
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclopentadiene-mediated hydride transfer from rhodium complexes†
C. L.
Pitman
,
O. N. L.
Finster
and
A. J. M.
Miller
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: ajmm@email.unc.edu
First published on 1st March 2016
Abstract
Attempts to generate a proposed rhodium hydride catalytic intermediate instead resulted in isolation of (Cp*H)Rh(bpy)Cl (1), a pentamethylcyclopentadiene complex, formed by C–H bond-forming reductive elimination from the fleeting rhodium hydride. The hydride transfer ability of diene 1 was explored through thermochemistry and hydride transfer reactions, including the reduction of NAD+.
Transition metal catalysts capable of selective hydride transfer to the enzyme cofactor nicotinamide adenine dinucleotide (NAD+) to form the 1,4-reduced product (1,4-NADH) are critical links between organometallic and enzymatic catalysis in emerging strategies in sustainable, enantioselective organic synthesis.1 Biocompatible catalytic routes for 1,4-NADH regeneration provide access to enzymatic hydride transfer reactivity without stoichiometric amounts of the complex molecule 1,4-NADH.2 Of the organometallic catalysts that have been shown to regenerate NADH, rhodium complexes have emerged as selective and efficient catalysts for reduction at the 4-position of nicotinamides, spurring innovation in tandem bio-organometallic catalysis (Scheme 1).1b
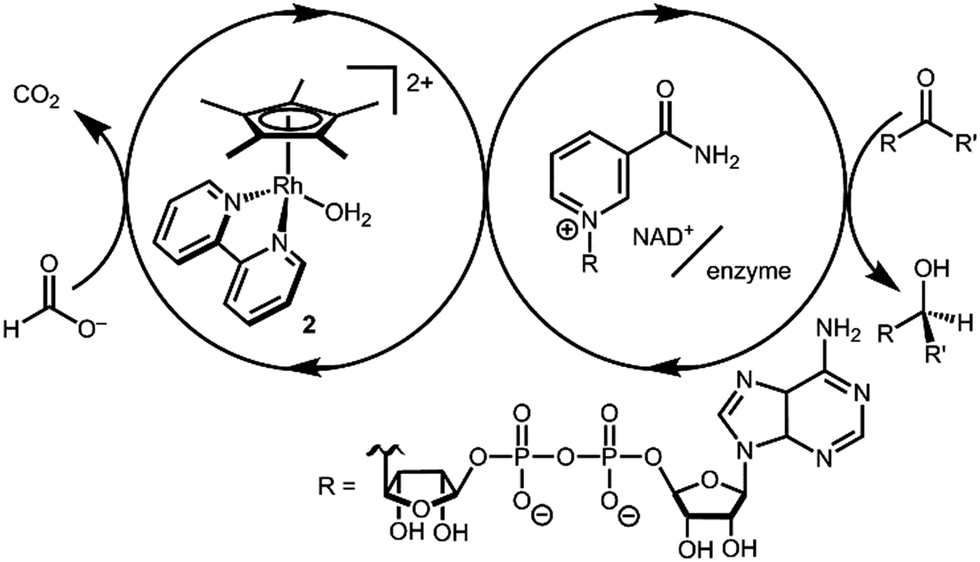 | ||
| Scheme 1 Tandem catalytic cycle for Rh, NAD+, and enzyme mediated reductions. | ||
In the presence of a precatalyst like [Cp*Rh(bpy)(OH2)]2+ (2; Cp* is pentamethylcyclopentadienyl and bpy is 2,2′-bipyridine), generation of 1,4-NADH can be accomplished using chemical reductants (e.g. formate) or by electrochemical methods (by 1H+/2e−). The mechanism is typically proposed to proceed via [Cp*Rh(bpy)(H)]+ (3) with selectivity directed by coordination of NAD+ to the Rh centre after an η5- to η3-Cp* ring slip.3 Drawing on this mechanism, Cp*Rh(bpy)-based catalysts have been applied in ketone and aldehyde reductions4 and hydrogen evolution.5
After considering the hydricity, or hydride donor ability, of the iridium analogues [Cp*Ir(bpy)(H)]+,6 we were interested in the comparison to rhodium hydride 3. Relatively few hydricity values have been determined in water, and these Rh complexes provided an opportunity to learn more about an important catalytic intermediate and add new data to the emerging area of aqueous hydricity.7
In order to determine the hydricity of 3, we first needed a preparative route for this proposed—but not previously isolated—intermediate. Reduction of [Cp*Rh(bpy)(Cl)][Cl] (4) in a pH 5 formate solution (following a procedure that cleanly generates the Ir analogue [Cp*Ir(bpy)(H)][PF6])8 produced a dark red solution from which a green solid precipitated on addition of [NH4][PF6]. Dissolution of the solids in CD3CN cleanly produced a red solution containing a new species. Surprisingly, the Cp* methyl resonances were inequivalent: two singlets (6H integration each) and a doublet (J = 6.2 Hz, 3H) were present in the aliphatic region, and a downfield quartet (δ 2.31, J = 6.2 Hz, 1H) indicated a pentamethylcyclopentadiene (Cp*H) fragment containing a new C–H bond (Fig. S4, ESI†).
An alternative procedure involving protonation of a reduced Cp*Rh(bpy) (5) species was also attempted. Reduction of chloride 4 by NaBH4 in 1 M NaOH led to precipitation of dark purple 5. Dropwise addition of a dilute solution of HCl·Et2O to an ethereal solution of 5 produced a Cp*H-containing product similar to the one described above.
Crystals suitable for X-ray diffraction were prepared by vapour diffusion of a solution of the Cp*H complex in DCM with pentane. The resulting molecular structure revealed the product to be (Cp*H)Rh(bpy)(Cl) (1), a Rh(I) complex containing a η4-pentamethylcyclopentadiene ligand with the new C–H bond endo with respect to the metal centre (Fig. 1). The long C1–C2 distance (1.517(2) Å) compared to the short C2–C3 (1.440(3) Å) distance confirms that the species is a diene. In contrast, the crystal structure of Cp*Rh(I) complex 5 shows only a 0.034 Å difference amongst the cyclopentadienyl C–C bonds.9 Aromaticity has clearly been broken with a C2′–C1–C2–C3 torsional angle of 31.9(2)° compared to 3.42° in 5. The bromide analogue (Cp*H)Rh(bpy)(Br) was isolated by Winkler, Gray and Blakemore during the preparation of this manuscript and is being investigated as a possible intermediate in H2 evolution in acetonitrile.10
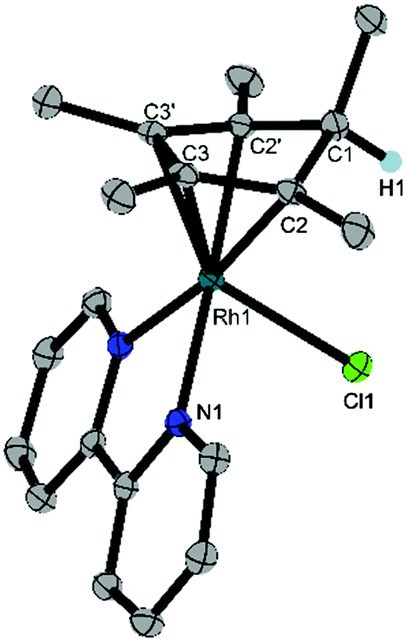 | ||
| Fig. 1 Structural representation of 1 with ellipsoids drawn at 50% of probability level (a mirror plane bisects the bpy ligand). A CH2Cl2 solvent molecule and hydrogens except H1 are omitted for clarity. Selected distances (Å) and angles (deg): C1–C2 1.517(2), C2–C3 1.440(3), C3–C3′ 1.430(4), Rh1–N1 2.1157(15), Rh1–Cl1 2.5440(6), C2′–C1–C2–C3 31.9(2). | ||
The structure of complex 1 yields clues about the probable mechanism of its formation. The endo orientation of the hydride is consistent with C–H bond-forming reductive elimination of Cp* and a Rh–H. Reductive elimination of Cp* with hydride ligands has been observed from Rh and Ir metal hydrides with dissociation of the free diene,11 and Cp*Rh(Cp*H) has been prepared.12 As shown in Scheme 2, a Rh hydride intermediate is also consistent with the observation that the Cp*H product is formed both by hydride transfer from formate and by protonation of 5.
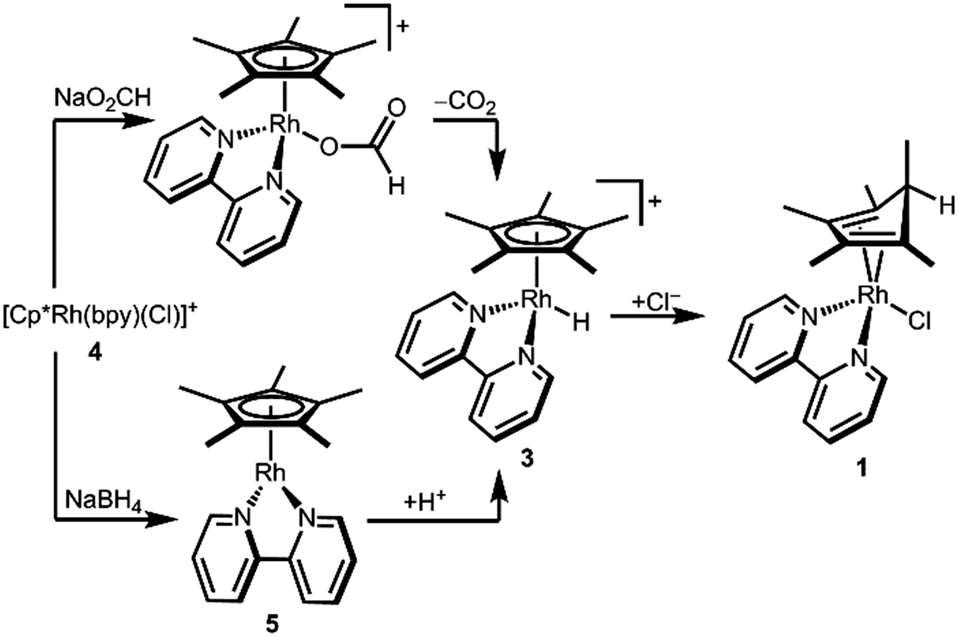 | ||
| Scheme 2 Alternative routes to diene 1. | ||
The intermediacy of a metal hydride was probed by low temperature NMR experiments. Indeed, protonation of 5 with HCl at 233 K allowed the observation of a Rh–H resonance by 1H NMR in a pre-cooled probe (δ −9.60, JRhH = 19.9 Hz, Fig. S5, ESI†), which converted to diene 1 upon warming.
Density functional theory (DFT) calculations are consistent with a Rh hydride intermediate that is unstable towards C–H reductive elimination. As illustrated in Scheme 3, reductive elimination of the Rh hydride to form the Cp*H complex is favourable by −4.1 kcal mol−1. In contrast, the Ir analogue [Cp*Ir(bpy)(H)]+, which has been isolated and structurally characterized,8 is predicted to be unfavourable to Cp*H elimination by 8.1 kcal mol−1. Interestingly, the only prior report of a similar bpy-supported Rh hydride complex is the methyl-substituted complex [Cp*Rh(6,6′-Me-bpy)(H)]+, which features steric bulk that might influence this equilibrium.13
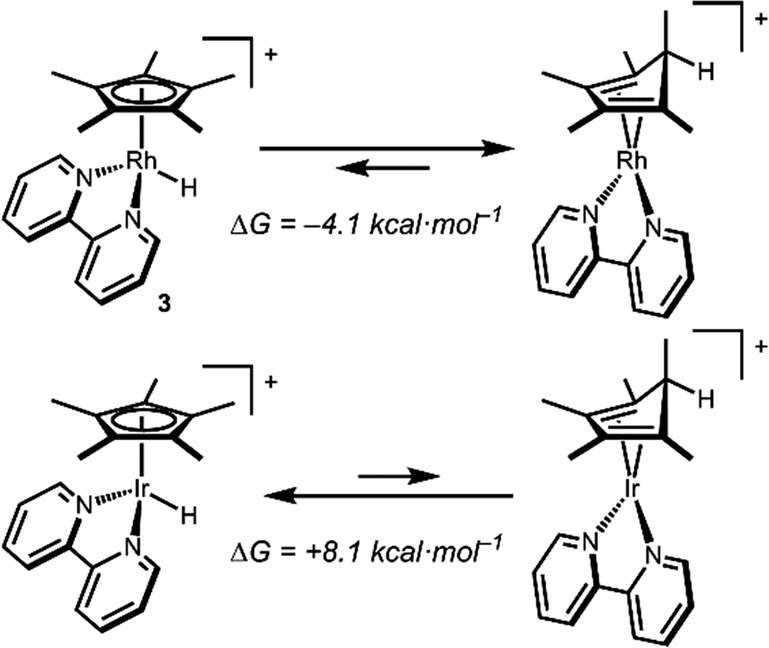 | ||
| Scheme 3 Free energies for reductive elimination from Cp*M–H (M = Rh, Ir) by DFT. | ||
The apparent instability of the Rh hydride intermediate with respect to reductive elimination raises questions about how Cp*Rh-based catalysts mediate hydride transfer reactions. Diene 1 could undergo hydride transfer indirectly via a Rh–H intermediate or via a C–H bond-breaking direct hydride transfer. The latter mechanism illustrates the similarity between diene 1 and a variety of transition metal complexes ligated by organic hydride donors and acceptors14 and non-innocent ligand backbones capable of de-aromatization.15
To better understand complex 2, we sought to measure the hydricity and establish hydride transfer reactivity. We focused on the closely related complex [(Cp*H)Rh(4,4′-COO-bpy)]− (1COO) due to its favourable solubility profile in water.16 For Ir–H complexes, carboxylate substitution has a very minor impact on hydricity,6 and with the additional distance to the substitution site, the impact on hydricity is expected to be similarly minor for (Cp*H)Rh complexes.
The hydricity (eqn (5)) was established by determining the pKa of 1COO (eqn (1)), the reduction potential of [Cp*Rh(bpy-COO)(OH)]− (eqn (2)) and the pKa of the Rh(III) aquo complex Cp*Rh(bpy-COO)(OH2) (2COO; eqn (3)). Combining these experimental values with the free energy of 2e− proton reduction (eqn (4))17 provides 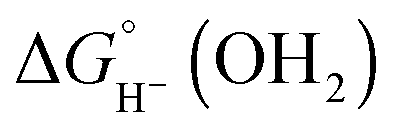 , the effective hydricity with the formation of an aquated product, according to eqn (6).
, the effective hydricity with the formation of an aquated product, according to eqn (6).
| 1COO ⇆ 5COO + H+ | (1) |
| 5COO + OH− ⇆ [Cp*Rh(bpy-COO)(OH)]− + 2e− | (2) |
| [Cp*Rh(bpy-COO)(OH)]− + H+ ⇆ 2COO | (3) |
| H+ + 2e− ⇆ H− | (4) |
| 1COO + H2O ⇆ 2COO + H− | (5) |
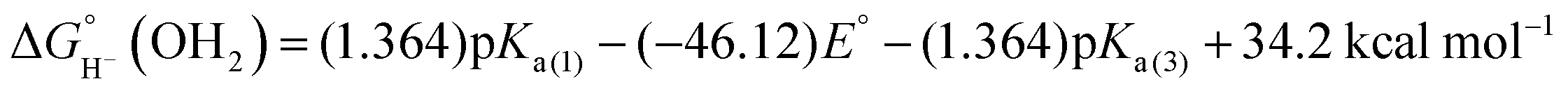 | (6) |
The reduction potential was measured by cyclic voltammetry (CV) in aqueous phosphate electrolyte. Above pH 9, the 2e− reduction of [Cp*Rh(bpy-COO)(OH)]− to [Cp*Rh(bpy-COO)]2− (5COO) is quasi-reversible (ΔEp = 30–80 mV in the pH range) and E1/2 shifts cathodically by 24.6 mV per pH unit, close to the ideal 29.5 mV per pH unit shift of a 1OH−/2e− process (Fig. S7, ESI†). Extrapolating this trend to pH 0 (the standard state in eqn (1)–(5)) provides the formal potential, E° = −0.25 V, for the reduction of the hydroxide complex.
To confirm the products of electrochemical reduction, controlled potential electrolysis (CPE) of [Cp*Rh(bpy-COO)(OH)]− was performed under basic conditions. CPE resulted in a midnight blue solution of 5COO after passing 2e− per Rh of charge. Upon addition of pD 7 0.1 M sodium phosphate buffer, the blue solution turned red and 1H NMR spectroscopy confirmed formation of 1COO, as indicated by the characteristic 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:3 pattern of the Cp* methyl resonances.
:6:3 pattern of the Cp* methyl resonances.
Diene 1COO has pKa < 10 based on a spectrophotometric titration adding acid to an aqueous solution of 5COO (Fig. S9, ESI†). The relative instability of these Rh species (vide infra) led us to carry out a complementary electrochemical titration by monitoring the growth of the oxidation of 5COO by CV as a function of solution pH, providing pKa > 8 (Fig. S10, ESI†). Each method provides a limiting value (see ESI† figure captions), and we, therefore, estimate that 1COO has pKa = 9 ± 1.
The Rh(III) species exists as the aquo Cp*Rh(bpy-COO)(OH2) (2COO), not the hydroxo complex, under the neutral, aqueous conditions of most catalysis.5a Incorporation of the pKa of the aquo complex (8.8 by spectrophotometric titration) accounts for this protonation state.
Based on the experimentally determined E° and pKa values, eqn (6) provides the aqueous hydricity of [(Cp*H)Rh(bpy-COO)]− to form aquo 2COO: 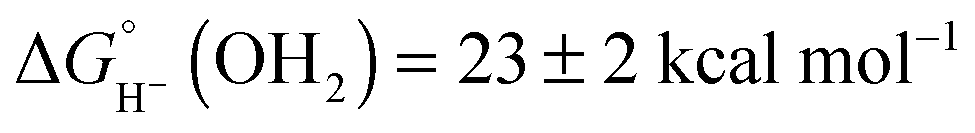 .
.
Hydride transfer to complex 2COO from species with 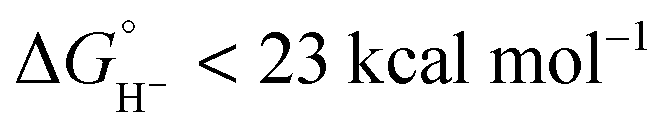 is expected to be favourable, and hydride transfer to unsubstituted 2 is expected to proceed with similar driving forces. As expected, [(C6Me6)Ru(bpy)(H)]+
is expected to be favourable, and hydride transfer to unsubstituted 2 is expected to proceed with similar driving forces. As expected, [(C6Me6)Ru(bpy)(H)]+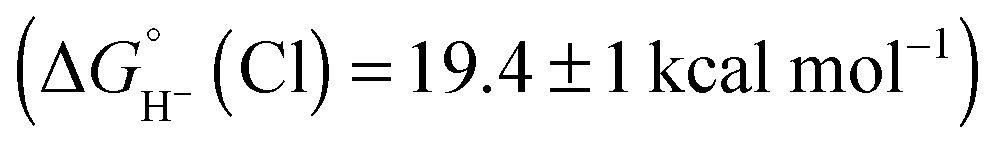 6 reacts with chloride 4 (Cl− is displaced in water5a) to produce the corresponding hydride transfer product 1 (Scheme 4). The product slowly decomposed, preventing the system from reaching equilibrium. Transfer does not occur from weaker hydride sources: combining [Cp*Ir(bpy-COO)(H)]−
6 reacts with chloride 4 (Cl− is displaced in water5a) to produce the corresponding hydride transfer product 1 (Scheme 4). The product slowly decomposed, preventing the system from reaching equilibrium. Transfer does not occur from weaker hydride sources: combining [Cp*Ir(bpy-COO)(H)]−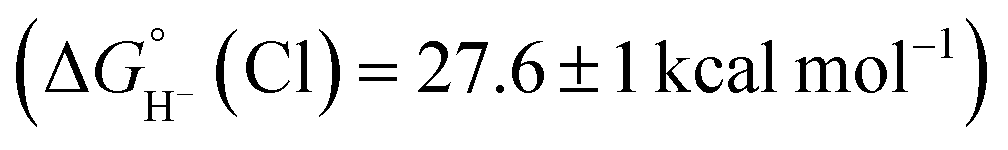 6 with 4 results in no reaction. In accord with the hydricity values, in the reverse reaction diene 1 reacted completely with [Cp*Ir(bpy-COO)(Cl)]− to form [Cp*Ir(bpy-COO)(H)]−.
6 with 4 results in no reaction. In accord with the hydricity values, in the reverse reaction diene 1 reacted completely with [Cp*Ir(bpy-COO)(Cl)]− to form [Cp*Ir(bpy-COO)(H)]−.
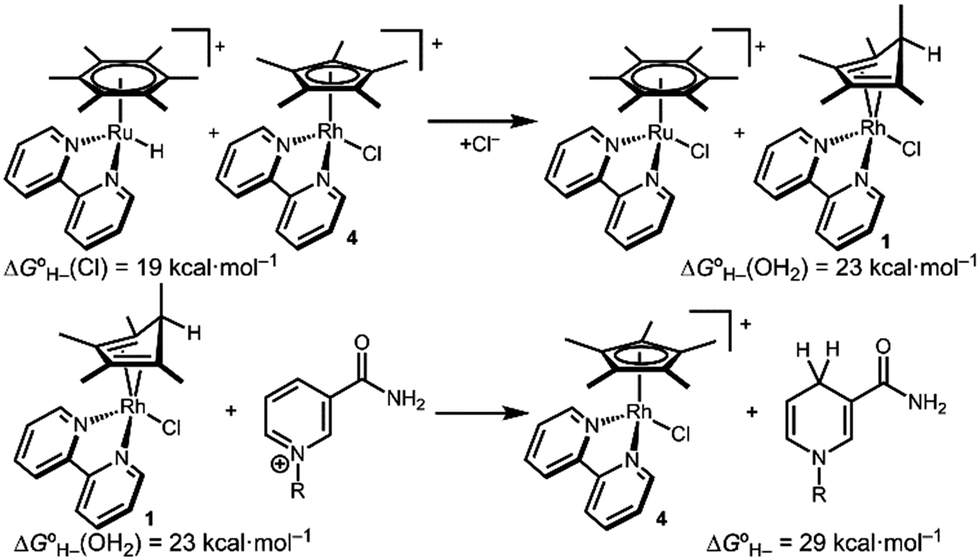 | ||
| Scheme 4 Selected hydride transfer reactions. | ||
After establishing the viability of diene complex 1 in hydride transfer reactions with transition metal complexes, we turned our attention to hydride transfer involving NAD+. The hydricity of 1,4-NADH is approximately 29 kcal mol−1 (see ESI†),18 so the Rh diene complex 1 should be sufficiently hydridic to reduce NAD+. A red solution of isolated 1 quickly turned yellow on addition of NAD+. 1H NMR spectroscopy confirmed consumption of 1 and selective production of 1,4-NADH within 15 minutes.
Finally, we assessed the viability of diene species 1 as an intermediate on the NAD+ reduction cycle by mimicking various chemical and electrochemical catalytic conditions typically employed. Reduction of 4 in D2O with 10 eq. formate forms the red hydride migrated complex 1 immediately, as judged by the appearance of a 6:6:3 pattern in the Cp* region. The same species is also formed upon reduction of chloride 4 at −0.64 V vs. NHE in pD 7 0.1 M phosphate buffer. Even treatment of aquo 2 with 1 atm H2 in pD 7 0.1 M phosphate buffer produced a diene complex.
The presence of 1 under catalytically relevant conditions indicates that it is a viable intermediate. Complex 1 is not the only Rh species in these solutions, however, and this species does not exhibit long term stability under aqueous conditions. Bubbles formed on the walls of NMR tubes containing 1 in neutral aqueous solutions, indicating H2 evolution. The Cp* methyl protons also scrambled H for D. Such scrambling has been observed for Cp* ligands and typically proceeds through a base-assisted mechanism via fulvene intermediates.19 We have also observed the per-deuteration of Cp* in [Cp*Ir(bpy-COO)(H)]− by 2H NMR and MS, but deuteration in the Ir manifold occurs over the course of weeks, while deuteration in the Rh manifold occurs over the course of hours. Broad resonances shifted slightly upfield of each proteo Cp*H signal appear quickly before the signals slowly disappear altogether.
Scheme 5 combines our new findings with Fish's original mechanistic proposal3 to construct an alternative mechanistic hypothesis. Starting from the aquo precatalyst 2, a 1H+/2e− reduction (either by a hydride donor, e.g. formate, or through reduced species 5) transiently produces metal hydride 3. Reductive elimination yields a (Cp*H)Rh moiety. The endo orientation of the proton seems to ideally position the C–H bond to deliver hydride to a bound substrate such as NAD+ ligating the Rh centre. Following hydride transfer, displacement of NADH by water regenerates the initial state of the catalyst. Several other mechanisms can be envisioned, such as hydride transfer via reversible access to the high energy hydride intermediate 3. The mechanism in Scheme 5 offers an alternative path for substrate binding without invoking an η5- to η3-Cp* ring slip.
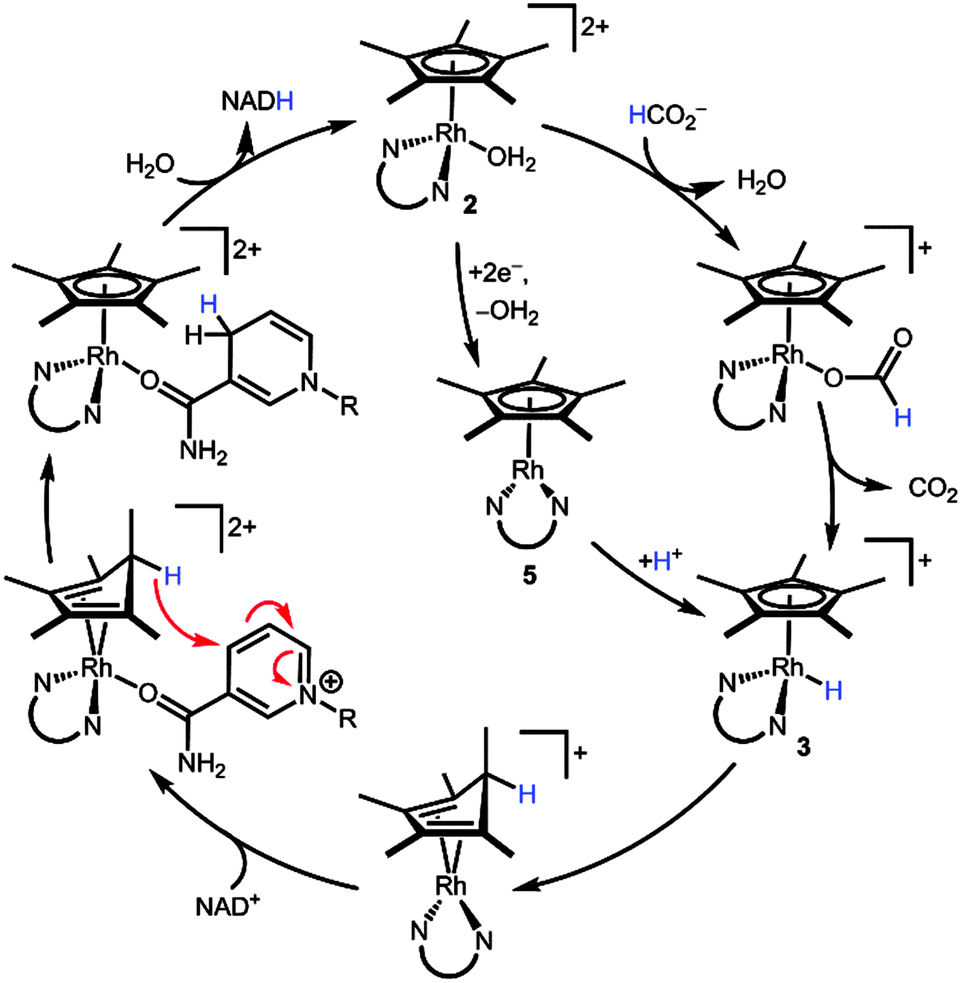 | ||
| Scheme 5 Proposed mechanism for the reduction of NAD+ through a [(Cp*H)Rh(bpy)]+ intermediate. N∪N is bpy. | ||
We have prepared a pentamethylcyclopentadiene complex of Rh that is a plausible intermediate in the selective catalytic reduction of NAD+ to 1,4-NADH. Hydricity measurements confirm that diene 1 is thermodynamically capable of hydride transfer to NAD+. A series of hydride transfer reactions to NAD+ and other transition metals are consistent with the hydricity value. This surprising ligand-based hydride transfer reactivity, involving the typically innocent pentamethylcyclopentadienyl ligand, suggests new pathways for Cp*Rh-catalyzed management of protons and electrons.
The authors gratefully acknowledge funding from NSF Center for Enabling New Technologies through Catalysis (CENTC), CHE-1205189, the University of North Carolina at Chapel Hill, and the Royster Society of Fellows (C. L. P.). We thank P. S. White for crystallographic support and K. R. Brereton for her assistance with DFT.
Notes and references
-
(a) J.-M. Fang, C.-H. Lin, C. W. Bradshaw and C.-H. Wong, J. Chem. Soc., Perkin Trans. 1, 1995, 967–978 RSC
; (b) H. C. Lo and R. H. Fish, Angew. Chem., Int. Ed., 2002, 41, 478–481 CrossRef CAS
- W. A. van der Donk and H. Zhao, Curr. Opin. Biotechnol., 2003, 14, 421–426 CrossRef CAS PubMed
- H. C. Lo, C. Leiva, O. Buriez, J. B. Kerr, M. M. Olmstead and R. H. Fish, Inorg. Chem., 2001, 40, 6705–6716 CrossRef CAS PubMed
-
(a) T. Ghosh, T. Slanina and B. König, Chem. Sci., 2015, 6, 2027–2034 RSC
-
(a) U. Kölle and M. Grätzel, Angew. Chem., Int. Ed. Engl., 1987, 26, 567–570 CrossRef
- C. L. Pitman, K. R. Brereton and A. J. M. Miller, J. Am. Chem. Soc., 2016, 138, 2252–2260 CrossRef CAS PubMed
-
(a) C. Creutz and M. H. Chou, J. Am. Chem. Soc., 2009, 131, 2794–2795 CrossRef CAS PubMed
- T. Abura, S. Ogo, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 4149–4154 CrossRef CAS PubMed
- J. D. Blakemore, E. S. Hernandez, W. Sattler, B. M. Hunter, L. M. Henling, B. S. Brunschwig and H. B. Gray, Polyhedron, 2014, 84, 14–18 CrossRef CAS
- L. M. A. Aguirre Quintana, S. I. Johnson, S. L. Corona, W. Villatoro, W. A. Goddard III, M. K. Takase, D. G. VanderVelde, J. R. Winkler, H. B. Gray and J. D. Blakemore, submitted.
-
(a) W. D. Jones, V. L. Kuykendall and A. D. Selmeczy, Organometallics, 1991, 10, 1577–1586 CrossRef CAS
-
(a) O. V. Gusev, L. I. Denisovich, M. G. Peterleitner, A. Z. Rubezhov, N. A. Ustynyuk and P. M. Maitlis, J. Organomet. Chem., 1993, 452, 219–222 CrossRef CAS
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568–1577 CrossRef CAS
-
(a) Y. Matsubara, S. E. Hightower, J. Chen, D. C. Grills, D. E. Polyansky, J. T. Muckerman, K. Tanaka and E. Fujita, Chem. Commun., 2014, 50, 728–730 RSC
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed
- 1COO may be ligated by H2O, but the solvent is assigned unity activity so the thermodynamics are not affected.
- S. J. Connelly, E. S. Wiedner and A. M. Appel, Dalton Trans., 2015, 44, 5933–5938 RSC
-
(a) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed
-
(a) J. W. Kang and P. M. Maitlis, J. Organomet. Chem., 1971, 30, 127–133 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR and UV-vis spectra. Hydricity determination and DFT details. CCDC 1447440 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00575f |
| This journal is © The Royal Society of Chemistry 2016 |