
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloidal bimetallic platinum–ruthenium nanoparticles in ordered mesoporous carbon films as highly active electrocatalysts for the hydrogen evolution reaction†
René
Sachse
 ab,
Denis
Bernsmeier
a,
Roman
Schmack
a,
Ines
Häusler
c,
Andreas
Hertwig
b,
Katrin
Kraffert
a,
Jörg
Nissen
d and
Ralph
Kraehnert
*a
ab,
Denis
Bernsmeier
a,
Roman
Schmack
a,
Ines
Häusler
c,
Andreas
Hertwig
b,
Katrin
Kraffert
a,
Jörg
Nissen
d and
Ralph
Kraehnert
*a
aTechnische Universität Berlin, Faculty II Mathematics and Natural Sciences, Institute of Chemistry, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: ralph.kraehnert@tu-berlin.de
bFederal Institute for Materials Research and Testing (BAM), Unter den Eichen 44-46, 12203 Berlin, Germany
cTechnische Universität Berlin, Faculty II Mathematics and Natural Sciences, Institute of Optics and Atomic Physics, Straße des 17. Juni 135, 10623 Berlin, Germany
dTechnische Universität Berlin, ZELMI, Straße des 17. Juni 135, 10623 Berlin, Germany
First published on 4th March 2020
Abstract
Hydrogen features a very high specific energy density and is therefore a promising candidate for clean fuel from renewable resources. Water electrolysis can convert electrical energy into storable and transportable hydrogen gas. Under acidic conditions, platinum is the most active and stable monometallic catalyst for the hydrogen evolution reaction (HER). Yet, platinum is rare and needs to be used efficiently. Here, we report a synthesis concept for colloidal bimetallic platinum–ruthenium and rhodium–ruthenium nanoparticles (PtRuNP, RhRuNP) and their incorporation into ordered mesoporous carbon (OMC) films. The films exhibit high surface area, good electrical conductivity and well-dispersed nanoparticles inside the mesopores. The nanoparticles retain their size, crystallinity and composition during carbonization. In the hydrogen evolution reaction (HER), PtRuNP/OMC catalyst films show up to five times higher activity per Pt than Pt/C/Nafion® and PtRu/C/Nafion® reference catalysts.
Introduction
Hydrogen plays an important role in many processes of the chemical industry, e.g. the synthesis of ammonia. In addition, hydrogen offers the highest specific energy density per mass of all chemicals1 and thus is an excellent medium for the storage and transport of energy. Electrocatalytic splitting of water is one possibility to produce hydrogen. For an efficient electrolysis, catalytically active electrode coatings are used to reduce the required overpotential. Under acidic conditions platinum based electrocatalysts are typically employed. However, Pt is an expensive and scarce element and it needs to be used efficiently.2,3 Typically, Pt nanoparticles (NP) are deposited onto conductive and porous carbon supports to increase the active surface area of the catalyst.4–6 However, these inks are based on binding agents like Nafion®, which can block pores and active sites.7,8 Ordered mesoporous carbon (OMC) coatings are well known for their high surface area and good electrical conductivity.9–11 Size and spacing of their mesopores are well-defined and controllable, making them ideal supports for catalyst nanoparticles.12,13Metallic nanoparticles feature a high surface area to volume ratio. In bimetallic nanoparticles the material's geometric and electronic properties can be modified.14,15 These modifications results in changed adsorption and reactivity behaviour.16,17
The combination of platinum–ruthenium was reported as a suitable candidate for bimetallic platinum-containing catalysts in the context of electrocatalyst.18,19 Using Ru@Pt core–shell nanoparticles, Elbert et al. and also J. X. Wang et al. showed enhanced Pt mass-activity compared to Pt and PtRu alloy catalyst in the hydrogen evolution and hydrogen oxidation reaction (HOR).20,21 The higher activity results from a higher specific platinum surface of the platinum shell. X. Wang et al. attributed performance improvements in Ru@Pt core–shell nanoparticles to a compressive strain effect that optimizes adsorption–desorption energetics of H intermediates.22 However, core–shell configurations are typically limited by their thermal stability as alloys are formed at higher temperatures (>500 °C).23
Earlier, we reported a synthesis of Nafion®-free coatings for highly efficient HER catalysis. The catalyst films were synthesized by molecular metal precursors, carbon precursors as well as pore templates and deposited directly on conductive substrates as porous Pt/OMC, Ru/OMC and PtRu/OMC coatings.24 However, in this approach the formation of the active particles as well as the graphitization of the film and the formation of the pore structure necessarily occur at the same time and the same conditions. Hence, particles' properties such as size and composition could not be controlled independently of the properties of the supporting carbon film.
Herein, we present an approach for the synthesis of bimetallic nanoparticles, supported on ordered mesoporous carbon. Following a modified route of the well-established Bönnemann system,25–27 preformed bimetallic colloids are employed as metal precursors. In this approach particle size and composition of the colloid determine the particle properties in the finally formed catalyst. Exemplarily, the systems PtRuNP/OMC as well as RhRuNP/OMC illustrate the synthesis approach. For all studied metal particles, the carbon coatings are highly uniform with templated three-dimensional mesopore structures. All films feature a good metal dispersion, a highly accessible internal surface and excellent electrical conductivity. When combined with the unique electronic properties of the combination Pt–Ru, the developed PtRuNP/OMC coatings outperform previously reported PtRu/OMC coatings as well as conventional Pt/C/Nafion® and PtRu/C/Nafion® in HER catalysis.
This contribution describes first the synthesis and properties of the bimetallic PtRuNP colloid. The deposited PtRuNP/OMC are characterized by ellipsometry, ellipsometric porosimetry (EP), sheet conductivity measurements, SAXS, SEM, STEM, WDX, XPS and XRD. Electrochemical properties and HER performance measured in a rotating disc electrode setup (RDE) are compared to monometallic PtNP/OMC, RuNP/OMC as well as commercial Pt/C/Nafion® and PtRu/C/Nafion®. Corresponding information on Rh and RhRuNP system are reported in the ESI.†
Experimental
Chemicals
For the synthesis of bimetallic PtRu nanoparticles, tetraoctylammonium bromide (TOAB, 98%) and potassium triethylborohydride solution (KBEt3H, 1 M in THF) were used from Sigma-Aldrich. Tetrahydrofuran (THF, ≥99.9%) was purchased from Carl Roth. Platinum(II) chloride (PtCl2, 99.99%) and ruthenium(III) chloride (RuCl3, 99.9%) from Alfa Aesar were used as metal precursors.The synthesis of mesoporous films containing metal nanoparticles was realized with 1,3-dihydroxybenzene (≥99%), Pluronic F-127 (PEO106–PPO70–PEO106, Mw = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1) and formaldehyde solution (37 wt% in H2O) procured from Sigma-Aldrich. Ethanol (EtOH, >99%, absolute) was purchased from VWR and tetrahydrofuran (THF, ≥99.9%) was bought from Carl Roth. 3 M hydro chloric acid (HCl) was prepared by diluting a 12 M HCl (Alfa Aesar) with MilliQ water (18 MOhm cm). All chemicals were used as received without any further purification. Pt on Vulcan (BASF, XC 72 R, Cat. No. 181, 10 wt%Pt) and platinum–ruthenium alloy on graphitized carbon (Sigma-Aldrich, 20% Pt, 10% Ru) were employed as reference catalysts.
600 g mol−1) and formaldehyde solution (37 wt% in H2O) procured from Sigma-Aldrich. Ethanol (EtOH, >99%, absolute) was purchased from VWR and tetrahydrofuran (THF, ≥99.9%) was bought from Carl Roth. 3 M hydro chloric acid (HCl) was prepared by diluting a 12 M HCl (Alfa Aesar) with MilliQ water (18 MOhm cm). All chemicals were used as received without any further purification. Pt on Vulcan (BASF, XC 72 R, Cat. No. 181, 10 wt%Pt) and platinum–ruthenium alloy on graphitized carbon (Sigma-Aldrich, 20% Pt, 10% Ru) were employed as reference catalysts.
Substrate pre-treatment
MeNP/OMC films were deposited on different substrates which were properly cleaned prior to synthesis. Three different classes of Si-wafers were obtained from University Wafers and are characterized by either a single or double side polishment with (100) orientation (ca. 600 μm thick) or an overall thickness of 50 μm. All silicon substrates were cleaned with ethanol and heated in air for 2 h at 600 °C prior to film deposition to ensure the same conditions for all substrates. Si-wafers were used for EP, SEM, physisorption, TEM, SAXS, WDX and XPS. SiO2-glass substrates (Science Services GmbH) for measurement of electrical conductivity were cleaned in ethanol prior to film deposition. For electrochemical measurement, glassy carbon substrates (GC, Sigradur G from HTW) were first polished with a 0.05 mm diamond dispersion and subsequently immersed two times in a mixture of water and ethanol for ultrasonication. Finally, the GC substrate was rinsed with ethanol.Synthesis of bimetallic PtRu-nanoparticles
Colloidal metal nanoparticles were synthesized under argon atmosphere in a glove box (H2O < 1 ppm, O2 < 1 ppm) as described by T. J. Schmidt27 (Scheme 1). In a typical synthesis 547 mg TOAB were dissolved in 1.2 ml THF. Subsequently, 1 ml potassium triethylborohydride solution was added under constant stirring leading to the formation of a white precipitate. After 1 hour, the solution was transferred into a freezer (0 °C) for 20 hours. The resulting white precipitate was filtered (syringe filter, 5.0 μm, PTFE) and washed with THF (0.55 ml). A second filtration (syringe filter, 0.2 μm, Nylon) was performed to obtain tetraoctylammonium hydridotriethylborat as a clear solution which was added under constant stirring to a suspension composed of 2.07 mg platinum(II) chloride and 13.11 mg ruthenium(III) chloride (metal ratio Pt:Ru = 0.14:0.86). Another 24 h of stirring yields a black colloidal solution.
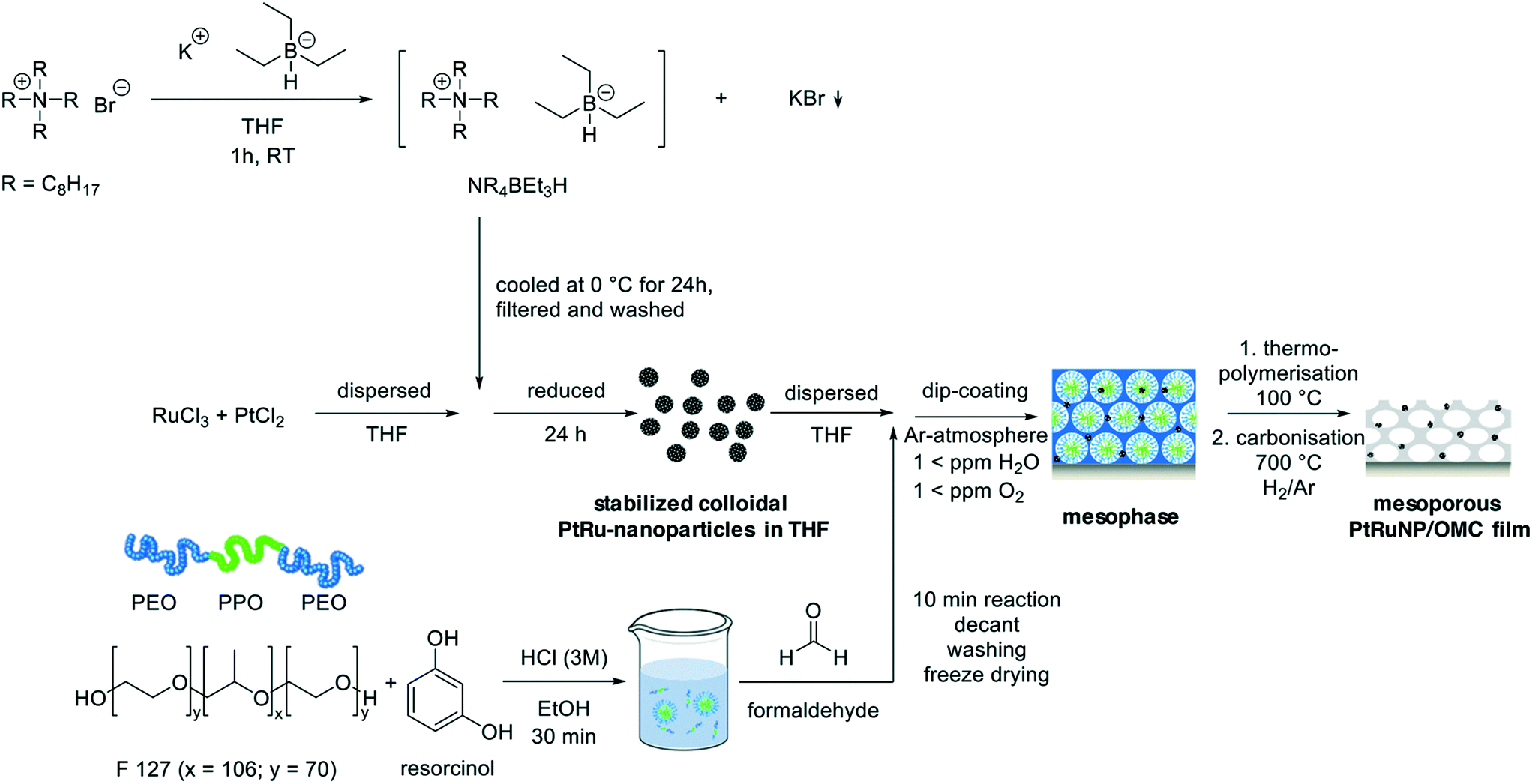 | ||
| Scheme 1 Illustration of the synthesis approach to mesoporous carbon films with incorporated bimetallic platinum–ruthenium nanoparticles. For the synthesis of bimetallic nanoparticles, an ammonium bromide is dissolved in THF. KBEt3H is added and a tetraoctylammonium hydridotriethylborat as reduction and stabilization agent obtained. This substance is added to a solution containing platinum chloride and ruthenium chloride in THF to yield platinum–ruthenium nanoparticles. A structure-directing triblock copolymer is mixed with the carbon precursor resorcinol under acidic conditions. The addition of formaldehyde starts a polycondensation reaction. After its separation the obtained polymer mixture is added to the PtRu colloid. A film is deposited onto substrates and after thermal treatments a mesoporous carbon film with integrated nanoparticles is obtained. | ||
Synthesis of PtRuNP containing mesoporous carbon films
Mesoporous carbon films with integrated nanoparticles were synthesized by dip-coating on different substrates (Scheme 1). In a typical synthesis 1101 mg 1,3-dihydroxybenzene and 300 mg Pluronic F-127 were dissolved in 4.5 ml EtOH. Subsequently 4.5 ml of 3 M HCl was added to the solution and mixed for 30 min. Afterwards, 1.21 ml of formaldehyde solution was added, and the reaction mixture was stirred for another 10 min. The obtained precipitate was separated from the mixture by centrifugation (10 min, 7500 rpm) and the top-layer was discarded. The precipitate was washed three times with MilliQ water and freeze dried for 12 h (−48 °C, 0.16 mbar). The obtained dry resin was completely dissolved in 2 ml THF under Ar-atmosphere and mixed with 8 ml of a filtered PtRuNP colloid solution (syringe filter, 0.2 μm, Nylon) for an hour. Films were deposited via dip-coating in Ar-atmosphere (H2O < 1 ppm, O2 < 1 ppm) and the coated substrates were dried at 100 °C in air for 12 h. Finally, the films were carbonized in a tube furnace for 3 h at 700 °C in a flowing H2/Ar-atmosphere (4 vol%H2 in 96 vol%Ar) with a heating ramp of 1 K min−1. Catalyst films were additionally treated in a tube furnace at 300 °C in air for 5 min to remove carbon residues from NP surfaces. A subsequent treatment in H2/Ar-atmosphere for 15 min at 350 °C was performed to reduce partially oxidized NP surfaces.Synthesis of reference catalyst
The Pt/C/Nafion® and PtRu/C/Nafion® reference catalysts were produced according to an established routine reported in literature.28,29 In this procedure 5 mg of Pt/Vulcan powder (10 wt%Pt) was dispersed in 3.98 ml of MilliQ water and 20 μl of a Nafion® solution (5%) was added. To this suspension 1 ml of isopropanol was added and sonicated for 15 min (Branson Sonifier, 6 W output power). The obtained ink was immediately deposited onto cleaned GC substrates via drop-casting and subsequently dried at 60 °C. For the PtRu/C/Nafion® reference catalyst 5 mg of PtRu/Vulcan powder (20 wt%Pt, 10 wt%Ru) was dispersed in 7.96 ml of MilliQ water and 20 μl of a Nafion® solution (5%) as well as 2 ml of isopropanol were added. The sonification and drop-casting procedures were carried out as for the Pt/C/Nafion® catalyst.Physicochemical characterization
SEM images were record on a JEOL 7401F at 10 kV and TEM images were collected on a FEI Tecnai G2 20 S-TWIN transmission electron microscope operated at 200 kV on deposited colloidal nanoparticles on carbon-coated copper grids and on fragments of film samples scraped off the substrates and deposited on carbon-coated copper grids. STEM/EDX investigations were carried out at a JEOL JEM 2200FS field-emission microscope operated at 200 kV. Images were evaluated with the ImageJ program, version 1.51 g (http://www.imagej.nih.gov/ij/). Amounts of Pt and Ru in the films, the mass depths, were calculated using the StrataGem film analysis software (v 4.8) based on wavelength dispersive X-ray (WDX) spectra analysed with a JEOL JXA-8530F electron microprobe at 10 kV.Films coated on thin silicon wafers (50 μm) were used for 2D-SAXS patterns recorded at the mySpot beamline at BESSY II synchrotron (Berlin, Germany) with a calibrated radiation energy of 12.518 keV. XRD data were collected on a Bruker D8 Advance (Cu Kα radiation) with gracing incident beam (1°). X-ray diffraction reflections were assigned using the Powder Diffraction Files database. XPS were recorded under ultra-high vacuum (2 × 10−9 bar) with an Omicron DAR 400 using monochromatic Al-Kα excitation at an EA 125X hemispherical energy analyser (Omicron). Overview scans (5 scans) were recorded with a pass energy of 100 eV and detail scans (20 scans) with a pass energy of 50 eV.
Two-point sheet conductivity measurements were performed with a Keithley Model 6517B Electrometer employing an 8 × 8 pin probe head with an altering polarity sequence of the pins. Kr physisorption were measured at 77 K with a Quantachrome Autosorb-iQ. Samples were degassed in vacuum at 150 °C for 2 h prior to sorption analysis. The surface area was evaluated with BET. Due to the low film volume, ellipsometric porosimetry (EP) was performed for the pore size distribution. An environmental cell was fixed on a variable angle spectroscopic ellipsometer (VASE) M2000DI (J. A. Woollam) with a spectral range of 193–1690 nm. 60° was used as incident angle and CompleteEASE software (v6.42) for data analysis. The ellipsometric model used, consist of a silicon/SiO2 (3 nm) layer and an anisotropic Bruggeman effective medium approximation (EMA) with Gaussian oscillators.30,31 Porosimetry measurements were carried out by the change of the relative humidity by mixing dry nitrogen gas and water saturated nitrogen gas controlled with two mass flow controllers. The flux was 2.5 L min−1 and a constant temperature of 23 °C was applied. The relative humidity was measured behind the cell for each water partial pressure. The change of the refractive index in dependence of the varying relative humidity was evaluated at 589 nm. Pore size distribution (PSD) were calculated by the procedure described by Boissiere et al.32,33
Electrochemical testing
Electrochemical measurements were performed by using a three-electrode disc setup (Pine MSR rotator, BioLogic SP-200 potentiostat). A reversible hydrogen electrode (RHE, Gaskatel, HydroFlex) was used as a reference, Pt gauze (Chempur, 1024 mesh cm2, 0.06 mm wire diameter, 99.9%) as counter electrode, and a coated GC disk as working electrode. All potentials are referenced to the RHE. GC disks were prepared by cutting 5 mm circular discs from larger PtRuNP/OMC coated GC substrates and mounted on a rotating disk shaft. GC disks rotated at a speed of 2000 rpm during EC testing and sulfuric acid employed as the electrolyte solution (0.5 M H2SO4, Fixanal, Fluka Analytical). The electrolyte solution was purged for 30 min with N2 prior to electrocatalytic testing. HER performance was studied by cyclic voltammetry in a potential range of 50 to −250 mV with a scan rate of 20 mV s−1. Impedance spectroscopy was measured in order to correct recorded cyclic voltammograms for ohmic losses. For correction of diffusional limitations, a Koutecky–Levich analysis of PtRuNP/OMC catalysts was performed with H2-saturated 0.5 M H2SO4 in a potential window between −50 and 400 mV with a scan rate of 10 mV s−1. The electrolyte was purged with H2 prior to each cycle with a certain rotation speed.Results and discussion
The first part analysis the colloidal bimetallic PtRu nanoparticles and compares them to monometallic Pt and Ru nanoparticles from the similar syntheses. The second part characterizes the morphology of a PtRuNP/OMC catalyst film and the influence of the carbonization atmosphere. The final part shows the electrocatalytic performance in the acidic hydrogen evolution reaction (HER) of PtRuNP/OMC with varying composition ratios of Pt to Ru in comparison to monometallic MeNP/OMC (Pt, Ru) catalysts and to a commercial Pt/C/Nafion® reference catalyst.Physicochemical properties of PtRuNP
Bimetallic PtRuNP were synthesized by dissolving platinum(II) chloride and ruthenium(III) chloride together in THF and reducing them simultaneously with tetraoctylammonium hydridotriethylborat in THF. Fig. 1 shows a TEM image (a) of bimetallic PtRuNP with a histogram of measured particle diameters (b) and XRD measurements (c) of monometallic PtNP, RuNP and bimetallic PtRuNP.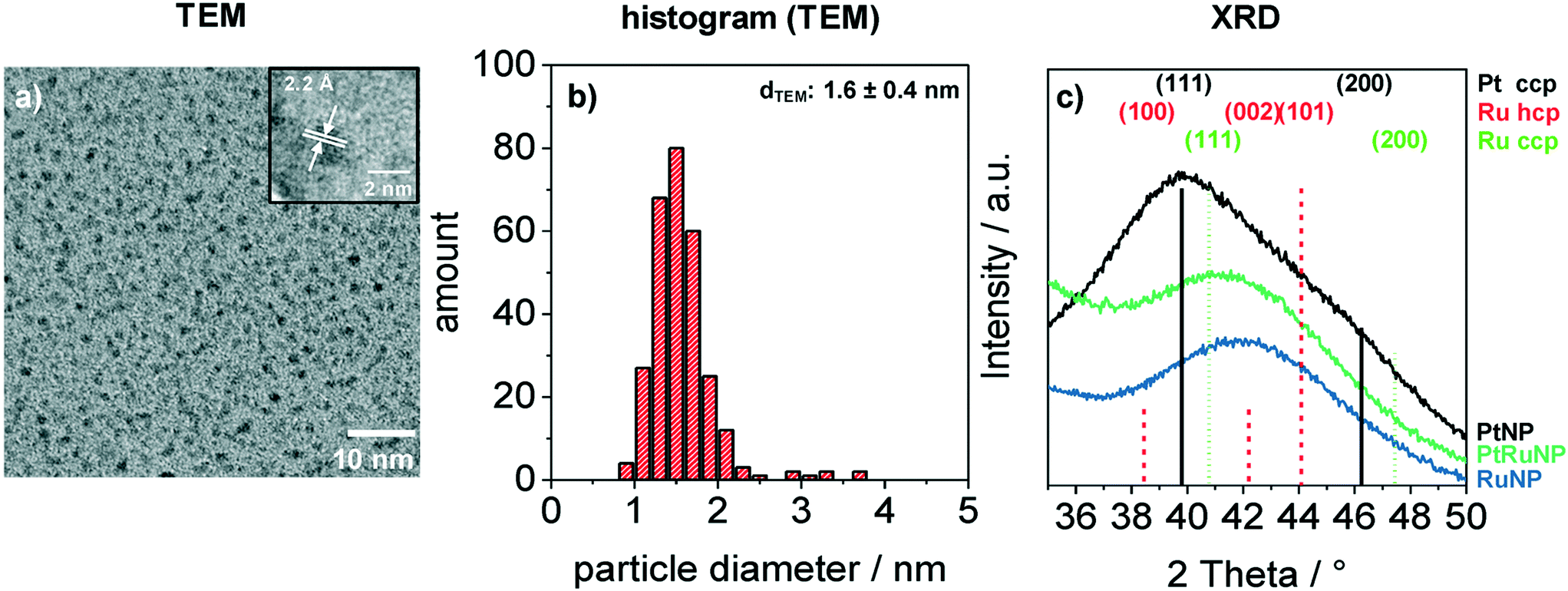 | ||
| Fig. 1 Characterization of stabilized colloidal platinum–ruthenium nanoparticles with TEM and XRD. a) TEM images of small bimetallic platinum–ruthenium nanoparticles. Inset (HR-TEM) shows a single nanoparticle with lattice planes which have a distance of 2.2 Å. A histogram (b) indicates an average diameter of 1.6 nm and a narrow size distribution. c) Grazing incidence XRD measurement of colloidal platinum, ruthenium und platinum–ruthenium nanoparticles with indicated reflections of cubic Pt (00-004-0802), cubic Ru (01-088-2333) and hexagonal Ru (00-006-0663). | ||
The TEM image (a) shows small nanoparticles (dark spots). Electron diffraction patterns in the high-resolution (HR) TEM image (Fig. 1a inset) suggests that the nanoparticles possess a high crystallinity. The regular distance of fringes amounts to 2.2 Å. This distance cannot be attributed to a single platinum or ruthenium plane due to their very similar lattice parameters. The histogram (b) of measured particles' diameters from TEM indicates the presence of monodisperse small nanoparticles with an average diameter of 1.6 nm. XRD measurement (Fig. 1c) of bimetallic PtRuNP shows a broad diffraction pattern which cannot be attributed to a single Pt or Ru crystal phase. In contrast, the diffraction patterns of monometallic PtNP and RuNP are related to cubic Pt and to a mixture of hexagonal as well as cubic Ru phases, respectively. The PtRuNP pattern shows a shift to lower 2 theta values in comparison to the pattern of pure RuNP. This shift can be an indication of alloyed PtRu nanoparticles and a change in the lattice parameter of the Ru content. The broad reflection indicates a small crystallite size. The obtained crystallite size from the Debye–Scherrer equation is 1.7 nm, which corresponds to the TEM analyses.
When ruthenium acetylacetonate (Ru(acac)3) is employed as precursor, TEM images (ESI† Fig. S1) reveal an aggregation of RuNP. Acetylacetonate ligand seem to prevent a successful stabilization. Detailed studies on the interaction of the protection shell (N(octyl)4Cl) with the colloidal metal core show that chloride ions form the centre part of a double layer, which interacts with the metal atoms as well as with the N(octyl)4+ groups.34
In summary, the colloid synthesis using platinum and ruthenium chloride yields small crystalline nanoparticles with an indication of alloy formation.
Physicochemical properties of PtRuNP/OMC
Films were deposited via dip-coating from a mixture of carbon precursors and colloidal PtRuNP in THF, as described in the experimental part. Fig. 2 shows structural features of a PtRuNP/OMC film carbonized in H2/Ar atmosphere at 700 °C. All films were subsequently heat-treated at 300 °C in air and additionally at 350 °C under H2/Ar. Fig. 2 presents a SEM image in cross-section mode (a), 2D-SAXS patterns in transition mode with a X-ray incident angle of 90° (b) and 10° (c), GI-XRD measurement (d) as well as XPS analyses in the Ru 3p region (e) and in the Pt 4f region (f).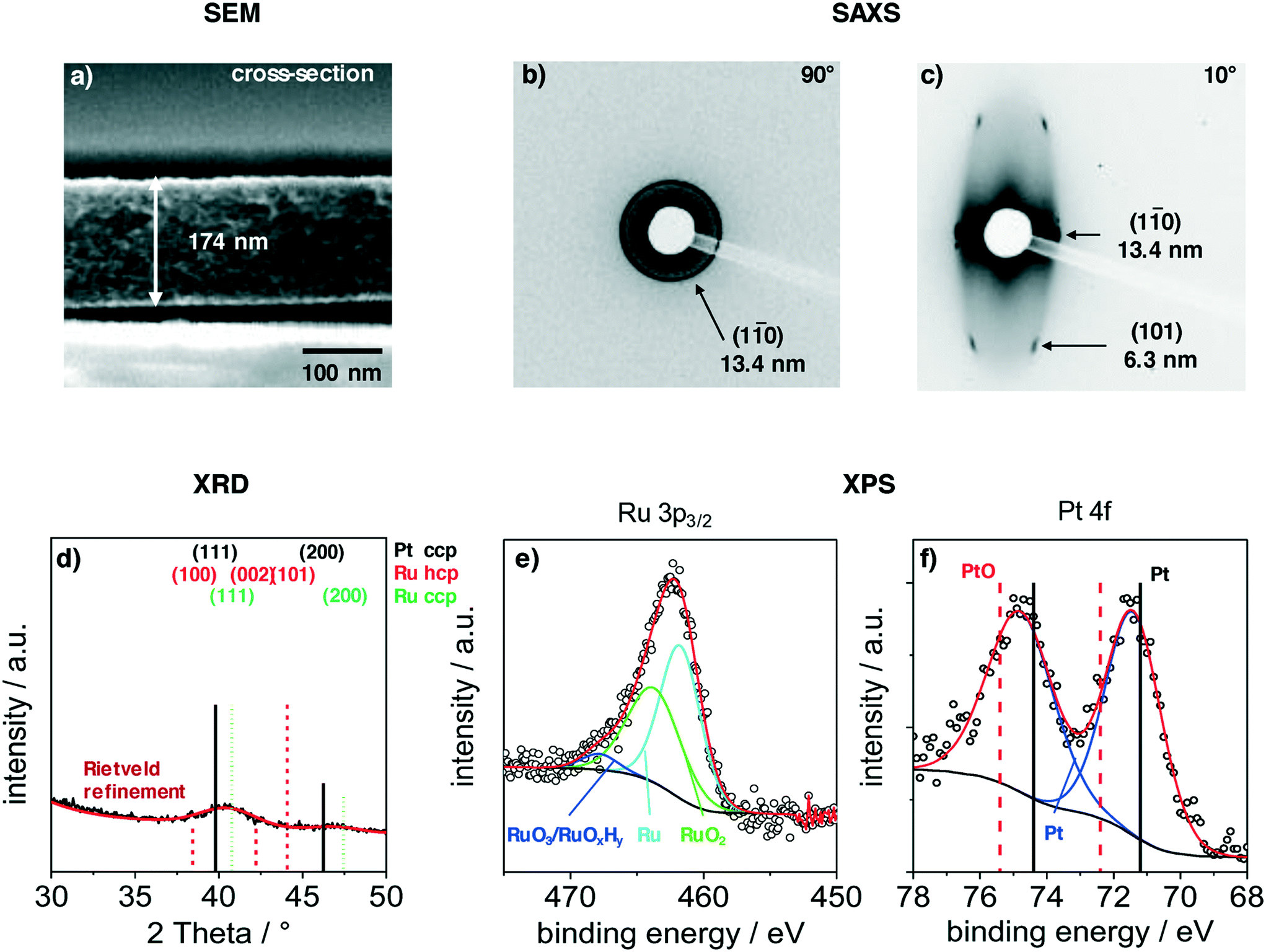 | ||
| Fig. 2 Characterization of a mesoporous carbon film with integrated platinum–ruthenium nanoparticles (0.3 wt%Pt and 2.2 wt%Ru) which was carbonized at 700 °C in H2/Ar, additionally heat-treated at 300 °C in air and reduced in H2/Ar at 350 °C. a) Cross-section SEM image of a mesoporous carbon film with a film thickness of 174 nm. SAXS patterns with incident angels of 90° (b) and 10° (c). GI-XRD pattern (d) with indicated reflections of cubic Pt (00-004-0802), cubic Ru (01-088-2333) and hexagonal Ru (00-006-0663) and a Rietveld refinement. Deconvoluted XPS spectra in the Ru 3p3/2 region (e) and Pt 4f region (f). | ||
The cross-section SEM image (Fig. 2a) shows a homogenous film thickness of about 174 nm. Mesopores are present throughout the film volume and have an ellipsoidal shape. SEM and TEM images of the film present the ordered mesopore structure (ESI† Fig. S2).
Modelling of spectroscopic ellipsometry measurements in a spectral range of 192–1697 nm gives a film thickness of about 177 nm and a metal volume fraction of 0.2 vol%Pt and 2.6 vol%Ru which correspond to 10 wt%Pt and 90 wt%Ru. A mesopore and porosity characterization was performed via ellipsometric porosimetry (EP). ESI† Fig. S3a shows the adsorption–desorption isotherm of the refractive index at a wavelength of 589 nm against the relative humidity (r.H.) of a PtRuNP/OMC film. The adsorption shows a slight increase of the refractive index up to ca. 70% r.H. and strong increase at higher r.H. (85%) due to the mesopore filling by capillary condensation. The PSD analysis (ESI† Fig. S3b) from the hysteresis shows mesopores with diameters from 2 to 9 nm for the adsorption with a maximum at 4.3 nm and mesopores with diameters of about 2 nm for the desorption. The difference of the pore radius from the adsorption and desorption branch suggests cylindrical pores and can be explained by the hemispherical meniscus which is formed during the vapor adsorption and desorption.35,36 Similar values for the pore radius from the adsorption are described in literature.9,37,38 The ratio of absorbed solvent volume to film volume indicates the accessible porous volume of the film which amounts to 45% (ESI† Fig. S3c).
SAXS measurements in transition mode were recorded to study the pore ordering in detail. A measurement with an X-ray incident angle of 90° (Fig. 2b) shows an isotropic ring which can be attributed to the (1–10) plane of a cubic pore system and a periodic distance of 13.4 nm. A SAXS pattern recorded in an angle of 10° (Fig. 2c) indicates an ellipsoidal ring with scattering spots on the ring. These can be attributed to the (1–10) and (101) planes with periodic distances of 13.4 nm and 6.3 nm, respectively. The periodic pore distance perpendicular to the substrate amounts to 3.5 nm. Accordingly, shrinkage of the film is about 75% perpendicular to the substrate due to the removal of the template and the carbonization. This phenomenon is typical for soft-templated films.9,39,40
A GI-XRD measurement (Fig. 2d) shows two broad reflections at 40.4° and 46.8°. These reflections are located between the main reflections of cubic Pt and cubic Ru. As the indicated reflection are rather proximate to each other, the observed signal cannot be attributed to one single phase. A refinement via Rietveld yields a crystallite size of 2.3 nm. The surface composition of the particles was studied with XPS in the regions of Ru 3p3/2 (Fig. 2e) and Pt 4f (Fig. 2b). The deconvoluted spectra in the Ru 3p3/2 region can be attributed to metallic Ru, RuO2 and to RuO3/RuOxHy species.41,42 The integration of the signals give a ratio of 52:43:5 for Ru:RuO2:RuO3/RuOxHy. The Pt 4f region shows a doublet at 71.5 eV (Pt 4f7/2) and 74.8 eV (Pt 4f5/2). This doublet is shifted by 0.5 eV to higher binding energies compared to metallic platinum.43,44 A deconvolution and integration of the signal for Pt and PtO is not possible due to the very low amount of Pt at the surface. The results suggest that Pt and Ru are alloyed, and that Ru is partially oxidized. The partially oxidized Ru surface can be an effect of the additionally treatment after carbonization. However, the surface of Ru particles partially oxidizes at room temperature in air.45
The BET surface area of a PtRuNP/OMC film carbonized at 700 °C in H2/Ar was determined by Kr physisorption. A film with a thickness of 150 nm had a surface area of 273 m2 m−2. WDX/StrataGem analysis reveals a film density of 1.3 g cm−3 and the resulting specific surface area amounts to 1349 m2 g−1. In literature there are similar surface areas reported for OMC materials.12,13,24,46 The geometric loading of the PtRuNP/OMC film was determined by WDX analysis by using StrataGem software. The mass depth of all components of this film amounts to 17.8 μg cm−2 carbon, 2.0 μg cm−2 oxygen, 0.06 μg cm−2 platinum and 0.45 μg cm−2 ruthenium. Therefore, the weight loadings for the film of Pt and Ru amounts to 0.3 wt%Pt and 2.2 wt%Ru. The metal weight loadings are 12 wt%Pt and 88 wt%Ru which are in a good agreement with the results of the ellipsometry analyses. For electrocatalysis, a crucial factor is the electrical conductivity. A 150 nm thick film has an electrical conductivity of 12.1 S cm−1. This value is about 1.5 times higher than for a previously reported commercial Pt/Vulcan catalyst.47
The nanoparticles inside the film volume were studied with TEM and STEM. Fig. 3 presents the results from a PtRuNP/OMC fragment. TEM images (Fig. 3a) shows small nanoparticles in the mesoporous carbon and the HR image (inset) reveals a high crystallinity of a nanoparticle after carbonization. The regular distance of fringes amounts to 2.2 Å. This distance corresponds to that of the colloidal nanoparticles (Fig. 1a) and cannot be attributed to a single metal phase. The TEM histogram (Fig. 3b) shows nanoparticles from 1 to 4 nm in diameter and a very small number of particles with diameters larger than 4 nm. The average diameter amounts to 1.6 ± 0.7 nm and is in a good agreement with Rietveld results. The particle size does not change during integration and carbonization of the film. The composition of PtRuNP was studied in detail with STEM-EDX. Fig. 3c shows a STEM image in dark field mode of a particle with a diameter of about 6 nm. An EDX line profile of this nanoparticle (Fig. 3d) detects a lower Pt and a higher Ru signal intensity and a rather homogenous distribution. EDX area scan of smaller particles (about 2 nm, Fig. 3e and f) detects Ru along with a small Pt signal. For a detailed analysis, EDX point-scans of smaller particle indicate small Pt signals for the Pt Mα and Lα lines (ESI† Fig. S4). However, due to the small amount of Pt (Pt = 0.06 μg cm−2; Ru = 0.45 μg cm−2) a quantification of Pt from EDX is not feasible.
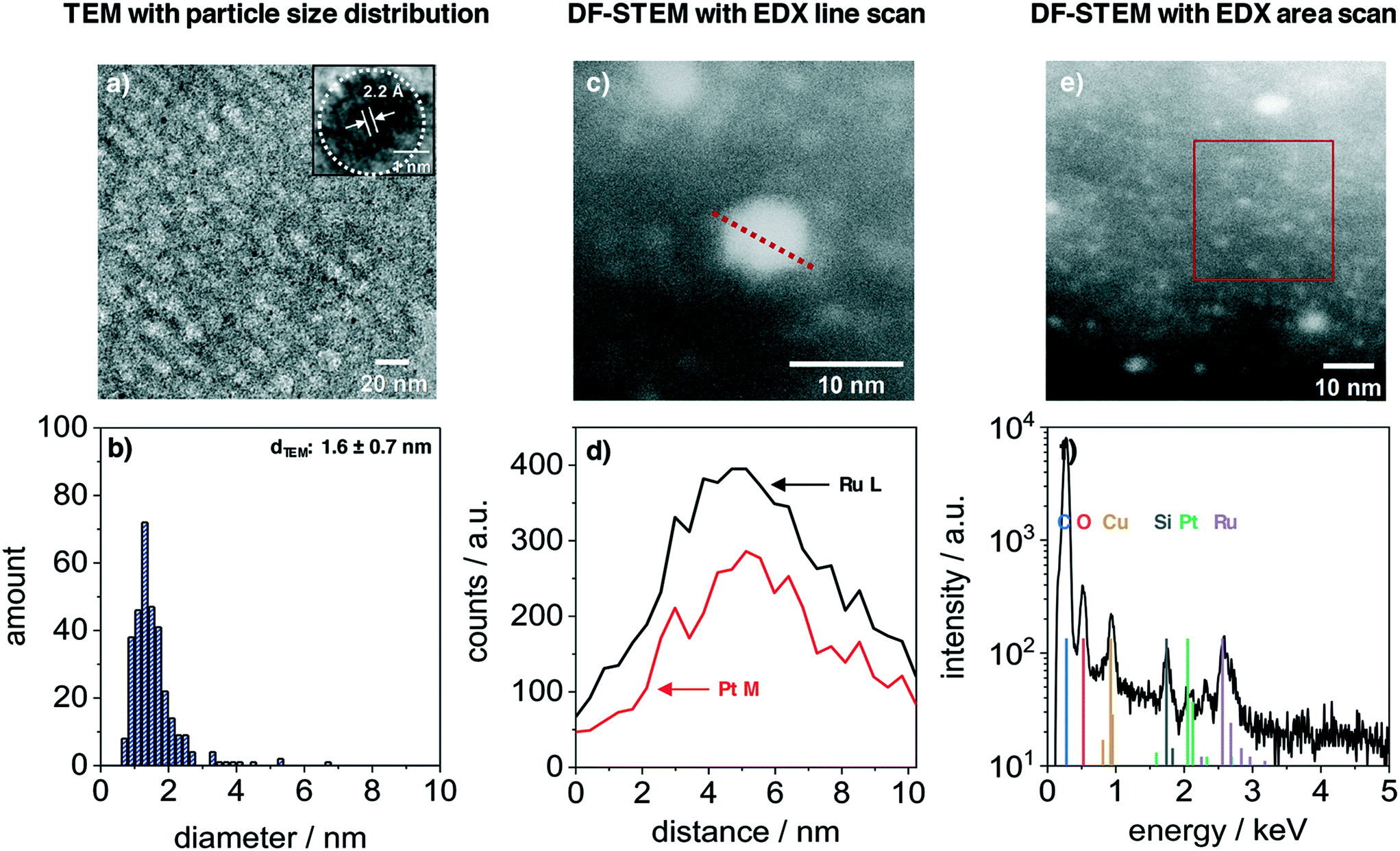 | ||
| Fig. 3 TEM and STEM-EDX characterization of platinum–ruthenium nanoparticles (0.3 wt%Pt and 2.2 wt%Ru) in a mesoporous carbon film which was carbonized at 700 °C in H2/Ar, additionally heat-treated at 300 °C in air and reduced in H2/Ar at 350 °C. a) TEM image displays small nanoparticles throughout the film volume and a HR-TEM image (inset) indicates lattice planes of nanoparticles. The particles have an average diameter of 1.6 nm (b). c) Dark-field scanning TEM image with EDX line scan of a particle with a diameter of about 6 nm. d) Corresponding EDX analysis of the EDX line scans c). e) DF-STEM image with EDX area scan of smaller particles. f) Corresponding EDX analysis of the EDX area scan in e). | ||
The atmosphere during carbonization has a considerable influence on the film morphology (ESI† Fig. S5). Using a H2/Ar atmosphere, the film exhibits a homogeneous mesopore structure at the surface. By using an Ar atmosphere instead, the nanoparticles undergo a growth in size and the film lose the meso-structure. If a N2 atmosphere is used, the nanoparticles also undergo a growth in size and the film surface has wormlike cavities. In this case, the nanoparticles inside and at the surface of the carbon film, catalyse the oxidative decomposition when contaminations of oxygen are present.48,49 The reductive H2/Ar atmosphere prevents on the one hand the growth of particles and the oxidative decomposition of the carbon film on the other hand.
In summary, the results indicate the successful synthesis of ordered mesoporous carbon films and the homogenous inclusion of nanoparticles throughout the film volume by using a H2/Ar atmosphere. The nanoparticles retain their crystalline structure after carbonization, and TEM and STEM-EDX analyses indicate the presence of PtRu alloy nanoparticles.
Electrochemical testing in the HER regime
The next part presents the electrochemical performance of monometallic PtNP/OMC, RuNP/OMC and bimetallic PtRuNP/OMC films in the hydrogen evolution reaction. Fig. 4 presents the electrochemical performance of a RuNP/OMC (1.9 μgRu cm−2), a PtNP/OMC (1.1 μgPt cm−2), a Pt0.1Ru0.9NP/OMC (0.14 μgPt cm−2 and 0.62 μgRu cm−2) and a Pt0.06Ru0.94NP/OMC (0.06 μgPt cm−2 and 0.45 μgRu cm−2) catalyst as well as commercial Pt/C/Nafion® (1.0 μgPt cm−2) and PtRu/C/Nafion® (1.0 μgPt cm−2 and 0.5 μgRu cm−2) reference catalysts in the HER. Physicochemical properties of the RuNP/OMC are shown in the ESI† (Fig. S6) and the PtNP/OMC is described in detail by Bernsmeier et al.38 The current responses in the HER region are shown for the 2nd (Fig. 4a) and 25th cyclic voltammogram (Fig. 4b), respectively. Fig. 4c represents the mass-based HER-activity obtained by dividing the current density recorded in the 25th CV at a potential of −50 mV with the WDX/StrataGem platinum mass. Prior to the electrochemical testing, MeNP/OMC catalysts were treated under mild conditions (300 °C) in air to remove carbon residuals from NP surfaces for an optimal catalytic activity and reduced afterwards at 350 °C in a H2/Ar atmosphere.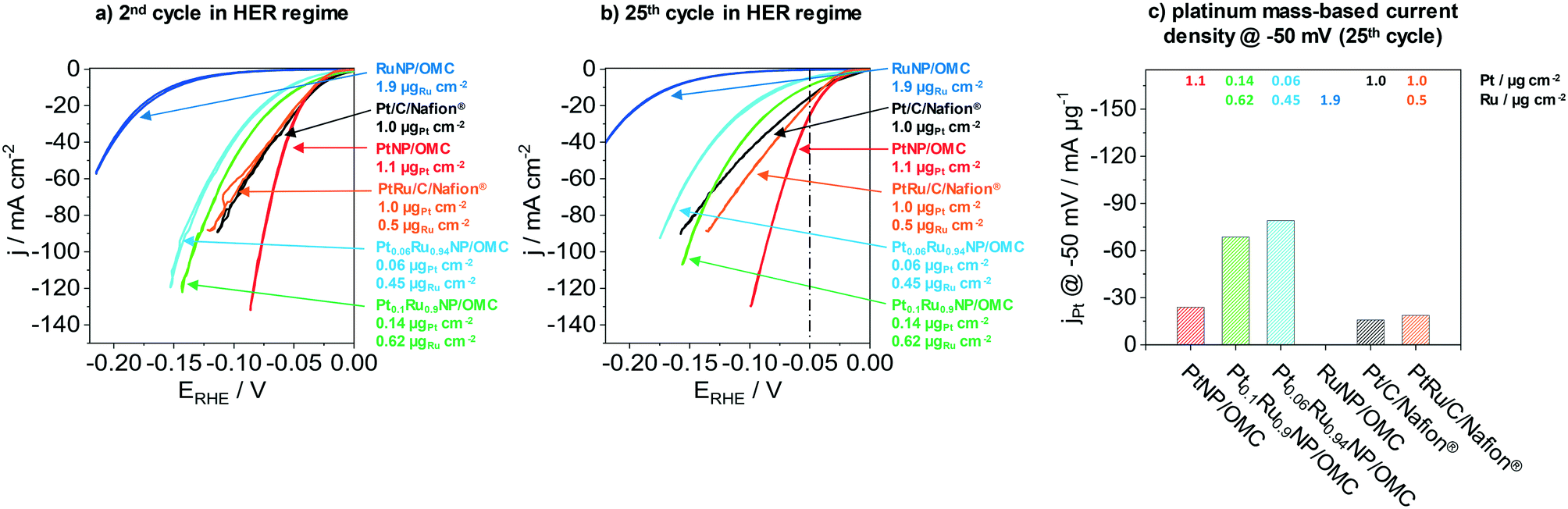 | ||
| Fig. 4 The electrocatalytic performance is illustrated for MeNP/OMC catalyst films and a commercial Pt/C/Nafion® catalyst. MeNP/OMC films carbonized for 3 h under a H2/Ar atmosphere followed by a subsequent heat-treatment at 300 °C in air and an additionally heat-treatment at 350 °C in H2/Ar. The shown Pt/C/Nafion® PtRu/C/Nafion® catalysts were prepared by a typical ink-cast procedure. Cyclic voltammograms (CV) were recorded in the HER regime of 50 mV to −250 mV with a scan rate of 20 mV s−1 in 0.5 M sulfuric acid being used as an electrolyte solution. 4a) shows the 2nd and 4b) the 25th cycle. The measured current density at −50 mV from the 25th CV were divided by the platinum mass in order to obtain the electrocatalytic platinum mass-based HER-activity (4c). | ||
RuNP/OMC shows the lowest current response during the 2nd cycle (Fig. 4a). The measured geometric current density at −50 mV vs. RHE gives a value of −0.5 mA cm−2. Platinum containing catalysts exhibit values of −8.5 mA cm−2, −12.5 mA cm−2 and −29.4 mA cm−2 at the same overpotential for Pt0.06Ru0.94NP/OMC, Pt0.1Ru0.9NP/OMC and PtNP/OMC, respectively. The commercial reference catalysts reach values of −24.9 mA cm−2 and −22.4 mA cm−2 for the Pt/C/Nafion® and PtRu/C/Nafion® respectively. The PtNP/OMC and both reference catalysts (Pt/C/Nafion® and PtRu/C/Nafion®) show similar activities at this potential. PtRuNP/OMC catalysts reach two to three times lower current densities than the pure platinum and the reference catalysts. However, the geometric platinum loadings of the PtRuNP/OMC catalyst are seven to seventeen times lower. The low current density of the RuNP/OMC catalyst is in agreement with literature which states that ruthenium suffers from higher overpotentials in comparison to platinum-based HER-catalysts.50
To gain further insights into mechanistic aspects and to develop a profound understanding of the obtained materials, the potential was plotted as a function of the logarithmic current density (ESI† Fig. S7). The obtained graphs show so called Tafel-plots with their slopes “b” utilized to identify the rate-determining step.51 Tafel slopes around 30 mV dec−1 are associated with limitations caused by chemical desorption of H2 (Tafel reaction, Hads + Hads ⇌ H2). Values of about 40 mV dec−1 refer to limitations induced by the electrochemical desorption of H2 from the active electrocatalytic centre (Heyrovsky reaction, Hads + H+ + e− ⇌ H2).51,52 Tafel slopes observed for both PtNP/OMC and Pt/C/Nafion® catalysts amount to about 30 mV dec−1 suggesting that both classes of material exhibit the same mechanistic behaviour. The rate-determining step for these catalysts can be assigned to the Tafel reaction. Platinum catalysts reported in literature present similar values for the Tafel slope tested in 0.5 M H2SO4.53,54 Tafel slopes from PtRuNP/OMC catalysts exhibit b values of about 40 mV dec−1 and the rate-determining step can be attributed to the Heyrovsky reaction. The PtRu/C/Nafion® reference catalyst show instead a b value of about 27 mV dec−1 and therefore has a similar behaviour as the pure platinum catalysts. A reason for the higher b values of PtRuNP/OMC catalysts compared to the reference PtRu/C/Nafion® can be the lower platinum content. Due to the mixture of platinum with ruthenium, less platinum active sites are adjacent. Furthermore, it is known that on Ru surface, the H2 formation is dominated by the Heyrovsky mechanism.55,56
Fig. 4b shows the current response after the 25th CV. All catalysts show a decrease in the current density compared to the 2nd CV. PtNP/OMC exhibits a value of −25.6 mA cm−2 and shows the lowest decrease of the current response (13%) at a potential of −50 mV. The Pt/C/Nafion® and PtRu/C/Nafion® reference catalysts reach values of −15.3 mA cm−2 and −18.7 mA cm−2 which correspond to a decrease of 39% and 17%, respectively. PtRuNP/OMC catalysts achieve 9.1 mA cm−2 (28% decrease) and −4.7 mA cm−2 (45% decrease) for the Pt0.1Ru0.9NP/OMC and Pt0.06Ru0.94NP/OMC. The current density of the RuNP/OMC shows a value of 0.4 mA cm−2 and therefore a similar degradation (18%) as the PtNP/OMC. The decrease of the current response of 13–45% during cycling can be related to organic impurities and the adsorption of sulfate ions, and is also reported for highly purified electrolytes.57 The lower current density of Pt/C/Nafion® and PtRu/C/Nafion® can be attributed to transport limitations and blocked active sites and pores due to the ionomer (Nafion®).8
Fig. 4c compares the platinum mass-based current density at a potential of −50 mV of the 25th cycle during CV. PtNP/OMC reaches a platinum mass-based current density of −23.9 mA μgPt−1 and shows a 1.5 times higher mass based current density than the commercial Pt/C/Nafion® catalyst (−15.9 mA μgPt−1). The PtRuNP/OMC catalysts show three to four times higher platinum mass-based current densities compared to the commercial PtRu/C/Nafion® catalyst (−18.7 mA μgPt−1) and reach values of −68.6 and −79.0 mA μgPt−1 for Pt0.1Ru0.9NP/OMC and Pt0.06Ru0.94NP/OMC, respectively. An additional comparison of a series of PtRuNP/OMC catalysts with constant Pt:Ru ratio but varied total amount of deposited metal shows that the catalytic performance scales in the studied range linearly with the amount of deposited metal (see ESI† Fig. S8).
The HER as well as the hydrogen oxidation reaction (HOR) are often limited by mass transport effects when platinum group metals are used under acidic conditions.58 A correction of the hydrogen diffusion limitations in RDE measurement for the HER/HOR was proposed by Zheng et al. and is based on a Koutecky–Levich evaluation.58Fig. 5 presents the polarization curves of Pt0.1Ru0.9NP/OMC (0.14 μgPt cm−2/0.62 μgRu cm−2) and Pt0.06Ru0.94NP/OMC (0.06 μgPt cm−2/0.45 μgRu cm−2) in H2 saturated 0.5 M H2SO4 at different rotation speeds as well as the Tafel plots of HER/HOR kinetic currents. The insets of Fig. 5a and c represent the Koutecky–Levich plots, where the reciprocal value of the measured current density is plotted as a function of the reciprocal square root of the rotation rate in order to obtain the diffusional limited current. The kinetic current can be determined by using the reversible Koutecky–Levich equation (1/j = 1/jD + 1/jK). Fig. 5b and d show Butler–Volmer fits of the kinetic current density based on this correction of diffusional limitations. Pt0.1Ru0.9NP/OMC and Pt0.06Ru0.94NP/OMC give exchange current densities of j0 = 0.33 mA cm−2 and j0 = 0.50 mA cm−2, respectively. In contrast to the uncorrected Tafel analysis (ESI† Fig. S7), the obtained value for Pt0.1Ru0.9NP/OMC (j0 = 1.18 mA cm−2) is about three to four times lower and Pt0.06Ru0.94NP/OMC (j0 = 0.66 mA cm−2) shows a similar exchange current density. For comparison, the exchange current density of the PtNP/OMC and the Pt/C/Nafion® catalyst amounts to 3.64 mA cm−2 (ref. 38) and 1.72 mA cm−2 respectively. The Pt mass-based exchange current density of Pt0.1Ru0.9NP/OMC (j0,m = 2.39 A mgPt−1) is similar to PtNP/OMC (j0,m = 2.28 A mgPt−1)38 and to a commercial Pt/Vulcan/Nafion® catalyst film (j0,m = 1.72 A mgPt−1)24 tested under identical conditions. Pt0.06Ru0.94NP/OMC has a three to four times higher Pt mass-based exchange current density (j0,m = 8.34 A mgPt−1). A PtRu/OMC catalyst prepared from a similar synthesis route employing dissolved noble metal ions as precursors shows a lower Pt mass-based exchange current density (6.46 A mgPt−1).24
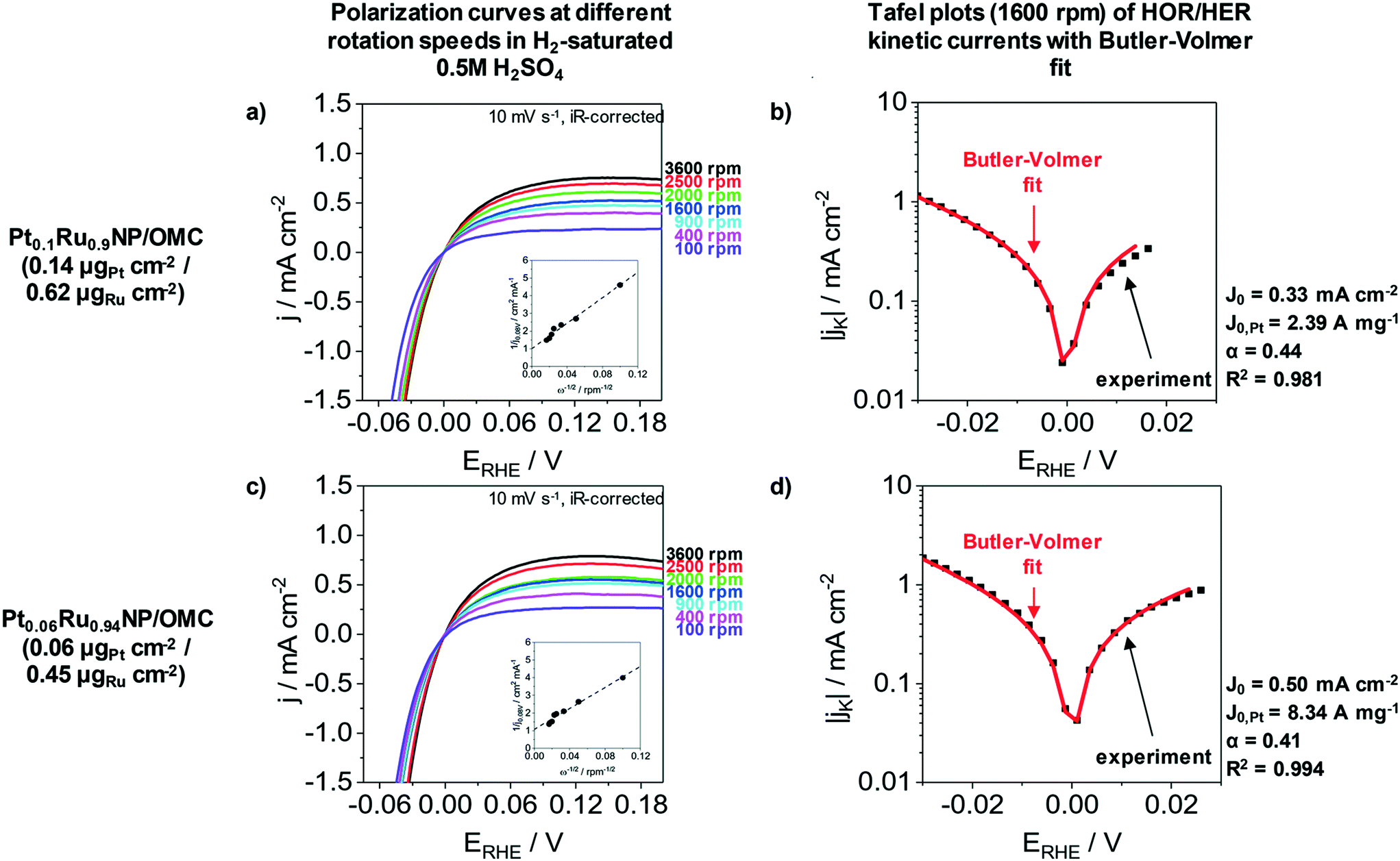 | ||
| Fig. 5 Correction of diffusional limitations for HER/HOR analysis of Pt0.1Ru0.9NP/OMC (a and b) catalyst film (WDX/StrataGem: 0.14 mgPt cm−2/0.62 mgRu cm−2) and a Pt0.06Ru0.94NP/OMC (c and d) catalyst film (WDX/StrataGem: 0.06 mgPt cm−2/0.45 mgRu cm−2) in H2-saturated 0.5 M H2SO4 at a scanning rate of 10 mV s−1. a) and c) polarization curves at different rotation speeds (100 to 3600 rpm). The insets show Koutecky–Levich plots (0.08 V vs. RHE). The slope yields a Bc0 value of 0.028 mA (cm2 rpm1/2)−1 for Pt0.1Ru0.9NP/OMC and 0.034 mA (cm2 rpm1/2)−1 for Pt0.06Ru0.94NP/OMC. The rotation speed dependence in both the HER and HOR branches is an indication for diffusional limitations. b) and d) Tafel plots of HER/HOR kinetic currents at a rotation speed of 1600 rpm with fittings of a Butler–Volmer equation. The geometric exchange current densities obtained from these fits amount to j0 = 0.33 mA cm−2 for Pt0.1Ru0.9NP/OMC and j0 = 0.50 mA cm−2 for Pt0.06Ru0.94NP/OMC. | ||
Other PtRu catalysts published in recent years show good HER activities at low platinum content. For example, nanoparticles with a PtRu shell on a WC core, tested in 0.1 M HClO4, need an overpotential of −38.2 mV to reach a current density of −10 mA cm−2 (mass loading: 13 μgPt cm−2 and 7 μgRu cm−2).59 For comparison, a commercial PtRu catalyst with a higher platinum loading (25.5 μgPt cm−2 and 25.5 μgRu cm−2) requires an overpotential of −42.4 mV in order to achieve the same current density under the same conditions.59Table 1 compares different Pt containing catalysts from literature tested in 0.5 M H2SO4. In most cases, the reported catalysts require significantly higher amounts of platinum to achieve similar performances. The large surface area of the mesoporous carbon matrix and the improved distribution of small alloy NPs, which provides an enhanced accessibility of active sites, can explain the increased catalytic activity of PtRuNP/OMC.
| Reference | Catalyst | Metal loading | Potential @ 10 mA cm−2 | Tafel slope | |
|---|---|---|---|---|---|
| This work | Pt0.3Ru0.7NP/OMC | 0.09 μgPt cm−2 | 0.14 μgRu cm−2 | −100.6 mV | 41 |
| Pt0.3Ru0.7NP/OMC | 0.20 μgPt cm−2 | 0.23 μgRu cm−2 | −59.9 mV | 42 | |
| Pt0.3Ru0.7NP/OMC | 0.44 μgPt cm−2 | 0.39 μgRu cm−2 | −42.3 mV | 37 | |
| Pt0.1Ru0.9NP/OMC | 0.14 μgPt cm−2 | 0.62 μgRu cm−2 | −43.7 mV | 42 | |
| Pt0.06Ru0.94NP/OMC | 0.06 μgPt cm−2 | 0.45 μgRu cm−2 | −55.3 mV | 41 | |
| Bernsmeier et al.24 | Pt/OMC | 1.2 μgPt cm−2 | — | −38.4 mV | 38 |
| PtRu/OMC | 0.2 μgPt cm−2 | 0.5 μgRu cm−2 | −36.3 mV | 38 | |
| This work | Pt/C/Nafion | 1.0 μgPt cm−2 | — | −30.9 mV | 30 |
| This work | PtRu/C/Nafion | 1.0 μgPt cm−2 | 0.5 μgRu cm−2 | −34.4 mV | 27 |
| Gao et al.68 | Pt/Vulcan/Nafion | 56.0 μgPt cm−2 | — | −33.5 mV | 30 |
| Li et al.69 | PtRu@RFCS-6 h | 0.71 μgPt cm−2 | 17.7 μgRu cm−2 | −19.7 mV | 27 |
| Li et al.70 | Pt ML/Ag NF/Ni foam | 0.55 μgPt cm−2 | — | −70 mV | 53 |
| Chen et al.71 | PtCoFe@CN | 13.1 μgPt cm−2 | — | −45 mV | 32 |
| Liu et al.72 | Pd@PdPt | 121.7 μgPt cm−2 | 444.2 μgPd cm−2 | −39 mV | 38 |
| Jiang et al.73 | Pt NC/N-graphene-2 | 5.6 μgPt cm−2 | — | −24 mV | 28 |
The carbonization in a reducing atmosphere (e.g. H2/Ar) might induce Pt-nanoclusters to appear on the surface of the PtRu alloy NPs.60 The electronic structure of the surface Pt-atoms is changed by neighbouring Ru-atoms, which weakens the hydrogen adsorption energy.19,61,62 Density functional (DFT) calculations of metal surfaces show synergistic effects (strain and ligand effects) when adjacent M-atoms are present alongside Pt-atoms.63–65 Ru modifies the d-band structure of Pt, resulting in a change in the Pt surface d-band width. The Pt d-band of PtRu is broader and lower in energy than that of Pt, and therefore has an optimal hydrogen binding energy closer to zero compared to Pt. Moreover, ruthenium acts as a promoter for the adsorption and dissociation of water, thus providing an excess of protons and electrons.66,67 Furthermore, RuOxHy species in PtRuNPs serve as electron- and proton-conducting hydrous oxides for the adsorbed hydrogen at Pt-active site.67 These explanation, are in a good agreement with our experimental data and the good performances as well as the high Pt-mass based exchange current densities of PtRuNP/OMC catalysts. Kinetic studies also show that the Heyrovsky mechanism predominates and H2 is generated via the electrochemical desorption.
Conclusions
We present a synthesis approach to bimetallic nanoparticles incorporated in ordered mesoporous carbon films with controllable and tunable particle composition. The synthesis approach relies on metal halogenates dissolved in a reduction agent which is simultaneously a stabilization agent. The stabilized nanoparticles are mixed with polymeric carbon precursors in the presence of a structure-directing agent. A carbonization under reductive atmosphere forms the mesoporous carbon films with incorporated bimetallic nanoparticles. The carbon films are prepared via dip-coating without the use of a binding agent which potentially blocks both mesopores and active sites. The mesoporous films show a high surface area, a good electrical conductivity and an improved distribution of nanoparticles in the carbon matrix. The PtRuNP retain their size, composition and crystalline structure during carbonization. PtRuNP/OMC films show high platinum mass-based activities in the acidic hydrogen evolution reaction (HER). The high HER performance is attributed to Pt-nanoclusters on the surface of the alloyed particles and the Ru-modified d-band structure of Pt combined with the high surface area and electrical conductivity. The proposed concept of bimetallic nanoparticles in ordered mesoporous carbon coatings enables the synthesis of a broad variety of bimetallic catalysts (e.g. RhRuNP/OMC) for all kind of reactions in electrochemical and heterogeneous catalysis.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work is funded by the European Metrology Research Programme (EMRP) 16ENG03 Hybrid metrology for thin films in energy applications (HyMET). The EMRP is jointly funded by the EMRP participating countries within EURAMET and the European Union. The authors thank ZELMI (TU-Berlin) for providing infrastructure for electron microscopy. We thank Stefan Rümmler for XPS measurements. SAXS measurements were conducted on beamline mySpot at BESSY II with assistance of Anke Kabelitz. Denis Bernsmeier acknowledges generous funding from VIP+ (BMBF) under contract FKZ 03VP05390. Ralph Kraehnert acknowledge generous funding from BMBF under contract FKZ 03EK3009.Notes and references
- M. Hirscher, Angew. Chem., Int. Ed., 2011, 50, 581–582 CrossRef CAS PubMed
.
- I. E. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed., 2011, 50, 1476–1477 CrossRef CAS PubMed
- S. Bai, C. Wang, M. Deng, M. Gong, Y. Bai, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 12120–12124 CrossRef CAS PubMed
- Z. Liu, X. Y. Ling, X. Su, J. Y. Lee and L. M. Gan, J. Power Sources, 2005, 149, 1–7 CrossRef CAS
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed
- M. Eguchi, K. Baba, T. Onuma, K. Yoshida, K. Iwasawa, Y. Kobayashi, K. Uno, K. Komatsu, M. Kobori, M. Nishitani-Gamo and T. Ando, Polymer, 2012, 4, 1645 Search PubMed
- S. Tanaka, Y. Katayama, M. P. Tate, H. W. Hillhouse and Y. Miyake, J. Mater. Chem., 2007, 17, 3639–3645 RSC
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed
- Z. Qiang, J. Xue, G. E. Stein, K. A. Cavicchi and B. D. Vogt, Langmuir, 2013, 29, 8703–8712 CrossRef CAS PubMed
- D. Bernsmeier, L. Chuenchom, B. Paul, S. Rümmler, B. Smarsly and R. Kraehnert, ACS Catal., 2016, 6, 8255–8263 CrossRef CAS
- D. Bernsmeier, M. Bernicke, E. Ortel, A. Bergmann, A. Lippitz, J. Nissen, R. Schmack, P. Strasser, J. Polte and R. Kraehnert, ChemElectroChem, 2017, 4, 221–229 CrossRef CAS
- F. Vigier, C. Coutanceau, F. Hahn, E. M. Belgsir and C. Lamy, J. Electroanal. Chem., 2004, 563, 81–89 CrossRef CAS
- P. J. Kulesza, I. S. Pieta, I. A. Rutkowska, A. Wadas, D. Marks, K. Klak, L. Stobinski and J. A. Cox, Electrochim. Acta, 2013, 110, 474–483 CrossRef CAS PubMed
- O. A. Petrii, J. Solid State Electrochem., 2008, 12, 609 CrossRef CAS
- J. E. Mueller, P. Krtil, L. A. Kibler and T. Jacob, Phys. Chem. Chem. Phys., 2014, 16, 15029–15042 RSC
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS
- W. J. Mitchell, J. Xie, T. A. Jachimowski and W. H. Weinberg, J. Am. Chem. Soc., 1995, 117, 2606–2617 CrossRef CAS
- K. Elbert, J. Hu, Z. Ma, Y. Zhang, G. Chen, W. An, P. Liu, H. S. Isaacs, R. R. Adzic and J. X. Wang, ACS Catal., 2015, 5, 6764–6772 CrossRef CAS
- J. X. Wang, Y. Zhang, C. B. Capuano and K. E. Ayers, Sci. Rep., 2015, 5, 12220 CrossRef CAS PubMed
- X. Wang, Y. Zhu, A. Vasileff, Y. Jiao, S. Chen, L. Song, B. Zheng, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2018, 3, 1198–1204 CrossRef CAS
- S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333 CrossRef CAS PubMed
- D. Bernsmeier, M. Bernicke, E. Ortel, R. Schmack, J. Polte and R. Kraehnert, J. Catal., 2017, 355, 110–119 CrossRef CAS
- H. Bönnemann, W. Brijoux, R. Brinkmann, R. Fretzen, T. Joussen, R. Köppler, B. Korall, P. Neiteler and J. Richter, J. Mol. Catal., 1994, 86, 129–177 CrossRef
- H. Bönnemann, G. Braun, W. Brijoux, R. Brinkmann, A. S. Tilling, K. Seevogel and K. Siepen, J. Organomet. Chem., 1996, 520, 143–162 CrossRef
- T. J. Schmidt, M. Noeske, H. A. Gasteiger, R. J. Behm, P. Britz, W. Brijoux and H. Bönnemann, Langmuir, 1997, 13, 2591–2595 CrossRef CAS
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765 CrossRef CAS PubMed
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef
- E. Ortel, A. Hertwig, D. Berger, P. Esposito, A. M. Rossi, R. Kraehnert and V.-D. Hodoroaba, Anal. Chem., 2016, 88, 7083–7090 CrossRef CAS PubMed
- D.-M. Rosu, E. Ortel, V.-D. Hodoroaba, R. Kraehnert and A. Hertwig, Appl. Surf. Sci., 2017, 421, 487–493 CrossRef CAS
- M. R. Baklanov, K. P. Mogilnikov, V. G. Polovinkin and F. N. Dultsev, J. Vac. Sci. Technol., B, 2000, 18, 1385–1391 CrossRef CAS
- C. Boissiere, D. Grosso, S. Lepoutre, L. Nicole, A. B. Bruneau and C. Sanchez, Langmuir, 2005, 21, 12362–12371 CrossRef CAS PubMed
- S. Bucher, J. Hormes, H. Modrow, R. Brinkmann, N. Waldöfner, H. Bönnemann, L. Beuermann, S. Krischok, W. Maus-Friedrichs and V. Kempter, Surf. Sci., 2002, 497, 321–332 CrossRef CAS
- D. G. Shamiryan, M. R. Baklanov, S. Vanhaelemeersch and K. Maex, Electrochem. Solid-State Lett., 2001, 4, F3–F5 CrossRef CAS
- P. Löbmann, J. Sol-Gel Sci. Technol., 2017, 84, 2–15 CrossRef
- J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Microporous Mesoporous Mater., 2009, 118, 218–223 CrossRef CAS
- D. Bernsmeier, R. Sachse, M. Bernicke, R. Schmack, F. Kettemann, J. Polte and R. Kraehnert, J. Catal., 2019, 369, 181–189 CrossRef CAS
- V. L. Chavez, L. Song, S. Barua, X. Li, Q. Wu, D. Zhao, K. Rege and B. D. Vogt, Acta Biomater., 2010, 6, 3035–3043 CrossRef CAS PubMed
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 RSC
- A. Arico, P. Creti, H. Kim, R. Mantegna, N. Giordano and V. Antonucci, J. Electrochem. Soc., 1996, 143, 3950–3959 CrossRef CAS
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS
- W.-D. Schneider and C. Laubschat, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 997 CrossRef CAS
- M. Romeo, J. Majerus, P. Légaré, N. Castellani and D. Leroy, Surf. Sci., 1990, 238, 163–168 CrossRef CAS
- M. Zhang, W. Chen, S.-J. Ding, X.-P. Wang, D. W. Zhang and L.-K. Wang, J. Vac. Sci. Technol., A, 2007, 25, 775–780 CrossRef CAS
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan and A. Stein, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS
- Y. L. Hsin, K. C. Hwang and C.-T. Yeh, J. Am. Chem. Soc., 2007, 129, 9999–10010 CrossRef CAS PubMed
- E. T. Turkdogan and J. V. Vinters, Carbon, 1972, 10, 97–111 CrossRef CAS
- J. P. Neeft, M. Makkee and J. A. Moulijn, Fuel, 1998, 77, 111–119 CrossRef CAS
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1971, 33, 351–378 CrossRef CAS
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed
- S. Fletcher, J. Solid State Electrochem., 2009, 13, 537–549 CrossRef CAS
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. D. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed
- Y. H. Li, P. F. Liu, L. F. Pan, H. F. Wang, Z. Z. Yang, L. R. Zheng, P. Hu, H. J. Zhao, L. Gu and H. G. Yang, Nat. Commun., 2015, 6, 8064 CrossRef CAS PubMed
- J. Wang, Z. Wei, S. Mao, H. Li and Y. Wang, Energy Environ. Sci., 2018, 11, 800–806 RSC
- Z. Chen, J. Lu, Y. Ai, Y. Ji, T. Adschiri and L. Wan, ACS Appl. Mater. Interfaces, 2016, 8, 35132–35137 CrossRef CAS PubMed
- B. Conway and L. Bai, J. Electroanal. Chem. Interfacial Electrochem., 1986, 198, 149–175 CrossRef CAS
- J. Zheng, Y. Yan and B. Xu, J. Electrochem. Soc., 2015, 162, F1470–F1481 CrossRef CAS
- S. T. Hunt, M. Milina, A. C. Alba-Rubio, C. H. Hendon, J. A. Dumesic and Y. Román-Leshkov, Science, 2016, 352, 974–978 CrossRef CAS PubMed
- R. Bavand, A. Korinek, G. A. Botton, A. Yelon and E. Sacher, J. Phys. Chem. C, 2017, 121, 23104–23119 CrossRef CAS
- J. Greeley and M. Mavrikakis, J. Phys. Chem. B, 2005, 109, 3460–3471 CrossRef CAS
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS
- M. E. Scofield, Y. Zhou, S. Yue, L. Wang, D. Su, X. Tong, M. B. Vukmirovic, R. R. Adzic and S. S. Wong, ACS Catal., 2016, 6, 3895–3908 CrossRef CAS
- W. F. Lin, M. S. Zei, M. Eiswirth, G. Ertl, T. Iwasita and W. Vielstich, J. Phys. Chem. B, 1999, 103, 6968–6977 CrossRef CAS
- C. L. Green and A. Kucernak, J. Phys. Chem. B, 2002, 106, 1036–1047 CrossRef CAS
- M.-R. Gao, J.-X. Liang, Y.-R. Zheng, Y.-F. Xu, J. Jiang, Q. Gao, J. Li and S.-H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS
- K. Li, Y. Li, Y. Wang, J. Ge, C. Liu and W. Xing, Energy Environ. Sci., 2018, 11, 1232–1239 RSC
- M. Li, Q. Ma, W. Zi, X. Liu, X. Zhu and S. Liu, Sci. Adv., 2015, 1, e1400268 CrossRef PubMed
- J. Chen, Y. Yang, J. Su, P. Jiang, G. Xia and Q. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 3596–3601 CrossRef CAS PubMed
- Y. Liu, S. Liu, Z. Che, S. Zhao, X. Sheng, M. Han and J. Bao, J. Mater. Chem. A, 2016, 4, 16690–16697 RSC
- B. Jiang, F. Liao, Y. Sun, Y. Cheng and M. Shao, Nanoscale, 2017, 9, 10138–10144 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02285f |
| This journal is © The Royal Society of Chemistry 2020 |