
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatial atmospheric pressure molecular layer deposition of alucone films using dimethylaluminum isopropoxide as the precursor†
Hardik
Jain
 ab,
Mariadriana
Creatore
b and
Paul
Poodt
*ab
ab,
Mariadriana
Creatore
b and
Paul
Poodt
*ab
aTNO/Holst Centre, 5656 AE Eindhoven, Netherlands. E-mail: paul.poodt@tno.nl
bDepartment of Applied Physics, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
First published on 21st April 2022
Abstract
Trimethylaluminum is the most used aluminum precursor in atomic and molecular layer deposition (ALD/MLD). It provides high growth-per-cycle (GPC), is highly reactive and is relatively low cost. However, in the deposition of hybrid alucone films, TMA tends to infiltrate into the films requiring very long purge steps and thereby limiting the deposition rate (nm s−1) of the process. From our previous studies, we know that dimethylaluminum isopropoxide (DMAI) could be a potential candidate to substitute TMA in alucone depositions as it does not seem to infiltrate into the films. In this study, we perform a more detailed investigation of MLD of alucone on an atmospheric pressure spatial MLD system using DMAI as the aluminum precursor. The effect of deposition temperature and reactant purge times on the overall GPC has been investigated and a decreasing GPC with increasing deposition temperature and increasing EG purge time has been observed. Furthermore, the DMAI alucone films have been compared for their chemical environment and degradation with the films prepared using TMA and EG, showing striking similarities between the two. The results demonstrate that DMAI can be used as an alternative precursor to TMA for MLD of alucone films and this work can be used as a guide for designing efficient MLD processes in the future.
Introduction
Molecular layer deposition (MLD), a thin film deposition technique analogous to atomic layer deposition (ALD), has gathered significant attention in the past few years. With all the advantages of ALD such as precise control over the growing film thickness, exceptional uniformity and excellent conformality, MLD further extends ALD by enabling the deposition of purely organic1–3 and hybrid organic–inorganic films.4–6 Owing to their own unique properties, MLD films have found potential applications in flexible electronics,7–9 catalysis,10 lithium-ion batteries,11 modifying desalination membranes,12 tuning the wettability of pharmaceutical drugs13 and as inhibitor films in area-selective deposition (ASD).14 In order to facilitate the industrial employment of MLD processes, high deposition rates and low-cost precursors, processes and equipment are required. Spatial ALD,15 a technique that is based on spatial separation of the reactants rather than temporal separation has been previously used to increase the deposition rates of many ALD processes.16–18 The high deposition rates are realized by employing very short cycle times which are mainly made possible by the fact that long purge steps are not necessary. Similarly, spatial MLD can also be used to increase the deposition rate of hybrid films and has been previously explored for purely organic MLD films.12,19 In addition to the choice of a reactor, the choice of precursors and co-reactants is of equal importance in determining the deposition rate20 of MLD processes.Amongst hybrid films, alucone films are one of the most widely studied examples. Alucone films have been realized by using various organic co-reactants (e.g. ethylene glycol,4 hydroquinone,21 glycerol,22 glycidol,23 1,4 butane diol,24 1,4-butynediol,24 and 4-mercaptophenol25) while the metal–organic precursor for aluminum employed in most of these examples is trimethylaluminum (TMA), as TMA is known to have a very high vapor pressure and a high reactivity towards most of the common reactants like water,26 H2O2,27 O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and ethylene glycol.4 It, therefore, requires very short exposure times to saturate the growth surface and appears to be suitable for upscaling ALD/MLD processes.29 However, a drawback of using TMA in high throughput MLD of alucone films is the fact that it tends to infiltrate into porous MLD films during deposition requiring very long purge steps to outgas. Thus, the purge step that is ideally redundant or very short in a spatial reactor becomes necessary and very long when it comes to alucone films prepared using TMA. This eventually leads to very long cycle times and very low deposition rates4,20,30 and although the deposition rates can be slightly improved by optimizing the process parameters,20 TMA-based MLD processes will be very challenging to employ in applications that require high deposition rates. This problem can be potentially solved by using aluminum precursors that do not infiltrate during deposition.
28 and ethylene glycol.4 It, therefore, requires very short exposure times to saturate the growth surface and appears to be suitable for upscaling ALD/MLD processes.29 However, a drawback of using TMA in high throughput MLD of alucone films is the fact that it tends to infiltrate into porous MLD films during deposition requiring very long purge steps to outgas. Thus, the purge step that is ideally redundant or very short in a spatial reactor becomes necessary and very long when it comes to alucone films prepared using TMA. This eventually leads to very long cycle times and very low deposition rates4,20,30 and although the deposition rates can be slightly improved by optimizing the process parameters,20 TMA-based MLD processes will be very challenging to employ in applications that require high deposition rates. This problem can be potentially solved by using aluminum precursors that do not infiltrate during deposition.
Several alternative aluminum precursors exist, for example, halide-based substitutes such as Al2Cl6.31 However the corrosive nature of hydrogen halides created as byproducts in the process can be detrimental to the reactor parts and the films themselves.32 Furthermore, these precursors also pose the risk of halide incorporation into the film.31 Other alternatives to TMA include amide based Al(NMe2)3 which is known to be highly reactive and suitable for oxides33 but it exists in its solid phase (m.p. = 82–84 °C) and has a very low vapor pressure (∼1 Torr @ 78 °C) compared to TMA (∼145 Torr @ 78 °C). Although [Al(NEt2)3]2 exists in its liquid phase at low temperatures, its low vapor pressure (∼0.045 Torr @ 80 °C) is still an issue.34 Amongst alkoxide-based alternatives, dimethylaluminum isopropoxide (DMAI) has been previously proved to be an effective alternative to TMA in depositing alumina films by CVD35,36 and ALD.37,38 DMAI is heteroleptic, has comparatively high vapor pressure (∼9 Torr @ 66.5 °C) and is known to exist as a dimer in the gas phase even at high temperatures39 which reduces its pyrophoricity. In our previous work,20 we briefly demonstrated the MLD of alucone using DMAI and ethylene glycol (EG) and also showed that DMAI, unlike TMA, shows a negligible influence of its purge time on the overall growth-per-cycle (GPC) probably because it does not infiltrate into the MLD films. The work showed DMAI's great potential for the high deposition rate MLD of alucones. However, we did not investigate the effect of other parameters like reactant exposure times, purge times and deposition temperature on alucone growth and did not evaluate if the properties of the resultant DMAI + EG films are similar to those of films prepared using TMA + EG.
Hence, in this work, we will investigate the spatial MLD of alucone using DMAI in more detail. We have investigated the growth of DMAI + EG alucone films and studied the impact of deposition temperature and EG purge time on the overall GPC of the process. To investigate the effect of the choice of precursor on film properties, we have made a detailed comparison of various properties between films prepared using DMAI and TMA including their ambient stability.
Experimental details
Materials and experimental methods
For the experimental work, trimethylaluminum (Akzo Nobel, semiconductor grade), dimethylaluminum isopropoxide (Strem Chemicals) and ethylene glycol (Sigma Aldrich, 99.8%) were used as reactants in this study. All three reactants were dosed using a dip-tube bubbler assembly. In the case of alucone films prepared using TMA and EG, the TMA bubbler was kept at room temperature while the EG bubbler was heated to 80 °C. The depositions carried out using DMAI and EG by keeping the DMAI bubbler at room temperature and the EG bubbler at 80 °C yielded films with a very low GPC. Hence, to increase the vapor pressure of the reactants, the DMAI bubbler was heated to 75 °C and the EG bubbler was heated to 100 °C. Partial pressures of the reactants were further set by adjusting the carrier and dilution flows. Double-side polished Si wafers were used as substrates in this work.All experiments in this work were performed using a rotary, atmospheric pressure spatial ALD/MLD setup described in detail before.29 A schematic of the setup is shown in Fig. 1. The exposure and purge times of the reactants at a given point on the substrate are calculated using eqn (1) where L is the length of the arc that the point traces in the reactant/purge zone at a radius r from the center of the injector head and f is the rotation frequency:
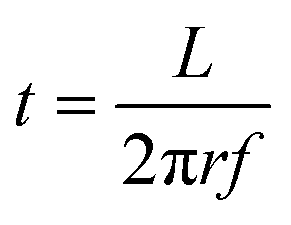 | (1) |
 | ||
| Fig. 1 (a) A schematic drawing of the bottom side of the spatial MLD injector head. (b) A schematic drawing of the entire reactor. The substrate is placed in between the stationary injector head and the rotary substrate table. Each rotation of the substrate table sequentially exposes the substrate to the precursor and the co-reactant and corresponds to one MLD cycle. The entire setup is placed inside an oven heated to the desired deposition temperature. | ||
For a fixed rotation frequency, a reactant's exposure and purge times are fixed as well and are co-related. In cases where an additional purge time for a reactant was required, we adopted a similar strategy used before,20 where the rotation was halted after the reactant's dose during which the stationary film is simply purged by the overhead N2. Once deposited, all film characterization studies were carried out ex situ.
Film thickness measurements
The film thicknesses were determined using a Horiba Jobin Yvon spectroscopic ellipsometer. Due to the limited stability of the alucone films in air,4,22 the measurements were performed within 15 min after deposition. The film thickness values were extracted using the Cauchy dispersion relation in the spectral range of 1–3.5 eV. A 3-layer stack model was used with the Si substrate as the bottom, a native SiO2 of 2 nm as the middle and the alucone film as the top layer.IR spectroscopy
Fourier transfer infrared spectroscopy (FTIR) of the alucone films was performed using a VERTEX 70 spectrometer from Bruker. The measurements were performed with as limited ambient exposure as possible (<15 min). Absorbance spectra were collected within a wavenumber range of 400–4000 cm−1 with a resolution of 4 cm−1 and averaged over 512 scans. The absorbance spectrum arising from the employed Si substrate was measured before each deposition and was accounted for while calculating the respective film absorbance spectrum. The baseline for each spectrum was corrected manually using the OriginPro software.X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) spectra were acquired using a Thermo Scientific KA1066 spectrometer equipped with a monochromatic Al Kα source (E = 1.487 keV). The survey spectra of the top surface of the films were acquired using a pass energy of 200 eV. In order to know the relative elemental composition and the environment within the bulk of the films, the films were sputtered using an ion gun with the lowest possible ion energy of 200 eV in several steps of 15 s each. High resolution spectra of Al 2p, O 1s and C 1s regions were collected at each step. The binding energies in the spectra were referenced to the adventitious C 1s peak at 284.8 eV to correct for any shifts. Furthermore, all of the acquired peaks were fitted with the Thermo Scientific™ Avantage™ software using Gaussian–Lorentzian line shapes and a ‘smart’ background.X-Ray reflectivity
X-ray reflectivity (XRR) analysis was performed using a PANalytical X'Pert PRO diffractometer. A Cu anode with Kα radiation of 1.54 Å wavelength was used with a filament current of 40 mA and a voltage of 45 kV. ω–2θ scans were acquired from 0.01° to 1.5° with a step size of 0.01°. The resultant data were analyzed using PANalytical's X'Pert Reflectivity software to extract the thicknesses and densities of the films. Although the exposure of the films to the ambiance was limited to less than 5 min before the start of the measurements, sample alignment and the measurements themselves were performed in an ambient atmosphere and the thicknesses and compositions of the measured films could have continuously changed during the measurements. In order to minimize the impact, we have only involved the first 4–5 fringes in the fitting during which the relative degradation in the films is believed to be minimal.Results and discussion
We have investigated the variation of film thickness with the number of MLD cycles at a constant substrate temperature of 200 °C and constant exposure times of DMAI and EG (380 ms) as shown in Fig. 2a. The carrier gas flows of DMAI and EG were set to 100 sccm each and were further diluted by additional 300 sccm of the inert gas (total flow = 400 sccm). The purge times of DMAI and EG were also fixed at ∼4.2 s and ∼1 s respectively. It can be observed that the film thickness increases linearly with the number of cycles indicating a GPC of ∼0.04 nm.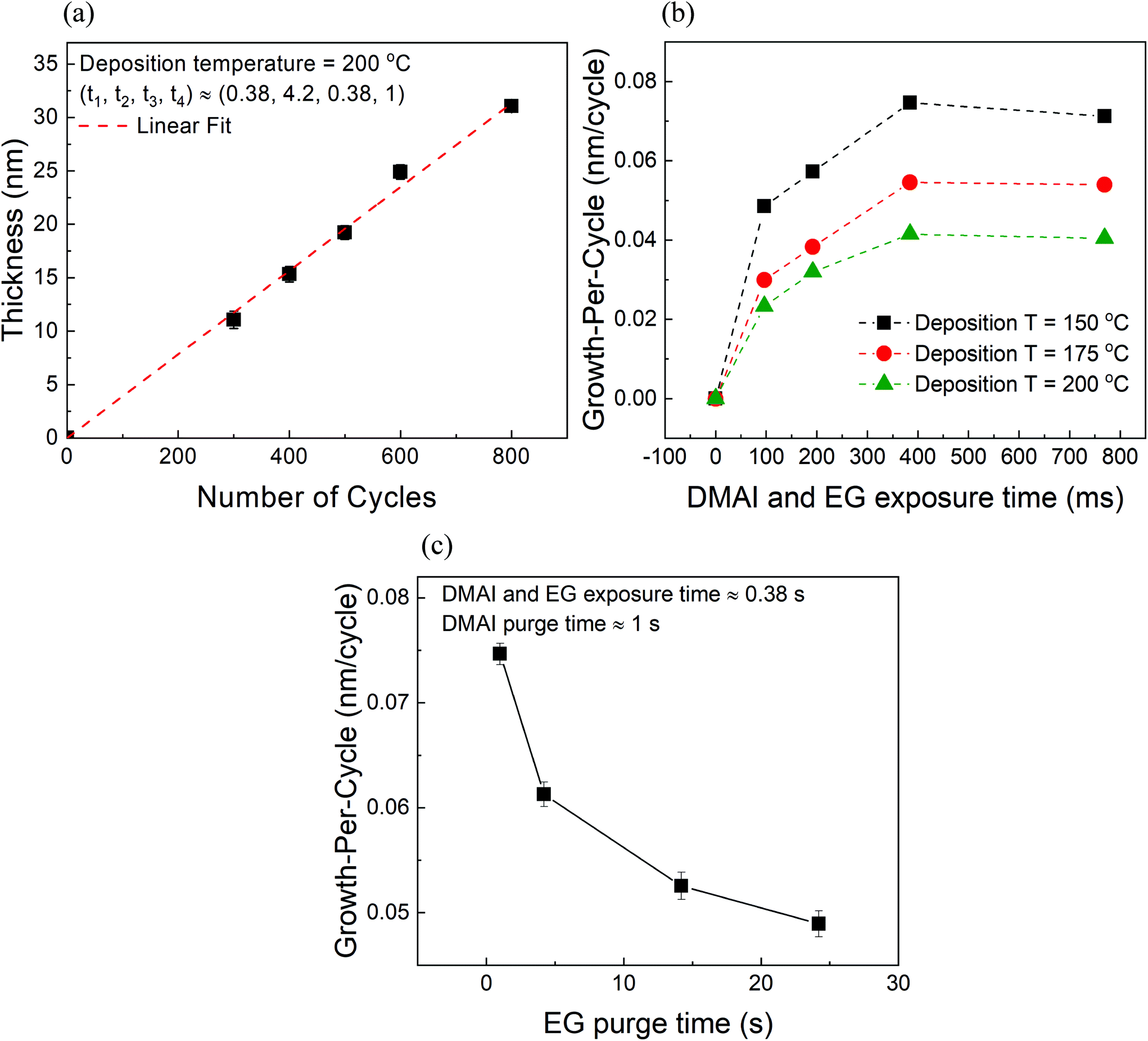 | ||
| Fig. 2 (a) Measured film thickness vs. the number of MLD cycles at 200 °C deposition temperature where t1 and t3 are the exposure times of DMAI and EG respectively and t2 and t4 are their respective purge times in seconds. (b) Growth-per-cycle (GPC) vs. reactant exposure time. (c) GPC as a function of EG purge time. In these experiments conducted at 150 °C, the DMAI and EG exposure time and the DMAI purge time were kept constant at 0.38 s, 0.38 s and 1 s respectively while the EG purge time was varied systematically. The DMAI and EG carrier and dilution flows were set at 100 and 300 sccm respectively. | ||
Fig. 2b shows the variation of growth-per-cycle (GPC) with respect to the exposure time of DMAI and EG at different substrate temperatures. It can be observed that the GPC at a given exposure time decreases with increasing deposition temperature. An ALD temperature window within which the GPC of a process remains relatively constant is not observed within the explored range of 150–200 °C. Although the temperature range explored here is relatively small, previously studied hybrid MLD processes also in most cases show a decreasing trend in the GPC with increasing deposition temperature.4–6,22 Therefore, it appears that the often reported ALD temperature window does not exist in the MLD of alucone. Furthermore, the GPC at each temperature also appears to increase with exposure time and at long enough exposure times it appears to saturate. However, it must be kept in mind that in these experiments, the individual purge times of the reactants are coupled to their exposure times and hence, one might not be able to see the effect of reactant purge times on the GPC exclusively. From our previous work, we know that providing longer purge times for DMAI has no significant effect on the overall GPC.20 In the present work, however, we found that providing longer purge times for EG led to a decrease in the GPC (Fig. 2c). While performing these experiments at a deposition temperature of 150 °C, the exposure times of DMAI and EG and the purge time of DMAI were kept constant while only the purge time of EG was varied systematically from ∼1 s to ∼24 s. It can be observed that the total GPC decreases with increasing EG purge time before appearing to reach a steady state value at longer purge durations.
One could possibly attribute the above observation to the infiltration and subsequent outgassing of EG from the alucone films as it is commonly known that MLD films are porous in nature and during their exposure steps reactants can infiltrate into the films subsequently taking a very long time to diffuse out.4,30 However, using an in situ quartz crystal microbalance (QCM), Dameron et al. have previously observed that unlike the excess mass gain observed during TMA's pulse attributed to its infiltration, EG's pulse showed no excess mass gain and they concluded that, unlike TMA, EG shows negligible or no infiltration into the alucone films.4 This was reconfirmed in our previous studies on alucone films prepared using TMA and EG where we also found that unlike TMA, the partial pressure of EG had a negligible influence on the overall GPC.20 Hence, infiltration of EG was found to be less likely to occur in TMA + EG films. The same can be concluded for the DMAI + EG films with an assumption that the alucone films prepared using DMAI have similar properties (for e.g. density, porosity) to those of films prepared using TMA and as one would observe ahead in the XRR studies, this assumption is a justified one. On the other hand, it has been previously observed in the case of zincone films that for very long exposure times of EG (∼4 s), the surface MLD reactions of EG are accompanied by its surface physisorption (Peng et al.).5 With the help of QCM studies, Peng et al. reported that the excess mass gain corresponding to EG physisorption was seen to decrease in the subsequent EG purge step. In the present case, although the exposure times of EG are much shorter than those employed by Peng et al., the employed partial pressure of EG is very high (∼4 torr). This can lead to excess surface adsorption (physisorption) of EG which can then demand long purge steps. In a case where the provided EG purge time is insufficient, it can lead to an additional adsorption component in the overall growth which would understandably decrease with increasing EG purge time as more and more of the excess EG desorbs from the surface thereby explaining the decrease in the GPC with EG purge time in Fig. 2c.
Chemical analysis
In order to compare the chemical architecture of the alucone films prepared using DMAI and TMA, we have recorded the IR absorbance spectra of the individual films as shown in Fig. 3. To have a negligible impact of EG adsorption or TMA infiltration on the bonding characteristics of the films, a comparison has been made between the films prepared using sufficient purges for both reactants. Also for the sake of comparison, the absorbance spectra have been normalized. At the outset, both films appear to have similar bonding features. The region between 800 and 950 cm−1 corresponds to the Al–O phonon mode which is slightly shifted to a higher frequency range when compared to the phonon modes observed in an alumina film40 but aligns well with the literature on alucone.4,22 The shoulders and peaks in the region corresponding to the C–O stretch are also present in either of the films in the IR range of ∼1000–1100 cm−1. One would also expect the existence of C–C peaks in the spectra attributable to EG and these peaks are indeed found to appear at ∼1140 cm−1 in both spectra. Furthermore, the expected CH2 vibration bands also appear at frequencies of around 1192 cm−1, 1255 cm−1 and 1354 cm−1 in each of the spectra. In the higher frequency region, it can again be observed that the absorbance spectra from both films overlap quite well reiterating a similar bonding environment. The peaks at frequencies of ∼2870 cm−1 and ∼2940 cm−1 which correspond to the symmetric and asymmetric stretching vibrations of CH2 respectively are present in the spectra of both films. A less intense peak around 2708 cm−1 attributable to the CH combination mode also shows up in either spectrum. Lastly, the broad absorbance bands observed at a frequency of ∼3400 cm−1 are due to OH stretching vibrations and likely arise due to the adsorption of ambient water or changes that occur within the films upon their short exposure to the ambiance while being transferred from the deposition chamber to the FTIR setup. Also observed is that the presence and positions of the peaks are quite similar to what has been previously reported on alucone films prepared on low-pressure temporal setups.4,22,41 Hence, these results show not only that the bonding environment of the films prepared using DMAI + EG is very similar to the one within a TMA + EG film but also that the alucone films prepared on an atmospheric pressure spatial MLD setup are comparable to the ones prepared on temporal setups in terms of their IR absorbances.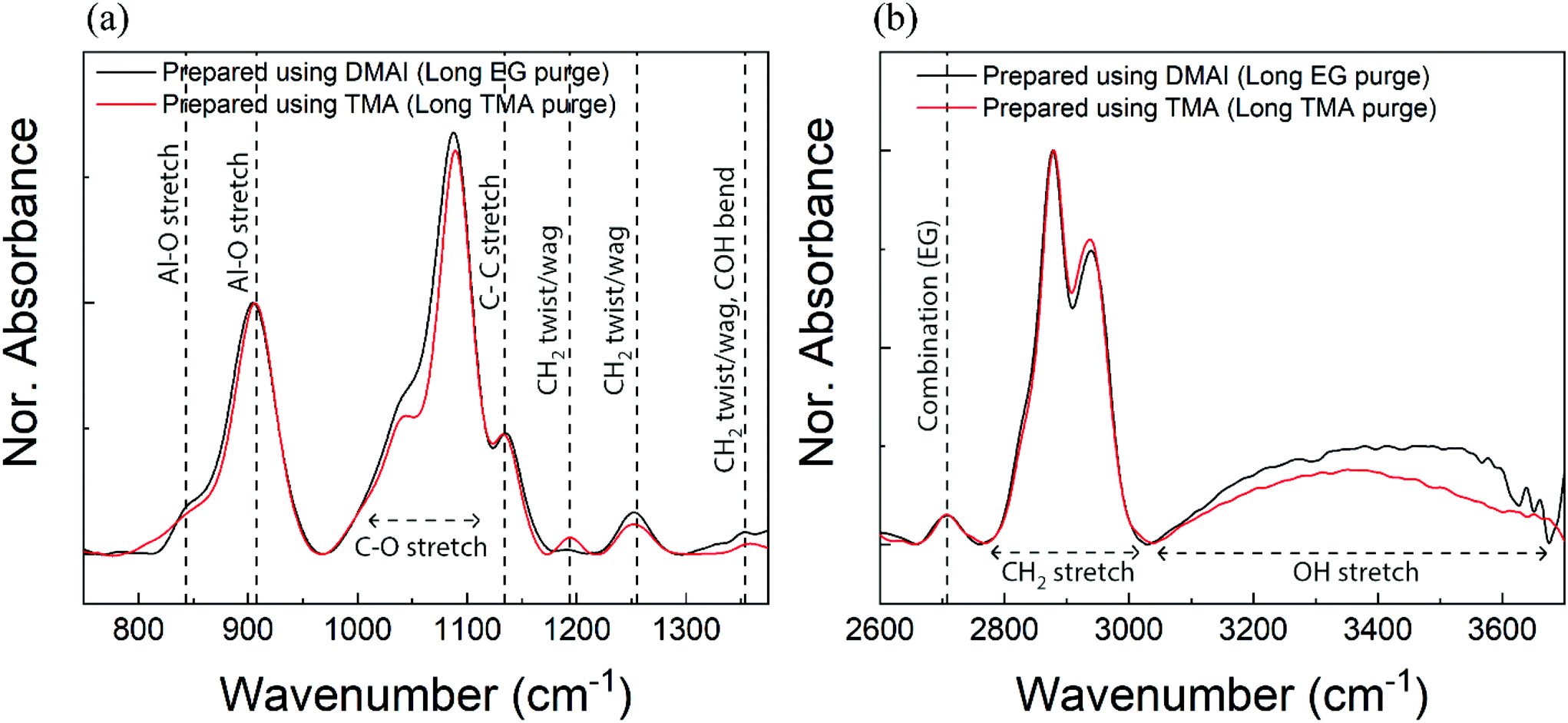 | ||
| Fig. 3 IR absorbance of the as-prepared films deposited using DMAI (black) and TMA (red) in the (a) low-frequency region and (b) high-frequency region (long EG purge = 24 s; long TMA purge = 98 s). | ||
X-ray photoelectron spectroscopy (XPS) was used to compare the relative elemental composition of the alucone films prepared using DMAI and TMA. The film prepared using DMAI and EG had a long enough EG purge step to avoid any influence of EG adsorption on the film composition. Similarly, the TMA + EG film was prepared using a very long TMA purge time to have a negligible CVD growth component in the film. The XPS survey scan of both films revealed a dominant presence of only 3 elements (Al, O, and C) in the top region of the films (see the ESI's section A†). All three elements are expected to be present in an alucone film. It must be noted that the films were momentarily exposed to the environment (<15 min) before the measurements and hence, the compositions in the top regions of the films might not be the best representatives of the compositions within their bulk. In order to sputter away the exposed top part and to know the composition within the bulk of the films, we have combined XPS analysis with sputtering. After 8 sputtering cycles of 15 s each, the acquired Al 2p, O 1s and C 1s spectra are provided in the ESI's section A.† The spectra corresponding to the Al environment within the individual films could each be simulated with a single peak at a binding energy of ∼74.8 eV which is very similar to the literature on alucone films.4,42 The O 1s spectra from both films could be deconvoluted into two subpeaks, the first of which is observed at ∼531 eV possibly corresponding to the Al–O environment and the other at ∼532 eV due to the C–O environment.42 The observed C 1s spectra could also be further deconvoluted into 3 subpeaks present at binding energies of ∼284.8 eV, ∼287 eV and ∼289 eV. The peak at the lowest energy could be attributed to C–C species whereas the one at ∼287 eV is often attributed to C–O species.4,42 Both carbon environments are expected in an alucone film. A minor peak at a binding energy of ∼289 eV is also observed and is commonly known to be due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. The origin of the CO environment is not obvious but it has been previously observed in the alucone films that were exposed to the ambient atmosphere.4 It is also worth noting that the profile of the Al 2p and O 1s spectra remained unchanged after each sputtering cycle whereas with the growing number of sputtering cycles, the C 1s spectra showed substantial modification in which the peaks at the lowest energy (∼284.8 eV) increased in their intensities while the ones at ∼287 eV decreased. The presence of the CO species in the unexposed bulk of our alucone films suggests either that the CO groups do not only arise due to ambient exposure but can also be a part of the film's growth chemistry or that the CO species are formed as a result of sputtering during XPS analysis.
O species. The origin of the CO environment is not obvious but it has been previously observed in the alucone films that were exposed to the ambient atmosphere.4 It is also worth noting that the profile of the Al 2p and O 1s spectra remained unchanged after each sputtering cycle whereas with the growing number of sputtering cycles, the C 1s spectra showed substantial modification in which the peaks at the lowest energy (∼284.8 eV) increased in their intensities while the ones at ∼287 eV decreased. The presence of the CO species in the unexposed bulk of our alucone films suggests either that the CO groups do not only arise due to ambient exposure but can also be a part of the film's growth chemistry or that the CO species are formed as a result of sputtering during XPS analysis.
The total area under each of the elemental regions acquired from both films remained constant through all the sputtering cycles suggesting no preferential sputtering. Hence, we were able to calculate the relative elemental composition of Al, O, and C within the film's bulk as shown in Fig. 4. It can be seen that after sputtering away the top part of the film, the relative composition of all three elements remained stable with increasing depth suggesting a homogeneous composition within the bulk of both films. Also, the relative elemental compositions of both films are very similar where the aluminum, oxygen and carbon contents of the DMAI + EG film are around 23%, 53% and 24% respectively while those of the TMA + EG film are around 24%, 53% and 23% respectively. The relative carbon content of both films is slightly higher than what is reported by Van de Kerckhove et al. (21%) in their alucone (TMA + EG) films.22 As one would observe ahead, MLD films are known to lose their carbon content upon ambient exposure and one of the reasons behind the slightly higher carbon content of our films compared to that reported by Van de Kerckhove et al.22 could be a difference in the amount of time the samples were exposed to the ambient atmosphere before their XPS measurements. Nevertheless, the fact that our alucone films prepared using DMAI and TMA have very similar carbon contents indicates that the higher carbon content in our films is probably not due to the use of a different precursor.
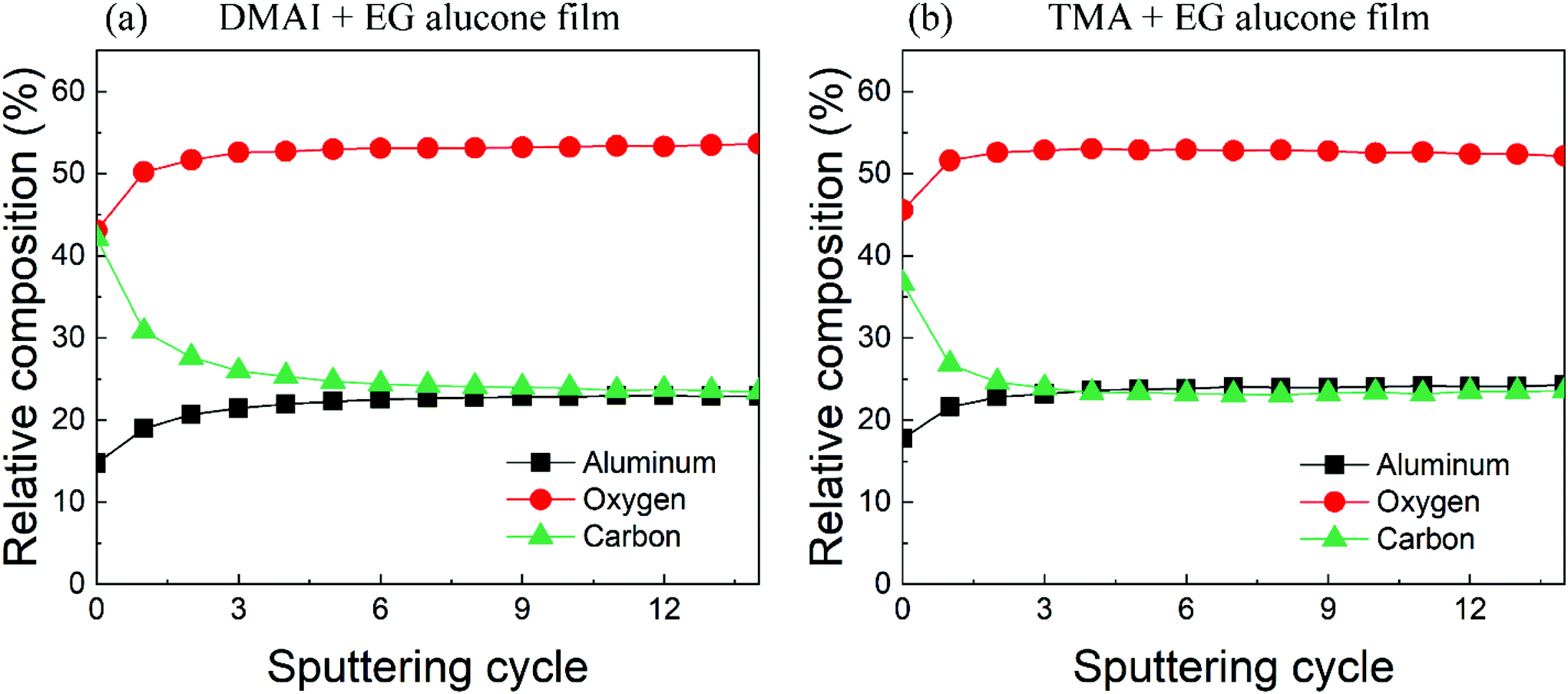 | ||
| Fig. 4 Relative elemental composition of the as-prepared alucone films deposited using (a) DMAI + EG and (b) TMA + EG. | ||
X-Ray reflectivity
X-ray reflectivity (XRR) was used to determine the densities of the films and as a complementary technique to spectroscopic ellipsometry to measure the film thicknesses. The extracted thicknesses of the films were found to be consistent with the results obtained from spectroscopic ellipsometry. The extracted density of the DMAI + EG film was found to be around 1.72 g cm−3 whereas that of the film prepared using TMA + EG was around 1.75 g cm−3, both being very close to other reports on the densities of alucone films.4 The measured and fitted intensities versus the incident angle are shown in Fig. 5. Since the number of fringes considered for the fitting was very limited, the derived densities may vary a little from the above mentioned values. However, the preliminary results presented here do indicate that the films prepared using either TMA or DMAI have very close densities.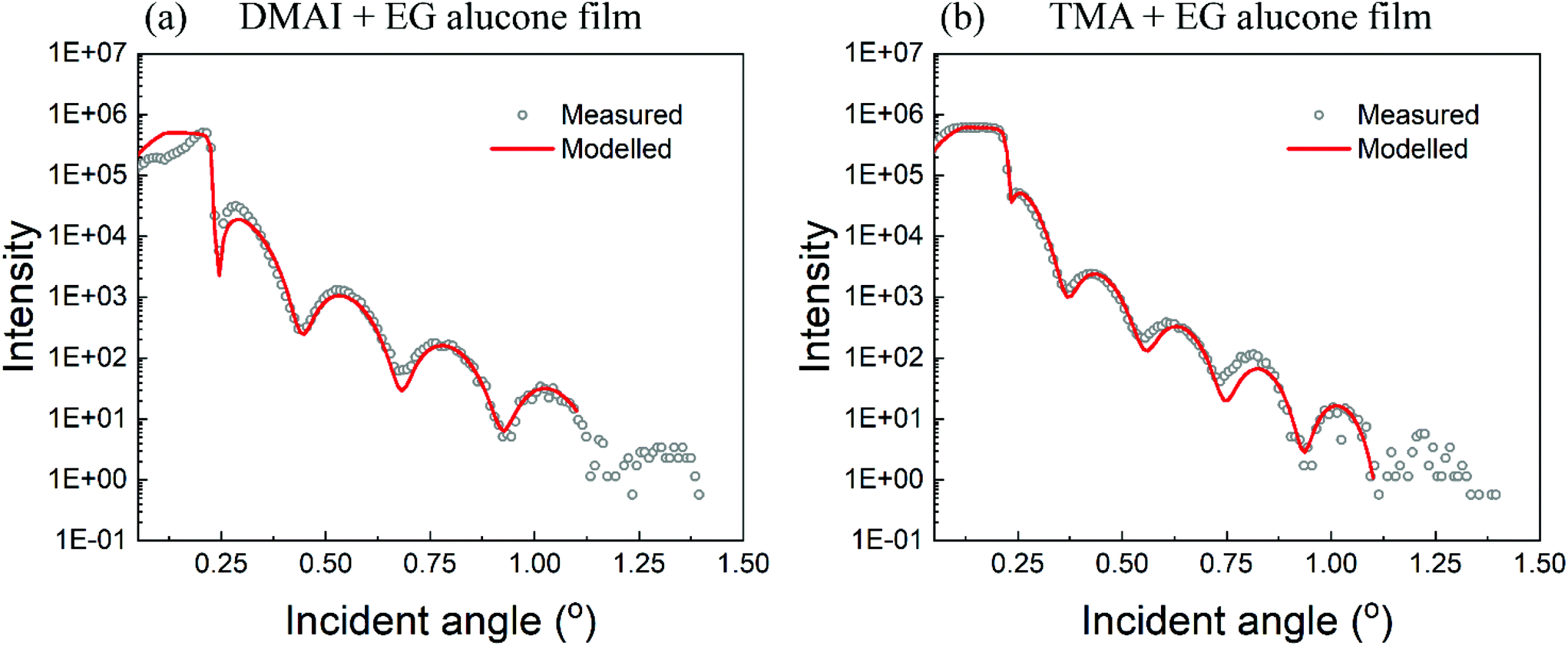 | ||
| Fig. 5 XRR scans of the alucone films prepared using (a) DMAI and (b) TMA. The fitted intensity is shown in red. The extracted density of the DMAI + EG film ≈ 1.72 g cm−3 and that of TMA + EG film ≈ 1.75 g cm−3. | ||
Degradation of the films upon ambient exposure
MLD films are known to degrade upon their exposure to the ambient atmosphere due to their reaction with moisture.4,43,44 Thus, we have investigated the change in the alucone film properties upon exposure to air for both DMAI and TMA based films (ESI's section B†). The FTIR spectra of both films after 24 h of ambient exposure, when compared to those of the unexposed films, reveal that mainly the peaks corresponding to carbon-containing environments (C–O, CH2) decrease in their intensities while the ones corresponding to the reaction of the films with ambiance or water adsorption45 (∼3400 cm−1) increase (ESI's section B, Fig. S3†). Similarly, the XPS studies show that upon ambient exposure the relative aluminum and oxygen contents of the films increase (∼29% and ∼63% respectively) while the carbon content of the films decreases to 7% (ESI's section B, Fig. S4†). The trends in bonding and compositional changes occurring in both films are in line with those reported earlier by others4,22 however with one difference that the alucone films prepared by Van de Kerckhove et al.22 had much more carbon left within their films (17%) after ambient exposure. It is very likely that the reported carbon content was measured at the film surface as it is much closer to the value we found on the surface of our films (ESI's section B, Fig. S4†). In contrast, the relative carbon content of 7% that we report is similar to that measured from the bulk of our films. Furthermore, the above changes are often accompanied by a change in the film thickness.4,22 So, we have also monitored the thickness of our alucone film prepared using DMAI over 72 h and found that the maximum reduction occurred within the first two hours of exposure after which the thickness seemed to have stabilized (ESI's section B, Fig. S5†). The final thickness after 72 h of exposure is around 74% of the original thickness which is slightly lower than what Dameron et al.4 reported (80%) but very similar to what Van de Kerckhove et al.22 reported (74%). To conclude, the bonding and compositional changes upon ambient exposure occurring in a film prepared using DMAI are very similar to the ones occurring in a TMA + EG film. One can also conclude that the alucone films prepared on an atmospheric pressure spatial MLD setup do not show any higher degree of instability in the ambient atmosphere than the ones prepared on conventional low-pressure temporal setups.Discussions and conclusions
Because of its high reactivity and volatility, TMA appears to be an ideal precursor for applications requiring large-scale and high deposition rate MLD of alucones. However, we have previously demonstrated that TMA infiltrates into the deposited MLD film and requires very long purge times to avoid CVD, hence resulting in very low deposition rates.20 To overcome the problem of long purge times required by TMA in alucone MLD, we showed that dimethylaluminum isopropoxide (DMAI) is an excellent alternative for TMA, as long purge times are no longer required.20 A possible explanation for this could be DMAI's larger molecular size and its ability to remain in its dimeric form even at higher temperatures.39 The larger molecular size (see the ESI's section C†), the higher molecular weight and a different molecular shape compared to TMA could essentially mean a smaller diffusion coefficient for DMAI46 and thus reduce its diffusivity within the alucone films. DMAI would find it even more difficult to infiltrate into the alucone films if the resultant alucone films are less porous. However, from the results of the XRR studies above, we discover that the DMAI + EG alucone films have a density almost equal to that of the TMA + EG films and very close to the literature value for the density of alucone films.4,22 It could also be the case that DMAI has very limited solubility in the alucone films and therefore, does not infiltrate. Although the exact reason is uncertain, the resulting significant reduction in the purge time of DMAI as compared to TMA will have a large positive impact on the overall cycle time and deposition rate of alucone films.We also observed that, unlike the TMA + EG process where the purge time of EG was found to be insignificant, in the case of the DMAI + EG process, the required purge time for EG was found to be much longer (>20 s). This can be explained by EG physisorption caused by the very high EG partial pressures used in this work. Nevertheless, the purge time required by the excess EG to desorb from the film surface is much less than what was needed by TMA to outgas from the films’ bulk and hence, even after considering the long EG purge time in calculations, the achievable alucone deposition rates using DMAI + EG are more than 40% higher than those obtained when using TMA. The deposition rate could possibly be further enhanced by adopting similar strategies that were used to improve the TMA's outgassing rate such as increasing the purge gas flow or increasing the deposition temperature which could lead to faster desorption of EG. Moreover, one can also optimize the EG partial pressure to reduce unnecessary adsorption.
Furthermore, the byproducts of an ALD/MLD reaction are often assumed to be highly volatile and inert during the film growth reactions. However, in some situations47 the byproducts might participate in the film growth reactions thereby hindering the growth process or altering the film's chemistry. It is worth noting that one of the byproducts of the reactions of DMAI and EG with the growth surface could be isopropanol (CH3CHOHCH3) which has a hydroxyl group (–OH). The hydroxyl group could compete with the hydroxyl groups of EG for adsorption on the surface sites or the MLD reactions. Since isopropanol is monodentate (only one hydroxyl group), it could also terminate a surface site and lead to low GPC and high carbon incorporation within the film. The observed similarity in the carbon content of the DMAI and TMA alucone films (Fig. 4) indicates that the participation of the isopropanol molecules in the overall growth process, if any, is minimal.
The film properties of the DMAI + EG alucone films were compared with those prepared using TMA. Using FTIR, it was confirmed that the alucone films possess very similar bonding characteristics irrespective of the employed metal precursor. Similar conclusions were also drawn when we compared the chemical environment and elemental composition of the films using X-ray photoelectron spectroscopy (TMA/DMAI + EG) and film densities using X-ray reflectivity. Upon comparing the film properties with those reported in the literature, most of which are based on low-pressure temporal setups, we also observed that the alucone films prepared on an atmospheric pressure spatial MLD setup do not differ significantly from the films prepared on low-pressure temporal setups. The ambient stability of the DMAI + EG alucone films was also investigated and we found that the films do show limited stability in an ambient atmosphere but which is very similar to that of the TMA + EG films. Thus, in conclusion, this work proves the combined ability of DMAI and an atmospheric pressure spatial MLD setup to produce alucone films with comparable properties to those of the films made using TMA and temporal setups but with much higher deposition rates.
Abbreviations
| MLD | Molecular layer deposition |
| ALD | Atomic layer deposition |
| CVD | Chemical vapor deposition |
| TMA | Trimethylaluminum |
| DMAI | Dimethylaluminum isopropoxide |
| EG | Ethylene glycol |
| GPC | Growth-per-cycle |
| FTIR | Fourier transform infrared |
| XPS | X-ray photoelectron spectroscopy |
| QCM | Quartz crystal microbalance |
Author contributions
H. J. performed all the experiments mentioned in this work. The FTIR measurements and XPS analysis were also performed by H. J. The manuscript was written with the help of P. P. and M. C.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Prof. Mariadriana Creatore acknowledges the NWO Aspasia Program. The authors also acknowledge the help they received from Ms Arbresha Muriqi and Dr Michael Nolan from the Tyndall National Institute, Ireland in terms of density functional theory calculations to better understand the reactions of DMAI with the growth surface. The authors would also like to thank Dr Gerben van Straaten and Dr Saravana Balaji Basuvalingam from the Eindhoven University of Technology for their respective help with the XRR and XPS measurements. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 765378.References
- H.-I. Shao, S. Umemoto, T. Kikutani and N. Okui, Polymer, 1997, 38, 459–462 CrossRef CAS.
- M. Putkonen, J. Harjuoja, T. Sajavaara and L. Niinistö, J. Mater. Chem., 2007, 17, 664–669 RSC.
- P. Sundberg and M. Karppinen, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- A. A. Dameron, D. Seghete, B. B. Burton, S. D. Davidson, A. S. Cavanagh, J. A. Bertrand and S. M. George, Chem. Mater., 2008, 20, 3315–3326 CrossRef CAS.
- Q. Peng, B. Gong, R. M. VanGundy and G. N. Parsons, Chem. Mater., 2009, 21, 820–830 CrossRef CAS.
- A. I. Abdulagatov, R. A. Hall, J. L. Sutherland, B. H. Lee, A. S. Cavanagh and S. M. George, Chem. Mater., 2012, 24, 2854–2863 CrossRef CAS.
- S.-H. Lee, H.-J. Jeong, K.-L. Han, G. Baek and J.-S. Park, J. Mater. Chem. C, 2021, 9, 4322–4329 RSC.
- M. Park, S. Oh, H. Kim, D. Jung, D. Choi and J.-S. Park, Thin Solid Films, 2013, 546, 153–156 CrossRef CAS.
- W. Xiao, D. Y. Hui, C. Zheng, D. Yu, Y. Y. Qiang, C. Ping, C. L. Xiang and Z. Yi, Nanoscale Res. Lett., 2015, 10, 130 CrossRef PubMed.
- P. Ingale, C. Guan, R. Kraehnert, R. Naumann d'Alnoncourt, A. Thomas and F. Rosowski, Catal. Today, 2021, 362, 47–54 CrossRef CAS.
- J. Liu and J. Wang, Front. Energy Res., 2021, 9, 1–8 Search PubMed.
- B. C. Welch, O. M. McIntee, T. J. Myers, A. R. Greenberg, V. M. Bright and S. M. George, Desalination, 2021, 520, 115334 CrossRef CAS.
- D. La Zara, F. Zhang, F. Sun, M. R. Bailey, M. J. Quayle, G. Petersson, S. Folestad and J. R. van Ommen, Appl. Mater. Today, 2021, 22, 100945 CrossRef.
- S. Lee, G. Baek, H. Kim, Y.-H. Kim and J.-S. Park, Dalton Trans., 2021, 50, 9958–9967 RSC.
- P. Poodt, D. C. Cameron, E. Dickey, S. M. George, V. Kuznetsov, G. N. Parsons, F. Roozeboom, G. Sundaram and A. Vermeer, J. Vac. Sci. Technol., A, 2012, 30, 010802 CrossRef.
- P. Poodt, A. Lankhorst, F. Roozeboom, K. Spee, D. Maas and A. Vermeer, Adv. Mater., 2010, 22, 3564–3567 CrossRef CAS PubMed.
- D. H. Levy, S. F. Nelson and D. Freeman, J. Disp. Technol., 2009, 5(12), 484–494 CAS.
- L. Hoffmann, K. O. Brinkmann, J. Malerczyk, D. Rogalla, T. Becker, D. Theirich, I. Shutsko, P. Görrn and T. Riedl, ACS Appl. Mater. Interfaces, 2018, 10, 6006–6013 CrossRef CAS PubMed.
- D. J. Higgs, J. W. DuMont, K. Sharma and S. M. George, J. Vac. Sci. Technol., A, 2018, 36, 01A117 CrossRef.
- H. Jain and P. Poodt, Dalton Trans., 2021, 50, 5807–5818 RSC.
- D. Choudhury, S. K. Sarkar and N. Mahuli, J. Vac. Sci. Technol., A, 2015, 33, 01A115 CrossRef.
- K. Van de Kerckhove, M. K. S. Barr, L. Santinacci, P. M. Vereecken, J. Dendooven and C. Detavernier, Dalton Trans., 2018, 47, 5860–5870 RSC.
- Y. Lee, B. Yoon, A. S. Cavanagh and S. M. George, Langmuir, 2011, 27, 15155–15164 CrossRef CAS PubMed.
- D. Choudhury, G. Rajaraman and S. K. Sarkar, J. Vac. Sci. Technol., A, 2018, 36, 01A108 CrossRef.
- G. Baek, S. Lee, J.-H. Lee and J.-S. Park, J. Vac. Sci. Technol., A, 2020, 38, 022411 CrossRef CAS.
- G. S. Higashi and C. G. Fleming, Appl. Phys. Lett., 1989, 55, 1963–1965 CrossRef CAS.
- J.-F. Fan, K. Sugioka and K. Toyoda, Jpn. J. Appl. Phys., 1991, 30, L1139–L1141 CrossRef CAS.
- J. B. Kim, D. R. Kwon, K. Chakrabarti, C. Lee, K. Y. Oh and J. H. Lee, J. Appl. Phys., 2002, 92, 6739–6742 CrossRef CAS.
- P. Poodt, A. Lankhorst, F. Roozeboom, K. Spee, D. Maas and A. Vermeer, Adv. Mater., 2010, 22, 3564–3567 CrossRef CAS PubMed.
- D. Seghete, R. A. Hall, B. Yoon and S. M. George, Langmuir, 2010, 26, 19045–19051 CrossRef CAS PubMed.
- L. Hiltunen, H. Kattelus, M. Leskelä, M. Mäkelä, L. Niinistö, E. Nykänen, P. Soininen and M. Tiittad, Mater. Chem. Phys., 1991, 28, 379–388 CrossRef CAS.
- K. E. Elers, T. Blomberg, M. Peussa, B. Aitchison, S. Haukka and S. Marcus, Chem. Vap. Deposition, 2006, 12, 13–24 CrossRef CAS.
- S. C. Buttera, D. J. Mandia and S. T. Barry, J. Vac. Sci. Technol., A, 2017, 35, 01B128 CrossRef.
- R. Katamreddy, R. Inman, G. Jursich, A. Soulet and C. Takoudis, J. Electrochem. Soc., 2006, 153, C701–C706 CrossRef CAS.
- W. Koh, S.-J. Ku and Y. Kim, Thin Solid Films, 1997, 304, 222–224 CrossRef CAS.
- B. W. Schmidt, W. J. Sweet, E. J. Bierschenk, C. K. Gren, T. P. Hanusa and B. R. Rogers, J. Vac. Sci. Technol., A, 2010, 28, 238–243 CrossRef CAS.
- W. Cho, K. Sung, K.-S. An, S. Sook Lee, T.-M. Chung and Y. Kim, J. Vac. Sci. Technol., A, 2003, 21, 1366–1370 CrossRef CAS.
- S. E. Potts, G. Dingemans, C. Lachaud and W. M. M. Kessels, J. Vac. Sci. Technol., A, 2012, 30, 021505 CrossRef.
- S. Y. Lee, B. Luo, Y. Sun, J. M. White and Y. Kim, Appl. Surf. Sci., 2004, 222, 234–242 CrossRef CAS.
- E. Levrau, K. Van De Kerckhove, K. Devloo-Casier, S. Pulinthanathu Sree, J. A. Martens, C. Detavernier and J. Dendooven, J. Phys. Chem. C, 2014, 118, 29854–29859 CrossRef CAS.
- J. W. Dumont and S. M. George, J. Phys. Chem. C, 2015, 119, 14603–14612 CrossRef CAS.
- E. Kazyak, M. Shin, W. S. Lepage, T. H. Cho and N. P. Dasgupta, Chem. Commun., 2020, 56, 15537–15540 RSC.
- L. Ghazaryan, E.-B. Kley, A. Tünnermann and A. Viorica Szeghalmi, J. Vac. Sci. Technol., A, 2013, 31, 01A149 CrossRef.
- J. Kint, F. Mattelaer, S. S. T. Vandenbroucke, A. Muriqi, M. M. Minjauw, M. Nisula, P. M. Vereecken, M. Nolan, J. Dendooven and C. Detavernier, Chem. Mater., 2020, 32, 4451–4466 CrossRef CAS.
- A. C. Dillon, A. W. Ott, J. D. Way and S. M. George, Surf. Sci., 1995, 322, 230–242 CrossRef CAS.
- J. Crank, The Mathematics of Diffusion, Clarendon Press, Oxford, 1975 Search PubMed.
- S. García-García, A. López-Ortega, Y. Zheng, Y. Nie, K. Cho, A. Chuvilin and M. Knez, Chem. Sci., 2019, 10, 2171–2178 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: A. Ex situ X-ray photoelectron spectroscopy. B. Degradation of the films upon ambient exposure. C. Molecular size comparison between DMAI and TMA. See DOI: https://doi.org/10.1039/d2dt00570k |
| This journal is © The Royal Society of Chemistry 2022 |