
Facile and expedient access to bis-coumarin–iminothiazole hybrids by molecular hybridization approach: synthesis, molecular modelling and assessment of alkaline phosphatase inhibition, anticancer and antileishmanial potential†
Aliya Ibrara,
Sumera Zaibb,
Imtiaz Khana,
Farukh Jabeencd,
Jamshed Iqbal*b and
Aamer Saeed*a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad-45320, Pakistan. E-mail: aamersaeed@yahoo.com; Fax: +92-51-9064-2241; Tel: +92-51-9064-2128
bCentre for Advanced Drug Research, COMSATS Institute of Information Technology, Abbottabad-22060, Pakistan. E-mail: drjamshed@ciit.net.pk
cFlorida Center for Heterocyclic Compounds, Department of Chemistry, University of Florida, Fl. 32611, USA
dCenter for Computationally Assisted Science and Technology, North Dakota State University, Fargo, ND 58102, USA
First published on 14th October 2015
Abstract
In the design of new drugs, the hybridization approach might allow obtaining molecules with improved biological activity with respect to the corresponding lead compounds. Thus, adopting this approach, a new series of novel bis-coumarin–iminothiazole hybrids was designed, synthesized and tested for their biological action against alkaline phosphatase, leishmaniasis and cancerous cells. The structures of the synthesized hybrid compounds (5a–m) were characterized and established by using spectro-analytical data. The synthesized analogues were evaluated against alkaline phosphatase where compound 5j was emerged as a potent inhibitor with IC50 value of 1.38 ± 0.42 μM. This inhibitory efficacy is two-fold higher as compared to the standard inhibitor. The synthesized compounds were also assayed for their anti-leishmanial potential against Leishmania major and compound 5i was observed as the lead candidate with 70.4% inhibition. The prepared compounds also showed cytotoxic behavior against kidney fibroblast (BHK-21) and lung carcinoma (H-157) cancer cell lines. Molecular docking of the synthesized library of iminothiazole derivatives against ALP was performed to delineate ligand–protein interactions at molecular level which suggested that the major interacting residues in the binding sites of the proteins might have an instrumental role in the inhibition of enzyme's function. Our results inferred that compounds 5j–m may serve as potential surrogates for the development of potent inhibitors of ALP.
Introduction
Alkaline phosphatase (ALP, E.C. 3.1.3.1.) is a non-specific phosphomonoester hydrolase that catalyzes the hydrolysis and transphosphorylation of a wide variety of organic monophosphates and regulates the functions of many biological systems.1–3 The most common occurrence of ALP in nature suggests its involvement in fundamental biochemical processes, however, there is no positive evidence regarding its physiological functions. The possible roles of ALP have been suggested as hydrolysis of phosphoesters, phosphate transferase activity, protein phosphatase activity, phosphate transport, modulation of organic cation transport, and involvement in cell proliferation.4,5 The biological action of the alkaline phosphatase in serum is associated with metabolic bone (hypophosphatasia) and liver diseases and also is used as a marker of osteoblastic differentiation.6 Alkaline phosphatases may potentially be employed as therapeutic agents and show several uses in clinical medicine and in biotechnology. The most common human cancers like breast, kidney, lung and prostate have a great potential to metastasize to bone, directing towards painful and fatal consequences.7 Bisphosphonates are widely used to reduce symptoms of osteolytic bone metastasis commonly seen in breast, kidney and lung cancer.8 High serum level of tissue non-specific alkaline phosphatases represents elevated osteoblast activity in patients with metastatic prostate cancer.9,10 The chemical agent that inhibit alkaline phosphatase enzyme may be useful to prevent increased osteoblast activity, commonly seen in bone metastasis.Cancer is a notably complex, widespread and lethal disease and at present one of the most leading causes of death accounting for 7.6 million (around 13% of all deaths) in 2008, that are projected to continue rising, with an estimated 13.1 million deaths in 2030.11 Cancer can affect almost every tissue lineage in the human body and poses great challenges to medical science. Most cancers are characterized by uncontrolled cell proliferation, lack of cell differentiation and loss of contact inhibition, which confers upon the tumor cell a capability to invade local tissues and metastasize.12,13 Nearly all cancers are caused by abnormalities in the genetic material of the transformed cells and involve deregulation of essential enzymes and other proteins controlling cell division and proliferation.14 Many efforts have been made to the discovery of new anticancer agents which is of a paramount importance.
Over three billion people are estimated to be at risk for the parasitic diseases such as malaria, trypanosomiasis and leishmaniasis.15 Leishmaniasis is a widespread parasitic disease that is caused by protozoan parasites of the genus Leishmania in tropical and subtropical areas in both the old and new worlds. According to recent World Health Organization reports, 88 countries are affected, comprehending 12 million infected people worldwide, with approximately 350 million people at risk. The incidence is increasing worldwide, with 1–2 million new cases registered annually, despite all efforts being made to fight the disease.15–18 It occurs in three major forms; cutaneous (CL), muco-cutaneous (MCL) and visceral leishmaniasis (VL), depending upon parasite species. L. donovani and L. infantum are major causative agents of VL, while L. major, L. tropica, L. aethiopica, L. braziliensis, L. panamensis, L. amazonensis and L. mexicana cause CL.19 L. braziliensis and L. panamensis are also agents of muco-cutaneous leishmaniasis. Therapy of patients with leishmaniasis is still a serious problem and urgent and sustained efforts are needed for the development of new, efficient, and safe drugs.
Heterocycles occupy a central position in organic chemistry20–22 and are an integral part of the chemical and life sciences. Heterocyclic ring systems have emerged as powerful scaffolds for many biological evaluations playing an important role in the design and discovery of new physiological/pharmacologically active molecules.23 Among them, 1,3-thiazole is an important and versatile structural analogue possessing a diverse spectrum of biological actions.24–30 On the other hand, as a fragrant compound, coumarin derivatives are known to possess multiple biological functions, including anti-HIV, anti-acetylcholinesterase, antifungal, antioxidant, antihelmintic, antitumor, antibacterial, antiviral, anticancer, and anticlotting activities, and find extensive application in pharmaceuticals and agrochemicals.31–35
The molecular hybridization strategy involving the integration of two or more pharmacophoric units in the same molecule, is a rationally attractive approach for the design and development of new bioactive agents.36,37 These combined pharmacophores probably offer some advantages such as in overcoming drug resistance38 as well as improving their biological potency.39 The literature survey revealed the biological importance of coumarin-hybrid compounds associated with an array of biological functions. These molecules possess coumarin and other heterocyclic and non-heterocyclic skeletons (Fig. 1).40 In view of literature findings and our continued interest aiming at developing new molecules as potent biological candidates,31,41,42 we designed and synthesized a new series of bis-coumarin–iminothiazole hybrids using molecular hybridization approach. The synthesized compounds were evaluated for their alkaline phosphatase inhibition, anticancer and antileishmanial potential. In addition, molecular docking studies of these compounds have also been carried out to gain further insights into the biological properties. The results of this study are presented in this paper.
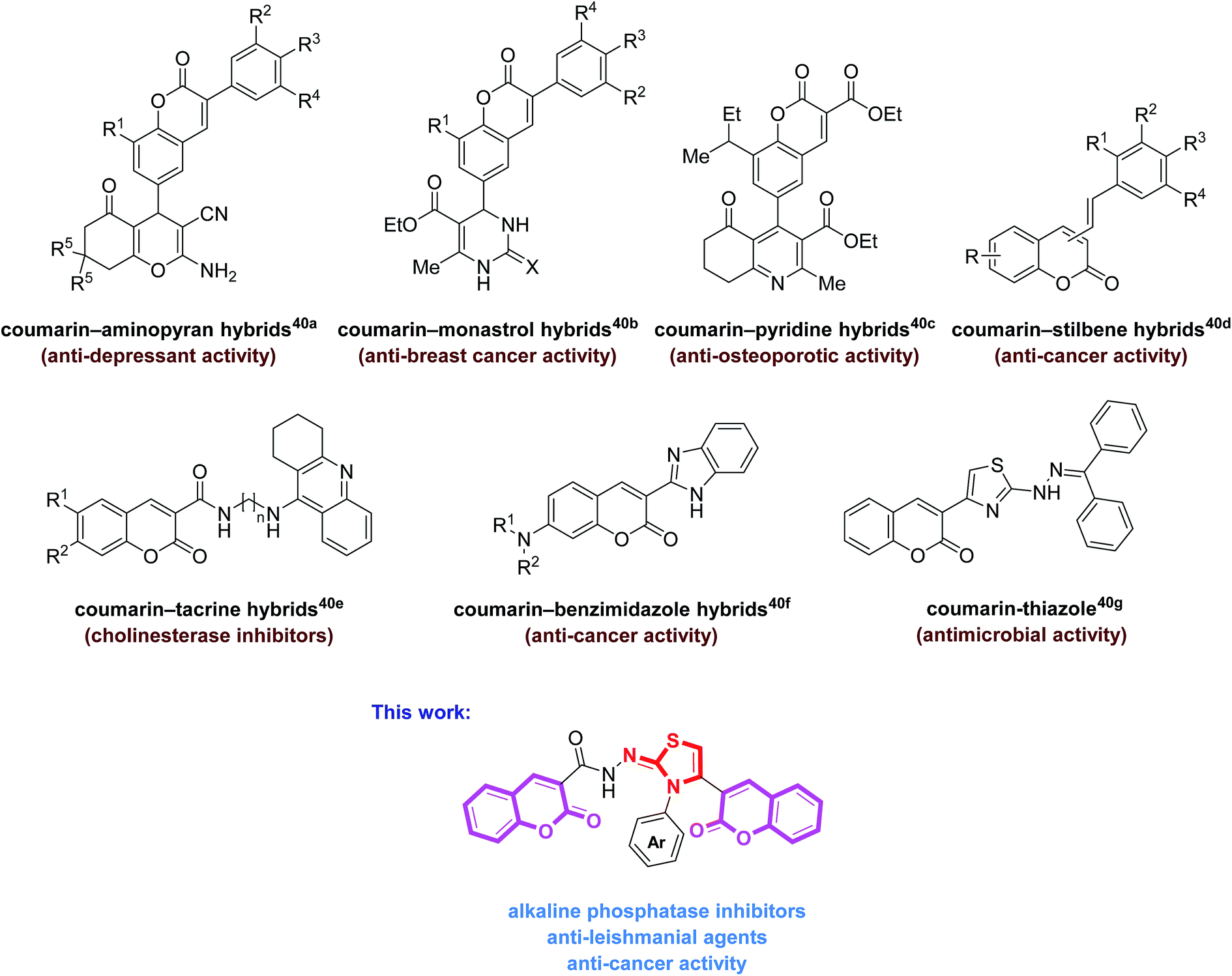 | ||
| Fig. 1 Examples of bioactive coumarin-hybrid motifs and our designed hybrid molecules. | ||
Results and discussion
Synthesis and characterization of bis-coumarin–iminothiazole hybrids
The reaction sequence employed for the synthesis of bis-coumarin–iminothiazole compounds (5a–m) is illustrated in Scheme 1. To a cold mixture of salicylaldehyde (1) and ethyl acetoacetate (2) was added catalytic amount of piperidine. The reaction mixture was stirred for 30 min at room temperature. The precipitated solid was filtered off and recrystallized (chloroform) to afford 3-acetyl-2H-chromen-2-one in good yield.43 The formation of 3-acetyl-2H-chromen-2-one was indicated by its IR spectrum where absorption bands observed at 1759 and 1705 cm−1 were attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of ketone and lactone functionalities. 1H NMR spectral data exhibited resonance for characteristic coumarin proton (H-4) at 8.66 ppm along with other aromatic protons at appropriate chemical shift values. Methyl protons were also observed as singlet at 2.10 ppm. 3-Acetyl-2H-chromen-2-one was also confirmed by 13C NMR data where characteristic carbonyl carbon (ketone) appeared at 197.3 ppm in addition to lactonic carbon resonating at 156.6 ppm.
O of ketone and lactone functionalities. 1H NMR spectral data exhibited resonance for characteristic coumarin proton (H-4) at 8.66 ppm along with other aromatic protons at appropriate chemical shift values. Methyl protons were also observed as singlet at 2.10 ppm. 3-Acetyl-2H-chromen-2-one was also confirmed by 13C NMR data where characteristic carbonyl carbon (ketone) appeared at 197.3 ppm in addition to lactonic carbon resonating at 156.6 ppm.
 | ||
| Scheme 1 Synthesis of bis-coumarin–iminothiazole hybrid compounds (5a–m). | ||
3-Acetyl-2H-chromen-2-one was brominated by refluxing with Br2 solution in chloroform.40 FTIR spectral data indicated the formation of compound (3) where stretching absorptions appeared at 1743 and 1716 cm−1 for ester and ketone carbonyls. 1H NMR exhibited characteristic signal for methylene carbon as singlet at 4.90 ppm. Furthermore, 13C NMR also confirmed the bromination of acetyl coumarin as methylene carbon resonated at 55.4 ppm.
Reaction of 3-(2-bromoacetyl)-2H-chromen-2-one (3) and substituted thiosemicarbazides (4a–m), synthesized previously,31 in methanol under basic conditions afforded title products (5a–m) in pleasing yields.44 The structural diversity on this scaffold was ensured by using a range of electron-rich and electron-deficient functional groups, attached to the thiazole core. The formation of thiazole derivatives (5a–m) was evidenced by IR spectroscopy where characteristic stretching absorptions observed in the range of 3410–3293 and 1629–1607 cm−1 were due to N–H and CN groups, respectively. In 1H NMR spectra, these compounds showed dominant signals in the range of 11.90–10.98 ppm, assigned to N–H moiety. 13C NMR spectra also confirmed the presence of CN group in the range of 159.7–157.2 ppm. The purity of the synthesized products (5a–m) was ascertained by elemental analysis.
Pharmacology
| Entry | Substituent (R) | ALP inhibition IC50 ± SEM (μM) |
|---|---|---|
| 5c | 4-Cl | 5.78 ± 0.92 |
| 5d | 2-OMe | 3.92 ± 0.79 |
| 5e | 3-OMe | 4.36 ± 0.83 |
| 5f | 2-Me | 3.62 ± 0.65 |
| 5g | 3-Me | 4.38 ± 0.72 |
| 5h | 4-Me | 3.47 ± 0.67 |
| 5i | 2,3-diMe | 2.59 ± 0.33 |
| 5j | 2,4-diMe | 1.38 ± 0.42 |
| 5k | 2,6-diMe | 1.79 ± 0.27 |
| 5l | 3,4-diMe | 1.39 ± 0.21 |
| 5m | 3,5-diMe | 1.41 ± 0.19 |
| KH2PO4 | — | 2.43 ± 0.04 |
Among the tested thiazole analogues, compound 5j with an IC50 value of 1.38 ± 0.42 μM emerged as the most active compound of the series. This two-fold strong inhibitory activity may be attributed to the two methyl groups present at 3- and 5-position of aryl ring directly attached to the thiazole core. A slight modification in substituent pattern on aryl ring gave compound 5l where substituents were present at 3- and 4-position. This compound also exhibits strong inhibitory activity (IC50 = 1.39 ± 0.21 μM) as compared to the standard drug. Moreover, a similar inhibitory potential was observed when methyl groups shift to 3- and 5-position at the aromatic ring as depicted by compound 5m (IC50 = 1.41 ± 0.19 μM). Compound 5k with methyl groups at 2- and 6-position also afforded potent inhibition, higher than the standard reference. Compound with methyl groups at 2- and 3-position of the aromatic ring maintained inhibitory potential comparable to standard drug. The other compounds in the series with mono substitution at various positions of aryl ring showed diminished activities. These substitutions include electron-withdrawing as well as electron-donating groups. Overall, within the synthesized series, derivatives bearing two methyl groups at variable positions of the aromatic ring attached to iminothiazole skeleton showed remarkable inhibitory potential and indicated the influence of steric factors on the modulation of alkaline phosphatase potential.
| Entry | Substituent (R) | Leishmaniasis (%inhibition) | |||
|---|---|---|---|---|---|
| 100 μM | 10 μM | 1 μM | 0.1 μM | ||
| 5c | 4-Cl | 40.6 ± 2.2 | 34.6 ± 1.4 | 31.7 ± 1.6 | 22.4 ± 1.4 |
| 5d | 2-OMe | 58.3 ± 2.1 | 56.8 ± 1.7 | 50.4 ± 2.2 | 45.5 ± 1.4 |
| 5e | 3-OMe | 59.4 ± 1.5 | 56.3 ± 1.3 | 52.9 ± 1.5 | 49.3 ± 1.7 |
| 5f | 2-Me | 52.2 ± 1.7 | 51.9 ± 2.3 | 48.6 ± 1.7 | 41.7 ± 2.2 |
| 5g | 3-Me | 62.7 ± 1.6 | 59.4 ± 1.2 | 56.3 ± 1.4 | 53.7 ± 1.2 |
| 5h | 4-Me | 59.0 ± 1.4 | 57.2 ± 2.1 | 54.1 ± 1.6 | 38.2 ± 1.3 |
| 5i | 2,3-diMe | 70.4 ± 2.2 | 66.2 ± 1.4 | 60.7 ± 1.5 | 53.9 ± 1.6 |
| 5j | 2,4-diMe | 60.6 ± 1.5 | 58.7 ± 1.5 | 54.4 ± 1.1 | 52.8 ± 1.7 |
| 5k | 2,6-diMe | 58.4 ± 1.4 | 56.2 ± 1.4 | 49.1 ± 1.7 | 43.2 ± 1.1 |
| 5l | 3,4-diMe | 53.2 ± 1.2 | 47.9 ± 3.7 | 43.2 ± 2.2 | 38.6 ± 1.3 |
| 5m | 3,5-diMe | 53.8 ± 2.1 | 49.7 ± 1.2 | 45.3 ± 1.3 | 36.4 ± 1.2 |
| Amphotericin B | — | 79.8 ± 1.8 | 76.3 ± 1.4 | 74.8 ± 2.7 | 69.9 ± 2.3 |
The careful examination of the anti-leishmanial activity results revealed that most of the compounds showed >50% inhibition whereas some of the tested iminothiazoles showed good inhibition of Leishmania major in vitro. Among all the compounds screened in the series, 5i showed highest inhibition of 70.4 ± 2.2% at 100 μM, whereas amphotericin B showed 79.8% inhibition. Compound 5i incorporates two methyl substituents on the aryl ring attached directly to thiazole ring. This compound also showed highest potential at variable concentrations. The activity was reduced when these dimethyl substituents were present at variable positions as depicted by compound 5j. Also, compound 5g with mono methyl substituent at meta-position also exhibited significant inhibition (62.7 ± 1.6 μM). Apart from these substituents, electron-rich (–OMe) as well as electron-poor (–Cl) groups were tolerated which showed moderate inhibition of Leishmania major. Least active compound among the series was 5c displaying 40.6 ± 2.2% inhibition at 100 μM.
Most of the compounds among thiazole series exhibited (>50%) inhibition for both cell lines with slightly different capacity due to their structure diversity in terms of attached functional groups with thiazole skeleton (Tables 3 and 4). Among the tested compounds, 5m showed the highest inhibition of 66.8 ± 1.1 and 64.2 ± 2.3% at 100 and 10 μM, respectively, against BHK-21 cells. This compound possesses dimethyl substituent at 3- and 5-position of the aryl ring at thiazole skeleton. A slight decrease in inhibition was observed when substitution pattern is changed to 2,4- or 3,4-positions as depicted by compound 5j and 5l, respectively. Several other substitutions including electron-donating and electron-withdrawing were also tolerated but afforded reduced inhibition. Compound 5k bearing dimethyl substitution at 2- and 6-position gave least inhibition of 37.8 ± 1.2% at 0.1 μM concentration. Compounds with only one methyl group at ortho- or para-position exhibited inhibition slightly greater than 50% at 100 and 10 μM.
| Entry | Substituent (R) | BHK-21 cell lines (%inhibition) | |||
|---|---|---|---|---|---|
| 100 μM | 10 μM | 1 μM | 0.1 μM | ||
| 5c | 4-Cl | 50.5 ± 1.4 | 48.3 ± 1.2 | 47.2 ± 1.8 | 41.8 ± 2.5 |
| 5d | 2-OMe | 59.4 ± 1.3 | 55.3 ± 1.7 | 52.8 ± 1.2 | 47.0 ± 1.7 |
| 5e | 3-OMe | 53.2 ± 1.7 | 50.2 ± 2.1 | 46.3 ± 1.8 | 45.5 ± 1.2 |
| 5f | 2-Me | 54.7 ± 1.2 | 51.8 ± 2.4 | 49.9 ± 1.6 | 47.2 ± 1.6 |
| 5g | 3-Me | 52.9 ± 1.3 | 50.7 ± 1.5 | 47.3 ± 2.3 | 45.1 ± 1.8 |
| 5h | 4-Me | 57.3 ± 1.5 | 54.8 ± 2.8 | 50.5 ± 1.7 | 48.7 ± 1.2 |
| 5i | 2,3-diMe | 57.2 ± 1.2 | 51.6 ± 2.5 | 47.2 ± 2.6 | 41.9 ± 3.3 |
| 5j | 2,4-diMe | 62.9 ± 3.2 | 61.1 ± 2.6 | 60.7 ± 1.8 | 56.4 ± 2.4 |
| 5k | 2,6-diMe | 54.7 ± 2.1 | 51.8 ± 2.4 | 44.3 ± 2.7 | 37.8 ± 1.2 |
| 5l | 3,4-diMe | 62.1 ± 1.8 | 61.8 ± 1.2 | 56.4 ± 2.9 | 52.9 ± 1.9 |
| 5m | 3,5-diMe | 66.8 ± 1.1 | 64.2 ± 2.3 | 59.4 ± 1.2 | 55.5 ± 2.7 |
| Vincristine | — | 74.5 ± 2.9 | 72.6 ± 3.1 | 70.9 ± 2.4 | 69.8 ± 1.9 |
| Entry | Substituent (R) | H-157 cell lines (%inhibition) | |||
|---|---|---|---|---|---|
| 100 μM | 10 μM | 1 μM | 0.1 μM | ||
| 5c | 4-Cl | 53.7 ± 1.6 | 50.3 ± 0.8 | 47.2 ± 2.7 | 44.7 ± 1.2 |
| 5d | 2-OMe | 54.4 ± 1.5 | 52.3 ± 1.3 | 49.4 ± 1.7 | 48.4 ± 1.2 |
| 5e | 3-OMe | 52.5 ± 0.7 | 49.8 ± 1.1 | 44.9 ± 1.2 | 35.2 ± 1.6 |
| 5f | 2-Me | 56.6 ± 1.4 | 52.4 ± 1.9 | 47.5 ± 2.8 | 44.3 ± 2.7 |
| 5g | 3-Me | 51.0 ± 1.3 | 44.2 ± 2.5 | 38.1 ± 2.4 | 32.8 ± 1.3 |
| 5h | 4-Me | 54.6 ± 1.8 | 51.8 ± 2.7 | 50.9 ± 1.1 | 47.6 ± 1.5 |
| 5i | 2,3-diMe | 54.4 ± 3.9 | 51.7 ± 1.7 | 47.2 ± 1.2 | 43.6 ± 1.3 |
| 5j | 2,4-diMe | 63.6 ± 2.0 | 61.4 ± 1.6 | 56.7 ± 2.4 | 52.1 ± 1.4 |
| 5k | 2,6-diMe | 57.9 ± 1.1 | 55.2 ± 0.4 | 51.6 ± 1.1 | 44.4 ± 1.6 |
| 5l | 3,4-diMe | 65.0 ± 1.8 | 57.3 ± 0.7 | 53.8 ± 2.6 | 45.5 ± 1.7 |
| 5m | 3,5-diMe | 63.0 ± 3.1 | 61.1 ± 1.2 | 57.4 ± 2.1 | 54.3 ± 2.5 |
| Vincristine | — | 74.5 ± 2.9 | 72.6 ± 3.1 | 70.9 ± 2.4 | 69.8 ± 1.9 |
In case of H-157 cell lines, among the tested thiazole hybrids, compound 5l was the most potent analogue with inhibition value of 65.0 ± 1.8% at 100 μM while 57.3 ± 0.7% at 10 μM. This activity may be attributed to the presence of dimethyl groups at 3- and 4-position of aryl ring attached to thiazole core. A slight downward shift in activity was observed when position of substituents is moved to 2- and 4-position or 3- and 5-position, respectively. Compounds 5j and 5m with dimethyl substituents at 2- and 4-position and 3- and 5-position showed 61.4 ± 1.6 and 61.1 ± 1.2% inhibitions at 10 μM, respectively as compared to vincristine (72.6 ± 3.1% at 10 μM). Least activity was observed in case of 5g which incorporates only one methyl group at meta-position.
In general, among the tested bis-coumarin–iminothiazole hybrids, the compounds with dual substitutions were found to possess highest activities and emerged as lead candidates. This pattern also suggests that the steric factors are generally more operative in modulating the activity profile as compared to the electronic properties. These results also speculate some further research to optimize bis-coumarin–iminothiazole template against a range of biological targets with promising inhibitory potential.
Molecular modelling
According to Lipinski's Ro5, most drug like molecules have molecular weight ≤ 500, logarithm of the octanol/water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) ≤ 5, total polar surface area (TPSA) < 140 Å2, number of hydrogen bond donors (HBD) ≤ 5 and hydrogen bond acceptor (HBA) ≤ 10.50 Further modifications in the Ro5 were made by Veber et al.49 who suggested the number of rotatable bond (NOR) of drug like molecule must be fewer or equal to 10.50 Molecules violating more than one of these rules may have problem with bioavailability.
P) ≤ 5, total polar surface area (TPSA) < 140 Å2, number of hydrogen bond donors (HBD) ≤ 5 and hydrogen bond acceptor (HBA) ≤ 10.50 Further modifications in the Ro5 were made by Veber et al.49 who suggested the number of rotatable bond (NOR) of drug like molecule must be fewer or equal to 10.50 Molecules violating more than one of these rules may have problem with bioavailability.
Structural analyses of the 3D generated model of the ALP showed that enzyme is constructed with two chains with the co-crystallized ligand in each chain, pocket was found around the co-crystallized ligand. After docking the standard drug in the pocket of both chains, the active site of the chain B was selected for further studies. Pocket was found to be a small cavity, mostly constructed with hydrophilic residues some of these are Tyr107, Lys108, Arg166, His153, Val89, His 320, Ser429, Thr431 and Gln317. Molecular docking studies were carried out to determine the probable binding modes of these compounds in the active sites of the enzyme, rat Intestinal Alkaline Phosphate (IALP) (PDB code: 4KJG). All the docked conformations of each ligands were analyzed. From the results of calculations, we chose variants with the minimal energy of the enzyme–inhibitor complex. All the preferred docked conformations formed one cluster inside the pocket as shown in Fig. 2.
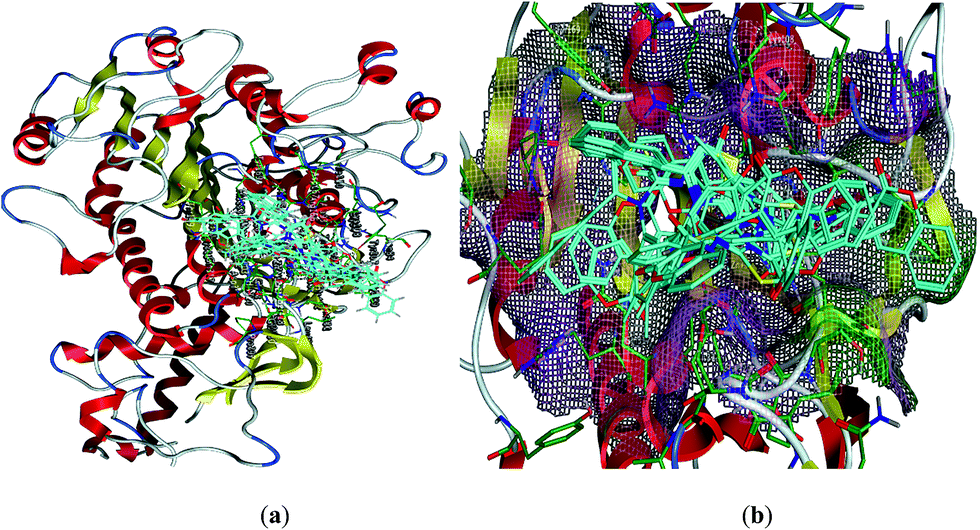 | ||
| Fig. 2 Superimposition of the ligands inside the active site; (a) overlaying of all the minimum energy docked poses inside the pocket. Ligands are shown in cyan color in stick mode while key residues are shown in green color in stick mode. Backbone receptor is shown in cartoon and ribbon form. (b) Close view of the active site, pocket is marked with surfaces. | ||
Almost all the compounds have similar binding interaction pattern. In all the ligands except 5c, 5f, 5h, 5i and 5m, the carbonyl oxygen present on the coumarin moiety formulated strong hydrogen bonding with the amino acid residues in active site, while phenyl ring of the same moiety established H–arene interaction in all the ligands except 5d and 5k. In addition to these, tremendous polar interactions also played important role for strong binding of the ligands inside the active site. Compounds 5j–m are found to be the most active compounds of the series and these compounds were also found to exhibit strong interaction with the amino acids residues inside the active site. Compound 5m inhibited the catalytic activities of the ALP by anchoring over active site through multiple polar interactions, a hydrophobic interaction and Pi–H interaction although it didn't establish hydrogen bonding like other three most active compounds 5j, 5k and 5l. Fig. 3 shows plausible binding modes for three most active representative compounds 5j, 5k and 5l of the series. While images of 2D interaction for all the ligands along with the detailed legend are shown in Fig. S1.† The binding free energies derived from docking (London dG ranging from −11.4376 to −7.4100 kcal mol−1) indicate that almost all the synthesized imino-thiazole derivatives exhibited stronger binding affinity for alkaline phosphatase (Table S2†). The poses are ranked by the scores from the GBVI/WSA binding free energy calculation in the “S” field. It is quite interesting to note that the binding free energy in “S” field is in quite narrow range from −6.0532 to −6.8090 kcal mol−1 (Table S2†). According to the computations performed, all the docked ligands are located in the active site of alkaline phosphatase (PDB code: 4KJG).51 Investigation of the binding mode analysis of the adopted conformations revealed that both the phenyl rings (coumarin moiety) are oriented almost in parallel to the main plane. This orientation of phenyl rings may lead to its interaction (H–arene) with the amino acid residues in the active site of alkaline phosphatase.
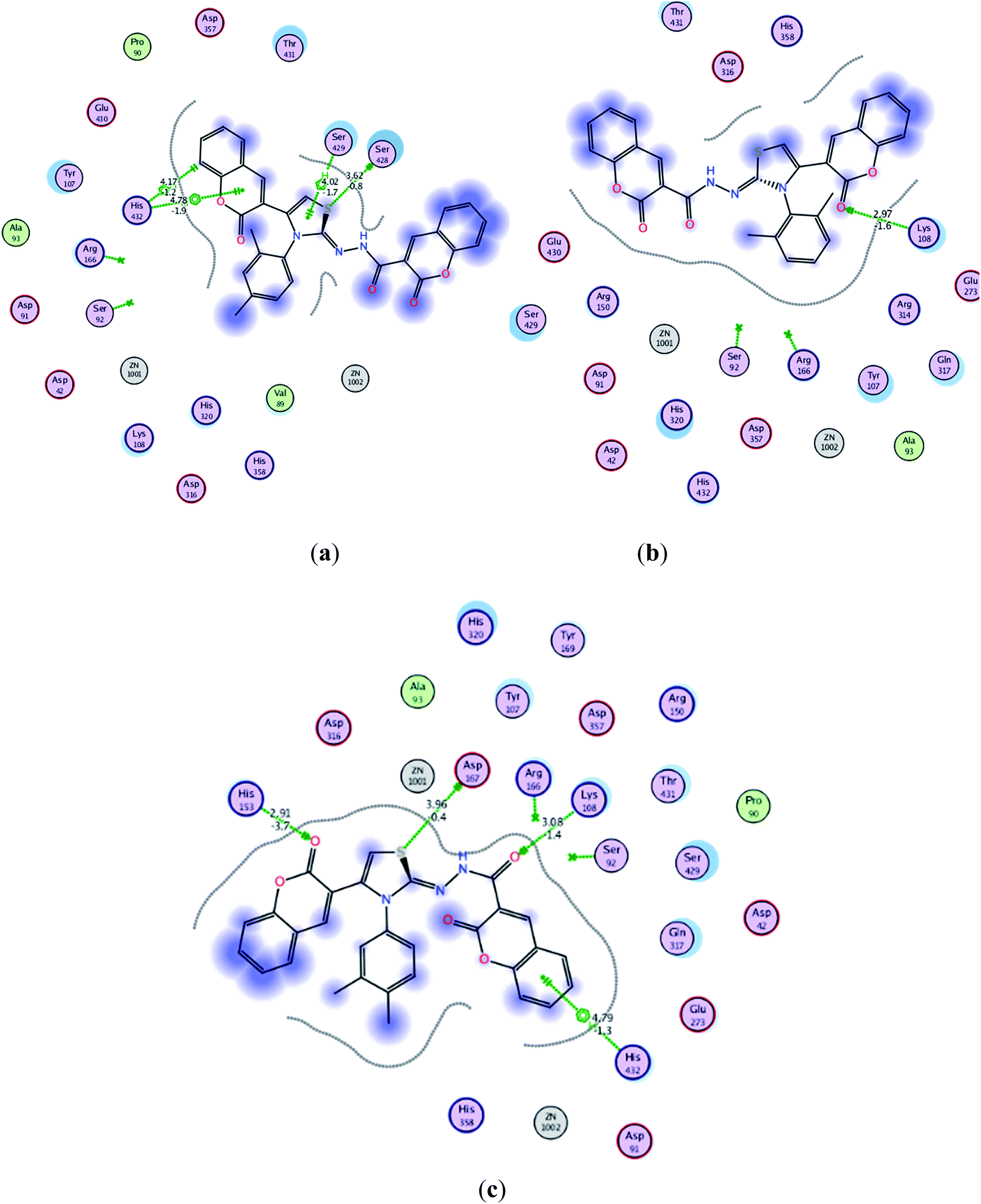 | ||
| Fig. 3 Interaction of ligands with the active site residues of receptor in 2D space; dotted green lines present hydrogen bonding and other interactions. Detailed description is given in the Fig. S1 in ESI† under the caption legend (a) compound 5j (b) compound 5k and (c) compound 5l. | ||
The calculated geometries of the inhibitor 5l in the enzyme's active site was analyzed to study the orientation of its docking pose and binding mode as shown in Fig. 4. As a result the positions of oxygen, it formed a strong H-bonding with the Asp167, His153 and Lys108 in addition to H–arene interaction of the phenyl ring with His432. Several strong polar interactions may reflect additional fixation of inhibitor 5l which occupied the enzyme's active site. Again binding of two phenyl rings through Pi–H interaction with key residues could be the strong reason of low energy and good activity of the inhibitor 5l (Fig. 3c and 4). Examination of the most preferred docked pose of inhibitor 5k (Fig. 3b) revealed that the oxygen atom of the carbonyl group established a strong hydrogen bond with Lys108. In addition to these, several polar interactions also played a significant role for strong binding, low binding energy and in turn good activity of the entire imino-thiazole library against ALP. The synthesized library 5a–m is composed basically of three phenyl rings, causing the whole structure to be quite rigid and bulky. The rigidity imparted the expanded orientation to the compounds which favored the maximum exposure of the ligand to active site residues leading to remarkable interactions.
 | ||
| Fig. 4 Docking pose of the ligand 5l inside the active site. Interaction of ligand 5l with the active site residues of receptor in 3D space; dashed blue lines represent hydrogen bonding while dotted yellow line represents H–arene interaction. | ||
It can be concluded from the docking computations that quite low binding free energy (−6.0532 to −6.8090 kcal mol−1) coupled with the multiple interactions with amino acids residues provided the rational for good activities of these inhibitors specially 5j–m (IC50 = 1.38 to 1.79 μM) which were noticed to be more potent as compared to standard drug (IC50 = 2.43 μM) against IALP. This is in accordance with findings that introducing coumarin moieties at the both terminal of the structure of thiazoles lead to greater strength in addition to imparting the flat, expanded orientation to the structures which consequently helped in strong binding. This can provide the rational for more effective binding through hydrogen bonding and arene–H interaction in the enzyme active site. These results proclaimed compounds 5j–m to be further developed as ALP inhibitors.
Conclusions
In summary, a series of novel bis-coumarin–iminothiazole hybrids (5a–m) has been prepared by the reaction of bromoacetyl coumarin and a range of thiosemicarbazide derivatives in methanol. The structural diversity of the synthesized derivatives was ensured by using a diverse range of substituents including electron-donating and electron-withdrawing groups on aryl part of thiosemicarbazide. The synthesized analogues were screened against alkaline phosphatase where 5j emerged as a potent inhibitor with an IC50 value of 1.38 ± 0.42 μM which is two-fold strong inhibition as compared to the standard inhibitor. The synthesized compounds were also assayed for their anti-leishmanial potential and compound 5i was found to be the lead candidate with 70.4 ± 2.2% inhibition at 100 μM. The prepared compounds also showed cytotoxic behavior against kidney fibroblast (BHK-21) and lung carcinoma (H-157) cell lines. Compound 5l showed dual inhibition (66.8 ± 1.1 and 65.0 ± 1.8%) against BHK-21 and H-157 cell lines, respectively. In general, among the tested bis-coumarin–iminothiazole hybrids, the compounds with dual substitutions were found to possess highest activities and emerged as lead candidates. This pattern also suggests that the steric factors are generally more operative in modulating the activity profile as compared to the electronic properties. The detailed binding mode analysis with docking simulation showed that the inhibitors could be stabilized in the active site through the formation of multiple interactions with catalytic residues and the establishment of hydrophobic contacts in a cooperative fashion. Finally, based on the activity findings and docking analysis, it could be proclaimed that the synthesized inhibitors could be further developed as novel and potent inhibitors of alkaline phosphatase.Experimental
General
Unless otherwise noted, all materials were obtained from commercial suppliers (Aldrich and Merck companies) and used without further purification. Thin layer chromatography (TLC) was performed on Merck DF-Alufoilien 60F254 0.2 mm precoated plates. Product spots were visualized under UV light at 254 and 365 nm. Melting points were recorded on a Stuart melting point apparatus (SMP3) and are uncorrected. Infra-red (IR) spectra were recorded on FTS 3000 MX, Bio-Rad Merlin (Excalibur model) spectrophotometer. 1H NMR spectra were recorded on a Bruker Avance (300 MHz) spectrometer. Chemical shifts (δ) are quoted in parts per million (ppm) downfield of tetramethylsilane, using residual solvent as internal standard (DMSO-d6 at 2.50 ppm). Abbreviations used in the description of resonances are: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), Ar (aromatic). Proton-decoupled 13C NMR spectra were recorded on a Bruker Avance (75 MHz) spectrometer using deuterated solvent as internal standard (DMSO-d6 at 39.52 ppm). The elemental analysis was performed on Leco CHNS-932 Elemental Analyzer, Leco Corporation (USA).Synthesis of 3-acetyl-2H-chromen-2-one
To a cold mixture of salicylaldehyde 1 (1.0 mol) and ethyl acetoacetate 2 (1.0 mol), catalytic amount of piperidine was added and reaction mixture was stirred for 30 min at room temperature. The solid separated was filtered and washed with ethanol. Recrystallization of the solid from chloroform afforded 3-acetyl-2H-chromene-2-one.43 Yield (93%): m.p.: 119–121 °C; Rf: 0.61 (20% EtOAc/hexane). The data for 3-acetyl-2H-chromene-2-one was consistent with literature.52Synthesis of 3-(2-bromoacetyl)-2H-chromen-2-one (3)
3-Acetyl-2H-chromene-2-one (10.0 mmol) was dissolved in dry chloroform and a solution of Br2 (10.0 mmol) in chloroform was added dropwise with continuous stirring and mixture was heated at reflux for 6–7 h. The progress of the reaction was monitored by thin layer chromatography (TLC). After completion of the reaction, the mixture was concentrated under reduced pressure, and washed with diethyl ether and recrystallized (70% CHCl3/EtOH) to afford 3-(2-bromoacetyl)-2H-chromen-2-one (3).40 Yield: (69%); m.p.: 161–163 °C; Rf: 0.26 (20% EtOAc/hexane). The data for 3 was consistent with those observed in the literature.52Synthesis of 1,4-disubstituted thiosemicarbazides (4a–m)
The carbohydrazide (6.8 mmol) was dissolved in methanol (30 mL), and a solution of substituted isothiocyanate (6.6 mmol) separately dissolved in methanol (10 mL) was added dropwise with continuous stirring. The reaction mixture was heated at reflux for 10–12 h and monitored by TLC. After consumption of the starting materials, the mixture was cooled to room temperature. The methanol was evaporated under reduced pressure leaving behind the crude product as oil that became solidified on cooling and was recrystallized (20% EtOAc/hexane) to yield thiosemicarbazides (4a–m).31 The spectroscopic data for thiosemicarbazides (4a–h) has previously been reported in our papers31 (Rf = 20% EtOAc/hexane).O), 1572, 1540 (CC), 1271 (CS); 1H NMR (300 MHz, DMSO-d6): δ 11.51 (s, 1H, NH–CO), 11.13 (s, 2H, NH–CS), 9.01 (s, 1H, H-4), 7.71–7.68 (m, 2H, ArH), 7.43–7.38 (m, 1H, ArH), 7.27–7.17 (m, 1H, ArH), 7.00–6.95 (m, 2H, ArH), 6.16–6.13 (m, 1H, ArH), 2.27 (s, 3H, CH3), 2.09 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 177.3, 164.4, 159.8, 152.7, 140.1, 136.9, 133.3, 130.8, 127.4, 125.1, 123.5, 120.7, 119.0, 117.3, 116.3, 114.1, 112.7, 22.2, 17.3; anal. calcd for C19H17N3O3S: C, 62.10; H, 4.64; N, 11.43; S, 8.71; found: C, 62.02; H, 4.58; N, 11.39; S, 8.65.O), 1569, 1528 (CC), 1269 (CS); 1H NMR (300 MHz, DMSO-d6): δ 11.54 (s, 1H, NH–CO), 11.10 (s, 2H, NH–CS), 9.13 (s, 1H, H-4), 7.73–7.69 (m, 2H, ArH), 7.43–7.40 (m, 2H, ArH), 6.97–6.95 (m, 1H, ArH), 6.67–6.63 (m, 1H, ArH), 6.13–6.11 (m, 1H, ArH), 2.27 (s, 3H, CH3), 2.02 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 178.0, 163.1, 159.8, 153.1, 143.3, 139.0, 132.3, 130.7, 129.1, 127.3, 126.1, 122.6, 120.3, 119.6, 118.2, 116.1, 114.3, 21.3, 17.8; anal. calcd for C19H17N3O3S: C, 62.10; H, 4.59; N, 11.40; S, 8.69; found: C, 62.03; H, 4.51; N, 11.31; S, 8.63.O), 1572, 1523 (CC), 1269 (CS); 1H NMR (300 MHz, DMSO-d6): δ 11.43 (s, 1H, NH–CO), 11.02 (s, 2H, NH–CS), 9.09 (s, 1H, H-4), 7.78–7.70 (m, 2H, ArH), 7.45–7.43 (m, 2H, ArH), 6.95–6.92 (m, 2H, ArH), 6.63–6.61 (m, 1H, ArH), 2.19 (s, 6H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 171.1, 161.9, 158.2, 153.0, 141.1, 138.4, 133.0, 128.1, 127.0, 125.1, 123.7, 121.0, 119.3, 116.7, 114.2, 23.9; anal. calcd for C19H17N3O3S: C, 62.09; H, 4.60; N, 11.39; S, 8.68; found: C, 62.01; H, 4.54; N, 11.35; S, 8.61.O), 1565, 1549 (CC), 1263 (CS); 1H NMR (300 MHz, DMSO-d6): δ 11.63 (s, 1H, NH–CO), 11.11 (s, 2H, NH–CS), 8.86 (s, 1H, H-4), 7.73–7.65 (m, 2H, ArH), 7.42–7.41 (m, 2H, ArH), 6.86 (m, 1H, ArH), 6.23–6.21 (m, 2H, ArH), 2.36 (s, 3H, CH3), 2.27 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 179.2, 163.9, 158.4, 152.2, 144.0, 139.8, 135.7, 133.8, 131.0, 129.3, 127.8, 125.5, 122.4, 119.1, 117.6, 116.6, 114.7, 22.3, 19.3; anal. calcd for C19H17N3O3S: C, 62.11; H, 4.62; N, 11.40; S, 8.67; found: C, 62.00; H, 4.52; N, 11.37; S, 8.61.O), 1562, 1529 (CC), 1259 (CS); 1H NMR (300 MHz, DMSO-d6): δ 11.48 (s, 1H, NH–CO), 11.09 (s, 2H, NH–CS), 9.09 (s, 1H, H-4), 7.79–7.66 (m, 2H, ArH), 7.42–7.39 (m, 2H, ArH), 6.69–6.66 (m, 3H, ArH), 2.27 (s, 6H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 175.6, 163.2, 157.4, 151.2, 140.8, 137.0, 131.2, 129.6, 127.8, 124.5, 123.1, 120.2, 118.8, 115.0, 113.5, 19.9; anal calcd for C19H17N3O3S: C, 62.08; H, 4.59; N, 11.38; S, 8.69; found: C, 62.03; H, 4.50; N, 11.33; S, 8.59.Synthesis of (E)-N′-(3-(substituted)-4-(2-oxo-2H-chromen-3-yl)thiazol-2(3H)-ylidene)-2-oxo-2H-chromene-3-carbohydrazide (5a–m)
To a mixture of substituted thiosemicarbazide (4a–m) (1.0 mmol) and 3-(2-bromoacetyl)-2H-chromen-2-one (3) (1.0 mmol) in methanol, anhydrous sodium acetate (4.0 mmol) was added and heated at reflux for 5–6 h. The reaction mixture was then cooled to room temperature and the solid product formed was filtered off, washed with water, dried (MgSO4), and recrystallized (EtOH) to afford title products (5a–m) in good yields44 (Rf = 20% EtOAc/hexane).O), 1629 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.23 (s, 1H, NH), 9.67 (s, 1H, H-4), 8.98 (s, 1H, H-4′), 7.96–7.79 (m, 4H, ArH), 7.43–7.40 (m, 5H, ArH), 7.23–7.21 (m, 1H, ArH), 7.01–6.86 (m, 2H, ArH), 6.66–6.60 (m, 1H, ArH); 13C NMR (75 MHz, DMSO-d6): δ 168.3, 159.1, 154.0, 151.2, 148.6, 143.2, 133.9, 131.6, 130.4, 129.1, 128.6, 127.3, 126.7, 125.3, 123.5, 121.2, 119.8, 118.0, 116.3, 112.5, 98.6; anal. calcd for C28H16ClN3O5S: C, 62.04; H, 2.96; N, 7.73; S, 5.90; found: C, 62.00; H, 2.90; N, 7.68; S, 5.87.O), 1619 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.48 (s, 1H, NH), 9.89 (s, 1H, H-4), 9.01 (s, 1H, H-4′), 8.01–7.93 (m, 4H, ArH), 7.42–7.09 (m, 5H, ArH), 7.03–6.95 (m, 2H, ArH), 6.90–6.86 (m, 1H, ArH), 6.65–6.63 (m, 1H, ArH); 13C NMR (75 MHz, DMSO-d6): δ 169.1, 158.3, 153.4, 150.1, 149.6, 142.0, 135.7, 133.0, 131.8, 130.6, 128.5, 127.0, 126.4, 125.1, 122.7, 121.3, 119.6, 118.1, 116.6, 114.8, 99.1; anal. calcd for C28H16ClN3O5S: C, 62.04; H, 2.96; N, 7.73; S, 5.90; found: C, 62.02; H, 2.88; N, 7.70; S, 5.85.O), 1612 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.48 (s, 1H, NH), 8.59 (s, 1H, H-4), 8.19 (s, 1H, H-4′), 7.89–7.80 (m, 4H, ArH), 7.68–7.61 (m, 4H, ArH), 7.48–7.36 (m, 4H, ArH); 13C NMR (75 MHz, DMSO-d6): δ 158.8, 158.7, 155.1, 154.9, 149.2, 149.1, 147.8, 135.6, 135.1, 131.5, 131.3, 125.6, 125.5, 123.2, 122.6, 118.6, 118.5, 118.3, 116.7; anal. calcd for C28H16ClN3O5S: C, 62.04; H, 2.96; N, 7.73; S, 5.90; found: C, 62.00; H, 2.90; N, 7.69; S, 5.87.O), 1615 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.90 (s, 1H, NH), 8.48 (s, 1H, H-4), 8.33 (s, 1H, H-4′), 8.32–8.25 (m, 2H, ArH), 7.90 (t, J = 8.7 Hz, 1H, ArH), 7.28–7.00 (m, 3H, ArH), 6.98–6.96 (m, 3H, ArH), 6.94–6.92 (m, 2H, ArH), 6.89–6.86 (m, 3H, ArH), 3.88 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 169.7, 159.2, 152.7, 147.3, 144.4, 138.4, 138.2, 136.6, 134.4, 132.1, 129.2, 128.9126.1, 125.2, 121.1, 119.6, 116.3, 113.2, 111.9, 111.5, 64.5; anal. calcd for C29H19N3O6S: C, 64.79; H, 3.54; N, 7.80; S, 5.96; found: C, 64.73; H, 3.49; N, 7.73; S, 5.91.O), 1623 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.67 (s, 1H, NH), 9.15 (s, 1H, H-4), 8.68 (s, 1H, H-4′), 8.38–8.27 (m, 2H, ArH), 7.73–7.62 (m, 4H, ArH), 7.30–7.26 (m, 1H, ArH), 7.10–6.99 (m, 4H, ArH), 6.38–6.31 (m, 2H, ArH), 3.65 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 169.7, 159.3, 157.1, 151.6, 150.7, 148.1, 143.9, 132.0, 130.7, 128.1, 126.1, 125.2, 124.6, 123.3, 121.7, 120.3, 119.9, 116.6, 111.6, 110.7, 95.8, 56.3; anal. calcd for C29H19N3O6S: C, 64.79; H, 3.54; N, 7.80; S, 5.96; found: C, 64.71; H, 3.50; N, 7.70; S, 5.89.O), 1609 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.70 (s, 1H, NH), 8.47 (s, 1H, H-4), 8.10 (s, 1H, H-4′), 7.43–7.41 (m, 4H, ArH), 7.26–7.14 (m, 4H, ArH), 7.00–6.81 (m, 4H, ArH), 2.30 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 167.2, 153.0, 149.6, 146.2, 144.8, 138.9, 137.0, 134.8, 131.7, 128.9, 127.5, 126.2, 120.7, 119.6, 116.4, 115.7, 114.6, 111.4, 21.0; anal. calcd for C29H19N3O5S: C, 66.77; H, 3.67; N, 8.05; S, 6.13; found: C, 66.68; H, 3.60; N, 8.01; S, 6.08.O), 1616 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.21 (s, 1H, NH), 9.43 (s, 1H, H-4), 8.86 (s, 1H, H-4′), 7.86–7.79 (m, 4H, ArH), 7.53–7.35 (m, 5H, ArH), 7.03–6.98 (m, 2H, ArH), 6.66–6.60 (m, 1H, ArH), 6.53–6.49 (m, 1H, ArH), 2.12 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 169.1, 158.3, 153.6, 151.2, 148.5, 137.2, 132.9, 131.2, 129.1, 128.6, 127.1, 126.0, 124.7, 122.5, 121.7, 119.2, 117.6, 116.1, 114.3, 111.5, 98.7, 20.1; anal. calcd for C29H19N3O5S: C, 66.77; H, 3.67; N, 8.05; S, 6.13; found: C, 66.70; H, 3.62; N, 8.00; S, 6.06.O), 1607 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.09 (s, 1H, NH), 9.68 (s, 1H, H-4), 8.94 (s, 1H, H-4′), 7.98–7.84 (m, 4H, ArH), 7.65–7.62 (m, 2H, ArH), 7.46–7.39 (m, 3H, ArH), 6.99–6.93 (m, 2H, ArH), 6.36–6.29 (m, 2H, ArH), 2.20 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 170.0, 159.1, 154.2, 150.2, 147.2, 139.0, 132.2, 131.5, 130.6, 128.4, 127.5, 125.9, 124.3, 123.3, 120.1, 118.8, 116.2, 114.6, 96.3, 23.9; anal. calcd for C29H19N3O5S: C, 66.77; H, 3.67; N, 8.05; S, 6.13; found: C, 66.69; H, 3.60; N, 8.01; S, 6.09.O), 1615 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.143 (s, 1H, NH), 9.10 (s, 1H, H-4), 8.69 (s, 1H, H-4′), 7.96–7.84 (m, 4H, ArH), 7.69–7.49 (m, 4H, ArH), 7.43–7.40 (m, 1H, ArH), 6.99–6.96 (m, 2H, ArH), 6.65–6.59 (m, 1H, ArH), 2.23 (s, 3H, CH3), 2.01 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 169.3, 159.7, 152.8, 150.3, 147.7, 141.1, 136.8, 131.3, 129.1, 128.4, 127.5, 126.6, 125.2, 124.7, 121.0, 120.0, 119.3, 118.3, 116.5, 114.7, 100.2, 23.5, 19.3; anal. calcd for C30H21N3O5S: C, 67.28; H, 3.94; N, 7.84; S, 5.98; found: C, 67.23; H, 3.90; N, 7.80; S, 5.93.O), 1612 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.02 (s, 1H, NH), 9.09 (s, 1H, H-4), 8.58 (s, 1H, H-4′), 7.93–7.87 (m, 2H, ArH), 7.83–7.74 (m, 2H, ArH), 7.61–7.59 (m, 1H, ArH), 7.37–7.19 (m, 4H, ArH), 6.96–6.91 (m, 1H, ArH), 6.73–6.65 (m, 2H, ArH), 2.34 (s, 3H, CH3), 2.02 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 168.1, 158.5, 152.0, 151.6, 148.3, 143.8, 139.1, 137.4, 136.3, 133.2, 131.6, 130.4, 129.1, 127.3, 126.7, 125.5, 121.2, 119.3, 117.1, 114.7, 100.1, 26.3, 15.8; anal. calcd for C30H21N3O5S: C, 67.27; H, 3.95; N, 7.83; S, 5.97; found: C, 67.19; H, 3.84; N, 7.76; S, 5.83.O), 1607 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.11 (s, 1H, NH), 9.39 (s, 1H, H-4), 9.00 (s, 1H, H-4′), 7.83–7.69 (m, 4H, ArH), 7.54–7.51 (m, 2H, ArH), 7.39–7.34 (m, 3H, ArH), 6.99–6.96 (m, 2H, ArH), 6.67–6.63 (m, 1H, ArH), 2.09 (s, 6H, 2 × CH3); 13C NMR (75 MHz, DMSO-d6): δ 169.9, 157.2, 151.4, 150.3, 149.0, 147.4, 133.5, 130.6, 129.1, 128.6, 126.9, 125.4, 123.2, 121.7, 119.1, 118.8, 116.3, 114.5, 99.0, 17.6; anal. calcd for C30H21N3O5S: C, 67.27; H, 3.95; N, 7.83; S, 5.97; found: C, 67.20; H, 3.89; N, 7.79; S, 5.86.O), 1629 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.14 (s, 1H, NH), 9.01 (s, 1H, H-4), 8.67 (s, 1H, H-4′), 7.97–7.92 (m, 1H, ArH), 7.81–7.69 (m, 2H, ArH), 7.63–7.57 (m, 1H, ArH), 7.48–7.20 (m, 4H, ArH), 7.12–7.02 (m, 1H, ArH), 6.99–6.93 (m, 1H, ArH), 6.89–6.80 (m, 2H, ArH), 2.22 (s, 3H, CH3), 2.10 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 168.1, 158.2, 151.1, 149.0, 147.7, 140.1, 137.8, 131.5, 130.2, 129.6, 127.3, 126.8, 125.2, 123.7, 121.0, 119.7, 118.4, 116.3, 115.1, 114.8, 99.0, 20.8, 18.1; anal. calcd for C30H21N3O5S: C, 67.26; H, 3.93; N, 7.81; S, 5.96; found: C, 67.20; H, 3.87; N, 7.75; S, 5.89.O), 1618 (CN); 1H NMR (300 MHz, DMSO-d6): δ 11.13 (s, 1H, NH), 9.10 (s, 1H, H-4), 8.89 (s, 1H, H-4′), 7.83–7.71 (m, 2H, ArH), 7.68–7.63 (m, 1H, ArH), 7.42–7.31 (m, 4H, ArH), 7.07–7.00 (m, 2H, ArH), 6.95–6.89 (m, 1H, ArH), 6.65–6.53 (m, 2H, ArH), 2.31 (s, 6H, 2 × CH3); 13C NMR (75 MHz, DMSO-d6): δ 169.2, 158.1, 154.0, 151.6, 147.2, 141.3, 137.7, 131.5, 129.0, 128.1, 126.7, 125.2, 123.1, 121.8, 120.5, 118.7, 116.9, 114.3, 96.9, 24.3; anal. calcd for C30H21N3O5S: C, 67.26; H, 3.93; N, 7.81; S, 5.96; found: C, 67.23; H, 3.85; N, 7.77; S, 5.83.Pharmacological protocols
| Inhibition (%) = [100 − (abs of test compound/abs of control) × 100] |
IC50 values of selected compounds exhibiting >50% activity at 0.5 mM were calculated after dilutions using computer program GraphPad, San Diego, California, USA.
Leishmanicidal assay
| Inhibition (%) = [100 − (abs of test compound/abs of control) × 100] |
Cytotoxicity assay
| Inhibition (%) = [100 − (abs of test compound/abs of control) × 100] |
Molecular docking
Acknowledgements
A. I is grateful to the Higher Education Commission of Pakistan for the financial support under Indigenous 5000 PhD Fellowship Program. We are thankful to Dr Charles Dennis Hall and Prof. Alan R. Katritzky (late) (University of Florida) for the MOE software license. The Department of Energy, United State of America, is gratefully acknowledged for support through Grant No. DE-SC0001717.References
- M. H. le Du and J. L. Millan, J. Biol. Chem., 2002, 277, 49808–49814 CrossRef CAS PubMed.
- L. Zhang, M. Balcerzak, J. Radisson, C. Thouverey, S. Pikula, G. Azzar and R. Buchet, J. Biol. Chem., 2005, 280, 37289–37296 CrossRef CAS PubMed.
- S. P. Coburn, J. D. Mahuren, M. Jain, Y. Zubovic and J. Wortsman, J. Clin. Endocrinol. Metab., 1998, 83, 3951–3957 CAS.
- D. Sarrouilhe, P. Lalegerie and M. Baudry, Biochim. Biophys. Acta, 1992, 1118, 116–122 CrossRef CAS.
- M. Muda, N. N. Rao and A. Torriani, J. Bacteriol., 1992, 174, 8057–8064 CAS.
- M. Li, W. Ding, B. Baruah, D. C. Crans and R. Wang, J. Inorg. Biochem., 2008, 102, 1846–1853 CrossRef CAS PubMed.
- G. R. Mundy, Nat. Rev. Cancer, 2002, 2, 584–593 CrossRef CAS PubMed.
- G. D. Roodman, N. Engl. J. Med., 2004, 350, 1655–1664 CrossRef CAS PubMed.
- C. J. Logothetis and S.-H. Lin, Nat. Rev. Cancer, 2005, 5, 21–28 CrossRef CAS PubMed.
- K. Jung, M. Lein, C. Stephan, K. von Hösslin, A. Semjonow, P. Sinha, S. A. Loening and D. Schnorr, Int. J. Cancer, 2004, 111, 783–791 CrossRef CAS PubMed.
- D. Sloane, Methods Mol. Biol., 2009, 471, 65–83 Search PubMed.
- M. Mareel and A. Leroy, Physiol. Rev., 2003, 83, 337–376 CrossRef CAS PubMed.
- J. Weschel, K. Haglund and E. M. Haugsten, Biochem. J., 2011, 437, 199–213 CrossRef PubMed.
- J. Cummings, T. H. Ward, M. Ranson and C. Dive, Biochim. Biophys. Acta, 2004, 1705, 53–66 CAS.
- WHO, Research Priorities for Chagas Disease, Human African Trypanosomiasis and Leishmaniasis, WHO Technical Report Series No. 975, 2012, p. 116 Search PubMed.
- C. R. Davies, R. P. Kaye, S. L. Croft and S. Sundar, BMJ [Br. Med. J.], 2003, 15, 377–382 CrossRef.
- P. Trouiller, E. Torreele, P. Olliaro, N. White, S. Foster, D. Wirth and B. Pécoul, Trop. Med. Int. Health, 2001, 6, 945–951 CrossRef CAS.
- J. Alvar, I. D. Vélez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. den Boer, PLoS One, 2012, 7, e35671 CAS.
- Working to Overcome the Global Impact of Neglected Tropical Diseases, First WHO Report on Neglected Tropical Diseases, ed. D. W. T. Compton and P. Peters, World Health Organization, Geneva, 2010 Search PubMed.
- (a) V. Polshettiwar and R. S. Varma, Curr. Opin. Drug Discovery Dev., 2007, 10, 723–737 CAS; (b) I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193–244 CrossRef CAS PubMed.
- (a) A. Padwa and S. K. Bur, Tetrahedron, 2007, 63, 5341–5378 CrossRef CAS PubMed; (b) I. Khan, A. Ibrar, W. Ahmed and A. Saeed, Eur. J. Med. Chem., 2015, 90, 124–169 CrossRef CAS PubMed.
- D. M. D'Souza and T. J. Muller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC.
- G. Eren, S. Unlu, M. T. Nunez, L. Labeaga, F. Ledo, A. Entrena, E. B. Lu, G. Costantino and M. F. Sahin, Bioorg. Med. Chem., 2010, 18, 6367–6376 CrossRef CAS PubMed.
- R. J. Gillespie, A. I. Cliffe, C. E. Dawson, C. T. Dourish, S. Gaur, P. R. Giles, A. M. Jordan, A. R. Knight, A. Lawrence, J. Lerpiniere, A. Misra, R. M. Pratt, R. S. Todd, R. Upton, S. M. Weiss and D. S. Williamson, Bioorg. Med. Chem. Lett., 2008, 18, 2920–2923 CrossRef CAS PubMed.
- V. Jaishree, N. Ramdas, J. Sachin and B. Ramesh, J. Saudi Chem. Soc., 2012, 16, 371–376 CrossRef CAS PubMed.
- A. T. Ricardo, A. Luz and D. P. Carlos, Bioorg. Med. Chem., 2003, 11, 2175–2182 CrossRef.
- A. Zablotskaya, I. Segal, A. Geronikaki, T. Eremkina, S. Belyakov, M. Petrova, I. Shestakova, L. Zvejniece and V. Nikolajeva, Eur. J. Med. Chem., 2013, 70, 846–856 CrossRef CAS PubMed.
- M. H. M. Helal, M. A. Salem, M. S. A. El-Gaby and M. Aljahdali, Eur. J. Med. Chem., 2013, 65, 517–526 CrossRef CAS PubMed.
- R. Romagnoli, P. G. Baraldi, M. K. Salvador, M. E. Camacho, D. Preti, M. A. Tabrizi, M. Bassetto, A. Brancale, E. Hamel, R. Bortolozzi, G. Basso and G. Viola, Bioorg. Med. Chem., 2012, 20, 7083–7094 CrossRef CAS PubMed.
- K. N. Venugopala, M. Krishnappa, S. K. Nayak, B. K. Subrahmanya, J. P. Vaderapura, R. K. Chalannavar, R. M. Gleiser and B. Odhav, Eur. J. Med. Chem., 2013, 65, 295–303 CrossRef CAS PubMed.
- (a) A. Saeed and A. Ibrar, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1801–1810 CrossRef CAS PubMed; (b) A. Raza, A. Saeed, A. Ibrar, M. Muddassar, A. A. Khan and J. Iqbal, ISRN Pharmacol., 2012, 2012, 1–11 CrossRef PubMed; (c) A. Saeed, P. A. Channar, Q. Iqbal and J. Mahar, Chin. Chem. Lett., 2015 DOI:10.1016/j.cclet.2015.09.011.
- O. Dömötör, T. Tuccinardi, D. Karcz, M. Walsh, B. S. Creaven and É. A. Enyedy, Bioorg. Chem., 2014, 52, 16–23 CrossRef PubMed.
- Y. Zhao, Q. Zheng, K. Dakin, K. Xu, M. L. Martinez and W.-H. Li, J. Am. Chem. Soc., 2004, 126, 4653–4663 CrossRef CAS PubMed.
- J. R. Hwu, S.-Y. Lin, S.-C. Tsay, E. de Clercq, P. Leyssen and J. Neyts, J. Med. Chem., 2011, 54, 2114–2126 CrossRef CAS PubMed.
- S.-C. Tsay, J. R. Hwua, R. Singha, W.-C. Huang, Y. H. Chang, M. H. Hsu, F.-K. Shieh, C.-C. Lin, K. C. Hwang, J. C. Horng, E. de Clercq, I. Vliegen and J. Neyts, Eur. J. Med. Chem., 2013, 63, 290–298 CrossRef CAS PubMed.
- K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422–487 CrossRef CAS PubMed.
- F. W. Muregi and A. Ishih, Drug Dev. Res., 2010, 71, 20–32 CAS.
- C. Hubschwerlen, J. L. Specklin, C. Sigwalt, S. Schroeder and H. H. Locher, Bioorg. Med. Chem., 2003, 11, 2313–2319 CrossRef CAS.
- R. Pingaew, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Med. Chem. Res., 2014, 23, 1768–1780 CrossRef CAS.
- (a) K. V. Sashidhara, R. K. Modukuri, S. Singh, K. B. Rao, G. A. Teja, S. Gupta and S. Shukla, Bioorg. Med. Chem. Lett., 2015, 25, 337–341 CrossRef CAS PubMed; (b) K. V. Sashidhara, S. R. Avula, K. Sharma, G. R. Palnati and S. R. Bathula, Eur. J. Med. Chem., 2013, 60, 120–127 CrossRef CAS PubMed; (c) K. V. Sashidhara, R. K. Modukuri, D. Choudhary, K. B. Rao, M. Kumar, V. Khedgikar and R. Trivedi, Eur. J. Med. Chem., 2013, 70, 802–810 CrossRef CAS PubMed; (d) F. Belluti, G. Fontana, L. D. Bo, N. Carenini, C. Giommarelli and F. Zunino, Bioorg. Med. Chem., 2010, 18, 3543–3550 CrossRef CAS PubMed; (e) Q. Sun, D.-Y. Peng, S.-G. Yang, X.-L. Zhu, W.-C. Yang and G.-F. Yang, Bioorg. Med. Chem., 2014, 22, 4784–4791 CrossRef CAS PubMed; (f) K. Paul, S. Bindal and V. Luxami, Bioorg. Med. Chem. Lett., 2013, 23, 3667–3672 CrossRef CAS PubMed; (g) R. Aggarwal, S. Kumar, P. Kaushik, D. Kaushik and G. K. Gupta, Eur. J. Med. Chem., 2013, 62, 508–514 CrossRef CAS PubMed.
- (a) I. Khan, S. Zaib, A. Ibrar, N. H. Rama, J. Simpson and J. Iqbal, Eur. J. Med. Chem., 2014, 78, 167–177 CrossRef CAS PubMed; (b) I. Khan, S. J. A. Shah, S. A. Ejaz, A. Ibrar, S. Hameed, J. Lecka, J. L. Millan, J. Sévigny and J. Iqbal, RSC Adv., 2015, 5, 64404–64413 RSC.
- (a) I. Khan, A. Ibrar, S. Zaib, S. Ahmad, N. Furtmann, S. Hameed, J. Simpson, J. Bajorath and J. Iqbal, Bioorg. Med. Chem., 2014, 22, 6163–6173 CrossRef CAS PubMed; (b) I. Khan, S. M. Bakht, A. Ibrar, S. Abbas, S. Hameed, J. M. White, U. A. Rana, S. Zaib, M. Shahid and J. Iqbal, RSC Adv., 2015, 5, 21249–21267 RSC; (c) A. Saeed, P. A. Channar, Q. Iqbal and J. Mahar, Chin. Chem. Lett., DOI:10.1016/j.cclet.2015.09.011.
- T. Somerai, L. Szilagyi and S. Horvath, Arch. Pharm., 1986, 139, 238–241 CrossRef PubMed.
- K. M. Dawood, H. Abdel-Gawad, H. A. Mohamed and F. A. Badria, Med. Chem. Res., 2011, 20, 912–919 CrossRef CAS.
- J. Iqbal, Anal. Biochem., 2011, 414, 226–231 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- M. Niks and M. Otto, J. Immunol. Methods, 1990, 130, 149–151 CrossRef CAS.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- K. Ghosh, T. D. Mazumde, R. Anumula, B. Lakshmaiah, P. P. Kumar, S. Singaram, T. Matan, S. Kallipatti, S. Selyam, P. Krishnamurthy and M. Ramarao, J. Struct. Biol., 2013, 184, 182–192 CrossRef CAS PubMed.
- N. Ingale, V. Maddi, M. Palkar, P. Ronad, S. Mamledesai, A. H. M. Vishwanathswamy and D. Satyanarayana, Med. Chem. Res., 2012, 21, 16–26 CrossRef CAS.
- K. Araki-Sasaki, S. Aizawa, M. Hiramoto, M. Nakamura, O. Iwase, K. Nakata, Y. Sasaki, T. Mano, H. Handa and Y. Tano, J. Cell. Physiol., 2000, 182, 189–195 CrossRef CAS.
- G. S. Longo-Sorbello, G. m. Say, D. Banerjee and J. R. Bertino, Cytotoxicity and cell growth assays, Cell Biology, Four-Volume Set: A Laboratory Handbook, 2005, vol. 1, pp. 315–324 Search PubMed.
- Molecular Operating Environment (MOE), 2013.08, Chemical Computing Group Inc., 1010, Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14900b |
| This journal is © The Royal Society of Chemistry 2015 |