
A synergic investigation of experimental and computational dual atom electrocatalysis for CO2 conversion to C1 and C2+ products
Saurabh Vinod
Parmar†
,
Damanpreet
Kaur†
and
Vidya
Avasare
 *
*
Department of Chemistry, Ashoka University, Sonipat, Haryana-131029, India. E-mail: vidya.avasare@ashoka.edu.in
First published on 14th June 2024
Abstract
This review delves into the promising field of dual atom catalysts (DACs) for the electrochemical reduction of CO2 to yield a diverse array of Cn (n = 1, 2, and 3) products. Through a comprehensive examination of recent advancements, mechanistic insights, and experimental findings, the review highlights the pivotal role of DACs in promoting selective and efficient eCO2RR. DACs, characterized by the synergistic interaction between two metal (homo/hetero) atoms, offer promising opportunities to address environmental challenges associated with CO2 emissions while simultaneously enabling the sustainable production of clean energy. The eCO2RR facilitates the formation of C1 to C2+ products including CO. CO is a key intermediate involved in C–C coupling reactions to form C2+ products.
Introduction
The ever-increasing levels of atmospheric CO2 pose one of the most pressing challenges. Anthropogenic activities, such as the burning of fossil fuels and industrial processes, have led to a significant increase in CO2 emissions, contributing to global warming and climate change.1–3 The International Energy Agency predicted that CO2 emissions will reach 40.3 Gt by 2030 and will grow to 50 Gt in 2050 if not restrained.4,5 Addressing this issue requires innovative strategies for mitigating CO2 levels while simultaneously transitioning towards a sustainable energy economy.6 One promising approach towards mitigating CO2 emissions is through its conversion to other valuable chemicals and fuels such as methane, methanol, ethanol, and other useful hydrocarbons. This approach not only alleviates the environmental burden but also creates opportunities for sustainable energy production and resource utilization.7–10 Various institutes and industries have been established with frequent government support for mitigation and conversion of CO2. In 2022, 1.66 Mt of methanol, a prominent future fuel, was produced from CO2 during the hydrogenation of CO by Methanex.11Heterogeneous catalysis plays a crucial role in CO2 reduction, offering efficient pathways for transforming CO2 into value-added chemicals.12,13 Unlike homogeneous catalysis, which involves catalysts and reactants in the same phase, heterogeneous catalysis utilizes solid catalysts that remain distinct from the reaction mixture. This distinction enables easier separation of the catalyst from the product, facilitating continuous operation of the same catalyst and reducing the environmental impact.14,15
While heterogeneous catalysis presents a promising avenue for the CO2 reduction reaction (CO2RR), traditional heterogeneous catalysts often suffer from limitations such as low selectivity, poor activity, and limited stability.16–18 To address these challenges, SACs have gained tremendous fame due to their exceptionally high catalytic performance and atom efficiency.19–24 However, SACs have inherent limitations, due to their reliance on a single active center. SACs often experience a decline in catalytic activity when faced with multi-intermediate reactions.25–31 In SACs, it is easy to optimize the catalyst for a single intermediate reaction but optimizing SACs according to distinct intermediates of a multi-step electrochemical reaction is unfeasible. Linear scaling relations, discussed by Norskov and coworkers and Sautet and coworkers, between the adsorption strength of reaction intermediates further hamper the activity of SACs (Fig. 1a).32–34
 | ||
| Fig. 1 (a) Effect of free energy change caused by the scaling relations between *CO and *CHO. (b) C2N supported heteronuclear transition-metal dimers in stabilizing *CHO. (c) The scaling relations between adsorption energies of *CHO and *CO.30 | ||
In the case of the electrocatalytic CO2 reduction reaction (eCO2RR), stronger binding of *CHO/*COH moieties reduces the free energy change of CH4 formation. However, according to the linear scaling relation, stronger binding of *CHO/*COH always follows up with an increase in *CO adsorption which suggests that the rate-determining step requires a high limiting potential.35–37 The structural difference between linear *CO and bent *CHO or *COH can help in the stabilization of one over the other. Since SACs are considered a bridge between the mono-nuclear homogeneous and heterogeneous catalysts, similar inspiration was drawn from homogeneous dimeric catalysts and bimetallic alloy catalysts forming dual atom catalysts (DACs). DACs are the combination of two SACs positioned adjacent or bonded to each other in three-dimensional space which results in increased metal loading, along with enhancement in top and bridge active sites for better flexibility and reactivity, where synergistic interaction between the two metal centers significantly enhances the overall catalytic activity.38–42 There are two types of DACs: (i) homonuclear DACs, which consist of two identical metal atoms (e.g., Pd–Pd and Cu–Cu) and (ii) heteronuclear DACs, which consist of two different types of metal atoms (e.g., Fe–Ni and Cu–Co).43–45 In the context of the eCO2RR, DACs provide C-affinity and O-affinity sites that strengthen *CHO, *COOH and *COH binding while *CO binding remains the same.
Li and coworkers demonstrated the advantages of DACs over SACs and nanoparticles (NPs) by synthesizing supported homonuclear Pd2DAC for the CO2RR and DFT calculations were used to compare the catalytic activity of Pd2-DAC, Pd1-SAC, and Pd-NPs.46 The direct relation between the limiting potential and *CO adsorption proved Pd2-DAC to be better than Pd1-SAC and Pd-NPs. The superiority of DACs over SACs in the CO2RR was also shown by Chen and co-workers by synthesizing heteronuclear Ni–Cu DACs. The synergistic effect of Ni and Cu decreased the HOMO–LUMO gap promoting *COOH adsorption compared to Ni SACs.47 Also the work carried out by Zhang and coworkers on in silico formation of urea from N2 and CO2 over a bimetallic catalyst gives further insight into the perks of the synergistic relation between the two metals. The proposed criterion tries to use this synergy to the fullest so that the bimetallic active sites have stabilizing interaction with the support layer. The metals need to perform a particular task; for example, the M1 site needs to have the ability to specifically adsorb N2, and *N2 should have a higher hydrogenation reaction barrier, while the M2 site needs to have the ability to adsorb CO2, and efficiently reduce CO2 to CO, which further desorbs easily. This endorses the superiority of DACs and the advantages of using bimetallic catalytic systems.48 In short, DACs possess all attributes of SACs in addition to the synergistic interaction of the two metals at active site, which significantly enhances the activity and efficiency. These advantages lead to the formation of unique structures and electronic properties that help to orchestrate rate-determining steps (RDSs) which were discussed by Warner and coworkers.49 The type of support used greatly affects the overall activity of DACs; for example, Ir1Mo1 DAC loaded on N-doped carbon favours the hydrogen evolution reaction (HER) while Ir1Mo1 DAC loaded on a TiO2 support catalyses the reverse process, hydrogen adsorption.50,51 This is possible only because of the change in the dominant metal active site with respect to the support. Thus, using DACs for multi-intermediate reactions such as the eCO2RR seems logical and has been found to show better results in the formation of C1 and C2 products from CO2 (Scheme 1).
 | ||
| Scheme 1 Plausible mechanism for the formation of C1 and C2 products by the eCO2RR using DACs. | ||
As efforts intensify to mitigate climate change and transition towards a low-carbon economy, the development of DACs for the eCO2RR holds immense promise. By harnessing the unique capabilities of DACs, researchers are ready to unlock new opportunities for sustainable energy generation and resource utilization, weaving a future where CO2 serves not as a pollutant but as a valuable feedstock for the production of essential chemicals and fuels. Han and Yang, in their review, discussed a bio-inspired self-assembly approach for the efficient synthesis of DACs using organic and inorganic porous materials as supports. This opens new avenues for the synthesis of active DACs for various applications including the eCO2RR using metal precursors having biomolecules and amino acids as ligands.52
Synthetic strategies to design and develop DACs
The synthesis of DACs involves the preparation of catalytic materials containing two same or different metal atoms in proximity. DACs are synthesized using various methods, each offering unique advantages, allowing control over catalyst composition, structure, and properties. Some commonly used synthetic strategies are given below:Wet chemical methods
Wet chemical methods, such as co-precipitation, impregnation, and sol–gel processes, are commonly used for the synthesis of DACs. These methods involve the reduction of metal precursors in solution followed by deposition onto a support material (Fig. 2). By controlling the reaction conditions, including pH, temperature, and reaction time, precise control over the composition and morphology of DACs can be achieved. | ||
| Fig. 2 General steps in the synthesis of DACs by wet chemical methods. | ||
Chen and coworkers synthesized a Cu–Pd atom pair on an alloy support by mixing aqueous solution of H2PdCl4 and Cu(NO3)2 with Te-nanowire solution at room temperature for 12 hours.53 The product was centrifuged and then dispersed into a 0.01 M NaOH solution and stirred for another 12 hours. The amount of Cu-doping was varied from sample to sample (0.02 wt%, 0.06 wt%, 0.16 wt%, and 0.20 wt% of Cu) by controlling the amount of Cu-precursor. The wt% of Cu in these samples was determined by coupled plasma mass spectroscopy (CP-MS).
Dinca and coworkers synthesized M3HAB (HAB = hexaiminobenzene) MOF by treating HAB·3HCl with ammoniacal solutions of CuSO4·5H2O in aqueous mixtures of water and dimethyl sulfoxide, heated at 60 °C under atmospheric conditions for 2 hours. It was found that oxygen from air is responsible for getting crystalline M3HAB and in the absence of air, the outcome is limited to the formation of amorphous grey powder of M3HAB.54 Chen and coworkers used this method to synthesize Cu–HAB and added it to a degassed methanol solution of SnCl2·2H2O. The mixture was heated and stirred until a purple-black CuSn–HAB powder was formed.55
Despite being straightforward and simple, this method has a few drawbacks. It lacks precise control over the dispersion and distribution of metal atoms on the support material which results in non-uniform atom distribution, leading to variations in catalytic activity and selectivity among DACs. The scalability of such a process will always be under scrutiny due to the possibility of agglomeration of metal atoms.
Pyrolysis
Reduction of organometallic precursors over a support is a common approach for the synthesis of DACs. This method involves subjecting precursors to high temperatures in an inert atmosphere, leading to the decomposition of the organometallic precursors over the support (Fig. 3). | ||
| Fig. 3 General steps followed in the synthesis of DACs by pyrolysis of organometallic precursors on a support. | ||
Zhang and coworkers developed a general strategy for preparing homonuclear and heteronuclear DACs by trapping their respective organometallic precursors in ZIF-8 (ZIF = Zeolitic Imidazolate Framework), followed by annealing under N2. This led to the formation of well-defined M1M2 DACs.56 The precursors were mostly bimetallic systems, showing a higher degree of DAC formation.
Zhao and coworkers synthesized Fe–Co DACs by pyrolysis of ferrocene and CoCl2 in varying ratios on an annealed ZIF-8 (NC) support. To their surprise, it was observed that at any given ratio of ferrocene to CoCl2, including bulk CoCl2, the wt% of Fe remained higher than that of Co in the formed DACs.57 This was happening because Fe atoms form a more stable structure in an N-doped carbon matrix than Co, leading to a much higher content of Fe.58,59
It is important to note that pyrolysis may also have drawbacks, such as the potential for incomplete decomposition of precursors, the formation of undesirable by-products or impurities, and the limited control over the dispersion and distribution of metal species on the support material. Addressing these challenges may require the optimization of pyrolysis conditions, the development of novel precursor compounds, or the use of additional post-treatment steps to enhance the performance of DACs synthesized via pyrolysis.
Impregnation
The impregnation method offers simplicity, versatility, and scalability to the DAC synthesis process. In this method, metal precursors are dissolved or dispersed in a suitable solvent and impregnated onto a support material, followed by drying and calcination to generate DACs. The impregnation method allows for the controlled deposition of metal species onto the support material, enabling the formation of well-defined dual-atom configurations. The impregnation method involves the selection of a suitable support material for DACs. Common support materials include metal oxides (e.g. TiO2, Al2O3, etc.) and or carbon-based materials such as activated carbon, carbon nanotubes, zeolites, etc. The choice of the support material depends on the desired catalytic activity, stability, and compatibility.60,61Metal precursors, typically metal salts or organometallic complexes, are dissolved in a suitable solvent to form a homogeneous solution. The selection of metal precursors depends on the desired composition and properties of DACs. Multiple metal precursors can be used to synthesize dual-atom configurations, with different metal combinations offering unique catalytic functionalities. The support material is then impregnated with the metal precursor solution, typically by incipient wetness impregnation or soaking.62 During impregnation, the support material is immersed in the precursor solution allowing the metal species to diffuse into the porous structure of the support material. The impregnation process is carried out under controlled conditions to ensure uniform deposition and saturation of metal species on the support material. Similarly, Li and coworkers synthesized Pd-DACs by impregnating a homogeneous Pd-complex on acetylene black. By replacing the counter anions, the solubility of the complex changed, leading to precipitation of the DACs.53 Overall, the impregnation method offers a versatile and effective approach for the synthesis of DACs, enabling the controlled deposition of metal species onto support materials and the formation of well-defined catalytic configurations.
Although the method is pretty straightforward, it may result in the occlusion of active sites within the porous structure of the support material, limiting their accessibility to reactant molecules. This can lead to mass transfer limitations and reduced catalytic efficiency, particularly for reactions involving bulky or sterically hindered substrates. Surface functionalization can enhance the stability and binding affinity of metal species, promoting stronger interactions and improved catalytic performance. This method typically does not involve surface functionalization of the support material, which may limit the interactions between metal species and the support surface leading to instability of the overall catalyst.
Atomic layer deposition (ALD)
ALD is a versatile thin-film deposition technique that allows precise control over the deposition of individual atomic layers onto a substrate. In the context of DAC synthesis, ALD offers a unique approach for the controlled deposition of metal atoms onto support materials with atomic precision (Fig. 4). By adjusting different parameters, such as the precursor concentration and deposition temperature, one can tailor the composition and structure of DACs for specific electrocatalytic applications.63,64 | ||
| Fig. 4 General procedure for synthesis of DACs by the ALD process. | ||
For example, Sun and coworkers used the ALD method to form Pt–Ru DACs on N-doped graphene (NGQ).65 They used trimethyl(methylcyclopentadienyl)platinum(IV) (MeCpPtMe3) as the platinum precursor and ethyl-substituted ruthenocene as the ruthenium precursor to be deposited on the NGQ. While ALD offers control over catalyst composition and structure, it also has several drawbacks. ALD's ability to create specific DACs might be limited by difficulty depositing certain metals and finding suitable precursor molecules for both metals.66–68 Achieving even distribution of both metals can be challenging, affecting performance.
DACs in eCO2RR
Electrochemical CO2 reduction utilizing DACs represents a promising avenue for mitigating carbon emissions and producing value-added chemicals ranging from simple molecules such as carbon monoxide (CO) and formate (HCOO−) to more complex products such as ethanol, methane, and ethylene. These products hold significant potential for various applications, including sustainable fuel sources, chemical synthesis, and energy storage. By harnessing the catalytic prowess of DACs, the eCO2RR stands poised as a sustainable strategy for addressing both environmental challenges and the growing demand for renewable energy sources.Continuing with the applications of DACs, we classify DACs based on the type of product formed from CO2 such as CO, CO2H, CH3OH, CH4, etc. along with C2+ hydrocarbons.
Carbon monoxide: a key intermediate in C2+ product formation
CO2 reduction is an alarming need in the present scenario. The conversion of CO2 to CO is an industrially and economically significant process as CO can serve as a building block in the industrial synthesis of organic and organometallic compounds.69–73 Syngas (CO + H2) is the key reagent in the thermocatalytic Fischer–Tropsch synthesis of value-added carbon products.74 It is also a preliminary step towards the synthesis of C1 or C2 products from carbon dioxide. CO can either be further reduced to form C1 products or undergo C–C coupling between different CO substrates to form C2 products (Scheme 2). | ||
| Scheme 2 Plausible mechanistic steps involved in CO mediated C–C coupling to form C2 products. | ||
Typically, CO2 to CO conversion occurs in three steps:75
| CO2 + H+ + e− → *COOH |
| *COOH + H+ + e− → *CO + H2O |
| *CO → CO + * |
The rate-determining step (RDS) involved in the formation of CO mainly depends on the intrinsic properties of the metals. For example, Fe has a high CO affinity that leads to high free energy for *CO desorption or increases the chances of CO poisoning, whereas, some metals such as Ir and Mo have sluggish kinetics for *COOH intermediate formation.47,48 Many homogeneous and heterogeneous catalysts have been explored to catalyze CO2 to CO formation. As discussed above, DACs have shown promising catalytic performance toward carbon dioxide utilization. Therefore, we discuss experimentally and theoretically explored dual-atom catalysts (DACs) for CO formation from CO2 (Table 1).
| Metal | Substrate | Electrolyte | Faradic efficiency | Current density | Potential | Ref. |
|---|---|---|---|---|---|---|
| Cu2 | Pd10Te3 | 0.2 M NaHCO3 | 92% | 8.6 mA cm−2 | −0.78 V | 76 |
| Ag2 | Graphene | 0.5 M KHCO3 | 93.4% | 11.8 mA cm−2 | −0.7 V | 77 |
| Pd2 | Acetylene black | 0.5 M KHCO3 | 98.2% | 6.76 mA cm−2 | −0.85 V | 79 |
| Co2 | Carbon nanotubes | 0.5 M KHCO3 | 91.76% | — | −0.7 V | 82 |
| Fe2 | N-doped carbon | — | >80% | −32.04 mA cm−2 | −1.0 V | 83 |
| Fe2 | N-doped porous carbon | 0.1 M KHCO3 | 96% | 2.9 mA cm−2 | −0.6 V | 85 |
| 94% | 3.5 mA cm−2 | −0.7 V | ||||
| Fe2 | Graphitic carbon nitride | 0.5 M KHCO3 | 98.6% | — | −0.4 V | 86 |
| Ni2 | N-doped carbon black | 0.5 M KHCO3 | >95% | 26.5 mA cm−2 | −0.6 to −1.0 V | 87 |
| Ni2 | N-rich MOF | 1.0 M KHCO3 | 94.3% | 150 mA cm−2 | — | 88 |
| Ni2 | C carriers and dicyandiamide | 1 M KOH | 99% | 1 A cm−2 | — | 89 |
| Ni2 | Carbon black | — | 98.9% | 23.46 mA cm−2 | −0.88 V | 90 |
| Ni–Fe | ZIF | 0.5 M KHCO3 | 98% | 7.4 mA cm−2 | −0.7 V | 75 |
| Ni–Fe | N doped C (ZIF) | 0.1 M KHCO3 | 97.8% | 18.6 mA cm−2 | −0.6 to −0.9 V | 94 |
| Ni–Fe | S, N doped C | — | 96% | 40.1 mA cm−2 | −0.9 V | 95 |
| Co–Ni | N doped porous C nanosheets | 0.1 M KHCO3 | 96.4% | 3.2 mA cm−2 | −0.48 V | 96 |
| Co–Ni | N doped C (ZIF-8) | 0.1 M KHCO3 | 95% | 13.34 mA cm−2 | −0.8 V | 97 |
| Ni–Cu | N doped C matrix (MOF: ZIF) | 0.5 M KHCO3 | 99.2% | 29.9 mA cm−2 | −0.79 V | 103 |
| Ni–Cu | N doped C matrix (ZIF-8) | 0.5 M KHCO3 | 97.7% | 13.7 mA cm−2 | −0.7 V | 105 |
| Ni–Cu | N doped graphene | 0.1 M KHCO3 | 98% | −18.2 mA cm−2 | −1.07 to −1.27 V | 106 |
| Fe–Cu | N doped C matrix (MOF: ZIF-8) | 0.1 M KHCO3 | 99% | 12.91 mA cm−2 | −0.8 V | 110 |
| Ni–Zn | N doped C | 0.5 M KHCO3 | 99% | −20 mA cm−2 | −0.8 V | 107 |
| Zn–Co | N doped C | Saturated KHCO3 | 93.2% | 26 mA cm−2 | −0.5 V | 108 |
| Cu–Zn | Pc-MOFs | 0.1 M KHCO3 | 88% | — | −0.7 V | 109 |
| Ni–Ag | N rich porous C | 0.1 M KHCO3 | 99.2% | 12.6 mA cm−2 | −0.8 V | 101 |
Cu and some noble metal-based catalysts such as Ag, Pd, and Pt have been known for the carbon dioxide reduction reaction (CO2RR) but have certain drawbacks such as higher overpotential and lower current density. Chen and coworkers reported copper-containing homonuclear DACs stabilized by Te defects on Pd10Te3 alloy nanowires using wet chemical methods with varying metal loadings.76 Combining results of characterization techniques such as X-ray absorption near-edge structure (XANES), Fourier-transformed (FT) k3 weighted spectra and wavelet transform (WT) plots, it was concluded that Cu existed as Cux and displayed atomic level dispersion in defects of Pd10Te3. The dispersion of Cux in these defects indicates the importance of defects in stabilizing the atomically dispersed Cu atoms. The Cux was protected by an oxygen atom as predicted by energy-dispersive X-ray spectroscopy (EDS) mapping. Theoretically, it was found that Cux and Cu4 displayed similar plots, thus x ≈ 4 (3, 4, 5). The calculations revealed that Cu4 had one Cu bonded to oxygen to form Cu1x+ which further binds to a neighboring Cu atom to form the (Cu10 – Cu1x+) atom pair, which directly participated in electrocatalysis, whereas the other two Cu atoms accommodated the defects around the vacancy occupying the O atom. The 0.10% Cu loading was found to be optimal delivering 18.74 mA cm−2 current density at −0.98 V (vs. RHE) as revealed by linear sweep voltammetry (LSV) curves. It shows high selectivity for the CO2RR vs. the HER with an efficiency of FECO = 92% at −0.78 V. The competing HER reaction significantly gets suppressed to FEH2 = 3%. Virtually unchanged current density and a small decline of FECO from 92% to 80% were observed after testing the sample at −0.78 V for 180 minutes. By increasing the metal loading, there was a sharp decrease in performance as it started forming CuOx and CuPdx whereas Cux species gradually disappeared. DFT calculations also confirmed that (Cu10–Cu1x+) displayed a lower free energy barrier (Fig. 5c) than the classic Cu metal and Cu embedded in Cu2O; thus, the catalyst's efficiency was enhanced for the eCO2RR by improvements in both kinetics and thermodynamics.
 | ||
| Fig. 5 (a) HAADF-STEM image of dual atom-Co/CNT. (b) Intensity profile obtained over the red line in (a). (c) FECO and FEH2 at different applied potentials. (d) Calculated Gibbs free-energy diagrams for CO2 electrocatalysis reduction to CO.82 | ||
In the interesting application of silver-based DACs, Zhou and coworkers reported a dual atom Ag2/graphene catalyst (Ag2–G) present in an AgN3–AgN3 coordination environment with 0.10% metal content as given by ICP data.77 Interestingly, they highlighted that the aromatic ligands of silver complexes provide the steric hindrance around Ag atoms to prevent them from agglomerating. The XRD pattern and aberration-corrected HAADF-STEM confirmed the uniform dispersion of Ag atoms. The XPS analysis suggested the valence state of Ag between 0 and +1 and WT-EXAFS (extended X-ray absorption fine structure) provided evidence for the presence of a bond between two Ag atoms. The electrochemical analysis manifested that the Ag2–G catalyst works efficiently in the wide potential range of −0.5 to −0.9 V reaching a maximum FECO of 93.4% at −0.7 V with a current density of 11.8 mA cm−2. Ag2–G exhibited excellent stability when tested for 36 h at −0.7 V. The DFT study also suggested that Ag2–G-DAC promoted CO2 adsorption and stabilized the intermediates by reducing the energy barriers. Thus, this study further highlights the superiority of DACs in the CO2RR.
Usually, Pd-SACs have been known for excellent HER as compared to FECO as it easily forms Pd–H species.78 Wang and coworkers used a palladium(II) precursor, a NO3− ion source, KPF6 and acetylene black to get an atomically dispersed Pd2DAC by the anion replacement deposition precipitation (ARDP) method; which achieved high FECO and interestingly exhibited superior performance.79 The X-ray photoelectron spectroscopy (XPS), aberration corrected-high angle annular dark field-scanning transmission electron microscopy (AC-HAADF-STEM) and EXAFS data reveal that Pd2DAC exhibited a Pd(II)N2O2 coordination structure. Surprisingly, the average Pd–Pd interatomic distance was much shorter (1.96 Å) than the theoretically reported distance of 2.85 Å. The electrochemical activity investigated by linear sweep voltammetry (LSV) demonstrated that Pd2DAC shows a larger current density as compared to Pd1SAC and PdNP/C in the potential range from −0.20 to −0.85 V with a maximum FECO of 98.2% at −0.85 V vs. RHE and displayed consistent 80+% FE up to −0.95 V. Pd2DAC shows partial current densities of CO ca. 6.76 mA cm−2 at −0.85 V vs. RHE, which remains unchanged even after testing for 12 hours, accounting for its excellent stability. Looking into kinetic insights, Tafel plots of Pd2DAC show a lower slope (ca. 148.9 mV dec−1), which proves that the rate-limiting step (RLS) was CO2 activation. Herein, the potential determining step (PDS) was the activation of CO2 to *COOH with the lowest PDS free energy of 1.28 eV. In the volcanic activity profile of UL (limiting potential) versus adsorption of *CO, Pd2 was close to the top of the volcano with optimized UL showing the highest catalytic performance following the Sabatier principle. The DFT study demonstrated that the Pd2DAC has a moderate adsorption strength of *CO which facilitates CO desorption. The synergic effect of dimeric Pd atoms efficiently performs the eCO2RR.
As discussed before, two neighboring metal atoms without any direct linkage can also introduce synergic effects like in DACs. In this context, Zeng and coworkers highlighted such interactions by preparing Pt/MoS2 having different Pt mass loadings of 0.2%, 1.0%, 5.0%, and 7.5%.80 The XANES and EXAFS data accounted for the atomic dispersion of Pt atoms throughout all the samples and it was shown that Pt atoms replaced the Mo atoms in MoS2 nanosheets. Every Pt atom with its corresponding S atom acts as the active catalytic site. The distance between the two nearest Pt atoms at 0.2% loading was more than 3 nm and this distance decreased with the increase in the metal loading, resulting in synergic effects being observed in 5.0% and 7.5% Pt-loaded samples. The TOF of 7.5% Pt/MoS2 was 162.5 h−1, 14.8 times higher than that of 0.2% Pt/MoS2. The major intermediate for 0.2% Pt/MoS2 was CH3OH* whereas for 7.5% Pt/MoS2, it was COOH* respectively. The DFT study elaborates that isolated 0.2% Pt/MoS2 favors the direct conversion of CO2 to methanol without forming formic acid whereas the neighboring Pt monomers favors methanol formation via the formic acid pathway. Thus, these synergic effects lead to enhancement in catalytic activity by altering the energy barriers and resulting in different reaction paths as well.
Although research on DACs in the electrochemical CO2RR is growing fast, many researchers have opted for inexpensive first-row transition metals such as Fe, Co, and Ni as the basis for their DACs. These first-row transition metals are preferred in DACs for several reasons, natural abundance, versatile catalytic activity, stability, etc. Also, these transition metals can be effectively supported on various materials such as carbon, zeolites, etc. enhancing catalyst dispersion and stability. This compatibility with support materials expands the versatility and applicability of DACs.
Owing to their poor catalytic performance, cobalt-based DACs have been less explored for the eCO2RR.81 However, rational design and fine-tuning of the local environment improve the catalytic performance of these Co catalysts. Wang and coworkers designed a dual atom Co-salophen (Co-DAC) catalyst by using a non-covalent anchoring strategy to disperse Co on carbon nanotube (CNT) surfaces.82 Uniform dispersion of Co dual atoms on CNTs was characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), HAADF-STEM, and Raman spectroscopy (Fig. 5a). The presence of 3.94 wt% of metallic Co in a hybrid material of dual atom-Co/CNT was confirmed by coupled plasma optical electron spectroscopy (CP-OES) and the interatomic distance between two metals was studied by calculating the distance (0.525 nm) between two spots in the HAADF image (Fig. 5b). The use of GC and NMR confirms the formation of CO and H2 and the absence of any liquid by-products. Co-DAC followed similar reaction kinetics to single atom-Co/CNT but outperformed single atom-Co/CNT in the entire potential range from −0.35 to −0.90 V showing the highest FECO of 91.76% at −0.7 V (Fig. 5c). The LSV curves account for 3.5 times higher current density and 3.9 times higher partial current density for dual atom-Co/CNT as compared to single atom-Co/CNT. It showed satisfactory stability when investigated electrochemically for 10 hours at −0.70 V (vs. RHE). Interestingly, a significant decrease in the Tafel slope of Co-DAC was observed due to the formation of intermolecular H-bonds between activated *CO2 and H2O, and a reduction in the kinetics of the conversion of *CO2 to *COOH was observed. The enhanced intrinsic activity of Co-DAC was indicated by a high TOF of 2056.7 h−1 at −0.90 V (vs. RHE). DFT calculations indicated the high adsorption energy of Co-DAC (3.58 eV). Furthermore, the presence of strong π–π stacking interactions between components of hybrid materials and the larger Fermi level of Co/CNT were observed in dual atom-Co/CNT. These factors contribute to the enhanced activity and stability of the catalyst compared to single atom-Co/CNT. The DFT predicted the conversion of CO2 to *COOH to be the RDS having a ΔG of 0.639 eV (Fig. 5d). This work demonstrated a synergistic effect between the Co–Co pair. One Co atom was primarily bound to H2O and afterward, the other one gets attached to CO2, thus forming an H-bond between CO2 and H2O, which weakens the C–O bond to promote the activation of CO2 and stabilizing the *COOH intermediate (ΔG = −0.509 eV). As per the theoretical study, the introduction of three explicit H2O molecules further decreases the free energy of *COOH and *CO formation. Thus, the synergic effect of two Co atoms proved to be beneficial for its catalytic activity enhancement in the CO2RR.
In addition to the aforementioned metals, extensive research has been conducted on DACs based on Fe and Ni. Considering Fe-based DACs for the CO2RR, the CO adsorption study on the active site of Fe depicts a strong interaction, leading to difficulty in *CO desorption. Therefore, rational catalyst design has been demonstrated to achieve promising catalytic performance. In this context, Lee and coworkers successfully synthesized Fe2–N6–C DAC with excellent FECO efficiency (>80%) using the template-assisted protocol under H2 pyrolysis conditions.83 The Fex–Ny–C configuration varied with the H2 concentration to get the desired results. The 5% and 10% concentrations of H2 provided ortho-Fe2–N6–C and para-Fe2–N6–C respectively. The HAADF-STEM and EDS mapping accounted for uniform distribution (Fig. 6a) with an average distance of 2.37 ± 0.31 Å and 2.67 ± 0.29 Å between Fe–Fe atoms of ortho-Fe2–N6–C and para-Fe2–N6–C respectively. The changes in local coordination of Fe are depicted by FT-EXAFS spectra (Fig. 6c). The XANES data depicted the oxidation state of Fe to be between 0 and +3.84 The LSV curves revealed that ortho-Fe2–N6–C exhibited higher current density and the lowest Tafel slope (120 mV dec−1) than para-Fe2–N6–C. Both ortho-Fe2–N6–C and para-Fe2–N6–C possessed energy efficiency (EE) values of over 50% in a potential window of −0.5 V to −0.9 V. The electrochemical performance of ortho-Fe2–N6–C attained the highest TOF of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 637 h−1 at 1.0 V and exhibited outstanding stability even after 21 hours of electrolysis. As per DFT, the 3d coupling between dual atomic Fe in ortho-Fe2–N6–C resulted in decreased orbital energy levels (d band center) as shown in Fig. 6b which facilitates *CO desorption. Moreover, the top site binding configuration of *CO on ortho-Fe2–N6–C provided moderate *CO binding energy and easy *CO desorption. The ortho-Fe2–N6–C turned out to be highly selective towards the CO2RR vs. the HER. In short, Fe2–N6–C was found to be an efficient catalyst for CO2RR than Fe1–N4–Cs, and the ortho-Fe2–N6–C outperforms the para-Fe2–N6–C (Fig. 6c).
637 h−1 at 1.0 V and exhibited outstanding stability even after 21 hours of electrolysis. As per DFT, the 3d coupling between dual atomic Fe in ortho-Fe2–N6–C resulted in decreased orbital energy levels (d band center) as shown in Fig. 6b which facilitates *CO desorption. Moreover, the top site binding configuration of *CO on ortho-Fe2–N6–C provided moderate *CO binding energy and easy *CO desorption. The ortho-Fe2–N6–C turned out to be highly selective towards the CO2RR vs. the HER. In short, Fe2–N6–C was found to be an efficient catalyst for CO2RR than Fe1–N4–Cs, and the ortho-Fe2–N6–C outperforms the para-Fe2–N6–C (Fig. 6c).
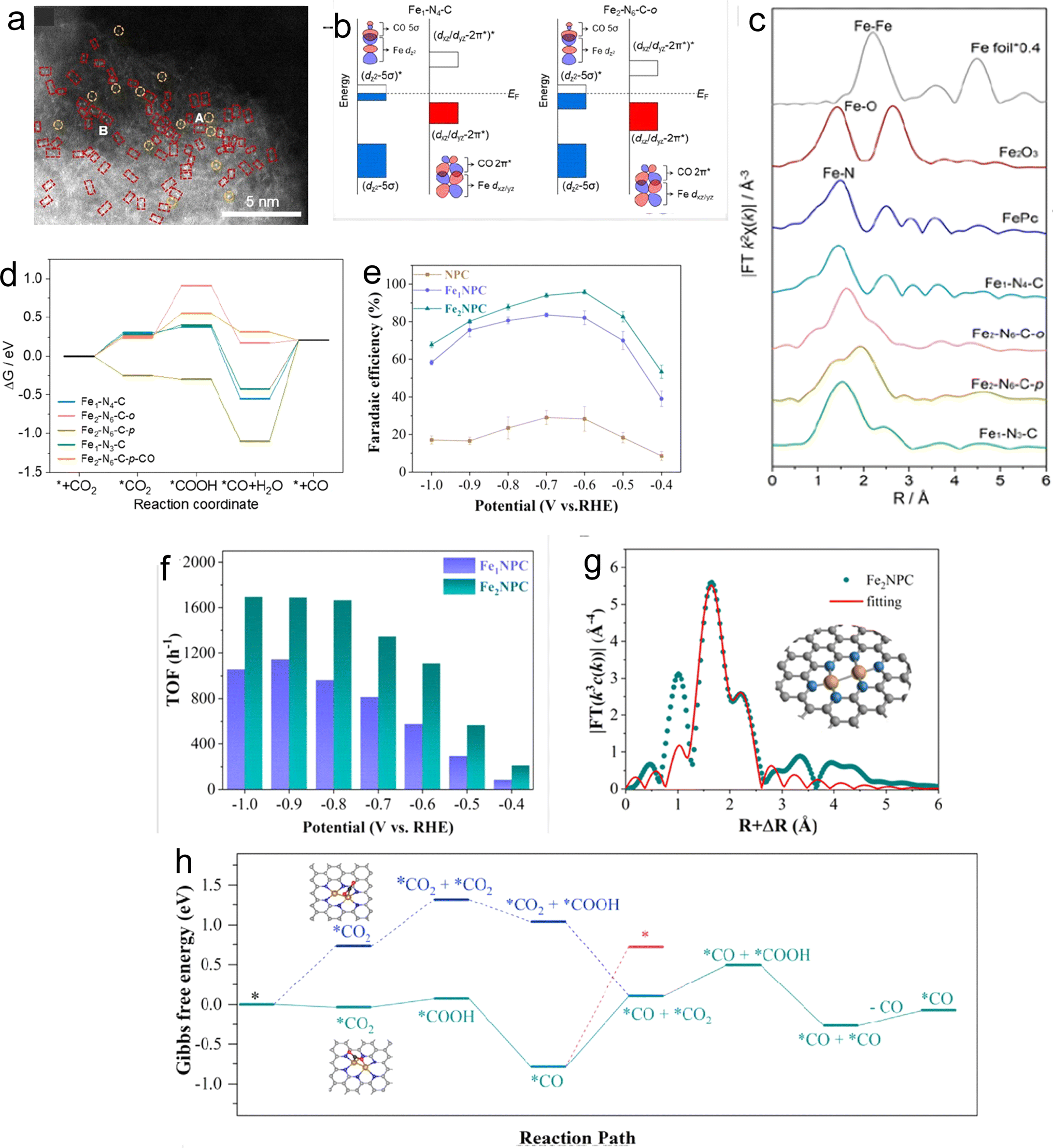 | ||
| Fig. 6 (a) Aberration-corrected (AC)-HAADF-STEM image of Fe2–N6–C-o (single sites and bimetallic Fe pairs are highlighted with yellow circles and red rectangles). (b) Orbital interaction between Fe-3d and adsorbed CO for Fe1–N4–C and Fe2–N6–C-o. (c) Fourier-transformed k2-weighted EXAFS spectra. (d) The free energy diagram of the CO2RR for Fe1–N4–C, Fe2–N6–C-o, Fe2–N6–C-p, Fe1–N3–C, and Fe2–N6–C-p–CO. Reprinted (adapted) with permission from ref. 83. Copyright© 2022, American Chemical Society. (e) FECO of NPC, Fe1NPC, and Fe2NPC. (f) TOF of Fe1NPC and Fe2NPC. (g) Corresponding EXAFS R-space fitting curves. (h) Calculated free energy for ECR on Fe2NPC. Reprinted (adapted) with permission from ref. 85. Copyright© 2022, American Chemical Society. | ||
Furthermore, Quan and coworkers successfully synthesized another type of iron DAC, Fe2NPC, on nitrogen-doped porous carbon by pyrolysis of Fe2(CO)9 and the ZIF-8 precursor with an Fe content of 0.34% as shown by ICP spectrometer measurements.85 The pore size distribution result demonstrated negligible effects of different iron precursors on the catalyst structure. The XPS data confirmed Fe–N bonds and Fe K-edge absorption indicated the valence state of Fe between +2 and +3. Furthermore, WT and EXAFS data revealed that diatomic Fe sites of Fe2NPC appeared in Fe2N6 coordination with an Fe–Fe bond length of 2.47 Å (Fig. 6g). On electrochemical analysis, Fe2NPC provided the highest FECO of 96.0% at −0.6 V and 94% at −0.7 V along with a TOF of 3721 h−1 at −0.6 V which was 1.8 times higher than that of Fe1NPC (Fig. 6d and e). The electrochemical impedance spectroscopy (EIS) and electrochemical surface area (ECSA) data disclosed that an improved intrinsic activity of active sites accelerates the performance of Fe2NPC. The catalyst displayed superior kinetics with a much smaller Tafel slope of 60 mV dec−1 and amazing stability even after 12 hours of electrolysis. DFT results revealed the binding of CO2 on Fe2N6 with a novel bridge-like strategy comprising two atoms O and C in CO2 that are bonded to two Fe atoms and this provided a facile pathway for CO desorption (0.9 eV). Thus, this work highlights the improved intrinsic activity of Fe-DACs through the bridge-like binding of CO2 on the catalyst surface.
Selectivity, activity, and electronic structure modulations play a key role in the rational designing of electrocatalysts for the CO2RR. Liaw and coworkers used practical in operando spectroscopy to illustrate the factors responsible for intermediate stabilization in DACs.86 The step thermolysis method was used for the synthesis of the Fe2/g-C3N4 precursor followed by pyrolysis with N doped carbon black to form g-C3N4 exempted Fe2DAC (Fig. 7a). The XAS, HAADF-STEM, and EPR analyses of both Fe2/g-C3N4 precursor and Fe2DAC were performed. AC-STEM images confirmed dual Fe sites. The average Fe–Fe distance was observed to be 3.01 Å and 2.71 Å for the Fe2/g-C3N4 precursor and Fe2DAC respectively and both displayed the ferric state. The FT-EXAFS, EPR, and AC-STEM data collectively indicated that the conservation of atomically dispersed Fe–Fe pairs in Fe2DAC was due to the template effect of the Fe2/g-C3N4 precursor. Fe2DAC exhibited a relatively better CO selectivity of 98–88% as compared to Fe-SAC (86–49%). Fe2DAC showed a high FECO of 98.6% at an over-potential of 400 mV with a TOF of 2.03 s−1. The CO selectivity gets retained even after 20 hours of continuous operation, which demonstrates the high stability of Fe2DAC. The charge transfer coefficient (α) of Fe2DAC was found to be 0.53, which highlighted that Fe2DAC catalyzed the CO2RR proceeded through the SET (single electron transfer) mechanism before the chemical protonation. The cation effect on the behavior of Fe2DAC was studied which interestingly induced a shift in the RDS with an increase in the cation size. The strong interaction between a large cation (Cs+) and anionic intermediates stabilizes the RDS enhancing the CO2 reduction. This highlighted the dominant role of thermodynamics in the CO2RR. Furthermore, the in operando electron paramagnetic resonance (EPR) and attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) measurements manifested that the thermodynamic stabilization was through the strong orbital interactions among the Fe-d orbitals, the s orbital of the alkali cation and the empty π* orbital of the surface-bound intermediates and proceeds further with the proposed mechanism given in Fig. 7b. Hence, this work promotes the idea of in operando techniques and also emphasizes the interesting cation effect on the performance of Fe2DAC favoring larger cations for efficient CO2RR.
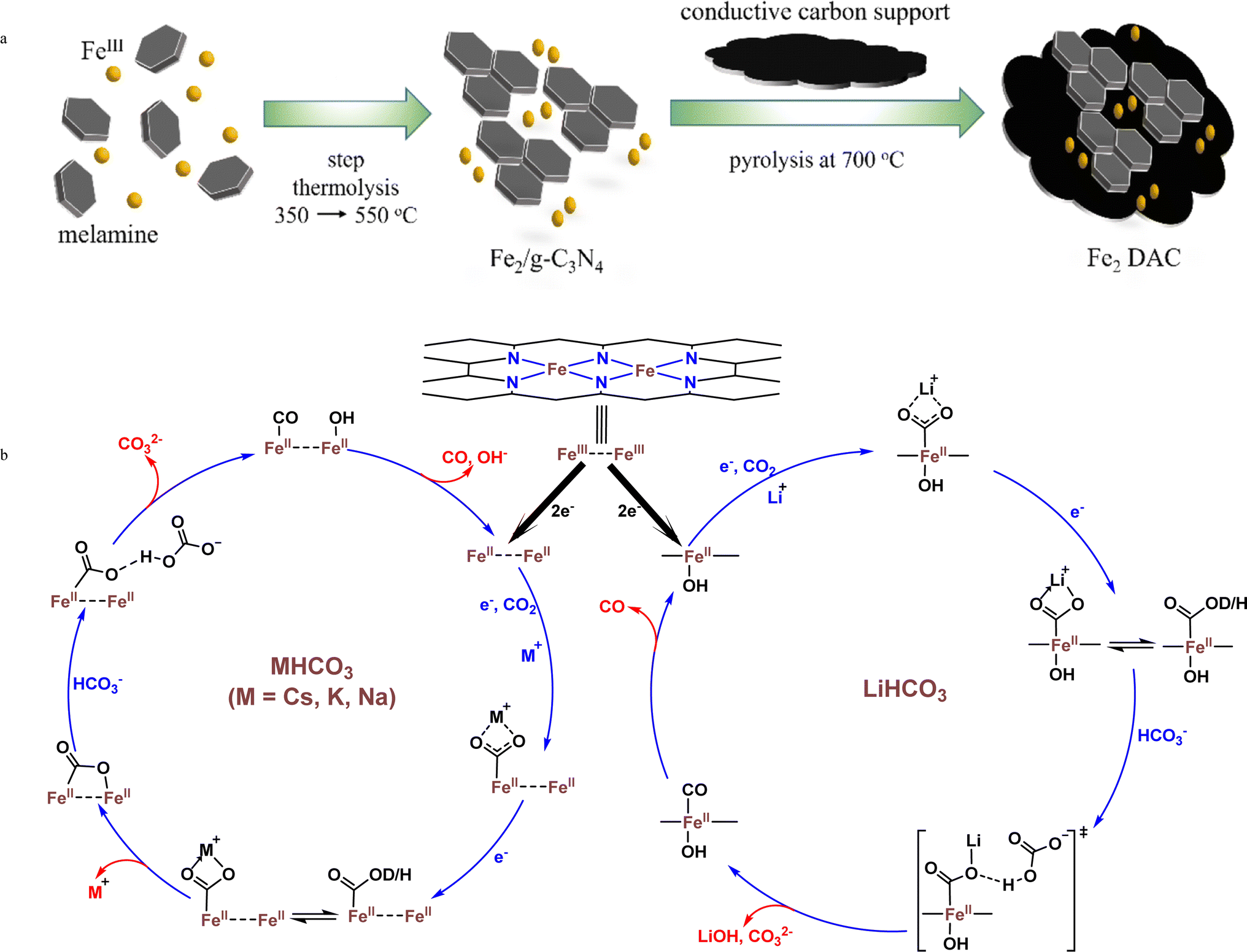 | ||
| Fig. 7 (a) Synthetic scheme of an atomically precise dual Fe2 catalyst (Fe2DAC) using the di-nuclear Fe2 site preservation strategy.86 (b) eCO2RR mechanism for Fe2DAC involving alkali metal bicarbonates. | ||
Apart from iron, nickel has been a promising candidate for the catalytic CO2RR. However, there is a risk of CO poisoning due to its high CO adsorption ability. To overcome this problem, Cao and coworkers adopted an in situ strategy to synthesize a Ni-based catalyst with different Ni and N loadings as modulation of N sources helped in the controllable synthesis of catalysts.87 As per the ICP-OES data, out of the varyingly synthesized Ni catalysts, Ni–N@C1 and Ni–N@C1-1% exhibited the highest Ni contents of 0.237% and 1.067% respectively. SEM characterization accounted for the spherical morphology of the carbon support and the +2-oxidation state of Ni was confirmed by XPS. Combined HAADF-STEM and EXAFS data provided strong evidence of ∼73% di-atomic Ni sites and the rest single-atomic sites (Fig. 8a and d). Di-atomic sites adopted the Ni2N6 environment whereas single-atomic sites adopted the NiN4 coordination environment. Electrolysis study of Ni–N@C1 reported 95–100% FECO as shown in Fig. 8b, in a potential range of −0.6 to −1.0 V vs. RHE, and the HER activity was highly suppressed with FEH2 less than 5%. It showed a high current density of 37.2 A mg−1 of Ni at −1.1 V. After 7 hours of electrolysis, FECO remained at 90% and it decreased to 80% after 10 hours. This suggested that the N/Ni ratio plays a significant role in the structure formation during pyrolysis and a moderate N/Ni ratio leads to materials with a large fraction of Ni2 sites.
 | ||
| Fig. 8 (a) Aberration-corrected HAADF-STEM image of Ni–N@C1. (b) Partial current density of CO. (c) Faradaic efficiencies of CO with different PANI/Ni feed ratios.87 (d) Fourier-transform of Ni K-edge EXAFS spectrum of Ni–N@C1. (e) High-resolution HAADF-STEM image of Ni2/NC. Reprinted (adapted) with permission from ref. 88. Copyright© 2021, American Chemical Society. (f) Free energy profiles of the electroreduction of CO2 to CO at Ni sites for Ni2–NxCy and Ni–N2C2. (g) EXAFS fittings and (inset) optimized model for Ni2–N3C4. Reprinted (adapted) with permission from ref. 90. Copyright© 2022, Wiley-VCH GmbH. (h) Proposed reaction pathways on O–Ni2–N6. | ||
Yao and coworkers presented further evidence of excellent electrocatalytic performance in nickel-based catalysts. Their work showcases the successful synthesis of a Ni2/NC catalyst through temperature-regulated pyrolysis, which has demonstrated remarkable efficacy in the eCO2RR with a FECO of 94.3% at a current density of 150 mA cm−2.88 Characterization data confirmed the Ni content of ∼0.18 wt% and a distance of ca. 2.63 Å between adjacent Ni atoms present in the Ni2–N6 environment and the HAADF-STEM image shown in Fig. 8d accounted for the uniform distribution of atoms. Furthermore, the electrochemical analysis demonstrated a CO current density of 141 mA cm−2 at −1.25 V vs. RHE and excellent catalyst stability for over 50 hours, with an efficiency of 91% at −0.6 V. Interestingly, operando XAFS and the operando XANES together identified initial and dynamic structural evolution of Ni2 sites, which were also confirmed using DFT calculations. The catalyst Ni2–N6 adsorbs the oxygen-relevant intermediates to form an oxygen-bridged O–Ni2–N6 site behaving as a key to excellent selectivity. Thus, operando techniques helped in proposing a reaction pathway of the eCO2RR catalyzed by Ni2–N6 as given in Fig. 8f.
Another notable contribution comes from Zhang and coworkers, who synthesized Ni2NC through the co-pyrolysis of Ni-citric acid adsorbed on C-carriers and dicyandiamide.89 The HAADF-STEM images confirmed Ni dual atomic sites with ∼0.25 nm distance between adjacent Ni atoms and the ICP data accounted for 0.29% metal loading. Interestingly, the in situ ESTEM and theoretical investigation suggested that dual atomic site formation is preferred at high temperatures during pyrolysis. The EXAFS data accounted for the valence state of Ni between 0 and +2 with a 2.51 Å bond length of the Ni–Ni bond and the presence of an N3–Ni–Ni–N3 coordination environment. This demonstrates an industrial-level catalytic performance showing 99% FECO with a current density of 1 A cm−2 exhibiting robust stability during 30 hours of electrolysis. The ab initio molecular dynamics (AIMD) study suggested that OHad-induced Ni sites (Ni2N6OH) have shown lower formation energy than Ni2N6 due to electron-rich microenvironment formation.
Further noteworthy research on Ni-DACs was conducted by Zhong and coworkers, who investigated three different DACs, Ni2–N7, Ni2–N5C2, and Ni2–N3C4 which were synthesized by temperature-controlled pyrolysis to regulate their coordination environments as it has been an effective strategy to optimize catalytic efficiency to reduce CO2.90 Herein, nitrogen contents often get decreased with an increase in pyrolysis temperature.91 Characterization techniques such as TEM, EDS, and aberration-corrected HAADF-STEM depict a large number of nickel atoms tethered on N-doped atoms with a Ni–Ni atomic distance of ∼3.1 Å. EXAFS fittings indicated the bridging of two Ni atoms by one N atom (Fig. 8g). Thus, Ni–NxCy shows a homogeneous distribution of C, N, and Ni atoms, and XANES data reported the valence states of Ni in the middle of 0 and +2. Ni2–N3C4 showed the best electrochemical activity with a maximum FECO of 98.9% at −0.88 V vs. RHE and FECO of 96.4% at −0.6 V along with a total current density of 151.26 mA cm−2. No obvious decay in the current density and FECO was observed even after 30 hours of electrolysis, leading to its robust stability. DFT calculations further provided insights into the reaction mechanism with the help of Gibbs free energies (Fig. 8e). The binding energy values illustrated that the *COOH and *CO intermediates get weakly bonded to Ni2–N7 and Ni2–N5C2, as compared to the moderate binding with Ni2–N3C4. In short, the superior performance of Ni2–N3C4 was attributed to its proper binding energies to *COOH and *CO intermediates. These results provided a facile strategy to construct DACs by modulating electronic structures via coordination environment engineering.
Along with the homonuclear Fe2 and Ni2 DACs, heteronuclear Ni and Fe DACs for the CO2RR were also explored by Zhao and coworkers by synthesizing an efficient CO2RR DAC featuring Fe and Ni metal sites anchored on nitrogenized carbon (Ni/Fe–N–C) by an ion-exchange strategy based on pyrolysis of Zn/Ni/Fe ZIF.75 This dual catalyst was successfully characterized by SEM, HAADF-STEM, EDS, and XANES techniques. As per LSV, dual atom-Ni/Fe–N–C outperforms single atom-Ni–N–C and Fe–N–C across the entire potential window from −0.4 to −1.0 V with a TOF of 7682 h−1, reaching a maximum FECO of 98% at −0.7 V and achieved a current density of 7.4 mA cm−2, and was found to be stable even after 30 hours of electrolysis. Tafel slopes indicated that the formation of surface adsorbed *COOH intermediate is the RDS and the catalyst Ni/Fe–N–C enhanced kinetics of this step. The DFT study suggested that the binding strength of *COOH and *CO on Ni/Fe–N–C is weak, which lowers the energy barrier with sufficiently higher selectivity for the CO2RR than for the HER.
Furthermore, Tang and coworkers used a computational approach applying the grand canonical potential kinetics (GCP-K) method92 for an N-doped graphene-based Fe&Ni@g-N6 dual catalyst and reported an interesting reaction mechanism for the CO2RR.93 They suggested that the first step has to be the adsorption of CO2 on the Fe–Ni center with a free energy change of −0.14 eV followed by the attack of the hydroxyl group on the activated *CO to form the *COOH intermediate via cis- and trans-pathways, which further undergoes facile dehydration to form the desired product, *CO. Typically, the last step should be the desorption of *CO but it was proposed that the Fe–Ni center had a high adsorption energy of −2.22 eV for *CO, making its desorption impossible. Thus, CO adsorbed on the catalyst surface passivates the Fe–Ni center. Interestingly, *CO (A) coordinated to FeNi@g-N6 behaved as a new catalyst to promote further adsorption of another CO2 molecule and formed a new *CO (B) coordinated to FeNi@g-N6. After the formation of *CO (A) and *CO (B) on FeNi@g-N6, one of the CO molecules undergoes facile desorption with a desorption energy of 0.85 eV. Further study of the kinetics of the competing reactions revealed the dominance of the eCO2RR over the HER.
In another report, Wu and coworkers discussed a ZIF–NC–Ni–Fe catalyst synthesized by developing a two-step approach for metal adsorption (i) by trapping metal ions in a nitrogen-doped micro-porous ZIF substrate and (ii) thermal activation instead of classical pyrolysis (Fig. 9a).94 HAADF-STEM data showed strong evidence of the presence of Ni and Fe in close-proximity (Fig. 9b). The DFT suggested three possible structures out of which 2N-bridged-(Fe–Ni)–N6 was found to be the most efficient as shown by the free energy diagram (Fig. 9c). ZIF–NC–Ni–Fe exhibited a maximum FECO of 97.8% at −0.6 V and TOF of 2210 h−1 at −0.9 V. It was proposed that the metal content was <2%. In general, the precursor mass ratio should be between 3:1 and 1:3 for Ni/Fe to achieve efficient results.
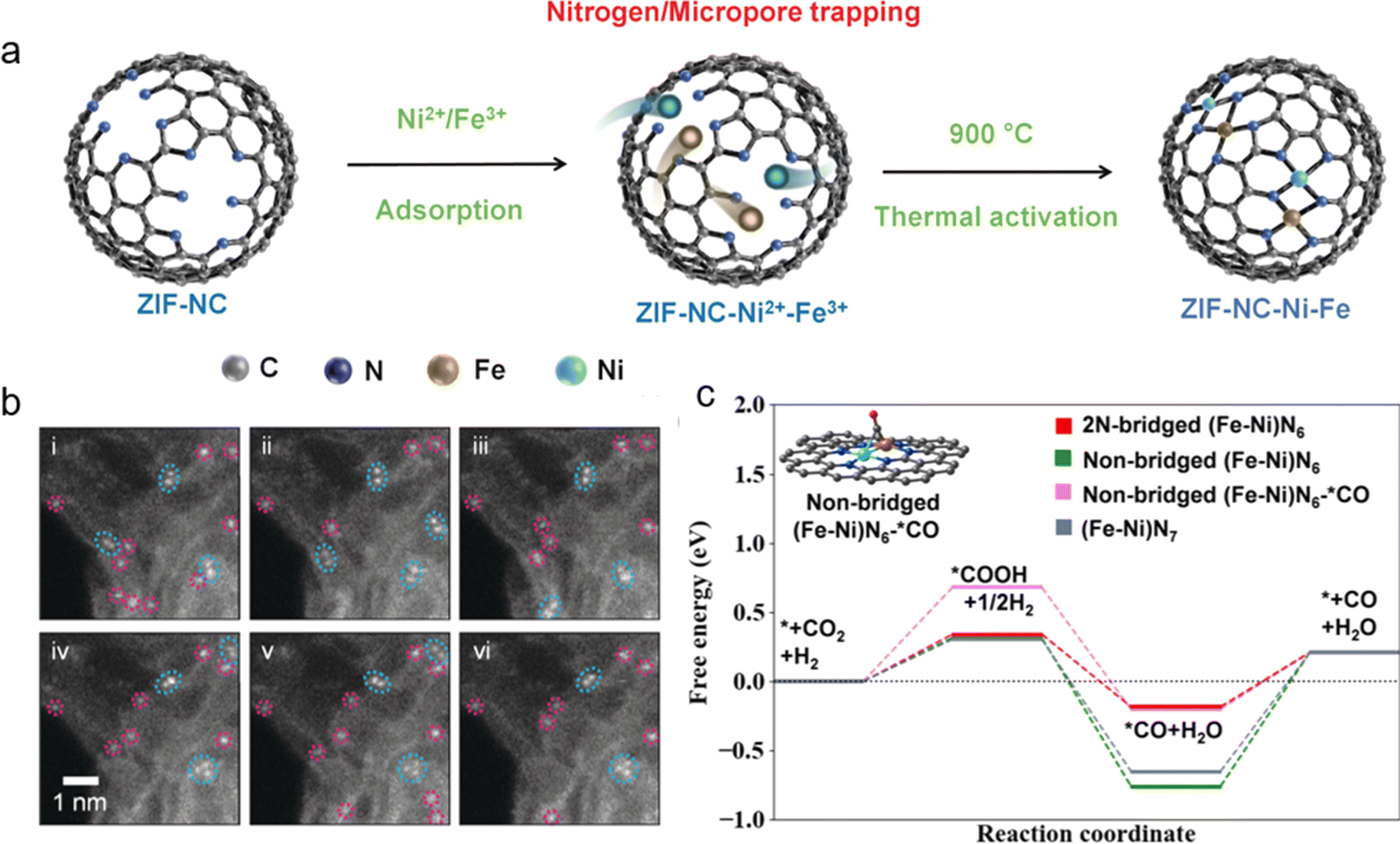 | ||
| Fig. 9 (a) Scheme of the possible formation of dual metal Ni–Fe sites in the ZIF–NC–Ni–Fe catalyst. (b) Multiple HAADF-STEM acquisitions, demonstrating metal atom pairs (cyan), and individual atoms (magenta). (c) Predicted free energy evolution of the CO2RR on various dual metal Fe–Ni sites. Reprinted (adapted) with permission from ref. 94. Copyright© 2022, Wiley-VCH GmbH. | ||
As discussed above, hetero-atom doping has helped in the enhancement of the DACs' activity. Most of the experiments emphasize the importance of N-doping. In contrast, Hu and coworkers manifested the positive effect of co-doping of S and N in a Fe–Ni DAC for the CO2RR process.95 The DFT study has helped understand the effect of S-coordination on regulating the d-band center of Fe–Ni DAC with a decrease in the ΔG value of the PDS. Further for investigating the result of DFT studies they fabricated S and N co-doped Fe–Ni–NSC DAC along with only N-doped Fe–Ni–NC DAC. AC-TEM and HAADF-STEM data accounted for the successful formation of dual sites along with mixed valence states between 0 and +2 for both Fe and Ni atoms. ICP-OES indicated 0.45 and 0.42 wt% of Ni and Fe respectively and the dual atoms are present in N2SFe–S–NiN2 coordination. The EXAFS data proved the absence of Fe–Fe and Ni–Ni bonds whereas the predicted Fe–Ni bond length was ∼2.4–2.5 Å. Electrochemical analysis gives an excellent catalytic activity between the potential window of −0.6 to −1.0 V with a maximum FECO of 96% at −0.9 V having a total current density of 40.1 mA cm−2 showing robust stability after 12 hours of testing. Furthermore, it was found that the S-coordinated DAC has a d-band center distant from the Fermi level as compared to only the N-doped one, making Fe in Fe–Ni–NSC lose fewer electrons than in Fe–Ni–NC. As a result, a weakening in CO2 adsorption was observed which decreases the ΔG value of the CO desorption step, PDS from 1.27 eV to 0.53 eV making CO desorption easier in Fe–Ni–NSC. Thus, this work highlights the importance of S doping in effectively increasing the catalytic activity for the eCO2RR.
After Ni–Fe, Ni–Co heteronuclear DACs were reported to be promising candidates for the eCO2RR by Zhuang and coworkers.96 The synthesis of N-bridged Co–N–Ni supported N-doped porous carbon nanosheets was performed (Fig. 10a), which showed a maximum of 96.4% FECO at −0.48 V and a high TOF of 2049 h−1 at a low over-potential of 370 mV. The characterization results from HAADF-STEM, AC-STEM, and EDS mapping showed about ∼70% dual sites with 0.24 wt% metal content with atomically dispersed atoms. The HAADF-STEM imaging showed the surface with interesting graphene-like wrinkles and ripples as depicted in Fig. 10b. The absence of direct bonding between Ni–Ni, Co–Co, and Ni–Co atoms was shown by EXAFS (Fig. 10e and f). The catalyst displayed excellent stability over 20 hours of electrolysis. The high performance of the catalyst was attributed to the electronic tuning effect of the N-bridged Co–N–Ni bimetallic sites. DFT methods illustrated the formation of *COOH as the potential limiting step (PLS). The Co/Co–N–Ni catalyst showed the lowest free energy of 0.56 eV for PLS (Fig. 10d). Thus, this work underlines the importance of electronic tuning in enhancing the synergistic effects of DACs.
 | ||
| Fig. 10 (a) The structure model of N-bridged Co–N–Ni bimetallic sites in Co–N–Ni/NPCNSs: Ni (cyan), Co (pink), N (blue) and C (gray). (b) HAADF-STEM image showing ripples. (c) FECO. (d) Free energy diagrams for the CO2RR to CO.95 (e and f) EXAFS spectra at the (e) Co K-edge and (f) Ni K-edge. | ||
In recent studies, Wang and coworkers used the pyrolysis-induced cation exchange method to synthesize Co–Ni–N–C with a high FECO of 95% at −0.8 V and high durability even after 40 hours of testing.97 Interestingly, to facilitate reaction activation, the microporosity of the catalyst served as the defect sites and played a crucial role in the CO2RR. The XPS results revealed that Ni atoms in Co–Ni–N–C behave as electron donors to the neighboring Co atoms.98 Computational study reported a Ni–Co bond distance of 2.4 Å and predicted two possible pathways: either the *CO gets desorbed from the active Co–Ni site or the second CO2 molecule gets reduced while the first CO molecule has already been bound to the active site, leading to passivation of the catalyst.
A wise combination of metals in heteronuclear DACs was proven to be crucial in enhancing the electrocatalytic activity (Fig. 11). Considering the Ni and Ag combination, the strong CO binding affinity of Ni poses a risk of CO poisoning or impedes CO desorption,99 while Ag demonstrates the capability to effectively suppress the HER albeit with a high energy barrier for the formation of *COOH.100 In light of this, Xu and colleagues devised atomically dispersed Ni–Ag DACs supported on defective nitrogen-rich porous carbon (Ni–Ag/PC–N) through cascade-anchored pyrolysis.101 The HAADF-STEM image confirmed uniform dispersion (Fig. 12d). The ICP-MS indicated Ni and Ag loadings of 0.46% and 6.60% respectively in Ni–Ag/PC–N. The XANES and EXAFS indicated the presence of the N3–Ni–Ag–N3 coordination motif in the DAC. The XPS results illustrated that the oxidation state of Ni falls within 0 and +1 andAg is in the +1 oxidation state. The electrocatalytic study exhibited an outstanding FECO of 99.2% at −0.8 V in an aqueous solution of 0.1 M KHCO3 as shown in Fig. 12e. The collaborative action of Ni and Ag in the DAC mitigated inherent challenges, thereby reducing the potential of the RDS and enhancing the catalytic activity and selectivity (Fig. 12f). Furthermore, the porous and defective nature of the support material contributed to augmenting an exceptional catalytic activity.102
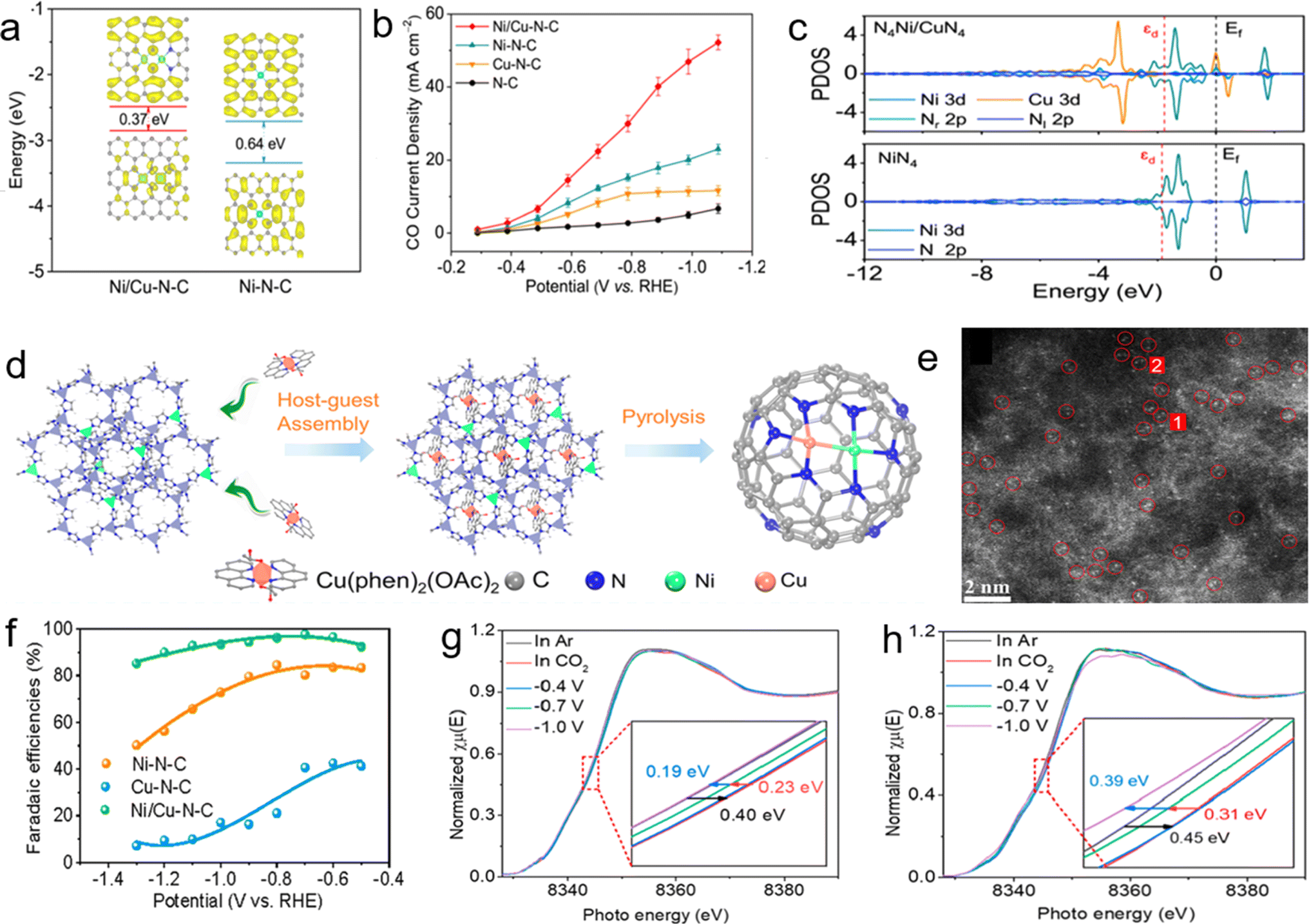 | ||
| Fig. 11 (a) HOMO and LUMO spin electron densities of the proposed Ni/Cu–N–C and Ni–N–C models. (b) CO partial current density of the prepared catalysts. (c) Calculated partial density of states (PDOS) for Ni of N4Ni/CuN4 and NiN4. Reprinted (adapted) with permission from ref. 103. Copyright© 2021, American Chemical Society. (d) Schematic illustration of the synthetic procedure of the diatomic Ni/Cu–N–C catalyst. (e) Aberration-corrected HAADF-STEM image of Ni/Cu–N–C. (f) Faradaic efficiency for CO production. (g and h) Normalized operando Ni K-edge XANES spectra for (g) Ni/Cu–N–C and (h) Ni–N–C at various biases (applied voltage vs. RHE).105 | ||
 | ||
| Fig. 12 (a) Magnified atomic-resolution HAADF-STEM image of the Ni–Zn atomic pair; individual Zn or Ni atoms are marked with red, orange, and green circles, respectively. (b) Comparison of FE of CO and H2. (c) The free energy of each intermediate state on the metal atom sites in Zn–N4–C, Ni–N4–C, and ZnNi–N6–C. Reprinted (adapted) with permission from ref. 107. Copyright© 2021, Wiley-VCH GmbH. (d) AC-HAADF-STEM images of Ni–Ag/PC–N. (e) CO faradaic efficiency of eCO2R from −0.7 V to −1.3 V vs. RHE. (f) Gibbs free energy diagram of the eCO2R to CO process.101 | ||
Individually Ni-SAC and Cu-SAC exhibit HER suppressing activity and they are kinetically not very active for the first electron–proton transfer step in the eCO2RR to form th *COOH intermediate. He and coworkers reported a novel Ni/Cu–N–C catalyst comprising ∼74% well-dispersed diatomic moieties with a distance of ∼2.6 Å between adjacent atoms.103 Further XPS elucidated the valence states, where peaks of Cu appeared between +1 and +2 states and peaks of Ni appeared between 0 and +2 states. Surprisingly, there was a downward and upward shift in the binding energy of Cu and Ni respectively, which suggested the interaction of metal atoms with each other and electronic structure modifications in the two metal atoms. Moreover, Ni/Cu–N–C showed a narrow HOMO–LUMO gap, which leads to enhanced catalytic activity (Fig. 11a). The catalyst demonstrated over 95% FECO from −0.39 to −1.09 V vs. RHE, bearing an outstanding FECO of 99.2% and current density of 29.9 mA cm−2 at −0.79 V, which was 1.97, 2.78 and 10.6 times higher than that of Ni–N–C, Cu–N–C, and N–C respectively (Fig. 11b). The catalyst exhibited excellent stability even after 60 hours of continuous electrolysis with a TOF of 3960 h−1 at −0.79 V. The DFT study revealed that adsorption was carried out on the Ni atom site and Cu was boosting its activity by narrowing the d band gap of Ni ions in Ni/Cu–N–C as shown by the partial density of states (PDOS) in Fig. 11c and proceeds with increased electron mobility.104 Thus this work also highlights the positive effect of electronic modulations of DACs as suggested in the previously discussed work of Hu and coworkers.95
A similar type of synergistic effect had been seen in the study by Chen and coworkers.105 The authors delved into mechanistic insights into the CO2RR catalyzed by different metal atom pairs Ni–M (M = Zn, Co, Cu, Cr, Mn, Ti, and V) using DFT methods. All the NiMN6 structures are found to be more stable as compared to the pristine NiN4 site due to the lowering of Gibbs free energy making *COOH formation exothermic. Thus, instead of *COOH formation, the *CO desorption step was the RDS. Concerning this RDS step only NiCuN6 (0.59 eV) and NiMnN6 (0.44 eV) proved to be more efficient than the other heteroatom DACs. Furthermore, on the comparison basis of the CO2RR vs. the HER, only NiCuN6 was found to be sufficiently selective for CO2 reduction. To validate the efficacy of the computational study, Ni/Cu–N–C was synthesized by a host–guest strategy (Fig. 11d).47 The HAADF-STEM data (Fig. 11e) showed a Ni–Cu distance of about 2.4 Å and the energy shifts in the XPS spectra of the catalyst accounted for strong Ni–Cu coupling interactions. The catalyst displayed a superior FECO of 97.7% with a current density of 13.7 mA cm−2 at −0.7 V and a high TOF of 20695 h−1 at −0.6 V (Fig. 11f). The Ni/Cu–N–C catalyst was found to be stable even after 60 hours of continuous electro-analysis. Operando characterization (Fig. 11g and h) displayed a decline in the FECO below −0.8 V due to less active over-reduced Ni species. Cu not only enhanced the catalytic activity of Ni but also protected Ni from being over-reduced. The weak electron distribution between Ni and Cu could be considered quasi-covalent bonding which built up the fundamental understanding of the synergistic effects between dual atoms.
Interestingly, Qiao and coworkers further described the dependence of synergistic effects on intermetallic distance.106 Primarily, DFT calculations were performed using four models of dNiCu-d with four different intermetallic distances, 2.6 Å, 4.1 Å, 4.9 Å, and 5.3 Å. During the study of the effect of intermetal interactions on electronic structures, it was observed that there is a negative correlation between the net electronic effect (|Δe|) and the inter-metallic distance (d). In the case of dNiCu-d (2.6 Å), the most electron depletion was found on Cu (≈0.08e) compared to Ni (≈0.02e). If the intermetallic distance increases, |Δe| becomes negligible. In this case, the threshold reaches 5.3 Å to stimulate effective intermetal electronic interactions. To verify DFT findings experimentally, a Ni–Cu DAC on N-doped graphene (NiCu–NC) was synthesized by pyrolyzing the precursors with Cu and Ni salts under an argon atmosphere. The ICP-MS confirms Ni and Cu metal loadings of ∼0.67 and 0.88 wt% in Ni–Cu DAC and HAADF-STEM images validate the presence of Ni–Cu bond with a distance of ∼0.51 nm. When the catalytic activity was tested in a H-cell using 0.1 M KHCO3 then ∼98% FECO at −1.07 V vs. RHE was observed with a TOF of 681 h−1 at −1.27 V. The experimental study also endorses the correlation between inter-metal distance and electrocatalytic performance.
Continuing the study on Ni-based heteronuclear DACs, Lu and coworkers investigated a Ni–Zn catalyst utilizing a competitive complexation approach using metal chlorides and chitosan.107 The ICP-MS analyses confirmed Ni and Zn loadings of 0.84 and 1.32 wt% respectively. XANES analysis revealed that both metals exhibited valence states ranging between +1 and +2. The EXAFS curves indicated a Ni–Zn–N6–C coordinating structure with a shorter interatomic distance of 2.5 Å that endorses stronger interatomic interactions with the lowest energy and highest fitting quality for NiZn–N6–C. The FT-EXAFS and WT-EXAFS spectra showed the presence of electronic interactions between Ni and Zn, which modulate the electronic structure of Ni–Zn–N6–C for enhanced intrinsic activity and selectivity during the CO2RR. Notably, electronic structural alterations were evidenced by the shifting of Ni and Zn K-edge towards lower energy indicating the loss of fewer electrons in NiZn–N6–C for Ni as well as Zn. The DFT study reveals that the free energy barrier for NiZn–N6–C is lower than that of their SACs as shown in Fig. 12c. The NiZn–N6–C catalyst effectively reduced the free energy for *COOH formation and increased free energy for *CO at a low overpotential of −0.05 V by facilitating the adsorption of C atoms on Ni and one of the O atoms on Zn, respectively. The catalyst exhibited exceptional performance, achieving a maximum FECO of 99% at −0.8 V, while maintaining FEH2 < 10% (Fig. 12b). Furthermore, the catalyst demonstrated a current density of −20 mA cm−2 and exhibited good stability for 28 hours. These results underscore the significance of mechanistic understanding and rational design in achieving efficient catalysis.
Further emphasizing Zn-based heteronuclear DACs, Gong and coworkers synthesized Zn/Co–N–C DAC by pyrolysis and post-acid treatment.108 HAADF-STEM accounted for uniform atomic distribution with 76.2% dual spots and 22.8% isolated spots. EXAFS and XANES data show the absence of any direct bonding between Zn and Co atoms as the distance between these two atoms was found to be 0.28 nm with an average valence state between 0 and +2 for both Co and Zn. The XPS data reveal 0.35 wt% of Zn and 0.72 wt% of Co in Zn/Co–N–C DAC. Electrochemical analysis of the catalyst in saturated KHCO3 aqueous solution shows an excellent FECO of 93.2% having a current density of 26 mA cm−2 at −0.5 V and an outstanding stability even after testing for 30 hours. Furthermore, the DFT study elaborates that the electronic effects of neighboring Zn and Co atoms decrease the energy barrier of both *COOH adsorption and *CO desorption steps, thus favoring the eCO2RR process. Hence, this work manifested the synergic electronic effects between Zn and Co in optimizing intermediate adsorption and thus enhancing the catalytic efficiency.
There is another piece of evidence of Zn-based heteronuclear DAC studied by Feng and coworkers containing phthalocyanine (Pc) based layered 2D conjugated MOFs (PcM–O8–M1, M = Cu or Zn, M1 = Cu or Zn) using DFT methods. The DFT study revealed that the formation of *COOH is the RLS. PcCu–O8–Zn was found to have low overpotential and was the most efficient for selective CO formation.109 To endorse the theoretical results, PcCu–O8–Zn1 DAC was synthesized by the solvothermal approach, which constitutes phthalocyanine-copper as a ligand and a zinc-bis(dihydroxy) complex as the linkage. The XRD pattern accounted for the layered MOF structure and the XAS and EXAFS data prove the presence of Cu and Zn in +2 states. At −0.7 V, FECO reaches up to 88% with a high TOF of 0.39 s−1 when tested electrochemically. It also exhibited the lowest Tafel slope of 125 mV dec−1 which demonstrates its faster kinetics and manifested good stability after 10 hours of testing. Operando XAS and SEIRAS analyses suggested that the eCO2RR occurs at the Zn metal center whereas Cu helps synergistically in proton–electron transfer during the reaction. Thus, this work sheds light on the importance of electrocatalytic applications of well-designed MOFs in the CO2RR process.
Further exploring the synergistic effects between dual metal atoms, He and coworkers synthesized N4Fe–CuN3 with a coordination environment by using a MOF assisted strategy.110 The HAADF-STEM images reveal that the inter-atomic distance is 0.26 nm. The XPS suggests the presence of 0.29% Fe and 0.18% Cu, and also during the formation of diatomic sites the valence state of Cu decreases whereas for Fe it increases. The K-edge XANES spectra of Fe and Cu indicated oxidation states of Fe between 0 and +3 and for Cu, it lies between 0 and +2. The EXAFS fitting results confirmed an average bond length of ∼2.0 Å in N4Fe–CuN3 (Fig. 13c and d). An electrocatalytic study showed an excellent FECO of 99.2% at −0.8 V (vs. RHE) with a TOF of 5047 h−1 (Fig. 13). The reduced energy barrier (0.22 eV) for the PLS (potential limiting step) that is *COOH formation was confirmed by DFT study. The shifting of the d band center closer to the Fermi level causes the weakening of the bonding interaction with *CO to facilitate the desorption of CO from the surface to inhibit CO poisoning which is generally shown by iron catalysts. This study illustrates that precisely constructed DAC exhibits synergic effects efficiently leading to proper positioning of the d band center.111
 | ||
| Fig. 13 (a) SEM image of the Fe–Cu catalyst. (b) TOF of Fe/Cu–N–C, Fe–N–C, and Cu–N–C at various applied potentials. (c) Fourier transformation of the EXAFS spectra in R space. (d) EXAFS fitting for Fe/Cu–N–C in R space.110 | ||
In another significant study on heteronuclear DACs, Yang and Liu studied four types of combinations of Fe, Ni, Co, and Cu based on different N-dopings namely M1M2-3A, M1M2-3S, M1M2-4A, and M1M2-4S through the DFT study.112 Interestingly it was found that the N4-coordinated DAC weakly adsorbs CO2 as compared to the N3-coordinated DAC. Usually, linear scaling relationships which are generally observed in the case of metal surfaces and SACs are expected to break in DACs. However, in this case, it was found to be the opposite for M1M2-3A, M1M2-4A, and M1M2-4S DACs, whereas the M1M2-3S type DAC breaks scaling relations as expected. A universal descriptor has been found by including intrinsic properties such as the number of d electrons, electronegativity of the metal, and coordinating atoms. Applying this descriptor, CoCu-3A, CoCo-3A, FeNi-3A, and FeFe-4S were found to have low limiting potentials. However, CoCo-3A exhibited excellent catalytic performance during the DFT study. It was also found that the formation of HCOOH was thermodynamically controlled, whereas, CO formation was kinetically controlled. The pH and applied potential also significantly contribute to the eCO2RR and have a great influence on the PDS and UL. Hence, this work highlights some major theoretical insights into the catalytic performance of Fe, Ni, Co, and Cu-based heteronuclear DACs.
Syngas synthesis
In the above electrocatalytic studies, CO was obtained as a major product along with H2 in minor quantities as a by-product. Syngas (CO + H2) is a highly valuable feedstock for many chemical reactions as well as in the production of fuels. To get selective formation of syngas, the fine-/tuning ratio of CO and H2 is required. This was successfully demonstrated by Zhang and coworkers by synthesizing porous carbon-supported NixFe1−x–NC catalysts by varying Fe and Ni contents.113 The XPS endorsed the presence of Fe(II) and Ni(II) and HAADF-STEM indicated atomically dispersed Ni and Fe in Ni0.5Fe0.5–NC. There was a significant increase in the FECO production as well as the current density of CO by increasing the x value from 0 to 0.08 in NixFe1−x–NC whereas by increasing Fe content, the current density of H2 production increased. Thus, by adjusting the x, the CO/H2 ratios were changed from 0.14 to 10.86 at −0.86 V vs. RHE. The CO/H2 ratio tuning could be achieved by modulating the nominal Ni/Fe ratio as well as by controlling the applied potentials. An eight-hour of catalyst durability test showed good stability of NixFe1−x–NC. DFT calculations indicated a difference in the adsorption preferences for Fe and Ni, where Ni proved to be effective for CO production and Fe proved to be effective for H2 production. Hence by combining the differently selective active sites, it is possible to achieve controllable catalytic performance to get syn gas.The Ni/SAC supported on N-doped carbon exhibited favorable activity to CO evolution whereas Co/SAC supported on N-doped carbon showed exclusive activity for the HER. Hence balancing Co/Ni in a suitable ratio for effective production of syngas is required. In this context, Chen and co-workers designed a CoNi–NC DAC for efficient syngas yield with a total current density >74 mA cm−2 at −1.0 V vs. RHE.114 The XANES data displayed 0 and +2 oxidation states of Co and Ni, respectively. Modulating the two metal sites for balanced CO2RR/HER for getting tunable CO2/H2 (0.25–3.3) without sacrificing the high total current densities is the key to efficient eCO2RR.
Lanthanides have also been incorporated in DACs in this context,115,116 Du and coworkers reported syngas synthesis using Zn and Ln DACs.117 The CO/H2 ratios by tuning the amount of metals in DACs have been successfully demonstrated. Impregnation and annealing were used to anchor metals on thin carbon nitride nanosheets. Two DACs were developed, ZnLa-1/CN with Zn:La close to 2:1, which was confirmed as per the local coordination data by EXAFS, and ZnLa-2/CN with Zn:La close to 1:2. Zn and La were responsible for CO and H2 production respectively. The catalyst showed a FEsyngas of 80% (Fig. 14c) and ZnLa-1/CN demonstrated FEH2 twice that of FECO exhibiting a CO/H2 ratio of 0.5 suitable for further methanol synthesis. ZnLa-1/CN exothermically favored (−0.94 eV) CO formation whereas ZnLa-2/CN showed an endothermic trend (0.72 eV) but both showed exothermic behavior of H2 formation (1.34 and 2.01 eV) respectively (Fig. 14d).
 | ||
| Fig. 14 (a) Plot depicting the current density of H2versus current density of CO. Reprinted (adapted) with permission from ref. 114. Copyright© 2020 Wiley-VCH GmbH. (b) AC HAADF-STEM images of dual atomic catalyst ZnLa-1/CN. (c) The FE and corresponding syngas ratio of ZnLa-2/CN. (d) Energy profile for the formation of CO and H2O.117 | ||
CO2 reduction to C1 hydrocarbons
In addition to CO, various other C1 products arise from the eCO2RR, encompassing formate, formic acid, methane, and methanol, among others. These products hold significant utility and importance across diverse applications.118 Formic acid, for instance, demonstrates broad synthetic versatility and serves as a prospective medium for hydrogen storage. Methanol stands out as a crucial liquid fuel and superior hydrogen storage material for a cleaner energy source, while methane fuels industrial operations and household power systems. The formation of these products predominantly hinges upon the interaction of CO with the metal catalytic site; weak binding may result in the desorption of CO, whereas strong metal binding facilitates further hydrogenation. Additionally, post-CO reduction, the relative adsorption strength of oxygen dictates whether methanol, methane, or formic acid predominates as the principal product. Formic acid production entails a 2-electron reduction process, whereas methanol and methane result from 6-electron and 8-electron pathways, respectively.119 In this exploration, we examined the existing dataset and observed a notable dearth of experimental research. Consequently, the documented data stand to offer significant utility in the strategic formulation and prediction of the efficacy and specificity of forthcoming DACs unearthed through ongoing research endeavors.CO2 reduction to formic acid
Formic acid has a high-profit value and low energy input. Some main group elements have been reported, which had shown a great selectivity for formate production from CO2.120 Tin was found to have good efficiency in converting CO2 to formate.121 Wei and coworkers reported that a Ni–Sn atomic pair catalyst supported on a hierarchical N-doped carbon nanosheet array developed by the impregnation and pyrolysis method resulted in an N4–Ni–Sn–N4 configuration.122 The HAADF-STEM image indicates the homogeneous dispersion of atoms (Fig. 15a). The authors proved the synergic effects between Ni–Sn in the DAC by studying the change in one metal after doping it with the other: an increased oxidation state of Sn (positively charged) after introducing Ni and a decreased oxidation state of Ni after introducing Sn. DFT calculations proposed both the metals as active sites, N4–*Ni–Sn–N4 as well as N4–Ni–*Sn–N4, where *Sn (ΔG = −0.05 eV) was thermodynamically more favorable for CO2 to *OCHO conversion than *Ni (ΔG = 0.57 eV) as shown in Fig. 15c and also predicted the possible reaction mechanism scheme. It was noticeable that the energy barrier for *OCHO (−0.05 eV) was much less than that for *COOH (1.36 eV) on *Sn, which was probably the reason for formate formation instead of CO. Due to the synergic effect of Ni over Sn *OCHO formation gets favored (−0.05 eV) as Sn-SAC didn't show equivalent activity (0.78 eV). The catalyst exhibited a FEformate of 86.1% at −0.82 V (vs. RHE) and TOF of 4752 h−1 with maximum formate production up to 36.7 mol h−1 gSn−1 (Fig. 15b). | ||
| Fig. 15 (a) HAADF-STEM image of NiSn-APC. (b) Faradaic efficiencies for formate at various applied potentials. (c) Free energy diagram for production of formate. Reprinted (adapted) with permission from ref. 122. Copyright© 2021 Wiley-VCH GmbH. (d) Eads of all reaction intermediates involved in the CO2 hydrogenation to HCOOH process on SnMN6/G. (e) Optimal reaction pathway for HCOOH synthesis on SnMN6/G. Reprinted (adapted) with permission from ref. 126. Copyright© 2023, American Chemical Society. (f) LH mechanism for CO2 activation. | ||
Further exploring the activity of Sn in formate formation, Chen and coworkers introduced a Cu–Sn DAC by annealing followed by an impregnation process over CeO2−x nanorods having abundant oxygen vacancies which stabilized the Cu and Sn atoms as revealed by (DFT+U) coulomb corrected DFT calculations.123,124 The data from XANES predicted a +4 state for Sn and Cu lies between the +1 and +2 state and it also showed Cu–O and Sn–O coordination, which was supported by DFT analysis and WT plots. Electrochemical testing using a H-cell (0.5 M KHCO3) indicated a current density of 76.2 mA cm−2 and FEHCOOH of 98.1% at −1.2 V vs. RHE. Further analysis using a flow cell displayed a TOF of 4320 h−1 and a remarkable HCOOH formation of 2324.7 μmol h−1 cm−2. DFT revealed the formation of a thermodynamically stable *OCHO intermediate on CuSn/CeO2−x as well as the low energy RDS to generate HCOOH (0.24 eV) and then to form CO (0.38 eV). This work also highlighted the importance of oxygen vacancies in the electrocatalytic CO2 reduction process.
Sn in selective HCO2H production, environment-friendly behavior, and low cost of Sn(II)-based catalysts make them attractive candidates for the eCO2RR.125 Ma and Chen theoretically studied a series of Sn–M DACs.126 DFT study was used to set up models for five systems of Sn–M (M = Fe, Co, Ni, Cu, Zn) on N-doped graphene. SnMN6/G configuration was found stable for all five systems as demonstrated by formation energy analysis. SnCuN6/G was found to be most stable (−7.14 eV) and SnCoN6/G has relatively weaker stability (−6.49 eV). ICOHP values revealed a direct relation between the atomic number and the bonding strength between Sn and the transition metal. Sn was the major active site and it depicted an electrophilic nature in all five whereas Co and Ni were behaving as nucleophilic sites and Fe, Cu, and Zn also showed an electrophilic nature. Adsorption energy values of CO2 and H2 were very close (Fig. 15d). The LH (Langmuir–Hinshelwood) mechanism was predicted as shown in Fig. 15f. Furthermore, as there were two possible intermediates, by attack of H* on the C atom in CO2, the HCOO* intermediate is formed whereas if H* attacked the O atom in CO2, formation of COOH* occurred. The adsorption strength and activation energies were calculated and HCOO* was found to be more favorable. The catalytic activity of SnZnN6/G was found to be optimal (Ea of the RDS = 0.98 eV) due to good charge transfer ability (Fig. 15e).
The excellent performance of tin as the active metal site for CO2 to formic acid formation has already been established. Recent reports by Cucinotta and coworkers showed the first principle study of SACs and DACs using tin disulfide monolayers (SnS2) as the support material, which also proved to be effective in formic acid production.127 Thus, these reports further encouraged the exploration of the combination of p-block and d-block elements in CO2RR activity.
After Sn, bismuth, a p-block element, was also found to have a high selectivity for formic acid. Bi has a lot of perks including sluggish HER performance over the eCO2RR enhancing its selectivity towards formate formation, along with environmental friendliness and low cost. Bi has a positive standard reduction potential (Bi3+/Bi, 0.308 V vs. SHE) and can be effectively used as a cathode catalytic material.128 The weak binding of Bi with the *OCHO intermediate impedes the formation of formic acid, but using a Bi-metal DAC can effectively work, providing suitable binding energy for O in *OCHO through the synergic effects of the DACs. Various reports have been found where Bi-doped materials, Bi nanosheets, and Bi alloys are proven to be efficient in formate production but the area of Bi-based SACs and DACs has still not been explored much.129–132
However, Li and Tang used computational methods to explore Bi DACs supported on N-doped graphene (g-N6) along with other elements.133 According to their study, Bi on N-doped graphene was found to be quite stable as depicted by the study of formation energy and dissolution potential values (Fig. 16a). In a homonuclear Bi2 DAC the distance between central metals was found to be 3.45 Å. The authors studied the limiting potentials of various metals for different pathways and Bi manifested good selectivity for the formic acid formation pathway compared to other metals as shown in Fig. 16b.
 | ||
| Fig. 16 (a) DFT-computed dissolution potential and formation energy of homonuclear DACs, where the red, blue, and green areas represent the unstable, quasi-stable, and stable DACs. (b) Thermodynamic limiting potentials toward C1 products CO (orange) and HCOOH (wine), as well as the competitive H2 product (olive) on homonuclear DACs.133 | ||
Furthermore, the binding energies of the critical intermediates on Bi2 DAC demonstrate the stability of the formate intermediate to get the desired result (Fig. 17). Thus, Bi-containing DACs have a great potential to reduce CO2 to formic acid selectively. The theoretical data provided by Li and Tang support a brighter future for Bi DACs.
 | ||
| Fig. 17 Binding energies of the critical intermediates on Bi2 DAC.133 | ||
The HCOO* intermediate involved in formic acid formation also proceeds towards methane and methanol formation. There are several instances where DACs provide methane and methanol efficiently from CO2.
CO2 reduction to methane and methanol
While Cu has shown promising performance in reducing CO2, it still faces challenges such as high overpotential and low efficiency, largely due to the competitive HER.134 Chen and coworkers studied a Cu dimer anchored on a C2N layer using spin-polarized DFT calculations in an acidic environment.135 Out of two predicted structures, Con-1 (Cu atoms on the same side) and Con-2 (Cu atoms on different sides), atomic relaxations revealed high stability of Con-2 over Con-1 and was thus used for further studies. The structural stability of Cu anchored on the C2N layer was confirmed by larger adsorption energy (−3.58 eV) than Cu cohesion energy (−3.38 eV). Moreover, PDOS accounted for the hybridization between Cu-3d and N-2p orbitals making the (Cu2@C2N) structure stable. On further exploring the reaction pathways (Fig. 18d), two paths were found but path 2 was proposed to be more favorable as the HCOO* formation step was exothermic (−0.20 eV) while COOH* formation is endothermic (0.18 eV). Thus, the most suitable pathway was:| CO2 → HCOO* → HCOOH* → H2COOH* → H2CO* → H2COH* → CH2* → CH3* → CH4 |
 | ||
| Fig. 18 Free energy profile for the CO2RR to CH3OH and CH4 on Cu2@C2N at (a) 0.00 V and (b) −0.23 V. Reprinted (adapted) with permission from ref. 135. Copyright© 2018, American Chemical Society. (c) Energy diagrams of CO production and further CO hydrogenation on different MN@2SV DACs at 0 V vs. RHE. (d) Proposed reaction paths for CO2 electrochemical reduction on Cu-based catalysts, producing methane (CH4) and methanol (CH3OH). (e) Screening of CO2 electroreduction intermediate (CO, COOH, CHO, H, O, and OH) adsorption for CO on different MN@2SV DACs. Reprinted (adapted) with permission from ref. 136. Copyright© 2015, American Chemical Society. | ||
HCOO* to HCOOH* was found as the PDS with a limiting potential of −0.23 V (Fig. 18a and b). The enhanced activity of Cu was attributed to its moderate interaction with intermediates resulting in low limiting potential. Cu had a higher energy barrier for the HER than CH4, and even the HER can be suppressed more by using non-aqueous solutions and adjusting the pH of the electrolyte.
Sun and coworkers pursued DFT screening using the VASP to investigate the vacancy effect of the graphene support using Fe dopant pairs and interestingly the adjacent single vacancy (Fe2@2SV) was the most effective for intermediate adsorption.136 Further screening of various metal dopant pairs was performed. Among these DACs, MnCu@2SV and NiCu@2SV were found to be highly selective for CH4 and CH3OH generation (Fig. 18e). CHO was found to be more stable than CO resulting in further CO reduction, and the free energy barrier of CO to CHO step was reported to be 0.71 eV and 0.64 eV on NiCu@2SV and MnCu@2SV respectively which was even less than that of the reported effective Cu(111) catalyst (Fig. 18c). Selective product formation is due to their different oxophilicities. This study predicts that graphene-based substrates work well for CH3OH formation.
Looking into the selectivity and efficiency insights, Shi and coworkers performed theoretical calculations using the DFT and VASP by making DAC pairs from metals such as Cu, Ru, and Fe for electrocatalysis.137 The pairs M1–M2 (Cu–Cu, Fe–Fe, Ru–Ru, Ru–Cu, Ru–Fe, and Cu–Fe) were anchored on monolayer graphitic single triazine-based C3N3 (g-CN) with large holes in the structure, where the M atoms got adsorbed and bonded to three N atoms. Out of all six pairs, only Cu–Fe maintained a flat surface, whereas the remaining five showed slight distortion in their structures. AIMD simulations indicated the high thermodynamic stability of these pairs under thermal conditions of the CO2RR. This study revealed the selectivity effect of the already discussed two pathways (formate and carboxyl).138 Except for RuRu@g-CN (0.31 eV), the other five were observed to follow the OCHO* intermediate pathway. The potential limiting step was either OCHO* to OCH2O* or *CO to *OCH and it was reported that a decrease in catalytic activity was observed with an increased pH value and only RuCu@g-CN and RuFe@g-CN were proved to have good selectivity (99.86% and 99.95% respectively) for the CO2RR vs. the HER. RuCu and RuFe DACs exhibited low limiting potentials of −0.40 V and −0.58 V respectively for the formation of CH4. Surprisingly a very interesting statement was proposed that the average of individual adsorption-free energies of homonuclear DACs is nearly equal to the adsorption-free energy of corresponding heteronuclear DACs.
Furthermore, Sarkar and Ghoshal reported theoretically analyzing Ru2 and Rh2 DACs dispersed on TiO2 and g-CN (g-CN being more efficient) supports but practically Rh is highly expensive and these dimers suffered from poor selectivity for the CO2RR than for the HER.139 Hence they replaced one noble metal from Ru2 DAC with a transition metal and designed M–Ru DACs (M = Sc to Zn) on g-CN support using the VASP. Thermodynamic stability was screened by comparing their average binding energies to average cohesive energies and it was found that except for Cu–Ru and Zn–Ru, all other DACs are stable. Comparison of DACs for the CO2RR and HER was performed based on Gibbs free energy values (Fig. 19a). Further calculations depicted that V–Ru had shown the most efficient active site for CO2 to CH4 conversion exhibiting a high theoretical FECH4 of 99.57%. Although three catalysts V–Ru, Cr–Ru, and Ti–Ru demonstrated excellent activity and selectivity for CH4 generation with low limiting potentials (UL) of −0.11 V, −0.27 V, and −0.29 V respectively (Fig. 19b), VRu@g-CN was found to be the best and promising catalyst.
 | ||
| Fig. 19 (a) Comparison between the free energy of hydrogen adsorption ΔG(*H) and initial protonation steps of CO2 to form *COOH/*HCOO intermediates ΔG(*COOH/*HCOO). (b) Correlation between the limiting potential (UL) of the CO2RR and the adsorption strength of CO2 (in eV) over MRu@g-CN (M = Sc–Ni) catalysts. Reprinted (adapted) with permission from ref. 139. Copyright© 2024, American Chemical Society. | ||
Owing to the renowned activity of Ni in the CO2RR, An and coworkers combined the DFT and CHE (computational hydrogen electrode) model to explore a Ni–Co DAC supported by an optimized structure of nitrogenated holey 2D graphene (C2N-h2D) and comparison with homonuclear Ni2 and Co2 dimers was performed.140 Two bonding arrangements were identified but only the asymmetric N2Ni–CoN2 configuration demonstrated superior performance as compared to the symmetric one, as indicated by geometric parameters. The strong binding strength of Ni–Co dimers thermodynamically supports the possibility of experimental synthesis. The bent shape of the adsorbed CO2 molecule (∠OCO < 180°) having a negative partial charge signifies sufficient activation of the CO2 molecule. The NiCo@C2N catalyst undergoes a formate formation pathway with a low energy barrier of 0.25 eV (via *HCOOH intermediate) instead of a high barrier of the *CHO intermediate to form CH4. The heteronuclear Ni–Co catalyst showed superior behavior to Ni2 and Co2 dimers (Fig. 20b–d) in CO2RR selectivity over the HER (Fig. 20e) and it has a comparatively low energy barrier for O* removal in the form of water and this superiority was attributed to the synergy of dual heteroatoms. This study also highlighted that the HER can be suppressed by increasing the pH up to 7 and by high CO2 pressure along with confirming the good impact of co-adsorbed *CO, *OH, or *H in CO2 reduction instead of the expected deactivation of metal sites by them.
 | ||
| Fig. 20 (a) DFT-optimized structures along the primary reaction pathway of the CO2RR toward CH4 formation, red rectangle: PDSs. (b–d) Free energy profiles of the CO2RR toward CH4 formation (b–d) and the HER (e) at 0 V (vs. RHE). Reprinted (adapted) with permission from ref. 140. Copyright© 2019, American Chemical Society. | ||
Further exploring suitable DACs for efficient methane formation from CO2, Mei and coworkers computationally studied twenty-one combinations of six different transition metals (Fe, Cu, Ni, Pd, Pt, and Au) anchored on an MOF-808-EDTA support, where metal atoms are stabilized by N atoms of EDTA ligands.141 Gibbs free energy for CO2 adsorption showed only MOF-808-EDTA-FeFe (−1.33 eV), MOF-808-EDTA-FePt (−0.47 eV), and MOF-808-EDTA-NiFe (−0.21 eV) to be exergonic and these catalysts exhibited a self-adaptive and flexible nature by adjusting their M–M, M–O, and M–C bond lengths. The Gibbs free energy comparison of H*, COOH*, and HCOO* intermediates proved the suppression of the HER over the CO2RR only in Fe–Fe (ΔGCOOH* = −1.04 eV and ΔGH* = −0.27 eV) and Fe–Pt (ΔGHCOO* = −1.35 eV and ΔGH* = −0.33 eV) catalysts. Metal atoms get reduced by electron transfer from the substrate and, further, these active centers transfer the electrons to CO2 resulting in bent CO2− and got activated. Calculations from the free energy profile demonstrated the formation of the COOH* intermediate (−1.04 eV) and CO* to CHO* was the RDS (+0.56 eV) in the case of the Fe–Fe catalyst whereas the Fe–Pt catalyst went through the formation of the HCOO* intermediate and the HCOOH* to CHO* step was the limiting step (+0.35 eV) (Fig. 21a and c). Thus, this study revealed mechanistic insights for the formation of C1 products and also accounted for the enzyme-like dynamic ability and adaptability which stabilized the reaction intermediates in DACs.
 | ||
| Fig. 21 (a) Gibbs free energy profiles for the electroreduction of CO2 to CH4, HCOOH, CO, and CH3OH over MOF-808-EDTA-FeFe. (b) Evolution of configurations with varying values of dFe–Fe (left Y axis) and θ2N–2N (right Y axis) in the CO2RR process. (c) Gibbs free energy profiles for the electroreduction of CO2 to CH4, HCOOH, CO, and CH3OH over MOF-808-EDTA-FePt. (d) Evolution of configurations with varying values of dFe–Pt (left Y axis) and θ2N–2N (right Y axis) in the CO2RR process. Reprinted (adapted) with permission from ref. 141. Copyright© 2023, American Chemical Society. | ||
Tandem catalysis142 is a good strategy in the case of multistep reactions to get complex products; however, in the case of the eCO2RR, random CO migration inhibits tandem catalysis. However, Zhuo and co-workers thought of a way out and reported atomically “inner tandem” catalysis for CO2 reduction by using computational studies.143 The authors designed cobalt-containing heteronuclear Co–N–M DACs (where M = Cr, Mn, Fe) through a N bridged structure. The catalyst successfully proceeded within a combinatorial pathway with Co as the active site and it outperformed traditional tandem catalysis in a way that it provided directional reasonable migration to intermediates. Co–N–Cr was found to be most efficient with the lowest thermodynamic energy barrier of 0.35 eV and kinetic barrier of 0.09 eV for intermediate migration for CH4 production. Thus, adjustable intermediate migration enhanced the activity of the process.
In another theoretical study, Wang and co-workers investigated twenty-six transition metal DACs anchored on 2D extended Pc for methane formation.144 The authors first examined the stability on the grounds of formation energy (Ef), the difference between binding and cohesive energy (ΔEb), and dissolution potential (UL), and 22 DACs were reported to be stable. Further investigations revealed that only 8 out of 22 DACs were found to be better in the CO2 adsorption study. Delving into the catalytic mechanism of Mo2-Pc, it was found that CO2 to CH4 reduction required six steps, an eight-electron pathway, and the O* species left behind were reduced to water in two steps. Mechanistic insights indicated *COOH to *CO conversion to be the PDS and the three dimers, Mo2-Pc, W2-Pc, and Ti2-Pc, had shown a small limiting potential (UL) of −0.57, −0.59, and −0.81 V vs. CHE respectively (Fig. 22). Further diving into the origin of CO2 activation, it was found that after adsorption of CO2 on the M2-Pc substrate e− gets transferred from M to CO2 and the antibonding orbital of CO2 gets shifted towards the Fermi level and it gets activated. Selectivity of the CO2RR concerning the HER had been checked and Mo2-Pc was found to be highly selective for CH4 formation from CO2. Thus, this study established thorough mechanistic insights into the eCO2RR process.
 | ||
| Fig. 22 Free-energy profiles for the CO2RR at zero applied voltage vs. CHE. (a–c) CO2RR to the CH4 product on (a) Mo2-Pc, (b) W2-Pc and (c) Ti2-Pc. (d) Limiting potential diagram of the CO2RR to CH4. The red dashed line is the measured UL (−0.84 V) of the CO2RR to CH4 on the Cu(211) surface. Reprinted (adapted) with permission from ref. 144. Copyright© 2024 Wiley-VCH GmbH. | ||
Manifesting the significance of DACs in the electrocatalytic reduction of CO2 to C1 products, Lu, and coworkers theoretically designed the FeCo–NC DAC along with the Fe–NC and Co–NC SAC for a thorough comparison.145 The authors reported better electron transfer ability, high CO2 adsorption energy (−0.64), and large deviation in the O–C–O bond angle (137°) for FeCo–NC, which promoted efficient CO2 activation. Furthermore, it demonstrated a feasible six-electron pathway for methanol formation and an eight-electron pathway for methane formation as shown in Fig. 23a and b. The intermediate CO formed here helped in orbital interactions between the d orbital of metal and 2π* orbital of CO which led to the formation of a d–2π* occupied bonding orbital below the Fermi level which in turn stabilized CO and further reduction was possible by this CO assisted FeCo–NC catalyst. Thus, this work theoretically emphasizes efficient electron transfer and orbital interactions in DACs which proved beneficial for the eCO2RR to get C1 products other than CO.
 | ||
| Fig. 23 (a) Free energy diagram of the CO2RR to CH3OH via the six-electron pathway. (b) Free energy diagram of the CO2RR to CH4via the eight-electron pathway. (c) Volcano relation between limiting potential for the CO2RR to CH3OH and CH4 and Eads(CO) on Fe–NC/Co–NC, FeCo–NC, and CO assisted FeCo–NC. Reprinted (adapted) with permission from ref. 145. Copyright© 2023 Wiley-VCH GmbH. | ||
As discussed in the CO formation, the inherent vacancy defects in the substrate proved to be effective for stable anchoring of metal atoms and may help synergistically in the electrocatalytic process.146 Tang and coworkers explored MoS2 with S vacancies as the support material and tested twenty-one metal dimer systems, including homonuclear as well as heteronuclear catalysts using DFT and machine learning (ML).147 All the DACs had shown high thermodynamic stability with negative formation energy ranging from −4.92 to −6.16 eV and AIMD simulations also accounted for the dynamic stability. It was found that CO2 binding energy lies between −0.45 and −0.98 eV for all the catalysts and MoS2–NiCr was screened to have the highest CO2 adsorption energy (−0.98 eV) resulting in the bent structure of CO2. In MoS2–NiCr two metal atoms were bound to the two oxygen atoms of CO2 whereas all other catalyst samples had binding between only one metal and oxygen. Only NiCr and CrCo had shown the desired low overpotential (0.58 and 0.44 eV respectively) for CH4 formation. So, further calculations were performed for these two only. Talking about the mechanistic insights, the *CHO intermediate pathway was more favorable than the *COH, and CH4 generation needed lower energy than other C1 products. In NiCr both metal centers played a key role whereas in CrCo only Cr was doing so. Both of these DACs have shown excellent catalytic performance. In the case of CrCo DAC, eCO2RR vs. HER details showed poor selectivity as compared to the HER. In ML analysis, seven basic parameters were considered out of which two were found to be highly crucial in the eCO2RR, which are intermetallic distance (dM−M) and the number of outer electrons of two metal atoms (Ne1 and Ne2). These data provided powerful atomic-level insights for designing DACs in the future.
Recently, Zhang and coworkers constructed a DFT-based active learning (AL) framework i.e. DFT and ML by selecting the best algorithm followed by three iteration loops, which predicted the limiting potential (UL) and uncertainty (σ).148,149 Once uncertainty σ ≤ 0.20 V, the iteration was terminated, and UL and descriptors were analyzed to find a correlation. A total of 282 possible catalysts (M1–M2–N6-Gra) were analyzed by considering the characteristics of elements as well as the synergic effects between the dual atoms to decide the descriptors (X). Transition metals were selected as M1, whereas p block metals as M2 keeping the coordination environments completely identical. 42 (M1–M2–N6-Gra) catalysts were filtered through the three iteration loops; among them 29 had HCOOH selectivity screening success >70% with errors below 0.20 V. On comparison with Bi (012), the five best candidates (Pt–Bi, Au–Bi, Hf–Pb, As–Sb, and Ni–Bi) were screened, out of which three candidates (Pt–Bi, Hf–Pb, and As–Sb) were kinetically and thermodynamically stable and it was concluded that large positive charge on the active metal site is effective. Thus, this study promotes the use of machine learning algorithms for material exploration for the eCO2RR.
Chen and coworkers studied a unique conformation of DACs on graphene, inverse sandwich structures previously known only in lanthanide–boron clusters.150,151 This inspired the authors to work on similar structures for transition metal DACs to understand the structure, stability, and mechanism of working. Extending their previous study on the eCO2RR, the authors studied five homonuclear M2DACs (M = Co, Ni, Rh, Ir, and Pt) on graphene.152 The results indicated that Ni, Rh, and Pt DACs efficiently converted CO2 to CH3OH while Co and Ir proved efficient in the formation of CH4.
The study also comprises 125 heteronuclear DACs, where one metal belongs to M (M = Co, Ni, Rh, Ir, and Pt) while the other metal, M′ belongs to Sc to Au. The binding energy calculations of these DACs with the defective graphene layer provided 88 stabilized DACs for further first principle molecular dynamics (FPMD) simulations. The FPMD study provided 31 structurally stable and rigid DACs with negative adsorption energies. Furthermore, the authors used ML using Gradient Boosting Regression (GBR) to unravel the catalytic performance of new DACs. All combinations of the 28 transition metals (Sc to Au) producing 784 DACs were studied using the binding and cohesion energy difference (ΔE) and limiting potentials (UL) as the descriptor (Fig. 24).
 | ||
| Fig. 24 ML-predicted heat map of UL (Volt) values for 784 MM′⊥gra. Reprinted (adapted) with permission from ref. 150. Copyright© 2023, American Chemical Society. | ||
Three DACs (RhPt, RhIr, and RuPt) were found to have UL values below 0.4 V making them the best candidates. This inference was also supported by the outcomes of FPMD simulations. This study presents a vast thorough analysis of TM-DACs and highlights the feasibility of their results being useful for the eCO2RR.
CO2 reduction to C2+ products
In addition to C1 products, some DACs have shown the formation of multi-carbon products. In particular, C2 and C2+ through tandem mechanisms are involved in the eCO2RR. In general, C–C coupling to get higher hydrocarbon derivatives is a difficult task because activation energy barriers for such conversions are very high, making the C–C coupling unfavorable.153 The *CO formation is the key intermediate to get multi-carbon products as the C–C coupling can go through the *CO–COH pathway as it is more favorable than the well-known *CO dimerization pathway. The kinetic barriers of C–C coupling can be decreased significantly by increasing the degree of intermediate hydrogenation. Copper is the only metal found to convert CO2 to multi-carbon products with decent efficiency.154–157 This research has opened doors for efficient conversion of CO2 to higher hydrocarbon-containing fuels in the future. By following this thought and the fact that DACs can show better per-atom efficiency than traditional heterogeneous catalysts, new Cu-based DACs were synthesized and studied for the CO2RR. Although multi-carbon products were detected in the Cu-catalyzed CO2RR, Cu-based DACs generating C2+ products effectively were a rare case.158–160 C2+ product formation requires the coupling of two reduced CO2 substrates such as *CO–*CO, *CO–*COH/*CHO, *COH/*CHO, and *COH/*CHO. The M–CO binding should not be too strong as it would undermine the C–C coupling step and if the M–CO binding is too weak then it would promote CO release. Thus, fine-tuning of DACs is a crucial process in increasing the efficiency of the formation of C2+ products.Han and coworkers successfully converted CO2 to higher derivatives of alcohols using nitrogen-doped graphene quantum dots on Cu nanorods (NGQ/Cu-nr).161 The XPS study of NGQ depicted the presence of three components of N present: pyridinic (63%), pyridonic (12%), and graphitic (25%). After the formation of NGQ/Cu-nr, the amount of pyridinic N was reduced to 41% while the pyridonic N was increased to 26% pointing towards the adsorption of Cu-atoms in the pockets containing pyridinic N to get 5 wt% of Cu loading. The reaction was performed in a flow cell with 1 M KOH as electrolyte and polytetrafluoroethylene (PTFE) membrane with 220 nm pore size. The FEC2+ for the CO2RR reached up to 80.4% with a current density of 282.1 mA cm−2 at −0.9 V vs. RHE (reversible hydrogen electrode) (Fig. 25). They converted this into a fuel cell system by adding IrO2 as the anode for the oxygen evolution reaction generating an efficiency of 51.6% at 3.6 V for the formation of C2+ alcohols, mostly ethanol and propanol.
 | ||
| Fig. 25 (a and b) The distribution of C2+ products at different potentials over NGQ/Cu-nr and Cu-nr. (c) Partial current density versus maximum C2+ alcohol FE for various catalysts. (d) FE of C2+ alcohols and ethylene on different catalysts at a potential of −0.9 V versus RHE. The stars show the corresponding FE ratios of alcohols to alkenes. (e) The current density and FE of C2+ products on NGQ/Cu-nr at −0.9 V vs. RHE with 100-hour potentiostatic electrolysis tests. Reproduced from ref. 161, Copyright© 2020, Wiley-VCH GmbH. | ||
The DFT analysis revealed a synergistic effect between Cu and the NGQ that boosted the C2+ alcohol production. Previous reports have shown the use of NGQ alone for the conversion of carbon dioxide to C2 products with high selectivity, but NGQ was kinetically inert due to low current density.162,163 On further delving into the mechanism the existence of the intermediate *C2H3O was found, which was responsible for the formation of ethylene as well as ethanol depending on the metal–O bond strength of the intermediate. This study highlighted the formation of *C2H3O supported by NGQ which made the process exergonic (−0.26 eV). The DFT and experimental study established that the lowering of the energy barrier below the standard ethylene formation barrier resulted in the formation of ethanol.
To achieve a high selectivity in the formation of ethylene only Cu was considered the active center. Zhang and coworkers utilized a unique strategy of using an iridium porphyrin-based metal–organic framework (MOF) as the support for Cu-SAs@Ir-PCN-222-PA (PCN = porous coordination network).164 Detailed characterization study revealed that there was no formation of homonuclear DACs of Cu+ and Cu2+ as there was no evidence of the presence of a Cu–Cu bond or Cu–Cu proximity (Fig. 26a). The Fourier transform extended X-ray absorption fine structure (FT-EXAFS) revealed the coordination of the Cu-atom with two oxygen and one nitrogen atom while the Ir-atom was locked in PCN-222 (Fig. 26b). Through ICP-OES, the Cu loading was determined to be 1.8 wt%.
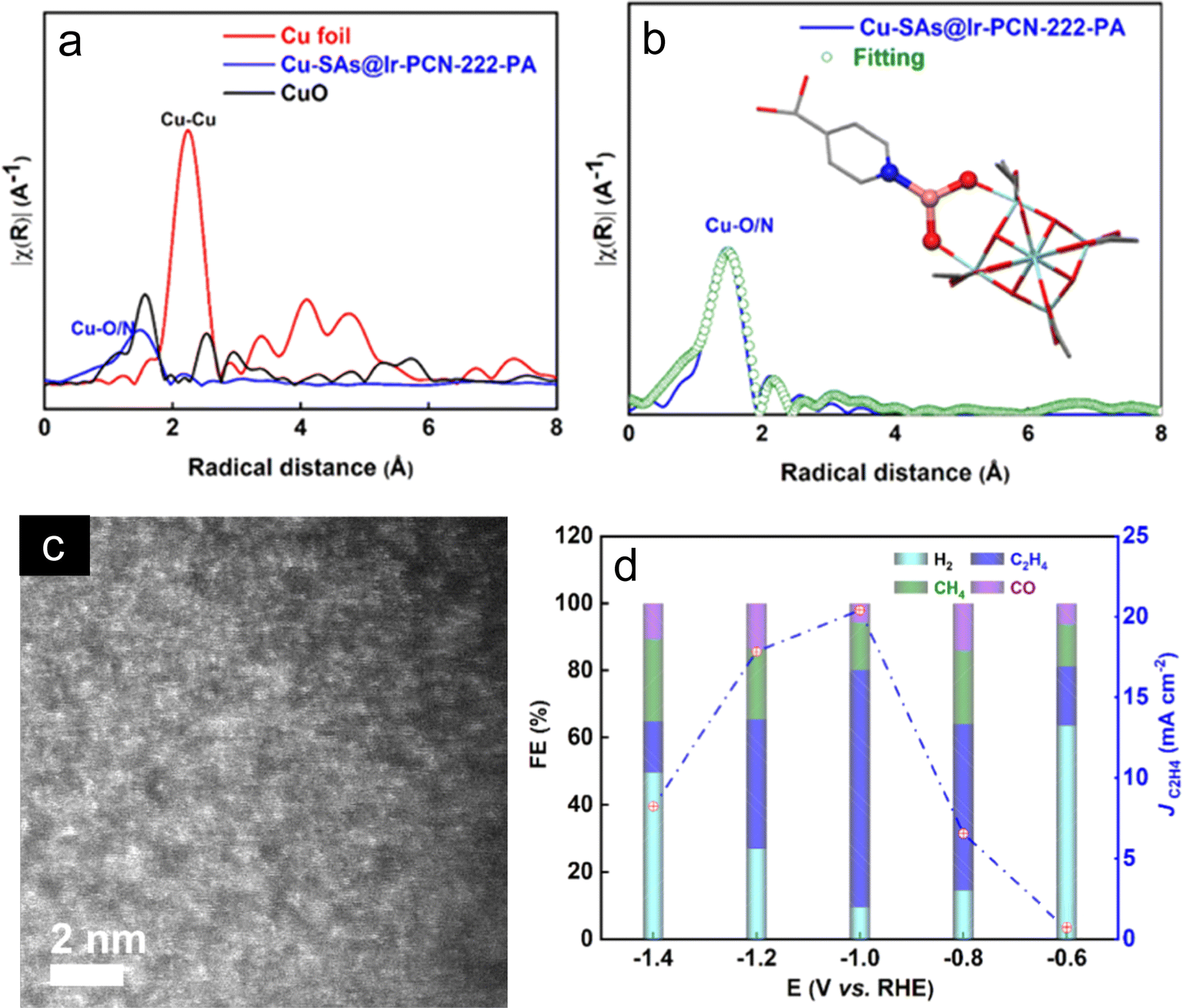 | ||
| Fig. 26 (a) EXAFS of Cu-SAs@Ir-PCN-222-PA, CuO, and Cu foil at the Cu K-edge and (b) EXAFS fitting curves from R space results of Cu-SAs@Ir-PCN-222-PA. (c) Aberration-corrected HAADF-STEM images of Cu-SAs@Ir-PCN-222-PA. (d) Faradaic efficiency (FE) and C2H4 partial current densities (red circle) on Cu-SAs@Ir-PCN-222-PA at various applied potentials. Reprinted (adapted) with permission from ref. 164. Copyright© 2024, American Chemical Society. | ||
The electrochemical catalytic study revealed the dominance of ethylene as the product between the voltage range of −0.8 V and −1.2 V with the best results (FEC2H4 = 70.9%) at −1.0 V vs. RHE at a current density of ca. 20 mA cm−2 (Fig. 26d). No alcohols or higher degree hydrocarbons were detected showing high selectivity of the electrocatalyst towards ethylene formation. When a similar reaction was performed using Ir-PCN-222 it displayed conversion of CO2 to CO which further reduced to CH4 without forming any C2+ products. The Fourier transform infrared spectroscopy (FTIR) analysis disclosed the formation of the Ir–CO bond (1918 cm−1) in addition to the Cu–CO bond (2040 cm−1) indicating the capability of Ir in CO2 reduction. Also, the IR band for C–O at 1542 cm−1 pointed towards the formation of a Cu–COCHO intermediate. This validated the involvement of both Ir and Cu in the formation of ethylene, endorsing the synergistic effects of dual atoms in the eCO2RR as discussed in previous studies.
The DFT was employed to understand the mechanism involved in the formation of ethylene and the role of iridium in the process. The studies revealed a synergistic behavior between copper and iridium in reducing CO2 to CO while in the case of Cu, protonation of *CO leads to *COH and desorption and diffusion of CO from the Ir-centre to the Cu-centre take place leading to the C–C coupling of CO and *COH yielding *COCHO. Furthermore, consecutive protonation and dehydration cycles lead to the formation of ethylene. It is worth noting that the mechanistic steps involved in Zhang and coworkers' study go through a different intermediate than the *C2H3O intermediate discussed by Han and coworkers for the formation of ethylene and ethanol indicating either interconnected or tandem pathways for the formation of similar products.
Ethanol is considered to be the most important C2 product due to its applications in energy and fuel and its liquid nature making transportation easy. Chen and coworkers worked on the electroreduction of CO2 (eCO2RR) to get higher yields of ethanol. The electrocatalyst Cu–Sn DAC (CuSn–HAB) was synthesized by using 2D-MOF of hexaiminobenzene (HAB), Cu2SO4, and SnCl2.61 CuSn–HAB, as the working electrode in a flow cell (1 M KOH as electrolyte), exhibited excellent performance toward the eCO2RR to ethanol conversion. The reaction was performed at a voltage as low as −0.57 V vs. RHE with a FEEtOH of 56% and a current density of 68 mA cm−2. These results were good but with the addition of the fact that no ethylene was formed throughout the reaction, the catalyst proved to be highly selective towards ethanol formation. Further exploration by the authors revealed that the catalyst without Sn, Cu–HAB, was only capable of forming CO at such low potentials promoting the role of Sn in further reduction of CO and C–C coupling of intermediates to form ethanol. The mechanistic investigation via DFT enlightened few key intermediates that significantly affect the selectivity of the product: (a) reduction of CO2 to CO on Cu while reduction to OCH2 on the Sn atom, (b) C–C coupling of CO and OCH2 on the Sn-atom and (c) further formation of thermodynamically favourable OCH2CH3 on the tin center. The ethylene formation was calculated to be 2.72 eV, thermodynamically unfavourable under the given conditions which align with the experimental proof.
Copper catalysts are known for efficient CO2RR but are challenged by high overpotential and low faradaic efficiency.165,166 Chen and coworkers performed computational analysis for a Cu-dimer on the C2N layer for the eCO2RR to study the effect of Cu-DAC on N-doped graphene.135 They proposed interconnected reaction pathways for the formation of methane, methanol, and ethylene (Scheme 1). The proposed CuSn DAC binds through the MOF structure within a proximity distance of 2 Å without forming a Cu–Sn bond (Fig. 27). The ethylene formation mechanism was studied at zero potential, and promising results were found for the eCO2RR with the most important step being CO–CO coupling with 0.76 eV energy. This proved that hydrogenation of *CO to *COH (1.16 eV) or *CHO (1.44 eV) was thermodynamically unfavorable. Their study of competing reactions revealed the HER to be at 0.71 eV, which is slightly lower than that of ethylene formation (Fig. 27). To resolve this, they proposed adjustment of the electrolyte pH and the use of non-aqueous solutions to suppress the HER process.167 This method of adjusting the pH to suppress or enhance the efficacy of the HER in DACs has been thoroughly studied by Fan and coworkers for tungsten-based DACs.168
 | ||
| Fig. 27 (a) AC-HAADF-STEM image. (b) Wavelet transform (WT) contour plots of EXAFS for CuSn–HAB at the copper K-edge. (c) Free energy profile for the CO2RR to C2H4 on Cu2@C2N at 0.00 and −0.76 V. (d) Free energy profile for the HER on Cu2@C2N at 0 and −0.71 V. Reprinted (adapted) with permission from ref. 135. Copyright© 2023, American Chemical Society. | ||
Following Chen's work, Zhang's group169 and Zheng's group170 performed the eCO2RR using a Cu-dimer on a MOF surface with quite discrete outcomes. Zhang and coworkers used boron-imidazolate framework (BIF) as the support. They formed two catalysts on MOF nanosheets: Cu2[BH(mim)3]2Cl2 (BIF-102, mim = 2-methylimidazole) and Cu2[BH(mim)3]2HCOO2 (BIF-103). The eCO2RR was performed in 0.5 M KHCO3 electrolyte to give a current density of 12.1 mA cm−2 at 0.94 V vs. RHE (Fig. 28). The FEC2+ for BIF-102 at −1.0 V vs. RHE was ca. 12% while that of BIF-103 was near 8%. Interestingly all these electrocatalytic reactions provide only ethylene with traces of ethane.
 | ||
| Fig. 28 (a) LSV curves of electrocatalysts in CO2 saturated 0.5 M KHCO3 electrolyte. (b) The FE of valuable gas and liquid products of BIF-102NSs electrocatalysts at different applied potentials. (c) The FEC2H4 of BIF-102NSs, BIF-103NSs, and BIF-104NSs electrocatalysts at different applied potentials. (d) Comparison of the CO2RR gas production activity between electrocatalysts at −1.0 V vs. RHE in CO2 saturated electrolyte. Reproduced from ref. 169, Copyright© 2021 Wiley-VCH GmbH. | ||
Zheng and coworkers synthesized Cu and CuNi SACs on the MOF surface similar to Zhang's work but on a C–N MOF (BTC MOF) through pyrolysis.170 The EXAFS proved the two metals to be in the vicinity of each other at ca. 1.8 Å, and also HAADF displayed the metals to form in pairs indicating the formation of dual SACs or DACs. Their results were much more promising as the FEC2+ for Cu-SAC was 91% constituting acetate (33%), ethylene (32%), ethanol (∼20%), and n-propanol (∼5%). These results were produced at a comparatively high potential of −1.66 V vs. RHE and a high current density of 90+ mA cm−2 (Fig. 29). These experimental results were further supported by a DFT study indicating a barrier of 0.54 eV for ethylene formation, displaying better performance than any SAC, thus indicating the presence of synergistic effects between the metals as in DACs. These results reported by Chen, Zhang, Zheng and their coworkers on Cu-SACs indicate that finely synthesized MOFs are a better option for a support at higher potentials. Thus, DACs with tuned MOFs can provide better results for C2+ product formations.
 | ||
| Fig. 29 (a) HAADF-STEM image of the dual Cu SAC. The dual Cu atomic sites are highlighted with red circles, and (b) Cu K-edge EXAFS fittings of dual Cu SAC. (c) Partial current densities on dual Cu SAC at different working potentials, and (d) faradaic efficiencies of C2+ products on dual Cu SAC at different potentials. Reprinted (adapted) with permission from ref. 170. Copyright© 2021, American Chemical Society. | ||
Mei and coworkers performed a computational study on a DAC–EDTA complex anchored on MOF-808 by using a series of metals (M1/M2) such as Fe, Cu, Ni, Pt, and Au as metals for DAC (MOF-808-EDTA-M1M2).141 It was found that the formation of MOF-808-EDTA-FeFe, MOF-808-EDTA-FePt, and MOF-808-EDTA-NiFe was thermodynamically favourable, with Gibbs free energy values of −1.33, −0.47, and −0.21 eV, respectively. All catalysts were studied for comparison of the CO2 to COOH/HCOO formation vs. the HER, with MOF-808-EDTA-FeFe and MOF-808-EDTA-FePt being better at the eCO2RR than at the HER (Fig. 30).
 | ||
| Fig. 30 (a and b) Calculated Gibbs free energy profiles for CO2 electroreduction to C2H6 (dark), C2H4 (red), and C2H5OH (navy) over MOF-808-EDTA-FeFe and MOF-808-EDTA-FePt, respectively. (c and d) Configuration evolution with varying values of dFe–Fe and dFe–Pt (left Y axis) and θ2N–2N (right Y axis), respectively in the CO2RR process. Reprinted (adapted) with permission from ref. 141. Copyright© 2023, American Chemical Society. | ||
Thus, these two catalysts were further explored for the CO2 reduction process, which revealed the inefficiency of the catalysts facilitate protonation of the *CO group to *COH (MOF-808-EDTA-FeFe ΔG = 1.9 eV; MOF-808-EDTA-FePt ΔG = 2.02 eV). This further proceeded through the *CHO–*CO coupling step to form ethanol, ethylene, and ethane. The energy barriers calculated at zero potential are given in Table 2.160 The DFT study endorsed that only MOF-808-EDTA-FeFe and MOF-808-EDTA-FePt catalysts were the most efficient for the eCO2RR to get ethanol and ethane.
| Catalyst | Desorption energy (G) | ||
|---|---|---|---|
| Ethanol | Ethene | Ethane | |
| MOF-808-EDTA-FeFe | 0.87 eV | 1.28 eV | 0.87 eV |
| MOF-808-EDTA-FePt | 0.95 eV | 1.66 eV | 0.66 eV |
Men and coworkers explored the catalytic potential of Ni2@C2N, Co2@C2N, and NiCo@C2N as representative models to examine how the cooperation of metallic dimer sites influences the eCO2RR using the DFT.140 They found that these dimer sites are stabilized within the 2D C2N thin layer through the creation of polarized and hybridized metal–N bonds, maintaining their structural integrity even at temperatures up to 500 K, particularly for NiCo@C2N and Co2@C2N. The collaborative action of metallic dimers was elucidated by examining the electronic resonance of 3d-states within Ni–Ni, Co–Co, and Ni–Co dimer sites, as well as their shortened metallic bond lengths. Interestingly, co-adsorbed species such as *H, *OH, and *CO did not deactivate the metallic dimer sites, thereby ensuring the availability of active sites for a continuous eCO2RR cycle and facilitating multiple competing reaction pathways. Among these catalysts, NiCo@C2N, with a limiting potential of −0.25 V, was identified as the most effective for catalyzing the CO2RR towards methane (CH4) formation, followed by Co2@C2N (UL = −0.30 V) and Ni2@C2N (UL = −0.67 V). Though the catalysts depicted good efficacy for CH4 formation, the formation of C2 products such as ethylene was relatively low due to the substantial energy required for intermediate formation across all three catalysts (Fig. 31). The high stability of *CO ruled out the possibility of C–C coupling products.
 | ||
| Fig. 31 (a) DFT-optimized structures along the primary reaction pathway of the CO2RR toward C2H4 formation. (b–d) Free energy profiles of the CO2RR toward C2H4 formation at 0 V (vs. RHE). (e) Two optimized isomeric structures of the COCOH intermediate. COCOHa: decoupled configuration; COCOHb: coupled configuration. Reprinted (adapted) with permission from ref. 142. Copyright© 2019, American Chemical Society. | ||
Singh and coworkers performed computational analysis on a series of metal DACs supported on N-doped graphene for the eCO2RR.171 This study constituted a comparative study of twenty-one DACs and their respective bulks. It was found that the DACs were more stable than the bulk. DFT calculations were performed to find an efficient descriptor for preliminary studies and rule out the DACs that didn't follow the given conditions. The first descriptor applied was the binding energy of the metal to *OH (G(*OH)), *H (G(*H)), and *CO (G(*CO)); strong binding (<−0.9 eV) indicates diminished CO2 hydrogenation efficiency of the DACs. Thus, Cr–Cr, Mn–Mn, and Cr–Mn DACs were ruled out due to strong G(*OH) and G(*H) binding energies. Similarly, more descriptors at a threshold of 0.75 eV, as given in Fig. 32, were applied to reach the best DACs that could perform the CO2RR to yield C2 products. Each bond in the structure describes a heterometal DAC while the lobes on the metal indicate a homonuclear DAC. The group further tracked the transition state for the coupling of *CO–*CHO in all the best catalysts, considering an energy barrier of 0.75 eV and below to be efficient for the coupling step.
 | ||
| Fig. 32 Every bond in the network describes a heteronuclear DAC while the lobes on metals describe a homonuclear DAC. The descriptors define the path while the catalysts above the threshold value are ruled out from further analysis. | ||
Thus, the reaction paths and the limiting potentials calculated showed nine promising catalysts with −0.80 V or higher limiting potential. In the coupling step, six DACs (Cr–Cu, Mn–Cu, Co–Ni, Co–Cu, Ni–Cu, and Co–Co) exhibited energy barriers lower than the threshold value of 0.75 eV. All of these six DACs have a higher selectivity for the C2 process between the HER and CO2RR compared to Cu(211)172,173 and Au(211).174 The AIMD results of the six DACs with the minimum limiting potential and acceptable energy barrier revealed that all of them exhibit high thermal stability at 500 K.
Similar work was conducted by Li and coworkers by performing a comprehensive computational study to examine the efficacy of twenty-six dimeric metal DACs. Through rigorous activity and selectivity screening, Mo2, W2, and Re2 DACs were found to show comparatively good activity among the 26 DACs for ethylene formation by the eCO2RR (Fig. 33). The FE is 68.57% for Mo2-DAC and ca. 100% for Re2-DAC, which is higher than that of the Cu(111) surface, with a higher limiting potential (UL) of −1.4 V to 0.8 V vs. RHE.144 Although the FE for ethylene formation is high, the high limiting potential confines the application of these DACs.
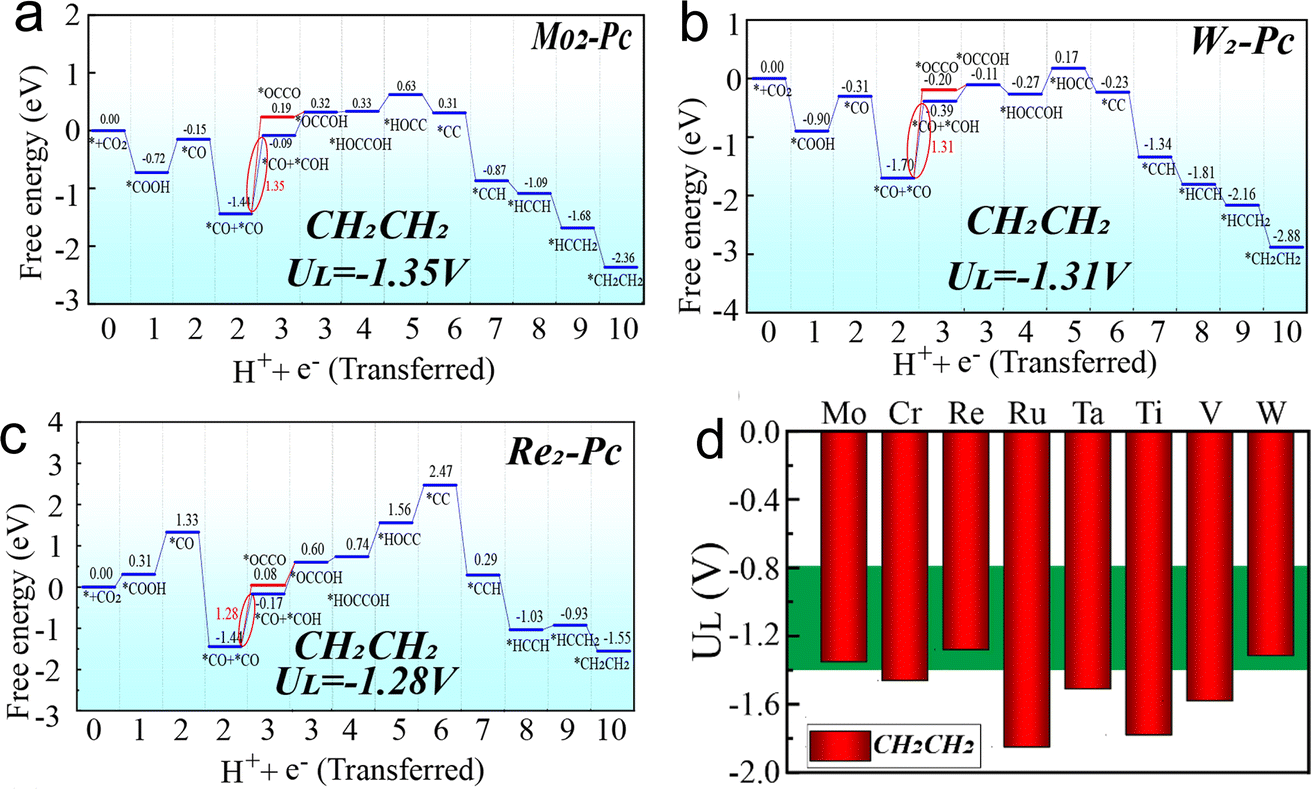 | ||
| Fig. 33 Free-energy profiles for the CO2RR at zero applied voltage vs. CHE for (a) Mo2-DAC, (b) W2-DAC and (c) Re2-DAC. (d) Histogram of limiting potential of CO2 reduced to CH2CH2. CO2 reduced to CH2CH2 on the Cu(111) surface is shaded green. Reproduced from ref. 144, Copyright© 2024 Wiley-VCH GmbH. | ||
While Singh and coworkers worked on the identification of metal DACs on N-doped holey graphene for the CO2RR for the formation of C2 products, a similar ab initio computational study by Zhao and co-workers signified the importance of substrates in DACs for CO2 reduction particularly for higher order products.175 The authors focused on eight different N-doped graphene (CxNy) surfaces (4N-V2, 5N-V3, 6N-V4(a), 6N-V4(b), 6N-V6, C2N, g-C3N4, and V6 = graphene) where the % N in the material was varied from 3.6–15.6%. Among all, the 6N-V6 support showed the least formation energy (Eform = 0.16 eV Å−1) and hence it was considered for further studies.
The binding energy of homonuclear transition metal DACs (Sc to Cu) on 6N-V6 was evaluated, which showed an increasing instability trend as the number of electrons on the metal center increased. The adsorption energy for single and double CO2 on the DACs was studied as given in Fig. 34, the early transition metal DACs showed higher binding energy which was found to be decreased across the period. However, the high activity of the Sc–Sc dimer can be related to the fact that the size of the DAC was larger than that of the hole available on the CxNy-sheet which caused Sc–Sc DAC to be positioned out of the plane.
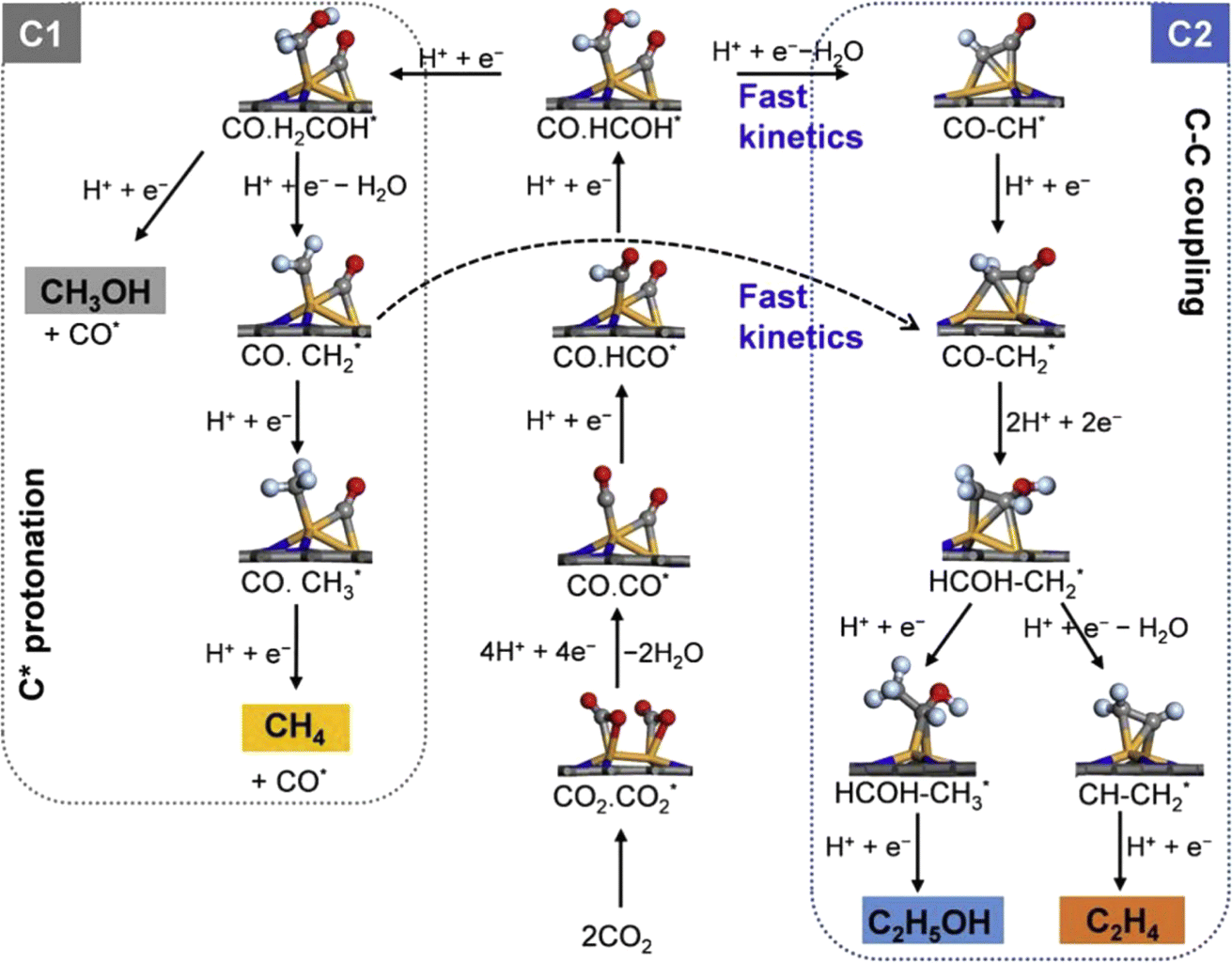 | ||
| Fig. 34 The CO2 reduction pathways to various C1 and C2 products on the supported Fe2 dimer.175 | ||
The hypothesis considered by the authors was that the DACs capable of activating two CO2 molecules simultaneously exhibit a better probability of forming C–C coupled products and this was a significant insight into getting C2+ products. Adsorption of CO, protonation of CO and the energy barrier of C–C coupling of protonated intermediates are considered the key steps of the eCO2RR (Fig. 14). Since the Fe2 dimer displayed good adsorption energy for both CO2 and also had enough experimental data to back the synthesis of similar DACs, it was considered for further analysis on the CxNy supports. The C1 and C2 product formation pathways were studied for the Fe2–CxNy catalysts. Among them, Fe2–C2N displayed great efficiency for ethanol formation with a barrier of ca. 0.94 eV. The adsorption competition between CO2 and H* species was studied to find 5N-V3, 6N-V6, C2N, g-C3N4, and V6 doped with the Fe-dimer to show better eCO2RR efficacy than 4N-V2, 6N-V4(a) and 6N-V4(b).
Summary
In conclusion, the exploration of DACs for the reduction of CO2 to various value-added products marks a significant advancement in the field of catalysis. Through comprehensive research efforts and meticulous investigations, DACs have demonstrated promising potential in facilitating the conversion of CO2 into a spectrum of desirable compounds including CO, formate, formic acid, methanol, methane, ethanol, ethene, and beyond. The versatility of DACs lies in their ability to finely tune catalytic properties through precise control over the composition, structure, and electronic properties of dual metal atoms. Throughout this review, numerous studies have showcased the remarkable performance of DACs in enhancing the selectivity, efficiency, and overall catalytic activity for the CO2 reduction reaction. Notably, DACs have shown superior behaviour compared to traditional single-metal catalysts, attributed to the synergistic effects arising from the interaction between two different metal atoms to facilitate coordination between oxygen and carbon atoms. The cooperative action of DACs not only promotes the activation of CO2 molecules but also facilitates their subsequent conversion into specific target products with high selectivity.Moreover, the insights gained from mechanistic studies, computational modelling, and experimental investigations have provided valuable guidance for the rational design and optimization of DACs tailored for specific CO2 reduction pathways. The development of DACs holds immense promise for addressing the pressing global challenge of CO2 emissions while simultaneously offering opportunities for the sustainable production of valuable chemicals and fuels.
Challenges and future scope
While research on DACs holds promise, several challenges and areas for future development warrant attention. Developing reliable and scalable synthesis methods for DACs remains a significant challenge. Additionally, characterizing DAC structures at atomic resolution poses technical difficulties due to their intricate nature and complexity in synthesis. The loading of active metals in DACs is generally low, leading to the production of low current density. Simple and large-scale synthesis of DACs with precise and uniform metal active centres is directly related to the practical value of DACs. Enhancing the activity and selectivity of DACs for specific reactions is crucial for practical applications. Therefore, developing and optimizing synthesis strategies to obtain high metal atom loadings and stable DACs is urgently needed.Looking ahead, DACs can reduce the use of noble metal bulk catalysts drastically as DACs only use 1–2 wt% of metal. This leads to a decrease in the cost of the catalyst thus making the whole reaction more economical. Although this is true, the synthesis of DACs is presently expensive due to the complex process and use of intricate techniques for synthesis and characterization. This limits the large-scale production and commercialization of DACs, which can be overcome in the future by finding new methods, precursors, and substrates for the efficient synthesis of DACs. Further research endeavours are warranted to delve deeper into the intricate mechanisms governing DAC-catalyzed CO2 reduction processes and to overcome remaining challenges such as improving stability, scalability, and economic viability while reducing the current density and working potential of the catalysts. With continued interdisciplinary efforts and innovative approaches, DACs are poised to play a pivotal role in advancing the frontier of CO2 utilization toward a greener and more sustainable future.
Data availability
This is a review article hence there is no experimental data associated with this article.Author contributions
VA ideated the work and VA, SVP and DK contributed in manuscript writing.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
VA acknowledges SERB-CRG (File No. CRG/2022/003758) for the funding. VA, SVP, and DK acknowledge the Department of Chemistry, Ashoka University, and Axis bank for the infrastructure and facilities. SVP and DK acknowledge Ashoka University for a PhD fellowship.References
- NASA, Evidence for Human-Caused Climate Change, retrieved April 12, 2024, https://science.nasa.gov/climate-change/ Search PubMed.
- S. Nagireddi, J. R. Agarwal and D. Vedapuri, ACS Eng. Au, 2024, 4, 22–48 CrossRef CAS.
- IEA, CCUS in Clean Energy Transitions, IEA, Paris, 2020, https://www.iea.org/reports/ccus-in-clean-energy-transitions, Licence: CC BY 4.0 Search PubMed.
- IEA, Global CO2 Emissions from Energy Combustion and Industrial Processes, 1900-2022, IEA, Paris, https://www.iea.org/data-and-statistics/charts/global-co2-emissions-from-energy-combustion-and-industrial-processes-1900-2022, License: CC BY 4.0 Search PubMed.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- H. Xing, S. Spence and H. Chen, Renewable Sustainable Energy Rev., 2020, 134, 110222 CrossRef CAS.
- R. Sen, A. Goeppert and G. K. Surya Prakash, Angew. Chem., Int. Ed., 2022, 61, e202207278 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J. P. Jones, G. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- M. Samolia and V. Avasare, ACS Appl. Energy Mater., 2023, 6, 10748–10760 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. MacDowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- https://www.methanex.com/news/release/methanex-reports-first-quarter-2023-results/accessed on 12th April 2024.
- D. H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- S. Fukuzumi and D. Hong, Eur. J. Inorg. Chem., 2014, 2014, 645–659 CrossRef CAS.
- H. Lee, X. Wu and L. Sun, Nanoscale, 2020, 12, 4187–4218 RSC.
- R. Luque and A. S. Burange, Heterogeneous Catalysis, American Chemical Society, 2022 Search PubMed.
- Heterogeneous Catalysts: Advanced Design, Characterization, and Applications, ed. W. Y. Teoh, A. Urakawa, Y. H. Ng and P. Sit, John Wiley & Sons, 2021, vol. 2 Search PubMed.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795–2806 RSC.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- H. Yook, J. Hwang, W. Yeo, J. Bang, J. Kim, T. Y. Kim, J.-S. Choi and J. W. Han, Adv. Mater., 2023, 35, 2204938 CrossRef CAS PubMed.
- Z. Li, R. Ge, J. Su and L. Chen, Adv. Mater. Interfaces, 2020, 7, 2000396 CrossRef CAS.
- X. Zheng, P. Li, S. Dou, W. Sun, H. Pan, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 2809–2858 RSC.
- J. Xi, H. S. Jung, Y. Xu, F. Xiao, J. W. Bae and S. Wang, Adv. Funct. Mater., 2021, 31, 2008318 CrossRef CAS.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC.
- B. Singh, M. B. Gawande, A. D. Kute, R. V. Varma, P. Fornasiero, P. McNeice, R. V. Jagadeesh, M. Beller and R. Zboril, Chem. Rev., 2021, 121, 13620–13697 CrossRef CAS PubMed.
- W. H. Li, J. Yang, D. Wang and Y. Li, Chem, 2022, 8, 119–140 CAS.
- G. Yasin, A. Kumar, S. Ajmal, M. A. Mushtaq, M. Tabish, A. Saad, M. A. Assiri, M. T. Nazir and Q. Zhuo, Coord. Chem. Rev., 2024, 501, 215589 CrossRef CAS.
- F. Yang and W. Xu, J. Mater. Chem. A, 2022, 10, 5673–5698 RSC.
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC.
- L. Li, K. Yuan and Y. Chen, Acc. Mater. Res., 2022, 3, 584–596 CrossRef CAS.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skulason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- F. Calle-Vallejo, D. Loffreda, M. Koper and P. Sautet, Nat. Chem., 2015, 7, 403–410 CrossRef CAS PubMed.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- C. Shi, H. A. Hansen, A. C. Lausche and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 4720–4727 RSC.
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1600463 CrossRef.
- W. Yang, Z. Jia, B. Zhou, L. Wei, Z. Gao and H. Li, Commun. Chem., 2023, 6, 6 CrossRef CAS PubMed.
- M. Huang, H. Meng, J. Luo, H. Li, Y. Feng and X.-X. Xue, J. Phys. Chem. Lett., 2023, 14, 1674–1683 CrossRef CAS PubMed.
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang, F. Lin, H. Luo, J. Li, M. Tong, E. Wang and S. Guo, Adv. Mater., 2021, 33, 2102576 CrossRef CAS PubMed.
- A. M. R. Zawadzki, A. J. Nielsen, R. E. Tankard and J. Kibsgaard, ACS Catal., 2024, 14, 1121–1145 CrossRef.
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2022, 12, 2102715 CrossRef CAS.
- Y. Li, Y. Li, H. Sun, L. Gao, X. Jin, Y. Li, L. V. Zhi, L. Xu, W. Liu and X. Sun, Nano-Micro Lett., 2024, 16, 139 CrossRef CAS PubMed.
- Y. Yang and S. Liu, J. Phys. Chem. C, 2024, 128, 6269–6279 CrossRef CAS.
- J. Sun, L. Tao, C. Ye, Y. Wang, G. Meng, H. Lei, S. Zheng, C. Xing, X. Tao, P. Wu, J. Chen, S. Du, D. Wang and Y. Li, J. Am. Chem. Soc., 2023, 145, 7113–7122 CrossRef CAS PubMed.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–113393 CrossRef CAS PubMed.
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 9661–9671 CrossRef CAS PubMed.
- K. Li, Y. Wang, J. Lu, W. Ding, F. Huo, H. He and S. Zhang, Cell Rep. Phys. Sci., 2023, 4, 101435 CrossRef CAS.
- Z. He, K. He, A. W. Robertson, A. I. Kirkland, D. Kim, J. Ihm, E. Yoon, G.-D. Lee and J. H. Warner, Nano Lett., 2014, 14, 3766–3772 CrossRef CAS PubMed.
- J. Shi, R. Li, J. Zhang, Y. Wang, W. Ma, Z. Yue, C. Jin, Y. Liu, L. Zheng, J. Bai, X. Li, K. Leng and Y. Qu, ACS Appl. Mater. Interfaces, 2024, 16, 889–897 CrossRef CAS PubMed.
- J. Fu, J. Dong, R. Si, K. Sun, J. Zhang, M. Li, N. Yu, B. Zhang, M. G. Humphrey, Q. Fu and J. Huang, ACS Catal., 2021, 11, 1952–1961 CrossRef CAS.
- X. Han and S. Yang, Chem, 2022, 8, 2584–2586 CAS.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- J.-H. Dou, L. Sun, Y. Ge, W. Li, C. H. Hendon, J. Li, S. Gul, J. Yano, E. A. Stach and M. Dinca, J. Am. Chem. Soc., 2017, 139, 13608–13611 CrossRef CAS PubMed.
- Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2023, 145, 26783–26790 CrossRef CAS PubMed.
- Y. X. Zhang, S. Zhang, H. Huang, X. Liu, B. Li, Y. Lee, X. Wang, Y. Bai, M. Sun, Y. Wu, S. Gong, X. Liu, Z. Zhuang, T. Tan and Z. Niu, J. Am. Chem. Soc., 2023, 145, 4819–4827 CrossRef CAS PubMed.
- X. Zhu, G. Liu, X. Tao, P. Huang, Q. Wang, G. Chen, J. Yang, L. Zhang and Y. Zhou, ACS Omega, 2023, 8, 41708–41717 CrossRef CAS PubMed.
- A. Mehmood, J. Pampel, G. Ali, H. Y. Ha, F. Ruiz-Zepeda and T.-P. Fellinger, Adv. Energy Mater., 2018, 8, 1701771 CrossRef.
- L. Li, C. Chen, R. Cao, Z. Pan, H. He and K. Zhou, Appl. Catal., B, 2020, 268, 118747 CrossRef.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- S. Zhang, Y. Wu, Y. X. Zhang and Z. Niu, Sci. China: Chem., 2021, 64, 1908–1922 CrossRef CAS.
- Y. Zhou, E. Song, W. Chen, C. U. Segre, J. Zhou, Y.-C. Lin, C. Zhu, R. Ma, P. Liu, S. Chu, T. Thomas, M. Yang, Q. Liu, K. Suenaga, Z. Liu, J. Liu. and J. Wang, Adv. Mater., 2020, 32, 2003484 CrossRef CAS PubMed.
- J. Fonseca and J. Lu, ACS Catal., 2021, 11, 7018–7059 CrossRef CAS.
- Z. Gao and Y. Qin, Acc. Chem. Res., 2017, 50, 2309–2316 CrossRef CAS PubMed.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T.-K. Sham, M. Gu, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- S. Y. Kim, B. J. Cha, S. Saqlain, H. O. Seo and Y. D. Kim, Catalysts, 2019, 9, 266 CrossRef CAS.
- C. Chen, M. Sun, K. Wang and Y. Li, SmartMat, 2022, 3, 533–564 CrossRef CAS.
- T. He, A. R. P. Santiago, Y. Kong, M. A. Ahsan, R. Luque, A. Du and H. Pan, Small, 2022, 18, 2106091 CrossRef CAS PubMed.
- X. Ma, J. Albertsma, D. Gabriels, R. Horst, S. Polat, C. Snoeks, F. Kapteijn, H. B. Eral, D. A. Vermaas, B. Mei, S. d. Beer and M. A. Veen, Chem. Soc. Rev., 2023, 52, 3741 RSC.
- P. Mathur, S. Chatterjee and V. D. Avasare, Adv. Organomet. Chem., 2007, 55, 201–277 CrossRef.
- A. Haynes, Acetic Acid Synthesis by Catalytic Carbonylation of Methanol, in Catalytic Carbonylation Reactions. Topics in Organometallic Chemistry, ed. M. Beller, Springer, Berlin, Heidelberg, 2006, vol 18 Search PubMed.
- I. Ojima, C.-Y. Tsai, M. Tzamarioudaki and D. Bonafoux, The Hydroformylation Reaction, in Organic Reactions, 2004 Search PubMed.
- J. October, K. Köhnke, N. Thanheuser, A. J. Vorholt and W. Leitner, Eur. J. Org Chem., 2022, 2022, e202201018 CrossRef.
- W. D. Shafer, M. K. Gnanamani, U. M. Graham, J. Yang, C. M. Masuku, G. Jacobs and B. H. Davis, Catalysts, 2019, 9, 259 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- J. Jiao, R. Lin, S. Liu, W. C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S. F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- Y. Li, C. Chen, R. Cao, Z. Pan, H. He and K. Zhou, Appl. Catal., B, 2020, 268, 118747 CrossRef CAS.
- Q. He, J. H. Lee, D. Liu, Y. Liu, Z. Lin, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Adv. Funct. Mater., 2020, 30, 2000407 CrossRef CAS.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 133, 13500–13505 CrossRef.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- Q. N. Zhan, T. Y. Shuai, H. M. Xu, Z. J. Zhanga and G. R. Li, J. Mater. Chem. A, 2023, 11, 7949–7986 RSC.
- L. Hong, X. Liu, B. Chi, G. Xiab and H. Wang, J. Mater. Chem. A, 2023, 11, 6321–6328 RSC.
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K. J. Noh, K. S. Lee and J. W. Han, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- X. Zhao, K. Zhao, Y. Liu, Y. Su, S. Chen, H. Yu and X. Quan, ACS Catal., 2022, 12, 11412–11420 CrossRef CAS.
- K. C. Li, Z. H. Wu, C. H. Ke, Y. C. Lee, J. F. Lee, J. M. Chen, S. C. Haw, F. T. Tsai and W. F. Liaw, J. Mater. Chem. A, 2023, 11, 2377–2390 RSC.
- M. J. Sun, Z. W. Gong, J. D. Yi, T. Zhang, X. Chen and R. Cao, Chem. Commun., 2020, 56, 8798–8801 RSC.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed.
- Q. Hao, H.-x. Zhong, J.-z. Wang, K.-h. Liu, J.-m. Yan, Z.-h. Ren, N. Zhou, X. Zhao, H. Zhang, D.-x. Liu, X. Liu, L.-w. Chen, J. Luo and X.-b. Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef.
- Y. N. Gong, C. Y. Cao, W. J. Shi, J. H. Zhang, J. H. Deng, T. B. Lu and D. C. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202215187 CrossRef CAS PubMed.
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed.
- R. Sundararaman, W. A. Goddard and T. A. Arias, J. Chem. Phys., 2017, 146, 114104 CrossRef PubMed.
- F. Li, H. Wen and Q. Tang, J. Mater. Chem. A, 2022, 10, 13266–13277 RSC.
- Y. Li, W. Shan, M. J. Zachman, M. Wang, S. Hwang, H. Tabassum, J. Yang, X. Yang, S. Karakalos, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2022, 61, e202205632 CrossRef CAS PubMed.
- K. Huang, R. Li, H. Qi, S. Yang, S. An, C. Lian, Q. Xu, H. Liu and J. Hu, ACS Catal., 2024, 14, 8889–8898 CrossRef CAS.
- J. Pei, T. Wang, R. Sui, X. Zhang, D. Zhou, F. Qin, X. Zhao, Q. Liu, W. Yan, J. Dong, L. Zheng, A. Li, J. Mao, W. Zhu, W. Chen and Z. Zhuang, Energy Environ. Sci., 2021, 14, 3019–3028 RSC.
- J. Chen, M. R. Ahasan, J. S. Oh, J. A. Tan, S. Hennessey, M. M. Kaid, H. M. Kaderi, L. Zhou, K. U. Lao, R. Wang and W. N. Wang, J. Mater. Chem. A, 2024, 12, 4601–4609 RSC.
- L. Qiu, F. Liu, L. Zhao, W. Yang and J. Yao, Langmuir, 2006, 22, 4480–4482 CrossRef CAS PubMed.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal., B, 2019, 241, 113–119 CrossRef CAS.
- H. Mistry, Y. W. Choi, A. Bagger, F. Scholten, C. S. Bonifacio, I. Sinev, N. J. Divins, I. Zegkinoglou, H. S. Jeon, K. Kisslinge, J. C. Yang, J. Rossmeisl and B. R. Cuenya, Angew. Chem., Int. Ed., 2017, 56, 11394–11398 CrossRef CAS PubMed.
- Z. Guo, H. Zhu, G. Yang, A. Wu, Q. Chen, Z. Yan, K. L. Fow, H. Do, J. D. Hirst, T. Wu and M. Xu, Chem. Eng., 2023, 476, 146556 CrossRef CAS.
- Y. Dong, Q. Zhang, Z. Tian, B. Li, W. Yan, S. Wang, K. Jiang, J. Su, C. W. Oloman, E. L. Gyenge, R. Ge, Z. Lu, X. Ji and L. Chen, Adv. Mater., 2020, 32, 2001300 CrossRef CAS PubMed.
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS.
- L. Zhao, Q. Wang, X. Zhang, C. Deng, Z. Li, Y. Lei and M. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 35888–35895 CrossRef CAS PubMed.
- X. Han, Z. Chen, W. Chen, C. Lv, Y. Ji, J. Li, W. C. Cheong, X. Lei, Q. Peng, C. Chen, D. Wang, C. Lian and Y. Li, Chem. Commun., 2021, 57, 1895–1898 RSC.
- D. Yao, C. Tang, X. Zhi, B. Johannessen, A. Slattery, S. Chern and S. Z. Qiao, Adv. Mater., 2023, 35, 2209386 CrossRef CAS PubMed.
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. Dong and X. Feng, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed.
- M. Feng, X. Wu, H. Cheng, Z. Fan, X. Li, F. Cui, S. Fan, Y. Dai, G. Leic and G. He, J. Mater. Chem. A, 2021, 9, 23817–23827 RSC.
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2021, 31, 2007423 CrossRef CAS.
- Y. Yang and S. Liu, J. Phys. Chem. C, 2024, 128, 6269–6279 CrossRef CAS.
- M. Zhang, Z. Hu, L. Gu, Q. Zhang, L. Zhang, Q. Song, W. Zhou and S. Hu, Nano Res., 2020, 13, 3206–3211 CrossRef CAS.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS PubMed.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- M. Zhu, C. Zhao, X. Liu, X. Wang, F. Zhou, J. Wang, Y. Hu, Y. Zhao, T. Yao, L. M. Yang and Y. Wu, ACS Catal., 2021, 11, 3923–3929 CrossRef CAS.
- Z. Liang, L. Song, M. Sun, B. Huang and Y. Du, Sci. Adv., 2021, 7, eabl4915 CrossRef CAS PubMed.
- N. Yusuf, F. Almomani and H. Qiblawey, Fuel, 2023, 345, 128178 CrossRef CAS.
- B. Xiong, Y. Yang, J. Liu, Z. Hua and Y. Yang, Fuel Process. Technol., 2022, 233, 107315 CrossRef CAS.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- J. Tian, R. Wang, M. Shen, X. Ma, H. Yao, Z. Hua and L. Zhang, ChemSusChem, 2021, 14, 2247–2254 CrossRef CAS PubMed.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed.
- H. Wang, N. Wen, Y. Wang, X. Jiao, Y. Xia and D. Chen, Adv. Funct. Mater., 2023, 33, 2303473 CrossRef CAS.
- B. Ren, E. Croiset and L. R. Sandoval, J. Catal., 2020, 383, 273–282 CrossRef CAS.
- F. Cheng, X. Zhang, K. Mu, X. Ma, M. Jiao, Z. Wang, P. Limpachanangkul, B. Chalermsinsuwan, Y. Gao, Y. Li, Z. Chen and L. Liu, Energy Technol., 2021, 9, 2000799 CrossRef CAS.
- F. Ma and X. Chen, ACS Appl. Nano Mater., 2023, 6, 16546–16554 CrossRef CAS.
- G. Chen, M. Buraschi, R. A. Heidous, S. Bonakala ,F. E. Mellouhi and C. S. Cucinotta, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-bk36r.
- J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2013, 135, 8798–8801 CrossRef CAS PubMed.
- K. Yao, H. Wang, X. Yang, Y. Huang, C. Kou, T. Jing, S. Chen, Z. Wang, Y. Liu and H. Liang, Appl. Catal., 2022, 311, 121377 CrossRef CAS.
- C. W. Lee, J. S. Hong, K. D. Yang, K. Jin, J. H. Lee, H.-Y. Ahn, H. Seo, N.-E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 CrossRef CAS.
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1801536 CrossRef.
- J. Zhang, K. Zhao, Y. Ma, W. Chen, X. Shi, C. Sun, J. Niu and Q. Zhang, Small Struct., 2023, 5, 2300323 CrossRef.
- F. Li and Q. Tang, J. Mater. Chem. A, 2021, 9, 8761–8771 RSC.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- J. Zhao, J. Zhao, F. Li and Z. Chen, J. Phys. Chem. C, 2018, 122, 19712–19721 CrossRef CAS.
- Y. Li, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2015, 5, 6658–6664 CrossRef CAS.
- S. Zhu, K. Wan, H. Wang, L. J. Guo and X. Shi, Nanotechnology, 2021, 32, 385404 CrossRef CAS PubMed.
- D. Ren, J. Fong and B. S. Yeo, Nat. Commun., 2018, 9, 925 CrossRef PubMed.
- S. Ghoshal and P. Sarkar, J. Phys. Chem. C, 2024, 128, 2392–2405 CrossRef CAS.
- Q. Huang, H. Liu, W. An, Y. Wang, Y. Feng and Y. Men, ACS Sustainable Chem. Eng., 2019, 7, 19113–19121 CrossRef CAS.
- W. Xue, J. Li, H. Huang, W. Zhang and D. Mei, Inorg. Chem., 2023, 62, 930–941 CrossRef CAS PubMed.
- N. Shindoh, Y. Takemoto and K. Takasu, Chem. - Eur. J., 2009, 15, 12168–12179 CrossRef CAS PubMed.
- Y. Liu, H. Chen, Y. Yang, C. Jiao, W. Zhu, Y. Zhang, X. Wu, J. Mao and Z. Zhuo, Energy Environ. Sci., 2023, 16, 5185–5195 RSC.
- J. Wang, M. Wu, B. Zhao, R. Wan and Z. Li, Int. J. Quantum Chem., 2024, 124, e27308 CrossRef CAS.
- S. Cao, S. Zhou, H. Chen, S. Wei, S. Liu, X. Lin, X. Chen, Z. Wang, W. Guo and X. Lu, Energy Environ. Mater., 2023, 6, e12287 CrossRef CAS.
- C. Tsai, H. Li, S. Park, J. Park, H. S. Han, J. K. Nørskov, X. Zheng and F. A. Pedersen, Nat. Commun., 2017, 8, 15113 CrossRef PubMed.
- H. Li, C. Deng, F. Li, M. Ma and Q. Tang, J. Mater. Inf., 2023, 3, 25 Search PubMed.
- H. Ding, Y. Shi, Z. Li, S. Wang, Y. Liang, H. Feng, Y. Deng, X. Song, P. Pu and X. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 12986–12997 CrossRef CAS PubMed.
- C. B. Wahl, M. Aykol, J. H. Swisher, J. H. Montoya, S. K. Suram and C. A. Mirkin, Sci. Adv., 2021, 7, eabj5505 CrossRef CAS PubMed.
- L. Yu, F. Li, J. Huang, B. G. Sumpter, W. E. Mustain and Z. Chen, ACS Catal., 2023, 13, 9616–9628 CrossRef CAS.
- W.-L. Li, T.-T. Chen, D.-H. Xing, X. Chen, J. Li and L.-S. Wang, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, E6972–E6977 CAS.
- L. Yu and F. Li, Phys. Chem. Chem. Phys., 2022, 24, 9842–9847 RSC.
- W. Yang, Z. Jia, B. Zhou, L. Chen, X. Ding, L. Jiao, H. Zheng, Z. Gao, Q. Wang and H. Li, ACS Catal., 2023, 13, 9695–9705 CrossRef CAS.
- L. Zaza, K. Rossi and R. Buonsanti, ACS Energy Lett., 2022, 7, 1284–1291 CrossRef CAS.
- M. Ebaid, K. Jiang, Z. Zhang, W. S. Drisdell, A. T. Bell and J. K. Cooper, Chem. Mater., 2020, 32, 3304–3311 CrossRef CAS.
- Y. Yang, Z. Tan, J. Zhang, J. Yang, R. Zhang, S. Wang, Y. Song and Z. Su, Green Energy Environ., 2023 DOI:10.1016/j.gee.2023.09.002.
- S. Ajmal, Y. Yang, M. A. Tahir, K. Li, A. Bacha, I. Nabi, Y. Liu, T. Wang and L. Zhang, Catal. Sci. Technol., 2020, 10, 4562–4570 RSC.
- Q. Qin, H. Suo, L. Chen, Y.-X. Wang, J.-Z. Wang, H.-K. Liu, S.-X. Dou, M. Lao and W.-H. Lai, Adv. Mater. Interfaces, 2024, 2301049 CrossRef CAS.
- H. Yang, S. Li and Q. Xu, Chin. J. Catal., 2023, 48, 32–65 CrossRef CAS.
- Z. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z.-L. Wang, Small, 2022, 18, 2107450 CrossRef CAS PubMed.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma, L. Zheng, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 16459 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai and A. Z. Yu, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840 CrossRef CAS PubMed.
- Q. Mo, S. Li, C. Chen, H. Song, Q. Gao and L. Zhang, ACS Sustainable Chem. Eng., 2024, 12, 6093–6101 CrossRef CAS.
- W. Ren, X. Tan, J. Qu, S. Li, J. Li, X. Liu, S. P. Ringer, J. M. Cairney, K. Wang, S. C. Smith and C. Zhao, Nat. Commun., 2021, 12, 1449 CrossRef CAS PubMed.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qina and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- K.-S. Kim, W. J. Kim, H.-K. Lim, E. K. Lee and H. Kim, ACS Catal., 2016, 6, 4443–4448 CrossRef CAS.
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan and X. Fan, Sci. Adv., 2020, 6, eaba6586 CrossRef CAS PubMed.
- P. Shao, W. Zhou, Q.-L. Hong, L. Yi, L. Zheng, W. Wang, H.-X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687 CrossRef CAS PubMed.
- S. Li, A. Guan, C. Yang, C. Peng, X. Lv, Y. Ji, Y. Quan, Q. Wang, L. Zhang and G. Zheng, ACS Mater. Lett., 2021, 3, 1729–1737 CrossRef CAS.
- D. Chen, Z. Chen, Z. Lu, J. Tang, X. Zhang and C. V. Singh, J. Mater. Chem. A, 2020, 8, 21241–21254 RSC.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- Y. Zhao, S. Zhou and J. Zhao, iScience, 2020, 23, 101051 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |