
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery and mechanistic study of a photocatalytic indoline dehydrogenation for the synthesis of elbasvir†
Hatice G.
Yayla
a,
Feng
Peng
b,
Ian K.
Mangion
b,
Mark
McLaughlin
b,
Louis-Charles
Campeau
b,
Ian W.
Davies
b,
Daniel A.
DiRocco
*b and
Robert R.
Knowles
*a
aDepartment of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: rknowles@princeton.edu
bDepartment of Process Chemistry, Merck & Co., Inc. Rahway, New Jersey 07065, USA. E-mail: daniel.dirocco@merck.com
First published on 7th December 2015
Abstract
Elbasvir is a potent NS5A antagonist for the treatment of chronic hepatitis C. A seemingly trivial indoline oxidation en route to the target compound was complicated by epimerization of a stereogenic hemiaminal center under most standard oxidation conditions. To address this issue, a novel visible light photoredox process for indoline oxidation was developed involving an iridium photosensitizer and environmentally-benign perester oxidant. The reaction was discovered through a high-throughput experimentation campaign and the optimized process was demonstrated on 100 g scale in flow to afford a key intermediate towards the target compound. A battery of kinetic, electrochemical, and spectroscopic studies of this process indicates a radical chain mechanism of dehydrogenation involving selective HAT from the substrate by an alkoxy radicals. Notably, isotope effects were used to validate the chain mechanism when quantum yield data proved ambiguous.
Introduction
More than 150 million people worldwide are infected with the hepatitis C virus (HCV), the leading cause of liver disease and liver transplantations.1 It is estimated that as many as five million of these people are co-infected with the human immunodeficiency virus (HIV), which typically leads to higher viral loads and results in accelerated disease progression.2 With limited treatment options for co-infected patients, HCV has become a leading cause of death for HIV patients. Elbasvir, an inhibitor of the HCV NS5A protein (Fig. 1), administered in combination with grazoprevir, an HCV protease inhibitor, has been clinically studied as a highly efficacious, and well tolerated oral regimen for the treatment of HCV infection, including patients with HIV co-infection (Fig. 1). During our development of a synthetic route to elbasvir, a seemingly trivial oxidation of chiral hemiaminal ether 1 to indole 2 proved capricious due to facile epimerization of the hemiaminal stereocenter.3,4 Following extensive screening, KMnO4 was found to be the only oxidant capable of converting 1 to 2 without loss of enantiopurity (Fig. 1).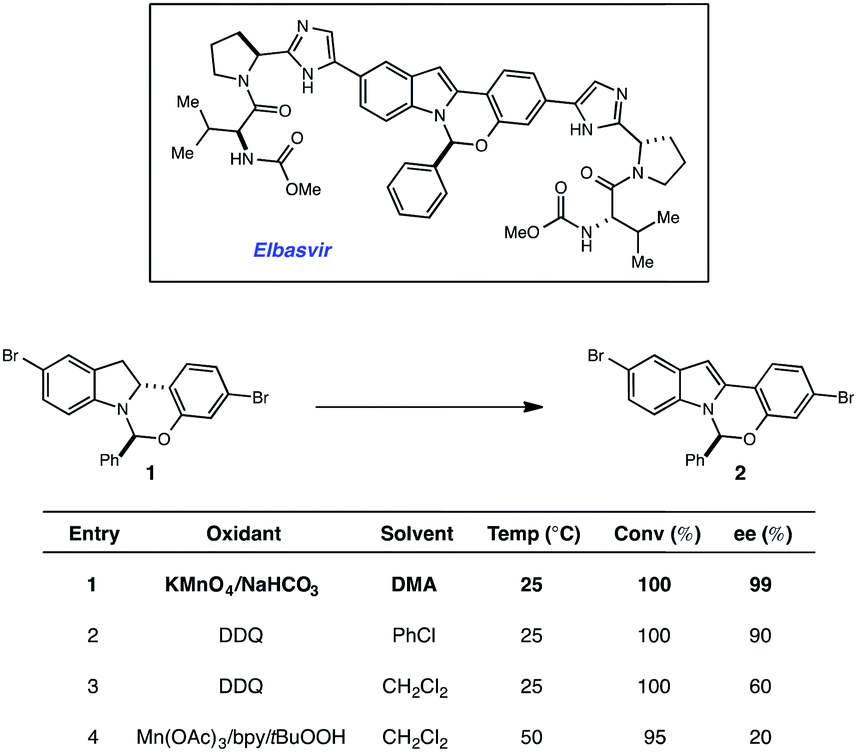 | ||
| Fig. 1 Permanganate-mediated dehydrogenation in the synthesis of elbasvir. | ||
Although KMnO4 is an effective oxidant for this transformation, subsequent generation of MnO2 as a by-product complicates isolation and is environmentally unsustainable for large-scale production. In the pursuit of an alternative method that would circumvent the use of stoichiometric metal oxidants, we elected to investigate the potential efficacy of a redox catalyst that could be turned over with a benign terminal oxidant. Visible-light photoredox catalysis5 has recently been shown to enable a variety of oxidative, reductive and redox neutral processes under mild conditions.6,7 Based on these findings, we elected to explore the viability of a photocatalytic process for the dehydrogenation of 1. Herein we describe the outcome of these studies and report a novel and scalable catalytic system for the oxidation of 1. A detailed mechanistic investigation established that indoline oxidation occurs through a radical chain process. Key evidence for the chain process was obtained through the combination of reaction progress kinetic analysis (RPKA) and kinetic isotope effects. We anticipate this protocol will prove useful for elucidating chain processes in photoredox reactions where quantum yield data alone was inconclusive. The results of these studies are described herein.
