
Selective production of 5-hydroxymethylfurfural from fructose in the presence of an acid-functionalized SBA-15 catalyst modified with a sulfoxide polymer†
Mariah R.
Whitaker
,
Aamena
Parulkar
 and
Nicholas A.
Brunelli
*
and
Nicholas A.
Brunelli
*
The Ohio State University, William G. Lowrie Department of Chemical and Biomolecular Engineering, 151 W. Woodruff Ave., Columbus, OH 43210, USA. E-mail: brunelli.2@osu.edu; Web: www.twitter.com/OSUChemEProfBru
First published on 27th September 2019
Abstract
Biomass is a renewable carbon feedstock that can be converted to 5-hydroxymethylfurfural (HMF), a useful platform chemical that can be modified to produce valuable chemicals and fuels. Previous research has shown that high HMF selectivity can be achieved in organic solvents such as dimethyl sulfoxide (DMSO) because of its capability to stabilize HMF in solution, but DMSO is an undesirable solvent to use industrially as product separation from the reaction solution is difficult. Surface functionalization of porous catalysts has been shown as a method to introduce solvent-effects at the surface of heterogeneous catalysts, thus avoiding the need for high boiling solvents like DMSO. Poly(ethylene sulfoxide) (PESO) is added to the surface of sulfonic acid (SA) functionalized SBA-15 silica to obtain the bifunctional catalyst SA-PESO-SBA-15. Co-localization of the sulfoxide polymer with sulfonic acid groups inside the catalyst pores (SA-PESO-SBA-15) increased HMF selectivity to 51% from 26% obtained by monofunctional SA-SBA-15 at 27% fructose conversion in water. Additionally, this bifunctional catalyst performs best in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) THF:water cosolvent, a more industrially preferred cosolvent system, obtaining 79% HMF selectivity at 87% fructose conversion. Overall, these materials are promising for the selective conversion of fructose to HMF.
:1 (w/w) THF:water cosolvent, a more industrially preferred cosolvent system, obtaining 79% HMF selectivity at 87% fructose conversion. Overall, these materials are promising for the selective conversion of fructose to HMF.
Nicholas Brunelli is an Assistant Professor of Chemical and Biomolecular Engineering (CBE) at The Ohio State University. He received his undergraduate degree in CBE from OSU (2004) and his doctoral degree in Chemical Engineering from the Caltech (2010) supported by a National Science Foundation Graduate Research Fellowship. Dr. Brunelli's current research program focuses on controlled synthesis of materials to create new materials and to better elucidate structure–function behavior in the field of catalysis. This research has been recognized with the 2018 ACS Influential Researcher Award, 2019 Lumley Research Award, 2019 AIChE Futures, and the National Science Foundation CAREER Award (2017). |
Design, System, ApplicationSelective 5-hydroxymethylfurfural (HMF) production from biomass in an economically viable process is a crucial challenge to be overcome on our way toward sustainable chemical and fuel production. Currently, the most efficient way to achieve high HMF selectivity at high conversions is utilize high boiling polar aprotic organic solvents such as dimethyl sulfoxide (DMSO). Here, we have designed a heterogeneous acid catalyst that incorporates a DMSO-like polymer (poly(ethylene sulfoxide) or PESO) within its pores to achieve similar solvation effects as DMSO at the molecular level within the nanocomposite catalyst. We find that co-localization of the DMSO-like polymer and sulfonic acid sites within the catalyst support is a crucial design aspect to making a selective catalyst. Since beneficial solvation interactions are designed into to catalyst structure, this catalyst has the potential to be used in presence of a basic heterogeneous catalyst to perform one-pot tandem reactions in water starting from the glucose isomerization to fructose, which is subsequently dehydrated to HMF selectively. |
1. Introduction
Sustainable production of chemicals and fuels can be achieved through utilizing biomass as a renewable carbon feedstock to produce compounds identified by the US Department of Energy to be most desirable such as 5-hydroxymethylfurfural (HMF).1–3 HMF can be produced from lignocellulose hydrolysis into C6 hexoses glucose and fructose that can further be dehydrated to produce HMF. However, the economic viability of biomass processing is hindered by several aspects, including the low selectivity for fructose dehydration to HMF, especially in water.1,4–6 Although water is a common solvent to use in sugar chemistry, HMF selectivity is low in water because HMF is rehydrated to form several byproducts.1 To prevent HMF rehydration and increase selectivity, organic solvents have been utilized either as a cosolvent7–10 or as an extracting solvent in a biphasic reactor to remove HMF from the aqueous reaction phase.11,12 Although organic solvents have been shown to be beneficial, they have some limitations that can increase processing costs.13 Thus, identifying methods to increase HMF selectivity in an economically viable way represents a critical research need.Organic solvents can be used to increase HMF selectivity significantly.10 By incorporating an organic solvent in water, the hydrogen bonding and dipole moments of the solvent change, affecting the thermodynamics of reactants, transition states, intermediates, and products. The different solvents can also impact the reaction rate, catalyst activity, and reaction mechanism.14 Water miscible organic solvents such as dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP), γ-valerolactone (GVL), and tetrahydrofuran (THF), are commonly used as cosolvents with water, which maintains fructose solubility while promoting HMF selectivity. Of these organic solvents, DMSO has been shown to obtain greater than 80% selectivity at high fructose conversions. DMSO has also been extensively studied to understand its unique properties. In DMSO, the mechanistic pathway that is selective for HMF is favored because the solvent increases the concentration of the β-fructofuranose isomer in solution.7,15–20 DMSO also promotes selectivity in that it stabilizes HMF through hydrogen bonding interactions thus minimizing rehydration reactions to organic acid byproducts such as levulinic acid and formic acid.21–24 Additionally, DMSO increases the reaction rate of acid catalyzed reactions by stabilizing initial and transition states.25,26 Finally, DMSO stably promotes the fructose dehydration reaction at typical reaction conditions and does not degrade in situ to strong acid species at these conditions.27 Although DMSO has been shown to increase fructose dehydration rate and increase HMF selectivity, the high boiling point of this solvent make for difficult and costly separation of HMF from the reaction mixture.9 The conditions (temperatures of 180 °C) required to separate DMSO would result in significant DMSO degradation.27 Therefore, it is advantageous to avoid the use of high-boiling organic solvents to reduce the energy costs for separation.
Current research has turned toward creating engineering solutions such as (a) heterogeneous catalysis design and (b) biphasic reactors to overcome this limitation while still leveraging the beneficial solvent interactions provided by organic solvents. One engineering solution involves designing heterogenous catalysts incorporating organic solvent-like functional groups on the catalyst surface and/or within its structure.11,28–31 For the selective dehydration of fructose to HMF, researchers have examined several materials, including organosilane functionalized mesoporous silica,30 and synthetic polymers including porous organic polymers (POPs)11,31 and polymer nanocomposites.28 Of these, functionalized mesoporous silica provides facile immobilization of various functional groups. Dumesic, Scott, and coworkers functionalized SBA-15 silica catalysts by co-condensation methods using a bifunctional organosilane containing a thioether and sulfonic acid functional group.29,30 For comparison, the thioether is oxidized to a sulfone to promote HMF selectivity similar to DMSO.30 These catalysts were used for fructose dehydration reactions preformed in a biphasic reactor. A biphasic reactor has an aqueous phase where the reaction takes place and an immiscible organic liquid phase that continuously extracts HMF from the aqueous layer limiting byproduct formation.11,12 Performing the dehydration reaction with these catalysts in a biphasic reactor further increases the HMF selectivity and yield.
Alternatively, synthetic organic polymers are able to achieve higher functional group loadings and have been used in the form of linear polymers,11 porous organic polymers (POPs),31 or polymer nanocomposites28 for biomass conversion reactions. Román-Leshkov et al. included the linear polymer poly(1-vinyl-2-pyrrolindinone) (PVP) in the aqueous phase of a biphasic reactor to mimic the organic solvent N-methyl-2-pyrrolidone (NMP) to increase HMF selectivity. By incorporating PVP and using 7:3 methyl isobutyl ketone:2-butanol as the immiscible extracting organic solvent, an HMF selectivity of 77% was obtained at 90% fructose conversion in presence of HCl catalyst.32 This result indicates that the presence of a polymer with organic solvent functionality could increase HMF selectivity. While PVP has been investigated for catalyst design, there is no literature investigating a DMSO-like polymer such as poly(ethylene sulfoxide) (PESO) as a component for catalyst design in increasing HMF selectivity.
In this work, we describe the synthesis and catalytic performance of a polymer nanocomposite incorporating a DMSO-like polymer poly(ethylene sulfoxide). A DMSO-like polymer is chosen because of the extensive evidence that DMSO increases the fructofuranose concentration,7,20 stabilizes HMF preventing rehydration reactions,21 and increases activity of acid catalysts.26 PESO is covalently bound to mesoporous SBA-15 silica via surface-initiated ring opening polymerization of ethylene sulfide. It is hypothesized that covalent immobilization of the polymer will increase the stability of the polymer nanocomposite catalyst. Secondary immobilization of 3-mercaptopropyl trimethoxysilane is done. The composite material is oxidized with 30% hydrogen peroxide to convert the immobilized sulfide polymer to poly(ethylene sulfoxide) and surface thiols to sulfonic acid groups. The bifunctional nanocomposite catalyst is tested in batch reactions using either water or 4:1 THF:water as the solvent. The bifunctional nanocomposite catalyst obtains 51% HMF selectivity as compared to 26% obtained by SA-SBA-15 at 27% conversion in water. Although this catalyst significantly improves selectivity in water, it performs best in 4:1 (w/w) of THF:water obtaining 79% HMF selectivity at 87% fructose conversion.
2. Experimental methods
2.1. Chemicals
The following chemicals are used as received, including: D-fructose (99%, Alfa Aesar); 5-hydroxymethylfurfural (98%, Ark Pharm Inc.); tetrahydrofuran (ACS grade stabilized, Macron Fine Chemicals); ethylene sulfide (98%, Sigma Aldrich); potassium tert-butoxide (>97%, TCI); 30% hydrogen peroxide (Sigma Aldrich); methanol (HPLC grade, VWR BDH Chemicals); mercaptopropyl trimethoxysilane (Gelest); concentrated sulfuric acid (ACS grade, Fisher); and concentrated hydrochloric acid (ACS grade 36.5–38%, VWR BDH Chemicals). De-ionized (DI) water is obtained from house supply that produces Milli-Q quality water. Toluene (ACS grade, VWR BDH Chemicals) is dried using an MBraun DriSolv System. Dimethyl sulfoxide (DMSO; ACS grade, Fisher) is dried over molecular sieves in a flame dried round bottom flask overnight before using in the synthesis of the unsupported polymer.2.2. Catalytic material synthesis
One gram of PES and 35 mL of THF are placed in a 100 mL round bottom flask. Hydrogen peroxide (33 mL of 30% wt, 262.24 mmol, 16 eq.) is added. The solution is stirred at 600 RPM for 24 hours. After 24 hours, the oxidized polymer poly(ethylene sulfoxide) (PESO) is isolated by vacuum filtration and is washed with 1 L of DI water, and dried in a Petri dish place inside a hood overnight.
SH-SBA-15 (13.5 g) is then suspended in aqueous sodium hydroxide (0.08 M, 325 mL) at 900 RPM for one minute to exchange the protons of the immobilized thiol groups with sodium cations. The solid is then filtered via vacuum filtration and thoroughly washed with DI water (1 L). The sulfur atoms immobilized on the silica surface are basic species that are able to catalyze a surface ring-opening polymerization reaction. The base treated SH-SBA-15 (8 g) is dried in a flask with a stir bar on a high vacuum line overnight at 90 °C. DMSO (200 mL) is added to the flask and the solid is suspended for an hour under nitrogen with the flask immersed in a cold-water bath. To attach PES at 40 wt% to SBA-15, 5.4 grams (5.3 mL) of ethylene sulfide is added dropwise via a syringe to the flask. For 80 wt% PES incorporation, 4 grams (3.96 mL) is added to 1 gram of base treated SH-SBA-15 in the same way in 25 mL of DMSO. The solution is stirred for 24 hours at room temperature. The reaction is quenched by adding methanol (100 mL). The solid is isolated by vacuum filtration and washed with 200 mL methanol. The solid (PES-SBA-15) is degassed at 10 mTorr and 80 °C overnight to remove excess methanol.
To produce the material with only the polymer (no acid sites; PESO-SBA-15), PES-SBA-15 is suspended in THF (same volume as 30% hydrogen peroxide added). Hydrogen peroxide (30% wt, 20 eq. of H2O2 to sulfide) is added to the reaction flask. The solution is stirred for 24 hours at 600 RPM at room temperature. The solid is filtered by vacuum filtration and washed with DI water (1 L). PESO-SBA-15 is degassed at 80 °C under reduced pressure. Exact masses and volumes used for the PESO-SBA-15 at 40 and 80 wt% PESO are outlines in Table S1 in ESI.†
For the bifunctional material containing the polymer and acid sites (SA-PESO-SBA-15), 3-MPTMS is grafted to PES-SBA-15 using the same procedure as discussed previously with the target loading of 1 mmol 3-MPTMS g−1 loading for PES-SBA-15 materials of 40 and 80 wt%. Exact masses used in this synthesis are detailed in Table S2 in ESI.† After grafting, SH-PES-SBA-15 is suspended in 50/50 volume mixture of THF and 30% hydrogen peroxide (20 eq.). Exact masses used in this synthesis are detailed in Table S3 in ESI.† The mixture is stirred for 24 hours at room temperature. The solid is filtered by vacuum filtration and washed with DI water (100 mL per gram catalyst). Once the solid is dry on the filter paper, it is suspended in 1 M sulfuric acid solution (25 mL per gram catalyst) for 30 minutes at room temperature. The solid is filtered with vacuum filtration and washed with 1 L of DI water.
2.3. Catalytic material characterization
The materials are characterized using many standard techniques, including Raman Spectroscopy, Thermogravimetric Analysis, Transmission FTIR and Attenuated Total Reflectance FTIR, and nitrogen physisorption. Raman Spectroscopy is performed using a Renishaw inVia Raman Microprobe at a wavelength setting of 633 nm, at 20% intensity for the Ar laser, at an objective setting of 50× for the microscope, and using a gradient of 1800. Thermogravimetric Analysis (TGA) is performed using a Netzsch Jupiter STA 449 F5 from 20–900 °C at a ramp rate of 20 K min−1 for 1:28 minutes under the constant purge of nitrogen and air at 20 mL min−1 each. Transmission and Attenuated Total Reflectance FTIR are performed on a ThermoScientific instrument using KBr pellets and a Smart iTX accessory. KBr pellets are prepared by diluting 1 mg of catalyst to a 100 mg mixture with KBr. The solid mixture is placed in a die and pressed into a pellet using a hydraulic press set at a pressure of 10000 psi for 3 hours. Nitrogen physisorption analysis is conducted using a Micromeritics Instrument. Before analysis, the polymer nanocomposite materials and bare SBA-15 are degassed for 24 hours at 40 °C.
As a control, 0.33 grams of the prepared Ellman's solution is diluted to a 20 grams solution with DI water. The diluted solution (3 mL in cuvette) is analyzed by UV-vis. The only absorbance peak observed is at 330 nm corresponding to DNTB. This spectrum is compared to that obtained from catalyst titration samples to determine the extent of NTB2− evolution. To titrate catalyst samples (SH-PES-SBA-15 or SA-PESO-SBA-15), 20 mg of catalyst solid is suspended in 10 mL 3 mM Ellman's solution for 24 hours. The solid is removed by vacuum filtration. The filtrate (0.33 grams) is diluted to 20 grams with DI water. The diluted solution (3 mL) is added to a cuvette, and the absorbance of the solution is collected at 412 nm using the buffer solution as the background. The concentration of surface thiols is calculated from the amplitude of the absorbance peak at 412 nm using the calibration curve. Duplicates are performed for each sample, and the average thiol loading is reported in mmol g−1.
2.4. Catalytic testing
All reactions are performed at 120 °C and an agitation rate of 420 RPM in presence of 2 mol% acid catalyst starting from a fructose concentration of 0.5 mmol per gram solution. Before adding the reaction mixture, the thick-walled glass pressure tube containing a magnetic stir bar is weighed with the cap and O-ring. Two grams of a stock solution in either water or 4:1 (w/w) THF:water is placed in the tube, before immersing the capped tube into an oil bath heated to 120 °C to a depth that the level of the oil bath is just above the liquid level of the reaction solution in tube. After the desired amount of time, the tube is removed from the oil bath and is immersed in an ice bath to quench the reaction. After the pressure tube has cooled to room temperature, the tube with the cap on is weighed to confirm that no mass has been lost. The reaction solution is diluted by 4 grams of DI water before the tube is capped and shaken vigorously. The diluted solution is filtered with a 0.22 μm syringe filter, and then 1.5 grams of the filtrate is placed in a vial and analyzed with High Performance Liquid Chromatography (HPLC). A Waters Sugar Pak 1 column equipped with the corresponding guard column is used to separate compounds at 70 °C, a 0.6 mL min−1 flow rate of DI water, and an injection volume of 10 μL. Fructose and HMF concentrations are determined by using Refractive Index and UV-vis detectors, respectively. For reactions performed in water, an aqueous stock solution of fructose at a concentration of 0.5 mmol per gram solution is prepared. Two grams of this solution is weighed into a 15 mL thick walled glass pressure tube sealed with a Teflon cap containing a back sealing FETFE O-ring. For reactions performed in 4:1 (w/w) THF:water, an aqueous stock solution of fructose at a concentration of 2.5 mmol per gram solution is prepared. This solution (0.4 grams) is diluted with 1.6 grams of THF in a 5 mL thick walled glass pressure tube sealed with a Teflon cap containing a back-sealing, THF resistant PTFE O-ring. It is necessary to prepare 4:1 (w/w) THF:water reaction solutions in this way as this mixture can separate into two liquid phases, making preparation of HPLC samples with accurate concentrations difficult.
2.5. Catalyst recycle tests
Three pressure tubes each containing 2 mol% SA-PESO-40-SBA-15 catalyst and 2 grams of 4:1 (w/w) THF:water containing fructose (0.5 mmol g−1 solution) are immersed in an oil bath pre-heated to 120 °C. After seven hours of reaction time, the pressure tubes are immersed in an ice bath to quench the reaction. After the reaction solution is diluted by four grams of DI water, the catalyst is removed using vacuum filtration. The filtrate (1.5 grams) is filtered again by passing it through a 0.22 μm syringe filter into a HPLC vial for analysis. The catalyst is then washed with 26 grams of DI water. The catalyst with the filter paper is dried in a Petri dish open to air inside a hood overnight. From one of the three pressure tubes, the post reaction material (used 1 time) is characterized, using TGA, nitrogen physisorption, and ATR-FTIR. The post reaction material in the other two pressure tubes are dried in a hood before using in a second 7 hour reaction in 4:1 (w/w) THF:water. This procedure is repeated two more times to collect material characterization data after three catalyst uses.
3. Results and discussion
3.1. Material synthesis and characterization of PESO
Poly(ethylene sulfoxide) (PESO) has been reported to be a potential polymer homologue of DMSO since it exhibits similar chemical properties.33 PESO is synthesized by the methods described in Scheme 1 and is characterized to determine the effectiveness of the oxidation method. Solubility issues make it difficult to characterize PESO with classical techniques such as Gel Permeation Chromatography (GPC) or NMR, but it has been determined that FTIR, Raman, and TGA can be effective. TGA curves are shown in Fig. 1 that correspond to PES and PESO. The TGA curve for PES is observed to have two mass losses. The first mass loss occurring around 350 °C corresponds to combustion of poly(ethylene sulfide) (PES). There is a second mass loss around 400 °C corresponds to oxidized sulfur that likely formed under TGA conditions, consistent with previous work.33 The TGA curve of the polymer after oxidation with 30% hydrogen peroxide has changed considerably with the first mass loss occurring around 200 °C. The second mass loss step at 400 °C is much larger indicating the presence of sulfoxide units. This suggests that the polymer has been transformed from PES to a new polymer that is oxidized.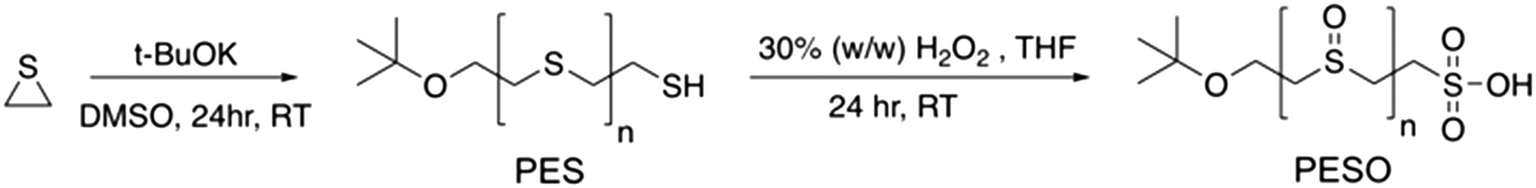 | ||
| Scheme 1 Synthesis of poly(ethylene sulfoxide) (PESO). | ||
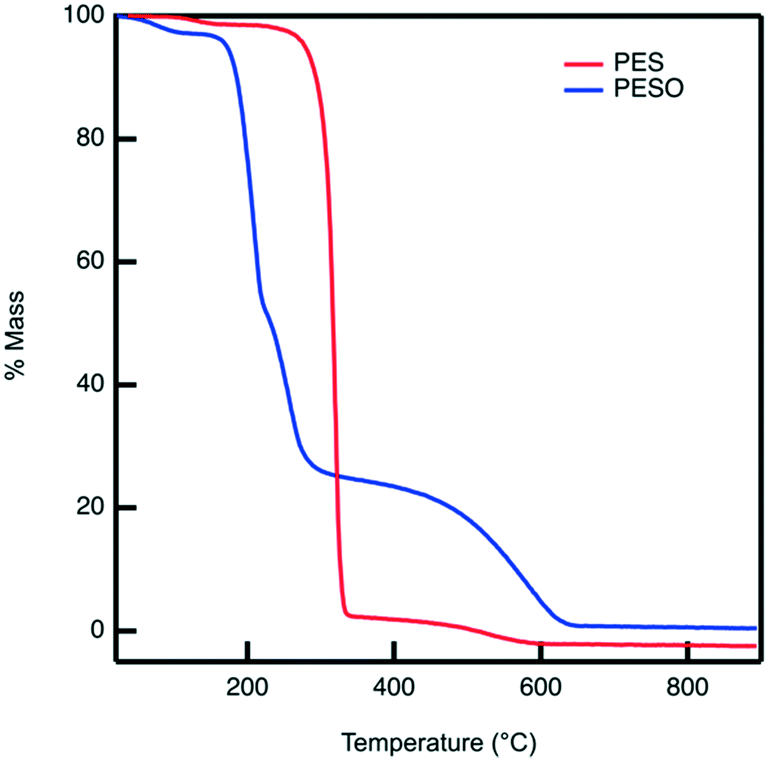 | ||
| Fig. 1 TGA curves for PES (red) and PESO (blue). Catalyst samples are degassed on a high vacuum line for two hours before doing the analysis. Dried catalyst (7 mg) is weighed in a tared aluminum oxide crucible before the analysis is performed from 20–900 °C at a ramp rate of 20 K min−1 under the constant purge of nitrogen and air at 20 mL min−1 each. | ||
The oxidized polymer could consist of sulfoxide, sulfone, and sulfonic acid functional groups. Since the synthesis procedure for the sulfide polymer involves a ring-opening polymerization of ethylene sulfide via potassium tert-butoxide, the terminal groups of PES are a tert-butyl group and a thiol group. Upon oxidation with 30% hydrogen peroxide, the thiol terminal groups are oxidized to sulfonic acid groups. The relative concentration of sulfoxide to sulfone and sulfonic acid groups in the polymer can be determined by transmission FTIR. The peaks observed in the ATR-FTIR spectrum of PESO in Fig. 2 are deconvoluted using a data plotting software. The peaks at 1024, 1257, and 1321 cm−1 correspond to the presence of sulfoxide, sulfonic acid, and sulfone functional groups, respectively. The relative percentage of each sulfinyl species is calculated by using extinction coefficients for the respective functional groups reported in literature.39,40 From the data shown in Fig. 2, the relative percentage of sulfinyl species obtained in PESO is 92% sulfoxide, 5% sulfonic acid, and 3% sulfone.
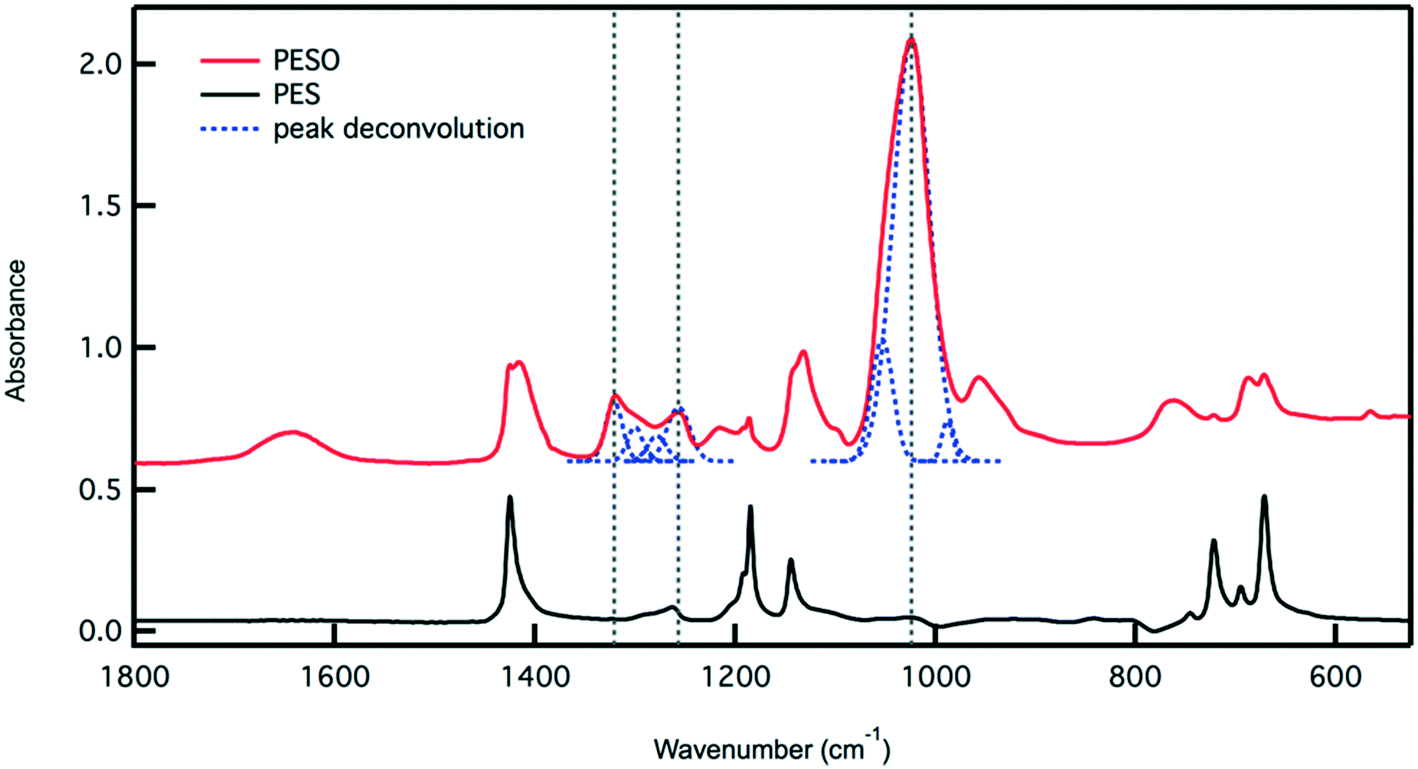 | ||
Fig. 2 Transmission IR spectrum for PESO and PES. The peaks in the PESO spectrum are deconvoluted and integrated. Taking into account the extinction coefficients of sulfoxide,40 sulfone and sulfonic acid,41 the relative percentage of each sulfinyl is calculated. The integrated peaks used for calculation occur at 1024 (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1257 (–SO3H), and 1321 (–SO2) cm−1. These peaks are denoted using a vertical dashed line. The peak deconvolution is also presented as a dashed line. O), 1257 (–SO3H), and 1321 (–SO2) cm−1. These peaks are denoted using a vertical dashed line. The peak deconvolution is also presented as a dashed line. | ||
3.2. Material synthesis and characterization of supported PESO
An organic–inorganic composite of PESO and SBA-15 is made in two manners: (1) a monofunctional material containing only PESO and (2) a bifunctional material with PESO and sulfonic acid sites. The benefit of incorporating PESO in SBA-15 is that this increases the efficiency of separating the polymer from the reaction solution. Indeed, the unsupported polymer often sticks to the filter paper when filtered. Initially, PESO is polymerized from the surface of SBA-15 at different weight percent of 20, 40, and 80 for use with homogeneous sulfuric acid for the fructose dehydration reaction in water.The composite materials are characterized with thermogravimetric analysis to quantify the amount organic content loaded onto the silica support. As shown in Fig. 3, the TGA curves for PES-SBA-15 and PESO-SBA-15 are observed to have mass loss at similar temperatures as unsupported PES and PESO, respectively. The ATR-FTIR spectra for these composite materials depicted in Fig. 4 also are similar to the IR spectrum for the unsupported polymers in Fig. 2. This suggests that the organic content present in SBA-15 indeed corresponds to PES and PESO. The FTIR spectra depicted in Fig. 4 show that the ratio of the intensities for the SO peak (1016–1030 cm−1) to the Si–O peak (1042 cm−1) decreases as the weight percent of polymer incorporation decreases. The sulfoxide peak is not visible in the spectra for PESO-20-SBA-15, because it is smaller than the broad Si–O peak, which ultimately overlaps the sulfoxide peak. The disappearance of peaks at 673 and 726 cm−1 that correspond to the sulfide polymer in the spectrum for PESO-20-SBA-15 suggests that the sulfide polymer is mostly oxidized.
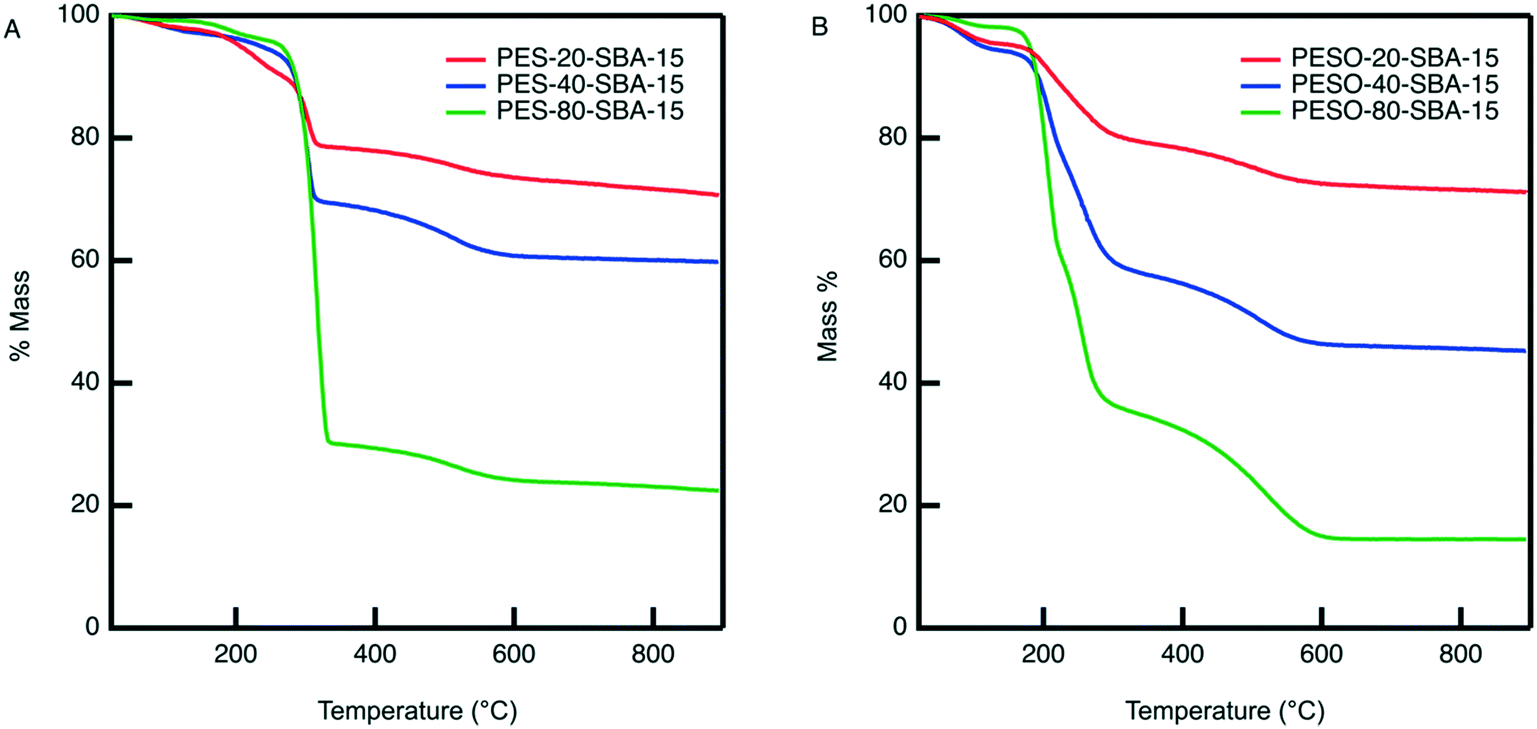 | ||
| Fig. 3 TGA curves of (A) PES-SBA-15 and (B) PESO-SBA-15 at different weight percent polymer incorporated into SBA-15. Catalyst samples are degassed on a high vacuum line for two hours before doing the analysis. Dried catalyst (7 mg) is weighed in a tared aluminum oxide crucible before the analysis is performed from 20–900 °C at a ramp rate of 20 K min−1 under the constant purge of nitrogen and air at 20 mL min−1 each. | ||
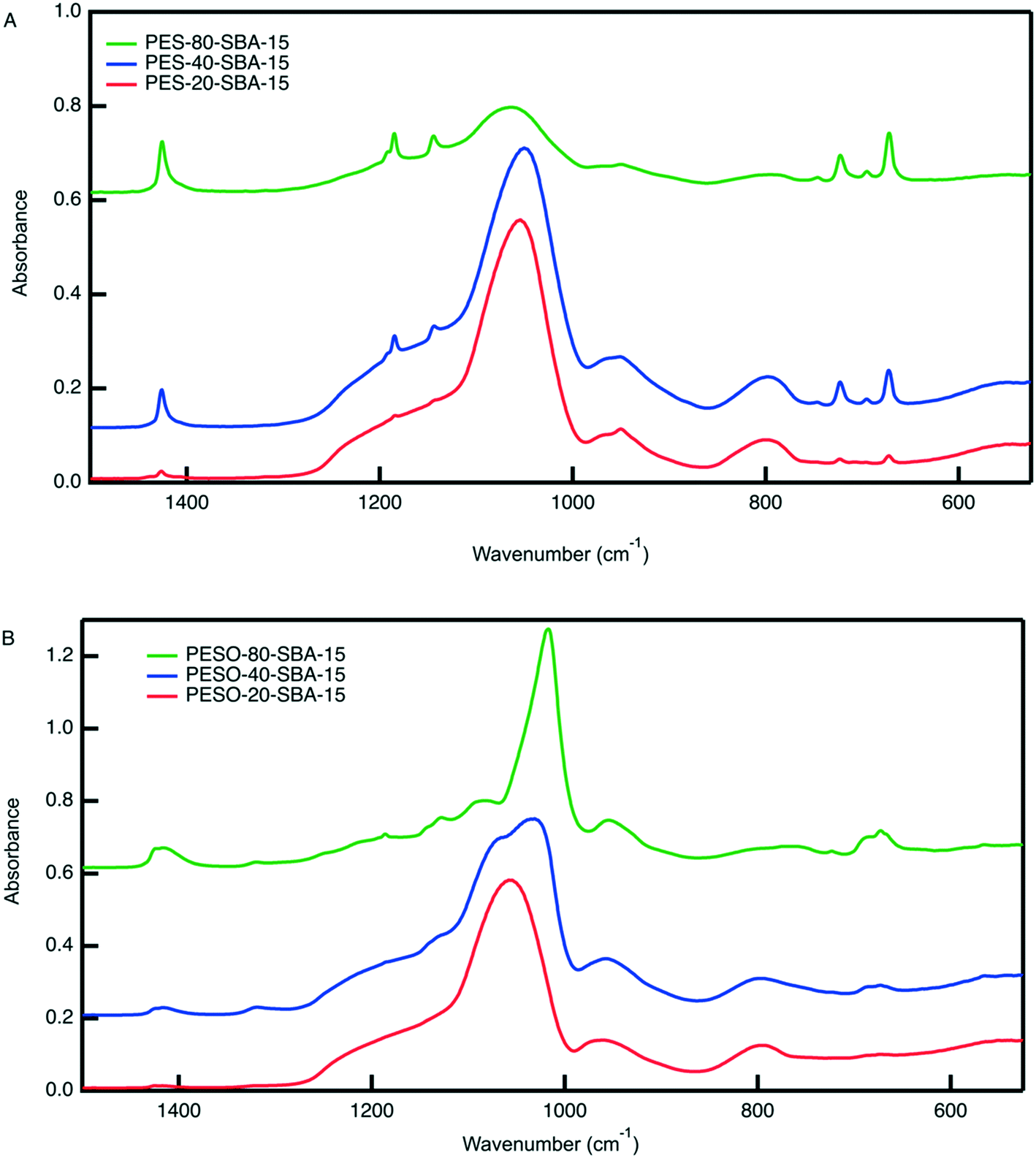 | ||
| Fig. 4 ATR-FTIR spectra for (A) PES-SBA-15 and (B) PESO-SBA-15 at different weight percent polymer in SBA-15. The Si–O peak that corresponds to the Si–O bonds in SBA-15 occurs at 1042 cm−1. The peaks unique to PES are located at the following wavenumbers: 672 (symmetric rocking, –CH2–), 696, 723, 1184 (asymmetric twisting, –CH2–), and 1425 cm−1 (symmetric stretching, –CH2–).42 The sulfoxide peak occurs in the range of 1016–1030 cm−1. | ||
These materials are also characterized by nitrogen physisorption to examine their textural properties. While bare SBA-15 has a BET surface area of 920 m2 g−1, incorporating PESO in its pores reduces its surface area, as summarized in Table 1. The isotherms for PESO-SBA-15 materials in Fig. S3† show that even at 80 wt% polymer incorporation, a hysteresis is observed, indicating that the material remains porous even at high polymer incorporation.
| Material | Weight percent organic content – TGA (%) | Calculated weight percent of PESO in SBA-15 (%) | Acid loading (mmol g−1) | BET surface area (m2 g−1 STP) | BdB-FHH pore size (nm) |
|---|---|---|---|---|---|
| Bare SBA-15 | — | — | — | 920 | 6.7 |
| PESO-20-SBA-15 | 24 | — | — | 340 | 5.6 |
| PESO-40-SBA-15 | 49 | — | — | 230 | 5.4 |
| PESO-80-SBA-15 | 84 | — | — | 100 | 5.4 |
| SA-PESO-40-SBA-15 | 48 | 42 | 0.38 | 220 | 5.2 |
| SA-PESO-80-SBA-15 | 77 | 75 | 0.10 | 81 | 4.4 |
The bifunctional composite materials (SA-PESO-SBA-15) are characterized using the same methods for the monofunctional materials. ATR-FTIR spectra in Fig. S4 and TGA curves in Fig. S5† for the bifunctional materials both show that PES is oxidized to PESO. Acid–base titration of the bifunctional catalyst materials confirms the presence of acidic sites. As listed in Table 1, SA-PESO-40-SBA-15 has a higher acid loading of 0.38 mmol g−1 than SA-PESO-80-SBA-15 at 0.10 mmol g−1 loading. The higher loading of PESO for SA-PESO-80-SBA-15 potentially decreased the grafting efficiency of the secondary immobilization of 3-MPTMS during the synthesis of this catalyst. Additionally, the weight percent of PESO after sulfonic acid immobilization is qualitatively estimated by subtracting the acid loading (mmol g−1 total) measured by acid/base titration from the organic composition obtained from TGA analysis, as summarized in Table 1.
To determine the efficiency in oxidizing surface thiols to sulfonic acid groups, Ellman's titration is performed on SH-PES-SBA-15 and SA-PESO-SBA-15. UV-vis spectroscopy is used to monitor the appearance of an absorbance peak at 412 nm, which corresponds to NTB2− production resulting from reaction between the Ellman's reagent and surface thiols. In Fig. S6.a,† SH-PES-40-SBA-15 is titrated three times resulting in an average thiol loading of 0.36 mmol g−1. This thiol loading is similar to the sulfonic loading of the oxidized material SA-PESO-40-SBA-15 quantified by acid/base titration. Furthermore, Ellman's titration of SA-PESO-40-SBA-15 does not result in NTB2− formation as evidenced by the absence of a peak at 412 nm in Fig. S6.b.† These results indicate that there are no detectible quantities of residual surface thiols in SA-PESO-SBA-15, suggesting that the thiols have been oxidized to sulfonic acid groups. To determine the porosity of these bifunctional composite catalysts, nitrogen physisorption is performed. The Type 4 isotherms in Fig. S7† show that the bifunctional composite materials also remain porous, with pore size and BET surface area decreasing with increasing polymer loading.
3.3. Catalytic testing with unsupported PESO in water
The effect of PESO on HMF selectivity in the presence of homogeneous sulfuric acid (2 mol%) is investigated in a batch reactor using water as a solvent. It is hypothesized that the DMSO-like polymer PESO will provide beneficial solvation interactions similar to DMSO to increase HMF selectivity. After allowing the reaction to proceed for 24 hours, the mixture composition is analyzed to determine fructose conversion and HMF selectivity, as shown in Fig. 5. As a baseline without PESO addition, it is determined that sulfuric acid (2 mol%) achieves HMF selectivity of 43% at a fructose conversion of 30%. With the presence of PESO (200 mg), it is found that the polymer with homogeneous sulfuric acid increases HMF selectivity to 53% at a comparable fructose conversion of 25% conversion. However, HMF selectivity is not further improved when the amount of PESO incorporated is increased to 400 mg.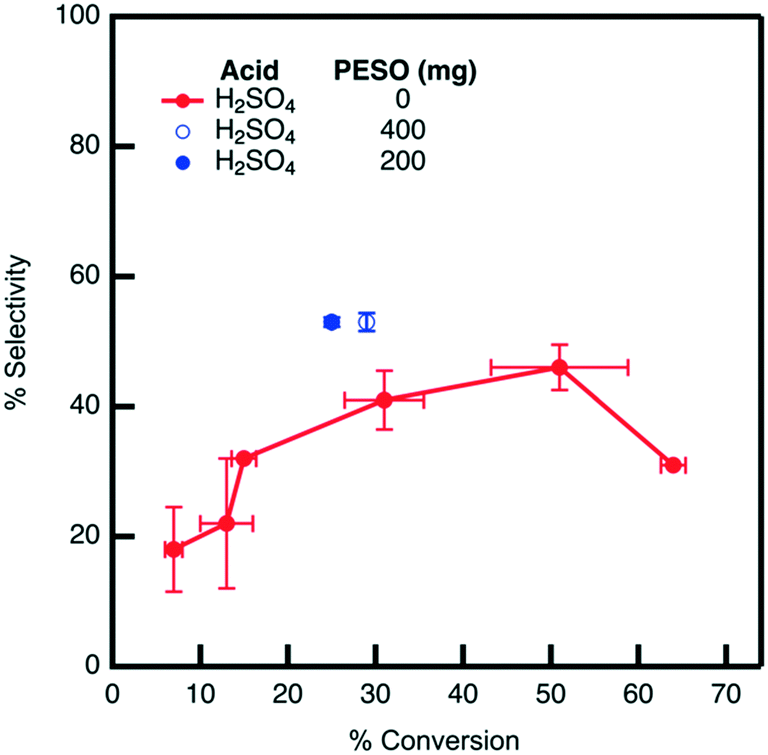 | ||
| Fig. 5 Selectivity versus conversion obtained by sulfuric acid alone (red) and in presence of PESO (blue) in water. The legend lists the mass of PESO incorporated in the reaction solution. All data points correspond to a reaction time of 24 hours. All reactions are performed at 120 °C and an agitation rate of 420 RPM starting from a fructose concentration of 0.5 mmol g−1 solution in water. | ||
3.4. Catalytic testing with supported PESO in water
The difficulty in separating unsupported PESO caused us to examine incorporating PESO on a porous silica support. The effect of PESO immobilized within the pores of SBA-15 silica (PESO-SBA-15) on HMF selectivity in presence of 2 mol% homogeneous sulfuric acid is examined for the fructose dehydration reaction in water. The total amount of PESO incorporated in the reaction solution is fixed to 50 mg for all PESO-SBA-15 materials tested. This requires adjusting the amount of catalyst added according to the percentage of organic provided by TGA. Thus, the effect of polymer loading on catalytic performance can be tested. As shown in Fig. 6, catalytic testing results indicate that the presence of the polymer nanocomposite has no effect on the rate at which homogeneous sulfuric acid catalyzes the reaction. In addition, the polymer nanocomposite material mostly does not increase HMF selectivity beyond what is obtained by homogeneous sulfuric acid alone, which is different than the 10% increase in selectivity that is obtained with unsupported PESO. One potential reasoning for this difference is that PESO supported on SBA-15 and sulfuric acid predominantly exist in separate domains in the reaction solution. The homogeneous sulfuric acid is freely suspended throughout the reaction solution while PESO is confined inside the pores of SBA-15, unlike for the unsupported PESO case where the polymer and sulfuric acid are suspended throughout the reaction solution. Therefore, it is hypothesized that sulfuric acid freely suspended through the reaction solution converts fructose before it can diffuse through the composite material to interact with the polymer confined inside the pores of SBA-15. Because of the absence of polymer-fructose interactions, fructose is not exposed to DMSO-like solvation interactions that promote the concentration of fructofuranose isomers, which is key to promoting HMF selectivity.7,28 Therefore, it is suggested that co-localization of the DMSO-like polymer PESO and sulfonic acid groups throughout the SBA-15 pore channels will increase the effectiveness of the polymer on increasing HMF selectivity.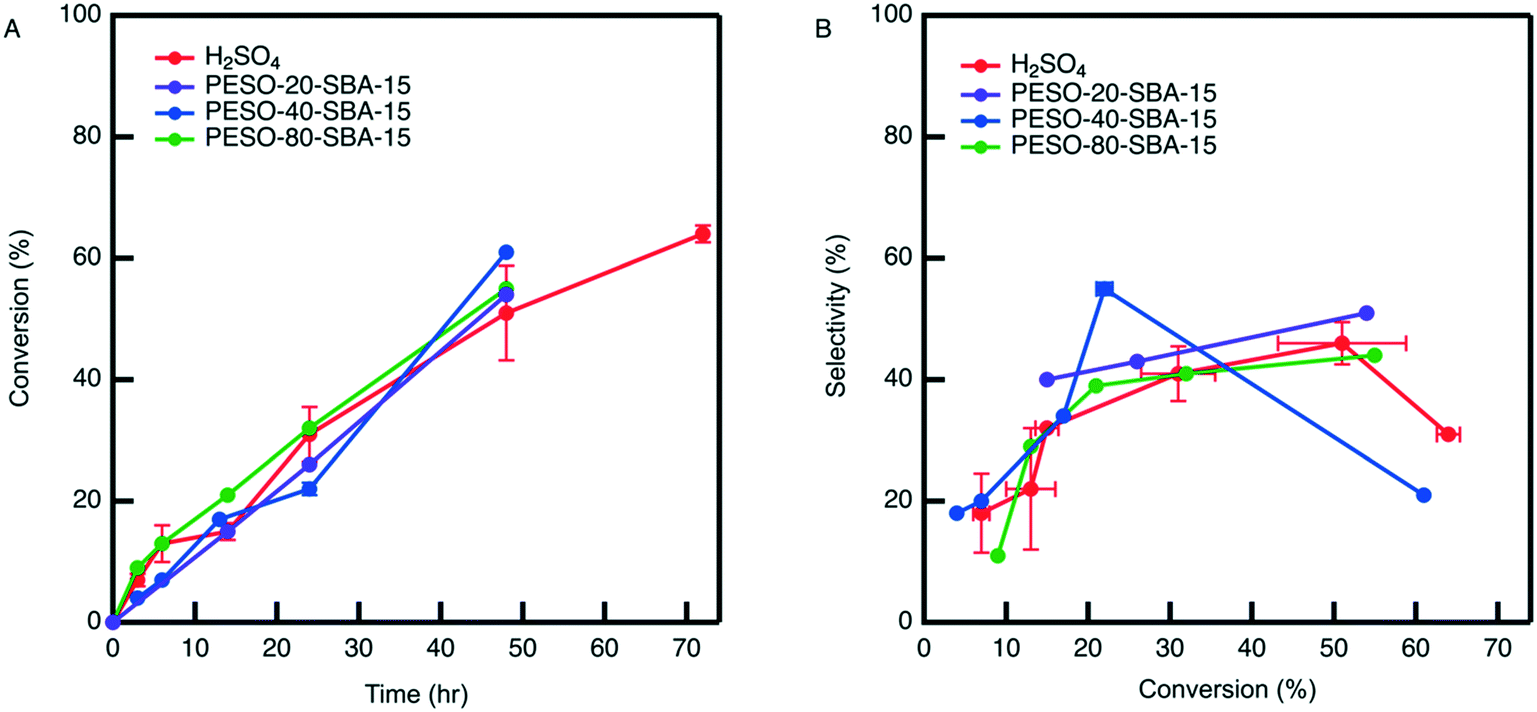 | ||
| Fig. 6 (A) Conversion versus time and (B) selectivity versus conversion results obtained by sulfuric acid alone and in presence of PESO-SBA-15 in water. The legend lists the different PESO-SBA-15 materials of different weight percent PESO. The red curve represents results obtained from homogenous sulfuric acid alone. All reactions are performed at 120 °C and an agitation rate of 420 RPM starting from a fructose concentration of 0.5 mmol g−1 solution in water. Total acid and PESO in the reaction are constrained to 2 mol% and 50 mg, respectively. | ||
To co-localize sulfoxide and sulfonic acid groups within the pores of SBA-15, a bifunctional polymer nanocomposite catalyst is synthesized by grafting sulfonic acid (SA) organosilanes in close proximity to PESO polymerized from the surface of SBA-15. For comparison, the catalytic activity of homogenous sulfuric acid and heterogeneous SA-SBA-15 is investigated. Although the two catalysts have similar catalytic activity, homogenous sulfuric acid obtains significantly higher HMF selectivity in water, as depicted in Fig. S8.†
When compared to SA-SBA-15 in Fig. 7, the co-localization of PESO (40 wt%) with sulfonic acid on SBA-15 increases the HMF selectivity two-fold. However, increasing the polymer weight percent to 80% does not further increase HMF selectivity compared to SA-PESO-40-SBA-15. Fig. 7A shows that the observed rate in presence of SA-PESO-40-SBA-15 is slower compared to that observed with SA-SBA-15 and SA-PESO-80-SBA-15. It is hypothesized that the presence of PESO in the catalyst pores increases time required for diffusion of fructose through the catalyst before reaching the acid sites as compared to SA-SBA-15, which does not have polymer present in its pores. More diffusion limitations exist for SA-PESO-40-SBA-15 as compared to SA-SBA-15 because of fructose interacting with PESO throughout the catalyst pore channels, thus slowing substrate diffusion through the particle. However, these diffusion effects are not observed with SA-PESO-80-SBA-15. It is suggested that these diffusion effects are not observed because initial immobilization of 80 wt% PES during synthesis made the second immobilization of 3-MPTMS unfavorable inside the pores, thus forcing the thiol containing organosilanes to attach solely to the outer surface of silica particles when synthesizing the SH-PES-80-SBA-15 precursor material. Thus, upon diffusion to the catalyst surface, fructose encounters surface sulfonic acid groups before interacting with PESO inside the catalyst pores. Ultimately, the results are similar to PESO-SBA-15 in presence of homogeneous sulfuric acid where sulfoxide and sulfonic acid groups are not co-localized within SBA-15 pores.
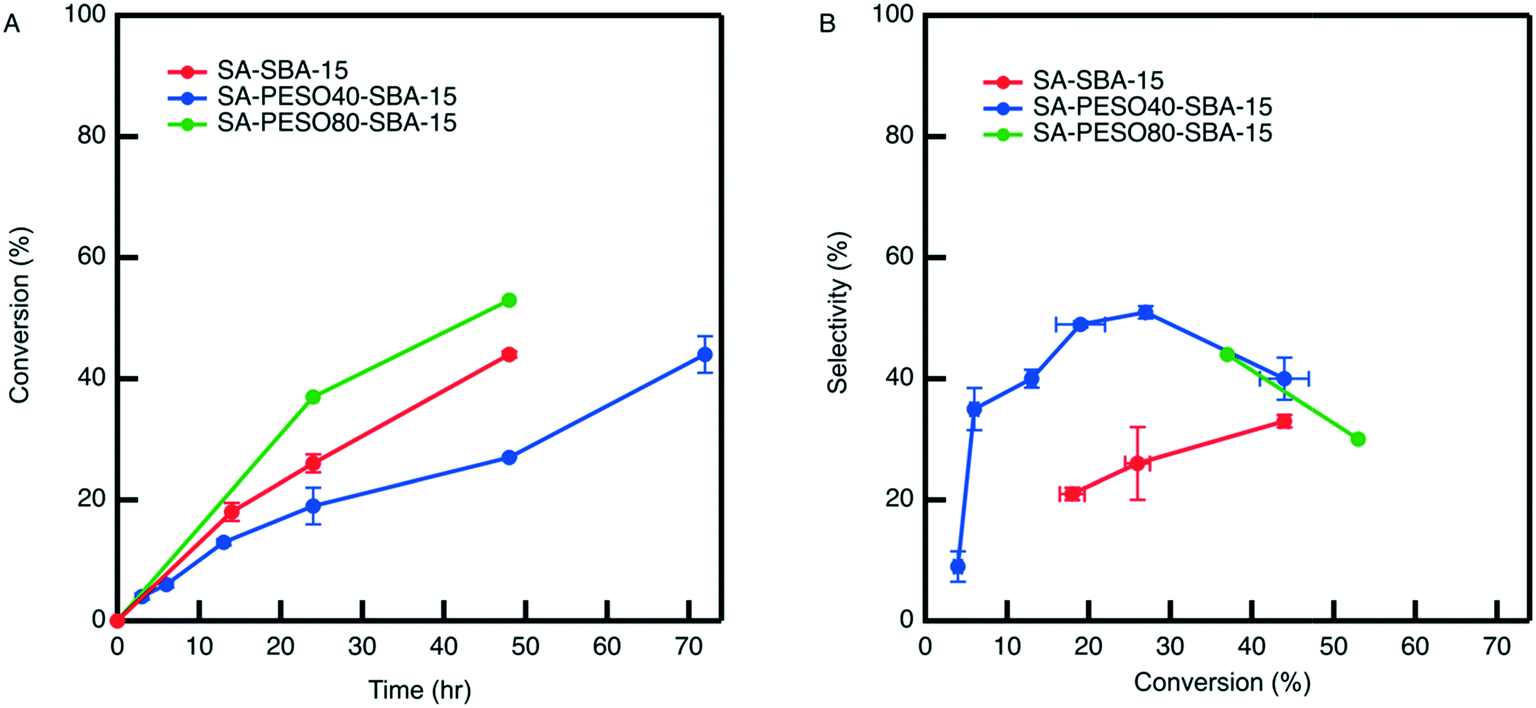 | ||
| Fig. 7 (A) Conversion versus time and (B) selectivity versus conversion results obtained by SA-PESO-SBA-15 of various weight percent PESO in water as a solvent. The red curve represents results obtained from SA-SBA-15 in water. All reactions are performed with 2 mol% acid at 120 °C and an agitation rate of 420 RPM starting from a fructose concentration of 0.5 mmol g−1 solution in water. | ||
One interesting aspect of the porous bifunctional polymer nanocomposite catalysts is that there is a significant drop in HMF selectivity past 30% fructose conversion in water. Mechanistic studies in water have shown that the HMF concentration reaches a maximum and then decreases at longer reaction times because it begins to rehydrate to form organic acids.7 Hence, it explains the reason for the drop in HMF selectivity in presence of the catalysts in water. Although selectivity is increased because of co-localized solvent-like molecules and acidic sites, once HMF diffuses out of the polymer nanocomposite catalyst it enters an aqueous environment and is subject to rehydration reactions.
3.5. Catalytic testing with supported PESO in non-aqueous solvents
Organic solvents limit HMF rehydration and other condensation reactions between reactants and products. THF is an attractive alternative in that its boiling point is lower than that of DMSO, ultimately requiring less energy to separate through distillation and is easily recyclable. To prevent HMF rehydration from occurring in the bulk solvent environment, the dehydration reaction is performed with SA-PESO-40-SBA-15 in a cosolvent of 4:1 (w/w) THF:water. The results depicted in Fig. 8 show that the cosolvent itself is not active for the dehydration, unlike what is observed in DMSO:water cosolvents. However, SA-PESO-40-SBA-15 obtains high catalytic activity and HMF selectivity in 4:1 THF:water, resulting in 79% HMF selectivity at 87% fructose conversion. The observed rate of reaction in presence of SA-PESO-40-SBA-15 in 4:1 THF:water is significantly faster than the rate observed in water. This is consistent with Mellmer et al. found that the rate constant of Brønsted acid catalyzed fructose dehydration in 4:1 THF:water is about 8 times the rate constant observed in water.26 In comparison to SA-SBA-15, the bifunctional catalyst obtains higher HMF selectivity after 6 hours of reaction time. SA-PESO-40-SBA-15 has a slower initial rate than SA-SBA-15 because of the diffusion limitations from the presence of PESO.
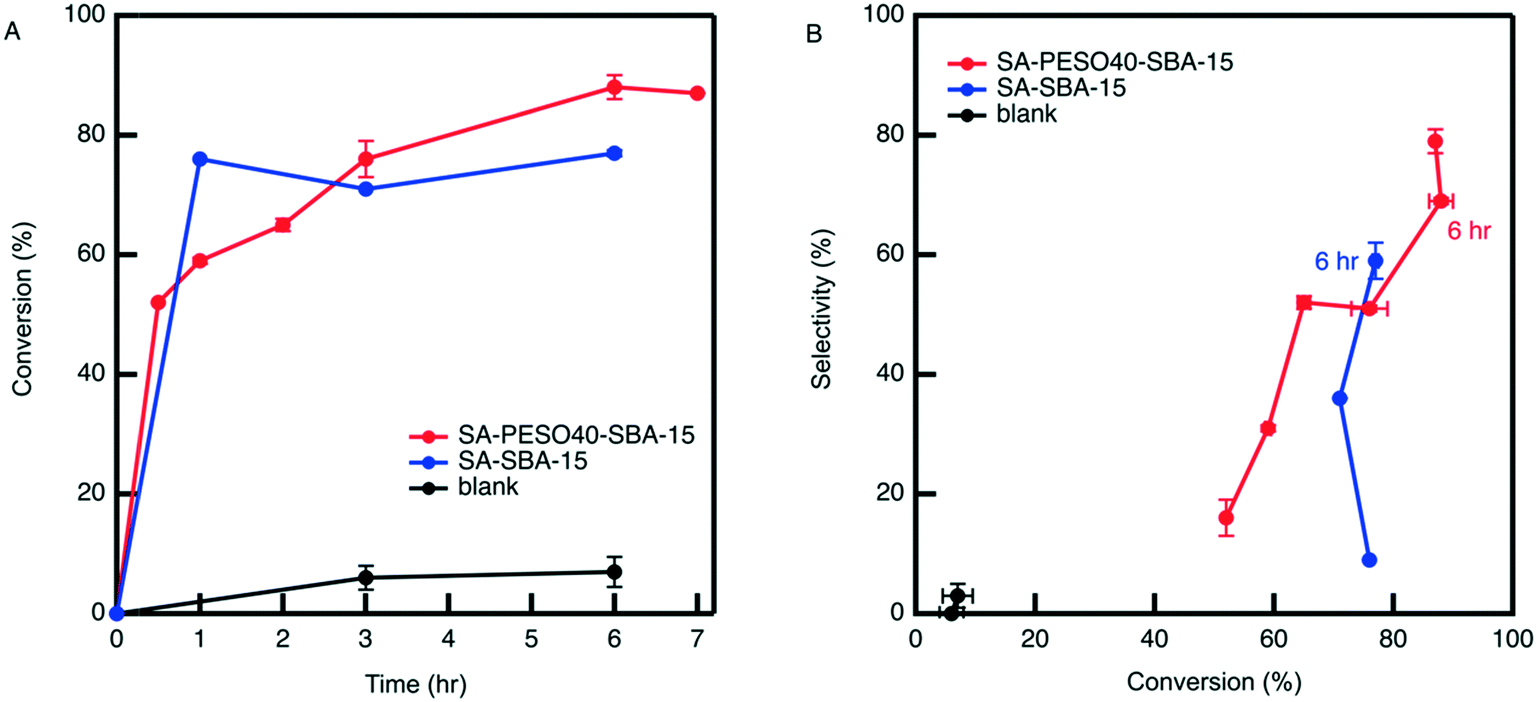 | ||
| Fig. 8 (A) Conversion versus time and (B) selectivity versus conversion results obtained by SA-PESO-40-SBA-15 (2 mol%) compared to that obtained by SA-SBA-15 and in absence of catalyst in 4:1 THF:water. All reactions are performed at 120 °C and an agitation rate of 420 RPM starting from a fructose concentration of 0.5 mmol g−1 solution in 4:1 THF:water. | ||
3.6. Catalyst reuse tests in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 (w/w) THF:water
:1 (w/w) THF:water
Since the best catalytic results are obtained with SA-PESO-40-SBA-15 in 4:1 THF:water, catalyst reuse tests are conducted in this solvent. A reaction time of 7 hours is used for each catalyst usage before filtering out the catalyst, washing it with DI water (26 g), and drying in the hood before the next use. The results depicted in Fig. 9 show fructose conversion and selectivity obtained for SA-PESO-40-SBA-15 after each use. Although fructose conversion decreases slightly and is maintained at 77%, HMF selectivity decreases after each use.
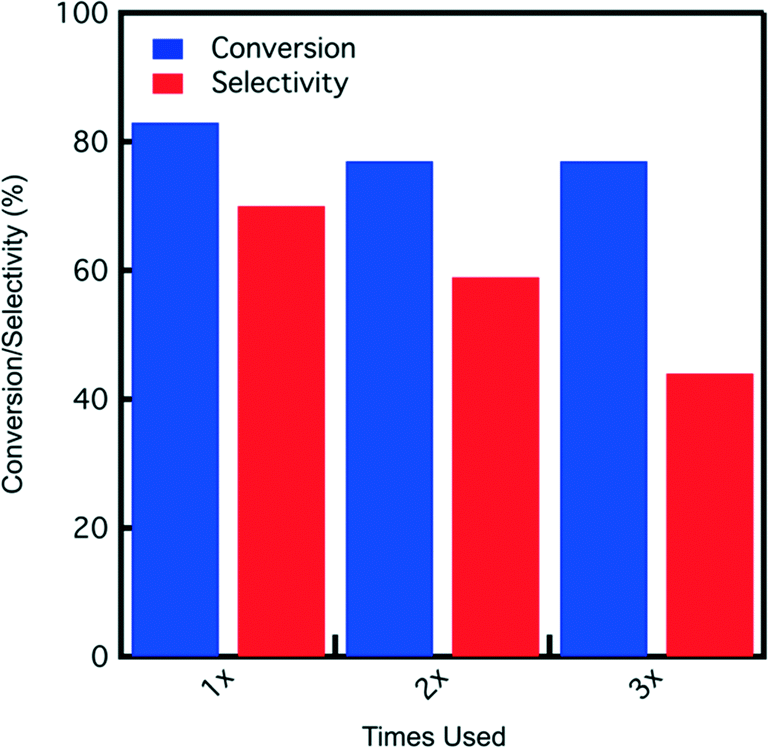 | ||
| Fig. 9 Fructose conversion and HMF selectivity obtained after several uses of SA-PESO-40-SBA-15 in 4:1 THF:water. All reactions are performed for 7 hours of reaction time at 120 °C and an agitation rate of 420 RPM starting from a fructose concentration of 0.5 mmol per gram solution. | ||
Post reaction material characterization is used to investigate the observed the changes in the catalytic performance of the bifunctional catalyst upon its reuse. Analysis with nitrogen physisorption indicates that the textural properties of the material remain similar after each cycle (Fig. S9, Table S2†), suggesting that the silica support remains stable under reaction conditions. ATR-FTIR analysis of the reused catalyst shows changes in surface functionality. The FTIR spectra in Fig. 10 of the catalyst material after each use show that the sulfoxide peak at 1029 cm−1 gradually decreases in intensity. Notably, the sulfone peak at 1450 cm−1 does not grow in intensity, indicating that the disappearance of the sulfoxide peak is not because of sulfoxide units being oxidized to sulfone groups. Therefore, the disappearance of the sulfoxide peak could potentially be attributed to leaching of PESO from the catalyst support.
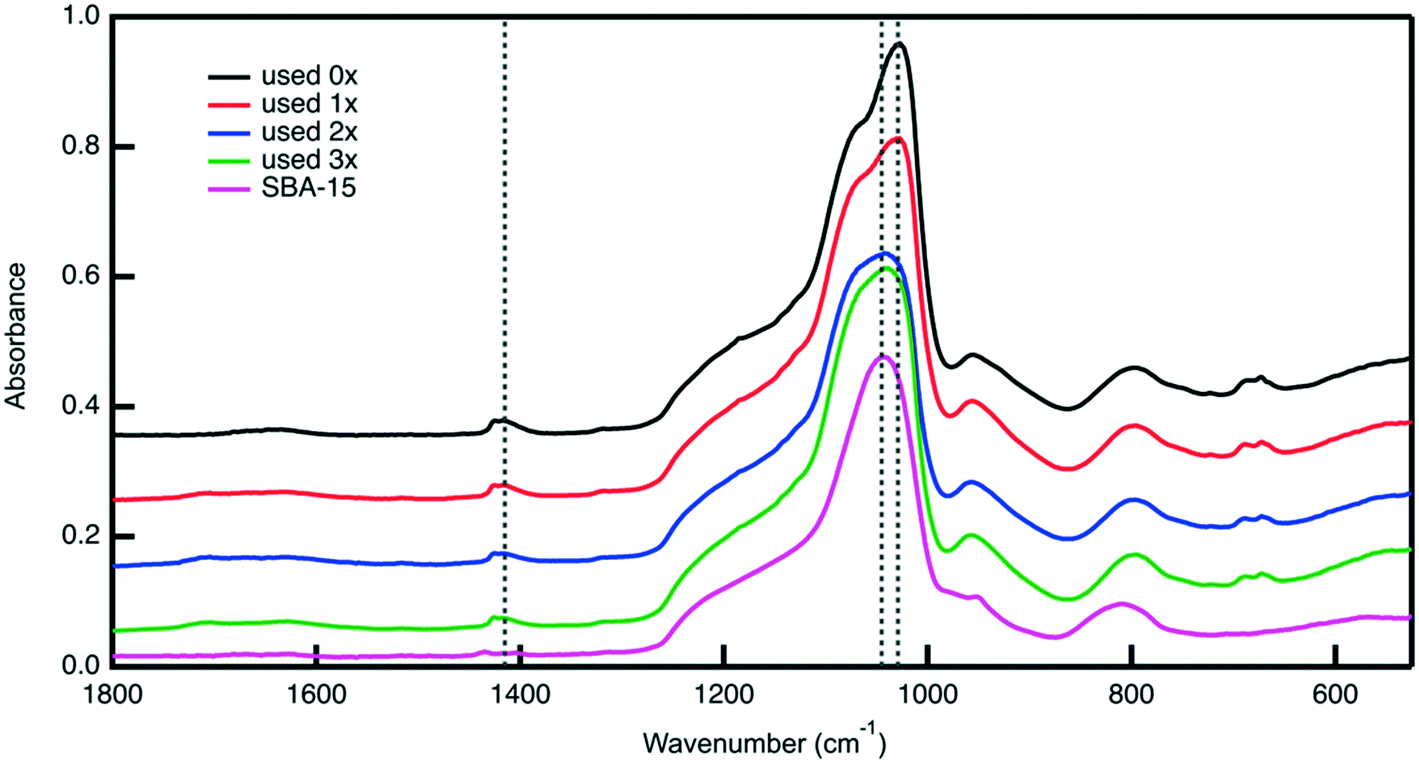 | ||
| Fig. 10 ATR-FTIR spectra of reused SA-PESO-40-SBA-15 compared to bare SBA-15. The sulfoxide, sulfone, and Si–O peaks are labelled by the dotted line occurring at 1029, 1415, and 1045 cm−1 respectively. | ||
TGA analysis of the catalyst after each reuse is shown in Fig. S9.† The mass loss percent between 200–400 °C decreases as the number of times used increases. This indicates that the sulfoxide polymer is leaching from SBA-15. Interestingly, after the first use, the residual mass percent decreases as compared to the zero times used material while the decrease in mass percent loss at 200–400 °C is observed. This suggests that some organic material is adsorbing to the catalyst while PESO is leaching. After using the catalyst in a reaction, the solid turns from white to brown, suggesting that the brown humins solids are adsorbed to the catalyst. Ultimately, the results of post reaction material characterization suggest that the loss of the sulfoxide polymer is potentially the main cause for the decrease in HMF selectivity after it is recycled. Previous work in our group has shown that incorporating a hydrophobic aryl group in the alkyl chain of the organosilane attaching the polymer to SBA-15 can minimize organosilane leaching.43 Such a strategy will be the subject of future work.
4. Conclusions
Co-localizing sulfonic acid groups and PESO within the pores of SBA-15 promoted HMF selectivity obtained in water from 26% to 51% as compared to monofunctional SA-SBA-15. Although the presence of PESO in the catalyst pores is the source of diffusion limitations resulting in slower observed rates, promoting interactions between the DMSO-like polymer and fructose is key to promoting HMF selectivity. Furthermore, SA-PESO-40-SBA-15 achieves the best results in 4:1 (w/w) THF:water obtaining 79% HMF selectivity at 87% fructose conversion in 7 hours. Improvement to the catalyst design, such as modifying the organosilane linker with a hydrophobic aryl group, could potential prevent PESO leaching from the surface and increase catalyst recyclability. Ultimately, the incorporation of PESO within the pores of acid functionalized SBA-15, introduces DMSO-like solvent effects that increases HMF selectivity in more economical solvents (water, 4:1 THF:water) avoiding the use of DMSO as a bulk solvent. Thus, this catalyst has the potential to be used in a catalyst system with a heterogeneous base for the one-pot tandem reaction to produce HMF selectively in water starting with glucose in presence of a heterogeneous base catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. R. W., A. P., and N. A. B. gratefully acknowledge the National Science Foundation (NSF 1605037 and 1653587) and the Ohio State University Institute for Materials Research (OSU IMR FG0211 and EMR-G00018) for their financial support.References
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- M. J. M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- S. Tareq, Y. H. T. Yap, T. A. Saleh, A. H. Abdullah, U. Rashid and M. I. Saiman, J. Mol. Liq., 2018, 271, 885–891 CrossRef CAS.
- A. A. Alshaheri, M. I. M. Tahir, M. B. A. Rahman, T. B. S. A. Ravoof and T. A. Saleh, Chem. Eng. J., 2017, 327, 423–430 CrossRef CAS.
- A. A. Alshaheri, M. I. M. Tahir, M. B. A. Rahman, T. Begum and T. A. Saleh, J. Mol. Liq., 2017, 240, 486–496 CrossRef CAS.
- H. Kimura, M. Nakahara and N. Matubayasi, J. Phys. Chem. A, 2013, 117, 2102–2113 CrossRef CAS PubMed.
- B. F. M. Kuster, Carbohydr. Res., 1977, 54, 177–183 CrossRef CAS.
- D. W. Brown, A. J. Floyd, R. G. Kinsman and Y. Roshan-ali, J. Chem. Technol. Biotechnol., 1982, 32, 920–924 CrossRef CAS.
- L. Shuai and J. S. Luterbacher, ChemSusChem, 2016, 9, 133–155 CrossRef CAS.
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS PubMed.
- Y. J. Pagán-Torres, T. Wang, J. M. R. Gallo, B. H. Shanks and J. A. Dumesic, ACS Catal., 2012, 2, 930–934 CrossRef.
- A. H. Motagamwala, K. Huang, C. T. Maravelias and J. A. Dumesic, Energy Environ. Sci., 2019, 12, 2212–2222 RSC.
- X. Guo, J. Tang, B. Xiang, L. Zhu and H. Yang, ChemCatChem, 2017, 9, 3218–3225 CrossRef CAS.
- M. S. Feather and J. F. Harris, Adv. Carbohydr. Chem. Biochem., 1973, 28, 161–224 CrossRef CAS.
- F. W. Lichtenthaler and S. Ronninger, J. Chem. Soc., Perkin Trans. 2, 1990, 2, 1489–1497 RSC.
- V. Nikolakis, S. H. Mushrif, B. Herbert, K. S. Booksh and D. G. Vlachos, J. Phys. Chem. B, 2012, 116, 11274–11283 CrossRef CAS PubMed.
- P. Dais and A. Perlin, Carbohydr. Res., 1987, 169, 159–169 CrossRef CAS.
- P. Dais and A. S. Perlin, Carbohydr. Res., 1985, 136, 215–223 CrossRef CAS.
- G. S. Svenningsen, R. Kumar, C. E. Wyman and P. Christopher, ACS Catal., 2018, 8, 5591–5600 CrossRef CAS.
- G. Tsilomelekis, T. R. Josephson, V. Nikolakis and S. Caratzoulas, ChemSusChem, 2014, 7, 117–126 CrossRef CAS PubMed.
- S. H. Mushrif, S. Caratzoulas and D. G. Vlachos, Phys. Chem. Chem. Phys., 2012, 14, 2637 RSC.
- J. Horvat, B. Klaic, B. Metelko and V. Sunjic, Croat. Chem. Acta, 1986, 59, 429–438 CAS.
- S. K. R. Patil, J. Heltzel and C. R. F. Lund, Energy Fuels, 2012, 26, 5281–5293 CrossRef CAS.
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53, 11872–11875 CrossRef CAS.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, P. Bai, K. Ma, M. Neurock and J. A. Dumesic, Nat. Catal., 2018, 1, 199–207 CrossRef CAS.
- M. R. Whitaker, A. Parulkar, P. Ranadive, R. Joshi and N. A. Brunelli, ChemSusChem, 2019, 12, 2211–2219 CrossRef CAS.
- R. Alamillo, A. J. Crisci, J. M. R. Gallo, S. L. Scott and J. A. Dumesic, Angew. Chem., 2013, 125, 10539–10541 CrossRef.
- A. J. Crisci, M. H. Tucker, J. A. Dumesic and S. L. Scott, Top. Catal., 2010, 53, 1185–1192 CrossRef CAS.
- A. J. Crisci, M. H. Tucker, M. Y. Lee, S. G. Jang, J. A. Dumesic and S. L. Scott, ACS Catal., 2011, 1, 719–728 CrossRef CAS.
- Q. Sun, S. Wang, B. Aguila, X. Meng, S. Ma and F.-S. Xiao, Nat. Commun., 2018, 9, 1–8 CrossRef.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- T. Oyama, K. Naka and Y. Chujo, Macromolecules, 1999, 32, 5240–5242 CrossRef CAS.
- T. Noguchi, Y. Hirai and M. Kirihara, Chem. Commun., 2008, 3040–3042 RSC.
- N. A. Brunelli, S. A. Didas, K. Venkatasubbaiah and C. W. Jones, J. Am. Chem. Soc., 2012, 134, 13950–13953 CrossRef CAS.
- N. Deshpande, E. H. Cho, A. P. Spanos, L.-C. Lin and N. A. Brunelli, J. Catal., 2019, 372, 119–127 CrossRef CAS.
- N. Deshpande, L. Pattanaik, M. R. Whitaker, C.-T. Yang, L.-C. Lin and N. A. Brunelli, J. Catal., 2017, 353, 205–210 CrossRef CAS.
- Y. Noda, K. Li, A. Engler, W. Elliott and R. M. Rioux, Catal. Sci. Technol., 2016, 6, 5961–5971 RSC.
- M. S. C. Flett, Spectrochim. Acta, 1962, 18, 1537–1556 CrossRef CAS.
- T. Cairns, G. Eglinton and D. T. Gibson, Spectrochim. Acta, 1964, 20, 31–43 CrossRef CAS.
- M. S. C. Flett, Spectrochim. Acta, 1962, 18, 1537–1556 CrossRef CAS.
- J. L. Koenig, Appl. Spectrosc. Rev., 1971, 4, 233–305 CrossRef CAS.
- N. Deshpande, PhD, The Ohio State University, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00093c |
| This journal is © The Royal Society of Chemistry 2020 |