
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHolding it together: noncovalent cross-linking strategies for ionogels and eutectogels
Matthew J.
Panzer

Department of Chemical & Biological Engineering, Tufts University, 4 Colby Street, Medford, MA 02155, USA. E-mail: matthew.panzer@tufts.edu
First published on 6th September 2022
Abstract
When a hydrogel simply won’t cut it – either because it dries out too quickly, or it does not tolerate more than roughly one volt when applied in an electrochemical device – where is the savvy materials researcher to turn? This is where two important classes of nonaqueous gel counterparts, known as ionogels and eutectogels, can truly shine. Replacing the aqueous liquid phase of a hydrogel with either an ionic liquid (IL) or a deep eutectic solvent (DES) allows one to realize an array of versatile gel electrolyte materials that offer outstanding nonvolatility, wider windows of electrochemical stability, reasonably high ionic conductivity, and nearly unlimited chemical design possibilities. In addition to choosing a specific IL or DES, there are a myriad of options when it comes to constructing a solid, three-dimensional, volume-spanning network (or scaffold) that will support the nonaqueous liquid phase of an ionogel or eutectogel. In this focused review, several recent approaches to forming these gels using noncovalent scaffold assembly and cross-linking are examined, and the primary noncovalent interactions responsible (e.g. hydrogen bonding, solvophobicity, coulombic interactions) are identified. Noncovalent scaffold assembly in nonaqueous, ion-dense electrolytes often leads to supramolecular gel materials that can exhibit extreme stretchability, good toughness, and an ability to self-heal in many cases. After reviewing several strategies that have been recently employed for creating ionogels and eutectogels, a brief inspection of some motivating noncovalently cross-linked scaffolds reported for hydrogels is presented with the hopes that these may provide inspiration for the future design of novel ionogels and eutectogels by the materials research community.
 Matthew J. Panzer | Matthew J. Panzer is a Professor and currently Director of Graduate Studies in the Department of Chemical & Biological Engineering at Tufts University. After earning an Honors BS degree in chemical engineering with distinction from the University of Delaware in 2002 and a PhD in chemical engineering at the University of Minnesota in 2007, he established his research laboratory at Tufts in 2009 following a two-year postdoctoral associate position at MIT. His research focuses primarily on the design of novel gel electrolytes featuring ionic liquids and deep eutectic solvents for electrochemical energy storage applications. |
1. Introduction
Ionic liquids and deep eutectic solvents are two kindred classes of next-generation liquid electrolyte materials that possess intriguing properties and suggest a myriad of possible applications, from energy storage to wearable sensors, electrochromic displays, gas separations, and even drug delivery.1 Ionic liquids (ILs) are molten salts at or near ambient temperature, and thus consist solely of cations and anions. The most common IL cation/anion charge stoichiometry is +1/−1. Although recognized as unique materials for over a century now,2,3 the number of literature reports on ILs really began to increase substantially around the late 1990s, following the development earlier that decade of several aprotic ILs that could be handled safely in air.4 Deep eutectic solvents (DESs), on the other hand, are generally formed by combining at least two different components and are therefore mixtures by definition.5 The most common DES type (“type III”)6 consists of an organic salt (hydrogen bond acceptor, HBA) paired with at least one charge neutral component (hydrogen bond donor, HBD) in a eutectic or near-eutectic composition, in order to obtain a substantially depressed melting point of the mixture. The salt concentration inherent to a DES is usually much higher (i.e. approximately 4–5 mol L−1) than that of a dilute electrolyte solution. Following the first DES report by Abbott7 in 2003, studies of DESs in the literature began to appear at a rapid pace since around 2010.8 Despite their inherent differences, both ILs and DESs share the following important features: (1) a high ion density, (2) low volatility, and (3) a wide degree of chemical tunability.9–13Supporting either an IL or a DES with a three-dimensional solid scaffold, such as a cross-linked polymer network, is a well-established method of obtaining freestanding viscoelastic composite materials whose mechanical properties can be controlled by design.1,14 In the case of ILs, these materials are known as ionogels (or ion gels, iongels, or sometimes ionic liquid gels).1,15–18 When the liquid component is a DES, these materials are referred to as eutectogels (or DES gels).19,20 Implicit in describing a material as either an ionogel or a eutectogel is usually the following: (1) a freestanding and/or non-flowing nature, i.e. within a tilted or inverted vial; and (2) a liquid component volume (or mass) fraction of ∼50% or higher.21 While the formation of covalent bonds between polymer chains throughout a 3D polymer network is a straightforward method of obtaining either class of these nonaqueous gel electrolytes, such scaffolds tend to produce brittle ionogels/eutectogels whose stretchability can be limited at high cross-link densities and do not self-repair/heal due to the irreversibility of breaking covalent bonds.20,22 By contrast, noncovalent cross-links (e.g. those formed by hydrogen bonds, coulombic interactions, solvophobicity, etc.) between polymer chains, low molecular weight gelators (LWMGs), or other colloidal nanostructures can enable much greater energy dissipation and can often self-heal or be readily reshaped due to the weaker, reversible nature of the cross-links.23 A diverse array of potential noncovalent interactions to draw from exists24 when designing ionogels and eutectogels; however, only a fraction of these have been utilized to date. Meanwhile, the current literature abounds with numerous examples of noncovalently cross-linked hydrogels that exhibit desirable physical attributes, such as: extreme stretchability, a self-healing capability, and even underwater adhesion.25–27 The goal of this focused review is to summarize the recent state of the field regarding ionogels and eutectogels that feature noncovalently cross-linked scaffolds. It is meant to be illustrative, rather than exhaustive. The focus is on materials selection and design, not on applications. It also seeks to draw inspiration from the comparatively broader hydrogel literature and suggest future noncovalent scaffold assembly approaches that may similarly be effective within ion-dense IL and/or DES environments.
1.1. Archetypical ILs
The most commonly studied ILs can by and large be grouped into two camps: those that are significantly water-miscible and those that are essentially water-immiscible.3 These are often referred to as “hydrophilic” and “hydrophobic” ILs, respectively; the extent of IL miscibility with water is naturally determined by the specific chemical structures of the ions. An alternative way to divide common ILs into two groups is based on their protic or aprotic chemical nature.28 Importantly, protic ILs (such as ethylammonium nitrate) generally exist in a state of dynamic equilibrium between protonated and deprotonated moieties, thus perpetuating a (potentially small) population of charge neutral species.29 To date, aprotic ILs have been utilized substantially more than protic ILs for creating ionogels and are thus the focus in this review. Chemical structures of some of the most common aprotic ILs reported in the literature for ionogels are shown in Fig. 1. Several excellent reviews of ILs/ionogels and their applications have been written in recent years, to which the reader can be referred to learn more.1,10,15,16,30,31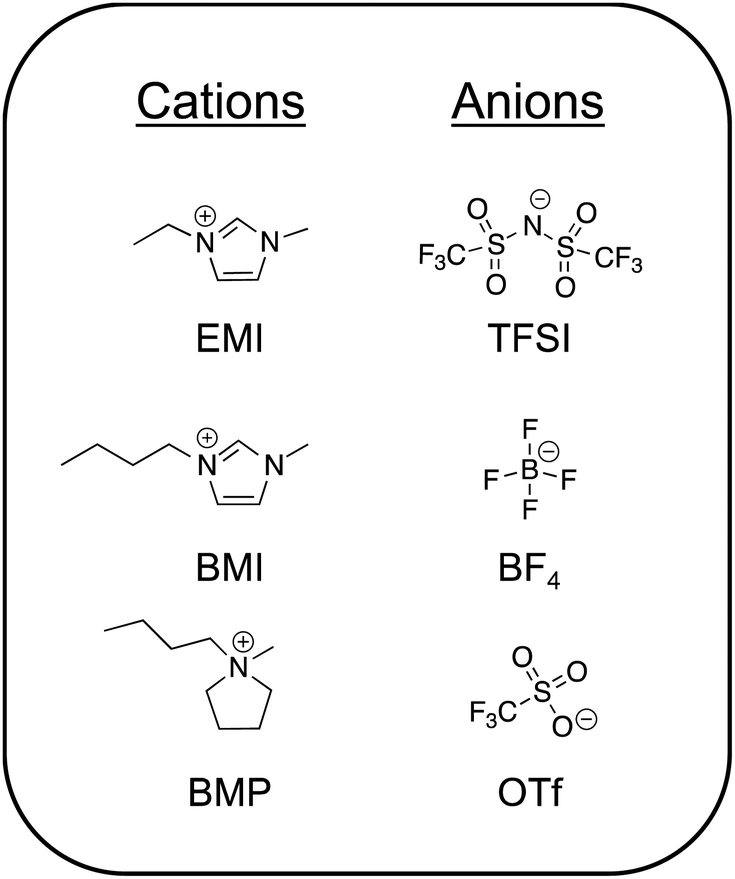 | ||
| Fig. 1 Chemical structures of common IL cations and anions found in ionogels. | ||
Aprotic ILs having fluorinated anions are generally preferred for electrochemical energy storage applications (e.g. batteries, supercapacitors) due to their lower viscosities (yielding higher ionic conductivities) and wide windows of electrochemical stability.16 Many of these ILs are essentially water-immiscible. An exemplar of this class is 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMI TFSI). In order to use an IL as a battery electrolyte, one typically dissolves an appropriate metal salt into it, often featuring the same anion (e.g. LiTFSI). Among the most common IL cations suggested for electrochemical devices are: pyrrolidinium, imidazolium, piperidinium, phosphonium, and ammonium.12,32 Popular anions for battery electrolyte applications are TFSI and its petit frère, bis(fluorosulfonyl)imide, FSI.33–35 More recently, asymmetric variations on this theme have also been explored.36 Aprotic ILs that exhibit a higher degree of water miscibility generally include those with non-fluorinated anions such as acetate (Ac) or ethyl sulfate.37,38
A less studied, but interesting area of IL research is the combination of two (or more) ILs.39 In some cases, lower melting eutectic compositions of such blends may offer advantages compared to either of their individual IL components.40–42 This is an area of great potential for future research, which dovetails nicely into the introduction of DESs in the following section.
1.2. Archetypical DESs
Unlike ILs, DESs are always mixtures that contain at least one charge neutral component, usually the HBD.43 The first reported DES,7 a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio mixture of urea:choline chloride, U:ChCl (2:1) (also known as reline), is one of the most studied to date. Urea, which melts at 134 °C, when combined under gentle heating and stirring with choline chloride (a solid up to its thermal decomposition near 300 °C), forms a homogeneous liquid mixture that exhibits a freezing point of 12 °C and possesses a high salt concentration/ionic species density of approximately 4.8 M. An extensive hydrogen bonding network between and among urea (HBD) molecules with the chloride anion and the hydroxyl group of the choline cation44 gives rise to the very deep melting point depression and low volatility of this archetypical DES. Indeed, hydrogen bonding plays a key role in virtually every deep eutectic mixture proposed to date.43,45,46 More recently, the naturally occurring zwitterion trimethylglycine (or simply, “betaine”) has been introduced as a versatile alternative HBA to the ubiquitous choice of ChCl.47 The reader is directed to several comprehensive reviews of DESs to learn more about these systems.6,9,43,48,49 Chemical structures of a few common DES components are shown in Fig. 2.
:1 molar ratio mixture of urea:choline chloride, U:ChCl (2:1) (also known as reline), is one of the most studied to date. Urea, which melts at 134 °C, when combined under gentle heating and stirring with choline chloride (a solid up to its thermal decomposition near 300 °C), forms a homogeneous liquid mixture that exhibits a freezing point of 12 °C and possesses a high salt concentration/ionic species density of approximately 4.8 M. An extensive hydrogen bonding network between and among urea (HBD) molecules with the chloride anion and the hydroxyl group of the choline cation44 gives rise to the very deep melting point depression and low volatility of this archetypical DES. Indeed, hydrogen bonding plays a key role in virtually every deep eutectic mixture proposed to date.43,45,46 More recently, the naturally occurring zwitterion trimethylglycine (or simply, “betaine”) has been introduced as a versatile alternative HBA to the ubiquitous choice of ChCl.47 The reader is directed to several comprehensive reviews of DESs to learn more about these systems.6,9,43,48,49 Chemical structures of a few common DES components are shown in Fig. 2.
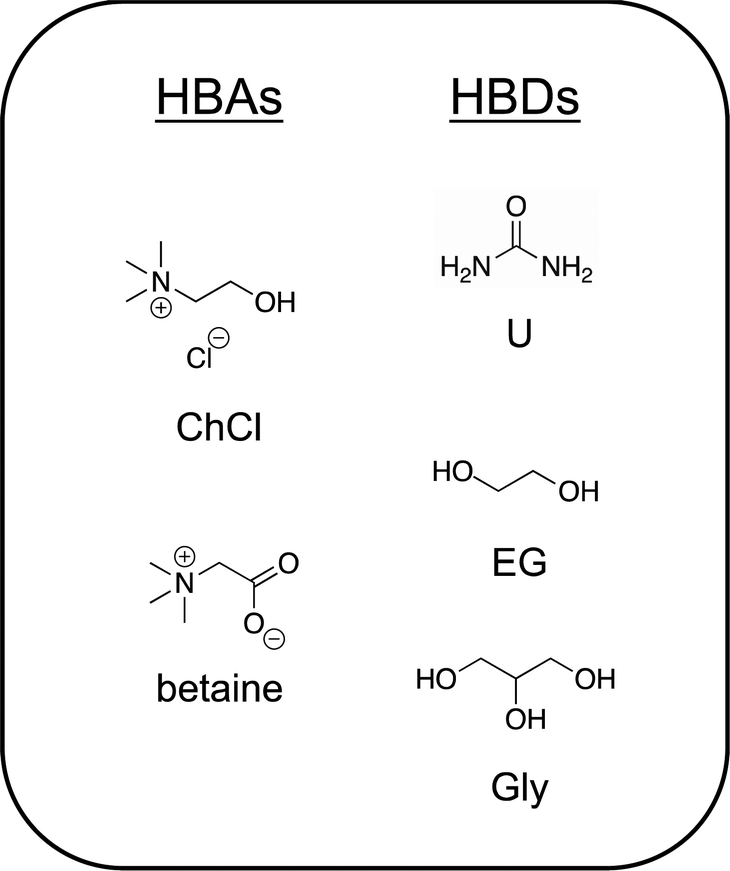 | ||
| Fig. 2 Chemical structures of common DES components, a combination of HBAs and HBDs. | ||
While there remains some uncertainty in the field regarding what criterion to use when labeling a liquid eutectic mixture as “deep” or not,5 it is important to recognize that no claims regarding a mixture's status as a DES should really be made without measuring and reporting accurate liquidus data for several HBD:salt compositions (i.e. providing an experimental solid–liquid phase diagram). Best practice would be to also include the raw data, such as differential scanning calorimetry (DSC) scans, that led to the proposed phase diagram.50,51 Repeatable DSC measurements on DESs can be challenging to obtain, however, due to the tendency of DESs to exhibit substantial supercooling.52 Indeed, one system that was widely accepted as being a “true DES” for many years (a 2:1 molar ratio of ethylene glycol (EG):choline chloride, EG:ChCl (2:1), or ethaline) was recently shown to have a very different actual eutectic mixture stoichiometry and a nearly ideal melting point depression upon performing a careful DSC study.50 Nevertheless, the ethaline mixture has been among the most popular for forming eutectogels to date, likely due to its low viscosity and high room temperature ionic conductivity as compared to other DESs.8
The vast majority of reported DESs are water-miscible, although the development of “hydrophobic DESs” has also been championed in recent years.53,54 It is also very likely that many reports of purported salt:HBD binary eutectic mixtures also contain small amounts of water, whether introduced intentionally or inadvertently. This is important inasmuch as water has been demonstrated, at low levels, to act as a co-HBD in certain DESs and can result in significant changes in mixture properties (such as viscosity).55,56 For the DES reline, it has been found that the original DES nanostructure is retained when adding up to approximately 40–50 wt% H2O.57 Therefore, some DESs can tolerate modest water contents without wholly losing their DES-like character. Besides intentionally adding a controlled amount of water to a DES, mixtures of different organic HBDs with a common salt58 (or vice versa59) as well as mixtures of two different DESs are so far underexplored in the field; these avenues should present a great opportunity to further tune the properties of these ion-dense, low cost electrolytes.
2. Noncovalent cross-linking approaches
There are many noncovalent intermolecular interactions that one could potentially exploit to form a cross-linked 3D scaffold that supports a significant amount of liquid electrolyte, thereby creating a freestanding gel. Some of the most common ones that have been used to create ionogels, eutectogels, or hydrogels to date include:• Hydrogen bonding
• van der Waals interactions
– Solvophobicity, π–π interactions, host–guest interactions
• Coulombic interactions
– Metal–ligand coordination, zwitterionic dipole–dipole.
In order for a solid “building block” (e.g. small molecule, polymer, or colloidal nanoparticle) to spontaneously assemble and form a 3D noncovalently cross-linked scaffold within an ionogel or eutectogel, a basic requirement is the existence of somewhat stronger self interactions (i.e. solid–solid) compared to interactions with the surrounding solvent species (i.e. solute–solvent). However, the self interactions must also not be too strong so as to cause aggregation/precipitation within the IL or DES, which would preclude the ability for the solid to be well-dispersed throughout the liquid and form a fractal, volume-spanning network. The noncovalent interaction that has been most commonly employed to form cross-links within ionogels/eutectogels to date is hydrogen bonding, either between polymer segments or LMWGs. While several studies have investigated colloidal silica-supported ionogels,60–62 these scaffolds are not a focus of this review. For some polymers that can form intra-/interchain crystalline domains that serve as noncovalent cross-linking points within a gel, these interactions may also often be a manifestation of hydrogen bonding (e.g. poly(vinyl alcohol), PVA,63 and poly(vinylidene fluoride), PVDF64). Another important noncovalent interaction is solvophobicity, which was the key driving force for the formation of the first triblock copolymer-supported ionogels.65,66 Intrinsic to solvophobic effects are the ubiquitous intermolecular attractive forces that scale with molecular polarizabilities and dipole moments (i.e. van der Waals interactions).67 Indeed, solvophobicity of a solid component within a liquid is a manifestation of comparatively stronger self-attraction, which may often – but not exclusively – be due to van der Waals forces. In the case of scaffold molecules that contain aromatic ring motifs, π–π interactions can also be another source of noncovalent assembly.68 Host–guest interactions, such as the complementary binding between β-cyclodextrin and adamantane,69 are often a combination of hydrogen bonding and/or van der Waals interactions between the host functional group and its guest. Coulombic (or charge-based) interactions are yet another potential important source of noncovalent cross-links.70 These are interactions involving atomic species or functional groups bearing fixed charges, including ion–ion interactions (such as metal ion–ligand coordination), ion–dipole interactions, as well as dipole–dipole assemblies due to fixed charge pairs (e.g. in the case of polyzwitterions).
The strengths of all of these noncovalent interactions can be expected to vary with the chemical details of the specific cross-linking units and of the liquid environment (e.g. charge densities, dipole moments, polarizabilities), as well as with temperature. Generally, noncovalent interaction energies near ambient conditions tend to be on the order of ∼5–30 kJ mol−1,27 or a few times the background molar thermal energy (2.5 kJ mol−1 at 25 °C), and they are approximately 1–2 orders of magnitude lower than typical covalent bond energies (e.g. C–H, ∼400 kJ mol−1). The density of ions present in the liquid phase and effective permittivity are also clearly important factors for determining the strength of noncovalent interactions that are driven by coulombic interactions in both ILs and DESs, which inherently possess high concentrations of ionic species.
3. Ionogels with noncovalent scaffolds
A large number of different ionogel manifestations have appeared in the literature over the past two decades. Indeed, several review articles on the topic have been published for the interested reader.1,15–18 Here, a select number of recent examples of ionogels that have been supported using different noncovalent cross-linking strategies is summarized, in order to provide a sense for what approaches have been pursued as of late. In several cases, multiple noncovalent interactions play a role in scaffold cross-linking; this is typical of many biological and synthetic supramolecular assemblies.71 Some general conclusions that can be drawn from the ionogel examples summarized in Table 1 are:
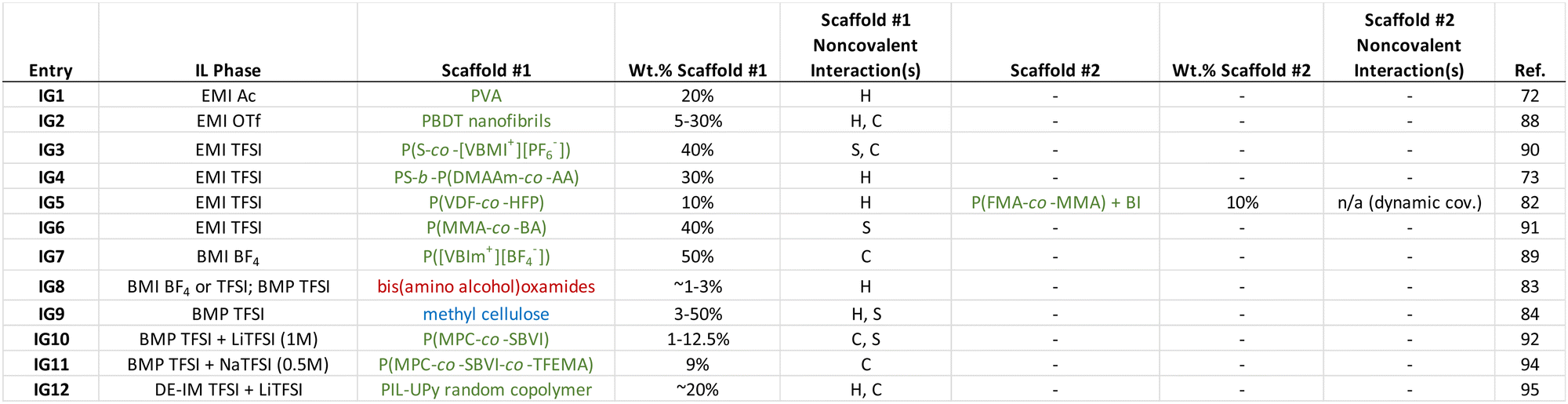
|
(1) Synthetic polymers have largely been the scaffold of choice for recently reported ionogels that feature noncovalent cross-links.
(2) Copolymers are often employed, as different monomers can play complementary roles within the scaffold (i.e. tuning solubility, relative backbone flexibility/stiffness, cross-link functionality).
(3) Hydrogen bonding and coulombic interactions are the two most common noncovalent cross-linking strategies employed for ionogels in recent years.
(4) While a typical ionogel polymer scaffold content is ∼10–40 wt%, some polymers can enable ionogel formation at contents as low as 1–5 wt%.
3.1. Hydrogen bonded ionogel scaffolds
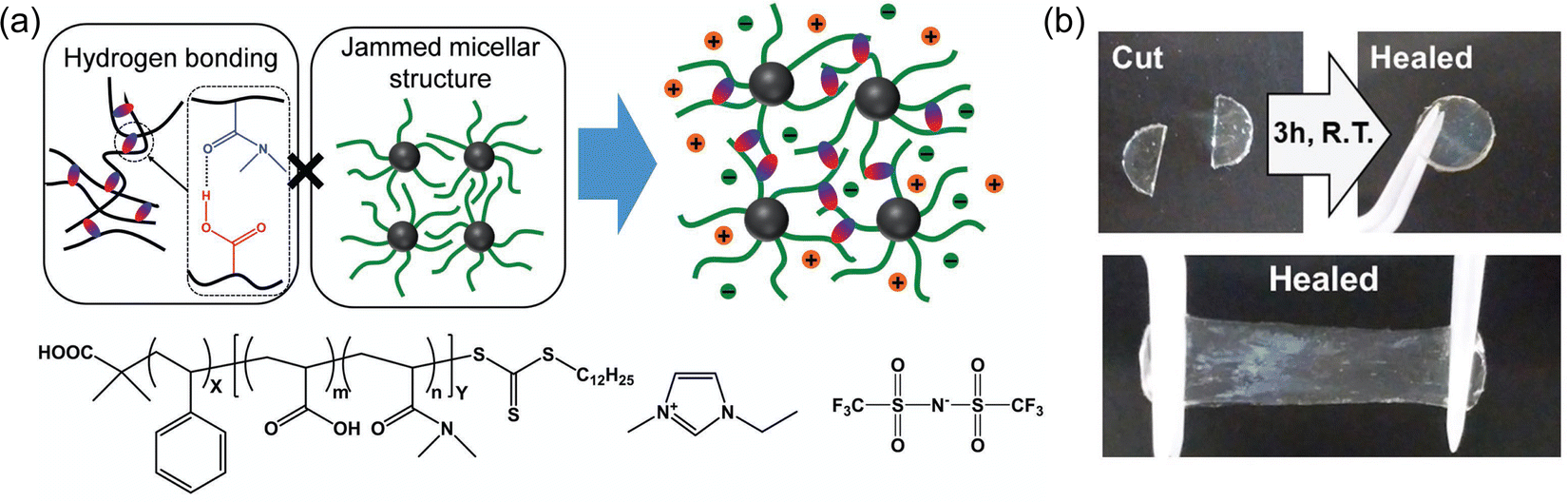
Leveraging extensive hydrogen bonding between commercially available poly(vinyl alcohol) (PVA) chains, Wang and coworkers72 successfully formed freestanding, stretchable ionogels using EMI TFSI (IG1, Table 1). The polymer network was formed by dissolving PVA into the IL while heating and stirring, followed by several freeze–thaw cycles. Inorganic phosphor nanoparticles were also incorporated into some of these ionogels, which allowed for visible gel color tunability. Due to the noncovalent hydrogen bonds that formed the cross-linked network, these ionogels were also able to self-heal (i.e. two pieces merged to become a single ionogel) in a matter of a few hours at ambient conditions (Fig. 3). | ||
| Fig. 3 (a) PVA-supported ionogels containing red or blue nanophosphors. (b) Connection of the two ionogels after 2 h. (c) Stretching of the merged ionogel after 4 h at ambient conditions. Reproduced with permission from ref. 72, copyright 2022 Wiley-VCH. | ||
In addition to homopolymers such as PVA that can promote cross-link formation though hydrogen bonding, copolymers have also been used to create noncovalent ionogels. Tamate et al. synthesized diblock copolymers via sequential reversible addition fragmentation chain transfer (RAFT) polymerization that consisted of an IL–phobic poly(styrene) (PS) block together with a hydrogen bonding block comprised of a statistical copolymer of N,N-dimethylacrylamide (DMAAm) and acrylic acid (AA).73 Ionogels were created by blending the copolymer into the IL (EMI TFSI) using a cosolvent mixture (50:50 v/v methanol:dichloromethane), which was subsequently removed by heating under vacuum (IG4). The assumption of such an approach, which is commonly employed to create ionogels when the (co)polymer is synthesized separately (as opposed to in situ synthesis within the IL, e.g. via free radical polymerization of monomers22,74–80), is that the cosolvent can be completely removed while the IL remains behind in its entirety. Nevertheless, the PS-b-P(DMAAm-co-AA) diblock copolymers of Tamate and coworkers were able to establish a three-dimensional ionogel scaffold by self-assembly. Solubility differences between the two blocks in the IL resulted in the formation of micellar structures with poly(styrene)-rich cores and hydrogen bonded cross-links formed between the P(DMAAm-co-AA) coronal chains (Fig. 4a). These transparent and stretchable ionogels could reach tensile fracture strains of 400% and were also able to self-heal within a few hours at room temperature (Fig. 4b).
 | ||
| Fig. 4 (a) Schematic illustration and chemical structures of diblock copolymer-supported ionogels. (b) Self-healing ability of a 30 wt% diblock copolymer-supported ionogel. Adapted from ref. 73 with permission, copyright 2018 Wiley-VCH. | ||
Another ionogel scaffold that leveraged hydrogen bonding to form noncovalent cross-links was a linear amphiphilic poly(urethane-urea) copolymer reported by Chen and Guo, which produced highly stretchable ionogels that could also be 3D-printed.81 While the majority of ionogels reported have utilized a single scaffold, Tang et al. employed two distinct/interpenetrating polymeric networks (IG5) to form ionogels supporting EMI TFSI (so-called “double network” gels).82 The first network consisted of commercially available poly(vinylidene fluoride-co-hexafluoropropylene), P(VDF-co-HFP). This copolymer forms crystalline regions between PVDF segments on neighboring chains, driven primarily by H–F hydrogen bonding interactions, which serve as physical cross-links. Its highly fluorinated structure facilitates good compatibility with EMI TFSI and other ILs having similarly fluorinated anions. The second network employed in this group of ionogels was realized by dynamic covalent cross-linking of poly(furan-2-ylmethyl methacrylate-co-methyl methacrylate), P(FMA-co-MMA), using controlled amounts of N,N′-(4,4′-diphenylmethane)bismaleimide (BI). The authors contended that heating the samples to 100 °C was sufficient to break some of the BI-enabled dynamic covalent bonds, which could then re-form at lower temperatures, providing this network with good healability.82
Hydrogen bonding between several bis(amino alcohol)oxamides, which can be effective LMWGs, was employed by Santic et al. to realize noncovalent ionogels using three different ILs (IG8).83 Typical of LMWG-supported ionogels, the authors found that gelation could be achieved at or below ∼1 wt% solids. Using a chemically-modified version of the most abundant biopolymer on Earth, Mantravadi and coworkers created ionogels of 1-butyl-1-methylpyrrolidinium (BMP) TFSI supported by methylcellulose (IG9).84 In this case, both hydrogen bonding and solvophobic interactions between methylcellulose chains played a role in forming a robust noncovalent scaffold. The group of D’Anna has also demonstrated biopolymer-supported ionogels,85 as well as ionogels formed using LMWGs of ammonium/phosphonium86 or imidazolium salts,87 within all of which hydrogen bonding was shown to be a key noncovalent interaction.
Self-assembled helical aggregates of rigid sulfo-aramid chains were used to support EMI trifluoromethanesulfonate (OTf) with as low as 5 wt% of the polyanionic scaffold poly(2,2′-disulfonyl-4,4′-benzidine terephthalamide), PBDT, as reported by Fox et al. (IG2).88 PBDT helix formation was largely driven by hydrogen bonding, while coulombic interactions between these helical structures due to the large sulfonate charge density on the polyanion likely also played an important role in forming a physically cross-linked network. These PBDT–IL composites were formed by casting from a dilute solution, using deionized water as the cosolvent. Notably, they exhibited some of the highest combinations of room temperature elastic modulus (∼GPa) and ionic conductivity (∼5 mS cm−1) among ionogels recorded to date.
3.2. Coulombic interactions within the ionogel scaffold
Noncovalent cross-links formed by coulombic interactions within the poly(IL), or PIL, scaffold consisting of poly(1-vinyl-3-butylimidazolium tetrafluoroborate), P([VBIm+][BF4−]) dispersed in BMI BF4 were demonstrated by Xiang and coworkers (IG7).89 Intuitively, such charge-based noncovalent linkages between adjacent PIL chains must be mediated by mobile BF4 anions. These ionogels also exhibited robust adhesion to glass, metal, and plastic surfaces. Using a slightly different IL monomer incorporated together with styrene as a statistical copolymer, Seo and Moon prepared EMI TFSI ionogels (IG3) supported by a noncovalent scaffold of poly(styrene-co-1-(4-vinylbenzyl)-3-methylimidazolium hexafluorophosphate), P(S-co-[VBMI+][PF6−]).90 Here, both solvophobic (i.e. PS regions) and coulombic (i.e. PIL regions) interactions were important for noncovalent cross-link formation. The authors also demonstrated successful ionogel formation using a P([VBMI+][PF6−]) homopolymer, although the elastic modulus of the copolymer-supported ionogel using the same solids content was approximately five times larger by comparison.3.3. Solvophobicity as a factor for ionogel scaffolds
As an exemplar of noncovalent scaffold assembly due mainly to solvophobic interactions, statistical copolymers of poly(methyl methacrylate-co-butyl acrylate), P(MMA-co-BA), were employed to create EMI TFSI ionogels by Kim et al. (IG6).91 The authors posited that the low glass transition temperature of the IL-phobic PBA regions allowed these ionogels to exhibit greater stretchability compared to similar copolymers featuring PS as the solvophobic component due to more effective stress dissipation (Fig. 5).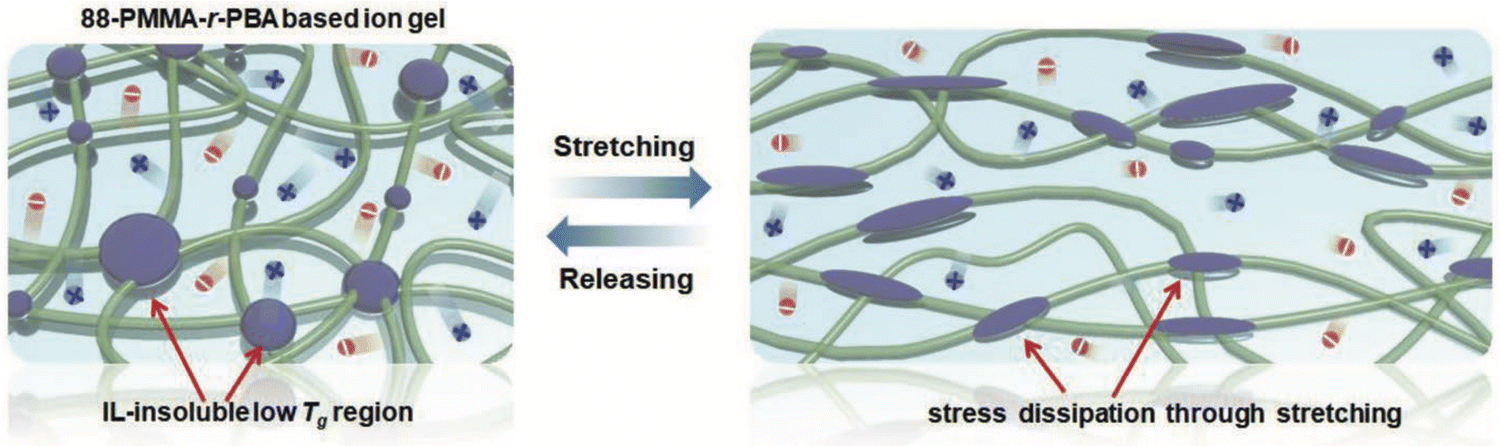 | ||
| Fig. 5 Schematic illustration of scaffold structure leading to highly stretchable ionogels supported by P(MMA-co-BA) copolymers in EMI TFSI. Adapted from ref. 91 with permission, copyright 2019 Wiley-VCH. | ||
3.4. Alkali metal salt/IL solutions in ionogels
With an eye toward using for ionogels as safer (nonflammable) battery electrolytes, alkali metal salt/IL solutions are commonly used.16 Here, the presence of Li+ or Na+ cations can enable additional noncovalent coulombic interactions between charged polymer chains. For example, the Panzer group demonstrated robust ionogel formation in 1 M LiTFSI/BMP TFSI solution via in situ UV-initiated free radical copolymerization of poly(2-methacryloyloxyethyl phosphorylcholine-co-sulfobetaine vinylimidazole), P(MPC-co-SBVI) (IG10).92 This particular fully zwitterionic (ZI) copolymer scaffold (Fig. 6) facilitated noncovalent cross-linking due to different types of coulombic interactions (i.e. MPC–Li+–MPC, versus SBVI–SBVI) interactions, which was also evidenced by the notably different solubilities of the two ZI monomers in the IL/salt solution.93 A similar class of ZI copolymer-supported ionogels was realized using a 0.5 M NaTFSI/BMP TFSI solution, even as the two ZI monomers were “diluted” within a linear terpolymer incorporating a third, non-ZI monomer: 2,2,2-trifluoroethyl methacrylate (TFEMA), (IG11).94 Enhanced SBVI–Na+ attraction within this IL electrolyte compared to the analogous Li+ system was also observed.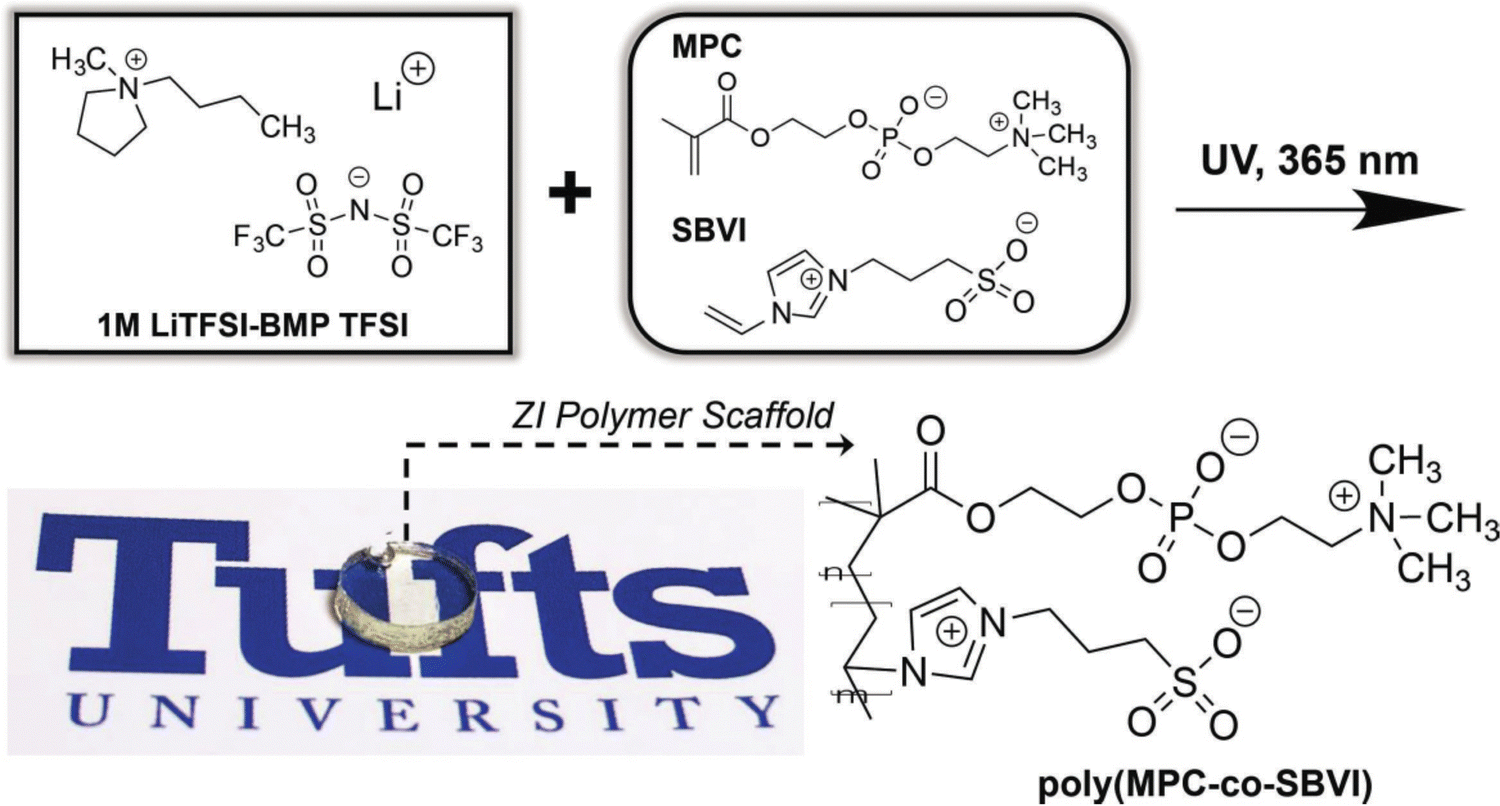 | ||
| Fig. 6 Schematic showing the chemical structures of IL electrolyte ions and two different zwitterionic (ZI) co-monomers (MPC, SBVI) employed to form fully ZI copolymer scaffolds via UV-initiated free radical copolymerization, yielding transparent and robust ionogels. Adapted from ref. 92 with permission, copyright 2018 Wiley-VCH. | ||
Ionogels with a LiTFSI/1,2-dimethyl-3-ethoxyethylimidazolium (DE-IM) TFSI solution supported by a PIL copolymer containing strongly hydrogen bonding ureido-pyrimidinone (UPy) moieties were reported by Guo et al. (IG12).95 Effective dimerization between UPy units on neighboring polymer chains due to four hydrogen bonds per pair was responsible for the majority of noncovalent cross-links within these flexible and self-healing ionogels. After synthesizing and purifying the copolymer, ionogel membranes were prepared by combining the copolymer, LiTFSI, and DE-IM TFSI in acetone, then casting a film that was subsequently dried under vacuum to remove the acetone cosolvent, and finally hot-pressed at 120 °C for 10 min (Fig. 7).
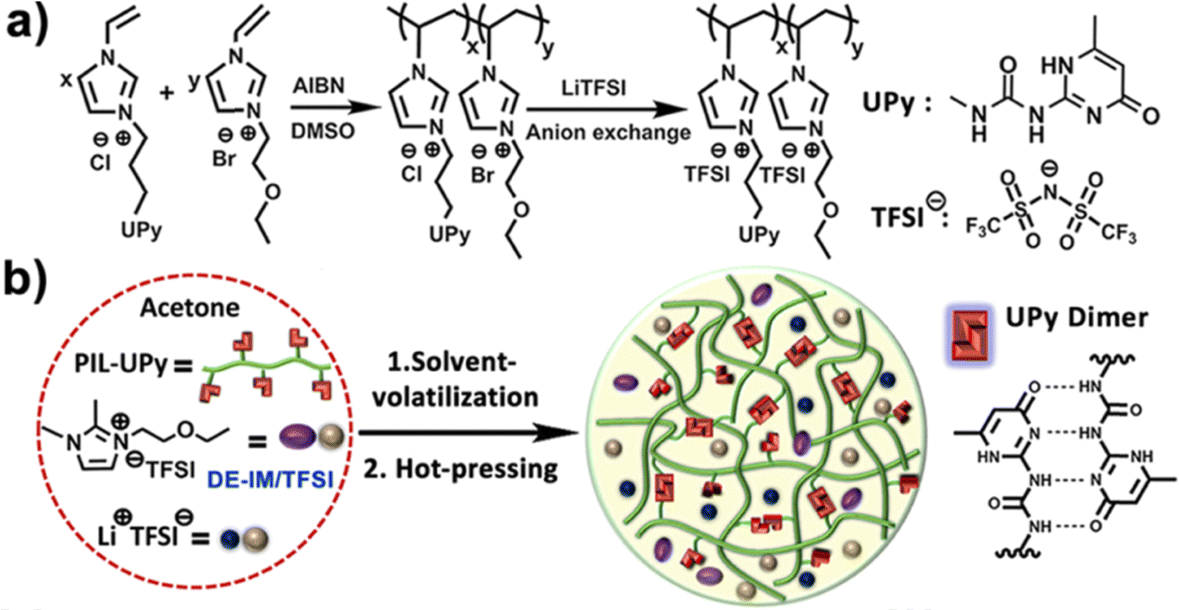 | ||
| Fig. 7 (a) Schematic illustration of the synthesis of PIL-UPy copolymers, and (b) process used to prepare ionogel membranes. Adapted from ref. 95 with permission, copyright 2019 American Chemical Society. | ||
4. Eutectogels with noncovalent scaffolds
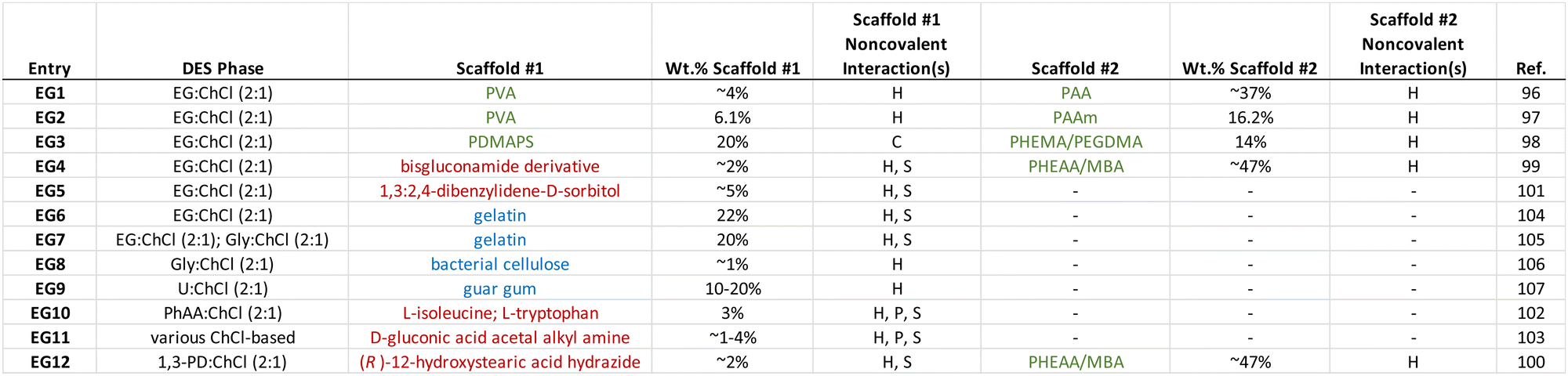
Similar to the case of ionogels, noncovalent interactions are also exploited to create eutectogels that can exhibit highly stretchable mechanical behavior and often a self-healing nature. The DES liquid phase, which constitutes the majority of the eutectogel by volume (or mass), typically possesses an inherently high ionic species density, which is comparable to that of a typical IL. It is also characterized by significant hydrogen bonding between its subcomponents (i.e. HBA and HBD) that leads to thermodynamically nonideal mixture behavior. Thus, it is perhaps not surprising to see that hydrogen bonding has also been the most commonly employed noncovalent scaffold assembly strategy for eutectogels reported to date. Some of the key takeaways from the eutectogel materials summarized in Table 2 include:
|
(1) Hydrogen bonding, often in conjunction with solvophobic effects, is the most often used approach for noncovalent eutectogel scaffold assembly.
(2) Low molecular weight gelators (LMWGs) and bio-derived polymers have been more frequently employed for eutectogels than for ionogels in recent years.
(3) The use of two distinct, complementary scaffolds (i.e. double networks) is also more common for eutectogels, as of late.
(4) The vast majority of eutectogels reported to date have employed ChCl-based DESs.
4.1. Hydrogen bonding for eutectogel formation
In 2021, Wang and coworkers96 reported the formation of noncovalently cross-linked eutectogels in EG:ChCl (2:1) containing a double network of two distinct scaffolds: the first being a PVA commercial polymer (which formed crystalline interchain domains, driven by hydrogen bonding interactions) and the second being a network of poly(acrylic acid), PAA, that can form hydrogen bonds both with PVA and with itself, which was synthesized in situ (EG1, Table 2). While eutectogels could also be formed using PVA alone, superior mechanical properties were achieved by casting a PVA/DES solution containing acrylic acid monomer, which was subsequently polymerized in situ to form the PAA second network (Fig. 8). The eutectogel formulation that exhibited the highest tensile strength (2.6 MPa) and fracture toughness (8.4 MJ m−3), denoted by the authors as PVA/PAA9, contained approximately 4 wt% PVA and 37 wt% PAA.
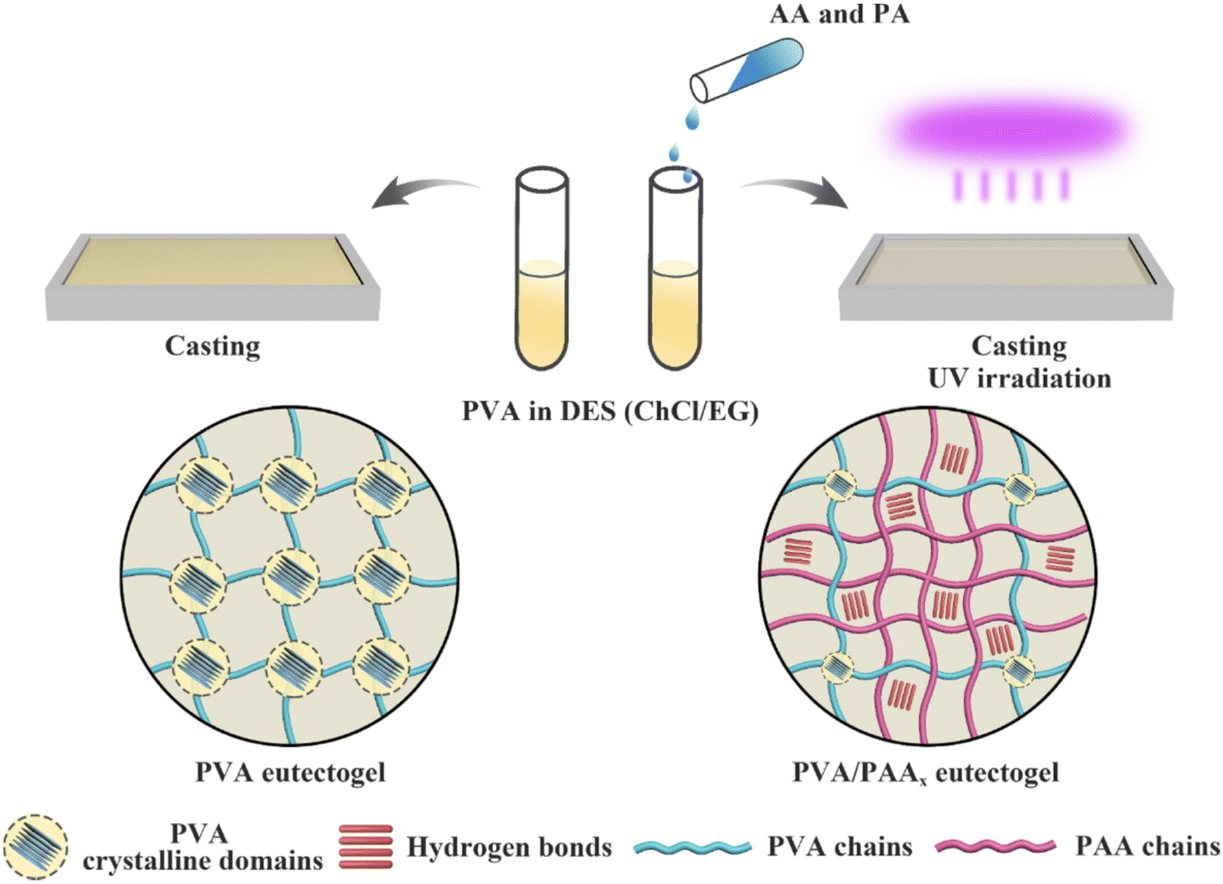 | ||
| Fig. 8 Schematic illustration for the synthesis of single network PVA (left) and double network PVA/PAA (right) eutectogels. Adapted from ref. 96 with permission, copyright 2021 American Chemical Society. | ||
A similar double network approach to forming eutectogels using EG:ChCl (2:1) was also reported earlier in 2021 by another group; in that case, the second network formed in situ via UV-initiated free radical polymerization was poly(acrylamide), PAAm (EG2).97 By heating these double network eutectogels to 130 °C, melt-injection molding and fiber extrusion of thermoplastic and self-healing composite gels could be realized. Coulombic interactions, meanwhile, played a supporting role in the double network eutectogels reported by Lan and coworkers (EG3).98 Here, a zwitterionic first network, poly(3-dimethyl(methacryloyloxyethyl)ammonium propane sulfonate), PDMAPS, was paired with a covalently cross-linked second network of poly(2-hydroxyethyl methacrylate), PHEMA. Both scaffolds were formed in situ within the EG:ChCl (2:1) DES through sequential UV-initiated polymerization steps (Fig. 9).
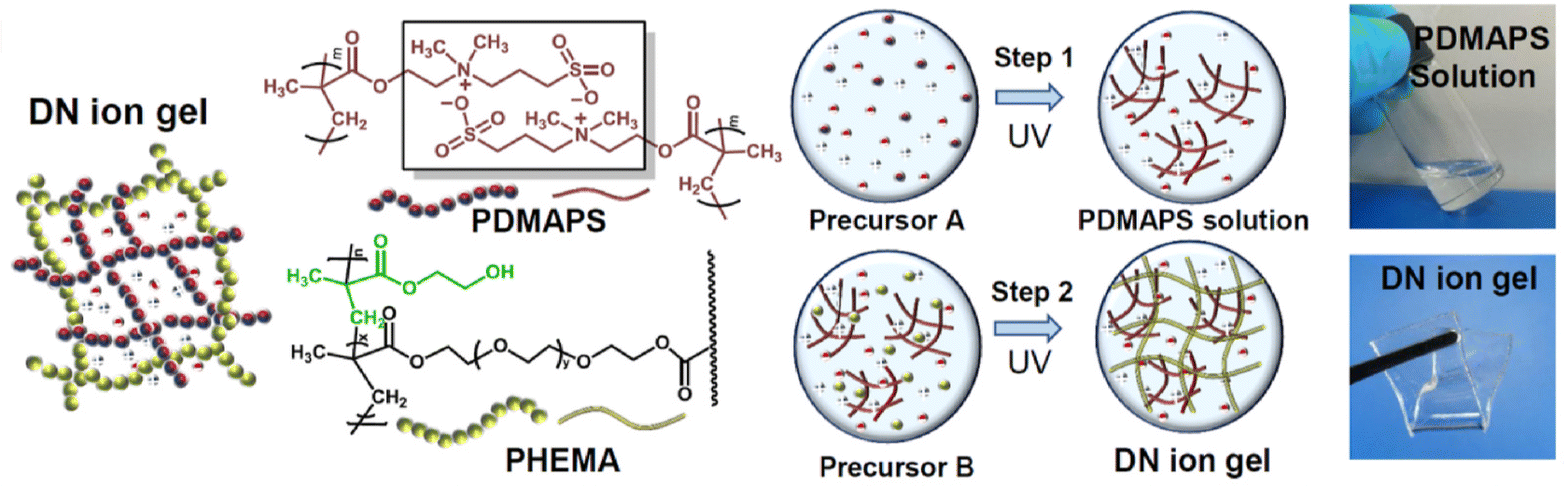 | ||
| Fig. 9 Schematic illustration of the chemical structures and formation strategy for double network eutectogels combining zwitterionic and covalently cross-linked hydrogen bonding scaffolds. Adapted from ref. 98 with permission, copyright 2021 Elsevier. | ||
4.2. LMWGs for eutectogels
Gelation can also be achieved by the addition of certain small molecule/low molecular weight gelators (LMWGs), some of which possess a surfactant nature, into DESs. Typically, a small amount (<5 wt%) of the LMWG is first dissolved into the DES at an elevated temperature; upon cooling to room temperature, self-assembly of a three-dimensional LMWG network can occur, leading to gelation. The major noncovalent interactions responsible for LMWG assembly in the formation of eutectogels are hydrogen bonding and solvophobic interactions, which often appear together. In 2021, the groups of Li and Liu reported two similar manifestations of “supramolecular-polymer double network” eutectogels that featured both a noncovalent LMWG first scaffold and a second, covalently cross-linked synthetic polymer scaffold.99,100 In each case, the LMWG was dissolved into the DES along with the monomer N-hydroxyethylacrylamide (HEAA) and a small amount of covalent cross-linker N,N′-methylenebis(acrylamide), MBA, at an elevated temperature. After the solution cooled and the first eutectogel network (LMWG) had been formed, the second network was created by in situ UV-initiated free radical copolymerization of HEAA and MBA to form a covalently cross-linked poly(N-hydroxyethylacrylamide) (PHEAA) synthetic polymer scaffold (Fig. 10). While both hydrogen bonds and solvophobic interactions drove the formation of the LMWG network, the authors suggested that additional hydrogen bonding between the two networks also contributed to the outstanding mechanical properties of these eutectogels. In one of the studies performed by this team, a series of bisgluconamide derivatives served as LMWGs to create eutectogels based on EG:ChCl (2:1) (EG4).99 For their second study, (R)-12-hydroxystearic acid hydrazide was used as the LMWG, while the DES of interest was a 2:1 molar ratio mixture of 1,3-propanediol (1,3-PD) with ChCl (EG12).100 In both cases, impressive eutectogel tensile fracture strains exceeding 4000% were achieved, as well as gels having a highly adhesive and self-healing nature.
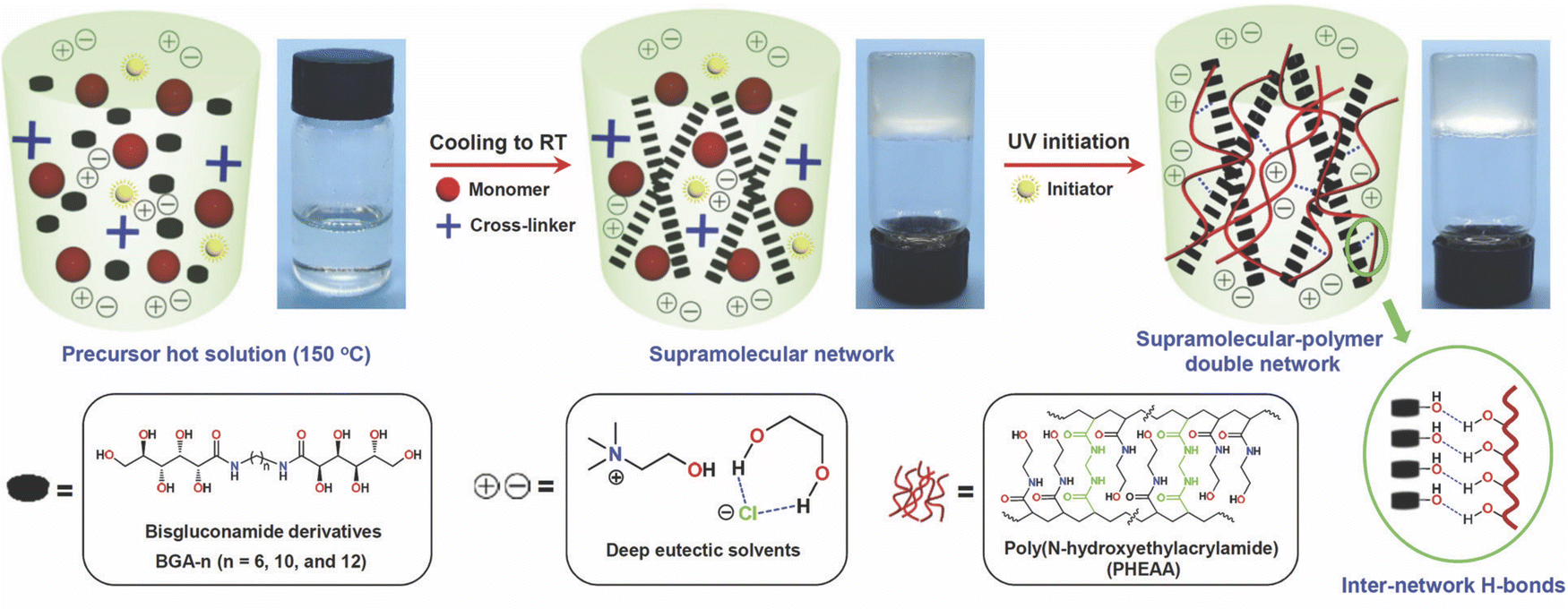 | ||
| Fig. 10 Schematic illustration of the synthesis process for supramolecular-polymer double network eutectogels based on a LMWG and a cross-linked synthetic polymer. Adapted from ref. 99 with permission, copyright 2021 Royal Society of Chemistry. | ||
Ruiz-Olles and coworkers reported LMWG-based single network eutectogels that employed the small molecule 1,3:2,4-dibenzylidene-D-sorbitol in EG:ChCl (2:1) (EG5) and a few other DESs.101 Gelation was achieved either by a heat/cool cycle or by ultrasonication; the latter approach was reported to significantly reduce eutectogel formation time from around 1 h to approximately 10 s. The L-amino acids isoleucine and tryptophan were reported to be effective LMWGs for the DES consisting of a 2:1 molar ratio of phenylacetic acid (PhAA) and ChCl by Marullo et al. (EG10).102 In the case of tryptophan, the authors suggested that π–π interactions amongst the LMWGs and with the PhAA HBD may also have contributed to the noncovalent assembly of these scaffolds.
Zhang and coworkers synthesized a series of LMWGs in the form of D-gluconic acid acetal alkyl amines having different alkyl chain lengths, and such materials were able to form eutectogels using several ChCl-based DESs (EG11).103 The authors reported that a delicate balance of noncovalent interactions among the LMWGs, including hydrogen bonding, π–π interactions, and solvophobic effects, led to the creation of 1D (fiber) assemblies and transparent eutectogels for EG:ChCl (2:1) and Gly:ChCl (2:1), while 2D (sheet) structures were formed within U:ChCl (2:1) that resulted in translucent gels (Fig. 11). These observations highlight the need to carefully consider the relative strengths and nature of scaffold–scaffold, DES–DES, and scaffold–DES interactions when designing future eutectogels with targeted properties.
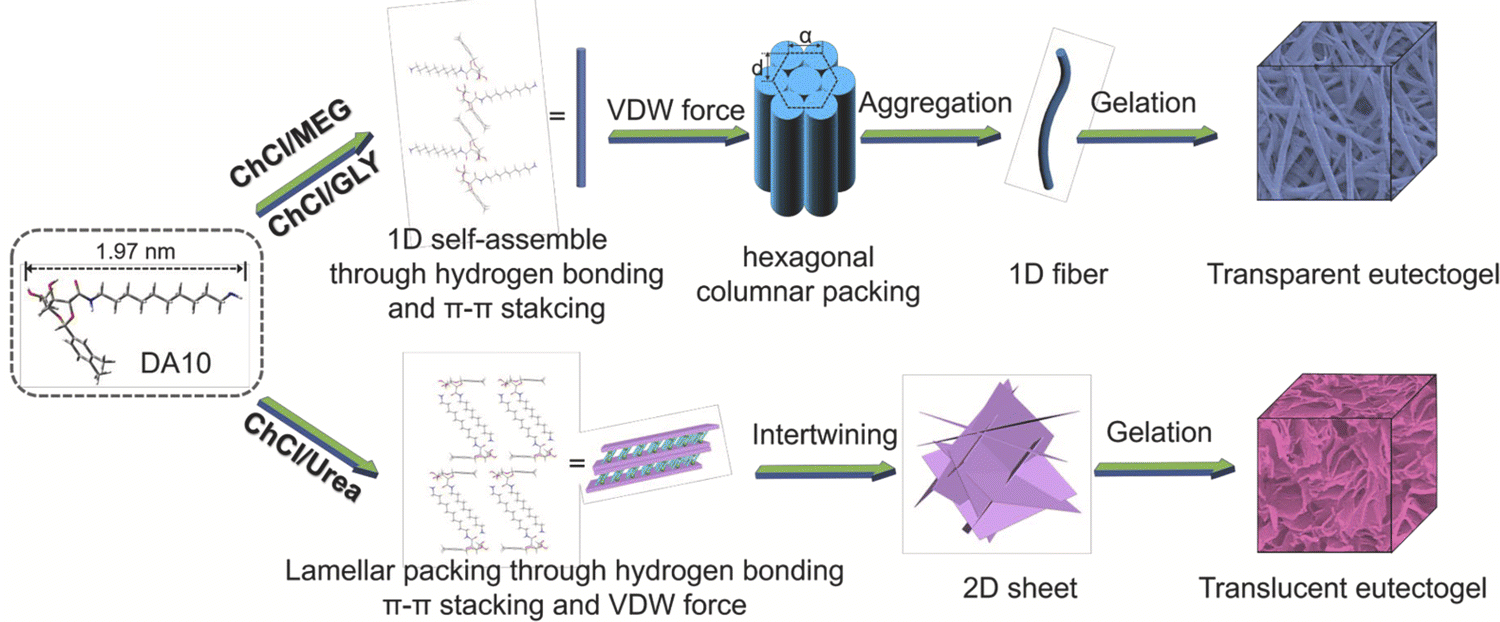 | ||
| Fig. 11 Proposed self-assembly mechanisms of a LMWG within different ChCl-based DESs. Adapted from ref. 103 with permission, copyright 2021 Elsevier. | ||
4.3. Bio-derived polymer eutectogel scaffolds
The use of bio-derived polymer scaffolds in eutectogels allows one to take advantage of both their typically biocompatible character and to exploit some of the inherent self-assembly strategies designed by nature (e.g. double/triple helix formation). In 2019, the Panzer group demonstrated the formation of thermoreversible eutectogels using gelatin as a biopolymer scaffold (EG6).104 While hydrogels containing similar gelatin contents were brittle and exhibited poor stretchability, it was observed that gelatin-supported eutectogels of EG:ChCl (2:1) could be repeatedly compressed up to at least 90% strain and were much tougher (Fig. 12). The DES environment therefore played a key role in modifying gelatin self-assembly upon cooling from a heated solution. It was posited that the number of triple helix cross-links was reduced in the eutectogel, while a greater number of noncovalent cross-links due to solvophobic interactions between gelatin chains was introduced. In a follow-up study, the same group reported that the intentional addition of a small amount of water (∼5–6 wt%) to EG:ChCl (2:1) or Gly:ChCl (2:1) DESs led to the creation of even tougher gelatin-supported eutectogels that also exhibited improved room temperature ionic conductivities (EG7).105
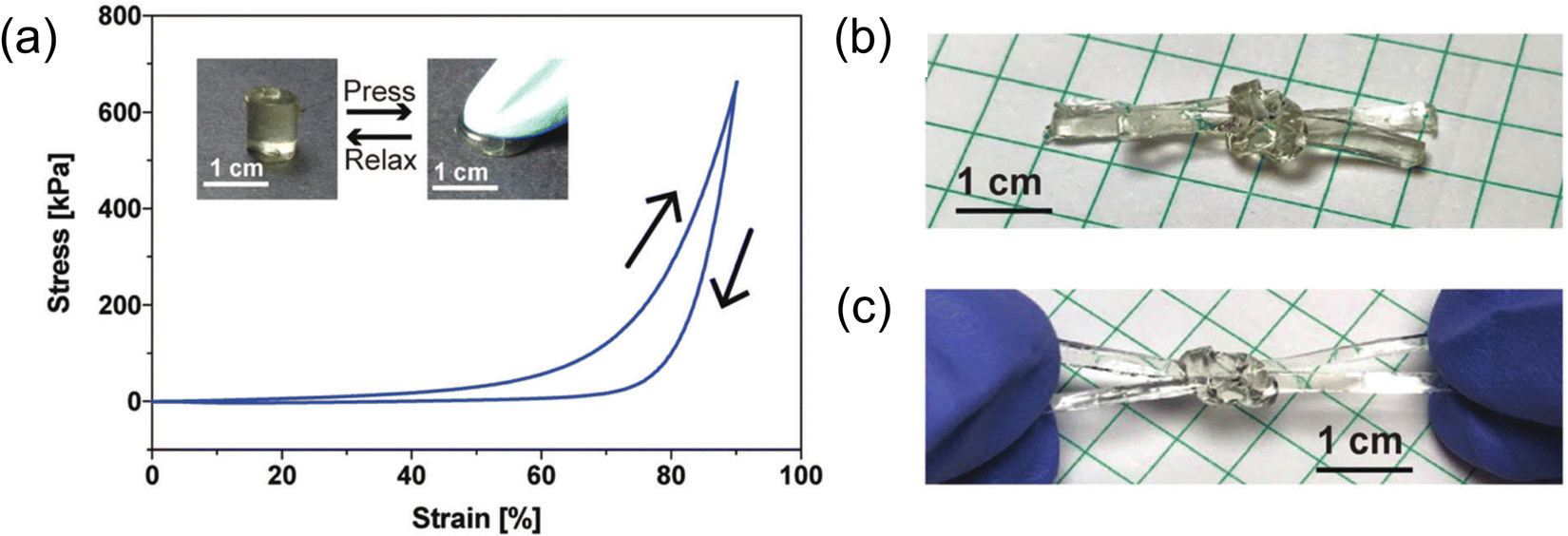 | ||
| Fig. 12 (a) Compressive stress–strain response of a 22 wt% gelatin-supported eutectogel with excellent shape recovery. (b) Square knot tied using two long, rectangular eutectogel strips. (c) Stretching the eutectogel square knot by hand. Adapted from ref. 104 with permission, copyright 2019 Royal Society of Chemistry. | ||
Smith and coworkers employed bacterial cellulose (BC) to created eutectogels using the Gly:ChCl (2:1) DES (EG8).106 Here, eutectogel materials were obtained by first creating BC-supported hydrogels, then solvent exchanging the water for 200-proof ethanol (i.e. alcogel formation), and finally equilibration of the alcogel in the DES and then removing any remaining ethanol by evaporation. Remarkably, BC-supported eutectogels containing as low as ∼1 wt% cellulose were reported. Hydrogen bonding among the BC microfibrils themselves and between the microfibrils and DES components were both found to be important for the scaffold self-assembly. Another biopolymer reported to form eutectogels is guar gum, a polysaccharide that is extracted from seeds of Cyamopsis tetragonoloba (EG9).107 Here, too, hydrogen bonding between the biopolymer scaffold and the DES components was key to the resulting eutectogel mechanical properties.
5. Inspiration from hydrogels containing noncovalent scaffolds
While many promising developments have been made in the design of novel ionogels and eutectogels in recent years, the fact remains that hydrogels dominate the scientific literature among all gel electrolyte materials. Indeed, a Web of Science database search for the topic “hydrogel” (in late April 2022) yielded over 35000 documents published just within the last 5 years. For comparison, a search for the terms “ionic liquid” and “gel” together over the same time span yielded 1300 results, while a search for “deep eutectic” and “gel” yielded approximately 150 items. Therefore, it stands to reason that the much broader hydrogel literature can likely provide inspiration for certain noncovalent scaffold designs that may translate well to new types of ionogels and eutectogels having desirable properties, such as the ability to self-heal. A comprehensive summary of all previous hydrogels that have employed noncovalent cross-linking strategies is beyond the scope of this review; rather, a few recent examples have been selected here to highlight some noteworthy approaches that should be of interest to researchers who are looking for new ways to design ionogels and/or eutectogels. For additional background, the reader is directed to several recent reviews of hydrogel materials.25,108–111
5.1. Hydrogen bonding strategies
Ultrastiff and tough supramolecular hydrogels featuring poly(methacrylamide-co-methacrylic acid)scaffolds, P(MAAm-co-MAA), were reported by Wang and coworkers in 2019.112 These materials, which exhibited Young's modulus values exceeding 200 MPa and tearing fracture energies above 20 kJ m−2, were synthesized by in situ copolymerization of methacrylamide (MAAm) and methacrylic acid (MAA) monomers in various molar ratios, and their outstanding mechanical properties were attributed to extensive hydrogen bonding between the co-monomers (Fig. 13). The authors also suggested that the hydrophobic methyl groups on the copolymer backbone served to enhance the stability of these dynamic hydrogen bonds while in the presence of a competitive hydrogen bond-forming species such as water. Indeed, they reported that the acrylate copolymer-supported hydrogel analogues were less stable when highly swollen with water. It appears likely, then, that methacrylate copolymers such as these may offer similar advantages for the development of highly stiff and tough eutectogels in the future, given the potential for hydrogen bonding competition from the DES components. They may also be advantageous for creating ionogels that feature water-miscible ILs (such as EMI ethyl sulfate), in which hydrogen bonding can also play an important role.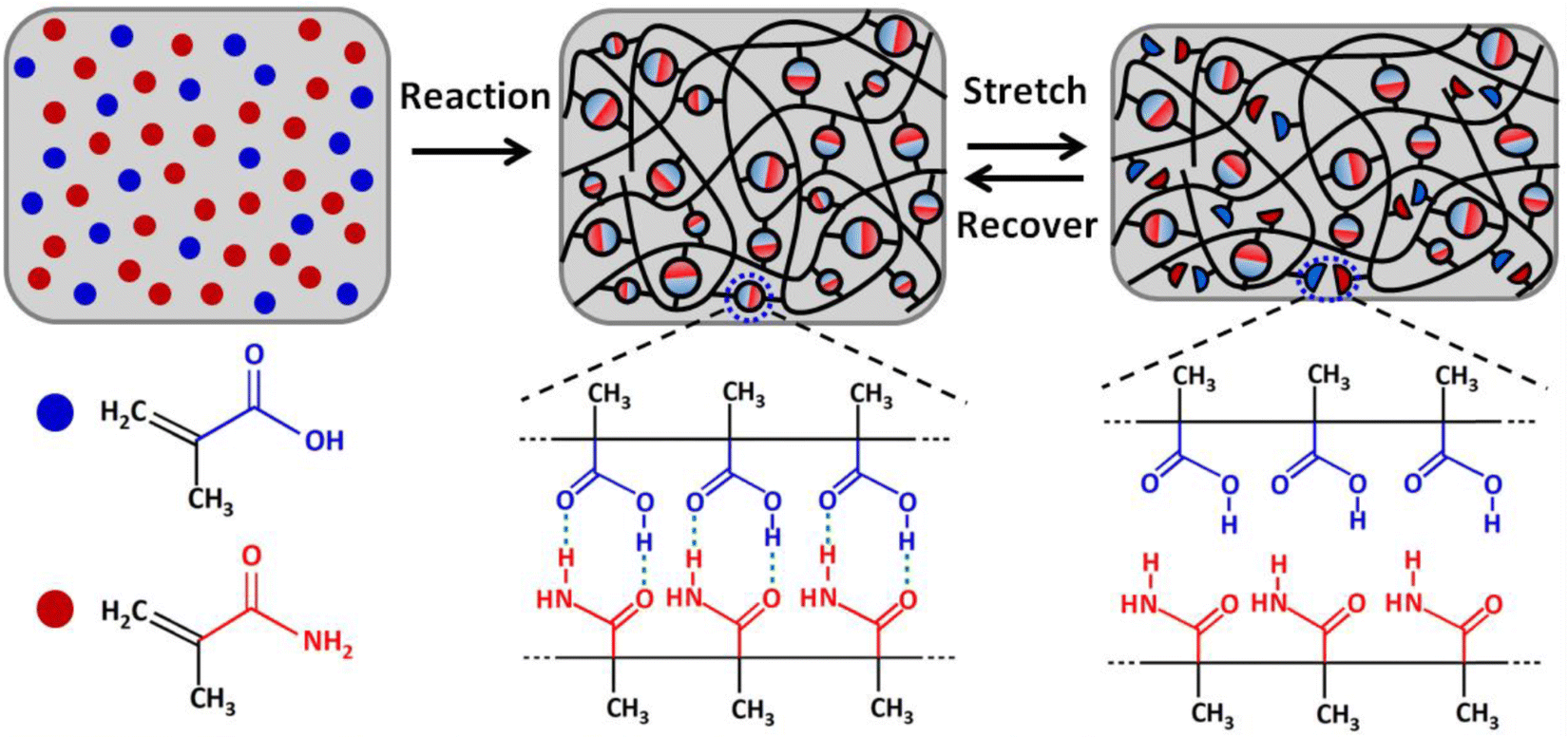 | ||
| Fig. 13 Schematic illustration of the formation of P(MAAm-co-MAA) scaffolds for realizing ultrastiff and tough hydrogels via robust hydrogen bonding. Adapted from ref. 112 with permission, copyright 2019 American Chemical Society. | ||
A combination of hydrogen bonding and coulombic interactions served to enable the formation of zwitterionic hydrogels described by Liu et al., which were supported by poly(acrylic acid-co-propylsulfonate dimethylammonium propylmethacrylamide) scaffolds, P(AA-co-PDP).113 By tuning the co-monomer ratio and total polymer content, highly stretchable hydrogels (>4000% strain) that also demonstrated an ability to self-heal were obtained. A related approach, reported by Zhang et al. in 2021, was the combination of PAA with a ZI small molecule (e.g. betaine, trimethylglycine) to form supramolecular hydrogels that exhibited strain-stiffening behavior and good adhesion to many different surfaces (Fig. 14).114 Such scaffolds may also prove to be useful for future eutectogels, including those that already contain a ZI component like betaine that serves as the DES HBA.
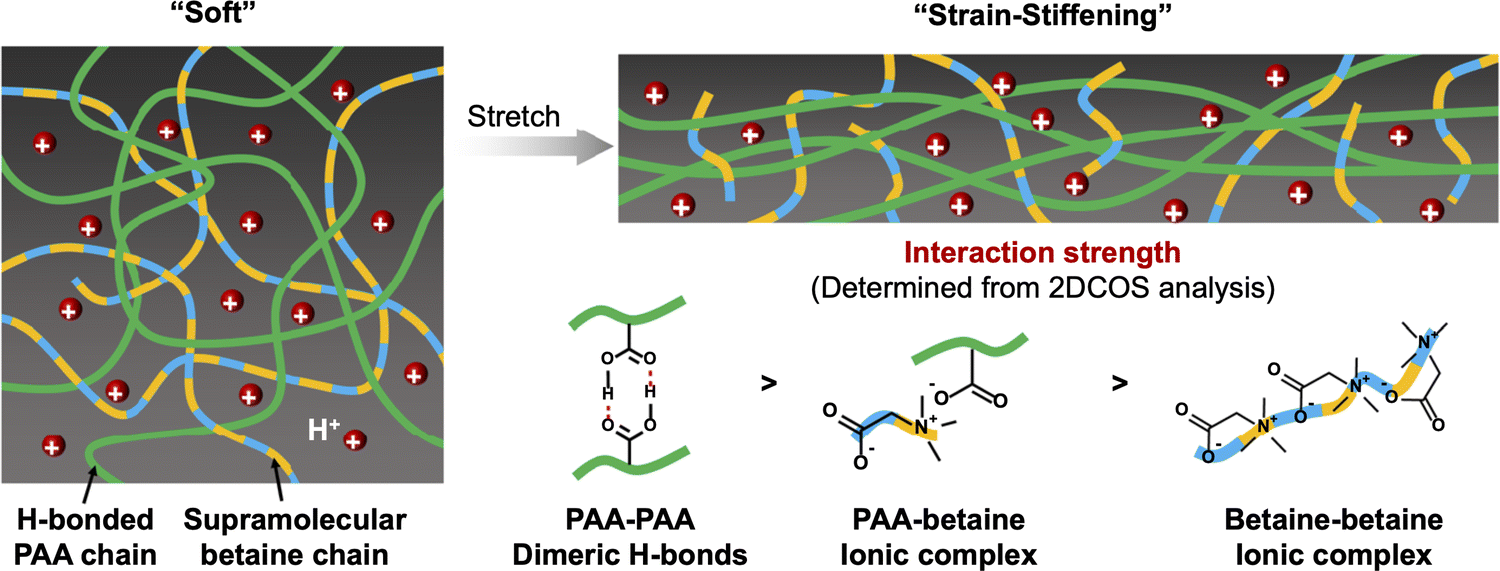 | ||
| Fig. 14 Schematic illustration of the strain-stiffening hydrogel scaffold consisting of a hydrogen bonding polymer (PAA) and zwitterionic small molecule (betaine) network. Adapted from ref. 114, published under an Open Access Creative Commons License. | ||
5.2. Other noncovalent assembly approaches
Metal–ligand coordination, a type of coulombic interaction, can be found in certain biological materials (including mussel cuticles115 and metalloproteins116), and it has been widely utilized to design hydrogels for practical applications such as wound healing dressings. A classic example is a calcium alginate hydrogel, in which negatively charged carboxylate ligands on two neighboring alginate biopolymer chains coordinate with/chelate a single Ca2+ divalent cation, thereby forming a robust, yet reversible, noncovalent cross-link.117 In a recent report, Jing and coworkers described the synthesis of a hybrid hydrogel scaffold consisting of a combination of nanochitin, PAA, and Al3+ cations.118 An abundance of carboxylate groups presented both on the biopolymer fibers (nanochitin) as well as on the PAA synthetic polymer facilitated the formation of metal–ligand coordination bonds between the scaffold materials; hydrogen bonding between the polymer chains also contributed to the gel mechanical properties. Zhang et al. employed a similar approach by copolymerizing acrylic acid and the ZI monomer DMAPS with AlCl3 added to a mixture of H2O and an IL (EMI Ac).119 These materials, dubbed “ionohydrogels,” thus leveraged a P(AA-co-DMAPS) scaffold that benefited from a combination of metal–ligand coordination, hydrogen bonds, and ZI coulombic interactions to generate a plethora of noncovalent cross-links (Fig. 15). Such hybrid gels may serve to marry desirable aspects of both hydrogels and ionogels together, thus blurring the line between these two historically distinct classes of materials.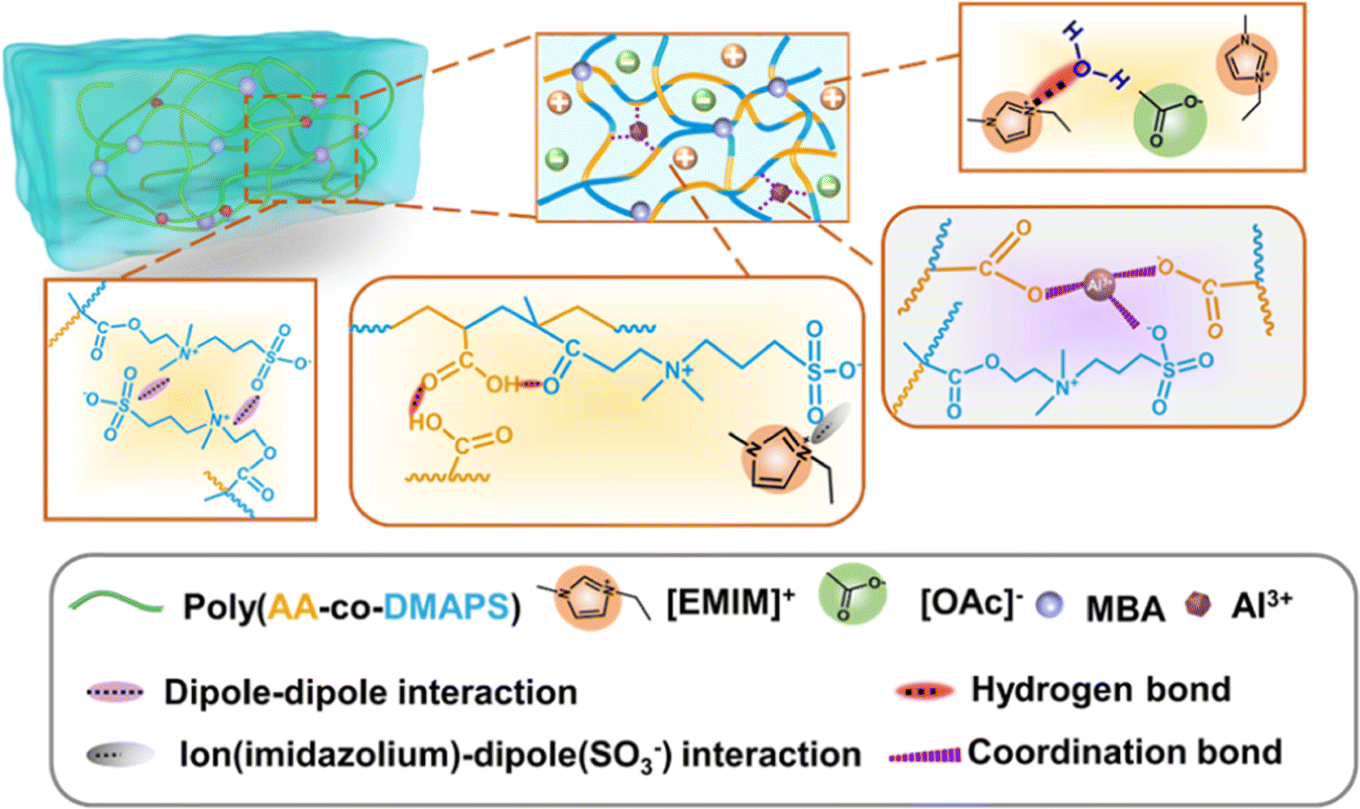 | ||
| Fig. 15 Schematic illustration of copolymer-supported ionohydrogels (i.e. IL + H2O mixture as the liquid phase) and the various noncovalent interactions present within these materials. Adapted from ref. 119 with permission, copyright 2022 American Chemical Society. | ||
Another useful noncovalent assembly strategy that has been used to realize supramolecular hydrogels is to leverage host–guest interactions (i.e. “molecular recognition”) between two or more subunits that utilizes some combination of hydrogen bonding/coulombic/van der Waals contributions and typically displays a large equilibrium binding constant. A recent example of a noncovalent hydrogel assembled largely due to host–guest interactions is the combination of a β-cyclodextrin polymer (P-CD) and an adamantane-modified PAA (PAA-Ad) reported by Hou and coworkers.120 Here, adamantane groups covalently attached to the PAA polymer played the role of “guests” that were tightly bound within the cavities presented by β-cyclodextrin unit “hosts” of P-CD (Fig. 16). Cyclodextrins have also been combined with IL-like dicationic imidazolium salts to realize supramolecular hydrogel networks.121,122 Another example of a host–guest interaction exploited for supramolecular hydrogel assembly was the use of a cucurbit[8]uril macrocyclic host molecule to bind two different guests at the same time within its barrel-shaped cavity, each from a distinct guest-bearing polymer chain.123
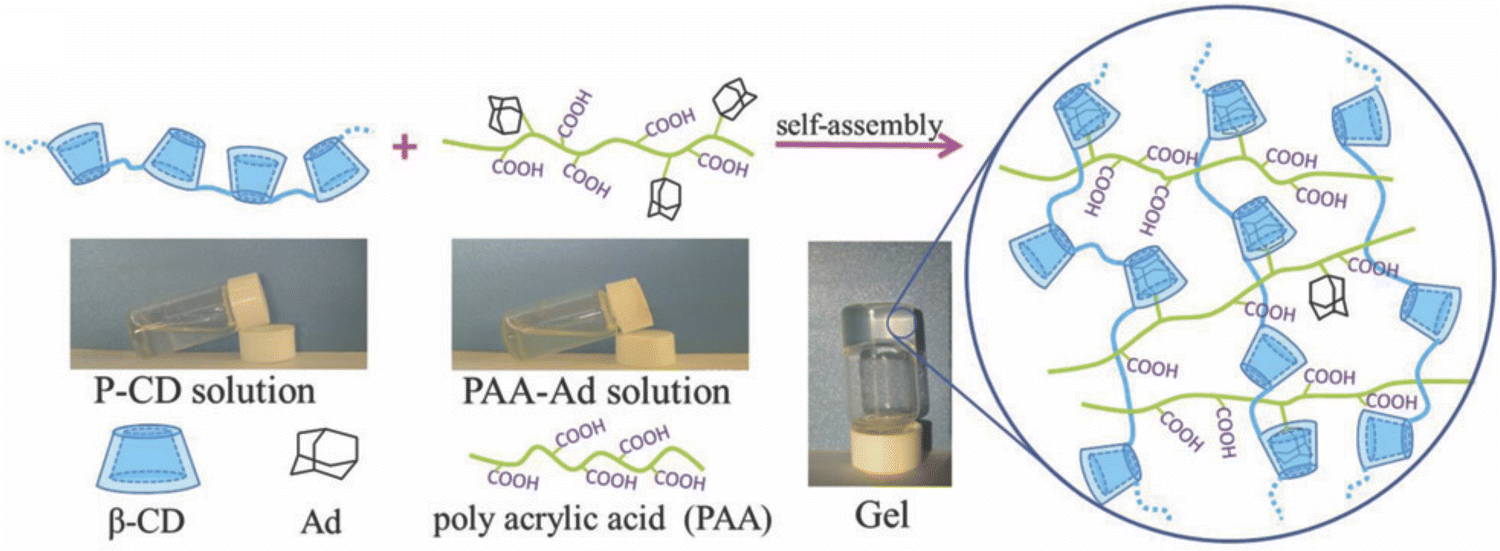 | ||
| Fig. 16 Schematic illustration of hydrogel formation via host–guest interactions between polymers bearing β-cyclodextrin and adamantane groups. Adapted from ref. 120 with permission, copyright 2019 Royal Society of Chemistry. | ||
While not yet extensively explored for the formation of hydrogels, the use of halogen bonding to realize noncovalent cross-links is also worthy of mentioning as an interesting strategy. Bertolani et al. created halogen bonding-based hydrogels via iodination of a human calcitonin-derived amyloidogenic peptide fragment, which served as a gelator.124 Meazza and coworkers125 demonstrated that halogen bonding between two complementary LMWGs (e.g. a bis(pyridyl urea) and 1,4-diiodotetrafluorobenzene) in methanol/H2O or DMSO/H2O mixtures led to the successful formation of “co-gels.” Since halogen bonds, in contrast to hydrogen bonds, tend to be hydrophobic by nature, this class of noncovalent interactions might prove to be useful for the design of future ionogels that feature water-immiscible ILs.
Finally, a clever strategy to leverage the inherent sensitivity of various noncovalent interactions to a change in the solvent environment was reported in 2020 by Xu et al., who demonstrated the formation of PVA-supported “exogels” that could be stretched to nearly 1000% strain prior to failure.126 Starting by dissolving the PVA scaffold in a good solvent (DMSO), the authors then performed a solvent exchange with water (a poorer solvent), which led to the assembly of a greater density of hydrogen bonded cross-links between PVA chains, and thus, robust hydrogels (Fig. 17). While this particular example utilized hydrogen bonding to form the gel scaffold, it stands to reason that other noncovalent interactions (coulombic, solvophobic, etc.), which will also be sensitive to the surrounding liquid properties (e.g. polarizability, dipolarity, etc.), could also be employed to form “exogels” whereby a scaffold dispersed in a good solvent is subsequently exchanged for an IL or DES as the poorer solvent. A high extent of removal of the original solvent can be promoted by repeated immersion of the gel in fresh IL/DES.
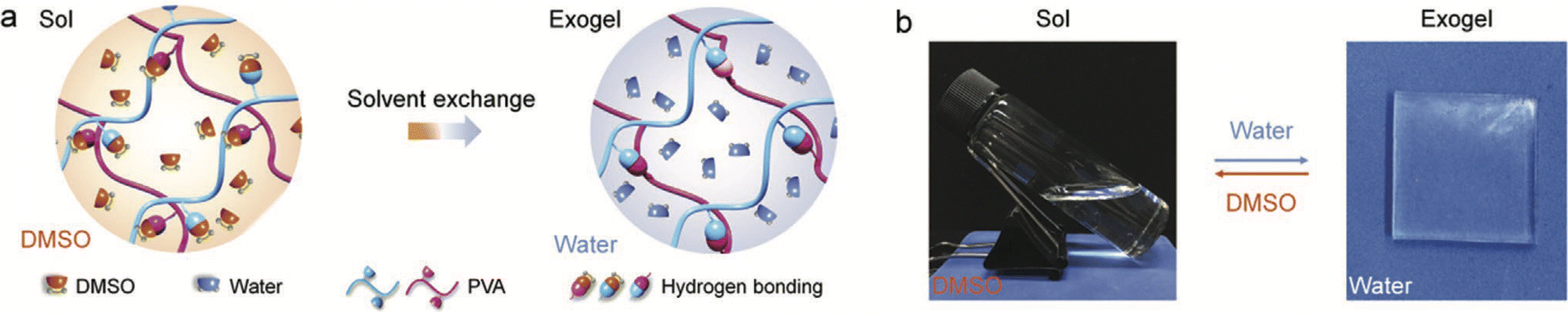 | ||
| Fig. 17 (a) Schematic illustration of PVA “exogel” formation using a solvent exchange strategy to replace a good solvent (DMSO) for the polymer with a poorer one (water), which promoted a greater number of interchain hydrogen bonding cross-links and robust hydrogel creation. (b) Reversible sol–gel transition of PVA upon exchanging solvents. Adapted from ref. 126 with permission, copyright 2020 Wiley-VCH. | ||
6. Conclusions and future opportunities
This review has sought to provide an overview of strategies that have been used to construct noncovalently cross-linked scaffolds for ionogels and eutectogels during the past several years. The inherently weaker and more reversible nature of noncovalent cross-links compared to covalent bonds can enable the development of nonaqueous gel electrolytes that possess outstanding stretchability and toughness, a capacity for self-healing, moderately high ionic conductivity, and superior nonvolatility in comparison to conventional hydrogel materials. The combination of two or more noncovalent interactions to form multiple cross-link types (e.g. hydrogen bonding along with coulombic and/or solvophobic effects) inside ionogels or eutectogels is an approach that can be used to more finely tune their mechanical behaviors, particularly if the relative densities of the different cross-link types within the gels can be varied. Importantly, there are currently many more reports of noncovalent scaffold assemblies for hydrogels in the literature than for ionogels and eutectogels combined. Therefore, great opportunities for translating some of these unique hydrogel approaches to the nonaqueous, ion-dense environments of ILs and DESs exist at the moment.When it comes to designing ionogels that are supported by noncovalently cross-linked scaffolds, synthetic (co)polymers have been the most popular choice as of late. Also noteworthy is the relatively small number of IL cations/anions that have been employed to create these ionogels (i.e. mostly 1,3-dialkylimidazolium or 1,1-dialkylpyrrolidinium cations with fluorinated anions, see Table 1). Since hydrogen bonding has already been shown to be an effective strategy of forming noncovalent cross-links within ILs, the investigation of additional bio-derived polymer scaffolds may be one worthy direction of study. Careful comparisons between any differences in scaffold self-assembly that are observed between ionogels featuring water-immiscible versus water-miscible ILs is another area that is ripe for fundamental exploration. Recently, zwitterionic functional groups have demonstrated an enticing ability to provide robust, noncovalent cross-links within ionogels due to coulombic and/or solvophobic interactions. Further studies to better understand the ramifications of choosing a specific ZI group chemistry for a given IL are thus of great interest.
In the area of eutectogels supported by noncovalent interactions within the scaffold, ChCl-based DESs (especially EG:ChCl (2:1), see Table 2) have been the primary focus in the literature to date. Thus, utilizing a greater diversity of DES chemistries for eutectogels, especially in the HBA identity (i.e. “beyond ChCl”), should be a priority for future investigations. Although both hydrogen bonding homopolymer networks and ZI scaffolds have each been employed in noncovalently cross-linked eutectogels, copolymers that combine both functionalities have been less explored to date. This could provide fertile ground for future eutectogel scaffold design; however, care should be taken to investigate the air stability of any eutectogels containing ZI functional groups, as most ZI moieties are highly hygroscopic. While small amounts of water in a eutectogel may, in some cases, provide an enhancement of ion transport and/or mechanical properties without disrupting the fundamental DES character, reproducibility could become an issue if the steady-state water content inside the gel is not controlled or quantified. Solvophobic interactions have also been underexplored for polymeric scaffold assembly in eutectogels up to this point. Here, synthetic copolymers that combine two or more monomers, each having a different extent of solubility in the DES, will likely provide a promising path to interesting new materials.
Comparatively, the use of metal–ligand coordination to form noncovalent cross-links in either ionogels or eutectogels has been fairly rare to date. This is likely because such an assembly strategy necessitates the addition of a multivalent salt (such as CaCl2 or AlCl3) whose presence may introduce undesirable anionic species into the final gel electrolyte material, as in the case of a >3 V class battery electrolyte. However, for the design of eutectogels that feature a ChCl-based DES, and thus will inherently contain a large concentration of chloride anions, this may not be a substantial concern. Manifestations of ionogels or eutectogels supported primarily by host–guest interactions or halogen bonding are also not yet commonly reported; it is expected that both of these strategies could lead to novel materials in the years ahead. In whatever manner they are created, ionogels and eutectogels that feature noncovalently cross-linked scaffolds can provide fertile ground for fundamental discovery in the realm of soft materials. Thanks to their superior resistance to volatilization, they will persist long after their hydrogel cousins have evaporated.
Abbreviations
| 1D | One-dimensional |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| 1,3-PD | 1,3-Propanediol |
| AA | Acrylic acid |
| Ac | Acetate anion |
| BA | Butyl acrylate |
| BC | Bacterial cellulose |
| BF4 | Tetrafluoroborate anion |
| BI | N,N′-(4,4′-Diphenylmethane)bismaleimide |
| BMI | 1-Butyl-3-methylimidazolium cation |
| BMP | 1-Butyl-1-methylpyrrolidinium cation |
| C | Coulombic interactions |
| ChCl | Choline chloride |
| DE-IM | 1,2-Dimethyl-3-ethoxyethylimidazolium cation |
| DES | Deep eutectic solvent |
| DSC | Differential scanning calorimetry |
| DMAAm | N,N-Dimethylacrylamide |
| DMAPS | 3-Dimethyl(methacryloyloxyethyl)ammonium propane sulfonate |
| DMSO | Dimethyl sulfoxide |
| EG | Ethylene glycol |
| EMI | 1-Ethyl-3-methylimidazolium cation |
| FSI | Bis(fluorosulfonyl)imide anion |
| Gly | Glycerol |
| H | Hydrogen bonding |
| HBA | Hydrogen bond acceptor |
| HBD | Hydrogen bond donor |
| HEAA | N-Hydroxyethylacrylamide |
| IL | Ionic liquid |
| LMWG | Low molecular weight gelator |
| MAA | Methacrylic acid |
| MAAm | Methacrylamide |
| MBA | N,N-Methylenebis(acrylamide) |
| MPC | 2-Methacryloyloxyethyl phosphorylcholine |
| OTf | Trifluoromethanesulfonate anion |
| P | π–π interactions |
| PAA | Poly(acrylic acid) |
| PAA-Ad | Adamantane-modified poly(acrylic acid) |
| P(AA-co-DMAPS) | Poly(acrylic acid-co-3-dimethyl(methacryloyloxyethyl) ammonium propane sulfonate) |
| P(AA-co-PDP) | Poly(acrylic acid-co-propylsulfonate dimethylammonium propylmethacrylamide) |
| PAAm | Poly(acrylamide) |
| PBA | Poly(butyl acrylate) |
| PBDT | Poly(2,2′-disulfonyl-4,4′-benzidine terephthalamide) |
| P-CD | β-Cyclodextrin polymer |
| P(DMAAm-co-AA) | Poly(N,N-dimethylacrylamide-co-acrylic acid) |
| PDMAPS | Poly(3-dimethyl(methacryloyloxyethyl) ammonium propane sulfonate) |
| PEGDMA | Poly(ethylene glycol)dimethacrylate |
| P(FMA-co-MMA) | Poly(furan-2-ylmethyl methacrylate-co-methyl methacrylate) |
| PhAA | Phenylacetic acid |
| PHEAA | Poly(N-hydroxyethylacrylamide) |
| PHEMA | Poly(2-hydroxyethyl methacrylate) |
| PIL | Poly(ionic liquid) |
| P(MAAm-co-MAA) | Poly(methacrylamide-co-methyl methacrylate) |
| P(MMA-co-BA) | Poly(methyl methacrylate-co-butyl acrylate) |
| P(MPC-co-SBVI) | Poly(2-methacryloyloxyethyl phosphorylcholine-co-sulfobetaine vinyl imidazole) |
| PS | Poly(styrene) |
| P(S-co-[VBMI+][PF6−]) | Poly(styrene-co-1-(4-vinylbenzyl)-3-methylimidazolium hexafluorophosphate) |
| PVA | Poly(vinyl alcohol) |
| P([VBIm+][BF4−]) | Poly(1-vinyl-3-butylimidazolium tetrafluoroborate) |
| PVDF | Poly(vinylidene fluoride) |
| P(VDF-co-HFP) | Poly(vinylidene-co-hexafluoropropylene) |
| RAFT | Reversible addition fragmentation chain transfer |
| S | Solvophobicity |
| SBVI | Sulfobetaine vinylimidazole |
| TFEMA | 2,2,2-Trifluoroethyl methacrylate |
| TFSI | Bis(trifluoromethanesulfonyl)imide anion |
| U | Urea |
| UPy | Ureido-pyrimidinone |
| UV | Ultraviolet |
| ZI | Zwitterionic |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Science Foundation (CBET-1802729) is gratefully acknowledged.References
- L. C. Tome, L. Porcarelli, J. E. Bara, M. Forsyth and D. Mecerreyes, Mater. Horiz., 2021, 8, 3239–3265 RSC.
- P. Walden, Bull. Acad. Imper. Sci., 1914, 8, 405–422 Search PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965–967 RSC.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- G. García, S. Aparicio, R. Ullah and M. Atilhan, Energy Fuels, 2015, 29, 2616–2644 CrossRef.
- Y. Kitazawa, K. Ueno and M. Watanabe, Chem. Rec., 2018, 18, 391–409 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- L. C. Tome and D. Mecerreyes, J. Phys. Chem. B, 2020, 124, 8465–8478 CrossRef CAS PubMed.
- L. Zhang, D. Jiang, T. Dong, R. Das, D. Pan, C. Sun, Z. Wu, Q. Zhang, C. Liu and Z. Guo, Chem. Rec., 2020, 20, 948–967 CrossRef CAS PubMed.
- N. Chen, H. Zhang, L. Li, R. Chen and S. Guo, Adv. Energy Mater., 2018, 8, 1702675 CrossRef.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- B. Joos, T. Vranken, W. Marchal, M. Safari, M. K. Van Bael and A. T. Hardy, Chem. Mater., 2018, 30, 655–662 CrossRef CAS.
- H. Qin and M. J. Panzer, ChemElectroChem, 2017, 4, 2556–2562 CrossRef CAS.
- S. F. Zopf, A. J. D’Angelo, H. Qin and M. J. Panzer, in Polymerized Ionic Liquids, ed. A. Eftekhari, Royal Society of Chemistry, 2018, pp. 381–415 Search PubMed.
- A. F. Visentin and M. J. Panzer, ACS Appl. Mater. Interfaces, 2012, 4, 2836–2839 CrossRef CAS.
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef CAS.
- B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818 CrossRef CAS PubMed.
- K. Xue, S. S. Liow, A. A. Karim, Z. Li and X. J. Loh, Chem. Rec., 2018, 18, 1517–1529 CrossRef CAS PubMed.
- K. Chen, Q. Lin, L. Wang, Z. Zhuang, Y. Zhang, D. Huang and H. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 9748–9761 CrossRef CAS PubMed.
- J. Yang, R. Bai, B. Chen and Z. Suo, Adv. Funct. Mater., 2020, 30, 1901693 CrossRef CAS.
- C. A. Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC ; discussion 81–96, 465–471.
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed.
- D. M. Correia, L. C. Fernandes, P. M. Martins, C. García-Astrain, C. M. Costa, J. Reguera and S. Lanceros-Méndez, Adv. Funct. Mater., 2020, 30, 1909736 CrossRef CAS.
- M. Forsyth, L. Porcarelli, X. Wang, N. Goujon and D. Mecerreyes, Acc. Chem. Res., 2019, 52, 686–694 CrossRef CAS PubMed.
- E. Frackowiak, G. Lota and J. Pernak, Appl. Phys. Lett., 2005, 86, 164104 CrossRef.
- H. Yoon, A. S. Best, M. Forsyth, D. R. MacFarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 4656–4663 RSC.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629–A1637 CrossRef CAS.
- M. Kerner, N. Plylahan, J. Scheers and P. Johansson, Phys. Chem. Chem. Phys., 2015, 17, 19569–19581 RSC.
- A. S. Shaplov, E. I. Lozinskaya, P. S. Vlasov, S. M. Morozova, D. Y. Antonov, P.-H. Aubert, M. Armand and Y. S. Vygodskii, Electrochim. Acta, 2015, 175, 254–260 CrossRef CAS.
- A. A. Fernandez, M. J. A. van Dongen, D. Blanco-Ania and P. H. J. Kouwer, RSC Adv., 2014, 4, 30267–30273 RSC.
- D. Dobler, T. Schmidts, C. Zinecker, P. Schlupp, J. Schafer and F. Runkel, AAPS PharmSciTech, 2016, 17, 923–931 CrossRef CAS PubMed.
- O. Stolarska, A. Pawlowska-Zygarowicz, A. Soto, H. Rodriguez and M. Smiglak, Carbohydr. Polym., 2017, 178, 277–285 CrossRef CAS PubMed.
- O. Stolarska, H. Rodríguez and M. Smiglak, Fluid Phase Equilib., 2016, 408, 1–9 CrossRef CAS.
- O. Stolarska, A. Soto, H. Rodríguez and M. Smiglak, RSC Adv., 2015, 5, 22178–22187 RSC.
- R. Lin, P.-L. Taberna, S. Fantini, V. Presser, C. R. Pérez, F. Malbosc, N. L. Rupesinghe, K. B. K. Teo, Y. Gogotsi and P. Simon, J. Phys. Chem. Lett., 2011, 2, 2396–2401 CrossRef CAS.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- A. H. Turner and J. D. Holbrey, Phys. Chem. Chem. Phys., 2019, 21, 21782–21789 RSC.
- Y. Zhang, D. Poe, L. Heroux, H. Squire, B. W. Doherty, Z. Long, M. Dadmun, B. Gurkan, M. E. Tuckerman and E. J. Maginn, J. Phys. Chem. B, 2020, 124, 5251–5264 CrossRef CAS PubMed.
- D. O. Abranches, L. P. Silva, M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, ChemSusChem, 2020, 13, 4916–4921 CrossRef CAS.
- D. V. Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef CAS.
- T. El Achkar, H. Greige-Gerges and S. Fourmentin, Environ. Chem. Lett., 2021, 19, 3397–3408 CrossRef CAS.
- V. Agieienko and R. Buchner, Phys. Chem. Chem. Phys., 2022, 24, 5265–5268 RSC.
- A. van den Bruinhorst, L. Kollau, M. C. Kroon, J. Meuldijk, R. Tuinier and A. C. C. Esteves, J. Chem. Phys., 2018, 149, 224505 CrossRef PubMed.
- D. Reuter, C. Binder, P. Lunkenheimer and A. Loidl, Phys. Chem. Chem. Phys., 2019, 21, 6801–6809 RSC.
- D. J. G. P. van Osch, L. F. Zubeir, A. van den Bruinhorst, M. A. A. Rocha and M. C. Kroon, Green Chem., 2015, 17, 4518–4521 RSC.
- C. Florindo, L. C. Branco and I. M. Marrucho, ChemSusChem, 2019, 12, 1549–1559 CrossRef CAS PubMed.
- C. D'Agostino, L. F. Gladden, M. D. Mantle, A. P. Abbott, E. I. Ahmed, A. Y. M. Al-Murshedi and R. C. Harris, Phys. Chem. Chem. Phys., 2015, 17, 15297–15304 RSC.
- E. O. Fetisov, D. B. Harwood, I. W. Kuo, S. E. E. Warrag, M. C. Kroon, C. J. Peters and J. I. Siepmann, J. Phys. Chem. B, 2018, 122, 1245–1254 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- M. B. Haider, D. Jha, R. Kumar and B. Marriyappan Sivagnanam, Int. J. Greenh. Gas Control, 2020, 92, 102839 CrossRef CAS.
- Z. Chen, T. L. Greaves, G. G. Warr and R. Atkin, Chem. Commun., 2017, 53, 2375–2377 RSC.
- K. Ueno, Polym. J., 2018, 50, 951–958 CrossRef CAS.
- K. Ueno and M. Watanabe, Langmuir, 2011, 27, 9105–9115 CrossRef CAS PubMed.
- K. Ueno, K. Hata, T. Katakabe, M. Kondoh and M. Watanabe, J. Phys. Chem. B, 2008, 112, 9013–9019 CrossRef CAS PubMed.
- T. Yan, Y. Zou, X. Zhang, D. Li, X. Guo and D. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 9856–9864 CrossRef CAS.
- T. Song, X. Zhang, Y. Li, K. Jiang, S. Zhang, X. Cui and L. Bai, Ind. Eng. Chem. Res., 2019, 58, 6887–6898 CrossRef CAS.
- T. P. Lodge and T. Ueki, Acc. Chem. Res., 2016, 19, 2107–2114 CrossRef PubMed.
- Y. He, P. G. Boswell, P. Buhlmann and T. P. Lodge, J. Phys. Chem. B, 2007, 111, 4645–4652 CrossRef CAS PubMed.
- J. F. Dobson and T. Gould, J. Phys. Condens. Matter, 2012, 24, 073201 CrossRef PubMed.
- L. M. de Espinosa, G. L. Fiore, C. Weder, E. Johan Foster and Y. C. Simon, Prog. Polym. Sci., 2015, 49-50, 60–78 CrossRef CAS.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- P. Wang, G. Li, W. Yu, C. Meng and S. Guo, Adv. Mater. Interfaces, 2022, 9, 2102426 CrossRef.
- R. Tamate, K. Hashimoto, T. Horii, M. Hirasawa, X. Li, M. Shibayama and M. Watanabe, Adv. Mater., 2018, 30, 1802792 CrossRef.
- M. A. B. H. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976–4983 CrossRef CAS PubMed.
- A. J. D'Angelo and M. J. Panzer, Chem. Mater., 2019, 31, 2913–2922 CrossRef.
- L. Rebollar and M. J. Panzer, ChemElectroChem, 2019, 6, 2482–2488 CrossRef CAS.
- M. E. Taylor and M. J. Panzer, J. Phys. Chem. B, 2018, 122, 8469–8476 CrossRef CAS PubMed.
- F. Lind, L. Rebollar, P. Bengani-Lutz, A. Asatekin and M. J. Panzer, Chem. Mater., 2016, 28, 8480–8483 CrossRef CAS.
- A. J. D'Angelo, J. J. Grimes and M. J. Panzer, J. Phys. Chem. B, 2015, 119, 14959–14969 CrossRef PubMed.
- A. F. Visentin, T. Dong, J. Poli and M. J. Panzer, J. Mater. Chem. A, 2014, 2, 7723–7726 RSC.
- L. Chen and M. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 25365–25373 CrossRef CAS PubMed.
- Z. Tang, X. Lyu, A. Xiao, Z. Shen and X. Fan, Chem. Mater., 2018, 30, 7752–7759 CrossRef CAS.
- A. Santic, M. Brinkkotter, T. Portada, L. Frkanec, C. Cremer, M. Schonhoff and A. Mogus-Milankovic, RSC Adv., 2020, 10, 17070–17078 RSC.
- R. Mantravadi, P. R. Chinnam, D. A. Dikin and S. L. Wunder, ACS Appl. Mater. Interfaces, 2016, 8, 13426–13436 CrossRef CAS PubMed.
- C. Rizzo, G. Misia, S. Marullo, F. Billeci and F. D'Anna, Green Chem., 2022, 24, 1318–1334 RSC.
- F. Billeci, F. D'Anna, H. Q. N. Gunaratne, N. V. Plechkova and K. R. Seddon, Green Chem., 2018, 20, 4260–4276 RSC.
- C. Rizzo, S. Marullo, P. R. Campodonico, I. Pibiri, N. T. Dintcheva, R. Noto, D. Millan and F. D’Anna, ACS Sustainable Chem. Eng., 2018, 6, 12453–12462 CrossRef CAS.
- R. J. Fox, D. Yu, M. Hegde, A. S. Kumbhar, L. A. Madsen and T. J. Dingemans, ACS Appl. Mater. Interfaces, 2019, 11, 40551–40563 CrossRef CAS PubMed.
- S. Xiang, F. Zheng, S. Chen and Q. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 20653–20661 CrossRef CAS.
- D. G. Seo and H. C. Moon, Adv. Funct. Mater., 2018, 28, 1706948 CrossRef.
- Y. M. Kim and H. C. Moon, Adv. Funct. Mater., 2020, 30, 1907290 CrossRef CAS.
- A. J. D'Angelo and M. J. Panzer, Adv. Energy Mater., 2018, 8, 1801646 CrossRef.
- M. E. Taylor, D. Clarkson, S. G. Greenbaum and M. J. Panzer, ACS Appl. Polym. Mater., 2021, 3, 2635–2645 CrossRef CAS.
- H. Qin and M. J. Panzer, Chem. Mater., 2020, 32, 7951–7957 CrossRef CAS.
- P. Guo, A. Su, Y. Wei, X. Liu, Y. Li, F. Guo, J. Li, Z. Hu and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 19413–19420 CrossRef CAS.
- Y. Wang, J. Wang, Z. Ma and L. Yan, ACS Appl. Mater. Interfaces, 2021, 13, 54409–54416 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, R. Plamthottam, M. Tebyetekerwa, J. Xu, J. Zhu, C. Zhang and T. Liu, Macromolecules, 2021, 54, 3832–3844 CrossRef CAS.
- J. Lan, B. Zhou, C. Yin, L. Weng, W. Ni and L.-Y. Shi, Polymer, 2021, 231, 124111 CrossRef CAS.
- K. Wang, H. Wang, J. Li, Y. Liang, X. Q. Xie, J. Liu, C. Gu, Y. Zhang, G. Zhang and C. S. Liu, Mater. Horiz., 2021, 8, 2520–2532 RSC.
- Y. Liang, K. Wang, J. Li, H. Wang, X. Q. Xie, Y. Cui, Y. Zhang, M. Wang and C. S. Liu, Adv. Funct. Mater., 2021, 31, 2104963 CrossRef CAS.
- J. Ruiz-Olles, P. Slavik, N. K. Whitelaw and D. K. Smith, Angew. Chem., Int. Ed., 2019, 58, 4173–4178 CrossRef CAS PubMed.
- S. Marullo, A. Meli, F. Giannici and F. D’Anna, ACS Sustainable Chem. Eng., 2018, 6, 12598–12602 CrossRef CAS.
- B. Zhang, H. Sun, Y. Huang, B. Zhang, F. Wang and J. Song, Chem. Eng. J., 2021, 425, 131518 CrossRef CAS.
- H. Qin, R. E. Owyeung, S. R. Sonkusale and M. J. Panzer, J. Mater. Chem. C, 2019, 7, 601–608 RSC.
- R. E. Owyeung, S. R. Sonkusale and M. J. Panzer, J. Phys. Chem. B, 2020, 124, 5986–5992 CrossRef CAS PubMed.
- C. J. Smith, 2nd, D. V. Wagle, N. Bhawawet, S. Gehrke, O. Holloczki, S. V. Pingali, H. O'Neill and G. A. Baker, J. Phys. Chem. B, 2020, 124, 7647–7658 CrossRef.
- J. Depoorter, A. Mourlevat, G. Sudre, I. Morfin, K. Prasad, A. Serghei, J. Bernard, E. Fleury and A. Charlot, ACS Sustainable Chem. Eng., 2019, 7, 16747–16756 CrossRef CAS.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Adv. Funct. Mater., 2020, 30, 2002759 CrossRef CAS.
- Y. Li, B. Xue and Y. Cao, ACS Macro Lett., 2020, 9, 512–524 CrossRef CAS.
- K. J. De France, E. D. Cranston and T. Hoare, ACS Appl. Polym. Mater., 2020, 2, 1016–1030 CrossRef CAS.
- C. Zhang, B. Wu, Y. Zhou, F. Zhou, W. Liu and Z. Wang, Chem. Soc. Rev., 2020, 49, 3605–3637 RSC.
- Y. J. Wang, X. N. Zhang, Y. Song, Y. Zhao, L. Chen, F. Su, L. Li, Z. L. Wu and Q. Zheng, Chem. Mater., 2019, 31, 1430–1440 CrossRef CAS.
- C. Liu, F. Li, G. Li, P. Li, A. Hu, Z. Cui, Z. Cong and J. Niu, ACS Appl. Mater. Interfaces, 2022, 14, 9608–9617 CrossRef PubMed.
- W. Zhang, B. Wu, S. Sun and P. Wu, Nat. Commun., 2021, 12, 4082 CrossRef CAS.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- J. M. Harrowfield, C. R. Chim., 2005, 8, 199–210 CrossRef CAS.
- C. Wang, T. Yokota and T. Someya, Chem. Rev., 2021, 121, 2109–2146 CrossRef CAS.
- X. Jing, P. Feng, Z. Chen, Z. Xie, H. Li, X.-F. Peng, H.-Y. Mi and Y. Liu, ACS Sustainable Chem. Eng., 2021, 9, 9209–9220 CrossRef CAS.
- X. Zhang, C. Cui, S. Chen, L. Meng, H. Zhao, F. Xu and J. Yang, Chem. Mater., 2022, 34, 1065–1077 CrossRef CAS.
- N. Hou, R. Wang, R. Geng, F. Wang, T. Jiao, L. Zhang, J. Zhou, Z. Bai and Q. Peng, Soft Matter, 2019, 15, 6097–6106 RSC.
- F. D'Anna, P. Vitale, S. Marullo and R. Noto, Langmuir, 2012, 28, 10849–10859 CrossRef PubMed.
- C. Rizzo, F. D'Anna, S. Marullo, P. Vitale and R. Noto, Eur. J. Org. Chem., 2014, 1013–1024 CrossRef CAS.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS.
- A. Bertolani, L. Pirrie, L. Stefan, N. Houbenov, J. S. Haataja, L. Catalano, G. Terraneo, G. Giancane, L. Valli, R. Milani, O. Ikkala, G. Resnati and P. Metrangolo, Nat. Commun., 2015, 6, 7574 CrossRef PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- L. Xu, S. Gao, Q. Guo, C. Wang, Y. Qiao and D. Qiu, Adv. Mater., 2020, 32, 2004579 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |