
Building in vitro models for mechanistic understanding of liver regeneration in chronic liver diseases
Khushi
Karnawat
 ,
Rithika
Parthasarathy
,
Mesevilhou
Sakhrie
,
Harikeshav
Karthik
,
Konatala Vibhuvan
Krishna
and
Gowri Manohari
Balachander
*
,
Rithika
Parthasarathy
,
Mesevilhou
Sakhrie
,
Harikeshav
Karthik
,
Konatala Vibhuvan
Krishna
and
Gowri Manohari
Balachander
*
School of Biomedical Engineering, Indian Institute of Technology (BHU) Varanasi, Varanasi-221005, India. E-mail: gowribalachander.bme@iitbhu.ac.in; Tel: +91 542-7165134
First published on 27th June 2024
Abstract
The liver has excellent regeneration potential and attains complete functional recovery from partial hepatectomy. The regenerative mechanisms malfunction in chronic liver diseases (CLDs), which fuels disease progression. CLDs account for 2 million deaths per year worldwide. Pathophysiological studies with clinical correlation have shown evidence of deviation of normal regenerative mechanisms and its contribution to fueling fibrosis and disease progression. However, we lack realistic in vitro models that can allow experimental manipulation for mechanistic understanding of liver regeneration in CLDs and testing of candidate drugs. In this review, we aim to provide the framework for building appropriate organotypic models for dissecting regenerative responses in CLDs, with the focus on non-alcoholic steatohepatitis (NASH). By drawing parallels with development and hepatectomy, we explain the selection of critical components such as cells, signaling, and, substrate-driven biophysical cues to build an appropriate CLD model. We highlight the organoid-based organotypic models available for NASH disease modeling, including organ-on-a-chip and 3D bioprinted models. With the focus on bioprinting as a fabrication method, we prescribe building in vitro CLD models and testing schemes for exploring the regenerative responses in the bioprinted model.
 Khushi Karnawat | Khushi Karnawat is pursuing her PhD at the IIT (BHU) Varanasi in Prof. Gowri's lab. She is working on developing patient-derived stem cell organoids to study the different stages of NASH pathogenesis leading to hepatocellular carcinoma. She graduated from Parul University in 2021 and subsequently completed her post-graduation at Sardar Patel University as a gold medallist in 2023 with specialization in biotechnology. |
 Rithika Parthasarathy | Rithika Parthasarathy received her MSci in Chemistry from the University of Nottingham in 2023 and is an incoming graduate student in the Chemistry PhD program at the University of Arizona. Her interests include bioconjugation and protein chemistry along with nanotechnology, including developing biochemical probes to investigate protein functions in disease mechanisms. |
 Mesevilhou Sakhrie | Mesevilhou Sakhrie is currently pursuing his PhD at the IIT (BHU) Varanasi in Prof. Gowri's lab. His field of expertise involves the fabrication of biomaterials and using microbial products in the application of chronic wound healing. He obtained his integrated master's degree in microbiology from the Central University of Rajasthan. |
 Harikeshav Karthik | Harikeshav is pursuing his BTech in Biomedical engineering at IIT (BHU) Varanasi. In Dr Gowri Balachander's lab he got to explore various concepts of tissue engineering and liver regeneration. In addition to his academic pursuits, Harikeshav engages himself in sports. |
 Konatala Vibhuvan Krishna | K. Vibhuvan Krishna is currently pursuing his BTech at IIT (BHU) Varanasi in Biomedical Engineering. His field of expertise includes modelling of biomedical devices using software programs like COMSOL Multiphysics and MATLAB. He is interested in data analytics as he believes the use of data science in biology is the key to many unanswered questions. |
 Gowri Manohari Balachander | Gowri M Balachander received her PhD from the Indian Institute of Science (IISc) Bangalore (2012) and did her post-doctoral fellowship at the National University of Singapore (NUS). She is currently an Assistant Professor at the Indian Institute of Technology (BHU) Varanasi in the School of Biomedical Engineering. She has worked extensively in developing organotypic models for breast cancer and liver and organ-on-a-chip models for NASH. She is passionate about the unsolved mysteries of liver regeneration and is setting up her lab and clinical collaborations to pursue her dream. |
1. Introduction
Chronic liver diseases (CLDs) are debilitating progressive diseases accounting for an estimated 4% of all deaths globally per year. CLDs are marked by a continuous deterioration of the liver via inflammation and regeneration of the liver parenchyma, leading to fibrosis and cirrhosis. The most common etiologies of CLDs include viral hepatitis, prolonged alcohol consumption, autoimmune disease, and genetic disorders. However, in recent years, non-alcoholic fatty liver disease (NAFLD), affecting over 25% of people globally, has been the primary contributor to CLDs.1–3 NAFLD, a hepatic manifestation of metabolic syndrome, is a progressive condition representing a wide spectrum of liver diseases marked by the accumulation of fat in hepatocytes termed as steatosis.4 These conditions range from simple steatosis to non-alcoholic steatohepatitis (NASH) with extreme cases leading to fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). NASH is currently the leading cause of HCC with around 2.6% of cases per annum.5 Research on the pathogenesis of NASH indicated that it is commonly associated with varying degrees of fibrosis, with many cases exhibiting advanced liver fibrosis in the F2 stage (portal fibrosis with few septa) or higher. With NASH being a progressive condition, analysis of sequential liver biopsy specimens suggests that the fibrosis stage F2 would progress to cirrhosis (F4) within 20 years in the case of NASH.6 The speed at which NASH progresses varies for each person owing to factors such as genetic and metabolic health, making it a highly individualized condition.The complex and heterogeneous nature of NASH and NAFLD has complicated the discovery of drug compounds as they involve multiple pathways and factors that influence disease progression. Despite these obstacles, many researchers have explored a wide range of therapeutic targets, and ongoing clinical trials continue to foster hope for the effective treatment of NASH with liver fibrosis. While various strategies have been employed to alleviate disease symptoms, it is crucial to emphasize that as of now, there are no curative therapies for CLDs.2
Preventative care, specifically lifestyle modification, has been mainly utilized to slow down disease progression and reduce complications. Weight loss through dietary modifications, physical exercise and in extreme cases bariatric surgery has shown significant improvement in histological studies. Irrespective of the method, 7–10% reduction in body weight has been recommended for NASH patients.7,8 A control study conducted by Ueno et al. showed that a combination of restricted diet and exercise for 3 months that resulted in a weight loss of 4.5–6.8 kg led to lower total cholesterol levels, which marked reduction in steatosis and improved liver function.9 However, only a small fraction of patients with NASH can sustain the weight loss necessary for optimal therapeutic outcomes, with approximately 50% of NASH patients failing to achieve a decrease in fibrosis.10 Drug development for NASH has been extensively explored in the last few years with numerous new and repurposed drugs currently in clinical trials, but with poor success.7 Elafibranor, a PPAR-α/PPAR-δ dual agonist, showed NASH resolution in Phase 2b trials but failed to show effective results in Phase 3 trials. Saroglitazar, a PPAR-α/PPAR-γ dual agonist, has shown good performance in Phase 2 trials and is awaiting results from Phase 3 trials. Obeticholic acid (OCA) is a semi-synthetic analogue of chenodeoxycholic acid, a bile acid that acts as a farnesoid X receptor agonist. OCA, developed by Intercept Pharmaceuticals Inc., was leading the field of NASH treatment until the FDA rejected the company's NDA for treatment due to several liver toxicity signals along with an increase in low-density lipoprotein (LDL) and total cholesterol levels.11 More recently, resmetirom, an oral thyroid hormone receptor beta-agonist, has shown very good performance in Phase 3 trials for the treatment of NASH with liver fibrosis. In a trial involving 966 adults with NASH and fibrosis stages of F1, F2 and F3, results indicated that those administered with 80 and 100 mg doses of resmetirom improved NASH and liver fibrosis by at least one stage.12 Recently in March 2024, the United States Food and Drug Administration (US FDA) has given accelerated approval for the use of resmetirom in noncirrhotic NASH patients with mild fibrosis, but along with diet and exercise.13
Histochemical studies investigating the pathogenesis of NASH have pointed towards the activation of stem cells in the regeneration process during fibrotic development and progression.14,15 Upon stimulation of the stem cells of the liver, hepatic progenitor cells (HPCs), a cascade follows, giving rise to the ductular reaction (DR). The DR is characterized by a reactive lesion consisting of expanding biliary ductules with a complex of stromal and inflammatory cells and is found in chronic liver diseases of all etiologies.16 A study conducted by Richardson et al. noted a strong positive correlation between the fibrosis stage and DR in individuals afflicted with NASH particularly within the context of portal fibrosis.17 Moreover, the expansion of HPCs exhibited a noteworthy correlation with both portal and lobular inflammation, with increased hepatocellular ballooning. Also, fat accumulation in hepatocytes significantly compromises hepatocyte replication, hindering the liver's natural ability to replace damaged or dying cells.18 This resultant impaired regeneration process further results in progression of liver damage in NASH and the growth of abnormal cells leading to HCC.
Liver regeneration has traditionally been explored in the context of partial hepatectomy (PHx) in rodents, wherein two-thirds of the liver is removed via surgery.19 This provokes a massive response followed by cellular proliferation to reach a stage where the functions of the liver are completely restored.20 In rodents, successful regeneration of the liver has been observed even after twelve sequential PHx procedures.21 The orchestration of the cellular regeneration following PHx in rodents is one of the most well studied phenomena of organ regeneration.22,23 Due to the successful restoration of liver functions after PHx, it has often been viewed through a positive lens. However clinical evidence from the last decade shows that regenerative responses in chronic liver diseases are different from the regenerative responses in PHx, and they can turn detrimental and further drive the disease progression.14,24 We lack clear mechanistic understanding of regenerative responses and still do not understand why an organ of such remarkable regenerative potential fails to regenerate in chronic liver diseases.25 Most of the studies done so far are based on animal models or histopathological evidence from clinical samples.17
Animal models have been instrumental in understanding the pathogenesis and progression of NASH26,27 and testing potential therapeutic interventions.28 The different types of animal models for NASH and the methods of NASH induction and their applications have been extensively reviewed elsewhere.29–31 Briefly, NASH is induced in the animals via one of the four following methods: (i) modified diet/nutrition, (ii) toxins, (iii) genetic modification and (iv) combination of diet and toxins. Despite their utility, animal models cannot fully replicate the exact etiology, pathogenesis, and severity of NASH progression in humans due to significant species differences in liver architecture, regenerative mechanisms, disease progression, inflammatory markers, metabolism rates, and drug responses as summarised in Table 1.29,30,32 Suitable models should exhibit obesity, insulin resistance, and systemic inflammation akin to those observed in humans and display features such as macrovesicular steatosis, lobular inflammation, hepatocellular ballooning (preferably with Mallory–Denk bodies), and hepatic fibrosis. Lesions should predominantly be centrilobular or panacinar, with perisinusoidal fibrosis starting in zone III and progressing to bridging fibrosis and nodule formation. Advanced fibrosis should predispose to hepatocellular carcinoma (HCC). Models should mirror the dietary, systemic, and histological characteristics of human disease, demonstrating activation of key pathways such as de novo lipogenesis, the unfolded protein response, oxidative stress, apoptosis, and fibrogenic pathways.33 The inaccurate representation of human CLD pathology in animal models is reflected in the high failure rates (up to 80%) in phase II/III clinical trials.34 Animal models also lack handle on experimental manipulation for mechanistic studies and the throughput for testing therapies. In the context of NASH, we aim to explain how we could build three-dimensional (3D) models that can be used to study liver regeneration.
| Model | Type | Limitations |
|---|---|---|
| Methionine and choline-deficient (MCD) diet | Diet | Animals show extensive weight loss and do not develop insulin resistance, fibrosis develops in the periportal region, does not induce characteristics of metabolic syndrome |
| High-fat diet (HFD) | Diet | Do not progress to advanced fibrosis and cirrhosis |
| Leptin deficiency (ob/ob mice) | Genetic | Fibrosis develops in the portal areas; might need additional dietary changes to induce NASH |
| CCl4 treatment | Toxin | Unnatural etiology and different from humans; can develop advanced fibrosis but spontaneous regression is observed after withdrawal of CCl4 treatment |
| STAM model (streptozotocin) + HFD | Combination of toxin and diet | Molecular mechanisms are different from humans. Toxicity of streptozotocin can alter disease etiology |
While there have been several in vitro models developed to mimic specific aspects of CLDs like NASH, there are hardly any models to recapitulate the regenerative responses observed in CLDs or at least PHx. One of the very first attempts in this regard is the work done by Chhabra et al., where they have developed vascularized hepatic tissues with PHH spheroids embedded in a gel with a single channel lined with HUVECs (Fig. 2D). Upon stimulation with IL1-β they observed the re-entry of hepatocytes into the cell cycle, mimicking the growth-factor triggered proliferative response in regeneration.35 Although the authors claim that the model mimics regenerative responses, it fails to capture the two critical components of regeneration such as injury and repair of damaged tissue. An ideal model for regeneration of normal liver should comprise a functioning mature liver tissue, a controllable injury and a measurable response and recovery of the mature tissue. While modelling regeneration in CLDs, the critical aspects of the disease should be modelled in the mature liver tissue, followed by a controllable injury and then assessment of recovery of the injured tissues. While modelling CLDs, the disease itself causes the death of hepatocytes, and therefore the injury can be controlled by the extent of disease induction in the model. Assessment of cellular and molecular responses in such models will provide great insights into understanding regeneration in CLDs and enable testing of potential drugs and therapies. We first introduce the important components of a mature liver tissue, such as cells, signaling molecules, and the extracellular matrix (ECM), and their changes in CLDs. We then provide a review of in vitro models for CLDs with the focus on NASH and then discuss more recent developments in 3D bioprinting. Then we provide critical insights into developing appropriate 3D models for liver regeneration.
2. Building tissue-like in vitro models
Within tissues, cells engage in biochemical and mechanical interactions with neighbouring cells and the extracellular matrix (ECM). These interactions create a 3D communication framework crucial for maintaining tissue integrity and homeostasis.36 Cellular processes like proliferation, migration, and apoptosis are regulated within this framework, which is influenced by the microenvironment.37 Developing a good in vitro model involves understanding and replicating the specific microenvironment of the target tissue, including cells and the extra-cellular matrix (ECM) matrix, with due attention to factors such as matrix elasticity and microarchitecture. Unlike traditional two-dimensional (2D) cultures, 3D cell cultures can be tailored to effectively recreate these physiological cell–cell and cell–ECM interactions, making them better mimics of real tissues. Consequently, 3D cultures find extensive application in diverse areas of cell biology, including tumor research, cell adhesion, cell migration, and epithelial morphogenesis.38The components of a 3D in vitro model include cells, signaling molecules and the scaffold. The selection of appropriate cell types is paramount, as different cell types have their distinct complementary functions in the tissues. Signaling molecules, such as growth factors and cytokines, or chemical gradients provide the necessary biochemical microenvironment to trigger cellular responses and drive processes like differentiation, proliferation, or migration. The scaffold or substrate serves as the physical support for the cells and can modulate their behaviour by presenting mechanical and structural cues. Altogether, these components are essential for creating in vitro models that recapitulate the complexity of tissues and organs, enabling researchers to understand disease mechanisms, test drugs, and develop regenerative therapies with higher fidelity to in vivo conditions.39
In the following section we explain the components of normal liver tissue, including cells, signaling molecules and the substrate, and the responses to PHx and in CLDs, with the focus on NASH. We envision to provide deeper insights into the choice of these components and the fabrication techniques to build appropriate in vitro models for deciphering regeneration in CLDs.
2.1. Cells
 | ||
| Fig. 1 Organization of the liver into the hexagonal units namely lobules. Each hepatic lobule is formed by a distinctive arrangement of 6 portal triads enclosing chords of hepatocytes running towards the central vein. The blood from the portal vein and hepatic artery perfuses the organ through the sinusoids. | ||
The choice of cells plays an important role in building an in vitro model. Each of these cells play an important role in regenerating the liver following PHx, which we have summarized briefly in the following section to provide a quick understanding. A detailed description has been provided in ref. 42.
Numerous growth factors and cytokines originate at the site of hepatectomy or infiltrate the liver through the circulatory system. They influence the proliferation, differentiation, activation, migration, and survival of cells in the regenerating liver. We have explained the most important growth factors and the cytokines at appropriate instances in Table 2, where we discuss cells and their roles in hepatic functions in an adult liver and responses in regeneration after PHx and in CLDs, and in Section 2.2, we elaborate the molecular signaling involved in liver growth, during regeneration and in CLDs. Overall, liver regeneration after PHx is a dynamic and intricate process that requires tight coordination between the different cells in the liver. The cells that are activated after PHx return to their basal state once the regeneration is complete. The excess fibrous matrix is resolved via the reprogramming of KCs and hepatic stellate cells. Such homeostatic responses are completely deregulated in CLDs.
| Cells | Responses to hepatectomy | Responses in chronic liver diseases |
|---|---|---|
| Hepatocytes | Following a partial hepatectomy, hepatocytes enter into the cell cycle and begin synthesizing DNA. More than 95% of the hepatocytes replicate within 48 hours after PHx52 | Hepatocyte proliferation is reduced and cells exhibit cell cycle arrest and stain positive for nuclear p2153 |
| Biliary epithelial cells (BECs) | Stimulated to divide and replenish the bile duct tissue lost after PHx. The BECs form a trough like structure and extend into the hepatic lobule called canals of Hering (CoH). CoH are postulated to house the liver stem cells that are capable of regenerating both the hepatocytes and BECs54,55 | Destruction of bile ducts and impaired bile flow or cholestasis has been observed in several CLD56 |
| Expansion of bile ducts with accompanying stromal and inflammatory cells called the ductular reaction (DR) is found in CLDs and represent the regenerative nodules in the liver. The abundance of DR correlates with severity of disease progression and higher risk of mortality53 | ||
| Kupffer cells (KCs) | KCs are the first immune cells to become activated after PHx due to the TLRs present on their surface that recognize DAMPs.57 KCs produce IL-6 and TNF-α that stimulate the hepatocytes to proliferate following hepatectomy.58 They release pro- and anti-inflammatory mediators, which attract other immune cells from the circulatory system into the liver to aid in tissue regeneration. They remove cellular waste and damaged hepatocytes, which is important for tissue repair. They are important for the resolution of fibrosis after an acute injury. KCs secrete various pro-resolution mediators that stimulate the expression of various matrix metalloproteinases (MMPs) and suppress the inhibitors of MMPS | KCs lose their tolerogenic potential and activate persistent inflammation in the liver. They secrete TGF-β1, which drives the myofibroblastic transformation of stellate cells and fibrosis in CLDs.59 KCs secrete TNF-α, IL-1, and MCP-1, which stimulate HSC proliferation. Kupffer cells secrete gelatinase and degrade type IV collagen, contributing to the development of fibrosis60 |
| Hepatic stellate cells (HSC) | They are activated in response to PHx and begin to produce ECM proteins.61 This helps to create a temporary scaffold to support the regenerating liver tissue. Once the liver has regenerated, the HSCs revert back to the state of being dormant.56 HSCs activated upon acute injury create a temporary scar of collagenous proteins at the injury site, safeguarding the liver from further harm. Moreover, they secrete cytokines and growth factors that stimulate the regeneration of hepatic epithelial cells42 | In response to liver injury from viral infection or hepatic toxins, HSCs transdifferentiate into activated myofibroblast-like cells upon receiving signals from damaged hepatocytes and KCs. In cases of chronic liver diseases, persistent activation of stellate cells leads to liver fibrosis, characterized by extensive scar formation and disruption of liver structure and function62 |
| Liver sinusoidal endothelial cells (LSECs) | LSECs produce growth factors and signaling molecules, which stimulate hepatocyte proliferation and regulate blood flow in the liver sinusoids.63 They are activated to divide and replace the lost sinusoidal endothelial cells and are essential for the successful regeneration of the liver after PHx64 | LSECs become dysfunctional: they deposit the basement membrane and lose their fenestrae, which impairs solute and molecular transport. This capillarization of the LSECs significantly contributes to hepatic insulin resistance and steatosis. Dysfunctional LSECs contribute to increased portal pressure in CLDs65 |
In CLDs such as fatty liver and NASH, loss of hepatocytes occurs due to lipotoxicity and the damage signals from the hepatocytes activate the non-parenchymal cells such as the KCs, hepatic stellate cells and LSECs. In contrast to PHx, restoration of functional hepatocytes via proliferation is not achieved, and liver functions progressively decline. The non-parenchymal cells that are activated are permanently turned on and continue to elicit wound healing responses that progressively damage the tissue microenvironment. For example, in later stages of NASH, the activated hepatic stellate cells continue to deposit the collagenous matrix and resolution of fibrosis is unachievable. Such a matrix is not conducive for hepatocytes to function normally and their functions decline exacerbating the decline in liver functions. This uncontrolled fibrosis progresses to cirrhosis and complete loss of hepatic functions and liver failure.
Although hepatocyte injury is common in PHx and CLDs, contrasting responses are observed with loss of homeostasis in CLDs. The functions and responses of the major cells in the liver to PHx and CLDs are summarised in Table 2.
It is critical to choose the right types of cells and induce their distinct responses to model CLDs. The above section provides an overview of the behaviour of different cell types and their responses in PHx and CLDs. For example, insulin transport to the hepatocytes occurs through the fenestrae in the LSECs. If the aim is to study the impaired insulin transport to the hepatocytes in fatty liver, a co-culture of hepatocytes and LSECs in which solutes pass through the fenestrations in LSECs is required. Upon simulation of fatty liver conditions, the loss of fenestrations in LSECs and formation of the basement membrane would be ideal to aptly mimic the impaired insulin transport in fatty liver.
As explained in Sections 2.1.1 and 2.1.2, multiple parenchymal and non-parenchymal cells in the liver are important for the pathogenesis of CLDs. While developing models it is important to incorporate different cell types to recapitulate events in CLDs. The use of primary cells is limited due to the following reasons: firstly, primary cells do not proliferate in vitro and often require repeated isolation, which has ethical concerns and incurs high costs; they might lose their characteristics upon in vitro culture and may behave differently and therefore allow a small period of experimental manipulation. On the other hand, cell lines are transformed and might be incapable of exhibiting true differentiated features. Due to these reasons, more often in vitro cultures tend to use induced pluripotent stem cells (iPSCs) and differentiate them into the specific cell types of interest. The differentiation is based on the factors that drive the differentiation of these cells from the progenitors during embryonic development. Knowing the embryonic development of liver cells is beneficial for devising experimental procedures to differentiate human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs) into specific liver cell types and for making useful modifications to the existing procedures when more than one cell type is being differentiated from the same cell source to be incorporated in the model. Also understanding the embryonic origin helps to understand the regeneration of these cell types in CLDs. In the following section, we provide a brief overview of the embryonic origins of these cells that might be useful for developing protocols for differentiation of iPSCs into various liver cell types and for tailoring the reported protocols.
The embryonic development of the liver starts from the emergence of the hepatic diverticulum from the ventral foregut endodermal tissue.41 Endodermal cells with sustained hepatic competence give rise to hepatoblasts.72 Hepatoblasts serve as the common precursor for hepatocytes and BECs.73 The non-parenchymal cells such as KCs, hepatic stellate cells, and LSECs within the context of the sinusoid are derived from mesodermal progenitors, collectively comprising the sinusoidal cell population.74 Lineage tracing experiments conducted for hepatic stellate cells in avian embryos have yielded empirical evidence that mesothelial cells originating from the proepicardium and septum transversum mesenchyme exhibit the potential to differentiate into both endothelial and hepatic stellate cells within the hepatic sinusoidal network. Recent investigations conducted in both human and murine subjects provide corroborative evidence supporting the mesothelial origin of hepatic stellate cells.41 In contrast, hepatic stellate cells within the human fetal liver exhibit the expression of CD34 and cytokeratin 7/8, suggesting that they might originate from the endoderm. The embryonic origin of hepatic stellate cells remains a matter of ambiguity due to the concurrent expression of marker genes representative of all three germ layers. Given their close physical adjacency and concurrent expression of angiogenic factors, it has been postulated that hepatic stellate cells and LSECs may share a common precursor.75 LSECs could be potentially derived from the omphalomesenteric veins, common cardinal veins, or posterior cardinal veins.76 KCs are derived from the primitive macrophages from the early erythro-myeloid progenitors in the yolk sac. These primitive macrophages migrate into the liver bud and differentiate into KCs upon receiving hepatic cues from the developing liver.77
2.2. Signaling
Signaling molecules can guide morphogenesis, cell fate decisions and differentiation. In adult tissues, signaling molecules are the main form of communication between the different types of cells to activate specific functions in response to stimuli. Regeneration after PHx is an intricately coordinated process mainly carried out through timed secretion of signaling molecules by the remnant cells. However, in the diseased state, the control of the secretion of signaling molecules is lost, which can cause persistent activation/deactivation of the concerned pathway leading to aberrant cellular responses.In a 3D model, signaling molecules can be deployed for two important purposes. Firstly, they can be used to differentiate iPSCs to derive the desired cell types to build the model. As iPSCs have the potential of embryonic stem cells, the choice of signaling molecules, model of delivery, timing and period of treatment are based on the expression of these molecules in the developing liver in the embryo. Secondly, signaling molecules can be used to mimic critical aspects of disease or conditions. For example, IL1β stimulation was adopted to mimic the regenerative conditions in a vascularized hepatic ensemble in vitro.35
In the following sections, we provide a brief description of the signaling molecules governing the differentiation of stem cells into hepatic cells and how these molecules and concerned signaling pathways are involved in CLDs.
Hepatocytes. The hepatoblasts, which are the common progenitor cells for hepatocytes and BECs, are derived from the endodermal cells from the ventral foregut and enter into a bed of vascularized ECM called the septum transversum mesenchyme (STM). The bone morphogenetic proteins (BMPs) from the STM and the fibroblast growth factor (FGF) signals from the developing heart drive the hepatic fate in the endodermal cells. These signaling molecules induce the expression of transcription factors FOXA and GATA, which induce hepatic competence or the ability to respond to hepatic inducing signals in the endodermal cells by occupying the albumin (Alb) gene enhancer region preceding liver specification.72 This leads to induction of hepatic gene expression, notably including α-fetoprotein (AFP), transthyretin, and hepatocyte nuclear factor-4α (HNF-4α). Subsequently, these cells express albumin and crucial transcription factors such as Hhex and Prox-1, which are critical for liver development.72 The principal transcription factors that exhibit expression within hepatocytes during their differentiation from hepatoblasts comprise FoxA1/2, HNF1α and β, HNF-4α, and HNF6. Additionally, oncostatin M originating from the hematopoietic compartment, hepatocyte growth factor (HGF), and glucocorticoid hormones play critical roles in the differentiation process of hepatocytes from hepatoblasts. Notably, the activation of Wnt signaling via β-catenin has been demonstrated to facilitate the maturation of hepatocytes.78 This information has been the basis of development of a protocol for the differentiation of iPSCs into mature hepatocytes.79
BECs. The transcription factors SOX9 and HNF6 are the earliest signals that drive the commitment of biliary fate in hepatoblasts lining the portal veins.72 Transforming growth factor-beta (TGF-β) facilitates the differentiation process of hepatoblasts into biliary cells while concurrently inhibiting the differentiation of hepatocytes. The Wnt signaling pathway provides support for the differentiation of cells towards the biliary lineage.78 iPSCs are differentiated into biliary lineage cells or cholangiocytes using these transcription factors.80,81
Non-parenchymal cells of the liver. VEGF promotes primitive sinusoidal endothelial morphogenesis. The involvement of Wnt signaling in sinusoidal growth is also noteworthy. The potential roles of Wnt/β-catenin and Pitx2 have been proposed in hepatic stellate cell differentiation and expansion during development.78 iPSCs are differentiated into LSECs82 and hepatic stellate cells based on these signaling molecules.83 KCs are derived from macrophage progenitors that do not rely on the MYB transcription factor for their development. These progenitors undergo differentiation into macrophages with liver-specific characteristics in response to signals and cues provided within the hepatic microenvironment, which has been used to differentiate iPSCs into mature Kupffer cells.77
The signaling molecules and associated pathways that work to regenerate the liver after PHx can also turn detrimental in CLDs. For example, IL-6 produced by KCs and other NPCs in response to PHx facilitates the commencement of liver regeneration to activate hepatocyte proliferation via the MAPK and STAT3 signaling pathways.86 IL-6 contributes to hepatoprotection by boosting anti-caspase regulators and reducing oxidative damage, thereby preventing apoptosis. Additionally, hepatocytes are shielded by IL-6 from apoptosis caused by Fas. By triggering the MAPK signalling pathway, IL-6 also encourages the mitogenesis of hepatocytes and growth of cells87 apart from playing a role in initiating and progressing liver regeneration; cytokines and growth factors also set off negative feedback loops that result in their own downregulation, which is required to inhibit the proliferation of hepatocytes to halt liver regeneration once the necessary functional liver mass is regenerated. IL-6 upregulates the expression of the suppressor of cytokine signalling (SOCS) protein SOCS3, which in turn suppresses the phosphorylation of STAT3 and decreases IL-6 signalling.88 However, persistent activation of IL-6 signaling fuels carcinogenesis and tumor formation in the liver.89 In Table 3, we have summarized the roles of a few other signaling pathways that are critical for liver development and regeneration, but can also turn detrimental and fuel disease progression in CLDs.
| Signaling pathways | Function in adult tissue | Response to hepatectomy | Response in chronic liver diseases |
|---|---|---|---|
| NOTCH | Plays a critical role in the development of IHBD78 | Jagged1 expression increases in the first 24 hours following PHx | Jagged1 gene mutations cause Alagille syndrome, characterized by loss of bile ducts leading to neonatal jaundice90 |
| Directs biliary commitment in hepatoblasts and morphogenesis of IHBD | Stimulates hepatocyte proliferation during the early phases of regeneration following PHx | Aberrant activation in cholangiocytes and hepatocytes can lead to cholangiocarcinoma (CCA) | |
| Aberrant expression in hepatocytes promotes their trans-differentiation into biliary cells | Crucial to maintain the pool of HPCs by inhibiting the differentiation of HPCs into mature hepatocytes | Prolonged activation in hepatocytes results in aggressive HCC | |
| Is activated in HPCs during liver repair, generating reactive cholangiocytes | Depending on the precise cellular environment, it can activate transcription of specific target genes that can both cause and prevent apoptosis91–93 | Activation in malignant cells can serve as cancer-initiating cells for HCC or CCA94 | |
| Hepatocellular damage inhibits Notch, promoting progenitor commitment towards hepatocyte lineage95 | |||
| Wnt/β-catenin | Essential for hepatic specification and contributes to hepatic morphogenesis | Growth factors, cytokines, and cell–cell interactions drive the activation of Wnt in hepatocytes following PHx, which drives their proliferation and differentiation | Involved in the pathophysiology and found to be dysregulated in a variety of ways in CLDs96 |
| Influences hepatoblast proliferation and survival in vivo and also in vitro | Inhibiting the Wnt/β-catenin signalling in rats hinders liver regeneration97 | Increased activation in CLDs is due to overexpression of Wnt ligands, reduced expression of Wnt pathway inhibitors and mutations in genes encoding Wnt signalling components85 | |
| Important for cholangiocyte differentiation of hepatoblasts72 | Involved in the activation and proliferation of oval cells, which are putative stem cells found in the rodent liver42 | Its persistent activation stimulates the activation of hepatic stellate cells leading to collagen deposition and fibrosis14 and can trigger the proliferation of hepatocytes fuelling the progression to HCC | |
| Crucial for hepatocyte maturation and hepatic zonation78,98 | |||
| TGF-β | Activates Smad2/3 and Smad4-dependent gene regulation, affecting MAP kinase signaling | Hepatic stellate cells secrete TGF-β after PHx, which in turn activates this signaling in hepatocytes | The main driver of fibrosis in CLDs |
| Deficient embryos in Smad2 +/−; Smad3 +/− exhibit hypoplastic livers due to defective β1-integrin and β-catenin signaling78 | It exerts its anti-proliferative effects on hepatocytes and halts their proliferation | Produced by inflammatory and immune cells, it directly promotes fibrogenesis by inducing the transcription of type I and III collagens through the Smad signaling pathway76 | |
| One of the main feedback mechanisms that prevent excessive proliferation of hepatocytes after PHx and an important regulator controlling the end of regeneration99 | Inhibiting TGF-β reduces NASH-induced fibrosis, especially when combined with interleukin (IL) signaling inhibition100 | ||
| Hippo-YAP/TAZ | Pivotal for liver development, cell fate determination, homeostasis, stem cell maintenance, epithelial to mesenchymal transition, and regeneration101 | Yap/Taz target genes increase hepatocyte proliferation and cell cycle progression | Upregulated in hepatocytes in CLDs |
| Cooperates to control the transcription of SOX9 and induces biliary fate in hepatoblasts102 | Inhibits cell death by upregulating genes that shield hepatocytes from stress-induced apoptosis103 | Implicated in liver fibrosis and tumorigenesis101 | |
| Is activated in BECs and hepatocytes in response to injury, facilitating differentiation and proliferation to maintain homeostasis101 | Activates reprogramming of metabolic pathways towards anaerobic glycolysis to facilitate nucleotide synthesis necessary for replication104 | Dysregulated Hippo signaling leads to dysfunctional bile duct formation implicated in several bile duct disorders105 | |
| Activation in NPCs may facilitate disease progression101 | |||
| HGF | Stimulates hepatocytes to produce inositol phosphate and raises intracellular calcium concentrations | A powerful mitogen stimulating the division of surviving hepatocytes commencing the process of regeneration | Beneficial for attenuating fibrosis in acute injury created by bile duct ligation due to its anti-apoptotic and proliferative roles106 |
| This triggers MAP kinase and phospholipase D, resulting in arachidonic acid release | It is sequestered to the ECM in the liver in its inactive form. A massive amount of HGF is released within an hour after PHx and is activated by urokinase plasminogen activator (uPA) | It is the primary cytokine secreted by the cancer associated fibroblasts that aid in self-renewal of the tumor initiating cells in HCC107 | |
| This series of events stimulates DNA and protein synthesis in hepatocytes, which leads to cell proliferation | Activated HGF binds to cMET on hepatocytes and activates STAT3, NF-κB, PI3K, ERK1/2 and β-catenin signaling pathways switching on several mechanisms that are essential for liver regeneration108 | ||
| Also promotes cell motility | Attracts HPCs to the site of injury so they can mature into adult hepatocytes and replace damaged tissue | ||
| Shields hepatocytes from apoptosis | Facilitates the uptake of nutrients and the generation of energy required to satisfy the needs of rapidly expanding tissue109 | ||
2.3. Scaffolds
Scaffolds provide the physical support for the three-dimensional (3D) growth of cells. They are mimics of the ECM in tissues. The structure of the ECM governs cell polarity and tissue architecture, which are critical determinants of cell functions. Hence the biochemical composition and the physical/mechanical properties of the ECM are specific to the organ and get remodelled in response to injuries and diseases and undergo gradual changes with age. We aim to provide a brief overview of the ECM and some of the critical physical properties of normal liver and the ECM changes in PHx and in CLDs, which could guide the choice of materials and fabrication techniques and offer important considerations for designing scaffolds for in vitro models for liver regeneration.Type I collagen, a major component of the adult ECM in the liver, increases during the progression of NAFLD and NASH. Collagen type I production remains an essential hallmark of fibrosis as its excessive generation and cross-linking can lead to a significant increase in matrix stiffness. Zheng et al. proposed that the accumulation of fat during the steatosis stage creates a proinflammatory milieu, leading to an increase in hepatic stellate cell activation, hence resulting in large deposits of collagen type 1 in NASH patients.112 In a study regarding ECM remodeling in liver fibrosis conducted by Baiocchini et al., it was discovered that the abundance of collagen type I, particularly the genes COL1A1 and COL1A2, was the major contributor to driving the excessive structural changes of the ECM, hence leading to progression of fibrosis.57 When measuring the stiffness of liver tissue of NAFLD patients using transient elastography, Mori et al. made a notable discovery demonstrating the greater presence of collagen type I and an increase in myofibroblasts. The study also reported a correlation between the increase in collagen deposits and the expression of α-smooth muscle actin (αSMA) as the disease progressed to fibrosis in NASH.113 Future work on NAFLD can explore a visual representation of the expression of αSMA using 3D models, within the context of liver stiffness and ECM remodeling. Such advances can help provide a deeper insight into NAFLD disease progression and potential biomarkers for intervention.
While in vitro models of portal hypertension have been significantly lacking in the current literature, certain in vivo models have been successfully used to mimic this condition. The partial portal vein ligation model has been widely explored to study the pathophysiology of portal hypertension since it is inexpensive, reproducible and can rapidly induce portal hypertension.122 Recently Ortega-Ribera et al. developed a microfluidic liver-on-a-chip device to mimic pathological and physiological pressures on primary liver sinusoidal endothelial cell (LSEC) culture. Primary LSECs were cultured using a microfluidic liver-on-a-chip device under normal or increased hydrodynamic pressure within a 1 to 12 mmHg pathophysiological range. RNA sequencing was used to identify pressure-sensitive genes, which were validated via liver biopsies of patients with CLDs with PH and those without PH. Transcriptomic analysis showed chromobox 7 (CBX7) as a pressure-sensitive key transcription factor. This study highlighted the harmful effect of pathological pressure on LSECs and identified the key proteins present in the blood of patients with CLDs allowing for the prediction of portal hypertension.123
However, in vitro models of PH need to be further developed, to provide more controlled and precise environments to study the underlying mechanisms of liver fibrosis. Such models would pave the way for researchers to replicate the cascade of reactions leading to fibrosis, including elevated blood flow resistance and liver tissue stiffness.
Bioinert hydrogels can provide scaffolding to cells; however, adding peptides to such hydrogels can help fulfil other cellular functions of the ECM. Certain cells adhering to the ECM molecules can be fixed onto ECM scaffolds that mimic the specific structures of the tissues or organs. For instance, RDG peptide sequences on ECM-mimetic hydrogels have been copolymerized with PEG-diacrylate to create an adherable hydrogel surface for cells, which has also been shown to promote cell differentiation and survival.127
3. In vitro models for chronic liver diseases
An in vitro model for a disease should recapitulate the essential features of the disease that would cater to the application of the model. The pathogenesis of NAFLD begins with fat accumulation as lipid droplets in hepatocytes due to insulin resistance, adipose derived and gut-derived factors. Beyond a limit the hepatocytes undergo death due to lipotoxicity. Dying hepatocytes release signaling molecules that activate inflammation and wound healing responses in the surrounding stroma. This activates the KCs, hepatic stellate cells and LSECs to participate in repairing the tissue. However, in progressive NAFLD, due to dysregulation of feedback mechanisms, the activated non-parenchymal cells lead to persistent inflammation and fibrosis, which cause the progression of NAFLD to NASH and further cirrhosis. Uncontrolled progression might lead to liver failure and/or hepatocellular carcinoma.128 Ideally, parenchymal and non-parenchymal cells interacting in a 3D space with flow are required for the recapitulation of essential features of NASH such as steatosis, hepatocellular injury, inflammation and fibrosis. This warrants the co-culture of all these cells in a microfluidics chip. However, simpler models with one or more cell types that recapitulate specific aspects of the disease have been developed, which have been instrumental in understanding the mechanisms of this disease. In this section we highlight the salient features of a few notable models for NAFLD and NASH ranging from simple single cell type-based organoid models to more sophisticated organ-on-a-chip models. An exhaustive collection of such models has been reviewed elsewhere.1293.1. Organoid models
Liver organoid models are usually developed using hepatocyte cell lines such as HepG2 or using primary human hepatocytes (PHH) with or without the incorporation of BECs and non-parenchymal cells such as KCs, hepatic stellate cells and LSECs.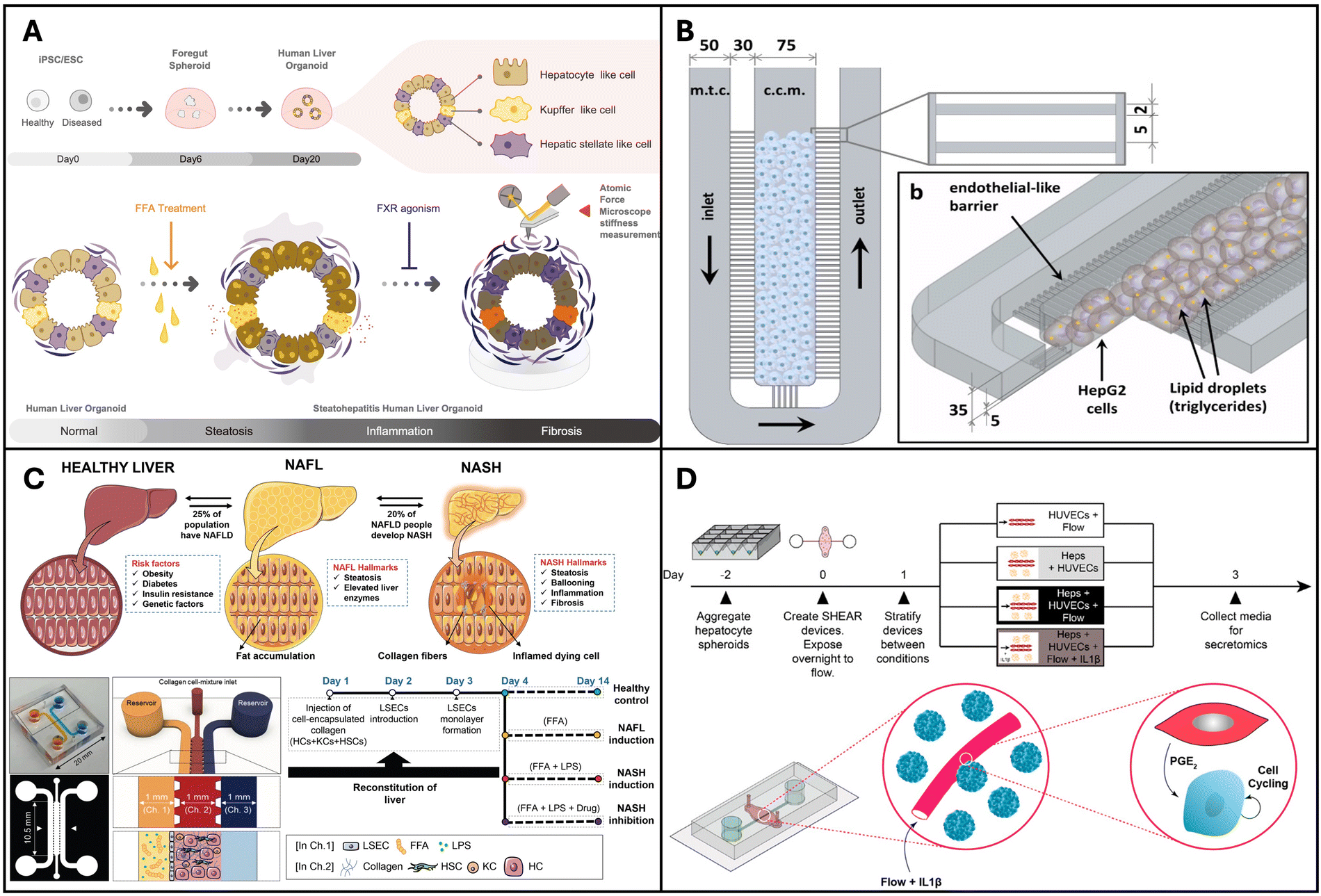 | ||
| Fig. 2 Organoids and organ-on-a-chip models for NAFLD and NASH. (A) Pluripotent stem cell lines used to create multi-cellular human liver organoids that are transcriptomically similar to tissues found in living organisms. Reproduced from ref. 136. Copyright 2019 Elsevier Inc. (B) Microfluidic chip microarchitecture with high-density hepatic cell culture containing lipid accumulation. Reproduced with permission from ref. 137 copyright 2016, Public Library of Science PLOS. (C) NASH on-chip model with an FFA reservoir chamber and an LSEC-coated inlet and outflow channel enclosing HCs, KCs, and hepatic stellate cells in a hydrogel channel. Reproduced with permission from ref. 138. Copyright 2020, Wolters Kluwer Health, Inc. (D) Vascularized hepatic model to study flow-dependent paracrine regeneration signals in response to cytokine and IL1(β) stimulation. Reproduced with permission from ref. 35. Copyright 2022, PNAS. | ||
3.2. Organ-on-a-chip devices
Organ-on-a-chip devices are more complex systems but have higher physiological relevance due to the following reasons: they can integrate multiple cell types achieve organ level and/or multi-organ level interactions while providing compartmentalization for each cell/tissue type that allows for individual assessment; physical cues such as shear stress can be integrated and controlled in these devices; in situ assessment is feasible, as they are miniature devices and have throughput and small-scale benefits.139Gori et al. designed a microfluidic platform for simulating non-alcoholic fatty liver disease (NAFLD), featuring a central culture chamber for HepG2/C3A seeding (Fig. 2B). This chamber was separated from the medium chamber by an array of 2-μm-wide microchannels, mimicking the endothelial barrier of the liver sinusoid to facilitate continuous nutrient diffusion and waste removal. NAFLD was induced via treatment with free fatty acids (FFAs) such as palmitic and oleic acids. The authors observed a gradual and chronic intracellular triglyceride accumulation in the microfluidic device, in contrast to the more acute triglyceride overload observed in the static culture. Moreover, a 48-hour FFA treatment resulted in reduced viability in static cultures, whereas hepatocytes in the microfluidic chip exhibited greater tolerance to the treatment.137 Similarly, Kostrzewski and colleagues replicated the NAFLD condition using a commercial apparatus, namely the LiverChip® platform developed by CN Bio Innovations in the UK. This platform comprises 12 individual bioreactors wherein the fluid is circulated through a collagen-coated scaffold by a pneumatically driven micro-pump (1 μL s−1). This setup facilitates the creation of 3D microtissues within the scaffold channels. Primary human hepatocytes (PHHs) were cultured in either a lean or fat medium (supplemented with 600 μmol L−1 FFA), and the progression of steatosis in the fat medium was observed and confirmed through oil-red O staining. These findings underscore the effectiveness of a 3D in vitro NAFLD model for drug screening, highlighting its potential for identifying compounds capable of preventing or reversing hepatic steatosis.140
Feaver et al. developed a shear device to mimic the hepatic sinusoidal flow. PHH, primary human macrophages and primary hepatic stellate cells were co-cultured on the device and NAFLD was induced using conditions that mimic human NASH. Using a multiomics approach the authors showed that the model faithfully mimicked the NASH patient tissues in vivo, which convincingly proves the applicability of this model for mechanistic studies and drug testing applications.141 The goal of in vitro models is to mimic tissues in vivo and this is one of the first models to demonstrate comparison with clinical data. More recently Freag et al. developed a NASH-on-a-chip model via the co-culture of four main liver cells, namely hepatocytes, KCs, LSEC and hepatic stellate cells, from primary human tissues (Fig. 2C). By inducing NASH, they were able to observe intracellular lipid accumulation, hepatocellular ballooning, inflammation and fibrosis in their NASH-on-a-chip model and demonstrated its drug testing capabilities.138
3.3. Organoid models for other chronic liver diseases
Wang et al. developed human expandable hepatic organoids (hEHOs) from human embryonic stem cells. These hEHOs had stable bipotent hepatoblast like progenitors, could be expanded for more than 20 passages and could be differentiated into both functional hepatic or cholangiocyte organoids. When transplanted into the injured livers of immunocompromised mice, these organoids could differentiate into mature hepatocytes and improve liver functions. By incorporating human fetal liver mesenchymal cells (hFLMCs) and treatment with ethanol, they could recapitulate the pathogenesis associated with ALD, particularly inflammation as shown by the elevated pro-inflammatory signaling of interleukin-1 (IL-1) and interleukins-17 (IL-17).142 The excellent differentiation and maturation potential of hEHOs could be extremely useful for studying disease mechanisms; however, the ethical implications still need to be deliberated. Akbari et al. reported generation of hepatic organoids from iPSCs using EpCAM positive endodermal cells as intermediates (eHEPO). The eHEPO organoids could be generated in 2 weeks and the authors showed that they could be expanded for more than a year. Using patient-specific iPSCs, they could develop organoids with mutations in the argininosuccinate synthetase (ASS1) enzyme to model citrullinemia. They could also reverse the disease by overexpression of the wild type gene.143 Such models with long-term expansion capabilities and genetic manipulation open an arena of opportunities for modelling chronic liver diseases as they can be monitored for slow changes in the disease phenotype and provide excellent opportunities for transplantation to supplement hepatocyte functions.4. 3D bioprinting
3D bioprinting is an advanced form of additive manufacturing (AM) that constructs tissues or organs layer by layer, following a bottom-up approach. By depositing cells and matrices in an automated precise manner mimicking the arrangement in vivo, the complex structure and functions of any tissue can be replicated.144 The intricate features of the tissue can be conceptualized using computer-aided design (CAD) software or extracted from medical images, such as magnetic resonance imaging (MRI) or computed tomography (CT) scans.145–147 This technology deals with living entities, such as cells and tissues, necessitating consideration of factors like material biocompatibility, cell sensitivity to printing methods, growth factor delivery, and perfusion. The automated nature of the process allows for precise cell patterning and controlled extracellular matrix (ECM) organization. The layer-by-layer construction of bio-printed tissues results in interconnected pores, providing an optimal environment for the perfusion of gases and nutrients and facilitating inter- and intra-cellular communication.146 These bio-printed tissues with enhanced intercellular communication can serve as valuable references for understanding in vivo physiology, particularly since animal models often fall short in predicting human pathophysiological responses.144Various 3D bioprinting methods, such as laser-assisted printing, inkjet printing, and extrusion-based printing, have been developed. Among these, both extrusion-based and inkjet printing methods involve the use of scaffolds as the supporting structures,145,146 with extrusion-based printing being the most commonly utilized in liver-related 3D printing studies.147
4.1. Bio-inks
They comprise two essential components: biomaterials and cells. In the actual printing process, these constituents are blended and printed in varying proportions and densities, undergoing further processing based on the applied printing method.147 The bio-inks serve the dual purpose of safeguarding cells from the external environment during printing and creating a microenvironment that mimics tissue conditions.148 The development of bio-inks places emphasis on printability, biocompatibility, and mechanical properties as crucial considerations.147 Bioinks offer the added benefits of tunable functionalities, including stimuli responsiveness and programmable properties.145 Once 3D analogues are printed using bio-inks, they undergo stabilization through thermal, ionic, chemical, or a combination of crosslinking methods.148 Alginate, GelMA, gelatin, collagen, and dECM, among others, are commonly employed as bioinks and use of CaCl2 and LAP + UV, temperature control, etc. are commonly used curing methods.147 Cell and matrix selection is a crucial aspect of bioink development, which is contingent upon the model's specific requirements, influencing various parameters in the bioink selection process. In vitro liver models encompass primary hepatocytes, hepatic cell lines derived from tumors or liver slices, and hepatic cells derived from stem cells.146 As explained in Section 2, various parenchymal and non-parenchymal cells have specific roles in NASH pathogenesis and the choice of cell types depends on the primary question to be addressed using the model.4.2. 3D Bioprinted models
Maji et al. developed a perfusable vascularized liver sinusoid model on a chip using a one-step bioprinting technique. They used a co-axial bioprinting method to construct a co-culture of hepatocytes and endothelial cells to resemble a liver sinusoid. It consisted of a central compartment housing a perfusable pre-vascular structure made with HUVEC cells, functioning as a sacrificial core, and an outer compartment composed of alginate and collagen accommodating HepG2 cells (Fig. 3C). This innovative perfusable 3D liver sinusoid-on-a-chip (LSOC-P) demonstrated significantly improved viability, proliferation, and the expression of liver-specific genes and proteins in hepatocytes, in comparison to a 3D model with a hepatocyte-based core/shell structure under static conditions and the conventional 2D sandwich culture system.149 Kizawa et al. 3D bioprinted spheroids with human primary hepatocytes and mouse fibroblasts on a needle array and placed it in a perfusion chamber for 4 days, which led to spheroid fusion and formation of a tissue-like construct with duct-like and sinusoid-like structures. The liver constructs could maintain their hepatic functions such as drug metabolism for more than 7 weeks.150 Primary human hepatocytes are conventionally cultured in a collagen sandwich configuration, which preserves their functions for less than a week. Such 3D bioprinted liver tissue constructs could be valuable for screening of drugs. Hiller et al. 3D printed optimized bio-inks comprising alginate, gelatin, and dECM using a pneumatic extrusion bioprinter (Fig. 3A). The HepaRG liver cells cultured on the bioprinted matrix maintained their viability and functionality and this construct was amenable to transduction with adeno-associated virus (AAV). Thus, the resulting bioprinted model was determined to be suitable for virus-assisted chronic liver diseases.151 Hong et al. showed that several microtissue spheroids can be produced using the bioprinting technique, which combines a microfluidic emulsification device and precursor cartridge having a coaxial nozzle with two different bioink compartments (Fig. 3B). These spheroids have a biomimetic structure that mimics the lobules of the liver, where endothelial (C3A) and hepatic cells (HepG2) are patterned. Compared to unstructured spheroids, they retain structural integrity better and allow for extended periods of high cell viability cultivation. Moreover, the liver markers MRP2, albumin, and CD31 are expressed at higher levels in the structured spheroids and the stable transplantation of structured microtissue spheroids was confirmed by in vivo experiments.152 | ||
| Fig. 3 3D Bioprinted liver models. (A) A microextrusion-based bioprinted model made up of mature HepaRG cells, alginate, gelatin, and human extracellular matrix. Reproduced with permission from ref. 151. Copyright 2018, MDPI. (B) Coaxially structured HepG2/C3A spheroids created using pre-set extrusion bioprinting and a microfluidic emulsification technology. Reproduced with permission from ref. 152. Copyright 2021, John Wiley and Sons. (C) Extrusion-based hollow bioprinted model consisting of 2 bioinks made via a coaxial nozzle such that the inner/core bioinks flow out to create lumen. Reproduced with permission from ref. 149. Copyright 2023, Elsevier. (D) A native liver lobule-like structured, extrusion-based bioprinted model incorporating two bioinks: HLC hepatocyte-like cells and HUVEC/HHSC human umbilical vascular endothelial cells/human hepatic stellate cells. Reproduced with permission from ref. 153. Copyright 2022, ACS Publication. | ||
Janani et al. bioprinted a human vascularized liver model. They developed two distinct bio-inks, parenchymal and non-parenchymal cells. They differentiated adipose-derived mesenchymal stem cells into hepatocyte-like cells (HLCs) and printed them alongside HUVECs and hepatic stellate cells into an alternating hexagonal pattern to resemble the chords of hepatocytes in the hepatic lobule (Fig. 3D). The bioprinted liver model developed a functional sinusoid lumen-like network and exhibited mature hepatic functions such as albumin production, urea production and cytochrome enzyme activity. They tested for hepatotoxic drugs and the model exhibited excellent drug testing capabilities. Such a platform could serve as an ideal physiological system for drug testing.153
Apart from the above-mentioned models, 3D bioprinted models are available commercially. Organovo® has bioprinted liver tissues with primary human hepatocytes co-cultured with liver endothelial cells, hepatic stellate cells, and HUVECs. Such engineered constructs exhibit mature hepatic functions and are amenable to implantation in animals.154
5. Building in vitro models for regeneration
As highlighted in Sections 3 and 4, the current in vitro liver models exhibit mature hepatic functions and can recapitulate the essential features of NASH such as steatosis, hepatocellular injury, inflammation, and fibrosis. While these might be important for drug testing applications, we are missing an essential feature of NASH progression, that is regeneration. As explained in the Introduction, the decline in regenerative potential of hepatocytes and the dysregulation of the regenerative pathways are major causes for the progression of disease and the development of irreversible fibrosis.14 Currently liver regeneration is looked mainly from the angle of PHx. In PHx, the regeneration occurs in remnant hepatocytes supported by normal non-parenchymal cells and the ECM. However, in CLDs like NASH regeneration of hepatocytes occurs in the presence of damaged hepatocytes, activated non-parenchymal cells, and more importantly the altered extracellular matrix. Each of these can affect regeneration of hepatocytes, which has to be studied in detail using experimental in vitro organotypic models. Most of the studies for regeneration in NASH are histopathological studies correlated with clinical outcomes.99 Although these models have provided great insights, we need in vitro organotypic models that recapitulate regenerative conditions in CLDs.An ideal model for regeneration should comprise a normal/diseased adult liver tissue, an insult with measurable damage, and withdrawal of the insult to measure recovery. The scheme for such a model is shown in Fig. 4. The primary mechanism of hepatocyte regeneration is via proliferation and only when the proliferation is blocked the hepatic progenitor cells (HPCs) are activated. Therefore, the hepatocytes in a normal liver construct should be capable of self-renewal and this proliferative capability and recovery can be studied when there is a controlled damage. The simplest way to mimic PHx in vitro on monolayers could be scratch injury to physically remove a part of the cell layer. In 3D cultures, controlled liver injury can be produced using physical methods such as laser ablation and radiofrequency ablation. Pharmacological agents such as acetaminophen, thioacetamide, ethanol and carbon tetrachloride could be used at different doses to assess the effects of these compounds on the differentiated tissues after washout. The extent of tissue damage could be assessed using the following parameters: expression of cell damage markers (activation of cellular caspases, flow-cytometry using annexin V/propidium iodide staining), apoptosis (esterase-based or ATP-based assay compatible with 3D samples and calcein-AM ethidium homodimer-1 assay (live/dead imaging)), loss of functionality, and loss of tissue integrity. The recovery of the tissues from the damage after washout could be assessed using the following parameters: the rate of proliferation (can be quantified using EdU assay, PicoGreen assay or cell nuclei staining via DAPI and Ki-67 immunostaining); restoration of cellular functionality (enzymatic cytochrome activity, albumin and urea production for hepatocytes, alkaline phosphatase activity and secretory functions of cholangiocytes) and the restoration of structural features and integrity of the tissues (hepatic canalicular network and biliary ducts). The spatial arrangement of newly produced cells and tissue integrity can be analysed via confocal Z-stack imaging and multiphoton microscopy and can be quantitively analysed using image analysis software programs like ImageJ, Imaris or CellProfiler. The kinetics of cellular, structural and functional recovery have to be assessed. The activation (exit from cellular quiescence) and proliferation of hepatocytes and the stem/progenitor cells at different stages of recovery have to be assessed for low- and high-dose injuries. This should be compared against the baseline levels to assess the extent of activation and proliferation of these cells in response to tissue injury. The ability of stem/progenitor cells to give rise to functional parenchymal cells can be evaluated using the labeling and dye-tracing method and compared to the baseline levels.
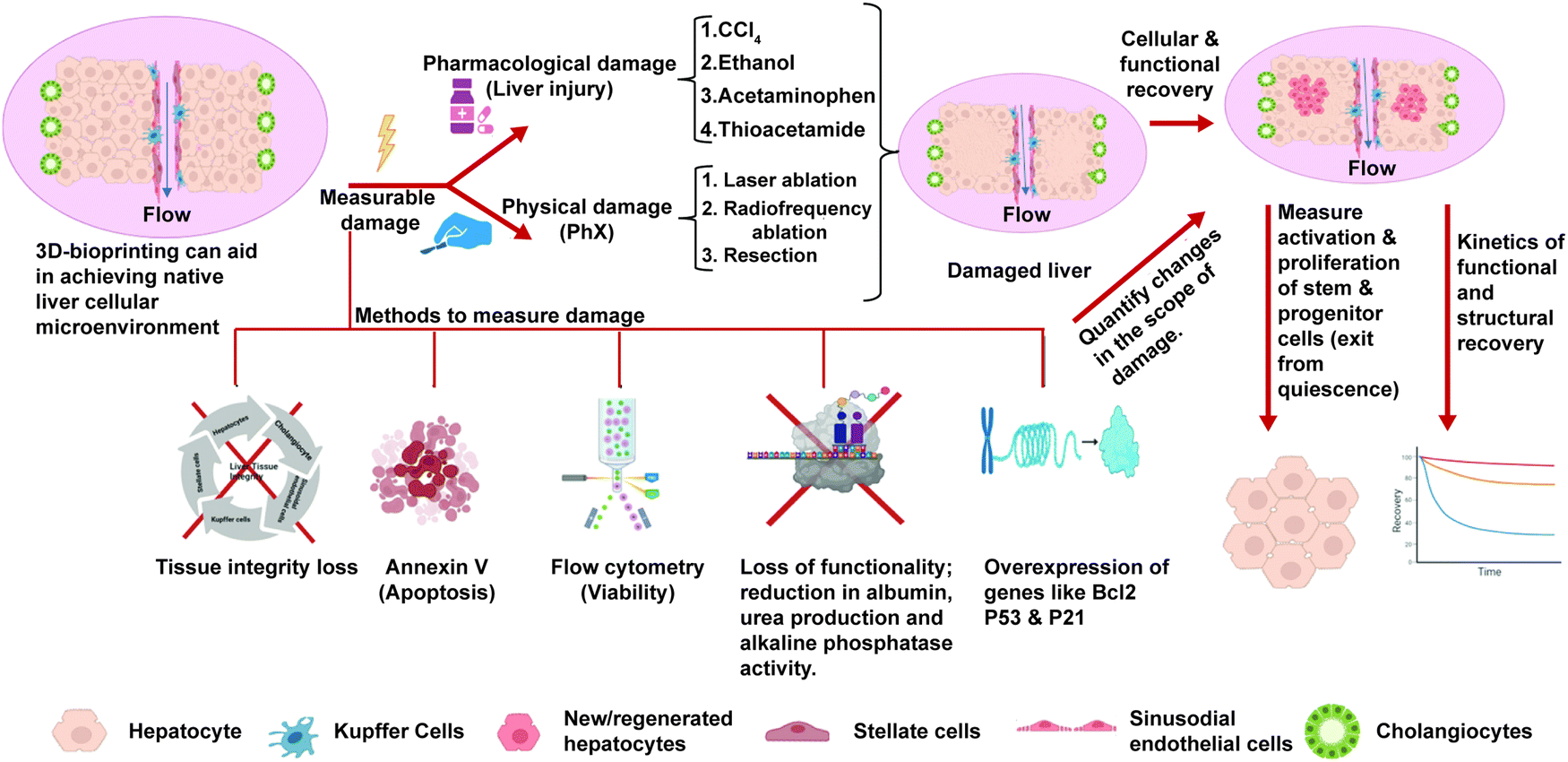 | ||
| Fig. 4 Ideal 3D bioprinted organotypic liver model for studying regeneration. The bioprinted model should have parenchymal and non-parenchymal cells in tissue-like cellular arrangements with molecular gradients and flow to exhibit mature hepatic functions. Controlled damage can be induced via physical and pharmacological insults and the extent of damage has to be measured. Once the insult is withdrawn, the regeneration and recovery of the tissue integrity, cellular composition and hepatic functions must be measured to assess the regenerative capabilities of the tissue. A similar approach can be used for organotypic NASH liver tissues to assess the regenerative capacity, study the molecular mechanisms and test drugs that affect regeneration. | ||
Studying the recovery of normal hepatocytes cells under controlled injury could be useful to understand regeneration in healthy hepatocytes. In such a model, the hepatocytes could be treated with free fatty acids, insulin and other metabolites to mimic NASH. A controlled injury on such an engineered NASH tissue could be used to study how the proliferative capacity of hepatocytes is affected. The severity of NASH could be incremented to understand the stage at which hepatocytes lose their proliferative potential. Once the extent of recovery can be measured, such models could be used to study the mechanisms of altered signalling pathways that mislead regeneration and manipulate these pathways to boost regeneration. Such models would be valuable for testing drugs that worsen tissue damage and screening compounds that can alleviate damage and promote regeneration.
Another critical aspect of regeneration is ECM remodeling. Regenerative mechanisms such as ductular reactions in CLDs misfire and fuel fibrosis.17 Non-resolving and progressive fibrosis is one of the main co-pathogenic mechanisms in CLD progression and is a critical determinant of mortality in NASH.155 The reciprocal interactions between the ECM and the cells maintain this state and resolution of fibrosis is the most difficult-to-achieve target of NASH therapy.7 Therefore it is important to select an appropriate material as a scaffold and account for ECM remodeling in the in vitro model for liver regeneration. The description of the ECM of normal liver and the changes in CLDs discussed in Sections 2.3.1 and 2.3.2 provide insights into choosing the scaffold material for developing the models to study ECM remodeling in CLDs in vitro. Natural ECMs such as collagens, laminins, fibronectins and their combinations that are present in the liver could be used as they would be a mimic of the native in vivo tissues. However, proteinaceous scaffold materials are expensive, difficult to handle, suffer from batch to batch variation, can themselves impart signaling in cells, and most importantly offer very limited manipulation of their mechanical properties. Synthetic materials do not have these limitations and are extensively used in building CLD models.32 However, when synthetic materials are chosen for chemical and mechanical tailoring, it must be ensured that these synthetic matrices allow matrix deposition and remodeling by the cells on them. Balachander et al. had developed mechanomimetic scaffolds using polycaprolactone (PCL) on which cells were able to deposit their own matrix and mimic tissues in vivo.156,157 They had also shown that electrospun fibrous PCL mats that mimicked the fibrous ECM in tissues allowed fibroblasts to adhere, deposit and remodel the matrix in a parallel patterned manner, which mimicked the aggressive desmoplastic stroma.158 Such matrices with tunable mechanical properties and amenable to ECM deposition and remodeling by the cells cultured on them are good choices as scaffold materials. ECM remodeling in 3D cultures can be assessed via techniques such as immunocytochemistry to stain for the specific ECM proteins and non-staining imaging techniques like second harmonic generation (SHG)159 and scanning electron microscopy160 to reveal the architecture and extent of fibrosis. Optical coherence tomography could be used to assess ECM remodeling in 3D cultures.161
As pathogenesis in CLDs involve BECs and non-parenchymal cells as explained in Section 2, it would be ideal to develop a co-culture model with these main cell types. One of the concerns in co-cultures is suboptimal heterotypic and homotypic interactions leading to phenotypic instability and misrepresentation of tissue properties. This could be avoided by employing 3D bioprinting. 3D bioprinting can be tailored to achieve optimal spatial homotypic and heterotypic interactions. Additionally, 3D printing can create tissue-specific ECM gradients and incorporation of flow can achieve gradients of oxygen, nutrients, and hormones. Incorporation of physical cues such as flow and molecular gradients would vastly increase the phenotypic relevance of such models, although this remains unexplored so far.
We believe that development of such in vitro liver constructs by 3D bioprinting will improve exponentially in the coming years due to advanced ongoing research in biomaterials, bioinks, cell biology and molecular medicine, which would help us uncover the puzzle of poor liver regeneration in NASH.
6. Conclusion
In this review, we aim to show the readers the dangers of increasing CLDs, particularly metabolic diseases such as NASH, and the lacunae in our understanding of the regenerative mechanisms in these diseases. We discuss the limitations of animal models and the need for suitable in vitro models. We discuss in detail how the components required to build an in vitro model, such as the cells, signaling molecules and the matrix, function in a mature liver, their responses in PHx to achieve complete functional recovery, and the deregulation of the same in CLDs, which leads to disease progression and carcinogenesis. We describe in vitro models that have been developed to model CLDs with a special focus on 3D bioprinting as a fabrication method. We explain how an ideal in vitro model could be developed by inciting a controlled injury in a mature liver tissue and measuring the responses and the rate of recovery to assess regeneration. We describe how 3D bioprinting is suited to build such models and how these models could be used to study molecular mechanisms and test drugs. The highly inter-disciplinary nature of this review has restricted us from providing detailed discussion on several interesting topics for which we have provided references wherein they are discussed in detail. We aim to introduce this novel concept to researchers and to steer them to develop meaningful in vitro models for liver regeneration.Data availability statement
This is a review article and we do not have any data availability report.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the Science and Engineering Research Board (SERB), Government of India (SRG/2023/001183) is gratefully acknowledged.References
- Y. Lee, Y. Cho, B.-W. Lee, C.-Y. Park, D. H. Lee, B.-S. Cha and E.-J. Rhee, Nonalcoholic Fatty Liver Disease in Diabetes. Part I: Epidemiology and Diagnosis, Diabetes, Metab. J., 2019, 43, 31, DOI:10.4093/dmj.2019.0011.
- S. Milić and D. Štimac, Nonalcoholic Fatty Liver Disease/Steatohepatitis: Epidemiology, Pathogenesis, Clinical Presentation and Treatment, Dig. Dis., 2012, 30, 158–162, DOI:10.1159/000336669.
- J. Maurice and P. Manousou, Non-alcoholic fatty liver disease, Clin. Med., 2018, 18, 245–250, DOI:10.7861/clinmedicine.18-3-245.
- P. Paschos and K. Paletas, Non alcoholic fatty liver disease and metabolic syndrome, Hippokratia, 2009, 13, 9–19 CAS.
- E. Dhamija, S. B. Paul and S. Kedia, Non-alcoholic fatty liver disease associated with hepatocellular carcinoma: An increasing concern, Indian J. Med. Res., 2019, 149, 9–17, DOI:10.4103/ijmr.IJMR_1456_17.
- A. M. Diehl and C. Day, Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis, N. Engl. J. Med., 2017, 377, 2063–2072, DOI:10.1056/NEJMra1503519.
- J. M. Fraile, S. Palliyil, C. Barelle, A. J. Porter and M. Kovaleva, Non-Alcoholic Steatohepatitis (NASH) - A Review of a Crowded Clinical Landscape, Driven by a Complex Disease, Drug Des., Dev. Ther., 2021, 15, 3997–4009, DOI:10.2147/DDDT.S315724.
- A. C. Sheka, O. Adeyi, J. Thompson, B. Hameed, P. A. Crawford and S. Ikramuddin, Nonalcoholic Steatohepatitis: A Review, JAMA, J. Am. Med. Assoc., 2020, 323, 1175–1183, DOI:10.1001/jama.2020.2298.
- T. Ueno, H. Sugawara, K. Sujaku, O. Hashimoto, R. Tsuji, S. Tamaki, T. Torimura, S. Inuzuka, M. Sata and K. Tanikawa, Therapeutic effects of restricted diet and exercise in obese patients with fatty liver, J. Hepatol., 1997, 27, 103–107, DOI:10.1016/S0168-8278(97)80287-5.
- G. Musso, M. Cassader, F. Rosina and R. Gambino, Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials, Diabetologia, 2012, 55, 885–904, DOI:10.1007/s00125-011-2446-4.
- S. A. M. Albhaisi and A. J. Sanyal, New drugs for NASH, Liver Int., 2021, 41, 112–118, DOI:10.1111/liv.14844.
- S. A. Harrison, P. Bedossa, C. D. Guy, J. M. Schattenberg, R. Loomba, R. Taub, D. Labriola, S. E. Moussa, G. W. Neff, M. E. Rinella, Q. M. Anstee, M. F. Abdelmalek, Z. Younossi, S. J. Baum, S. Francque, M. R. Charlton, P. N. Newsome, N. Lanthier, I. Schiefke, A. Mangia, J. M. Pericàs, R. Patil, A. J. Sanyal, M. Noureddin, M. B. Bansal, N. Alkhouri, L. Castera, M. Rudraraju and V. Ratziu, A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis, N. Engl. J. Med., 2024, 390, 497–509, DOI:10.1056/NEJMoa2309000.
- FDA Approves First Treatment for Patients with Liver Scarring Due to Fatty Liver Disease, (n.d.). https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-patients-liver-scarring-due-fatty-liver-disease (accessed June 13, 2024).
- C. Zhu, I. Tabas, R. F. Schwabe and U. B. Pajvani, Maladaptive regeneration—the reawakening of developmental pathways in NASH and fibrosis, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 131–142, DOI:10.1038/s41575-020-00365-6.
- A. Bria, J. Marda, J. Zhou, X. Sun, Q. Cao, B. E. Petersen and L. Pi, Hepatic progenitor cell activation in liver repair, Liver Res., 2017, 1, 81–87, DOI:10.1016/j.livres.2017.08.002.
- V. L. Gadd, R. Skoien, E. E. Powell, K. J. Fagan, C. Winterford, L. Horsfall, K. Irvine and A. D. Clouston, The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease, Hepatology, 2014, 59, 1393–1405, DOI:10.1002/hep.26937.
- M. M. Richardson, J. R. Jonsson, E. E. Powell, E. M. Brunt, B. A. Neuschwander-Tetri, P. S. Bhathal, J. B. Dixon, M. D. Weltman, H. Tilg, A. R. Moschen, D. M. Purdie, A. J. Demetris and A. D. Clouston, Progressive Fibrosis in Nonalcoholic Steatohepatitis: Association With Altered Regeneration and a Ductular Reaction, Gastroenterology, 2007, 133, 80–90, DOI:10.1053/j.gastro.2007.05.012.
- T. Roskams, Progenitor cell involvement in cirrhotic human liver diseases: from controversy to consensus, J. Hepatol., 2003, 39, 431–434, DOI:10.1016/S0168-8278(03)00333-7.
- D. Palmes and H. U. Spiegel, Animal models of liver regeneration, Biomaterials, 2004, 25, 1601–1611, DOI:10.1016/S0142-9612(03)00508-8.
- G. K. Michalopoulos and M. DeFrances, Liver Regener., 2005, 101–134, DOI:10.1007/b99968.
- D. J. Ingle and B. L. Baker, Histology and Regenerative Capacity of Liver Following Multiple Partial Hepatectomies, Exp. Biol. Med., 1957, 95, 813–815, DOI:10.3181/00379727-95-23371.
- N. Fausto, J. S. Campbell and K. J. Riehle, Liver regeneration, Hepatology, 2006, 43(2 Suppl 1), 45–53, DOI:10.1002/hep.20969.
- G. K. Michalopoulos, Liver regeneration, J. Cell. Physiol., 2007, 213, 286–300, DOI:10.1002/jcp.21172.
- S. Kaur, H. Siddiqui and M. H. Bhat, Hepatic Progenitor Cells in Action Liver Regeneration or Fibrosis?, Am. J. Pathol., 2015, 185, 2342–2350, DOI:10.1016/j.ajpath.2015.06.004.
- C. Zhu, I. Tabas, R. F. Schwabe and U. B. Pajvani, Maladaptive regeneration—the reawakening of developmental pathways in NASH and fibrosis, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 131–142, DOI:10.1038/s41575-020-00365-6.
- A. M. Diehl, Lessons from animal models of NASH, Hepatol. Res., 2005, 33, 138–144, DOI:10.1016/j.hepres.2005.09.022.
- A. Cast, M. Kumbaji, A. D. Souza, K. Rodriguez, A. Gupta, R. Karns, L. Timchenko and N. Timchenko, Liver Proliferation Is an Essential Driver of Fibrosis in Mouse Models of Nonalcoholic Fatty Liver Disease, Hepatol. Commun., 2019, 3, 1036–1049, DOI:10.1002/hep4.1381/suppinfo.
- H. H. Hansen, M. Feigh, S. S. Veidal, K. T. Rigbolt, N. Vrang and K. Fosgerau, Mouse models of nonalcoholic steatohepatitis in preclinical drug development, Drug Discovery Today, 2017, 22, 1707–1718, DOI:10.1016/j.drudis.2017.06.007.
- P. K. Santhekadur, D. P. Kumar and A. J. Sanyal, Preclinical models of non-alcoholic fatty liver disease, J. Hepatol., 2018, 68, 230–237, DOI:10.1016/j.jhep.2017.10.031.
- D. H. Ipsen, J. Lykkesfeldt and P. Tveden-Nyborg, Animal Models of Fibrosis in Nonalcoholic Steatohepatitis: Do They Reflect Human Disease, Adv. Nutr., 2020, 11, 1696–1711, DOI:10.1093/advances/nmaa081.
- C. Z. Larter and M. M. Yeh, Animal models of NASH: Getting both pathology and metabolic context right, J. Gastroenterol. Hepatol., 2008, 23, 1635–1648, DOI:10.1111/j.1440-1746.2008.05543.x.
- A. Paradiso, M. Volpi, C. Rinoldi, N. Celikkin, N. Contessi Negrini, M. Bilgen, G. Dallera, F. Pierini, M. Costantini, W. Święszkowski and S. Farè, In vitro functional models for human liver diseases and drug screening: beyond animal testing, Biomater. Sci., 2022, 11, 2988–3015, 10.1039/d1bm01872h.
- S. C. L. Sanches, L. N. Z. Ramalho, M. J. Augusto, D. M. Da Silva and F. S. Ramalho, Nonalcoholic Steatohepatitis: A Search for Factual Animal Models, BioMed Res. Int., 2015, 574832, DOI:10.1155/2015/574832.
- A. Gough, A. Soto-Gutierrez, L. Vernetti, M. R. Ebrahimkhani, A. M. Stern and D. L. Taylor, Human biomimetic liver microphysiology systems in drug development and precision medicine, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 252–268, DOI:10.1038/s41575-020-00386-1.
- A. Chhabra, H.-H. Greco Song, K. A. Grzelak, W. J. Polacheck, H. E. Fleming, C. S. Chen, S. N. Bhatia, S. Duncan and N. Parashurama, A vascularized model of the human liver mimics regenerative responses, ( 2022) Search PubMed.
- H. K. Kleinman, D. Philp and M. P. Hoffman, Role of the extracellular matrix in morphogenesis, Curr. Opin. Biotechnol, 2003, 14, 526–532, DOI:10.1016/j.copbio.2003.08.002.
- M. J. Bissell, D. C. Radisky, A. Rizki, V. M. Weaver and O. W. Petersen, The organizing principle: Microenvironmental influences in the normal and malignant breast, Differentiation, 2002, 70, 537–546, DOI:10.1046/j.1432-0436.2002.700907.x.
- Air–liquid interface Growth medium Apoptosis/anoikis Aggregation Polarization Differentiation, 2007. https://www.nature.com/reviews/molcellbio.
- E. Knight and S. Przyborski, Advances in 3D cell culture technologies enabling tissue-like structures to be created in vitro, J. Anat., 2015, 227, 746–756, DOI:10.1111/joa.12257.
- L. Cordero-Espinoza and M. Huch, The balancing act of the liver: tissue regeneration versus fibrosis, J. Clin. Invest., 2018, 128, 85–96, DOI:10.1172/JCI93562.
- A. Chaudhuri, Embryonic development of liver and its future implications, in Human Fetal Growth and Development: First and Second Trimesters, Springer International Publishing, 2016, pp. 331–345 DOI:10.1007/978-3-319-14874-8_26.
- G. K. Michalopoulos, Liver regeneration, J. Cell. Physiol., 2007, 213, 286–300, DOI:10.1002/jcp.21172.
- P. Bedossa and V. Paradis, Liver extracellular matrix in health and disease, J. Pathol., 2003, 200, 504–515, DOI:10.1002/path.1397.
- N. Li and J. Hua, Immune cells in liver regeneration, Oncotarget, 2017, 8, 3628–3639, DOI:10.18632/oncotarget.12275.
- I. N. Crispe, Hepatic T cells and liver tolerance, Nat. Rev. Immunol., 2003, 3, 51–62, DOI:10.1038/nri981.
- A. Castellaneta, A. Di Leo, R. Francavilla, M. Margiotta, M. Barone, A. Amoruso, L. Troiani, A. W. Thomson and A. Francavilla, Functional modification of CD11c+ liver dendritic cells during liver regeneration after partial hepatectomy in mice, Hepatology, 2006, 43, 807–816, DOI:10.1002/hep.21098.
- M. Minagawa, H. Oya, S. Yamamoto, T. Shimizu, M. Bannai, H. Kawamura, K. Hatakeyama and T. Abo, Intensive expansion of natural killer T cells in the early phase of hepatocyte regeneration after partial hepatectomy in mice and its association with sympathetic nerve activation, Hepatology, 2000, 31, 907–915, DOI:10.1053/he.2000.5850.
- L. M. Polimeno Margiotta, L. Marangi, T. Lisowskyl, A. Azzarone, E. Lerardi MA Frassanito and A. Francavilla Flwm, Molecular mechanisms of augmenter of liver regeneration as immunoregulator: its effect on interferon-y expression in rat liver, 2000.
- F. Tamura, A. Masuhara, I. Sakaida, E. T. S. U. O. Fukumoto and T. Nakamura A N D, K. Okita, FK506 promotes liver regeneration by suppressing natural killer cell activity, n.d.
- B. V. Martin-Murphy, D. J. Kominsky, D. J. Orlicky, T. M. Donohue and C. Ju, Increased susceptibility of natural killer T-cell-deficient mice to acetaminophen-induced liver injury, Hepatology, 2013, 57, 1575–1584, DOI:10.1002/hep.26134.
- F. Fasbender, A. Widera, J. G. Hengstler and C. Watzl, Natural killer cells and liver fibrosis, Front. Immunol., 2016, 7, 19, DOI:10.3389/fimmu.2016.00019.
- S. Yagi, M. Hirata, Y. Miyachi and S. Uemoto, Liver regeneration after hepatectomy and partial liver transplantation, Int. J. Mol. Sci., 2020, 21(21), 8414, DOI:10.3390/ijms21218414.
- M. M. Richardson, J. R. Jonsson, E. E. Powell, E. M. Brunt, B. A. Neuschwander-Tetri, P. S. Bhathal, J. B. Dixon, M. D. Weltman, H. Tilg, A. R. Moschen, D. M. Purdie, A. J. Demetris and A. D. Clouston, Progressive Fibrosis in Nonalcoholic Steatohepatitis: Association With Altered Regeneration and a Ductular Reaction, Gastroenterology, 2007, 133, 80–90, DOI:10.1053/j.gastro.2007.05.012.
- R. Saxena and N. Theise, Canals of Hearing: Recent Insights and Current Knowledge, Semin. Liver Dis., 2004, 24, 43–48, DOI:10.1055/s-2004-823100.
- J. Chen, L. Chen, M. A. Zern, N. D. Theise, A. M. Diehl, P. Liu and Y. Duan, The diversity and plasticity of adult hepatic progenitor cells and their niche, Liver Int., 2017, 37, 1260–1271, DOI:10.1111/liv.13377.
- G. K. Michalopoulos and B. Bhushan, Liver regeneration: biological and pathological mechanisms and implications, Nat. Rev. Gastroenterol. Hepatol., 2021, 18, 40–55, DOI:10.1038/s41575-020-0342-4.
- A. Baiocchini, C. Montaldo, A. Conigliaro, A. Grimaldi, V. Correani, F. Mura, F. Ciccosanti, N. Rotiroti, A. Brenna, M. Montalbano, G. D’Offizi, M. R. Capobianchi, R. Alessandro, M. Piacentini, M. E. Schininà, B. Maras, F. Del Nonno, M. Tripodi and C. Mancone, Extracellular matrix molecular remodeling in human liver fibrosis evolution, PLoS One, 2016, 11(3), e0151736, DOI:10.1371/journal.pone.0151736.
- N. Fausto, J. S. Campbell and K. J. Riehle, Liver regeneration, J. Hepatol., 2012, 57, 692–694, DOI:10.1016/j.jhep.2012.04.016.
- G. Kolios, V. Valatas and E. Kouroumalis, Role of Kupffer cells in the pathogenesis of liver disease, 2006. https://www.wjgnet.com1007-9327/12/7413.asp.
- S. L. Friedman, F. J. Roll, J. Boules, D. M. Arenson and D. M. Bissel, Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix, J. Biol. Chem., 1989, 264, 10756–10762, DOI:10.1016/s0021-9258(18)81686-6.
- N. Faust0 and N. Fausto, Liver regeneration, J. Hepotol., 2000, 32, 19–31 CrossRef PubMed.
- T. Higashi, S. L. Friedman and Y. Hoshida, Hepatic stellate cells as key target in liver fibrosis, Adv. Drug Delivery Rev., 2017, 121, 27–42, DOI:10.1016/j.addr.2017.05.007.
- S. A. Mao, J. M. Glorioso and S. L. Nyberg, Liver regeneration, Transl. Res., 2014, 163, 352–362, DOI:10.1016/j.trsl.2014.01.005.
- M. Klaas, T. Kangur, J. Viil, K. Mäemets-Allas, A. Minajeva, K. Vadi, M. Antsov, N. Lapidus, M. Järvekülg and V. Jaks, The alterations in the extracellular matrix composition guide the repair of damaged liver tissue, Sci. Rep., 2016, 6, 27398, DOI:10.1038/srep27398.
- A. Hammoutene and P. E. Rautou, Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease, J. Hepatol., 2019, 70, 1278–1291, DOI:10.1016/j.jhep.2019.02.012.
- I. Kaur, A. Vasudevan, P. Rawal, D. M. Tripathi, S. Ramakrishna, S. Kaur and S. K. Sarin, Primary Hepatocyte Isolation and Cultures: Technical Aspects, Challenges and Advancements, Bioengineering, 2023, 10, 131, DOI:10.3390/bioengineering10020131.
- S. Berardis, C. Lombard, J. Evraerts, A. El Taghdouini, V. Rosseels, P. Sancho-Bru, J. J. Lozano, L. van Grunsven, E. Sokal and M. Najimi, Gene Expression Profiling and Secretome Analysis Differentiate Adult-Derived Human Liver Stem/Progenitor Cells and Human Hepatic Stellate Cells, PLoS One, 2014, 9, e86137, DOI:10.1371/journal.pone.0086137.
- E. Pfeiffer, V. Kegel, K. Zeilinger, J. G. Hengstler, A. K. Nüssler, D. Seehofer and G. Damm, Featured Article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells, Exp. Biol. Med., 2015, 240, 645–656, DOI:10.1177/1535370214558025.
- M. Werner, S. Driftmann, K. Kleinehr, G. M. Kaiser, Z. Mathé, J.-W. Treckmann, A. Paul, K. Skibbe, J. Timm, A. Canbay, G. Gerken, J. F. Schlaak and R. Broering, All-In-One: Advanced preparation of Human Parenchymal and Non-Parenchymal Liver Cells, PLoS One, 2015, 10, e0138655, DOI:10.1371/journal.pone.0138655.
- W. Dong, A. Lu, J. Zhao, S. Yin, B. Ou and H. Feng, An efficient and simple co-culture method for isolating primary human hepatic cells: Potential application for tumor microenvironment research, Oncol. Rep., 2016, 36, 2126–2134, DOI:10.3892/or.2016.4979.
- V. Kegel, D. Deharde, E. Pfeiffer, K. Zeilinger, D. Seehofer and G. Damm, Protocol for Isolation of Primary Human Hepatocytes and Corresponding Major Populations of Non-parenchymal Liver Cells, J. Visualized Exp., 2016, 19, e53069, DOI:10.3791/53069.
- F. P. Lemaigre, Mechanisms of Liver Development: Concepts for Understanding Liver Disorders and Design of Novel Therapies, Gastroenterology, 2009, 137, 62–79, DOI:10.1053/j.gastro.2009.03.035.
- V. A. McLin and A. M. Zorn, Molecular control of liver development, Clin. Liver Dis., 2006, 10, 1–25, DOI:10.1016/j.cld.2005.10.002.
- A. M. Zorn, Chapters Cellular and nuclear reprogramming Ectoderm specification and differentiation Endoderm specification and differentiation Epigenetics Genomics and proteomics Germ cell and somatic stem cell biology in reproduction Manufacturing Mesoderm specification and differentiation Niche biology, homing, and migration Renewal Stem cell immunology Therapeutic prospects Tissue engineering Liver development, n.d. https://www.stembook.org/node/512.
- C. Yin, K. J. Evason, K. Asahina and D. Y. R. Stainier, Hepatic stellate cells in liver development, regeneration, and cancer, J. Clin. Invest., 2013, 123, 1902–1910, DOI:10.1172/JCI66369.
- H. Nonaka, M. Tanaka, K. Suzuki and A. Miyajima, Development of murine hepatic sinusoidal endothelial cells characterized by the expression of hyaluronan receptors, Dev. Dyn., 2007, 236, 2258–2267, DOI:10.1002/dvdy.21227.
- F. Tasnim, J. Xing, X. Huang, S. Mo, X. Wei, M. H. Tan and H. Yu, Generation of mature kupffer cells from human induced pluripotent stem cells, Biomaterials, 2019, 192, 377–391, DOI:10.1016/j.biomaterials.2018.11.016.
- D. Shin and S. P. S. Monga, Cellular and molecular basis of liver development, Comp. Physiol., 2013, 3, 799–815, DOI:10.1002/cphy.c120022.
- I. H. Park, N. Arora, H. Huo, N. Maherali, T. Ahfeldt, A. Shimamura, M. W. Lensch, C. Cowan, K. Hochedlinger and G. Q. Daley, Disease-Specific Induced Pluripotent Stem Cells, Cell, 2008, 134, 877–886, DOI:10.1016/j.cell.2008.07.041.
- F. Sampaziotis, M. C. De Brito, P. Madrigal, A. Bertero, K. Saeb-Parsy, F. A. C. Soares, E. Schrumpf, E. Melum, T. H. Karlsen, J. A. Bradley, W. T. H. Gelson, S. Davies, A. Baker, A. Kaser, G. J. Alexander, N. R. F. Hannan and L. Vallier, Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation, Nat. Biotechnol., 2015, 33, 845–852, DOI:10.1038/nbt.3275.
- M. Ogawa, S. Ogawa, C. E. Bear, S. Ahmadi, S. Chin, B. Li, M. Grompe, G. Keller, B. M. Kamath and A. Ghanekar, Directed differentiation of cholangiocytes from human pluripotent stem cells, Nat. Biotechnol., 2015, 33, 853–861, DOI:10.1038/nbt.3294.
- B. K. Gage, J. C. Liu, B. T. Innes, S. A. MacParland, I. D. McGilvray, G. D. Bader and G. M. Keller, Generation of Functional Liver Sinusoidal Endothelial Cells from Human Pluripotent Stem-Cell-Derived Venous Angioblasts, Cell Stem Cell, 2020, 27, 254–269, DOI:10.1016/j.stem.2020.06.007.
- J. Vallverdú, R. A. Martínez García de la Torre, I. Mannaerts, S. Verhulst, A. Smout, M. Coll, S. Ariño, T. Rubio-Tomás, B. Aguilar-Bravo, C. Martínez-Sánchez and D. Blaya, Directed differentiation of human induced pluripotent stem cells to hepatic stellate cells, Nat. Protoc., 2021, 16, 2542–2563 CrossRef PubMed.
- E. Arriazu, M. R. De Galarreta, F. J. Cubero, M. Varela-Rey, M. P. P. De Obanos, T. M. Leung, A. Lopategi, A. Benedicto, I. Abraham-Enachescu and N. Nieto, Extracellular matrix and liver disease, Antioxid. Redox Signaling, 2014, 21, 1078–1097, DOI:10.1089/ars.2013.5697.
- F. Böhm, U. A. Köhler, T. Speicher and S. Werner, Regulation of liver regeneration by growth factors and cytokines, EMBO Mol. Med., 2010, 2, 294–305, DOI:10.1002/emmm.201000085.
- L. I. Kang, W. M. Mars and G. K. Michalopoulos, Signals and cells involved in regulating liver regeneration, Cells, 2012, 1, 1261–1292, DOI:10.3390/cells1041261.
- I. V. Kholodenko and K. N. Yarygin, Cellular Mechanisms of Liver Regeneration and Cell-Based Therapies of Liver Diseases, BioMed Res. Int., 2017, 8910821, DOI:10.1155/2017/8910821.
- S. Hora and T. Wuestefeld, Liver Injury and Regeneration: Current Understanding, New Approaches, and Future Perspectives, Cells, 2023, 12(17), 2129, DOI:10.3390/cells12172129.
- D. Schmidt-Arras and S. Rose-John, IL-6 pathway in the liver: From physiopathology to therapy, J. Hepatol., 2016, 64(6), 1403–1415, DOI:10.1016/j.jhep.2016.02.004.
- Y. Kodama, M. Hijikata, R. Kageyama, K. Shimotohno and T. Chiba, The role of notch signaling in the development of intrahepatic bile ducts, Gastroenterology, 2004, 127, 1775–1786, DOI:10.1053/j.gastro.2004.09.004.
- J. Lu, Y. Zhou, T. Hu, H. Zhang, M. Shen, P. Cheng, W. Dai, F. Wang, K. Chen, Y. Zhang, C. Wang, J. Li, Y. Zheng, J. Yang, R. Zhu, J. Wang, W. Lu, H. Zhang, J. Wang, Y. Xia, T. M. De Assuncao, N. Jalan-Sakrikar, R. C. Huebert, B. Zhou and C. Guo, Notch Signaling Coordinates Progenitor Cell-Mediated Biliary Regeneration Following Partial Hepatectomy, Sci. Rep., 2016, 6, 22754, DOI:10.1038/srep22754.
- N. Tanimizu and A. Miyajima, Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors, J. Cell Sci., 2004, 117, 3165–3174, DOI:10.1242/jcs.01169.
- F. Geisler and M. Strazzabosco, Emerging Roles of Notch Signaling in Liver Disease, Hepatology, 2015, 61(1), 382–392, DOI:10.1002/hep.27268.
- C. Morell, R. Fiorotto, L. Fabris and M. Strazzabosco, Notch signalling beyond liver development: emerging concepts in liver repair and oncogenesis, Clin. Res. Hepatol. Gastroenterol., 2013, 37(5), 447–454, DOI:10.1016/j.clinre.2013.05.008.
- C. M. Morell, R. Fiorotto, L. Fabris and M. Strazzabosco, Notch signalling beyond liver development: Emerging concepts in liver repair and oncogenesis, Clin. Res. Hepatol. Gastroenterol., 2013, 37(5), 447–454, DOI:10.1016/j.clinre.2013.05.008.
- M. J. Perugorria, P. Olaizola, I. Labiano, A. Esparza-Baquer, M. Marzioni, J. J. G. Marin, L. Bujanda and J. M. Banales, Wnt–β-catenin signalling in liver development, health and disease, Nat. Rev. Gastroenterol. Hepatol., 2019, 16, 121–136, DOI:10.1038/s41575-018-0075-9.
- T. H. Kim, W. M. Mars, D. B. Stolz, B. E. Petersen and G. K. Michalopoulos, Extracellular matrix remodeling at the early stages of liver regeneration in the rat, Hepatology, 1997, 26, 896–904, DOI:10.1002/hep.510260415.
- J. Behari, The Wnt/β-catenin signaling pathway in liver biology and disease, Expert Rev. Gastroenterol. Hepatol., 2010, 4, 745–756, DOI:10.1586/egh.10.74.
- S. J. Forbes and P. N. Newsome, Liver regeneration-mechanisms and models to clinical application, Nat. Rev. Gastroenterol. Hepatol., 2016, 13, 473–485, DOI:10.1038/nrgastro.2016.97.
- R. F. Schwabe, I. Tabas and U. B. Pajvani, Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis, Gastroenterology, 2020, 158, 1913–1928, DOI:10.1053/j.gastro.2019.11.311.
- J. O. Russell and F. D. Camargo, Hippo signalling in the liver: role in development, regeneration and disease, Nat. Rev. Gastroenterol. Hepatol., 2022, 19(5), 297–312, DOI:10.1038/s41575-021-00571-w.
- N. Wu, Q. Nguyen, Y. Wan, T. Zhou, J. Venter, G. A. Frampton, S. Demorrow, D. Pan, F. Meng, S. Glaser, G. Alpini and H. Bai, The Hippo signaling functions through the Notch signaling to regulate intrahepatic bile duct development in mammals, Lab. Invest., 2017, 97, 843–853, DOI:10.1038/labinvest.2017.29.
- A. R. Kim, J. I. Park, H. T. Oh, K. M. Kim, J. H. Hwang, M. G. Jeong, E. H. Kim, E. S. Hwang and J. H. Hong, TAZ stimulates liver regeneration through interleukin-6–induced hepatocyte proliferation and inhibition of cell death after liver injury, FASEB J., 2019, 33, 5914–5923, DOI:10.1096/fj.201801256RR.
- J. H. Koo and K. L. Guan, Interplay between YAP/TAZ and Metabolism, Cell Metab., 2018, 28, 196–206, DOI:10.1016/j.cmet.2018.07.010.
- D. Yimlamai, B. H. Fowl and F. D. Camargo, Emerging evidence on the role of the Hippo/YAP pathway in liver physiology and cancer, J. Hepatol., 2015, 63, 1491–1501, DOI:10.1016/j.jhep.2015.07.008.
- J. L. Xia, C. Dai, G. K. Michalopoulos and Y. Liu, Hepatocyte growth factor attenuates liver fibrosis induced by bile duct ligation, Am. J. Pathol., 2006, 168, 1500–1512, DOI:10.2353/ajpath.2006.050747.
- E. Y. T. Lau, J. Lo, B. Y. L. Cheng, M. K. F. Ma, J. M. F. Lee, J. K. Y. Ng, S. Chai, C. H. Lin, S. Y. Tsang, S. Ma, I. O. L. Ng and T. K. W. Lee, Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling, Cell Rep., 2016, 15, 1175–1189, DOI:10.1016/j.celrep.2016.04.019.
- Y. Zhao, W. Ye, Y. D. Wang and W. D. Chen, HGF/c-Met: A Key Promoter in Liver Regeneration, Front. Pharmacol., 2022, 13, 808855, DOI:10.3389/fphar.2022.808855.
- J. P. Iredale, A. Thompson and N. C. Henderson, Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation, Biochim. Biophys. Acta, Mol. Basis Dis., 1832, 2013, 876–883, DOI:10.1016/j.bbadis.2012.11.002.
- R. McClelland, E. Wauthier, J. Uronis and L. Reid, Gradients in the Liver's Extracellular Matrix Chemistry from Periportal to Pericentral Zones: Influence on Human Hepatic Progenitors, Tissue Eng., Part A, 2008, 14, 59–70, DOI:10.1089/ten.a.2007.0058.
- T. Kietzmann, Metabolic zonation of the liver: The oxygen gradient revisited, Redox Biol., 2017, 11, 622–630, DOI:10.1016/j.redox.2017.01.012.
- X. Zheng, W. Liu, J. Xiang, P. Liu, M. Ke, B. Wang, R. Wu and Y. Lv, Collagen I promotes hepatocellular carcinoma cell proliferation by regulating integrin β1/FAK signaling pathway in nonalcoholic fatty liver, Oncotarget, 2017, 8, 95586–95595, DOI:10.18632/oncotarget.21525.
- M. Mori, H. Fujii, T. Ogawa, S. Kobayashi, S. Iwai, H. Morikawa, M. Enomoto, A. Tamori, A. Sawada, S. Takeda and N. Kawada, Close correlation of liver stiffness with collagen deposition and presence of myofibroblasts in non-alcoholic fatty liver disease, Hepatol. Res., 2011, 41, 897–903, DOI:10.1111/j.1872-034X.2011.00842.x.
- E. Geervliet and R. Bansal, Matrix metalloproteinases as potential biomarkers and therapeutic targets in liver diseases, Cells, 2020, 9(5), 1212, DOI:10.3390/cells9051212.
- R. F. Schwabe, I. Tabas and U. B. Pajvani, Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis, Gastroenterology, 2020, 158, 1913–1928, DOI:10.1053/j.gastro.2019.11.311.
- A. Mallat and S. Lotersztajn, Cellular Mechanisms of Tissue Fibrosis. 5. Novel insights into liver fibrosis, Am. J. Physiol.: Cell Physiol., 2013, 305, 789–799, DOI:10.1152/ajpcell.00230.2013.-Liver.
- M. J. P. Arthur, Matrix Degradation in the Liver, 1990.
- P. Ruiz-Blázquez, V. Pistorio, M. Fernández-Fernández and A. Moles, The multifaceted role of cathepsins in liver disease, J. Hepatol., 2021, 75, 1192–1202, DOI:10.1016/j.jhep.2021.06.031.
- T. Zuo, Q. Xie, J. Liu, J. Yang, J. Shi, D. Kong, Y. Wang, Z. Zhang, H. Gao, D.-B. Zeng, X. Wang, P. Tao, W. Wei, J. Wang, Y. Li, Q. Long, C. Li, L. Chang, H. Ning, Y. Li, C. Cui, X. Ge, J. Wu, G. Li, X. Hong, X. Yang, E. Dai, F. He, J. Wu, Y. Ruan, S. Lu and P. Xu, Macrophage-Derived Cathepsin S Remodels the Extracellular Matrix to Promote Liver Fibrogenesis, Gastroenterology, 2023, 165, 746–761, DOI:10.1053/j.gastro.2023.05.039.
- M. McConnell and Y. Iwakiri, Biology of portal hypertension, Hepatol. Int., 2018, 12, 11–23, DOI:10.1007/s12072-017-9826-x.
- M. Lunova, S. Frankova, H. Gottfriedova, R. Senkerikova, M. Neroldova, J. Kovac, E. Kieslichova, V. Lanska, E. Sticova, J. Spicak, M. Jirsa and J. Sperl, Portal hypertension is the main driver of liver stiffness in advanced liver cirrhosis, Physiol. Res., 2021, 563–577, DOI:10.33549/physiolres.934626.
- B. Castaneda, W. Debernardi-Venon, J. C. Bandi, V. Andreu, S. Pérez-Del-Pulgar, E. Moitinho, P. Pizcueta and J. Bosch, The role of portal pressure in the severity of bleeding in portal hypertensive rats, Hepatology, 2000, 31, 581–586, DOI:10.1002/hep.510310306.
- M. Ortega-Ribera, A. Gibert-Ramos, L. Abad-Jordà, M. Magaz, L. Téllez, L. Paule, E. Castillo, R. Pastó, B. de Souza Basso, P. Olivas, L. Orts, J. J. Lozano, R. Villa, J. Bosch, A. Albillos, J. C. García-Pagán and J. Gracia-Sancho, Increased sinusoidal pressure impairs liver endothelial mechanosensing, uncovering novel biomarkers of portal hypertension, JHEP Rep., 2023, 5(6), 100722, DOI:10.1016/j.jhepr.2023.100722.
- E. A. Aisenbrey and W. L. Murphy, Synthetic alternatives to Matrigel, Nat. Rev. Mater., 2020, 5, 539–551, DOI:10.1038/s41578-020-0199-8.
- M. B. Van Eldijk, C. L. McGann, K. L. Kiick and J. C. M. Van Hest, Elastomeric polypeptides, Top. Curr. Chem., 2012, 310, 71–116, DOI:10.1007/128_2011_205.
- M. Conley Wake, P. K. Gupta and A. G. Mikos, Original Contribution Fabrication Of Pliable Biodegradable Polymer Foams To Engineer Soft Tissues, 1996.
- K. A. Kyburz and K. S. Anseth, Synthetic Mimics of the Extracellular Matrix: How Simple is Complex Enough, Ann. Biomed. Eng., 2015, 43, 489–500, DOI:10.1007/s10439-015-1297-4.
- G. Parthasarathy, X. Revelo and H. Malhi, Pathogenesis of Nonalcoholic Steatohepatitis: An Overview, Hepatol. Commun., 2020, 4, 478–492, DOI:10.1002/hep4.1479.
- M. J. Ramos, L. Bandiera, F. Menolascina and J. A. Fallowfield, In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications, iScience, 2022, 25, 103549, DOI:10.1016/j.isci.2021.103549.
- P. Pingitore, K. Sasidharan, M. Ekstrand, S. Prill, D. Lindén and S. Romeo, Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis, Int. J. Mol. Sci., 2019, 20, 1629, DOI:10.3390/ijms20071629.
- M. Kozyra, I. Johansson, Å. Nordling, S. Ullah, V. M. Lauschke and M. Ingelman-Sundberg, Human hepatic 3D spheroids as a model for steatosis and insulin resistance, Sci. Rep., 2018, 8, 14297, DOI:10.1038/s41598-018-32722-6.
- M. Duriez, A. Jacquet, L. Hoet, S. Roche, M. D. Bock, C. Rocher, G. Haussy, X. Vigé, Z. Bocskei, T. Slavnic, V. Martin, J. C. Guillemot, M. Didier, A. Kannt, C. Orsini, V. Mikol and A. C. Le Fèvre, A 3D human liver model of nonalcoholic steatohepatitis, J. Clin. Transl. Hepatol., 2020, 8, 359–370, DOI:10.14218/JCTH.2020.00015.
- S. McCarron, B. Bathon, D. M. Conlon, D. Abbey, D. J. Rader, K. Gawronski, C. D. Brown, K. M. Olthoff, A. Shaked and T. D. Raabe, Functional Characterization of Organoids Derived From Irreversibly Damaged Liver of Patients With NASH, Hepatology, 2021, 74, 1825–1844, DOI:10.1002/hep.31857.
- Y. Guan, D. Xu, P. M. Garfin, U. Ehmer, M. Hurwitz, G. Enns, S. Michie, M. Wu, M. Zheng, T. Nishimura, J. Sage and G. Peltz, Human hepatic organoids for the analysis of human genetic diseases, JCI Insight, 2017, 2(17), e94954, DOI:10.1172/JCI.INSIGHT.94954.
- F. Sampaziotis, M. C. De Brito, P. Madrigal, A. Bertero, K. Saeb-Parsy, F. A. C. Soares, E. Schrumpf, E. Melum, T. H. Karlsen, J. A. Bradley, W. T. H. Gelson, S. Davies, A. Baker, A. Kaser, G. J. Alexander, N. R. F. Hannan and L. Vallier, Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation, Nat. Biotechnol., 2015, 33, 845–852, DOI:10.1038/nbt.3275.
- R. Ouchi, S. Togo, M. Kimura, T. Shinozawa, M. Koido, H. Koike, W. Thompson, R. A. Karns, C. N. Mayhew, P. S. McGrath, H. A. McCauley, R. R. Zhang, K. Lewis, S. Hakozaki, A. Ferguson, N. Saiki, Y. Yoneyama, I. Takeuchi, Y. Mabuchi, C. Akazawa, H. Y. Yoshikawa, J. M. Wells and T. Takebe, Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids, Cell Metab., 2019, 30, 374–384, DOI:10.1016/j.cmet.2019.05.007.
- M. Gori, M. C. Simonelli, S. M. Giannitelli, L. Businaro, M. Trombetta and A. Rainer, Investigating nonalcoholic fatty liver disease in a liver-on-a-chip microfluidic device, PLoS One, 2016, 11(17), e0159729, DOI:10.1371/journal.pone.0159729.
- M. S. Freag, B. Namgung, M. E. Reyna Fernandez, E. Gherardi, S. Sengupta and H. L. Jang, Human Nonalcoholic Steatohepatitis on a Chip, Hepatol. Commun., 2021, 5, 217–233, DOI:10.1002/hep4.1647.
- N. Azizipour, R. Avazpour, D. H. Rosenzweig, M. Sawan and A. Ajji, Evolution of biochip technology: A review from lab-on-a-chip to organ-on-a-chip, Micromachines, 2020, 11, 1–15, DOI:10.3390/mi11060599.
- T. Kostrzewski, T. Cornforth, S. A. Snow, L. Ouro-Gnao, C. Rowe, E. M. Large and D. J. Hughes, Three-dimensional perfused human in vitro model of non-alcoholic fatty liver disease, World J. Gastroenterol., 2017, 23, 204–215 CrossRef PubMed.
- R. E. Feaver, B. K. Cole, M. J. Lawson, S. A. Hoang, S. Marukian, B. R. Blackman, R. A. Figler, A. J. Sanyal, B. R. Wamhoff and A. Dash, Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis, JCI Insight, 2016, 1, 0–18, DOI:10.1172/jci.insight.90954.
- S. Wang, X. Wang, Z. Tan, Y. Su, J. Liu, M. Chang, F. Yan, J. Chen, T. Chen, C. Li, J. Hu and Y. Wang, Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury, Cell Res., 2019, 29, 1009–1026, DOI:10.1038/s41422-019-0242-8.
- S. Akbari, G. G. Sevinç, N. Ersoy, O. Basak, K. Kaplan, K. Sevinç, E. Ozel, B. Sengun, E. Enustun, B. Ozcimen, A. Bagriyanik, N. Arslan, T. T. Önder and E. Erdal, Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling, Stem Cell Rep., 2019, 13, 627–641, DOI:10.1016/j.stemcr.2019.08.007.
- S. V. Murphy and A. Atala, 3D bioprinting of tissues and organs, Nat. Biotechnol., 2014, 32, 773–785, DOI:10.1038/nbt.2958.
- K. A. Deo, K. A. Singh, C. W. Peak, D. L. Alge and A. K. Gaharwar, Bioprinting 101: Design, Fabrication, and Evaluation of Cell-Laden 3D Bioprinted Scaffolds, Tissue Eng., Part A, 2020, 26, 318–338, DOI:10.1089/ten.tea.2019.0298.
- X. Ma, X. Qu, W. Zhu, Y. S. Li, S. Yuan, H. Zhang, J. Liu, P. Wang, C. S. E. Lai, F. Zanella, G. S. Feng, F. Sheikh, S. Chien and S. Chen, Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2206–2211, DOI:10.1073/pnas.1524510113.
- B. Jin, Y. Liu, S. Du, X. Sang, H. Yang and Y. Mao, Current trends and research topics regarding liver 3D bioprinting: A bibliometric analysis research, Front. Cell Dev. Biol., 2022, 10, 1047524, DOI:10.3389/fcell.2022.1047524.
- J. H. Park, J. Jang, J. S. Lee and D. W. Cho, Current advances in three-dimensional tissue/organ printing, Tissue Eng. Regen. Med., 2016, 13, 612–621, DOI:10.1007/s13770-016-8111-8.
- S. Maji, M. Lee, J. Lee, J. Lee and H. Lee, Development of lumen-based perfusable 3D liver in vitro model using single-step bioprinting with composite bioinks, Mater. Today Bio., 2023, 21, 100723, DOI:10.1016/j.mtbio.2023.100723.
- H. Kizawa, E. Nagao, M. Shimamura, G. Zhang and H. Torii, Scaffold-free 3D bio-printed human liver tissue stably maintains metabolic functions useful for drug discovery, Biochem. Biophys. Rep., 2017, 10, 186–191, DOI:10.1016/j.bbrep.2017.04.004.
- T. Hiller, J. Berg, L. Elomaa, V. Röhrs, I. Ullah, K. Schaar, A. C. Dietrich, M. A. Al-Zeer, A. Kurtz, A. C. Hocke, S. Hippenstiel, H. Fechner, M. Weinhart and J. Kurreck, Generation of a 3D liver model comprising human extracellular matrix in an alginate/gelatin-based bioink by extrusion bioprinting for infection and transduction studies, Int. J. Mol. Sci., 2018, 19(10), 3129, DOI:10.3390/ijms19103129.
- G. Hong, J. Kim, H. Oh, S. Yun, C. M. Kim, Y. M. Jeong, W. S. Yun, J. H. Shim, I. Jang, C. Y. Kim and S. Jin, Production of Multiple Cell-Laden Microtissue Spheroids with a Biomimetic Hepatic-Lobule-Like Structure, Adv. Mater., 2021, 33(36), e2102624, DOI:10.1002/adma.202102624.
- G. Janani, S. Priya, S. Dey and B. B. Mandal, Mimicking Native Liver Lobule Microarchitecture In Vitro with Parenchymal and Non-parenchymal Cells Using 3D Bioprinting for Drug Toxicity and Drug Screening Applications, ACS Appl. Mater. Interfaces, 2022, 14, 10167–10186, DOI:10.1021/acsami.2c00312.
- 3D bioprinted therapeutic liver tissue - Organovo, Inc., (n.d.). https://organovo.com/3d-bioprinted-therapeutic-liver-tissue/(accessed April 5, 2024).
- T. Higashi, S. L. Friedman and Y. Hoshida, Hepatic stellate cells as key target in liver fibrosis, Adv. Drug Delivery Rev., 2017, 121, 27–42, DOI:10.1016/j.addr.2017.05.007.
- G. M. Balachander, S. A. Balaji, A. Rangarajan and K. Chatterjee, Enhanced Metastatic Potential in a 3D Tissue Scaffold toward a Comprehensive in Vitro Model for Breast Cancer Metastasis, ACS Appl. Mater. Interfaces, 2015, 7, 27810–27822, DOI:10.1021/acsami.5b09064.
- G. M. Balachander, B. Rajashekar, P. M. Sarashetti, A. Rangarajan and K. Chatterjee, MiRNomics Reveals Breast Cancer Cells Cultured on 3D Scaffolds Better Mimic Tumors in Vivo than Conventional 2D Culture, ACS Biomater. Sci. Eng., 2018, 4, 116–127, DOI:10.1021/acsbiomaterials.7b00694.
- G. M. Balachander, P. M. Talukdar, M. Debnath, A. Rangarajan and K. Chatterjee, Inflammatory Role of Cancer-Associated Fibroblasts in Invasive Breast Tumors Revealed Using a Fibrous Polymer Scaffold, ACS Appl. Mater. Interfaces, 2018, 10, 33814–33826, DOI:10.1021/acsami.8b07609.
- L. Gailhouste, Y. Le Grand, C. Odin, D. Guyader, B. Turlin, F. Ezan, Y. Désille, T. Guilbert, A. Bessard, C. Frémin, N. Theret and G. Baffet, Fibrillar collagen scoring by second harmonic microscopy: A new tool in the assessment of liver fibrosis, J. Hepatol., 2010, 52, 398–406, DOI:10.1016/j.jhep.2009.12.009.
- O. Ohtani, The Maceration Technique in Scanning Electron Microscopy of Collagen Fiber Frameworks: Its Application in the Study of Human Livers, Arch. Histol. Cytol., 1992, 55, 225–232, DOI:10.1679/aohc.55.Suppl_225.
- R. L. Blackmon, R. Sandhu, B. S. Chapman, P. Casbas-Hernandez, J. B. Tracy, M. A. Troester and A. L. Oldenburg, Imaging Extracellular Matrix Remodeling in Vitro by Diffusion-Sensitive Optical Coherence Tomography, Biophys. J., 2016, 110, 1858–1868, DOI:10.1016/j.bpj.2016.03.014.
| This journal is © The Royal Society of Chemistry 2024 |