
Nickel-catalyzed synthesis of Zn(I)–Zn(I) bonded compounds†
Shengjie
Jiang
a,
Yanping
Cai
a,
Ambre
Carpentier
b,
Iker
del Rosal
 b,
Laurent
Maron
*b and
Xin
Xu
*a
b,
Laurent
Maron
*b and
Xin
Xu
*a
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 215123 Suzhou, P. R. China. E-mail: xinxu@suda.edu.cn
bLPCNO, CNRS & INSA, Université Paul Sabatier, 135 Avenue de Rangueil, 31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 16th November 2021
Abstract
This work reports the first catalyzed synthesis of d-block metal–metal bonded complexes. The treatment of terminal zinc hydrides [LZnH] [L = CH3C(2,6-iPr2C6H3N)CHC(CH3)(N(CH2)nCH2PR2); n = 1, 2; R = Ph, iPr] in the presence of 5 mol% Ni(CO)2(PPh3)2 afforded Zn(I)–Zn(I) bonded compounds [L2Zn2] in high isolated yields with concomitant elimination of dihydrogen. Stoichiometric reactions, kinetic studies and DFT calculations were conducted to elucidate the reaction mechanism.
Catalytic reactions that enable bond formation play a pivotal role in synthetic chemistry.1 In this respect, transition metal-catalyzed C–C bond-forming reactions are pervasive in the synthesis of organic molecules and macromolecules.2–5 Bond formation between other p-block elements through catalytic processes has also been well-established;6 the generation of homonuclear B–B,7 Si–Si,8 and P–P9 bonds by metal-catalyzed dehydrocoupling reactions are representative examples. In contrast, catalyzed synthesis of d- and s-block metal–metal bonds is unknown.
The study of metal–metal bonding is of fundamental importance for furthering our understanding of chemical bonds, catalysis, and bioinorganic chemistry as well as metal surface chemistry.10,11 To date, all reported d- or s-block metal–metal bonded complexes have been prepared via stoichiometric reactions, such as reduction, ligand redistribution, alkane/salt elimination, etc. Among these methods, reducing reactions with alkali metal-based reductants provide major access to low-valent metal–metal bonded compounds.12 However, this method is disfavored because of potential safety hazards and the formation of stoichiometric metal halide by-products.
We herein found that a commercially available nickel(0) reagent, Ni(CO)2(PPh3)2, was an efficient catalyst for the synthesis of Zn(I)–Zn(I) bonded compounds from Zn(II) hydrides, with concomitant elimination of H2 as the only by-product. This is the first example of the catalyzed synthesis of d-block metal–metal bonded compounds. To shed light on the catalysis mechanism, we isolated and characterized a series of Zn/Ni heterometallic intermediates and conducted NMR kinetic studies.
In our prior work, we found that a palladium(0) reagent Pd(PPh3)4 could induce stoichiometric dehydrocoupling of NNP ligand-based zinc hydrides to form the corresponding trimetallic products;13 however, this reagent is not applicable to a catalytic reaction. Encouraged by this result, some other widely used Pd and Ni reagents, such as Pd(OAc)2, Pd(dba)2 (dba = dibenzylideneacetone), Pd2(dba)3, (C3H5)PdCl, Ni(COD)2, and NiCl2(PPh3)2, were also screened under catalytic conditions (5 mol% catalyst loading, 60 °C, and 24 h), but none were effective. Gratifyingly, when a commercially available Ni(0) catalyst, Ni(CO)2(PPh3)2, was applied under the given conditions, Zn(II) hydride 1a was completely transformed into Zn(I)–Zn(I) bonded compound 2a (an 85% isolated yield) with the release of H2, as evidenced by the corresponding NMR reaction (Scheme 1). Moreover, such a Ni-catalyzed dehydrocoupling reaction could also tolerate other tridentate NNP ligand-based zinc hydrides, affording the corresponding dizinc complexes (Scheme 1). However, different reaction times were required for 2b (13 h, 83%) and 2c (24 h, 64%) to achieve a satisfactory yield, indicating that the length of the pendant arm significantly affects the rate of the catalytic reaction. When using (Dipp-nacnac)ZnH14 (Dipp-nacnac = [{(2,6-iPr2C6H3)N(Me)C}2CH]) as a substrate, however, no dehydrocoupling product was obtained, even under harsher conditions (10 mol% [Ni] catalyst, 80 °C, and 36 h). Hence, it is inferred that the pendant phosphine arm on the β-diketiminato ligand framework plays a crucial role for this catalytic dehydrogenation reaction.
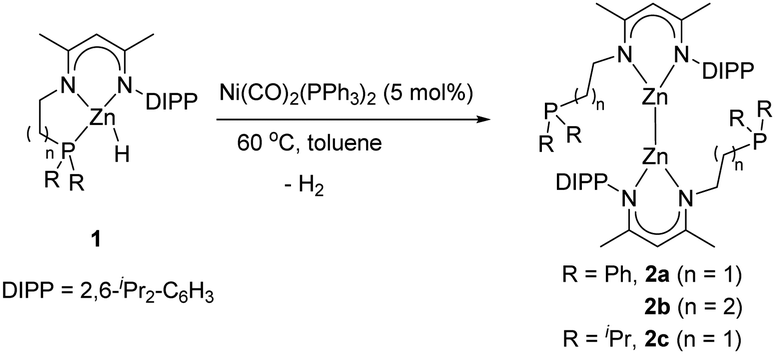 | ||
| Scheme 1 Catalytic dehydrocoupling of zinc hydrides with Ni(CO)2(PPh3)2. | ||
Thermal dehydrogenation of metal hydrides to form the corresponding metal–metal bonded complexes has been reported for group 12 elements.15 Thus, a control experiment in which zinc hydrides 1 were treated under typical reaction conditions (60 °C, 24 h) in the absence of a nickel catalyst was also examined, which did not give any dehydrocoupling product and complex 1 remained unreacted. This was further confirmed by DFT calculations, which showed an inaccessible energy barrier up to 35.1 kcal mol−1 (for details, see Fig. S50 in the ESI†).
Complex 2a was comprehensively characterized by elemental analysis, multinuclear NMR spectroscopy, and single crystal X-ray diffraction. The molecular structure of 2a (Fig. 1) showed a homonuclear bimetallic framework with a direct Zn–Zn bond. The bond length of Zn1–Zn2 was found to be 2.371(1) Å, comparable with those in other reported Zn–Zn bonded compounds supported by β-diketiminato ligands, i.e., (Dipp-nacnac)2Zn2 (2.3586(7) Å)16 and (Mes-nacnac)2Zn2 (2.3813(8) Å) (Mes-nacnac = [{(2,4,6-Me3C6H2)N(Me)C}2CH]).17 The coordination geometry of each zinc atom (ΣZn1ZnNN = 359.7(2)° and ΣZn2ZnNN = 359.9(2)°) is planar tricoordinate with a pendant phosphine arm becoming dissociated from zinc during the course of the reaction. Each zinc atom is located on the plane of the delocalized monoanionic β-diketiminato ligand and two ligands are arranged in a nearly orthogonal orientation with a N1–Zn1–Zn2–N3 torsion angle of 81.9(2)°. Both complexes 2b and 2c showed similar spectroscopic properties and structural data to those of 2a, with selected data shown in Table 1 for comparison (for details including molecular structures, see the ESI†).
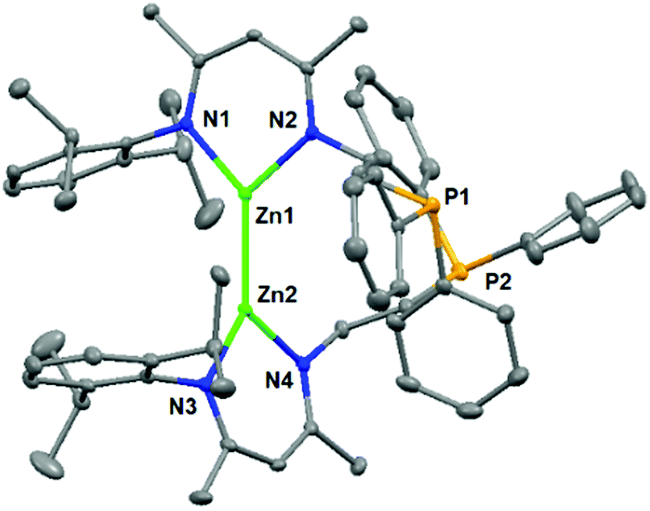 | ||
| Fig. 1 Molecular structure of complex 2a. Hydrogen atoms are omitted for clarity, and displacement ellipsoids are drawn at the 30% probability level. | ||
| Compounds | 2a | 2b | 2c |
|---|---|---|---|
| a Bond lengths in Å, angles in deg, and NMR in ppm (C6D6). | |||
| Zn1–Zn2 | 2.371(1) | 2.334(1) | 2.342(2) |
| Zn1–N1 | 1.987(2) | 1.985(2) | 1.979(2) |
| Zn1–N2 | 2.007(2) | 1.968(2) | 1.987(2) |
| Zn2–N3 | 1.986(2) | 1.984(2) | 1.972(2) |
| Zn2–N4 | 2.000(2) | 1.977(2) | 1.991(2) |
| ΣZn1ZnNN | 359.7(2) | 358.1(2) | 359.4(3) |
| ΣZn2ZnNN | 359.9(2) | 359.0(3) | 359.3(4) |
| N1–Zn1–Zn2–N3 | 81.9(2) | 133.5(2) | 83.4(1) |
| 31P{1H} | −20.5 | −16.9 | 0.6 |
| 31P{1H} of the ligand | −21.5 | −16.6 | −1.5 |
The bonding properties of Zn(I)–Zn(I) bonded compound 2a were further studied by DFT calculations. Natural bonding orbital (NBO) analysis found a Zn–Zn bond that was fully covalent and, as expected, not polarized (50–50%). This bond involved sp hybrid orbitals on each Zn (78%s + 22%p), which has a lower s character compared with the reported Zn–Zn bonds,16,17 and appeared to be the HOMO−2 molecular orbitals (see Fig. S51 in the ESI†). The Wiberg bond index (WBI) was 0.97 in line with a highly covalent Zn–Zn bond.
To gain more insights into this Ni-catalyzed dehydrocoupling reaction, stoichiometric reactions of zinc hydrides 1 with the Ni(0) reagent Ni(CO)2(PPh3)2 were performed (Scheme 2). Initially, the reaction of 1a with the Ni reagent in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio gave the hydride-bridged heterobimetallic [Zn–H–Ni] complex 3 in 61% isolated yield, which was unambiguously characterized by elemental analysis, multinuclear NMR spectroscopy, and single crystal X-ray diffraction. The solid-state structure of complex 3 (Fig. 2) showed that Zn–H was bound to the Ni center [Zn1–Ni1 2.348(1) Å, Zn1–H1 1.68(3) Å, Ni1–H1 1.58(4) Å, and Zn1–H1–Ni1 92.0(2)°] in an η2-fashion, indicating a stretched σ–Zn–H complex.18 The coordination sphere of the nickel center was completed by one pendant PPh2 group [Ni1–P1 2.183(1) Å], one PPh3 ligand [Ni1–P2 2.180(1) Å] and one CO ligand [Ni1–C32 1.727(4) Å]. In the solution-state 1H NMR spectrum, the bridged hydride signal was clearly observed at a high field at δ -3.47 ppm.19 The reaction of zinc hydride 1c with Ni(CO)2(PPh3)2 (1 equiv.) produced the analogous [Zn–H–Ni] complex 4 in 73% yield (Scheme 2). Complex 4 features similar spectroscopic properties and structural data to those of 3, with the molecular structure shown in Fig. S33 in the ESI.† In contrast, the reaction of zinc hydride 1b with one equivalent of Ni(CO)2(PPh3)2 under the same conditions afforded several undefined species.
:1 molar ratio gave the hydride-bridged heterobimetallic [Zn–H–Ni] complex 3 in 61% isolated yield, which was unambiguously characterized by elemental analysis, multinuclear NMR spectroscopy, and single crystal X-ray diffraction. The solid-state structure of complex 3 (Fig. 2) showed that Zn–H was bound to the Ni center [Zn1–Ni1 2.348(1) Å, Zn1–H1 1.68(3) Å, Ni1–H1 1.58(4) Å, and Zn1–H1–Ni1 92.0(2)°] in an η2-fashion, indicating a stretched σ–Zn–H complex.18 The coordination sphere of the nickel center was completed by one pendant PPh2 group [Ni1–P1 2.183(1) Å], one PPh3 ligand [Ni1–P2 2.180(1) Å] and one CO ligand [Ni1–C32 1.727(4) Å]. In the solution-state 1H NMR spectrum, the bridged hydride signal was clearly observed at a high field at δ -3.47 ppm.19 The reaction of zinc hydride 1c with Ni(CO)2(PPh3)2 (1 equiv.) produced the analogous [Zn–H–Ni] complex 4 in 73% yield (Scheme 2). Complex 4 features similar spectroscopic properties and structural data to those of 3, with the molecular structure shown in Fig. S33 in the ESI.† In contrast, the reaction of zinc hydride 1b with one equivalent of Ni(CO)2(PPh3)2 under the same conditions afforded several undefined species.
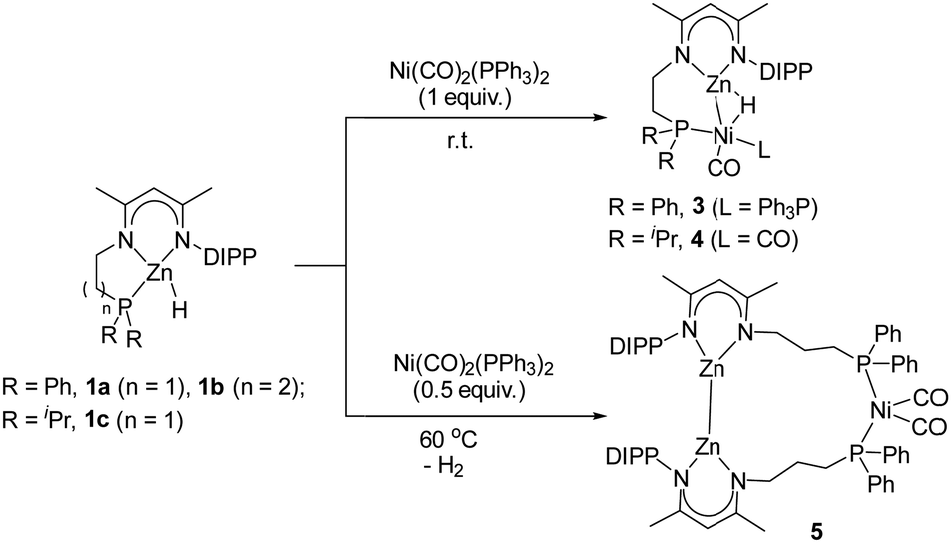 | ||
| Scheme 2 Stoichiometric reactions of zinc hydrides 1 with Ni(CO)2(PPh3)2. | ||
 | ||
| Fig. 2 Molecular structure of complex 3. Hydrogen atoms (except Zn–H) are omitted for clarity, and displacement ellipsoids are drawn at the 30% probability level. | ||
Subsequently, the treatment of zinc hydride 1b with a 0.5 molar equivalent Ni reagent was also investigated (Scheme 2), yielding complex 5 as a colorless solid in 67% isolated yield with the concomitant release of the H2 and PPh3 ligands. However, the analogous product could not be obtained for zinc hydride 1a or 1c under the same conditions, highlighting that the length of the pendant phosphine arm plays a crucial role in the stability of reaction intermediates. Single crystals of complex 5 were obtained from a layered toluene/hexane solution at room temperature. The solid-state structure of complex 5 (Fig. 3) showed a thirteen-membered metallacycle containing a Zn–Zn bond with a bond length of 2.378(1) Å, comparable with other unsupported Zn(I)–Zn(I) bonds.20 The excessively long Ni⋯Zn distances (>6.9 Å) also rule out the Ni⋯Zn interaction. The four-coordinate nickel center was found to be completed by pairs of phosphine ligands [Ni1–P1 2.217(1) Å and Ni1–P2 2.213(2) Å] and CO ligands [Ni1–C65 1.777(3) Å and Ni1–C66 1.770(3) Å]. During the course of this reaction, dehydrocoupling of the Zn–H to the Zn–Zn bond occurred. Notably, all three isolated heterometallic complexes 3, 4, and 5 enabled the formation of the final Zn–Zn bonded complex 2 under the above catalytic conditions (for more details, see the ESI†), suggesting their possible involvement in the catalytic cycle.
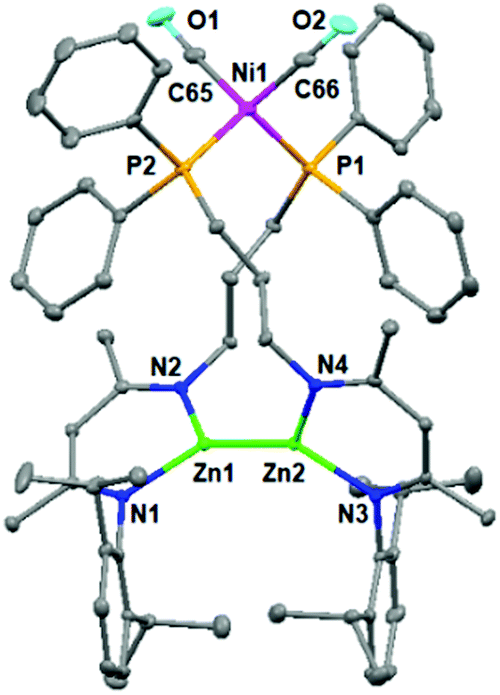 | ||
| Fig. 3 Molecular structure of complex 5. Hydrogen atoms are omitted for clarity, and displacement ellipsoids are drawn at the 30% probability level. | ||
Kinetic studies of the catalytic dehydrocoupling reactions were also performed by varying the catalyst loading in the range of 5–15 mol% and monitoring by 1H NMR spectra (for details, see the ESI†). In all cases, zero-order kinetic behaviors were observed on zinc hydride 1a. As a result, a first-order dependence on [Ni(CO)2(PPh3)2] was obtained (Fig. S44 in the ESI†). The replacement of substrate 1a (Zn–H) with the deuterium-labeled complexes 1a–d (Zn–D) resulted in a kinetic isotope effect (KIE) of 1.04(5) (Fig. S39 in the ESI†), suggesting that Zn–H bond cleavage is unlikely to be involved in the rate-determining step of catalysis. The activation parameters for the current reaction were accessed through Eyring analyses at five different temperatures from 50 °C to 70 °C (ΔH‡ = 14.4(4) kcal mol−1, ΔS‡ = −37.8(13) cal mol−1 K−1, and ΔG‡ = 25.7(8) kcal mol−1 at 298.2 K) (Fig. S49 in the ESI†). The large negative value of ΔS‡ is indicative of an associative mechanism.
Based on the above investigation, we proposed a plausible mechanistic framework for the Ni-catalyzed Zn–Zn bond formation as shown in Scheme 3. The reaction is initiated by the coordination of the Zn–H bond to the Ni center to give a σ–Zn–H complex, similar to the transition metal-catalyzed dehydrocoupling of p-block compounds.6,21 The resultant complex then reacts with another molecule of zinc hydride, possibly to produce the hydride-bridged [Zn–Ni–Zn] species 6 with the release of CO ligands, which can also be obtained from the reaction of zinc hydride with Ni(COD)2.13 In addition, complex 6 can also be used as a catalyst for the current transformation when placed under a CO atmosphere (see the ESI,† for details). Complex 6 is unstable in the presence of CO and undergoes H2 elimination19 to form the heterotrimetallic [Zn–Zn][Ni] complex via the inferred intermediate 7. The direct conversion of complex 4 to 7 is unlikely to occur due to a zero-order kinetic behavior observed for LZnH (1). Further reaction of the [Zn–Zn][Ni] complex with zinc hydride produces the Zn–Zn bonded product 2, with the regeneration of the σ–Zn–H complex to complete the catalytic cycle.
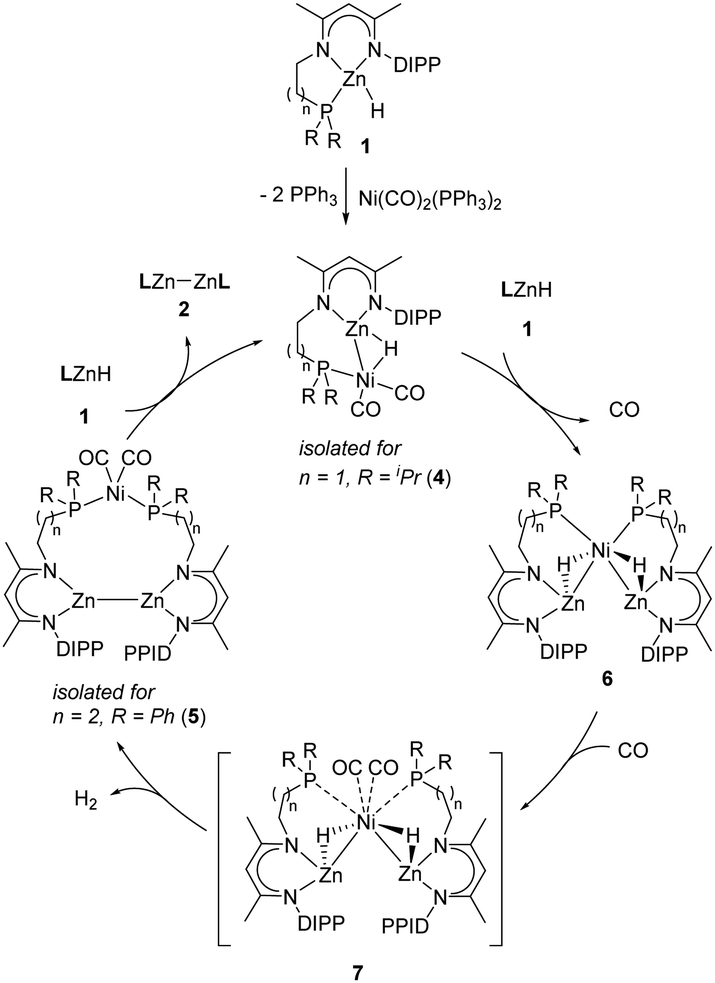 | ||
| Scheme 3 Plausible mechanistic framework for Ni-catalyzed synthesis of Zn–Zn bonded compounds. | ||
In summary, the nickel-catalyzed dehydrocoupling of zinc hydrides to Zn–Zn bonded compounds was successfully achieved. Mechanistic studies revealed that the catalytic reaction is initiated by the formation of a σ–Zn–H complex, similar to the transition metal-catalyzed synthesis of p-block element–element bonds. We propose that the NNP ligand employed in this study plays a crucial role for the reaction, because such a bifunctional ligand brings together the zinc hydride substrate and the transition metal catalytic center, thus facilitating the formation of a Ni⋯H–Zn σ-complex and the following dehydrocoupling reaction. This work realizes the first example of d-block metal–metal bond formation in a catalytic fashion and may offer a new pathway to construct a broader range of metal–metal bonds.
This work was supported by the National Natural Science Foundation of China (21871204). L. M. is a member of the Institut Universitaire de France. The authors acknowledge the HPCs CALcul en Midi-Pyrénées (CALMIP-EOS grant 1415).
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Cornils, W. A. Herrmann, H.-W. Zanthoff and C.-H. Wong, Catalysis from A to Z: A Concise Encyclopedia; Fourth Edition. Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed.
- D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540 CrossRef CAS.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS.
- (a) K. Soga and T. Shiono, Prog. Polym. Sci., 1997, 22, 1503 CrossRef CAS; (b) S. Mecking, Angew. Chem., Int. Ed., 2001, 40, 534 CrossRef CAS.
- (a) M. S. Hill, Struct. Bonding, 2010, 136, 189 CrossRef CAS; (b) E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817 CrossRef CAS PubMed.
- (a) H. Braunschweig and F. Guethlein, Angew. Chem., Int. Ed., 2011, 50, 12613 CrossRef CAS PubMed; (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed.
- J. Y. Corey, Adv. Organomet. Chem., 2004, 51, 1 CrossRef CAS.
- (a) J. D. Masuda, A. J. Hoskin, T. W. Graham, C. Beddie, M. C. Fermin, N. Etkin and D. W. Stephan, Chem. – Eur. J., 2006, 12, 8696 CrossRef CAS PubMed; (b) R. J. Less, R. L. Melen, V. Naseri and D. S. Wright, Chem. Commun., 2009, 4929 RSC.
- S. T. Liddle, Molecular Metal-Metal Bonds. Compounds, Synthesis, Properties, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed.
- (a) P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef CAS PubMed; (b) D. S. Nesterov, O. V. Nesterova and A. J. L. Pombeiro, Coord. Chem. Rev., 2018, 355, 199 CrossRef CAS.
- (a) S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754 CrossRef CAS PubMed; (b) A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Río and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 693 CrossRef CAS PubMed; (c) T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844 CrossRef CAS PubMed; (d) B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717 CrossRef PubMed.
- (a) M. Chen, S. Jiang, L. Maron and X. Xu, Dalton Trans., 2019, 48, 1931 RSC; (b) S. Jiang, M. Chen and X. Xu, Inorg. Chem., 2019, 58, 13213 CrossRef CAS PubMed.
- J. Spielmann, D. Piesik, B. Wittkamp, G. Jansen and S. Harder, Chem. Commun., 2009, 3455 RSC.
- (a) Z. Zhu, M. Brynda, R. J. Wright, R. C. Fischer, W. A. Merrill, E. Rivard, R. Wolf, J. C. Fettinger, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2007, 129, 10847 CrossRef CAS PubMed; (b) P. Jochmann and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 9831 CrossRef CAS PubMed; (c) J. Intemann, P. Sirsch and S. Harder, Chem. – Eur. J., 2014, 20, 11204 CrossRef CAS PubMed.
- Y. Wang, B. Quillian, P. Wei, H. Wang, X.-J. Yang, Y. Xie, R. B. King, P. V. R. Schleyer, H. F. Schaefer III and G. H. Robinson, J. Am. Chem. Soc., 2005, 127, 11944 CrossRef CAS PubMed.
- S. Schulz, D. Schuchmann, U. Westphal and M. Bolte, Organometallics, 2009, 28, 1590 CrossRef CAS.
- (a) O. Ekkert, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2016, 55, 16031 CrossRef CAS PubMed; (b) M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348 RSC.
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373 CrossRef CAS PubMed.
- (a) A. Grirrane, I. Resa, A. Rodríguez and E. Carmona, Coord. Chem. Rev., 2008, 252, 1532 CrossRef CAS; (b) T. Li, S. Schulz and P. W. Roesky, Chem. Soc. Rev., 2012, 41, 3759 RSC.
- R. Waterman, Chem. Soc. Rev., 2013, 42, 5629 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2099545–2099551 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05719g |
| This journal is © The Royal Society of Chemistry 2021 |