DOI:
10.1039/C4QO00306C
(Review Article)
Org. Chem. Front., 2015,
2, 179-190
Catalytic enantioselective organic transformations via visible light photocatalysis
Received
21st November 2014
, Accepted 5th December 2014
First published on 10th December 2014
Abstract
In this minireview, catalytic enantioselective transformations via visible light photocatalysis are discussed. High enantioselectivities are achieved from the combination of visible light photocatalysis with chiral organocatalysts. Considering the huge progress made in both asymmetric synthetic chemistry and visible light photocatalysis chemistry, more new chiral catalysts with varieties of transition-metals will be developed and engaged in the field of asymmetric photoreactions.
 Chengfeng Wang | Chengfeng Wang was born in Anhui province, China. She received a BSc in Chemistry from Anhui Normal University (2013) and now is pursuing a Master's degree in the laboratory of Professor Zhan Lu at Zhejiang University. She is currently conducting asymmetric reactions utilizing visible light. |
 Zhan Lu | Zhan Lu was born in 1981 in the Zhejiang Province. He received a BSc (2003) and a Ph.D. degree (2008) under the supervision of Professor Shengming Ma in Chemistry from Zhejiang University. After postdoctoral research with Professor Shannon S. Stahl and Professor Tehshik P. Yoon at University of Wisconsin-Madison, he returned to Zhejiang University in 2013 and joined the chemistry faculty. Currently his research interests focus on radical chemistry, transition-metal catalysis, and new methodologies for asymmetric synthesis. |
1. Introduction
Racemic mixtures, consisting of two enantiomers which have the same chemical structure but which may differ in biological activities such as pharmacology, toxicology, pharmacokinetics, metabolism etc., are utilized as drugs and used to occupy more than half a percent of the sale of drugs.1 Thalidomide is an example that illustrates this difference in which the (R)-enantiomer acts as a sedative in contrast to deformities caused by the (S)-enantiomer. Due to unpredictable factors in the presence of mixtures of isomers, it is necessary to obtain enantiopure molecules. To date, a huge number of natural or artificial chiral molecules, which have been isolated, synthesized, and identified, have had an impact in different research areas such as chemistry, biology, pharmacology, materials science and so on.2 Among a variety of different strategies, catalytic asymmetric transformation is one of the most efficient methods.3
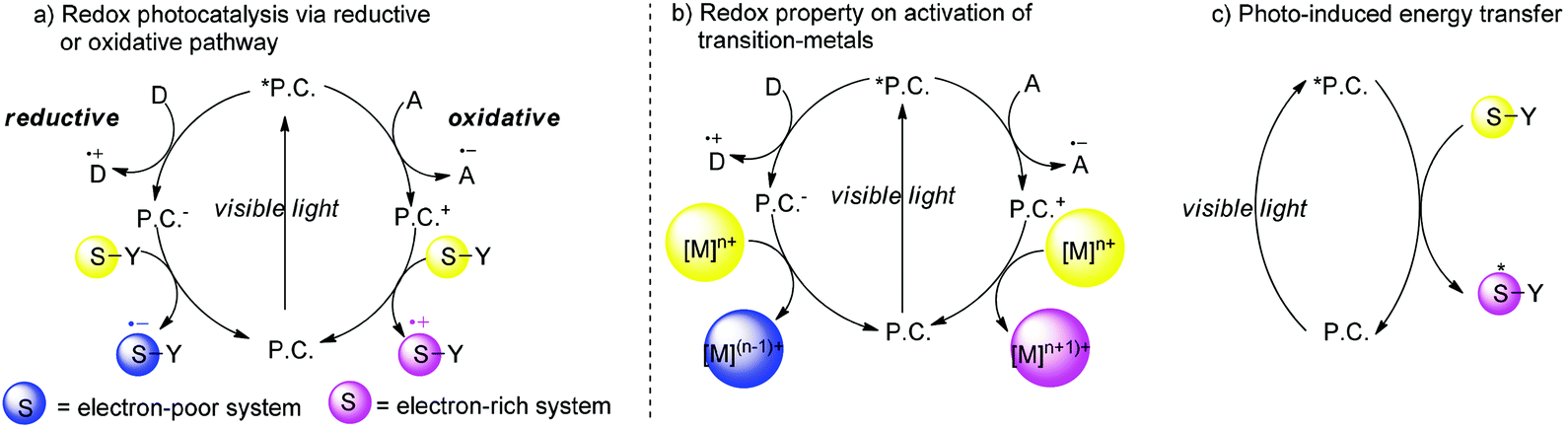
In the past seven years, there has been a considerable interest in synthetic photochemistry in which visible light can be utilized as the energy source by using transition metal chromophores.4 In the working model, transition metal complexes or organic molecules (P.C.) as photo-sensitizers absorb visible light to generate photoexcited species which usually have a long life time and remarkable redox properties that may engage in a single-electron transfer (SET) process with organic substrates. Because of this unique property, redox transformation of photoexcited species (*P.C.) may conduct reductive quenching or oxidative quenching. In a reductive quenching cycle, photoexcited species *P.C. could be reduced by the electron donor (D) to produce the reductive species P.C.− which have strong reductive potential and undergo a single electron reduction pathway. The electron-poor substrates abstract an electron from P.C.− to generate the more active radical anion S˙− which undergoes further organic transformations, simultaneously P.C. are regenerated and also the reduced species which subsequently give an electron to P.C. and then regenerate P.C. back into the cycle. Alternatively in an oxidative quenching cycle, single electron oxidation of photoexcited species *P.C. by the electron acceptor (A) yields radical anion A˙+ and the oxidized species P.C.+ which plays a role as an oxidant to capture an electron from the electron-rich substrates. Alternatively the visible light photocatalysis could also involve transition-metals in which P.C.+ could oxidize [M]n+ to a higher valence state [M](n+1)+ while P.C.− could also reduce [M]n+ to a lower valence state [M](n−1)+ instead of working directly with the substrates (Scheme 1b). The organic transformations could be also initiated by energy transfer rather than by a single electron-transfer mechanism (Scheme 1c). When the triplet energy of the catalyst is similar to the triplet energy of the substrate, the energy transfer from the photoexcited species *P.C. to the substrate could occur to generate the excited state of the substrate which could undergo further organic transformations. Due to uncontrollable background reactions, although enantioselective photochemical reactions have been studied by Kuhn since 1930s, utilizing a photocatalyst with light to create highly enantioselective chiral molecules is still the Holy Grail of photosynthesis.5 Recently, catalytic asymmetric organic transformations combined with visible light photocatalysis have been reported in several cases. There are two reaction models: (1) Radicals or radical ions generated directly by photoreactions under the irradiation of visible light could react with chiral intermediates generated by asymmetric catalysts with other active substrates to create a new chiral center; (2) The chiral catalysts directly combined with substrates to form electron-pair intermediates which could absorb visible light and undergo further transformations. Because of high activation energy for radicals or radical ions, it is difficult to control the selectivity of asymmetric visible light photocatalyzed reactions. In this minireview, we summarize homogeneous catalytic asymmetric transformations utilizing visible light photocatalysis including chiral photocatalysts and racemic photocatalysts with chiral organocatalysts, chiral Brønsted acids or chiral Lewis acids.
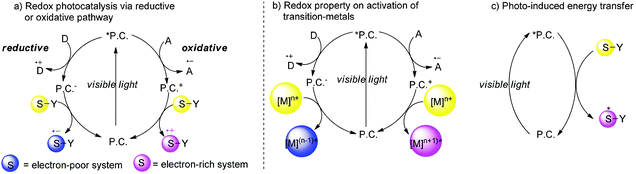 |
| | Scheme 1 Redox properties of visible light photocatalysis. | |
2. Chiral-at-metal visible light photocatalysis
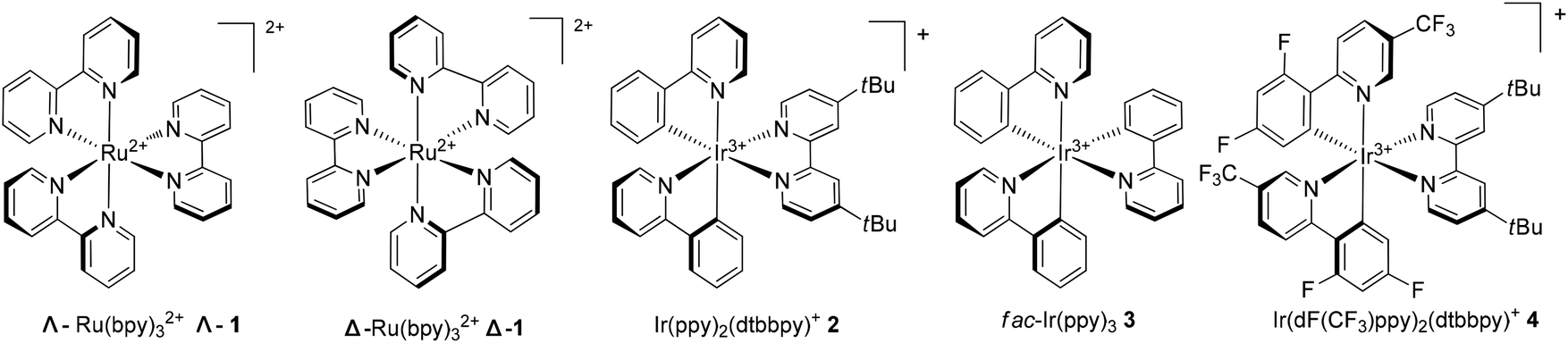
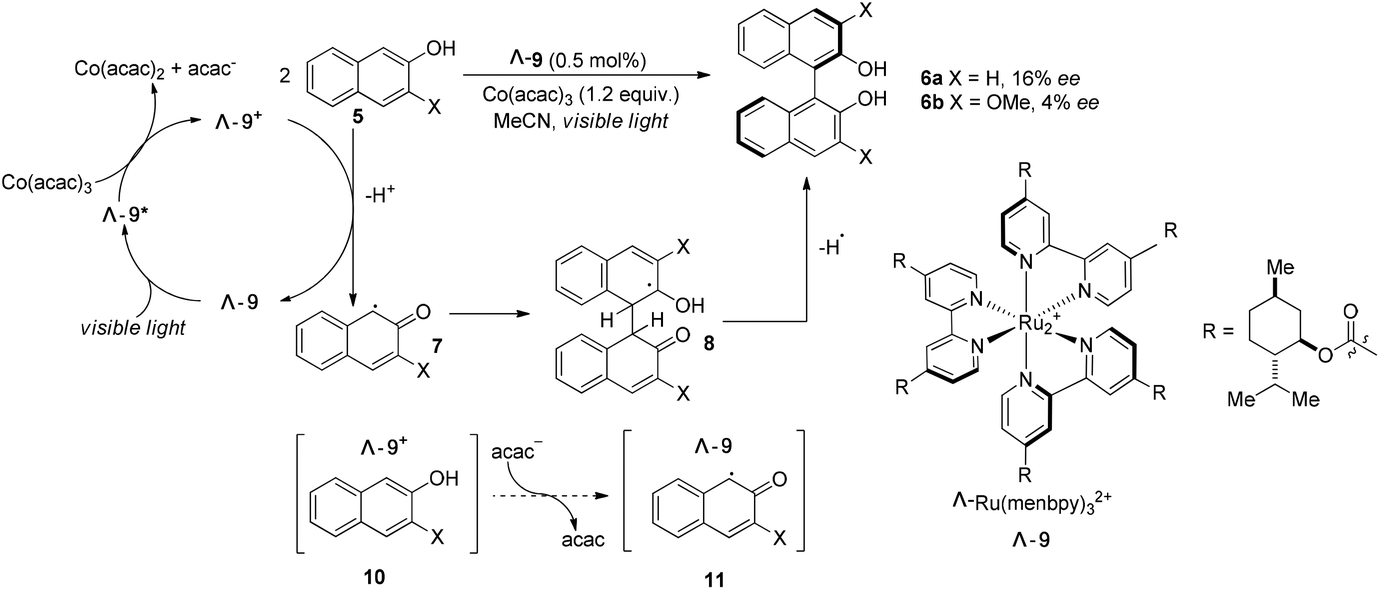
The Ru(bpy)32+ (1) complex possesses two enantiomers, a right-handed (Δ) helix and a left-handed (Λ) helix, depending on the chelation of the achiral ligands with the central metal around the C3 symmetric axis (Scheme 2).6 Chiral-at-metal complexes have been used to build a chiral environment to induce asymmetric organic reactions.7 Although visible light-driven transition metal complexes have been widely used in organic transformations, the applications of chiral complexes are quite limited. In the presence of a chiral menthol group on the bipyridine ligand, Λ-Ru(menbpy)32+ (Λ-9) could be easily obtained by column chromatography due to the different properties of the diastereoisomers.8 Under irradiation by visible light, complex Λ-9 was found to be capable of catalyzing the oxidative dimerization of naphthols 5 to yield 1,1′-bi-2-naphthol (BINOL) 6a in 16% ee or 3-methoxy-2-naphthol 6b in 4% ee (Scheme 3). Naphthols 5 are sufficiently electron-rich to be oxidized by the oxidative state Λ-Ru(menbpy)33+ (Λ-9+), which could be obtained from the visible-light photoexcited state Λ-9* using stoichiometric Co(acac)3 as a terminal oxidant, then deprotonated to yield α-carbonyl radical intermediate 7. The intermolecular radical coupling reaction of 7 with naphthols afforded the binaphthol radicals which could undergo aromatization and deprotonation to furnish the binaphthols.
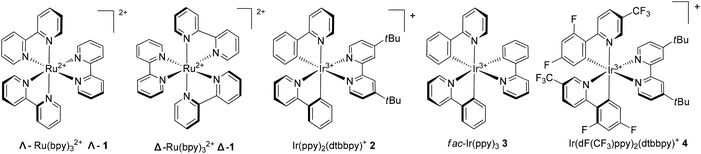 |
| | Scheme 2 Several commonly used transition-metal photocatalysts. | |
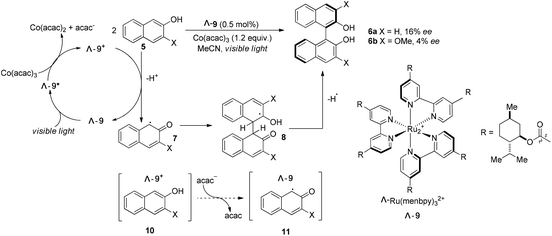 |
| | Scheme 3 Visible light promoted asymmetric dimerization of naphthols with catalyst Λ-9. | |
A general feature of photoredox catalysts reflected in this reaction might suggest that the photocatalysts typically do promote the generation of reactive radical species, however, they rarely serve to affect the step of bond-forming transformation. We propose that the chiral ion pair complexes 10 and 11 might be formed during the transformation (Scheme 3), however, these proposed complexes were not tight enough and easily released the two partners without any further interaction. Ohkubo's efforts might enlighten other scientists on the challenge of utilizing visible light to realize catalytic asymmetric photoreactions.
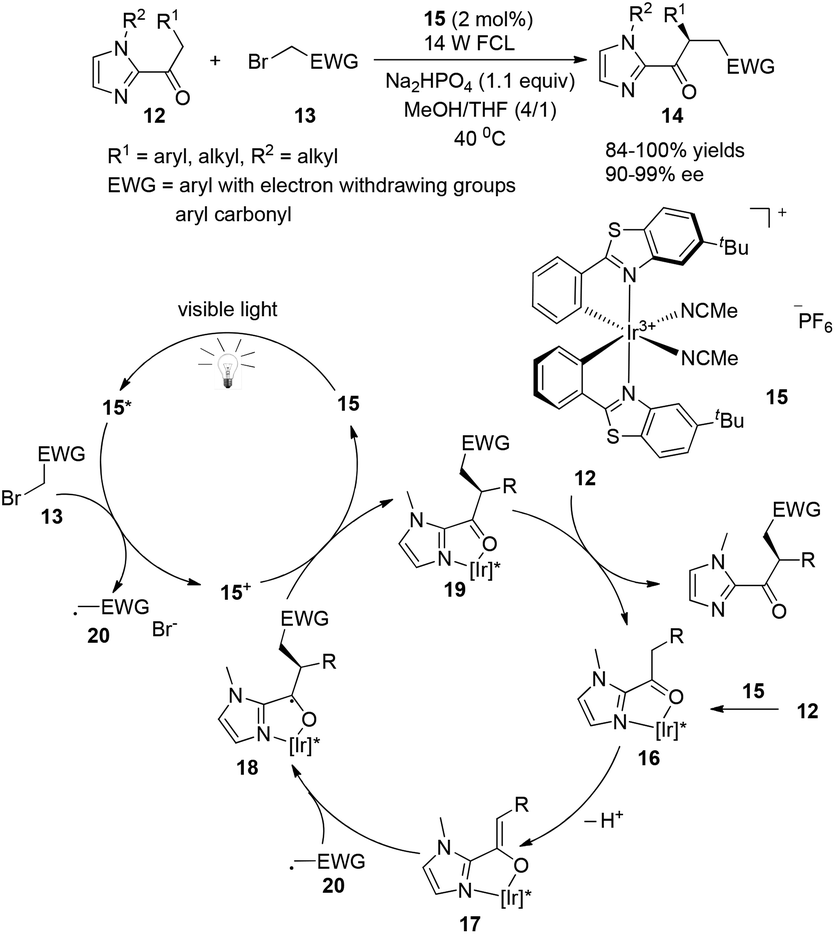
A very recent report presented by Meggers successfully demonstrated that chiral-at-metal visible light photoactivated sensitizers could realize the highly enantioselective α-benzylation of activated ketones (Scheme 4).9 The chiral iridium complexes are demonstrated to absorb visible light (425 nm) and have excellent redox properties. The key intermediate 16 could be formed by the coordination of iridium metal with an oxygen atom from the carbonyl group and a nitrogen atom from the imidazole group, converting to 18 by deprotonation and asymmetric radical addition of an electron withdrawing benzyl radical. The reaction afforded the α-benzylated ketones 14 in excellent yields (84–100%) and ee (90–99%). This unique example could be a landmark for designing chiral photosensitizers for catalytic visible-light-induced reactions.
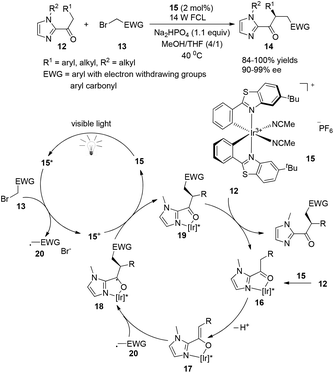 |
| | Scheme 4 Asymmetric α-benzylation of 2-acylimidazoles. | |
3. Chiral organocatalysts with hydrogen-bonding
3.1 Chiral thioureas
Amines are commonly used sacrificially to offer an electron to visible light photocatalysts, leading to amino cation radicals. This active amino cation radical will either deprotonate to generate an α-amino radical as a good radical nucleophile or lose a hydrogen atom to deliver iminium ions which are potentially capable of nucleophilic addition reactions, accelerating the development of diversified α-functionalization reactions of amines as well as asymmetric organic transformations.
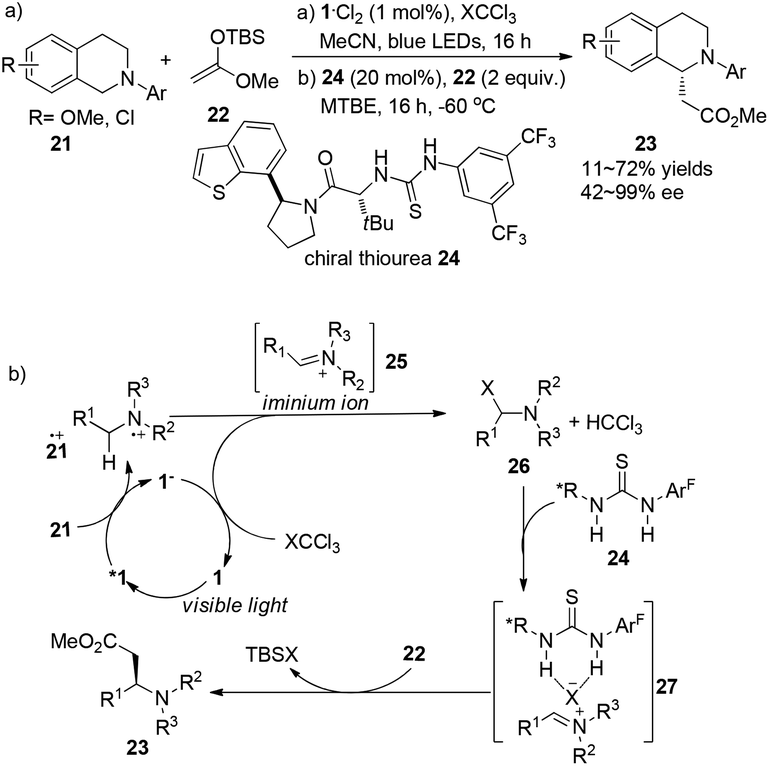
A report by Jacobsen and Stephenson indicated the feasibility of using a chiral thiourea as an anion-binding catalyst combined with a photoredox catalyst to conduct asymmetric C–H functionalization of N-aryltetrahydroisoquinoline.10 With the employment of 1·Cl2 (1 mol%) as the photocatalyst and thiourea 24 (20 mol%) as the organocatalyst under the irradiation of blue LED, N-aryltetrahydroisoquinolines 21 could undergo single electron oxidation by photoexcited *Ru(bpy)32+ (*1), followed by nucleophilic addition of the resulting iminium ion with silyl ketene acetals to give α-alkylated tetrahydroisoquinolines 23 in 11–72% yields and 42–99% ee (Scheme 5a). Single-electron reduction of XCCl3 by Ru(bpy)3+ (1−) followed by fragmentation delivered an X anion which could combine with an iminium cation to form intermediate 26, and regenerated the photocatalyst. A variety of chiral thioureas were screened as chiral anion-binding catalysts11 and 24 was quite suitable for the anion under the catalytic conditions to produce a chiral tight ion pair 27 which was subsequently attacked by silyl ketene acetal to afford α-esterificated tetrahydroisoquinoline derivatives 23 with moderate to excellent enantioselectivities (Scheme 5b). During this process, a highly variable enantioselectivity can be observed via the formation of different tight ion pairs among which Cl− from CCl4 contributes to more favourable interactions of the chloride-bonded thiourea catalyst in the enantioselectivity-determining transition state. Besides, solubility properties of the iminium ion 25 might be enhanced if PF6− existed in the reaction, enabling the racemic background reaction to a greater extent. For an appreciable application of this enantio-induction, a two-stage protocol was required involving a solvent change from acetonitrile to methyl tert-butyl ether and a reaction temperature decrease to −60 °C. High enantioselectivity was obtained in this reaction with limited substrates.
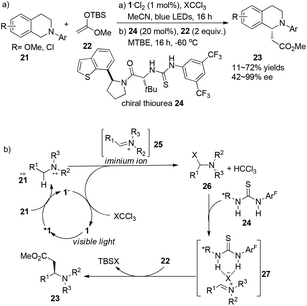 |
| | Scheme 5 (a) Asymmetric α-photoalkylation of N-aryl-tetrahydroisoquinoline with chiral thiourea 24. (b) Proposed mechanism through hydrogen–halogen bonding. | |
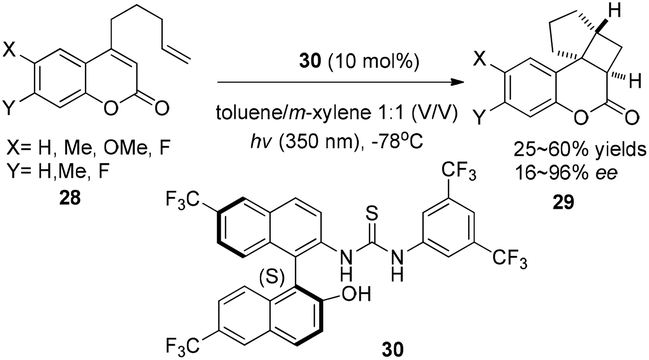
Using a chiral thiourea organocatalyst under irradiation by UV, Sibi and Sivaguru have demonstrated an electron or energy free process in the enantioselective intramolecular [2 + 2] photocycloaddition of 4-alkenyl-substituted coumarins (28), which leads to the corresponding products 29 in 25–60% yields with 16–96% ee in toluene–m-xylene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Scheme 6).12 The hydrogen-bonds between hydrogen of the chiral thiourea 30 and oxygen of the carbonyl moiety of 4-alkenyl-substituted coumarins play a key role to create chirality.
:1) (Scheme 6).12 The hydrogen-bonds between hydrogen of the chiral thiourea 30 and oxygen of the carbonyl moiety of 4-alkenyl-substituted coumarins play a key role to create chirality.
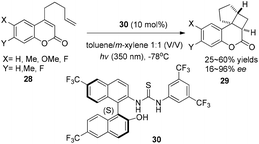 |
| | Scheme 6 Enantioselective [2 + 2] cycloadditions of coumarins with chiral thiourea 30 under UV irradiation. | |
3.2 Chiral thioxanthones
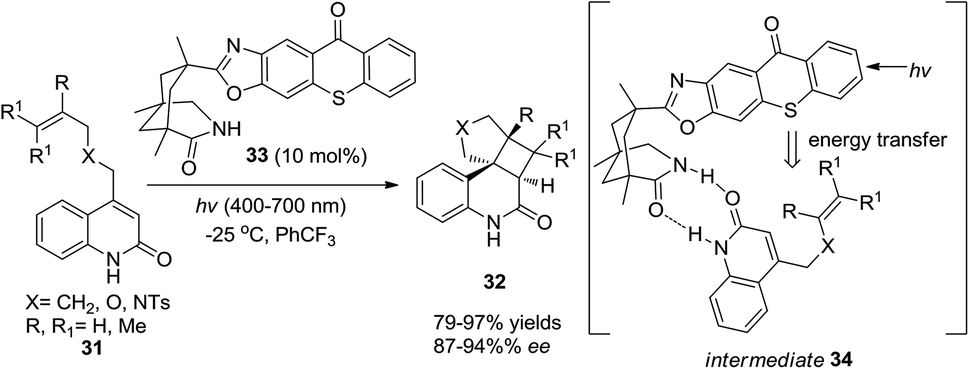
Bach and co-workers reported a catalytic loading of chiral thioxanthone (10 mol%) as an organocatalyst to conduct asymmetric intramolecular [2 + 2] cycloadditions of quinolones under visible light irradiation (Scheme 7).13 Chiral thioxanthone (33) could absorb visible light and then transmit the energy to quinolones (31) through triplet energy transfer14 which has not yet been confirmed but is apparently revealed in the photostability studies where the triplet state of thioxanthone acts less vigorously towards hydrogen abstraction15 than the triplet state of xanthone derivatives in a photocatalytic cycle. Due to the double hydrogen-bonding effect between chiral thioxanthone and quinolones, high enantioselectivity was achieved in 87–94% ee.
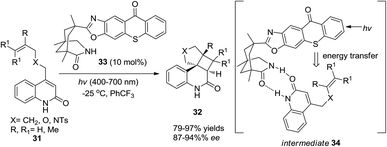 |
| | Scheme 7 Asymmetric intramolecular [2 + 2] photocycloaddition reaction with chiral thioxanthone 33. | |
3.3 Chiral phosphoric acids
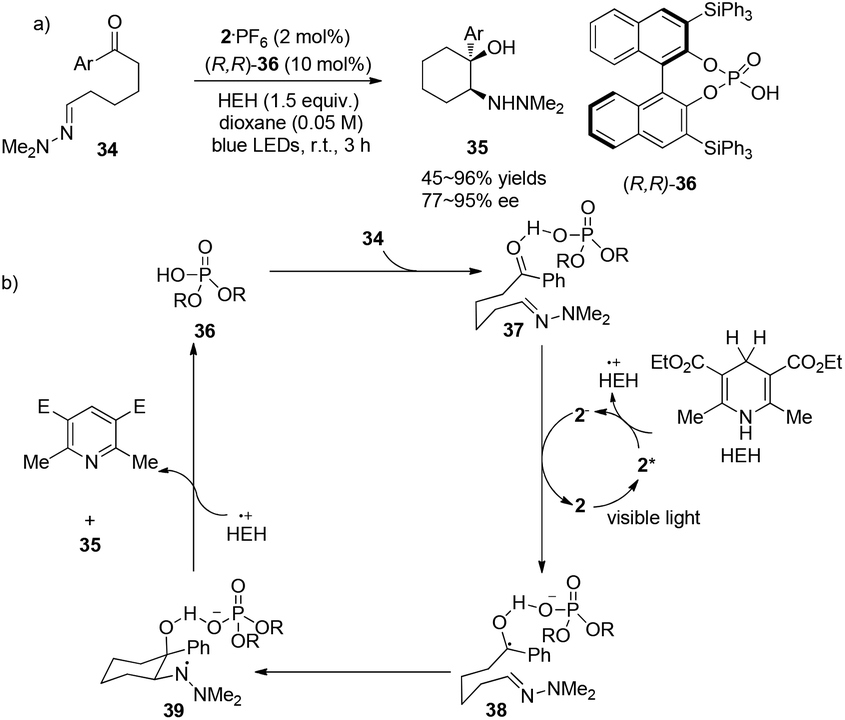
In a conceptually distinct contribution, Brønsted acids can also be utilized together with a visible light photocatalyst to promote asymmetric aza-pinacol photoreactions between a ketone and a hydrazone efficiently via a proton-coupled electron transfer (PCET) event as reported by Knowles.16 With chiral phosphoric acid (R,R)-36 (10 mol%) and 2·PF6 (2 mol%), 34 could undergo radical cyclization to convert to cyclic amino alcohol derivatives 35 in 45–96% yields with highest ees up to 95% (Scheme 8a). Hantzsch ester has been employed not only as a single-electron reductant but also as a hydrogen source. The ketone group, which is a good hydrogen-bond acceptor activated by (R,R)-36, could trap an electron from photoreduced species 2− to afford the radical intermediate 38. Intramolecular radical cyclization of 38 could afford amino-radical intermediate 39 which then receives a hydrogen atom from the oxidized HEH to furnish the final product and regenerate the phosphoric acid (Scheme 8b). Chirality of this reaction is introduced through activation of the aryl carbonyl group by a hydrogen-bonding effect with chiral phosphoric acid. This work demonstrates the concerted PCET activation as a potential approach to further development of asymmetric radical chemistry.
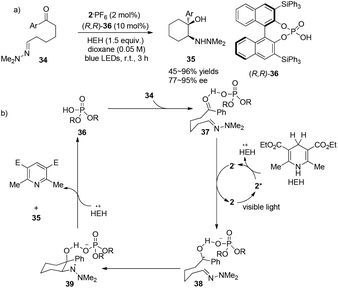 |
| | Scheme 8 (a) Chiral phosphoric acid catalyzed intramolecular cyclizations via visible light photocatalysis. (b) Proposed mechanism. | |
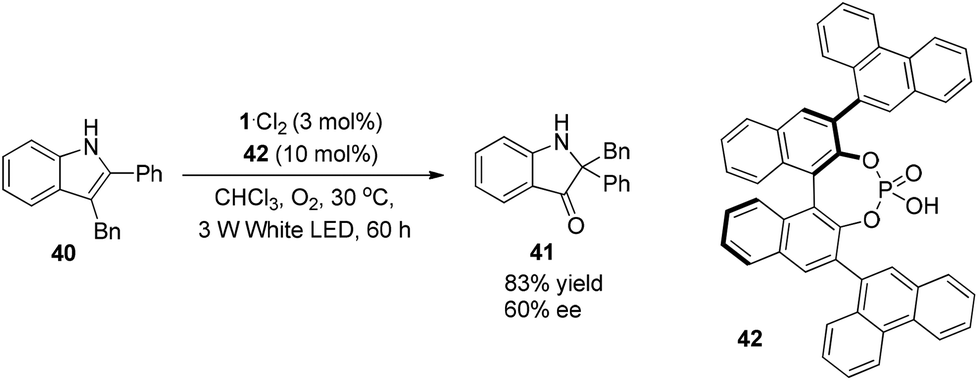
A photo-induced aerobic oxidation–semipinacol rearrangement reaction of indoles was presented recently by Lu and Xiao (Scheme 9).17 The asymmetric reaction could be achieved by using chiral phosphoric acid 42 as a Brønsted acid, giving the 2,2-disubstituted indolin-3-one in 83% yield and 60% ee.
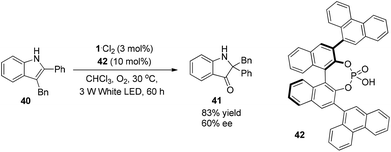 |
| | Scheme 9 Asymmetric aerobic oxidation–semipinacol rearrangement reaction of indoles. | |
4. Chiral N-heterocyclic carbenes
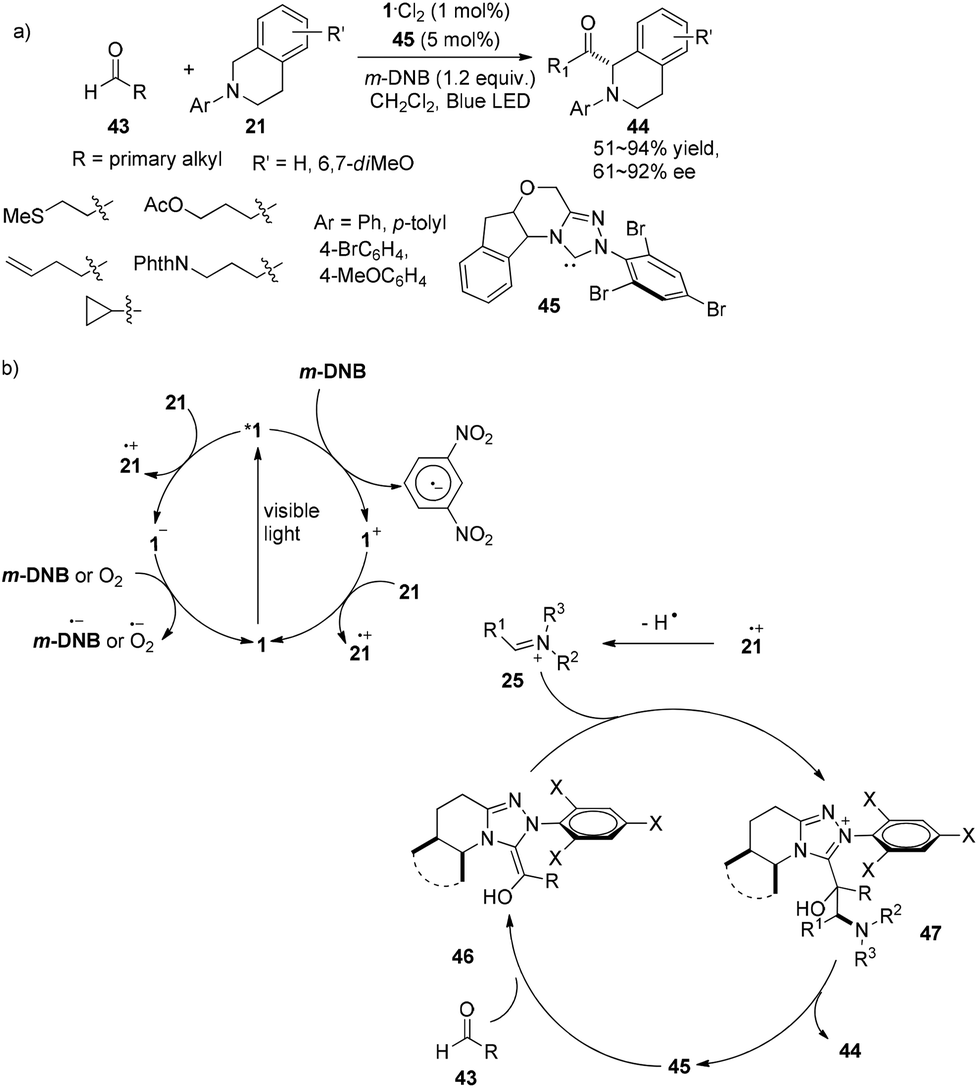
In 2012, Rovis introduced N-heterocyclic carbenes (NHCs) as organocatalysts for photocatalytic reactions and successfully operated the asymmetric α-acylation of N-phenyltetrahydroisoquinoline.18 A catalyst system containing 5 mol% of chiral NHC 45, 1 mol% of 1·Cl2 and 1.2 equiv. of m-dinitrobenzene (m-DNB) was capable of effecting reactions between N-phenyltetrahydroisoquinoline 21 and aldehydes 43 upon irradiation by a 15 W blue LED to afford α-acylated N-phenyltetrahydroisoquinoline products 44 in 51–94% yields and 59–92% ee (Scheme 10a). The reaction could tolerate alkenes, cyclopropanes and a variety of functionalized groups with protecting groups, such as thiols, alcohols and amines. Stoichiometric m-DNB serves as an oxidative quencher of photoexcited *1 resulting in Ru(bpy)33+ (1+) during which trace oxygen might act as a terminal oxidant. N-Phenyltetrahydroisoquinoline 21 could be oxidized by both photoexcited *1 and oxidized species 1+ followed by hydrogen atom abstraction to convert to electrophilic iminium ion 25. Condensation of the chiral NHC with aldehydes would form the highly nucleophilic Breslow intermediates 46 which could undergo nucleophilic addition of iminium ion 25 to give the intermediate adduct 47. Release of the NHC from 47 could afford α-acylated N-phenyltetrahydroisoquinolines and recycle the NHC (Scheme 10b). In this manner, an activated sp3 C–H bond can be converted to a C–C bond with no pre-activated substrates, providing new access to enantioselective α-acylated N-aryl-tetrahydroisoquinolines.
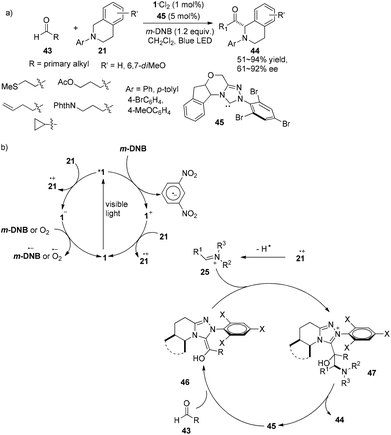 |
| | Scheme 10 (a) Asymmetric α-acylation of N-aryl-tetrahydroisoquinoline using a combination of a chiral NHC and a visible light photocatalyst. (b) Proposed mechanism. | |
5. Chiral amines
5.1 Chiral quaternary ammonium salts
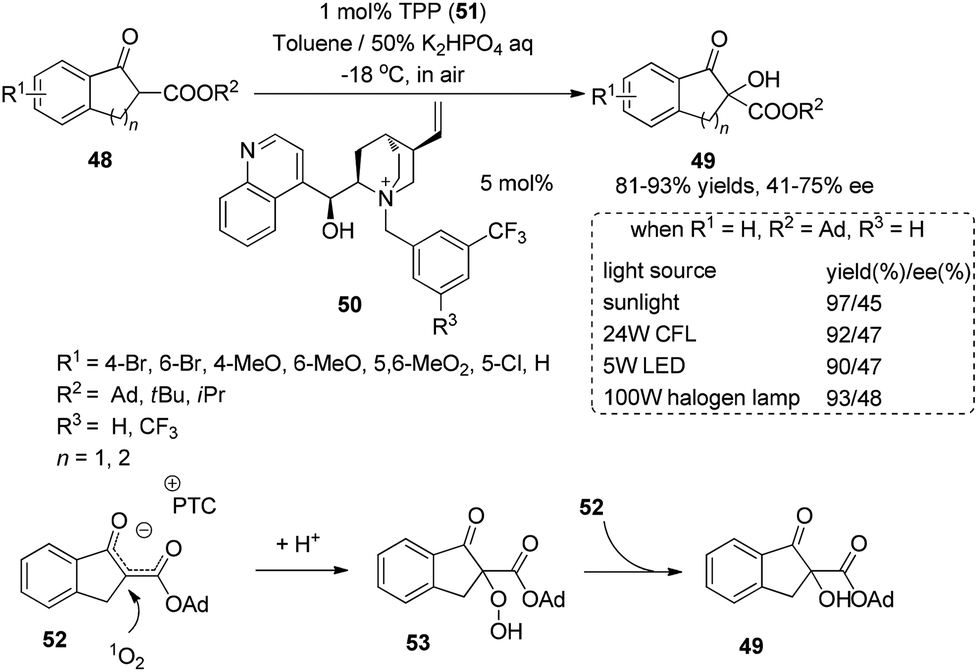
Chiral phase-transfer catalysts have received much attention for asymmetric organic transformations. Under irradiation with sunlight, the combination of a chiral phase-transfer catalyst with photosensitizer TPP could promote the oxidation of a 1,3-dicarbonyl compound to afford the hydroxylation product in 97% yield and 45% ee (Scheme 11).19 The chiral counter ion pair 52 formed in the reaction could be oxidized by singlet oxygen and then be protonated to afford a relatively stable hydroperoxide 53. The mechanism study showed that the hydroperoxide might react with ion pair 52 and be utilized as an oxidant to form the final product 49.
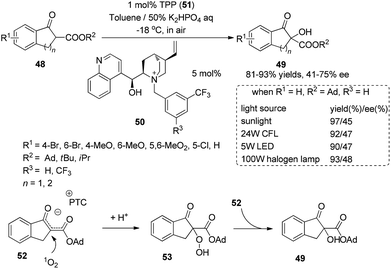 |
| | Scheme 11 Enantioselective α-photooxygenation of β-keto esters. | |
5.2 Chiral tertiary amines
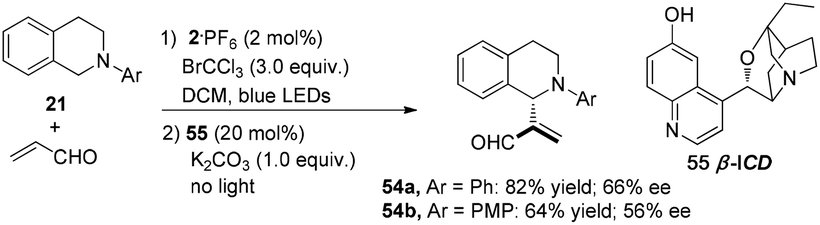
Xiao and co-workers reported an alternative dual catalysis system for the direct sp3 C–H acroleination of N-aryl-tetrahydroisoquinoline using 2·PF6 (2 mol%) as a photocatalyst, BrCCl3 (3.0 equiv.) as the oxidant under irradiation by blue light in cascade with addition of β-isocupreidine 55 (β-ICD, 20 mol%) as a chiral nucleophilic organoamine catalyst.20N-Aryl-tetrahydroisoquinoline 21 reacted with acrolein to afford 54a in 82% yield with 66% ee and 54b in 64% yield with 56% ee, respectively (Scheme 12).
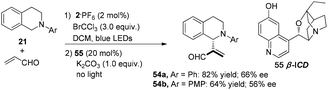 |
| | Scheme 12 Asymmetric α-alkylation of N-aryl-tetrahydroisoquinoline. | |
5.3 Chiral secondary amines
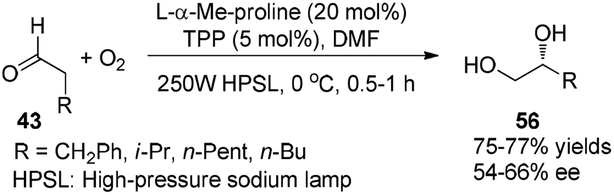
Córdova reported the direct amino acid-catalyzed asymmetric α-photooxygenation of aldehydes in 2004.21 When TPP was used as a photosensitizer under irradiation by a 250 W high-pressure sodium lamp, the aldehydes could be oxidized by molecular dioxygen and then be reduced by NaBH4 to give 1,2-dihydroxylation products in moderate yields and ee (Scheme 13).
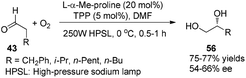 |
| | Scheme 13 Enantioselective α-photooxygenation of aldehydes. | |
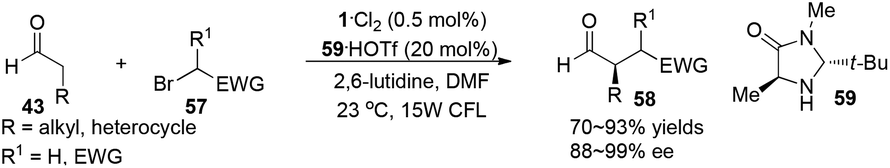
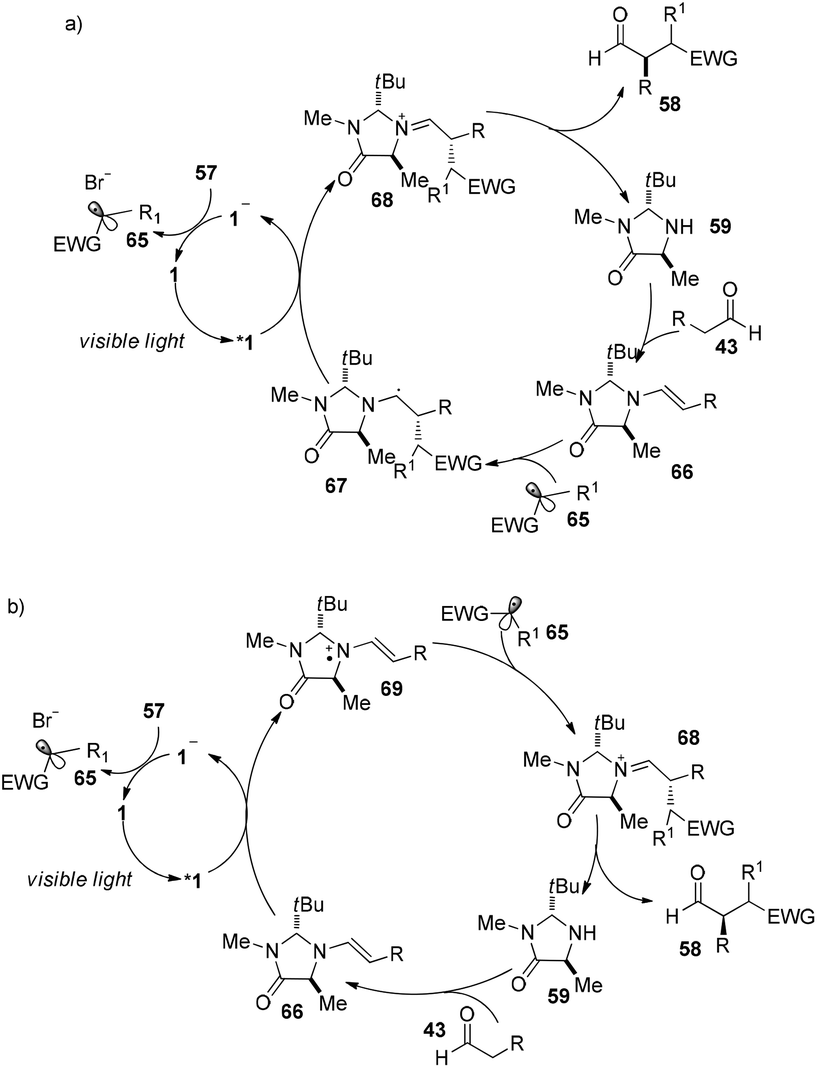
Direct asymmetric alkylation of aldehydes with alkyl bromides bearing electron-withdrawing functional groups was carried out in MacMillan's laboratory with a combination of 1·Cl2 (0.5 mol%) and chiral imidazolidinone 59 (20 mol%).22 The reaction could offer various α-alkylaldehydes derivatives 58 in 70–93% yields and 88–99% ee without any strong oxidant under visible-light irradiation from a 15 W CFL (CFL = compact fluorescent light) at room temperature (eqn (1)). An electrophilic alkyl radical is formed through single-electron oxidation of the electron-deficient alkyl bromide by 1− and fragmentation. Simultaneously, enamine intermediate 66 generated from interaction of chiral imidazolidinone with aldehyde reacts with alkyl radical 65 to form α-radical amine intermediates 67, which are subject to oxidization by photo-induced complex *1 to yield the iminium ion which could then be hydrolyzed to yield products 58 (Scheme 14a). However, there is a puzzle in the above catalytic cycle of which intermediates come into being first: electron-withdrawing alkyl radicals or 1−. In fact, the reduction potential of *1 (E1/2*II/I = +0.77 V vs. SCE) indicates its ability to oxidize enamine intermediates 66.23 There is another possible catalytic cycle (Scheme 14b). Both organocatalysis and visible light photocatalysis cycles could be initiated by the direct oxidation of enamine 66 by *1.
| | 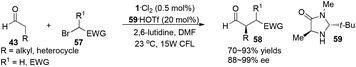 | (1) |
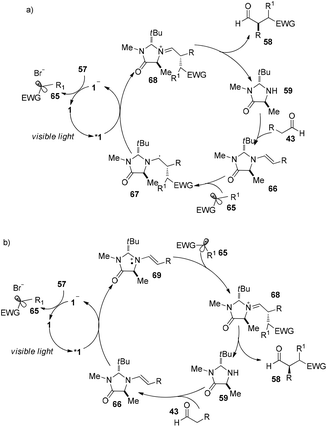 |
| | Scheme 14 (a) Proposed mechanism. (b) Alternatively proposed mechanism. | |
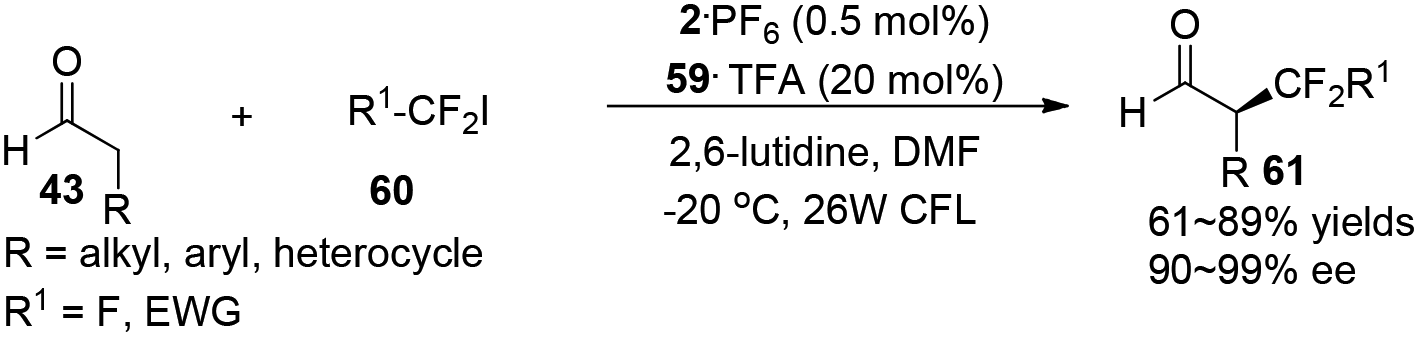
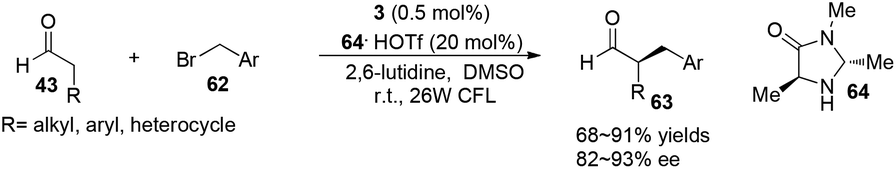
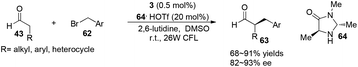
α-Perfluoromethylation and α-benzylation of aldehydes can also be realized in a conceptually similar manner.24 Under visible-light irradiation, 0.5 mol% of 2·PF6 with 20 mol% of 59·TFA or 0.5 mol% of [fac-Ir(ppy)3] 3 with 20 mol% of 64·HOTf were employed to catalyze the corresponding reactions, affording α-trifluoromethyl aldehydes 61 in 61–89% yields with 90–99% ee or α-benzylation of aldehydes 63 in 72–94% yields with 82–93% ee, respectively (eqn (2) and (3)).
| | 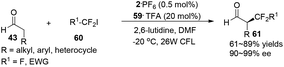 | (2) |
| |  | (3) |
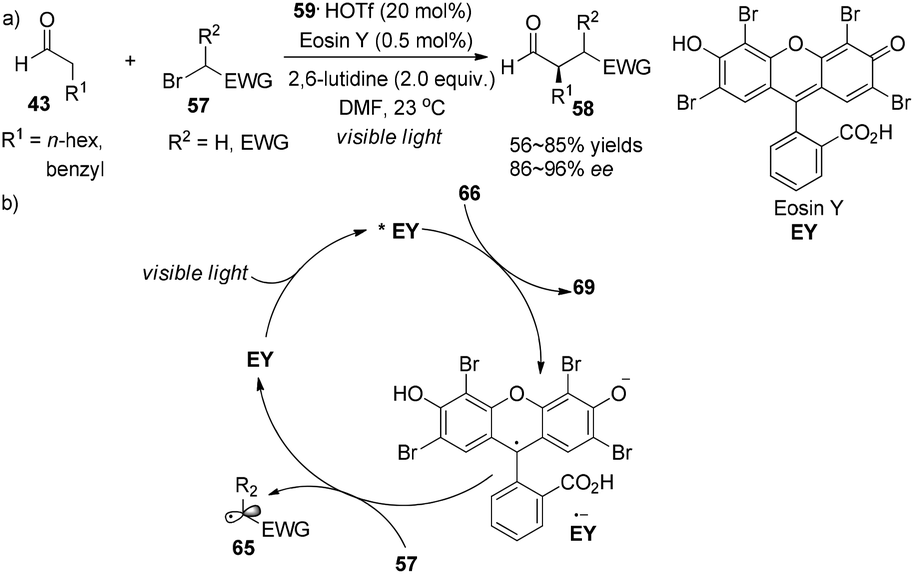
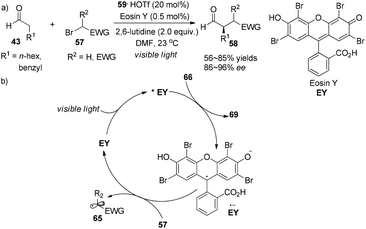
Alternatively, the photoredox organocatalysis strategy was demonstrated by Zeitler to access similar products.25 Readily accessible xanthene dye Eosin Y (EY) (0.5 mol%) serves as organic photo-sensitizer in concert with imidazolidinone 59 (20 mol%) to effect α-alkylation of aldehydes 43 with electrophilic alkyl bromides 57, giving the adducts 58 in 56–85% yields with 86–96% ee (Scheme 15a). In the photoredox cycle, as an analogue of the transition-metal photocatalyst 1, the redox property of EY (*EY (E1/2(*EY/EY˙− = +0.83 V vs. SCE) or (EY˙− (E1/2(EY/EY˙− = −1.06 V vs. SCE))) is sufficiently capable of conducting SET processes (Scheme 15b). Ferroud et al. also achieved enantioselective synthesis of α-alkylated aldehydes using Rose Bengal (RB) as a photosensitizer.26
 |
| | Scheme 15 (a) Asymmetric α-alkylation of aldehyde with Eosin Y. (b) Proposed mechanism. | |
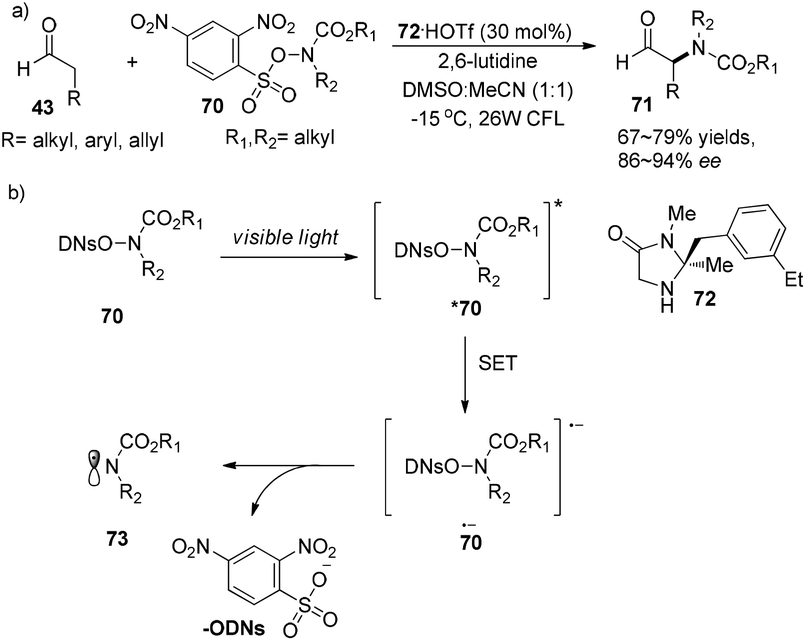
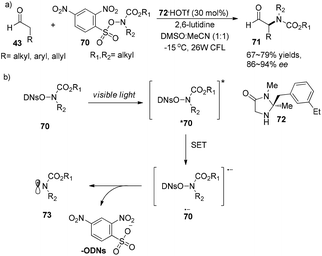
Later on, MacMillan and co-workers developed a new methodology for the asymmetric α-amination of aldehydes.27 Sulfonyl-protected hydroxyamides 70 are not only amido radical sources but also good photosensitizers which can absorb visible light to become photoexcited state species *70. Photoexcited species *70 can capture an electron, then eliminate the 2,4-dinitrophenylsulfate ion to give amido radicals 73. The reaction could proceed with chiral secondary amine 72·HOTf (30 mol%) as an organocatalyst, affording α-amino aldehydes 71 in 67–79% yields with 86–94% ee (Scheme 16a). The mechanism is similar to the previous reported cycle (Scheme 14) with the difference in the generation of amido radicals (Scheme 16b).
 |
| | Scheme 16 (a) Asymmetric α-amination of aldehydes via iminium ion. (b) Proposed mechanism. | |
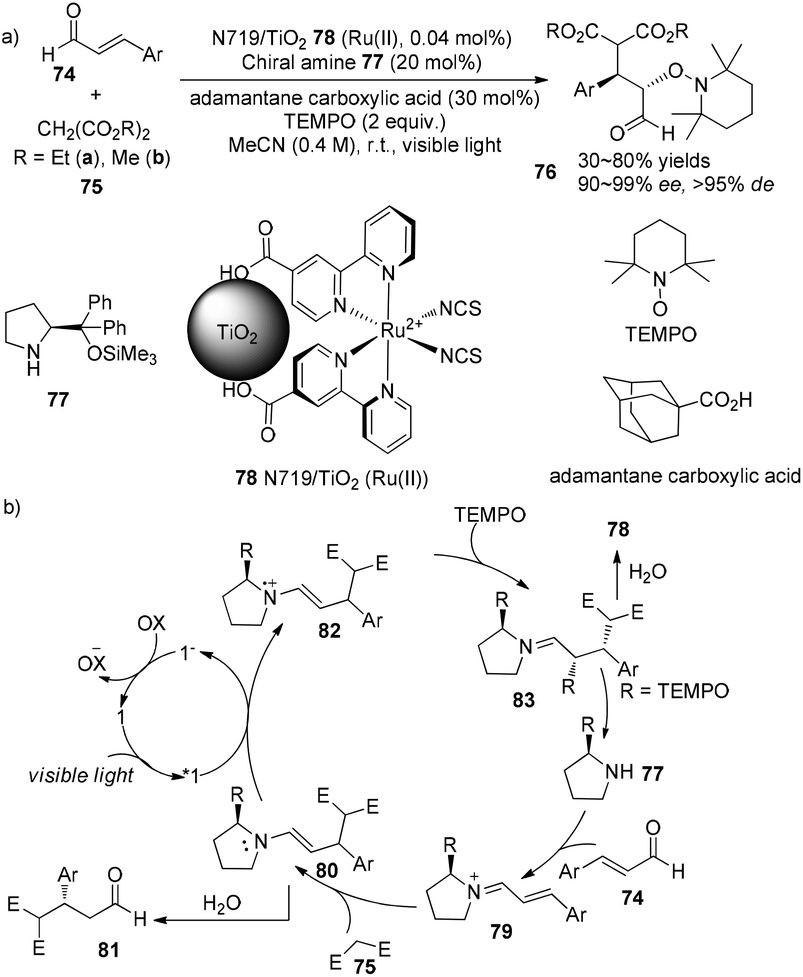
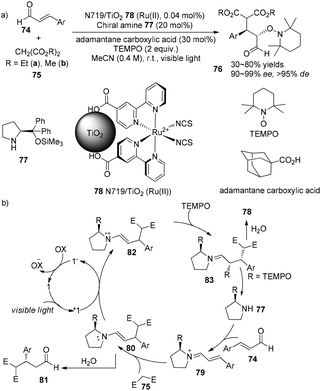
An enantioselective tandem Michael addition–oxyamination reaction of α,β-unsaturated aldehydes with diethyl malonate and TEMPO was demonstrated by Jang using (S)-2-[diphenyl(trimethylsilyloxy)methyl]pyrrolidine 77 (20 mol%) as an organocatalyst combined with N719/TiO2 Ru(II) 78 (0.04 mol%) as photosensitizer to prepare α,β-substituted aldehydes 74 in 53–80% yields with 96–99% ee and >95% de (Scheme 17a).28 Using dimethyl malonate instead of diethyl malonate, the addition gave a decreased yield and selectivity (30% yield and 90% ee). Side-products 81 can also be obtained through hydrolysis of enamine intermediate 80. Enantioselectivity of this reaction is controlled by chiral pyrrolidine29 while diastereoselectivity is controlled by N719/TiO2 Ru(II), respectively.
 |
| | Scheme 17 (a) Enantioselective tandem Michael addition–oxidation of aldehydes. (b) Proposed mechanism. | |
5.4 Chiral primary amines
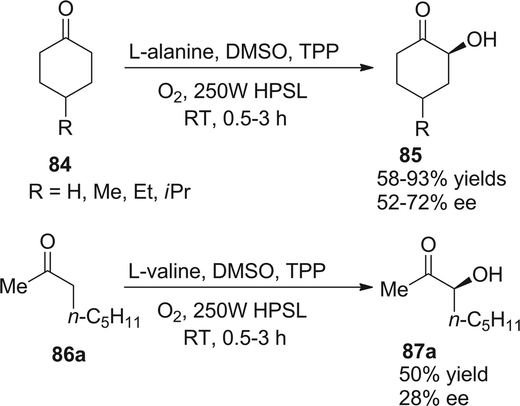
In 2004, Córdova demonstrated a similar protocol to achieve asymmetric α-photooxygenation of cyclic ketones in combination with photosensitizer TPP and natural amino acids, yielding α-hydroxylated ketones with moderate ee (Scheme 18).30 The acyclic ketone showed a slightly low reactivity and the enantioselectivity was compromised.
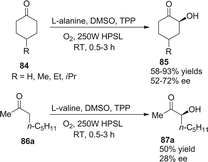 |
| | Scheme 18 Enantioselective α-photooxygenation of ketones. | |
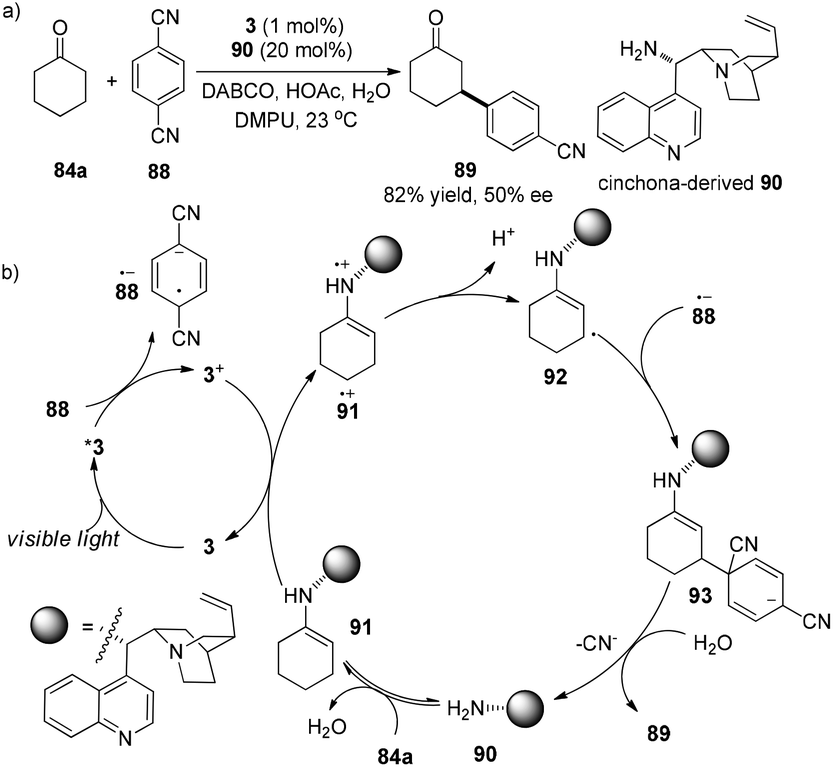
MacMillan and co-workers developed an elaborate dual catalytic model to promote β-arylation of aldehydes or cyclic ketones with cyanoarenes.31 In the photosensitizer Ir(ppy)3 (3)-catalyzed reaction, isopropyl benzylamine or azepene were utilized as the organocatalysts for aldehydes or cyclic ketones, respectively. Using cinchona-derived organocatalyst 90 (20 mol%) in conjunction with 3 (1 mol%), the asymmetric reaction of cyclohexanone with 1,4-dicyanobenzene gave the chiral product 89 in 82% yield and 50% ee (Scheme 19a). 1,4-Dicyanobenzene 88 functions as an oxidant of photo-induced excited complex *3 to yield 3+ which oxidizes electron-rich enamine 91 to the enamine radical cation 91˙+. Due to the weak allylic C–H bond, deprotonation of radical cation 91˙+ at the β-position of the carbonyl group can occur to form cyclohexenyl radical 92. Radical coupling of 92 with radical anion 88˙− gives rise to anion 93 which can undergo elimination of cyano anion in the presence of water to deliver the desired product 89 and regenerate amine catalyst 90 (Scheme 19b).
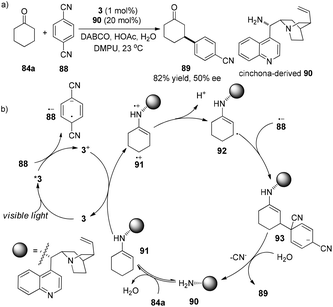 |
| | Scheme 19 (a) Direct asymmetric β-arylation of cyclohexane. (b) Proposed mechanism. | |
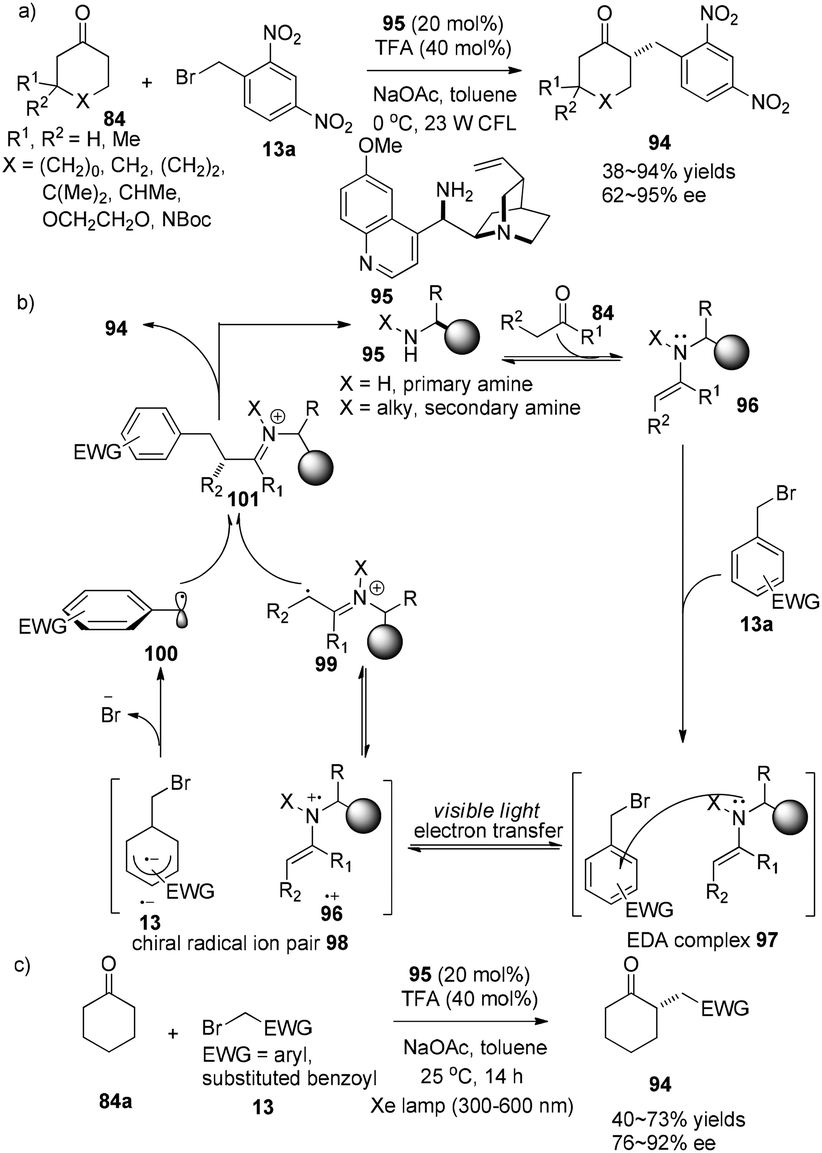
The Melchiorre group demonstrated a chiral ion pair driven asymmetric α-alkylation of cyclic ketones. The easily obtained quinidine-derived primary amine 95 (20 mol%) is involved in both activation of carbonyl compound 13 and the enantioselectivity-defining event to achieve enantioselective α-alkylcycloketones 94 in 38–94% yields with 74–95% ee (Scheme 20a).32 Initially, the electron donor–acceptor (EDA) complex 97 is formed via aggregation of aryl bromides with electron-rich enamine. Single electron transfer inside this EDA complex 97 from nitrogen to alkyl bromide affords a chiral radical ion pair 98 which is a combination of aryl radical anion and amino radical cation. Then facile fragmentation of Br− from 13˙+ followed by coupling with amino radical cation 99 affords imine cation 101 which is known to easily release α-alkylcycloketone 94 (Scheme 20b).
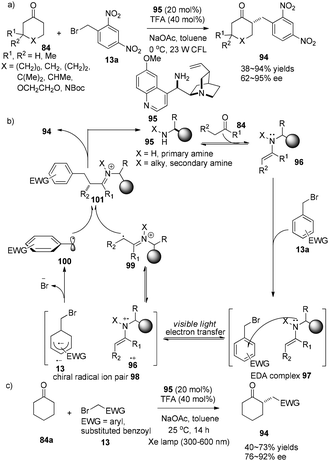 |
| | Scheme 20 (a) and (c) Asymmetric catalytic α-alkylation of cycloketones with alkyl halides. (b) Proposed mechanism. | |
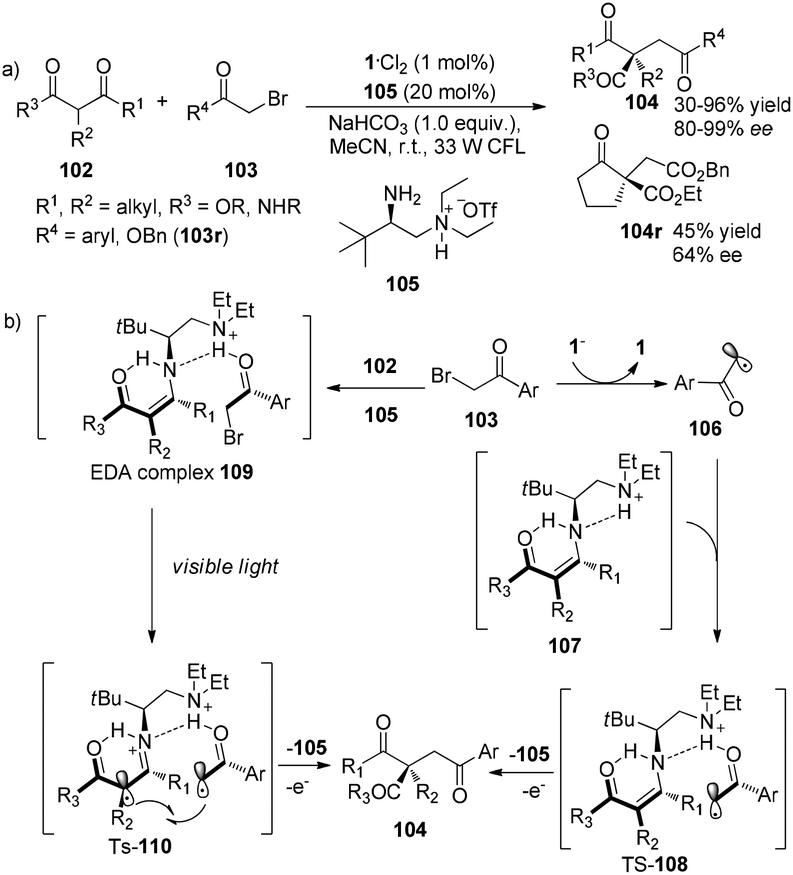
Subsequent to Melchiorre’s report, Luo and his co-workers have revealed that chiral primary amines do really help to construct chiral quaternary carbon centers in asymmetric enamine-based transformation of β-ketocarbonyls.33 The reaction of β-ketocarbonyls and α-bromocarbonyls employing 1·Cl2 (1 mol%) and chiral diamine salt 105 (20 mol%) as co-catalyst afforded α-alkylated β-ketonyls in 30–96% yields with 64–99% ee (Scheme 21a). There are two possible transition intermediates: the proposed major intermediate TS-108 could be formed in the photoredox pathway and the proposed minor intermediate TS-110 could be generated in the electron donor–acceptor (EDA) pathway. In the photoredox pathway, the acetophenone radicals formed by photoreduction of 2-bromoacetophenones could be bonded with the enamine intermediates 107, generated from condensation of the chiral primary amine catalyst with β-ketocarbonyls 102, via hydrogen-bonding between hydrogen of enamines and keto moiety of acetophenone radicals, forming the transition state TS-108. Intramolecular radical addition of TS-108 followed by release of an electron and 105 afforded products 104. While in the EDA pathway, 2-bromoacetophenones could interact directly with β-ketocarbonyls catalyzed by chiral amine to afford the EDA complexes 109 which could undergo intramolecular electron transfer from enamines to 2-bromoacetophenones under the irradiation of a 33 W CFL to give the transition state TS-110 (Scheme 21b). Intramolecular radical–radical coupling followed by hydrolysis of this transition state would achieve final product 104. Benzyl bromoacetate which has lower electron-withdrawing property of ester moiety compared with keto moiety on 2-bromoacetophenones was also suitable for this transformation to give the desired product, however, in decreased yield and enantioselectivity. This result shows that the weaker hydrogen-bonding between carbonyl and protonated tertiary amine, results in poorer reactivity and enantioselectivity.
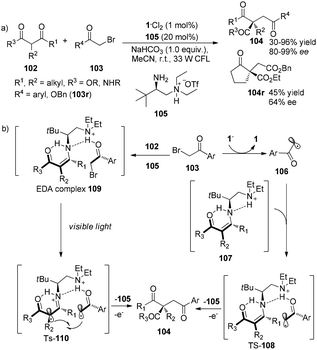 |
| | Scheme 21 (a) Enantioselective α-photoalkylation of β-ketocarbonyls with a combination of chiral primary amine and photocatalyst. (b) Proposed mechanism. | |
6. Chiral Lewis acids
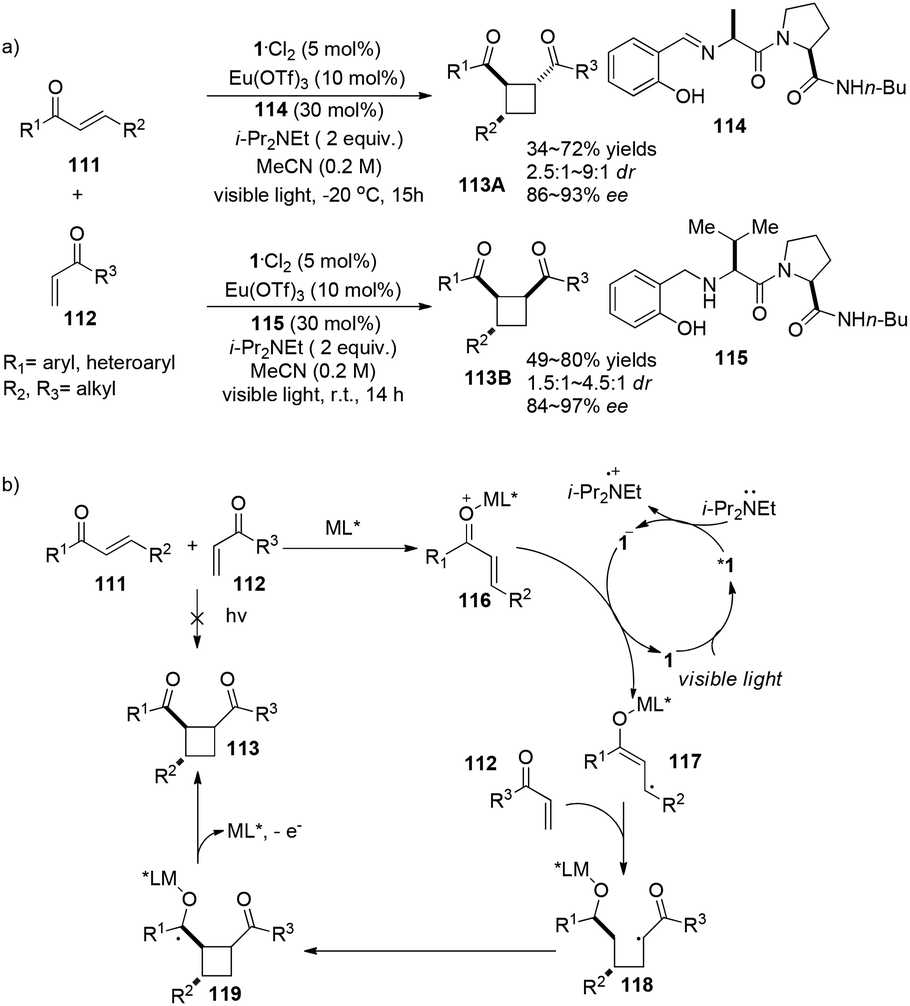
The Yoon group has developed a new strategy to construct chiral cyclobutanes via highly enantioselective [2 + 2] photocycloadditions of α,β-unsaturated ketones involving a visible light photocatalyst and stereo-controlling chiral Lewis acids.34 Employing 5 mol% of 1·Cl2 as the visible light photocatalyst, 10 mol% of Eu(OTf)3 as the Lewis acid, 30 mol% of peptide ligand 93 as the chiral ligand in conjunction with 2 equiv. of i-Pr2NEt, the reaction of aryl enones 111 with alkyl vinyl ketones 112 gave trans-cyclobutanes 113A in 34–72% yields and 86–93% ee (Scheme 22a). Additionally, when ligand 115 is used instead of 114, cis-cyclobutanes 113B are obtained in 49–80% yield with 84–97% ee. Radical 117 derived from single-electron reduction of activated aryl enone by the excited state complex *1 adds to enone 112 to form a chiral Lewis acid mediated radical 118 which subsequently undergoes intermolecular cyclization before elimination of cyclobutane 113 (Scheme 22b). Their research suggests that enantioselectivity can be controlled by a chiral Lewis acid mediated substrate complex.
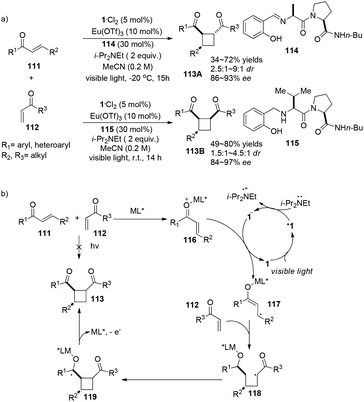 |
| | Scheme 22 a) Europium and ruthenium co-catalyzed asymmetric [2 + 2] photocycloaddition. (b) Proposed mechanism. | |
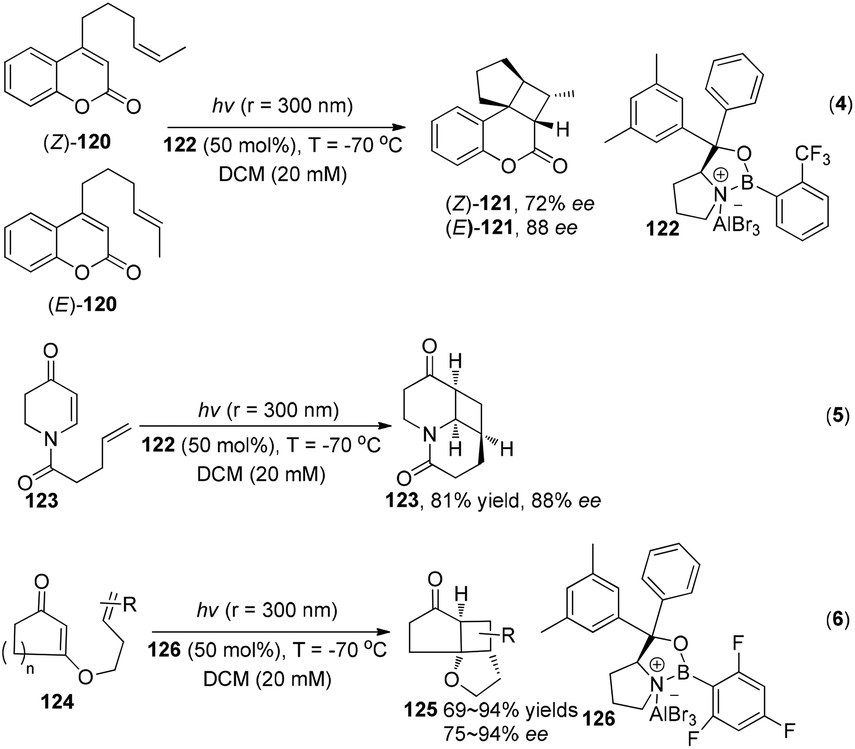
While under the irradiation of ultraviolet light, a series of interesting results using chiral Lewis acids were reported by Bach and his co-workers to successfully construct with high enantioselectivity cyclobutanes through asymmetric intramolecular [2 + 2] photocycloadditions (Scheme 23).35
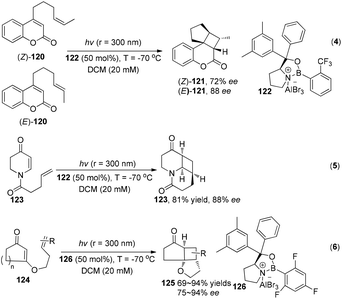 |
| | Scheme 23 Enantioselective Lewis acid catalyzed intramolecular [2 + 2] photocycloaddition under UV irradiation. | |
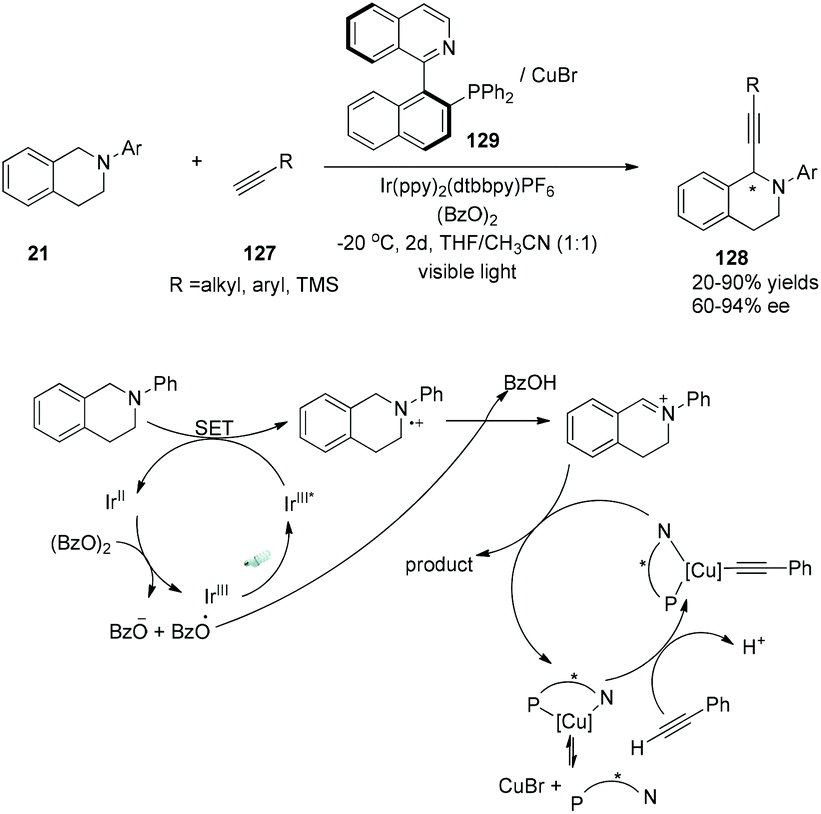
Very recently, a visible-light-induced asymmetric cross-dehydrogenative coupling (CDC) reaction of N-aryl-tetrahydroisoquinolines with alkynes was demonstrated by Li and his co-workers (Scheme 24).36 In the reactions, N-aryl-tetrahydroisoquinolines could be oxidized by the photoexcited sensitizer using benzoyl peroxide as the terminal oxidant, undergoing deprotonation to yield the imine intermediate. A chiral Cu–QUINAP–acetylide species was formed from copper-mediated deprotonation of alkynes nucleophilically added to the imine intermediate, resulting in the formation of chiral α-alkynylation N-aryl-tetrahydroisoquinolines in 20–90% yields and 60–94% ee.
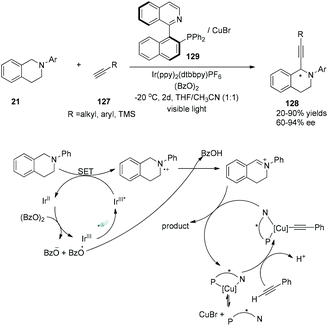 |
| | Scheme 24 Asymmetric cross-dehydrogenative coupling (CDC) reaction of N-aryl-tetrahydroisoquinolines with alkynes. | |
7. Asymmetric cross-coupling reactions
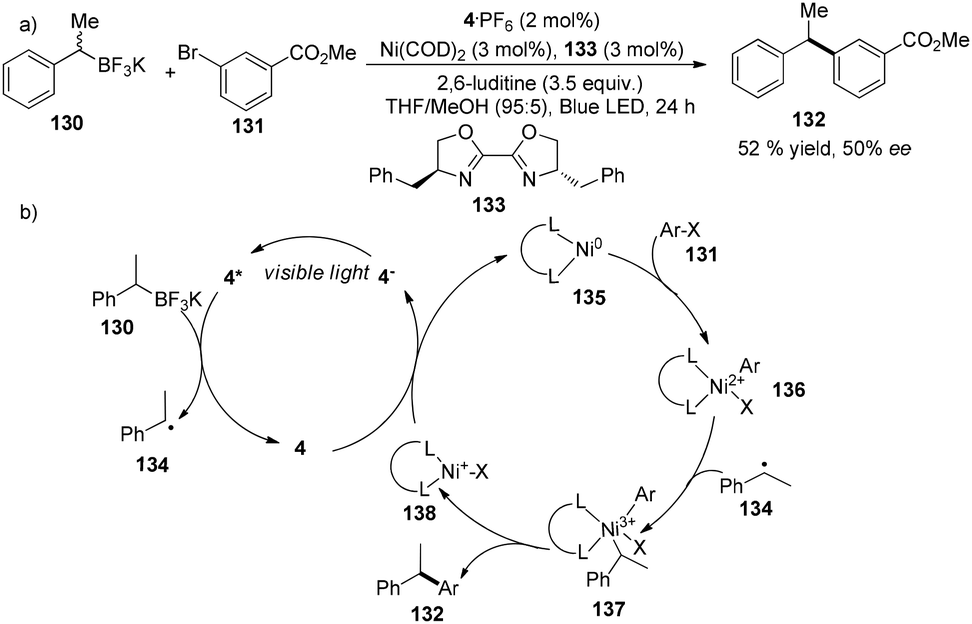
A very recent report from the Molander group revealed that asymmetric Suzuki cross-coupling of potassium methyl benzyltrifluoroborate with methyl 3-bromobenzoate could be facilitated by a combination of 4·PF6 (2 mol%), Ni(COD)2 (3 mol%) and chiral ligand 133 (3 mol%) to yield methyl 3-(1-phenylethyl)benzoate 132 in 52% yield with 50% ee (Scheme 25a).37 Under irradiation by visible light, photoexcited *4 accepted an electron from 130 to become more reductive species 4−. Classic oxidative addition of Ni(COD)2 with aryl halide can deliver a Ni(II) species 136 that can intercept a benzyl radical to afford high valent Ni(III) species 137. Subsequent reductive elimination of high valent Ni(III) species 137 would furnish the cross-coupling product, meanwhile giving Ni(I) species 138 which could undergo single-electron reduction by 4− to regenerate the Ni(0) catalyst (Scheme 25b). Compared to traditional cross-coupling, this photoredox cross-coupling could dramatically promote single-electron transmetallation instead of two-electron transmetallation. Although there is only one example with moderate yield and enantioselectivity, their findings would significantly provide a new access to catalytic asymmetric cross-coupling reactions using sp3-carbon as the partner.
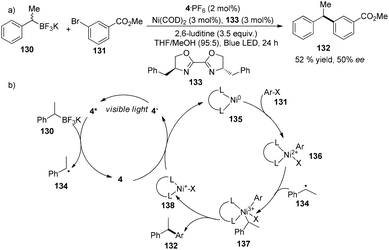 |
| | Scheme 25 (a) Nickel and iridium co-catalyzed asymmetric Suzuki cross-coupling reaction. (b) Proposed mechanism. | |
8. Conclusions
Under irritation with visible light, highly enantioselective transformations could be successfully achieved by combination of visible light photocatalysts and other chiral catalysts, such as thioureas, amines, carbenes, Lewis acids, phosphoric acids. Mild conditions, such as room temperature, no strong base or acid, low energy light source, are utilized in most of reactions which could tolerate variously functionalized groups, decrease environment pollution, save energy and realize sustainable chemistry. Meggers's landmark work on designing a visible-light-induced chiral photocatalyst might make a huge improvement in asymmetric phototransformations. Another interesting initial result for the asymmetric cross-coupling reaction for CSp3–CSp2 bond formation would explore a new strategy to regenerate transition-metal catalysts by photoredox single-electron transmetallation. Although this type of reaction is so far limited, considering the huge progress made in asymmetric catalysis chemistry, more new organocatalysts and varieties of transition-metals with novel chiral ligands will be developed and engaged in the field of asymmetric photoreactions.
Acknowledgements
Financial support from the National Science Foundation of China (21472162), the “Thousand Youth Talents Plan”, the Starting Funds from Zhejiang University.
Notes and references
-
(a) W. Walther and T. Netscher, Chirality, 1996, 8, 397 CrossRef CAS;
(b) K. M. Rentsch, J. Biochem. Biophys. Methods, 2002, 54, 1 CrossRef CAS;
(c) N. M. Davies and X.-W. Teng, Adv. Pharm., 2003, 1, 242 Search PubMed.
- For some selected reviews on applications of chiral molecules, see:
(a) Á. M. Montaña and C. Batalla, Curr. Med. Chem., 2009, 16, 2235 CrossRef;
(b) B. Kasprzyk-Hordern, Chem. Soc. Rev., 2010, 39, 4466 RSC.
- For some selected books on asymmetric catalysis, see:
(a)
E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999, vol. I–III Search PubMed;
(b)
A. Berkessel and H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to Application in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed;
(c)
P. I. Dalko, in Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007, vol. 1–4 Search PubMed. For some selected reviews on asymmetric catalysis, see:
(d) P. M. Pihko, Angew. Chem., Int. Ed., 2004, 43, 2062 CrossRef CAS PubMed;
(e) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS PubMed;
(f) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed;
(g) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed;
(h) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561 CrossRef CAS PubMed; G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2011, 111, PR284 Search PubMed;
(i) C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930 CrossRef CAS PubMed.
- For selected reviews on organic transformations via visible light photocatalysis, see:
(a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed;
(b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed;
(c) J. M. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC;
(d) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef;
(e) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 (
Angew. Chem.
, 2012
, 124
, 6934
) CrossRef CAS PubMed;
(f) L. Shi and W.-J. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC;
(g) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed;
(h) T. P. Yoon, ACS Catal., 2013, 3, 895 CrossRef CAS PubMed;
(i) Y.-M. Xi, H. Yi and A.-W. Lei, Org. Biomol. Chem., 2013, 11, 23878 Search PubMed;
(j) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorious, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed;
(k) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed. For some selected pioneer works on organic transformations via visible light photocatalysis, see:
(l) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed;
(m) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS PubMed;
(n) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed.
-
(a) W. Kuhn and E. Braun, Naturwiss., 1929, 17, 207 Search PubMed;
(b) P. Wessig, Angew. Chem., Int. Ed., 2006, 45, 2168 CrossRef CAS PubMed;
(c) R. Neier, Science, 2014, 344, 368 CrossRef CAS PubMed.
- M. Chavarot, S. Ménage, O. Hamelin, F. Charnay, J. Pécaut and M. Fontecave, Inorg. Chem., 2003, 42, 4810 CrossRef CAS PubMed.
- For selected reviews on chiral-at-metal complexes, see:
(a) J.-L. Pierre, Coord. Chem. Rev., 1998, 178–180, 1183 CrossRef CAS;
(b) U. Knof and A. von Zelewsky, Angew. Chem., Int. Ed., 1999, 38, 302 CrossRef;
(c) H. Brunner, Angew. Chem., Int. Ed., 1999, 38, 1194 CrossRef CAS;
(d) P. D. Knight and P. Scott, Coord. Chem. Rev., 2003, 242, 125 CrossRef CAS;
(e) C. Ganter, Chem. Soc. Rev., 2003, 32, 130 RSC;
(f) J. Lacour and V. Hebbe-Viton, Chem. Soc. Rev., 2003, 32, 373 RSC;
(g) M. Fontecave, O. Hamelin and S. Ménage, Top. Organomet. Chem., 2005, 15, 271 CrossRef CAS;
(h) E. B. Bauer, Chem. Soc. Rev., 2012, 41, 3153 RSC;
(i) E. C. Constable and P. A. Gale, Chem. Soc. Rev., 2013, 42, 1427 RSC;
(j) L. Gong, M. Wenzel and E. Meggers, Acc. Chem. Res., 2013, 46, 2635 CrossRef CAS PubMed.
-
(a) T. Hamada, H. Ishida, S. Usui, Y. Watanabe, K. Tsumura and K. Ohkubo, J. Chem. Soc., Chem. Commun., 1993, 909 RSC;
(b) T. Hamada, H. Ishida, S. Usui, K. Tsumura and K. Ohkubo, J. Mol. Catal., 1994, 88, L1 CrossRef CAS.
- H.-H. Huo, X.-D. Shen, C.-Y. Wang, L.-L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100 CrossRef CAS PubMed.
- G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112 RSC.
- For selected recent reviews, see:
(a) K. Bark and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef PubMed;
(b) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed.
- N. Vallavoju, S. Selvakumar, S. Jockusch, M. P. Sibi and J. Sivaguru, Angew. Chem., Int. Ed., 2014, 53, 5604 CrossRef CAS PubMed.
- R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368 CrossRef CAS PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS PubMed.
-
(a) P. J. Wagner and B.-S. Park, Org. Photochem., 1991, 11, 227 CAS;
(b) E. C. Lathioor and W. J. Leigh, Photochem. Photobiol., 2006, 82, 291 CrossRef CAS PubMed.
- L. J. Rono, H. G. Yalya, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735 CrossRef CAS PubMed.
- W. Ding, Q.-Q. Zhou, J. Xuan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Tetrahedron Lett., 2014, 55, 4648 CrossRef CAS PubMed.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef CAS PubMed.
- M.-M. Lian, Z. Li, Y.-C. Cai, Q.-W. Meng and Z.-X. Gao, Chem. – Asian J., 2012, 7, 2019 CrossRef CAS PubMed.
- Z.-J. Feng, J. Xuan, S.-D. Xia, W. Ding, W. Guo, J.-R. Chen, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Org. Biomol. Chem., 2014, 12, 2037 CAS.
- A. Córdova, H. Sundén, M. Engqvist, I. Ibrahem and J. Casas, J. Am. Chem. Soc., 2004, 126, 8914 CrossRef PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed.
- I. Tabaković, Electrochem. Acta., 1995, 40, 2809 CrossRef.
-
(a) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS PubMed;
(b) H.-W. Shih, M. N. V. Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600 CrossRef CAS PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed.
- K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293 RSC.
- G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521 CrossRef CAS PubMed.
- H.-S. Yoon, X.-H. Ho, J. Jang, H.-J. Lee, S.-J. Kim and H.-Y. Jang, Org. Lett., 2012, 14, 3272 CrossRef CAS PubMed.
- C. Palomo and A. Mielgo, Angew. Chem., Int. Ed., 2006, 45, 7876 CrossRef CAS PubMed.
- H. Sundén, M. Engqvist, J. Casas, I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2004, 43, 6532 CrossRef PubMed.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed.
- E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438 RSC.
- Y.-B. Zhu, L. Zhang and S.-Z. Luo, J. Am. Chem. Soc., 2014, 136, 14642 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef CAS PubMed.
-
(a) H. Guo, E. Herdtweck and T. Bach, Angew. Chem., Int. Ed., 2010, 49, 7782 CrossRef CAS PubMed;
(b) R. Brimioulle and T. Bach, Science, 2013, 342, 840 CrossRef CAS PubMed;
(c) R. Brimioulle and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 12921 CrossRef CAS PubMed.
- I. Perepichka, S. Kundu, Z. Hearne and C.-J. Li, Org. Biomol. Chem., 2015, 13, 447 CAS.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.