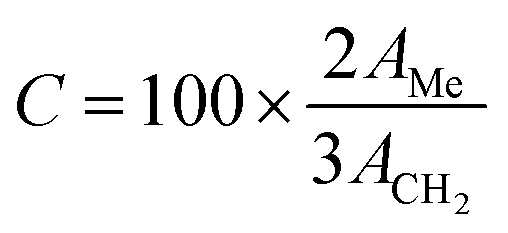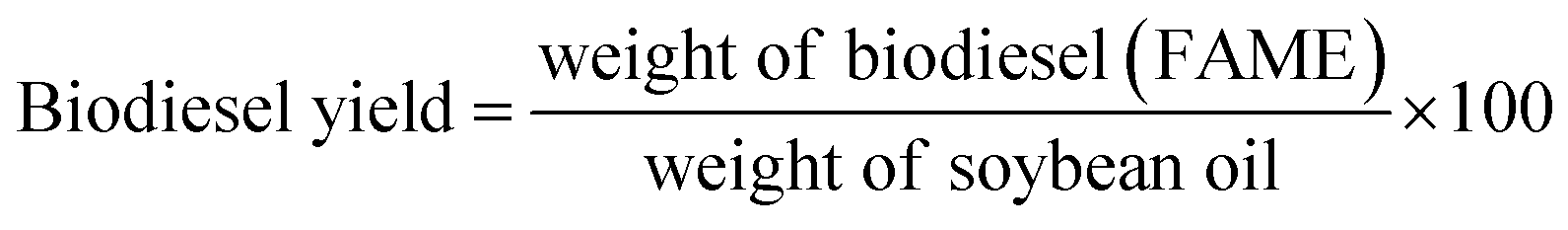DOI:
10.1039/C8RA02397B
(Paper)
RSC Adv., 2018,
8, 20131-20142
Waste snail shell derived heterogeneous catalyst for biodiesel production by the transesterification of soybean oil†
Received
19th March 2018
, Accepted 1st May 2018
First published on 4th June 2018
Abstract
A waste snail shell (Pila spp.) derived catalyst was used to produce biodiesel from soybean oil at room temperature for the first time. The snail shell was calcined at different temperatures of 400–1000 °C. The synthesized catalysts underwent XRD, SEM, TEM, EDS, FTIR, XRF, TG/DTA and N2 adsorption–desorption isotherm (BET) analysis. The major component CaO was determined at a calcination temperature of 900 °C as depicted in the XRD results. 100% conversion of soybean oil to methyl ester biodiesel was obtained, as confirmed by 1H NMR. A biodiesel yield of 98% was achieved under optimized reaction conditions such as a calcination temperature of 900 °C, a catalyst loading of 3 wt%, a reaction time of 7 h and a methanol to oil ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and biodiesel conversion was confirmed by FT-NMR and IR spectroscopies. A total of 9 fatty acid methyl esters (FAMEs) were identified in the synthesized biodiesel by the retention time and fragmentation pattern data of GC-MS analysis. The catalyst was recycled 8 times without appreciable loss in its catalytic activity. A high biodiesel yield of 98% was obtained under these optimised conditions. The catalyst has the advantage of being a waste material, therefore it is easily prepared, cost free, highly efficient, biogenic, labor effective and environmentally friendly, making it a potential candidate as a green catalyst for low cost production of biodiesel at an industrial scale.
:1, and biodiesel conversion was confirmed by FT-NMR and IR spectroscopies. A total of 9 fatty acid methyl esters (FAMEs) were identified in the synthesized biodiesel by the retention time and fragmentation pattern data of GC-MS analysis. The catalyst was recycled 8 times without appreciable loss in its catalytic activity. A high biodiesel yield of 98% was obtained under these optimised conditions. The catalyst has the advantage of being a waste material, therefore it is easily prepared, cost free, highly efficient, biogenic, labor effective and environmentally friendly, making it a potential candidate as a green catalyst for low cost production of biodiesel at an industrial scale.
1. Introduction
Currently, biomass gains remarkable attention for being a source of both renewable energy and platform chemicals. Biodiesel, which mainly consists of fatty acid methyl esters, has many advantages for the environment compared with fossil fuels such as low sulfur content, low toxicity and low emissions of CO2 and CO, as well as being biodegradable and renewable in nature.1–3 Nowadays, biodiesel is produced using homogeneous catalysts such as potassium and sodium hydroxide because of their high accessibility and manageable cost. Reaction kinetics and the mass transfer of triglycerides in the transesterification reaction have been studied to optimise the conditions of the process in order to decrease the total biodiesel cost.4–6 Nevertheless, this process still has terrible limitations that lead to an increase in the total cost of biodiesel production. By using a heterogeneous catalyst in the transesterification process these problems can be reduced. The main advantages of using a heterogeneous catalyst are that it is environmentally friendly, non-corrosive and easy to separate from liquid products and it has fewer disposal problems and higher selectivity and activity.7,8
Heterogeneous catalysts are of various types such as acids, bases, and enzymes. Usually a base catalyst is preferred in the transesterification reaction as acid and enzyme catalysts require high operating conditions (high temperature, high methanol to oil ratio and long reaction time) for the production of biodiesel. However, each type of catalyst has their own specific characteristics that could be exploited in the transesterification reaction. For this reason, to date, substantial developments have been observed in supported catalysts: Zn@CaO,9 CaO@SnO2,10 CaO@La2O3,11 hydroxyapatite-encapsulated γ-Fe2O3 nanoparticles (HAp-γ-Fe2O3),12 Fe3O4@HKUST-1,13 Fe3O4@silica,14 WO3@ZrO2,15 lipase/GO–Fe3O4,16 and Cu/ZnO.17 However, the preparation of such catalysts involves several steps that make the processes longer and complex. High chemical costs further increase the total cost of preparation. Therefore, to reduce the total biodiesel preparation cost, waste material usage as a catalyst is a wise choice. Several waste materials such as egg, oyster, crab and snail shells, chicken manure and animal bone have been used as a source of CaO catalyst for the production of biodiesel from various vegetable oils.18–21 CaO is one of the most promising catalysts among the heterogeneous catalysts, because of its high catalytic activity in the transesterification reaction, ease in production process, low solubility in methanol and biodiesel and low cost.22–25
Use of waste materials as a catalyst source not only reduces the total cost of biodiesel production but is also associated with the capability of recycling the natural mineral sources, resulting in making the process green and environmentally friendly. Maneerung et al.26 used chicken manure to extract CaO and convert it to an active catalyst at a calcination temperature of 850 °C. Rezaei et al.27 prepared CaO from mussel shells to produce biodiesel from soybean oil. A biodiesel yield of 94% was obtained under optimized conditions such as a methanol to oil ratio of 24:1, temperature of 60 °C, time of 8 h, catalyst loading of 12 wt% and at a calcination temperature of 1050 °C. Viriya-empikul et al.28 synthesized CaO from the industrial waste shells of Meretrix venus clams, eggs and golden apple snails and compared the catalytic activities during the transesterification reaction to produce biodiesel from palm olein oil. The results revealed that CaO derived at a calcination temperature of 800 °C from snail and egg shells provides a high biodiesel yield of 94%. Joshi et al.29 derived CaO from chicken egg shells and impregnated the catalyst with various metal oxides such as ZnO, MnO2 and Fe2O3 to investigate the catalytic activity in the transesterification of jatropha oil and karanja oil. Waste cooking oil was used to produce biodiesel using chicken manure by Maneerung et al. and a FAME yield of 90% was obtained with the optimum reaction parameters of catalyst loading at 7.5 wt%, methanol:oil ratio of 15:1, temperature of 65 °C and time of 7 h.26 The performance of mollusk and egg shell derived CaO catalysts used in transesterification reactions has been studied by many researchers. In most of the studies a high biodiesel yield of more than 94% is reported using vegetable oils at reaction temperatures of 60–65 °C after 3 h with a methanol:oil molar ratio of 6:1 to 12:1.22,30 In order to attain a reaction temperature of 60–65 °C during the transesterification reaction, heating is often required and most of the suggested heating methods from the studies are an oil/water bath, ultrasound radiation or a microwave which all need an electricity supply that increases the total cost of the biodiesel. Interestingly, Reddy et al.2 reported that a biodiesel yield of 99% can be successfully produced from soybean oil and poultry fat at room temperature using a CaO catalyst of size 20 nm and with a 90 m2 g−1 surface area, while the yield was 2% with the commercial CaO catalyst having a low surface area of 1 m2 g−1. However, the study also reported that an inert atmosphere should be maintained for catalyst storage and whilst performing the transesterification reaction.2 This method may have the potential to attract the biodiesel production industry. However, it has the following difficulties to overcome: complex production methods and the use of a highly priced catalyst. Therefore, it is absolutely essential to search for alternative sources of more active yet stable CaO catalysts which can perform the transesterification reaction at ambient conditions.
Snails belong to the phylum Mollusca, and have the second largest number of species inhabiting the earth after arthropods with more than one hundred thousand described species.31–35 Freshwater snail shells are available in abundant amounts worldwide and belong to the classes Gastropoda and Bivalvia.31 Freshwater snails are edible and play vital roles in ecosystems since they serve as food for crabs, fish, birds, humans etc. and their productivity is associated with the various food chains.36 With changes in environmental conditions and geographical location, the shell morphometry and body size of snails within a population changes.37–39 This prompted researchers to investigate the characteristics of snail shells. In Eastern and Northeast India, people use snails as a food source and an abundant amount of shells are going to waste.40 Therefore, present studies aim to make use of these waste shells for the production of biodiesel. Freshwater snail shells (Pila species; Family: Ampullariidae (Pilidae) Gray, 1847) are widely available in Northeast India.40 Hence, the use of waste shells may help the development on an industrial level in this region.
Recently, our research group has reported the application of M. acuminata peel ash as a solid catalyst in the production of biodiesel.41 The present study aims at using a green, economical and waste derived CaO catalyst prepared from Pila spp. snail shells as a cheap, easily available, non-toxic and biodegradable solid catalyst for the production of biodiesel at room temperature from soybean oil. To the best of our knowledge there is no report found where the shells of edible freshwater snails (Pila spp.), locally called “chengkawl bial” in Mizoram (Northeast India), were used as a heterogeneous catalyst for transesterification of soybean oil to biodiesel. We also report here a comprehensive study of the snail shells (Pila spp.) following characterisation by powder X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), EDS, FTIR, XRF, TG/DTA and N2 adsorption–desorption isotherm (BET) analysis.
2. Experimental methodology
2.1 Materials
Soybean oil was purchased from the local market in Silchar, Assam, India and its physical properties and compositions are listed in Table 1 of the ESI.† Waste snail shells (Pila spp.) were procured from Mizoram, India. Methanol of analytical reagent (AR) grade was acquired from Merck, India. Double distilled water was used throughout the experiments. Bromothymol blue (pKa = 7.2), phenolphthalein (pKa = 9.8), indigo carmine (pKa = 12.2), 2,4-dinitroaniline (pKa = 15.0) and 4-nitroaniline (pKa = 18.4) were purchased from Sigma Aldrich.
2.2 Catalyst preparation
The collected snail shells were washed several times with distilled water to remove unwanted materials and were dried in an oven at 100 °C for 12 h. The snail shells were then ground to a fine powder in a mortar and pestle (outer diameter of 4 inch and 3 inch high), sieved (mesh sizes of 125–250 μm) and calcined in a muffle furnace for 4 h at various temperatures in the range of 400–1000 °C. After calcination, the catalyst was stored in a desiccator to avoid contact with air.
2.3 Characterization of the catalysts
XRD, TEM, SEM, EDS, FTIR, XRF and BET analyses were performed to evaluate the structure and morphology of the uncalcined and calcined snail shells. XRD measurements were carried out on a Bruker AXS D8-Advance powder X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å) with a scan speed of 2° min−1. Scanning electron microscopy coupled with EDS (model no. JEOL JSM-7600F) was used to determine the size, shape and elemental composition of the catalyst. SEM images were obtained on a FEI Quanta 200 F using a tungsten filament doped with lanthanum hexaboride (LaB6) as an X-ray source, fitted with an ETD detector with a high vacuum mode using secondary electrons and an acceleration tension of 30 kV. Samples were analyzed by spreading them on carbon tape. Transmission electron microscopy images were obtained on a JEOL, JEM2100 microscope. N2 adsorption–desorption isotherms were obtained with a Micromeritics ASAP 2010 surface area and porosity analyzer. Functional groups were identified in the samples using FTIR analysis and IR spectra were recorded in the range 400–4000 cm−1 with a 3000 Hyperion FTIR spectrometer (Bruker, Germany). Catalyst constituents were determined using an S4 Pioneer Energy Dispersive X-ray fluorescence spectrometer. Thermal transitions of the catalyst were determined using TG/DTA (model no. STA 409 Netzsch Geratebau GMBH, Germany) under a nitrogen flow at a pressure of 1.5 bar and a flow rate of 2 L h−1.
In order to determine the H range of basic sites for each catalyst, Hammett indicator experiments were performed. The Hammett indicators used were bromothymol blue (pKa = 7.2), phenolphthalein (pKa = 9.8), indigo carmine (pKa = 12.2), 2,4-dinitroaniline (pKa = 15.0) and 4-nitroaniline (pKa = 18.4). Anhydrous ethanol was used during the experiment. Approximately 50 mg of the catalyst was mixed with 10 mL of an anhydrous ethanol solution of the Hammett indicator, shaken and was then allowed to equilibrate for 2 h. Afterwards, a color change in the solution was observed. When the solution exhibited a color change it signified that the basic strength of the catalyst was higher than the indicator used. However, when the solution showed no color change the basic strength of the catalyst was lower than the indicator used.42
2.4 Transesterification reactions and methods of analysis
Transesterification of soybean oil was carried out by mixing 7 g of soybean oil, 2.02 mL of methanol and 210 mg of catalyst in a 50 mL round bottom flask on a magnetic stirrer. Reaction parameters such as a methanol to oil ratio of 4:1 to 12:1, catalyst (CaO) concentration of 2–3.5 wt% and reaction time of 3 h to 8 h were tested during experimentation. The speed of the magnetic stirrer was kept constant at 800 rpm. All reactions were carried out at ambient temperature (28 °C) and pressure. The progress of the reaction was monitored using thin layer chromatography (TLC). Once the reaction was complete (i.e. all the starting material was converted to products, as indicated by TLC), the obtained biodiesel was analyzed by NMR. After completion of the reaction was confirmed by TLC, the mixture was transferred to a separating funnel and kept for 3 h to separate the lighter phase (biodiesel) from the denser phase (glycerol). Then centrifugation was carried out at 4000 rpm for 7 min to separate out the catalyst and the biodiesel. The collected catalyst was then washed with water, dried in an oven at 120 °C and finally the catalyst was regenerated by calcination at 900 °C. The process of regeneration of the catalyst was repeated for successive cycles. The obtained biodiesel composition was analyzed with GC-MS.
2.5 Fatty acid methyl ester product analysis
Once the transesterification reaction was completed, the biodiesel obtained was analyzed by NMR spectroscopy. 1H NMR spectra were recorded on a Bruker Avance II (400 MHz) spectrometer using tetramethylsilane (TMS) as an internal reference. The conversion percentage of transesterification was determined using the integrated area covered by the signals of the methylene and methoxy protons.43 The relevant signals chosen for integration were those of the methoxy groups in the FAME (singlet, 3.6 ppm) and those of the α-CH2 protons present in all the triglyceride derivatives (triplet, 2.3 ppm) of the soybean oil. Biodiesel conversion was determined by the equation derived by Knothe and Kenar (eqn (1)),44| | 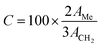 | (1) |
where C indicates the triglyceride to biodiesel (fatty acid methyl ester) conversion percentage (%), AMe denotes the integration value of the methyl esters, and ACH2 is the integration value of the methylene protons. The factor of 2 in the numerator ascribes to the proton number in methylene and the 3 in the denominator ascribes to the proton number in the methyl ester. The biodiesel yield was calculated by the equation given by Leung and Guo (eqn (2)).45| | 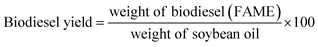 | (2) |
Biodiesel formation was also confirmed by FTIR analysis using a Bruker, Germany 3000 Hyperion Microscope with Vertex 80. The obtained product (biodiesel) was then analysed with GC-MS and the spectra were recorded on an Agilent 7890-GC, Jeol AccuTOF GCV-MS (GC-MS conditions for fatty acid analysis are given in the ESI, S7†). The oil conversion and composition of FAME contained in the final product were further analyzed by GC-MS. Compounds were identified by comparing the retention time and mass spectra with a library of data of the mass spectra of compounds. Methyl hexanoate was used as an analytical standard and FAME composition was quantified by the internal standard method. The calibration curve was obtained using samples with a fixed mass of an internal standard and various masses of biodiesel dissolved in a constant volume.
3. Results and discussion
3.1 Catalyst characterization
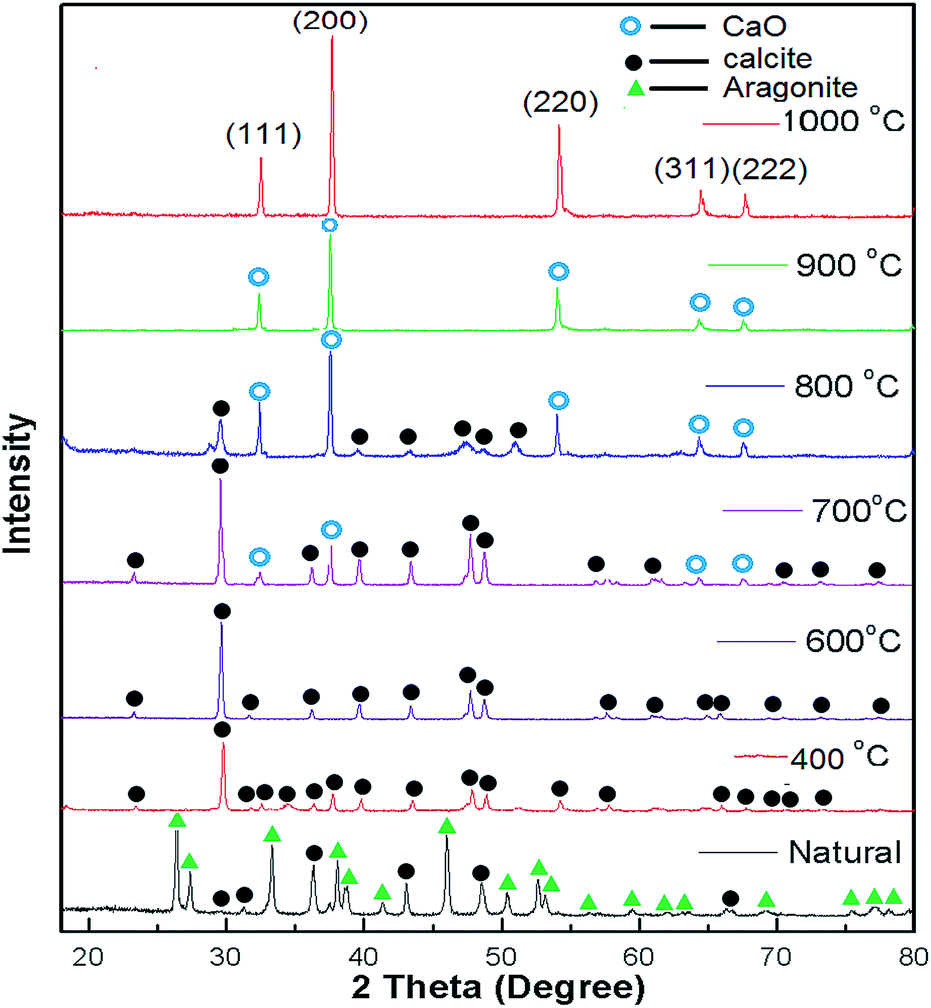
XRD patterns of the uncalcined (natural) snail shells and calcined snail shells (CSS) were recorded and analyzed. From Fig. 1, it can be observed that CaCO3 is the major component of the uncalcined snail shells and is in two crystalline phases, aragonite and calcite, confirmed from the reported data of CaCO3 (JCPDS file no. 41-1475 and 29-0306).46 CaCO3 (aragonite and calcite) was converted into calcite at a calcination temperature of 400 °C and retained in that phase up to 600 °C. A CaO minor phase at 700 °C and a major phase at 800 °C were observed.46 Calcium carbonate finally completely decomposed to CaO at a calcination temperature of 900 °C. It was concluded from the graph that at a calcination temperature of more than 800 °C, all the CaCO3 was converted to CaO. This was also confirmed by TGA analysis as shown in Fig. 2. Intensity peaks of CaO, determined at temperatures of 900 °C and 1000 °C, at 2 theta values of 32.37, 37.55, 54.03, 64.37 and 67.65 correspond to the (111), (200), (220), (311) and (222) planes, respectively, which match well with the reported data of CaO (JCPDS file no. 48-1467), confirming the formation of CaO. The intense and sharp peak of the calcined snail shells revealed the crystalline nature of the catalyst. The results are well matched with the paper reported by Hu et al.46
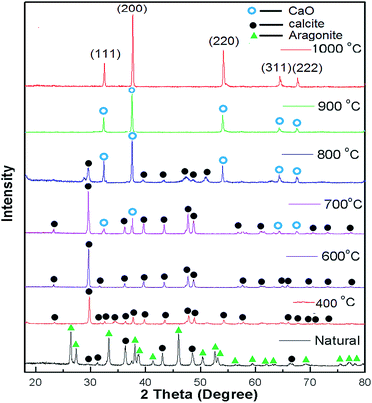 |
| | Fig. 1 XRD patterns of natural snail shells and calcined snail shells of different temperatures ranging between 400 to 1000 °C for 4 h. | |
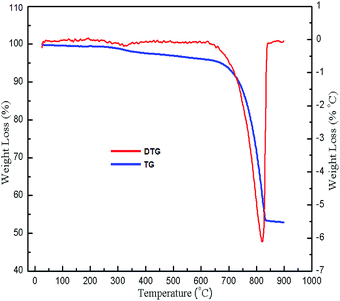 |
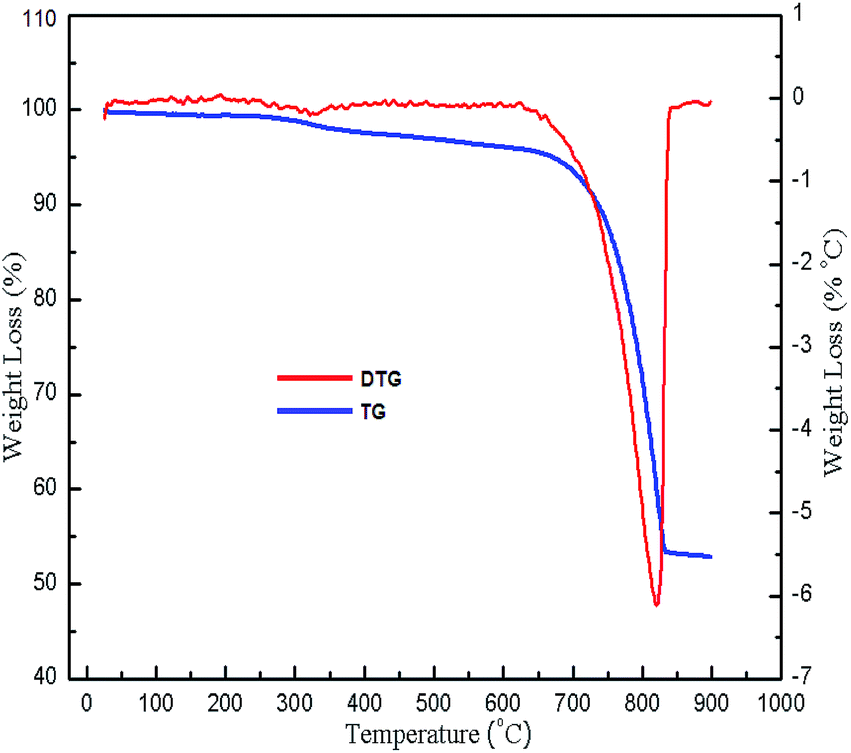
| | Fig. 2 TG/DTG of snail shells. | |
The elemental compositions of the uncalcined snail shells and the CSS catalyst (after calcination at 900 °C) were determined using XRF. Calcium was found to be the major component in both the snail shells and CSS catalyst. In addition, trace elements (in ppm range) such as V, Cr, Ni, Cu, Ga, Zn, Rb, Sr, Y, Mo, Ba, Ce, Zr and Pb were also observed in both the samples. The composition of the snail shells and catalysts is shown in Tables 1 and 2, respectively.
Table 1 Composition of snail shells
| Sl. no. |
Compound formula |
Weight (%) |
| 1 |
CaCO3 |
97.14 |
| 2 |
SiO2 |
0.628 |
| 3 |
SrO |
0.457 |
| 4 |
Fe2O3 |
0.409 |
| 5 |
Na2O |
0.336 |
| 6 |
MnO |
0.308 |
| 7 |
K2O |
0.212 |
| 8 |
Al2O3 |
0.180 |
| 9 |
MgO |
0.152 |
| 10 |
Cr2O3 |
0.083 |
| 11 |
SO3 |
0.073 |
| 12 |
CuO |
0.015 |
| 13 |
TiO2 |
0.005 |
| 14 |
P2O5 |
0.004 |
Table 2 Composition of calcined snail shells calcined at 900 °C for 4 h
| Sl. no. |
Compound formula |
Weight (%) |
| 1 |
CaO |
98.017 |
| 2 |
SiO2 |
0.467 |
| 3 |
Fe2O3 |
0.357 |
| 4 |
MnO |
0.2431 |
| 5 |
MgO |
0.182 |
| 6 |
Na2O |
0.170 |
| 7 |
SrO |
0.1603 |
| 8 |
Al2O3 |
0.1301 |
| 9 |
SO3 |
0.0605 |
| 10 |
P2O5 |
0.054 |
| 11 |
K2O |
0.074 |
| 12 |
Cr2O3 |
0.035 |
| 13 |
CuO |
0.011 |
| 14 |
TiO2 |
0.004 |
TG/DSC analysis was carried out with the aim to determine the temperature at which CaCO3 decomposes to CaO. The weight loss of the snail shells was observed in two stages as shown in Fig. 2: in the first stage, 5.026% weight loss occurred between the temperature range 50–672.5 °C due to the evaporation of water and the decomposition of organic material; in the second stage, 43% weight loss occurred between the temperature range 672.5–830 °C due to the decomposition of CaCO3 to CaO with the release of CO2.18,30 The results of the TG analysis were supported by DSC analysis results. The endothermic peak at 823 °C (ESI, Fig. 1†) indicated the decomposition of CaCO3 to CaO.47 There was no weight loss above the temperature of 840 °C. Thus CaCO3 might have decomposed to CaO at a temperature of 840 °C.
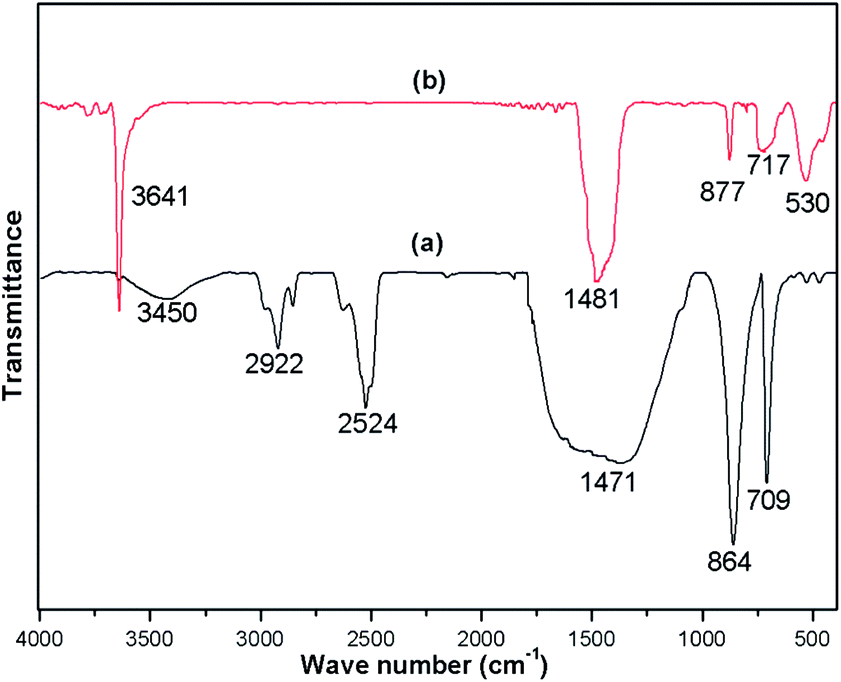
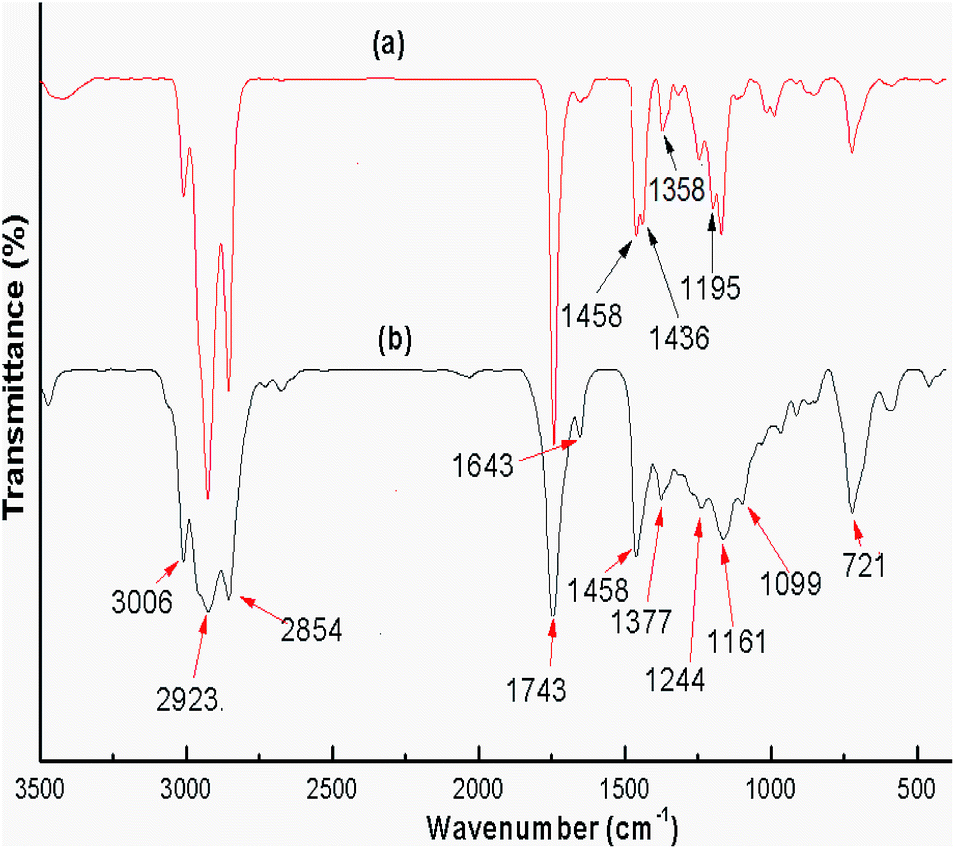
FTIR spectra were recorded for both the uncalcined snail shell powder and for the calcined snail shells at 900 °C to determine the absorption bands of the materials present. The FTIR spectra of the uncalcined snail shells and calcined shells are shown in Fig. 3(a) and (b), respectively. For the uncalcined snail shells (Fig. 3(a)), the major absorption band found at 1471 cm−1 can be ascribed to the asymmetric stretching of CO32− molecules and the other two major absorption bands observed at 864 and 709 cm−1 can be ascribed to the out-of-plane band and the in-plane band modes of vibration for the CO32− molecules.29 These results attributed to the presence of CaCO3 in the snail shells. The calcined snail shells lost the carbonate ion resulting in a shift in the absorption bands corresponding to CO32− to high energy (i.e. 1481, 877 and 530 cm−1), as shown in Fig. 3(b). This is due to a decrement in the reduced mass of the functional group associated with the CO32− ions.29 The absorption bands of the organic matter obtained for the uncalcined shells at 2524, 2858 and 2922 cm−1 completely vanished after calcination of the shells at 900 °C. The presence of a water molecule in the snail shells (uncalcined) was confirmed from the broad peak which appeared at 3450 cm−1, and the appearance of a new peak at 3641 cm−1 in the calcined shell spectrum indicated the formation of Ca(OH)2 from the airing of CaO. The disappearance of moderate to weak signals and the shifting of absorption bands corresponding to CO32− after calcination of the snail shells confirmed the decomposition of CaCO3 to CaO. IR absorption bands in the uncalcined and calcined snail shell spectra agreed well with the reported literature.29
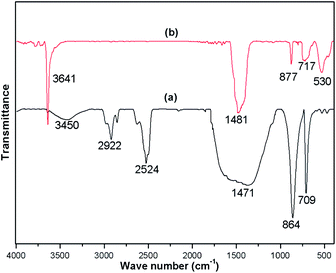 |
| | Fig. 3 FTIR of (a) snail shells (b) snail shells calcined at 900 °C for 4 h. | |
Scanning electron microscopy (SEM) was carried out to study the morphology of the snail shells and the snail shells calcined at 900 °C. Natural snail shells (Fig. 4(a) and (b)) exhibited irregular sizes of rod and spherical particles and also displayed layers of rod-like structures.46 After calcination of the snail shells at 900 °C, irregular sizes of semi-spherical particles18 and, more importantly, porous surfaces were observed from the images in Fig. 4(c).29,46 The porous surface obtained after calcination may be due to the release of water and gaseous CO2 during decomposition of CaCO3 to CaO.46,48 Gaseous water and CO2 act as porogens. The presence of porosity on the surface of the catalyst increases the BET surface area that leads to an increase in the catalytic activity.46
 |
| | Fig. 4 SEM images of (a) and (b) uncalcined and (c) calcined snail shells as well as (d) the EDS spectra of calcined shells at 900 °C for 4 h. | |
EDS analysis of snail shells at various calcination temperatures such as 600, 700, 800, and 900 °C was carried out to determine the elements present in the samples. The major elements Ca, O and C were found in all the samples. Apart from these, other elements such as C, Si, Mg and Al were also detected. These components were also observed in the XRF analysis of the catalyst. The carbon percentage decreases and the calcium percentage increases with an increase in the calcination temperature due to the release of CO2 during decomposition of CaCO3 to CaO, as illustrated from the EDS spectra (ESI, Fig. 2†). From the SEM-EDS image in Fig. 4(d), an abundant amount of calcium is observed and a very small carbon percentage of 4.6% remained after calcination of the snail shells at 900 °C. EDS mapping obtained from calcination temperatures of 600–900 °C are shown in the ESI in Fig. 3–6.†
TEM analysis results for uncalcined snail shells and for snail shells calcined at 900 °C are shown in Fig. 5(a) and (b), respectively. Fig. 5(a) illustrates that the natural snail shell particle shapes were spherical and rod shaped. The agglomerations of the particles after calcination of the shells can be seen in Fig. 5(c) and the final shape remains spherical and rod shaped, which is confirmed in Fig. 5(b).
 |
| | Fig. 5 TEM images of (a) uncalcined and (b) and (c) calcined snail shells. | |
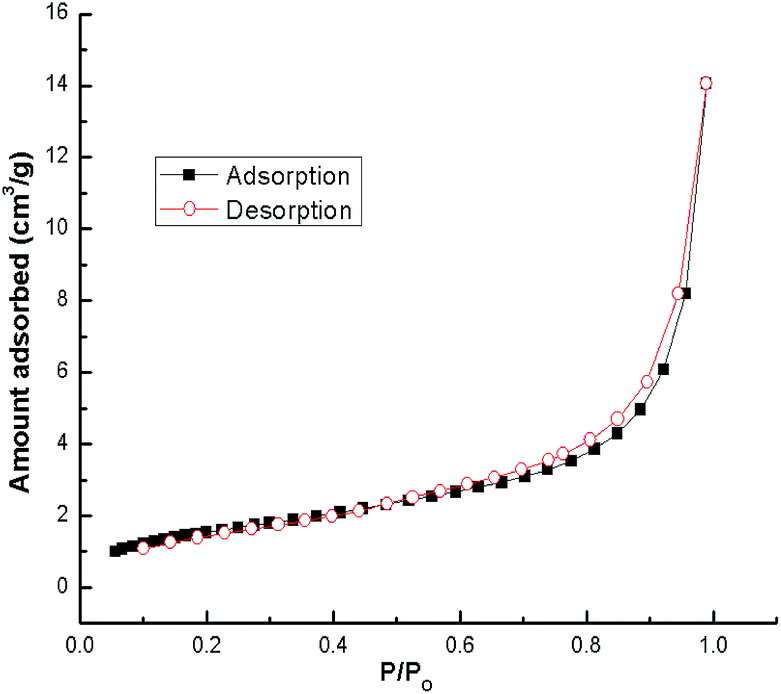
N2 adsorption–desorption isotherms for snail shells calcined at 900 °C are shown in Fig. 6. The results confirmed that the snail shell derived catalyst (CaO) conforms to the type-IV isotherm indicating mesoporous material characteristics. Moreover, a shifting of the hysteresis loop to high P/Po values (>0.8) indicated that the mesopores present in the sample are of larger sizes.49 A similar trend (type-IV isotherm) was found in other catalysts as well (ESI Fig. 7–9†). The BET surface area and pore volume of the samples with different calcination temperatures are shown in Table 3. From the XRD results of natural shells and calcined shells at 400–600 °C, the phases of calcium carbonate were aragonite, calcite and calcite, respectively, which means that CO2 is not released from CaCO3. From the calcination temperature of 700 °C, CO2 starts to be released from CaCO3 and is fully converted to CaO at 900 °C. TGA results also support the XRD results. Thus an increase in surface area correlates with the weight loss steps in the TGA curve.47,59 Therefore, the specific surface area increases with an increase in temperature. Moreover, for the catalyst calcined at 700–800 °C, an increase in the surface area was probably due to a modification of the sample composition during calcination. Above a calcination temperature of 800 °C, an increase in the BET surface area may be because of the crystal growth of calcium oxide. Pore volume increased during calcination of the shells (Table 3) at a higher temperature which can be ascribed to the development of porosity in the calcined shells. The formation of pores in the prepared catalyst was caused by the evolution of gaseous carbonization products (CO2 in our case) and was partly due to the formation of CaO.47,54 A similar result has also been discussed by other researchers.29,46,52,54 The increase in surface area with temperature was also reported by Boro et al.47 Thus, the BET surface area increases with an increase in the calcination temperature as a result of an increase in porosity on the surface of the samples.29,47 The specific surface area decreased to 4.3 m2 g−1 (Table 3) during calcination at 1000 °C due to the sintering effect from prolonged heating at a higher temperature.54 Reddy et al.2 reported a low yield of 2% biodiesel obtained with a commercial CaO catalyst with a surface area of 1 m2 g−1. In addition, a biodiesel yield of 96% was achieved from soybean oil at room temperature using CaO derived from egg shells with a BET surface area of 1.8 m2 g−1, (catalyst loading of 5.8 wt% and for 9 h) by Piker et al.1 However, in the present work, 100% conversion of soybean oil to biodiesel was achieved at room temperature which may be attributed to the higher BET surface area of our catalyst, as the higher surface area of the catalyst increases the interaction due to the availability of higher basic centers which makes the reaction faster, resulting in increased rates of transesterification.1
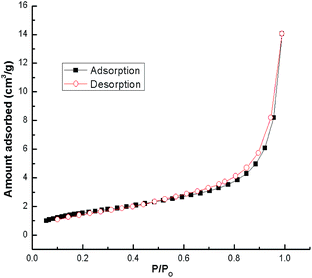 |
| | Fig. 6 N2 adsorption–desorption analysis of snail shells calcined at 900 °C for 4 h. | |
Table 3 BET analysis of calcined and uncalcined shells
| Sl. no. |
Cal. temp. (°C) |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore diameter (Å) |
Basic strength (H−) |
| 1 |
Natural snail shell |
2.7 |
0.00848 |
184 |
H− < 7.2 |
| 2 |
700 |
3.7 |
0.012 |
164 |
7.2 < H− < 9.8 |
| 3 |
800 |
5.3 |
0.0217 |
158 |
9.8 < H− < 12.2 |
| 4 |
900 |
7 |
0.0312 |
148 |
12.2 < H− < 15 |
| 5 |
1000 |
4.3 |
0.017 |
160 |
9.8 < H− < 12.2 |
The shell (calcined at 900 °C) basic strength (H−) obtained was 12.2 < H− < 15 and is considered as a strong base for transesterification reaction. The value is higher than that reported by Kawashima et al.,50 Joshi et al.29 and is similar to that reported by Yoosuk et al.51 The basic strength is directly related to the surface area since an increase in the surface area increases the basic strength of the catalyst, as shown in Table 3. The basic site of the CaO catalyst is generally known to be the active species in heterogeneous transesterification and the number of basic sites should depend on the surface area.28
3.2 Optimisation of several reaction parameters for transesterification methods
A series of transesterification reactions were performed using the snail shell catalyst in order to obtain optimized reaction conditions by varying the different parameters. The reusability of CaO was also studied under optimized reaction conditions. Several studies reported moderate to high biodiesel yields under optimum reaction parameters using waste shell derived catalysts as shown in Table 4. However there is no report where a waste freshwater snail shell derived catalyst was used to produce biodiesel at room temperature (28 °C) from soybean oil.
Table 4 Comparison of snail shell with other reported biomass based heterogeneous catalysts