Results and discussion
Reaction discovery and optimization
High-throughput experimentation was utilized to evaluate a series of photocatalysts and benign terminal oxidants for the dehydrogenation of 1 (Fig. 2). An initial round of experimentation identified both nitromethane and tert-butylperacetate (tBPA) as competent stoichiometric oxidants, with the most active catalyst identified as the heteroleptic IrIII complex [Ir(dF-CF3-ppy)2(dtbpy)](PF6) (Fig. 2, D7, F7). The modest yields in our screening efforts are attributable to post-reaction decomposition, which can be minimized by shortening reaction duration. We and others have previously shown that peresters can be used as efficient electron acceptors in photoredox-catalyzed processes.8 Moreover, their ease of handling, high availability, low cost and benign decomposition products make them suitable for large-scale manufacturing.9 As a result, we elected to move forward with perester-based systems for further optimization.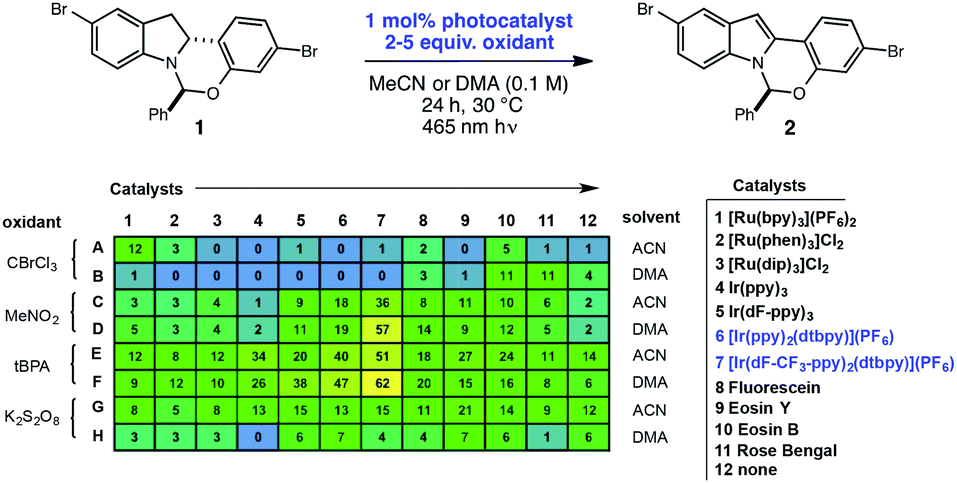 | ||
| Fig. 2 Identification of a photoredox-mediated dehydrogenation via high-throughput experimentation. Reactions performed on 2.5 μmol scale using 465 nm LEDs. Numbers in table refer to assay yields based on HPLC analysis with an internal standard. | ||
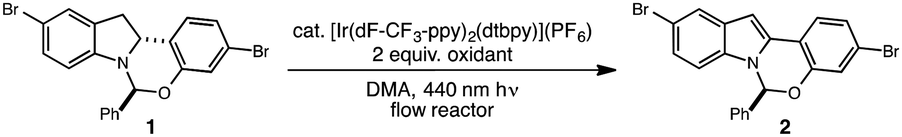
Many photocatalytic processes are rate-limited by the light flux of the reactor; as such, a flow reactor can be used to increase the surface area of the solution and thereby increase light flux and reaction rate.10–16 A series of experiments were performed in a simple flow reactor, built according to the basic design principles of Booker-Milburn using 440 nm LEDs as the light source.17 Residence times (τ) between 30 and 60 minutes led to full conversion at catalyst loadings as low as 0.1 mol% using tert-butylperbenzoate (tBPB) as the oxidant (Table 1, entries 1–2). It was shown that tBPA requires a longer τ and leads to more significant erosion of enantiopurity in product 2 (Table 1, entry 3 vs. entry 4). Slight erosion of optical purity is observed at higher temperatures (Table 1, entries 5–6); however, maintaining the reactor jacket temperature below 0 °C provided consistently high enantiopurity (Table 1, entries 7, 1). This process was demonstrated on a lab scale at a throughput of 0.04 mol h−1 (τ = 60 min). After 5 h, ∼100 grams was processed and isolated by direct crystallization upon the addition of water, providing the desired indole in 85% isolated yield and 99.8% ee (Table 1, entry 8).18 Notably, the photo-mediated reaction compares favorably to the use of permanganate from a process perspective, as the simple workup and direct crystallization of the product from the reaction media results in >60% decrease in the process mass intensity for the step.19
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | [1]/M | Cat. loading | Oxidant | t/min | Ta/°C | Yieldb (%) | eec (%) |
| a Refers to jacket temperature of reactor. b Assay yield based on volumetric HPLC analysis. c Enantiomeric excess determined by chiral HPLC analysis. d Isolated yield on 100 g scale. | |||||||
| 1 | 0.5 | 0.1 mol% | tBPB | 60 | −10 | 94 | 99.7 |
| 2 | 0.5 | 0.1 mol% | tBPB | 30 | 0 | 93 | 99.9 |
| 3 | 0.4 | 0.2 mol% | tBPB | 45 | 0 | 90 | 99.5 |
| 4 | 0.4 | 0.2 mol% | tBPA | 90 | 0 | 89 | 99.1 |
| 5 | 0.2 | 0.5 mol% | tBPB | 60 | 25 | 84 | 96.7 |
| 6 | 0.2 | 0.2 mol% | tBPA | 120 | 25 | 88 | 95.0 |
| 7 | 0.4 | 0.2 mol% | tBPB | 45 | −10 | 86 | 99.8 |
| 8 | 0.5 | 0.1 mol% | tBPB | 60 | −5 | 85d | 99.8 |
Mechanism
With these optimized photocatalytic conditions in hand, we sought to better understand the mechanism of oxidation. A significant challenge in the development of this indoline dehydrogenation was the lability of the hemiaminal stereocenter of 1, which readily epimerizes with many common oxidants such as 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ), tetracyanoethylene and Mn(OAc)3. A likely pathway for epimerization with single-electron oxidants involves the formation of the radical cation 3, which could undergo reversible β-scission and erode the stereochemical integrity of the hemiaminal center (Fig. 3).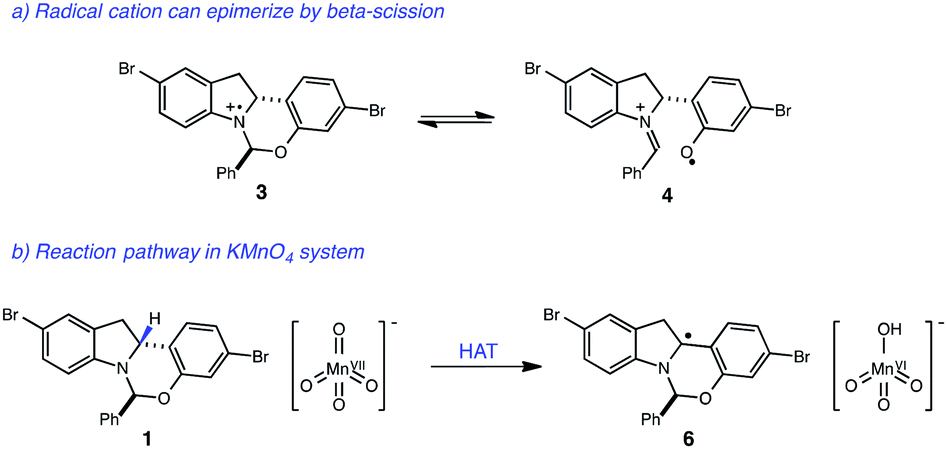 | ||
| Fig. 3 (a) Proposed pathway for epimerization of starting material. (b) Proposed mechanism for KMnO4-mediated oxidation of indoline core. | ||
Given that both the KMnO4 and the photoredox/perester systems led to retention of enantiopurity, we questioned whether there was a fundamental connection between the operative mechanisms of these reactions. Specifically, it is well documented that KMnO4 can act as a hydrogen atom abstractor for benzylic C–H bonds.20 We therefore postulated that the KMnO4 oxidation may proceed via the formation of α-amino radical 6 rather than radical cation 3. We anticipated that the hemiaminal stereocenter of intermediate 6 would be configurationally stable, providing a rationale for retention of enantiopurity. Consistent with this view, oxidation of labeled indoline 1b is markedly slower, suggestive of a rate-determining C–H abstraction step (Fig. 4). Furthermore, isotopologue 1a performed identically to the original substrate 1 with no loss of either the deuterium label or of optical purity, consistent with the proposal above.
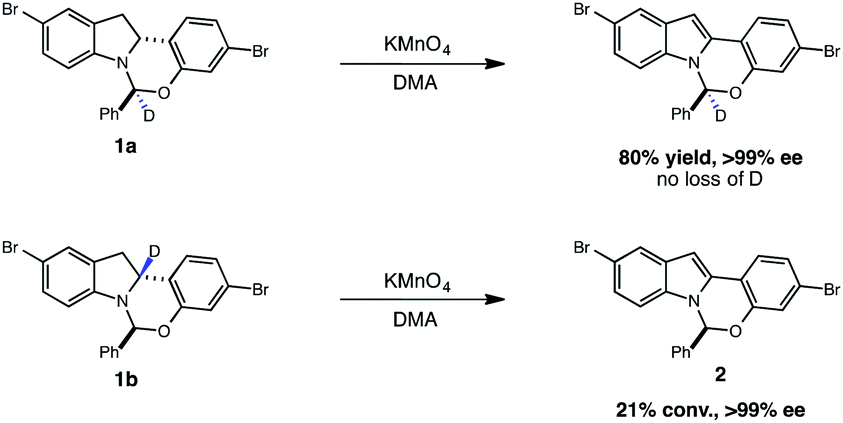 | ||
| Fig. 4 Isotope-dependence of indoline oxidation with potassium permanganate. | ||
Extending this line of thinking, we reasoned that indoline oxidation using the photoredox/perester system may also proceed via a hydrogen atom transfer (HAT) process. Specifically, one-electron reduction of the perester results in O–O bond fragmentation to form an alkoxy radical and carboxylate anion.21–23 Alkoxy radicals are classical H-atom abstractors, known to readily abstract weak C–H bonds.24,25 Therefore, hydrogen atom transfer from 1 to the tBuO˙ radical formed upon reduction of tBPB would also furnish radical intermediate 6, identical to the presumed intermediate generated in permanganate oxidations (Fig. 5). Additionally, we wondered whether it was possible for tBuO˙ radicals to initiate a radical chain process wherein HAT from 1 is a propagation step, as outlined in Fig. 6. In recent years, several examples of photoredox catalysts acting as radical chain initiators have appeared in the literature, most notably in the recent work of Yoon.26–29 In the following sections, our efforts to validate the proposed mechanism and evaluate each elementary step in this reaction sequence are described.
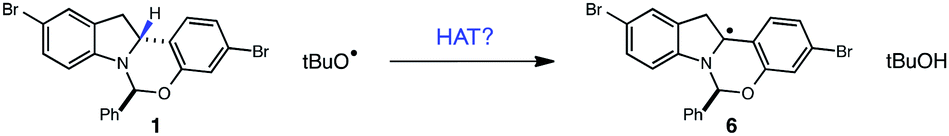 | ||
| Fig. 5 Proposed indoline oxidation pathway in the perester/photoredox oxidation. | ||
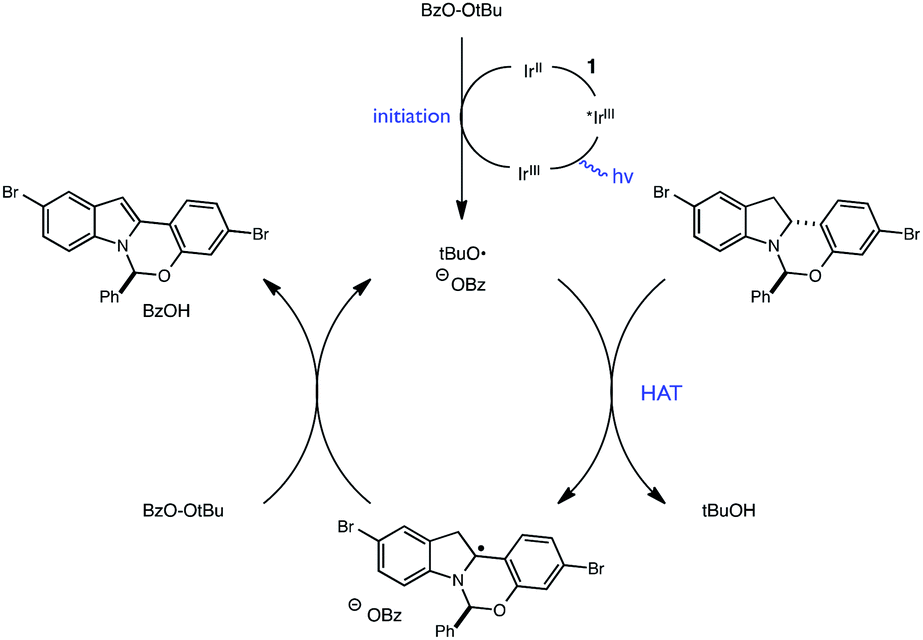 | ||
| Fig. 6 Proposed chain mechanism. | ||
Using this kinetic assay, the quantum yield of indoline oxidation was evaluated. Quantum yields that exceed 1 are often taken as evidence for a radical chain pathway. The quantum yield at any point in the reaction can be defined as the ratio between reaction rate (M s−1) and photon flux (Einsteins L−1 s−1).32 The photon flux at 402 nm through our reactor was assessed to be 2.5 × 105 Einsteins L−1 s−1, using potassium ferrioxolate as a chemical actinometer.33 The wavelength of the emission band for the LEDs was determined (402 nm) using a UVN-SR fiber optic spectrometer.34 Quantum yield information across the entire range of conversion was then obtained by plotting the ratio of this rate data to photon flux versus indoline concentration (Fig. 7). At the outset of the reaction, the quantum yield is ∼2 and then decreases as the reaction progresses.35 While this finding is consistent with a radical chain mechanism, it is not unequivocal evidence. Specifically, non-chain photocatalytic pathways that generate two reactive intermediates from a single excitation event may also produce two equivalents of product, assuming perfect quantum efficiency. To distinguish between these possibilities we turned to the kinetic isotope effect studies described below.
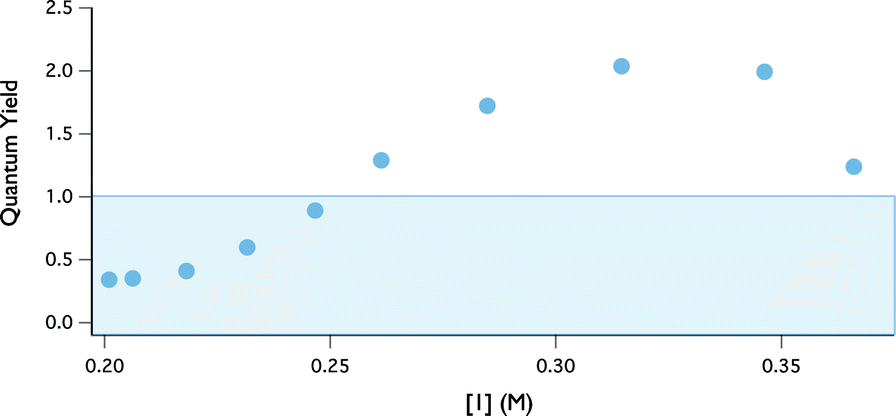 | ||
| Fig. 7 Quantum yield as a function of reaction progress using 0.1 mol% [Ir(dF-CF3-ppy)2(dtbpy)](PF6), 2 equiv. tBPB, 1 equiv. 1 in DMA. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of isotopologues react together in the same reaction flask)39 in a radical chain will reflect almost exclusively on the selectivity for H/D cleavage in the propagation steps, through which the majority of the starting material is consumed. By extension, as the slower steps such as initiation and termination contribute minimally towards the overall degree of starting material consumption, their impact on the intermolecular KIE is expected to be minimal. Moreover, assuming a constant mechanism of C–H/D cleavage, one would expect the competition KIE to be invariant over the entire course of the reaction.
:1 mixture of isotopologues react together in the same reaction flask)39 in a radical chain will reflect almost exclusively on the selectivity for H/D cleavage in the propagation steps, through which the majority of the starting material is consumed. By extension, as the slower steps such as initiation and termination contribute minimally towards the overall degree of starting material consumption, their impact on the intermolecular KIE is expected to be minimal. Moreover, assuming a constant mechanism of C–H/D cleavage, one would expect the competition KIE to be invariant over the entire course of the reaction.
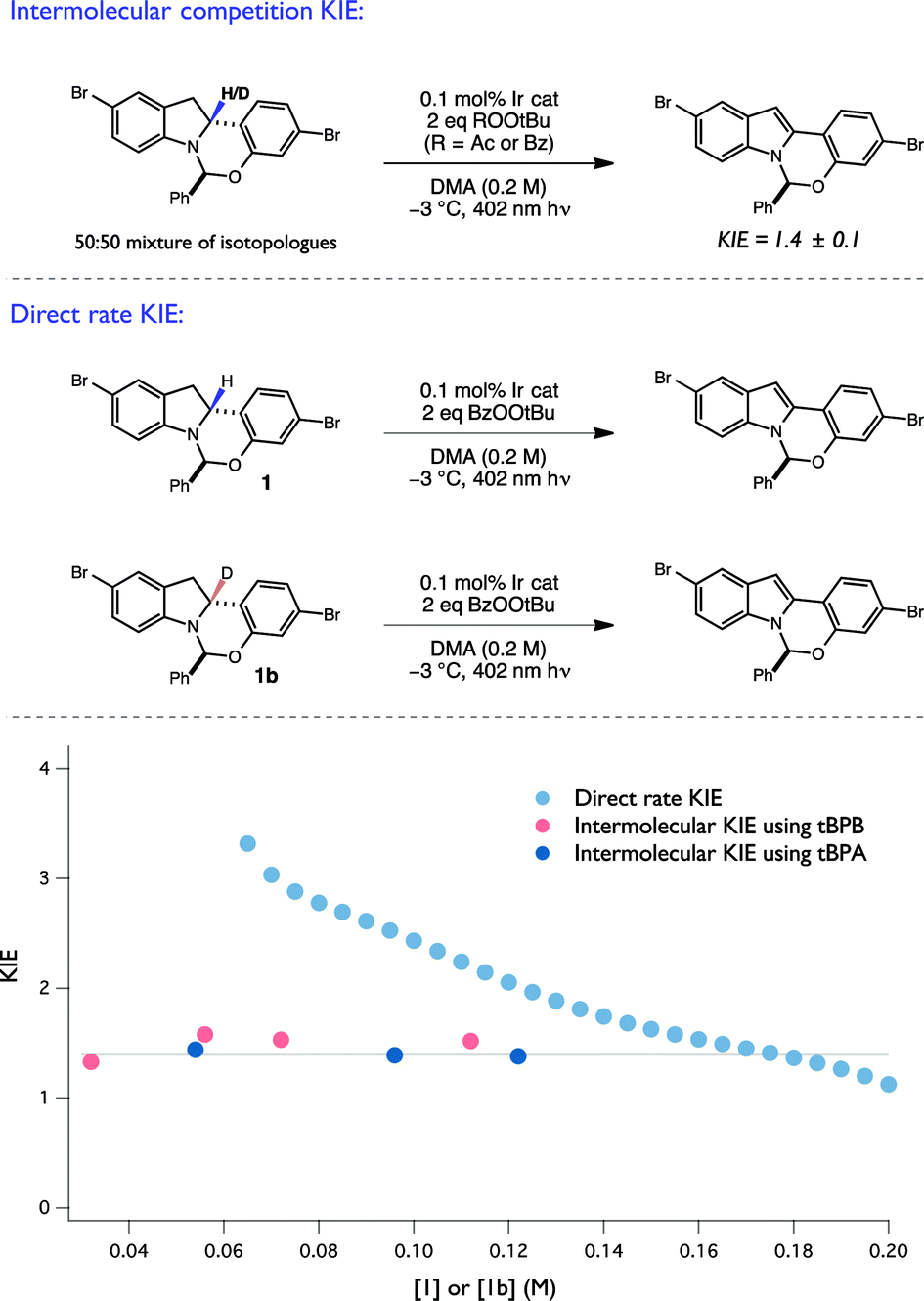 | ||
| Fig. 8 Summary of KIE experiments with 1b. | ||
To evaluate the intermolecular competition KIE, a 1:1 mixture of 1 and 1b were oxidized together under standard conditions in the same reaction flask and the degree of isotopic enrichment in the recovered starting material was measured as a function of conversion, resulting in a KIE of 1.4 ± 0.1 (Fig. 8). Importantly, the measured KIE was constant across a large range of conversion, consistent with an invariant mechanism of isotopic discrimination in the C–H/D cleavage event. Moreover, this value of 1.4 is consistent with previously reported KIE values for tBuO˙ radical-mediated H-atom abstraction from amine α C–H bonds.25,40 Lastly, competition KIEs using a structurally-distinct perester oxidant, tPBA, were also found to provide an identical KIE, consistent with the generation of a common tBuO˙ radical intermediate when either oxidant is used.
Next, the direct rate KIE was determined using RPKA, wherein two independent reactions, one with protiated and the other with deuterated substrate, were run in parallel. The ratio of the graphical rate equations for the two reactions was then plotted as a function of starting material concentration (Fig. 8). Remarkably, the direct rate KIE was observed to increase markedly as a function of substrate conversion. As the competition KIE results suggest that the selectivity for H/D abstraction remains constant over this range of substrate concentrations, this unusual feature is consistent with a mechanism wherein the relative contributions of any isotopically sensitive initiation and termination steps (vide infra) to the overall rate evolve as a function of reaction progress, and by extension with the operation of a radical chain process.
This hypothesis was further bolstered by investigation of H/D KIE at the C-3 position of the indoline. Competition experiments between 1 and 1c resulted in no measurable KIE at C-3 (kH/kD = 0.98 ± 0.02), consistent with an irreversible and selectivity-determining C–H cleavage at C-2 during propagation (Fig. 9). However, when a direct rate KIE for these isotopologues was measured using RPKA (Fig. 9), a substantial inverse KIE (∼0.7) was present across a wide range of substrate concentrations. This unusual kinetic feature is also consistent with a radical chain mechanism. Specifically, as the rate constants for termination steps are found in the denominator of chain rate laws, termination steps exhibiting normal KIEs are expected to result in overall inverse KIEs. As disproportionation of two radical intermediates is a common mechanism of chain termination, we believe that this inverse KIE is reporting on the isotopic sensitivity of H-atom abstraction from the C-3 position of radical 6 by a tBuO˙ radical. Taken together with the quantum yield findings, we believe that these complex kinetic isotope effects provide a convincing argument for the presence of a radical chain mechanism. More broadly, these experiments present a novel kinetic framework for evaluating radical chain mechanisms involving HAT steps when quantum yield data proves ambiguous. They also demonstrate that caution must be exercised in extrapolating information from direct rate KIE data for chain processes, as the measured values can be highly sensitive to the extent of reaction progress.
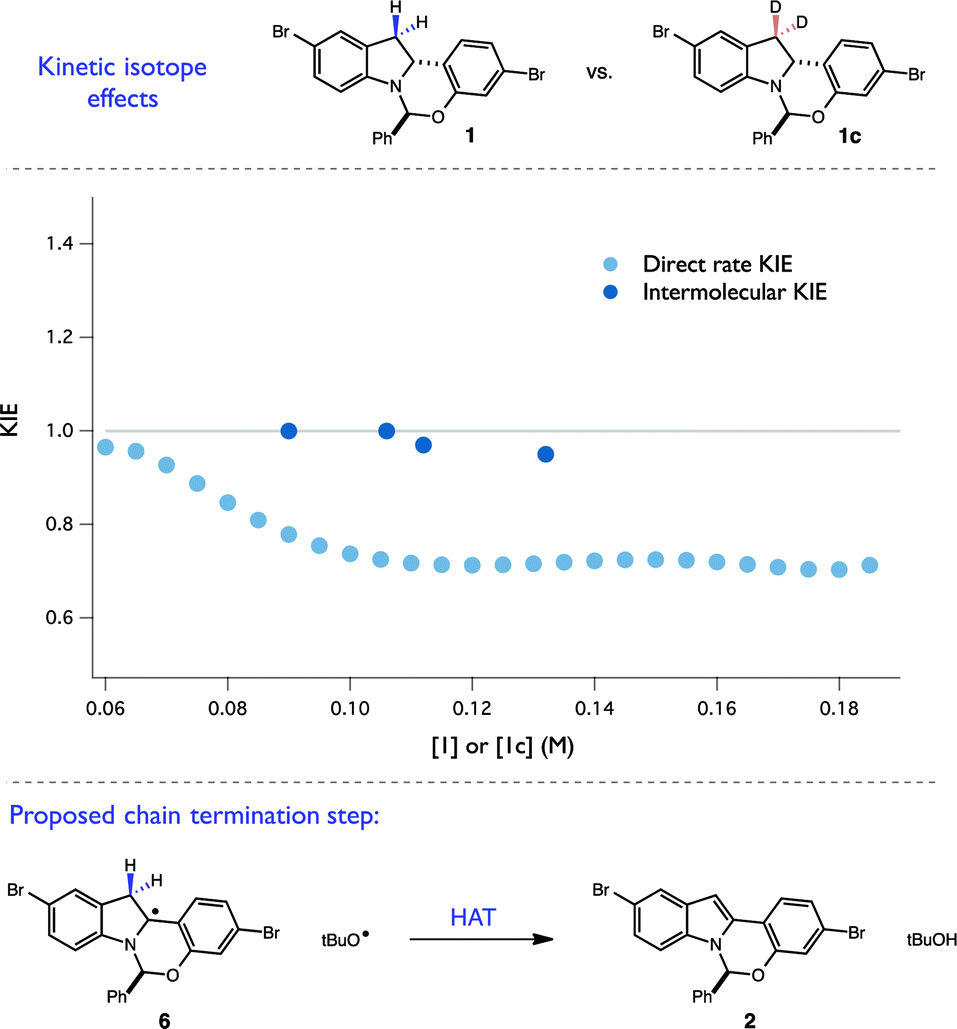 | ||
| Fig. 9 Summary of KIE experiments with 1c. | ||
Despite the presence of several weak C–H bonds, the KIE studies above suggest that HAT between tBuO˙ radical and indoline 1 to form radical 6 is highly selective for the C-2 position. As the rates of HAT reactions are known to scale with the thermodynamic driving force of the abstraction event, we questioned whether there was a connection between the C–H BDFEs of 6 and the observed site-selectivity.41,42 Indeed, DFT calculations (UB3LYP/6-311+G(d,p)) (Fig. 10) predict the C-2 methine to be the weakest C–H bond in the molecule, rationalizing the observed site selectivity.
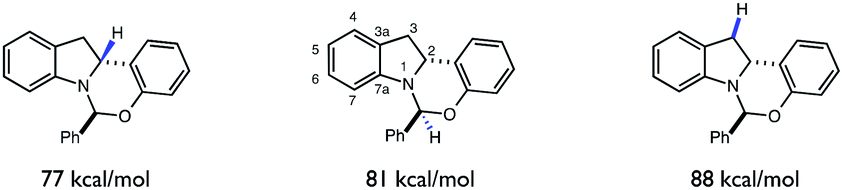 | ||
| Fig. 10 Uncorrected bond energies for C–H bonds of 1 (UB3LYP/6-311+g(d,p)). | ||
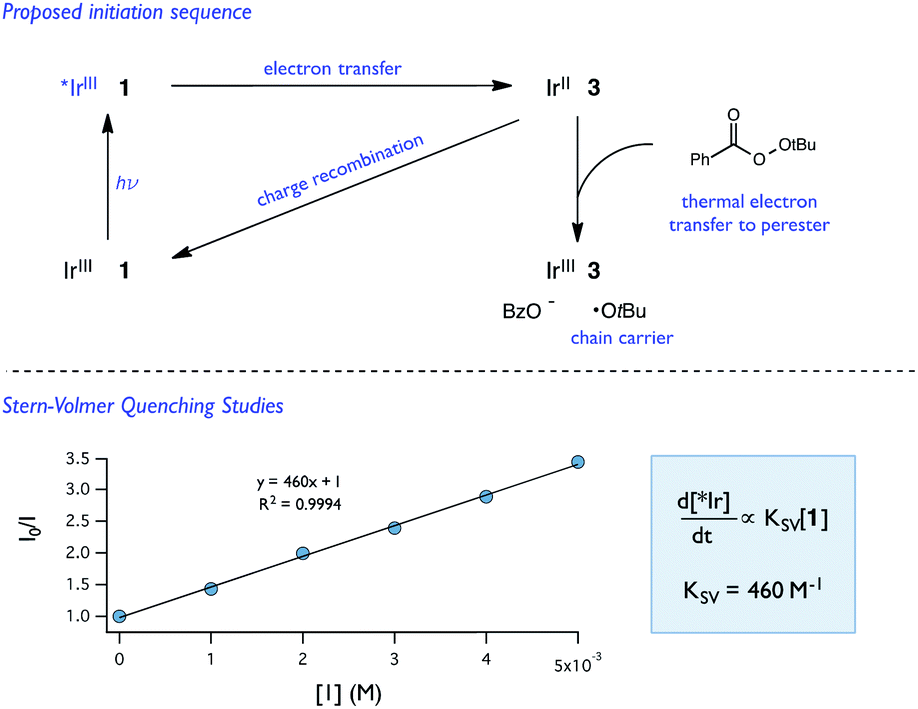 | ||
| Fig. 11 Proposed initiation sequence and Stern–Volmer quenching studies of 1 and [Ir(dF-CF3-ppy)2(dtbpy)](PF6). | ||
While Stern–Volmer studies are routinely employed to evaluate the rates of excited-state electron transfer events, kinetic evaluation of thermal electron transfers between ground state redox partners is more challenging. We were, nonetheless, able to obtain evidence for the viability of the perester reduction by IrIIvia cyclic voltammetry. Specifically, voltammograms of the Ir catalyst alone exhibit a reversible peak (E1/2 = −1.72 V vs. Fc) for the IrII/III couple (Fig. 12). However, upon addition of perester, the IrII/III couple becomes irreversible and a large catalytic wave is observed (Fig. 12). These peak features can be attributed to the reduction of IrIII on the surface of the electrode to form IrII, followed by reduction of a perester molecule in solution by the nascent IrII complex. The resulting IrIII species continues to cycle on the electrode in this fashion, giving rise to the observed catalytic current. The observation of a catalytic wave secures the viability of this elementary step and suggests that it can be plausibly incorporated into the photocatalytic mechanism. While this experimental technique is commonly used to evaluate charge transfer steps in homogeneous electrocatalysis,46,47 it has, to our knowledge, not been used as a means to study thermal electron transfers in the contemporary synthetic photoredox literature. We anticipate that this simple framework will become a general and useful approach to examine ‘dark’ electron transfers with reduced and oxidized forms of photocatalysts.
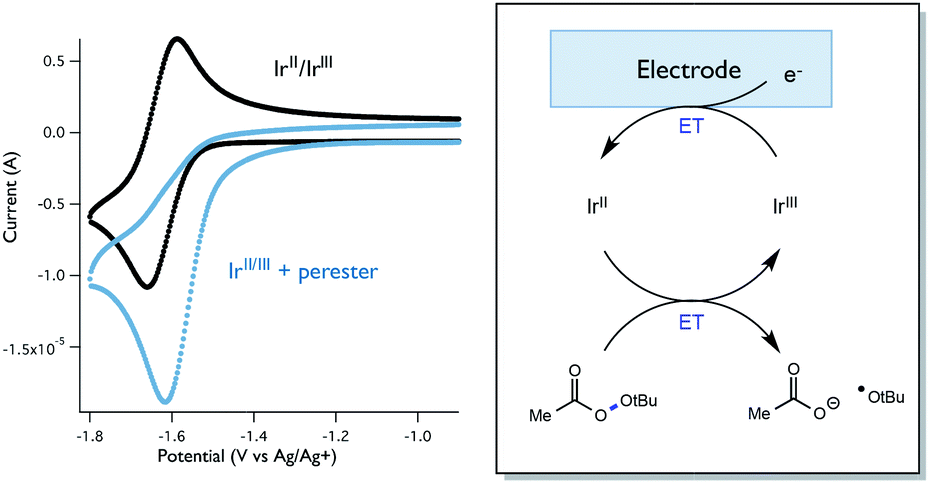 | ||
| Fig. 12 Voltammetric evidence for catalytic perester reduction by IrII. | ||
Conclusions
Herein we have reported the discovery and study of a photoredox-mediated indoline dehydrogenation for the synthesis of elbasvir. Multi-parallel experimentation identified the visible-light photoredox catalyst [Ir(dF-CF3-ppy)2(dtbpy)](PF6) and the environmentally-benign oxidant tert-butylperbenzoate as the optimal catalyst/oxidant combination, forming the product in high yields with no loss of optical purity. Using a flow reactor, ∼100 g of indoline substrate could be processed in just a few hours, providing one of the first demonstrations of photoredox catalysis in the production of a pharmaceutical intermediate. We anticipate that this demonstration will provide further support for the notion that contemporary photoredox catalysis can be carried out efficiently on process-relevant scales.A detailed mechanistic investigation revealed that this photocatalytic indoline oxidation occurs through a chain process involving a selective and irreversible hydrogen atom abstraction from the C-2 position of 1 by an alkoxy radical. This finding rationalizes the ability of these photocatalytic conditions to maintain the stereochemical integrity of the hemiaminal ether stereocenter, which readily epimerizes using most other oxidation protocols. Evidence for a chain process was secured through a series of complex kinetic isotope studies enabled by reaction progress kinetic analysis and quantum yield experiments. Notably, we expect that this collection of KIE experiments may serve as a general kinetic framework for investigating and elucidating chain mechanisms in situations where quantum yield data proves ambiguous. Additionally, we employed voltammetric techniques to demonstrate the kinetic feasibility of a modestly endergonic thermal electron transfer event between the reduced ground state of the Ir photocatalyst and the perester substrate. We anticipate that this simple protocol will become a general and useful approach to examine ‘dark’ electron transfers with reduced and oxidized photocatalysts. Taken all together, these studies provide useful new tools for studying the mechanisms of photoredox processes relevant to organic synthesis, and in particular for distinguishing between chain and non-chain processes where more classical methods fail.
Acknowledgements
Barry Rand and Tae-Wook Koh are gratefully acknowledged for assisting with measurement of LED emission spectra. We thank Donald V. Conway (Merck) and Michael K. Wismer (Merck) for design and fabrication of the flow reactor. We also thank Yi Ning Ji Chen for helpful discussions on RPKA. R. R. K. is a Fellow of the Alfred P. Sloan Foundation. We are also thank Lotus Separations for purification of the deuterated compounds 1b and 1c.Notes and references
- World Health Organization Fact Sheets: Hepatitis C, http://www.who.int/mediacentre/factsheets/fs164/en/, (accessed July 9, 2015).
- C. A. Coburn, P. T. Meinke, W. Chang, C. M. Fandozzi, D. J. Graham, B. Hu, Q. Huang, S. Kargman, J. Kozlowski, R. Liu, J. A. McCauley, A. A. Nomeir, R. M. Soll, J. P. Vacca, D. Wang, H. Wu, B. Zhong, D. B. Olsen and S. W. Ludmerer, ChemMedChem, 2013, 8, 1930–1940 CrossRef CAS PubMed
.
- H. Li, C.-Y. Chen, H. Nguyen, R. Cohen, P. E. Maligres, N. Yasuda, I. Mangion, I. Zavialov, M. Reibarkh and J. Y. L. Chung, J. Org. Chem., 2014, 79, 8533–8540 CrossRef CAS PubMed
- I. K. Mangion, C.-Y. Chen, H. Li, P. Maligres, Y. Chen, M. Christensen, R. Cohen, I. Jeon, A. Klapars, S. Krska, H. Nguyen, R. A. Reamer, B. D. Sherry and I. Zavialov, Org. Lett., 2014, 16, 2310–2313 CrossRef CAS PubMed
- For reviews on photoredox catalysis:
(a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed
-
(a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed
- For a recent example of photoredox catalysis applied towards the synthesis of a pharmaceutical target: J. J. Douglas, K. P. Cole and C. R. J. Stephenson, J. Org. Chem., 2014, 79, 11631–11643 CrossRef CAS PubMed
-
(a) D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed
- While all peroxides have an associated risk, the use of low temperatures (<0 °C) and continuous flow conditions minimize this risk. At ∼100 °C, tert-butylperbenzoate has been shown to undergo exothermic decomposition, see: S.-Y. Cheng, J.-M. Tseng, S.-Y. Lin, J. P. Gupta and C.-M. Shu, J. Therm. Anal. Calorim., 2008, 93, 121–126 CrossRef CAS
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem.–Eur. J., 2014, 20, 10562–10589 CrossRef CAS PubMed
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144–4147 CrossRef CAS PubMed
- F. R. Bou-Hamdan and P. H. Seeberger, Chem. Sci., 2012, 3, 1612–1616 RSC
- R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 4140–4143 CrossRef CAS PubMed
- Z. J. Garlets, J. D. Nguyen and C. R. J. Stephenson, Isr. J. Chem., 2014, 54, 351–360 CrossRef CAS PubMed
- D. Cantillo, O. de Frutos, J. A. Rincón, C. Mateos and C. O. Kappe, Org. Lett., 2014, 16, 896–899 CrossRef CAS PubMed
- Z. He, M. Bae, J. Wu and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 14451–14455 CrossRef CAS PubMed
- B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed
- For another recent report of a large-scale photoredox reaction, see: J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919–6 CrossRef PubMed
- PMI is a measure of the overall efficiency of the process. It is defined as the ratio between the total mass of reagent input and the total mass of product output. See ESI† for a comparative PMI analysis of both oxidation protocols (KMnO4vs. photoredox oxidation).
-
(a) K. A. Garder, L. L. Kuehnert and J. M. Mayer, Inorg. Chem., 1997, 36, 2069–2078 CrossRef
- Example of PCET-mediated reductive cleavage of peroxides: C. Costentin, V. Hajj, M. Robert, J.-M. Saveant and C. Tard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8559–8564 CrossRef CAS PubMed
- S. Antonello, F. Formaggio, A. Moretto, C. Toniolo and F. Maran, J. Am. Chem. Soc., 2001, 123, 9577–9584 CrossRef CAS PubMed
- R. Baron, A. Darchen and D. Hauchard, Electrochim. Acta, 2006, 51, 1336–1341 CrossRef CAS
- M. Finn, R. Friedline, N. K. Suleman, C. J. Wohl and J. M. Tanko, J. Am. Chem. Soc., 2004, 126, 7578–7584 CrossRef CAS PubMed
- D. Griller, J. A. Howard, P. R. Marriott and J. C. Scaiano, J. Am. Chem. Soc., 1981, 103, 619–623 CrossRef CAS
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC
- For other examples in photoredox catalysis with high reported quantum yields:
(a) Ł. Woźniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678–5681 CrossRef PubMed
- For examples in photoredox catalysis where a radical chain mechanism is invoked:
(a) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed
- For an elegant mechanistic study detecting key radical intermediates in a photoredox system, see: N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed
- For perspectives on RPKA, see:
(a) J. S. Mathew, M. Klussmann, H. Iwamura, F. Valera, A. Futran, E. A. C. Emanuelsson and D. G. Blackmond, J. Org. Chem., 2006, 71, 4711–4722 CrossRef CAS PubMed
- Notably, at very low loadings, an induction period emerges. This is likely due to trace inhibitors in solution. Furthermore, a “same excess” experiment was performed to validate that there was no kinetically significant catalyst deactivation occurring under the standard reaction conditions (0.1 mol% Ir). See ESI† for more details.
-
P. Klan and J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice, Wiley, Chichester, UK, 2009, p. 110 Search PubMed
-
(a) C. G. Hatchard and C. A. Parker, Proc. R. Soc. A, 1956, 235, 518–536 CrossRef CAS
- 60% of the photons emitted from the LEDs are emitted are between 397 nm and 407 nm. See ESI† for an emission spectrum of the LEDs used.
- It should also be noted that as the reaction progresses, the quantum yield eventually drops below 1. This does not necessarily represent a change in mechanism, but rather reflects the decline in reaction rate as starting material is depleted. It is well established that reactions with long chain lengths (>100) can exhibit quantum yields below the classic threshold of 1. For an example, refer to: J. E. Argüello, A. B. Peñéñory and R. A. Rossi, J. Org. Chem., 2000, 65, 7175–7182 CrossRef
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed
-
(a)
J. K. Kochi, Free Radicals, Wiley-Interscience, New York, 1973 Search PubMed
- For elegant kinetic studies that showcase the contribution of specific termination step on the overall rate, see:
(a) C. Walling and V. Kurkov, J. Am. Chem. Soc., 1966, 88, 4727–4728 CrossRef CAS
-
L. Melander and W. H. Saunders Jr, Reaction Rates of Isotopic Molecules, John Wiley and Sons Inc., New York, 1980, p. 95 Search PubMed
- J. M. Tanko, R. Friedline, N. K. Suleman and N. Castagnoli, J. Am. Chem. Soc., 2001, 123, 5808–5809 CrossRef CAS PubMed
-
(a) J. P. Roth, J. C. Yoder, T.-J. Won and J. M. Mayer, Science, 2001, 294, 2524–2526 CrossRef CAS PubMed
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1938, 34, 11–24 RSC
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS
- For an example where the Ir photocatalyst sensitizes substrates via energy transfer, see: Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed
- The presence of a hyperconjugative KIE during one-electron oxidation of 1 was also investigated with a Stern–Volmer luminescence quenching experiment and no significant KIE was observed. For experimental details, refer to the ESI†.
-
J.-M. Saveant, Elements of molecular and biomolecular electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, Wiley-Interscience, Hoboken, NJ, 2006, ch. 4 Search PubMed
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed
- Y. Fu, L. Liu, H.-Z. Yu, Y.-M. Wang and Q.-X. Guo, J. Am. Chem. Soc., 2005, 127, 7227–7234 CrossRef CAS PubMed
- S. J. Konezny, M. D. Doherty, O. R. Luca, R. H. Crabtree, G. L. Soloveichik and V. S. Batista, J. Phys. Chem. C, 2012, 116, 6349–6356 CAS
-
E. T. Denisov, T. G. Denisova and T. S. Pokidova, Handbook of Free Radical Initiators. Wiley – Interscience, Hoboken, NJ, 2003, p. 772 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data for all new compounds. See DOI: 10.1039/c5sc03350k |
| This journal is © The Royal Society of Chemistry 2016 |