
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing the PD-L1 interface peptide for positron emission tomography imaging of the PD-1 immune checkpoint†
Kuan
Hu
 a,
Lin
Xie
a,
Masayuki
Hanyu
a,
Yiding
Zhang
a,
Lingyun
Li
b,
Xiaohui
Ma
c,
Kotaro
Nagatsu
a,
Hisashi
Suzuki
a,
Weizhi
Wang
*b and
Ming-Rong
Zhang
*a
a,
Lin
Xie
a,
Masayuki
Hanyu
a,
Yiding
Zhang
a,
Lingyun
Li
b,
Xiaohui
Ma
c,
Kotaro
Nagatsu
a,
Hisashi
Suzuki
a,
Weizhi
Wang
*b and
Ming-Rong
Zhang
*a
aDepartment of Advanced Nuclear Medicine Sciences, National Institute of Radiological Sciences, National Institutes for Quantum and Radiological Science and Technology, Chiba, 263-8555, Japan. E-mail: zhang.ming-rong@qst.go.jp
bSchool of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: wangwz@bit.edu.cn
cDepartment of Vascular Surgery, General Hospital of People's Liberation Army, Beijing 100853, P. R. China
First published on 27th August 2020
Abstract
Interface peptides that mediate protein–protein interactions (PPI) are a class of important lead compounds for designing PPI inhibitors. However, their potential as precursors for radiotracers has never been exploited. Here we report that the interface peptides from programmed death-ligand 1 (PD-L1) can be used in positron emission tomography (PET) imaging of programmed cell death 1 (PD-1) with high accuracy and sensitivity. Moreover, the performance differentiation between murine PD-L1 derived interface peptide (mPep-1) and human PD-L1 derived interface peptide (hPep-1) as PET tracers for PD-1 unveiled an unprecedented role of a non-critical residue in target binding, highlighting the significance of PET imaging as a companion diagnostic in drug development. Collectively, this study not only provided a first-of-its-kind peptide-based PET tracer for PD-1 but also conveyed a unique paradigm for developing imaging agents for highly challenging protein targets, which could be used to identify other protein biomarkers involved in the PPI networks.
Introduction
Radiodiagnostics, such as PET imaging, has emerged as a vital component in precision medicine.1–7 Several radiotracers have been used extensively for patient stratification and prognosis assessment in clinical settings. Besides, numerous radiotracers are being studied in clinical trials or are under pre-clinical evaluation. Especially, peptide-based radiotracers have aroused escalating interest due to their low immunogenicity and good drug metabolism and pharmacokinetics (DMPK) properties.8–10 Moreover, peptides could be easily modified to improve their pharmacological properties.11,12 Tremendous efforts have been invested in developing peptide radiotracers. Many previous studies focused on endogenous ligands of pathogenic receptors or antigens, which were then properly reformed or modified (Scheme 1A) to obtain a panel of renowned tracers for tumor imaging or radiotherapy,9 such as 68Ga(177Lu)-dotatate,13111indium pentetreotide,14 and 68Ga-PSMA-I&T.15 However, the majority of pathogenic biomarkers are not associated with endogenous ligand(s), making the discovery of peptide radiotracers for these targets a formidable challenge. Recently, high-throughput screening techniques (HTS, e.g. phage display and PepArray) have achieved some success in the development of peptide tracers for membrane receptors and antigens in tumor cells.11 However, these HTS methods depend heavily on the construction and validation of a complex library of target compounds, leading to the moderate adaptability and flexibility of using this method for the dection of some targets.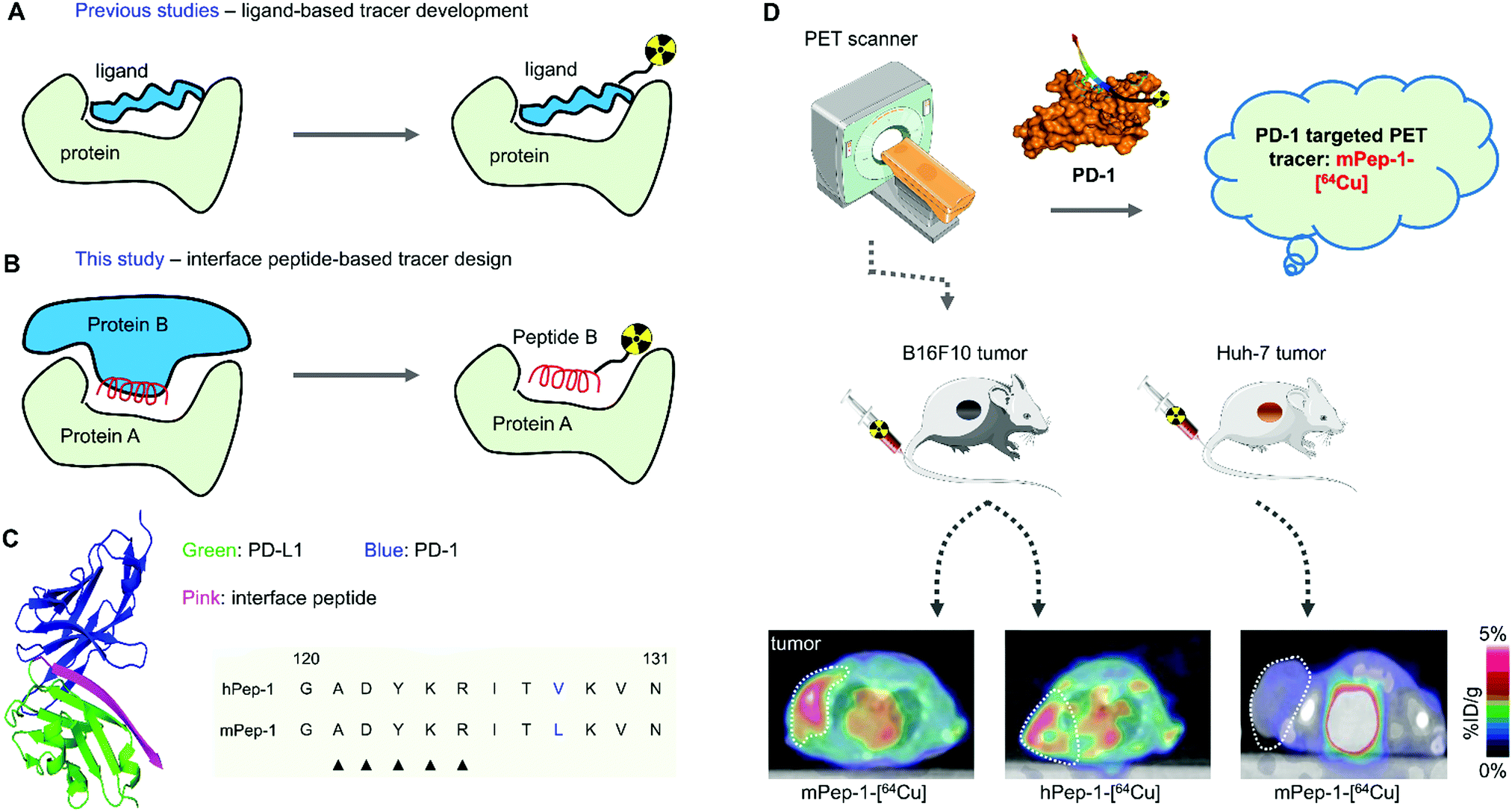 | ||
| Scheme 1 Strategies for peptide-based radiotracer development. (A) Previous studies focused on using radiotracer-integrated peptide ligands to track protein targets. (B) Conceptual diagram of interface peptide-based tracer design. The core component of the interface peptide-based tracer design is represented by the key sequence in protein B at the binding interface of a PPI, which is a potential ligand (inhibitor) for protein A. When truncated from the parental protein, the peptide sequence from protein B can be developed as radiotracers for tracking protein A. (C) Representative co-crystal structure of PD-1 and PD-L1. Two interface peptides (mpep-1 and hpep-1) were derived from mPD-L1 and hPD-L1, respectively. The black triangles indicate the “hot spots” that mediate the interactions between PD-1 and PD-L1. (D) The study plan of this work. Two kinds of tumors were used for PET study. Two tracers, mPep-1-[64Cu] and hPep-1-[64Cu], were evaluated for their capacity in imaging of PD-1. | ||
On the other hand, the interface peptide strategy is a straightforward and compelling method for obtaining ligand peptides of protein targets that are involved in protein–protein interactions (PPIs).16–18 Closely related to structure-based drug design, the rationale underscoring the interface peptide strategy is based on leveraging short peptides derived from the binding interface of PPIs as potent ligands for their protein counterparts. The feasibility of this strategy has been demonstrated for several targets, such as HIV-1 gp41 and p53-MDM2.19–21 Even though the interface peptide strategy has never been used for radiotracer development, considering that many disease biomarkers are involved in PPI networks, we hypothesized that interface peptides could be used as valuable precursors for radiotracer development (Scheme 1B).
Programmed death-1 (PD-1), binding to programmed death-ligand 1 (PD-L1), could negatively modulate the antitumor immune response.22 Analysis of clinical outcomes suggested that the expression level of PD-1 in tumor-infiltrating lymphocytes (TILs) is one of the critical indicators for predicting responses to immune checkpoint inhibitors.23,24 Therefore, non-invasive tracking of the spatiotemporal expression of PD-1 in malignancies is especially important. Several PD-1 targeting immunoradiotracers have been reported to date. However, due to their unfavourable pharmacokinetics and limited tumor tissue penetration, their application as optimal imaging agents in clinical practice is rather limited.25–33 Based on high-resolution co-crystal complex analysis, the interaction between PD-1 and PD-L1 is considered to meet the criteria of a typical PPI.34,35 In this study, we reported the development of short peptide-based PET tracers for PD-1 using the interface peptides from PD-L1. Specifically, two dodecapeptides derived from the binding interface of PD-L1 were modified and labeled with 64Cu for PET imaging of PD-1 in TILs from a murine melanoma model. Finally, the characteristics and imaging capacity of the tracers were compared.
Experimental
Materials and methods
All chemicals were purchased from Wako Pure Chemical Industries (Osaka, Japan) or Sigma-Aldrich (St Louis, MO, USA). 64Cu was produced at the National Institute of Radiological Science (Chiba, Japan) with 98% radionuclidic purity in house. All radio-HPLC analyses for the radiotracers were performed using a JASCO HPLC system (JASCO, Tokyo, Japan) coupled with a YMC-Triat-C18 column (4.6 mm i.d. × 150 mm, 5 μm, Waters, Milford, MA, USA). A flow rate of 1 mL min−1 was used. The gradient started with 90% solvent A (0.1% trifluoroacetic acid [TFA] in water) and 10% solvent B (0.1% TFA in acetonitrile [MeCN]), and 20 min later ended with 0% solvent A and 100% solvent B. The effluent radioactivity was measured using a NaI (TI) scintillation detector system (Ohyo Koken Kogyo, Tokyo, Japan). A 1480 Wizard autogamma counter (PerkinElmer, Waltham, MA, USA) was used to measure the radioactivity, as expressed in counts of radioactivity per minute (CPM), accumulating in cells and animal tissues. A dose calibrator (IGC-7 Curiemeter; Aloka, Tokyo, Japan) was used for the other radioactivity measurements. The PD-1/PD-L1 blockade Bioassay kit was purchased from Promega Corporation (Catalog No. J1250). Concanavalin A was purchased from Sigma-Aldrich (St Louis, MO, USA).Preparation of m(h)Pep-1-DOTA
Peptides m(h)Pep-1-DOTA were synthesized manually by an Fmoc-based solid-phase peptide synthesis strategy,36,37 as illustrated in Scheme S1 (ESI†). 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) was used as a chelator for 64Cu labeling and a PEG linker was used to space the peptide and DOTA. For DOTA conjugation, DOTA was preactivated by N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC·HCl, 2 mg) at a molar ratio for DOTA/EDC·HCl/N-hydroxysuccinimide (NHS) of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:4 in dimethyl sulfoxide (DMSO, 2 mL) for 3 h. Then the m(h)Pep-1 peptide conjugated resin (100 mg; 0.37 mmol g−1) was suspended in 1 mL of N-methyl-2-pyrrolidone (NMP). Afterward, the activated DOTA and 100 μL of DIEA were coadded to the resin. The reaction mixture was gently bubbled with N2 gas for 2 h. Finally, DOTA-PEG3-m(h)Pep-1 was cleaved from the resin and purified by high-performance liquid chromatography (HPLC). The collected product eluents were lyophilized and dissolved in H2O at a concentration of 1 mg mL−1 for use in radiolabeling reactions.
:5:4 in dimethyl sulfoxide (DMSO, 2 mL) for 3 h. Then the m(h)Pep-1 peptide conjugated resin (100 mg; 0.37 mmol g−1) was suspended in 1 mL of N-methyl-2-pyrrolidone (NMP). Afterward, the activated DOTA and 100 μL of DIEA were coadded to the resin. The reaction mixture was gently bubbled with N2 gas for 2 h. Finally, DOTA-PEG3-m(h)Pep-1 was cleaved from the resin and purified by high-performance liquid chromatography (HPLC). The collected product eluents were lyophilized and dissolved in H2O at a concentration of 1 mg mL−1 for use in radiolabeling reactions.
64Cu labeling
The labeling method is the same as that which we reported previously. Briefly, mPep-1 or hPep-1 peptide was radiolabeled with 64CuCl2. Twenty μg of peptide in 10 μL of 0.1 M sodium acetate buffer (pH = 4.1) was reacted with ∼370 MBq (10 mCi) of neutralized 64CuCl2 solution at 80 °C for 10 min. After incubation, the reaction mixture was purified by RP-HPLC.In vitro stability assays
The stability of the radiotracers in saline and in mouse serum was tested using the same protocol as that which we reported before. Ten microliters (∼100 μCi) of radiotracers were added to 90 μL of mouse serum (freshly prepared) and incubated at 37 °C with slight agitation for a designated time. Aliquots were taken out at each time point and 100 μL of MeCN and water (1:1, v/v) was added. Then the mixture was centrifuged for 10 min at 10000 rpm. The supernatant was analyzed using RP-HPLC. For RP-HPLC, the analysis used an additional guard column (Phenomenex Security Guard 3.00 mm I.D.) for the protection of the C-18 column. For the stability in saline, the radiotracers were incubated in saline (>95%, v), and then aliquots were taken out at each time point for RP-HPLC analysis.
Luciferase based PD-1/PD-L1 blockade assay
Checkpoint blockade was assayed by the PD-1/PD-L1 blockade luciferase reporter assay (Promega). Briefly, PD-L1 APC/CHO-K1 stably expressing cells were thawed in Ham's F-12 media containing 10% FBS. The cells were plated in a tissue culture treated, white 384 well plate at 5000 cells per 10 mL per well and allowed to incubate overnight. The following day, PD-1 expressing effector cells (Jurkat cells) in RPMI1640 with 1% FBS were plated separately at a concentration of 10000 cells per 20 mL per well. The effector cells were then treated with a 10-point serial dilution of vehicle, mPep-1, or hPep-1 for 30 min at 37 °C before being transferred to the plate containing the CHO cells. The coculture was then incubated at 37 °C for 6 h in a tissue culture incubator. Following incubation, the Bio-Glow reagent was added to the wells in a 1:1 ratio and incubated for 10 min at room temperature. Luminescence was measured from the top down on a Biacore plate reader with an integration time of 0.5 s. Data were analyzed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA).
Cell line and animals
The mouse melanoma cell line B16F10 and human hepatocellular carcinoma Huh-7 were maintained and passaged in a humidified CO2 incubator (37 °C/5% CO2) and the cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum and 0.5% penicillin/streptomycin.Male BALB/c nude−/− mice and C57BL/6J Jms mice (7 weeks old) were purchased from Japan SLC (Shizuoka, Japan). All animals received humane care, and the Animal Ethics Committee of the National Institute of Radiological Sciences approved all experiments. All experiments were carried out according to the recommendations of the Committee for the Care and Use of Laboratory Animals, National Institute of Radiological Sciences, and all animal studies were approved by the Animal Ethics Committee of National Institute for Quantum and Radiological Science and Technology. A tumor-bearing mouse model using C57BL/6J Jms was prepared via a left flank subcutaneous injection or hind leg subcutaneous injection of 100 μL of cells (1 × 106 cells per mouse). Tumor-bearing mice were used for studies when the tumor diameters reached 3–5 mm. A tumor-bearing mouse model using BALB/c nude−/− mice was prepared via a left flank subcutaneous injection of 100 μL of cells (5 × 106 cells per mouse). Tumor-bearing mice were used for studies when the tumor diameters reached 3–5 mm.
Cellular uptake and inhibition experiment
Murine T cells were obtained from the spleen of C57BL/6J mice using the MagniSort™ Mouse T cell Enrichment Kit (Invitrogen). After separation, the T cells were maintained in RPMI 1640 basal medium supplemented with 10% FBS (37 °C/5% CO2). One day before radiolabeling, the T cells were transferred to 12 well plates with 5 × 104 cells per well. Radiolabeled tracers in medium (740 KBq/1 mL) were added to each well, and the plates were incubated at 37 °C for 5 min, 10 min, 20 min, 40 min, and 60 min. After incubation, the T cells were gently collected into tubes by centrifugation, and then washed with PBS twice. The radioactivity in each tube was measured with an autogamma counter. For binding inhibition, unlabeled mPep-1-DOTA (2 mM) was added to each well. The same procedures as those in the tracer uptake assay were performed to obtain the inhibition rate at respective time points.Ex vivo biodistribution
A saline solution of tracer (1.85 MBq/100 μL) was injected into B16F10-bearing C57BL/6J jms mice via the tail vein. Three mice were sacrificed by cervical dislocation at 5 min, 20 min, 40 min, and 100 min after injection of mPep-1-[64Cu] or hPep-1-[64Cu]. Major organs, including the heart, liver, lungs, spleen, pancreas, kidneys, stomach, muscle, small intestine, intestinal lymph node, testes, bone, brain, blood, and the tumor were quickly harvested and weighed. The radioactivity in these organs was measured using the autogamma counter. The results are expressed as the percentage of injected dose per gram of wet tissue (% ID g−1). All radioactivity measurements were decay-corrected.Small-animal PET study
PET scans were conducted using an Inveon PET scanner (Siemens Medical Solutions, Knoxville, TN, USA), which provides 159 transaxial slices with 0.796 mm (center-to-center) spacing, a 10 cm transaxial field of view, and a 12.7 cm axial field of view. All list-mode acquisition data were sorted into three-dimensional (3D) sinograms, which were then Fourier rebinned into two-dimensional (2D) sinograms (frames × min: 4 × 1, 8 × 2, 8 × 5). Tumor-bearing B16F10/6J Jms or BALB/c nude−/− mice were kept in the prone position under anesthesia with 1–2% (v/v) isoflurane during the scan. The tracers (10–17 MBq/100–200 μL) were injected via a preinstalled tail vein catheter. Immediately after the injection, a dynamic scan in 3D list mode was acquired for 60 min (n = 3). Maximum intensity projection (MIP) images were obtained for C57BL/6J tumor-bearing mice. PET dynamic images were reconstructed by filtered back projection using Hanning's filter with a Nyquist cutoff of 0.5 cycle per pixel, which was summed using analysis software (ASIPro VM, Siemens Medical Solutions). Volumes of interest, including the spleen, muscle and tumor, were placed using the ASIPro software. The radioactivity was decay-corrected for the injection time and expressed as the percent of the total injection dose per gram tissue (% ID g−1).In vitro autoradiography
Frozen tumors from C57BL/6J Jms were trimmed with a razor blade and mounted in a cryostat chuck. Tissue sections were cut at a thickness of 10 μm using a cryostat (Bright OTF5000) and thaw mounted onto Superfrost® slides. Two consecutive sections were placed on each slide. The B16F10 tumor was cut into 10 μm sections and stored at 80 °C until they were used for the experiment. The tumor sections were preincubated with Tris–HCl buffer (pH 7.4, 50 mM), MgCl2 (1.2 mM), and CaCl2 (2 mM) solution for 20 min at ambient temperature, followed by incubation with mPep-1-[64Cu] or hPep-1-[64Cu] (7.4 MBq/200 mL) for 30 min at ambient temperature. For blocking studies, unlabeled mPep-1 (5 mM) was added to the incubation solution in advance to determine the specificity of radioligand binding. After incubation, tumor sections were washed with Tris buffer two times for 2 min and dipped in cold distilled water for 10 s. The tumor sections were dried with cold air and then placed on imaging plates (BASMS2025, GE Healthcare, NJ) for 20 min. Autoradiograms were obtained and ROIs were carefully drawn with the reference of naked-eye observation. The radioactivity was expressed as photostimulated luminescence values per unit area (PSL mm−2) and measured using a Bio-Imaging analyzer system (BAS5000, Fujifilm).Simulation of peptide structure
The URL for the PEP-FOLD program is https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PEP-FOLD3. The simulation of the peptides’ secondary structure followed the instructions of the constructors. The settings for the simulations include: Run label, PEPFOLD; Number of simulations, 100; sort models by sOPEP, and no demonstration mode was used.Statistics
All measurements are expressed as means ± standard deviations (SD). Statistical analyses were performed using the GraphPad Prism 8 software.Results
Interface peptide selection
From the crystal complex of human PD-1/PD-L1 (PDB: 4ZQK)38 and murine PD-1/PD-L1 (PDB: 3BIK),39 highly conserved residues for mediating the PPIs were observed within the remarkably similar binding interface of the PPIs.38–42 Additionally, the majority of the interacting residues were gathered at the C terminal of the extracellular IgV domain of PD-L1, spanning from the tyrosine at position 112 to the asparagine at position 131. Moreover, a contiguous segment comprised of five residues located at position 121 to 125 was reported to dominate the specific binding.40 Therefore, taking into account the structural characteristics and biophysical properties of the interface peptide, a peptide sequence stretching from Gly120 to Asn131, i.e. GADYKRIT(V/L)KVN, was selected as the interface peptide (Scheme 1C).43 Compared to the full-length sequence from Tyr112 to Asn131, the shorter peptide would be more stable in vivo. A residue variation at position 128 (V to L) was intentionally preserved between the two peptides, which would allow us to compare the differences in imaging capacity of the interface peptides from different species.Interface peptide synthesis and radiolabeling
The interface peptides, hPep-1 and mPep-1, were obtained by manual solid-phase peptide synthesis (Fig. 1A). Radionuclide 64Cu (t1/2 = 12.7 h, β+ = 17%, β− = 39%, EC = 43%, Emax = 0.656 MeV) is an attractive nuclide for PET study due to its comparatively long half-life and ease of labeling.44 To label 64Cu within the peptides, the chelator DOTA was connected to the N terminal of the peptides (Fig. 1B). To avoid any DOTA-related perturbation of the mode of peptide binding, a poly(ethylene glycol)3 (PEG3) linker was used to create space between the peptide and DOTA. Radiolabelling was then performed according to the method reported previously.45,46 As shown in Table 1, the labeling of both of the peptides achieved high radiochemical yields (>90%). Consequently, the molar activities for hPep-1-[64Cu] and mPep-1-[64Cu] were 53.4 ± 4.7 GBq μmol−1 and 55.4 ± 7.3 GBq μmol−1, respectively. mPep-1-[64Cu] showed a longer retention time (tR = 7.83 min) in the high-performance liquid chromatogram (HPLC) column than hPep-1-[64Cu] (tR = 7.48 min), indicating that mPep-1-[64Cu] was more hydrophobic than hPep-1-[64Cu] (Fig. S1, ESI†). This phenomenon could be attributed to the fact that Leu is more hydrophobic than Val. The radiochemical purities of both the tracers were above 95% after purification.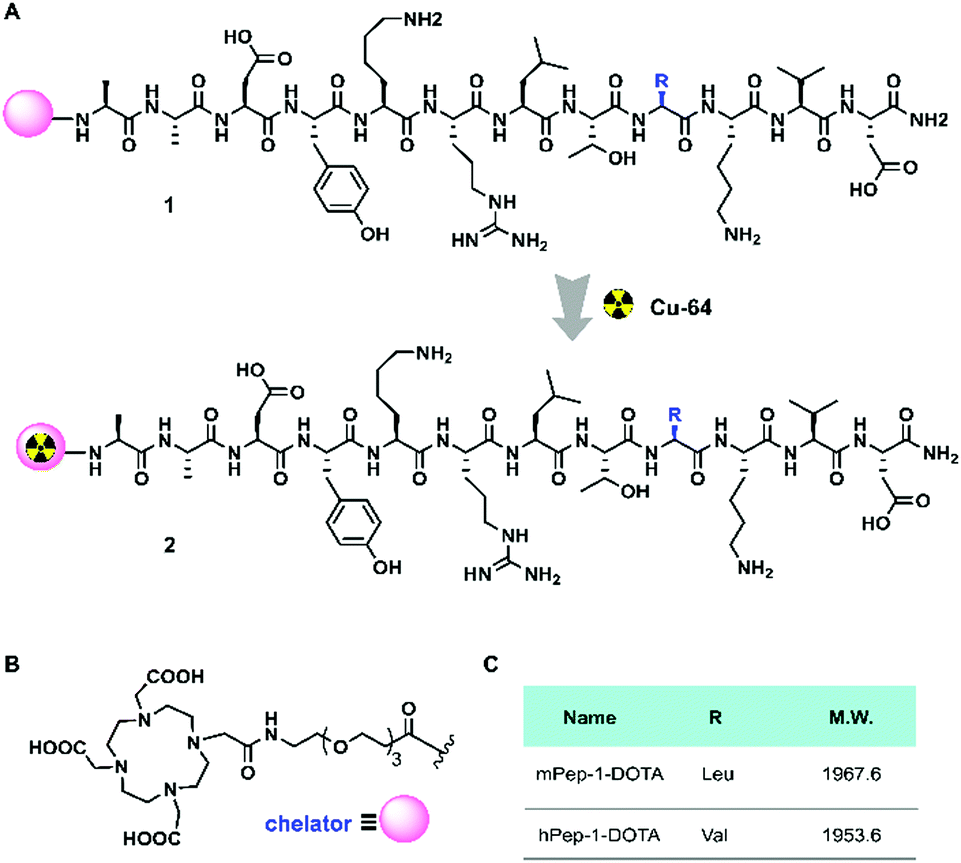 | ||
| Fig. 1 Chemical structures of the tracers. (A) 64Cu labeling reaction of hPep-1-DOTA and mPep-1-DOTA. (B) Chemical structure of the chelator and spacer. (C) The R residues and molecular weight (M.W.) of hPep-1 and mPep-1, respectively. | ||
| Tracers | hPep-1-[64Cu] | mPep-1-[64Cu] |
|---|---|---|
| a The radiochemical purity was determined by HPLC under conditions as follows: YMC-Triat-C18 column (4.6 mm i.d. × 150 mm, 5 μm); solvent gradient of 10–90% acetonitrile (0.1% trifluoroacetic acid (TFA)), 20 min; flow rate of 1 mL min−1. | ||
| Radiochemical yield (%) | >90 | >90 |
| Molar activity (GBq μmol−1) | 53.3 ± 4.7 | 55.4 ± 7.3 |
| Radiochemical puritya (%) | >95 | >95 |
| Retention time (tR) | 7.48 min | 7.83 min |
Both tracers showed good stability with no decomposition or 64Cu detachment after 24 hours of incubation in saline at 37 °C. In addition, >90% of the intact tracers were retained after the 24 hour incubation period. The stability of the tracers in mouse serum was also investigated and higher stability was observed in both mPep-1-[64Cu] and hPep-1-[64Cu], as >90% of the intact tracers were retained at 1 hour post-incubation (Fig. S2, ESI†).
In vivo PET imaging of PD-1
The murine melanoma B16F10-bearing C57BL/6J mouse is a suitable model for PET imaging study for PD-1, as the tumor-infiltrating lymphocytes in these mice overexpress mPD-1.25,47 Dynamic PET was performed for one hour following intravenous injection to monitor the tumor engagement and biodistribution of our tracers. Representative PET images coregistered with computed tomography (CT) images for mice with inoculated tumors (at 20 and 40 min p.i.) in the left-armpits are shown in Fig. 2A and B. Specifically, mPep-1-[64Cu] exhibited higher tumor uptake than hPep-1-[64Cu] at 20 and 40 min following injection. This phenomenon could be attributed to the higher binding ability of mPep-1-[64Cu] to PD-1 than that of hPep-1-[64Cu]. Both tracers were shown to mainly accumulate in the kidney (K) and bladder (B), suggesting that our tracers were renally clearable. Strong radioactivity was observed in the bladder, indicating that clearance of the tracers was dominated by the kidney–bladder metabolic pathway (Fig. S3, ESI†). Additionally, PET signal quantification was performed in the region of interest (ROI) and the resulting time–activity curves (TAC) from 0–60 min p.i. were plotted as Fig. 2C and D. Both tracers reached the highest tumor uptake at ∼8 min p.i., followed by a gradual decline in radioactivity in the tumors. Finally, mPep-1-[64Cu] revealed higher accumulation in both the tumor and the spleen (an immune cell-rich organ) than hPep-1-[64Cu]. | ||
| Fig. 2 PET-CT co-registered images and region of interest (ROI) curves of selected organs in B16F10-bearing C57BL/6J mice. Coronal and axial PET-CT images of mice post-injection of (A) mPep-1-[64Cu] and (B) hPep-1-[64Cu] at 20 min and 40 min. Areas within the white dashed circles indicate the tumors. (C) Tumor and muscle and (D) spleen uptake were estimated from the ROI of PET-CT images; for each curve the error bars represent standard deviations (SD), n = 3. | ||
To validate the above observations, mice bearing B16F10 tumors in both hind legs were imaged with our tracers. Similarly, mPep-1-[64Cu] showed higher uptakes in both the tumor and spleen than hPep-1-[64Cu] at 20 min and 40 min p.i. of the tracers (Fig. S4A and B, ESI†). Both tracers showed a heterogeneous distribution in tumors, revealing that most of the tracers were located at the periphery of the tumors, which was consistent with the TIL heterogeneity in the tumor microenvironment.48 Besides, the TAC of the tumor and spleen from the PET images also confirmed higher accumulation of mPep-1-[64Cu] in the tumor than that of hPep-1-[64Cu] (Fig. S4C and D, ESI†).
To verify that our tracers were PD-1 specific, we performed a blocking study in C57BL/6J mice bearing B16F10 tumor cells. The blocking study was performed by co-injection of mPep-1-[64Cu] and mPep-1-DOTA (5 mg kg−1) via the tail vein. As a result, an obvious decrease of tracer uptake in the tumor was observed at both 20 min and 40 min post-injection (Fig. S5A and B, ESI†). Afterward, the % ID g−1 values of the tumor, muscle, and spleen were quantified based on the PET images. The blocking results in a consecutive ∼1.5% ID g−1 reduction in the tumor from 5 min to 60 min post-injection (Fig. S5C, ESI†). Moreover, a more pronounced reduction of tracer uptake elicited by mPep-1-DOTA blocking was observed in the spleen (Fig. S5D, ESI†), in good agreement with the fact that the spleen is an important part of the immune system. No significant difference in muscle uptake between blocking and non-blocking mice was observed due to the very low background uptake in this organ. To further validate the radiotracers as being immune-cell specific, an immunodeficient mouse model was selected for PET imaging study. Balb/c nude mice inoculated with hepatoblastoma Huh-7 cells were imaged at one hour after intravenous injection of our tracers. Both tracers showed no tumor-specific uptake in Huh-7 tumors (Fig. S6A, ESI†). Furthermore, no statistically significant difference between mPep-1-[64Cu] and hPep-1-[64Cu] in tumor uptake at various time points was observed (Fig. S6B, ESI†). Taken together, the PET imaging studies demonstrated that mPep-1-[64Cu] outperformed hPep-1-[64Cu] in mapping PD-1 in B16F10 tumors. Besides, our tracers showed high reliability and reproducibility for imaging tumors at various locations in mice, suggesting their potential utility in the tracking of PD-1 in both primary and metastasis tumors.
Ex vivo biodistribution
The specific uptake of the tracers was further investigated via the ex vivo biodistribution. The biodistribution data in tumors and selected organs at 5 min, 20 min, 40 min, and 100 min p.i. of the tracers are summarized in Tables S1 and S2 (ESI†). These data were consistent with the PET imaging results shown in Fig. 2C and D. The uptakes of mPep-1-[64Cu] in tumors were 5.52 ± 1.66, 4.48 ± 0.82, and 4.08 ± 0.99 (mean % ID g−1 ± SD) at 5 min, 20 min, and 40 min p.i., respectively. Meanwhile, the tumor uptakes of hPep-1-[64Cu] were 3.37 ± 1.06, 2.60 ± 0.28, and 1.79 ± 0.04 at the same respective time points (Fig. 3A). The radioactivity in the spleen also varied significantly between the two tracers, with a 1.5–2 fold higher uptake of mPep-1-[64Cu] observed in the spleen compared to that of hPep-1-[64Cu] across all studied time points (Fig. 3B). The tumor to muscle (T/M) and tumor to blood (T/B) ratios for both tracers were calculated and are presented in Fig. 3C and D, respectively. mPep-1-[64Cu] showed higher T/M and T/B ratios at 0–40 min p.i., indicating a superior tumor to background contrast of mPep-1-[64Cu] than hPep-1-[64Cu], further indicating that mPep-1-[64Cu] was preferable due to its high sensitivity for visualizing small tumor lesions. The continuous increment of the T/M and T/B ratios along the postinjection time suggested that the clearance rate of the tracers in tumors was slower than those in the muscle and blood, indicating the specific binding of the tracers to PD-1. Notably, low radioactivities were exhibited in the lung and liver, suggesting the superior metabolic profiles of the tracers.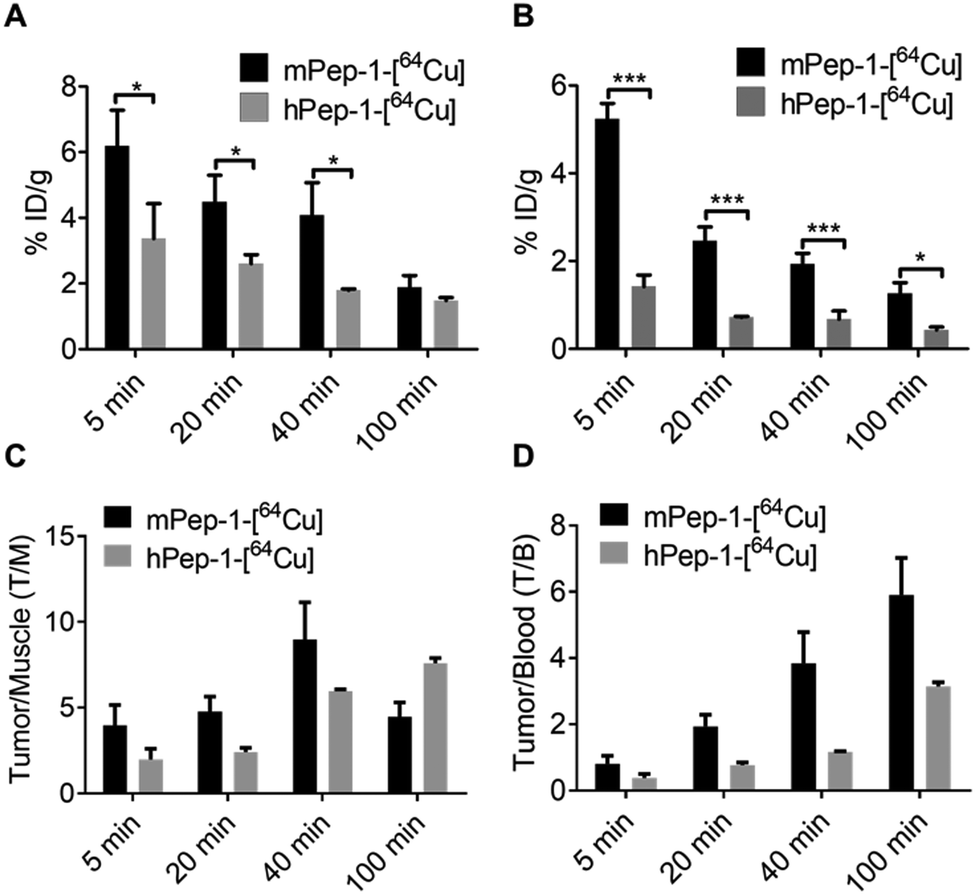 | ||
| Fig. 3 Ex vivo biodistribution, and tumor to muscle and tumor to blood ratios of the tracers in B16F10-bearing C57BL/6J mice. Uptake of the tracers in the tumor (A) and spleen (B) p.i. at 5 min, 20 min, 40 min, and 100 min, three mice for each time point. Unpaired Student's t-test was used for data comparisons. * < 0.05, *** < 0.001. (C) Tumor to muscle ratio of the tracer uptake. (D) Tumor to blood ratio of the tracer uptake. For all panels, the data represent mean ± SD, n = 3. | ||
Autoradiography
In addition, in vitro autoradiography (ARG) was used to further verify the specific binding of the tracer to PD-1. Tumor tissue sections from the same mouse bearing a B6F10 tumor were used for the ARG study. The tumor tissue section stained with mPep-1-[64Cu] exhibited significantly stronger radioactivity (1400 PSL mm−2, PSL; photostimulated luminescence) than that stained with hPep-1-[64Cu] (600 PSL mm−2) (Fig. 4A and B). When co-stained with excess mPep-1-DOTA, the binding of mPep-1-[64Cu] to the tissue section was drastically decreased to approximately half of the original intensity. These data unambiguously demonstrated that the binding of mPep-1-[64Cu] to B16F10 tumor tissues was PD-1 specific.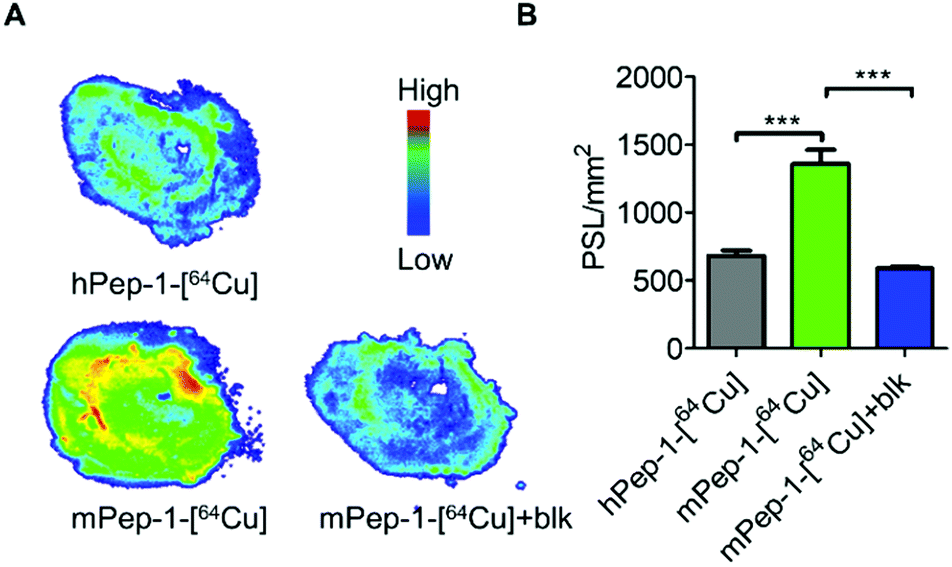 | ||
| Fig. 4 In vitro autoradiography. (A) Representative ARG images of the B16F10 tumor sectional tissue slides, blk: block. (B) Quantification of tracer binding as photo-stimulated luminescence per square millimeter (PSL mm−2, mean ± SD) in Fig. 4A. Student's t-test for unpaired measurements; n = 3, *** < 0.001. | ||
Investigation of the differentiation between mPep-1 and hPep-1
The aforementioned results have demonstrated that mPep-1-[64Cu] performed better as a radiotracer for PET imaging of PD-1 than hPep-1-[64Cu]. This differentiation could surprisingly be attributed to a sole residue variation (V128L), even though residue 128 is displayed in an out-of-interface protrusion in the co-crystal structure of PD-1 and PD-L1 (Fig. 5A),38,43 which has been identified as a non-critical site for PD-1 binding.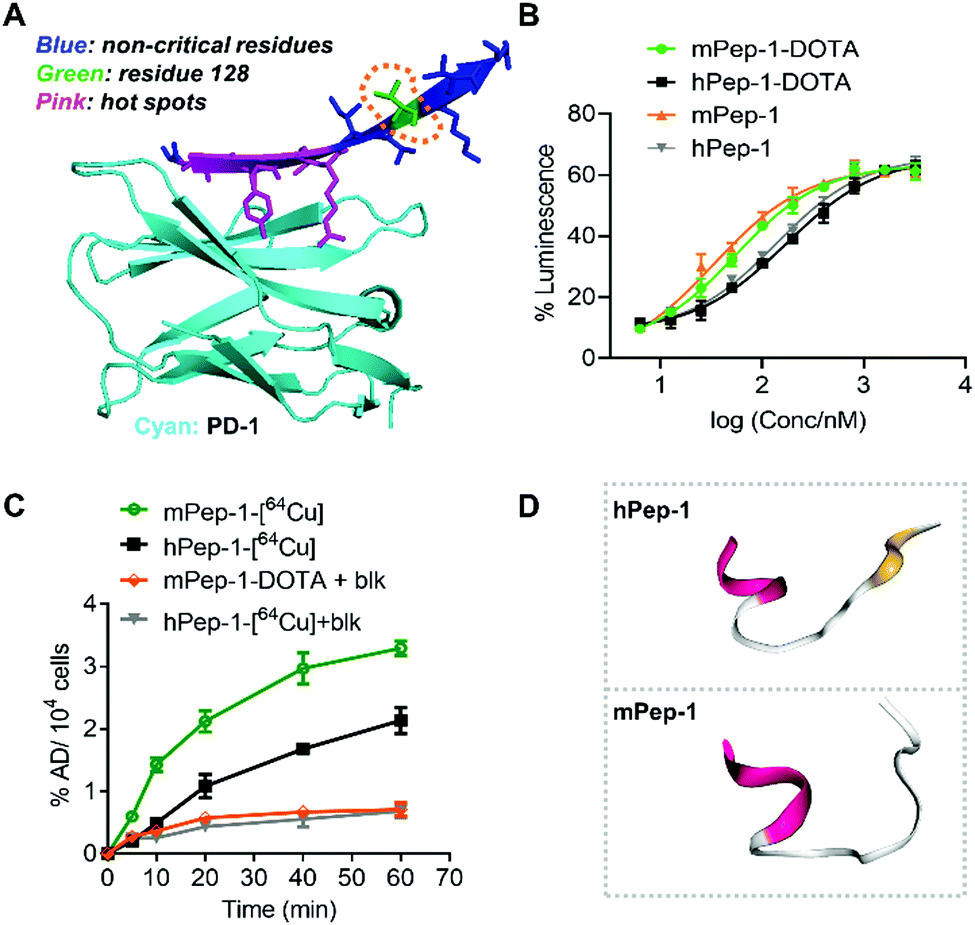 | ||
| Fig. 5 Binding affinity, cellular uptake, and simulated secondary structures of hPep-1 and mPep-1. (A) A representative illustration of the binding interface consisting of PD-1 and the interface peptide from PD-L1. The structure was reconstructed based on a PD-1/PD-L1 complex structure (ID: 3bik) in the PDB database. (B) Response curves for a PD-1/PD-L1 checkpoint luciferase reporter assay (RE-NFAT) used to examine the effects on the signaling pathway. Data represent three biological replicates, mean ± SD, n = 3. (C) Uptake of tracers in C57BL/6J mouse splenic cells (with Concanavalin A induced expression of PD-1). (D) Simulated structures of hPep-1 and mPep-1 by an online peptide structure prediction tool (PEP-FOLD3.5, https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/). | ||
To investigate the origin of the difference in performance of the two tracers, the binding ability of mPep-1-DOTA and hPep-1-DOTA to PD-1 was assessed by a PD-1/PD-L1 blockade luciferase reporter assay.43 The measurement of luminescence against doses indicated that mPep-1-DOTA exhibited lower EC50 (53.88 ± 3.37 nM) than hPep-1-DOTA (EC50 = 170.40 ± 3.39 nM). Furthermore, mPep-1 and hPep-1 without DOTA displayed similar EC50 (33.13 ± 7.08 nM and 136.3 ± 4.11 nM, respectively) to mPep-1-DOTA and hPep-1-DOTA correspondingly, suggesting negligible interference of the PEG3-DOTA tail with the binding mode of mPep-1 (Fig. 5B). The specific uptake of the radiotracers by PD-1 positive splenic cells (with Concanavalin A induced expression of PD-1) was further examined.25 As shown in Fig. 5C, the uptake of mPep-1-[64Cu] in splenic cells continuously increased from 0 min to 60 min, reaching ∼3.3% administration dose per 104 (AD% per 104) cells. In contrast, the uptake of hPep-1-[64Cu] was much lower than that of hPep-1-[64Cu], showing ∼2.1% AD per 104 cells at 60 min. Moreover, the uptake of both peptides was effectively inhibited by co-incubation with an excess of mPep-1-DOTA (blk), indicating the specific binding of mPep-1 to PD-1 (Fig. 5C). It is worth noting that some non-specific binding of the tracers to splenic cells exists, as ∼20% (incubation for 60 min) of the uptake cannot be blocked.
Taken together, the two short peptides showed significant differences in binding affinities to PD-1, which was probably the major differentiating factor leading to their varying performance in PET imaging. The difference in their binding affinities could be attributed to the fact that neither mPep-1 nor hPep-1 maintains its original β-sheet conformation when truncated from the protein folding environment. While Leu and Val exhibit different conformation inducing propensities, their presence can lead to the formation of different secondary structures of the peptides. This assumption was supported by the simulated structures of hPep-1 and mPep-1,49–52 in which the N-sides adopt similar α-helices, while the C sides assemble into a sheet-like conformation in hPep-1 and a random coil in mPep-1 (Fig. 5D). These results also highlighted the importance of peptide stapling technologies in enforcing the native protein folding structures of the short peptides.36,53,54
Discussion
In this study, we demonstrated that the interface peptide from mPD-L1 is a potential imaging agent for mPD-1 in the PD-1/PD-L1 PPI model. Although the short interface peptide departs from its original folding environment, it is shown to retain satisfactory specific binding to its original target. The PET imaging results confirmed the efficacy of the radiotracers in mapping of PD-1 in immune healthy mice. This study conveyed a unique paradigm for developing imaging agents for highly challenging targets, such as membrane receptors and antigens. The approach illustrated here is straightforward and easy to follow, further promoting its adaptability for use in other targets that are involved in PPIs to help identify them as biomarkers for various diseases.Moreover, a sole residue difference at position 128 between mPep-1-[64Cu] and hPep-1-[64Cu] contributed to the unexpected differentiations in their imaging capacities. As opposed to the previous conclusion that residue 128 is noncritical for PD-1 binding, our findings indicate that this site may be a determinant of varying imaging performance of the radiotracers, eliciting further interest in optimizing this residue to enhance the PD-1 binding ability of the tracer. Our findings also highlight the importance of in vivo PET imaging as a companion diagnostic in drug development, as it could provide a more comprehensive assessment of the pharmacological properties of the candidate drugs.
Although immunoradiotracers that target PD-1 have been intensively examined in preclinical and clinical studies, these tracers made from large molecular weight monoclonal antibodies yet show inherent limitations regarding their DMPK profiles, such as slow pharmacokinetics and metabolizing rate in vivo as well as poor target-to-nontarget contrast. In an overview of the reported immunoradiotracers (Table S3, ESI,† entries 1–10), most of them necessitate a waiting time exceeding 24 hours to gain the optimal PET images.25–33,55–57 The major clearance organs for immunoradiotracers are the liver and kidney, where high radiation exposure may cause organ dysfunction. As vital indicators of the image quality, the tumor to blood and tumor to muscle ratios of immunoradiotracers are usually compromised due to the unfavorable pharmacokinetics. In stark comparison, the peptide-based radiotracers mPep-1-[64Cu] (entry 11) display distinctly different DMPK profiles. mPep-1-[64Cu] accumulates in the targets shortly after injection in vivo due to its small size, which further imparts deep tissue penetration. Moreover, the physical half-life of mPep-1-[64Cu] in vivo is estimated to be hours, giving it fast clearance from non-targeted tissues and finally allowing it be excreted from the body in several hours, which could mitigate nonspecific radiation exposure and result in satisfactory image quality, verified by the high tumor to blood and tumor to muscle ratios.
In general, seeking peptide radiotracers that can be translated to clinical uses remains a formidable challenge. Recently, concomitant with the elegant application of novel peptide modification methodologies in tracer development, great success has been witnessed in developing peptide-based PET imaging agents, exemplified by the PD-L1 targeting macrocyclic peptide WL12.58–60 WL12 possesses an N-methylated backbone with 14 amide bonds and 1 thioether bond, conferring its high stability in vivo, as macrocyclization and N-methylation prevent the peptide from undergoing enzymatic degradation by multiple enzymes. WL12 was discovered by an extensive screening of a large library of peptides, and the success of this peptide benefits from the rational design. In contrast, our m(h)Pep-1 peptides are derived from an “interface peptide strategy”, which fundamentally relies on the structures of protein complexes. Complementary to the “HTS”, our strategy conveys a facile way to seek lead peptides that can target aberrant PPIs. However, unlike WL12, the interface peptides usually display sub-optimal performance. Further optimization of the interface peptide sequences, such as cyclization, N-methylation, multimerization, etc., is necessary to improve the target specificity and binding affinity. In the future, the combination of “interface peptide” with “rational optimization” would be a prevalent mode for peptide-based tracer development, as the interface peptide strategy does the “first-step” in seeking lead compounds while rational optimization gains the “best” performance of the tracers.
Conclusions
In conclusion, we performed a proof-of-concept study in exploiting the interface peptide in PD-L1 for the development of PET tracers for PD-1. mPep-1-[64Cu], the first reported peptide-based PET tracer for imaging PD-1, convincingly mapped PD-1 in a mouse model. Further investigations should be performed to evaluate the efficacy of the tracer in humanized animal models. Besides, improvement of the overall performance of the tracer by introducing chemical modifications, such as macrocyclization or PEGylation, should be considered to further promote the utility of the tracer in clinical settings.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the staff of the National Institutes for Quantum and Radiological Sciences and Technology (QST) for their support in cyclotron operation, radioisotope production, radiosynthesis, and animal experiments. We sincerely thank the financial support from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government (Basic Research B: No. 17H04267, Grant-in-Aid for Young Scientists: No. 19K17156, Grant-in-Aid for Research Activity Start-up: No. 18H06217), National Institutes for Quantum and Radiological Science and Technology of Japan (Presidential strategic fund for budding research).Notes and references
- P. Lambin, R. T. Leijenaar, T. M. Deist, J. Peerlings, E. E. De Jong, J. Van Timmeren, S. Sanduleanu, R. T. Larue, A. J. Even and A. Jochems, Radiomics: the bridge between medical imaging and personalized medicine, Nat. Rev. Clin. Oncol., 2017, 14, 749 CrossRef.
- A. A. Lammertsma, Forward to the Past: The Case for Quantitative PET Imaging, J. Nucl. Med., 2017, 58, 1019–1024 CrossRef.
- M. Baumann, M. Krause, J. Overgaard, J. Debus, S. M. Bentzen, J. Daartz, C. Richter, D. Zips and T. Bortfeld, Radiation oncology in the era of precision medicine, Nat. Rev. Cancer, 2016, 16, 234–249 CrossRef PubMed.
- N. Pillarsetty, K. Jhaveri, T. Taldone, E. Caldas-Lopes, B. Punzalan, S. Joshi, A. Bolaender, M. M. Uddin, A. Rodina, P. Yan, A. Ku, T. Ku, S. K. Shah, S. Lyashchenko, E. Burnazi, T. Wang, N. Lecomte, Y. Janjigian, A. Younes, C. W. Batlevi, M. L. Guzman, G. J. Roboz, J. Koziorowski, P. Zanzonico, M. L. Alpaugh, A. Corben, S. Modi, L. Norton, S. M. Larson, J. S. Lewis, G. Chiosis, J. F. Gerecitano and M. P. S. Dunphy, Paradigms for Precision Medicine in Epichaperome Cancer Therapy, Cancer Cell, 2019, 36, 559.e557–573.e557 CrossRef.
- M. Momcilovic, A. Jones, S. T. Bailey, C. M. Waldmann, R. Li, J. T. Lee, G. Abdelhady, A. Gomez, T. Holloway, E. Schmid, D. Stout, M. C. Fishbein, L. Stiles, D. V. Dabir, S. M. Dubinett, H. Christofk, O. Shirihai, C. M. Koehler, S. Sadeghi and D. B. Shackelford, In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer, Nature, 2019, 575, 380–384 CrossRef PubMed.
- Q. Wang, Y. Wang, J. Ding, C. Wang, X. Zhou, W. Gao, H. Huang, F. Shao and Z. Liu, A bioorthogonal system reveals antitumour immune function of pyroptosis, Nature, 2020, 579, 421–426 CrossRef PubMed.
- K. Hu, Z. Yang, L. Zhang, L. Xie, L. Wang, H. Xu, L. Josephson, S. H. Liang and M.-R. Zhang, Boron agents for neutron capture therapy, Coord. Chem. Rev., 2020, 405, 213139 CrossRef.
- M. Fani and H. R. Maecke, Radiopharmaceutical development of radiolabelled peptides, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39(suppl. 1), S11–S30 CrossRef PubMed.
- M. Fani, H. R. Maecke and S. M. Okarvi, Radiolabeled peptides: valuable tools for the detection and treatment of cancer, Theranostics, 2012, 2, 481–501 CrossRef CAS PubMed.
- X. Sun, Y. Li, T. Liu, Z. Li, X. Zhang and X. Chen, Peptide-based imaging agents for cancer detection, Adv. Drug Delivery Rev., 2017, 110–111, 38–51 CrossRef CAS.
- A. Henninot, J. C. Collins and J. M. Nuss, The current state of peptide drug discovery: back to the future?, J. Med. Chem., 2017, 61, 1382–1414 CrossRef.
- A. F. Räder, M. Weinmüller, F. Reichart, A. Schumacher-Klinger, S. Merzbach, C. Gilon, A. Hoffman and H. Kessler, Orally active peptides: is there a magic bullet?, Angew. Chem., Int. Ed., 2018, 57, 14414–14438 CrossRef.
- L. Bodei, V. Ambrosini, K. Herrmann and I. Modlin, Current Concepts in 68Ga-DOTATATE Imaging of Neuroendocrine Neoplasms: Interpretation, Biodistribution, Dosimetry, and Molecular Strategies, J. Nucl. Med., 2017, 58, 1718–1726 CrossRef CAS.
- T. A. Hope, J. Calais, L. Zhang, W. Dieckmann and C. Millo, 111In-pentetreotide scintigraphy vs. 68Ga-DOTATATE PET: Impact on Krenning Scores and Effect of Tumor Burden, J. Nucl. Med., 2019, 60, 1266–1269 CrossRef CAS.
- S. Schmuck, C. A. von Klot, C. Henkenberens, J. M. Sohns, H. Christiansen, H.-J. Wester, T. L. Ross, F. M. Bengel and T. Derlin, Initial experience with volumetric 68Ga-PSMA I&T PET/CT for assessment of whole-body tumor burden as a quantitative imaging biomarker in patients with prostate cancer, J. Nucl. Med., 2017, 58, 1962–1968 CrossRef CAS PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Structure-based design of inhibitors of protein–protein interactions: mimicking peptide binding epitopes, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- M. W. Peczuh and A. D. Hamilton, Peptide and protein recognition by designed molecules, Chem. Rev., 2000, 100, 2479–2494 CrossRef CAS PubMed.
- N. London, B. Raveh, D. Movshovitz-Attias and O. Schueler-Furman, Can self-inhibitory peptides be derived from the interfaces of globular protein–protein interactions?, Proteins, 2010, 78, 3140–3149 CrossRef CAS.
- J. Münch, L. Ständker, K. Adermann, A. Schulz, M. Schindler, R. Chinnadurai, S. Pöhlmann, C. Chaipan, T. Biet and T. Peters, Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide, Cell, 2007, 129, 263–275 CrossRef PubMed.
- J. T. Nguyen, C. W. Turck, F. E. Cohen, R. N. Zuckermann and W. A. Lim, Exploiting the basis of proline recognition by SH3 and WW domains: design of N-substituted inhibitors, Science, 1998, 282, 2088–2092 CrossRef PubMed.
- H. Imataka, A. Gradi and N. Sonenberg, A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation, EMBO J., 1998, 17, 7480–7489 CrossRef PubMed.
- D. M. Pardoll, The blockade of immune checkpoints in cancer immunotherapy, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef.
- S. Muenst, S. Soysal, F. Gao, E. Obermann, D. Oertli and W. Gillanders, The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer, Breast Cancer Res. Treat., 2013, 139, 667–676 CrossRef.
- J. M. Taube, A. Klein, J. R. Brahmer, H. Xu, X. Pan, J. H. Kim, L. Chen, D. M. Pardoll, S. L. Topalian and R. A. Anders, Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti–PD-1 therapy, Clin. Cancer Res., 2014, 20, 5064–5074 CrossRef PubMed.
- A. Natarajan, A. T. Mayer, L. Xu, R. E. Reeves, J. Gano and S. S. Gambhir, Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes, Bioconjugate Chem., 2015, 26, 2062–2069 CrossRef CAS PubMed.
- M. Hettich, F. Braun, M. D. Bartholoma, R. Schirmbeck and G. Niedermann, High-Resolution PET Imaging with Therapeutic Antibody-based PD-1/PD-L1 Checkpoint Tracers, Theranostics, 2016, 6, 1629–1640 CrossRef CAS PubMed.
- E. L. Cole, J. Kim, D. J. Donnelly, R. A. Smith, D. Cohen, V. Lafont, P. E. Morin, R. Y. C. Huang, P. L. Chow, W. Hayes and S. Bonacorsi, Radiosynthesis and preclinical PET evaluation of 89Zr-nivolumab (BMS-936558) in healthy non-human primates, Bioorg. Med. Chem., 2017, 25, 5407–5414 CrossRef CAS PubMed.
- Y. Du, X. Liang, Y. Li, T. Sun, Z. Jin, H. Xue and J. Tian, Nuclear and Fluorescent Labeled PD-1-Liposome-DOX-(64)Cu/IRDye800CW Allows Improved Breast Tumor Targeted Imaging and Therapy, Mol. Pharmaceutics, 2017, 14, 3978–3986 CrossRef CAS.
- C. G. England, E. B. Ehlerding, R. Hernandez, B. T. Rekoske, S. A. Graves, H. Sun, G. Liu, D. G. McNeel, T. E. Barnhart and W. Cai, Preclinical Pharmacokinetics and Biodistribution Studies of 89Zr-Labeled Pembrolizumab, J. Nucl. Med., 2017, 58, 162–168 CrossRef CAS PubMed.
- A. Natarajan, A. T. Mayer, R. E. Reeves, C. M. Nagamine and S. S. Gambhir, Development of Novel ImmunoPET Tracers to Image Human PD-1 Checkpoint Expression on Tumor-Infiltrating Lymphocytes in a Humanized Mouse Model, Mol. Imaging Biol., 2017, 19, 903–914 CrossRef.
- C. G. England, D. Jiang, E. B. Ehlerding, B. T. Rekoske, P. A. Ellison, R. Hernandez, T. E. Barnhart, D. G. McNeel, P. Huang and W. Cai, (89)Zr-labeled nivolumab for imaging of T-cell infiltration in a humanized murine model of lung cancer, Eur. J. Nucl. Med. Mol. Imaging, 2018, 45, 110–120 CrossRef PubMed.
- A. Natarajan, C. B. Patel, F. Habte and S. S. Gambhir, Dosimetry Prediction for Clinical Translation of (64)Cu-Pembrolizumab ImmunoPET Targeting Human PD-1 Expression, Sci. Rep., 2018, 8, 633 CrossRef PubMed.
- X. Guo, H. Zhu, T. Liu, X. Xu, Y. Kong, S. Yao, X. Sheng and Z. Yang, Development of (99m)Tc-conjugated JS001 antibody for in vivo mapping of PD-1 distribution in murine, Bioorg. Med. Chem. Lett., 2019, 29, 2178–2181 CrossRef PubMed.
- A. Domling and T. A. Holak, Programmed death-1: therapeutic success after more than 100 years of cancer immunotherapy, Angew. Chem., Int. Ed., 2014, 53, 2286–2288 CrossRef PubMed.
- M. Konstantinidou, T. Zarganes-Tzitzikas, K. Magiera-Mularz, T. A. Holak and A. Domling, Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies?, Angew. Chem., Int. Ed., 2017, 57, 4840–4848 CrossRef PubMed.
- K. Hu, H. Geng, Q. Zhang, Q. Liu, M. Xie, C. Sun, W. Li, H. Lin, F. Jiang, T. Wang, Y.-D. Wu and Z. Li, An In-tether Chiral Center Modulates the Helicity, Cell Permeability, and Target Binding Affinity of a Peptide, Angew. Chem., Int. Ed., 2016, 55, 8013–8017 CrossRef PubMed.
- K. Hu, C. Sun, D. Yang, Y. Wu, C. Shi, L. Chen, T. Liao, J. Guo, Y. Liu and Z. Li, A precisely positioned chiral center in an i, i + 7 tether modulates the helicity of the backbone peptide, Chem. Commun., 2017, 53, 6728–6731 RSC.
- K. M. Zak, R. Kitel, S. Przetocka, P. Golik, K. Guzik, B. Musielak, A. Domling, G. Dubin and T. A. Holak, Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1, Structure, 2015, 23, 2341–2348 CrossRef.
- D. Y.-w. Lin, Y. Tanaka, M. Iwasaki, A. G. Gittis, H.-P. Su, B. Mikami, T. Okazaki, T. Honjo, N. Minato and D. N. Garboczi, The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3011–3016 CrossRef PubMed.
- S. Tan, D. Chen, K. Liu, M. He, H. Song, Y. Shi, J. Liu, C. W. Zhang, J. Qi, J. Yan, S. Gao and G. F. Gao, Crystal clear: visualizing the intervention mechanism of the PD-1/PD-L1 interaction by two cancer therapeutic monoclonal antibodies, Protein Cell, 2016, 7, 866–877 CrossRef.
- X. Cheng, V. Veverka, A. Radhakrishnan, L. C. Waters, F. W. Muskett, S. H. Morgan, J. Huo, C. Yu, E. J. Evans, A. J. Leslie, M. Griffiths, C. Stubberfield, R. Griffin, A. J. Henry, A. Jansson, J. E. Ladbury, S. Ikemizu, M. D. Carr and S. J. Davis, Structure and interactions of the human programmed cell death 1 receptor, J. Biol. Chem., 2013, 288, 11771–11785 CrossRef.
- Z. Na, S. P. Yeo, S. R. Bharath, M. W. Bowler, E. Balikci, C. I. Wang and H. Song, Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab, Cell Res., 2017, 27, 147–150 CrossRef PubMed.
- R. J. Boohaker, V. Sambandam, I. Segura, J. Miller, M. Suto and B. Xu, Rational design and development of a peptide inhibitor for the PD-1/PD-L1 interaction, Cancer Lett., 2018, 434, 11–21 CrossRef CAS PubMed.
- C. J. Anderson and R. Ferdani, Copper-64 radiopharmaceuticals for PET imaging of cancer: advances in preclinical and clinical research, Cancer Biother. Radiopharm., 2009, 24, 379–393 CrossRef CAS PubMed.
- K. Hu, M. Hanyu, L. Xie, Y. Zhang, K. Nagatsu, H. Suzuki and M.-R. Zhang, Correction: Developing native peptide-based radiotracers for PD-L1 PET imaging and improving imaging contrast by pegylation, Chem. Commun., 2019, 55, 4273 RSC.
- K. Hu, J. Shang, L. Xie, M. Hanyu, Y. Zhang, Z. Yang, H. Xu, L. Wang and M.-R. Zhang, PET Imaging of VEGFR with a Novel 64Cu-Labeled Peptide, ACS Omega, 2020, 5, 8508–8514 CrossRef PubMed.
- M. Tomita, H. Yasui, K. Higashikawa, K. Nakajima, H. Takakura, T. Shiga, Y. Kuge and M. Ogawa, Anti PD-1 treatment increases [18F]FDG uptake by cancer cells in a mouse B16F10 melanoma model, EJNMMI Res., 2018, 8, 82 CrossRef PubMed.
- S. A. Weiss, S. W. Han, K. Lui, J. Tchack, R. Shapiro, R. Berman, J. Zhong, M. Krogsgaard, I. Osman and F. Darvishian, Immunologic heterogeneity of tumor-infiltrating lymphocyte composition in primary melanoma, Hum. Pathol., 2016, 57, 116–125 CrossRef PubMed.
- J. Maupetit, P. Derreumaux and P. Tuffery, PEP-FOLD: an online resource for de novo peptide structure prediction, Nucleic Acids Res., 2009, 37, W498–W503 CrossRef PubMed.
- P. Thevenet, Y. Shen, J. Maupetit, F. Guyon, P. Derreumaux and P. Tuffery, PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides, Nucleic Acids Res., 2012, 40, W288–W293 CrossRef PubMed.
- Y. Shen, J. Maupetit, P. Derreumaux and P. Tufféry, Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction, J. Chem. Theory Comput., 2014, 10, 4745–4758 CrossRef CAS PubMed.
- J. Maupetit, P. Derreumaux and P. Tuffery, A fast method for large-scale de novo peptide and miniprotein structure prediction, J. Comput. Chem., 2010, 31, 726–738 CAS.
- K. Hu, F. Yin, M. Yu, C. Sun, J. Li, Y. Liang, W. Li, M. Xie, Y. Lao, W. Liang and Z.-G. Li, In-Tether Chiral Center Induced Helical Peptide Modulators Target p53-MDM2/MDMX and Inhibit Tumor Growth in Stem-Like Cancer Cell, Theranostics, 2017, 7, 4566–4576 CrossRef CAS PubMed.
- Y. H. Lau, P. De Andrade, Y. T. Wu and D. R. Spring, Peptide stapling techniques based on different macrocyclisation chemistries, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- K. Broos, Q. Lecocq, G. Raes, N. Devoogdt, M. Keyaerts and K. Breckpot, Noninvasive imaging of the PD-1:PD-L1 immune checkpoint: embracing nuclear medicine for the benefit of personalized immunotherapy, Theranostics, 2018, 8, 3559–3570 CrossRef CAS PubMed.
- A. N. Niemeijer, D. Leung, M. C. Huisman, I. Bahce, O. S. Hoekstra, G. A. M. S. van Dongen, R. Boellaard, S. Du, W. Hayes, R. Smith, A. D. Windhorst, N. H. Hendrikse, A. Poot, D. J. Vugts, E. Thunnissen, P. Morin, D. Lipovsek, D. J. Donnelly, S. J. Bonacorsi, L. M. Velasquez, T. D. de Gruijl, E. F. Smit and A. J. de Langen, Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer, Nat. Commun., 2018, 9, 4664 CrossRef CAS.
- H. Huang, H. Zhu, Q. Xie, X. Tian, X. Yang, F. Feng, Q. Jiang, X. Sheng and Z. Yang, Evaluation of 124I-JS001 for hPD1 immuno-PET imaging using sarcoma cell homografts in humanized mice, Acta Pharm. Sin. B, 2020, 10, 1321–1330 CrossRef.
- S. Chatterjee, W. G. Lesniak, M. S. Miller, A. Lisok, E. Sikorska, B. Wharram, D. Kumar, M. Gabrielson, M. G. Pomper, S. B. Gabelli and S. Nimmagadda, Rapid PD-L1 detection in tumors with PET using a highly specific peptide, Biochem. Biophys. Res. Commun., 2017, 483, 258–263 CrossRef CAS.
- D. Kumar, A. Lisok, E. Dahmane, M. D. McCoy, S. Shelake, S. Chatterjee, V. Allaj, P. Sysa-Shah, B. Wharram, W. G. Lesniak, E. Tully, E. Gabrielson, E. M. Jaffee, J. T. Poirier, C. M. Rudin, J. V. Gobburu, M. G. Pomper and S. Nimmagadda, Peptide-based PET quantifies target engagement of PD-L1 therapeutics, J. Clin. Invest., 2019, 129, 616–630 CrossRef PubMed.
- W. G. Lesniak, R. C. Mease, S. Chatterjee, D. Kumar, A. Lisok, B. Wharram, V. R. Kalagadda, L. A. Emens, M. G. Pomper and S. Nimmagadda, Development of [(18)F]FPy-WL12 as a PD-L1 Specific PET Imaging Peptide, Mol. Imaging, 2019, 18, 1–9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00070a |
| This journal is © The Royal Society of Chemistry 2020 |