
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction and speciation studies of new diglycolamides for selective recovery of americium by solvent extraction†
Filip Kolesar ab,
Cécile Marie*c,
Laurence Berthonc,
Karen Van Heckea,
Ken Vergutsa,
Thomas Cardinaelsb and
Koen Binnemansb
ab,
Cécile Marie*c,
Laurence Berthonc,
Karen Van Heckea,
Ken Vergutsa,
Thomas Cardinaelsb and
Koen Binnemansb
aBelgian Nuclear Research Center (SCK CEN), Institute for Nuclear Energy Technology, Boeretang 200, 2400 Mol, Belgium
bKU Leuven, Department of Chemistry, Celestijnenlaan 200F, P. O. Box 2404, 3001 Leuven, Belgium
cCEA, DES, ISEC, DMRC, Univ Montpellier, Marcoule, France. E-mail: cecile.marie@cea.fr
First published on 31st March 2025
Abstract
Much research has gone into the development of extraction processes capable of separating minor actinides from highly active raffinates generated by the PUREX process. In particular, the separation of americium from curium remains challenging because of the similarity of their chemical properties. A new class of diglycolamide extractants called “unsymmetrical” diglycolamides (UDGAs), which contain at least two different alkyl chains, have recently been shown to have potential for improving the Am/Cm selectivity. However, the influence of the alkyl chains on the extraction efficiency and selectivity, and the formed complexes are not fully understood yet. For this purpose, using the AmSel system as reference, six UDGAs were studied by performing both extraction experiments and speciation studies, and compared to the benchmark extractant TODGA. The tested UDGAs all contain two dodecyl chains on one of the amide nitrogen atoms, and either two n-propyl, isopropyl, n-butyl, isobutyl, or n-pentyl chains, or a piperidine group on the opposing nitrogen atom. The results show that all of the tested UDGAs have equal or higher distribution ratios than TODGA, with the isopropyl derivative showing the most efficient extraction of Ln(III) and An(III). Selectivity for curium over americium was equal or higher in comparison with TODGA, with isopropyl and piperidine giving the highest separation factors, followed by pentyl, and the remaining UDGAs showing similar selectivity to TODGA. Speciation experiments of the complexes formed in the organic phase with neodymium were performed using FTIR, ESI-MS, and UV-vis spectrometry. This revealed very similar spectra for all of the diglycolamides, indicating no difference in the extraction mechanism.
Introduction
A potential solution for the growing global stock of spent nuclear fuel is the implementation of a multi-recycling partitioning and transmutation strategy.1 This would involve the removal of actinides from the spent fuel, decreasing the heat load and radiotoxicity levels of the remaining waste allowing it to be stored more efficiently in deep underground repositories.2 Of the actinides, uranium and plutonium can be separated through the PUREX process and ongoing research is aiming to adapt this process for the treatment of neptunium as well.3 The long half-life of the most common americium isotopes and their high heat-load make americium the most worthwhile candidate for transmutation.4 However, this would require the separation of americium from the fission products present in the spent fuel, the lanthanides in particular, as these can act as neutron poisons and inhibit the transmutation process. Additionally, the presence of curium greatly complicates the target fabrication and any handling of these targets due to its neutron radiation, as this requires extensive shielding.4 The shorter half-lives of Cm, as well as its much lower concentrations in spent fuel as opposed to Am (more than 10 times lower concentration for Cm than for Am5) means that storing the Cm in underground repositories together with the fission products is considered a preferable strategy.4 For this purpose, an extraction system capable of separating americium not only from the fission products, but also from curium, is required.An important step in the development of such extraction systems was the development of diglycolamide extractants, with N,N,N′,N′-tetraoctyldiglycolamide (TODGA, see Fig. 1a) being the most intensively investigated one.6–9 These ligands have good extraction properties in the highly acidic conditions of PUREX raffinates, and show a good affinity towards trivalent lanthanides and actinides. A characteristic phenomenon of these extractants is the formation of a third phase when the solubility of the metal-DGA complex in the organic diluent is exceeded, so addition of a phase modifier such as tributyl phosphate (TBP) or 1-octanol is necessary.10,11 A number of different extraction processes have been developed based on diglycolamide extractants, including i-SANEX12 for minor actinides (Am and Cm) recovery and EURO-EXAM for selective americium partitioning.13,14 A third system based on TODGA, also for the selective separation of americium, is the Americium Selective extraction process (AmSel).15 This process involves an initial co-extraction of Am, Cm, and Ln fission products with TODGA, followed by selective stripping with a hydrophilic BTBP or BTPhen complexant, typically 3,3′,3′′,3′′′-([2,2′-bipyridine]-6,6′-diylbis(1,2,4-triazine-3,5,6-triyl))tetrabenzenesulfonate (SO3-Ph-BTBP, see Fig. 1b). The separation is achieved through the combination of a hard O-donor extractant (TODGA) and a soft N-donor complexant (SO3-Ph-BTBP), in a so-called “push–pull system”.16 Due to the lanthanide/actinide contraction, trivalent ions become harder with increasing mass number, leading to preferential coordination with TODGA, whereas the softer ions with lower mass number prefer coordination with SO3-Ph-BTBP. The separation between lanthanides and actinides is further increased as a result of a stronger covalent character of the bond between the N-donor atoms and the actinides' 5f orbitals as opposed to the lanthanides' 4f orbitals.17 The combination of two ligands with inverse selectivity, i.e., the lipophilic TODGA with a preference for Cm over Am (SFCm/Am = 1.6) and the hydrophilic SO3-Ph-BTBP with a selectivity for Am over Cm (SFCm/Am = 1.6) yields a combined separation factor of 2.5 for the AmSel process.15 This system benefits from a highly water-soluble complexant that performs well in acidic conditions. However, due to the presence of sulfur, the system is not compliant with the CHON principle. This principle requires that only carbon, hydrogen, oxygen, and nitrogen atoms are present in the extraction system, allowing entire decomposition and limiting the generation of residual solid waste.18 Furthermore, the AmSel system is hindered by a low Am/Cm separation factor that makes upscaling to a continuous process difficult.
 | ||
| Fig. 1 Structures of the extractants of the AmSel process: (a) TODGA and (b) SO3-Ph-BTBP. | ||
So far, within the context of Am–Cm separation, diglycolamides have been considered primarily as extracting agents meant to co-extract Am, Cm, and the Ln, and research trying to improve the separation factor mainly focused on Am-selective nitrogen-donor compounds. The possibility of improving the selectivity of Am-extracting processes through modification of the diglycolamide extractant has therefore been inadequately explored. Nevertheless, some work has already been performed investigating the influence of DGA structure on its extraction properties, either to improve lanthanide–actinide or intralanthanide selectivity, or to affect other properties such as third-phase formation. Shortening the alkyl chains below five carbon atoms leads to diglycolamide extractants that are water-soluble.19 One such extractant, N,N,N′,N′-tetraethyldiglycolamide (TEDGA), is used as a hydrophilic masking agent in the EXAm process, where it enhances the selectivity for americium when combined with the organic extractants DMDOHEMA and HDEHP.20 The addition of TEDGA to the system increases the SFCm/Am from 1.6 to 2.5, which is on par with the AmSel process. Experiments investigating the effect of the alkyl chains' length on DGA performance were able to determine a trend whereby longer alkyl chains correlate with lower extraction of metal ions.21–23 The most common explanations given for this observation are the increased lipophilicity of the molecules hindering its approach of the organic-aqueous interphase where the complex must be formed, and the increase in steric hindrance making complexation more difficult.21,23,24 When extractions are performed with 2,2′-oxybis(N,N-di-n-decylpropanamide) (mTDDGA) which is methylated on the two carbons of the central skeleton, large differences in extraction behavior are observed for different diastereomers, further highlighting the importance of steric hindrance.25
Based on the theory that steric hindrance hinders complexation, it is expected that increased branching will lead to lower extraction strength, especially when the branched chains are close to the central diglycolamide skeleton. This is indeed observed for N,N,N′,N′-tetra-2-ethylhexyldiglycolamide (TEHDGA) which, despite having the same number of carbon atoms in its chains as TODGA, shows much weaker extraction.22,26,27 Similar observations were also made for diglycolamides containing butyl chains in various conformations, where branched butyl chains resulted in lower extraction strength.19,23 It was further shown that branching on the α or β position decreases the extraction efficiency, whereas branching at further positions showed little influence on the extraction strength but did still affect intra-lanthanide separation.28
A more recent development was the synthesis of “unsymmetrical” diglycolamides (UDGAs), of which the state of the art is discussed in more detail in our previous work.29 These are molecules that keep the same central diglycolamide skeleton, but have at least two different types of alkyl chains grafted onto the amide functions. This allows fine-tuning of the steric hindrance around the complexing center. Two types of such extractants can be identified: type 1 unsymmetrical diglycolamides contain on each side two identical alkyl chains while the two sides differ from each other (Fig. 2 top), and type 2 unsymmetrical diglycolamides contain on each side two different alkyl chains while the two sides are identical to each other (Fig. 2 bottom).30 Both of these types contain only one mirror plane, as opposed to the symmetrical diglycolamides that contain two. Furthermore, diglycolamides with three or four different types of alkyl chains contain no mirror planes and are fully unsymmetrical. Of the type 2 unsymmetrical diglycolamides N,N′-dimethyl-N,N′-dioctyldiglycolamide (DMDODGA) is the most intensively studied, mainly for the extraction of lanthanides or uranyl.31–33 One study also investigated the extraction of the actinides Np(V), U(VI), Am(III), and Pu(IV) by this diglycolamide.34 These studies showed that due to the decrease in steric hindrance, DMDODGA is a far more efficient extractant than TODGA, but retained good solubility in the organic phase because of the two long octyl chains. Its selectivity for Cm over Am has not yet been determined, but one study showed it was possible to separate the lanthanides Nd(III) and Dy(III) from the fission products Fe(III) and Ni(II) with DMDODGA.35 The higher extraction efficiency observed for Dy(III) over Nd(III) indicates some intra-lanthanide selectivity is retained after shortening the alkyl chains.
 | ||
| Fig. 2 General structures of type 1 and type 2 unsymmetrical diglycolamides. | ||
The type 1 unsymmetrical diglycolamides have been well studied by, amongst others, Ravi et al.36–38 who mainly focused on separating actinides and lanthanides from other fission products. By tuning the lengths of the various alkyl chains, they were able to obtain extractants with good selectivity, that did not form a third phase at elevated metal and nitric acid concentrations.39 By fixing on one side of the diglycolamide two dodecyl chains, modifier-free extraction was found to be possible when the alkyl groups on the other side were varied from butyl to decyl, with the best solubility found for the decyl variant.37 High extraction efficiency for the lanthanide Eu(III) was also observed for diglycolamides that combined straight alkyl chains (hexyl and octyl) with cyclic alkyl moieties grafted onto the amidic nitrogen (pyrrolidinyl, piperidinyl, morpholinyl).40 However, these extractants have not yet been tested on systems containing both americium and curium. Another investigated approach is elongating the alkyl chains on one side and shortening them on the other side of the diglycolamide extractant.30,38 This resulted in extractants that, while still soluble in the organic phase, showed significantly stronger extraction than what is observed for symmetrical diglycolamides with the same total number of carbons in the alkyl chains. Surprisingly, an improvement in intra-lanthanide separation was also observed.30
Follow-up experiments during the European GENIORS project tested three extractants of this type for americium–curium separation in combination with the hydrophilic complexant N,N,N′N′-tetrakis[(6-carboxypyridin-2-yl)methyl]ethylenediamine (TPAEN), the complexant used in the EURO-EXAM process.13 N,N-diisopropyl-N′,N′-didodecyldiglycolamide (iPDdDGA, see Fig. 3d) proved the most promising one of these extractants, showing a SFCm/Am value of 6.8 in combination with TPAEN, a nearly 50% improvement over the 4.3 value observed with TODGA in combination with the same complexant.41 In our recent study, iPDdDGA was further investigated in combination with the SO3-Ph-BTBP complexant of the AmSel system, and showed a general improvement of SFCm/Am from 2.5 up to 3.0.29 However, the strong increase in extraction efficiency, with distribution ratios 10–100 times higher than those observed for TODGA, could hinder the stripping process, especially during the recovery of Cm and Ln during solvent regeneration. Furthermore, no explanation for the increase in selectivity could be provided based on extraction experiments alone.
 | ||
| Fig. 3 Structures of the unsymmetrical diglycolamides investigated in this work: (a) PDdDGA, (b) BDdDGA, (c) PnDdDGA, (d) iPDdDGA, (e) iBDdDGA, and (f) pipDdDGA. | ||
Therefore, in order to better understand the influence of the diglycolamide structure on the extraction strength and selectivity, five new unsymmetrical diglycolamide structures of the first type were investigated in the present work. The structures all contain on one side two dodecyl alkyl chains and on the other side propyl, butyl, isobutyl, or pentyl chains, or a piperidinyl group (see Fig. 3). Additionally, both iPDdDGA and TODGA were included in the study as reference molecules. During extraction experiments, both the extraction step and stripping step in combination with SO3-Ph-BTBP were investigated, which was followed by speciation experiments using ESI-MS, FTIR, and UV-vis spectroscopy.
Experimental
Chemicals
Of the diglycolamide extractants, TODGA (>99%) was acquired from Inabata Pharmasynthese S.A.S. (Saint-Pierre-Les-Elbeuf, France), iPDdDGA (>99%) was acquired from Diverchim CDMO (Roissy-en-France, France), and N,N-piperdinyl-N′,N′-didodecyldiglycolamide (pipDdDGA, see Fig. 3f) (>95%) was acquired from AtlanChim pharma (Saint-Herblain, France). The remaining extractants were synthesized according to literature.30 A detailed synthesis procedure, as well as characterization data, can be found in the ESI (see S1).† 2-Methyltetrahydrofuran (>99.0%), dipropylamine (synthesis grade), dibutylamine (synthesis grade), diisobutylamine (99%), and 1-octanol (ACS grade) were acquired from Sigma-Aldrich (Steinheim, Germany). Didodecylamine (>97.0%) and dipentylamine (>98.0%) were acquired from TCI Europe N.V. (Paris, France). The coupling agent (1-cyano-2-ethoxy-2-oxoethylideneaminooxy)di-methylamino-morpholino-carbenium hexafluorophosphate (COMU) (98%) was acquired from Apollo Scientific (Manchester, UK). Diglycolic anhydride (>97.0%) and n-dodecane were acquired from Thermo Scientific (Waltham, MA, USA). HNO3 solutions were prepared from 68% Normapur grade HNO3 acquired from VWR (Radnor, PA, USA). SO3-Ph-BTBP (>98%) was acquired in free acid form from Technocomm Ltd (Edinburgh, UK).Methods
| Element | Concentration (g L−1) |
|---|---|
| La | 0.289 |
| Ce | 0.600 |
| Pr | 0.262 |
| Nd | 1.02 |
| Sm | 0.184 |
| Eu | 0.039 |
| Gd | 0.031 |
| Y | 0.098 |
Equal volumes of organic and aqueous phases were combined in Eppendorf safe-lock 2 mL microcentrifuge tubes, which were shaken at a fixed temperature of 20 °C using an IKA Vibrax with custom cooling cell. The samples were shaken for 15 minutes during extraction experiments, and 30 minutes during stripping experiments, as these times were shown to be more than sufficient to achieve equilibrium. Afterwards, the samples were centrifuged and the two phases separated with Eppendorf micropipettes. The organic phases were not pre-equilibrated with acid, but both aqueous and organic phases were titrated to determine the equilibrium acid concentration, with the results reported in the ESI (see S2).† Aliquots of both the aqueous and organic phases were analyzed with gamma spectrometry (to measure 152Eu and 241Am), alpha spectrometry (to measure 241Am and 244Cm), and ICP-OES (to measure the inactive lanthanides). The samples were diluted according to necessity (see below). For speciation experiments, extractions were performed with a simplified starting solution containing 10 mmol L−1 of Nd(III), without any radiotracer, in 3 mol L−1 HNO3. An identical organic phase was used as during extraction experiments.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10000 with acetonitrile. These were then injected into the electrospray source at a rate of 180 μL h−1 with a Cole Palmer syringe infusion pump. The samples were measured in positive ionization mode with a Bruker Daltonics microOTOF-Q II quadrupole TOF mass spectrometer. The capillary voltage of the spectrometer was set at −4500 V and the end-plate offset voltage was set at −500 V. The samples were dried and nebulized with nitrogen gas, the gas pressure was set at 0.3 bar, the temperature at 200 °C, and the flow rate at 4.0 L min−1. Spectra were recorded over an m/z range from 40 to 3000. Collision induced dissociation experiments were performed using nitrogen as collision gas. Typical collision energies varied between 15 and 50 eV and were optimized for individual ions. The spectra were analyzed with DataAnalysis 4.0 software.
:10000 with acetonitrile. These were then injected into the electrospray source at a rate of 180 μL h−1 with a Cole Palmer syringe infusion pump. The samples were measured in positive ionization mode with a Bruker Daltonics microOTOF-Q II quadrupole TOF mass spectrometer. The capillary voltage of the spectrometer was set at −4500 V and the end-plate offset voltage was set at −500 V. The samples were dried and nebulized with nitrogen gas, the gas pressure was set at 0.3 bar, the temperature at 200 °C, and the flow rate at 4.0 L min−1. Spectra were recorded over an m/z range from 40 to 3000. Collision induced dissociation experiments were performed using nitrogen as collision gas. Typical collision energies varied between 15 and 50 eV and were optimized for individual ions. The spectra were analyzed with DataAnalysis 4.0 software.Results and discussion
Extraction
In the first step of the AmSel process, a co-extraction of Am, Cm, and lanthanides from a highly acidic HAR solution with TODGA takes place. In the following extraction experiment it was examined whether UDGAs would be a suitable replacement for TODGA in this first step. For this, extractions were performed with 0.1 mol L−1 solutions prepared with the various diglycolamides in n-dodecane with 5 vol% 1-octanol, and a feed solution containing lanthanides (see Table 1) and 241Am, 244Cm, and 152Eu radiotracer in 3 mol L−1 HNO3. Distribution ratios for Am, Cm, and Eu are shown in Fig. 4, and data for lanthanides is shown in Fig. 5. | ||
| Fig. 4 Distribution ratios for extraction of Eu, Am, and Cm with diglycolamides. Aqueous phase: 18 mmol L−1 Ln(III) + 50 kBq mL−1 of 152Eu, 241Am, and 244Cm each dissolved in HNO3 solution with initial concentration 3 mol L−1. Organic phase: 0.1 mol L−1 diglycolamide extractant dissolved in n-dodecane + 5 vol% 1-octanol. Shaking time: 15 min. Temperature: 20 °C. | ||
 | ||
| Fig. 5 Distribution ratios for extraction of lanthanides with diglycolamides. Aqueous phase: 18 mmol L−1 Ln(III) + 50 kBq mL−1 of 152Eu, 241Am, and 244Cm each dissolved in HNO3 solution with initial concentration 3 mol L−1. Organic phase: 0.1 mol L−1 diglycolamide extractant dissolved in n-dodecane + 5 vol% 1-octanol. Shaking time: 15 min. Temperature: 20 °C. | ||
Very efficient extraction of Am, Cm, and Eu was observed for all of the tested diglycolamides. Of the new diglycolamides, nearly all showed much stronger extraction behavior compared to TODGA. Most showed distribution ratios in excess of 1000. Above this value, it becomes difficult to accurately measure distribution ratios as the remaining activity in the aqueous phase at equilibrium approaches the limit of detection. Only PnDdDGA showed distribution ratios comparable to TODGA. While it can be concluded that all diglycolamides show sufficient extraction to replace TODGA in the first step of the AmSel process, their relative extraction strength and Am/Cm selectivity could not be determined from the tracer experiment due to the high extraction efficiency.
Of the lanthanides, only La, Ce, and Pr showed distribution ratios that were sufficiently low to be measured. As Eu ratios were too high to measure with either gamma spectrometry, or ICP-OES, a comparison between the two methods could not be made. In the case of pipDdDGA, only distribution ratios for La and Ce could be determined, while for iPDdDGA the lanthanides are extracted to such an extent that their remaining concentrations in the aqueous phase at equilibrium fall below the limit of detection (1 mg L−1). Nevertheless, data measured for La and Ce indicate a general trend whereby shorter alkyl chains correspond with higher distribution ratios. The strongest extraction was observed for pipDdDGA (29 carbon atoms in alkyl chains) followed by PDdDGA (30 carbons in alkyl chains). After these, the highest distribution ratios were measured for BDdDGA and iBDdDGA (containing 32 carbons in the alkyl chains) which showed similar values to each other. This was followed by PnDdDGA (containing 34 carbons in the alkyl chains). This trend of higher distribution ratios for shorter alkyl chains can generally be explained by the decrease of steric hindrance, which makes complexation more favorable.23,43 Interestingly, slightly higher distribution ratios were found for La, Ce, and Pr extraction with PnDdDGA than for TODGA, despite the latter having fewer carbon atoms (32 instead of 34) in its alkyl chains. This might be related to the asymmetry of the diglycolamide extractants. While very long chains give a similar degree of steric hindrance (some studies showing only small differences between octyl and decyl chains37), differences between short alkyl chains have a much more pronounced effect. Such effects were already observed for the type 2 UDGAs, and have been attributed to this lowering of overall steric hindrance.23,43,44 This could also explain the very strong extraction observed for the UDGAs in this study, as higher distribution ratios were obtained than would be expected based on solely the number of carbon atoms in the alkyl chains. Whereas N,N,N′,N′-tetrahexyldiglycolamide (with only 24 carbon atoms) gives distribution ratios similar to TODGA, the UDGAs containing 32 carbon atoms give exponentially higher distribution ratios.45 This highlights one of the benefits of unsymmetrical diglycolamides, the steric hindrance of the extractants can be significantly decreased, resulting in a stronger extractant, while retaining a good solubility of the extractants in the organic phase.
The effect of branching is not clear from the data. In the case of BDdDGA and iBDdDGA, branching does not affect the distribution ratios to a significant extent. However, in the case of PDdDGA and iPDdDGA, the distribution ratios being above the limit of quantification for iPDdDGA suggests significantly stronger extraction than for PDdDGA. This might be a result of the position of the branching: in the case of iBDdDGA the branching is on the β-carbon whereas for iPDdDGA the branching is on the α-carbon. The proximity of the branching to the diglycolamide core, and the length of the branching alkyl chains have previously been found to be of importance.28,46 In the case of TEHDGA, containing ethyl branches on the β carbon, much lower distribution ratios are observed than for the unbranched TODGA. Such a decrease is not observed for methyl branches on this position, as shown in this work as well as in literature.43 For the type 2 UDGAs it was also found that a 2-ethylhexyl chain affects the extraction strength much more significantly than an isobutyl chain.43 For type 2 UDGAs, similarly to iPDdDGA and PDdDGA in this study, higher distribution ratios were also found for variants with branched propyl/butyl groups than for variants bearing linear propyl/butyl chains.24,46 This appears to be in contrast to the systematic study of Stamberga et al. where generally higher distribution ratios were found for extractants with the branching point further away from the complexing core, and the highest distribution ratios were found for linear extractants.28 A possible explanation for these observations is that only bulky groups were tested, with the smallest branched group being a 2-ethylhexyl. Extractants with alkyl chains of five or fewer carbons were not tested.
Stripping
The aim of this study was to find an extractant that shows better selectivity for Am over Cm than TODGA, but does not show the same excessive extraction strength that iPDdDGA shows, as this can complicate the stripping step. To further assess the selectivity of the new DGA extractants, stripping was performed by contacting the organic phases, loaded with elements of interest in the previous extraction experiment, with fresh aqueous phases containing 10 mmol L−1 of SO3-Ph-BTBP dissolved in 0.3 mol L−1 of HNO3. These conditions were chosen based on experiences with iPDdDGA.29 The distribution ratios for the Am stripping step are shown in Fig. 6A for 241Am, 244Cm, and 152Eu radiotracer, and Fig. 7 for the lanthanides. | ||
| Fig. 6 Distribution ratios for stripping of Eu, Am, and Cm with SO3-Ph-BTBP. Organic phase: 0.1 mol L−1 diglycolamide extractant dissolved in n-dodecane + 5 vol% 1-octanol, loaded from a solution containing 18 mmol L−1 Ln(III) (A) or 10−5 mol L−1 Eu(III) (B) + 50 kBq mL−1 of 152Eu, 241Am, and 244Cm each dissolved in 3 mol L−1 HNO3 solution. Aqueous phase: 10 mmol L−1 SO3-Ph-BTBP dissolved in 0.3 mol L−1 HNO3 solution. Shaking time: 30 min. Temperature: 20 °C. | ||
 | ||
| Fig. 7 Distribution ratios for stripping of lanthanides with SO3-Ph-BTBP. Organic phase: 0.1 mol L−1 diglycolamide extractant dissolved in n-dodecane + 5 vol% 1-octanol, loaded from a solution containing 18 mmol L−1 Ln(III) + 50 kBq mL−1 of 152Eu, 241Am, and 244Cm each dissolved in 3 mol L−1 HNO3 solution. Aqueous phase: 10 mmol L−1 SO3-Ph-BTBP dissolved in 0.3 mol L−1 HNO3 solution. Shaking time: 30 min. Temperature: 20 °C. | ||
During the stripping step, it was observed that for iPDdDGA and pipDdDGA, an emulsion-like third phase had formed and a full separation of the phases was not possible even after centrifugation. No third phase was observed for the other diglycolamide extractants. Typically, a third phase is observed when the amount of formed complex exceeds the solubility in the diluent, causing the phases to split.47 It is therefore usually found at elevated acid and/or metal concentrations, making the extraction stage more susceptible to the phenomenon than the stripping stage. As the acid and metal ion concentration are lower in the stripping step than in the extraction step, it is very unusual to observe third-phase formation in this step. Further investigation revealed that the addition of SO3-Ph-BTBP had no influence on the formation of this third phase, and that the third phase could also be observed during the extraction step if the HNO3 concentration of the feed solution was 1 mol L−1 or lower. The formation of a third phase could also be prevented by lowering the Ln concentration to 1 mmol L−1, or by increasing the 1-octanol concentration to 10 vol%. Analysis of the phases by FTIR and ESI-MS showed that a majority of complex was present in the emulsion, confirming that it is indeed a third phase. A possible explanation for this behavior could be the formation of different species at lower acid concentrations, with a lower solubility in the organic phase causing the phases to split. However, this could not be confirmed by speciation techniques including ESI-MS and FTIR. Alternatively, the increase in acidity might help solubilize the formed complexes, so that a difference in supramolecular organization of the organic phases at different acid concentrations is the explanation for the occurrence of a third phase at lower nitric acid concentrations. Further investigation into the supramolecular structure (e.g. by using small angle neutron scattering) could provide more insight into this occurrence. For iPDdDGA and pipDdDGA, the extractions were repeated with a feed solution containing a lower Ln concentration of 10−5 mol L−1 Eu as well as 241Am, 244Cm, and 152Eu radiotracer in order to verify whether a third phase would appear during stripping if a lower metal concentration is used. The organic phases were then contacted again with a stripping solution containing 10 mmol L−1 of SO3-Ph-BTBP dissolved in 0.3 mol L−1 of HNO3. No third-phase formation was observed under these conditions, the distribution ratios for the tracers are presented in Fig. 6B.
A comparison of TODGA and the UDGAs shows that despite the high distribution ratios observed during the extraction step, efficient stripping of Am can still be obtained with 10 mmol L−1 of SO3-Ph-BTBP. As both DAm and DCm values were found to be below 1, a lower concentration of BTBP and/or higher HNO3 concentration would be required to obtain selective stripping of americium. A comparison of the distribution ratios reveals the same general trend as was previously found during the extraction step, i.e., higher distribution ratios were found for DGAs with shorter alkyl chains. TODGA again showed the lowest distribution ratios of all tested DGAs, despite having fewer carbon atoms in its alkyl chains than PnDdDGA. Distribution ratios measured for iPDdDGA and pipDdDGA were more than an order of magnitude higher than those observed for the other DGAs. However, these distribution ratios cannot be directly compared with the other tested extractants because of the lower lanthanide concentration used for these two extractants. A lower concentration of extractable metal ions results in a higher free ligand concentration, which itself increases the distribution ratio.
Good separation factors between Am and the lanthanides were observed for all of the diglycolamides. SFEu/Am increases with the distribution ratios, with TODGA showing a value of around 300, PnDdDGA, iBDdDGA, BDdDGA, and PDdDGA showing a value of around 400. For iPDdDGA and pipDdDGA, with different extraction conditions, SFEu/Am around 1.0 × 103 were observed. No significant differences were observed in the separation factors between lanthanum and americium, with all measured values ranging between 12 and 14. This separation was previously found to be mainly influenced by the SO3-Ph-BTBP concentration, and can be further optimized.29 All of the new diglycolamides showed equal or better selectivity for americium over curium than what is observed for TODGA.15 TODGA, iBDdDGA, BDdDGA, and PDdDGA showed similar separation factors of 2.5. PnDdDGA showed a separation factor of 2.8, and iPDdDGA and pipDdDGA both showed a separation factor of 3.0. Again, some caution should be used when comparing separation factors at different extraction conditions. However, data from a previous study on iPDdDGA extraction showed similar SFCm/Am values despite using higher Ln concentrations.29 The current data suggests that PnDdDGA is the most interesting extractant for further testing, as it shows higher selectivity for curium over americium than TODGA, but does not share the same excessive extraction strength as iPDdDGA. Moreover, PnDdDGA did not form a third phase at higher metal concentrations when 5 vol% of 1-octanol was used as a phase modifier.
Speciation
During extraction experiments, a large difference in extraction strength was observed between the different diglycolamides. In order to better understand which complexes are formed between the lanthanides and the various UDGA extractants, the organic phases were characterized by several spectroscopic techniques. To do this, new extractions were performed in which a feed solution with 10 mmol L−1 of Nd(NO3)3 was dissolved in 3 mol L−1 HNO3. The same organic phases as in the extraction experiments was used, i.e., 0.1 mol L−1 DGA in n-dodecane + 5 vol% 1-octanol. The organic phases were then studied by Fourier-transform infrared spectroscopy (FTIR), ultraviolet-visible spectroscopy (UV-vis), and electrospray ionization mass spectrometry (ESI-MS).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O–C stretches of all the tested diglycolamides are shown in Table 2.
O and C–O–C stretches of all the tested diglycolamides are shown in Table 2.
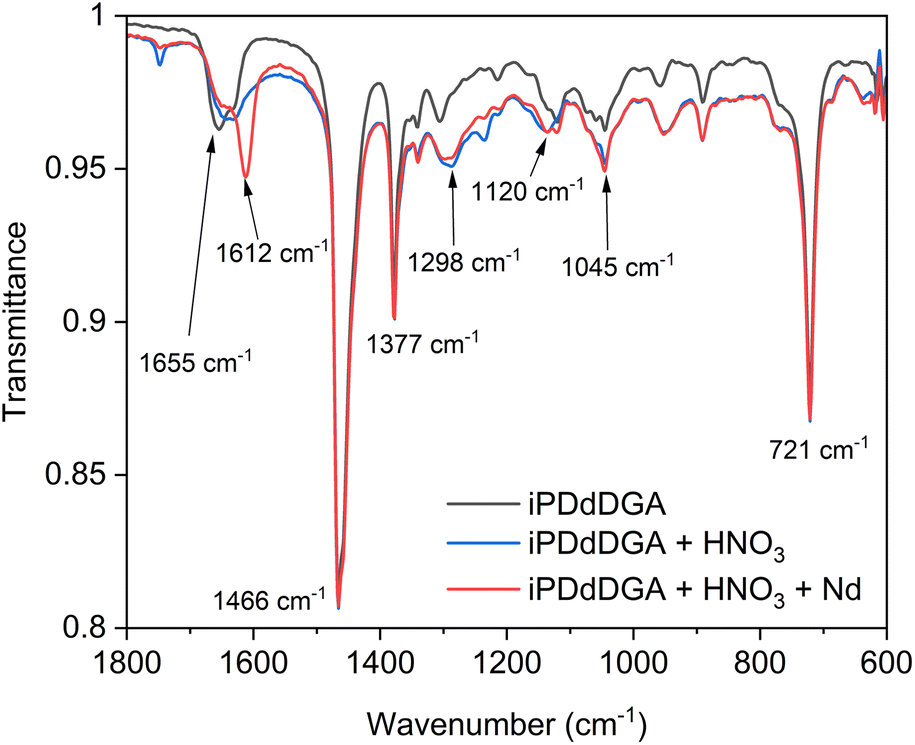 | ||
| Fig. 8 FTIR spectra obtained for the iPDdDGA organic phase (0.1 mol L−1 DGA in n-dodecane + 5 vol% 1-octanol) before extraction, after contact with 3 mol L−1 HNO3, and after contact with 10 mmol L−1 Nd(NO3)3 dissolved in 3 mol L−1 HNO3. [H+]org = 0.36 mol L−1, [Nd3+]org = 10 mmol L−1. | ||
O and C–O–C bands before and after extraction of Nd with various diglycolamides
| CO band free ligand (cm−1) |
CO band Nd complex (cm−1) |
C–O–C band free ligand (cm−1) | C–O–C band Nd complex (cm−1) | |
|---|---|---|---|---|
| TODGA | 1657 | 1614 | 1122 | 1124 |
| iPDdDGA | 1655 | 1612 | 1120 | 1120/1136 |
| PDdDGA | 1659 | 1614 | 1122 | 1124 |
| iBDdDGA | 1659 | 1612 | 1120 | 1120 |
| BDdDGA | 1655 | 1614 | 1124 | 1124 |
| PnDdDGA | 1657 | 1614 | 1124 | 1124 |
| pipDdDGA | 1657 | 1616 | 1122 | 1130 |
The most notable difference between free and complexed ligand is found for the CO stretching band that is present at 1655 cm−1 for the free ligand.8,48,49 When the solvent is contacted with nitric acid, a band displacement towards lower wavenumbers can be observed. This weakening of the CO bond is a result of protonation of the carbonyl oxygen at elevated nitric acid concentration.50 When the ligand is contacted with both acid and Nd(III), a new large band can be observed at 1612 cm−1, with a smaller free CO band at 1654 cm−1. This represents a red-shift of 42 cm−1, indicating a similar coordination with the Ln(III) ion as has already been observed for TODGA.48,49,51 A similar shift was observed for all of the tested diglycolamides, ranging from 41 cm−1 and 47 cm−1, which suggests that there are no significant differences in the bond strength between the neodymium ion and the extractant in the formed complexes. There also does not appear to be a correlation between the observed distribution ratios and the shift, with the largest shift found for iBDdDGA and the smallest shift found for BDdDGA. It can also be noted that no splitting of the carbonyl band can be observed despite the different symmetry shown by iPDdDGA. Such a split has previously been observed for the unsymmetrical DMDODGA extractant and was explained by the difference in inductive effects from the different chemical environment near the two carbonyl groups.32 This could also explain why such a split was only seen for type 1 UDGAs and not for type 2 UDGAs, even when the same substituents are used.
Apart from the carbonyl stretching band, a number of other bands can be recognized. 1466 cm−1 and 1377 cm−1 represent C–H and C–N stretching bands. Around 1300 cm−1 a broad band can be observed after extraction of acid that corresponds with N–O vibrations of nitrates from HNO3 and Nd(NO3)3. Similarly, a peak observed at 1045 cm−1 also represents the N–O vibrations of HNO3.49,52 The band observed at 1120 cm−1 corresponds with the ether C–O–C vibration, and a second peak can be observed to grow in at 1136 cm−1 when both HNO3 and Nd(III) is extracted with iPDdDGA. When only HNO3 is extracted, a single band is observed at 1136 cm−1, indicating this shift might represent interaction between HNO3 and the ether oxygen. This shift is also observed for pipDdDGA, but not for TODGA or the other tested diglycolamides. It is worth noting that iPDdDGA and pipDdDGA were the only diglycolamides for which a third phase was observed during the stripping step and showed the strongest extraction of all tested diglycolamides.
:3 M:L stoichiometry.53 Our results for TODGA-Nd complexes corroborate these results and indicate the same ML3 complex structure. The spectra for the different UDGA-Nd complexes show a great degree of similarity and no significant differences can be observed for either the hypersensitive band measured around 582 nm, nor the non-hypersensitive transition band measured at 736 nm (4I9/2 → 4F7/2 + 2S3/2), which was previously also found to be influenced by complexation with metal ions.54 This further indicates that the unsymmetrical diglycolamides form the same ML3 complexes as the symmetrical TODGA.
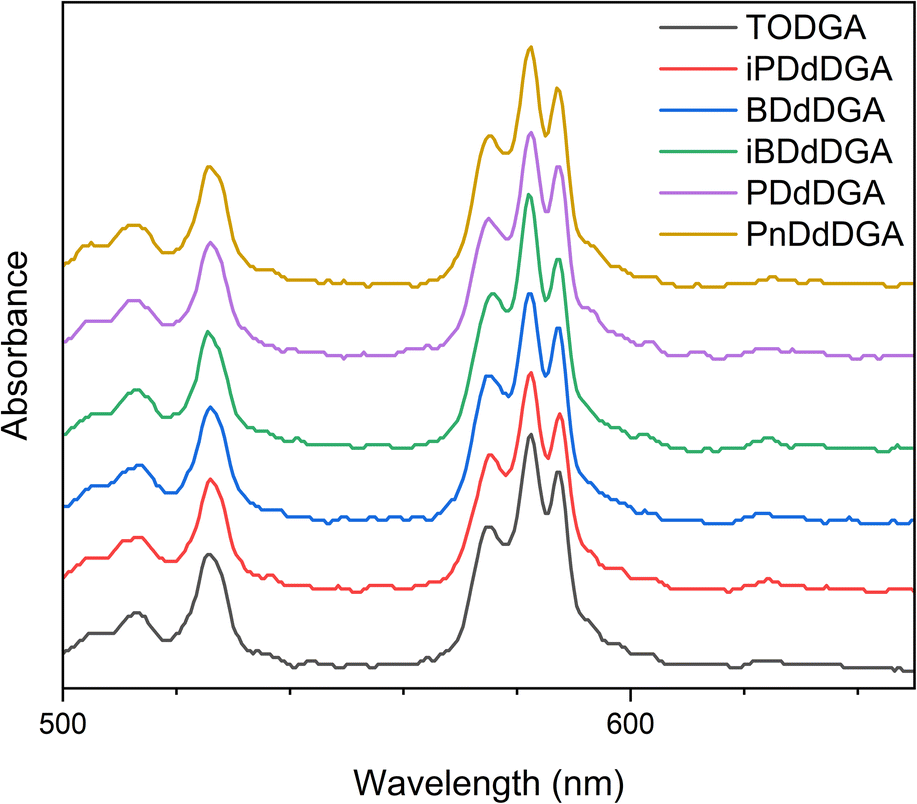 | ||
| Fig. 9 UV-vis spectra of the organic phases containing 0.1 mol L−1 of diglycolamide extractant in n-dodecane + 5 vol% 1-octanol after contact with 10 mmol L−1 Nd(NO3)3 in 3 mol L−1 HNO3. | ||
 | ||
| Fig. 10 ESI-MS spectrum of the organic phase containing 0.1 mol L−1 of iPDdDGA in n-dodecane + 5 vol% 1-octanol after contact with 10 mmol L−1 Nd(NO3)3 in 3 mol L−1 HNO3. Organic phases diluted 1:10000 in acetonitrile. | ||
| Assignment | TODGA | iPDdDGA | BDdDGA | iBDdDGA | PDdDGA | PnDdDGA | pipDdDGA |
|---|---|---|---|---|---|---|---|
| LH+ | 582.5 | 553.7 | 581.5 | 581.5 | 553.5 | 609.6 | 537.5 |
| L2Ca2+ | 600.5 | 572.5 | 600.5 | 600.5 | 573.0 | — | 556.5 |
| LNa+ | 603.5 | 575.5 | 603.5 | 603.5 | 575.5 | 631.6 | 559.5 |
| L3Nd3+ | 628.8 | 600.1 | 628.5 | 628.5 | 600.5 | 656.5 | 584.4 |
| L2Nd(NO3)2+ | 683.5 | — | 681.4 | 683.4 | 655.4 | — | 639.4 |
| L4Nd3+ | 822.3 | — | — | — | — | 860.4 | — |
| L3Ca2+ | 891.2 | 849.2 | 891.3 | 891.2 | 849.2 | 933.3 | 825.2 |
| L3Nd(NO3)2+ | 974.2 | 931.2 | 973.7 | 973.7 | 931.7 | 1015.8 | 907.7 |
| L2Na+ | 1184.0 | 1128.0 | 1184.0 | 1184.0 | 1128.0 | 1240.1 | 1095.9 |
| L4Nd(NO3)2+ | 1264.0 | 1207.0 | 1263.0 | 1263.0 | 1206.9 | 1319.1 | 1174.9 |
| L2Nd(NO3)2+ | 1426.9 | 1372.9 | 1428.9 | 1428.9 | 1372.9 | 1485.0 | 1340.8 |
Conclusion
A systematic study is presented on the influence of the alkyl chain length on the extraction properties of unsymmetrical diglycolamide extractants. Six UDGAs and TODGA were compared in both extraction and stripping tests, as well as in speciation experiments. The results show a significant increase in extraction efficiency for most UDGAs compared to TODGA. This shows that DGA extractants with different alkyl chains, where one side is shortened and the other lengthened, allow for an optimization of the steric hindrance, which determines extraction strength, while retaining good solubility in organic diluents. Furthermore, PnDdDGA, iPDdDGA, and pipDdDGA showed slightly higher curium/americium separation factors than TODGA when combined with SO3-Ph-BTBP. Despite iPDdDGA and pipDdDGA showing very large distribution ratios during the loading step, a third-phase was formed during the stripping step, making PnDdDGA the most promising extractant for further development. Analysis of the formed complexes with FTIR, ESI-MS, and UV-vis spectrometry did not show any significant differences in the complexes formed in the organic phase with the various diglycolamide extractants, implying that the extraction equilibrium is similar, regardless of the difference in alkyl chains. As no differences could be observed in the extraction mechanism, the higher extraction efficiency might be explained by the supramolecular organization of the extractants. The formation of a third phase for iPDdDGA and pipDdDGA during the stripping step, rather than during the loading step where a higher acid concentration is used, is not yet understood.Data availability
The data supporting this article has been included in the ESI.† This contains additional synthesis/characterization information, titration data, as well as additional FTIR, UV-vis, and ESI-MS spectra of the Nd-UDGA complexes.Author contributions
Filip Kolesar: conceptualization, investigation, formal analysis, visualization, writing – original draft. Cécile Marie: conceptualization, supervision, writing – review & editing. Laurence Berthon: conceptualization, supervision, writing – review & editing. Karen Van Hecke: supervision, writing – review & editing. Ken Verguts: supervision, writing – review & editing. Koen Binnemans: supervision, writing – review & editing. Thomas Cardinaels: funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Tomas Opsomer for his advice and assistance regarding the synthesis of the DGA compounds. The authors would also like to thank the LCIS and LILA laboratories at CEA for their assistance during experiments. The authors acknowledge the SCK CEN Academy for providing funding for a PhD fellowship and the FPS Economy for the support via the Energy Transition Fund. The authors also acknowledge ENEN for their financial assistance through the ENEN2plus mobility grant.Notes and references
- T. Kooyman, Ann. Nucl. Energy, 2021, 157, 108239 CAS.
- RED-IMPACT Impact of Partitioning, Transmutation and Waste Reduction Technologies on the Final Nuclear Waste Disposal Synthesis Report, Forschungszentrum Jülich GmbH, Jülich, Germany, 2008 Search PubMed.
- R. S. Herbst, P. Baron and M. Nilsson, in Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, eds. K. L. Nash and G. J. Lumetta, Woodhead Publishing, 2011, pp. 141–175, DOI:10.1533/9780857092274.2.141.
- T. Kooyman, L. Buiron and G. Rimpault, Ann. Nucl. Energy, 2018, 112, 748–758 CrossRef CAS.
- E. C. Buck, B. D. Hanson and B. K. McNamara, in Energy, Waste and the Environment: a Geochemical Perspective, ed. R. Gieré and P. Stille, Geological Society of London, 2004, vol. 236, p. 0 Search PubMed.
- Y. Sasaki, Y. Sugo, S. Suzuki and S. Tachimori, Solvent Extr. Ion Exch., 2001, 19, 91–103 CrossRef CAS.
- S. A. Ansari, P. N. Pathak, V. K. Manchanda, M. Husain, A. K. Prasad and V. S. Parmar, Solvent Extr. Ion Exch., 2005, 23, 463–479 CrossRef CAS.
- Y. Sasaki, P. Rapold, M. Arisaka, M. Hirata, T. Kimura, C. Hill and G. Cote, Solvent Extr. Ion Exch., 2007, 25, 187–204 CrossRef CAS.
- P. N. Pathak, S. A. Ansari, S. V. Godbole, A. R. Dhobale and V. K. Manchanda, Spectrochim. Acta, Part A, 2009, 73, 348–352 CrossRef CAS PubMed.
- G. Modolo, H. Asp, C. Schreinemachers and H. Vijgen, Solvent Extr. Ion Exch., 2007, 25, 703–721 CrossRef CAS.
- D. Magnusson, B. Christiansen, J. P. Glatz, R. Malmbeck, G. Modolo, D. Serrano-Purroy and C. Sorel, Solvent Extr. Ion Exch., 2009, 27, 26–35 CrossRef CAS.
- D. Whittaker, M. Sarsfield, R. Taylor, D. Woodhead, K. Taylor, M. Carrott, C. Mason, H. Colledge, R. Sanderson, B. Keywood, A. Bragg, C. White and C. Maher, Prog. Nucl. Energy, 2023, 166, 104956 CrossRef CAS.
- C. Marie, P. Kaufholz, V. Vanel, M.-T. Duchesne, E. Russello, F. Faroldi, L. Baldini, A. Casnati, A. Wilden, G. Modolo and M. Miguirditchian, Solvent Extr. Ion Exch., 2019, 37, 313–327 CrossRef CAS.
- N. Boubals, C. Wagner, T. Dumas, L. Chanèac, G. Manie, P. Kaufholz, C. Marie, P. J. Panak, G. Modolo, A. Geist and P. Guilbaud, Inorg. Chem., 2017, 56, 7861–7869 CAS.
- C. Wagner, U. Müllich, A. Geist and P. J. Panak, Solvent Extr. Ion Exch., 2016, 34, 103–113 CrossRef CAS.
- A. Geist, J.-M. Adnet, S. Bourg, C. Ekberg, H. Galán, P. Guilbaud, M. Miguirditchian, G. Modolo, C. Rhodes and R. Taylor, Sep. Sci. Technol., 2021, 56, 1866–1881 CrossRef CAS.
- C. Musikas, G. Le Marois, R. Fitoussi and C. Cuillerdier, in Actinide Separations, American Chemical Society, 1980, vol. 117, ch. 10, pp. 131–145 Search PubMed.
- J. N. Mathur, M. S. Murali and K. L. Nash, Solvent Extr. Ion Exch., 2001, 19, 357–390 CAS.
- S. Chapron, C. Marie, G. Arrachart, M. Miguirditchian and S. Pellet-Rostaing, Solvent Extr. Ion Exch., 2015, 33, 236–248 CrossRef CAS.
- M. Miguirditchian, V. Vanel, C. Marie, V. Pacary, M.-C. Charbonnel, L. Berthon, X. Hérès, M. Montuir, C. Sorel, M.-J. Bollesteros, S. Costenoble, C. Rostaing, M. Masson and C. Poinssot, Solvent Extr. Ion Exch., 2020, 38, 365–387 CAS.
- A. Sengupta, A. Bhattacharyya, W. Verboom, S. M. Ali and P. K. Mohapatra, J. Phys. Chem. B, 2017, 121, 2640–2649 CAS.
- Y. Sasaki, Y. Sugo, K. Morita and K. L. Nash, Solvent Extr. Ion Exch., 2015, 33, 625–641 CAS.
- E. A. Mowafy and D. Mohamed, Sep. Purif. Technol., 2014, 128, 18–24 CAS.
- H. Du, X. Peng, Y. Cui and G. Sun, Solvent Extr. Res. Dev., Jpn., 2020, 27, 81–89 CrossRef CAS.
- B. Verlinden, A. Wilden, K. Van Hecke, R. J. M. Egberink, J. Huskens, W. Verboom, M. Hupert, P. Weßling, A. Geist, P. J. Panak, R. Hermans, M. Verwerft, G. Modolo, K. Binnemans and T. Cardinaels, Solvent Extr. Ion Exch., 2023, 41, 59–87 CrossRef CAS.
- P. Deepika, K. N. Sabharwal, T. G. Srinivasan and P. R. Vasudeva Rao, Solvent Extr. Ion Exch., 2010, 28, 184–201 CrossRef CAS.
- S. Panja, P. K. Mohapatra, S. C. Tripathi, G. D. Dhekane, P. M. Gandhi and P. Janardan, Sep. Sci. Technol., 2013, 48, 2179–2187 CAS.
- D. Stamberga, M. R. Healy, V. S. Bryantsev, C. Albisser, Y. Karslyan, B. Reinhart, A. Paulenova, M. Foster, I. Popovs, K. Lyon, B. A. Moyer and S. Jansone-Popova, Inorg. Chem., 2020, 59, 17620–17630 CAS.
- F. Kolesar, K. Van Hecke, K. Verguts, C. Marie, L. Berthon, K. Binnemans and T. Cardinaels, ACS Omega, 2024, 9, 48336–48349 CAS.
- G. Mossand, PhD dissertation, Université Grenoble Alpes, 2017.
- Y. Liu, C. Zhao, Z. Liu, Y. Zhou, C. Jiao, M. Zhang, H. Hou, Y. Gao, H. He and G. Tian, J. Radioanal. Nucl. Chem., 2020, 325, 409–416 CAS.
- Y. Liu, C. Zhao, Z. Liu, S. Liu, Y. Zhou, C. Jiao, M. Zhang, Y. Gao, H. He and S. Zhang, RSC Adv., 2022, 12, 790–797 CAS.
- Z. Wei, C. Lu, Y. Zhou, C. Jiao, M. Zhang, H. Hou, Y. Gao and G. Tian, J. Radioanal. Nucl. Chem., 2020, 326, 583–589 CAS.
- Q. Liu, J. Zhou, L. Zhu, Y. Zhang, D. Li, S. Yang and G. Tian, Solvent Extr. Ion Exch., 2020, 38, 485–495 CAS.
- H. Narita and M. Tanaka, Solvent Extr. Res. Dev., Jpn., 2013, 20, 115–121 CAS.
- J. Ravi, K. A. Venkatesan, M. P. Antony, T. G. Srinivasan and P. R. Vasudeva Rao, J. Environ. Chem. Eng., 2013, 1, 690–695 CAS.
- J. Ravi, K. A. Venkatesan, M. P. Antony, T. G. Srinivasan and P. R. Vasudeva Rao, Radiochim. Acta, 2014, 102, 609–617 CAS.
- J. Ravi, K. A. Venkatesan, M. P. Antony, T. G. Srinivasan and P. R. Vasudeva Rao, Sep. Sci. Technol., 2016, 51, 32–40 CAS.
- J. Ravi, K. A. Venkatesan, M. P. Antony, T. G. Srinivasan and P. R. Vasudeva Rao, J. Radioanal. Nucl. Chem., 2013, 295, 1283–1292 CrossRef CAS.
- B. G. Tokheim, S. S. Kelly, R. C. Ronald and K. L. Nash, J. Radioanal. Nucl. Chem., 2020, 326, 789–800 CrossRef CAS.
- S. Bourg and C. Marie, GENIORS Project Report: WPSAR 5, Report D11.5, CEA, 2020 Search PubMed.
- G. Modolo, A. Wilden, P. Kaufholz, D. Bosbach and A. Geist, Prog. Nucl. Energy, 2014, 72, 107–114 CrossRef CAS.
- E. A. Mowafy, A. Alshammari and D. Mohamed, Radiochem, 2021, 63, 734–740 CrossRef CAS.
- G. Sun, X. Cai, J. Han, Y. Li, Y. Zhang, T. Yang and Y. Cui, J. Radioanal. Nucl. Chem., 2016, 308, 753–757 CrossRef CAS.
- X. Peng, M. Zhang, J. Yin, H. Zhao, J. Su, Y. Cui, X. Jiang and G. Sun, J. Rare Earths, 2025, 43(4), 815–821 CAS.
- E. A. Mowafy, A. Alshammari and D. Mohamed, Solvent Extr. Ion Exch., 2021, 40, 387–411 Search PubMed.
- K. Rama Swami, K. A. Venkatesan and M. P. Antony, Ind. Eng. Chem. Res., 2018, 57, 13490–13497 CAS.
- S. Murakami, M. Matsumiya, Y. Sasaki, S. Suzuki, S. Hisamatsu and K. Takao, Solvent Extr. Ion Exch., 2017, 35, 233–250 CAS.
- L. Berthon, A. Paquet, G. Saint-Louis and P. Guilbaud, Solvent Extr. Ion Exch., 2021, 39, 204–232 CAS.
- R. J. Ellis and M. R. Antonio, Langmuir, 2012, 28, 5987–5998 CAS.
- P. Narayanan, K. R. Swami, T. Prathibha and K. A. Venkatesan, J. Mol. Liq., 2020, 314, 113685 CAS.
- J. H. Yang, Y. Cui, G. X. Sun, Y. Nie, G. M. Xia and G. X. Zheng, J. Serb. Chem. Soc., 2013, 78, 93–100 CAS.
- T. Prathibha, S. Kumar, S. Chandra, S. Maji and N. Ramanathan, Inorg. Chim. Acta, 2023, 548, 121396 CAS.
- E. Campbell, V. E. Holfeltz, G. B. Hall, K. L. Nash, G. J. Lumetta and T. G. Levitskaia, Solvent Extr. Ion Exch., 2018, 36, 331–346 CAS.
- K. Schug and H. M. McNair, J. Sep. Sci., 2002, 25, 759–766 Search PubMed.
- A. Paquet, PhD dissertation, Université Montpellier, 2019.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00815h |
| This journal is © The Royal Society of Chemistry 2025 |