| Oil |
Catalyst |
Methanol:oil |
Temperature (°C) |
Catalyst amount (wt%) |
Time (h) |
Conversion (%) |
Reference |
| Sunflower |
Oyster shell |
9:1 |
60 |
2 |
4 |
89.2 |
Correia et al.25 |
| Soybean |
Chicken manure |
15:1 |
65 |
7.5 |
3 |
90 |
Maneerung et al.26 |
| Sunflower |
Crab shell |
6:1 |
60 |
3 |
3 |
83.1 |
Correia et al.18 |
| Sunflower |
Egg shell |
9:1 |
60 |
3 |
3 |
97.75 |
Correia et al.18 |
| Palm |
Obtuse horn |
12:1 |
65 |
5 |
6 |
86.75 |
Lee et al.53 |
| Palm |
Hydrated lime |
15:1 |
65 |
5 |
2 |
97 |
Roschat et al.52 |
| Used frying oil |
Exoskeleton of a Mollusk |
10:1 |
60 |
4 |
5 |
92 |
Agrawal et al.58 |
| Palm |
Freshwater bivalve mussel |
10:1 |
60 |
4 |
5 |
90 |
Madhuvilakku et al.48 |
| Waste frying oil |
Snail shells |
8.45:1 |
60 |
2 |
7 |
87.59 |
Birla et al.30 |
| Micro algal |
Angel wing shell (Cyrtopleura costata) |
150:1 |
65 |
9 |
1 |
84.11 |
Syazwani et al.59 |
| Palm |
Waste venus clam |
15:1 |
65 |
5 |
6 |
97 |
Syazwani et al.54 |
| Waste frying oil |
Natural white bivalve clam shell |
18:1 |
65 |
8 |
3 |
95.84 |
Girish et al.57 |
| Soybean |
Egg shell |
9:1 |
65 |
3 |
3 |
95 |
Wei et al.19 |
| Mustard |
Turbonilla striatula shell |
9:1 |
65 |
3 |
6 |
93 |
Boro et al.47 |
| Soybean |
Egg shell |
6:1 |
RT |
5.8 |
11 |
96 |
Piker et al.1 |
| Soybean |
Mussel shell |
24:1 |
60 |
12 |
8 |
94 |
Rezaei et al.27 |
| Soybean |
Snail shells |
6:1 |
RT |
3 |
7 |
100 |
This work |
3.2.1 Effect of calcination temperature.
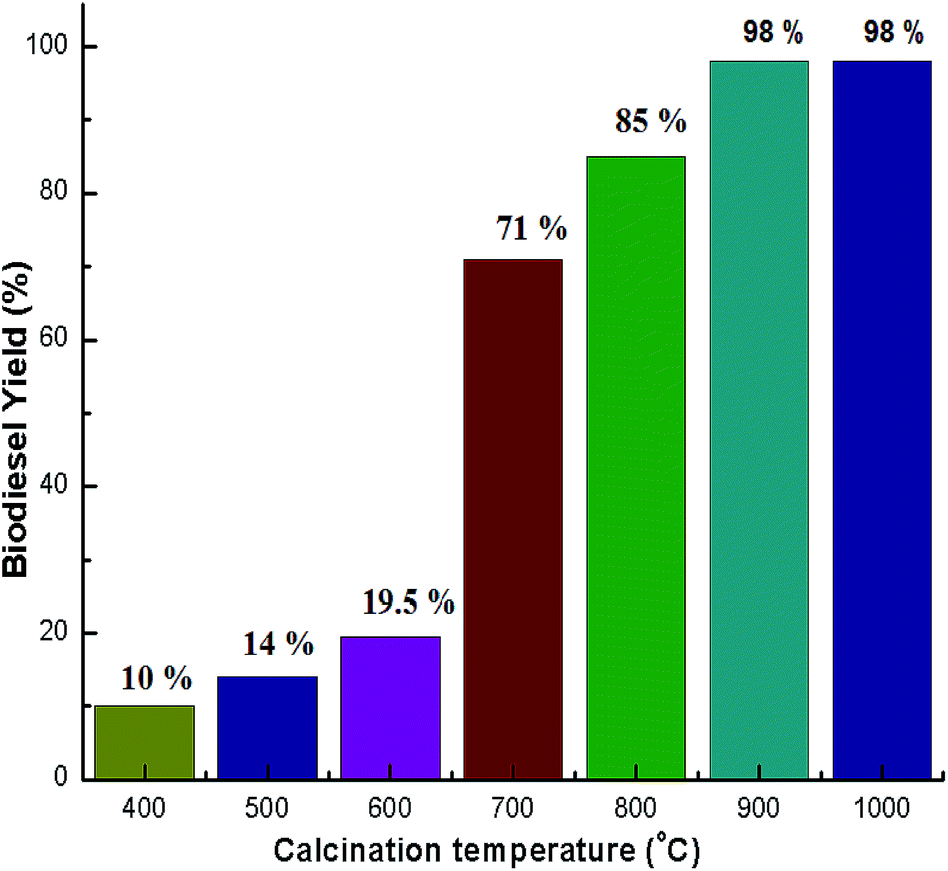
Calcination was carried out at several temperatures from 400–1000 °C to activate the catalyst. The activity of the CaO derived snail shell catalyst was assessed by performing transesterification of soybean oil to produce biodiesel at room temperature. The experimental results showed that the snail shells calcined above 800 °C that were used as a catalyst attained a biodiesel yield of 85–98%. Viriya-empikul et al.28 produced a biodiesel yield of 90% after calcination of egg shell at 800 °C. Birla et al.30 obtained 87.28% yield using a calcination temperature of 900 °C. With a calcination temperature of 700 °C, a biodiesel yield of 71% was observed.52 However, the activity of the catalyst was not much improved when the calcination temperature was below 600 °C. A biodiesel yield of below 20% was achieved when the calcination of the snail shells was below 600 °C. The biodiesel yields at different calcination temperatures are shown in Fig. 7. The variation noticed in catalytic activity of the snail shells at different temperatures matched with the variation in the XRD patterns (Fig. 1). Very low yields of biodiesel were obtained when the calcination of the snail shells was below 600 °C because these temperatures were not sufficient for the formation of CaO. Therefore, very low catalytic activity was observed at 600 °C. Snail shells calcined above 800 °C exhibited the best catalytic activity, whereas those calcined at 700 °C exhibited moderate activity due to the presence of CaCO3 as the major phase and CaO as the minor phase. Hence, it was confirmed that a snail shell derived catalyst contains CaO as the active phase. Since the snail shell catalyst was active at 900 °C and at 1000 °C it shows the exact similar performance, 900 °C is the optimized calcination temperature. In order to optimize the other reaction parameters, the snail shell derived catalyst calcined at 900 °C was used. Similar findings are also reported by Wei et al.19
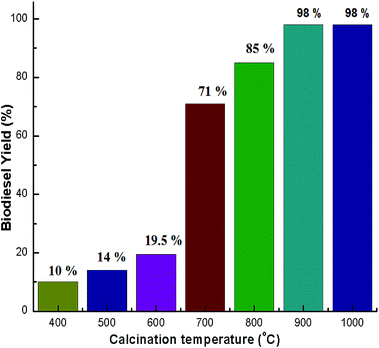 |
| | Fig. 7 Effect of calcination temperature on biodiesel yield obtained under optimised conditions: 6:1 methanol to oil ratio, 3.5 wt% catalyst loading, time 7 h and temperature of 28 °C (room temperature). | |
3.2.2 Effect of catalyst loading.
When the catalyst loading was less than 1%, a high biodiesel yield was never achieved, however a high amount of catalyst loading makes the reactant and catalyst mixture too viscous which leads to problems with mixing and, as a result, high power is required to adequately mix the solution.22 To minimize these problems, the optimum effect of catalyst loading on the yield of biodiesel was determined by varying the percentage of catalyst (2 wt%, 2.5 wt%, 3 wt% and 3.5 wt%) in the transesterification of soybean oil. The biodiesel yield obtained using a 6:1 methanol to oil ratio after 7 h at room temperature was analyzed. The biodiesel yield increases with catalyst loading from 2 to 2.5 wt% and up to 3 wt%, but it decreases beyond 3 wt% (Fig. 8). During transesterification, methanol is converted to the more reactive methoxide ions (nucleophile) by the active sites of CaO. This nucleophile attacks the carbonyl carbon (electrophile) of the glyceride molecules to form biodiesel. Thus, an increase in catalyst amount increases the total number of active sites, resulting in an increase in biodiesel conversion.52,53 However, catalyst loading beyond 3 wt% decreased the biodiesel yield due to the reaction mixture viscosity increasing, which led to poor diffusion of the reactants, resulting in a decrease in the biodiesel yield.26,30,53,54 The yield at a catalyst loading beyond 3 wt% also decreased, which may be due to the attainment of mass transfer limitation (rate determining step) between the reactant and catalyst.19 Therefore, the highest biodiesel yield was found at an optimum catalyst loading of 3 wt%. Similar results (3 wt%) are also reported by Boro et al.47 However, their reaction accelerates with temperature.
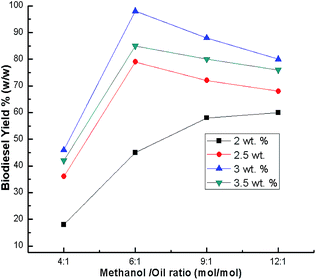 |
| | Fig. 8 Effect of catalyst loading on biodiesel yield after 7 h. | |
3.2.3 Effect of methanol to oil ratio and reaction time.
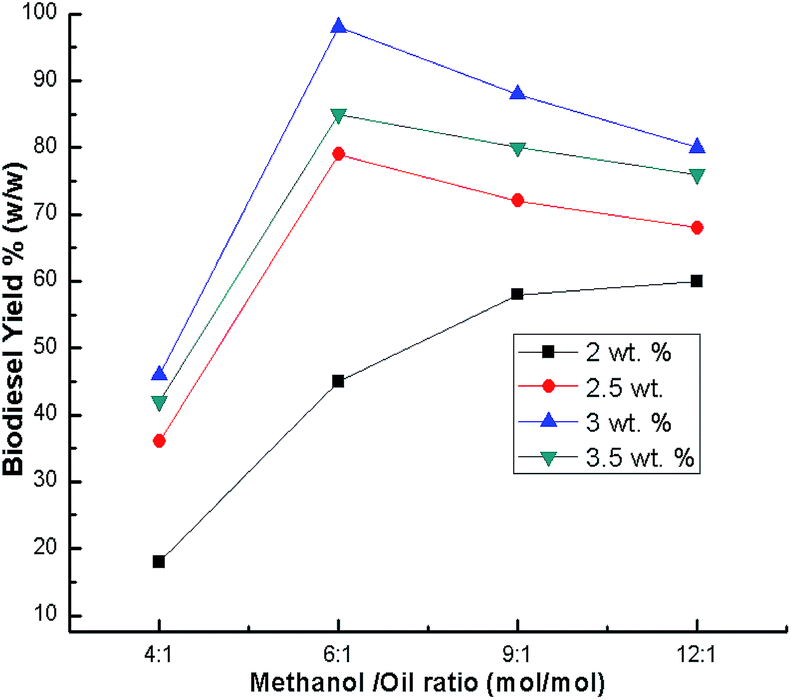
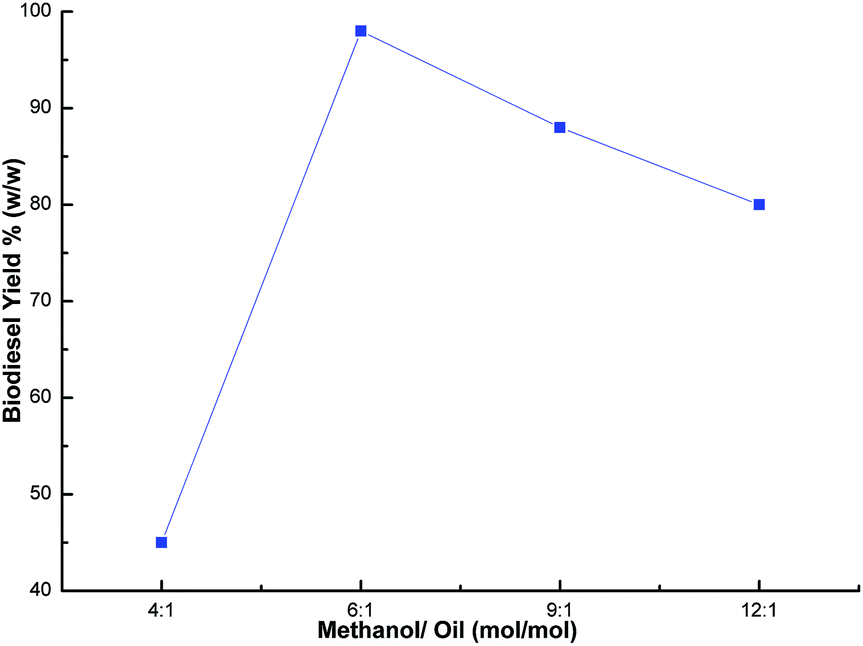
The methanol to oil ratio is another important factor which affects the biodiesel yield. Stoichiometrically, 3 mol of methanol is required to one mol of glyceride.55 In order to keep the equilibrium on the right side of the reaction, a slightly higher amount of methanol was needed. Thus, this parameter was optimized by carrying out the transesterification reaction with various methanol to oil ratios (1:4, 1:6, 1:9 and 1:12) using a catalyst loading of 3% and a time of 7 h at room temperature. The best biodiesel yield of 98% was obtained with the optimum methanol to oil ratio (6:1), whereas the biodiesel yield deteriorated beyond the methanol to oil ratio of 6:1 (Fig. 9) due to the excess methanol dissolving the glycerol and a back reaction may take place which hinders the reaction between the methanol with the catalyst and the oil.26,48,56 Thus, the methanol to oil ratio of 6:1 was the optimal ratio for the biodiesel yield of 98%. In order to optimize the reaction time, reactions were carried out with different time intervals (3 h, 5 h, 7 h and 8 h) using optimized catalyst loading (3 wt%) and the methanol to oil ratio of 6:1 at room temperature. The effect of reaction time was investigated and experimental results revealed that the yield increased with time, and achieved the highest yield of 99% after 7 h as shown in Fig. 10. The yield deteriorated slightly after 7 h because hydrolysis of esters may start to occur with a further increase in the reaction time, which results in more fatty acids forming soap.57 Additionally, a back reaction may take place after reaching the equilibrium since the reaction is reversible, subsequently decreasing the yield.54
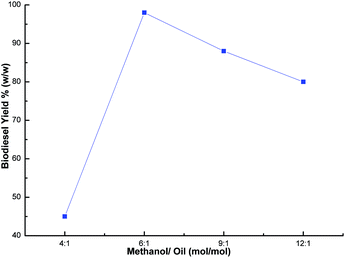 |
| | Fig. 9 Effect of methanol/oil ratio on biodiesel yield. | |
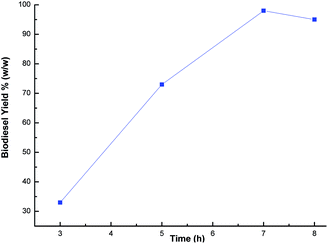 |
| | Fig. 10 Effect of time on biodiesel yield. | |
An optimum biodiesel yield of 98% was obtained with reaction parameters such as a methanol to oil ratio of 6:1, catalyst loading of 3 wt% and reaction time of 7 h. The results of this study were compared with the previous study as shown in Table 4. Under our optimized reaction conditions, 100% conversion of soybean oil to biodiesel was achieved without the requirement of any external heating which showed the catalyst’s superiority among the other listed catalysts.
3.3 Characterisation of FAME
3.3.1 Analysis of 1H-NMR.
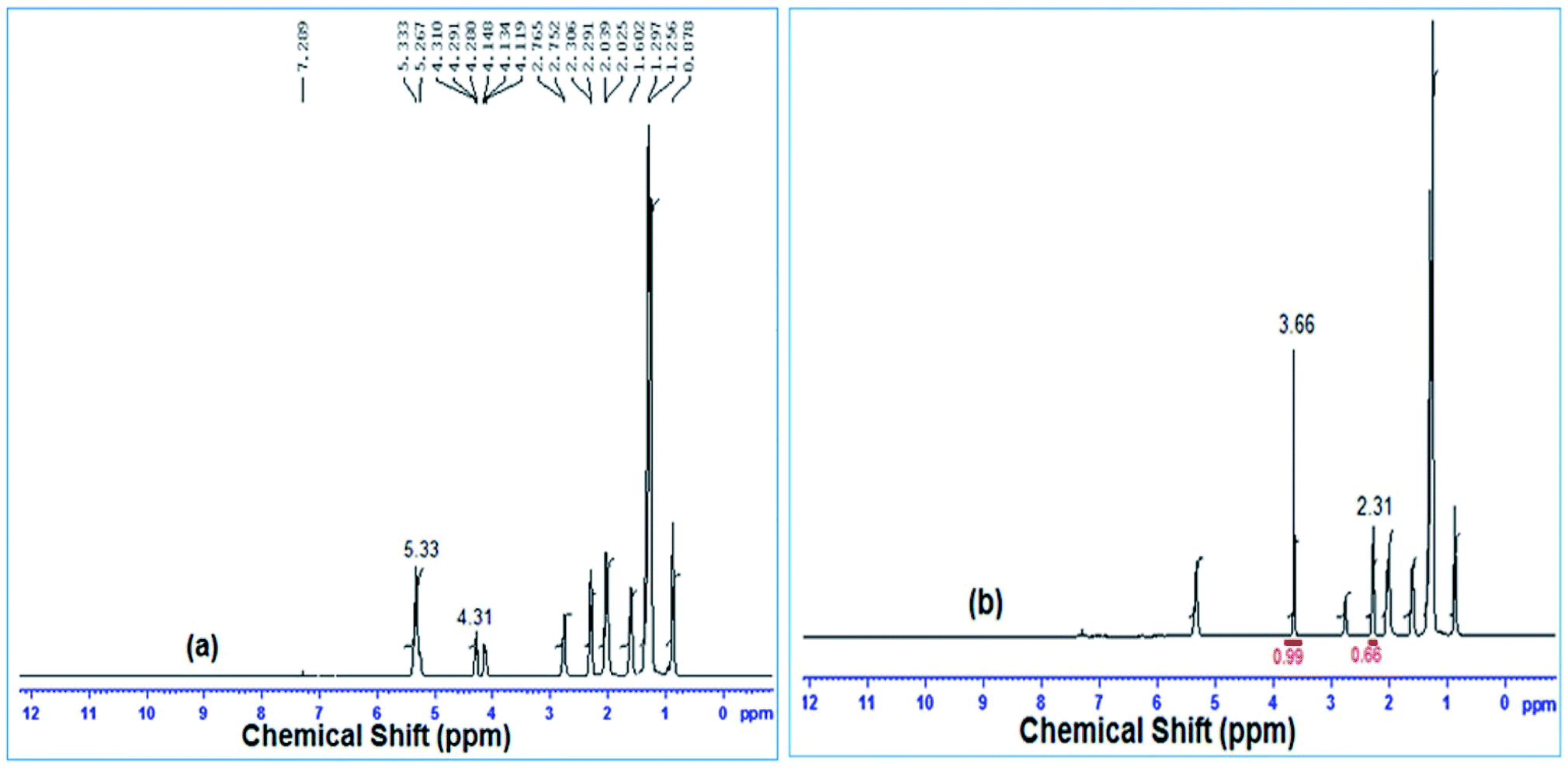
Biodiesel conversion from soybean oil using a snail shell derived catalyst was determined by 1H-NMR spectrometry and the spectra are depicted in Fig. 11. In the 1H NMR spectra, a methoxy proton characteristics peak was noticed at 3.66 ppm as a singlet and α-CH2 proton characteristic peaks were obtained as a triplet at 2.31 ppm, as shown in Fig. 11(b). The appearance of these two distinct characteristic peaks in the 1H NMR spectra (Fig. 11(b)) confirmed the formation of methyl ester after the transesterification of soybean oil to fatty acid methyl ester (FAME) by the snail shell derived heterogeneous catalyst. From Fig. 11(a), multiplets at 4.31 and 5.33 indicated the existence of glyceridic protons and olefinic hydrogen. The formation of biodiesel (FAME) was also confirmed by the absence of the signal at 4.31 ppm and the presence of a sharp peak at 3.66 ppm as shown in Fig. 11(b). The conversion of soybean oil to biodiesel was determined by the ratio of the integrated areas below the peaks at 3.66 ppm (methoxy protons) and 2.31 ppm (α-CH2 protons), as described by Gelbard et al.43 and 100% conversion was found by eqn (2). The results were well matched with the reported data by Gohain et al.60
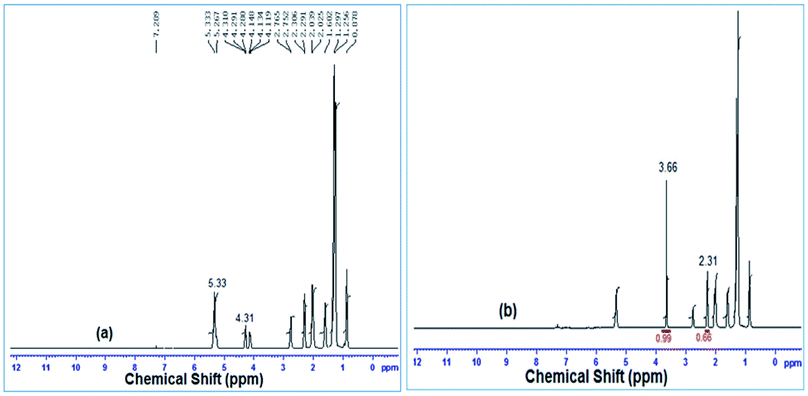 |
| | Fig. 11
1H-NMR spectroscopy of (a) fresh soybean oil and (b) soybean oil biodiesel. | |
3.3.2 FTIR of biodiesel.
Refined soybean oil and its products (biodiesel) obtained after transesterification under optimized conditions, such as a catalyst loading of 3 wt%, methanol to oil ratio of 6:1 and reaction time of 7 h, were investigated using FTIR spectroscopy and the spectra are illustrated in Fig. 12. Biodiesel and soybean oil were chemically similar resulting in relatively small differences observed between the spectra. The peaks in the region from 1800–1700 cm−1 ascribed to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O were observed in biodiesel as well as in soybean oil due to presence of CO in both samples.61 The spectra obtained in the range 1500–900 cm−1 is known as the fingerprint region which discriminates biodiesel from its source (soybean oil). In the biodiesel spectrum (Fig. 12(a)), the peak at 1436 cm−1 corresponds to the asymmetric stretching of –CH3 but the peak was absent in the spectrum of soybean oil (Fig. 12(b)).62 The glycerol group O–CH2 (mono, di and triglycerides) stretching was attributed to the absorbance peak at 1377 cm−1 which was found in soybean oil (Fig. 12(b)) but was absent in the biodiesel spectrum (Fig. 12(a)).63 The absorbance peak obtained at 1195 cm−1, attributed to the stretching of O–CH3, confirmed the formation of biodiesel, but this peak was absent in the spectrum of soybean oil.
O were observed in biodiesel as well as in soybean oil due to presence of CO in both samples.61 The spectra obtained in the range 1500–900 cm−1 is known as the fingerprint region which discriminates biodiesel from its source (soybean oil). In the biodiesel spectrum (Fig. 12(a)), the peak at 1436 cm−1 corresponds to the asymmetric stretching of –CH3 but the peak was absent in the spectrum of soybean oil (Fig. 12(b)).62 The glycerol group O–CH2 (mono, di and triglycerides) stretching was attributed to the absorbance peak at 1377 cm−1 which was found in soybean oil (Fig. 12(b)) but was absent in the biodiesel spectrum (Fig. 12(a)).63 The absorbance peak obtained at 1195 cm−1, attributed to the stretching of O–CH3, confirmed the formation of biodiesel, but this peak was absent in the spectrum of soybean oil.
