
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical stability of poly(phenylene oxide)-based ionomers in an anion exchange-membrane fuel cell environment†
Sapir
Willdorf-Cohen‡
a,
Abhishek N.
Mondal‡
 a,
Dario R.
Dekel
*ac and
Charles E.
Diesendruck
*bc
a,
Dario R.
Dekel
*ac and
Charles E.
Diesendruck
*bc
aThe Wolfson Department of Chemical Engineering, Technion-Israel Institute of Technology, Haifa 3200003, Israel. E-mail: dario@technion.ac.il
bSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 3200008, Israel. E-mail: charles@technion.ac.il
cThe Nancy & Stephan Grand Technion Energy Program (GTEP), Technion-Israel Institute of Technology, Haifa 3200003, Israel
First published on 5th September 2018
Abstract
In recent years, intense research interest has been focused towards the development of anion exchange membrane fuel cells (AEMFCs) due to their potential to circumvent the need for expensive platinum catalysts, tackling the high cost that impedes mass commercialization of fuel cells. However, AEMFCs are not yet practical due to the low chemical stability of the quaternary ammonium (QA) cationic groups during cell operation. Several functionalized polymers for anion exchange membranes (AEMs), including substituted poly(phenylene oxide) (PPO), have been proposed as suitable ionomeric materials, as they present good stability in strong alkaline solutions. However, while they perform well in ex situ stability tests in aqueous solutions, they still present limited performance during AEMFC operation. As the current density in the fuel cell increases, more water is consumed at the cathode side, reducing the hydration level and, in turn, increasing the nucleophilicity of OH− and its capability to attack the QA groups. Here, using our recently reported ex situ stability protocol that simulates the low-hydration environment of an AEMFC during operation, the alkaline stability of PPO-based anion exchange ionomers is measured and compared. Good agreement with previously studied QA molecules tested using the same protocol was found. Yet, the degradation processes in these ionomers are further accelerated compared to the small QA molecules as a consequence of the lower polarity of the polymer environment, which further increases the hydroxide reactivity. This study demonstrates the competence of this new ex situ stability protocol to test not only QA molecules, but also ionomers and membranes, showing alkaline stability results that are comparable to those obtained in real AEMFC tests.
1. Introduction
Anion exchange membrane fuel cells (AEMFCs) have been attracting significant attention in the field of energy conversion and storage devices.1–5 AEMFCs, working in an alkaline environment, allow the use of metals outside the platinum group as electrocatalysts, significantly reducing cost.6–10 AEMFCs operate with a solid electrolyte, allowing much simpler operation and improved performance stability.11 However, there are still a few challenges facing this technology including the development of high performance HOR catalysts,10,12,13 as well as the decrease in cell performance due to the carbonation process while using ambient air as a feed gas (containing CO2).14–19 Yet, the most important challenge in the development of high performance AEMFCs is the issue of chemical stability of anion exchange membranes (AEMs) in an alkaline environment. Given the potential for future commercialization of the technology, AEMs should be stable for several thousands of hours of operation at high current rates. Yet, to this day, AEMs undergo fast and continuous chemical degradation during cell operation.17 Although new cationic functional groups20–24 have been developed and show good stability in alkaline solutions, AEMFC stability has not yet been improved.2 It was recently reported that a combination of a high pH environment and low hydration level could explain why anion conducting polymers degrade during cell operation.25 In most ex situ studies, alkaline aqueous solutions at high temperatures have been used to simulate the AEMFC environment. In this case, 6 or more water molecules are present per hydroxide, enough to fill the first solvation sphere of the OH− ion.26 In many cases, quaternary ammonium (QA) salts show perfect stability in these tests, but in operational AEMFCs, where microsolvation is reduced, they fail rapidly.27 In an operating AEMFC, water is quickly consumed at the cathode side, producing a significant water gradient through the membrane. As the operating current of the AEMFC increases, the water gradient through the cell increases, and the degradation around the cathode is accelerated.2,25,28–30Taking these effects into consideration, we have developed a new protocol for the ex situ testing of QAs, which better represents the environment in an operating AEMFC. In our methodology, unsolvated hydroxide is prepared in dry DMSO and further separated from its counter-cation by coordination, leaving the anion “free” to attack the QA cation.27–29 Using this protocol, trimethylbenzyl ammonium (TMBA), one of the most comprehensively studied QAs,20,31,32 was shown to be a leader in terms of stability. Yet, while we have studied several QAs at different hydration levels (λ) and temperatures, these are not the only parameters affecting the kinetics of this reaction. The diffusion rate of the ions in the membrane, as well as the environment polarity, affect the nucleophilicity of the OH− as well as its conductivity, an important parameter to achieve high power densities in AEMFCs.33–36 In AEMs, these parameters are defined by the chemistry of the ionomer backbone. AEMs have been prepared where QAs were tethered into different polymer backbone chemistries such as poly(vinyl alcohol),37,38 polysulfones39–42 and poly(tetrafluoroethylene).33,34 Poly(phenyl oxide) (PPO)40,43–49 has also been extensively studied as a backbone for AEMs, since it presents good mechanical properties and high alkaline stability. In addition, PPO synthesis and substitution is relatively simple.50,51
Several research groups have synthesized and characterized PPO-based AEMs when TMBA is incorporated into the polymer structure (PPO–TMA) and a wide variety of conditions and methods were employed to evaluate their chemical stabilities in alkaline medium (Table 1). More importantly, all the reported ex situ stability studies in the literature were performed in aqueous solutions.40,44–49,52 In these studies, the hydration number (λ – number of water molecules per hydroxide) varies from 6 to 56 (corresponding to 10 and 1 M, respectively).40,44–49,52 As λ is reduced, the polymer degradation is accelerated, but not by a simple multiplication of the hydroxide concentration. For instance, Liu et al.46 conducted tests for PPO–TMA at 80° with λ = 28, 11 and 6 (2, 5 and 10 M). If the reaction kinetics followed a simple change in concentration (assuming first order in the hydroxide), the percentage of degradation should simply increase by 2.5 and 5 times, but it increases by ca. 5 and 20 times respectively, indicating that a change in reactivity also occurred. Given the low water concentration around the cathode of the AEMFC, studies in very dry environments need to be carried out in order to verify the real chemical stability of AEMs.25,27–30 In this study, we expand our previously described protocol28 to test the stability of the QA in an AEM backbone.
| OH− conc. [M] | λ | T [°C] | Method | t [h] | Deg.c [%] | Ref. |
|---|---|---|---|---|---|---|
| a λ is the hydration number (number of water molecules per OH−) calculated from the reported OH− concentration. b The decrease in hydroxide conductivity of AEMs was measured after the alkaline stability test. c Reported degradation in the measured properties. d Room temperature. | ||||||
| 1 | 56 | 80 | 1H-NMR | 192 | 85 | 44 |
| 2 | 28 | 60 | 1H-NMR | 168 | 15 | 45 |
| 2 | 28 | 80 | Conductivityb | 200 | ∼5 | 46 |
| 5 | 11 | Conductivityb | ∼24 | |||
| 10 | 6 | Conductivityb | ∼92 | |||
| 1 | 56 | 80 | Conductivityb | 100 | 40 | 47 |
| 3 | 19 | r.t.d | Conductivityb | 250 | 40 | 40 |
| 1 | 56 | 60 | IEC | 240 | 30 | 48 |
| 1 | 56 | 80 | Conductivityb | 80 | 60 | 49 |
2. Experimental
2.1 General
NBS (N-bromosuccinimide) and 2,2′-azobis-isobutyronitrile (AIBN) were obtained from Alfa Aesar U.K. and Glentham Life Sciences Ltd., respectively. Dry DMSO-d6 (99.96%, ca. 60 ppm of water) was obtained from Cambridge Isotope Laboratories. All other chemicals were purchased from Sigma-Aldrich and used without further purification unless noted. All NMR spectra were recorded using an AVANCE II 300, 400 or 500 MHz Bruker spectrometer at the Technion NMR facilities. Stability tests were performed in an MBraun MS-Unilab Pro SP glovebox, with a nitrogen atmosphere with less than 0.1 ppm water and O2.2.2 Synthesis of Br-PPO (Scheme 1)
A round-bottom flask (250 mL) was charged with chlorobenzene (100 mL). PPO (12 g, ca. 100 mmol of monomer) was slowly added and stirred until completely dissolved. NBS (7.12 g, 40 mmol) and AIBN (0.5 g, 3 mmol) were added to the solution, which was heated under reflux for 3 h. The reaction mixture was cooled to room temperature and the product precipitated by pouring the mixture into a 10-fold excess of ethanol. The polymer was recovered by filtration and washed several times with ethanol followed by drying in a vacuum oven overnight at 60 °C. The polymer was reprecipitated from chloroform/ethanol for further purification. The white powder was dried under vacuum overnight to get the desired Br-PPO with a degree of bromination of 25%, according to 1H NMR spectroscopy (see the ESI†).2.3 Preparation of QA functionalized polymers
Trimethylamine and triethylamine were used to functionalize the Br-PPO to achieve PPO–TMA and PPO–TEA, respectively. The functionalized polymers were synthesized via the Menshutkin reaction (Scheme 2).53 NMP was added to a round-bottom flask (25 mL). Br-PPO (0.5 g) was slowly added and stirred until completely dissolved. Trimethylamine in ethanol (4.2 M, 0.6 mL) was added dropwise and the solution was further stirred for 22 hours at room temperature. The dark brown coloured polymer solution was cast on a clean glass plate and dried at 60 °C for 24 h on top of a hotplate. To ensure complete solvent removal, the obtained polymer was further dried under high vacuum for 3 days, until no solvent peaks were seen by NMR. The polymer functionalized with triethylamine (TEA) was synthesized in a similar manner, using pure TEA instead of trimethylamine in ethanol. In this case, a material with partial substitution was used as the polymer with full replacement was insoluble in the test solution.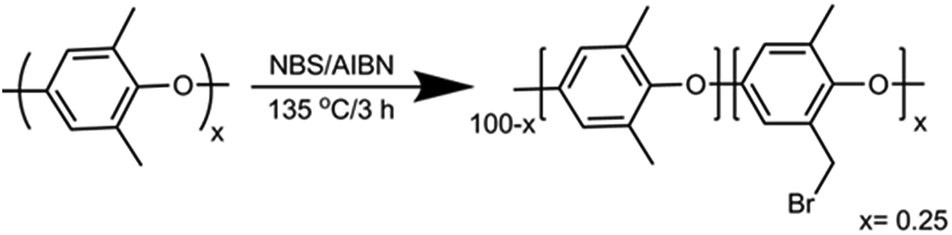 | ||
| Scheme 1 Synthesis of partially brominated PPO (Br-PPO). | ||
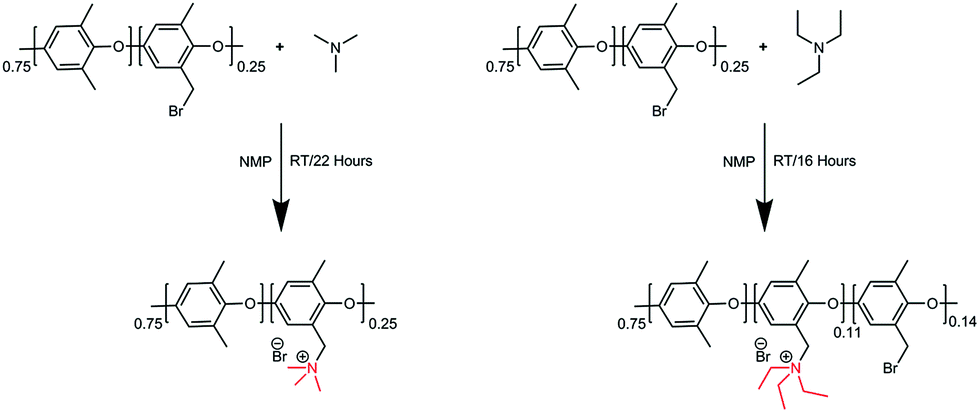 | ||
| Scheme 2 Preparation of PPO–TMA and PPO–TEA polymers. | ||
2.4 Preparation of a water-free hydroxide solution
The dry potassium hydroxide solution was prepared as recently reported by Dekel et al.28,29 In a three-necked flask, 18-crown-6 (CE) was heated to 60 °C under argon. Potassium metal, after washing with hexane, was slowly added to the flask to produce a blue solution via an exothermic reaction. This solution was slowly titrated using precise additions of Milli-Q water, until the colour changed to brown. The dry potassium hydroxide in crown ether (CE–K)+ OH− was cooled down to room temperature and transferred into the glovebox, where it was kept in a closed flask. The solution was prepared by weighing the (CE–K+) OH− in the glovebox and dissolving it in dry DMSO-d6 (500 μL) to form the stock solution (0.6 M).2.5 Kinetic studies of the QA functionalized polymers
The QA functionalized polymers (0.035 mmol of QA) were dissolved in DMSO-d6 (450 μL) in the glovebox and brought out in a closed syringe. For experiments under anhydrous conditions (λ = 0), the (CE–K+) OH− stock solution (0.05 mL) was added to a screw cap NMR tube in the glovebox and then, the QA solution (0.45 mL) was injected just before starting the NMR measurements.For experiments with λ > 0 (λ = 4 and 8), the amount of DMSO-d6 used to dissolve the ionomer was slightly reduced according to the required water volume, which was placed into the screw-cap NMR tube together with the (CE–K+) OH− stock solution (0.05 mL) before the addition of the ionomer solution, to achieve the desired λ. The aromatic signals were integrated over time to measure the conversion.
3. Results & discussion
In previous studies, we have shown that trimethylbenzyl ammonium (TMBA) and triethylbenzyl ammonium (TEBA) salts degrade rapidly in the presence of OH− at room temperature as the water content is reduced.29 When the number of water molecules solvating the hydroxide increases, its nucleophilicity and basicity are reduced and the QA degradation is significantly hindered.28 These results indicated that the currently used aqueous alkali ex situ tests to measure AEM stability may lead to false positive stability results where anion conducting polymers may appear to be alkali stable, but would not survive the unique conditions in an operating AEMFC. Here, our new ex situ test protocol that combines aggressive alkalinity and very low water content is used, for the first time, to test AEMs, in order to further clarify the importance of the combination of these effects on the chemical stability of QAs in ionomers. Two QA-grafted PPOs are tested: PPO–TMA and PPO–TEA. The degradation of PPO–TMA as a function of time (0.06 M OH−, room temperature) with different hydration numbers (λ) is shown in Fig. 1.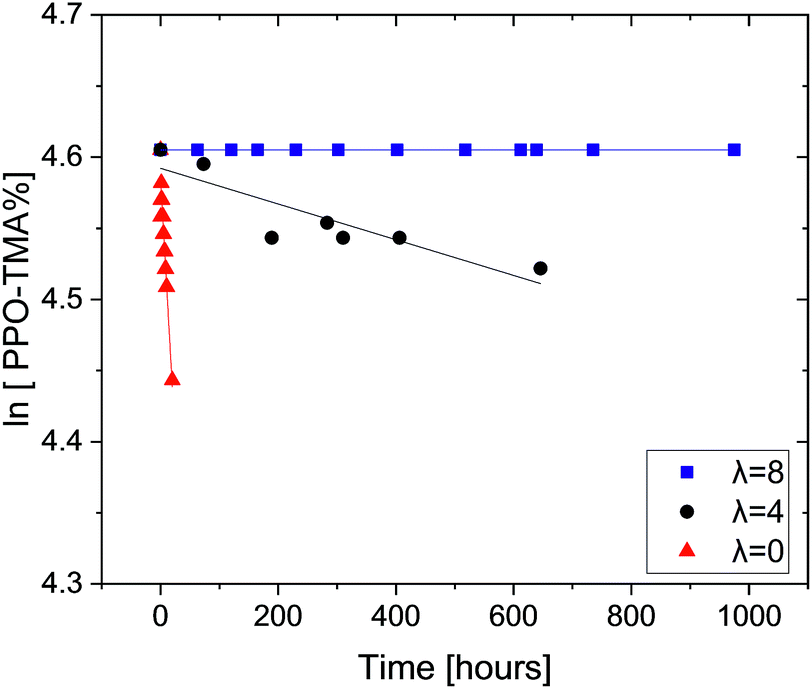 | ||
| Fig. 1 Remaining PPO–TMA fractions as a function of time with hydration number λ = 0, 4 and 8, in 0.06 M OH− DMSO-d6 solutions. All tests were done at room temperature. | ||
As expected, the decomposition rate of PPO–TMA increases as the water content is reduced. While this ionomer is completely stable when λ = 8, it degrades slowly at λ = 4 and rapidly in the absence of water (λ = 0). A pseudo first-order reaction is assumed since the hydroxide is in large excess. The experimental degradation data shown in Fig. 1 were fitted to the following linear equation: ln[QA] = ln[QA]0 + kt, where k is the degradation rate constant, and [QA] and [QA]0 are the QA concentrations at a given time t and initial time. The corresponding half-lives were calculated using the relationship t1/2 = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2k−1. Table 2 summarizes the degradation rates and the calculated half-lives for 25% PPO–TMA in 0.06 M OH−. In order to obtain a meaningful comparison with TMBA (small molecule), the rate was multiplied by 10 (assuming first order in hydroxide) to obtain the rate in 0.6 M OH−, which was used in our previous study.24
2k−1. Table 2 summarizes the degradation rates and the calculated half-lives for 25% PPO–TMA in 0.06 M OH−. In order to obtain a meaningful comparison with TMBA (small molecule), the rate was multiplied by 10 (assuming first order in hydroxide) to obtain the rate in 0.6 M OH−, which was used in our previous study.24
| λ | PPO–TMA in 0.06 M OH− | PPO–TMA in 0.6 M OH− | TMBA in 0.6 M OH− (ref. 28 and 29) | |||
|---|---|---|---|---|---|---|
| k × 10−3 [h−1] | Half-life [h] | Estimated k [h−1] | Half-life [h] | k × 10−4 [h−1] | Half-life [h] | |
| a Half-life estimated assuming a maximum degradation of 1% after 975 h. | ||||||
| 0 | 7.3 | 94.9 | 0.073 | 9.95 | 63.8 | 109 |
| 4 | 0.17 | 4077.3 | 0.0017 | 407.7 | 2.8 | 2460 |
| 8 | ∼0 | >34000a |
∼0 | >67000a |
∼0 | >34000 |
No degradation was observed at λ = 8 in the ionomer or QA with the same TMBA functional group. This again suggests that at adequate hydration levels, the QA can become completely stable at room temperature.25,29 The half-lives of PPO–TMA with λ = 0 and 4 are lower than the values measured previously for TMBA by approximately an order of magnitude at the same OH− concentration. This surprising acceleration in the decomposition rate can be rationalized considering the polar environment of the hydroxide. In both cases, both reactants are ionic in nature. In the small molecule QA study, both ions interacted with the DMSO environment, a solvent with a high dielectric constant.28 In the macromolecule, the polymer chain takes up a large hydrodynamic volume around the QAs, and therefore, when the OH− ionically interacts with the QA, its polar environment is reduced compared to the DMSO. Given that the reactants are charged and the product neutral, the reduction in polarity accelerates the reaction, hastening the degradation processes.54
Finally, we also tested PPO–TEA in order to further support these observations. PPO–TEA contains QAs which can be compared with TEBA, which has been tested by the same protocol in previous studies.28,29 Although both PPO–TEA and PPO–TMA present the same PPO backbone, their QAs degrade differently: PPO–TMA undergoes an SN2 hydroxide attack whereas PPO–TEA decomposes by an E2 (Hofmann elimination) mechanism (Scheme 3). In the SN2 mechanism, the carbon–nitrogen bond electrons move towards the nitrogen while the OH− forms a new bond with the α carbon, producing benzyl alcohol and trimethylamine. Meanwhile, in the Hofmann degradation mechanism, the OH− acts as a base and abstracts the β-hydrogen to the nitrogen. Fig. 2 summarizes the degradation of PPO–TEA measured in 0.06 M OH− at room temperature using different λ values.
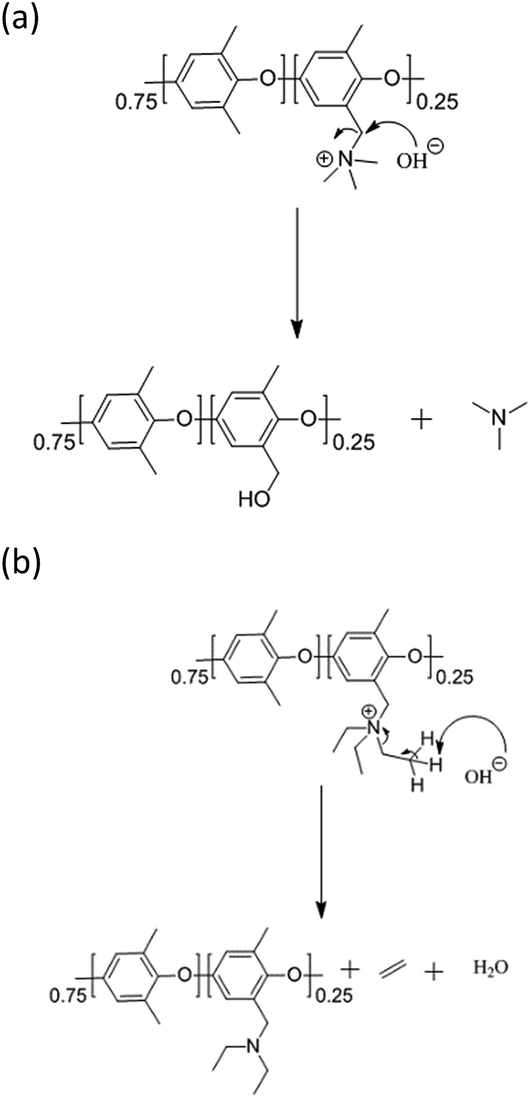 | ||
| Scheme 3 (a) PPO–TMA degradation through the SN2 mechanism, and (b) PPO–TEA degradation through the E2 (Hofmann elimination) mechanism. | ||
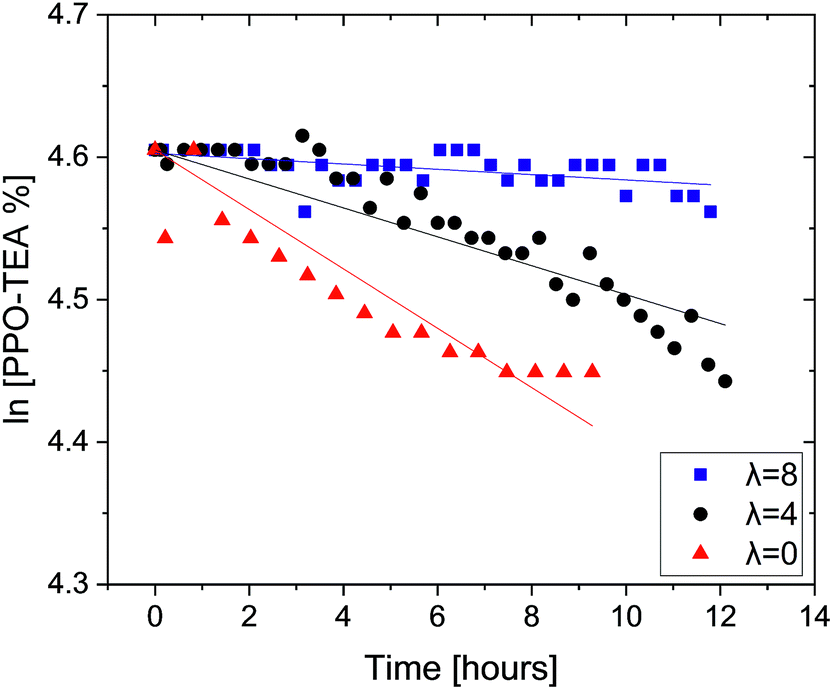 | ||
| Fig. 2 Remaining PPO–TEA fractions as a function of time with hydration number λ = 0, 4 and 8, in 0.06 M OH− DMSO-d6 solutions. All tests were done at room temperature. | ||
As expected, the decomposition rate of PPO–TEA increases as the water content is reduced, following the trend observed for PPO–TMA. In addition, PPO–TEA clearly decomposes much faster than PPO–TMA, by ca. an order of magnitude, following the same trend previously observed in TMBA and TEBA salts.28,29 In order to compare the decomposition rate of PPO–TEA with that of TEBA, a factor of 22.7 was used to correct for QA and OH− concentrations, assuming first order in each of them. Table 3 summarizes the degradation rates and the calculated half-lives, including the results for TEBA from previous studies for comparison.28,29
| λ | 11% PPO–TEA in 0.06 M OH− | 25% PPO–TEA in 0.6 M OH− | TEBA in 0.6 M OH− | |||
|---|---|---|---|---|---|---|
| k [h−1] | Half-life [h] | Estimated k [h−1] | Estimated half-life [h] | k [h−1] | Half-life [h] | |
| 0 | 0.0209 | 33.2 | 0.47 | 1.46 | >3.35 | <0.2 |
| 4 | 0.0102 | 68.3 | 0.23 | 3.01 | 0.0872 | 8 |
| 8 | 0.0019 | 364.8 | 0.04 | 16.21 | 0.0024 | 292 |
Looking initially at λ = 0, an estimated 50% decomposition is reached at ca. 1.9 h if PPO–TEA is at the same concentration as in our previous studies with TEBA. Meanwhile, in the small molecule, the first NMR measurement already showed full decomposition. Nevertheless, looking at λ = 4 and 8, the macromolecular PPO–TEA again decomposes faster compared to TEBA. One can conclude that, as for PPO–TMA, the ionomer also decomposes faster due to the same polar environment. Yet, for λ = 0, it seems that an initial inhibition effect is observed, perhaps due to the time required for anion exchange to occur before the elimination reaction can take place.
The results presented here indicate another factor affecting the kinetics of QA decomposition by hydroxide. The results support the view that even in a less polar polymer environment, water has an inhibiting effect, playing a role in the stability of the QAs.25 Given that these experiments were carried out at room temperature, and optimal AEMFCs operate at higher temperatures, we also tested these membranes at 80 °C. As expected, PPO–TEA decomposed completely with λ = 0 and even with λ = 16 after 24 h. PPO–TMA was tested for longer periods, and, after 1 week, had decomposed by 96% at λ = 0, and by almost 80% at λ = 16, indicating that these polymers are clearly not yet optimal for AEMFC operation at such high temperatures (see the ESI†).
The results of this work indicate that not only are there clear differences between our dry-hydroxide protocol and previous aqueous studies, but also that, while the trends between small molecules and ionomers seem to be the same, the polymer backbone, in addition to the hydration level and temperature, can have a significant effect on the kinetics of the QA decomposition reaction by changing the polar environment in which these chemical reactions occur. Therefore, it is highly likely that the polymer backbone choice, in addition to determining the ionic conductivity and mechanical properties, also directly affects the lifetime of the AEM.
