
Self-assembled polymeric micelles as amphiphilic particulate emulsifiers for controllable Pickering emulsions
Feng
Wang
ab,
Juntao
Tang
a,
Hui
Liu
 *ab,
Guipeng
Yu
a and
Yingping
Zou
a
*ab,
Guipeng
Yu
a and
Yingping
Zou
a
aCollege of Chemistry and Chemical Engineering, Central South University, 932 South Lushan Road, Changsha 410083, Hunan, P. R. China. E-mail: liuhui@csu.edu.cn
bHunan Provincial Key Laboratory of Efficient and Clean Utilization of Manganese Resources, Central South University, Changsha 410083, Hunan, P. R. China
First published on 25th December 2018
Abstract
Amphiphilic Pickering particles are of immense importance because of their dominant ability to affect the emulsion stability and type. Among the present amphiphilic particulate emulsifiers to prepare controllable Pickering emulsion, there is increasing evidence that self-assembled polymeric micelles are comparatively effective and they have gained attention recently. In this review, the important progress in self-assembled micelles based on amphiphilic copolymers for controllable Pickering emulsions is systematically summarized. A self-assembled micelle is used to design a controllable amphiphilic polymeric precursor and then self-assembles into the Pickering particle. We highlight block and random copolymer micelles that have been successfully employed in controlling Pickering emulsions, which aims to draw more attention to these self-assembled Pickering particles.
 Feng Wang | Feng Wang earned his Bachelor of Engineering in Chemical Engineering and Technology at the Anhui University of Technology in 2016. In the same year, he commenced his Master degree in Chemical Engineering under the guidance of Prof. Hui Liu at Central South University. His current research interests involve designing and developing stimuli-responsive macromolecular materials for applications of surface physical chemistry. |
 Juntao Tang | Juntao (Reynard) Tang works at the College of Chemistry and Chemical Engineering in Central South University, China as an associate professor. He received his PhD degree in Chemical Engineering under the guidance of Prof. Michael Tam at the University of Waterloo Canada in 2016 and continued the one-year post-doctoral research. His current research interests involve designing and developing functional nanomaterials derived from sustainable resources for advanced applications. |
 Hui Liu | Hui Liu is a professor of chemical engineering at Central South University. He received his PhD degree in 2007 from Central South University. After postdoctoral research with Prof. J. H. Yi (Central South University) and Prof. A. Heise (Dublin City University), he joined Central South University as an associate professor in 2011. His current research interests include the design and application of stimuli-responsive polymers (brushes), particulate emulsifiers, versatile functionalization of interfaces, and nanocomposite materials. |
 Guipeng Yu | Guipeng Yu received his PhD degree at the Dalian University of Technology in 2009, and then joined Central South University as an associate professor. In 2016, he became a member of the group of Prof. Dr Markus Antonietti and Jiayin Yuan at the Max-Planck-Institute of Colloids and Interfaces. In 2017, he returned to Central South University and is leading a research group in the department of chemistry. His research is focused on porous materials and their potential in (photo) catalysis, gas storage/separation and energy applications. |
 Yingping Zou | Yingping Zou is a full professor in Central South University. She received her PhD degree from the Institute of Chemistry, Chinese Academy of Sciences in 2008, and then performed her postdoctoral research at Laval University. She did her visiting research at Stanford University from 2012 to 2013 and in the Silicon Valley and Stanford University from 2014.8 to 2014.12. Her present research field is organic polymers for optoelectronic applications. She has published more than 100 research papers and more than 10 invention patents. |
1. Introduction
Empirically, Pickering emulsions are emulsions that are stabilized by fine micro- or nanoparticles.1–5 They have been extensively studied and optimized in recent years due to their properties such as better stability against coalescence and Ostwald ripening, as well as due to progress in materials chemistry that allows us to create more types and quantities of Pickering particles.6–11 Pickering emulsions not only retain several of the basic properties of conventional emulsions but also open up the application market of Pickering particles in medicine, food, and cosmetics. In many cases, for instance, emulsions stabilized by biomass-based particles are biologically compatible and environmentally friendly. This makes them a promising and attractive alternative in many industries seeking to avoid the use of traditional surfactants.11,12 Several reviews on the classification or application of Pickering particles have been published online.6,8–10 Nevertheless, a supplementary or updated review is still needed to summarize controllable Pickering emulsions.A great deal of work has been done to prepare and characterize controllable Pickering emulsions. Generally, the control of Pickering emulsions involves control over emulsion type, emulsion stability, emulsion phase inversion, and emulsifying efficiency. The emulsion type is controlled by the compositions of the emulsion phases and the wettability of the interfacial emulsifiers. A water-soluble hydrophilic emulsifier orients the emulsification process towards an oil-in-water (O/W) emulsion type, and a hydrophobic emulsifier gives a water-in-oil (W/O) emulsion. More specifically, according to Young's equation,11 O/W emulsions are favored when the contact angle in water, θw, is smaller than 90°. Conversely, θw > 90° favors W/O emulsions. Multiple emulsions are both O/W and W/O emulsions combined in the same droplets. The internal W/O droplets of a W/O/W multiple emulsion should be stabilized by means of hydrophobic particles, whereas the external surface of the W/O/W droplets should be covered by hydrophilic particles.11 In addition, equivalent wetting of the particle in either phase (θw = 90°) results in particle instability at the interface.8 Optimum stability of Pickering emulsions is ensured when the contact angle is close to 90° but retains some degree of preferential wettability for one phase over the other. Therefore, a phase inversion can be triggered by changes of the wetting behavior (the amphiphilicity of particles). Several other parameters such as the relative water and oil contents, the components of the two liquid phases, and the concentration of particles, can also induce emulsion phase inversion and control the type-morphology of the emulsion.9,13 The rapid development in materials technology has increased the variety of amphiphilic particles available; in particular, emulsions stabilized by amphiphilic polymer-modified nanoparticles14–22 have been successfully applied for tuning phase inversion and reversible emulsification–demulsification. Meanwhile, many researchers have started to design amphiphilic Pickering particles via the self-assembly of amphiphilic copolymers, and significant progress has been made in the preparation and application of a variety of self-assembled polymeric micelles.23–25 Moreover, the desirable amphiphilicity of polymeric micelles could cover the range from superhydrophobic to superhydrophilic. Therefore, the self-assembled amphiphilic nanomicelles used as particulate emulsifiers are a new and promising pathway for controllable Pickering emulsions.
A self-assembled polymeric micelle is used to design a controllable amphiphilic polymeric precursor and then self-assembles into the Pickering particle, and this facile strategy of controlling amphiphilicity has been extensively employed in controlling Pickering emulsions. This review attempts to summarize the important achievements of self-assembled micelles based on amphiphilic block and random copolymers for controllable Pickering emulsions. We aim to draw more attention to these easily accessible self-assembled Pickering particles, which are very likely to raise interest in translational amphiphilic copolymer research and self-assembled object application. Finally, we conclude our personal perspectives on the opportunities and challenges of self-assembled micellar emulsifiers.
2. Self-assembled micelles based on block copolymers
Over the past decade, micellar particles self-assembled from amphiphilic copolymers have displayed better architectural and amphiphilic tailorability at the molecular level than those prepared by other methods. The desirable structures of polymeric micelles could cover the range from unimolecular micelles26 to side chain-grafted micelles,27 and even anisotropic Janus micelles.28–30 Self-assembled polymeric micelles based on block and random copolymers have been recognized as an excellent model to elucidate the relationship between the emulsifying performance and structure of Pickering particulate emulsifiers.Since the pioneering work regarding linear triblock copolymer micelles as pH-responsive amphiphilic emulsifiers to stabilize 1-undecanol-in-water emulsions,31 interest in the surface activity and emulsifying performance of the self-assembled polymeric micelles has steadily gained increasing momentum. Recently, block copolymer micelles have shown great success in stabilizing the oil–water interface and controlling the stability of Pickering emulsions. The groups led by Prof. S. P. Armes conducted extensive and systematic work on Pickering emulsions in which self-assembled objects based on amphiphilic block copolymers were employed as particulate emulsifiers.32–42 RAFT-mediated polymerization-induced self-assembly (PISA) was used to prepare amphiphilic micellar nanoparticles which were subsequently evaluated as emulsifiers for the stabilization of Pickering emulsions. They found that linear poly(glycerol monomethacrylate)-b-poly(2-hydroxypropyl methacrylate) (PGMA-b-PHPMA) diblock copolymer micelles exhibited unusual thermo-responsive behavior (Fig. 1).32–35 During cooling from 21 to 4 °C, degelation of the micelle produced a free-flowing fluid with low viscosity, accompanied by a worm-to-sphere transition. Thompson and coworkers33,34 demonstrated that worm-like and spherical micelles or vesicles self-assembled from linear PGMA-b-PHPMA could not survive the high-shear conditions for efficient homogenization of the oil and aqueous phases. Whereas the as-prepared emulsions were still stable on account of the block copolymer acting as a polymeric surfactant, in which individual chains rather than particles were adsorbed at the oil–water interface. Particle dissociation during emulsification was attributed to the weakly hydrophobic character of the PHPMA unit. Covalent stabilization of these self-assembled objects could be readily achieved by the addition of ethylene glycol dimethacrylate (EGDMA) as the cross-linker during the PISA synthesis. The obtained cross-linked PGMA58-b-PHPMA350-b-PEGDMA20 block copolymer vesicles were sufficiently robust to act as Pickering emulsifiers, producing stable O/W emulsions for a range of model oils (Fig. 2).33,35 As shown in Fig. 3, TEM studies confirmed that the resulting cross-linked spherical or worm-like micelles survived emulsification and produced genuine Pickering emulsions (Fig. 3C and D). Alternatively, stabilization could be achieved by either replacing or supplementing the PHPMA block with the more hydrophobic poly(benzyl methacrylate) (PBzMA). The resulting linear spherical or worm-like micelles also survived emulsification (Fig. 3E and F) and produced stable n-dodecane-in-water Pickering emulsions.34 Hence chemical cross-linking was not mandatory for the Pickering system where sufficient stabilization could be conferred flexibly by introducing stronger hydrophobic interactions among the core-forming blocks.
 | ||
| Fig. 1 Thermoresponsive aqueous solution behavior of PGMA-b-PHPMA micelles. Reprinted with permission from American Chemical Society (ref. 32), Copyright (2012). | ||
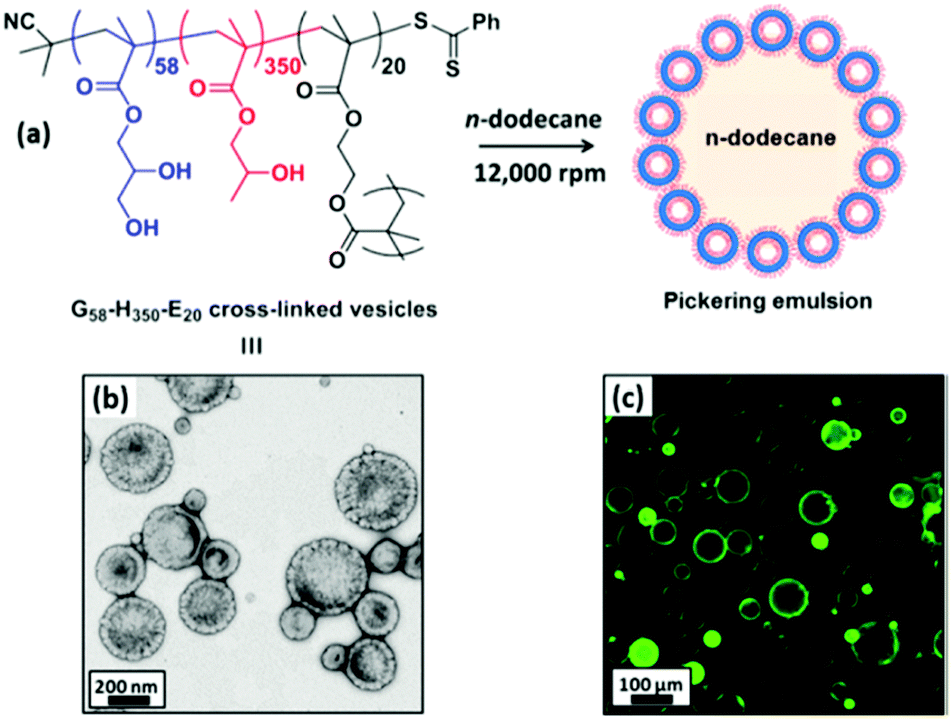 | ||
| Fig. 2 (a) Schematic representation of the preparation of Pickering emulsions using cross-linked PGMA58-b-PHPMA350-b-PEGDMA20 vesicles. (b) TEM image of cross-linked vesicles. (c) Fluorescence micrograph of colloidosomes obtained from a Pickering emulsion precursor prepared using fluorescein-labelled vesicles. Reprinted with permission from American Chemical Society (ref. 33 and 35), Copyright (2014). | ||
 | ||
| Fig. 3 TEM images of the edge and surface of various microcapsules prepared at 20 °C using: (A) linear G100-H200 spheres, (B) linear G45-H145 worms, (C) crosslinked G100-H200-E20 spheres, (D) crosslinked G45-H100-E10 worms, (E) linear G51-B250 spheres and (F) linear G37-H60-B30 worms. Reprinted with permission from Royal Society of Chemistry (ref. 34), Copyright (2014). | ||
After that, Armes et al.39,40 reported that the intelligent combination of hydrophilic and hydrophobic self-assembled block copolymer micelles facilitated the preparation of stable Pickering double emulsions using either water or oil as the continuous phase (Fig. 4). Specifically, fluorescence microscopy images showed that water-in-n-dodecane-in-water double emulsions were successfully formed. Fluorescein-labelled aqueous droplets were observed within oil droplets distributed in an aqueous continuous phase. Moreover, the addition of an oil-soluble Nile Red dye to n-dodecane confirmed that oil existed in the form of droplets (Fig. 4). Spherical nanomicelles self-assembled from poly(stearyl methacrylate)-b-poly(N-2-(methacryloyloxy)ethyl pyrrolidone) (PSMA-b-PNMEP) block copolymer via RAFT-mediated PISA had also been prepared, with a constant PSMA block length.41 PSMA14-b-PNMEP49 with an average diameter of 25 nm was used as a particulate emulsifier. As shown in Fig. 5, a hand-shaking W/O emulsion could be inverted to O/W type after high-speed homogenization. Further investigation suggested that high shear rates led to in situ inversions from the initial hydrophobic PSMA14-b-PNMEP49 spheres to hydrophilic PNMEP49-b-PSMA14 spheres.
 | ||
| Fig. 4 Schematic representation of the preparation of (a) W/O/W and (b) O/W/O Pickering double emulsions using the judicious combination of hydrophilic and hydrophobic block copolymer micelles. Fluorescence microscopy images confirm the successful formation of W/O/W Pickering double emulsions where (c) the aqueous phase is labelled with fluorescein and (d) the n-dodecane phase is labelled with Nile Red. Reprinted with permission from Elsevier (ref. 40), Copyright (2016). | ||
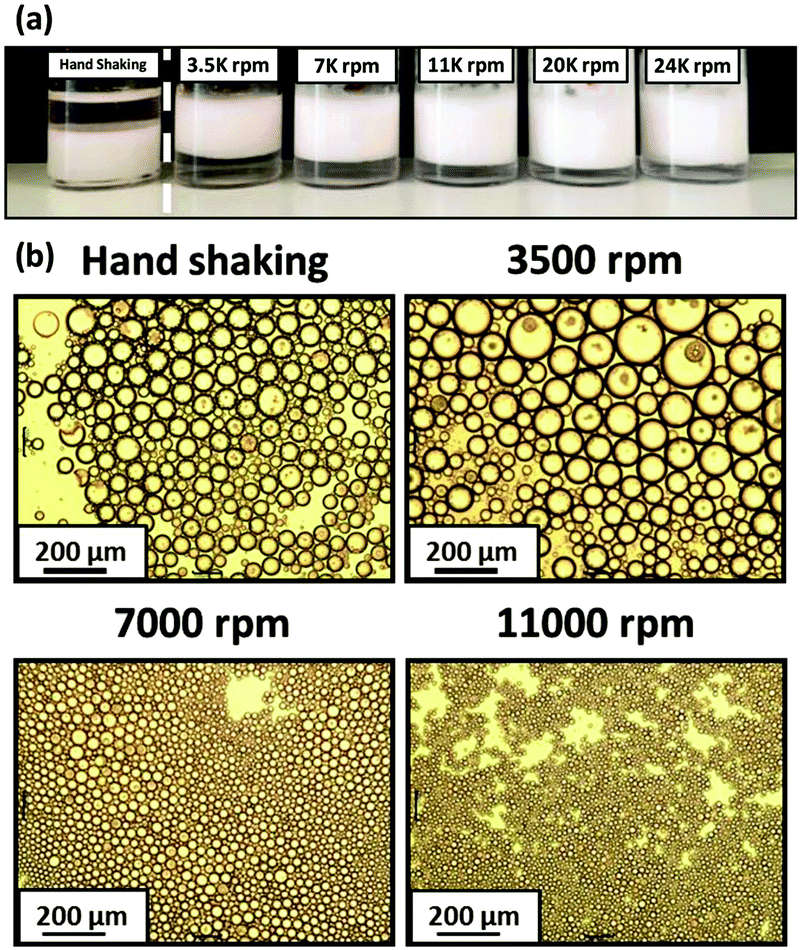 | ||
Fig. 5 (a) Digital photographs obtained for the Pickering emulsions prepared using 1.0% w/w PSMA14-b-PNMEP49 nanoparticles at various shear rates. O/W emulsions are formed in all cases, except when hand-shaking is used; this latter approach results in a W/O emulsion instead. (b) Optical microscopy images recorded for the droplets prepared via hand-shaking, or via homogenization at 3500 rpm, 7000 rpm or 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (scale bar = 200 μm), Reprinted with permission from Royal Society of Chemistry (ref. 41), Copyright (2016). 000 rpm (scale bar = 200 μm), Reprinted with permission from Royal Society of Chemistry (ref. 41), Copyright (2016). | ||
Tsarkova et al.43 demonstrated the interface-templated crystallization of biocompatible amphiphilic micelles of poly(ethylene oxide)-b-poly(ε-caprolactone) (PEO-b-PCL) block copolymers in the presence of high volume fractions of toluene, a good solvent for the PCL block. This crystallization behavior allowed multiple triggers in shaping the curvature of the oil/water interface toward the formation of stable multiple emulsions and the occurrence of phase inversion. The interfacial curvature (emulsion type) could be tuned by the volume fraction of the hydrophobic block or by the temperature trigger, and the latter affects both the solubility of the PEO block in water and the semicrystalline state of the PCL block. As could be observed from fluorescence microscopy and visual appearance in Fig. 6, a fast phase inversion from a water-continuous-phase emulsion to water-in-toluene emulsion was induced by thermal shock. The long-term stability of the emulsion droplets was ascribed to the multilamellar structure of alternating semicrystalline PCL and amorphous PEO in the interfacial layer, and the thermo-induced phase inversion was attributed to conformational changes of PEO and PCL blocks.
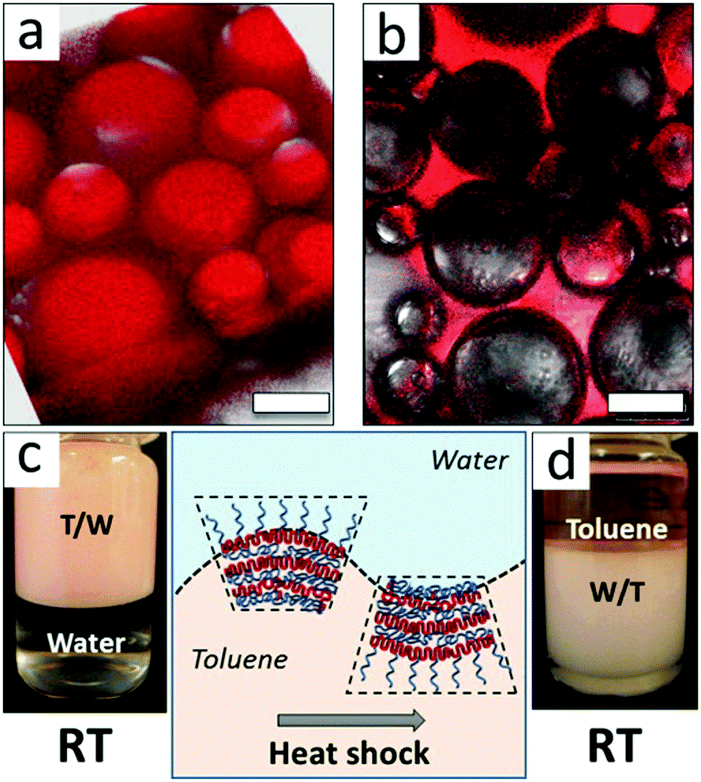 | ||
| Fig. 6 Confocal fluorescence micrographs and photos of emulsions stabilized with PEO113-b-PCL79 at room temperature (a and c) and the same emulsion after heat shock (b and d). Scale bar is 100 μm. Sketches of the temperature triggered conformational change of the PEO113-b-PCL79 interfacial layer. Reprinted with permission from American Chemical Society (ref. 43), Copyright (2016). | ||
In addition to the linear block polymers as stated above, the core cross-linked star (CCS) block polymers based on poly(N,N-dimethylacrylamide) (PDMA) as the high-polarity arm and poly(2-methoxyethylacrylate) (PMEA) as the low-polarity arm were self-assembled in water to form micelles.44 The composition and polarity of polymers significantly affected the emulsifying performance. The relatively high polarity facilitated the formation of O/W emulsions and lowering the polarity to a certain degree enhanced the stability of the emulsions. Moreover, multiple emulsions were gradually produced when the polarity was gradually lowered and the oil fraction in the emulsion samples was gradually increased, and at the same time the emulsions also became increasingly unstable.
3. Self-assembled micelles based on random copolymers
Now, researchers have a very clear understanding of the self-assembly mechanism of block copolymers.45–47 More importantly, block copolymers are not only structurally regular but their assembly morphology can usually be precisely controlled or even predicted by molecular parameters.23,48 However, the synthetic path of block copolymers is cumbersome and time consuming, and it is often necessary to sequentially control the polymerization or the post polymerization treatment. Compared with block copolymers, random copolymers have complex chain sequences and wide adjustable parameters, and their preparation is relatively simple (usually, one-step copolymerization of different monomers). Considerable random copolymers have been designed and synthesised to meet various requirements, and their self-assemblies are also expected if appropriate designs of the polymers were considered.48–53With this consideration, a series of polymeric micelles self-assembled from amphiphilic random copolymers were employed as amphiphilic particulate emulsifiers in recent years.54–65 The correlation between the oil–water interfacial property and emulsifying performance of polymeric micelles was deeply investigated. It was found that self-assembled micelles based on random copolymers were interfacially active. Different from the clear core–shell structure of block copolymer micelles, for random copolymer micelles, there were hydrophilic segments inside the micellar hydrophobic core while hydrophobic segments also existed in the micellar hydrophilic surface. Zhu and coworkers54 synthesized a series of random copolymer poly(acrylic acid-co-styrene) (P(AA-co-St)) and block copolymer poly(acrylic acid)-b-polystyrene (PAA-b-PSt) with similar chemical composition but different chain microstructure. The self-assembly behaviors of random and block copolymers in selective solvent were investigated. Polymeric micelles based on P(AA-co-St) and PAA-b-PSt with about 50 mol% hydrophilic AA unit were chosen as the model to explore the influence of micellar structure on emulsifying performance. For PAA-b-PSt micelles (B48), stable W/O emulsions could only be obtained when the pH was lower than 5. In contrast, P(AA-co-St) micelles (R49) had an excellent emulsifying performance at pH 4–10, and the pH-induced phase inversion was observed at pH higher than 6 (Fig. 7). Compared with PAA-b-PSt self-assembled micelles, P(AA-co-St) micelles had better interfacial performance and were more tailorable and controllable. The existing hydrophobic segment at the micelle peripheries provided interfacial activity for P(AA-co-St) micelles, and thus enhanced their emulsifying efficiency.
 | ||
| Fig. 7 Digital photos of toluene-water emulsions stabilized by (a) B48 micelles and (b) R49 micelles at various pH values before and 3 days after homogenization. SEM image of a polymerized water-in-styrene emulsion stabilized by (c) B48 micelles and (d) R49 micelles at pH = 4; and a styrene-in-water emulsion stabilized by R49 micelles at (e) pH = 7 and (f) pH = 10. All emulsions are homogenized with equal volume of micellar aqueous dispersion and toluene or styrene (containing 2 wt% V65) at 8000 rpm for 2 min. Micellar concentrations were 2.0 mg mL−1 with 0.1 M NaCl. Reprinted with permission from the PCCP Owner Societies (ref. 54), Copyright (2016). | ||
A one-step generation of stable multiple Pickering emulsions using nanomicelles as the sole emulsifier was reported (Fig. 8).55 These nanomicelles were self-assembled from an amphiphilic random copolymer poly(dodecyl acrylate-co-acrylic acid) (PDAA). The emulsifying performance of PDAA nanomicelles at different pH values demonstrated that multiple emulsions with long-term stability could only be stabilized by PDAA nanoparticles at pH 5.5, indicating that the surface wettability of PDAA nanoparticles played a crucial role in determining the type and stability of the resulting Pickering emulsions. Additionally, the polarity of oil did not affect the emulsifying performance of PDAA nanoparticles, and a wide variety of oils could be used as the oil phase to prepare multiple emulsions.
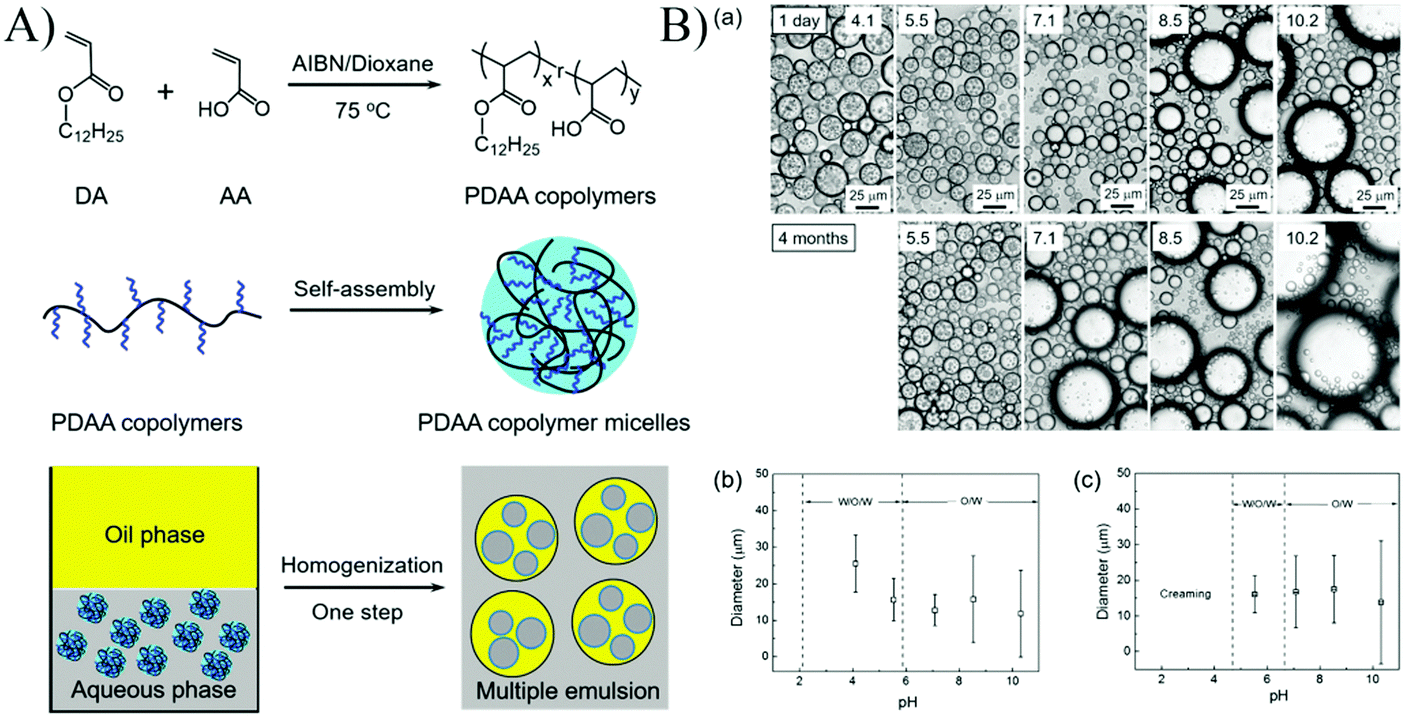 | ||
| Fig. 8 (A) Schematic illustration of the synthesis, and self-assembly process of the PDAA copolymers, and the application of using the PDAA micelles as a stabilizer to form multiple emulsions. (B) (a) The microscopy images of emulsion droplets stabilized by PDAA0.44 micelles at different pH values, and the emulsion droplet diameter after (b) 24 h and (c) 4 months in an ambient environment. The concentration of the PDAA0.44 micelles aqueous dispersion was 2 mg mL−1. The mean diameters were determined by analysis of more than 300 emulsion droplets. Reprinted with permission from Royal Society of Chemistry (ref. 55), Copyright (2016). | ||
Liu et al.58 prepared a series of photo-cross-linkable and pH-responsive micelles using amphiphilic random copolymers poly(7-(4-vinylbenzyloxyl)-4-methylcoumarin-co-acrylic acid) (PVMAA). Polymeric micelles based on PVMAA with about 12 mol% hydrophobic composition were chosen as the model to investigate the influence of photo-cross-linking on the emulsifying performance. The larger the shrinkage degree by photo-cross-linking (SDC), the lower the emulsifying efficiency. Additionally, the emulsifying efficiency of micelles increased with increasing pH. However, as the pH elevated over 8, the stability of the emulsions decreased due to the disintegration of the micelles (Fig. 9). Micelles with SDC of 95% kept their structural integrity due to the cross-linking, resulting in lower emulsifying efficiency of the micelles and worse stability of the emulsions. After that, another photo-cross-linkable and pH-responsive coumarin-containing self-assembled micelle poly (2-(dimethylamino)ethyl methacrylate-co-methyl methacrylate-co-7-(4-vinyl benzyloxyl)-4-methylcoumarin) (PDMV) was prepared by our group (Fig. 10).60 Similarly, the larger the SDC, the lower the emulsifying efficiency. The tertiary amine groups of terpolymers underwent inversion from protonation to deprotonation with increasing pH, causing the observed o/w to w/o phase inversion. Macrophase separation and demulsification were observed under strong alkaline conditions.
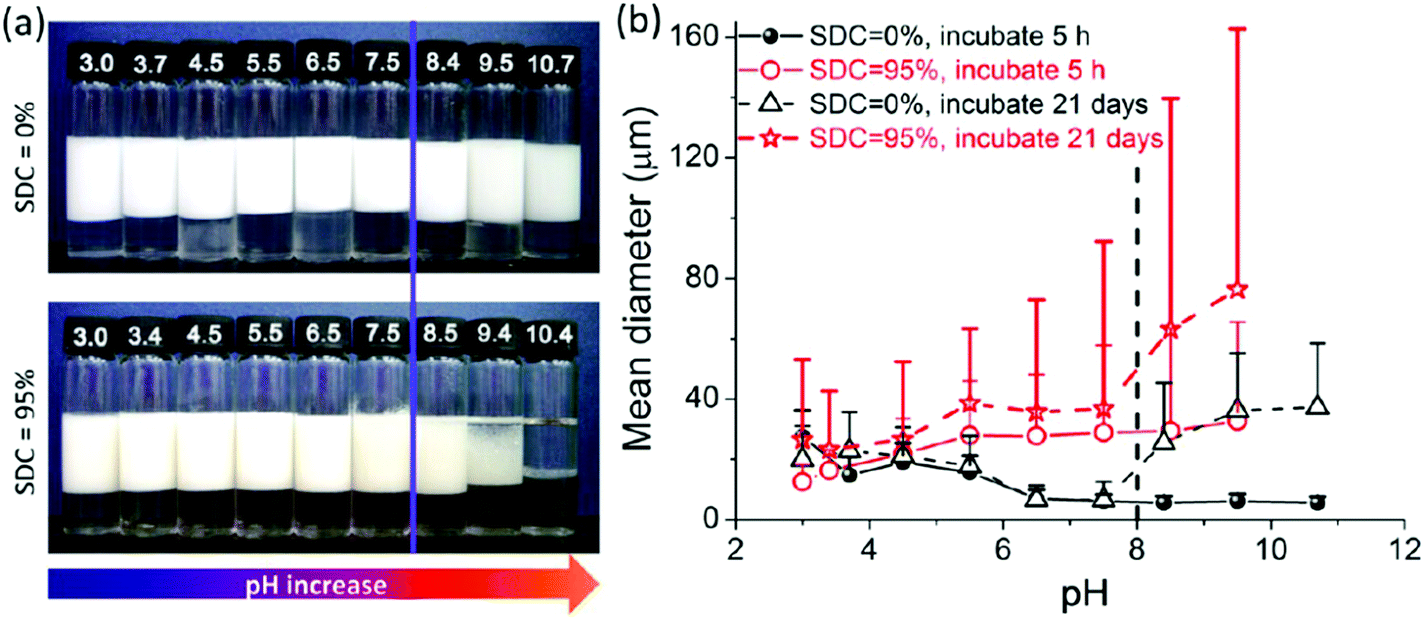 | ||
| Fig. 9 Properties of emulsions stabilized by PVMAA12 micelles with SDC of 0% and 95% at various pHs. (a) Appearance of the batches of toluene-in-water emulsions incubated 3 weeks after homogenization with equal volumes of toluene and micelle aqueous solution (4 mL/4 mL); the micelle concentrations were 2.0 mg mL−1 with 10 mM NaCl. (b) Plots of the statistical size of emulsion droplets against pH increase with different incubation times. The mean diameters of the emulsion droplets from the corresponding emulsions in (a) were counted through the optical microscope images. Reprinted with permission from American Chemical Society (ref. 58), Copyright (2014). | ||
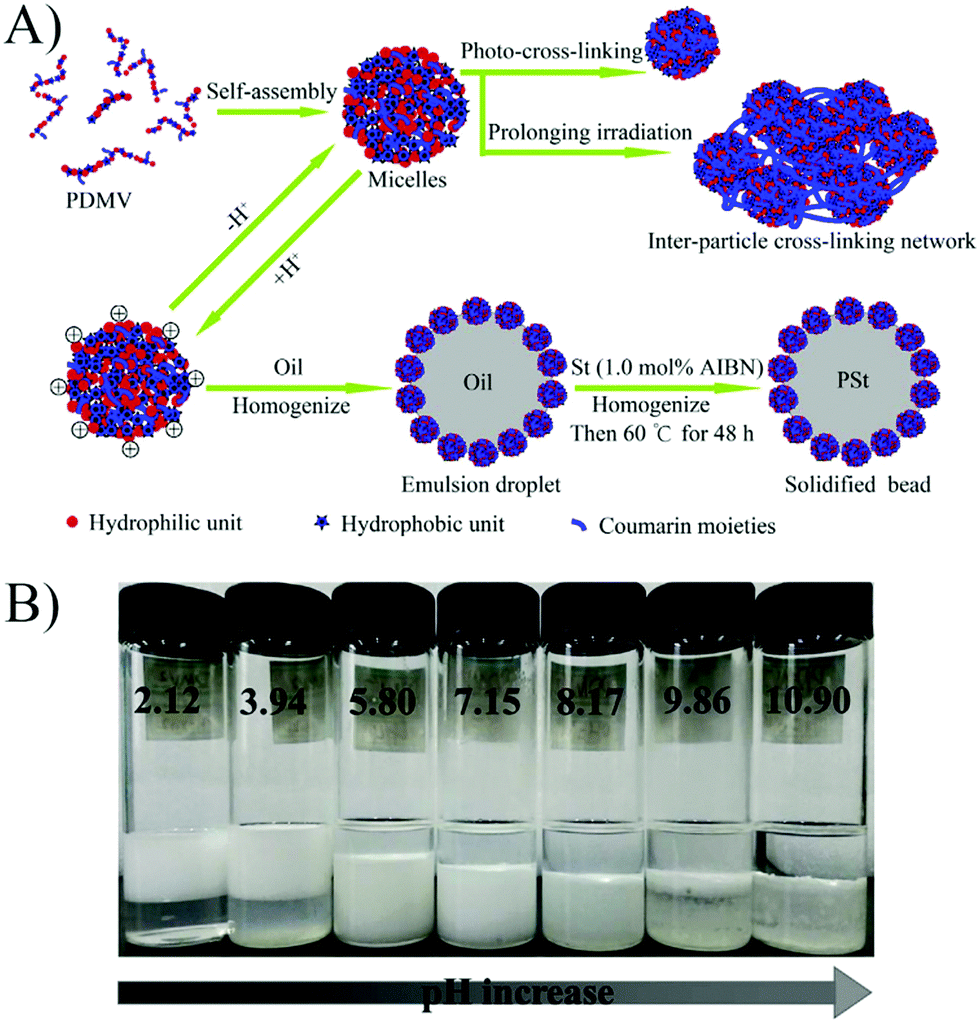 | ||
| Fig. 10 (A) Schematic illustration of self-assembly, photo-cross-linking, emulsification, and solidifying of the emulsion droplet. (B) Emulsifying performance of the PDMV15 micelles with various pHs. All the batches of emulsions were incubated for ten days after homogenization with equal volumes of toluene and micelle aqueous solution (4 mL/4 mL). All the micelle concentrations are 2.5 mg mL−1 with 10 mM NaCl. Reprinted with permission from Elsevier (ref. 60), Copyright (2017). | ||
Branched random copolymers have received much attention because of their attractive features such as multiple end groups, low viscosity, improved solubility, and three-dimensional spherical structure.66–69 Considering the unique structure and properties of branched copolymers, the self-assembled micelles of branched copolymers would exhibit different structure and interfacial properties from that of linear copolymers at the oil–water interface. Several studies have reported the emulsifiability of branched random copolymer micelles, and elucidated the difference between linear and branched copolymer micelles on stimuli-responsive emulsifying performance.70–72 For example, Pickering particles based on the self-assembled micelles of amphiphilic branched random poly(styrene-co-maleic anhydride) (BPSMA) have been successfully prepared to investigate controllable Pickering emulsions (Fig. 11).72 By comparing the structure and emulsifying performance of the branched copolymer micelles with that of the corresponding linear ones (LPSMA), it was found that the branched structure of the copolymer played an important role in emulsification. The emulsifying performance of BPSMA micelles was superior to that of LPSMA micelles due to the much better structural stability of BPSMA micelles, which was reflected by the emulsifying efficiency and the long-term stability of the emulsions (Fig. 11). The hydrolyzing of maleic anhydride and ionization of carboxyl to some extent could be postponed by the introduction of a branched structure in the copolymers.
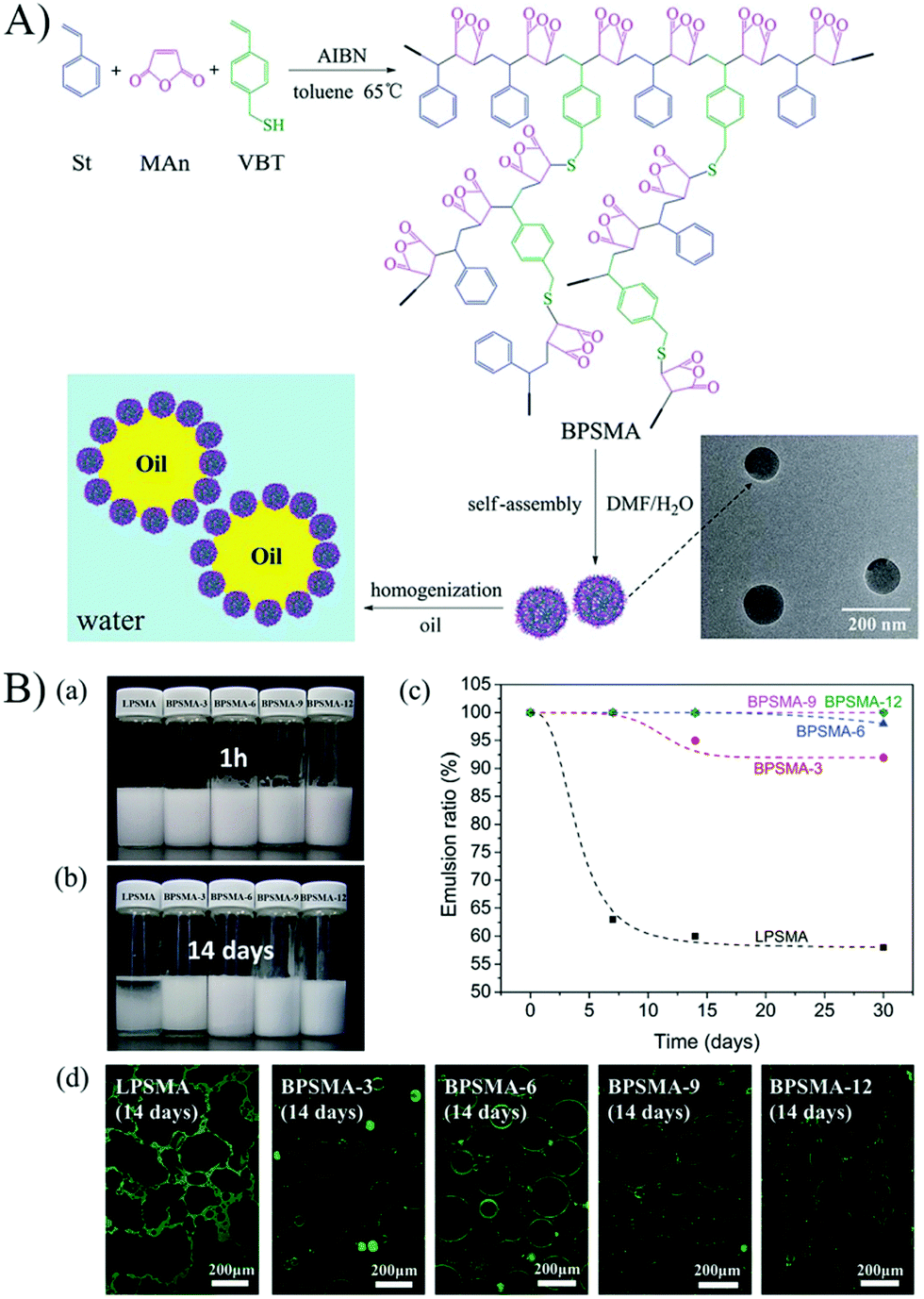 | ||
| Fig. 11 (A) Schematic illustration of the preparation and self-assembly of BPSMA, and the use of the self-assembled micelles as particulate emulsifiers to stabilize O/W Pickering emulsions. (B) (a and b) Appearance of the white oil/water emulsions stabilized by LPSMA and BPSMA micelles with different incubation times after homogenization, 1 h (a) and 14 days (b). (c) Emulsion ratio of the white oil/water emulsions stabilized by LPSMA and BPSMA micelles over time. (d) CLSM images of the white oil/water emulsions stabilized by the LPSMA and BPSMA micelles labelled by RhB (green) at 14 days after homogenization. All the emulsions were prepared with the volume ratio of oil to water phase as 5:1, and the micelle concentration was 0.75 mg mL−1. Reprinted with permission from Royal Society of Chemistry (ref. 72), Copyright (2015). | ||
4. Conclusion and perspective
With the rapid development of materials technology, more and more micro- or nanoparticles as the main components of Pickering emulsions have been synthesized and functionalized. In particular, the precise design of well-defined polymeric architectures with stimuli-responsive functionality is driven by the need for controllable material properties. Under such circumstances, polymeric micelles self-assembled from amphiphilic block or random copolymers have been successfully employed in the control of stability and type of Pickering emulsions. The vital advantages of using self-assembled polymeric micelles as Pickering particles are that the preparation process of these smart copolymers is simple, and the amphiphilicity of the resultant self-assembled objects is easy to control. Moreover, copolymers with stimuli-responsive attributes play a critical role in emulsifying performance and oil–water interfacial behavior. Considering these feature, these self-assembled micelles have great potential to replace traditional emulsifiers, just as they can work independently in more and more instances.In general, the combined examples cited in this review firstly highlight the recent progress in self-assembled polymeric micelles as amphiphilic particulate emulsifiers. It is clear that the copolymer composition can be used to adjust the micelle amphiphilicity and particle surface wettability. Partial wetting by water and oil required for adsorption at the oil/water interface is determined by hydrophilic or hydrophobic functional groups. Thus, most polymer-containing nanoparticles can be used to prepare controllable Pickering emulsions as a result of the amphiphilic and responsive functional groups of polymeric chains. This review has shown that self-assembly of amphiphilic polymers is one of the most promising and attractive ways to fabricate these functional particles. In our on-going polymer science, we can continue to explore and optimize these self-assembled particles by tailoring the properties of monomers or polymers. For example, we can synthesize cross-linkable monomers and highly branching copolymers to modify the particles or allow self-assembly into desired aggregates. In addition, the facile access to self-assembled polymeric micelles provides translational opportunities in a variety of research areas, especially in biomedical applications such as drug delivery, bioreactors, biosensing, and bioimaging.
Many researchers are focusing on self-assembled micelles as soft templates for different nanomaterials, such as mesoporous nanoparticles, hollow nanoparticles, and nanotubes, which might provide a new pathway and opportunity for versatile micellar emulsifiers. However, there are still challenges in this area. For example, how to recover micellar emulsifiers? Novel polymeric micelles are expected to be prepared for degradable functional emulsions, and even clinical emulsion applications. For example, the expected micelles as templates are employed as emulsion formulations to prepare degradable drug delivery materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (Grant No. 21376271), the Fundamental Research Funds for the Central Universities of Central South University (No. 2017zzts783), and the Hunan Provincial Science and Technology Plan Project, China (No. 2016TP1007).References
- S. U. Pickering, J. Chem. Soc., 1907, 91, 2001–2021 RSC
.
- W. Ramsden, Proc. Chem. Soc., 1903, 72, 156–164 CAS
- E. Dickinson, J. Sci. Food Agric., 2013, 93, 710–721 CrossRef CAS PubMed
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100, 503–546 CrossRef
- S. Lam, K. P. Velikov and O. D. Velev, Curr. Opin. Colloid Interface Sci., 2014, 19, 490–500 CrossRef CAS
- J. Wu and G.-H. Ma, Small, 2016, 12, 4633–4648 CrossRef CAS PubMed
- M. Rayner, D. Marku, M. Eriksson, M. Sjöö, P. Dejmek and M. Wahlgren, Colloids Surf., A, 2014, 458, 48–62 CrossRef CAS
- J. Tang, P. J. Quinlan and K. C. Tam, Soft Matter, 2015, 11, 3512–3529 RSC
- Z. Wang and Y. Wang, Materials, 2016, 9, 903 CrossRef PubMed
- C. C. Berton-Carabin and K. Schroën, Annu. Rev. Food Sci. Technol., 2015, 6, 263–297 CrossRef CAS PubMed
- Y. Chevalier and M.-A. Bolzinger, Colloids Surf., A, 2013, 439, 23–34 CrossRef CAS
- A. Schrade, K. Landfester and U. Ziener, Chem. Soc. Rev., 2013, 42, 6823–6839 RSC
- A. Kumar, S. Li, C.-M. Cheng and D. Lee, Ind. Eng. Chem. Res., 2015, 54, 8375–8396 CrossRef CAS
- M. Williams and S. P. Armes, Langmuir, 2014, 30, 2703–2711 CrossRef CAS PubMed
- K. Y. Yoon, Z. Li, B. M. Neilson, W. Lee, C. Huh, S. L. Bryant, C. W. Bielawski and K. P. Johnston, Macromolecules, 2012, 45, 5157–5166 CrossRef CAS
- M. Liu, X. Chen, Z. Yang, Z. Xu, L. Hong and T. Ngai, ACS Appl. Mater. Interfaces, 2016, 8, 32250–32258 CrossRef CAS PubMed
- Y. Qian, Q. Zhang, X. Qiu and S. Zhu, Green Chem., 2014, 16, 4963–4968 RSC
- M. Ranka, H. Katepalli, D. Blankschtein and T. A. Hatton, Langmuir, 2017, 33, 13326–13331 CrossRef CAS PubMed
- K. S. Silmore, C. Gupta and N. R. Washburn, J. Colloid Interface Sci., 2016, 466, 91–100 CrossRef CAS PubMed
- J. Tang, M. F. X. Lee, W. Zhang, B. Zhao, R. M. Berry and K. C. Tam, Biomacromolecules, 2014, 15, 3052–3060 CrossRef CAS PubMed
- J. Tang, R. M. Berry and K. C. Tam, Biomacromolecules, 2016, 17, 1748–1756 CrossRef CAS PubMed
- J. Glasing, P. G. Jessop, P. Champagne and M. F. Cunningham, Polym. Chem., 2018, 9, 3864–3872 RSC
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS
- X.-Q. Liu, C.-Y. Sun, X.-Z. Yang and J. Wang, Part. Part. Syst. Charact., 2013, 30, 211–228 CrossRef CAS
- J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7129 RSC
- L. Cheng, G. Hou, J. Miao, D. Chen, M. Jiang and L. Zhu, Macromolecules, 2008, 41, 8159–8166 CrossRef CAS
- C. Ma, X. Bi, T. Ngai and G. Zhang, J. Mater. Chem. A, 2013, 1, 5353–5360 RSC
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed
- Y. Chen, Macromolecules, 2012, 45, 2619–2631 CrossRef CAS
- J. W. Kim, J. Cho, J. Cho, B. J. Park, Y.-J. Kim, K.-H. Choi and J. W. Kim, Angew. Chem., Int. Ed., 2016, 55, 4509–4513 CrossRef CAS PubMed
- S. Fujii, Y. Cai, J. V. M. Weaver and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 7304–7305 CrossRef CAS PubMed
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. Ian Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450–12453 CrossRef CAS PubMed
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 RSC
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed
- K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk, J. A. Lane, M. J. Derry and S. P. Armes, Chem. Sci., 2015, 6, 4207–4214 RSC
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC
- K. L. Thompson, J. A. Lane, M. J. Derry and S. P. Armes, Langmuir, 2015, 31, 4373–4376 CrossRef CAS PubMed
- K. L. Thompson, C. J. Mable, J. A. Lane, M. J. Derry, L. A. Fielding and S. P. Armes, Langmuir, 2015, 31, 4137–4144 CrossRef CAS PubMed
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS
- V. J. Cunningham, S. P. Armes and O. M. Musa, Polym. Chem., 2016, 7, 1882–1891 RSC
- S. L. Rizzelli, E. R. Jones, K. L. Thompson and S. P. Armes, Colloid Polym. Sci., 2016, 294, 1–12 CrossRef CAS
- A. Manova, J. Viktorova, J. Köhler, S. Theiler, H. Keul, A. A. Piryazev, D. A. Ivanov, L. Tsarkova and M. Möller, ACS Macro Lett., 2016, 5, 163–167 CrossRef CAS
- X. Shi, M. Miao and Z. An, Polym. Chem., 2013, 4, 1950–1959 RSC
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS
- A. H. Groschel, A. Walther, T. I. Lobling, F. H. Schacher, H. Schmalz and A. H. E. Muller, Nature, 2013, 503, 247–251 CrossRef PubMed
- L. Li, K. Raghupathi, C. Song, P. Prasad and S. Thayumanavan, Chem. Commun., 2014, 50, 13417–13432 RSC
- X. Zhu and M. Liu, Langmuir, 2011, 27, 12844–12850 CrossRef CAS PubMed
- X. Liu, J.-S. Kim, J. Wu and A. Eisenberg, Macromolecules, 2005, 38, 6749–6751 CrossRef CAS
- M. Chen, M. Sun and X. Liu, J. Dispersion Sci. Technol., 2016, 37, 900–907 CrossRef CAS
- Y. Hirai, T. Terashima, M. Takenaka and M. Sawamoto, Macromolecules, 2016, 49, 5084–5091 CrossRef CAS
- M. Shibata, M. Matsumoto, Y. Hirai, M. Takenaka, M. Sawamoto and T. Terashima, Macromolecules, 2018, 51, 3738–3745 CrossRef CAS
- Y. Zhu, C. Yi, Q. Hu, W. Wei and X. Liu, Phys. Chem. Chem. Phys., 2016, 18, 26236–26244 RSC
- Y. Zhu, J. Sun, C. Yi, W. Wei and X. Liu, Soft Matter, 2016, 12, 7577–7584 RSC
- Y. Zhu, Q. Hu, W. Wei, C. Yi and X. Liu, Colloids Surf., A, 2016, 504, 358–366 CrossRef CAS
- M. Chen, Z. Geng, M. Sun, X. Liu and S. Chen, J. Dispersion Sci. Technol., 2014, 35, 757–764 CrossRef CAS
- C. Yi, J. Sun, D. Zhao, Q. Hu, X. Liu and M. Jiang, Langmuir, 2014, 30, 6669–6677 CrossRef CAS PubMed
- F. Wang and H. Liu, J. Phys. Chem. C, 2018, 122, 3434–3442 CrossRef CAS
- F. Wang, Y. Ouyang, H. Zou, Z. Yang and H. Liu, Colloids Surf., A, 2017, 535, 274–282 CrossRef CAS
- F. Wang, X. Yu and H. Liu, Colloid Polym. Sci., 2018, 296, 385–392 CrossRef CAS
- X. Liu, C. Yi, Y. Zhu, Y. Yang, J. Jiang, Z. Cui and M. Jiang, J. Colloid Interface Sci., 2010, 351, 315–322 CrossRef CAS PubMed
- J. Sun, C. Yi, W. Wei, D. Zhao, Q. Hu and X. Liu, Langmuir, 2014, 30, 14757–14764 CrossRef CAS PubMed
- C. Yi, Y. Yang, Y. Zhu, N. Liu, X. Liu, J. Luo and M. Jiang, Langmuir, 2012, 28, 9211–9222 CrossRef CAS PubMed
- F. Wang, X. Yu, Z. Yang, H. Duan, Z. Zhang and H. Liu, J. Phys. Chem. C, 2018, 122, 18995–19003 CrossRef CAS
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS PubMed
- S. Motokucho, H. Yamada, Y. Suga, H. Morikawa, H. Nakatani, K. Urita and I. Moriguchi, Polymer, 2018, 145, 194–201 CrossRef CAS
- Y. Zhou, W. Huang, J. Liu, X. Zhu and D. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS PubMed
- W. Wei, T. Wang, J. Luo, Y. Zhu, Y. Gu and X. Liu, Colloids Surf., A, 2015, 487, 58–65 CrossRef CAS
- C. Yi, N. Liu, J. Zheng, J. Jiang and X. Liu, J. Colloid Interface Sci., 2012, 380, 90–98 CrossRef CAS PubMed
- W. Wei, T. Wang, C. Yi, J. Liu and X. Liu, RSC Adv., 2015, 5, 1564–1570 RSC
| This journal is © the Partner Organisations 2019 |