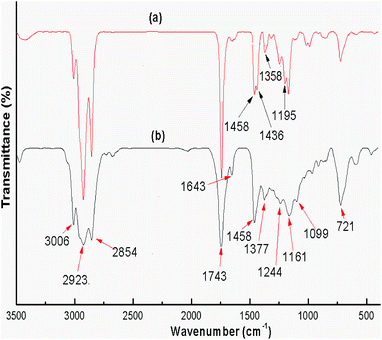 |
| | Fig. 12 FT-IR spectra of (a) soybean oil biodiesel and (b) soybean oil. | |
3.3.3 Physico-chemical properties of soybean oil biodiesel.
The physico-chemical properties of soybean oil biodiesel were determined as per ASTM standard and compared with petrodiesel, as depicted in Table 5.
Table 5 Physical properties of soybean oil biodiesel
| Physical properties |
Soybean oil biodiesel |
Petrodiesel |
ASTM standards |
| Kinematic viscosity (cst at 40 °C) |
6.098 |
2.83 |
ASTM D 445 |
| Cloud point |
0 °C |
6.4 |
ASTM D 97 |
| Pour point |
−2 °C |
3 |
ASTM D 2500 |
| Density (g cm−3) |
0.877 |
0.835 |
ASTM D 1448-1972 |
| Flash point (°C) |
150 |
70 |
ASTM D 7215 |
| Cetane number |
55 |
48 |
ASTM D 6890 |
| Copper strip corrosion |
1(a) |
1(a) |
ASTM D 130 |
3.3.4 GC-MS of biodiesel.
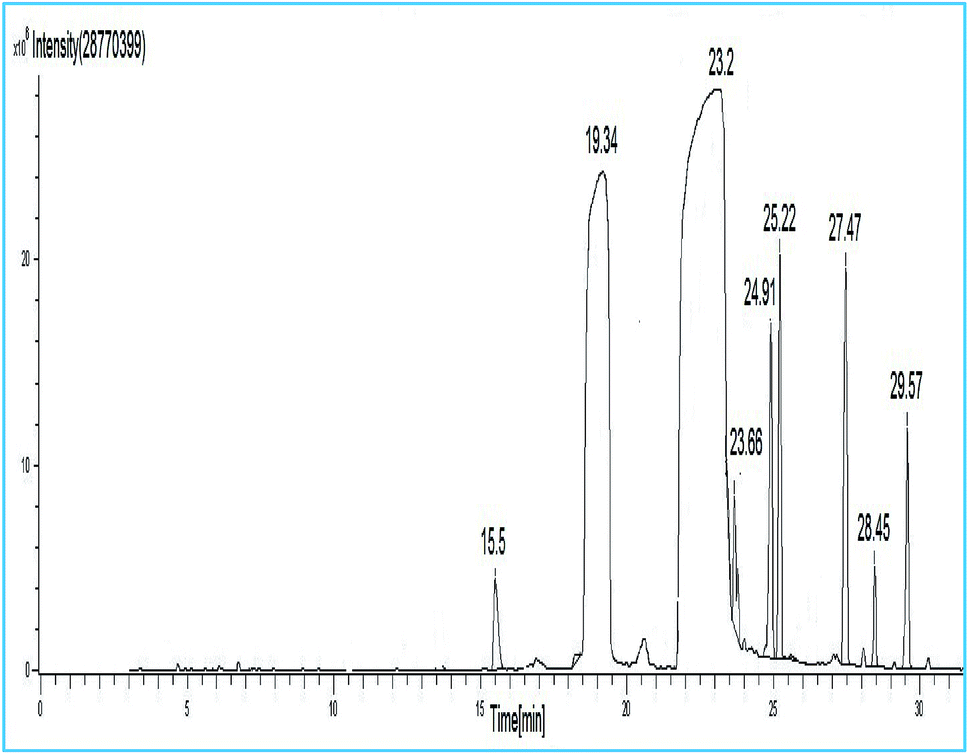
The synthesized soybean oil biodiesel composition was determined by GC-MS studies and gas chromatography as shown in Fig. 13. Furthermore, FAME content was estimated quantitatively by comparing fatty acid methyl ester peak areas to the internal standard (methyl heptadecanoate, C17:0) peak areas obtained by GC-MS. The soybean oil biodiesel composition, studied using GC-MS with respect to the retention time (R.t.), is summarized in Table 6. It was noticed from the GC-MS results that the major FAMEs were methyl-octadecadienoate (C18:2, linoleic acid, 48.94%), methyl-hexadecanoate (C16:0, palmitic acid, 17%), methyl-docosanoate (C22:0, docosanic acid, 9.49%) and methyl nonadecanoate (C20:0, nonadecanoic acid, 7.84%).
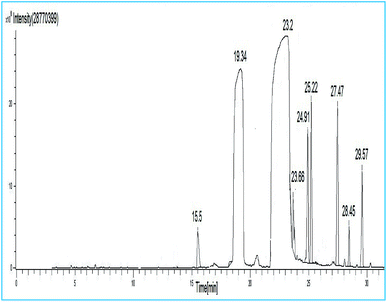 |
| | Fig. 13 GC-MS spectrum of soybean oil biodiesel. | |
Table 6 Chemical composition of soybean oil biodiesel
| Peak no. |
R.t. (min) |
Identified compounds |
Corresponding acid |
| 1 |
15.5 |
Methyl tetradecanoate |
C14:0 |
| 2 |
19.34 |
Methyl hexadecanoate |
C16:0 |
| 3 |
23.2 |
Methyl-octadeca-14,17-dieonate |
C18:2 |
| 4 |
23.66 |
Methyl octadeca-9-12-dienoate |
C18:2 |
| 5 |
24.91 |
Methyl-11-eicosenoate |
C20:1 |
| 6 |
25.22 |
Methyl nonadecanoate |
C20:0 |
| 7 |
27.47 |
Methyl docosanoate |
C22:0 |
| 8 |
28.45 |
Methyl tricosanoate |
C24:0 |
| 9 |
29.57 |
Methyl tetracosanoate |
C24:0 |
3.3.5 Reusability.
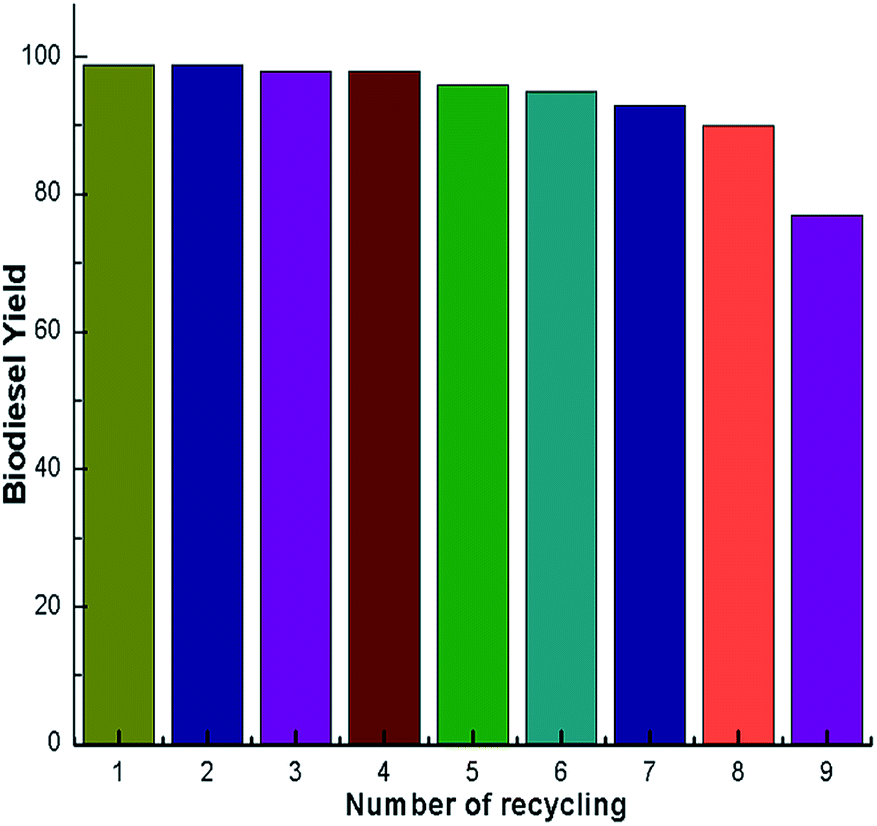
A catalyst reusability test is very important as it shows the advantage of using a catalyst to reduce the production cost for its use in industrial applications. The reusability of the synthesized calcined snail shell catalyst was studied with the optimized conditions and the results are illustrated in Fig. 14. After each cycle the catalyst was decanted from the reaction mixture by centrifugation, washed with distilled water and was dried overnight in an oven at 100 °C. Furthermore, the catalyst was reactivated by calcination at 900 °C for 4 h.29 Subsequently a new reaction was carried out with fresh reactants. The experimental results, shown in Fig. 14, illustrated that an acceptable biodiesel yield was obtained up to the 8th cycle. Beyond that a remarkable decrease in yield was observed which may be because of pore filling and surface poisoning by the triglycerides and glycerol leading to a decrease in the number of active sites of the catalyst, thereby reducing the biodiesel yields. The catalyst loading was adjusted after each reuse to the optimum reaction conditions. However, there was some loss of catalyst in each cycle due to leaching during the washing stage. This may also be the reason for the gradual reduction of yield after each catalytic cycle.
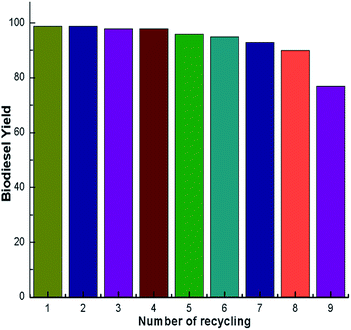 |
| | Fig. 14 Reusability of nanoparticles for the transesterification reaction. Operating conditions: catalyst loading of 3 wt% (w/w); methanol to oil ratio of 6:1 (m/m); reaction time of 7 h. | |
4. Conclusion
A green, ecofriendly, economical, nontoxic, biodegradable and reusable solid catalyst was developed by simple calcination of waste snail shells (Pila spp.). The calcined snail shells with a higher surface area (7 g m−2) exhibited high activity in the transesterification of soybean oil to biodiesel at room temperature. A high biodiesel yield of 98% was achieved under the optimized conditions, such as a calcination temperature of 900 °C, catalyst loading of 3 wt%, reaction time of 7 h and a methanol to oil ratio of 6:1. In the 8th cycle, a biodiesel yield of 91% was obtained; however, the yield drastically decreased in 9th cycle to 77%. Therefore the catalyst can be used up to 8th cycle without much loss in its catalytic activity. The production of biodiesel without external heating makes the process simple and contributes to the economy and ecofriendly nature of our protocol. Converting a waste material (snail shell) into a valuable product with minimal use of energy during the transesterification reaction and the material’s recyclability makes our method economic and environmentally benign. In addition to biodiesel production, these types of environmentally-friendly waste material derived chemicals have the potential to be efficient reagents and catalysts for diverse chemical transformations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Science and Engineering Research Board (SERB), New Delhi for financial support (Grant No. SB/FT/CS-103/2013 and SB/EMEQ-076/2014). We also acknowledge SAIF IIT Bombay, SAIF NEHU, SAIF IIT Roorkee, IIT Guwahati Chemical Engineering Department and Guwahati University for analysis studies.
References
- A. Piker, B. Tabah and N. Perkas, Fuel, 2016, 182, 34–41 CrossRef CAS.
- C. Reddy, V. Reddy, R. Oshel and J. G. Verkade, Energy Fuels, 2006, 20, 1310–1314 CrossRef CAS.
- S. Chatterjee, Dhanurdhar and L. Rokhum, Renewable Sustainable Energy Rev., 2017, 72, 560–564 CrossRef CAS.
- B. Likozar, A. Pohar and J. Levec, Fuel Process. Technol., 2016, 142, 326–336 CrossRef CAS.
- B. Likozar and J. Levec, Fuel Process. Technol., 2014, 122, 30–41 CrossRef CAS.
- B. Likozar and J. Levec, Appl. Energy, 2014, 123, 108–120 CrossRef CAS.
- J. P. C. Evangelista, T. Chellappa, A. C. F. Coriolano, V. J. Fernandes, L. D. Souza and A. S. Araujo, Fuel Process. Technol., 2012, 104, 90–95 CrossRef CAS.
- N. C. Om Tapanes, D. A. Gomes Aranda, J. W. D. Mesquita Carneiro and O. A. Ceva Antunes, Fuel, 2008, 87, 2286–2295 CrossRef CAS.
- D. Kumar and A. Ali, Energy Fuels, 2013, 27, 3758–3768 CrossRef CAS.
- W. Xie and L. Zhao, Energy Convers. Manage., 2013, 76, 55–62 CrossRef CAS.
- H. V. Lee, J. C. Juan and Y. H. Taufiq-Yap, Renewable Energy, 2015, 74, 124–132 CrossRef CAS.
- W. Xie, Y. Han and S. Tai, Fuel, 2017, 210, 83–90 CrossRef.
- W. Xie and F. Wan, Fuel, 2018, 220, 248–256 CrossRef CAS.
- Y. D. Chiang, S. Dutta, C. T. Chen, Y. T. Huang, K. S. Lin, J. C. S. Wu, N. Suzuki, Y. Yamauchi and K. C. W. Wu, ChemSusChem, 2015, 8, 789–794 CrossRef CAS PubMed.
- A. Guldhe, P. Singh, F. A. Ansari, B. Singh and F. Bux, Fuel, 2017, 187, 180–188 CrossRef CAS.
- W. Xie and M. Huang, Energy Convers. Manage., 2018, 159, 42–53 CrossRef CAS.
- B. Gurunathan and A. Ravi, Bioresour. Technol., 2015, 190, 424–428 CrossRef CAS PubMed.
- L. M. Correia, R. M. A. Saboya, N. de Sousa Campelo, J. A. Cecilia, E. Rodrguez-Castelln, C. L. Cavalcante and R. S. Vieira, Bioresour. Technol., 2014, 151, 207–213 CrossRef CAS PubMed.
- Z. Wei, C. Xu and B. Li, Bioresour. Technol., 2009, 100, 2883–2885 CrossRef CAS PubMed.
- Y. C. Sharma, B. Singh and J. Korstad, Energy Fuels, 2010, 24, 3223–3231 CrossRef CAS.
- S. H. Y. S. Abdullah, N. H. M. Hanapi, A. Azid, R. Umar, H. Juahir, H. Khatoon and A. Endut, Renewable Sustainable Energy Rev., 2017, 70, 1040–1051 CrossRef CAS.
- M. Kouzu, T. Kasuno, M. Tajika, Y. Sugimoto, S. Yamanaka and J. Hidaka, Fuel, 2008, 87, 2798–2806 CrossRef CAS.
- X. Liu, H. He, Y. Wang, S. Zhu and X. Piao, Fuel, 2008, 87, 216–221 CrossRef CAS.
- P. L. Boey, G. P. Maniam and S. A. Hamid, Chem. Eng. J., 2011, 168, 15–22 CrossRef CAS.
- L. M. Correia, N. D. Sousa Campelo, D. S. Novaes, C. L. Cavalcante, J. A. Cecilia, E. Rodríguez-Castellón and R. S. Vieira, Chem. Eng. J., 2015, 269, 35–43 CrossRef CAS.
- T. Maneerung, S. Kawi, Y. Dai and C. H. Wang, Energy Convers. Manage., 2016, 123, 487–497 CrossRef CAS.
- R. Rezaei, M. Mohadesi and G. R. Moradi, Fuel, 2013, 109, 534–541 CrossRef CAS.
- N. Viriya-empikul, P. Krasae, B. Puttasawat, B. Yoosuk, N. Chollacoop and K. Faungnawakij, Bioresour. Technol., 2010, 101, 3765–3767 CrossRef CAS PubMed.
- G. Joshi, D. S. Rawat, B. Y. Lamba, K. K. Bisht, P. Kumar, N. Kumar and S. Kumar, Energy Convers. Manage., 2015, 96, 258–267 CrossRef CAS.
- A. Birla, B. Singh, S. N. Upadhyay and Y. C. Sharma, Bioresour. Technol., 2012, 106, 95–100 CrossRef CAS PubMed.
- C. Lydeard, R. H. Cowie, W. F. Ponder, A. E. Bogan, P. Bouchet, S. A. Clark, K. S. Cummings, T. J. Frest, O. Gargominy, D. G. Herbert, R. Hershler, K. E. Perez, B. Roth, M. Seddon, E. E. Strong and F. G. Thompson, BioScience, 2004, 54, 321–330 CrossRef.
- M. Halwart, Int. J. Pest Manage., 1994, 40, 199–206 CrossRef.
- H. Madsena and N. M. Hung, Acta Trop., 2015, 141, 372–384 CrossRef PubMed.
- D. Vergaraa, J. A. Fuentesa, K. S. Stoya and C. M. Livelya, Molluscan Res., 2016, 37, 120–132 CrossRef.
- P. Shejwal, D. Wagh and M. Patil, Journal of Entomology and Zoology Studies, 2016, 4, 252–255 Search PubMed.
- G. Potkamp, M. J. A. Vermeij and B. W. Hoeksema, Contrib. Zool., 2017, 86, 111–144 Search PubMed.
- G. S. Kumar, E. K. Girija, M. Venkatesh, G. Karunakaran, E. Kolesnikov and D. Kuznetsov, Ceram. Int., 2017, 43, 3457–3461 CrossRef CAS.
- E. J. Kistner and M. F. Dybdahl, Evol. Ecol., 2013, 3, 424–436 CrossRef PubMed.
- C. Saha, S. Pramanik, J. Chakraborty, S. Parveen and G. Aditya, Journal of Entomology and Zoology Studies, 2016, 4, 490–497 Search PubMed.
- S. Ghosh, C. Jung and V. B. Meyer-Rochow, Ann. Aquac. Res., 2016, 3, 1024–1030 Search PubMed.
- G. Pathak, D. Das, K. Rajkumari and L. Rokhum, Green Chem., 2018, 20, 2365–2373 RSC.
- B. Yoosuk, P. Udomsap, B. Puttasawat and P. Krasae, Chem. Eng. J., 2010, 162, 135–141 CrossRef CAS.
- G. Gelbard, O. Brès, R. M. Vargas, F. Vielfaure and U. F. Schuchardt, J. Am. Oil Chem. Soc., 1995, 72, 1239–1241 CrossRef CAS.
- G. Knothe and J. A. Kenar, Eur. J. Lipid Sci. Technol., 2004, 106, 88–96 CrossRef CAS.
- D. Y. C. Leung and Y. Guo, Fuel Process. Technol., 2006, 87, 883–890 CrossRef CAS.
- S. Hu, Y. Wang and H. Han, Biomass Bioenergy, 2011, 35, 3627–3635 CrossRef CAS.
- J. Boro, A. J. Thakur and D. Deka, Fuel Process. Technol., 2011, 92, 2061–2067 CrossRef CAS.
- R. Madhuvilakku, R. Mariappan, S. Jeyapal, S. Sundar and S. Piraman, Ind. Eng. Chem. Res., 2013, 52, 17407–17413 CrossRef CAS.
- M. C. Manique, L. V. Lacerda, A. K. Alves and C. P. Bergmann, Fuel, 2017, 190, 268–273 CrossRef CAS.
- A. Kawashima, K. Matsubara and K. Honda, Bioresour. Technol., 2009, 100, 696–700 CrossRef CAS PubMed.
- B. Yoosuk, P. Udomsap, B. Puttasawat and P. Krasae, Chem. Eng. J., 2010, 162, 135–141 CrossRef CAS.
- W. Roschat, T. Siritanon, B. Yoosuk and V. Promarak, Energy Convers. Manage., 2016, 108, 459–467 CrossRef CAS.
- S. L. Lee, Y. C. Wong, Y. P. Tan and S. Y. Yew, Energy Convers. Manage., 2015, 93, 282–288 CrossRef CAS.
- O. N. Syazwani, S. H. Teo, A. Islam and Y. H. Taufiq-Yap, Process Saf. Environ. Prot., 2017, 105, 303–315 CrossRef CAS.
- Y. Tang, M. Meng, J. Zhang and Y. Lu, Appl. Energy, 2011, 88, 2735–2739 CrossRef CAS.
- M. Yadav, V. Singh and Y. C. Sharma, Energy Convers. Manage., 2017, 148, 1438–1452 CrossRef CAS.
- N. Girish, S. P. Niju, K. M. Meera Sheriffa Begum and N. Anantharaman, Fuel, 2013, 111, 653–658 CrossRef CAS.
- S. Agrawal, B. Singh and Y. C. Sharma, Ind. Eng. Chem. Res., 2012, 51, 11875–11880 CrossRef CAS.
- O. N. Syazwani, U. Rashid and Y. H. Taufiq Yap, Energy Convers. Manage., 2015, 101, 749–756 CrossRef.
- M. Gohain, A. Devi and D. Deka, Ind. Crops Prod., 2017, 109, 8–18 CrossRef CAS.
- I. P. Soares, T. F. Rezende, R. C. Silva, E. V. R. Castro and I. C. P. Fortes, Energy Fuels, 2008, 22, 2079–2083 CrossRef CAS.
- M. Yadav, V. Singh and Y. C. Sharma, Energy Convers. Manage., 2017, 148, 1438–1452 CrossRef CAS.
- M. A. Dubé, S. Zheng, D. D. McLean and M. Kates, J. Am. Oil Chem. Soc., 2004, 81, 599–603 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02397b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Samuel Lalthazuala
Rokhum
b and
Samuel Lalthazuala
Rokhum