4. Conclusions
To conclude, we have shown in this work the critical effect of hydration levels on the chemical stability of anion exchange ionomers for use as AEMs in AEMFCs. Using a recently developed ex situ technique that simulates the true alkaline and dry aggressive environment of an AEMFC during operation, the ionomers were found to degrade rapidly when the hydration levels were very low (λ ≤ 4). These results are consistent with in situ AEMFC longevity tests, where performance stability, up to now, has been shown to be very low, even when ‘stable’ QAs are used. This happens because, under the measurement conditions, the first solvation shell of OH− is not completely hydrated. However, when five or more water molecules per hydroxide ion are available for solvation, the OH− is less nucleophilic.28 We observed similar degradation trends when comparing different QAs in ionomers as for QA molecules. The measured half-lives of the ionomers studied were shown to be even shorter compared to those measured for free QA molecules, probably due to the decreased polar environment during the nucleophilic/basic attack by the charged OH− ions on the charged QAs. This indicates that the polymer backbone, in addition to affecting the conductivity and mechanical properties, also provides a polar environment for the chemical degradation reaction, affecting the lifetime of the membrane. More importantly, the results show that above a critical hydration level, the ionomers tested here can be quite stable. We strongly believe that the results of this study will help develop the chemistry necessary for the vital breakthrough in the development of anion conducting materials for long stable operation of AEMFCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded by the Nancy & Stephan Grand Technion Energy Program (GTEP); by the European Union's Horizon 2020 research and innovation program [grant No. 721065]; by the Ministry of Science, Technology & Space of Israel through the M-Era.NET Transnational Call 2015, NEXTGAME project [grant No. 3-12940], through the Israel-Germany Batteries Collaboration Call 2017 [German grant No. 2675], and through grant No. 3-12948; by the Israel Science Foundation (ISF) [grant No. 1481/17]; by the 2nd Israel National Research Center for Electrochemical Propulsion (INREP2-ISF); by the Russell-Berrie Nanotechnology Institute, Technion; by the Israel Innovation Authority through the KAMIN program [grant No. 60503]; and by the Ministry of National Infrastructure, Energy and Water Resources of Israel [grant No. 3-13671]. This research was also partially carried out within the framework of the UConn-Technion Energy Collaboration initiative, supported by the Satell Family Foundation, the Maurice G. Gamze Endowed Fund (at the American Technion Society), Larry Pitt and Phillis Meloff, and The Eileen and Jerry Lieberman UConn/Israel Global Partnership Fund.References
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7(10), 3135–3191 RSC.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef.
- T. J. Omasta, X. Peng, H. A. Miller, F. Vizza, L. Wang, J. R. Varcoe, D. R. Dekel and W. E. Mustain, J. Electrochem. Soc., 2018, 165(15), J3039–J3044 CrossRef.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377(1–2), 1–35 CrossRef.
- O. Z. Sharaf and M. F. Orhan, Renewable Sustainable Energy Rev., 2014, 32, 810–853 CrossRef.
- H. A. Miller, A. Lavacchi, F. Vizza, M. Marelli, F. Di Benedetto, F. D'Acapito, Y. Paska, M. Page and D. R. Dekel, Angew. Chem., Int. Ed., 2016, 55(20), 6004–6007 CrossRef PubMed.
- H. A. Miller, F. Vizza, M. Marelli, A. Zadick, L. Dubau, M. Chatenet, S. Geiger, S. Cherevko, H. Doan, R. K. Pavlicek, S. Mukerjee and D. R. Dekel, Nano Energy, 2017, 33, 293–305 CrossRef.
- M. Alesker, M. Page, M. Shviro, Y. Paska, G. Gershinsky, D. R. Dekel and D. Zitoun, J. Power Sources, 2016, 304, 332–339 CrossRef.
- X. Peng, T. J. Omasta, J. M. Roller and W. E. Mustain, Front. Energy, 2017, 11(3), 299–309 CrossRef.
- E. S. Davydova, S. Mukerjee, F. Jaouen and D. R. Dekel, ACS Catal., 2018, 8(7), 6665–6690 CrossRef.
- C. Lafforgue, M. Chatenet, L. Dubau and D. R. Dekel, ACS Catal., 2018, 8(2), 1278–1286 CrossRef.
- A. K. Mohanty, S. Devaraju, N. Kim and H. Paik, Solid State Ionics, 2018, 314, 46–56 CrossRef.
- S. Maurya, J. H. Dumont, C. N. Villarrubia, I. Matanovic, D. Li, S. Y. Kim, S. Noh, J. Han, C. Bae, H. A. Miller, C. H. Fujimoto and D. R. Dekel, ACS Catal., 2018, 8, 9429–9439 CrossRef.
- U. Krewer, C. Weinzierl, N. Ziv and D. R. Dekel, Electrochim. Acta, 2018, 263, 433–446 CrossRef.
- N. Ziv, W. E. Mustain and D. R. Dekel, ChemSusChem, 2018, 11(7), 1136–1150 CrossRef PubMed.
- N. Ziv and D. R. Dekel, Electrochem. Commun., 2018, 88, 109–113 CrossRef.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef.
- J. A. Wrubel, A. A. Peracchio, B. N. Cassenti, T. D. Myles, K. N. Grew and W. K. S. Chiu, ECS Trans., 2017, 80(8), 989–1003 CrossRef.
- J. Peng, A. L. Roy, S. G. Greenbaum and T. A. Zawodzinski, J. Power Sources, 2018, 380, 64–75 CrossRef.
- K. M. Hugar, H. A. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137(27), 8730–8737 CrossRef PubMed.
- M. Hirsch, S. Dhara and C. E. Diesendruck, Org. Lett., 2016, 18(5), 980–983 CrossRef PubMed.
- S. Aharonovich, N. Gjineci, D. R. Dekel and C. E. Diesendruck, Synlett, 2018, 29(10), 1314–1318 CrossRef.
- B. Zhang, R. B. Kaspar, S. Gu, J. Wang, Z. Zhuang and Y. Yan, ChemSusChem, 2016, 9(17), 2374–2379 CrossRef PubMed.
- A. G. Wright and S. Holdcroft, ACS Macro Lett., 2014, 3(5), 444–447 CrossRef.
- C. E. Diesendruck and D. R. Dekel, Curr. Opin. Electrochem., 2018, 9, 173–178 CrossRef.
- D. Marx, A. Chandra and M. E. Tuckerman, Chem. Rev., 2010, 110(4), 2174–2216 CrossRef PubMed.
- S. Pusara, S. Srebnik and D. R. Dekel, J. Phys. Chem. C, 2018, 122(21), 11204–11213 CrossRef.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29(10), 4425–4431 CrossRef.
- D. R. Dekel, S. Willdorf, U. Ash, M. Amar, S. Pusara, S. Dhara, S. Srebnik and C. E. Diesendruck, J. Power Sources, 2018, 375, 351–360 CrossRef.
- D. R. Dekel, I. G. Rasin, M. Page and S. Brandon, J. Power Sources, 2018, 375, 191–204 CrossRef.
- K. M. Meek and Y. A. Elabd, Macromolecules, 2015, 48(19), 7071–7084 CrossRef.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2(41), 17314–17320 RSC.
- J. Ponce-González, D. K. Whelligan, L. Wang, R. Bance-Soualhi, Y. Wang, Y. Peng, H. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9(12), 3724–3735 RSC.
- L. Wang, J. J. Brink, Y. Liu, A. M. Herring, J. Ponce-González, D. K. Whelligan and J. R. Varcoe, Energy Environ. Sci., 2017, 10(10), 2154–2167 RSC.
- J. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6(10), 1089–1093 CrossRef.
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, Polym. Chem., 2011, 2(8), 1641 RSC.
- W. Lu, Z. G. Shao, G. Zhang, Y. Zhao and B. Yi, J. Power Sources, 2014, 248, 905–914 CrossRef.
- T. H. Tsai, A. M. Maes, M. A. Vandiver, C. Versek, S. Seifert, M. Tuominen, M. W. Liberatore, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51(24), 1751–1760 CrossRef.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(7), 2490–2495 CrossRef PubMed.
- P. F. Msomi, P. Nonjola, P. G. Ndungu and J. Ramonjta, J. Appl. Polym. Sci., 2018, 135(10), 45959 CrossRef.
- F. X. Zhang, H. M. Zhang and C. Qu, J. Mater. Chem., 2011, 21(34), 12744–12752 RSC.
- N. Li, Q. Zhang, C. Wang, Y. M. Lee and M. D. Guiver, Macromolecules, 2012, 45(5), 2411–2419 CrossRef.
- L. Wu, T. Xu and W. Yang, J. Membr. Sci., 2006, 286(1–2), 185–192 CrossRef.
- H.-S. Dang and P. Jannasch, Macromolecules, 2015, 48(16), 5742–5751 CrossRef.
- Y. Zhu, Y. He, X. Ge, X. Liang, M. A. Shehzad, M. Hu, Y. Liu, L. Wu and T. Xu, J. Mater. Chem. A, 2017, 6, 527–534 RSC.
- L. Liu, X. Chu, J. Liao, Y. Huang, Y. Li, Z. Ge, M. A. Hickner and N. Li, Energy Environ. Sci., 2018, 11(2), 435–446 RSC.
- N. Li, M. D. Guiver and W. H. Binder, ChemSusChem, 2013, 6(8), 1376–1383 CrossRef PubMed.
- C. G. Arges, L. Wang, M.-S. Jung and V. Ramani, J. Electrochem. Soc., 2015, 162(7), F686–F693 CrossRef.
- N. Li, Y. Leng, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2013, 135(27), 10124–10133 CrossRef PubMed.
- C. W. Chen, I. H. Lin, C. C. Lin, J. L. Lin and M. Horie, Polymer, 2013, 54(21), 5684–5690 CrossRef.
- D. M. White and C. M. Orlando, ACS Symp. Ser., 1975, 6, 178–184 CrossRef.
- K. H. Gopi, S. G. Peera, S. D. Bhat, P. Sridhar and S. Pitchumani, Int. J. Hydrogen Energy, 2014, 39(6), 2659–2668 CrossRef.
- S. Arava and C. E. Diesendruck, Synthesis, 2017, 49, 3535–3545 CrossRef.
- A. Parker, Jpn. Chem. Rev., 1969, 69(1), 1–32 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of polymers and numerical data. See DOI: 10.1039/c8ta05785k |
| ‡ Sapir Willdorf-Cohen and Abhishek N. Mondal contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |