DOI:
10.1039/C9QO01438A
(Review Article)
Org. Chem. Front., 2020,
7, 896-904
Recent progress in C(aryl)–C(alkyl) bond cleavage of alkylarenes
Received
3rd December 2019
, Accepted 22nd February 2020
First published on 26th February 2020
Abstract
Cleavage of the C(aryl)–C(alkyl) σ-bond is important both in academy and industry as it holds the potential to provide straightforward access to a variety of targets from readily available chemical feedstocks such like alkylarenes. However, C(aryl)–C(alkyl) bonds of alkylarenes remain intact under most conventional reaction conditions. In this review, we highlight the results of recent C(aryl)–C(alkyl) bond cleavages that have been induced by homogeneous transition metal reagents, by oxidative cleavages, and by rearrangement.
1. Introduction
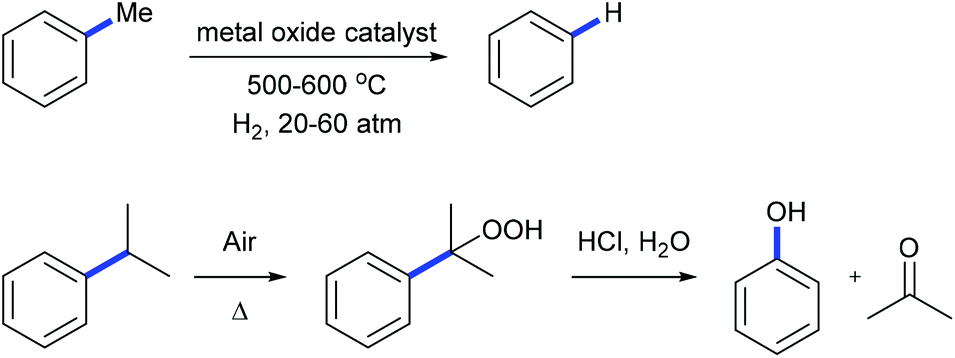
Alkylarenes are products from the coal or petrochemical industries. Transformations of these chemicals by cleaving the C(aryl)–C(sp3) σ-bond provide straightforward access to a variety of feedstocks and therefore are important both in academy and industry. Toluene hydrodealkylation is one of the industrial application examples for C(aryl)–C(sp3) bond cleavage. Toluene and hydrogen are compressed to pressures between 20 and 60 atmospheres and are heated to temperatures between 500 and 600 degrees centigrade in the presence of a metal oxide catalyst leading to the formation of benzene via C(aryl)–C(sp3) σ-bond cleavage (Scheme 1, above).1 Another example is the cumene–phenol process (or the Hock process) which produces over nine million tonnes of phenol per year.2 Oxidation of cumene under heating forms a cumene radical which bonds with an oxygen molecule to give cumene hydroperoxide. The hydroperoxide is then hydrolysed in an acidic medium (the Hock rearrangement) to give phenol and acetone (Scheme 1, below). Several modifications using a catalytic amount of N-hydroxyphthalimide or derivatives to perform this reaction under mild conditions are also known.3
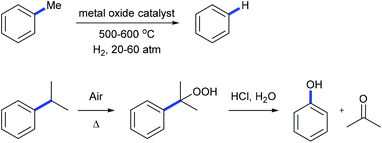 |
| | Scheme 1 Hock rearrangement and hydrogenation of toluene. | |
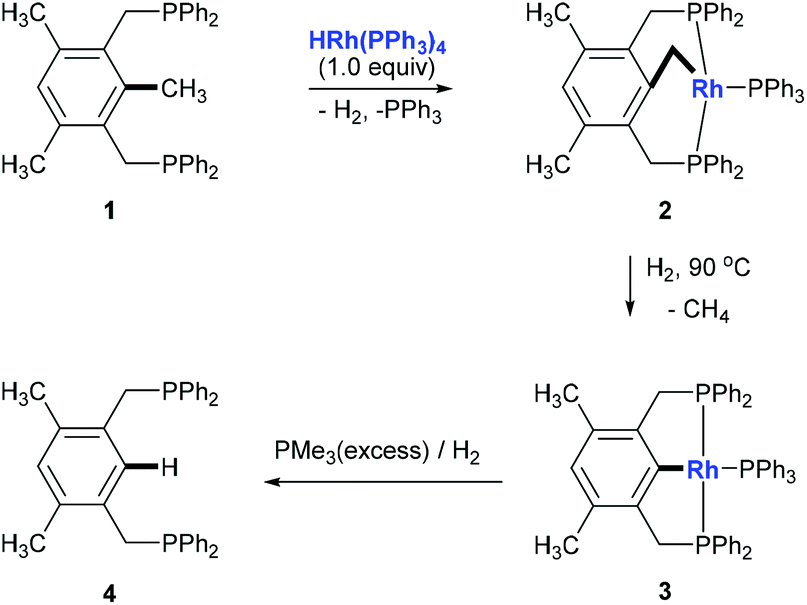
Although over the past few decades, several strategies towards C–C bond cleavage,4 such as using a small ring system to release the ring strain,5 using the driving force of the aromaticity in a prearomatic system,6 or using a carbonyl group or a cyano group to activate,7 have been established, transformations directly from non-polar, unstrained, unactivated alkylarenes remain rare because of the inertness and thermodynamic stability of the C(aryl)–C(sp3) bond in alkylarenes resulting from the high bond dissociation energy. Moreover, the C(aryl)–C(sp3) bonds in alkylarenes are surrounded by more C–H and C–C bonds and thus suffer from steric hindrances. In 1993, Milstein and co-workers reported the first example of insertion into an unstrained C(aryl)–C(sp3) σ-bond of a diphosphinoxylene in solution with the assistance of metal precoordination without using strain relief or aromatization as a driving force (Scheme 2, 1 to 4).8 The competing C–H activation process (1 to 2) was observed. Later, a catalytic version of this process was reported by using [{Rh(coe)2Cl}2] (coe = cyclooctene) either under H2 pressure or with an excess of triethoxysilane with 4–106 turnovers.9
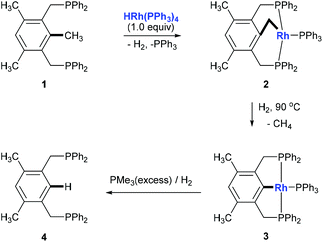 |
| | Scheme 2 C(aryl)–C(sp3) bond cleavage by rhodium. | |
Replacing one of the phosphine ligands with a less steric hindered nitrogen ligand provided a lower activation barrier of the C–C insertion than of the C–H insertion and allowed the C(aryl)–C(sp3) insertion to take place even at −30 °C.10 Other transition metal complexes such as platinum, ruthenium, osmium and nickel complexes towards C(aryl)–C(sp3) bond cleavage have also been demonstrated using similar PCP systems.11
Recently, the combination of homogeneous transition metal reagents with the assistance of monodentate ligands has provided a powerful tool for the cleavage of C(aryl)–C(alkyl) σ-bonds in alkylarenes via a direct or stepwise C–C bond insertion pathway. Moreover, several strategies using oxidative cleavage or rearrangement as a driving force to cleave the inert C(aryl)–C(sp3) bonds of alkylarenes have also been demonstrated. In this mini-review, we will focus on the discussion of these recent publications and four parts will be included. In the first part, we will briefly summarize the cleavage of C(aryl)–C(sp3) bonds via a C(aryl)–C(sp3) insertion using homogeneous transition metal reagents in the presence of monodentate ligands. In the second part, we will discuss the cleavage of C(aryl)–C(alkyl) bonds of alkylarenes, except for methylarenes, through the combination of an oxidative amination and a rearrangement in the absence of any directing groups. In the third part, we will introduce the recent results in the cleavage of C(aryl)–C(methyl) bonds using tandem oxidation/decarboxylative transformations. Finally, miscellaneous C(aryl)–C(sp3) bond cleavage of alkylarenes via alkyl or aryl migrations will be highlighted.
2. Monodentate ligand chelation-assisted C(aryl)–C(sp3) bond cleavage
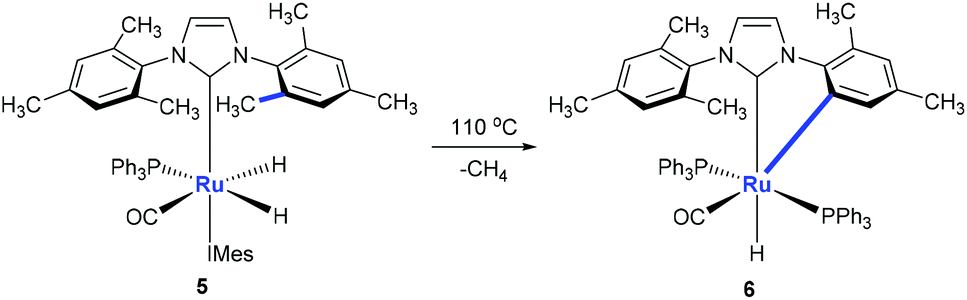
Monodentate ligands such as N-heterocyclic carbenes (NHCs) could assist the C(aryl)–CH3 bond cleavage. Whittlesey and co-workers reported a cleavage of the C(aryl)–CH3 bond of 1,3-dimesitylimidazol-2-ylidene (IMes) in the presence of Ru(PPh3)3(CO)H2 (Scheme 3).12 The methyl group on ruthenium complex 5 is eliminated with the hydride to release methane forming ruthenium complex 6 at the same time.
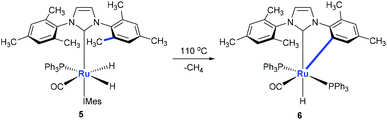 |
| | Scheme 3 Activation of the C(aryl)–CH3 bond in N-heterocyclic carbene using ruthenium complexes. | |
Using osmium-amido complex 7, Esteruelas and co-workers demonstrated that an isopropyl group of 1,3-bis(2,6-diisopropylphenyl)imidazolylidene could be activated and it involves a β-elimination, an imine dissociation, a C(aryl)–CH3 bond activation, a reductive elimination, and ammonia coordination processes (Scheme 4).13
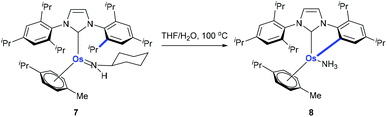 |
| | Scheme 4 Activation of the C(aryl)–iPr bond in N-heterocyclic carbene using an osmium complex. | |
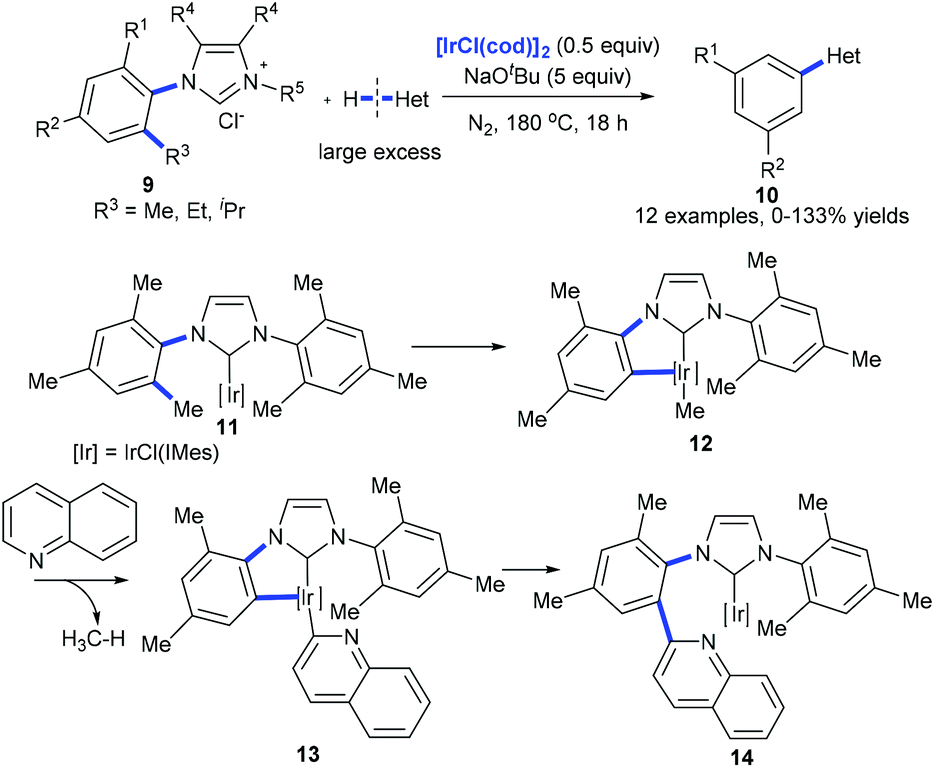
Recently, Tobisu et al. demonstrated the iridium-mediated arylation of quinolones with 9. The cleavage of a nonpolar, unstrained C(aryl)–C(alkyl) bond on N-heterocyclic carbene iridium complex 11 was proposed to occur generating complex 12 that reacted with quinolones with the release of methanes. The subsequent reductive elimination of 13 forms complex 14, which would lead to the coupling product 10 (Scheme 5).14
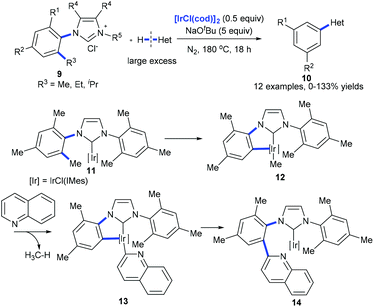 |
| | Scheme 5 Iridium-mediated arylation of various heteroarenes by activation of the C(aryl)–alkyl bond in N-heterocyclic carbene complexes of iridium. | |
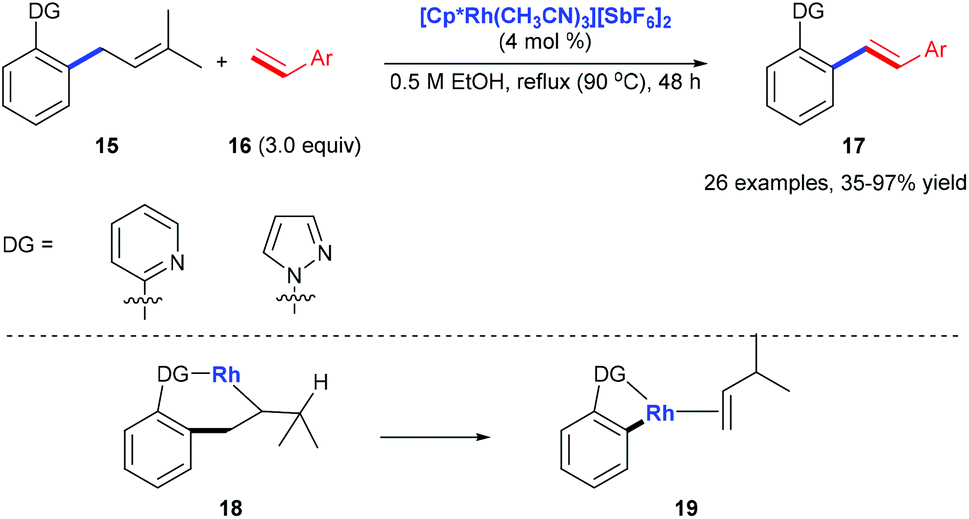
Pyridyl or pyrazolyl groups were also demonstrated as nitrogen monodentate ligands for C(aryl)–C(allyl) bond cleavage. Kakiuchi and co-workers reported a transformation of allyl groups in allylbenzenes 15 into alkenyl groups 17via a rhodium-catalysed C–C bond cleavage.15 Pyridyl and pyrazolyl groups in allylbenzenes work as a directing group for the β-carbon elimination of alkyl rhodium complex 18 to form 19 (Scheme 6).
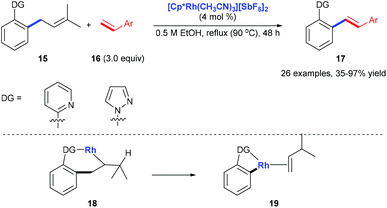 |
| | Scheme 6 Deallylative alkenylation of allylbenzene derivatives 15 with styrene derivatives 16. | |
3. C(aryl)–C(sp3) bond cleavage via tandem oxidative amination/rearrangement
3.1. Tandem oxidative amination/rearrangement mediated by DDQ and azide
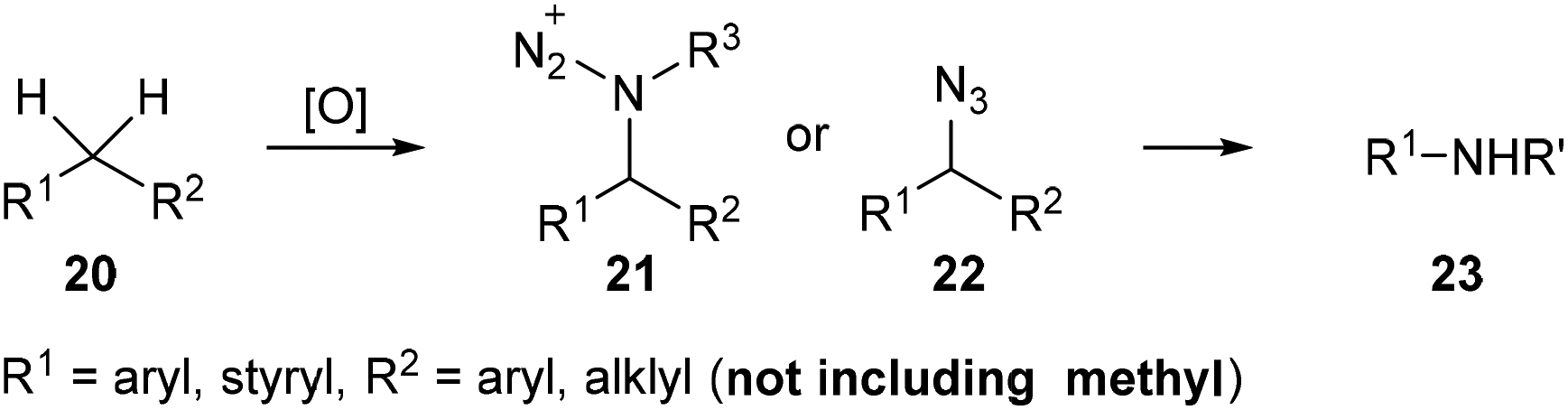
Nitrogen containing organic compounds such as amides and amines are important moieties in organic syntheses and in biological and pharmaceutical compounds. Recently, Jiao et al. developed a tandem oxidative substitution/rearrangement strategy to prepare amides or amines 23 from alkylarenes 20via amination intermediates 21 and 22 (Scheme 7). Besides, the substituent on the α-methyl on the alkylarenes is necessary for these reactions, while methylbenzene derivatives are inert under these reaction conditions.
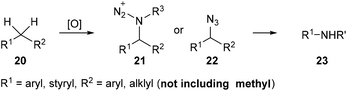 |
| | Scheme 7 Transformation of alkylarenes into amides or amines via a tandem oxidative substitution/rearrangement reaction. | |
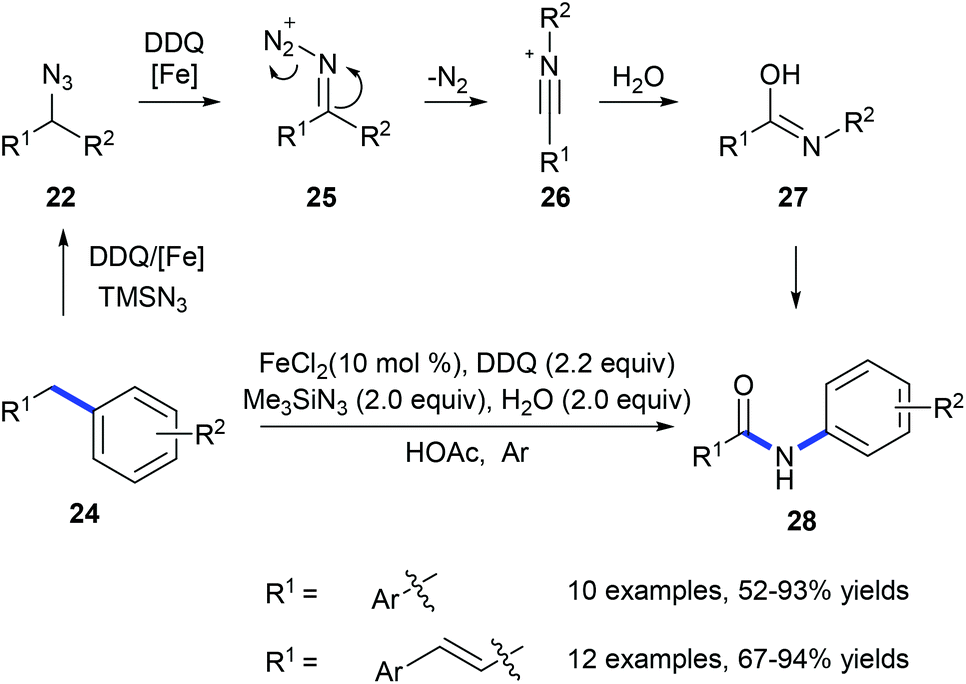
In 2011, Jiao and co-workers reported a transformation of benzyl hydrocarbons 24 into the corresponding amides 28 using a tandem iron-catalyzed oxidative substitution/rearrangement reaction (Scheme 8).16 In wet protonic acid, iron-assisted oxidative addition with TMSN3 in the presence of DDQ formed 25 which subsequently undergoes a highly chemoselective Beckmann rearrangement followed by hydrolysis resulting in the formation of amide 28. Six- and seven-membered rings could also be converted into the corresponding lactams in good yields. However, in the case of the reaction of fluorene, phenanthridine was produced suggesting that the desired lactam undergoes dehydration.
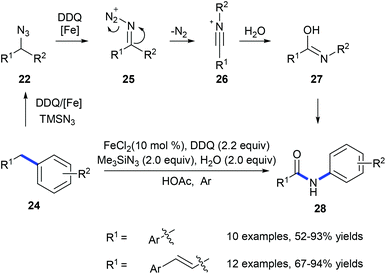 |
| | Scheme 8 Transformation of the benzyl hydrocarbons 24 into amides 28. | |
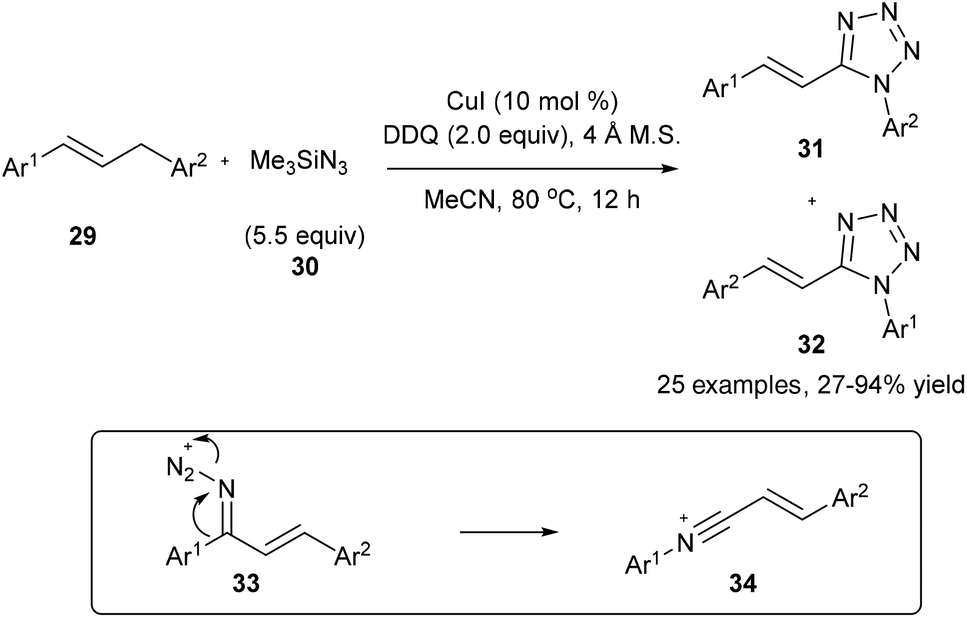
On the other hand, on using a copper catalyst in the presence of dry acetonitrile, the oxidative substitution of 29 or electron-rich alkylarenes could also lead to Beckmann-type aryl migration from the carbon atom to the nitrogen atom to generate intermediate 34; the following nucleophilic addition and cyclization with another azide finally lead to the desired tetrazole product 31 or 32 in good yields (Scheme 9).17
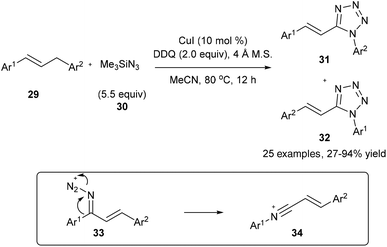 |
| | Scheme 9 Transformation of 29 into tetrazoles 31 and 32. | |
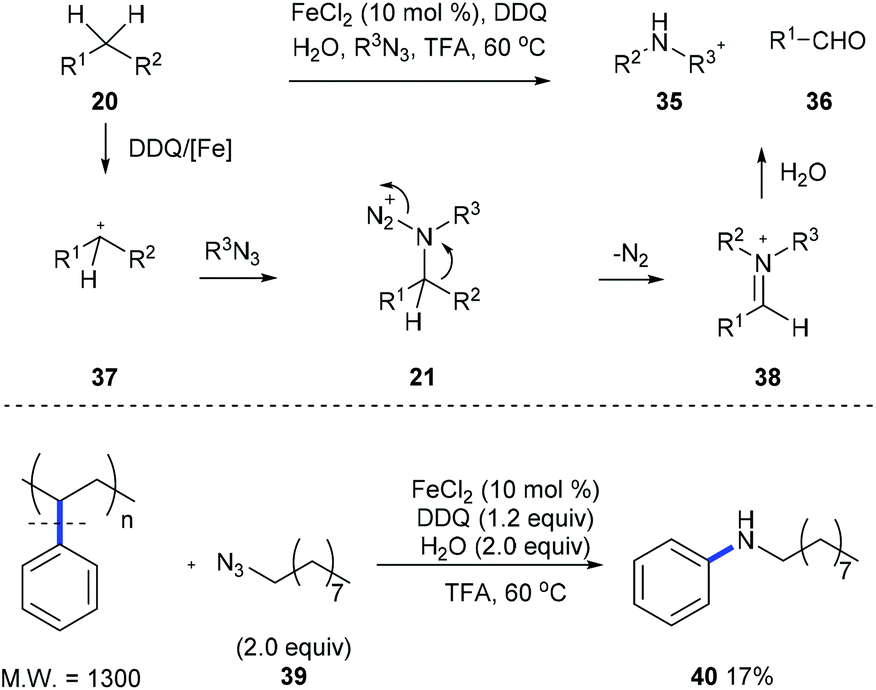
Later, Jiao et al. developed an iron-catalyzed cleavage of unactivated C(aryl)–C(alkyl) bonds on diarylmethanes or alkylarenes 20 using stable long chain alkyl azides to form arylamines 35 and aldehyde 36 (Scheme 10).18 Intermediate 21, generated from the nucleophilic attack of 37 by organic azide, is proposed, and it undergoes a Schmidt-type rearrangement involving the release of nitrogen and the trans-aryl group migration and leads to the formation of iminium 38. When a mixture of alkyl benzenes was subjected to the standard reaction conditions, a single N-alkylaniline was obtained in a moderate yield demonstrating the potential for applying this method to the conversion of a crude mixture of benzyl hydrocarbons from the oil and coal industry. Moreover, a commercially available polystyrene sample and waste polystyrene foam were both used to test the potential of this method in the degradation of polymers and they performed well, giving N-nonylaniline 40 in 17% yield.
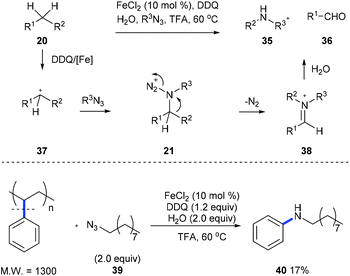 |
| | Scheme 10 Direct synthesis of arylamines through C(aryl)–C(sp3) bond cleavage. | |
The same group also reported an electrochemical C–C amination of alkylarenes 41 for the synthesis of anilines 43 using graphite plates as electrodes in an undivided cell without any external catalyst or oxidant (Scheme 11).19 The substrate is first oxidized on the anode generating a radical cation which is deprotonated to give a benzyl radical. The benzyl radical further undergoes oxidation forming an intermediate cation which could be nucleophilically attacked by organic azide 42 producing the Schmidt-type intermediate. This method is efficient for a variety of alkylarenes except for substrates with strong electron-withdrawing groups.
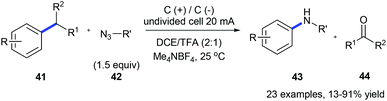 |
| | Scheme 11 Electrochemically oxidative C(sp2)–C(sp3) bond cleavage of alkylarenes for aniline synthesis. | |
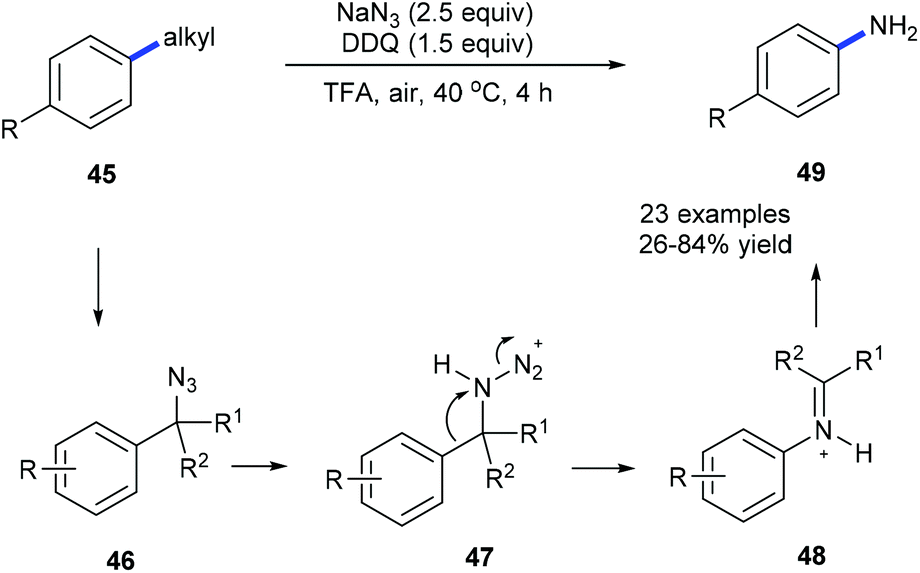
Using NaN3, DDQ and TFA, anilines 49 could be prepared from the corresponding alkylarenes 45 or benzyl alcohols under mild conditions via a dealkylating C–C amination (Scheme 12).20a Alkyl groups such as ethyl, i-propyl, benzyl, cyclohexyl, and n-butyl could be selectively cleaved and replaced by amine. Secondary benzyl alcohols could also be converted into the corresponding substituted anilines. A Schmidt-type rearrangement pathway including intermediates 46–48 was proposed. Utilization of oxygen as an alternative environmentally benign oxidant is also developed. To demonstrate the utility of this method the depolymerization of lignin was also studied by using model compounds obtaining the desired products in moderate yields.
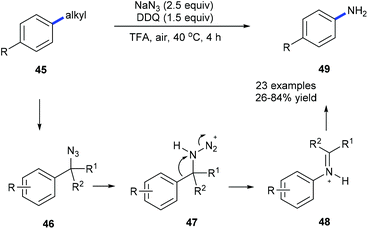 |
| | Scheme 12 Transformation of alkylbenzene derivatives 45 into anilines 49 through site-directed C(aryl)–C(sp3) bond cleavage. | |
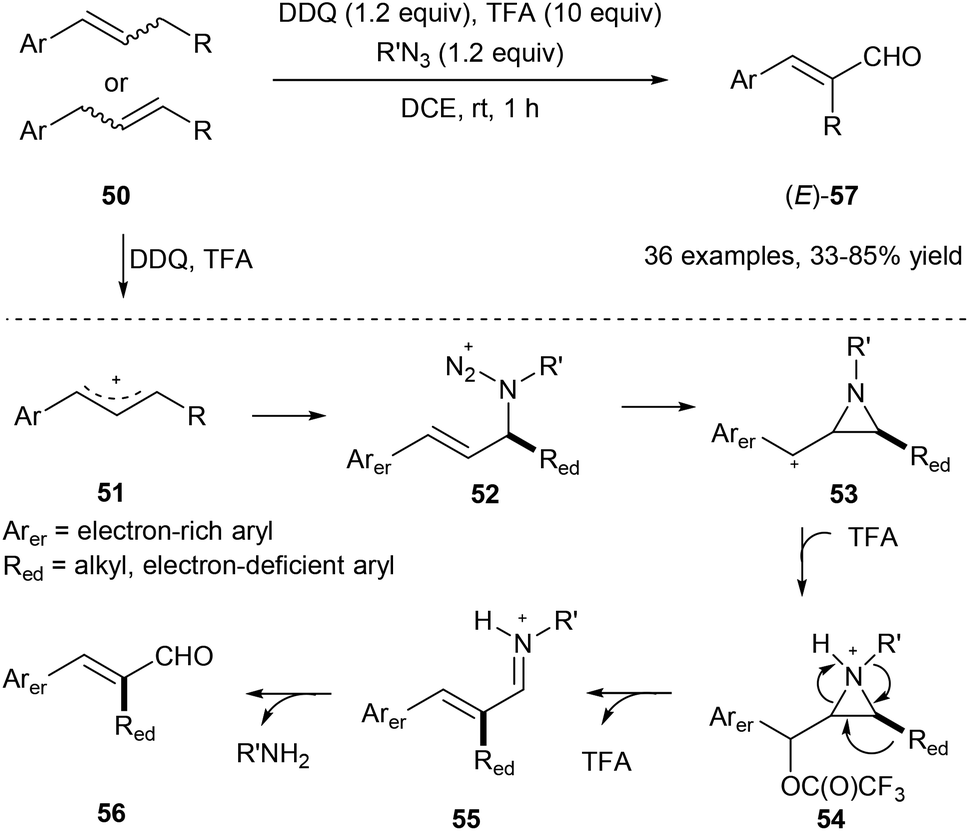
Using organic azide, DDQ and TFA, the allylic C(aryl)–C(alkyl) bond of 50 could be cleaved under metal-free conditions via a 1,2-aryl or alkyl migration producing the corresponding E-cinnamyl aldehydes 57 regio- and stereoselectively (Scheme 13).21 Alkyl azides work as a traceless reagent in this transformation by generating an azido cation intermediate 52 and finally turning into alkyl amines. In this transformation, E products are obtained in all cases, regardless of the geometry of the starting olefins.
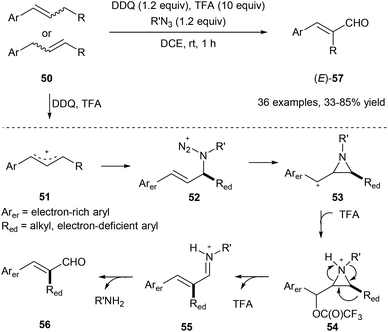 |
| | Scheme 13 Transformation of 50 into cinnamyl aldehydes 57 through selective allylic C–C bond cleavage. | |
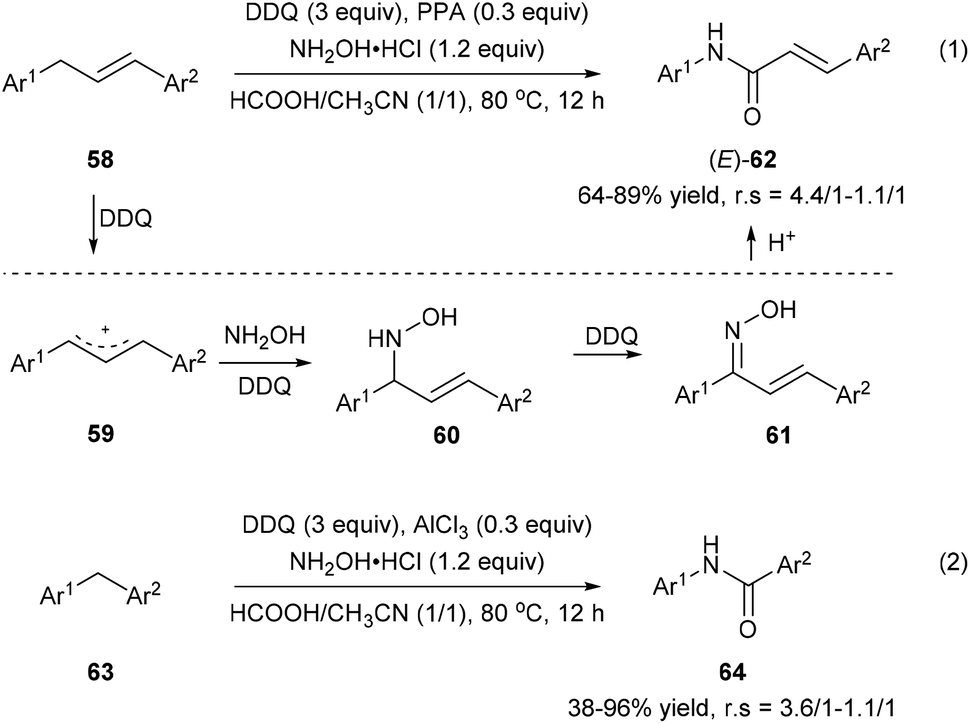
Zhang et al. also developed an oxidative amination/rearrangement of 1,3-diarylpropenes 58 or diarylmethanes 63 utilizing hydroxylamine as a nucleophile in the presence of DDQ and an acid (PPA or AlCl3). The initial single-electron transfer process between 58 and DDQ produces benzyl cation 59 followed by the attack of hydroxylamine giving rise to 60. Oxidation of 60 by DDQ affords ketoximes 61, which convert into the corresponding amides 62 and 64via a Beckmann rearrangement in the presence of an acid with 38–96% yields and 4.4/1–1.1/1 regioselectivities (Scheme 14).22
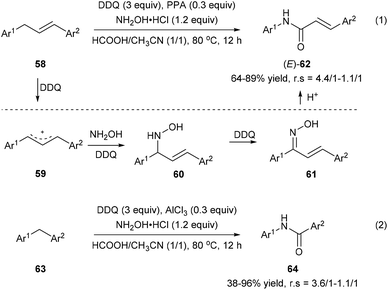 |
| | Scheme 14 Transformation of 1,3-diarylpropenes 58 and diarylmethanes 63 into amides via oxidative amination/rearrangement. | |
3.2. One-pot oxygenation/amination/rearrangement using nitromethane
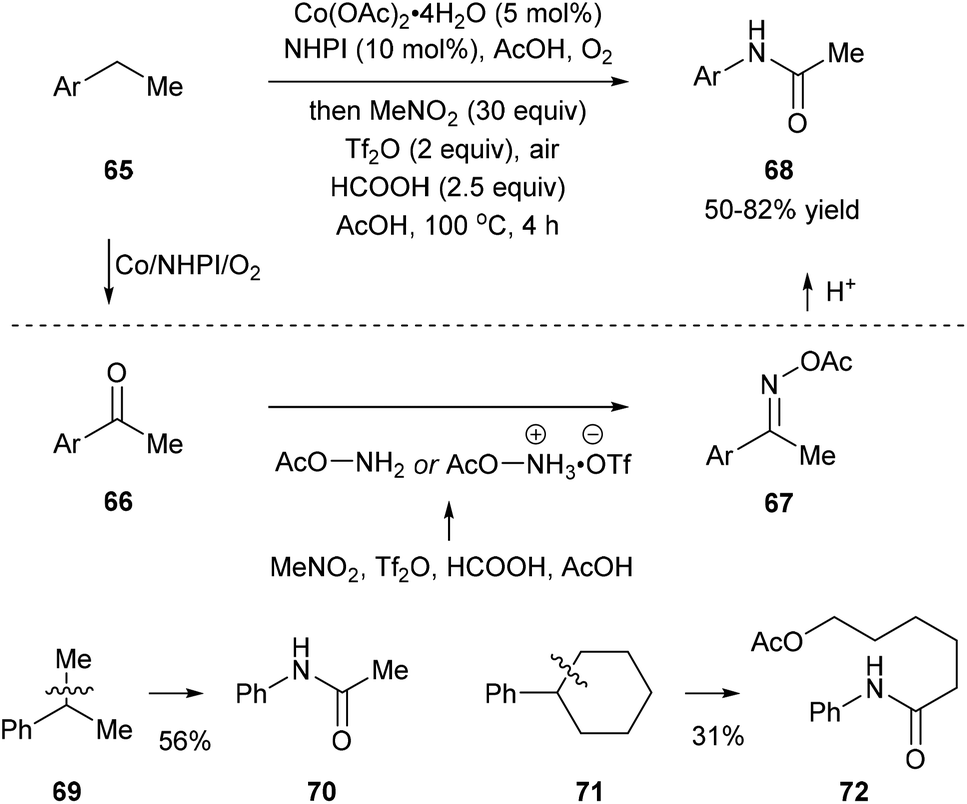
Recently, Jiao and co-workers reported an activation of nitromethane that utilizes triflic anhydride, formic acid, and acetic acid, and this method provides a nitrogen donor instead of azides for the amination of ketones or aldehydes. When they subjected ethylbenzenes 65 to the classic oxidation conditions using the Co/NHPI/O2 system, the corresponding ketones 66 were generated in situ, and the following addition of triflic anhydride, formic acid, and acetic acid afforded 67. Hydrolysis of 67 gave rise to the corresponding dealkylated amides 68. When cumene 69 and cyclohexylbenzene 71, important feedstock materials, were subjected to the protocol, secondary amides 70 and 72 were obtained through oxidative β-scission of alkyl chains. Reactions with substituted methylarenes were also investigated but they provided primary benzamides without C(aryl)–C(alkyl) bond cleavage (Scheme 15).23
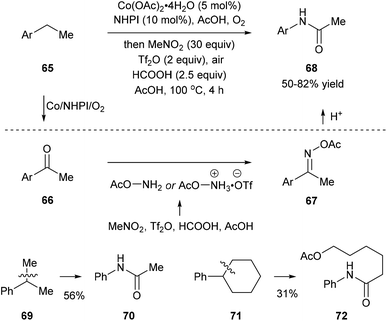 |
| | Scheme 15 Preparation of amides via a one-pot oxygenation/amination/rearrangement of alkylarenes using nitromethane. | |
4. Cleavage of the C(aryl)–CH3 bond via tandem oxidation/decarboxylative transformations
Using amination/rearrangement strategies, great success has been achieved in the scission of the C(aryl)–C(alkyl) bond in ethylarenes, cumene, cyclohexylbenzenes, etc., but demethylative C(aryl)–CH3 bond cleavage of inert methylarenes has been less reported.
Kim and co-workers reported a one-pot demethylative coupling reaction between inert methylarenes 73 and benzenes using substoichiometric silver nitrate in the presence of an excess amount of the potassium persulfate oxidant under heating. Observations of benzyl hydrogen sulfate, benzaldehyde, and benzoic acid by GC analysis, control experiments, and detection of carbon dioxide by GC-MS analysis suggest that this process involves a radical oxidation of methylarenes 73 to substituted benzoic acids 74 and a following decarboxylative coupling with benzenes. A two-electron oxidative transfer Ag(I)/Ag(III) system for the generation of the free radical SO4˙− is proposed based on the studies by XANES and EXAFS analyses (Scheme 16).24
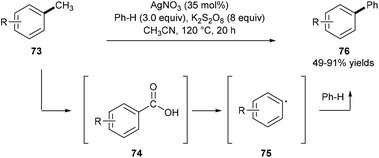 |
| | Scheme 16 Demethylative coupling between methylarenes 73 and benzenes. | |
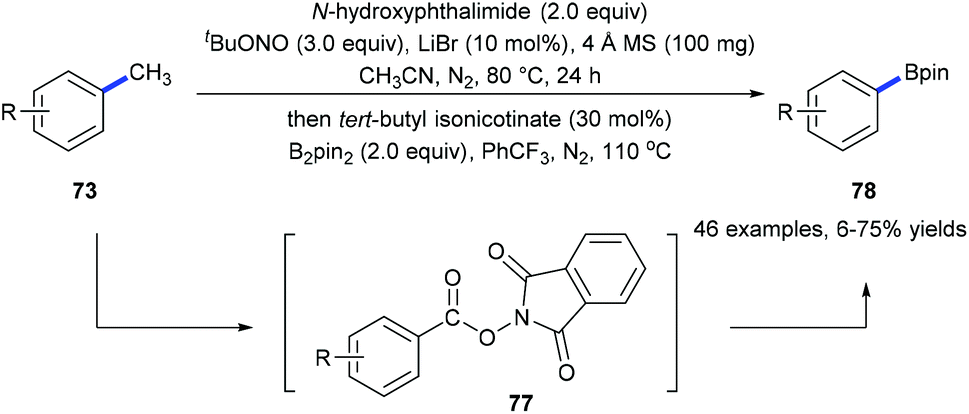
Recently, we have developed a site-selective C(aryl)–CH3 bond cleavage/borylation reaction25 by using a sequential tandem strategy (Scheme 17).26 Methyl groups of a variety of arenes 73 and biologically active natural products could be selectively cleaved and replaced by boryl groups under directing group-free and transition metal-free conditions. An N-hydroxyphthalimide-ester 77 was isolated from the first step of the standard conditions. It was proposed to be the key intermediate which converted into the corresponding boronate 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25a through a decarboxylative borylation (Scheme 17).
25a through a decarboxylative borylation (Scheme 17).
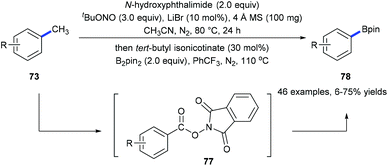 |
| | Scheme 17 Sequential radical demethylative C(aryl)–C(CH3) bond cleavage and borylation. | |
5. Miscellaneous C(aryl)–C(alkyl) bond cleavage via alkyl or aryl migrations
Miscellaneous C(aryl)–C(alkyl) bond cleavage of alkylarenes involving alkyl or aryl migrations has been reported by using a hypervalent iodine(III) reagent or a cationic N-heterocyclic carbene copper catalyst giving the corresponding product in good yields.
5.1 Hypervalent iodine(III)-mediated rearrangement
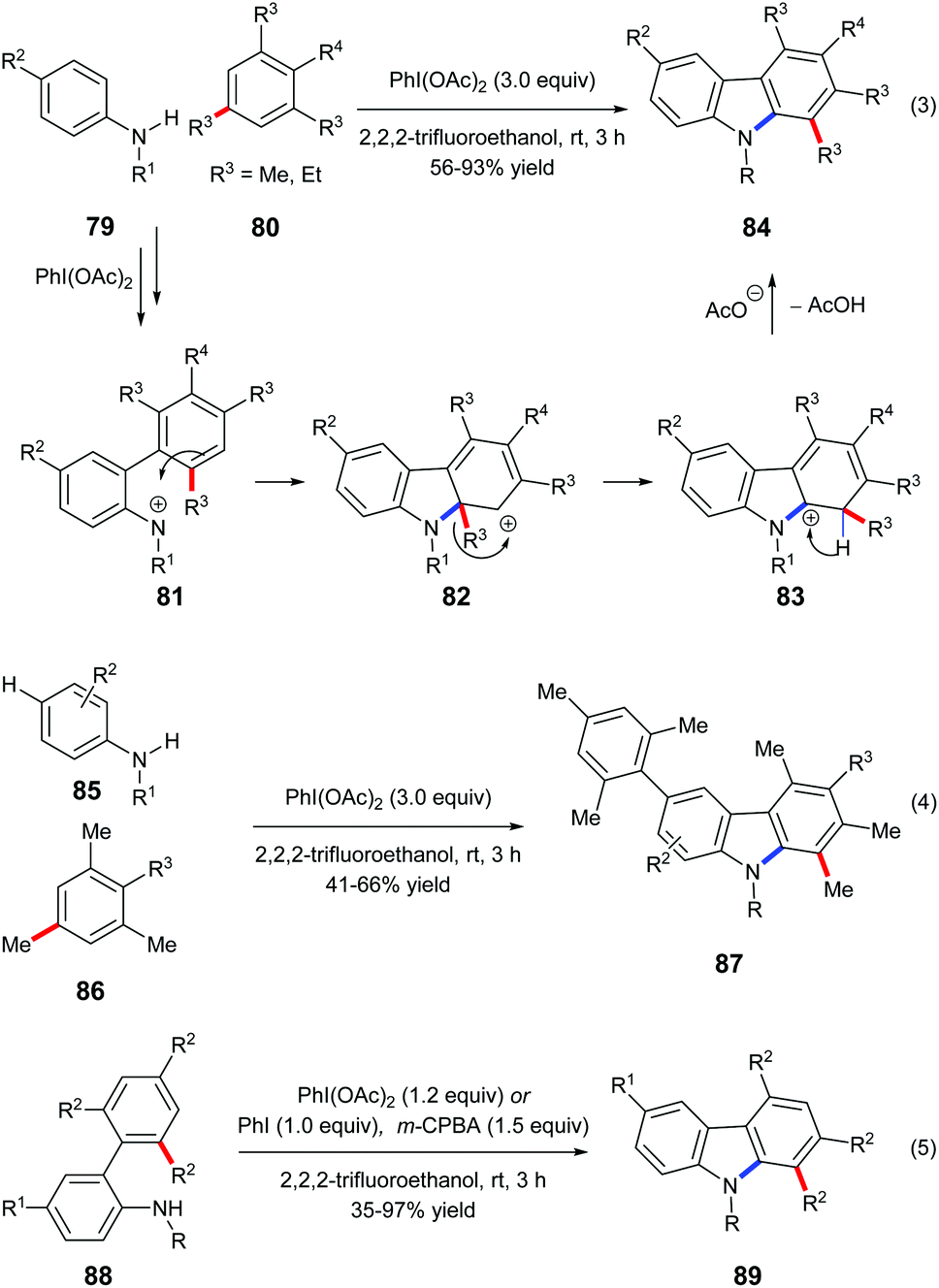
Mal et al. reported a one-pot carbazole synthesis via an intermolecular annulation between aryl sulfonamides 79 and substituted mesitylenes or 1,3,5-triethylbenzene 80. The hypervalent iodine(III) reagent PhI(OAc)2 prompted the formation of nitrenium ion 81 from sulphonamide, and the following electrophilic aromatic substitution led to carbenium intermediate 82 which underwent alkyl migration forming a more stable cationic intermediate 83 (Scheme 18, eqn (3)).27a When N-sulfonylanilides 85 with hydrogen at the para-position to the sulphonamide group were subjected to the standard conditions, para-arylated carbazoles 87 were obtained (Scheme 18, eqn (4)). An intramolecular reaction of 88 has also been reported by the same research group (Scheme 18, eqn (5)).27b
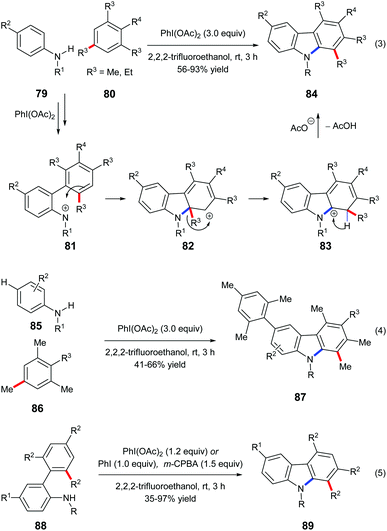 |
| | Scheme 18 PhI(OAc)2 prompted alkyl migration. | |
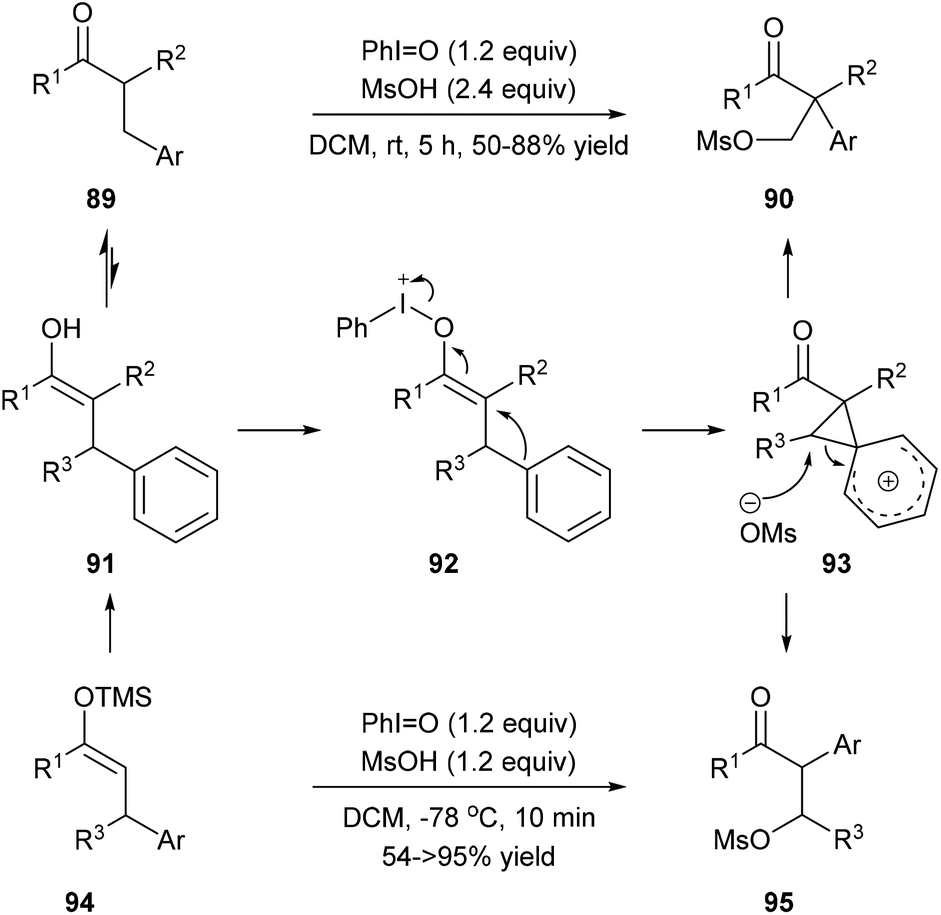
Maulide and co-workers found that a hypervalent iodine(III) reagent could induce a C(aryl)–C(alkyl) bond cleavage α-arylation of dicarbonyl compounds 89 that possess an aryl group in the β-position of ketones or ketone-derived silyl enol ethers 94 that feature an aryl group in the allylic position (Scheme 19). They proposed that a fragmentation of enolonium 92 could be triggered by a nucleophilic attack of the neighbouring arene to generate phenonium intermediate 93. Ring opening of 93 by weakly nucleophilic triflate accounts for the formation of the C(aryl)–C(alkyl) bond cleavage products 90 and 95.28
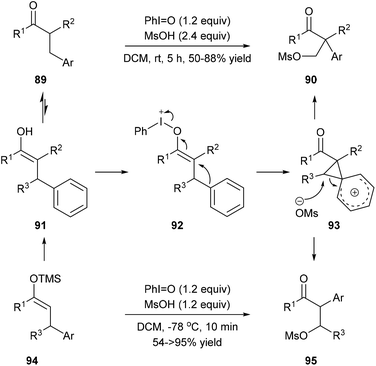 |
| | Scheme 19 Hypervalent iodine(III) induced aryl migration. | |
5.2 Copper-catalysed domino rearrangement
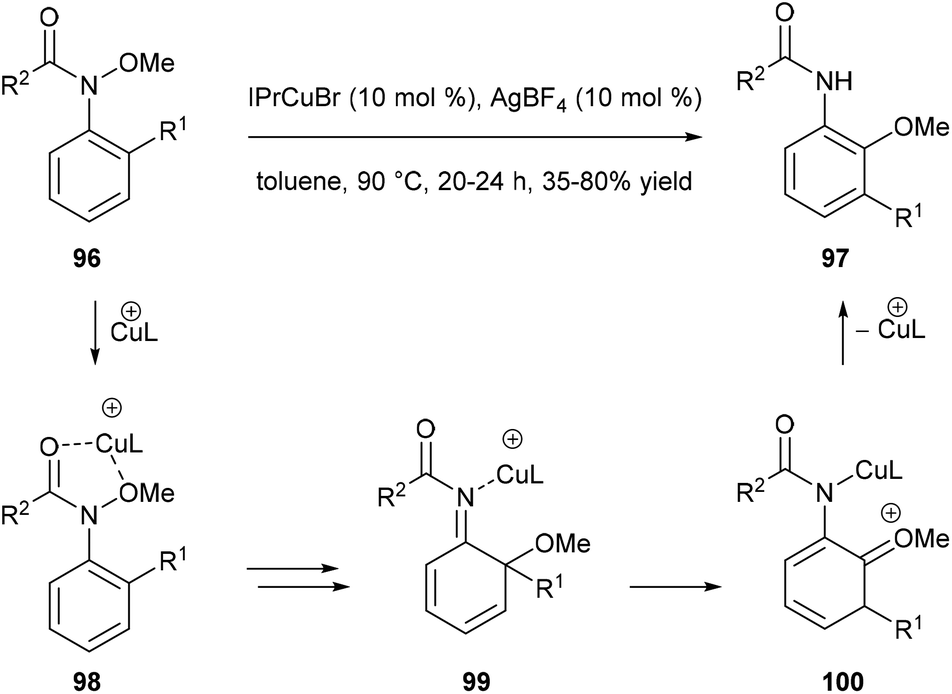
Nakamura and co-workers reported a catalytic domino rearrangement of N-methoxyanilines 96 that bear an electron-donating alkyl group (Scheme 20). The cationic copper catalyst was found to participate in the [1,3]-migration of the methoxy group from 98 to 99 and the [1,2]-migration of the alkyl group from the ortho to the meta position (from 99 to 100). This provides a method for the preparation of multisubstituted 2-aminophenol derivatives 97 from readily accessible N-methoxyanilines 96.29
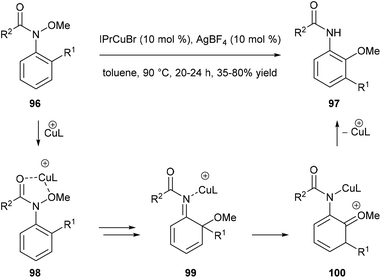 |
| | Scheme 20 Copper catalysed domino rearrangement of N-methoxyanilines 96. | |
6. Summary and outlook
The C(aryl)–C(alkyl) σ-bond in alkylarenes is one of the least reactive functional groups; however, its cleavage could be realized by elaborating the reaction system. In this mini-review, we have highlighted the advances in the cleavage of C(aryl)–C(alkyl) σ-bonds of alkylarenes using a transition-metal insertion in the presence of directing groups, a tandem oxidative amination/rearrangement, tandem oxidation/decarboxylative transformations, and alkyl or aryl migrations. The development of new strategies towards C(aryl)–C(alkyl) bond cleaving functionalization may lead to the invention of new selective and efficient processes for the utilization of alkylarenes which are available or chemical raw materials from the coal and petrochemical industries.
Conflicts of interest
There are no conflicts to declare.
Note from RSC Publishing
This article was originally published with the incorrect page numbers. This is the corrected, final version. The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.
Acknowledgements
We thank the National Natural Science Foundation of China (21602001, 21922109, 21672196, and 21831007), the Start-Up Funding from Nanjing Tech University (39837134), and the Fundamental Research Funds for the Central Universities of China (WK2060190086) for financial support.
Notes and references
-
R. Schmidt, et al., Hydrocarbons, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2015 Search PubMed.
-
(a) “Phenol”:
M. Weber, M. Weber and M. Kleine-Boymann, UllmannÏs Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed;
(b) Noncatalyzed radical chain oxidation: cumene hydroperoxide:
M. Weber, J.-B. G. Daldrup and M. Weber, Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives, First Edition, ed. S. S. Stahl and P. L. Alsters, Wiley-VCH, Weinheim, 2016 Search PubMed.
-
(a) R. Nakamura, Y. Obora and Y. Ishii, Selective One-Pot Synthesis of Various Phenols from Diarylethanes, Chem. Commun., 2008, 3417–3419 RSC;
(b) K. Chen, P. Zhang, Y. Wang and H. Li, Metal-free Allylic/Benzylic Oxidation Strategies with Molecular Oxygen: Recent Advances and Future Prospects, Green Chem., 2014, 16, 2344–2374 RSC;
(c) E. M. Yarkina, E. A. Kurganova, A. S. Frolov, N. V. Lebedeva and G. N. Koshel, Aerobic Liquid-Phase Oxidation of para-t-Butylcumene to Hydroperoxide, Pet. Chem., 2019, 59, 1245–1248 CrossRef CAS.
-
(a)
C–C Bond Activation, ed. G. Dong, Springer-Verlag, Berlin, Heidelberg, 2014 Search PubMed;
(b) Catalysis in C–C activation:
H. Li and Z.-J. Shi, Homogeneous Catalysis for Unreactive Bond Activation, ed. Z.-J. Shi, Wiley-VCH, Weinheim, 2016 Search PubMed;
(c)
Cleavage of Carbon–Carbon Single Bonds by Transition Metals, ed. M. Murakami and N. Chatani, Wiley-VCH Verlag GmbH, Weinheim, 2016 Search PubMed.
-
(a) K. C. Bishop III, Transition Metal Catalyzed Rearrangements of Small Ring Organic Molecules, Chem. Rev., 1976, 76, 461–486 CrossRef;
(b) M. Rubin, M. Rubina and V. Gevorgyan, Transition Metal Chemistry of Cyclopropenes and Cyclopropanes, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed;
(c) T. Seiser and N. Cramer, Enantioselective Metal-Catalyzed Activation of Strained Rings, Org. Biomol. Chem., 2009, 7, 2835–2840 RSC;
(d) A. Masarwa and I. Marek, Selectivity in Metal-Catalyzed Carbon-Carbon Bond Cleavage of Alkylidenecyclopropanes, Chem. – Eur. J., 2010, 16, 9712–9721 CrossRef CAS PubMed;
(e) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Cyclobutanes in Catalysis, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS PubMed;
(f) C. Aïssa, Transition-Metal-Catalyzed Rearrangements of Small Cycloalkanes: Regioselectivity Trends in β-Carbon Elimination Reactions, Synthesis, 2011, 21, 3389–3407 CrossRef;
(g) B.-L. Lu, L. Dai and M. Shi, Strained Small Rings in Gold-Catalyzed Rapid Chemical Transformations, Chem. Soc. Rev., 2012, 41, 3318–3339 RSC;
(h) D. J. Mack and J. T. Njardarson, Recent Advances in The Metal-Catalyzed Ring Expansions of Three- and Four-Membered Rings, ACS Catal., 2013, 3, 272–286 CrossRef CAS;
(i) I. Marek, A. Masarwa, P. O. Delaye and M. Leibeling, Selective Carbon-Carbon Bond Cleavage for the Stereoselective Synthesis of Acyclic Systems, Angew. Chem., Int. Ed., 2015, 54, 414–429 CrossRef CAS PubMed;
(j) T. Kondo, Ruthenium- and Rhodium-Catalyzed Strain-Driven Cleavage and Reconstruction of the C–C Bond, Eur. J. Org. Chem., 2016, 1232–1242 CrossRef CAS;
(k) G. Fumagalli, S. Stanton and J. F. Bower, Recent Methodologies that Exploit C–C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed.
-
(a) R. C. Hemond, R. P. Hughes and H. B. Locker, Competitive C-H and C-C Activation in the Reaction of Pentamethylcyclopentadiene with Decacarbonyldimanga-nese, Organometallics, 1986, 5, 2391–2392 CrossRef CAS;
(b) R. H. Crabtree, R. P. Dion, D. J. Gibboni, D. V. McGrath and E. M. Holt, Carbon-Carbon Bond Cleavage In Hydrocarbons By Iridium Complexes, J. Am. Chem. Soc., 1986, 108, 7222–7227 CrossRef CAS;
(c) H. Suzuki, Y. Takaya and T. Takemori, Selective Carbon-Carbon Bond Cleavage of Cyclopentadiene on a Trinuclear Ruthenium Pentahydride Complex, J. Am. Chem. Soc., 1994, 116, 10779–10780 CrossRef CAS;
(d) C. T. To and K. S. Chan, Carbon–Carbon Bond Activation by Group 9 Metal Complexes, Eur. J. Org. Chem., 2019, 6581–6591 CrossRef CAS.
-
(a) M. E. van der Boom and D. Milstein, Cyclometalated Phosphine-Based Pincer Complexes: Mechanistic Insight in Catalysis, Coordination, and Bond Activation, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed;
(b) C.-H. Jun, C. W. Moon, H. Lee and D.-Y. Lee, Chelation-Assisted Carbon–Carbon Bond Activation By Rh(I) Catalysts, J. Mol. Catal. A: Chem., 2002, 189, 145–156 CrossRef CAS;
(c) C.-H. Jun and J. H. Lee, Application of C–H and C–C Bond Activation in Organic Synthesis, Pure Appl. Chem., 2004, 76, 577–587 CAS;
(d) M. Tobisu and N. Chatani, Catalytic Reactions Involving The Cleavage Of Carbon–Cyano And Carbon–Carbon Triple Bonds, Chem. Soc. Rev., 2008, 37, 300–307 RSC;
(e) J. E. M. N. Klein and B. Plietker, Iron-Catalysed Carbon-Carbon Single Bond Activation, Org. Biomol. Chem., 2013, 11, 1271–1279 RSC;
(f) F. Chen, T. Wang and N. Jiao, Recent Advances in Transition-Metal-Catalyzed Functionalization of Unstrained Carbon–Carbon Bonds, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed;
(g) A. Dermenci, J. W. Coe and G. Dong, Direct Activation Of Relatively Unstrained Carbon–Carbon Bonds In Homogeneous Systems, Org. Chem. Front., 2014, 1, 567–581 RSC;
(h) M. H. Shaw and J. F. Bower, Synthesis And Applications of Rhodacyclopentanones Derived from C–C Bond Activation, Chem. Commun., 2016, 52, 10817–10829 RSC;
(i) D.-S. Kim, W.-J. Park and C.-H. Jun, Metal–Organic Cooperative Catalysis in C–H and C–C Bond Activation, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed.
-
(a) B. Rybtchinski and D. Milstein, Metal Insertion into C−C Bonds in Solution, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef;
(b) M. Gozin, A. Weisman, Y. Ben-David and D. Milstein, Activation of a Carbon-carbon Bond in Solution by Transition-metal Inserion, Nature, 1993, 364, 699–701 CrossRef CAS.
- S.-Y. Liou, M. E. van der Boom and D. Milstein, Catalytic Selective Cleavage of a Strong C–C Single Bond by Rhodium in Solution, Chem. Commun., 1998, 687–688 RSC.
- M. Gandelman, A. Vigalok, L. J. W. Shimon and D. Milstein, A PCN Ligand System. Exclusive C-C Activation with Rhodium(I) and C-H Activation with Platinum(II), Organometallics, 1997, 16, 3981–3986 CrossRef CAS.
-
(a) M. E. van der Boom, H.-B. Kraatz, L. Hassner, Y. Ben-David and D. Milstein, Carbon-Carbon vs Carbon-Hydrogen Bond Activation by Ruthenium(II) and Platinum(II) in Solution, Organometallics, 1999, 18, 3873–3884 CrossRef CAS;
(b) R. M. Gauvin, H. Rozenberg, L. J. W. Shimon and D. Milstein, Synthesis and Structure of New Osmium-PCP Complexes. Osmium-Mediated C-C Bond Activation, Organometallics, 2001, 20, 1719–1724 CrossRef CAS;
(c) M. E. van der Boom, S.-Y. Liou, L. J. W. Shimon, Y. Ben-David and D. Milstein, Nickel Promoted C–H, C–C and C–O Bond Activation in Solution, Inorg. Chim. Acta, 2004, 357, 4015–4023 CrossRef CAS;
(d) M. E. van der Boom, H.-B. Kraatz, Y. Ben-David and D. Milstein, Activation of a Non-Strained C-C Bond with Platinum(II), Chem. Commun., 1996, 2167–2168 RSC.
-
(a) R. F. R. Jazzar, S. A. Macgregor, M. F. Mahon, S. P. Richards and M. K. Whittlesey, C-C and C-H Bond Activation Reactions in N-Heterocyclic Carbene Complexes of Ruthenium, J. Am. Chem. Soc., 2002, 124, 4944–4945 CrossRef CAS PubMed;
(b) R. A. Diggle, S. A. Macgregor and M. K. Whittlesey, Computational Study of C-C Activation of 1,3-Dimesitylimidazol-2-ylidene (IMes) at Ruthenium: The Role of Ligand Bulk in Accessing Reactive Intermediates, Organometallics, 2008, 27, 617–625 CrossRef CAS.
- T. Bolano, M. L. Buil, M. A. Esteruelas, S. Izquierdo, R. Lalrempuia, M. Olivan and E. Onate, C-C Bond Activation of the NHC Ligand of an Osmium-Amido Complex, Organometallics, 2010, 29, 4517–4523 CrossRef CAS.
- S. Sakurai and M. Tobisu, Iridium-Mediated Arylation of Quinoline via the Cleavage of Carbon−Carbon and Carbon−Nitrogen Bonds of 1,3-Dimesitylimidazol-2-ylidene, Organometallics, 2019, 38, 2834–2838 CrossRef CAS.
- S. Onodera, S. Ishikawa, T. Kochi and F. Kakiuchi, Direct Alkenylation of Allylbenzenes via Chelation-Assisted C−C Bond Cleavage, J. Am. Chem. Soc., 2018, 140, 9788–9792 CrossRef CAS PubMed.
- C. Qin, W. Zhou, F. Chen, Y. Ou and N. Jiao, Iron-Catalyzed C-H and C-C Bond Cleavage: A Direct Approach to Amides from Simple Hydrocarbons, Angew. Chem., Int. Ed., 2011, 50, 12595–12599 CrossRef CAS PubMed.
- F. Chen, C. Qin, Y. Cui and N. Jiao, Implanting Nitrogen into Hydrocarbon Molecules through C-H and C-C Bond Cleavages: A Direct Approach to Tetrazoles, Angew. Chem., Int. Ed., 2011, 50, 11487–11491 CrossRef CAS PubMed.
- C. Qin, T. Shen, C. Tang and N. Jiao, FeCl2-Promoted Cleavage of the Unactivated C-C Bond of Alkylarenes and Polystyrene: Direct Synthesis of Arylamines, Angew. Chem., Int. Ed., 2012, 51, 6971–6975 CrossRef CAS PubMed.
- Y. Adeli, K. Huang, Y. Liang, Y. Jiang, J. Liu;, S. Song, C.-C. Zeng and N. Jiao, Electrochemically Oxidative C−C Bond Cleavage of Alkylarenes for Anilines Synthesis, ACS Catal., 2019, 9, 2063–2067 CrossRef CAS.
-
(a) J. Liu, X. Qiu, X. Huang, X. Luo, C. Zhang, J. Wei, J. Pan, Y. Liang, Y. Zhu, Q. Qin, S. Song and N. Jiao, From Alkylarenes to Anilines via Site-directed Carbon–carbon Amination, Nat. Chem., 2019, 11, 71–77 CrossRef CAS PubMed;
(b) A. Xia, X. Qi, X. Mao, X. Wu, X. Yang, R. Zhang, Z. Xiang, Z. Lian, Y. Chen and S. Yang, Metal-Free Aerobic Oxidative Selective C−C Bond Cleavage in Heteroaryl-Containing Primary and Secondary Alcohols, Org. Lett., 2019, 21, 3028–3033 CrossRef CAS PubMed.
- J. Liu, X. Wen, C. Qin, X. Li, X. Luo, A. Sun, B. Zhu, S. Song and N. Jiao, Oxygenation of Simple Olefins through Selective Allylic C-C Bond Cleavage: A Direct Approach to Cinnamyl Aldehydes, Angew. Chem., Int. Ed., 2017, 56, 11940–11944 CrossRef CAS PubMed.
- J. Qiu and R. Zhang, DDQ-Promoted Direct Transformation of Benzyl Hydrocarbons to Amides via Tandem Reaction of the CDC Reaction and Beckmann Rearrangement, Org. Biomol. Chem., 2013, 11, 6008–6012 RSC.
- J. Liu, C. Zhang, Z. Zhang, X. Wen, X. Dou, J. Wei, X. Qiu, S. Song and N. Jiao, Nitromethane as A Nitrogen Donor in Schmidt-Type Formation of Amides and Nitriles, Science, 2020, 367, 281–285 CrossRef CAS PubMed.
- A. K. Singh, M.-G. Kim, H.-J. Lee, R. Singh, S. H. Cho and D.-P. Kima, Direct Aryl-Aryl Coupling without Pre-Functionalization Enabled by Excessive Oxidation of Two-Electron Ag(I)/Ag(III) Catalyst, Adv. Synth. Catal., 2018, 360, 2032–2042 CrossRef CAS.
-
(a) W.-M. Cheng, R. Shang, B. Zhao, W.-L. Xing and Y. Fu, Isonicotinate Ester Catalyzed Decarboxylative Borylation of (Hetero)Aryl and Alkenyl Carboxylic Acids through N-Hydroxyphthalimide Esters, Org.
Lett., 2017, 19, 4291–4294 CrossRef CAS PubMed;
(b) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Decarboxylative Borylation, Science, 2017, 356, eaam7355 CrossRef PubMed;
(c) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Photoinduced Decarboxylative Borylation of Carboxylic Acids, Science, 2017, 357, 283–286 CrossRef CAS PubMed;
(d) D. Hu, L. Wang and P. Li, Decarboxylative Borylation of Aliphatic Esters under Visible-Light Photoredox Conditions, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS PubMed.
- P.-F. Dai, X.-S. Ning, H. Wang, X.-C. Cui, J. Liu, J.-P. Qu and Y.-B. Kang, Cleavage of C(aryl)-CH3 Bonds in the Absence of Directing Groups under Transition Metal Free Conditions, Angew. Chem., Int. Ed., 2019, 58, 5392–5395 CrossRef CAS PubMed.
-
(a) S. Maiti and P. Mal, Dehydrogenative Aromatic Ring Fusion for Carbazole Synthesis via C−C/C−N Bond Formation and Alkyl Migration, Org. Lett., 2017, 19, 2454–2457 CrossRef CAS PubMed;
(b) A. Bal, S. Maiti and P. Mal, Iodine(III)-Enabled Distal C-H Functionalization of Biarylsulfonanilides, J. Org. Chem., 2018, 83, 11278–11287 CrossRef CAS PubMed.
- J. Li, A. Bauer, G. D. Mauro and N. Maulide, α-Arylation of Carbonyl Compounds through Oxidative C-C Bond Activation, Angew. Chem., Int. Ed., 2019, 58, 9816–9819 CrossRef CAS PubMed.
- Y. Ishida, I. Nakamura and M. Terada, Copper-Catalyzed Domino [1,3]/[1,2] Rearrangement for the Efficient Synthesis of Multisubstituted ortho-Anisidines, J. Am. Chem. Soc., 2018, 140, 8629–8633 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
Yan-Biao
Kang
*a and
Yan-Biao
Kang