | ||
Scheme 2 Scope of o-acylaniline derivatives. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) In CH3CN at 90 °C. In CH3CN at 90 °C. | ||
Interestingly, vinyl ketone also gave 2-acylindoles 3e in 71% yield. Substituted aryl and heteroaryl ketone derived o-acylanilines led to the formation of 3f–3h in excellent yield. The formation and structure of 3g was unambiguously confirmed by single crystal X-ray analysis. Next, methyl- and chloro-substituted o-acylanilines were treated with 1a to afford 3i and 3j in 84 and 93% yields, respectively. Similarly, phenyl, and pyrenyl substituted 2-acylindoles 3k and 3l were achieved in good yields. Importantly, heteroaryl, pyridyl containing 2-acylindoles 3m were synthesized in 80% yield. Besides, N-methyl and allyl substituted aniline derivatives were also successfully converted to indole derivatives 3n and 3o.
Consequently, diverse substituted α-hydroxydimethylacetal derivatives 1 were examined under optimized conditions with o-aminobenzophenone. α-Hydroxydimethylacetal derived from methoxy- and fluoro-substituted acetophenone afforded products 3p–3r in excellent yields. Electron withdrawing CF3- and nitro-substituted 2-benzoylindoles 3s and 3t were achieved in 84% yield. The replacement of phenyl with furan, a heteroaryl moiety, also gave 3u in 85% yield. Furthermore, α-hydroxydimethylacetal derived from an aliphatic aldehyde underwent a smooth reaction to furnish 3v and 3w in >85% yield.
Having successfully studied the α-hydroxyaldehyde equivalent, we next directed our attention to the study of α-hydroxyketones. In this context, we chose benzoin 4a as a suitable substrate. The reaction of 2a and 4a under optimized conditions with 1 N HCl did not furnish the cyclized product. Instead, the formation of Heyns adduct 5 was observed in 70% yield (Scheme 3). To force the cyclization, conc. HCl was employed, which also gave 5 in 87% yield. In addition, all other attempts to obtain the trapped product was unsuccessful.
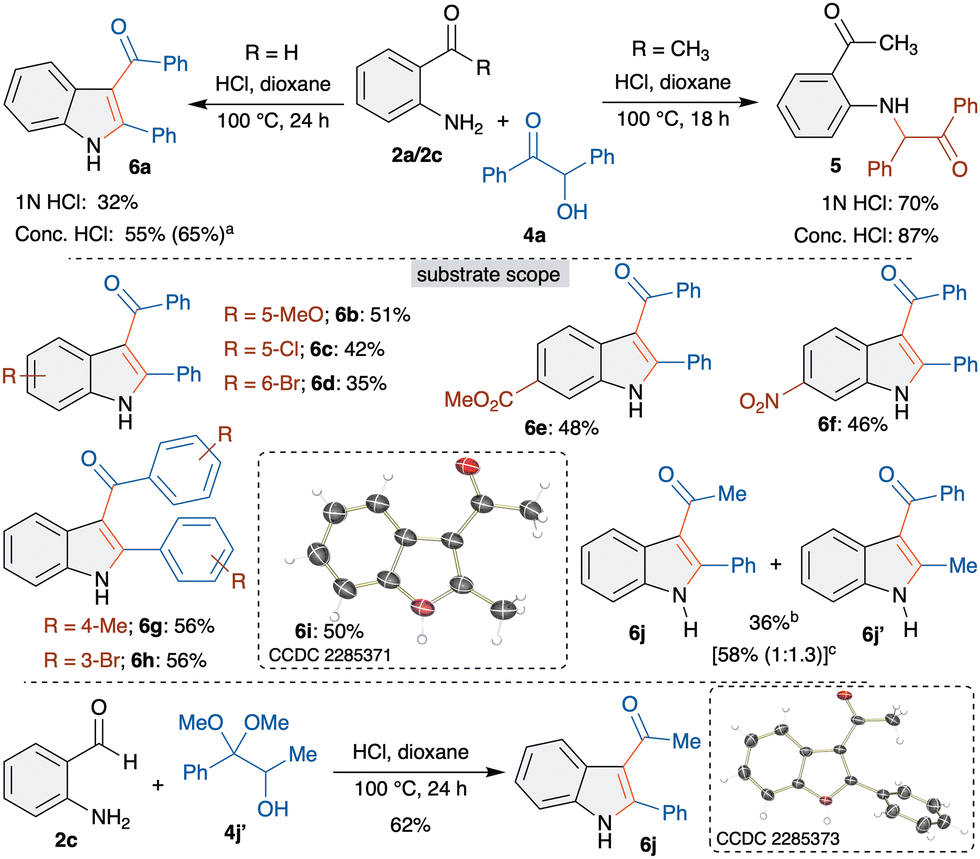 | ||
| Scheme 3 Synthesis of 3-acylindoles 6 and substrate scope. aAt 120 °C. bOnly 6j was observed. cTFA (0.5 equiv.) at 130 °C, 12 h. | ||
Based on this observation, we rationalized that developing steric crowd in the cyclization step possibly hinders the trapping. Hence, we envisaged to employ o-aminobenzaldehyde 2c with benzoin 4a. Interestingly, when benzoin 4a was treated with 2c in the presence of 1 N HCl, to our delight, the formation of the trapped product, 3-acylindole derivative 6a, was observed in 32% yield (Scheme 3). It is important to mention that the formation of 6a is accompanied by C–C bond cleavage and selective acyl migration. Having identified the unique reactivity, next, conditions were optimized to improve the yield of 6a (see supporting information for more details). Interestingly, increasing the concentration of acid gave 6a in 55% yield. The best yield of 65% was observed when 2c and 4a were treated with conc. HCl at 120 °C for 24 h.
After successfully optimizing the synthesis of 3-acylindoles 6, scope and limitations were investigated. Initially, various substituted o-aminobenzaldehydes 2 were examined. 5-Methoxy, 5-chloro and 4-bromo substituted 2-aminobenzaldehydes gave 3-acylindoles 6b–6d in moderate yields (Scheme 3). The reduction in the yields is due to the poor cyclization, which led to the isolation of the corresponding Heyns adducts (∼38% yield). Electron withdrawing methoxycarbonyl and nitro substituents were well tolerated under optimized conditions to afford 3-acylindoles 6e and 6f in good yields. Next, substituted benzoins were examined and all of them afforded 3-acylindoles 6g and 6h in 56% yield. Interestingly, use of acetoin also led to the formation of 6i in 50% yield. The structure of 6i was unambiguously confirmed by single crystal X-ray analysis. On the other hand, reaction of a mixed benzoin derivative, α-hydroxypropiophenone, with 2e furnished 6j in 36% yield. Attempts to increase the yield with TFA gave a regioisomeric mixture of 6j and 6j′ in 58% yield in a 1:1.3 ratio. The formation of 6j′ could be explained through the in situ isomerization of α-hydroxypropiophenone to α-hydroxyphenylacetone followed by an interrupted Heyns rearrangement. Based on our earlier observation and to avoid regioselectivity issues, α-hydroxydimethylketal 4j′ was synthesized and treated with 2c. The reaction afforded 6j as the sole product in 62% yield, which was confirmed by X-ray analysis. This proves that α-hydroxydimethyl(ac)ketals are efficient equivalents for α-hydroxycarbonyls.
Consequently, we focused our attention on the synthetic applicability of the developed method. At first, to demonstrate the synthetic viability, a gram scale reaction was performed with 1a and o-aminobenzophenone 2f, which led to the formation of 3f in comparable yield (see ESI† for more details). Subsequently, cyclopenta[a]indole 7, a higher cyclic analogue, was achieved from 3c in two steps. Propargylation of 3c with propargyl bromide, TBAB and NaOH followed by reaction with TiCl4 and tBuNH2 in toluene furnished 7 in 66% yield (Scheme 4).10
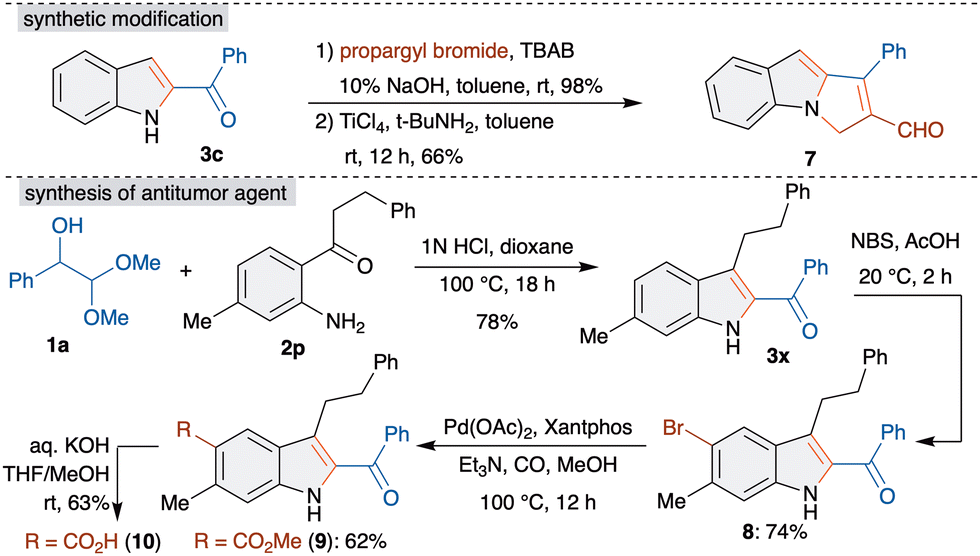 | ||
| Scheme 4 Synthetic applications. | ||
Furthermore, the development of a short synthesis of antitumor agent 10 was investigated.11 The sequence started with the reaction of 1a with 2p under optimized conditions, which afforded the key intermediate 3x in 78% yield. C5-bromination of 3x to 8 followed by palladium catalyzed methoxycarbonylation with Pd(OAc)2, Xantphos and Et3N in methanol under a carbon monoxide atmosphere at 100 °C gave the methoxycarbonylated product 9 in 62% yield. The final hydrolysis of the ester in 9 with aq. KOH gave 10 in 62% yield. This represents the four step synthesis of antitumor agent 10.
We next proposed a possible mechanism for the synthesis of 2- and 3-acylindoles. As shown in Scheme 5, reaction of 1 with acid would generate oxonium species A, which upon reaction with 2 would generate an imine B. Imine-enamine tautomerism of B would give aminoenol intermediate C, a Heyns intermediate. The intramolecular trapping of C with carbonyl would afford D. Next, D can undergo acid promoted dehydration to give E, which can undergo aromatization via two pathways based on R and R2. When R = H and R2 ≠ H, aromatization with loss of proton would furnish 3. On the other hand, when R ≠ H and R2 = H, aromatization with selective acyl migration occurs to give 6.
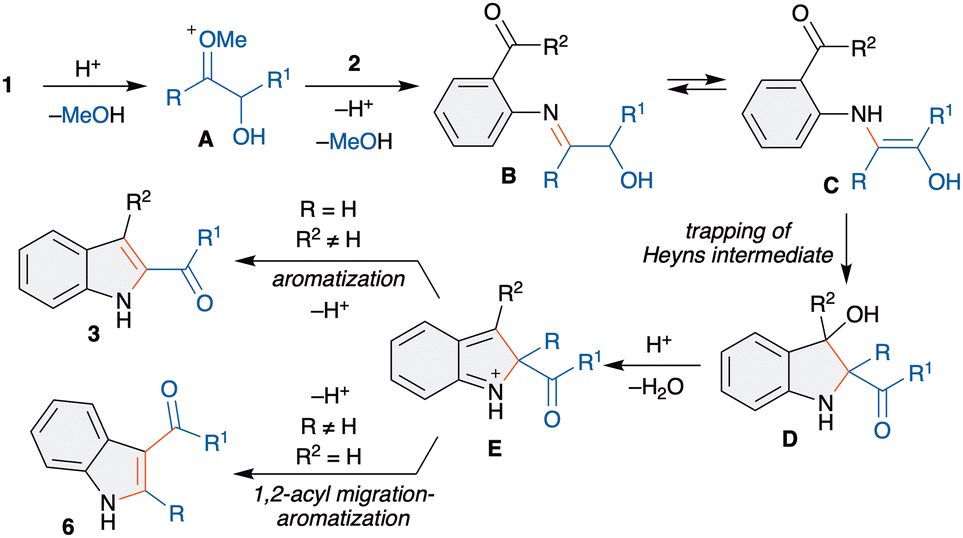 | ||
| Scheme 5 Plausible mechanism. | ||
In conclusion, we have developed an acid promoted efficient and expedited synthesis of 2-acyl and 3-acylindole derivatives from o-acyl/formylanilines and the α-hydroxycarbonyl equivalent. This method involves an interrupted Heyns rearrangementand the in situ generation and intramolecular trapping of aminoenol with tethered carbonyl, followed by aromatization with or without 1,2-acyl migration. The developed method allows the synthesis of diverse substituted 2-acyl and 3-acylindoles in good to excellent yields. Importantly, the present method offers excellent regiocontrol, good functional group tolerance, and gram-scale synthesis and allows the synthesis of higher analogues of indoles and an anti-tumor agent.
We thank DST-SERB, New Delhi, India (Project no. SB/SJF/2020-21/15) for financial support through the SwarnaJayanti Fellowship. M. A. thanks the Indian Institute of Technology Madras (IITM) for the fellowship. We thank Dr S. Krishnaswamy and Dr Ramkumar for single crystal analysis. We also thank DST and the SC-XRD lab, SAIF, IITM for providing X-ray facility.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. W. Gribble, Indole Ring Synthesis: From Natural Products to Drug Discovery, Wiley, 2016 CrossRef; (b) A. Brancale and R. Silvestri, Med. Res. Rev., 2007, 27, 209 CrossRef CAS PubMed; (c) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (d) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694 RSC.
- (a) S. Mahboobi, S. Teller, H. Pongratz, H. Hufsky, A. Sellmer, A. Botzki, A. Uecker, T. Beckers, S. Baasner, C. Schächtele, F. Überall, M. U. Kassack, S. Dove and F.-D. Böhmer, J. Med. Chem., 2002, 45, 1002 CrossRef CAS PubMed; (b) M. A. Tarselli, K. M. Raehal, A. K. Brasher, J. M. Streicher, C. E. Groer, M. D. Cameron, L. M. Bohn and G. C. Micalizio, Nat. Chem., 2011, 3, 449 CrossRef CAS PubMed; (c) Z.-X. Xi, X.-Q. Peng, X. Li, R. Song, H.-Y. Zhang, Q.-R. Liu, H.-J. Yang, G.-H. Bi, J. Li and E. L. Gardner, Nat. Neurosci., 2011, 14, 1160 CrossRef CAS PubMed; (d) E. Yamuna, R. A. Kumar, M. Zeller and K. J. Rajendra Prasad, Eur. J. Med. Chem., 2012, 47, 228 CrossRef CAS PubMed; (e) M. Morales and A. Bonci, Nat. Med., 2012, 18, 504 CrossRef CAS PubMed; (f) C. D. Rosenbaum, S. P. Carreiro and K. M. Babu, J. Med. Toxicol., 2012, 8, 15 CrossRef CAS PubMed; (g) A. Casapullo, G. Bifulco, I. Bruno and R. Riccio, J. Nat. Prod., 2000, 63, 447 CrossRef CAS PubMed; (h) B. P. Smart, R. C. Oslund, L. A. Walsh and M. H. Gelb, J. Med. Chem., 2006, 49, 2858 CrossRef CAS PubMed.
- (a) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (b) L. Ackermann, Synlett, 2007, 507 CrossRef CAS; (c) T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 2001, 2491 RSC; (d) D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195 CrossRef CAS PubMed.
- (a) A. K. Clarke, H. E. Ho, J. A. Rossi-Ashton, R. J. K. Taylor and W. P. Unsworth, Chem. – Asian J., 2019, 14, 1900 CrossRef CAS PubMed; (b) V. Fathi Vavsari, A. Nikbakht and S. Balalaie, Asian J. Org. Chem., 2022, 11, e202100772 CrossRef CAS; (c) G. Li, X. Huang and L. Zhang, Angew. Chem., Int. Ed., 2008, 47, 346 CrossRef CAS PubMed; (d) Y. Tokimizu, S. Oishi, N. Fujii and H. Ohno, Angew. Chem., Int. Ed., 2015, 54, 7862 CrossRef CAS PubMed; (e) K. Alex, A. Tillack, N. Schwarz and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 2304 CrossRef CAS PubMed; (f) O. Leogane and H. Lebel, Angew. Chem., Int. Ed., 2008, 47, 350 CrossRef CAS PubMed; (g) B. Zhou, Z. Wu, D. Ma, X. Ji and Y. Zhang, Org. Lett., 2018, 20, 6440 CrossRef CAS PubMed; (h) X. Yan, C.-F. Liu, X.-T. An, X.-M. Ge, Q. Zhang, L.-H. Pang, X. Bao and C.-A. Fan, Org. Lett., 2021, 23, 8905 CrossRef CAS PubMed; (i) F. Jafarpour, M. Ghasemi, F. Mohaghegh, S. Asgari and A. Habibi, Org. Lett., 2019, 21, 10143 CrossRef CAS PubMed.
- (a) R. Bernini, G. Fabrizi, A. Sferrazza and S. Cacchi, Angew. Chem., Int. Ed., 2009, 48, 8078 CrossRef CAS PubMed; (b) Z.-B. Zhang, Y. Yang, Z.-X. Yu and J.-B. Xia, ACS Catal., 2020, 10, 5419 CrossRef CAS; (c) T. Nanjo, S. Yamamoto, C. Tsukano and Y. Takemoto, Org. Lett., 2013, 15, 3754 CrossRef CAS PubMed; (d) A. Gogoi, S. Guin, S. K. Rout and B. K. Patel, Org. Lett., 2013, 15, 1802 CrossRef CAS PubMed; (e) Q. Shi, P. Li, X. Zhu and L. Wang, Green Chem., 2016, 18, 4916 RSC; (f) Z.-W. Zhang, H. Xue, H. Li, H. Kang, J. Feng, A. Lin and S. Liu, Org. Lett., 2016, 18, 3918 CrossRef CAS PubMed; (g) Z. Wang, Z. Yin and X.-F. Wu, Org. Lett., 2017, 19, 4680 CrossRef CAS PubMed; (h) J. Zhou, J. Li, Y. Li, C. Wu, G. He, Q. Yang, Y. Zhou and H. Liu, Org. Lett., 2018, 20, 7645 CrossRef CAS PubMed; (i) J. Liu, W. Wei, T. Zhao, X. Liu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2016, 81, 9326 CrossRef CAS PubMed; (j) A. Jaiswal, A. K. Sharma and K. N. Singh, Org. Biomol. Chem., 2020, 18, 1623 RSC.
- (a) W.-C. Gao, S. Jiang, R.-L. Wang and C. Zhang, Chem. Commun., 2013, 49, 4890 RSC; (b) B. V. S. Reddy, M. R. Reddy, Y. G. Rao, J. S. Yadav and B. Sridhar, Org. Lett., 2013, 15, 464 CrossRef CAS PubMed; (c) J. Gong, K. Hu, Y. Shao, R. Li, Y. Zhang, M. Hu and J. Chen, Org. Biomol. Chem., 2020, 18, 488 RSC; (d) T. Vivekanand, T. Sandhya, P. Vinoth, S. Nagarajan, C. U. Maheswari and V. Sridharan, Tetrahedron Lett., 2015, 56, 5291 CrossRef CAS.
- (a) T. M. Wrodnigg and B. Eder, The Amadori and Heyns Rearrangements: Landmarks in the History of Carbohydrate Chemistry or Unrecognized Synthetic Opportunities? in Glycoscience: Epimerisation, Isomerisation and Rearrangement Reactions of Carbohydrates, ed. A. E. Stütz, Springer Berlin Heidelberg, Berlin, Heidelberg, 2001, pp. 115 Search PubMed; (b) T. M. Wrodnigg, C. Kartusch and C. Illaszewicz, Carbohydr. Res., 2008, 343, 2057 CrossRef CAS PubMed; (c) T. M. Wrodnigg and A. E. Stütz, Angew. Chem., Int. Ed., 1999, 38, 827 CrossRef CAS.
- (a) J. Huang, G.-X. Li, G.-F. Yang, D.-Q. Fu, X.-K. Nie, X. Cui, J.-Z. Zhao and Z. Tang, Chem. Sci., 2021, 12, 4789 RSC; (b) G. Li, L. Tang, H. Liu, Y. Wang, G. Zhao and Z. Tang, Org. Lett., 2016, 18, 4526 CrossRef CAS PubMed; (c) X.-k Nie, Y. Chen, S.-q Zhang, X. Cui, Z. Tang and G.-x Li, Org. Lett., 2022, 24, 2069 CrossRef CAS PubMed; (d) L. Li, S. Zhang, X. Deng, G. Li, Z. Tang and G. Zhao, Org. Lett., 2021, 23, 6819 CrossRef CAS PubMed; (e) J.-x Liang, G.-b Yang, Y.-p Zhang, D.-d Guo, J.-z Zhao, G.-x Li and Z. Tang, Org. Chem. Front., 2020, 7, 3242 RSC; (f) J. Huang, J.-f Chen, X. Cui, J.-z Zhao, Z. Tang and G.-x Li, J. Org. Chem., 2022, 87, 3311 CrossRef CAS PubMed.
- S. Panda and P. Ghorai, Angew. Chem., Int. Ed., 2023, 62, e202306179 CrossRef CAS PubMed.
- G. Abbiati, A. Casoni, V. Canevari, D. Nava and E. Rossi, Org. Lett., 2006, 8, 4839 CrossRef CAS PubMed.
- L. You, Z. Xu, C. Punchihewa, D. M. Jablons and N. Fujii, Mol. Cancer Ther., 2008, 7, 1633 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2285371–2285373. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04144a |
| This journal is © The Royal Society of Chemistry 2023 |