DOI:
10.1039/D0PY01085E
(Paper)
Polym. Chem., 2021,
12, 1549-1561
An AIE-driven fluorescent polysaccharide polymersome as an enzyme-responsive FRET nanoprobe to study the real-time delivery aspects in live cells†
Received
29th July 2020
, Accepted 17th December 2020
First published on 18th December 2020
Abstract
We report aggregation induced emission (AIE) driven polysaccharide polymersomes as fluorescence resonance energy transfer (FRET) nanoprobes to study their intracellular enzyme-responsive delivery by real-time live-cell confocal microscopy bio-imaging techniques. An AIE active tetraphenylethylene (TPE) optical chromophore and plant-based vesicular directing hydrophobic unit were grafted on clinically relevant polysaccharide-dextran via enzyme-cleavable aliphatic ester chemical linkages. The TPE-tagged dextran self-assembled as 180 ± 20 nm blue-luminescent polymersomes in aqueous medium and exhibited excellent encapsulation capabilities for water soluble Rose Bengal (RB) and water insoluble Nile red (NR) fluorophores. The selective photoexcitation of the TPE chromophore enabled the FRET process between the TPE donor and RB (or NR) acceptor molecule in <50 Å Förster distance afforded by the polymersome. The FRET probe was very stable under extracellular conditions and it exclusively underwent lysosomal esterase enzymatic biodegradation at the intracellular compartments to release RB. The enzyme-trigger enabled the FRET probe to function as an extracellular turn-ON → intracellular turn-Off red-fluorescent signal (Probe-1). In this process, the AIE self-emission was also simultaneously restored on the TPE chromophore (blue-luminescent, Probe-2) followed by the isolation of donor and acceptor in the cytosol. As a result, this new design enabled the visualization of real-time enzyme-responsive delivery by monitoring the dual fluorescent signals from both the polymer host (blue) and encapsulated guest (red) in a single nano-platform. In vitro cytotoxicity studies established that the polymersome probe was non-toxic to cells up to 300 μg mL−1. Lyso-tracker staining experiments supported the FRET probe internalization in the lysosomal compartments for enzymatic-biodegradation. Live cell confocal microscopy with selective photo-excitation was used to directly monitor the enzyme-responsive FRET action in human breast cancer MCF 7 and wild-type mouse embryonic fibroblast cell lines (WT-MEFs). It was found that the tailor-made polymersome FRET probe was efficient to deliver the loaded cargo in <3 h in live cells which predicts the usefulness of the probe in biomedical research.
Introduction
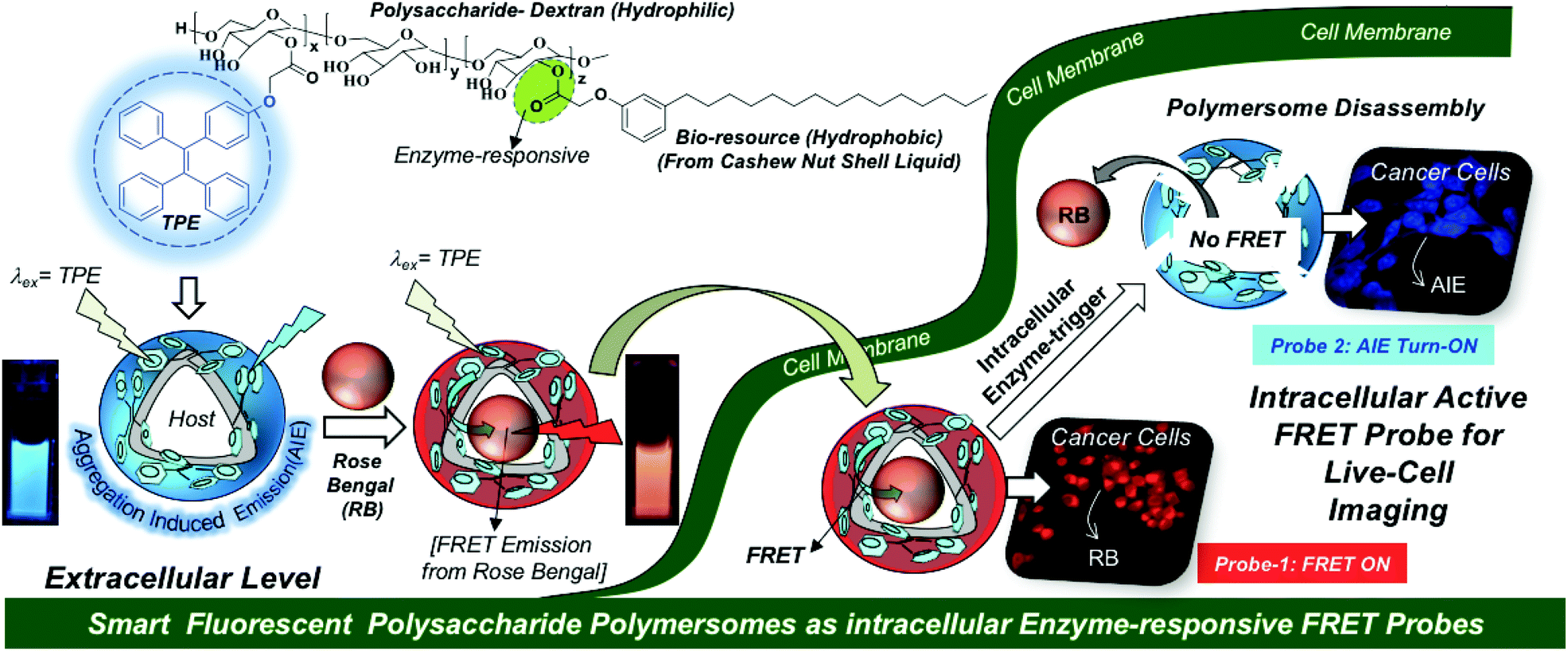
Fluorescent polymer nano-assemblies are dual-purpose biomaterials for delivering therapeutic drugs (or genes) while simultaneously probing their function in intracellular compartments or tumor micro-environments by bioimaging techniques.1–3 Amphiphilic block copolymer nanoparticles,4,5 unimolecular micelles,6 dendrimers,7 supramolecular nano-assemblies,8 polymersomes (or polymer vesicles)9,10etc., are some of the most important fluorescent macromolecular nano-assemblies that are explored in cancer therapy. Among them, polymersomes are unique self-assembled nano-scaffolds and they possess well-defined compartmentalization of the hydrophilic core and hydrophobic layer for loading both water soluble and insoluble drugs and fluorescent molecules, respectively.11,12 These unique features of the polymersome nano-assemblies make them distinct nano-scaffolds for loading wide ranges of cargoes irrespective of their solubility parameters.13–19 Aggregation induced emission (AIE) is a newly emerging photophysical phenomenon and it is an excellent system to exhibit strong fluorescence characteristics in aqueous medium.20–22 AIE-activated polymersomes were reported to study self-assembly aspects or CO2 capture in polypeptoids,23 polyacrylate and POSS block copolymers,24,25 poly(trimethylenecarbonate)s,26etc.; however, they have been rarely explored in the biomedical field. Hence, the development of new AIE-assisted stimuli-responsive fluorescent polymersomes would provide new opportunities for drug delivery to cancer cells.
To accomplish the above task, we have chosen a polysaccharide platform since it is largely in abundance in nature and possesses excellent biocompatibility under physiological conditions for biomedical reserach.27–30 Unlike micellar nanoparticles;31–33 the development of polysaccharide polymersomes was found to be very challenging task due to the requirement of a high degree of cooperativity between the rigid hydrophilic polysaccharide backbone and flexible hydrophobic units to produce vesicular nano-assemblies. Dextran-poly(γ-bezyl L-glutmate) block copolymers34 and dextran-graft-poly(L-lactides)35 are some of the important examples that were reported to self-assemble as polymersomes. Our research group has developed bio-resource based polysaccharide polymersomes by conjugating the plant-based hydrophobic unit 3-pentadecyl phenol (PDP, from the cashew nut shell liquid)36,37 as a vesicular directing component on the dextran backbone to bring the appropriate geometry for polymersome self-assembly in aqueous medium.38 The PDP hydrophobic handle is very versatile in producing enzymes,39,40 pH41 and GSH responsive42 polymersomes depending upon the choice of the chemical linkages chosen for the stimuli-responsiveness at the cellular level. These polysaccharide polymersomes were found to be efficient hosts for delivering water-soluble doxorubicin, water insoluble camptothecin, and also metal based cisplatin anticancer drugs together in a single nanoplatform.43 Metastasis-suppression phase-III drug MLN8237 was also successfully delivered to selectively inhibit Aurora kinase A mediated anchorage-independent growth in 3D breast cancer cells.44 In this present investigation, the AIE concept was cleverly employed in the dextran–PDP system to structurally engineer the first examples of fluorescent polysaccharide polymersomes. The AIE-driven polymersome was employed as a fluorescence resonance energy transfer (FRET)45–47 donor along with acceptor-fluorophores such as rose Bengal and Nile red to build next generation smart enzyme-responsive FRET nanoprobes. The proof-of-concept of a fluorescent polymersome FRET probe was successfully demonstrated in live cell bio-imaging and this new concept is shown in Fig. 1. In vitro cellular uptake studies in the breast cancer MCF 7 cell line and the wild-type mouse embryonic fibroblast cell line (WT-MEFs) were carried out to study the internalization of the FRET probes in the lysosomal compartments and their enzyme responsiveness by live-cell confocal microscope imaging. Custom designed fluorescent polysaccharide polymersomes are a new entry in the literature; thus, they would be very useful for drug delivery application and diagnostics for long term impact in cancer research.
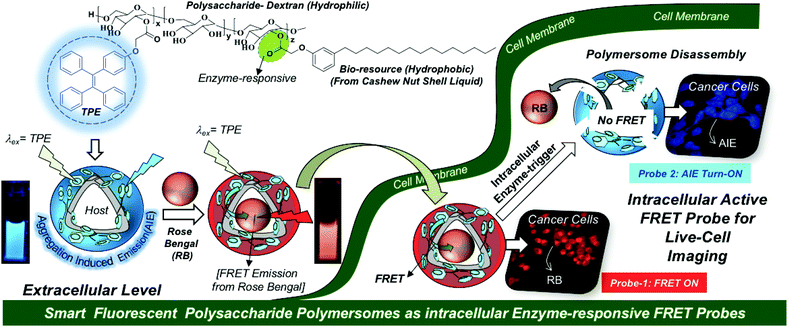 |
| | Fig. 1 Development of AIE-active polysaccharide polymersomes and their enzyme-responsive FRET fluorescent probes for monitoring intracellular level delivery in live cells. | |
Results and discussion
Synthesis and self-assembly of AIE polymersomes
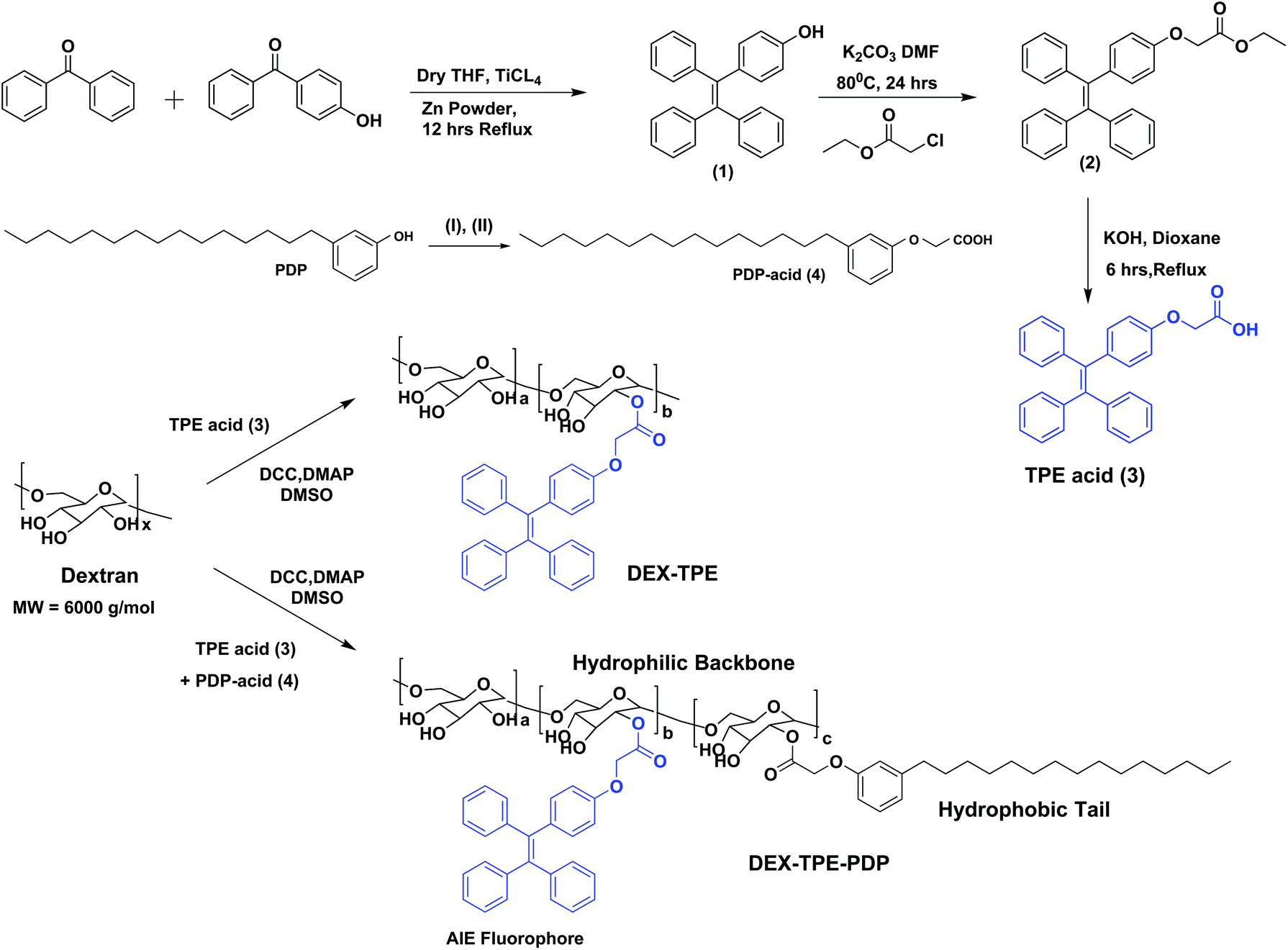
The aggregation induced emission (AIE) fluorophore tetraphenylethylene (TPE) was designed with a carboxylic acid functionality as shown in Scheme 1. Benzophenone and 4-hydroxy benzophenone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio) were coupled using TiCl4 and zinc powder to obtain 4-hydroxyl tetraphenylethylene (1). Compound (1) was then further alkylated using ethyl chloroacetate to get the TPE carboxylic ester (2) which was hydrolyzed to yield the TPE-carboxylic acid (3). 3-Pentadecylphenol from the cashew nut shell liquid was substituted using ethyl chloroacetate followed by hydrolysis to get the PDP carboxylic acid (4).38 TPE acid (3) and PDP acid (4) were anchored simultaneously on the dextran (polysaccharide, MW = 6000 g mol−1) backbone in a one pot esterification reaction to yield TPE-conjugated amphiphilic dextran. The structure of the polymer was characterized by 1H-NMR and the details are given in Fig. S1 in the ESI.† The substitution of the TPE unit on the dextran backbone was evident from the presence of peaks from 6.6 to 7.2 ppm with respect to the aryl-protons. Furthermore, the aryl proton ‘f’ in the TPE unit was shifted to the downfield region with respect to the formation of ester linkages. The PDP aromatic protons merged with TPE units in the polymer spectrum, whereas the peaks with respect to the alkyl tail were distinctly visible in the region 0.8 to 1.5 ppm. The degree of substitution of PDP and TPE units on the dextran backbone was determined by comparing the integration values of the anomeric proton with PDP or TPE protons. Degrees of substitutions were obtained as 2% and 5% for TPE and PDP units in the dextran backbone, respectively. To estimate the degree of TPE substitution, absorbance spectroscopy is also employed (see Fig. S2 in the ESI†). The absorbance technique showed the TPE content as 1.96% which is matching with the NMR based value of 2.0%. Substitution of a higher amount of aryl units (>10%) produced the water insoluble dextran sample; thus, the total amount of TPE + PDP was maintained <10% in order to make modified dextran useful for biomedical application.
:1 mole ratio) were coupled using TiCl4 and zinc powder to obtain 4-hydroxyl tetraphenylethylene (1). Compound (1) was then further alkylated using ethyl chloroacetate to get the TPE carboxylic ester (2) which was hydrolyzed to yield the TPE-carboxylic acid (3). 3-Pentadecylphenol from the cashew nut shell liquid was substituted using ethyl chloroacetate followed by hydrolysis to get the PDP carboxylic acid (4).38 TPE acid (3) and PDP acid (4) were anchored simultaneously on the dextran (polysaccharide, MW = 6000 g mol−1) backbone in a one pot esterification reaction to yield TPE-conjugated amphiphilic dextran. The structure of the polymer was characterized by 1H-NMR and the details are given in Fig. S1 in the ESI.† The substitution of the TPE unit on the dextran backbone was evident from the presence of peaks from 6.6 to 7.2 ppm with respect to the aryl-protons. Furthermore, the aryl proton ‘f’ in the TPE unit was shifted to the downfield region with respect to the formation of ester linkages. The PDP aromatic protons merged with TPE units in the polymer spectrum, whereas the peaks with respect to the alkyl tail were distinctly visible in the region 0.8 to 1.5 ppm. The degree of substitution of PDP and TPE units on the dextran backbone was determined by comparing the integration values of the anomeric proton with PDP or TPE protons. Degrees of substitutions were obtained as 2% and 5% for TPE and PDP units in the dextran backbone, respectively. To estimate the degree of TPE substitution, absorbance spectroscopy is also employed (see Fig. S2 in the ESI†). The absorbance technique showed the TPE content as 1.96% which is matching with the NMR based value of 2.0%. Substitution of a higher amount of aryl units (>10%) produced the water insoluble dextran sample; thus, the total amount of TPE + PDP was maintained <10% in order to make modified dextran useful for biomedical application.
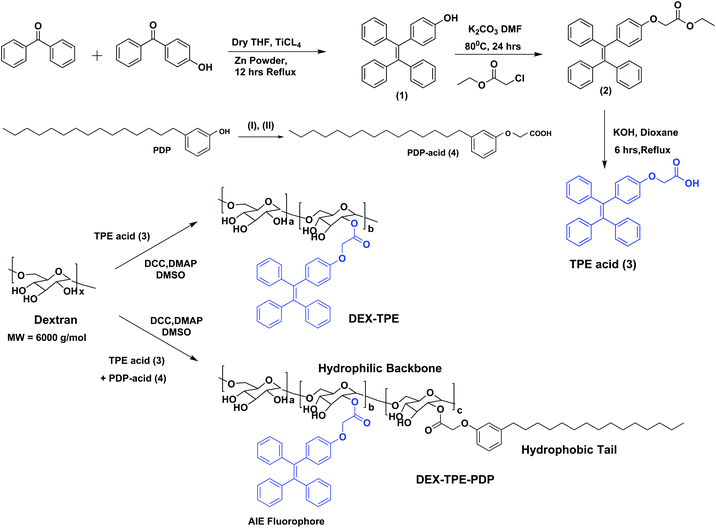 |
| | Scheme 1 Synthesis of TPE-acid, PDP-acid, PDP + TPE-conjugated amphiphilic dextran and TPE-conjugated dextran. The synthesis of PDP acid is done following the procedure in our earlier report.38 The reagents used are: (I) ethyl chloroacetate, K2CO3, DMF, 80 °C, 24 hours and (II) KOH, dioxane, 25 °C, 6 hours. | |
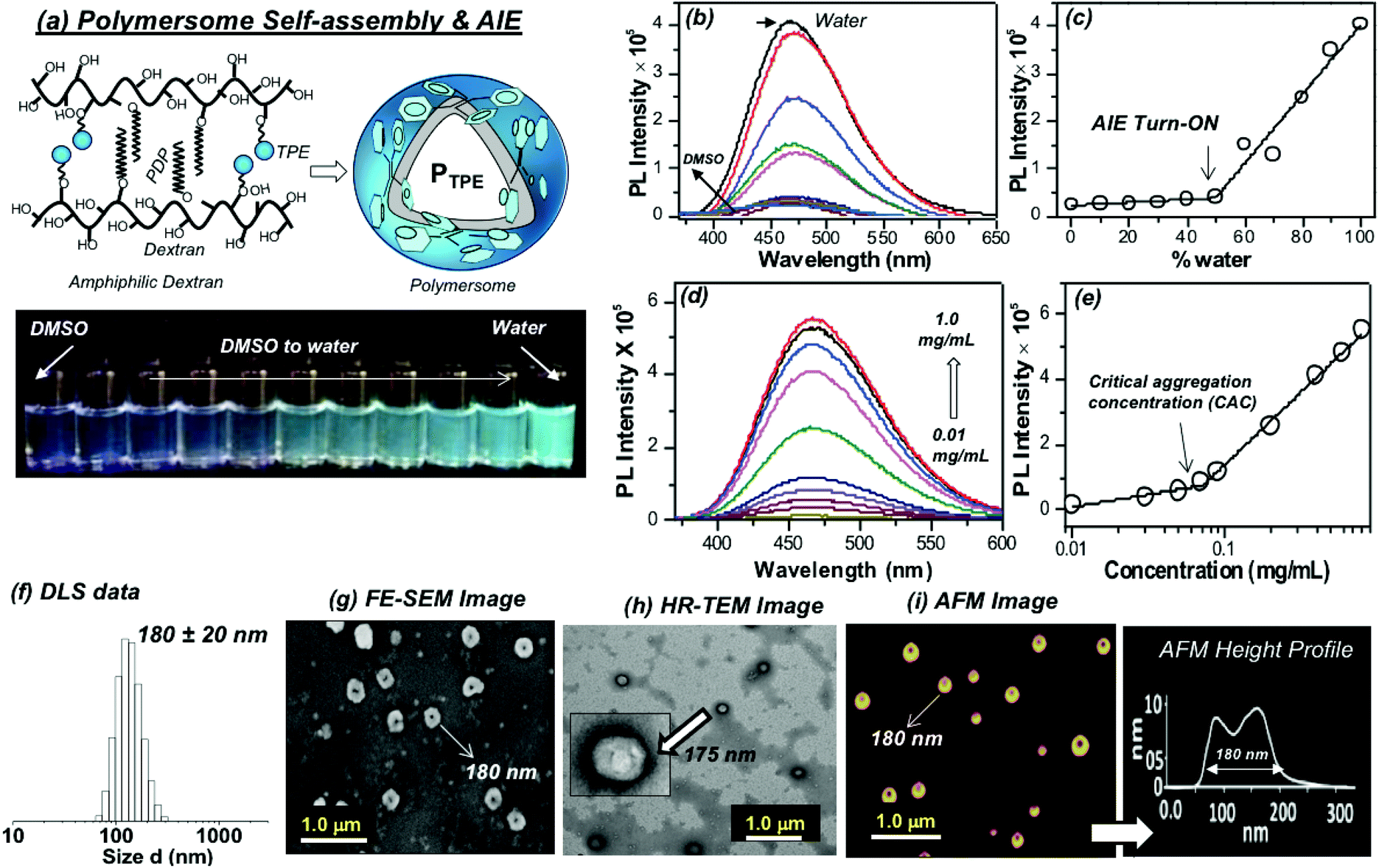
The self-assembly of the TPE and PDP conjugated dextran derivative in aqueous medium enabled the inter-digitization of the PDP hydrophobic tail to produce bi-layer type self-assembly which subsequently yielded the polymersomes.38,48 The rigid-TPE aryl chromophore occupies the hydrophobic layer created by the vesicular-assemblies and exhibits aggregation induced emission (AIE) with strong blue-luminescence as shown in Fig. 2a. The photographs of the polymer solution in Fig. 2a showed the appearance of strong blue-emission with respect to AIE phenomena. The emission spectra of the polymer solution (O.D. = 0.1, λexc = 340 nm) in water + DMSO solvent mixtures showed a strong blue-fluorescence in water with emission maxima at 460 nm with respect to the AIE from TPE (see Fig. 2b). The plot of PL intensity versus the solvent composition in Fig. 2c showed a clear break point at 50% of DMSO + water composition. The absorbance maxima of the TPE chromophore did not show any change in this solvent composition (see Fig. S3 in the ESI†); thus, the variation in the PL intensity in Fig. 2b was not due to variation in the molar absorbtivity of the TPE and it was primarily driven by the AIE process. The concentration dependent fluorescence measurement in Fig. 2d enabled the determination of the critical aggregation concentration (CAC) of the polymersomes to be 0.09 mg mL−1 as shown in Fig. 2e. To study the effect of temperature on the stability of dextran polymersomes, the sample was subjected to variable temperature fluorescence measurement ranging from 25 °C to 70 °C. The emission intensity in the spectra of the dextran polymersomes in Fig. S4 ESI† was found to decrease with the increase in the temperature and the break point was observed at 45 °C which is much above the physiological temperature 37 °C. Thus, these polymersomes have very good stability at physiological temperature.The dynamic light scattering (DLS) histogram of the polymersome showed monomodal distribution with a size of 165 ± 20 nm (see Fig. 2f). Field-emission scanning electron microscopy (FESEM, in Fig. 2g) images showed the formation of spherical objects with a distinct core in the middle with respect to vesicular nano-assemblies. The high resolution transmission electron microscopy (HR-TEM) image further validates the existence of spherical vesicular self-assemblies with a distinct layer (HR-TEM, Fig. 2h). The average size of the polymersomes was estimated to be 170 ± 10 nm from the TEM images. The expanded images showed the wall thickness of the polymersomes in the range of 4.7 to 5.0 nm in Fig. 2h. The wall thickness matches with our earlier work on the non-fluorescent dextran polymersomes having the same PDP units as vesicular directing agents with different stimuli such as pH and disulfide.38–40 Thus, the TPE conjugation in the dextran backbone did not alter the morphologies of the polymersomes. Earlier work from our group resolved the single crystal structure of the PDP-hydrophobic tail and the hydrophobic layer inter-digitization was determined to be 2.9 nm.48 Therefore, the wall thickness of the AIE-polymersomes was attributed to the existence of at least two hydrophobic layers. The atomic force microscopy (AFM) image depicts the formation of a donut-shaped morphology with respect to the polymersomes and the height profiles (see Fig. 2i) showed two hump shapes (M-shaped) as typically observed for polymersomes with respect to the size of 180 nm. The dialyzed aqueous solution of the polymersomes was directly drop-cast on the mica plates to record the AFM images. The slow drying of the sample makes the middle portion of the polymersomes compress to exhibit an M-shaped curvature. One would anticipate that the thickness at the middle of the polymersomes in the M-shaped profile should be equivalent to the twice of the wall thickness. In the present AFM image the middle portion is <10 nm which is almost equivalent to or less than twice the wall thickness measured using the HR-TEM images. Hence, the M-shape in the height profiles of the AFM images is typical of polymerosomes (for more images see Fig. S5 in the ESI†). The polymersome sizes obtained from the DLS, electron and atomic force microscopy images are matched very well which confirmed the existence of the polymersome nano-assemblies. To further validate the structural engineering of the present polymersome design, a dextran derivative was synthesized only with TPE units (without the PDP unit, see Scheme 1). This dextran-TPE derivative (without PDP substitution) was found to self-assemble as nano-particles rather than polymersomes (see Fig. S6 in the ESI†). In the AFM image, these nanoparticles typically appeared as spherical smooth surfaces (see Fig. S6†). Furthermore, the TPE-conjugated nanoparticle showed relatively low AIE properties compared to TPE + PDP-conjugated polymersomes (see Fig. S6†). Variable temperature DLS studies were done for the TPE−polymersome sample (shown in Fig. S7 in ESI†). The DLS histograms did not show any variation in size and the polymers typically possessed sizes <200 nm which confirmed their high thermal stability. These control experiments confirmed that the PDP conjugation is essential to produce polymersome nano-assemblies and also the polymersomes exhibited better AIE properties than nanoparticles in the dextran backbone. Hereafter, the dextran polymersome is referred to as “PTPE”.
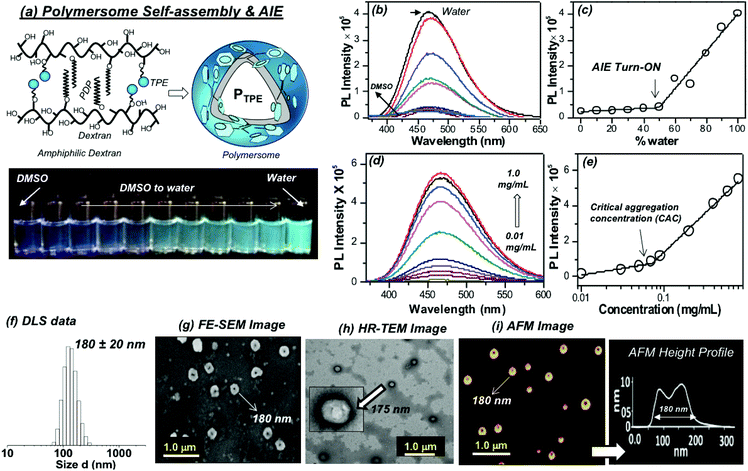 |
| | Fig. 2 (a) Schematics showing the self-assembly of the dextran derivative into polymersomes (PTPE). The vials in the photographs were taken for PTPE polymer solution in various DMSO + water compositions upon photo-excitation. (b) Solvent composition dependent emission spectra of PTPE in different water + DMSO mixtures from 0 to 100%. (c) Plot of PL intensity of PTPE at 460 nm with respect to the percent of water in a water + DMSO solvent mixture. (d) Concentration dependent emission spectra of PTPE in water. (e) Plot of the PL intensity of PTPE at 460 nm with respect to the concentration of the polymer. (f) Dynamic light scattering (DLS) histograms of the polysaccharide polymersome PTPE. (g) Field-emission scanning electron microscopy (FE-SEM) image of PTPE. (h) High-resolution transmission electron microscopy (HR-TEM) image of PTPE. (i) Atomic force microscopy (AFM) image and the height profile of PTPE. The concentration of the polymer was maintained as 0.1 mg mL−1. Emission spectra were recorded using λexc = 340 nm with 2 nm slit widths for excitation and emission. | |
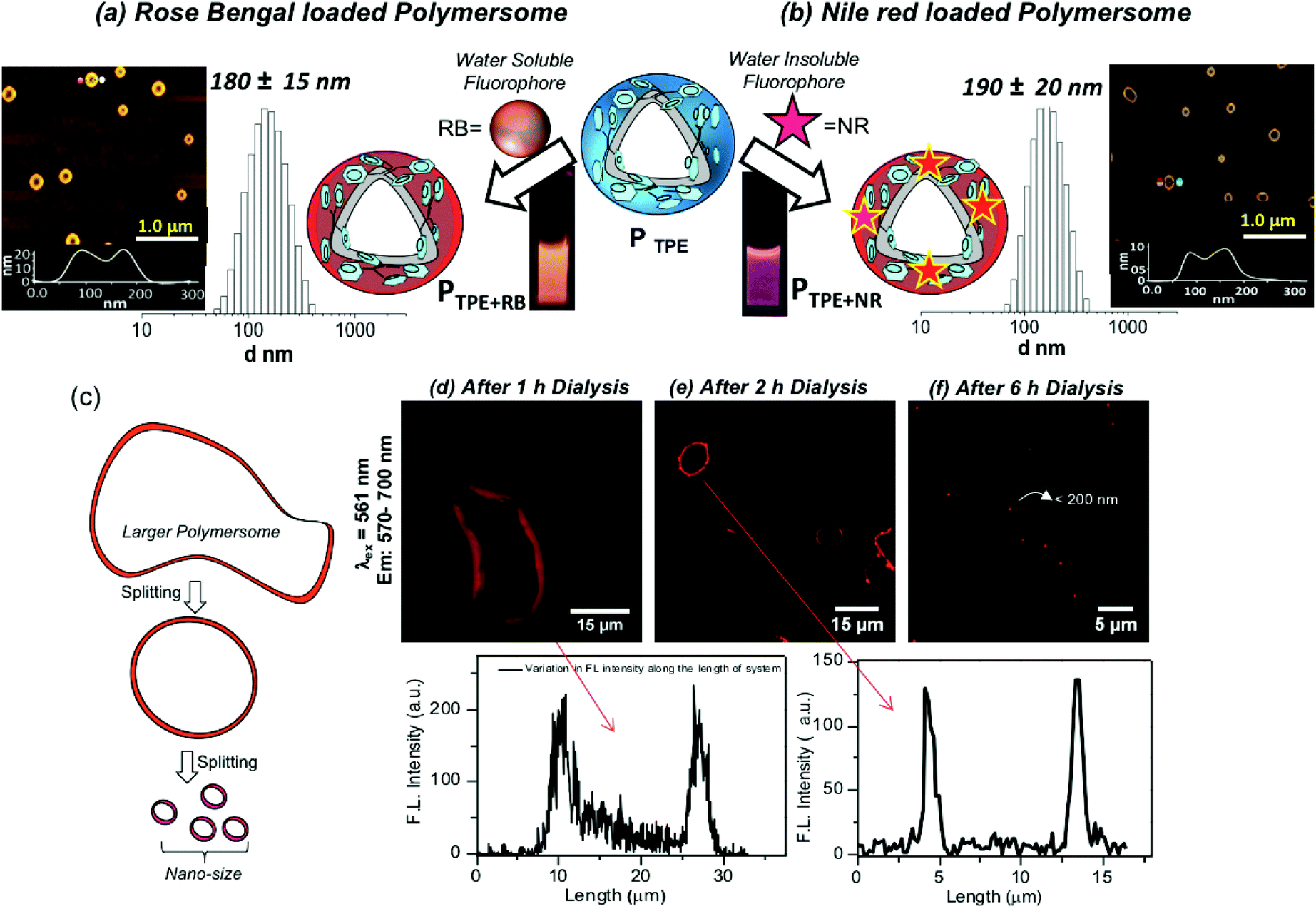
The water soluble fluorophore Rose Bengal (RB) was encapsulated in the PTPE by the dialysis method using a semi-permeable membrane having MWCO = 1000 g mol−1 following our earlier reported procedure.38–44 Both the polymer and RB were taken in a DMSO + water mixture and the solution was dialyzed with continuous replenishing of fresh water in the reservoir. In this process, the non-encapsulated RB molecules will be removed from the reservoir and the dialysis continued for more than 48 until the reservoir is free from the RB dye. This method ensures that only the encapsulated RB in the polymersome stays in the dialysis membrane. A similar method was employed for the NR encapsulation; however, NR being a water insoluble dye, the precipitate corresponding to un-encapsulated dye is filtered and removed. The RB and NR loaded polymersomes are referred to as PTPE+RB and PTPE+NR, respectively. The dye loading content (DLC) and dye loading efficiency (DLE) for these dyes were found to be 1.5% and 65% for PTPE+RB, and 1.8% and 55% for PTPE+NR, respectively. DLS histograms of PTPE+RB and PTPE+NR loaded polymersomes in Fig. 3a and b were found to be 180 ± 20 nm and 190 ± 20, respectively. AFM images of the PTPE+RB and PTPE+NR showed the existence of polymersome geometry with the average size of 185 ± 20 nm. The comparison of the polymersome images in the nascent (Fig. 2i) and dye loaded samples (Fig. 3a and b) suggested that the loading of the fluorophores does not alter the shape and size of the dextran polymersomes.
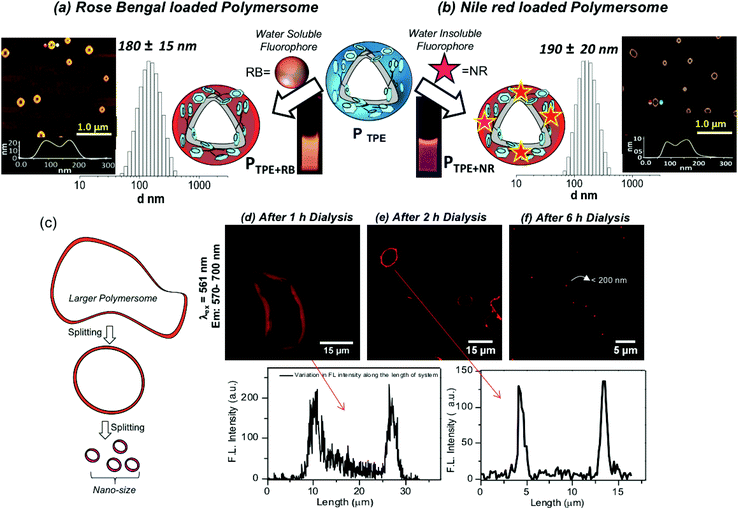 |
| | Fig. 3 (a) Encapsulation of the water soluble rose bengal (RB) fluorophore in the polymersome nano-assemblies. DLS histograms and AFM images are shown for the RB encapsulated sample PTPE+RB. (b) Encapsulation of the water insoluble Nile red (NR) fluorophore in the polymersome nano-assemblies. The DLS histograms and AFM images are shown for the NR encapsulated sample PTPE+NR. (c) Diagram showing the polymersome formation in the dialysis process. (d) Confocal images of the 1 h sample and its corresponding intensity plot. (e) Confocal images of the 2 h aliquot sample and its corresponding intensity plot. (f) Confocal images of the 6 h sample. | |
Polymersomes (or vesicles) are typically produced from the large mother vesicles (or larger polymersomes) produced at the beginning by the folding of the bi-layer membranes. The larger polymersomes subsequently undergo splitting in the solution in order to minimize the surface tension acting on it and the process continues till the end of the formation of the thermodynamically stable smaller polymersomes (see Fig. 3c).48 A experiment was carried out to capture the RB localization in the polymersomes during the dialysis process. Aliquots were taken at different time intervals of 1 h, 2 h and 6 h and drop-cast on a glass substrate for confocal microscopy imaging. The confocal images were recorded by exciting RB using a laser at 561 nm and collecting the signals at 570–700 nm and the images are shown in Fig. 3d–f. The images at 1 h clearly showed the existence of large mother vesicles of 150 micrometers and clear red signals from the vesicles (Fig. 3d). The FL intensity of the polymersomes recorded for this image indicates the presence of RB in the polymersomes. The inner cavity is clearly visible in the polymersome nano-assemblies. It is very difficult to exactly locate the RB molecules in the inner cavity from the hydrophobic wall of the polymersomes. With the increase in the dialysis time (2 h), the giant mother vesicles turned into micron-sized polymersomes as shown in Fig. 3e and the spherical morphology and wall of the polymersomes are clearly evolved. At 6 h dialysis, the size of the polymersome became very tiny (<200 nm) and it is impossible to locate the inner cavity from the wall thickness (due to the size limitation for the analysis of nano-assemblies by confocal microscopy). The above observation is clearly evident for the existence of fluorescent dextran polymersomes and also confirms the RB encapsulation.
Polymersome FRET probes
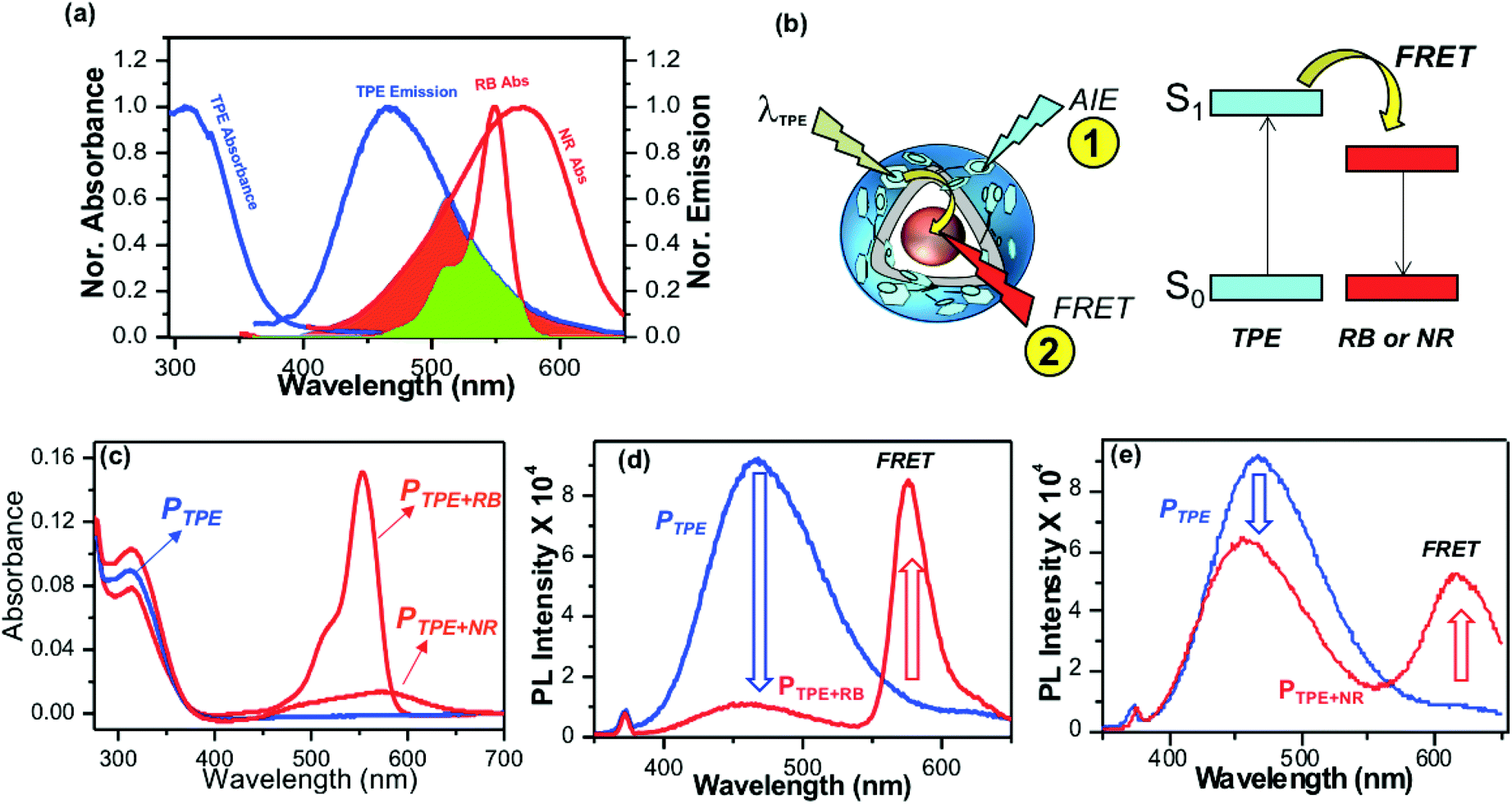
Polymersomes were encapsulated with the water soluble fluorophore dye Rose Bengal (RB) and the water insoluble dye Nile Red (NR) to study the FRET between the TPE donor and encapsulated cargoes as the acceptor. The emission spectrum of the TPE polymersome PTPE was overlaid with the absorption spectrum of RB loaded polymersomes PRB to determine the overlap integral for the FRET process as shown in Fig. 4a (see Fig. S8,† for more details). Absorption spectra of RB and NR chromophores showed 60% and 40% overlap with the emission spectra of PTPE. Fluorescent polymersomes having the encapsulated fluorophore molecules were expected to undergo two different types of photophysical processes as shown in Fig. 4b: (i) self-emission of the TPE chromophore via AIE (process 1), and (ii) fluorescence resonance energy transfer (FRET) from TPE to cargo chromophores and resulting FRET emission from the encapsulated molecules (process 2). A large wavelength difference between the emission spectra of the donor and acceptor is very much useful for the confocal microscopy imaging of FRET probes in biological systems. Upon the photo-excitation of the donor, this would facilitate the probing of the FRET emission from the acceptor without overlapping with the self-emission of the donor. Fig. S8† shows that both RB and NR have more than 150 to 180 nm red-shift in their emission spectra compared to that of the blue-emission from PTPE. Hence, both PTPE+RB and PTPE+NR are very good donor–acceptor pairs to exhibit the FRET process and one could easily trace the occurrence of FRET distinctly in confocal microscopy imaging. To study the FRET process, the optical density (OD) of TPE was maintained as 0.1 in PTPE, PTPE+RB, PTPE+NR as shown in Fig. 4c. Upon photo-excitation (at λex = 340 nm, TPE absorption maxima), PTPE showed strong blue-emission at 460 nm with respect to AIE in the aqueous medium (see Fig. 4d). The selective TPE photo-excitation of the PTPE+RB polymersome showed the disappearance of AIE-fluorescence (>95%) and exhibited significant emission from RB at 590 nm (see Fig. 4d). This strong emission at 590 nm was attributed to the excitation energy transfer from TPE (donor) to RB (acceptor) in PTPE+RB polymersomes. Similarly, the PTPE and PTPE+NR polymersomes were excited at TPE and their spectra are shown in Fig. 4f. The PTPE+NR polymersome showed FRET emission at 620 nm; however, the energy transfer from TPE to NR was not complete. The polymersome retained a significant amount of AIE fluorescence and showed only <20% reduction in its self-emission in PTPE+NR (in Fig. 4f) unlike its RB counterpart PTPE+RB (in Fig. 4d). Since the NR occupied in the layer of the polymersome, one would anticipate better interaction between the TPE and NR chromophores. Interestingly, the RB in the core of the polymersome exhibited better excitation energy harvesting efficiency compared to that of the NR. To further understand this observation, the polymersomes PTPE+RB and PTPE+NR were excited at their acceptor absorbance maxima and their emission spectra were compared with FRET emissions (for TPE excitation) in Fig. S7 in the ESI.† This experiment showed that almost 80% of the total emission possible from the RB chromophore was achieved through the FRET process in PTPE+RB. This further supports the efficient energy transfer process from the TPE donor to the RB acceptor. On the other hand, only less than <50% of the NR emission is observed for the FRET process in PTPE+NR indicating partial energy transfer between TPE and NR chromophores. These experiments suggest that RB is a better acceptor for the TPE chromophore in the polymersome. To elucidate the FRET phenomena in the PTPE+RB and PTPE+NR polymersomes, time-resolved fluorescence decay measurements were carried out by the time-correlated single photon counting (TCSPC) method to determine the Förster distance and FRET efficiencies following the reported procedures.49–52 TCSPC decay profiles for PTPE+RB and PTPE+NR are shown in Fig. S9 in the ESI† (excitation LED source 340 nm). The PTPE polymersome exhibited single exponential decay with a lifetime value as τ = 1.12 ns (see the table in Fig. S9†). The lifetime of the TPE chromophore significantly reduced in the presence of acceptor molecules RB and NR. For example, τ = 0.32 and τ = 0.87 ns were obtained for TPE in the presence of RB and NR in PTPE+RB and PTPE+NR, respectively. The reduction in the τ-value of the TPE is relatively less by the NR chromophore, indicating their weak bimolecular interaction between NR and TPE in the polymersome. On the other hand, RB significantly reduced the lifetime value for the TPE which is direct evidence for the strong interaction between the TPE and RB chromophores in the polymersome. The FRET efficiency was found to be 72% for the PTPE+RB pair and the foster distances (R0 = 15.67 Å) and the distance between two chromophores (R = 13.41 Å) which are below the <50 Å Förster distance required for FRET process. Based on the above analysis, it may be concluded that PTPE+RB is an excellent FRET probe with excellent photo-excitation energy transfer efficiency in aqueous medium; thus, this probe is chosen for further studies.
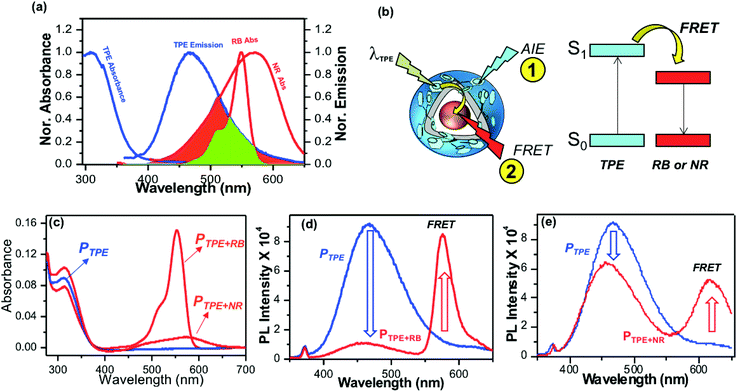 |
| | Fig. 4 (a) FRET overlap between PTPE and RB and NR fluorophores. The emission spectrum of AIE−polymersome PTPE is merged with the absorbance spectra of RB and NR. (b) Schematic representation for the occurrence of FRET between the TPE donor and RB acceptor. (c) Absorbance spectra of PTPE, PTPE+RB, PTPE+NR in aqueous medium. The OD of the TPE chromophore was maintained as 0.1. (d) Emission spectra of PTPE and PTPE+RB recorded at TPE excitation λexc = 340 nm. (e) Emission spectra of PTPE and PTPE+NR recorded at TPE excitation λexc = 340 nm. | |
Enzyme-trigger, cytotoxicity and cellular uptake
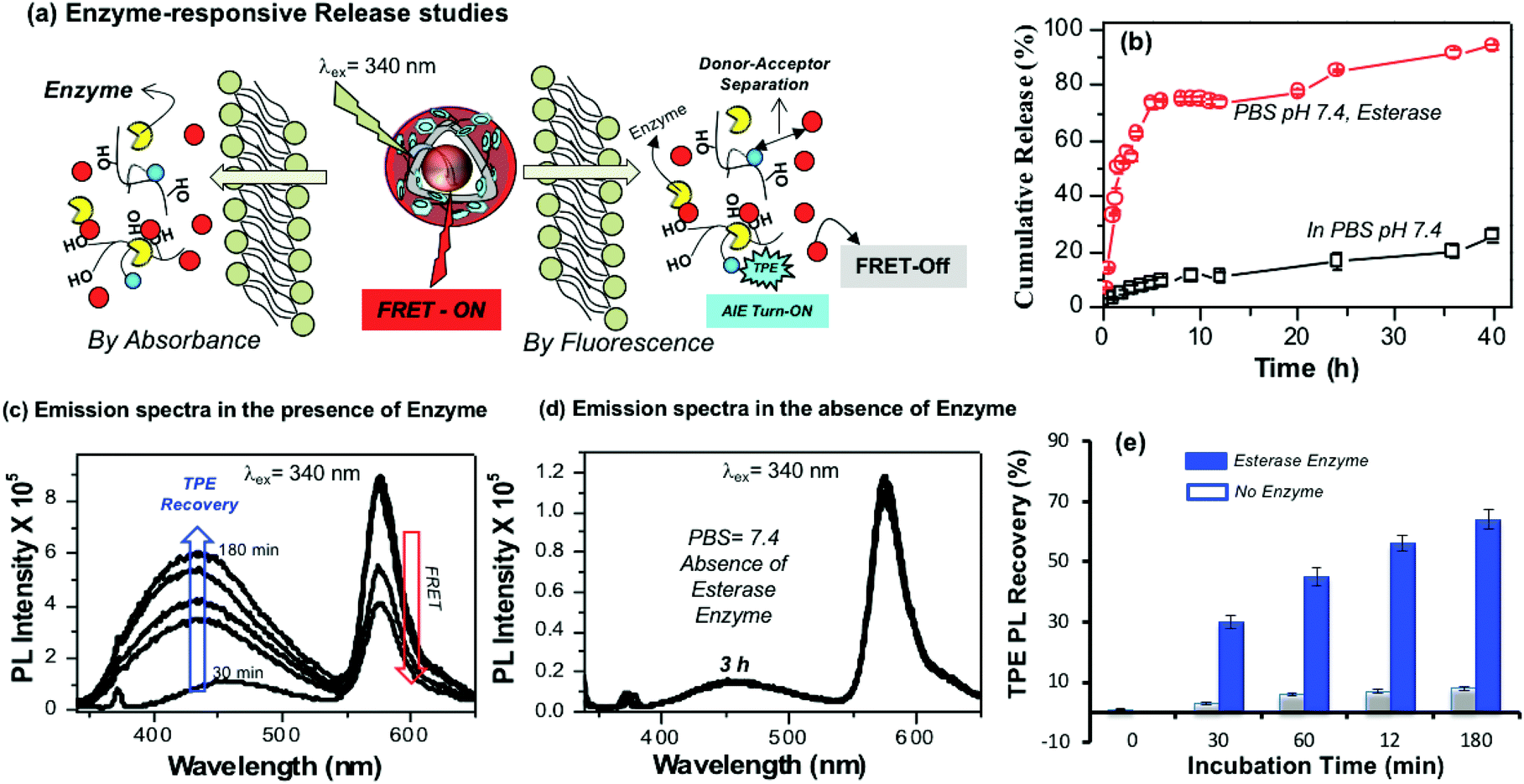
The aliphatic ester linkages that connect the PDP unit on the dextran backbone were susceptible to biodegradation by lysosomal enzymes at the intracellular level.38–40,53 The above photophysical studies (in Fig. 3 and 4) revealed that the tailor-made fluorescent polysaccharide polymersome is unique to exhibit FRET with RB. Therefore, enzyme-trigger could be employed to disassemble the polymersome via biodegradation of the aliphatic ester linkages that connect the hydrophobic PDP in the hydrophilic dextran backbone as shown in Fig. 5a. The dialysis method was employed to study the RB release kinetics from PTPE+RB in the presence of horse liver esterase enzyme in PBS buffer at 37 °C. A control experiment was also done without the enzyme in PBS buffer at 37 °C. The RB release kinetics in PTPE+RB could be studied by both absorbance and fluorescence techniques. The cumulative drug release determined by absorbance spectroscopy was plotted against the incubation time and is shown in Fig. 5b. It was observed that PTPE+RB polymersomes in PBS at pH 7.4 exhibited slight leaching (<15%) due to the influence of ions present in PBS. In the presence of enzyme, the polymersomes underwent enzymatic-disassembly and released >95% of the RB. These studies confirmed that the RB guest molecules could be readily released by enzyme-triggers from the polymersome host. A new model compound was synthesised by conjugating PDP-acid with D-glucose by a multi-step organic route and the details are given in Scheme S1 in the ESI.† The PDP acid has reactivity to couple in the D-glucose-acetal (compound 5)54 both at the 2 and 3-positions; however, these products are very close in polarity and the purification was very tedious. We were fortunate enough to isolate the 3-substituted ester (compound 6) in good quantity (35%) and in high purity. We could also isolate the 2,3-bis-subtituted derivative (compound 7) in a small amount. It was very difficult to isolate the 2-subsituted ester since it was mixed with other products during column chromatography. NMR structural analyses of these two compounds are shown in Fig. S10–S13 in the ESI.† Furthermore, compound 6 was de-protected to give 3-sustituted D-glucose-ester (compound 8) which was subjected to esterase enzyme-responsive studies following the literature procedure55 in 10% acetone in PBS buffer. In the absence of the enzyme (acetone + PBS), compound 8 did not show any change for 24 h. On the other hand compound 8 underwent enzymatic degradation in the presence of esterase enzyme within 12 h of incubation. NMR analysis in Fig. S14† showed that the 3(C–H) proton in the ester vanished completely after 24 h of incubation in the presence of enzymes. This experiment confirmed that the aliphatic ester linkage employed in the polymersome is enzyme-responsive for drug delivery application.
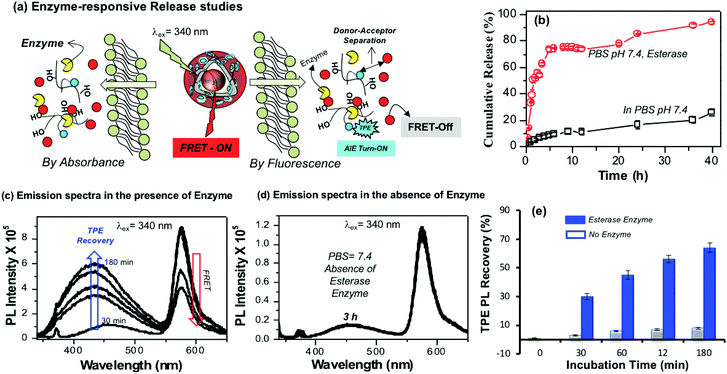 |
| | Fig. 5 (a) Enzyme-trigger disassembly of polymersomes monitored by absorbance (left) and FRET emission by fluorescence spectroscopy (right). (b) In vitro enzyme-responsive delivery of RB from the polymersomes in the presence of horse-liver esterase enzyme and in the absence of enzyme in PBS at 37 °C. (c) Emission spectra of TPE fluorescence recovery in PTPE+RB polymersome in the presence of enzyme in PBS pH 7.4. (d) Emission spectra PTPE+RB polymersome in the absence of enzyme in PBS pH 7.4. (e) Bar diagram showing the emission intensity recovery of TPE fluorophores in the presence or absence of enzymes at various incubation times. For all the studies the PTPE+RB polymersome concentration was maintained as 0.1 mg mL−1, slit width = 2 nm and λexc = 340 nm. | |
Enzyme-trigger experiments were also monitored to study the FRET process in PTPE+RB as shown in Fig. 5a. At the intracellular level, the enzymatic-disassembly results in the separation of the TPE donor and RB acceptor and increases the distance between these two chromophores as shown in Fig. 5a. This separation would directly influence the functioning of the FRET process and the polymersome PTPE+RB is expected to become FRET Turn-Off → Turn-On. The polymersomes were subjected to esterase treatment and the change in emission intensity was monitored over a period of time (up to 180 minutes). The influence of FRET emission upon exposure to esterase enzyme in PTPE+RB is shown Fig. 5c. Two important observations are very clear from the spectra: (i) the AIE from the TPE (self-emission) was re-gained in 3 h, and (ii) the FRET emission from the RB gradually decreased with the increase in the incubation time. In the absence of the enzyme, the PTPE+RB polymersomes exhibited excellent stability in PBS at pH 7.4 and there are no changes in the RB FRET emission (see Fig. 5d). This suggested that the polymersome, PTPE+RB FRET probe became Turn-On → Turn-Off exclusively by the enzymatic-disassembly process (Probe-1). The extent of TPE emission recovery was plotted against the incubation time and is shown in Fig. 5e. There is no change in the TPE emission recovery in the absence of the enzyme; however, the significant increase in the TPE emission could be noticed in presence of enzymes (Probe-2). Therefore, the present polymersome provides two fluorescence signals as the output to monitor the real-time enzyme-trigger disassembly of the polymersomes: the disappearance of the red-fluorescent signals from the RB FRET emission as a measure of extracellular turn-On → intracellular turn-Off FRET probe PTPE+RB and the appearance of a blue-fluorescent signal from the TPE as a measure of extracellular turn-Off → intracellular turn-On AIE probe PTPE+RB. It is important to mention that the present investigation is one of the first examples to report the construction of the FRET probes between TPE and RB chromophores (for nay types of nano-systems); thus, it has significant importance for biomedical application.
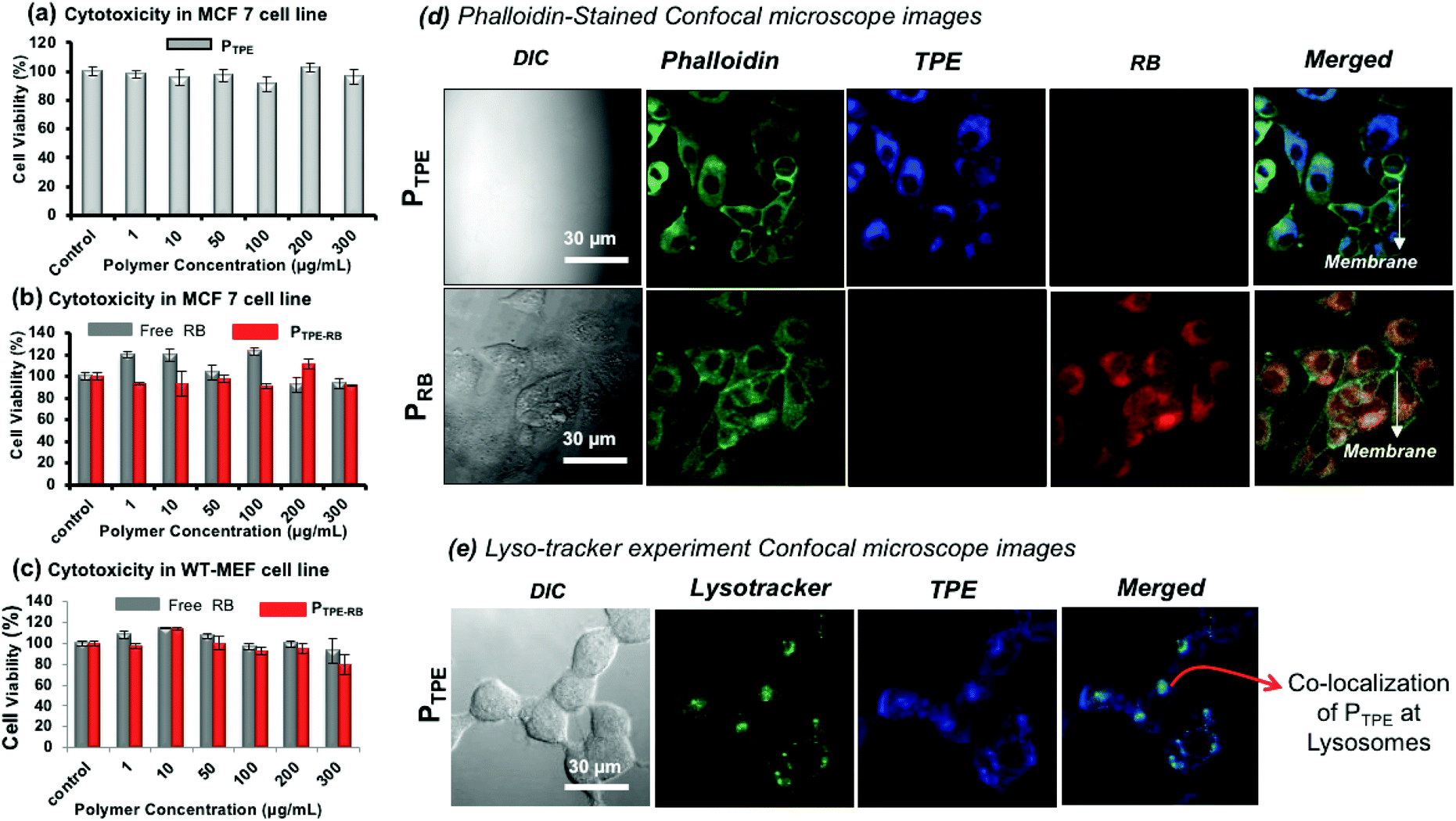
MTT assay was employed to study the cytotoxicity of the polymersomes (PTPE) in human breast cancer cells (MCF 7) and wild type mouse embryonic fibroblast (WT-MEF) cell lines. The polymer was highly biocompatible with both cancer and normal cells up to a concentration of 300 μg mL−1 (Fig. 6a) and it was also non-toxic to WT-MEF cells up to 80 μg mL−1 (see Fig. S15 in the ESI†). PTPE+RB also showed excellent biocompatibility in MCF 7and WT-MEF cells (Fig. 6b and c). The cells were viable up to 300 μg mL−1 concentration of RB, ensuring the compatibility of this system for bio-imaging applications in both cancer and healthy cell lines. To visualize the intracellular distribution of polymersomes, the MCF 7 cells were stained with phalloidin-488 which binds to actin to mark the cytoskeletal network of the cells and these images are shown in Fig. 6d. The cell morphology could be easily visualized using a 488 nm excitation laser source. The cells appeared healthy as is evident from a uniformly stained cytoskeleton and distinguishably illuminated plasma membrane (as shown in the phalloidin panel for both PTPE and PRB). Furthermore, the cells treated with PTPE alone showed a bright blue fluorescence emerging solely from the cytoplasm when excited with a 405 nm laser. The nucleus appeared visibly black amidst a well-lit cytoplasm, which strengthened our belief that nanoprobes reside in the cytoplasm without any nuclear breach (as shown in the TPE panel of PTPE). Similarly, cells treated with polymersomes loaded with rose bengal (PRB) gave intense red fluorescence emission coming only from the cytoplasm when excited using a 561 nm laser (as shown in the RB panel of PRB). Furthermore, the merged images revealed two important points: (i) no nanocarrier absorption upon the plasma membrane of the cells as the plasma membrane appeared prominently green fluorescent, and (ii) there was significant polymersome localization inside the cytoplasm as is evident from the regions of cyan blue fluorescence (corresponding to the overlap of PTPE with phalloidin) and the regions of yellow fluorescence (corresponding to the overlap of PRB with phalloidin). To understand the cellular uptake via the lysosomal pathway, PTPE (2.0 μM) was incubated in breast cancer cell lines (MCF 7) for 4 hours at 37 °C and subjected to confocal assisted live cell imaging (see Fig. 6e). The cells were stained with lysotracker green DND 26 which could selectively mark the acidic lysosomes of the cell. The TPE and lysotracker green were excited with 405 and 488 nm lasers, respectively, and were imaged in the blue channel (420–600 nm) and green channel (510–600 nm), respectively. The colocalization of the dark green color from lysotracker, and the blue color from TPE yielded the cyan color as shown in Fig. 6e. This validates that the polymersomes are getting degraded in the lysosomal compartment of the cells to release the loaded cargoes.
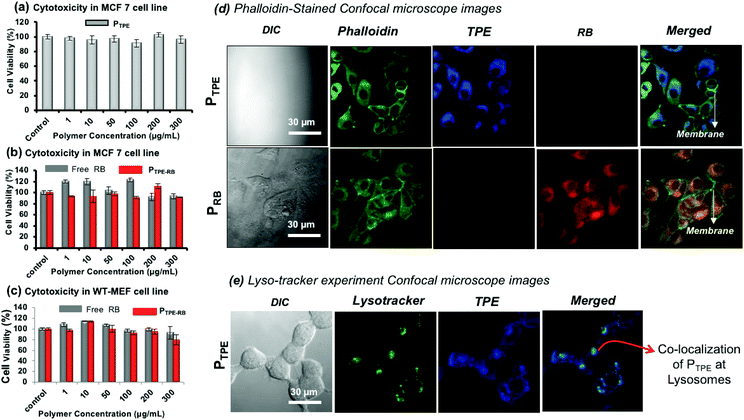 |
| | Fig. 6 (a) In vitro cytotoxicity data of PTPE in the MCF 7 breast cancer cell line. (b) In vitro cytotoxicity data of PTPE+RB and free RB in the MCF 7 breast cancer cell line. (c) In vitro cytotoxicity data of PTPE+RB and free RB in the WT-MEF cell line. (d). Fixed-cell confocal microscopy imaging of PTPE and PRB in the MCF 7 breast cancer cell line. The cells were stained with phallaoidin-488 to track the intracellular localization of the polymersomes. (e) Live-cell confocal microscopy imaging of PTPE+RB in the MCF 7 breast cancer cell line. The lysosomes were stained with Lysotracker green DND-26. The emission from TPE was observed in the blue channel and the emission from RB was observed in the red channel, and the merged image shown is the overlap of the three channels as mentioned. For lysotracker experiment, the cells were incubated for 3 hours. | |
Polymersome FRET probe at the cellular level
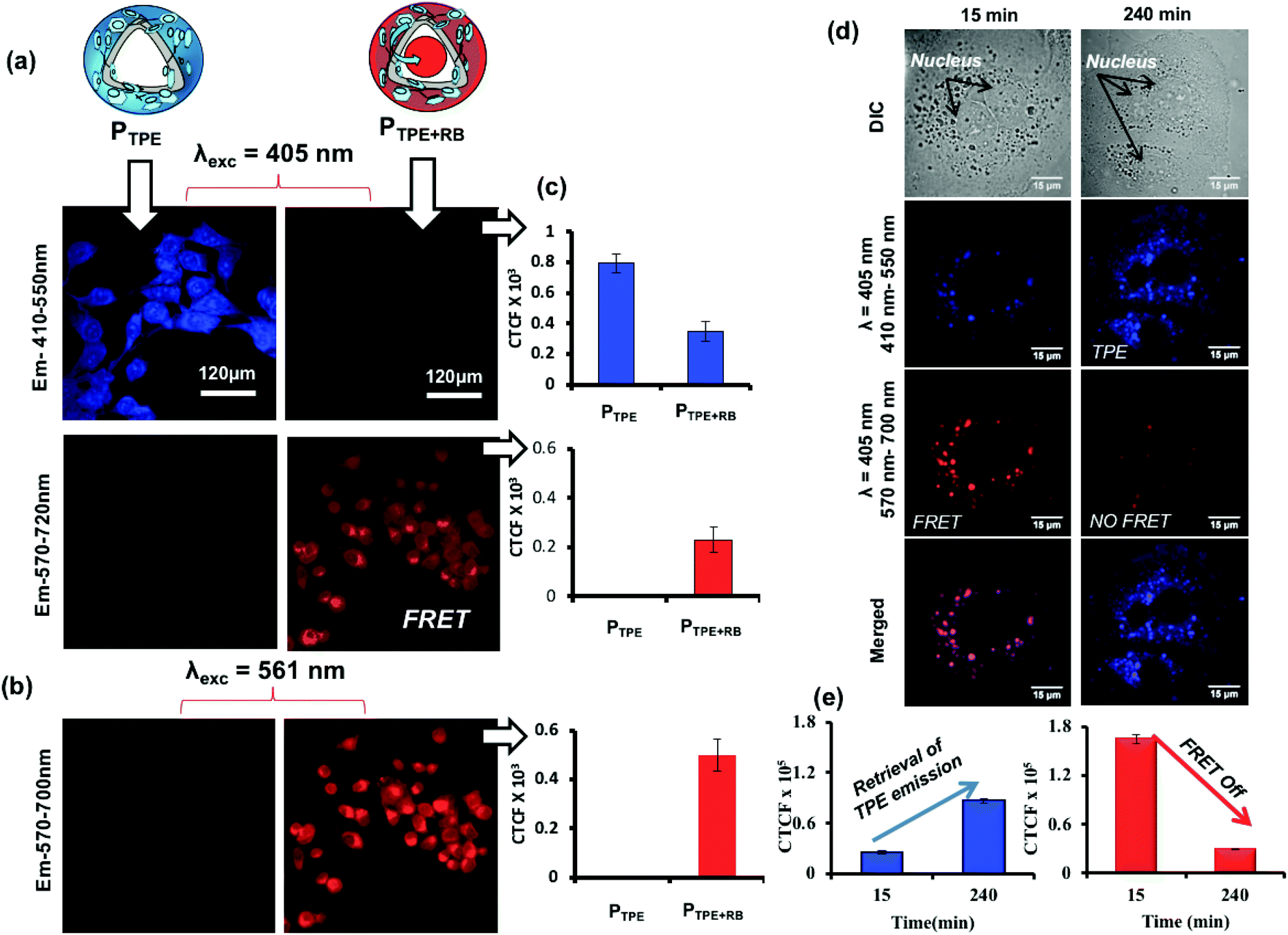
To study the polymersome FRET probe action, both breast cancer MCF 7 cell lines and WT-MEF cell lines were chosen and the FRET process was investigated both in fixed and live-cell imaging protocols. The fixed cell confocal imaging technique was first used to assess the FRET phenomenon in PTPE+RB polymersomes. MCF 7 cells were treated with PTPE+RB and PTPE and the images are shown in Fig. 7. Imaging was done on a confocal assisted microscope using 405 nm and 561 nm lasers as excitation sources. Two exclusive and decently separated collection channels were created to collect the emission intensity coming from both TPE and RB in the blue channel (410–550 nm, see Fig. 7a) and the red channel (570–700 nm, see Fig. 7b), respectively. Firstly, to establish the FRET mechanism, the TPE chromophore in PTPE was excited using a 405 nm laser and emission was collected in both red and blue channels as described above. An intense blue emission was seen coming from the TPE chromophore exclusively in the blue channel (Fig. 7a). Corrected total cell fluorescence (CTCF) values were estimated and plotted as shown in Fig. 7c. The quantitative description of this observation can be made in the adjacent bar diagram showing 0.8 × 103 CTCF in the blue channel vs. an almost negligible intensity in the red channel (see parallel bar diagrams in Fig. 7c). A realization of FRET between TPE and RB can be made with the PTPE+RB system. The TPE chromophore in PTPE+RB on excitation with a 405 nm laser showed a decreased fluorescence intensity in the blue channel (of 0.3 × 103 CTCF as opposed to 0.8 × 103 CTCF in PTPE) with a concomitant increase of fluorescence intensity in the red channel (0.3 × 103 CTCF). This fluorescence intensity in the red channel can be attributed to FRET emission which was completely absent in the control PTPE. The efficiency of FRET can also be calculated by selectively exciting RB in PTPE+RB using a 561 nm laser and collecting its self-emission in the red channel (see Fig. 7b) and quantified using CTCF in the adjacent bar diagram (see Fig. 7c). A 0.5 × 103 CTCF as self-emission vs. 0.3 × 103 CTCF of FRET emission can be seen in the red channel accounting for roughly 50% FRET efficiency. Time-dependent FRET was studied on PTPE+RB in fixed MCF 7 cell lines at two far separated time points. MCF 7 cells were treated with PTPE+RB having 5 μM concentration of rose bengal and incubated for two different time periods of 15 minutes and 240 minutes. At definite time points, the cells were fixed and imaged using confocal microscopy. The cellular images are shown in Fig. 7d. The first panel in the figure represents the cellular uptake for 15 minutes of incubation time where strong red emission was observed when excited with a TPE excitation source (405 nm) which can be attributed to FRET between TPE and RB. The merged image shows a magenta colour due to the co-localization of TPE and RB. The second panel shows the cellular uptake at 4 hours of incubation time wherein a strong blue emission was observed when excited at the TPE excitation (405 nm) but a faint emission was observed in the red channel suggesting disassembly, leading to the disappearance of FRET. CTCF plots further corroborate with this observation in Fig. 7e. There is a significant increase in the TPE emission from 15 minutes to 4 hours with a concomitant decrease in the emission of rose bengal when excited at TPE (405 nm). The FRET stays operational in a time window of 15 minutes to 4 h and then decays gradually in response to cellular enzymes.
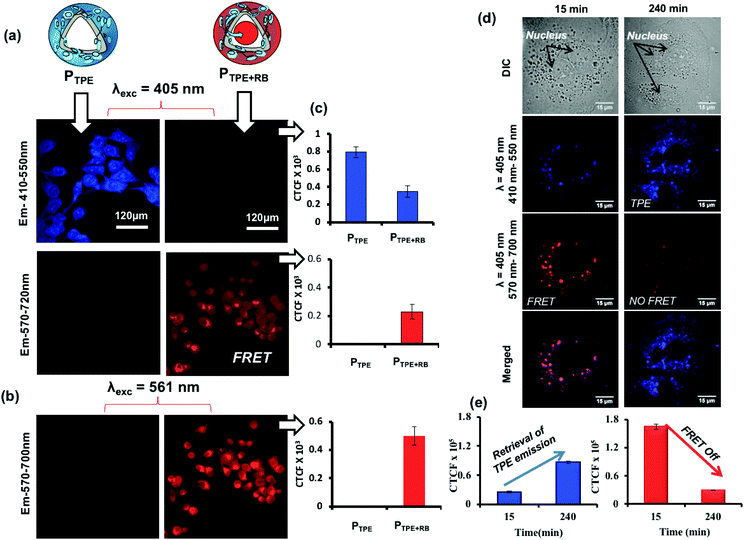 |
| | Fig. 7 (a) Confocal microscopy images for PTPE and PTPE+RB showing self and FRET emission when excited at 405 nm in the MCF 7 cell line. (b) Images for PTPE+RB showing self-emission when excited at 561 nm. (c) CTCF intensities for self and FRET emission of PTPE and PTPE+RB. (d) Time-dependent confocal microscopy images of PTPE+RB polymersomes at various incubation times in the MCF 7 breast cancer cell line. (e) Bar diagram showing the CTCF for the emission recovery of TPE and RB at various incubation times. | |
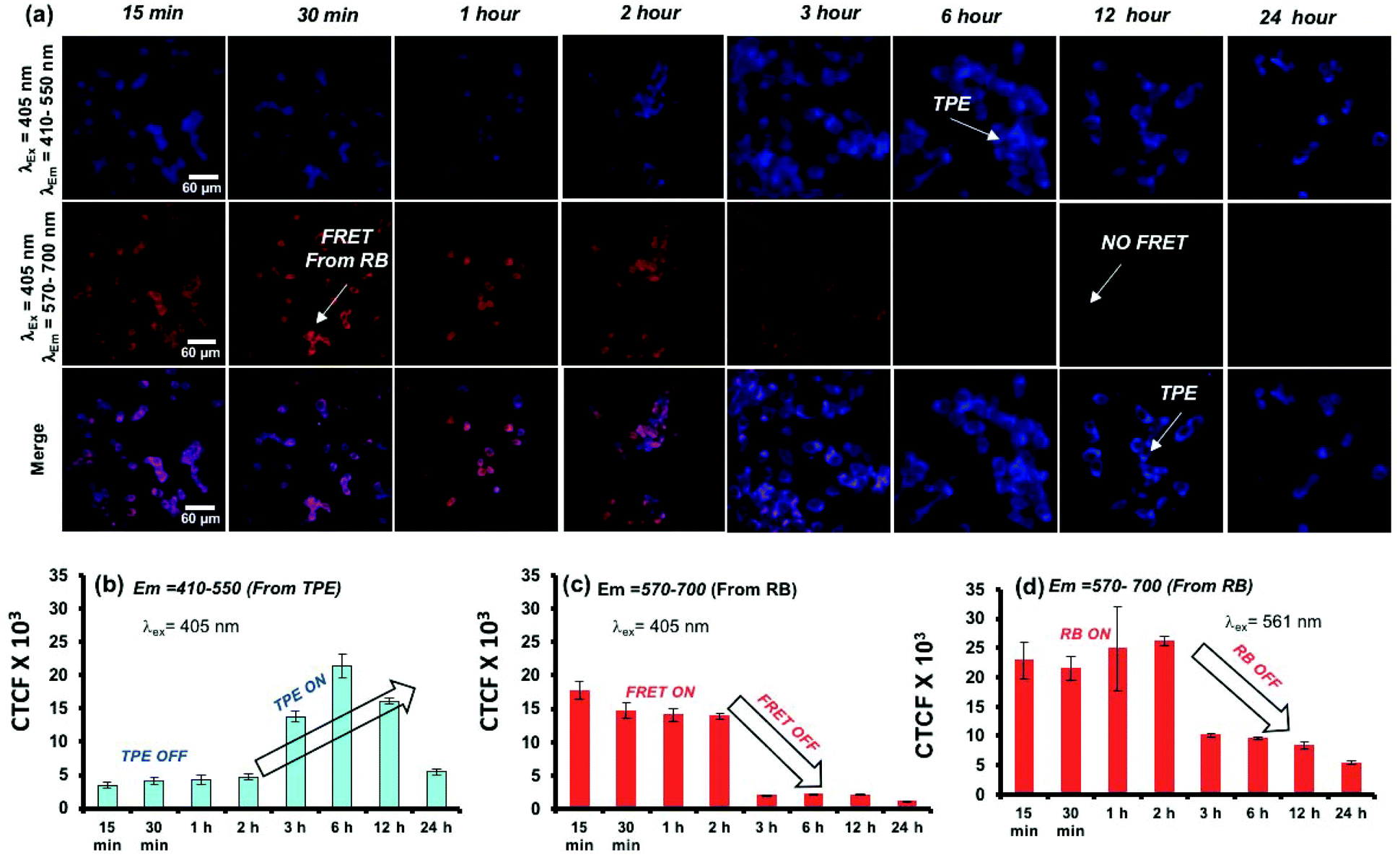
A live-cell confocal microscope study is employed for WT-MEF cell lines to directly visualize and quantify the intracellular release kinetics of the polymersome nano-assemblies. WT-MEF cells were treated with polymersomes by varying the incubation time in a four well live cell chamber and were imaged using a confocal instrument equipped with a stage incubator which was maintained at 37 °C and 5% CO2 throughout the experiment. The live cell imaging set-up, equipped with an incubator, allowed us to capture images continuously from 1 min to 45 min using a single well of a 4 well live cell chamber; however, for a longer incubation time (1 h and above), PTPE+RB was pre-incubated in live-cell chambers prior to imaging. Live cell images recorded at various time intervals are shown in Fig. 8a. The TPE chromophore was excited with a 405 nm laser and the images were captured in the range of 410–550 nm and 570–700 nm for the TPE and RB emission, respectively. The first and second rows showed TPE and RB emission and the merged image in the last row depicts the co-localization of both TPE and RB chromophores together at the intracellular level. At the intracellular level, the enzymes in the lysosomal compartment are expected to induce FRET Turn-On → Turn-Off properties in in vitro (as evident from the studies in Fig. 5). There are two observations drawn from the confocal images: (i) the TPE emission was initially low up to 2 h and it increases with the increase in the incubation time. The initial decrease of TPE emission is attributed to the partial transfer of the excitation energy form the TPE to RB for the FRET process. (ii) The FRET emission intensity from RB was stable up to 2 h; however, it decreased drastically with the increase in the incubation time. This observation can be attributed to the stable FRET process between the two chromophores for the initial 2 hours at the intracellular level. A decrease in FRET emission or an increase in TPE emission after 2 h signifies the enzymatic-disassembly of the polymersomes at the intracellular level. After 2 h, the merged images did not show red-FRET emission from RB and only blue-colour self-emission from TPE was observed. This trend is attributed to the disassembly of the polymersome at the cellular level and separation of RB from TPE. Due to this separation, TPE could not transfer the excitation energy to RB and the FRET signals are turned-Off (please note that λex is 340 nm for TPE). In Fig. 8b, the CTCF of TPE self-emission increased almost 3–4 fold with the increase in time up to 12 h, whereas the excretion of the polymer chains accounts for the decrease in TPE amount at longer incubation (24 h). The dextran-TPE polymer self-assembled as 180–200 nm size polymersomes and therefore they cannot be readily excreted by p-glycoprotien pumps (5 nm size pores) that are inherent to cells to clear the foreign bodies from the cytosol. This is the basic mechanism and foundation for the localization of nano-drugs at the cellular compartments. Therefore the internalization of the polymersomes retained its original self-assembled form until they were disassembled by the enzymes in the lysosoal compartments into individual chains. The disassembled polymer chains having TPE units no longer have the ability to withhold in the cytosol and are typically known to excrete from the cytosol. A similar observation was reported by us recently in the FRET probes and DOX release kinetics of the biodegradable L-aspartic acid based polyester nano-assemblies.52,53 Therefore, the decrease in the TPE emission with increasing incubation time is attributed to the above-mentioned enzymatic-disassembly of polymersomes followed by the clearance of the individual chains by the cell machinery. In Fig. 8c, the FRET emission from the RB fluorophore was not disturbed (2 h) until the disassembly of the polymersome occurred. Independently, the RB chromophore in PTPE+RB was also excited at 561 nm with a laser and RB self-emission was collected from 570–700 nm (see Fig. S16 in the ESI†). The CTCF data from this experiment for the self-emission of the RB fluorophore are shown in Fig. 8d. The decrease in the RB emission after 2 h is further supporting the enzymatic-disassembly. These live-cell experiments provided evidence for the enzymatic-disassembly of the polymersomes and their ability to function as useful Turn-On or Turn-Off fluorescent probes. The robustness of the custom designed polymersomes has been tested in three different conditions: (i) selective photo-excitation in cuvettes in PBS at pH = 7.4 in a spectrophotometer, (ii) selective photo-excitation in live-cell confocal microscopy imaging, and (iii) the selective photo-excitation in fixed-cell confocal microscope imaging. All three experiments provided evidence for the enzymatic-disassembly of the polymersomes and their ability to function as useful FRET probes. Finally, the scope of this polysaccharide polymersome design is not restricted only to the few fluorophore combinations; in general, it can be expanded to many other anticancer drugs or fluorescent molecules which will be the focus of the future research.
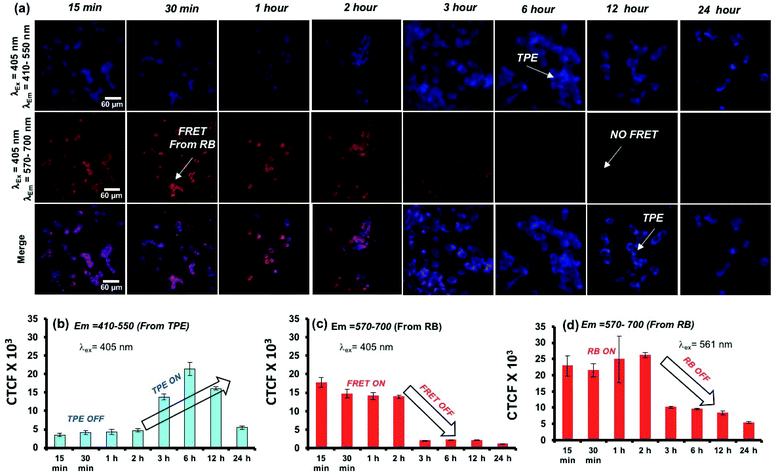 |
| | Fig. 8 (a) Live-cell confocal microscopy images for PTPE+RB polymersomes at various incubation times in the WT-MEF cell line. (b) Bar diagram showing the CTCF for the emission recovery of TPE at various incubation times followed by the selective photo-excitation of TPE using a 405 nm laser. (c) Bar diagram showing the CTCF for the RB FRET emission at various incubation times followed by the selective photo-excitation of TPE using a 405 nm laser. (d) Bar diagram showing the CTCF for the emission of RB at various incubation times followed by the selective photo-excitation of RB using a 561 nm laser. The images were recorded using the RB concentration at 5.0 μM in PTPE+RB in 25000 cells. | |
Conclusions
In summary, the present investigation reports novel TPE tagged dextran based polymersomes which exhibited a remarkable and distinct photophysical phenomenon such as AIE and FRET depending upon the nascent stage or the guest molecule it encapsulated. Fluorescent dextran based polymersomes were custom designed by substituting 3-pentadecylphenol (a vesicular director) and an AIE chromophore TPE to the sugar backbone. The system self-assembled in water to form polymersomes which was confirmed via DLS, electron microscopy, and atomic force microscopy techniques. These fluorescent polymersomes showed excellent encapsulation efficiencies for both water soluble and insoluble dyes. These fluorescent polymersomes could function as bio-imaging agents with both water soluble (rose bengal) and water insoluble (Nile red) dyes. The polymersome could stabilize these dyes in the hydrophilic core and hydrophobic layer respectively and showed enzyme responsive energy transfer between host (TPE tagged polymersomes) and guest molecules (RB). Rose bengal showed an efficient energy transfer phenomenon both in in vitro and cellular experiments. A time dependent decrease in FRET was observed upon enzymatic action under cellular conditions using live cell imaging. These systems are self-reporter in nature and reveal important information regarding the structural integrity of the nano-carriers. A positive readout of FRET describes the intactness of the nano-carriers, whereas a decrease in FRET readout indicates disassembly at the cellular level and release of payload. Using this phenomenon host–guest chemistry, important cellular events were monitored and demonstrated in various cell lines. Such systems hold tremendous potential in the design of futuristic nano medicine-cum-bioimaging agents.
Experimental
The materials used and methods employed for the analytical characterization, and the synthesis of TPE-acid, PDP-acid, TPE-PDP-substituted dextran, TPE-substituted dextran, PDP-substituted D-glucose model compound and other intermediate molecules are given in the ESI.† The details of the fluorophore encapsulation, characterization of polymersomes, release kinetics, and biological experimental procedures are also given in the ESI.† Fig. S17–S22† correspond to the NMR and mass data for the compounds and intermediates.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the research grant from the Science and Engineering Research Board (SERB) (project code CRG/2019/000496), New Delhi, INDIA; and the Indian Council for Medical Research, project number 35/03/2019-NANO/BMS. We thank IISER Pune Microscopy Facility for cellular imaging. We would also like to thank Dr Asha S. K. CSIR-NCL Pune for providing access to TEM and fluorescence lifetime measurements.
Notes and references
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- H. S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699–4718 RSC.
- X. Xu, R. Liu and L. Li, Chem. Commun., 2015, 51, 16733–16749 RSC.
- M. P. Robin, S. A. M. Osborne, Z. Pikramenou, J. F. Raymond and R. K. O'Reilly, Macromolecules, 2016, 49, 653–662 CrossRef CAS.
- S. M. Parke, S. Tanaka, H. Yu, E. Hupf, M. J. Ferguson, Y. Zhou, K. Naka and E. Rivard, Macromolecules, 2019, 52, 7477–7488 CrossRef CAS.
- Y. Huang, F. Qiu, D. Chen, L. Shen, S. Xu, D. Guo, Y. Su, D. Yan and X. Zhu, Small, 2017, 13, 1604062–1604072 CrossRef.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS.
- G. Stephenson, R. M. Parker, Y. Lan, Z. Yu, O. A. Scherman and C. Abell, Chem. Commun., 2014, 50, 7048–7051 RSC.
- T. Thambi, J. H. Park and D. S. Lee, Biomater. Sci., 2016, 4, 55–69 RSC.
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049 CrossRef CAS.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- I. Dewald and A. Fery, Adv. Mater. Interfaces, 2016, 4, 1600317 CrossRef.
- P. V. Pawar, S. V. Gohil, J. P. Jain and N. Kumar, Polym. Chem., 2013, 4, 3160–3170 RSC.
- A. Feng and J. Yuan, Macromol. Rapid Commun., 2014, 35, 767–779 CrossRef CAS.
- S. Iqbal, M. Blenner, A. Alexander-Bryant and J. Larsen, Biomacromolecules, 2020, 21, 1327–1350 CrossRef CAS.
- F. Meng, Z. Zhong and F. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
- X. Hu, Y. Zhang, Z. Xie, X. Jing, A. Belloti and Z. Gu, Biomacromolecules, 2009, 10, 197–209 CrossRef.
- X. Yi, D. Zhao, Q. Zhang, J. Xu, G. Yuan, R. Zhuo and F. Li, Polym. Chem., 2016, 7, 5966–5977 RSC.
- M. Anas, S. Jana and T. K. Mandal, Polym. Chem., 2020, 11, 2889–2903 RSC.
- J. Shi, Y. Li, Q. Li and Z. Li, ACS Appl. Mater. Interfaces, 2018, 10, 12278–12294 CrossRef CAS.
- J. Mei, I. Nelson, C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- X. Tao, H. Chen, S. Trepout, J. Cen, J. Ling and M.-H. Li, Chem. Commun., 2019, 55, 13530–13533 RSC.
- D. Zhang, Y. Fan, H. Chen, S. Trepout and M.-H. Li, Angew. Chem., Int. Ed., 2019, 58, 10260–10265 CrossRef CAS.
- X. Wang, Y. Yang, Y. Zuo, F. Yang, H. Shen and D. Wu, Chem. Commun., 2016, 52, 5320–5323 RSC.
- N. Zhang, H. Chen, Y. Fan, L. Zhou, S. Trepout, J. Guo and M.-H. Li, ACS Nano, 2018, 12, 4025–4035 CrossRef CAS.
- S. Mizrahy and D. Peer, Chem. Soc. Rev., 2012, 41, 2623–2640 RSC.
- Y. Wen and J. W. Oh, Macromol. Rapid Commun., 2014, 35, 1819–1832 CAS.
- Q. Liu, B. Duan, X. Xu and L. Zhang, J. Mater. Chem. B, 2017, 5, 5690–5701 RSC.
- A. Basu, K. R. Kunduru, E. Abtew and A. J. Domb, Bioconjugate Chem., 2015, 26, 1396–1412 CrossRef CAS.
- Y.-Z. Du, Q. Weng, H. Yuan and F.-Q. Hu, ACS Nano, 2010, 4, 6894–6902 CrossRef CAS.
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS.
- Y.-L. Li, L. Zhu, Z. Liu, R. Cheng, F. Meng, J.-H. Cui, S.-H. Ji and Z. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914–9918 CrossRef CAS.
- C. Schatz, S. Louguet, J.-F. Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS.
- W. Wang, S. Liu, Y. Huang, X. Jing and Z. Xie, J. Mater. Chem. B, 2015, 3, 5753–5759 RSC.
- C. Voirin, S. Caillol, N. V. Sadavarte, B. V. Tawade, B. Boutevin and P. P. Wadgaonkar, Polym. Chem., 2014, 5, 3142–3162 RSC.
- V. S. Balachandran, S. R. Jadhav, P. K. Vemula and G. John, Chem. Soc. Rev., 2013, 42, 427–438 RSC.
- P. S. Pramod, K. Takamura, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Biomacromolecules, 2012, 13, 3627–3640 CrossRef CAS.
- P. S. Pramod, R. Shah, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Nanoscale, 2014, 6, 11841–11855 RSC.
- P. S. Pramod, N. U. Deshpande and M. Jayakannan, J. Phys. Chem. B, 2015, 119, 10511–10523 CrossRef CAS.
- P. S. Pramod, R. Shah and M. Jayakannan, Nanoscale, 2015, 7, 6636–6643 RSC.
- N. U. Deshpande and M. Jayakannan, Biomacromolecules, 2017, 18, 113–126 CrossRef CAS.
- N. U. Deshpande and M. Jayakannan, Biomacromolecules, 2018, 19, 3572–3585 CrossRef.
- S. Inchanalkar, N. U. Deshpande, V. Kasherwal, M. Jayakannan and N. Balasubramanian, Mol. Pharmaceutics, 2018, 15, 3046–3059 CrossRef CAS.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4569 CrossRef CAS.
- L. L. Lock, Z. Tang, D. Keith, C. Reyes and H. Cui, ACS Macro Lett., 2015, 4, 552–555 CrossRef CAS.
- S. Acikgoz, G. Aktas, M. N. Inci, H. Altin and A. Sanyal, J. Phys. Chem. B, 2010, 114, 10954–10960 CrossRef CAS.
- S. Kashyap and M. Jayakannan, J. Phys. Chem. B, 2012, 116, 9820–9831 CrossRef CAS.
- H. Sahoo, J. Photochem. Photobiol., 2011, 12, 20–30 CrossRef CAS.
- B. Kulkarni and M. Jayakannan, ACS Biomater. Sci. Eng., 2017, 3, 2185–2197 CrossRef CAS.
- S. Saxena and M. Jayakannan, Biomacromolecules, 2017, 18, 2594–2609 CrossRef CAS.
- S. Saxena, A. Pradeep and M. Jayakannan, ACS Appl. Bio Mater., 2019, 2, 5245–5262 CrossRef CAS.
- S. Saxena and M. Jayakannan, Biomacromolecules, 2020, 21, 171–187 CrossRef CAS.
- A. V. Demchenko, P. Pornsuriyasak and C. De Meo, J. Chem. Educ., 2006, 83, 782–784 CrossRef CAS.
- E. M. Sanford and T. L. Smith, J. Chem. Educ., 2008, 85(7), 944–945 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C NMR, HRMS and MALDI-TOF spectra of the compounds and polymers, absorbance and emission spectral details, enzyme-responsiveness release data, FRET overlap, confocal images, and MALDI-TOF and HRMS spectra. See DOI: 10.1039/d0py01085e |
| ‡ These authors have contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*