
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultivalent glycocyclopeptides: conjugation methods and biological applications
David
Goyard
 ,
Angela Martin-Serrano
Ortiz
,
Didier
Boturyn
and
Olivier
Renaudet
*
,
Angela Martin-Serrano
Ortiz
,
Didier
Boturyn
and
Olivier
Renaudet
*
Univ. Grenoble Alpes, CNRS, DCM UMR 5250, F-38000 Grenoble, France. E-mail: olivier.renaudet@univ-grenoble-alpes.fr
First published on 4th October 2022
Abstract
Click chemistry was extensively used to decorate synthetic multivalent scaffolds with glycans to mimic the cell surface glycocalyx and to develop applications in glycosciences. Conjugation methods such as oxime ligation, copper(I)-catalyzed alkyne–azide cycloaddition, thiol–ene coupling, squaramide coupling or Lansbury aspartylation proved particularly suitable to achieve this purpose. This review summarizes the synthetic strategies that can be used either in a stepwise manner or in an orthogonal one-pot approach, to conjugate multiple copies of identical or different glycans to cyclopeptide scaffolds (namely multivalent glycocyclopeptides) having different size, valency, geometry and molecular composition. The second part of this review will describe the potential of these structures to interact with various carbohydrate binding proteins or to stimulate immunity against tumor cells.
 David Goyard | David Goyard obtained his PhD in organic chemistry in 2011 under the supervision of Dr J. P. Praly. He then joined R. Roy's research group at UQAM (Montreal, Canada) for an eighteen months postdoc. In September 2015 he started a second postdoc with O. Renaudet until September 2019. He was appointed as CNRS chargé de recherche in October 2019. His research interests focus on carbohydrate chemistry and the design of glycodendrimers to study multivalent carbohydrate protein interactions. |
 Angela Martin-Serrano Ortiz | Ángela Martín-Serrano Ortiz received her BSc in Chemistry from University of Castilla-La Mancha (2011) and her MSc in Biological Analysis and Laboratory Diagnosis from University of Granada (2012). Then, she joined the Allergy Group at Biomedical Research Institute of Málaga (IBIMA) and she did a stay at Royal Institute of Technology (KTH). After obtaining her PhD from the University of Málaga (2018), she joined BioNanoDen Group at University of Alcalá. Currently, she is member of MultiGlyco team at University of Grenoble-Alpes, where she works on the synthesis and preclinical evaluation of antibody recruiting glycodendrimers for cancer and infectious diseases treatment. |
 Didier Boturyn | Didier Boturyn received his MS degrees (1993) in Molecular Chemistry from the University of Grenoble in France. He then performed PhD research on the detection of DNA apurinic sites (1993–1996). After postdoctoral studies concerning bleomycin derivatives at the University of Virginia in USA with Professor Sidney Hecht (1997–1998), he got a position at the CNRS at the Department of Molecular Chemistry (DCM) in Professor Pascal Dumy group. He was promoted to Senior researcher in 2011 and then became Director of the DCM in 2021. His research interests include the synthesis and characterization of biomolecular systems for biological applications. |
 Olivier Renaudet | Olivier Renaudet obtained a PhD in Molecular Chemistry at the University of Grenoble in 2002, then he pursued postdoctoral researches at the University of Bern with Prof. J.-L. Reymond. After that, he moved back to Grenoble where he was promoted Assistant Professor in 2004 then Full Professor in 2011 at the Département de Chimie Moléculaire. He leads the MultiGlyco team which focuses on multidisciplinary projects aiming at developing antibody-recruiting glycodendrimers against tumors and pathogens. He was awarded by the Institut Universitaire de France (2011) and the European Research Council with the Consolidator and Proof of Concept grants (2015, 2020, 2022). |
1. Introduction
A myriad of molecular scaffolds such as cyclodextrins, calixarenes, fullerenes, polymers, dendrimers, peptides or nanoparticles was developed for diverse applications in glycoscience.1–3 Their utilization as a carrier to conjugate glycans was largely explored to provide multivalent glycosylated structures having enhanced binding avidity and selectivity towards carbohydrate-binding proteins due to the “glycoside cluster effect”.4,5 Cyclopeptides have also found an important place in this area over the past two decades. Originally used to expose peptide fragments for mimicking natural proteins,6,7 they were proven to be attractive for in vivo applications due to their conformational stability, improved resistance against enzymatic degradations and lack of immunogenicity. Taking advantage of these favorable features, cyclopeptides were more recently decorated with a variety of glycans with the aim to decipher, stimulate or inhibit multivalent biological processes.8 To achieve this purpose, the chemical strategy should be as modular as possible to provide a diversity of scaffold structures and thus identify the optimal spatial arrangement of glycan with respect to the targeted carbohydrate-binding protein. The synthesis of cyclopeptides, which first requires the solid-phase assembly of linear peptide precursors, clearly fulfils this criteria. Aminoacids with functionalizable side chain such as Lys, Asp or Glu can thus be incorporated at defined positions in the peptide sequence, thereby providing a total control of the valency as well as distances between glycans in the cyclopeptide.9,10 In addition, introducing β-turn inducers (i.e. Gly/Pro or Pro/D-pro) in the peptide sequence allows both the conformational stability of the scaffold in solution and the spatial orientation of glycans.Beside this, click chemistry undoubtedly had a huge positive impact in this field.11,12 The development of a large variety of conjugation methodologies has not only facilitated the synthesis of the first generations of multivalent glycosylated scaffolds but it has also spectacularly increased the molecular diversity that is now accessible. By incorporating “clickable” chemical functions at defined positions within the peptide sequence, cyclopeptides can be easily conjugated in aqueous conditions with unprotected glycans bearing complementary chemical functions to provide multivalent systems. In addition, the utilization of iterative protocols or successive orthogonal click reactions enables the preparation of more complex glycosylated systems with tunable biological properties, by combining multivalent scaffolds with similar or different structural features to a central cyclopeptide.13,14 This review aims at highlighting the recent advances in this field. The diversity of conjugation procedures available for the construction of cyclopeptides displaying covalently attached glycans through linear or branched linkers, namely glycoclusters and glycodendrimers respectively, will be described in the first part of this review. The second part will emphasize the biological applications that are currently being developed with these multivalent glycocyclopeptides, with a particular focus on antiadhesive and immunotherapeutic tools.
2. Conjugation methodologies
Click chemistry has become an essential tool to construct biomolecular systems with polyfunctional molecules which have often uncompatible chemical reactivity.15 The large variety of reactions available today rely on the regioselective incorporation of highly reactive functional groups in both reacting partners, i.e. glycans and peptides in the present case, to ligate them chemoselectively under mild experimental conditions. Among click reactions, oxime ligation (OL), copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC), thiol–ene coupling (TEC), thiol–chloroacetyl coupling (TCC), squaramide coupling (SqC) or Lansbury aspartylation have been explored as single methods to construct simple multivalent glycocyclopeptides as well as in multiple combinations to assemble more sophisticated biomolecular systems.Oxime ligation (OL)
Oxime ligation is a highly attractive reaction due to its excellent efficiency and compatibility with a large set of biomolecules. This reaction is generally quantitative and is carried out in aqueous media, making it suitable for the conjugation of unprotected glycans and peptides.16 The oxime ether bond results from the reaction between an oxyamine and an aldehyde (or a ketone) function which react under mild acidic conditions (pH 3–5)17 with high chemoselectivity and minor risk of side reaction. Moreover, oxime ether was shown to be stable in different chemical conditions18 as well as in vivo.19OL was first exploited for neoglycopeptides by taking advantage of the reducing end of carbohydrates that reacts as an electrophile20–22 with peptides bearing an aminooxy group. Nevertheless, this strategy implies the sugar ring-opening,23 a non-natural conformation that can affect the expected biological interaction.24 To overcome this limitation, several groups have developed synthetic strategies to incorporate aminooxy function at the reducing end of glycans by a stereoselective reaction based on phase transfer catalysis (Scheme 1A)25,26 or by glycosylation upon activation with a fluoride (Scheme 1B).27,28 These methodologies provided a library of aminooxy glycans 1–11 with a well-defined anomer configuration that is maintained upon condensation with the peptide bearing the complementary chemical group.
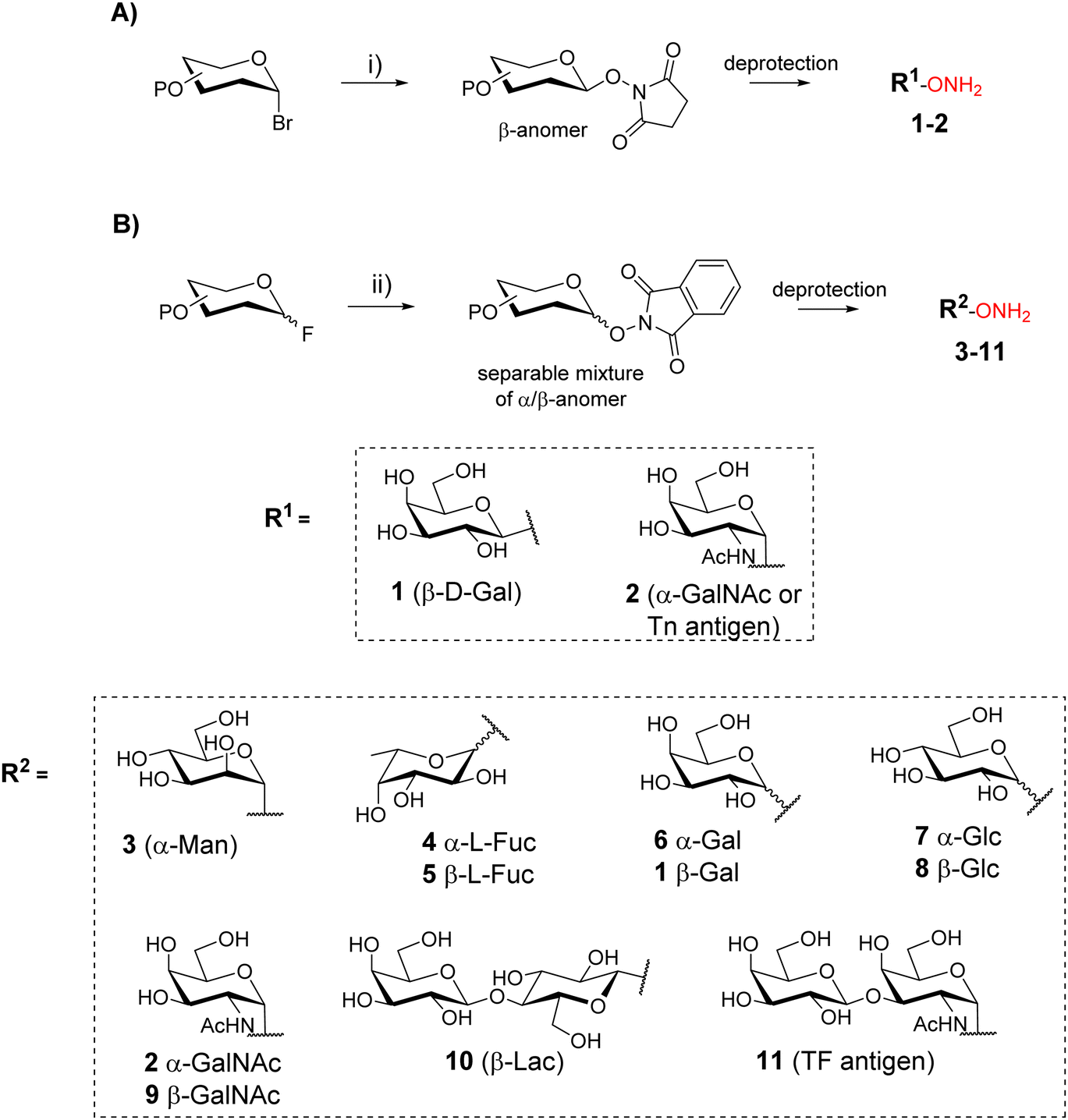 | ||
| Scheme 1 Strategies for the synthesis of aminooxy glycans 1–11: (A) using phase transfer catalysis from glycosyl bromides; (B) using glycosylation from glycosyl fluorides. Reagents and conditions: (i) NHS, TBAHS, CH2Cl2, Na2CO3 1 M; (ii) PhthOH, BF3·Et2O, TEA, CH2Cl2. Abbreviations: NHS: N-hydroxysuccinimide; TBAHS: tetra-butyl-ammonium-hydrogen-sulfate; PhthOH: N-hydroxyphthalimide; TEA: triethylamine; Gal: galactose; Glc: glucose; GalNAc: N-acetylgalactosamine; Man: mannose, Fuc: fucose, Lac: lactose; TF: Thomsen–Friedenreich. | ||
Functionalization of peptides with aldehyde or ketone functions can be performed using a variety of methodologies.29 For example, a site-specific introduction of aldehyde groups into recombinant proteins was reported using an hexapeptide containing a Cysteine (Cys) residue which can be transformed into a formylglycine (fGly) by a formylglycine-generating enzyme to provide the modified protein 12 (Scheme 2A).30
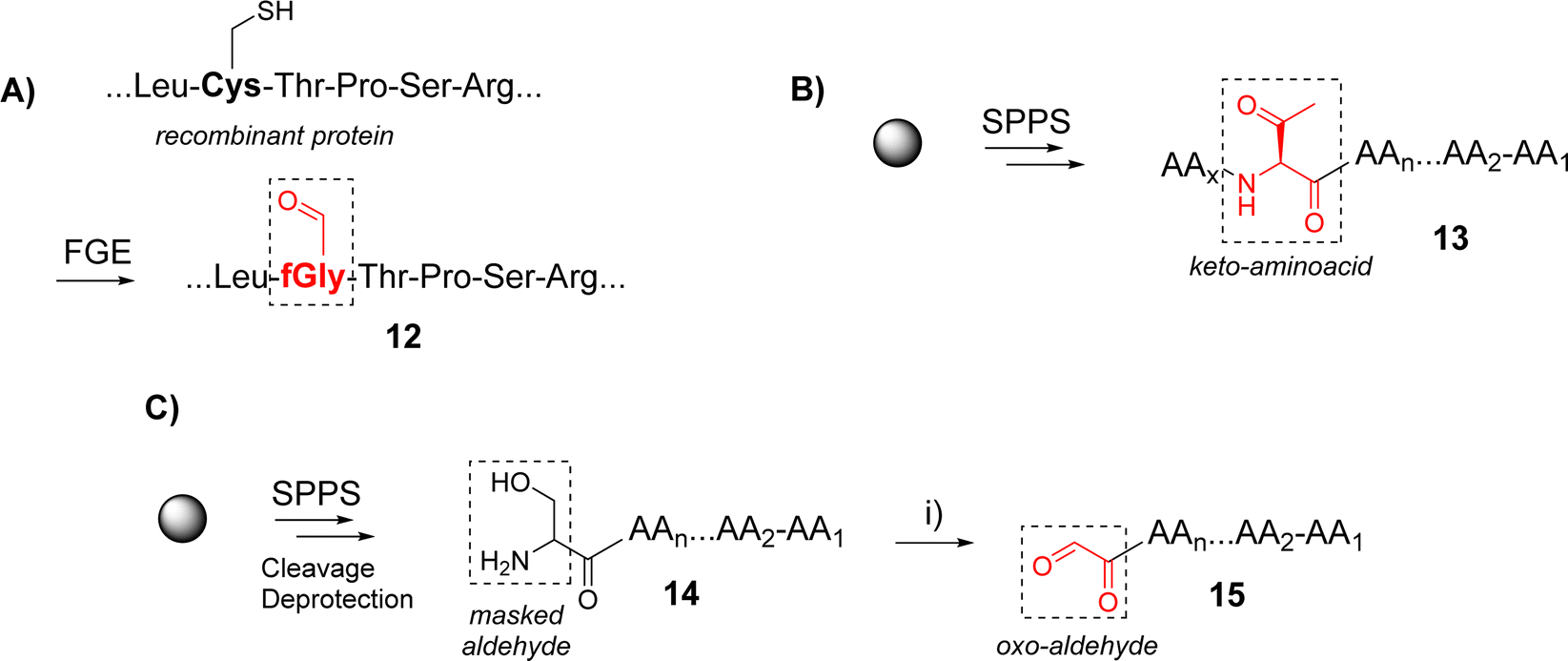 | ||
| Scheme 2 Example of methods to incorporate an aldehyde or a ketone function into a peptide sequence: (A) using an enzymatic process; (B) using the incorporation of a keto-amino acid; (C) using a periodic acid mediated serine oxidation. Reagents and conditions: (i) NaIO4, H2O. Abbreviations: FGE: formyl-generating enzyme; SPPS: solid-phase peptide synthesis. | ||
Chemical strategies based on solid-phase peptide synthesis (SPPS) can also allow the incorporation of a keto-amino acid at a defined position in a peptide sequence (13, Scheme 2B),26 or of a Serine (Ser) residue at N-terminus of a peptide (14) to provide an oxo-aldehyde function upon treatment with sodium periodate (15, Scheme 2C).31 While being largely exploited for different purposes,32,33 the latter strategy needs to be carefully controlled to avoid side oxidations of methionine, tryptophan and histidine.34 Most importantly, the oxime bond formed from an oxo-aldehyde, namely keto-oxime, is much more stable than an oxime ether resulting for the condensation with an aliphatic aldehyde, especially under biological conditions.16 This latter strategy using oxo-aldehydes is then particularly attractive to produce a wide range of multimeric glycocyclopeptides.
The first example of synthesis of multivalent glycocyclopeptides by OL was described in 2003 by Renaudet and Dumy.35 In this study, tetravalent structures were prepared from a cyclodecapeptide scaffold 16 (Scheme 3) bearing four Ser attached to Lysine (Lys) side chain in alternated position. After treatment with sodium periodate, the resulting scaffold bearing 4 oxo-aldehydes 17 was reacted with diverse aminooxy glycans in sodium acetate buffer (pH 4). Interestingly, much faster reactions were observed with alpha glycans compared to beta (1 h vs. 24 h, respectively) to achieve a quantitative conversion. The tetravalent glycoclusters 18–22 were finally isolated by RP-HPLC in 70–80% yield. NMR studies indicated that oxo-aldehydes provided oxime-ether derivatives as stereochemically homogeneous products whereas aliphatic aldehydes rather led to a E/Z mixture.
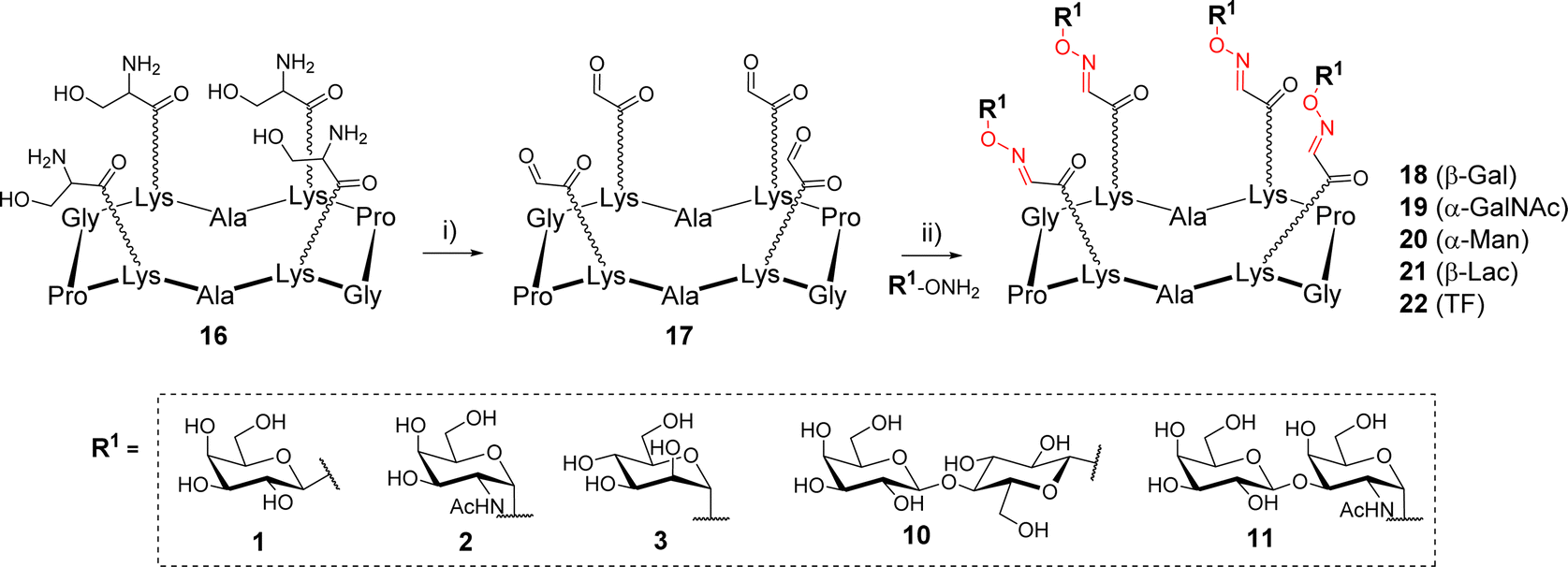 | ||
| Scheme 3 Assembly of tetravalent glycoclusters 18–22 by oxime ligation. Reagents and conditions: (i) NaIO4, H2O, r.t., 30 min, 83%; (ii) AcONa (pH 4), r.t., 1–24 h, 70–80%. | ||
The same strategy was used in other studies with the same efficiency,36,37 however significant differences of reactivity were observed with cyclopeptides displaying methylketone moieties.38
Copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC)
The copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC)39,40 was also widely used for the construction of biomolecular systems including multivalent glycosylated structures.41 This reaction occurs at room temperature using both organic and protic polar solvents such as alcohol or water, which are fully compatible with carbohydrates and peptides. In addition, the CuAAC reaction results from partners bearing an azide and an alkyne group to provide a very stable 1,2,3-triazole regioisomer exclusively. However, CuAAC requires special attention as the copper(I) catalyst can be trapped by chelating aminoacids and promote the formation of reactive oxygen species (ROS) responsible for biomolecular damages.42 Since 2002, significant methodological advances were carried out concerning the copper catalytic system.43 Depending on the solvents used in the reaction, a wide variety of copper source (Cu salts, complexes, nanoparticles, and metallic Cu) can be used as a pre-catalyst to provide the active Cu(I) species. Stabilization of the Cu(I) using tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) or other derivatives also has the ability to prevent the catalyst oxidation to Cu(II), which greatly improves the reaction outcome.CuAAC was used successfully by several groups for the synthesis of a variety of cyclopeptide-based glycoclusters and glycodendrimers. In a first study, tetravalent structures were prepared from the cyclodecapeptide 23 displaying four azido groups (Scheme 4A).44 These chemical functions were introduced in the peptide sequence by SPPS using Lys modified at their side chain (also called N-Fmoc-ε-azidonorleucine) by diazotransfer reaction.45
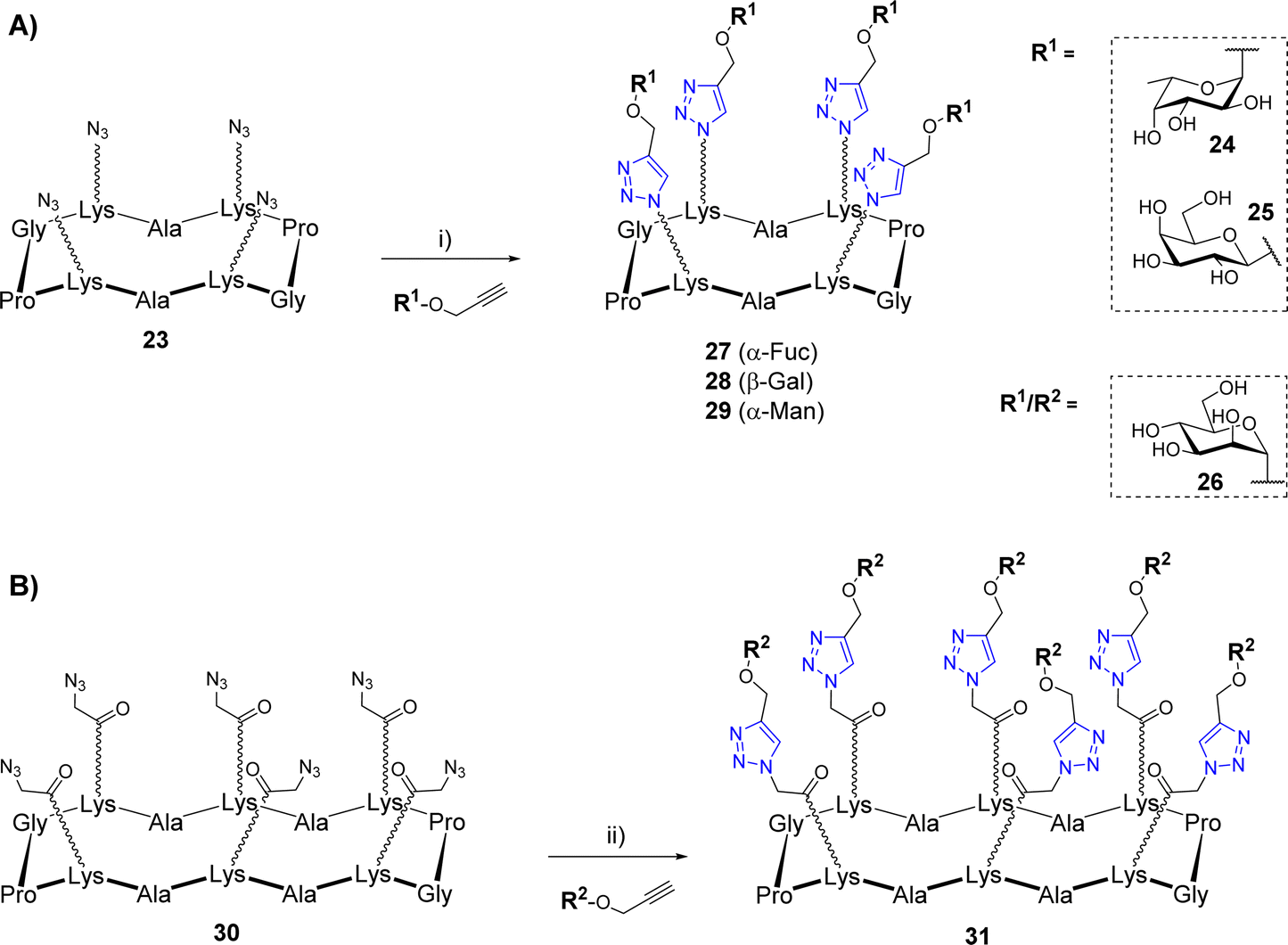 | ||
| Scheme 4 Assembly of glycoclusters by CuAAC: (A) from the tetravalent scaffold 23; (B) from the hexavalent scaffold 30. Reagents and conditions: (i) Cu micropowder, tBuOH/AcONH4 (pH 7.4), r.t., 3 h, 70–75%; (ii) CuSO4·5H2O, THPTA, sodium ascorbate, PBS (pH 7.4), r.t., 1 h, 85%. | ||
The coupling reactions were carried out with several propargyl glycans 24–26 using copper micropowder to afford the tetravalent structures 27–29 in 70–75% yields after HPLC purification. Of note, no trace of incomplete reaction was observed thus confirming a comparable efficiency of CuAAC to OL. In another study, Pifferi et al. described the synthesis of hexavalent glycoclusters from the tetradecapeptide 30 bearing an azido linker at the Lys side chains (Scheme 4B).46 In this case, the use of copper sulfate, sodium ascorbate and THPTA was preferred for the conjugation which allowed the formation of the mannosylated cluster 31 with higher yields (85%) than previous conditions. Similarly, the Li group has reported the synthesis of a tetravalent mannosylated cluster that was obtained from a cyclodecapeptide having four Lys functionalized with alkyne instead of azido group.47 The coupling reaction with azidoethyl mannoside was preformed under ultrasonic irradiation with Cu(OAc)2 and sodium ascorbate to generate Cu(I) in situ, however the expected tetravalent cluster was obtained with moderate yield (44%).
Other conjugations
Fan and coworkers synthesized galactosylated cyclopeptides using squaramide coupling (SqC). They designed a series of structures of different sizes to identify optimized ligands for the cholera toxin, a protein composed of five recognition domains specific for β-Gal.48Linear peptide precursors alternating various amino acids with L-Lys were synthesized on solid support using the standard Fmoc/tBu strategy and an allyl ester as protecting group of the C-terminal end (Scheme 5). After Fmoc and allyl removal, the peptides were head-to-tail cyclized on the solid support, cleaved and purified to obtain three cyclopeptides in 24–30% yield after HPLC purification. These peptides (such as 32) were further functionalized with PEGylated linkers using successive squaramide coupling steps. A first conjugation of the Lys side chain with the squarate activated linker 33 was carried out and followed by Boc removal by acidolysis. These two reactions were repeated successively until reaching the desired linker which was finally terminated with squarate activated galactosylated linker 34 using similar conditions. A total of twelve pentavalent structures such as 35 were thus prepared with yield over 70% after RP-HPLC purification. Subsequent competitive binding assays revealed IC50 in submicromolar range for the larger cyclopeptides having the shorter linkers.
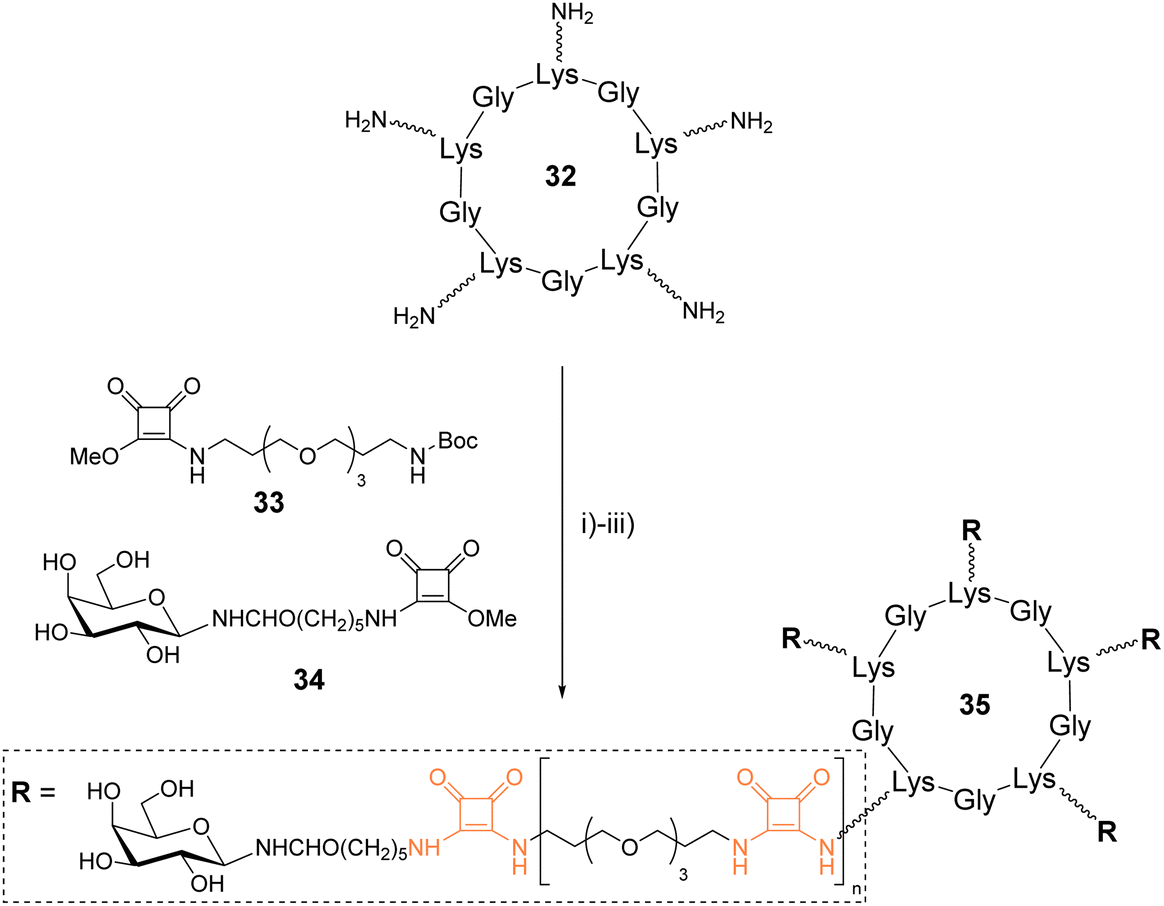 | ||
| Scheme 5 Synthesis of glycoclusters using squaramide coupling. Reagents and conditions: (i) 33, NaHCO3, H2O/CH3OH; (ii) TFA/CH2Cl2; (iii) 34, NaHCO3, H2O/CH3OH, approx. 70%. | ||
In 2004, Wittmann and co-workers identified high affinity ligands for wheat-germ agglutinin (WGA) from a one-bead one-compound library of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 440 cyclopeptides composed of alternated L- and D-amino acids with variable side chains and displaying up to six βGlcNAc through an urethane bond.49 On the basis of this report, the same group further designed a new cyclooctapeptide functionalized with four αGlcNAc units instead of βGlcNAc to change the glycan unit orientation and having a shorter and more rigid linker than previously.50 The cyclopeptide 36, prepared from a TentaGel resin, displays four adjacent or alternate D-2,4-diaminobutyric acids (D-Dab) as branching residues for the glycan units after amine deprotection (Scheme 6).
440 cyclopeptides composed of alternated L- and D-amino acids with variable side chains and displaying up to six βGlcNAc through an urethane bond.49 On the basis of this report, the same group further designed a new cyclooctapeptide functionalized with four αGlcNAc units instead of βGlcNAc to change the glycan unit orientation and having a shorter and more rigid linker than previously.50 The cyclopeptide 36, prepared from a TentaGel resin, displays four adjacent or alternate D-2,4-diaminobutyric acids (D-Dab) as branching residues for the glycan units after amine deprotection (Scheme 6).
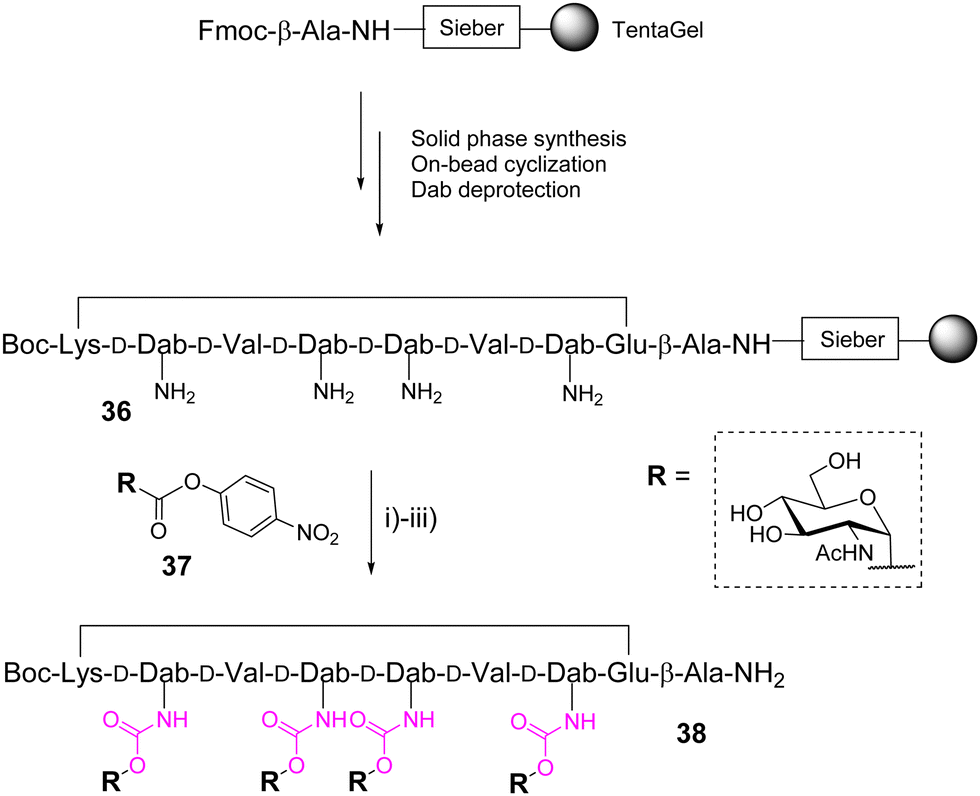 | ||
| Scheme 6 On-bead synthesis of a glycosyl carbamate-cyclopeptide 38 using glycosyl p-nitrophenyl carbonate 37. Reagent and conditions: 37, DIPEA, NMP; (ii) TFA/TIS/CH2Cl2 (1:1:98); (iii) NaOMe, MeOH/CHCl3, 15%. Abbreviations: DIPEA: diisopropylethylamine; NMP: N-methylmorpholine. | ||
The conjugation was carried out on the solid support with an excess of the acetylated glycosyl p-nitrophenyl carbonate 37 in presence of DIPEA in NMP. The total conversion of free amines was confirmed by colorimetric tests on the resin beads.51,52 After cleavage followed by deacetylation and RP-HPLC purification, the corresponding tetravalent glycopeptide 38 was obtained in 15% yield based on the resin loading. Competitive enzyme-linked lectin assays finally indicated a significant improvement for 38 over the best glycopeptides identified from the previous library with longer and more flexible linkers.49 This result was supported by X-ray crystallography and NMR spectroscopy experiments which indicate an optimized conformational pre-organization compatible with multivalent interactions with WGA.
In 2003, Otha et al. used a chemoenzymatic approach to synthesized glycotentacles composed of different amino acids and displaying three GM3 [i.e., Neu5Acα(2,3)Galβ(1,4)Glc], a well-known trisaccharide ligand of the hemagglutinin (HA) of influenza virus.53 The design was based on the structural feature of HA which is trimeric protein having a total of three binding sites for sialylated ligands separated by 40–50 Å. Three linear peptides of 18–22 aminoacids, each containing three Gln, were synthesized using the Fmoc-Asp-ODmab strategy54 then cyclized either on the solid support or in solution to afford cyclopeptides such as 39 (Scheme 7).
 | ||
| Scheme 7 Chemoenzymatic synthesis of a trivalent GM3-based cyclopeptide as blocker of influenza virus hemagglutinin. Reagents and conditions: (i) 40, Transglutaminase; (ii) α-2,3-(N)-sialyltransferase, CMP-Neu5Ac. | ||
After cleavage, deprotection and purification by gel filtration and preparative HPLC, the Gln side chain of the cyclopeptides were conjugated with a large excess of 6-aminohexyl lactoside 40 using transglutaminase from guinea pig liver. In each case, this enzymatic transamination reaction was found incomplete and led to unseparable mixtures of mono-, bi- and tri-functionalized cyclopeptides. The final incorporation of sialic acid units was carried out with CMP-Neu5Ac and the recombinant rat a-2,3-(N)-sialyltransferase to provide GM3 conjugates such as 41. Overall yields after the two enzymatic steps and purification by lectin-based affinity chromatography, gel filtration and HPLC were 8–10% for monovalent, 7–20% for divalent and 7–33% for trivalent structures. Further biological assays and structural studies revealed that the amino acid composition influences the glycocylcopeptide conformation in solution, the spatial orientation of GM3, and therefore the affinity to the influenza virus hemagglutinin.
The Danishefsky group designed a 14-amino acids cyclopeptide displaying a cluster of a high mannose glycan expressed in the silent region of the gp120 glycoprotein of HIV.55 The scaffold 42 is composed of two Pro-pro β-turns and three Asp to attach glycans in a controlled spatial orientation using the Lansbury aspartylation (Scheme 8).56
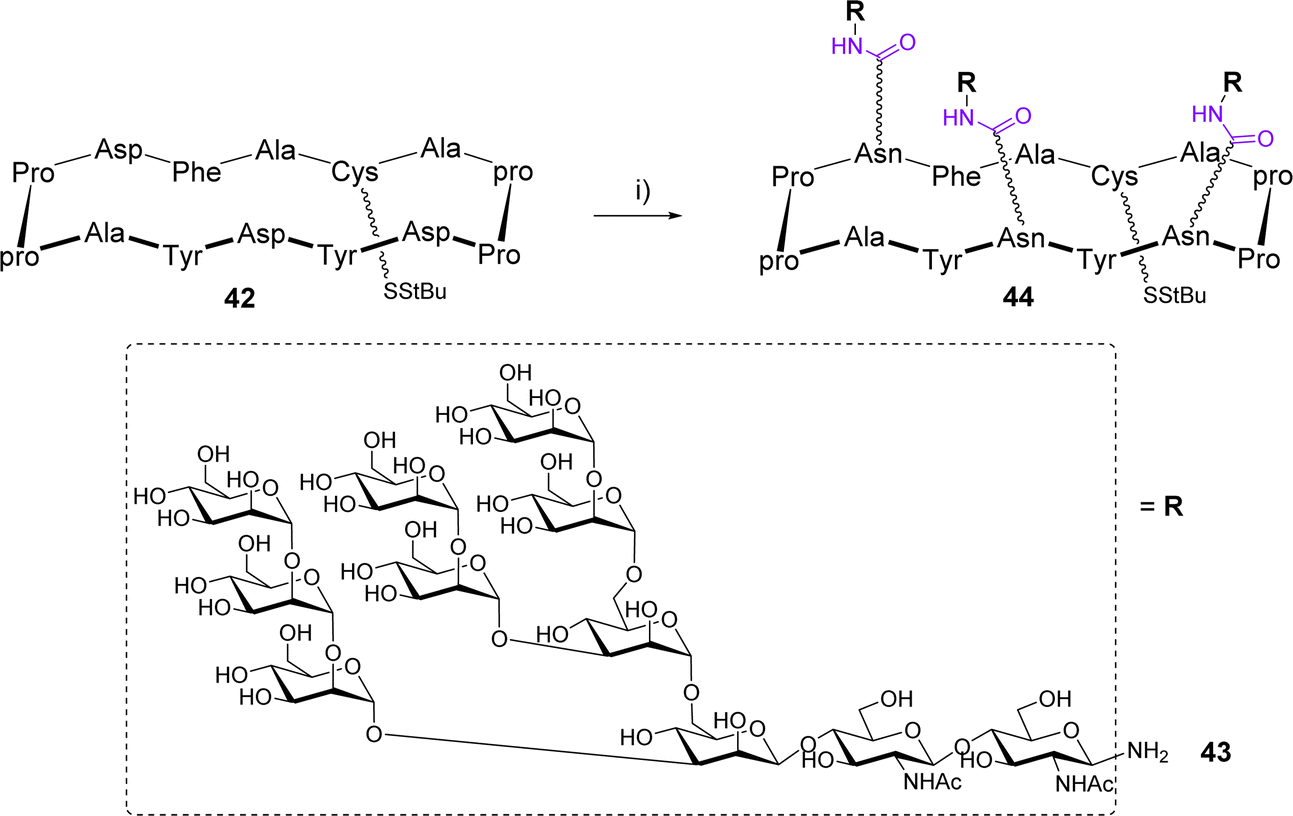 | ||
| Scheme 8 Conjugation of high-mannose glycan 43 by Lansbury aspartylation. Reagents and conditions: (i) HATU/DIPEA/DMSO then 43. Abbreviations: HATU: hexafluorophosphate azabenzotriazole tetramethyl uronium; DMSO: dimethylsulfoxide. | ||
This reaction was carried out in DMSO after a pre-activation step of the Asp side chain with HATU in the presence of DIPEA. The glycosylamine 43 was immediately added to mixture and the trivalent compound 44, obtained as a mixture with the divalent compound was isolated in 45% yield after HPLC purification. Finally, while SPR experiments confirmed the efficient binding with the human 2G12 antibody, the conjugation of 44 to a lipoprotein carrier from Neisseria meningitidis and subsequent immunization of guinea pigs and rhesus macaques failed to induce neutralizing antibodies against HIV.57 A comparable conjugation approach was reported by the Kunz group for the preparation of multivalent cyclooctapeptides as potential E-selectin ligands.58
The Nishimura group has reported the one-pot synthesis of cyclic peptides composed of several copies of the Ala–Thr–Ala tripeptide sequence 45 having the disaccharide β-D-galactosyl-(1–3)-α-D-N-acetylgalactosamine (also called TF antigen) attached to the Thr side chain (Scheme 9).59
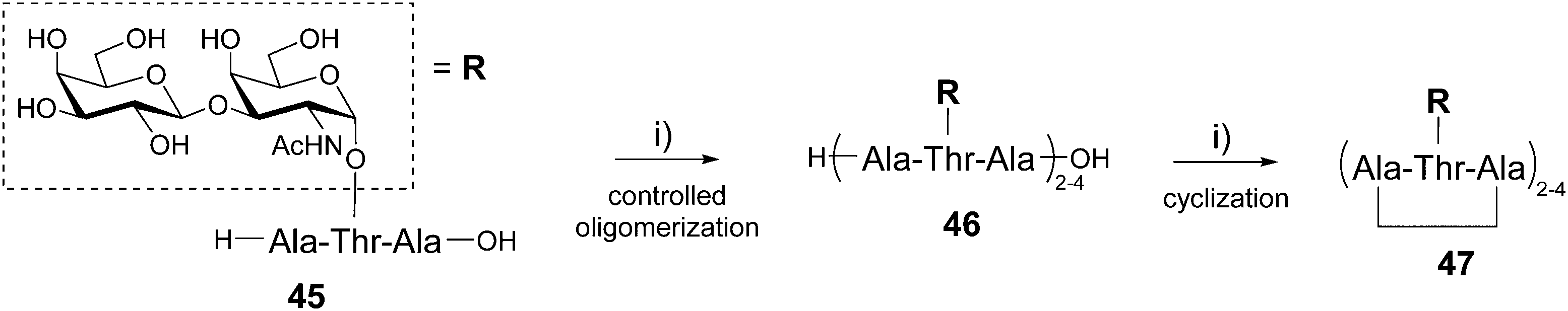 | ||
| Scheme 9 Synthesis of di-, tri- and tetravalent glycocyclopeptides 47 using a one-pot oligomerization/cyclization protocol. Reagents and conditions: (i) DPPA, TEA, DMF, 0 °C. | ||
This glycosylated tripeptide is a repeating sequence found in the antifreeze glycoproteins which possess thermal hysteresis activity allowing fishes to survive in freezing conditions.60 The glycosylated tripeptide 4561 was first oligomerized in a controlled manner using a polymerization promotor (diphenylphosphorylazide, DPPA) and triethylamine in DMF. This reaction can be performed in the presence of free hydroxyls and provided a mixture of linear peptides 46 formed by 2, 3 and 4 repeating motifs, the tetrameric structure being the major product. The cyclization step was finally carried out by diluting the crude mixture in DMF and adding DPPA and TEA. The reaction occurred quantitatively to afford the corresponding cyclic peptides presenting from 2 to 4 copies of the disaccharide 47 that were isolated by ion exchange chromatography (59% overall yield). These compounds have shown ice-growth inhibitory effect and high thermal hysteresis value, in particular for the smaller divalent structure.
Successive ligation strategies
Due to the robustness and versatility of OL, new assembly methods based on successive oxime bond formation were developed to combine other biomolecules to a cluster of glycans, thus providing more complex structures dedicated to diverse biological applications that will be described in the next part. Such an approach involving more than one conjugation step, requires the utilization of orthogonal protecting groups, either at the aminooxy or at the aldehyde function. However, both cases would require regioselective protecting groups whose removal conditions could be problematic if an oxime linkage is already present. Rose and co-workers suggested the use of a Ser as a masked oxo-aldehyde to avoid the fastidious manipulation of protecting groups.32 Beside this, another limitation of successive OL reactions could arise from an undesirable side reaction between an oxime bond and free aminooxy-containing compound required to generate the next one.62 Such a trans-oximation reaction is favoured by temperature and acidic catalysis, however, it was demonstrated that it does not occur on keto-oxime. These different studies converge towards the fact that successive oxime ligations should be performed from oxo-aldehydes generated by oxidative cleavage of Ser.On the basis of this conclusion, a cyclopeptide 48 composed of six Lys, four functionalized with Ser and two with an aminooxy linker was conjugated to different peptides having oxo-aldehyde at the N-terminus (Scheme 10).63,64
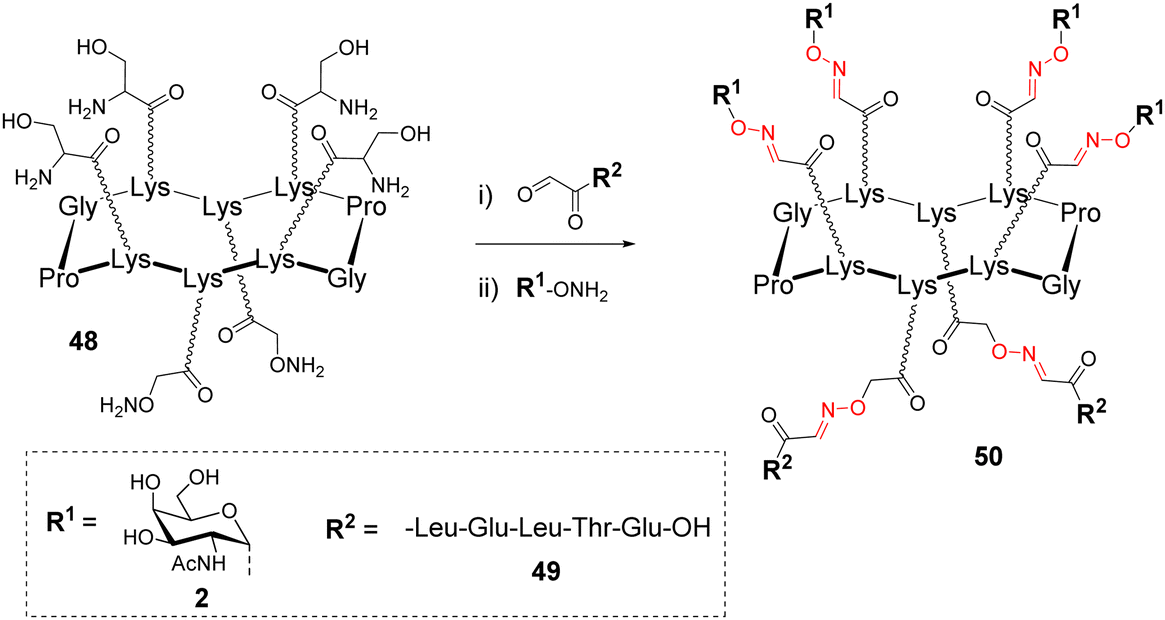 | ||
| Scheme 10 Functionalization of the second domain of a cyclopeptide scaffold 50 using successive oxime ligation. Reagents and conditions: (i) 0.1% TFA in CH3CN/H2O (1:1), 37 °C, o/n then NaIO4, H2O, r.t., 1 h, 95%; (ii) 0.1% TFA in CH3CN/H2O (1:1), 37 °C, o/n, 80%. | ||
After this first oxime conjugation with peptide 49, which was performed in a solution of acetonitrile/water containing 0.1% of trifluoroacetic acid (TFA), the crude mixture was treated with sodium periodate to generate four oxo-aldehydes. After purification, the resulting compound was successively conjugated with the aminooxy glycan 2 to afford 50 in excellent yield.
In other studies, OL was explored to increase the number of glycans presented at the surface of the cyclopeptide.65,66 To this aim, the upper domain of the cyclopeptide 51 displaying four oxo-aldehydes was functionalized with the cyclopeptide 52 containing four Ser and one aminooxy linker thus providing the dendritic structure 53 displaying sixteen Ser at the periphery (Scheme 11).
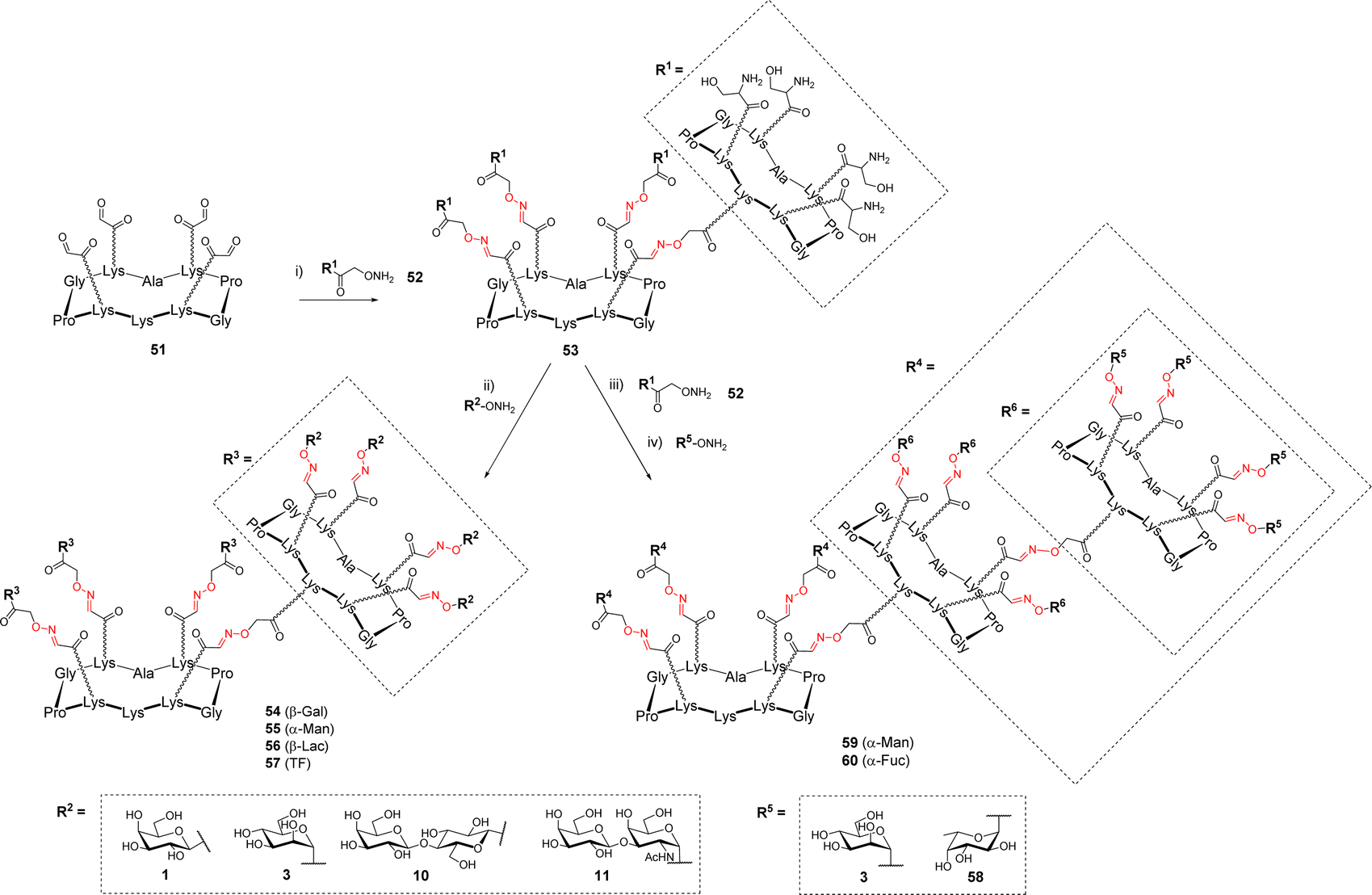 | ||
| Scheme 11 Extension of the cyclopeptide valency from 16 to 64 by iterative oxime ligation. Reagents and conditions: (i) 0.1% TFA in H2O, 37 °C, 4 h, acetone quenching; (ii) NaIO4, H2O, r.t., 30 min then 0.1% TFA in H2O, 37 °C, 4 h, 65–88%; (iii) 0.1% TFA in H2O, 37 °C, 1 h, acetone quenching then NaIO4, H2O, r.t., 30 min, 88%; (iv) 0.1% TFA in H2O, 37 °C, 4 h, 83–85%. | ||
The excess of 52 was quenched with acetone then the crude mixture was subsequently converted into oxo-aldehydes under standard conditions. The final conjugation with aminooxy glycans afforded hexadecavalent glycodendrimers 54–57. HPLC analysis indicated complete conversion and no difference of reactivity was observed compared to the preparation of the more simple tetravalent clusters. While NMR spectrum clearly showed the expected characteristic signals for oxime and anomer protons, the main problem reported by the authors was the fragmentation of oxime ether linkage during MS analysis, which significantly complicates the structural analysis of such compounds.
Due to the remarkable efficiency of this assembly method, the same group went one step further by conjugating another ‘layer’ of the scaffold 52 to 53 (Scheme 11).67 This reaction provided compound showing 64 Ser that was converted into 59–60 following the same process including successive oxidative cleavage of Ser and oxime ligation. These 64-valent glycodendrimers 59–60 with molecular weights of approximately 38 kDa were finally obtained after 3 iterative steps in 83–85% yield. Here again, the final characterization was proved difficult by MS due to the presence of 84 oxime linkages in these structures. However, circular dichroism and NMR experiments successfully confirmed the complete functionalization with 64 glycans and their perfect monodisperisty.
Similarly to OL, a successive CuAAC/CuAAC strategy was designed by Ribeiro et al. to synthesize an octavalent structure displaying both L-fucose and N-acetylneuraminic acid (Scheme 12).68
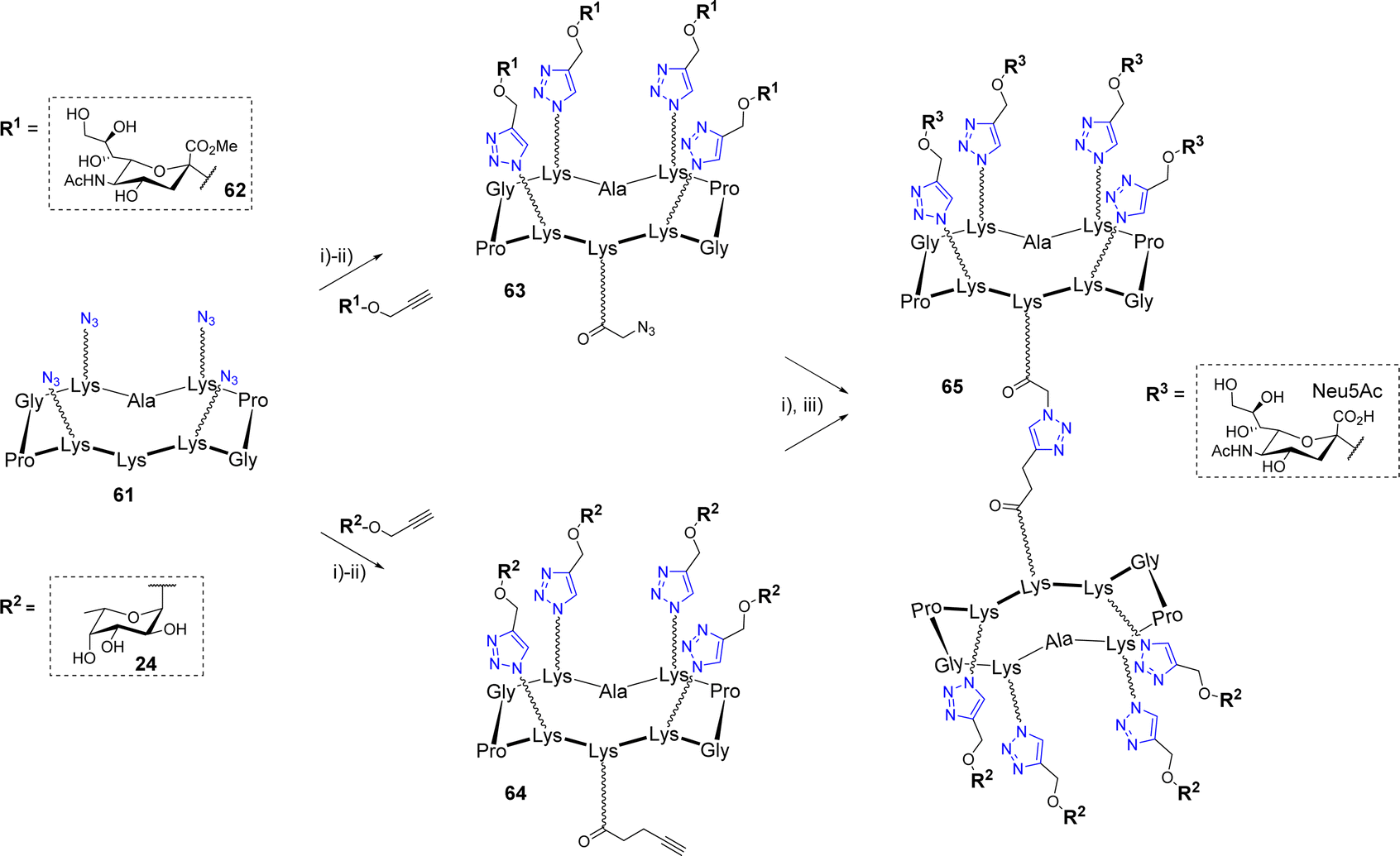 | ||
| Scheme 12 Synthesis of fucosylated and sialylated heteroglycocluster 65. Reagents and conditions: (i) CuSO4·5H2O, THPTA, sodium ascorbate, DMF/PBS (pH 7.5) (1:1), r.t., 1 h, 79% for 63, 85% for 64, 71% for 65; (ii) N-succinimidyl azidoacetate or N-succinimidyl pentynoate, DIPEA, DMF, r.t., 2 h, quant.; (iii) LiOH, r.t., 2 h, quant. | ||
Such multivalent compounds displaying different glycan units, namely heteroglycoclusters (hGCs), are subjected to increasing interests in glycosciences69–72 but remains difficult to synthesize using standard chemical methods. First, azido-functionalized scaffold 61 was conjugated with propargyl α-Neu5Ac 62 on one hand and α-Fuc 24 on the other hand. The resulting conjugates were elongated on their remaining free Lys by either an azido or alkyne linker to afford 63 and 64, respectively. These two compounds were finally combined by CuAAC to obtain the hGC 65 displaying a different saccharide on both faces. This new compound was used as a chemical tool to build a multilayer film composed of alternative layers of glycoclusters and a Janus neolectin having carbohydrate-binding domains specific for both Fuc and Neu5Ac.
Wang et al. also exploited a double CuAAC strategy to conjugate a peptide fragment of the tetanus toxoid and mimics of the Man9GlcNAc2 epitope of the HIV-neutralizing antibody 2G12 to a cyclodecapeptide (Scheme 13).73
 | ||
| Scheme 13 Structure of an oligomannose-based glycocluster prepared by successive CuAAC reactions. Reagents and conditions: (i) 67, CuSO4, sodium ascorbate, t-BuOH/H2O (1:1), approx. 90%; (ii) 69, 0.05 M NaHCO3, CH3CN/MeOH (1:1); (iii) 70, CuSO4, sodium ascorbate, t-BuOH/H2O (1:1), 70%. | ||
The first conjugation between scaffold 66 displaying four alkyne groups and the oligomannoside 67 was carried out using copper sulfate and sodium ascorbate to afford the tetravalent structure 68 in 90% yield. The two pending Lys below the scaffold were successively functionalized with the azido linker 69 then conjugated with the alkyne peptide 70 using the same CuAAC conditions. Further binding studies between diverse glycosylated scaffolds and 2G12 confirmed the interest of the multivalent presentation of glycans and suggested that the final compound 71 could be used as a potential vaccine candidate against HIV-1. To our knowledge, no immunological study has been reported so far.
Orthogonal ligation strategies
Most of the methods described above present an excellent orthogonal reactivity74,75 that revealed ideal to increase the molecular diversity of multivalent systems. After having demonstrated the compatibility of OL and CuAAC and the possibility to use them in a sequential one-pot process,76,77 Thomas et al. exploited this dual strategy to synthesize hGCs. To this aim, the authors synthesized scaffold 72 having two azides and two aldehydes in an alternative fashion (Scheme 14).78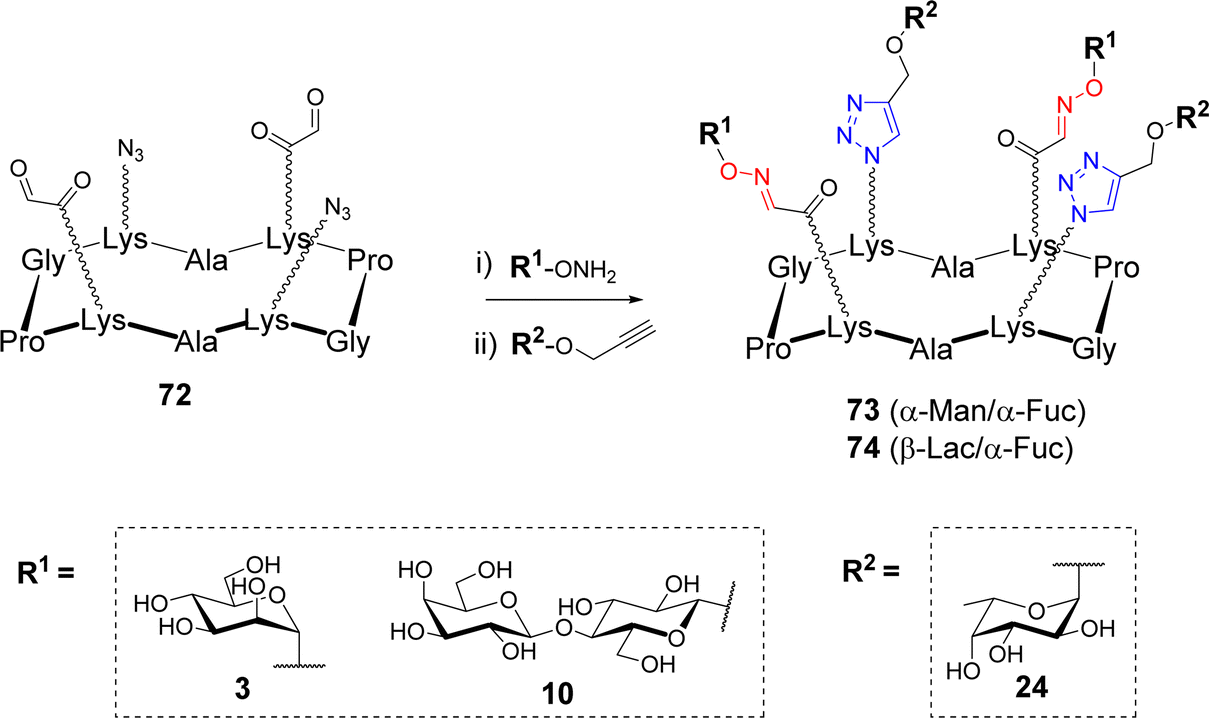 | ||
| Scheme 14 Synthesis of 2:2 tetravalent heteroglycoclusters using OL and CuAAC. Reagents and conditions: (i) 0.1% TFA in H2O; (ii) Cu micropowder, t-BuOH, AcONH4 100 mM pH 7.4 (1:1, v/v), conv. rate 83–93% (calculated from the crude analytical HPLC profile). | ||
Aminooxy 3, 10 and propargyl glycosides 24 were grafted in one-pot via OL and CuAAC respectively. A library of tetravalent 2:2 hGCs 73–74 exposing diverse combinations of α-L-Fuc, α-Man and β-Lac was thus obtained with excellent conversion rate (approx. 90%). The authors also reported a 3:1 series combining the same glycans but with a different distribution at the surface of the scaffold. No difference in reactivity was observed, thereby confirming the efficiency of this method to build hGCs with well-defined sugar positioning.
Multiple ligations are also very attractive for the construction of larger glycodendrimers.13 The dual CuAAC/OL sequence was applied to the synthesis of GalNAc-functionalized glycodendrimers.79 In this study, the authors investigated the efficiency of both convergent and divergent processes which are classically used for the synthesis of dendrimers.80–82 On one hand, scaffold 75 was first conjugated with propargyl α-GalNAc 76 by CuAAC (Scheme 15).
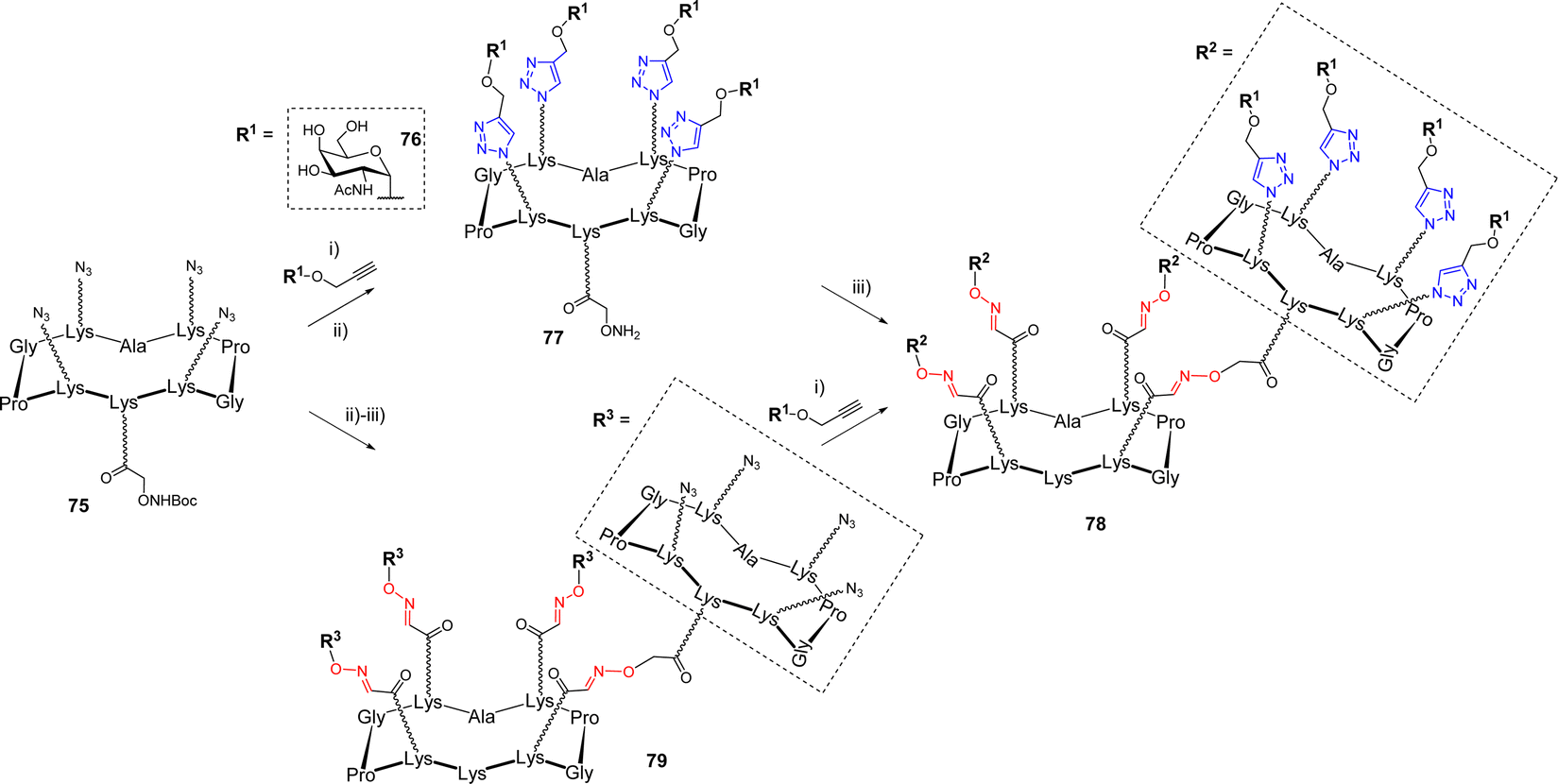 | ||
| Scheme 15 Convergent synthesis of the hexadecavalent GalNAc glycodendrimer 78 by CuAAC/OL. Reagents and conditions: (i) CuSO4·5H2O, THPTA, sodium ascorbate, DMF/PBS buffer (pH 7.5) (1:1), r.t., 2 h, 70% (divergent synthesis); (ii) TFA/H2O (6:4), r.t., 1 h, 77% over two steps for 77; (iii) 51, 0.1% TFA in H2O, 37 °C, 45 min, 90% for 78; 85% for 79. | ||
After Boc-deprotection of the cluster, the compound 77 was engaged in an OL reaction with the aldehyde-bearing scaffold 51, yielding the hexadecavalent dendrimer 78. On the other hand, the divergent sequence first proceeded with the oxime ligation between unprotected scaffolds 75 and 51, followed by CuAAC with propargyl glycoside 76 to simultaneously functionalize all sixteen positions, leading to the expected compound 78. Both strategies proceeded with good yields, highlighting the versatility of this dual ligation sequence. Of note, the authors reported that attempts to use a convergent strategy using OL failed due to the formation of a mixture of side products, presumably due to the presence of an excess aminooxylated building block that led to transoximation reactions.
The synthesis of more complex structures could be achieved using a comparable divergent protocol. Pifferi et al. prepared heterovalent glycodendrimers, displaying both Tn and TF tumor-associated antigens (Scheme 16).83
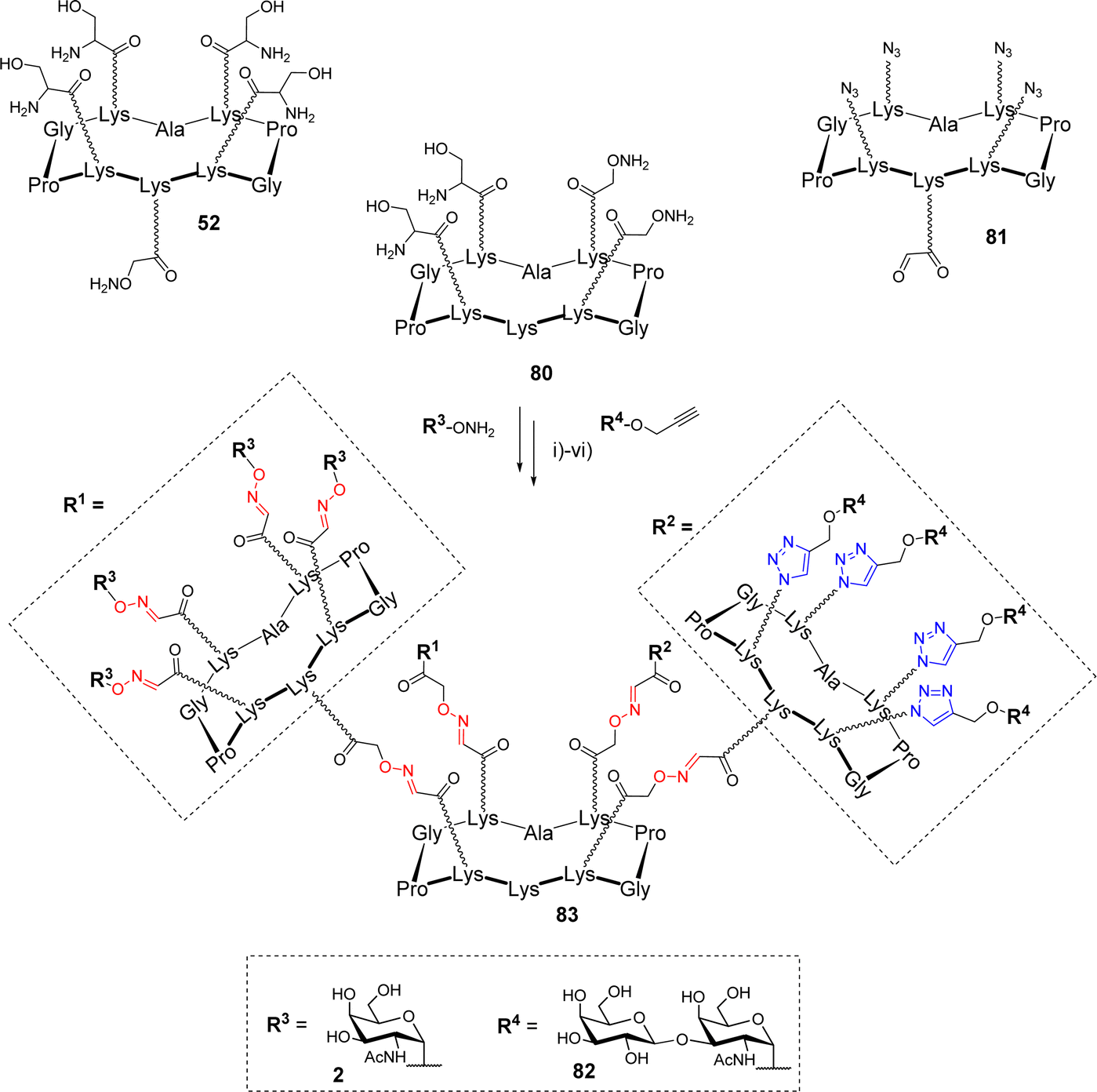 | ||
| Scheme 16 Synthesis of heterovalent glycodendrimers as epitope carriers for antitumor vaccines. Reagents and conditions: (i) 0.1% TFA in H2O/CH3CN (1:1), 37 °C, 30 min, 75%; (ii) NaIO4, H2O, r.t., 40 min, 70%; (iii) 0.1% TFA in H2O/CH3CN (1:1), 37 °C, 30 min, 85%; (iv) NaIO4, H2O, r.t., 40 min, 78%; (v) 0.1% TFA in H2O/CH3CN (1:1), 37 °C, 30 min; (vi) CuSO4, THPTA, sodium ascorbate, PBS (pH 7.4, 10 mM), r.t., 90 min, 55% (over two steps). | ||
The authors used scaffold 80, functionalized with two masked aldehydes and two oxyamines, as the central core. This compound was first conjugated through the aldehyde function of the cyclopeptide 81, then oxidative cleavage of Ser generated the two remaining oxo-aldehydes on the central core that underwent a second oxime ligation with 52. Subsequent ligation with aminooxy Tn 2, followed by CuAAC reaction with propargyl TF 82 afforded hexadecavalent glycodendrimer 83 exposing two different epitopes in an 8:8 ratio. The same strategy was employed to generate a similar compound where each peripheral cyclopeptide is functionalized with two copies of each sugar allowing the study of epitope clustering in the recognition process by antibodies.
Glycosyl thiols represent useful building blocks for the preparation of glycoclusters and glycodendrimers.84 After a previous study in which tetravalent structures were prepared by thiol–ene coupling (TEC),85 hGCs showing 4:2 combinations of glycosyl thiols have been reported.86 The scaffolds 84 and 85 equipped with different regioselective distribution of chloroacetyls and allyloxycarbonyls groups were successively conjugated with β-GlcNAc thiol 86 and α-Man thiol 87 using the TEC and thiol–chloroacetyl couplings (TCC) (Scheme 17).
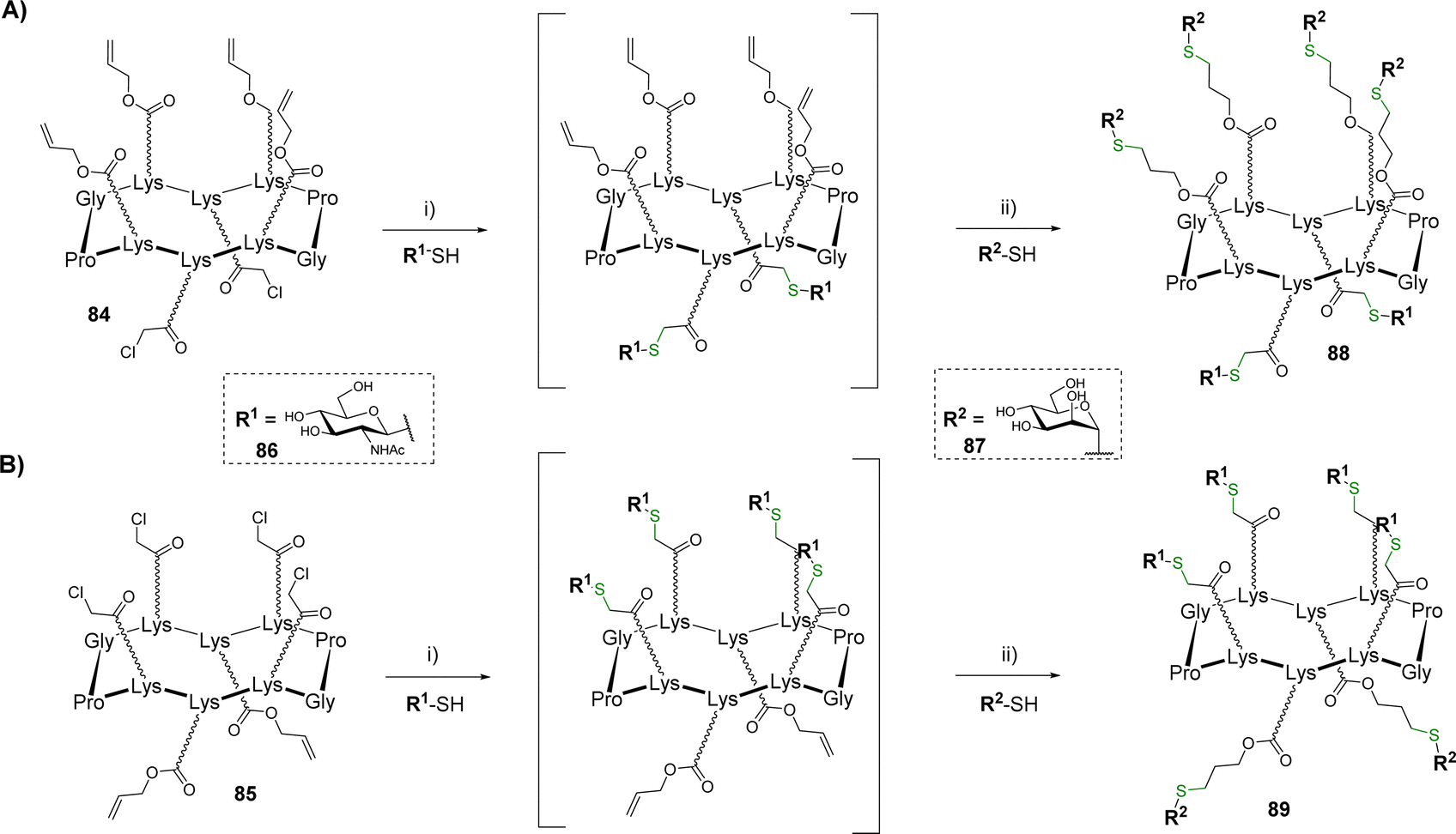 | ||
| Scheme 17 Synthesis of heteroglycoclusters by thiol–ene and thiol–chloroacetyl couplings. (A) Functionalization of the bottom domain as the first step; (B) functionalization of the upper domain as the first step. Reagents and conditions: (i) NaH, DMF, r.t., 1 h; (ii) DPAP, DMF/H2O, irrad. 365 nm, r.t., 45 min, 78% for 88, 77% for 89 over two steps. | ||
To prevent side reaction and avoid intermediate purification, the authors demonstrated that a one-pot protocol can be followed if the thiol–chloroacetyl coupling is performed as the first conjugation step. This reaction was carried out from scaffolds 84 (Scheme 17A) or 85 (Scheme 17B) and β-GlcNAc thiol 86 using NaH in DMF. The reaction was found quantitative in both cases within one hour. In this condition, the remaining excess of 86 spontaneously forms a disulfide adduct which makes the crude mixture directly usable without purification for the next TEC. After neutralization of the crude mixture with hydrochloric acid, this reaction occurred with α-Man thiol 87 using 2,2-dimethoxy-2-phenylacetophenone (DPAP) under irradiation at 365 nm. The resulting hGCs 88–89 were finally obtained in 77–78% after HPLC purification.
The same group reported the synthesis of more complex biomolecular systems using triple orthogonal conjugations. After a first study in which a cyclodecapeptide was successfully functionalized with peptides, glycans and oligonucleotides following a one-pot protocol involving oxime ligation, CuAAC and thiol–maleimide coupling,87 hGCs alternating α-Neu5Ac and β-GlcNAc were reported (Scheme 18).88
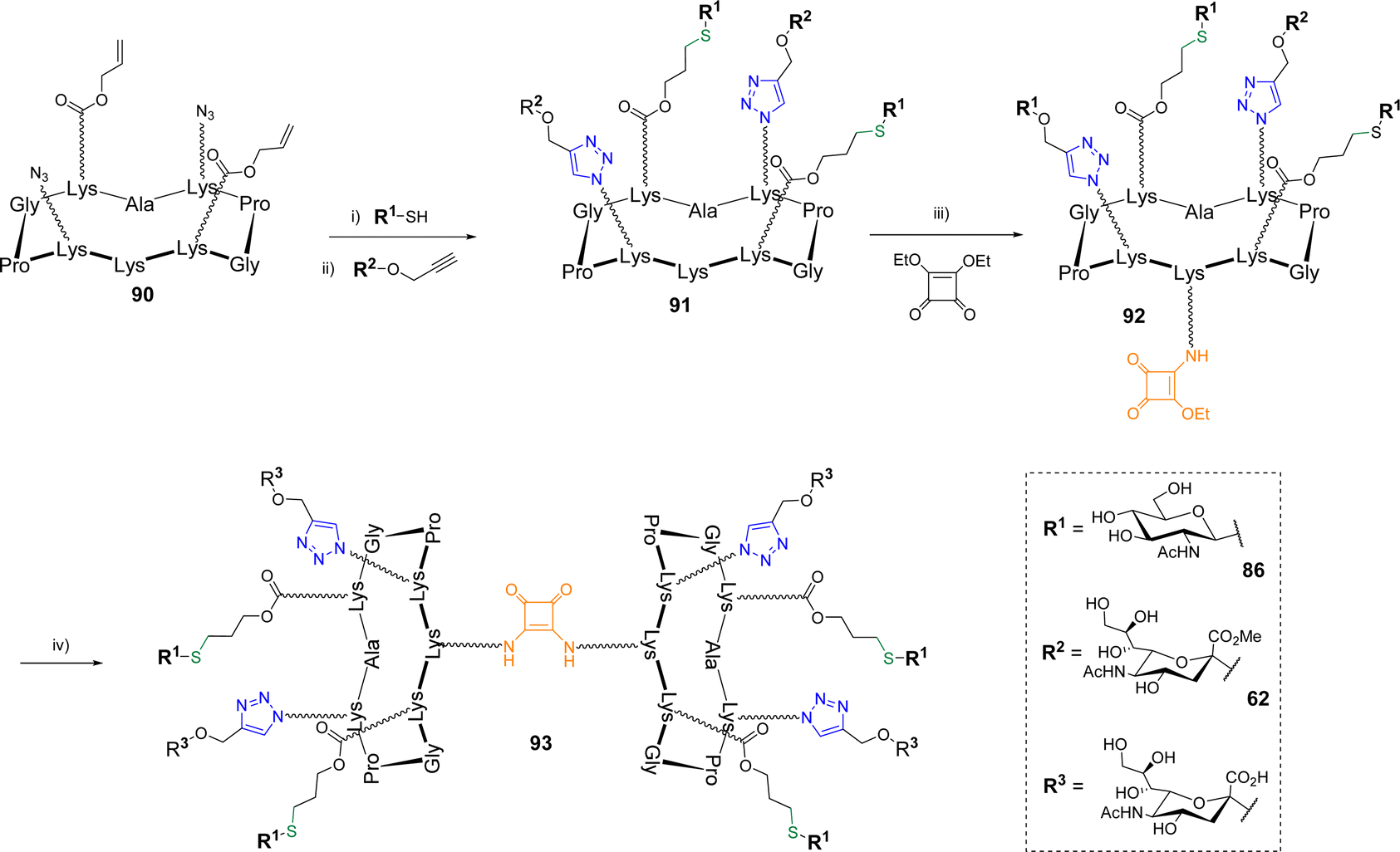 | ||
| Scheme 18 Synthesis of heterovalent structures using CuAAC, TEC and SqC. Reagents and conditions: (i) DPA, DMF/H2O, irrad. 365 nm, r.t., 45 min; (ii) Cu micropowder, PBS (pH 7.4), r.t., 45 min, 71% over two steps; (iii) DMF, DIPEA (pH 8.5), r.t., 4 h, 87%; (iv) 91, Na2CO3 (pH 9.5), 37 °C, 24 h, 67%. | ||
β-GlcNAc thiol 86 was first conjugated by TEC to scaffold 90 bearing two azides and two Alloc using DPAP under irradiation at 365 nm as reported above. No significant difference of reactivity was mentioned (70% yield). The conjugation of the resulting divalent structure with propargyl α-Neu5Ac 62 protected as a methyl ester was next performed by CuAAC with Cu micropowder in PBS to afford the heterovalent compound 91 in 71% yield. Of note, the authors highlighted difficulties when fully unprotected propargyl sialoside was used, which indicates that the free carboxylic acid negatively interfere in the CuAAC reaction. Finally, the pending amine of hGC 91 was involved in a two step self-condensation reaction using SqC to extend the valency to eight glycan units. The reaction was carried out with diethyl squarate which was first conjugated to 91 in DMF with DIPEA (pH 8.5) to provide compound 92. The second amidation reaction was performed between 91 and 92 in an aqueous solution of sodium carbonate (pH 9.5) to allow both the formation of the octavalent compound and the simultaneous deprotection of the methyl esters. The hGC 93 was finally obtained in a 67% yield after semi-preparative HPLC purification. Other hGCs combining diverse glycans (GlcNAc, Neu5Ac, Gal) were obtained with similar efficiency following the same strategy thus highlighting its versatility to extend the diversity of synthetic hGCs in a rapid and controlled manner.
The same group went one step further in the multi-click strategies by preparing heterovalent structures decorated with four different glycans with the aim to provide chemical tool giving access to heterogenous structures as found in the glycocalyx.89 Starting with a cyclodecapeptide scaffold 94 containing regioselectively functionalized Lys with oxo-aldehyde, Aloc, azide and chloroacetyl groups, four glycans were conjugated using OL, TEC, CuAAC and TCC (Scheme 19).
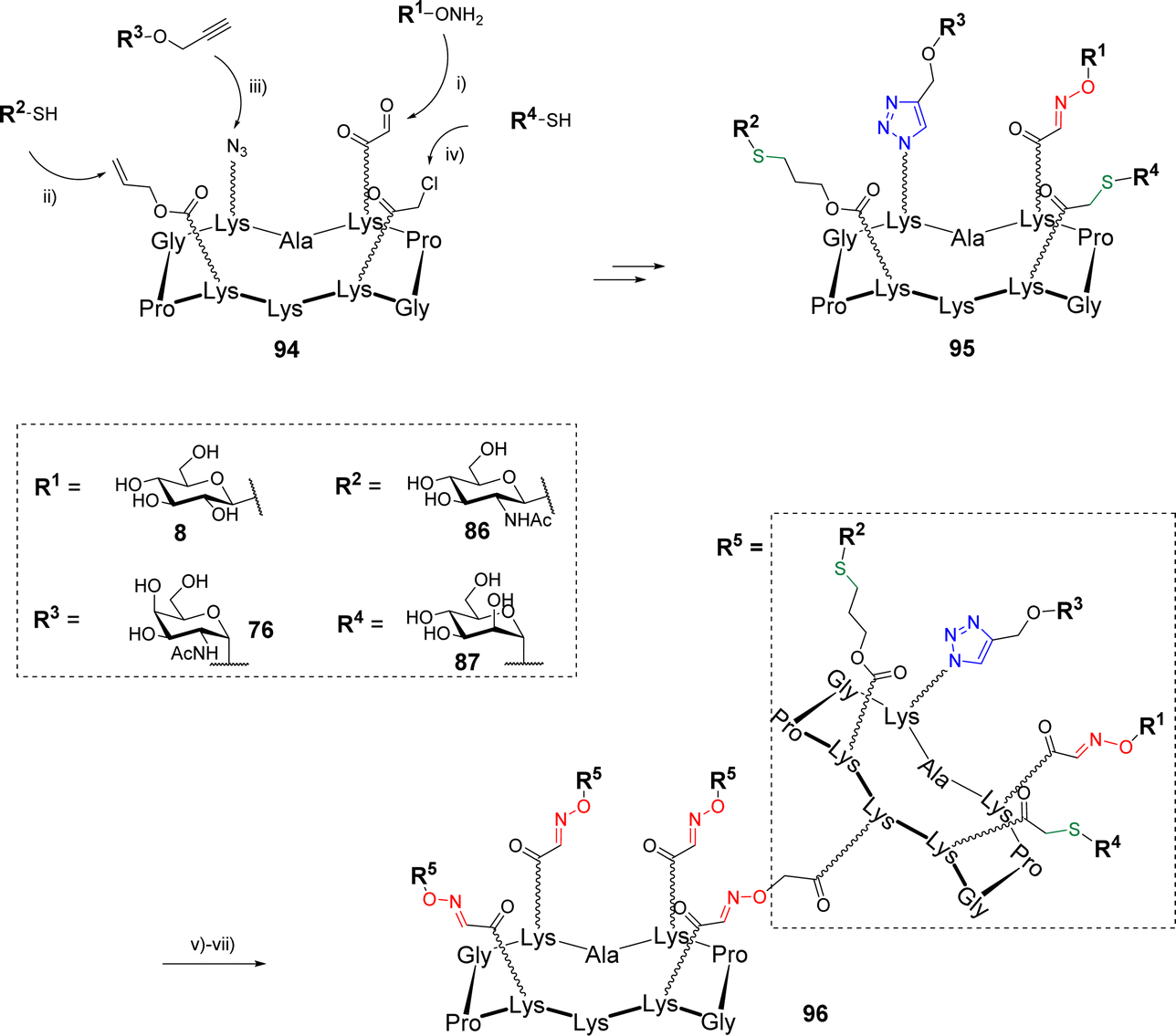 | ||
| Scheme 19 Sequential one-pot synthesis of the heterovalent glycocluster 95 using four conjugation strategies and assembly of the glycodendrimer 96. Reagents and conditions: (i) 0.1% TFA in H2O, r.t., 30 min; (ii) DPAP, DMF/H2O, irrad. 365 nm, r.t., 30 min; (iii) CuSO4, THPTA, sodium ascorbate, DMF/PBS (1:1, pH 7.4), r.t., 1 h; (iv) KI, DMF/H2O (1:1), DIPEA, r.t., 1 h; 47% over four steps; (v) N-succinimidyl (Boc-aminooxy)acetate, DIPEA, DMF; (vi) 50% TFA in CH2Cl2, r.t., 30 min, 93% over two steps; (vii) 51, 0.1% TFA in H2O, 37 °C, 30 min, 87%. | ||
Once having determined the optimal sequence and experimental conditions to avoid side reactions, the assembly process was carried out using a sequential one-pot approach by successive addition of the functionalized glycans 8, 86, 76 and 87 and adjustment of the reaction medium (i.e., pH and solvent). Intermediate HPLC analyses indicated that each reaction was both clean and quantitative until the last step leading to the heterovalent compound 95. The overall one-pot strategy was found to take a total of 3 hours instead of 4 days with the stepwise strategy including HPLC purification of each coupling intermediate, a single purification step and a global yield of 47% (vs. 41% by the stepwise strategy). Finally, the free amine pointing below the scaffold 95 was used to graft several linkers for further conjugations by OL or disulfide bond formation. Among other examples, the authors have treated 95 with the succinimidyl ester of Boc-aminooxy acetic acid to give a free aminooxy group after acidolysis and used for a final ligation on the tetravalent scaffold 51 (Scheme 19) by OL. The resulting hexadecavalent structure 96 displaying four different glycans was obtained in 87% yields after HPLC purification that demonstrates the large molecular diversity that is now accessible with orthogonal click reactions.
3. Biological applications
Lectin binding with low molecular weight glycoclusters
Lectins are proteins of non-immune origin, with no catalytic activity, that can reversibly bind carbohydrates.90 These carbohydrate-binding proteins can be found in all living organisms, however, they play different roles depending on whether they are expressed by plants, bacteria or animals. In most cases, lectins have a relatively weak affinity for their monovalent ligand (low millimolar to high micromolar) but this low affinity is often counterbalanced by their oligomeric quaternary structure that allows them to simultaneously bind several carbohydrate units. This multivalent binding can lead to a much stronger affinity, several orders of magnitude higher than the monovalent interaction.4,5,91In humans, lectins were found to be involved in a wide variety of biological processes. For example, the asialoglycoprotein receptor (ASGP-R) is uniquely expressed on the surface of hepatic cells and recognizes terminal Gal and GalNAc.92,93 Its natural function is to remove asialylated glycoproteins from circulation but its precise localization has been used to target hepatocytes.94,95 Delangle and coworkers have exploited this feature to deliver copper chelating glycocyclopeptide into hepatocytes.37 The redox properties of copper indeed make it a toxic element when its homeostasis is disturbed. Therefore, the development of intracellular copper chelators is of major interest to counterbalance its accumulation for patients suffering from the Wilson disease.96 To this aim, the authors synthesized cyclodecapeptides with two independent faces (Fig. 1).
 | ||
| Fig. 1 General structure of tetravalent ligands designed to target hepatocytes. Abbreviation: lys: D-lysine residue. | ||
The upper face displays GalNAc residues in a tetravalent manner to ensure an efficient recognition by the ASGP-R. The bottom face, exhibiting two Cys side chains that were oxidized as a disulfide bridge, acts as a precursor of the copper complexing site. The addition of a fluorophore on a D-lysine side chain (compound 97) allowed to confirm the uptake in hepatocytes by flow cytometry. It was also demonstrated that once confronted to the reductive intracellular environment, the disulfide bridge of 97 was cleaved into an efficient copper chelating agent. Finally, the importance of the rigid and well-defined cyclopeptide scaffold was also assessed since it was found to be internalized more efficiently than the other flexible structures. Additional binding experiments with HepG2 human heptic cells were next performed to evaluate the influence of the ligand valency and the anomer configuration of GalNAc. Among the series of the tested compounds (mono to tetravalent conjugates, various scaffolds, α- or β-GalNAc), the tetravalent structure displaying α-GalNAc 98 revealed as the best targeting ligand for ASGP-R.97
Many microorganisms, like bacteria and fungi, use lectins to adhere to the host glycocalix and to trigger the first step of infection.98 For example, Pseudomonas aeruginosa produces two lectins, LecA and LecB, which play a key role in the bacterial adhesion to host cells and in the biofilm formation. LecA and LecB are tetrameric proteins containing a total of four binding sites for Gal and Fuc, respectively, and thus represent key targets to develop antiadhesive agents as an alternative to antibiotic treatments.99–101 Other fucose-binding proteins have been identified in pathogens such as Burkholderia ambifaria and Aspergillus fumigatus. Among them, B. ambifaria BambL102 and A. fumigatus AFL (FleA)103 have the peculiarity of including the repetition of similar β-sheets. These so-called β-propeller lectins104 are able to bind six fucose residues with high affinity. In order to prevent pathogen adhesion to host cells, the fucose-based glycomimetic 99 was synthesized and evaluated for its binding to different fucose-specific lectins (Fig. 2).105
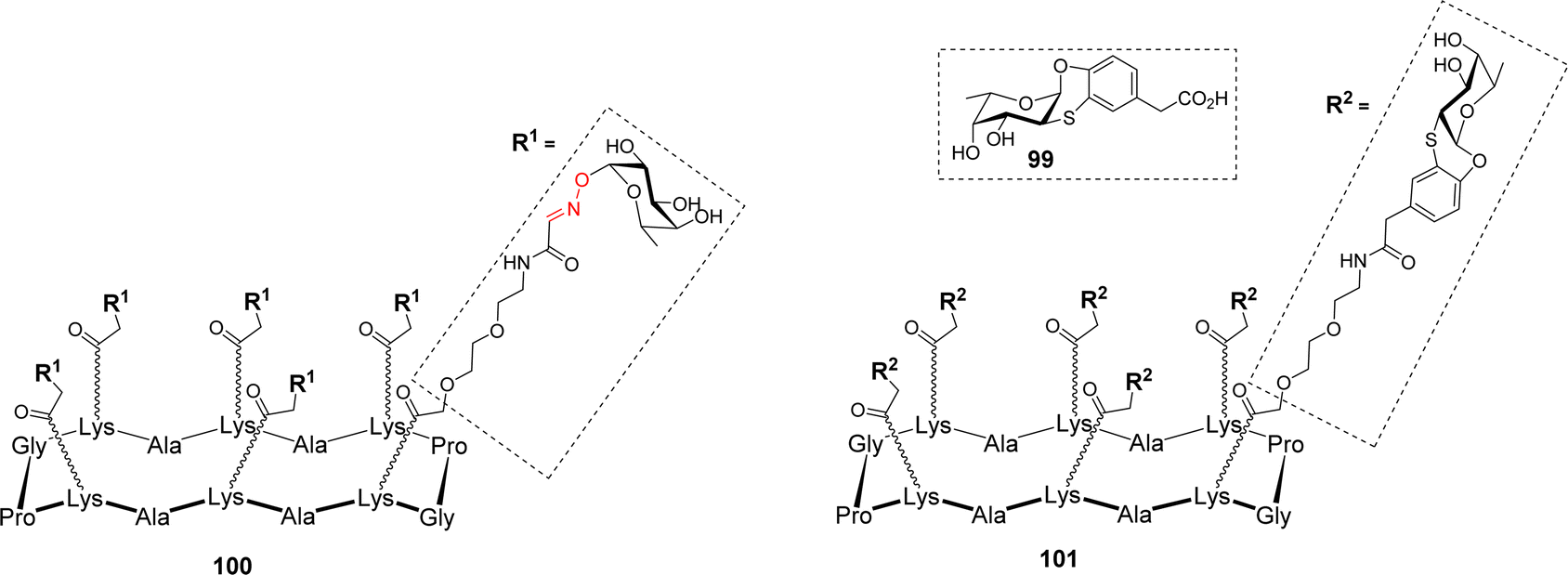 | ||
| Fig. 2 Structure of hexavalent glycoclusters displaying fucose (100) or a fucose mimetic (101). | ||
Interestingly, this glycomimetic bound to BambL and AFL the same way as the natural ligand with an unprecedented selectivity towards these β-propeller lectins. The authors definitely showed that 99 did not bind to other fucose-specific C-type lectins such as LecB or the human DC-SIGN. Capitalizing on this feature, multivalent versions of fucosylated conjugates were designed. Hexavalent glycoclusters 100 and 101, displaying α-Fuc or the glycomimetic 99 respectively, were prepared to match the six binding sites of the targeted proteins (Fig. 2).106 As predicted, Isothermal Titration Calorimetry (ITC) experiments with compound 100 showed no measurable binding to LecB, thus confirming that even at high local concentration, the epitopes cannot reach the binding pocket of LecB. In addition, these experiments indicated that hexavalent compounds 100 and 101 bind very strongly to both BambL (Kd = 16.8 and 13.8 nM) and AFL (Kd = 18.5 and 44 nM) with similar affinities albeit displaying different epitopes. Stoichiometries showed an approximate ratio of 1:1 and binding enthalpies that suggest at least four of the compounds’ epitopes are involved in the binding. Molecular modeling confirmed that a single hexameric compound cannot reach the six binding sites of one BambL trimer and although the distances are compatible between ligand and protein, the orientation of the epitopes is not favourable to induce a real chelate complex. Altogether, these data showed that these glycoclusters were the best-known ligands for BambL and AFL and provided an unprecedented selectivity toward β-propeller lectins.
Lectin binding with glycodendrimers
Cyclodecapeptide scaffolds with extended valency were used by Berthet et al. for studying the binding to the fucose-specific LecB lectin from P. aeruginosa.107 To evaluate the importance of the valency and the sugar headgroup, a series of structures were designed with α-Fuc and β-Fuc (Fig. 3).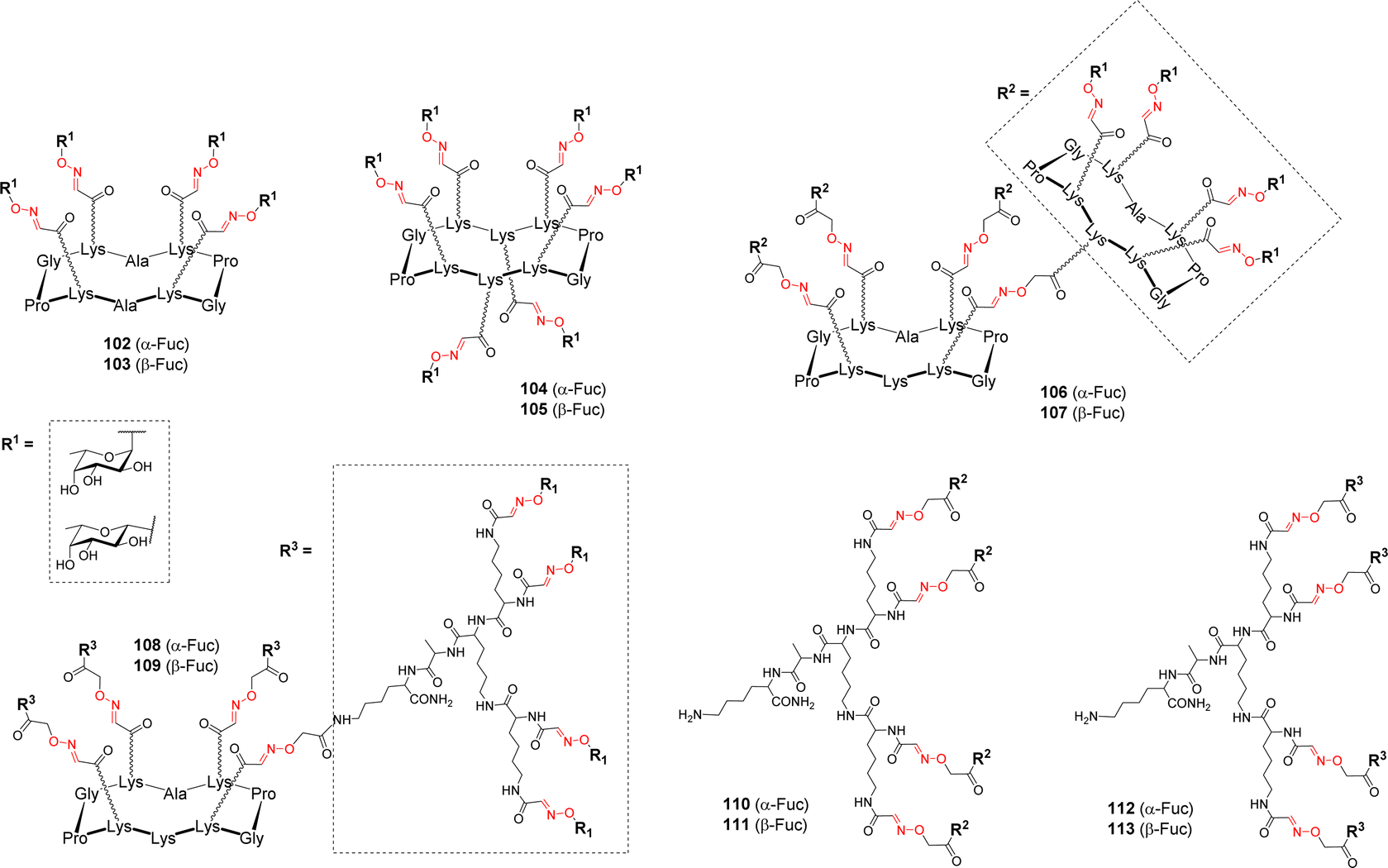 | ||
| Fig. 3 Structures of tetra-, hexa- and hexadecavalent fucosylated conjugates tested with LecB. | ||
Tetravalent (102–103), hexavalent (104–105) and hexadecavalent structures composed of diverse arrangements of cyclopeptide and/or polylysine dendron (106–113) were prepared using the successive OL as described above (Scheme 11). Competitive Enzyme-linked Lectin Assay (ELLA) and ITC experiments were next performed with LecB. The tetra- and hexavalent clusters did not show any significant binding enhancement compared to αMeFuc, regardless of the anomeric configuration. However, the hexadecavalent dendrimers exhibited IC50 in the nanomolar range, which could be expected for αFuc compounds, but more surprising in the case of the β anomers. Compound 108, consisting on a central cyclopeptide core functionalized with α-fucosylated dendrons was proved to be the most potent ligand (IC50 = 0.6 nM) compared to other hexadecavalent structures and it also showed the highest α selectivity. By contrast, no significant impact of the scaffold structure was observed in the β-Fuc series. In order to probe the thermodynamic parameters of their binding, several compounds were tested in ITC experiments. Stoichiometry data showed that each compound can bind between 3 and 6 lectin monomers. Compound 108 showed the lowest dissociation constant (Kd = 28 nM) while its β anomer analogue displayed a Kd ten times higher confirming that the α configuration is better suited for entry in the binding pocket of the protein. Overall, this study showed that even a low affinity ligand can reach high affinity if displayed on a scaffold with suitable valency and geometry. The strong binding enhancements obtained with the best glycodendrimer 108 make it a promising candidate to further investigate its potential anti-adhesive effect, however inhibition and dispersion of the P. aeruginosa biofilm using this compound has not been reported so far.
Based on these encouraging results, and considering that P. aeruginosa produces another lectin implicated in biofilm formation, namely the galactose-specific LecA, Goyard et al. exploited the versatility conferred by the cyclodecapeptide to design hGCs, potentially capable to simultaneously bind both LecA and LecB.108 Several heterovalent structures have been designed towards diverse carbohydrate-binding proteins so far.109–113 These studies emphasized the complexity of the cluster effect and confirmed that homovalent compounds reflect only partially glycocalyx heterogeneity, and obliviate the fact that other sugar units may participate in additional biological processes. The review by Jiménez Blanco et al. raised the question of the effect of functional promiscuity of glycoligands on different heteroglycocompounds and the fact that the complexity may create “messiness or noise in the processes they participate in”.69 The pioneering work of Reymond and coworkers, who designed dendrimers decorated with both α-Fuc and β-Gal to overcome the bacteria's antibiotic resistance, prompted the authors to address the question of this “heteroglycocluster effect”.114 To this aim, new heteroglycodendrimers 114–117 displaying eight copies of two different sugars (Fig. 4) were prepared using the dual OL/CuAAC strategy described above (Scheme 16).
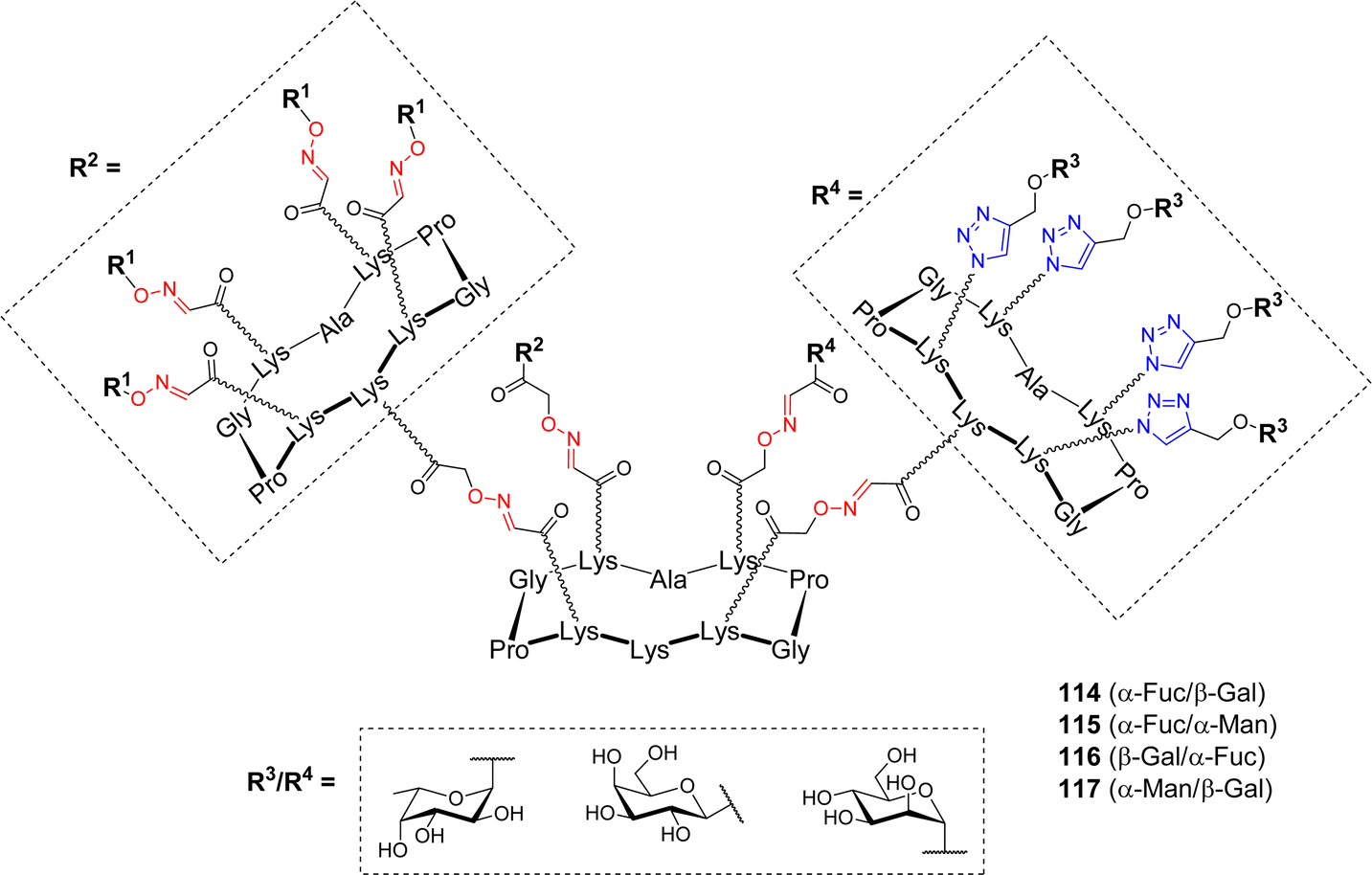 | ||
| Fig. 4 Structures of hGCs 114–117 with dual affinities for LecA and LecB. | ||
These heterovalent molecules, both bearing a lectin's natural ligand (either α-Fuc or β-Gal) and a mismatching sugar (α-Man), were evaluated by ITC for their binding affinity toward LecA and LecB in comparison with homovalent structures. Comparable affinities toward LecB were measured for compounds 114–116 displaying eight copies of α-L-Fuc, regardless of the mismatching sugar (Kd = 92, 85 and 118 nM respectively). These values were approximately two times higher than that of the fucosylated hexadecavalent compound 106 (Kd = 44 nM). Furthermore, stoichiometries indicated that the binding mode is the same for both compounds and that the presence of a mismatching sugar did not hinder the accessibility to the carbohydrate binding site. In the case of LecA, the same trend was observed. Heterovalent compounds 114, 116 and 117 bearing eight β-Gal moieties showed similar Kd values (34, 35 and 21 nM, respectively) than the corresponding hexadecavalent molecule (Kd = 14 nM). Stoichiometries were multiplied by a factor of two (n = 0.16 for heterovalent, n = 0.09 for homovalent) showing that the presence of the mismatching sugar did not impede on the binding. Overall, this study demonstrated that in the case of these bacterial lectins, crowding a multivalent ligand with a non-participating epitope did not negatively impact its efficiency. Compounds displaying eight copies of both α-Fuc and β-Gal could be evaluated for their simultaneous binding to both LecA and LecB and anti-adhesive potential.
The question of glycoligands promiscuity was also addressed by Mellet et al.115 They prepared mannosylated glycodendrimers (similar to 106 but having triazole linkages instead of oxime ethers, Fig. 3) that were used to trigger selective inhibition of lysosomal glucocerebrosidase or α-mannosidase depending on valency and topology. The authors were able to demonstrate that multivalent structures behaved as inhibitors instead of substrates of these biologically relevant enzymes. Considering the promiscuity of carbohydrate receptors at the surface of cells, amalgamating lectin-mediated uptake and glycosidase activity using a single multivalent compound could prove particularly appealing in the context of pathologies such as lysosomal storage disorders.
Burkholderia cenocepacia is another opportunistic bacterium responsible for severe pulmonary infections that are exacerbated in immunocompromised patients.116 Its multidrug resistance and ability to form biofilms make its infections difficult to treat. This pathogen produces three soluble lectins, among which BC2L-A,117 which assembles as a dimer and was proposed to bind oligomannose-type N-glycan structures in order to adhere to host tissues. In an effort to develop high affinity ligands for this lectin, Pifferi et al. synthesized a library of mannosylated glycodendrimers with various structural topologies and valencies ranging from 4 to 24.46 Different scaffolds composed of a tetradecacyclopeptide core combined with a decacyclopeptide (118) or a polylysine dendron (119) were prepared by CuAAC (Scheme 4 and Fig. 5).
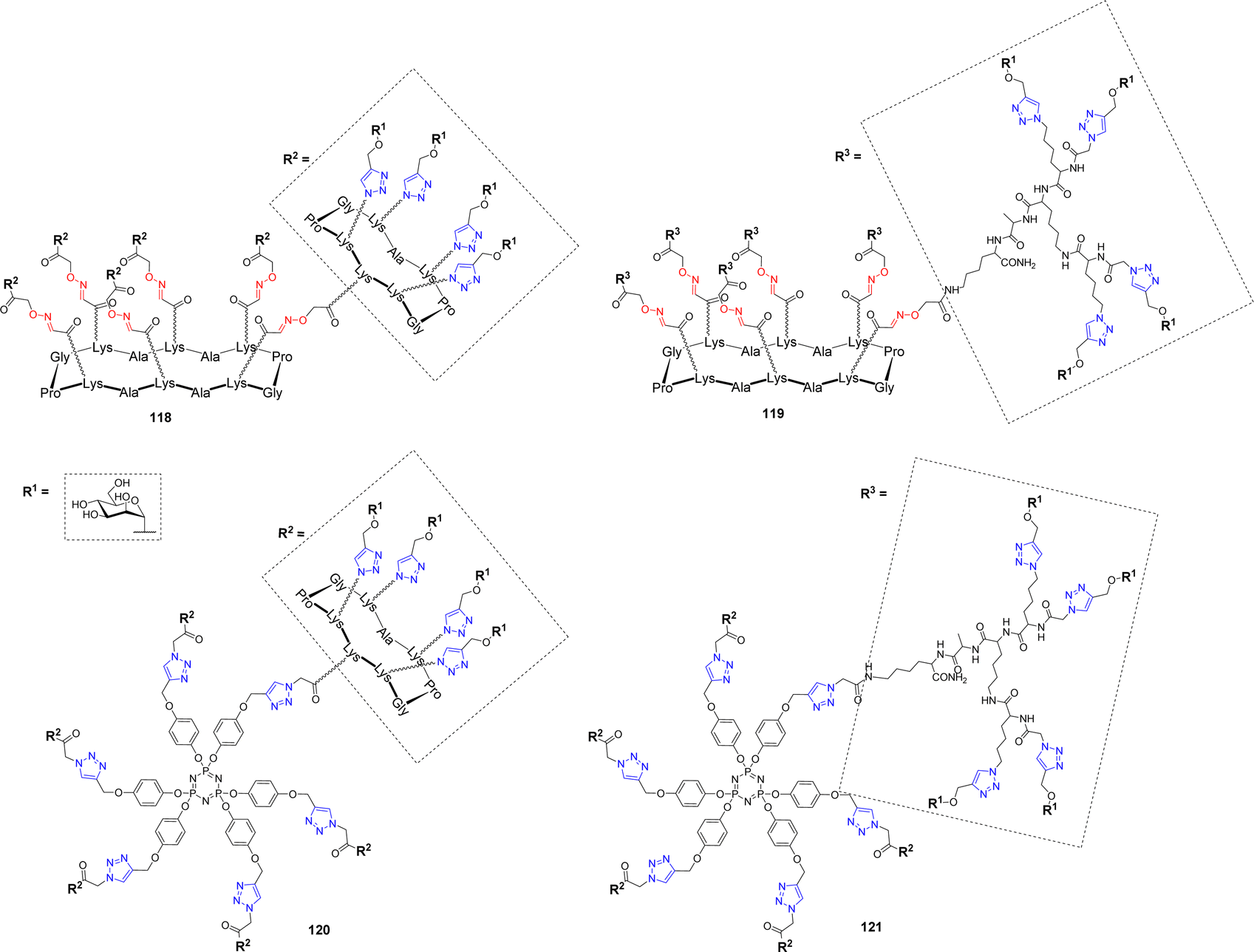 | ||
| Fig. 5 Structures of tetracosavalent mannosylated glycodendrimers 118–121 evaluated as BC2L-A ligands. | ||
To extend the diversity of the library, a cyclotriphosphazene core, which is characterized by the orientation of three grafting sites from either sides of the planar phosphazene118 was also decorated with the same peptides (120–121). ITC experiments were next performed to evaluate their binding with BC2L-A. As it is often the case with multivalent ligands, data showed an enthalpy-driven binding, counterbalanced by a strong entropy barrier, induced by the loss of flexibility upon lectin binding. Tetra- and hexavalent clusters 29 and 31 showed modest affinity enhancement compared to monovalent αMeMan but stoichiometries indicated that each sugar residue is available for binding and that the molecules can accommodate from 4 to 6 lectin monomers. Surprisingly, multimerization of the glycoclusters to form tetracosavalent dendrimers (118–121) did not lead to a significantly strengthened binding. Kd values for these dendrimers ranged between 200 and 250 nM, versus 200–500 nM for the tetra- and hexavalent compounds. Furthermore, despite their structural differences, both in core and peripheral scaffolds, these glycodendrimers exhibited very similar Kd values. However, the construction 121, displaying α-Man through the polylysine dendron and built around the cyclotriphosphazene core stands out with a Kd of 51 nM making it a potential candidate in an anti-adhesive approach.
Velasco-Torrijos et al. used identical scaffolds to develop multivalent adhesion inhibitors of the fungal pathogen Candida albicans to human buccal epithelial cells.119 This opportunistic pathogenic yeast is the most prevalent cause of fungal infections worldwide, particularly in hospital-acquired infections. Compound 122 (Fig. 6) had been previously reported as a successful inhibitor120 and was therefore further functionalized with an alkyne handle to allow conjugation on multivalent azidated scaffolds.
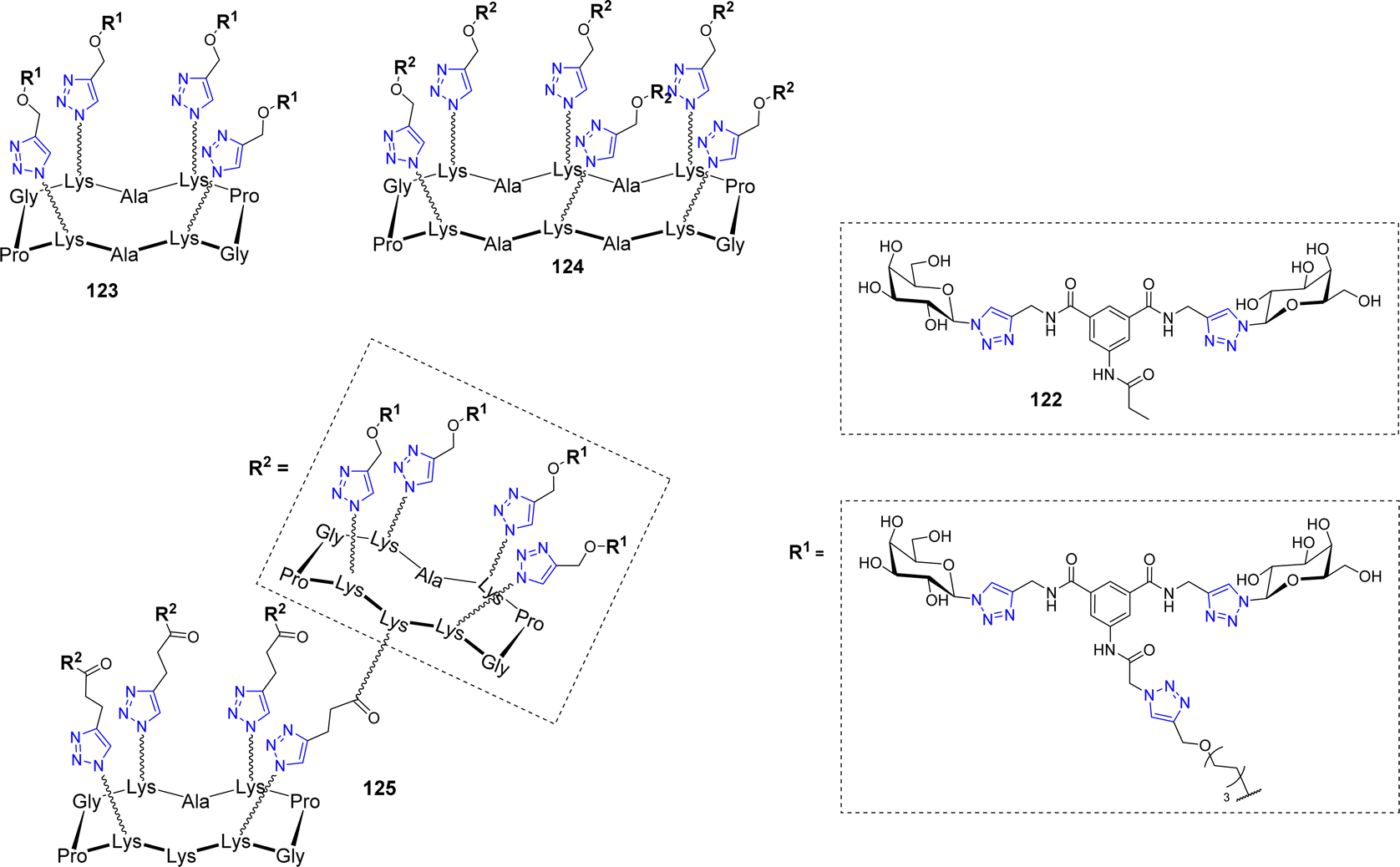 | ||
| Fig. 6 Structure of the bis-galactoside inhibitor 122 of Candida albicans adhesion and its multivalent conjugates 123–125. | ||
As opposed to the previously mentioned lectins, the adhesin triggering C. albicans infections has not been isolated or crystallized, therefore, no quantitative affinity experiments could be conducted. However, using both exclusion and competition assays, the inhibition of adhesion could be evaluated and showed a beneficial effect for glycoclusters 123–125 but not for glycodendrimers, which didn’t outperform the lead compound 122. These glycoclusters and glycodendrimers were also assayed in biofilm inhibition experiments and interestingly, none of them were able to reduce biofilm formation more efficiently than compound 122. This study not only highlighted the fact that increasing valency does not necessarily leads to improving biological activity but also that, if administered after pathogen adhesion, anti-adhesive agents may fail at biofilm dispersion, which is a crucial effect to achieve in the case of opportunistic pathogens.
Multivalent microarrays with lectins
The conformational rigidity and the geometrical features of multivalent glycocyclopeptides offer the possibility to prepare multivalent microarrays for studying the interaction with lectins with higher sensitivity than using single glycans. Moreover, much lower quantities are required to determine binding affinity compared with other physicochemical techniques such as SPR or ITC. Immobilizing multivalent ligands on glass slide allows the precise control of both surface density and three dimensional orientation of glycans, which is more relevant for the binding evaluation than surfaces displaying a monovalent layer of glycans.121–125 In a first study, a tetravalent cyclopeptide-based cluster of αGalNAc has been immobilized by oxime ligation on an aldehyde SiO2 slide and the interaction was compared with an analogue cyclopeptide presenting a unique αGalNAc unit (Fig. 7A).126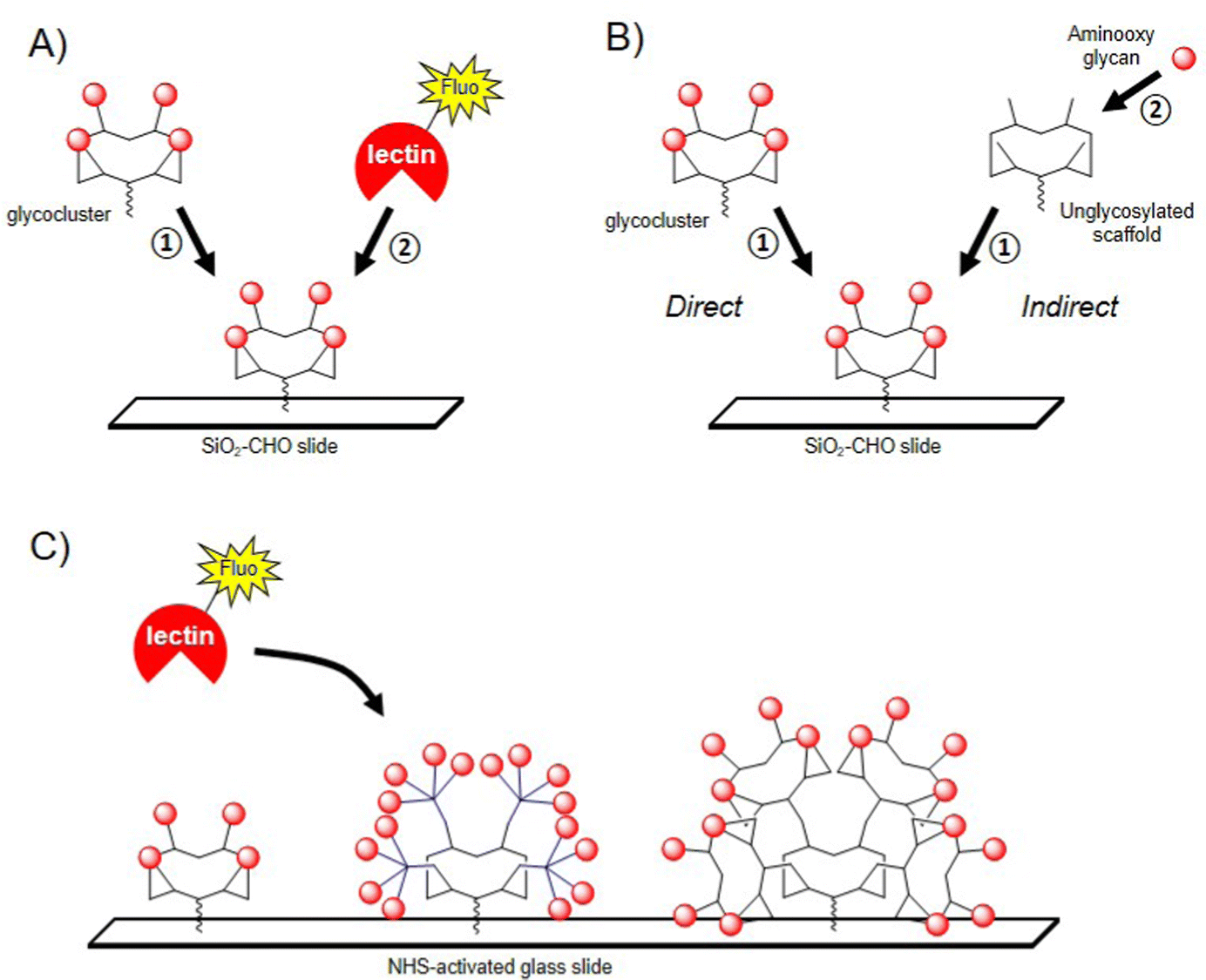 | ||
| Fig. 7 (A) Immobilization of glycoclusters by oxime ligation (1) and incubation with a fluorescent lectin (2); (B) direct (1) and indirect (2) approach for the preparation of glycocluster microarray; (C) glycodendrimer microarray for screening and measuring binding constants. | ||
After incubation with the FITC-labelled Helix pomatia agglutinin (HPA), the fluorescence reading revealed a strong signal intensity for the tetravalent ligand whereas negligible fluorescence was observed for the monovalent control even at higher immobilization concentration. This result was in good agreement with previous studies using other surfaces127 and clearly indicated the importance of the clustered presentation of GalNAc for the interaction with HPA, presumably due to the higher local concentration as well as a more favourable spatial orientation of glycan units. In the same study, the authors evaluated the on-chip assembly of glycoclusters using oxime ligation, called indirect approach (Fig. 7B). The choice of this ligation strategy was dictated by the fact that oxime bond formation is highly reproducible and quantitative under conditions compatible with microarray technology. To this aim, a scaffold presenting four copies of Ser was first grafted to the surface by oxime ligation before being treated with sodium periodate to provide clusters of glyoxoaldehydes (Fig. 7B).126 Aminooxy-Lac was successively spotted at defined position on the surface in conditions compatible with oxime ligation then the fluorescent lectin PNA from Arachis hypogae (peanut) was incubated after washing steps. When compared with the lactosylated glycocluster directly immobilized on the slide, the binding efficiency with the glycocluster obtained from the indirect approach revealed similar, thus demonstrating for the first time the potential of glycocluster fabrication on-chips, from simple building blocks and chemical procedures.
The same group further reported the preparation of glycodendrimers microarrays to measure interactions of multivalent structures having different αGalNAc valency and structural features with HPA (Fig. 7C).128 The authors decided to immobilize glycodendrimers with low surface density to avoid intermolecular chelation and to have access to intramolecular interaction processes. One monovalent, two tetravalent and four hexadecavalent structures having alternate combination of cyclopeptides and peptide dendrons (analogue structures depicted in Fig. 3) and a free Lys group were spotted at low concentration (100 μM to 0.03 μM) on NHS-activated glass slides using a piezoelectric microspotter to obtain spots with 200 μm diameter. Binding assays were first performed with fluorescent HPA to measure Kd and to identify the best ligand for the lectin. As expected, it was observed that structures with lower valency (from 1 to 4 GalNAc units) could not be measured due to low binding affinity. However, three hexadecavalent glycodendrimers revealed nanomolar affinity (Kd = 12–24 nM) thus confirming multivalent interactions with HPA. Of note, the hexadecavalent ligand composed of cyclopeptides both at the central core and at the periphery showed a Kd = 98 nM, thus indicating that ligands with identical valency but having one order of magnitude difference in affinity can be discriminated using this technology. Similar observations were reported more recently with the same compounds and lectin by BioLayer Interferometry.129,130 This study clearly highlights the interest of glycodendrimer microarrays for the screening of multivalent ligands in parallel with lectins using low quantity of materials and rapid experimental procedures.
Antitumoral synthetic vaccines
Human cancers overexpress aberrant glycans called Tumor-Associated Carbohydrate Antigens (TACAs), which are associated with tumor progression and malignancy.131–133 Despite these structures represent key structural elements in the design of antitumoral synthetic vaccines, they need to be associated with an antigenic structure to produce a T cell dependent immunological response with memory cells induction and to elicit high affinity binding with antibodies.134,135 For example, a classical approach to improve immunological response is glycan conjugation to carrier proteins, which allows CD4+ T cells activation and so, antibody switching from low-affinity IgM to higher affinity IgG and memory cell stimulation. Nevertheless, protein/glycan conjugation ratio is difficult to control and carrier proteins are often immunogenic by themselves. Vaccines based on well-defined multivalent scaffolds such as cyclopeptides represent an alternative to avoid these difficulties: (i) they allow simultaneous presentation of B and T cell epitopes to promote a strong and long-lasting immune response; (ii) their conjugation can be easily controlled by click chemistry, which allows the incorporation of a defined number of antigenic carbohydrates and peptides; (iii) multivalent presentation of carbohydrates increases antibody affinity by means of synergistic interaction with different IgG molecules.A first generation of fully synthetic vaccines using cyclopeptides was developed in 2005136 on the basis of the Multiple Antigenic Peptide reported by Leclerc group.137 A cyclopeptide was designed to present four copies of GalNAc as Tn antigen analogue (B cell epitope) and one peptide fragment from poliovirus (PV103–115) as CD4+ T cell epitope (126, Fig. 8A).
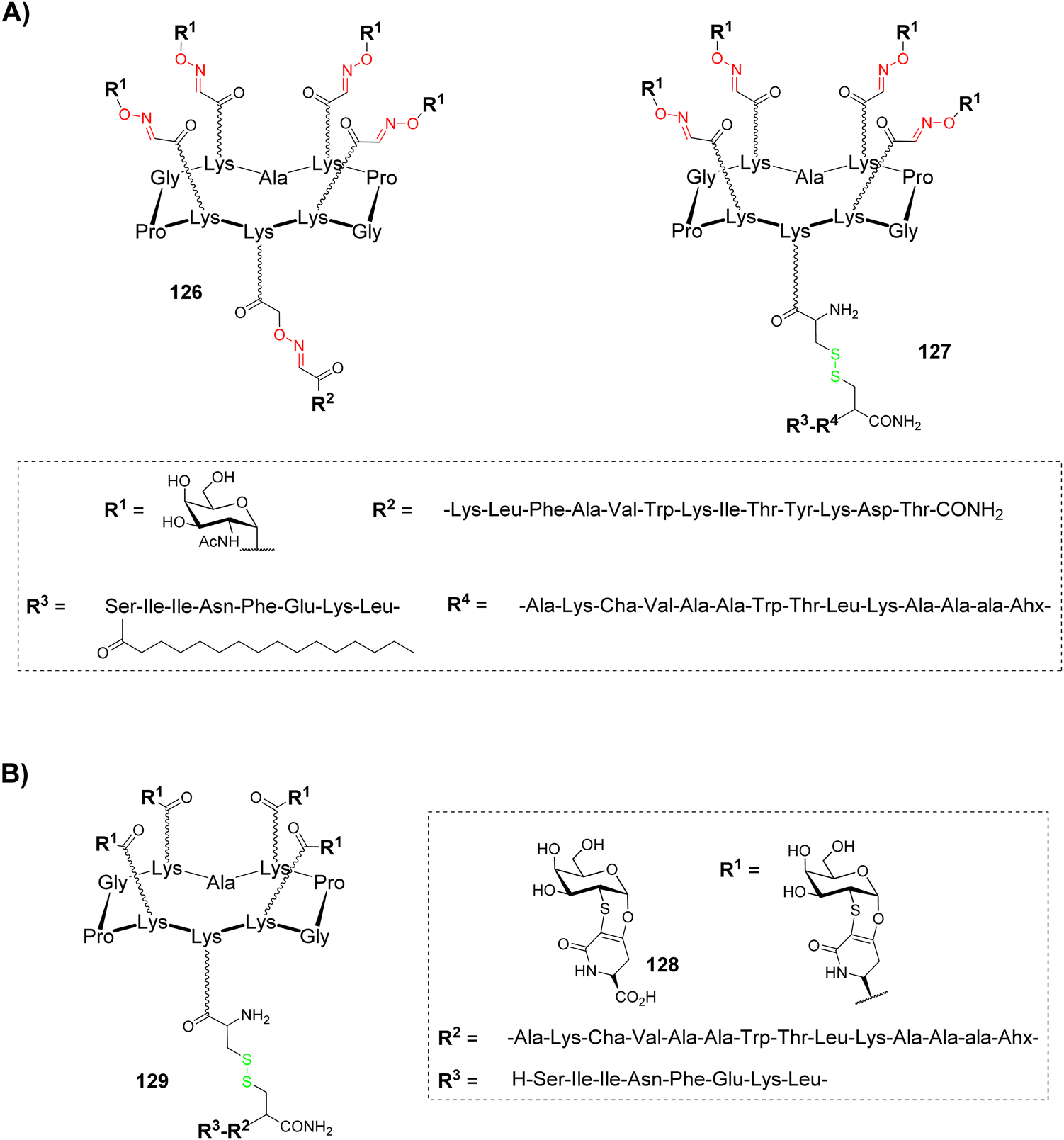 | ||
| Fig. 8 (A) Structure of two and four component antitumoral fully synthetic vaccine candidates based on cyclopeptide scaffolds (R1: Tn analogue; R2: PV peptide; R3: palmitoyl-OVA peptide; R4: PADRE peptide); (B) structure of an antitumoral vaccine candidate using a Tn mimetic (R1: Tn mimetic; R2: PADRE peptide; R3: OVA peptide). | ||
This vaccine candidate was prepared using the double oxime ligation procedure described above (Scheme 10). Immunological studies in mice have demonstrated the ability of this compound to elicit IgG antibodies which recognized the native Tn antigen expressed on human Jukart tumor cell line. This study has also highlighted the absence of significant immunogenicity of the cyclopeptide itself, thus confirming its potential to be used as a carrier for immunological applications.
In order to improve the immunological properties of the previous construct by synergistic induction of antibody, helper and cytotoxic T-cell responses and to reduce toxicity associated to external adjuvants, the same group designed a more complete vaccine candidate 127 (Fig. 8A) combining four components within a single molecule:138 (i) a clustered Tn analogue as B cell epitope, (ii) a PADRE peptide as CD4+ T cell epitope (Fig. 8A), (iii) a OVA257–264 peptide as CD8+ cytotoxic T lymphocyte (CTL) epitope and (iv) a palmitic acid as internal adjuvant. The previous Tn analogue was bound to the RAFT cyclopeptide by oxime ligation while the lipopeptidic chain was bound to the lower part of the scaffold through disulfide bond formation. Immunological evaluation on MO5/BALB/c mouse model showed high production of antibodies recognizing human MCF7 tumor cells as well as T cell stimulation. More interestingly, a complete survival rate and significant reduction of tumor growth was observed after 90 days,139 thus validating the synergic effect of covalently combining four different components in a single fully synthetic cyclopeptide-based vaccine candidate. Other structures by the same authors were further reported and have confirmed the previous observations.140
Glycan analogues, namely glycomimetics, are typically developed to improve in vivo stability of tumor associated antigens while preserving their recognition properties.141 The Nativi group has described a Tn antigen mimetic 128 (Fig. 8B),142 which was further exploited as key structural element for immunological applications.143 With the aim to evaluate its utilization in antitumoral synthetic vaccine, the Tn mimetic was conjugated by amide coupling to the upper face of a cyclodecapeptide scaffold (129, Fig. 8B) containing the PADRE peptide (CD4+ Th cell epitope) in line with the OVA257–264 peptide (CD8+ CTL epitope).144 This vaccine candidate 129 was administrated together with CpG1826 adjuvant on B10.D1 mice for in vivo evaluation. The results first showed a strong and long-lasting IgG/IgM response against the native form of the Tn antigen. In addition, in a group of ten mice inoculated with tumors then vaccinated with 129, seven were still alive after 60 days while none of the mice survived in the control group. Further experiments have suggested that the antitumoral protection is mainly due to B cells.
Due to the heterogeneity of the glycocalyx expressed by tumour cells, hGCs bearing different TACAs have found recent interest to induce the production of antibodies with multiple selectivity allowing tumor targeting at different progression stages. Different hGCs with high valency and bearing both Tn and TF antigens were reported recently (Fig. 9).83
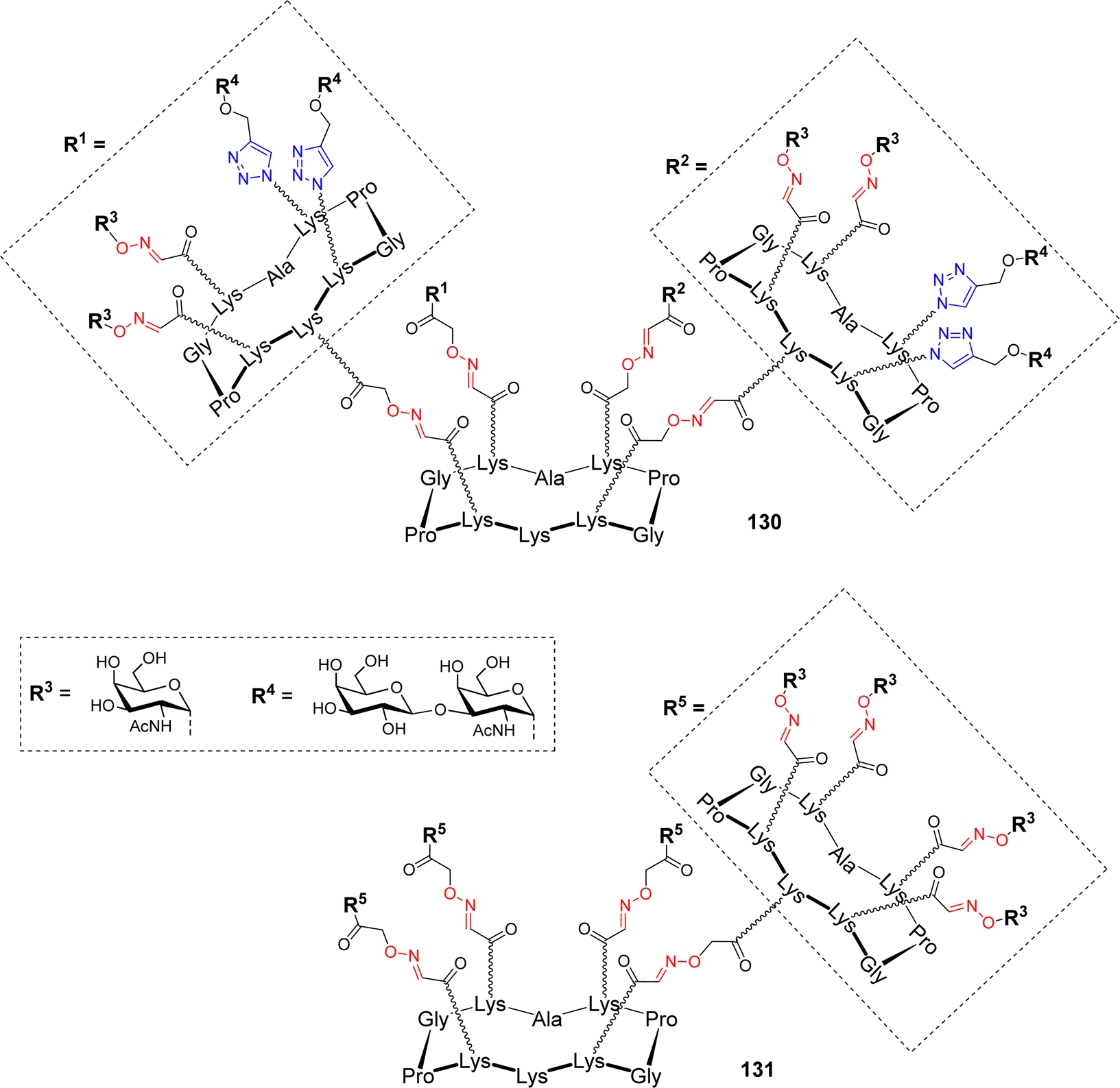 | ||
| Fig. 9 Homo- and heterovalent glycosylated platforms as carrier for antitumoral synthetic vaccines. | ||
Two hexadecavalent cyclodecapeptides 130 (Fig. 9) and 83 (Scheme 16) decorated with both Tn and TF epitopes in different topological arrangement have been assembled using OL and CuAAC. Binding experiments with the 9A7 Tn-specific monoclonal antibody (mAb) revealed that the structure 83 with separated Tn/TF presentation is recognized similarly to the homovalent structure 131 displaying only the Tn antigen (Fig. 9), and with higher efficiency than the structure with alternated distribution of antigens 130. The authors deduced that the presence of TF antigen in 131 does not interfere in the binding with 9A7, making this structure an attractive carrier for further conjugation with T cell epitopes.
In a more recent study, the influence of the nature and the sugar antigen valency were investigated.145 Tetra- and hexadecavalent vaccine candidates bearing an αGalNac 132–133 as a simple analogue or native Ser-linked Tn antigen 134–135 were synthesized (Fig. 10).
 | ||
| Fig. 10 Structure of tetra- and hexadecavalent vaccine candidates based on α-GalNAc or the native Tn antigen. | ||
These B cell epitopes were conjugated by disulfide bond formation with the chimeric peptide composed of the two ovalbumin fragments OVA323–339 and OVA257–264 as CD4+ and CD8+ T cell epitopes respectively. In vivo immunological evaluation on C57BL/6 mice using QS-21 as adjuvant showed the higher capacity of the hexadecavalent constructs displaying α-GalNAc 133 and the native Tn 135 to induce high IgG titers compared to the structures having lower valency 132 and 134. However, significant differences have been observed between the hexadecavalent structures since 133 elicited high IgG1 and IgG2c isotype titers whereas 135 rather promotes IgG2b subtype. Additional experiments with 133 and QS-21 as adjuvant revealed that generated antibodies are able to bind MCF7 cancer cells expressing Tn as well as strong CD4+ and CD8+ cellular responses. The authors concluded that the utilization of vaccine candidates based on GalNAc instead of the native Tn antigen are significantly easier to synthesized and that a high valency presentation of carbohydrate antigens is an important parameter to boost B cell activation and antibody production.
Antibody-recruiting glycodendrimers
An innovative immunotherapeutic approach was reported in the last decade to fight cancer. It was demonstrated that bifunctional molecules called Antibody Recruiting Molecules (ARMs) can both recruit endogenous antibodies present in the blood stream and bind cancer cells selectively to stimulate their immune-mediated destruction.146–148 To this aim, ARMs should contain a tumor binding module (TBM) to interact with a specific tumor associated protein and an antibody binding module (ABM) to bind to endogenous antibodies. This molecular combination allows the formation of a ternary complex between the ARM, the endogenous antibodies and the cancer cell, which leads to the activation of antitumoral cytotoxicity. In this context, cyclodecapeptides have been exploited to construct antibody-recruiting glycodendrimers (ARGs).149,150 Tetra- and hexadecavalent rhamnosylated structures 136–137 and a cRGD-based cluster 138 were first synthesized as ABMs and TBM respectively (Fig. 11)151 and their binding ability was investigated with endogenous antibodies and cancer cell lines.149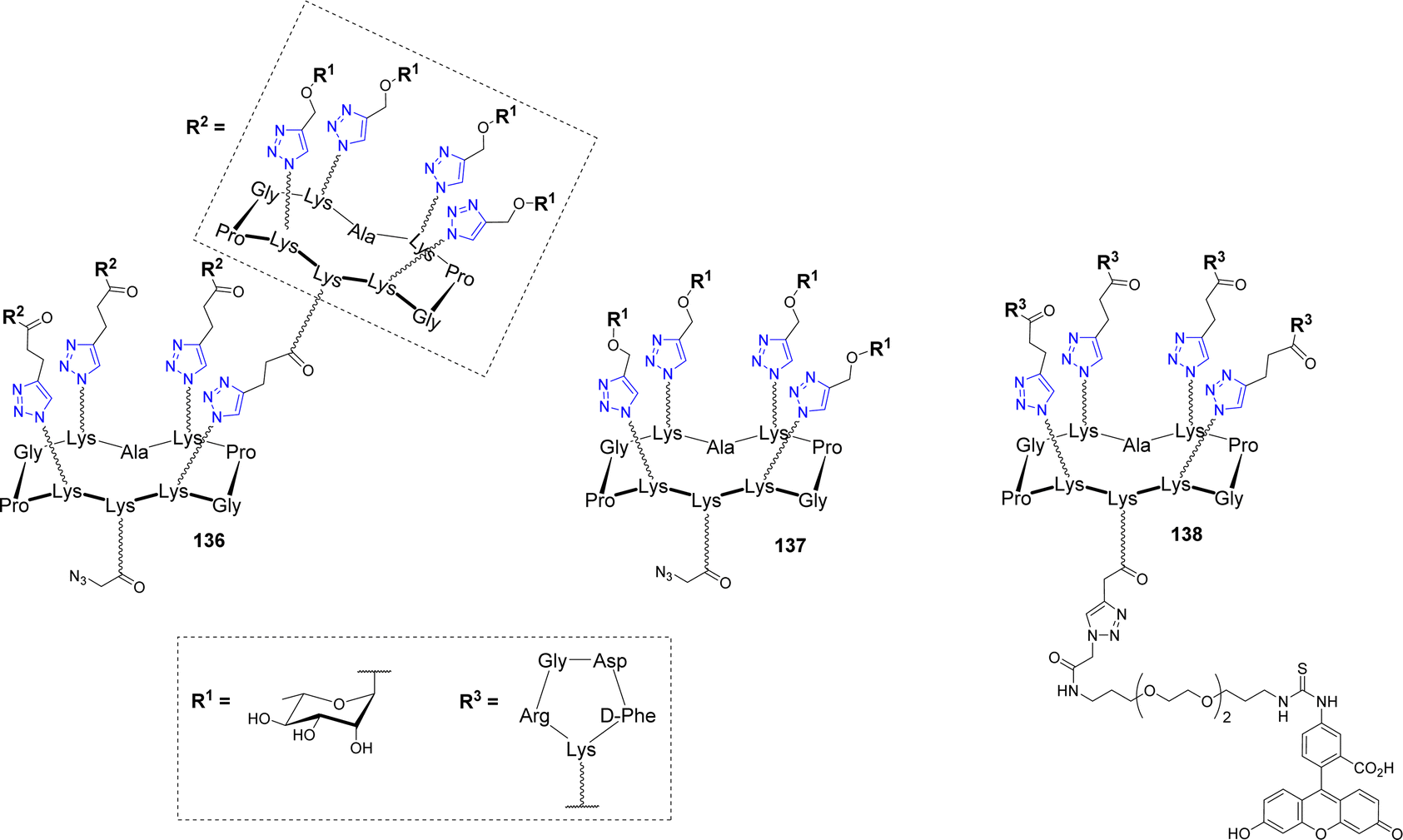 | ||
| Fig. 11 Structure of the Antibody Binding Modules 136–137 and the Tumor Binding Module 138. | ||
As expected, ELISA experiments confirmed the higher binding efficiency of the hexadecavalent ABM 136 with natural IgM present in human serum compared to the tetravalent control 137. In addition, flow cytometry experiments with labelled TBM 138 indicated the selective binding to αvβ3 integrin overexpressed by M21 tumor cell line while low interaction was observed with the monovalent cRGD. Once the crucial effect of multivalent presentation being demonstrated for both ABM and TBM, the authors conjugated these structures covalently in a single ARG molecule 139 (Fig. 12) by CuAAC.
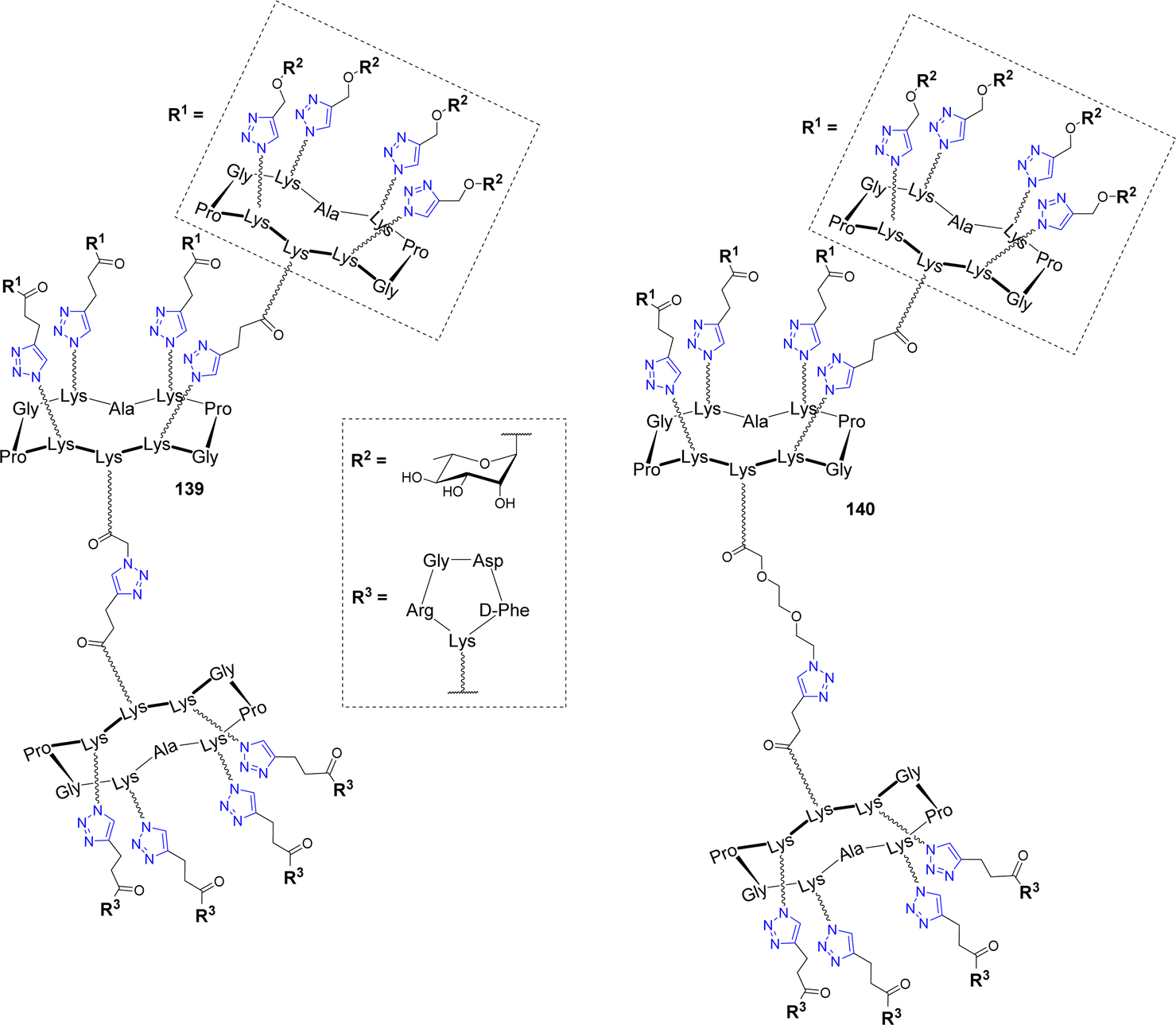 | ||
| Fig. 12 Structure of Antibody-Recruiting Glycodendrimers to stimulate immune-mediated cytotoxicity against cancer cells. | ||
Flow cytometry and confocal microscopy experiments have not only shown that the recognition properties of ABM and TBM are conserved, but they also demonstrated that the ARG 139 induces the formation of the ternary complex between M21 cells and IgM which is the prerequisite for immune stimulation. The same group further prepared a series of ARGs varying in the ABM geometry and the linker between ABM and TBM (140) and they studied their ability to stimulate immune mediated cytotoxicity of tumor cell lines.150 Flow cytometry and confocal microscopy experiments have first highlighted that the ABM architecture strongly influences on ternary complex formation. More importantly, a fluorescence-based cell viability assay allowed to identify by flow cytometry the ARG architecture 140 as the best candidate, since it has indeed the ability to induce 40–57% cytotoxicity. Moreover, it was found that spacer incorporation between ABM and TBM is an important parameter to increase cytotoxicity.
In another approach,152 the best ABM 136 (Fig. 11) was conjugated to the glycocalyx of BT-549 cancer cells by means of a metabolic labelling strategy using copper-free strain-promoted azide-alkyne cycloaddition.153,154 This cell surface modification was proved to induce antibody recruitment and cell cytotoxicity.
4. Conclusion & perspectives
The development of conjugation methodologies together with the ease of varying cyclopeptide composition with orthogonally protected and functionalizable aminoacids have significantly facilitated the chemical access of a diversity of multivalent structures having tailor-made structural and biological properties. Oxime ligation, copper(I)-catalyzed alkyne–azide cycloaddition, thiol–ene, thiol–chloroacetyl and squaramide couplings among other methodologies are now available to explore the structural requirements (i.e. geometry, size, valency, composition) for developing high affinity structures for biological targets such as bacterial lectins, cellular receptors or antibodies. These conjugation methods were widely exploited in stepwise or orthogonal one-pot approaches to access a wide variety of monodisperse homo- and heterovalent glycoclusters and glycodendrimers, ranging from 1 to 38 kDa, in excellent yields. The efficiency of these strategies have clearly pushed the structural limits that are now chemically accessible.While the potential of these structures to interact with lectins or to stimulate immunity against tumor cells was clearly demonstrated, the selectivity remains the major issue to overcome. The utilization of advanced computational modelling methods155 is undoubtedly a key tool to achieve this purpose by driving the design of glycomimetics or non-carbohydrate drug-like lectin inhibitors capable to target secondary binding pockets of these proteins.156,157 Besides this, the heterogenous expression of glycans at the surface of both normal and cancer cells is a crucial aspect to consider, making the design of multivalent systems much more complex than the simple multimerization of glycans on a scaffold. Due to the development of multi-click conjugation methods and glycodendrimer arrays, heterovalent structures combining diverse glycans in a variety of tridimensional geometry are now accessible to be screened as cell surface glycocalyx mimics. These structures indeed represent credible leads to understand how heteromultivalent binding is involved in complex biological recognition processes. Furthermore, by combining these different tools with the utilization of orthogonal click conjugations will definitely allow the development of innovative biomolecular systems with precise biological properties and high selectivity to reach clinical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of the reported studies was supported by CNRS, Université Grenoble Alpes, ICMG FR 2607, the French ANR projects VacSyn (ANR-12-JS07-0001-01), Glyco@Alps (ANR-15-IDEX-02), Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003) and the European Research Council Consolidator Grant “LEGO” (647938) and Proof of Concept Grant “THERA-LEGO” (963862).References
- O. Renaudet and R. Roy, Chem. Soc. Rev., 2013, 42, 4515–4517 RSC.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- Y. Kim, J. Y. Hyun and I. Shin, Chem. Soc. Rev., 2021, 50, 10567–10593 RSC.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- P. E. Dawson and S. B. H. Kent, J. Am. Chem. Soc., 1993, 115, 7263–7266 CrossRef CAS.
- M. Mutter, P. Dumy, P. Garrouste, C. Lehmann, M. Mathieu, C. Peggion, S. Peluso, A. Razaname and G. Tuchscherer, Angew. Chem., Int. Ed. Engl., 1996, 35, 1482–1485 CrossRef CAS.
- M. C. Galan, P. Dumy and O. Renaudet, Chem. Soc. Rev., 2013, 42, 4599–4612 RSC.
- Y. Singh, G. T. Dolphin, J. Razkin and P. Dumy, ChemBioChem, 2006, 7, 1298–1314 CrossRef CAS PubMed.
- D. Boturyn, E. Defrancq, G. T. Dolphin, J. Garcia, P. Labbe, O. Renaudet and P. Dumy, J. Pept. Sci., 2008, 14, 224–240 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- R. Sharma, K. Naresh, Y. M. Chabre, R. Rej, N. K. Saadeh and R. Roy, Polym. Chem., 2014, 5, 4321–4331 RSC.
- G. C. Daskhan, N. Berthet, B. Thomas, M. Fiore and O. Renaudet, Carbohydr. Res., 2015, 405, 13–22 CrossRef CAS PubMed.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. – Eur. J., 2014, 20, 34–41 CrossRef CAS.
- W. P. Jencks, J. Am. Chem. Soc., 1959, 81, 475–481 CrossRef CAS.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS.
- E. H. Nardin, J. M. Calvo-Calle, G. A. Oliveira, P. Clavijo, R. Nussenzweig, R. Simon, W. Zeng and K. Rose, Vaccine, 1998, 16, 590–600 CrossRef CAS.
- Y. Zhao, S. B. H. Kent and B. T. Chait, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1629–1633 CrossRef CAS.
- M. R. Carrasco, R. T. Brown, I. M. Serafimova and O. Silva, J. Org. Chem., 2003, 68, 195–197 CrossRef CAS PubMed.
- M. B. Thygesen, H. Munch, J. Sauer, E. Cló, M. R. Jørgensen, O. Hindsgaul and K. J. Jensen, J. Org. Chem., 2010, 75, 1752–1755 CrossRef CAS PubMed.
- F. Peri, P. Dumy and M. Mutter, Tetrahedron, 1998, 54, 12269–12278 CrossRef CAS.
- C. Jiménez-Castells, B. G. de la Torre, D. Andreu and R. Gutiérrez-Gallego, Glycoconjugate J., 2008, 25, 879–887 CrossRef.
- S. Cao, F. D. Tropper and R. Roy, Tetrahedron, 1995, 51, 6679–6686 CrossRef CAS.
- E. C. Rodriguez, L. A. Marcaurelle and C. R. Bertozzi, J. Org. Chem., 1998, 63, 7134–7135 CrossRef CAS PubMed.
- O. Renaudet and P. Dumy, Tetrahedron Lett., 2001, 42, 7575–7578 CrossRef CAS.
- V. Duléry, O. Renaudet, C. Philouze and P. Dumy, Carbohydr. Res., 2007, 342, 894–900 CrossRef PubMed.
- O. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS PubMed.
- J. Chen, W. Zeng, R. Offord and K. Rose, Bioconjugate Chem., 2003, 14, 614–618 CrossRef CAS PubMed.
- T. R. Branson, T. E. McAllister, J. Garcia-Hartjes, M. A. Fascione, J. F. Ross, S. L. Warriner, T. Wennekes, H. Zuilhof and W. B. Turnbull, Angew. Chem., Int. Ed., 2014, 53, 8323–8327 CrossRef CAS PubMed.
- J. M. Hooker, A. P. Esser-Kahn and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 15558–15559 CrossRef CAS PubMed.
- O. Renaudet and P. Dumy, Org. Lett., 2003, 5, 243–246 CrossRef CAS.
- O. Renaudet and P. Dumy, Tetrahedron Lett., 2004, 45, 65–68 CrossRef CAS.
- A. M. Pujol, M. Cuillel, O. Renaudet, C. Lebrun, P. Charbonnier, D. Cassio, C. Gateau, P. Dumy, E. Mintz and P. Delangle, J. Am. Chem. Soc., 2011, 133, 286–296 CrossRef CAS PubMed.
- O. Renaudet and P. Dumy, Bioorg. Med. Chem. Lett., 2005, 15, 3619–3622 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- A. Dondoni, Chem. – Asian J., 2007, 2, 700–708 CrossRef CAS PubMed.
- E. Lallana, R. Riguera and E. Fernandez-Megia, Angew. Chem., Int. Ed., 2011, 50, 8794–8804 CrossRef CAS PubMed.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- I. Bossu, N. Berthet, P. Dumy and O. Renaudet, J. Carbohydr. Chem., 2011, 30, 458–468 CrossRef CAS.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS.
- C. Pifferi, D. Goyard, E. Gillon, A. Imberty and O. Renaudet, ChemPlusChem, 2017, 82, 390–398 CrossRef CAS.
- Y.-X. Chen, L. Zhao, Z.-P. Huang, Y.-F. Zhao and Y.-M. Li, Bioorg. Med. Chem. Lett., 2009, 19, 3775–3778 CrossRef CAS PubMed.
- Z. Zhang, J. Liu, C. L. M. J. Verlinde, W. G. J. Hol and E. Fan, J. Org. Chem., 2004, 69, 7737–7740 CrossRef CAS PubMed.
- V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2004, 43, 900–903 CrossRef CAS PubMed.
- D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Möller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132, 8704–8719 CrossRef CAS PubMed.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS.
- W. S. Hancock and J. E. Battersby, Anal. Biochem., 1976, 71, 260–264 CrossRef CAS PubMed.
- T. Ohta, N. Miura, N. Fujitani, F. Nakajima, K. Niikura, R. Sadamoto, C.-T. Guo, T. Suzuki, Y. Suzuki, K. Monde and S.-I. Nishimura, Angew. Chem., Int. Ed., 2003, 42, 5186–5189 CrossRef CAS.
- W. C. Chan, B. W. Bycroft, D. J. Evans and P. D. White, J. Chem. Soc., Chem. Commun., 1995, 2209–2210 RSC.
- I. J. Krauss, J. G. Joyce, A. C. Finnefrock, H. C. Song, V. Y. Dudkin, X. Geng, J. D. Warren, M. Chastain, J. W. Shiver and S. J. Danishefsky, J. Am. Chem. Soc., 2007, 129, 11042–11044 CrossRef CAS PubMed.
- S. T. Cohen-Anisfeld and P. T. Lansbury, J. Am. Chem. Soc., 1993, 115, 10531–10537 CrossRef CAS.
- J. G. Joyce, I. J. Krauss, H. C. Song, D. W. Opalka, K. M. Grimm, D. D. Nahas, M. T. Esser, R. Hrin, M. Feng, V. Y. Dudkin, M. Chastain, J. W. Shiver and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15684–15689 CrossRef CAS PubMed.
- H. Herzner and H. Kunz, Carbohydr. Res., 2007, 342, 541–557 CrossRef CAS PubMed.
- M. Hachisu, H. Hinou, M. Takamichi, S. Tsuda, S. Koshida and S.-I. Nishimura, Chem. Commun., 2009, 1641–1643 RSC.
- R. Peltier, M. A. Brimble, J. M. Wojnar, D. E. Williams, C. W. Evans and A. L. DeVries, Chem. Sci., 2010, 1, 538–551 RSC.
- Y. Tachibana, N. Matsubara, F. Nakajima, T. Tsuda, S. Tsuda, K. Monde and S.-I. Nishimura, Tetrahedron, 2002, 58, 10213–10224 CrossRef CAS.
- G.-A. Cremer, N. Bureaud, D. Lelièvre, V. Piller, F. Piller and A. Delmas, Chem. – Eur. J., 2004, 10, 6353–6360 CrossRef CAS PubMed.
- E. Garanger, D. Boturyn, O. Renaudet, E. Defrancq and P. Dumy, J. Org. Chem., 2006, 71, 2402–2410 CrossRef CAS PubMed.
- K. Křenek, R. Gažák, G. C. Daskhan, J. Garcia, M. Fiore, P. Dumy, M. Šulc, V. Křen and O. Renaudet, Carbohydr. Res., 2014, 393, 9–14 CrossRef PubMed.
- O. Renaudet, D. Boturyn and P. Dumy, Bioorg. Med. Chem. Lett., 2009, 19, 3880–3883 CrossRef CAS.
- I. Bossu, M. Šulc, K. Křenek, E. Dufour, J. Garcia, N. Berthet, P. Dumy, V. Křen and O. Renaudet, Org. Biomol. Chem., 2011, 9, 1948–1959 RSC.
- B. Thomas, N. Berthet, J. Garcia, P. Dumy and O. Renaudet, Chem. Commun., 2013, 49, 10796–10798 RSC.
- J. P. Ribeiro, S. Villringer, D. Goyard, L. Coche-Guerente, M. Höferlin, O. Renaudet, W. Römer and A. Imberty, Chem. Sci., 2018, 9, 7634–7641 RSC.
- J. L. Jiménez Blanco, C. Ortiz Mellet and J. M. García Fernández, Chem. Soc. Rev., 2013, 42, 4518–4531 RSC.
- C. Müller, G. Despras and T. K. Lindhorst, Chem. Soc. Rev., 2016, 45, 3275–3302 RSC.
- H.-K. Choi, D. Lee, A. Singla, J. S.-I. Kwon and H.-J. Wu, Glycobiology, 2019, 29, 397–408 CrossRef CAS PubMed.
- M. González-Cuesta, C. Ortiz Mellet and J. M. García Fernández, Chem. Commun., 2020, 56, 5207–5222 RSC.
- J. Wang, H. Li, G. Zou and L.-X. Wang, Org. Biomol. Chem., 2007, 5, 1529–1540 RSC.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- M. Galibert, P. Dumy and D. Boturyn, Angew. Chem., Int. Ed., 2009, 48, 2576–2579 CrossRef CAS PubMed.
- M. Galibert, L. Sancey, O. Renaudet, J.-L. Coll, P. Dumy and D. Boturyn, Org. Biomol. Chem., 2010, 8, 5133–5138 RSC.
- B. Thomas, M. Fiore, I. Bossu, P. Dumy and O. Renaudet, Beilstein J. Org. Chem., 2012, 8, 421–427 CrossRef CAS PubMed.
- B. Thomas, C. Pifferi, G. C. Daskhan, M. Fiore, N. Berthet and O. Renaudet, Org. Biomol. Chem., 2015, 13, 11529–11538 RSC.
- M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593–4609 RSC.
- M. A. Mintzer and M. W. Grinstaff, Chem. Soc. Rev., 2011, 40, 173–190 RSC.
- R. Roy and T. C. Shiao, Chem. Soc. Rev., 2015, 44, 3924–3941 RSC.
- C. Pifferi, B. Thomas, D. Goyard, N. Berthet and O. Renaudet, Chem. – Eur. J., 2017, 23, 16283–16296 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573–586 RSC.
- M. Fiore, N. Berthet, A. Marra, E. Gillon, P. Dumy, A. Dondoni, A. Imberty and O. Renaudet, Org. Biomol. Chem., 2013, 11, 7113–7122 RSC.
- M. Fiore, G. C. Daskhan, B. Thomas and O. Renaudet, Beilstein J. Org. Chem., 2014, 10, 1557–1563 CrossRef PubMed.
- M. Galibert, O. Renaudet, P. Dumy and D. Boturyn, Angew. Chem., Int. Ed., 2011, 50, 1901–1904 CrossRef CAS PubMed.
- G. C. Daskhan, C. Pifferi and O. Renaudet, ChemistryOpen, 2016, 5, 477–484 CrossRef CAS PubMed.
- B. Thomas, M. Fiore, G. C. Daskhan, N. Spinelli and O. Renaudet, Chem. Commun., 2015, 51, 5436–5439 RSC.
- A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Essentials of Glycobiology, Cold Spring Harbor (NY), Cold Spring Harbor Laboratory Press, 4th edn, 2022 Search PubMed.
- M. Reynolds and S. Pérez, C. R. Chim, 2011, 14, 74–95 CrossRef CAS.
- G. Ashwell and J. Harford, Annu. Rev. Biochem., 1982, 51, 531–554 CrossRef CAS PubMed.
- O. Khorev, D. Stokmaier, O. Schwardt, B. Cutting and B. Ernst, Bioorg. Med. Chem., 2008, 16, 5216–5231 CrossRef CAS PubMed.
- J. Wu, M. H. Nantz and M. A. Zern, FBL, 2002, 7, 717–725 Search PubMed.
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139 CrossRef.
- P. Delangle and E. Mintz, Dalton Trans., 2012, 41, 6359–6370 RSC.
- M. Monestier, P. Charbonnier, C. Gateau, M. Cuillel, F. Robert, C. Lebrun, E. Mintz, O. Renaudet and P. Delangle, ChemBioChem, 2016, 17, 590–594 CrossRef CAS PubMed.
- A. Imberty and A. Varrot, Curr. Opin. Struct. Biol., 2008, 18, 567–576 CrossRef CAS PubMed.
- E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Pérez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Biol., 2002, 9, 918–921 CrossRef CAS.
- A. Imberty, M. Wimmerová, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- L. Gajdos, M. P. Blakeley, M. Haertlein, V. T. Forsyth, J. M. Devos and A. Imberty, Nat. Commun., 2022, 13, 194 CrossRef CAS.
- A. Audfray, J. Claudinon, S. Abounit, N. Ruvoën-Clouet, G. Larson, D. F. Smith, M. Wimmerová, J. Le Pendu, W. Römer, A. Varrot and A. Imberty, J. Biol. Chem., 2012, 287, 4335–4347 CrossRef CAS PubMed.
- J. Houser, J. Komarek, N. Kostlanova, G. Cioci, A. Varrot, S. C. Kerr, M. Lahmann, V. Balloy, J. V. Fahy, M. Chignard, A. Imberty and M. Wimmerova, PLoS One, 2013, 8, e83077 CrossRef PubMed.
- F. Bonnardel, A. Kumar, M. Wimmerova, M. Lahmann, S. Perez, A. Varrot, F. Lisacek and A. Imberty, Structure, 2019, 27, 764–775.e763 CrossRef CAS PubMed.
- B. Richichi, A. Imberty, E. Gillon, R. Bosco, I. Sutkeviciute, F. Fieschi and C. Nativi, Org. Biomol. Chem., 2013, 11, 4086–4094 RSC.
- D. Goyard, V. Baldoneschi, A. Varrot, M. Fiore, A. Imberty, B. Richichi, O. Renaudet and C. Nativi, Bioconjugate Chem., 2018, 29, 83–88 CrossRef CAS PubMed.
- N. Berthet, B. Thomas, I. Bossu, E. Dufour, E. Gillon, J. Garcia, N. Spinelli, A. Imberty, P. Dumy and O. Renaudet, Bioconjugate Chem., 2013, 24, 1598–1611 CrossRef CAS PubMed.
- D. Goyard, B. Thomas, E. Gillon, A. Imberty and O. Renaudet, Front. Chem., 2019, 7, 666 CrossRef CAS PubMed.
- M. Gómez-García, J. M. Benito, R. Gutiérrez-Gallego, A. Maestre, C. O. Mellet, J. M. G. Fernández and J. L. J. Blanco, Org. Biomol. Chem., 2010, 8, 1849–1860 RSC.
- M. I. García-Moreno, F. Ortega-Caballero, R. Rísquez-Cuadro, C. Ortiz Mellet and J. M. García Fernández, Chem. – Eur. J., 2017, 23, 6295–6304 CrossRef PubMed.
- T. K. Lindhorst, K. Bruegge, A. Fuchs and O. Sperling, Beilstein J. Org. Chem., 2010, 6, 801–809 CrossRef PubMed.
- S. P. Vincent, K. Buffet, I. Nierengarten, A. Imberty and J.-F. Nierengarten, Chem. – Eur. J., 2016, 22, 88–92 CrossRef CAS PubMed.
- M. Abellán Flos, M. I. García Moreno, C. Ortiz Mellet, J. M. García Fernández, J.-F. Nierengarten and S. P. Vincent, Chem. – Eur. J., 2016, 22, 11450–11460 CrossRef.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- M. González-Cuesta, D. Goyard, E. Nanba, K. Higaki, J. M. García Fernández, O. Renaudet and C. Ortiz Mellet, Chem. Commun., 2019, 55, 12845–12848 RSC.
- E. Mahenthiralingam, A. Baldwin and C. G. Dowson, J. Appl. Microbiol., 2008, 104, 1539–1551 CrossRef CAS.
- E. Lameignere, L. Malinovská, M. Sláviková, E. Duchaud, E. P. Mitchell, A. Varrot, O. Šedo, A. Imberty and M. Wimmerová, Biochem. J., 2008, 411, 307–318 CrossRef CAS PubMed.
- L. Abbassi, Y. M. Chabre, N. Kottari, A. A. Arnold, S. André, J. Josserand, H.-J. Gabius and R. Roy, Polym. Chem., 2015, 6, 7666–7683 RSC.
- H. Martin, D. Goyard, A. Margalit, K. Doherty, O. Renaudet, K. Kavanagh and T. Velasco-Torrijos, Bioconjugate Chem., 2021, 32, 971–982 CrossRef CAS PubMed.
- H. Martin, M. M. Govern, L. Abbey, A. Gilroy, S. Mullins, S. Howell, K. Kavanagh and T. Velasco-Torrijos, Eur. J. Med. Chem., 2018, 160, 82–93 CrossRef CAS PubMed.
- N. Parera Pera, H. M. Branderhorst, R. Kooij, C. Maierhofer, M. van der Kaaden, R. M. J. Liskamp, V. Wittmann, R. Ruijtenbeek and R. J. Pieters, ChemBioChem, 2010, 11, 1896–1904 CrossRef CAS PubMed.
- A. Goudot, G. Pourceau, A. Meyer, T. Gehin, S. Vidal, J.-J. Vasseur, F. Morvan, E. Souteyrand and Y. Chevolot, Biosens. Bioelectron., 2013, 40, 153–160 CrossRef CAS PubMed.
- A. Angeli, M. Li, L. Dupin, G. Vergoten, M. Noël, M. Madaoui, S. Wang, A. Meyer, T. Géhin, S. Vidal, J.-J. Vasseur, Y. Chevolot and F. Morvan, ChemBioChem, 2017, 18, 1036–1047 CrossRef CAS.
- H. S. Kim, J. Y. Hyun, S.-H. Park and I. Shin, RSC Adv., 2018, 8, 14898–14905 RSC.
- M. Mende, A. Tsouka, J. Heidepriem, G. Paris, D. S. Mattes, S. Eickelmann, V. Bordoni, R. Wawrzinek, F. F. Fuchsberger, P. H. Seeberger, C. Rademacher, M. Delbianco, A. Mallagaray and F. F. Loeffler, Chem. – Eur. J., 2020, 26, 9954–9963 CrossRef CAS.
- A. Hoang, E. Laigre, D. Goyard, E. Defrancq, F. Vinet, P. Dumy and O. Renaudet, Org. Biomol. Chem., 2017, 15, 5135–5139 RSC.
- O. Renaudet and P. Dumy, Org. Biomol. Chem., 2006, 4, 2628–2636 RSC.
- E. Laigre, C. Tiertant, D. Goyard and O. Renaudet, ACS Omega, 2018, 3, 14013–14020 CrossRef CAS PubMed.
- E. Laigre, D. Goyard, C. Tiertant, J. Dejeu and O. Renaudet, Org. Biomol. Chem., 2018, 16, 8899–8903 RSC.
- L. Picault, E. Laigre, E. Gillon, C. Tiertant, O. Renaudet, A. Imberty, D. Goyard and J. Dejeu, Glycobiology, 2022, 32(10), 886–896 Search PubMed.
- S. Hakomori, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10231–10233 CrossRef CAS PubMed.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef CAS PubMed.
- D. Feng, A. S. Shaikh and F. Wang, ACS Chem. Biol., 2016, 11, 850–863 CrossRef CAS PubMed.
- F. Peri, Chem. Soc. Rev., 2013, 42, 4543–4556 RSC.
- C. Pifferi, N. Berthet and O. Renaudet, Biomater. Sci., 2017, 5, 953–965 RSC.
- S. Grigalevicius, S. Chierici, O. Renaudet, R. Lo-Man, E. Dériaud, C. Leclerc and P. Dumy, Bioconjugate Chem., 2005, 16, 1149–1159 CrossRef CAS PubMed.
- R. Lo-Man, S. Bay, S. Vichier-Guerre, E. Dériaud, D. L. Cantacuzène and C. Leclerc, Cancer Res., 1999, 59, 1520–1524 CAS.
- O. Renaudet, L. BenMohamed, G. Dasgupta, I. Bettahi and P. Dumy, ChemMedChem, 2008, 3, 737–741 CrossRef CAS PubMed.
- I. Bettahi, G. Dasgupta, O. Renaudet, A. A. Chentoufi, X. Zhang, D. Carpenter, S. Yoon, P. Dumy and L. BenMohamed, Cancer Immunol. Immunother., 2008, 58, 187 CrossRef.
- O. Renaudet, G. Dasgupta, I. Bettahi, A. Shi, A. B. Nesburn, P. Dumy and L. BenMohamed, PLoS One, 2010, 5, e11216 CrossRef PubMed.
- C. Nativi, F. Papi and S. Roelens, Chem. Commun., 2019, 55, 7729–7736 RSC.
- J. Jiménez-Barbero, E. Dragoni, C. Venturi, F. Nannucci, A. Ardá, M. Fontanella, S. André, F. J. Cañada, H.-J. Gabius and C. Nativi, Chem. – Eur. J., 2009, 15, 10423–10431 CrossRef PubMed.
- M. Manuelli, S. Fallarini, G. Lombardi, C. Sangregorio, C. Nativi and B. Richichi, Nanoscale, 2014, 6, 7643–7655 RSC.
- B. Richichi, B. Thomas, M. Fiore, R. Bosco, H. Qureshi, C. Nativi, O. Renaudet and L. BenMohamed, Angew. Chem., Int. Ed., 2014, 53, 11917–11920 CrossRef CAS PubMed.
- C. Pifferi, A. Ruiz-de-Angulo, D. Goyard, C. Tiertant, N. Sacristán, D. Barriales, N. Berthet, J. Anguita, O. Renaudet and A. Fernández-Tejada, Chem. Sci., 2020, 11, 4488–4498 RSC.
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 1139–1151 CrossRef CAS PubMed.
- A. Uvyn and B. G. De Geest, ChemBioChem, 2020, 21, 3036–3043 CrossRef CAS PubMed.
- S. Achilli, N. Berthet and O. Renaudet, RSC Chem. Biol., 2021, 2, 713–724 RSC.
- B. Liet, E. Laigre, D. Goyard, B. Todaro, C. Tiertant, D. Boturyn, N. Berthet and O. Renaudet, Chem. – Eur. J., 2019, 25, 15508–15515 CrossRef CAS PubMed.
- B. Todaro, S. Achilli, B. Liet, E. Laigre, C. Tiertant, D. Goyard, N. Berthet and O. Renaudet, Biomater. Sci., 2021, 9, 4076–4085 RSC.
- D. Boturyn, J.-L. Coll, E. Garanger, M.-C. Favrot and P. Dumy, J. Am. Chem. Soc., 2004, 126, 5730–5739 CrossRef CAS.
- D. Goyard, P. I. Diriwari and N. Berthet, RSC Med. Chem., 2022, 13, 72–78 RSC.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- H. Wang and D. J. Mooney, Nat. Chem., 2020, 12, 1102–1114 CrossRef CAS PubMed.
- S. Perez and O. Makshakova, Chem. Rev., 2022 DOI:10.1021/acs.chemrev.2c00060.
- E. Shanina, S. Kuhaudomlarp, E. Siebs, F. F. Fuchsberger, M. Denis, P. da Silva Figueiredo Celestino Gomes, M. H. Clausen, P. H. Seeberger, D. Rognan, A. Titz, A. Imberty and C. Rademacher, Commun. Chem., 2022, 5, 64 CrossRef CAS.
- R. Wawrzinek, E.-C. Wamhoff, J. Lefebre, M. Rentzsch, G. Bachem, G. Domeniconi, J. Schulze, F. F. Fuchsberger, H. Zhang, C. Modenutti, L. Schnirch, M. A. Marti, O. Schwardt, M. Bräutigam, M. Guberman, D. Hauck, P. H. Seeberger, O. Seitz, A. Titz, B. Ernst and C. Rademacher, J. Am. Chem. Soc., 2021, 143, 18977–18988 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |