
Poly(ε-lysine) and its derivatives via ring-opening polymerization of biorenewable cyclic lysine
Maosheng
Li
and
Youhua
Tao
 *
*
Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Street 5625, Changchun 130022, People's Republic of China. E-mail: youhua.tao@ciac.ac.cn
First published on 20th November 2020
Abstract
Poly(ε-lysine) (ε-PL) is an unusual cationic, naturally-occurring biopolymer furnished by microorganism fermentation processes. ε-PL has attracted extensive attention in areas from food and cosmetic additives to the pesticide and pharmaceutical industries, owing to its excellent biocompatibility and antimicrobial activity. However, the lack of molecular weight and structural diversity of ε-PL from the biosynthetic route appears to be an important limitation, leading to the composition and properties of the prepared polymers being difficult to regulate for further applications. Recently, we demonstrated an efficient chemosynthetic approach that relied on ring-opening polymerization of lysine derived ε-lactams to provide ε-PL with diverse molecular weights. In the following mini-review, the recent progress on the chemosynthesis of poly(ε-lysine) and its derivatives via ring-opening polymerization of biorenewable cyclic lysine has been summarized. Moreover, the future development of the chemosynthesis of poly(ε-lysine) and its derivatives will be discussed.
 Maosheng Li | Dr Maosheng Li received his Ph.D. in Polymer Chemistry and Physics in 2020 from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, under the direction of Professor Youhua Tao. He is currently a research assistant at Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. His research interests focus on sustainable polymer synthesis, including the efficient use of amino acid resources, and the design and synthesis of organocatalysts for polymerizations. |
 Youhua Tao | Prof. Youhua Tao received his Ph.D. in Polymer Chemistry in 2008 from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, under the direction of Professor Xianhong Wang. After postdoctoral stays (2008–2013) with Professor Masami Kamigaito (Nagoya University, Japan) and Professor Christopher N. Bowman (University of Colorado at Boulder, U.S.A.), he accepted a full professor appointment at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. The main focus of his current research is new polymerization reactions of amino acid monomers. |
1 Introduction
Poly(ε-lysine) (ε-PL) is a naturally-occurring functionalized poly(amino acid), comprising an amide linkage between the α-carboxyl groups and ε-amino groups of lysine and remaining α-amino groups in the side chain. Natural ε-PL is secreted by microorganisms and is usually composed of 25 to 35 residues of L-lysine.1–3 Due to its unique structure, ε-PL exhibits plenty of properties, such as biocompatibility, biodegradability, water-solubility, thermostability, edibility, and antibacterial activity.4–8 What's more, it also displays remarkable endotoxin-selective removal, antiobesity activity and cell adhesion, inhibits pancreatic lipase activity, and prevents oral bacterial toxin production.9,10 Therefore, ε-PL has been extensively applied in food and cosmetic additives, antibacterial fibers, pesticides, and pharmaceuticals.8,11–15 At the current industrial scale, ε-PL is mainly produced by microorganisms via fermentation, which yields the target polymer with a low molecular weight (Mn) of around 3 kDa.6,8,15 Although this approach is promising, the lack of molecular weight and structural diversity of ε-PL from the biosynthetic route appears to be an important limitation, resulting in the composition and properties of the prepared polymers being difficult to regulate.Chemosynthetic procedures are considered as a powerful strategy to furnish polymers with controllable and diverse chemical structures.16 In particular, lysine, which serves as the constitutional unit of ε-PL, can be efficiently and cheaply obtained from crop waste (e.g. straw and waste molasses) via aerobic fermentation.16–18 It is no doubt that devising a viable chemosynthetic route that can transform low-cost lysine into high-value ε-PL with diverse Mn and composition is of great significance. In this premise, plenty of attempts have been performed in the past few decades, but the results have not always been satisfactory.19,20 Fully chemosynthetic approaches are more challenging than biotechnological protocols, and are limited to oligomer synthesis. Among these approaches, iterative synthesis methods, which are generally carried out on a solid-phase support, are the most obvious approaches to prepare oligomers of ε-PL. However, this route is not easily applicable to the synthesis of large amounts of ε-PL as required for applications in polymer science and material science due to the limited loading of solid-phase and complicated multistep protocols.
Considering the structural similarity of ε-PL and polycaprolactam, as well as the successful experience of polycaprolactam in industrial production,21–24 we foresaw that the large-scale synthesis of ε-PL through ring-opening polymerization (ROP) may be an ideal route. Since 2015, we have demonstrated an effective chemical strategy for the preparation of ε-PL, relying on ring-opening polymerization of a protected cyclic lysine monomer (ε-lactam) derived from lysine (Fig. 1).17,18,25–29 This approach, focused on cyclization/ring-opening polymerization of lysine, not only opens the door for feasible large-scale production of ε-PL, but also provides novel access to the production of other functionalized poly(amino acids), such as poly(γ-glutamate) from glutamic acid.30
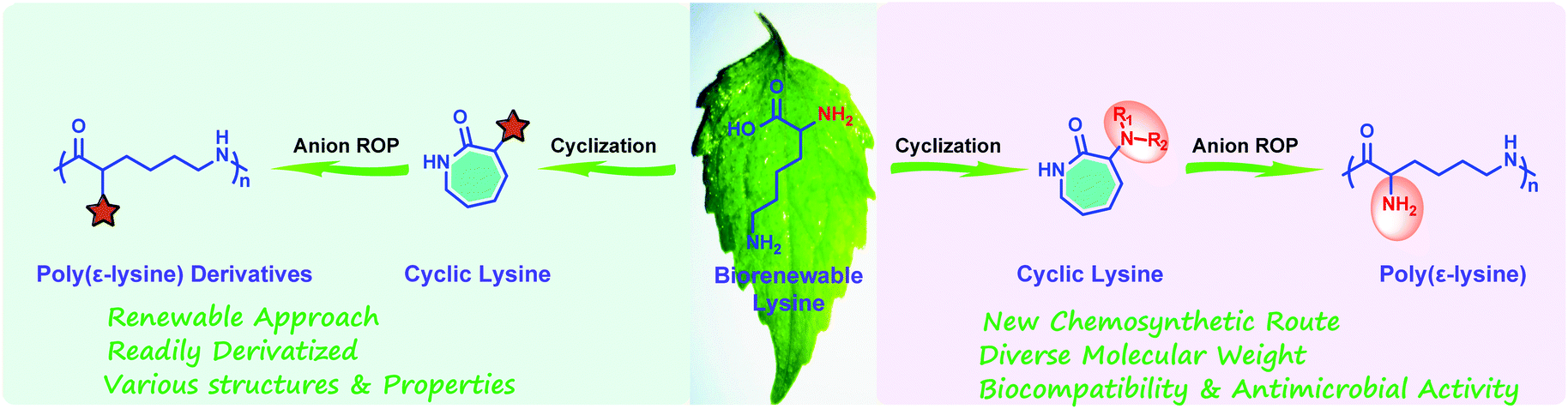 | ||
| Fig. 1 Chemosynthesis of poly(ε-lysine) and its derivatives via ring-opening polymerization of biorenewable cyclic lysine. | ||
In this mini-review, we thus intend to briefly introduce the history and properties of ε-PL, and then summarize our recent progress in the chemosynthesis of poly(ε-lysine) and its derivatives via ring-opening polymerization of biorenewable cyclic lysine (Fig. 1), finally emphasizing the remaining challenges and promising perspectives in the relevant fields.
2 History and properties of poly(ε-lysine)
Historically, microbial poly(ε-lysine) was accidentally discovered in 1977,1 when Shima and Sakai were screening Dragendorff-positive substances in the culture filtrate of actinomycetes isolated from soil. After identifying Streptomyces albulus as the original strain2 and lysine as the constitutional unit of ε-PL,3 the authors also demonstrated that ε-PL showed both antimicrobial and antiphage activity,31,32 which prompted them to optimize the culture conditions of Streptomyces albulus for ε-PL production. Since then, with the evolution of genetic engineering, bioinformatics, and advanced instruments, extensive investigations have been carried out to exploit microbial ε-PL, ranging from the screening of new ε-PL-producing strains to the development of efficient fermentation processes (Fig. 2). Readers who are interested in the advanced progress of microbial ε-PL could turn to related summaries.8,9,12,13,15 | ||
| Fig. 2 Diagram for the biosynthesis of poly(ε-lysine) via fermentation. | ||
The ε-PL from the biosynthetic route, containing between 25 and 35 lysine residues, is a light yellow powder with strong hygroscopicity and slightly bitter taste. FTIR analysis of ε-PL shows two strong absorption peaks at 1680–1640 cm−1 and 1580–1520 cm−1.3,33,34 Unlike its poly(α-lysine) (α-PL) analogue, no well-defined advanced structural information has been reported for ε-PL in solution or the solid state. The ε-PL exhibits excellent water solubility but is slightly soluble in ethanol. The isoelectric point of ε-PL with 25–35 residues is about pH 9.0, and the pKa is 7.6. According to reports, even after boiling at 100 °C for 30 minutes or autoclaving at 120 °C for 20 minutes, the ε-PL solution remains stable with intact polymer chains, even at pH 3.0. Therefore, ε-PL can inhibit heat-resistant bacteria and can be heat-treated after being added. Since ε-PL is a mixture, it does not have a fixed melting point and starts to soften and decompose above 250 °C.8
ε-PL is a cationic polymer material in water owing to its positively charged α-amino groups.8 The antibacterial mechanism of ε-PL is mainly manifested in the destruction of the microbial cell membrane structure through electrostatic adsorption, resulting in the interruption of material, energy and information transfer in cells, eventually leading to cell death.32,35,36 This is based on the cationic nature of ε-PL, which gives rise to outer membrane peeling and abnormal cytoplasmic distribution observed under electron microscopy. This could also be the reason for the distinction in minimum inhibitory concentrations (MICs) for bacteria, fungi and yeasts, which have different cell wall compositions and cell surface conditions. The ε-PL with a molecular weight between 3600–4300 Da has the best antibacterial activity, while when the molecular weight is lower than 1300 Da (about nine lysine residues), ε-PL will lose its antibacterial activity.32 Owing to the above physicochemical properties, ε-PL is widely used in applications ranging from food and cosmetic preservatives to agricultural and medical germifuga.
3 Chemosynthesis of poly(ε-lysine)
3.1 Design and synthesis of monomers
Inspired by the ring-opening polymerization of caprolactam,22,23 we envisioned synthesizing the seven-membered cyclic lysine monomer from lysine and polymerizing it to attain linear ε-PL.25 Initially, commercially available L-lysine monohydrochloride was used as a raw material and the α-amino-ε-caprolactam (M1) was prepared via esterification of L-lysine monohydrochloride with methanol in the presence of thionyl chloride, followed by intramolecular cyclization catalyzed by sodium hydroxide in dilute solution (Scheme 1A). During later research, the esterification reaction conditions were further optimized using cheap and non-toxic concentrated sulfuric acid instead of thionyl chloride.28 Obviously, a protective group of the α-amino group is necessary, as linear ε-PL cannot be prepared via direct polymerization of α-amino-ε-caprolactam. Thus, classical protecting groups, such as Boc, Cbz and phthaloyl (Pht), were adopted to acylate α-amino-ε-caprolactam, producing M2, M3 and M4 (Scheme 1B). However, preliminary polymerization of lactams M2 and M3 indicated that undesired proton abstractions from the α-amide would happen, resulting in double anionic propagation species. Due to the markedly more electron-deficient character for lactam M4, the Pht protective group was also unstable and was easily attacked by the anionic species, generating branched polymer chains during polymerization.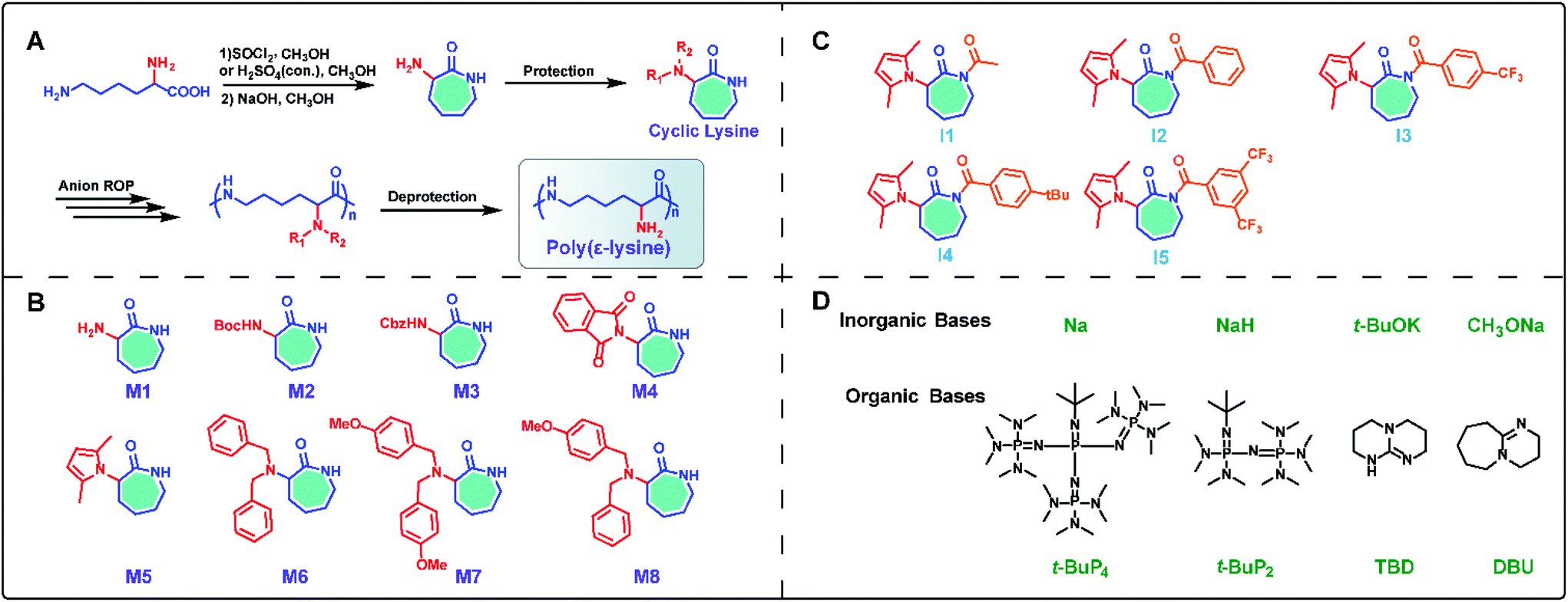 | ||
| Scheme 1 (A) The concept of the chemical approach for the preparation of poly(ε-lysine). Scope of the designed monomers (B), co-initiators (C), and catalysts including inorganic bases and organic bases (D) for possible polymerization in this review. | ||
Consequently, our synthesis access for linear ε-PL has to focus on the choice of protective groups, because we believe that the α-amino group interferes with the key polymerization step. An adequate α-amino-protecting group must possess these characteristics: (a) dual protection of amino functions; (b) less steric hindrance; (c) thermo-stability and chemical inertness during polymerization; (d) good solubility of the protected monomers and polymers in organic solvents; and (e) ability to be removed under mild conditions.25
Based on the above rules, we developed an uncommon amino protecting group, 2,5-dimethylpyrrole, which has been successfully used to synthesize the desired linear ε-PL (vide infra). The 2,5-dimethylpyrrole protected α-amino-ε-caprolactam (M5) was synthesized by p-toluenesulfonic acid (TsOH) catalyzed condensation reaction between the amine group in lactam M1 and 2,5-hexadiione. Further investigations (vide infra) also verified the availability of dual protection by benzyl (Bn) and/or methoxybenzyl (PMB) (M6–M8 in Scheme 1B). These monomers were achieved by coupling of lactam M1 and two equivalent relevant benzyl chlorides under basic conditions.
3.2 Mechanism considerations for ring-opening polymerization
It is well-known that the mechanism of anion ring-opening polymerization of lactam relies on nucleophilic attack of the activated monomer to the polymer chain end, which is normally named as the activated monomer mechanism.22,23 ROP of cyclic lysine monomers also follows this mechanism, and the driving force of ring-opening polymerization mainly comes from the strain release on the lactam ring.The anionic ROP of lysine derived lactams is started by a two-component catalyst system, containing lactamate anions, and N-acyllactam activators (co-initiator). As shown in Scheme 2A, firstly, the lactamate anion is generated by the proton transfer of a lactam monomer under highly basic conditions. The initiation step refers to the nucleophilic attack of the lactamate anion on the endocyclic carbonyl group in the activator, N-acyllactam, followed by proton transfer from another lactam monomer molecule to the resulting amidate anion, which gives the lactamate anion again. The propagation step also undergoes a similar reaction process with nucleophilic attack toward the acyllactam-type propagation chain end and the following proton transfer. Although not using an additional activator will result in slow initiation (Scheme 2B), the polymerization of lactam will also provide activated imide structures in situ through the nucleophilic attack by the lactamate anion on the monomer.
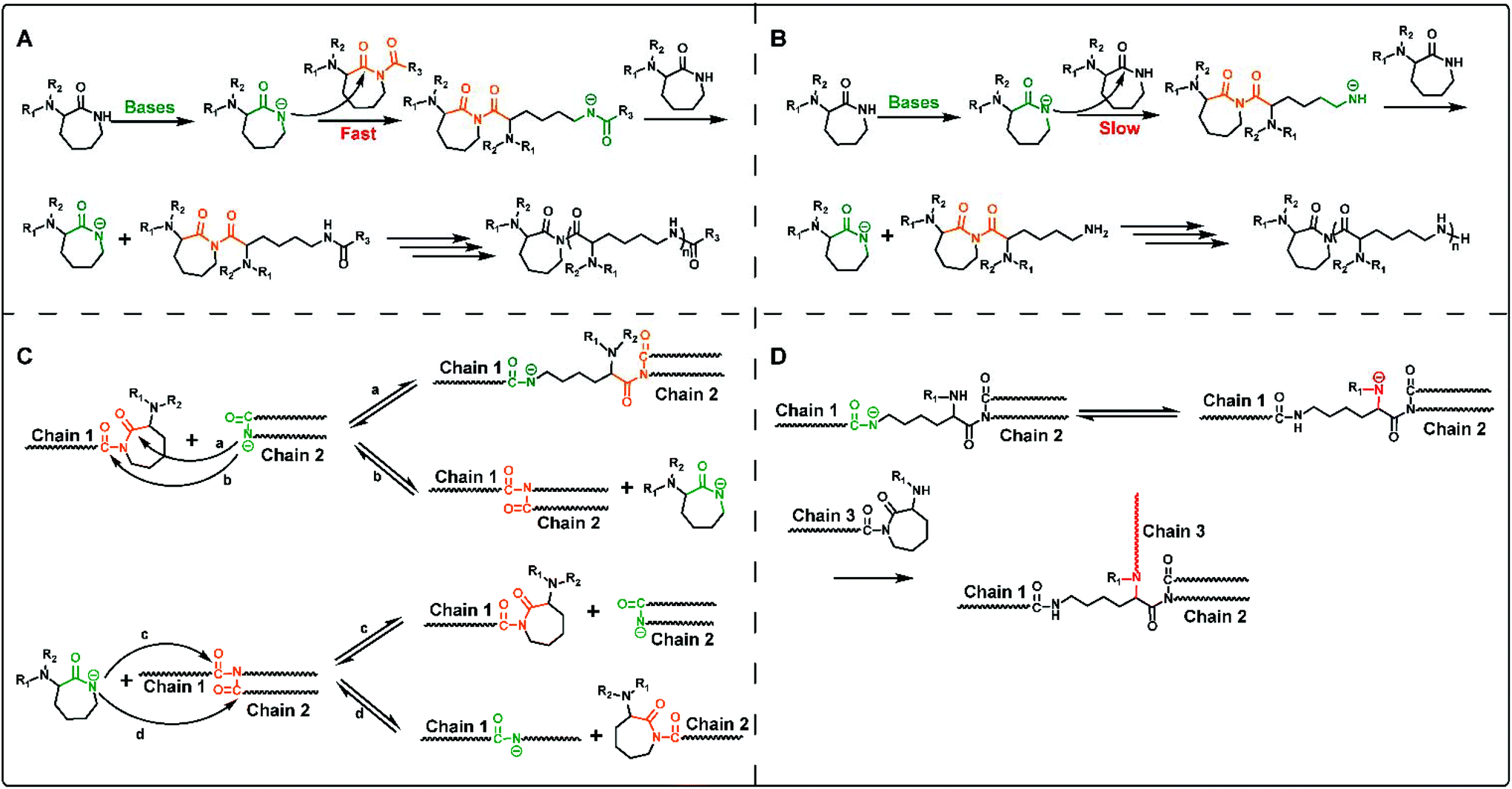 | ||
| Scheme 2 The activated monomer mechanism of ring-opening polymerization with additional co-initiators (A) and without additional co-initiators (B). (C) The transamidation reactions during polymerization. (D) Proposed branching process using mono amino-protective monomers (M1 and M2). | ||
In addition, the transamidation reactions involving proton transfer from lactamate anions to the polyamide chains as shown in Scheme 2C also proceed especially in the later stage of polymerization, resulting in the broadening of the molecular weight distribution of the final polymers. When polymerizing mono amino-protective monomers (e.g. M2 and M3), the transamidation reactions will form amide N anions along the polymer chain, which may produce imide groups and generate polymer branching during ROP (Scheme 2D). This is the reason why dual protection of α-amino-ε-caprolactam is a prerequisite for the synthesis of the desired linear ε-PL.
3.3 Polymerizations mediated by Na, NaH and alkoxide
The polymerization of monomer M5 was firstly performed in bulk using sodium as a catalyst, giving rise to a hard, tough, and light-colored mass polymer of M5.25 Consistent with Na/M5 feed ratios ranging from 0.5 to 10%, the resultant polymers at 260 °C were attained in 62–85% yield, and the molecular weight of the polymers can be regulated from 2.5 to 49.2 kDa as the Na/M5 feed ratio decreased, demonstrating the potential to diversify the Mn of ε-PL via a chemosynthesis approach. Attempts at conducting the polymerizations at 240 °C produced polymers with lower yields, which indicated that the lower temperature would increase the viscosities and reduce the mobility of the growing chain in bulk, thereby only achieving lower conversions of monomers. When prolonging the polymerization time, the Mn of the afforded polymer obviously decreased and the molecular weight distribution broadened, suggesting that the transamidation reactions would occur readily on continuous polymerization at elevated temperatures. As shown in Fig. 3, the structures of the resulting polymers are clearly characterized by the 1H NMR and 13C NMR spectra. The NMR spectra verified the stability of 2,5-dimethylpyrrole groups towards the initiating and propagating species under these experimental conditions. The advisable design of the 2,5-dimethylpyrrole group was further evidenced by the excellent solubility of the obtained polymers in common solvents, such as THF, CHCl3, CH2Cl2, and DMF.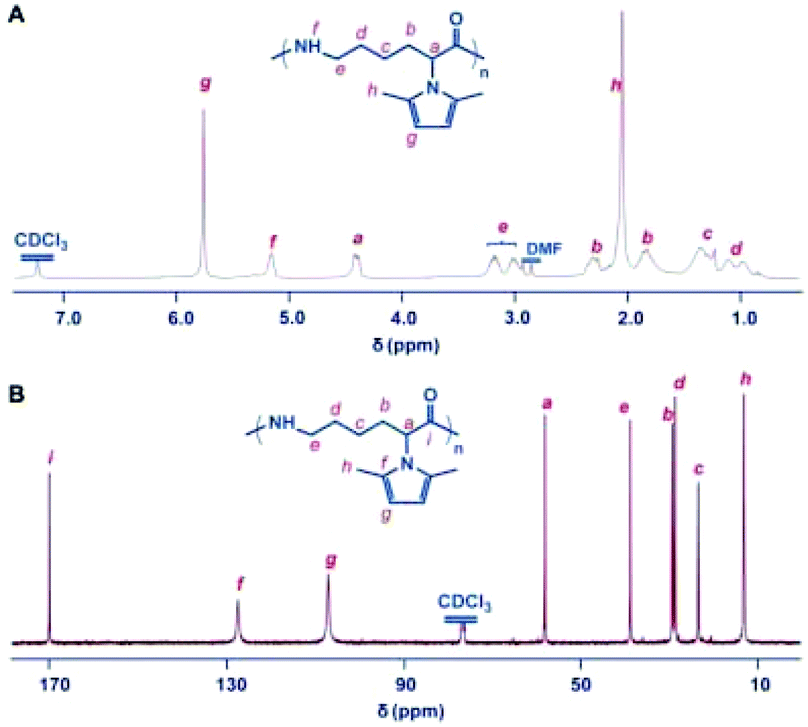 | ||
| Fig. 3 Structure characterization of the polymer of the 2,5-dimethylpyrrole protected α-amino-ε-caprolactam monomer. (A) 1H NMR spectrum of the polymer. (B) 13C NMR spectrum of the polymer. The polymer was obtained with sodium (0.5%) in bulk at 260 °C. All spectra were measured in CDCl3 at room temperature. Reproduced from ref. 25 with permission from the Royal Society of Chemistry, copyright 2015. | ||
The ring-opening polymerization of ε-lactam monomer M5 under bulk conditions was also investigated by using other inorganic bases, such as NaH, CH3ONa, and t-BuOK.28 When the polymerizations were carried out at 180 °C for 3 h with monomer/base/co-initiator (M/B/I) = 20/1/1, the yields of the polymerizations varied from 95% to 55%, and the rate of polymerization increased in the following order: t-BuOK < CH3ONa < NaH, suggesting high catalyst dependence of the polymerization reactivity. Moreover, upon increasing the M5/NaH/co-initiator ratio, higher molecular weight polymers (Mn reached 44.1 kDa) were synthesized. Interestingly, the polymerization mediated by these inorganic bases could proceed at observably lower temperature compared to using sodium. This is probably because of the better dispersibility of these inorganic bases compared with sodium in bulk as well as the employment of co-initiator, and consequently leading to rapid and sufficient initiation of the polymerization.
3.4 Polymerizations mediated by organocatalysts
In spite of the significant progress in inorganic base-mediated ROP of lactam in bulk, the synthesis of ε-PL still faces some formidable challenges, which include the high polymerization temperature (≥180 °C) and significantly compromise the protecting group scope owing to the low polymerizability and high melting point of ε-lactam derivatives. In consideration of energy-saving production processes, the polymerization conducted at temperatures ranging from 100 to 140 °C is industrially favorable. Inspired by these challenges, we aimed to improve upon the harsh reaction conditions and devise a new polymerization approach towards readily removable protecting groups of lysine-derived ε-lactams. To establish a new method, the following two challenges need to be addressed: (1) the polymerization must proceed in solution as bulk polymerization requires high temperatures, due to the high melting point of ε-lactam monomers, and (2) the catalysts must feature potent deprotonating capability and good solubility in organic solvents.26 Guided by the above working hypotheses as well as the remarkable progress in organocatalysis ROP of lactams, we anticipated that organic superbases could be used as excellent deprotonating agents,37–52 thereby realizing effective ROP of lysine derived ε-lactams affording linear ε-PL under mild conditions.Initially, organic superbase t-BuP4 [1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino) phosphoranylidenamino]-2λ5,4λ5-catenadi-(phosphazene)]45,46 was tested in the polymerization of monomer M5 with monomer/base/co-initiator (M/B/I) = 50/1/1 at 140 °C employing different aprotic organic solvents, such as DMSO, diethylene glycol dimethyl ether (DME), anisole (ANI), isopropylbenzene (IPB), and tert-butylbenzene (TBB).26 The polymerization conducted in ANI exhibited the optimal results, affording the targeted polymer with good monomer conversion (60%) and higher molecular weight (Mn = 12.1 kDa). Corresponding to the sodium-catalyzed ROP system, the Mn of the yielded polymers can be regulated by varying the ratio of t-BuP4/M5 or co-initiator/M, and the highest monomer conversion could arrive at 98%. The ROP behavior influenced by co-initiators with different steric bulk and electronic effect was also probed (Scheme 1C), revealing that co-initiators with less steric bulk and that are electron-deficient could be beneficial to the efficient ROP process. Surprisingly, the t-BuP4 mediated ROP could proceed even at 60 °C, despite lower conversion and molecular weight. Other commercial organic superbases were also examined (Scheme 1D),28 indicating that the basicity of the organic bases plays a key role in polymerization reactivity, which increased in the following order: DBU < TBD < t-BuP2 < t-BuP4. Further NMR studies (Fig. 4) verified the significant deprotonating capability of t-BuP4 from monomer M5, as evidenced by the distinct shift of the proton and phosphorus signals after mixing 1 equiv. each of t-BuP4 and M5, thereby supporting the anionic ROP mechanism (Scheme 2).
 | ||
Fig. 4 (A) 1H NMR spectra (toluene-d8, 25 °C) of the reaction between t-BuP4 and M5 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). (B) 31P NMR spectra (toluene-d8, 25 °C) of the reaction between t-BuP4 and M5 (1:1). Reproduced from ref. 26 with permission from the American Chemical Society, copyright 2017. :1). (B) 31P NMR spectra (toluene-d8, 25 °C) of the reaction between t-BuP4 and M5 (1:1). Reproduced from ref. 26 with permission from the American Chemical Society, copyright 2017. | ||
Encouraged by the above results, we further evaluated the ROP of other ε-lactam monomers with various protecting groups (M6–M8, in Scheme 1B) using t-BuP4 as a catalyst under mild reaction conditions. The ROP of these monomers also exhibited good to excellent monomer conversions and yielded targeted polymers with the expected structures, proved by the 1H NMR and 13C NMR characterizations.
3.5 Deprotection
The deprotection of the 2,5-dimethylpyrrole amino protecting groups of the ROP products was conducted by reacting with hydroxylamine/triethylamine in refluxed THF under a nitrogen atmosphere to provide the corresponding ε-PL with a moderate yield (20–30%).25 The structures of the chemosynthetic ε-PL were confirmed by the 1H NMR (Fig. 5A) and 13C NMR (Fig. 5C) characterizations, showing similar peak profiles to that of the biosynthetic ε-PL (Fig. 5B and D). It should be noted that when the polymerization was performed under high temperature and strong basicity conditions, racemization of α-H would occur, as evidenced by the broader proton peaks and multiple carbon peaks in the NMR spectra. Furthermore, the MALDI-TOF-MS spectrum of the chemosynthetic ε-PL consists of an array of peaks separated by a 128.2 Da interval corresponding to the lysine unit, and also indicated that the polymer contains an NH2 unit at the initiating terminal and a –COOH unit at the capping terminal (Fig. 6). These results illustrated that the ε-PL obtained through ROP possessed highly controlled terminal groups as well as a nearly perfect main-chain structure. In addition, the PMB amino protecting groups of the polymers could be more readily undergo hydrogenolysis in trifluoroacetic acid (TFA) furnishing ε-PL in 45% yield.26 Notably, ε-PL featuring diverse molecular weight ranging from 1.9 to 9.5 kDa can be readily prepared by the above process, demonstrating the superiority of the chemosynthetic strategy. The pKa of the resulting ε-PL was about 7.5 measured by potentiometric titration, which is close to that of biosynthetic ε-PL (pKa = 7.6). Compared with the [α]25D = +23° for biosynthetic ε-PL, the optical activity of the resulting ε-PL showed [α]25D = 0°, again indicating an inevitable racemization process under the current polymerization conditions.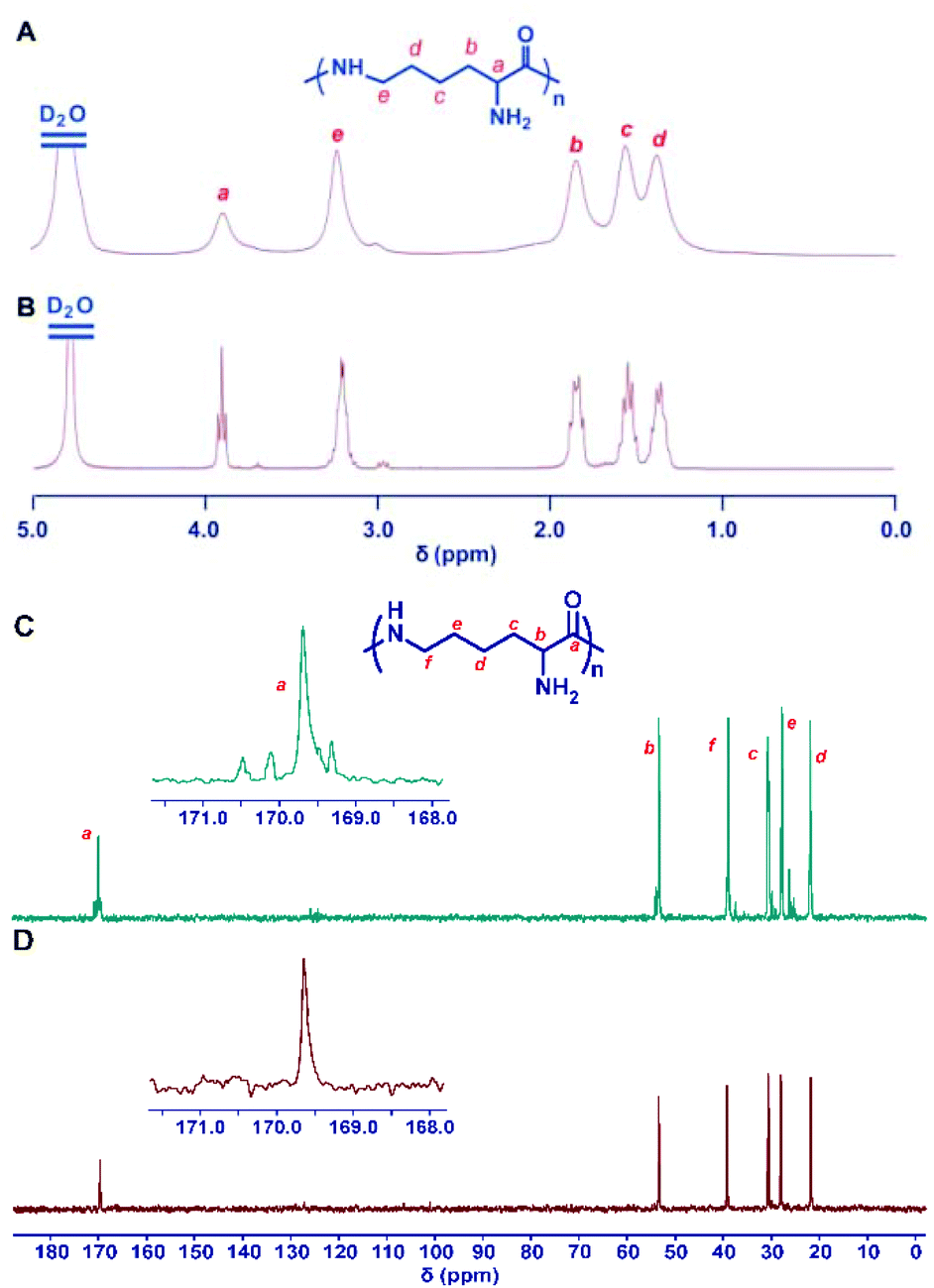 | ||
| Fig. 5 Structure characterization of ε-PL. 1H NMR spectra of chemosynthetic ε-PL (A) and biosynthetic ε-PL (B). 13C NMR spectra of chemosynthetic ε-PL (C) and biosynthetic ε-PL (D). All spectra were measured in D2O, pH 2 at room temperature. Reproduced from ref. 25 with permission from the Royal Society of Chemistry, copyright 2015. | ||
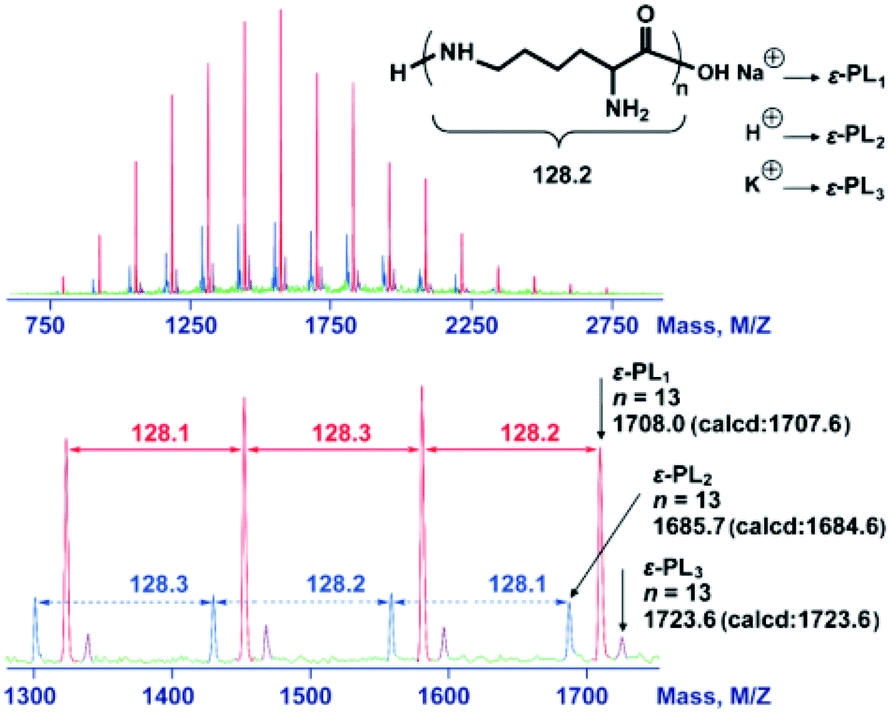 | ||
| Fig. 6 MALDI-TOF-MS spectra of the chemosynthetic ε-PL. The sample was obtained by ROP followed by the removal of the protecting group. The red peaks correspond to ε-PL1. The blue peaks correspond to ε-PL2. The purple peaks correspond to ε-PL3. Reproduced from ref. 25 with permission from the Royal Society of Chemistry, copyright 2015. | ||
3.6 Biocompatibility
To evaluate the biocompatibility of the chemosynthetic ε-PL, we investigated the cytotoxicity of the synthesized ε-PL by the standard MTT assay with L929 cells as the test cell strain (Fig. 7).25 The α-PL synthesized by ROP of NCA monomers53–62 and biosynthetic ε-PL were used as controls. Similar to the biosynthetic ε-PL, no significant toxicity was observed from chemosynthetic ε-PL treated cells at concentrations up to 0.5 mg mL−1 after 48 h of incubation, indicating the high compatibility of the chemosynthetic ε-PL (Fig. 7A and B). Moreover, to further show the advantages of this new chemosynthetic approach and to understand whether the increase in the molecular weight of ε-PL changed its biocompatibility, chemosynthetic ε-PL samples featuring different molecular weights were selected for the MTT cytotoxicity assay. The ε-PL sample with 9.5 kDa displayed no obvious toxicity at concentrations up to 0.25 mg mL−1 for 48 h incubation (Fig. 7C and D). The excellent cell compatibility as well as the diverse molecular weights endow chemosynthetic ε-PL with tremendous potential in the application of gene and drug delivery systems.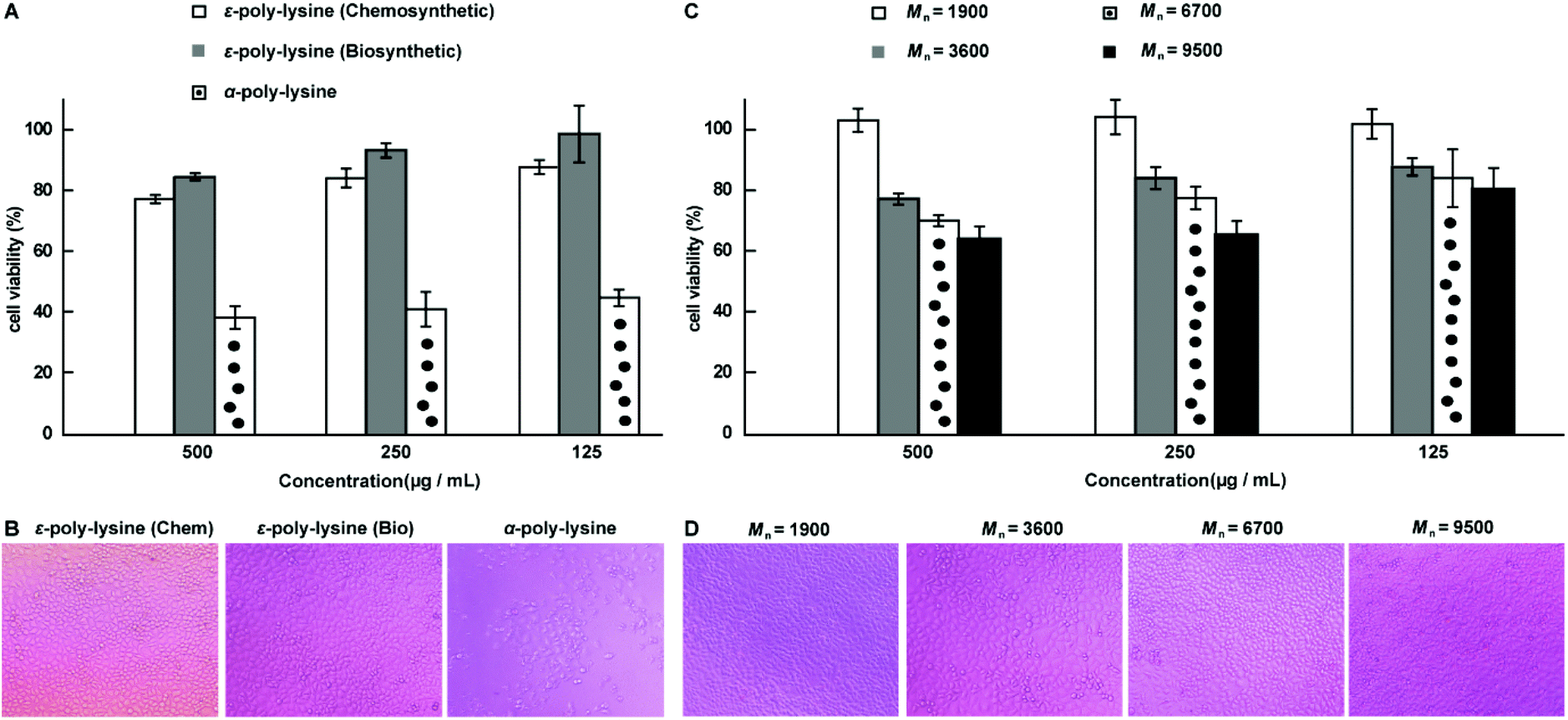 | ||
| Fig. 7 MTT cytotoxicity assay. (A) Viability of L929 cells, as determined by the MTT assay following 48 hours of treatment with ε-PL and α-PL, separately. (B) Microscopy images of L929 cells treated with 0.5 mg mL−1 of polymer. (C) MTT was used to detect the toxicity of the different ε-PLs as a function of molecular weight at various concentrations. (D) Microscopy images of L929 cells treated with 0.25 mg mL−1 of ε-PL. The MTT cytotoxicity assay showed no toxicity to L929 cells at concentrations of 0.125 to 0.25 mg mL−1 of ε-PL with varying molecular weights. The molecular weight values of ε-PL are estimated from the values of their corresponding acetylated versions after subtraction of the acetyl groups. Reproduced from ref. 25 with permission from the Royal Society of Chemistry, copyright 2015. | ||
4 Derivatives of poly(ε-lysine)
Functional polyamides that feature tunable mechanical properties, low toxicity, therapeutic potential, and structural similarity to peptides (amide bonds) exhibit enormous potential in the application of biomedical materials and other fields.22–24,47,63–79 We have developed a cyclization reaction of biorenewable lysine to yield ε-lactam monomer with a protected α-amino group. ROP of the obtained monomer, followed by deprotection, afforded polyamides bearing pendant amines [poly(ε-lysine)].17,25,26,28 To further achieve diversity of the structure and performance of polyamides, we employed similar cyclization of lysine derivatives to prepare a range of cyclic lysine monomers bearing pendant benzyl-protected hydroxyl, allyloxy, and oligo-ethylene glycol groups by further evolving the method we reported previously (Scheme 3).27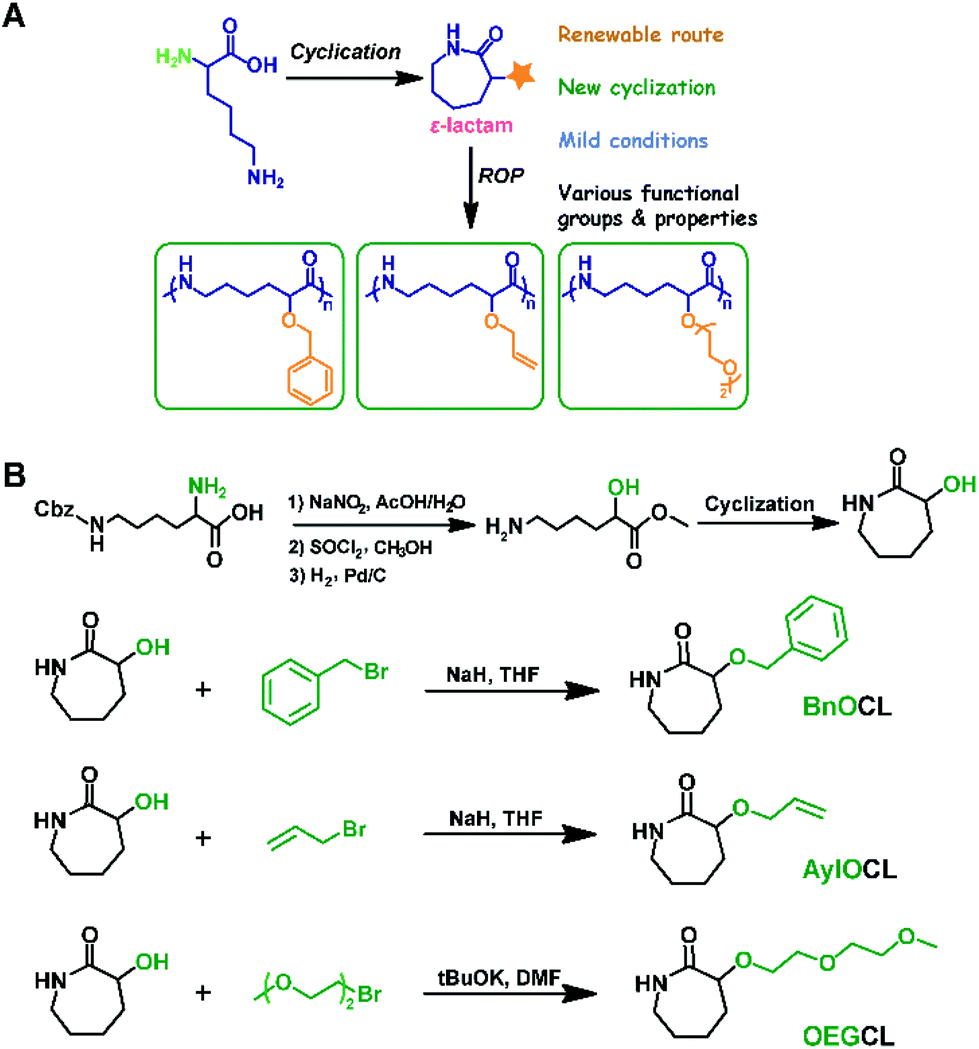 | ||
| Scheme 3 (A) Lysine derivative cyclization and organocatalytic ring-opening polymerization for the synthesis of functional polyamides. (B) Design and synthesis of functional cyclic lysine monomers. Reproduced from ref. 27 with permission from the American Chemical Society, copyright 2018. | ||
The ROP of the lysine derived ε-lactam monomers were efficiently conducted using superbase t-BuP4 as a catalyst in THF at room temperature, and provided a set of functional polyamides with Mn up to 47.7 kDa. Indeed, the very mild polymerization condition did not interfere with the above functional groups. The expected structures of the functional polyamides were verified by the 1H NMR, 13C NMR and MALDI-TOF-MS spectrum. The benzyl groups of the poly(BnOCL) were readily removed through Pd/C-catalyzed hydrogenolysis, furnishing polyamides bearing a pendant hydroxyl group (poly(HOCL)). The resultant allylated polyamides (poly(AylOCL)) further allowed facile incorporation of a wide range of functional pendants onto polyamides by thiol–ene click reactions. The oligo-ethylene glycol functional polyamides demonstrated semi-crystallinity, and had minimal cytotoxicity to HeLa cells. Also, the self-assembly of amphiphilic poly(OEGCL) into micelles (Fig. 8A) was identified by transmission electron microscopy (Fig. 8B) and dynamic light scattering (Fig. 8C). Collectively, these results suggested their great potential for biomedical applications.
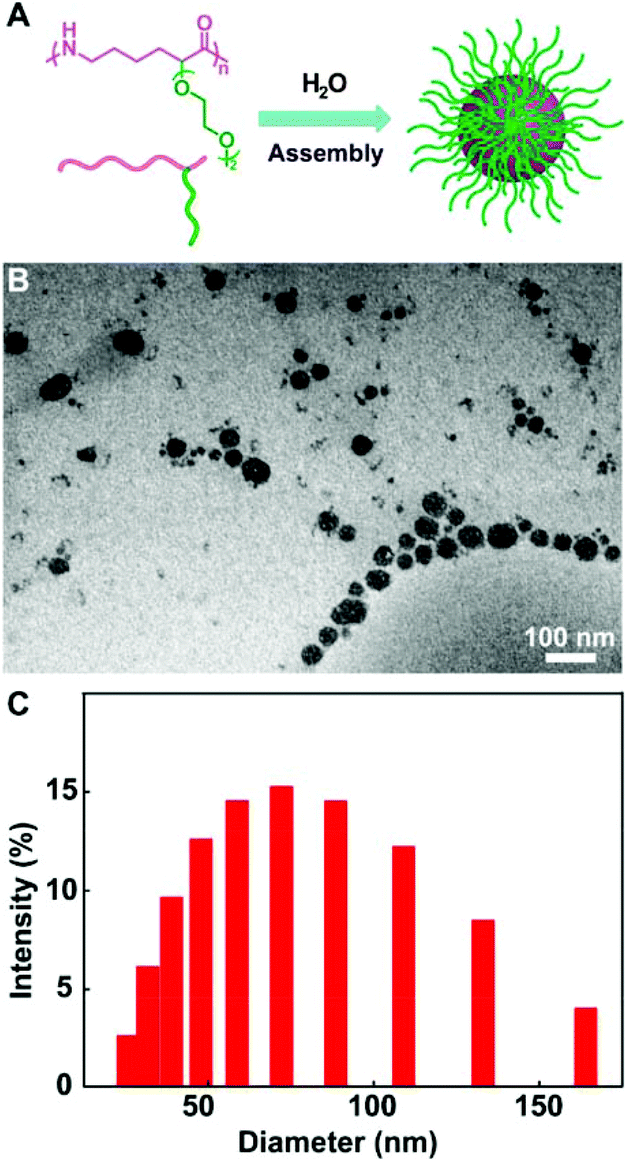 | ||
| Fig. 8 Self-assembly of amphiphilic poly(OEGCL) into micelles. (A) Illustration on the formation of micelles. (B) TEM image of the micelle sample drop deposited from H2O onto a carbon-coated copper grid; scale bar = 100 nm. (C) Size distribution of micelles measured by DLS. Reproduced from ref. 27 with permission from the American Chemical Society, copyright 2018. | ||
5 Conclusions and perspectives
In summary, we reviewed our recent progress in the chemosynthesis of poly(ε-lysine) and its derivatives with diverse molecular weights and structures, relying on cyclization of lysine, protection, ring-opening polymerization, and deprotection. With the growing demand of poly(ε-lysine) in various applications, the chemosynthetic route that can transform low-cost lysine to high-value ε-PL is of great interest. Although significant progress has been achieved in this area, the chemosynthesis of ε-PL still suffers from some formidable challenges, which has led to industrial activity remaining in the preliminary stages. The protecting groups and the catalysts are the two main themes in this area. The complete deprotection of 2,5-dimethylpyrrole or PMB groups faces poor to moderate yield (20–30% and 45%, respectively). Further optimizing the structures of the protecting groups as well as the deprotection conditions to provide ε-PL with excellent yields is highly desired. The ROP catalyzed by current inorganic bases still requires high polymerization temperature (≥180 °C) and significantly compromises the protecting group scope. The efficient ROP proceeded under mild conditions has to employ the costly phosphazene t-BuP4. We expect that the combination of low-cost metal salts and organic bases to realize efficient ROP of lysine-derived ε-lactams is promising.80,81 It is also a feasible approach to devise some amino functional groups without deprotection and obtain derivatives with similar functions to ε-PL. Considering the structural similarity of ε-PL and poly(caprolactam), we can foresee the copolymerization of caprolactam and cyclic lysine monomers to prepare antimicrobial fibers82 and films,83,84 which will attract extensive attention in both academia and industry. More commonly, the concept of utilizing the cyclization/ring-opening polymerization of a lysine derivative to afford high-value functionalized poly(amino acid) materials is universal and valuable to synthesize other poly(amino acids), such as poly(γ-glutamate)30 from glutamic acid that is often inaccessible to typical chemosynthetic methods.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grants 91856113, 51873211, 22001243, and 52073274).References
- S. Shima and H. Sakai, Agric. Biol. Chem., 1977, 41, 1807–1809 CAS.
- S. Shima and H. Sakai, Agric. Biol. Chem., 1981, 45, 2497–2502 CAS.
- S. Shima and H. Sakai, Agric. Biol. Chem., 1981, 45, 2503–2508 CAS.
- C. Zhou, P. Li, X. Qi, A. R. M. Sharif, Y. F. Poon, Y. Cao, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Biomaterials, 2011, 32, 2704–2712 CrossRef CAS.
- L. Mondragón, N. Mas, V. Ferragud, C. de la Torre, A. Agostini, R. Martínez-Máñez, F. Sancenón, P. Amorós, E. Pérez-Payá and M. Orzáez, Chem. – Eur. J., 2014, 20, 5271–5281 CrossRef.
- G. N. Roviello, S. Di Gaetano, D. Capasso, S. Franco, C. Crescenzo, E. M. Bucci and C. Pedone, J. Med. Chem., 2011, 54, 2095–2101 CrossRef CAS.
- M. Moccia, G. N. Roviello, E. M. Bucci, C. Pedone and M. Saviano, Int. J. Pharm., 2010, 397, 179–183 CrossRef CAS.
- S. B. Bankar and R. S. Singhal, RSC Adv., 2013, 3, 8586–8603 RSC.
- I.-L. Shih, M.-H. Shen and Y.-T. Van, Bioresour. Technol., 2006, 97, 1148–1159 CrossRef CAS.
- J. Hiraki, T. Ichikawa, S.-I. Ninomiya, H. Seki, K. Uohama, H. Seki, S. Kimura, Y. Yanagimoto and J. W. Barnett, Regul. Toxicol. Pharmacol., 2003, 37, 328–340 CrossRef CAS.
- A. K. Pandey and A. Kumar, Process Biochem., 2014, 49, 496–505 CrossRef CAS.
- S. C. Shukla, A. Singh, A. K. Pandey and A. Mishra, Biochem. Eng. J., 2012, 65, 70–81 CrossRef CAS.
- T. Yoshida and T. Nagasawa, Appl. Microbiol. Biotechnol., 2003, 62, 21–26 CrossRef CAS.
- I. L. Shih, Y. T. Van and M. H. Shen, Mini-Rev. Med. Chem., 2004, 4, 179–188 CrossRef CAS.
- Z. Xu, Z. Xu, X. Feng, D. Xu, J. Liang and H. Xu, Appl. Microbiol. Biotechnol., 2016, 100, 6619–6630 CrossRef CAS.
- Z. An, C. Chen, J. He, C. Hong, Z. Li, Z. Li, C. Liu, X. Lv, A. Qin, C. Qu, B. Z. Tang, Y. Tao, X. Wan, G. Wang, J. Wang, K. Zheng and W. Zou, Acta Polym. Sin., 2019, 50, 1083–1132 Search PubMed.
- Y. Tao, Acta Polym. Sin., 2016, 47, 1151–1159 Search PubMed.
- X. S. Chen, G. Q. Chen, Y. H. Tao, Y. Z. Wang, X. B. Lv, L. Q. Zhang, J. Zhu, J. Zhang and X. H. Wang, Acta Polym. Sin., 2019, 50, 1068–1082 Search PubMed.
- D. R. S. Kushwaha, K. B. Mathur and D. Balasubramanian, Biopolymers, 1980, 19, 219–229 CrossRef CAS.
- C. H. Ho, E. Odermatt, I. Berndt and J. C. Tiller, J. Polym. Sci. A Polym. Chem., 2008, 46, 5053–5063 CrossRef CAS.
- W. E. Hanford and R. M. Joyce, J. Polym. Sci., 1948, 3, 167–172 CrossRef CAS.
- K. Hashimoto, Prog. Polym. Sci., 2000, 25, 1411–1462 CrossRef CAS.
- Handbook of Ring-opening Polymerization, Copyright ©, ed. P. Dubois, O. Coulembier and J. M. Raquez, Wiley–VCH Verlag GmbH & Co. KGaA, 2009 Search PubMed.
- K. Marchildon, Macromol. React. Eng., 2011, 5, 22–54 CrossRef CAS.
- Y. Tao, X. Chen, F. Jia, S. Wang, C. Xiao, F. Cui, Y. Li, Z. Bian, X. Chen and X. Wang, Chem. Sci., 2015, 6, 6385–6391 RSC.
- J. Chen, M. Li, W. He, Y. Tao and X. Wang, Macromolecules, 2017, 50, 9128–9134 CrossRef CAS.
- W. He, Y. Tao and X. Wang, Macromolecules, 2018, 51, 8248–8257 CrossRef CAS.
- J. Lian, J. Chen, Y. Tao and X. Wang, Synthetic Polymer Chemistry: Innovations and Outlook, The Royal Society of Chemistry, 2020, pp. 243–263 Search PubMed.
- W. He and Y. Tao, Acta Polym. Sin., 2020, 51, 1083–1091 Search PubMed.
- A. Ogunleye, A. Bhat, V. U. Irorere, D. Hill, C. Williams and I. Radecka, Microbiology, 2015, 161, 1–17 CrossRef CAS.
- S. Shima, Y. Fukuhara and H. Sakai, Agric. Biol. Chem., 1982, 46, 1917–1919 CAS.
- S. Shima, H. Matsuoka, T. Iwamoto and H. Sakai, J. Antibiot., 1984, 37, 1449–1455 CrossRef CAS.
- S. Jia, B. Fan, Y. Dai, G. Wang, P. Peng and Y. Jia, Food Sci. Biotechnol., 2010, 19, 361–366 CrossRef CAS.
- S. Maeda, K.-K. Kunimoto, C. Sasaki, A. Kuwae and K. Hanai, J. Mol. Struct., 2003, 655, 149–155 CrossRef CAS.
- M. Hyldgaard, T. Mygind, B. S. Vad, M. Stenvang, D. E. Otzen and R. L. Meyer, Appl. Environ. Microbiol., 2014, 80, 7758–7770 CrossRef.
- T. Bo, P.-P. Han, Q.-Z. Su, P. Fu, F.-Z. Guo, Z.-X. Zheng, Z.-L. Tan, C. Zhong and S.-R. Jia, Food Control, 2016, 61, 123–134 CrossRef CAS.
- B. List, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- M. Fèvre, J. Vignolle, Y. Gnanou and D. Taton, in Polymer Science: A Comprehensive Reference, ed. M. Möller, Elsevier, Amsterdam, 2012, pp. 67–115 Search PubMed.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- S. Naumann and A. P. Dove, Polym. Chem., 2015, 6, 3185–3200 RSC.
- S. Naumann and A. P. Dove, Polym. Int., 2016, 65, 16–27 CrossRef CAS.
- S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Macromol. Rapid Commun., 2018, 39, 1800485 CrossRef.
- A. A. Kolomeitsev, I. A. Koppel, T. Rodima, J. Barten, E. Lork, G.-V. Röschenthaler, I. Kaljurand, A. Kütt, I. Koppel, V. Mäemets and I. Leito, J. Am. Chem. Soc., 2005, 127, 17656–17666 CrossRef CAS.
- S. Naumann, S. Epple, C. Bonten and M. R. Buchmeiser, ACS Macro Lett., 2013, 2, 609–612 CrossRef CAS.
- M. Li, Y. Tao, J. Tang, Y. Wang, X. Zhang, Y. Tao and X. Wang, J. Am. Chem. Soc., 2019, 141, 281–289 CrossRef CAS.
- X. Chen, H. Lai, C. Xiao, H. Tian, X. Chen, Y. Tao and X. Wang, Polym. Chem., 2014, 5, 6495–6502 RSC.
- S. Liu, T. Bai, K. Ni, Y. Chen, J. Zhao, J. Ling, X. Ye and G. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15478–15487 CrossRef CAS.
- B. Lin and R. M. Waymouth, Macromolecules, 2018, 51, 2932–2938 CrossRef CAS.
- J. U. Pothupitiya, R. S. Hewawasam and M. K. Kiesewetter, Macromolecules, 2018, 51, 3203–3211 CrossRef CAS.
- C. Deng, J. Wu, R. Cheng, F. Meng, H.-A. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330–364 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC.
- Z. Song, Z. Tan and J. Cheng, Macromolecules, 2019, 52, 8521–8539 CrossRef CAS.
- H. Lai, X. Chen, Q. Lu, Z. Bian, Y. Tao and X. Wang, Chem. Commun., 2014, 50, 14183–14186 RSC.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 CrossRef CAS.
- J. Yuan, Y. Sun, J. Wang and H. Lu, Biomacromolecules, 2016, 17, 891–896 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS.
- H. Zhang, J. Chen, X. Zhang, C. Xiao, X. Chen, Y. Tao and X. Wang, Biomacromolecules, 2017, 18, 924–930 CrossRef CAS.
- S. Wang, W. He, C. Xiao, Y. Tao and X. Wang, Biomacromolecules, 2019, 20, 1655–1666 CrossRef CAS.
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Macromolecules, 2014, 47, 8247–8254 CrossRef CAS.
- C. Bakkali-Hassani, D. Tunc, K. Roos, M. Planes, P. Lecomte and S. Carlotti, Macromolecules, 2017, 50, 175–181 CrossRef CAS.
- C. Bakkali-Hassani, M. Planes, K. Roos, A.-L. Wirotius, E. Ibarboure and S. Carlotti, Eur. Polym. J., 2018, 102, 231–237 CrossRef CAS.
- H. Bouchékif, D. Tunc, C. Le Coz, A. Deffieux, P. Desbois and S. Carlotti, Polymer, 2014, 55, 5991–5997 CrossRef.
- A. Sehlinger, P.-K. Dannecker, O. Kreye and M. A. R. Meier, Macromolecules, 2014, 47, 2774–2783 CrossRef CAS.
- S. Oelmann and M. A. R. Meier, Macromol. Chem. Phys., 2015, 216, 1972–1981 CrossRef CAS.
- M. Winnacker, J. Sag, A. Tischner and B. Rieger, Macromol. Rapid Commun., 2017, 38, 1600787 CrossRef.
- T. Ageyeva, I. Sibikin and J. Karger-Kocsis, Polymers, 2018, 10, 357 CrossRef.
- I. Sibikin and J. Karger-Kocsis, Adv. Ind. Eng. Polym. Res., 2018, 1, 48–60 Search PubMed.
- M. Winnacker and J. Sag, Chem. Commun., 2018, 54, 841–844 RSC.
- M. Li, F. Cui, Y. Li, Y. Tao and X. Wang, Macromolecules, 2016, 49, 9415–9424 CrossRef CAS.
- A. Sathyan, R. C. Hayward and T. Emrick, Macromolecules, 2019, 52, 167–175 CrossRef CAS.
- Y. Tao, Z. Wang and Y. Tao, Biopolymers, 2019, 110, e23288 CrossRef.
- L. Guo and D. Zhang, J. Am. Chem. Soc., 2009, 131, 18072–18074 CrossRef CAS.
- L. Guo, S. H. Lahasky, K. Ghale and D. Zhang, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef CAS.
- X. Zhang, S. Wang, J. Liu, Z. Xie, S. Luan, C. Xiao, Y. Tao and X. Wang, ACS Macro Lett., 2016, 5, 1049–1054 CrossRef CAS.
- S. Wang, Y. Tao, J. Wang, Y. Tao and X. Wang, Chem. Sci., 2019, 10, 1531–1538 RSC.
- S. Naumann, P. B. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS.
- M. L. McGraw and E. Y. X. Chen, Macromolecules, 2020, 53, 6102–6122 CrossRef CAS.
- Y. Qin, in Medical Textile Materials, ed. Y. Qin, Woodhead Publishing, 2016, pp. 145–160 Search PubMed.
- P. K. Dutta, S. Tripathi, G. K. Mehrotra and J. Dutta, Food Chem., 2009, 114, 1173–1182 CrossRef CAS.
- S. Quintavalla and L. Vicini, Meat Sci., 2002, 62, 373–380 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |