
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lanthanide promoted nickel catalysts for the integrated capture and conversion of carbon dioxide to methane via metal carbonates†
Christopher J.
Koch
 ,
Zohaib
Suhail
,
Prince
,
Anushan
Alagaratnam
,
Matthew
Coe
,
Alain
Goeppert
and
G. K. Surya
Prakash
*
,
Zohaib
Suhail
,
Prince
,
Anushan
Alagaratnam
,
Matthew
Coe
,
Alain
Goeppert
and
G. K. Surya
Prakash
*
Loker Hydrocarbon Research Institute, Department of Chemistry, University of Southern California, 837 Bloom Walk, Los Angeles, CA 90089-1661, USA. E-mail: gprakash@usc.edu
First published on 13th September 2024
Abstract
An integrated CO2 capture and conversion system utilizing metal hydroxide salts has been developed to capture CO2 from various sources including air in the form of carbonate salts and convert them directly into a synthetic fuel; methane. Nickel catalysts have previously been shown to convert carbonate salts, such as K2CO3 and Na2CO3, to methane. However, the productivity of these systems was rather modest in comparison to other catalysts based on ruthenium metal. With the help of lanthanide promoters, the methane productivity of nickel catalysts has been greatly improved. For the most part, the catalytic performance of the lanthanide promoted nickel catalysts followed the lanthanide contraction trend, i.e. the smaller the atomic size of the lanthanide, the higher the methane yield. Furthermore, the lanthanide promoted nickel catalysts are also stable under the alkaline conditions employed, maintaining their activity over five cycles of integrated CO2 capture and conversion. Lastly, the lanthanide promoted nickel catalysts were demonstrated to be more economical compared to ruthenium- and unpromoted nicked-based catalysts.
Sustainability spotlightCarbon dioxide (CO2) is a key greenhouse gas contributing to climate change. However, unlike other greenhouse gases, CO2 is reactive, especially when viewed as a Lewis acid. Utilizing a metal hydroxide base allows for CO2 to be captured as a carbonate salt for easy and safe transport. These carbonate salts can then be converted to other materials and fuels such as methane in an integrated carbon capture and conversion process. Carbon capture and its conversion to useful products is an important avenue to reach the 2050 goals set by the Paris Agreement. By converting carbon dioxide into methane, a main component of natural gas, this process can be directly integrated into existing electrical power and heat generation systems which are large emitters of CO2. The methane can also be used as a convenient hydrogen carrier. This work addresses the UN's Sustainable Development Goals 7 (Affordable and Clean Energy), 9 (Industry, Innovation and Infrastructure) and 13 (Climate Action). |
Introduction
Carbon capture and sequestration (CCS) and carbon capture and utilization (CCU) have been identified as pivotal technologies to help countries meet their carbon emission reduction goals.1,2 Providing new pathways for utilizing CO2 as a synthetic reagent with green hydrogen would allow the production of abundant, renewable and sustainable carbon fuels and chemical feedstocks.3–7 Identifying avenues to generate renewable methane utilizing carbon capture and conversion technologies would allow for the system to be carbon neutral and for methane to be synthesized on demand and stored for later use.8,9 This could be especially helpful as a large portion of the worldwide heating and electricity generation is currently powered by natural gas.10 To make these carbon capture technologies more economical and viable for renewable methane production, integration of the CO2 capture and conversion steps, which is known as integrated carbon capture and conversion (ICCC) would be advantageous.11–13Both amines and hydroxide salts have been utilized as capture agents in ICCC applications.14–17 Primary and secondary amines are promising agents for capturing carbon dioxide.18–21 However, they are often volatile and can be prone to oxidative degradation, which can limit the practicality of these systems.22–25 To circumvent these issues, additional synthetic strategies are often employed by either grafting the amine on a support or synthetically designing more stable amines.19,26–29 Hydroxide assisted systems are also promising for the capture of carbon dioxide from various sources including the air.30 However, a large portion of reported ICCC reactions is performed in organic solvents. The solubility of the hydroxide salt and subsequent carbonate salt in these organic solvents is often more limited, when compared to amine-based systems potentially hampering their applicability. The hydroxide base systems can also have issues with leaching from the catalyst when incompatible metal additives, such as calcium, are utilized.31 This can be circumvented by modifying the design of the catalyst.32,33
Many ICCC systems have been based on homogenous catalytic systems that used primarily ruthenium catalysts.34–36 However, the utilization of homogenous catalysts has often problems associated with the scalability of the system. Thus, employing heterogenous catalysts for ICCC technologies could be greatly beneficial. Heterogeneous catalysis has been studied to a lesser extent than its homogeneous counterpart for ICCC technologies.37 On the other hand, heterogenous catalysts have been utilized industrially for carbon dioxide conversion from pure CO2 streams and those systems could have applications in ICCC technologies.38–43
Materials referred to as dual functional (also often called bifunctional) containing both a heterogenous hydrogenation catalyst and an alkaline or alkaline earth metal to capture CO2 have been designed for ICCC. They are generally operated in a flow system where CO2 is first captured and then in a second step converted to methane with the mixed-in catalyst.44–49 Studies with these systems often utilize a pure CO2 stream during the capture. However, the concentration of CO2 in these systems can also be simulated to mimic flue gas or atmospheric conditions.50–52 In these cases, an inert gas is often utilized to act as a carrier gas.53–55 This is to circumvent issues of catalyst deactivation and oxidation that could occur if atmospheric air or industrial gases were utilized during the CO2 adsorption step as the active metal for the conversion of CO2 is also interacting with the gas mixture. During the hydrogenation phase the catalyst would then have to be reduced again in each consecutive CO2 capture/hydrogenation cycle. In bifunctional materials a considerable amount of potentially costly catalyst is also tied up with the capture media even when it is not being used for reduction. Therefore, there are benefits of having the ability to decouple the CO2 capture agent and the hydrogenation catalyst. It allows for relatively inexpensive materials to be used for the capture in a liquid aqueous phase and then contact the carbonates obtained after CO2 capture with the catalyst in another vessel, where it can be maintained in an active state.
Systems that utilize amines as capture agents with heterogenous catalytic systems have also been shown to have good conversion to methane and methanol. In such systems, studied for example by Heldebrant et al., precious metals are often utilized for the conversion of the CO2/capture species.56–58 Utilizing more abundant metals would render the system economically more viable. Hydroxide-assisted systems have also been developed and shown for the conversion of the capture products, carbonate salts, to methane.31 In these systems, a nickel-based catalyst was able to undergo five cycles of capture/hydrogenation with no loss in activity. A ruthenium catalyst was initially more active for the hydrogenation reaction, displaying higher reaction rates. However, this catalyst was not stable under the alkaline reaction conditions used and lost 70% of its activity after the initial hydrogenation reaction.59 Thus, enhancing the productivity of the hydroxide-assisted system but maintaining the recyclability of the system is important to improve the process. Overall, a process capturing CO2 from any source including air and converting the carbonate intermediates directly to methane as depicted in Fig. 1 is proposed.
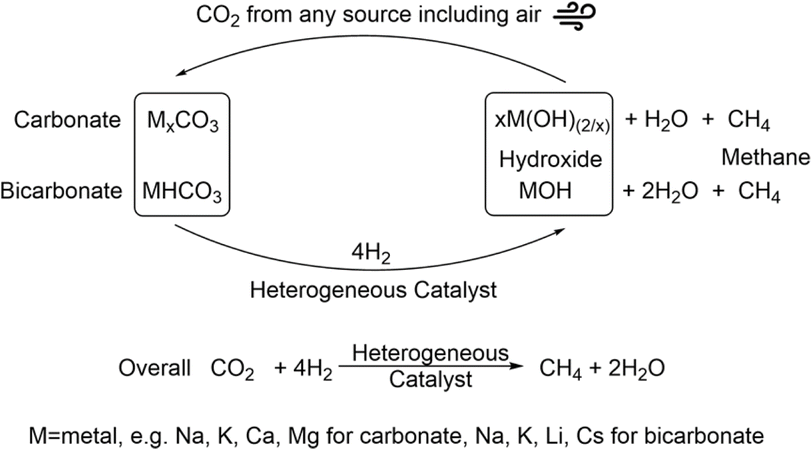 | ||
| Fig. 1 CO2 capture with a metal hydroxide and hydrogenation of the obtained carbonate/bicarbonate to methane with concurrent recycling of the base. | ||
Lanthanide promoters have previously been reported to improve the CO2 hydrogenation productivity to methane and methanol.32,60–64 Often, these improvements followed the lanthanide contraction, where smaller ionic sizes typically lead to higher productivity.64 This effect is often experienced at relatively low weight concentrations of the lanthanide when compared to the active metal, allowing for small amounts of lanthanide metals to be used to achieve large improvements to the system.60,65
Herein, we report a great gain in methane productivity over nickel-catalysts promoted with lanthanides. These catalysts are stable under alkaline conditions and can maintain activity for at least five reaction cycles. While all the lanthanides tested led to higher conversions of carbonate salts to methane, ytterbium displayed the greatest enhancement.
Experimental
Materials and methods
All experiments were carried out under an inert atmosphere (with N2 or Ar) using standard Schlenk techniques. Nickel nitrate hexahydrate (Ni(NO3)2·6H2O, 99.9% purity) and cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 98.0% purity) were purchased from Alfa Aesar. Gallium nitrate (Ga(NO3)3·9H2O) was purchased from Sigma Aldrich (99.9% purity). Yttrium (Y(NO3)3·6H2O), lanthanum (La(NO3)3·6H2O), cerium (Ce(NO3)3·6H2O), praseodymium (Pr(NO3)3·6H2O), neodymium (Nd(NO3)3·6H2O), samarium (Sm(NO3)3·6H2O), gadolinium (Gd(NO3)3·6H2O), dysprosium (Dy(NO3)3·6H2O), and ytterbium (Yb(NO3)3·5H2O) nitrate were all purchased from Sigma Aldrich (99% purity). α-Alumina (Al2O3), Aeroxide AluC, was obtained from Evonik. Potassium, rubidium, lithium, and sodium carbonate purchased from Sigma Aldrich had a purity of 97% or higher and were used without further purification. Commercial 5% Ru/Al2O3 and 5% Rh/Al2O3 were purchased from Alfa Aesar and were used without further activation or purification. D2O (CIL, D-99.9%), toluene-d8 (CIL, D-99.5%), and imidazole (Sigma Aldrich, 99%) were used as received. 13C-labelled potassium carbonate was purchased from stable isotopes with a purity of 98.7%. 1H and 13C NMR spectra were recorded on 400, 500 or 600 MHz, Varian NMR spectrometers. 1H and 13C NMR chemical shifts were determined relative to the residual solvent signals. The gas mixtures were analyzed using a Thermo Finnigan gas chromatograph (column: Supelco, Carboxen 1010 plot, 30 m × 0.53 mm) equipped with a TCD detector (CO detection limit: 0.099 v/v%). CO2 (Gilmore, instrument grade) and H2 (Gilmore, ultra-high pure grade 5.0) were used as received.Caution: Reactions are associated with H2 gas. They should be carefully handled inside proper fume hoods without any flame, spark, or static electricity sources nearby.
Catalyst synthesis
To prepare the catalysts, the corresponding metal nitrates were first dissolved in 100 mL H2O. After that the support was added to this solution and stirred for 5 hours. Water was then removed with a rotavapor and the obtained solid dried overnight in an oven at 120 °C in air. The dried material was then calcinated by heating it in air from room temperature to 700 °C at a rate of 5.8 °C min−1 and maintaining it at 700 °C for 2 hours before letting it cool back down to room temperature. This calcination temperature was chosen following previous reports, but more optimized calcination and activation conditions may improve the yield.65–68Metal concentrations in the catalysts were calculated based on the activated species. For example, 12% by weight of nickel (wt%) in the catalyst named 12% Ni/Al2O3. 1 gram of catalyst would thus be composed of 0.88 g Al2O3 and 0.12 g Ni. The 0.12 g of Ni corresponds to 2.04 mmol of nickel, which is equivalent to 0.59 g of Ni(NO3)2·6H2O, the nitrate used for the preparation in this example. Similarly, this method was utilized for catalysts containing promoters. For example, for a 12% Ni/3% Yb/Al2O3 catalyst, when 1 gram of catalyst was synthesized, the catalyst contained 0.85 g Al2O3, 0.12 g Ni, and 0.03 g Yb. In this scenario, 0.59 g of Ni(NO3)2·6H2O is still utilized. Additionally, 0.03 g of Yb is equivalent to 0.17 mmol of Yb, requiring 77.9 mg of Yb(NO3)3·5H2O to be added.
Catalyst activation
The catalyst was crushed and sieved to a size of 250 micrometers or less. The sieved material was then activated in a tubular quartz reactor placed in a tubular furnace (Lindberg Blue). Nitrogen was flown through the catalyst at a rate of 35 mL min−1 for 30 minutes at room temperature. After that a mixture of hydrogen/nitrogen (35 mL min−1 and 35 mL min−1, respectively) was flown through the catalyst while it was heated to 700 °C (5.8 °C min−1) and held at that temperature for 2 hours.65–68 The catalyst was then allowed to cool down and was stored in an inert atmosphere for later use.Hydrogenation of carbonates to methane
All carbonate and bicarbonate salts were purchased with a purity of 97% or higher from Sigma Aldrich. The activated catalyst was weighed in an atmosphere of argon and then transported to a nitrogen chamber. There, 10 mmol of carbonate was mixed with DI water as the solvent. The catalyst, solvent (water), and carbonate salt were placed in a borosilicate vial. This vial was then placed in a 125 mL Hastelloy Parr reactor that was sealed in the nitrogen chamber. The Parr reactor was pressurized with hydrogen (UHP). Then, the reactor was transferred to a pre-heated aluminum block, heated to the desired temperature and held at that temperature for the duration of the reaction. At the end of the reaction, the reactor was cooled to room temperature, the pressure was released, and the solvent was separated from the catalyst via decanting. A portion of the gas mixture was released into a gas collection bag for gas chromatography (GC) analysis. The yield was then computed by integration of the gas peaks from the GC analysis. A sample calculation is provided in the ESI (see eqn (S2)†).Air capture of CO2 with potassium hydroxide
To capture CO2 from air (∼420 ppm CO2) 11 mmol of potassium hydroxide (Sigma Aldrich, 97%) was dissolved in 11 mL of DI H2O in a vial. The vial was sealed and ambient air from the lab was then flown through the vial at the rate of 300 mL min−1. The CO2 capture from air was run for 48 hours. Afterwards, imidazole was added as an internal standard to a 1 mL aliquot of the capture solution. This aliquot was analyzed by 13C NMR with D2O as the deuterated solvent. The amount of CO2 was quantified through 13C NMR analysis. The remaining solution was used for the hydrogenation reaction.Capture from pure CO2
A known amount of alkali hydroxide (KOH) was dissolved in DI water (10 mL) in a vial with a magnetic stir bar. The gases inside the vial were then removed under vacuum. CO2 was subsequently added while stirring the solution at 800 rpm for 3 h and maintaining the CO2 pressure inside the reactor at 1 psi above atmospheric pressure. The amount of CO2 captured was calculated both through the volume of CO2 added and through gravimetric analysis of the solutions before and after the capture.Recycling experiments
Once the carbonate hydrogenation reaction according to the method described above was complete, the reactor was cooled down to room temperature and the pressure released. Part of the pressure was released into a collection bag for gas chromatography analysis. The reactor was then transferred to a nitrogen chamber and opened. The liquid in the reactor was separated from the catalyst by decantation and placed in a 100 mL round bottom flask. After evacuation, the obtained liquid was re-used for CO2 capture following the same conditions and parameters as detailed in the previous section entitled “Capture from pure CO2,” vide supra. The amount of CO2 captured was measured by both the volume of CO2 added and also by weight (gravimetrically). The liquid was then placed back in the reactor with the catalyst that was utilized in the previous cycle. The hydrogenation reactions were then performed again with the conditions detailed above.Powder X-ray diffraction (XRD)
Powder XRD analysis was performed on a sixth generation Rigaku Miniflex powder diffractometer. The catalyst was wet loaded onto a sample plate and then dried of any solvent. The scan was set from 10°–90° at a scan rate of 3° min−1. The resulting spectra were processed on the PDXL software.Scanning electron microscopy (SEM)
Scanning Electron Microscopy (SEM) images and energy dispersive X-ray spectroscopy (EDS) images were obtained from a JEOL JSM-7001F electron microscope with an acceleration voltage of 18 keV.X-ray fluorescence (XRF)
X-Ray Fluorescence (XRF) was conducted on a Bruker Tiger S8 instrument. The X-ray source is rhodium leading to residual rhodium signals, which are labelled in the spectrum. The spectra were all collected between 0–60 keV. The weight percentages of the metals were calculated using the Bruker software and all errors of the measurements are also included. The calculations were based on the Kα peak, except for ytterbium as the Ni Kα peak overlaps with the Yb Kα. Thus, the concentrations were based on the Kβ peaks for all of the nickel–ytterbium catalyst.Catalyst cost assessment
The cost analysis of the different catalytic processes was estimated using the CatCost program v.1.1.0 in Microsoft Excel v.16.80 developed by the Energy Material Network from the Department of Energy.67 The bulk costs of the chemical materials were calculated according to the method outlined in the ESI† and compared to prices from AliBaba. All values are adjusted to 2022 dollars (USD) using the Chemical Producer Price Index from the US Bureau of Labor Statistics.Results and discussion
Effect of promoter on carbonate salt reduction
At first, several types of catalysts were screened for the conversion of carbonate salts to methane in an aqueous solution. Each catalyst was tested with 10 mmol of K2CO3 in 10 mL water for 24 hours at a pressure of 50 bar H2, 225 °C and 300 mg of catalyst. The results of these experiments are shown in Table 1. Besides methane production in mmol, methane productivity in gmethane h−1 kgcat−1 was calculated as well to provide additional avenues for comparison. 5% Ru/Al2O3 is well reported as a catalyst efficient for producing methane from CO2 in the Sabatier process.40,68–70 It was similarly active for the conversion of potassium carbonate to methane, achieving a 100% yield and a productivity of 22.3 gmethane h−1 kgcat−1. 5% Rh/Al2O3, on the other hand, performed poorly in the conversion of potassium carbonate to methane, achieving only a 14% yield and a productivity of 3.12 gmethane h−1 kgcat−1. Earth abundant metals, such as nickel and cobalt, can also be utilized for the conversion of potassium carbonate to methane. 25% Ni/Al2O3 was able to achieve a yield of 64% and a productivity of 14.3 gmethane h−1 kgcat−1. Decreasing the metal concentration to 12% Ni/Al2O3 resulted in a yield of 12% and a productivity of 2.67 gmethane h−1 kgcat−1. Changing the support to silica decreased the performance of the catalyst slightly. 12% Ni/SiO2 resulted in a yield of 10% and a productivity of 2.23 gmethane h−1 kgcat−1. A silica support is nevertheless not ideal as it can react with alkali to form alkali silicate (e.g. sodium silicate with sodium hydroxide) which can diminish the catalytic activity.71–73 12% Co/Al2O3 was able to achieve a 11% yield and a productivity of 2.45 gmethane h−1 kgcat−1, similar to 12% Ni/Al2O3. Changing the support to silica with cobalt also resulted in a lower yield of 9% and a productivity of 2.01 gmethane h−1 kgcat−1 as well as the production of some CO2. Additionally, 7% Co/11% Ga/SiO2 was tested for the conversion of K2CO3 to determine if the alloy may exhibit reactivity similar to that of nickel–gallium alloys, although only CO2 was detected.74–76 For all the catalyst tested, the liquid phase of the reaction did not have any additional products.| Catalyst | Products | Methane productivity (gmethane h−1 kgcat−1) | |||
|---|---|---|---|---|---|
| CO2 (mmol) | CO (mmol) | CH4 (mmol) | CH4 yield (%) | ||
| a Conditions: 10 mmol K2CO3, 10 mL DI H2O, 225 °C, 300 mg catalyst, 50 bar H2 at room temperature, 24 hours. Yields calculated from the gas phase by gas chromatography are within ±5% error. | |||||
| 25% Ni/Al2O3 | 0 | 0 | 6.4 | 64 | 14.3 |
| 5% Ru/Al2O3 | 0 | 0 | 10 | 100 | 22.3 |
| 5% Rh/Al2O3 | 0 | 0 | 1.5 | 15 | 3.33 |
| 12% Ni/Al2O3 | 0 | 0 | 1.2 | 12 | 2.67 |
| 12% Ni/3% Y/Al2O3 | 0 | 0 | 6.9 | 69 | 15.4 |
| 12% Ni/3% La/Al2O3 | 0 | 0 | 6.2 | 62 | 13.8 |
| 12% Ni/3% Ce/Al2O3 | 0 | 0 | 4.8 | 48 | 10.7 |
| 12% Ni/3% Pr/Al2O3 | 0 | 0 | 6.3 | 63 | 14.0 |
| 12% Ni/3% Nd/Al2O3 | 0 | 0 | 6.3 | 63 | 14.0 |
| 12% Ni/3% Sm/Al2O3 | 0 | 0 | 6.4 | 64 | 14.3 |
| 12% Ni/3% Gd/Al2O3 | 0 | 0 | 6.9 | 69 | 15.4 |
| 12% Ni/3% Dy/Al2O3 | 0 | 0 | 6.9 | 69 | 15.4 |
| 12% Ni/3% Yb/Al2O3 | 0 | 0 | 7.0 | 70 | 15.6 |
| 12% Ni/3% Cu/Al2O3 | 0 | 0 | 1.2 | 12 | 2.67 |
| 12% Co/Al2O3 | 0 | 0 | 1.1 | 11 | 2.45 |
| 12% Ni/SiO2 | 0 | 0 | 1.0 | 10 | 2.23 |
| 12% Co/SiO2 | 1.4 | 0 | 0.9 | 9 | 2.01 |
| 7% Co/11% Ga/SiO2 | 1.8 | 0 | 0 | 0 | 0 |
Due to the promising activity that the non-noble metal catalyst based on nickel showed for the conversion of potassium carbonate to methane, Cu and several types of lanthanide promoters were added to this catalyst in an effort to improve the productivity of the system. In general, 3 wt% of the promoters was added. At first, a Cu promoted 12% Ni/3% Cu/Al2O3 catalyst was tested for the methanation of K2CO3, leading to 12% methane yield and a productivity of 2.67 gmethane h−1 kgcat−1, which was similar to the results achieved with the unpromoted 12% Ni/Al2O3 catalyst. Afterwards, a series of lanthanide promoters were investigated following reports showcasing significant improvements in reactivity with such promoters.32,77–81 Indeed, when these elements were added to the nickel-based catalyst the methane productivity jumped from 2.67 gmethane h−1 kgcat−1 to between 10.7 and 15.6 gmethane h−1 kgcat−1; representing a surprising 4 to 6 fold increase! For the most part, the results obtained with the lanthanide promoters followed the lanthanide contraction trend, where the smaller the atomic size of the lanthanide, the better the performance of the catalyst, as shown in Fig. 2. The lanthanum promoted catalyst, 12% Ni/3% La/Al2O3 led to a yield of 62% and a productivity of 13.8 gmethane h−1 kgcat−1. The cerium promoted catalyst yield of 48% and productivity of 10.7 gmethane h−1 kgcat−1, were lower than the ones for the lanthanum-promoted catalyst, constituting somewhat of an outlier in the lanthanide promotion trend. Cerium acting like an outlier is likely due to the metal promoter being not fully reduced to Ce during the catalyst activation step. CeO2 is observed in the XRD of the catalyst before and after the reaction (Fig. S10 and S11†). This correlates with previous reports where CeO2 is shown to not fully reduce at the activation temperature employed here and could explain why the 12% Ni/3% Ce/Al2O3 did not follow the lanthanide contraction trend.84 However, it should be noted that even though 12% Ni/3% Ce/Al2O3 had a lower activity it was still four times as effective as the unpromoted 12% Ni/Al2O3 catalyst. Neodymium and praseodymium promoted catalysts were both able to produce similar yields of 63% and a productivity of 14.0 gmethane h−1 kgcat−1. The samarium promoted catalyst produced a yield of 64% and a productivity of 14.3 gmethane h−1 kgcat−1. Gadolinium, dysprosium, and yttrium promoted catalysts were also able to achieve a yield of 69% and a productivity of 15.4 gmethane h−1 kgcat−1. The ytterbium promoted catalyst produced the highest yield of 70% with a productivity of 15.6 gmethane h−1 kgcat−1.
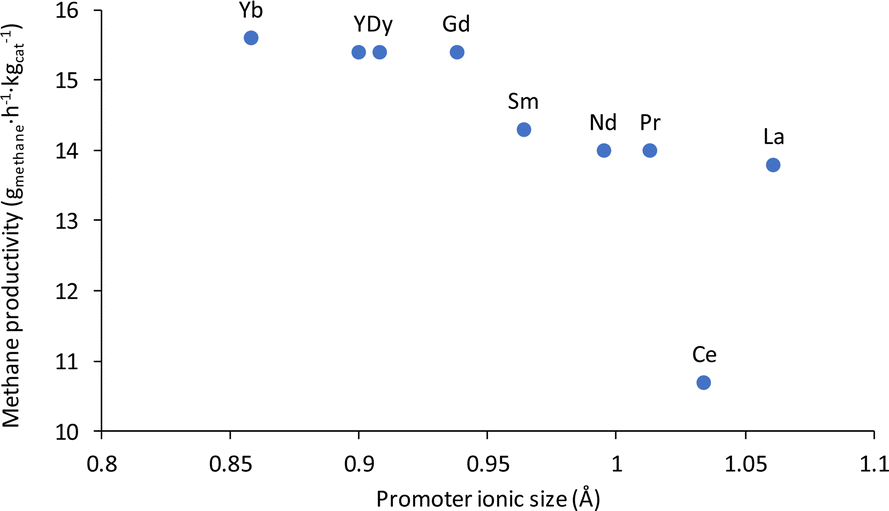 | ||
| Fig. 2 Methane productivity compared to the ionic size of the lanthanide promoter on 12% Ni/Al2O3.82,83 | ||
Following the lanthanide screening, the reaction scope of various carbonate salts was explored with the 12% Ni/3% Yb/Al2O3 catalyst that gave the highest methane yield with K2CO3. The results of this screening are shown in Table 2. Potassium carbonate achieved the highest methane yield of 70%. However, other carbonate salts were also converted to methane in varying degrees. Sodium carbonate led to a 50% yield. Other carbonate salts were not as efficient. Lithium, cesium, and magnesium carbonate all produced similar methane yields of 30, 34, and 30%, respectively. Calcium carbonate was also tested but gave a conversion of only 2%. From all the carbonates in this study, calcium carbonate had the lowest solubility in water (please see ESI†), which could be part of the much lower methane yield observed.
| Carbonate salt | Productsb | Methane productivity (gmethane h−1 kgcat−1) | |
|---|---|---|---|
| CH4 (mmol) | CH4 yield (%) | ||
| a Conditions: 10 mmol carbonate salt, 10 mL DI H2O, 225 °C, 300 mg 12% Ni/3% Yb/Al2O3, 50 bar H2 at room temperature, 24 hours. b No CO or CO2 detected in any of the reactions. Yields calculated from the gas phase by gas chromatography are within ±5% error. | |||
| Li2CO3 | 3.0 | 30 | 6.68 |
| Na2CO3 | 5.0 | 50 | 11.1 |
| K2CO3 | 7.0 | 70 | 15.6 |
| Cs2CO3 | 3.4 | 34 | 7.57 |
| MgCO3 | 3.0 | 30 | 6.68 |
| CaCO3 | 0.2 | 2 | 0.45 |
The carbonates utilized in this study have decomposition temperatures significantly higher than the reaction temperature (please see ESI for decomposition temperatures of the carbonates†). Calcium carbonate, which exhibits the lowest decomposition temperature at around 600 °C also led to the lowest methane yield.59 Therefore it does not seem that the decomposition temperature of the carbonate salt utilized plays a significant role in the reactivity of the carbonate. This is further supported by the results in Table 1 where CO2 was not detected in reactions conducted with the nickel and nickel–lanthanide catalysts. It would be expected that if the decomposition of the carbonates occurred CO2 would be released first. It should also be noted that the trend in carbonate conversion to methane was similar to previously reported systems.31,59
Effect of nickel and promoter loading on the carbonate salt reduction
To improve the methane productivity further, the weight percentages of nickel and ytterbium were increased as shown in Table 3, for a composition of 33% Ni/8% Yb/Al2O3 and 50% Ni/12.5% Yb/Al2O3. For comparison, the catalysts without ytterbium, 33% Ni/Al2O3 and 50% Ni/Al2O3, were synthesized and tested as well for a reaction duration of 24 hours. The effect of reaction time (6, 12, and 24 hours) was also tested on the Yb containing catalysts (Fig. 3). The 5% Ru/Al2O3 catalyst had its highest methane yield after 24 h. At the same time, the productivity of 5% Ru/Al2O3 was the lowest at 22.3 gmethane h−1 kgcat−1 after 24 h, compared to productivities of 42.3 and 68.6 gmethane h−1 kgcat−1 after 12 and 6 h, respectively. Nevertheless, as already mentioned, although 5% Ru/Al2O3 was very productive, this type of catalyst was not stable over several hydrogenation cycles under the reaction conditions.59 The 12% Ni/3% Yb/Al2O3 catalyst was able to achieve a productivity of 15.6 gmethane h−1 kgcat−1 in 24 hours, which was an improvement from the productivity of 2.67 gmethane h−1 kgcat−1 of the unpromoted 12% Ni/Al2O3 catalyst (Fig. 4). However, decreasing the reaction time to 12 and 6 hours resulted in productivities 11.6 and 9.80 gmethane h−1 kgcat−1, respectively. This trend was somewhat opposite to the behaviour of the 5% Ru/Al2O3 catalyst and catalysts containing higher Ni/Yb loadings discussed hereafter. Upon increasing the metal weight content, greater productivities were achieved. Utilizing the 33% Ni/8% Yb/Al2O3 catalyst a productivity of 18.1 gmethane h−1 kgcat−1 was obtained in 24 hours, which was an improvement over the productivity of 14.7 achieved by the unpromoted 33% Ni/Al2O3 catalyst. Decreasing the reaction time to 12 hours resulted in a productivity of 19.2 gmethane h−1 kgcat−1. Further decreasing the reaction time to 6 h led to a productivity of 21.4 gmethane h−1 kgcat−1. Increasing the metal concentrations of the catalyst to higher levels in 50% Ni/12.5% Yb/Al2O3 further increased the productivity of the process. Over 24 hours on 50% Ni/12.5% Yb/Al2O3, the productivity was 22.3 gmethane h−1 kgcat−1, which corresponds to full conversion of the carbonate salt to methane and was on par with the 5% Ru/Al2O3 catalyst. The productivity of the unpromoted 50% Ni/Al2O3 catalyst was lower, achieving a productivity of only 17.8 gmethane h−1 kgcat−1. Decreasing the reaction time to 12 and 6 hours resulted in productivities of 25.6 and 28.5 gmethane h−1 kgcat−1 respectively. Since the 50% Ni/12.5% Yb/Al2O3 catalyst was able to achieve quantitative yields in 24 h, it was used in further testing.| Catalyst | Time (h) | Productsb | Methane productivity (gmethane h−1 kgcat−1) | |
|---|---|---|---|---|
| CH4 (mmol) | CH4 yield (%) | |||
| a Conditions: 10 mmol K2CO3, 10 mL DI H2O, 225 °C, 300 mg catalyst, 50 bar H2 at room temperature. b No CO or CO2 detected in any of the reactions. Yields calculated from the gas phase by gas chromatography are within ±5% error. | ||||
| 5% Ru/Al2O3 | 24 | 10 | 100 | 22.3 |
| 5% Ru/Al2O3 | 12 | 9.5 | 95 | 42.3 |
| 5% Ru/Al2O3 | 6 | 7.7 | 77 | 68.6 |
| 12% Ni/Al2O3 | 24 | 1.2 | 12 | 2.67 |
| 12% Ni/3% Yb/Al2O3 | 24 | 7.0 | 70 | 15.6 |
| 12% Ni/3% Yb/Al2O3 | 12 | 2.6 | 26 | 11.6 |
| 12% Ni/3% Yb/Al2O3 | 6 | 1.1 | 11 | 9.80 |
| 33% Ni/Al2O3 | 24 | 6.6 | 66 | 14.7 |
| 33% Ni/8% Yb/Al2O3 | 24 | 8.1 | 81 | 18.1 |
| 33% Ni/8% Yb/Al2O3 | 12 | 4.3 | 43 | 19.2 |
| 33% Ni/8% Yb/Al2O3 | 6 | 2.4 | 24 | 21.4 |
| 50% Ni/Al2O3 | 24 | 8.0 | 80 | 17.8 |
| 50% Ni/12.5% Yb/Al2O3 | 24 | 10 | 100 | 22.3 |
| 50% Ni/12.5% Yb/Al2O3 | 12 | 5.8 | 58 | 25.6 |
| 50% Ni/12.5% Yb/Al2O3 | 6 | 3.2 | 32 | 28.5 |
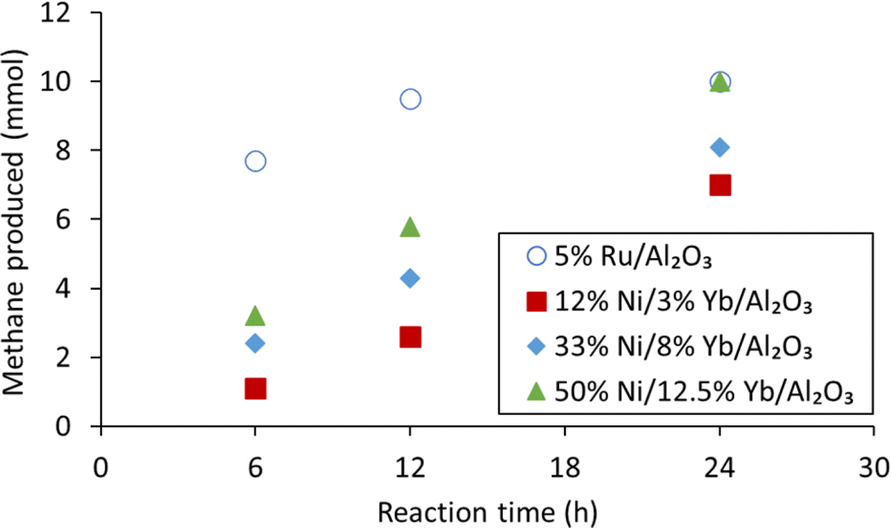 | ||
| Fig. 3 Methane production from K2CO3 on catalysts with various metal loadings of nickel and ytterbium in comparison to the commercial 5% Ru/Al2O3 catalyst after 6, 12 and 24 h reaction. Conditions: 10 mmol K2CO3, 10 mL DI H2O, 225 °C, 300 mg catalyst, 50 bar H2 at room temperature. | ||
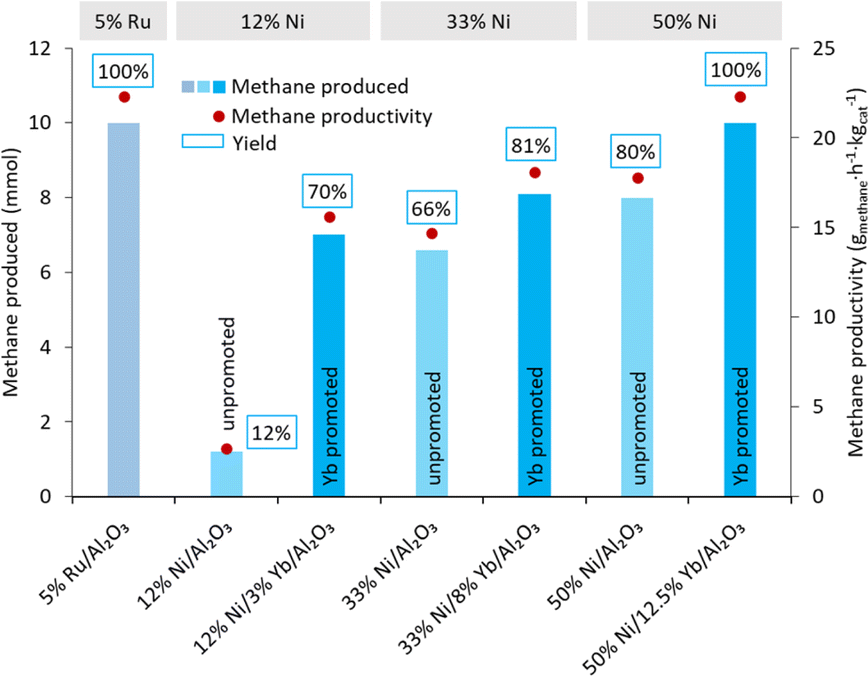 | ||
| Fig. 4 Methane production from K2CO3 on Yb promoted Ni/Al2O3 catalysts compared to unpromoted Ni/Al2O3 as a function of metal loading. Conditions: 10 mmol K2CO3, 10 mL DI H2O, 225 °C, 300 mg catalyst, 50 bar H2 at room temperature, 24 h. | ||
The 50% Ni/12.5% Yb/Al2O3 catalyst was used to convert several carbonate salts to methane and the results were compared to those with the 12% Ni/3% Yb/Al2O3 catalyst from Table 2. Indeed, all the carbonate salts did show improvements compared to the 12% Ni/3% Yb/Al2O3 catalyst as shown in Fig. 5. Using potassium carbonate as the reagent, the yield of methane improved from 70% with 12% Ni/3% Yb/Al2O3 to 100% with 50% Ni/12.5% Yb/Al2O3. 13C-labelled potassium carbonate was also used as a reagent in this reaction and produced 13CH4. The methane yield with K2CO3 and K213CO3 were the same at 100%. The reaction conditions were also modified to investigate the upper limits of methane productivity that can be achieved with the 50% Ni/12.5% Yb/Al2O3 catalyst. With 50 mg catalyst under 50 bar H2, 225 °C, and 6 hours a yield of 11% was achieved, which resulted in a methane productivity of 58.8 gmethane h−1 kgcat−1 (Table S1†). When sodium carbonate was used, a large increase in the yield of methane was observed, from 50% with the 12% Ni/3% Yb/Al2O3 to 100% with the 50% Ni/12.5% Yb/Al2O3. The methane yield with lithium carbonate also greatly improved from 30% to 73% with 12% Ni/3% Yb/Al2O3 and 50% Ni/12.5% Yb/Al2O3, respectively. Cesium carbonate experienced a slight increase in yield from 34% to 56% with the 50% Ni/12.5% Yb/Al2O3 catalyst. With MgCO3 a mild yield improvement from 30 to 46% was measured. Although a yield increase was also observed with calcium carbonate, the overall conversion remained low with the 50% Ni/12.5% Yb/Al2O3 catalyst.
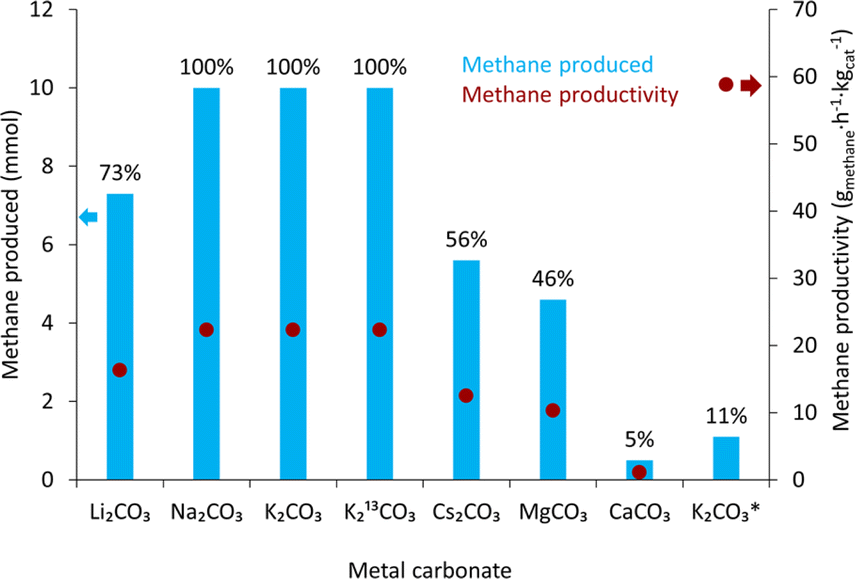 | ||
| Fig. 5 Conversion of various carbonate salts to methane over the 50% Ni/12.5% Yb/Al2O3. Conditions: 10 mmol carbonate salt, 10 mL DI H2O, 225 °C, 300 mg 50% Ni/12.5% Yb/Al2O3, 50 bar H2 at room temperature, 24 hours. *Conditions: 6 hours, 50 mg 50% Ni/12.5% Yb/Al2O3, 275 °C, 50 bar H2, 10 mL H2O. No CO or CO2 detected in any of the reactions. Productivity is shown with the red dots. The reaction yields in % are displayed above the blue columns. Yields calculated from the gas phase by gas chromatography are within ±5% error. | ||
Catalyst recyclability
The recyclability of the catalyst was tested as well to determine if the catalyst showed signs of deactivation or if the base underwent any side reaction over five cycles of reactivity. Utilizing 300 mg of the 50% Ni/12.5% Yb/Al2O3, five cycles of capture/hydrogenation were conducted, and the results are shown in Fig. 6. In the first cycle, 4 mmol of KOH were dissolved in 10 mL H2O and the solution subjected to a pure CO2 stream for 3 hours. The obtained media was then reacted with 50 bar H2 at 225 °C for 24 hours, producing 4 mmol of CH4. After reaction, the liquid media was separated from the catalyst and subjected to CO2 capture to determine if the base, KOH, had been regenerated. 4 mmol of CO2 were captured in the form of potassium bicarbonate, meaning that the base KOH had been fully regenerated and that the base was not undergoing a parasitic side reaction. Afterwards, the capture solution was reunited with the catalyst and the methanation conducted once more. These steps were repeated over five cycles of reactivity during which no loss in activity of the catalyst was observed. The metals on the catalyst did not seem to have oxidized as seen in the XRD pattern (Fig. S9†) and regeneration of the base was observed. This indicates that the catalyst was stable under these alkaline conditions and did not seem to participate in parasitic side reactions with the base, which was successfully regenerated for CO2 capture in subsequent cycles.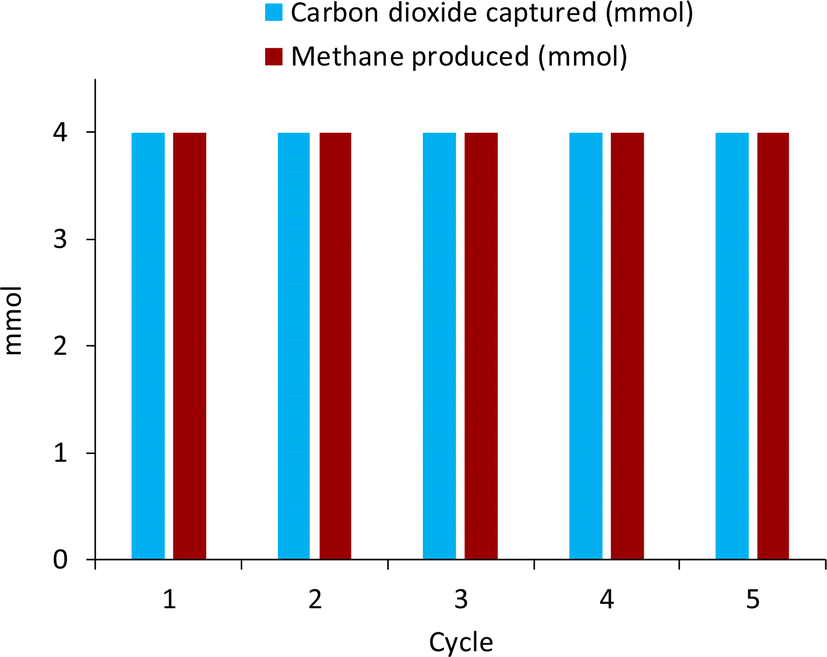 | ||
| Fig. 6 Recycling of the 50% Ni/12.5% Yb/Al2O3 catalyst over five cycles of reactivity. Conditions: 10 mL DI H2O, 225 °C, 300 mg 50% Ni/12.5% Yb/Al2O3, 50 bar H2 at room temperature, 24 hours. 4 mmol KOH used in first cycle. Yields calculated from the gas phase by gas chromatography are within ±5% error. | ||
The recyclability of nickel-based catalysts under alkaline conditions was previously demonstrated with unpromoted Ni/Al2O3 catalysts.31 However, when a similar study was undertaken with Ru/Al2O3 catalysts, the catalyst deactivated rapidly over the course of the reaction. A 70% loss in reactivity was observed after a single cycle.59 It was determined that the alkaline conditions interacted with the Ru-based catalyst leading to the formation of ruthenium oxide as observed by XRD of the catalyst post-reaction. This was proposed as a reason for the rather rapid decrease in activity in the case of Ru/Al2O3. Nickel-based catalysts and lanthanide promoted nickel catalysts presented here did not display a similar deactivation and the reactivity remained constant throughout five reaction cycles. Of course, extensive additional work will be needed to assess the long-term stability and robustness of the lanthanide promoted nickel catalysts. This and many other factors will determine the potential of these catalysts in a practical application.85,86
CO2 capture from air and integrated conversion to methane
Potassium hydroxide was also used to capture CO2 from air (∼420 ppm CO2). 11 mmol of KOH in 11 mL of water (1 M solution) captured 5.5 mmol of CO2 in the form of K2CO3. The potassium carbonate was then converted to methane over 50% Ni/12.5% Yb/Al2O3 in quantitative yields. Fig. 7 showcases this process' ability to capture CO2 from the atmosphere for the conversion to methane.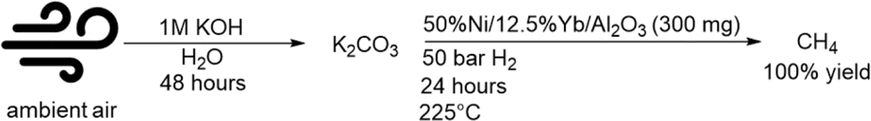 | ||
| Fig. 7 Direct air capture with KOH and conversion to methane over 50%Ni/12.5%Yb/Al2O3. | ||
Catalyst cost analysis
A cost analysis was performed to compare the economic viability and sustainability of the various nickel catalysts tested using the CatCost program developed by the Energy Material Network from the U. S. Department of Energy (Table 4).67 More details about this analysis can be found in the ESI.† The chosen variable for comparison is the cost of the catalyst needed in USD to produce 1 kg of methane. All catalysts were assumed to be reused for a three-year lifetime with one reaction cycle per day. This simplified analysis assumed only the input cost of the catalyst to produce methane for the ICCC to methane process. Utilizing a commercially available 5% Ru/Al2O3 catalyst in this process resulted in a cost of $7.51cat kgmethane−1, which was significantly more expensive than the nickel catalysts. The ytterbium promoter, despite the added metal to the nickel catalyst composition, decreased the cost relative to the respective unpromoted nickel catalysts. The 12% Ni/3% Yb/Al2O3 catalyst was the most cost-effective at $0.05cat kgmethane−1. On the other hand, the unpromoted 12% Ni/Al2O3 was the most expensive nickel catalyst at $0.30cat kgmethane−1. The 33% Ni/8% Yb/Al2O3 and 50% Ni/12.5% Yb/Al2O3 catalysts also differed in cost per kilogram produced compared to their respective unpromoted counterparts by approximately $0.02cat kgmethane−1. These results indicate that despite the added cost of the ytterbium for all promoted catalysts the process was more economical than with the unpromoted nickel and commercial ruthenium counterparts under these conditions. Furthermore, as discussed earlier the ruthenium catalyst is not as recyclable as the nickel and nickel–ytterbium catalysts. Extra costs from the energy and materials needed to collect deactivated ruthenium and reactivate it for further cycles render it less green and sustainable than the recyclable nickel–lanthanide catalyst. From an environmental and sustainability point of view, the carbon footprint associated with extracting and purifying lanthanides from the environment or recycling them must also be considered.87 A life cycle analysis (LCA) of the catalyst production process and the methane production from CO2via metal carbonates will thus be needed for a more complete picture and comparison with other routes/catalysts. Potentially, the carbon emissions from the mining and processing of lanthanide metals could also be captured and converted into methane using the process described here. While this would not render the process carbon neutral, it would at least employ/recycle the CO2 one more time before being emitted to the environment.| Catalyst | Cost ($cat kgmethane−1) |
|---|---|
| a Reference conditions: 10 mmol KOH, 10 mL DI H2O, 225 °C, 300 mg catalyst, 50 bar H2 at room temperature, 24 hours. The cost was taken over a three-year lifetime of the catalyst with one reaction cycle per day (total of 1095 reaction cycles). | |
| 50% Ni/12.5% Yb/Al2O3 | 0.07 |
| 50% Ni/Al2O3 | 0.09 |
| 33% Ni/8.5% Yb/Al2O3 | 0.07 |
| 33% Ni/Al2O3 | 0.09 |
| 12% Ni/3% Yb/Al2O3 | 0.05 |
| 12% Ni/Al2O3 | 0.30 |
| 5% Ru/Al2O3 | 7.51 |
Conclusions
Previous methods of hydroxide-assisted integrated capture and conversion of carbon dioxide to methane have utilized nickel and ruthenium catalysts. Methods based on nickel catalysts have been reported with relatively low productivity rates. Although ruthenium catalysts had a higher productivity, they also exhibited limited stability under alkaline conditions and were prone to deactivation. In the present report, the utilization of lanthanide promoted nickel catalysts led to a substantial increase in methane productivities with values up to 58.8 gmethane h−1 kgcat−1 for 50% Ni/12.5% Yb/Al2O3. Up to 100% conversion of the carbonate to methane was also achieved. The activity enhancement imparted by the addition of lanthanides was most pronounced at lower Ni/lanthanide loadings on the catalyst. For the most part, the catalytic performance of the lanthanide promoted nickel catalysts followed the lanthanide contraction trend, i.e. the smaller the atomic size of the lanthanide, the higher the methane yield.Furthermore, the lanthanide promoted nickel catalysts were able to maintain their reactivity over five cycles of reaction. The integrated direct air capture and conversion to methane was also achieved in quantitative yields. The promoted nickel catalysts were shown to be more cost-effective and sustainable than ruthenium and the unpromoted nickel catalysts. Overall, the results obtained demonstrate the positive effect of lanthanide promotion of nickel-based catalysts on methane productivity and reaction time for metal carbonate hydrogenation.
Data availability
Additional data supporting this article have been included as part of the ESI.†Author contributions
Christopher J. Koch: investigation, methodology, formal analysis, validation, writing – original draft preparation, visualization. Zohaib Suhail: investigation, validation, writing – original draft preparation. Prince: investigation, validation. Anushan Alagaratnam: investigation. Matthew Coe: investigation. Alain Goeppert: conceptualization, supervision, project administration, writing – review & editing, visualization. G. K. Surya Prakash: conceptualization, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Support of our work by the Loker Hydrocarbon Research Institute at the University of Southern California is gratefully acknowledged. We would like to thank the National Science Foundation (award: CHE-2018740) for the purchase of the diffractometer used to obtain powder diffraction patterns reported here. We would also like to thank the Center of Excellence for Nano Imaging (CNI) at the University of Southern California for microscope time and access to the XRF.Notes and references
- H. Pilorgé, N. McQueen, D. Maynard, P. Psarras, J. He, T. Rufael and J. Wilcox, Environ. Sci. Technol., 2020, 54, 7524–7532 CrossRef PubMed.
- A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- European Commission, Implementing the REPOWER EU Action Plan: Investment Needs, Hydrogen Accelerator and Achieving the Bio-Methane Target, Brussels, 2022 Search PubMed.
- M. C. Bacariza, D. Spataru, L. Karam, J. M. Lopes and C. Henriques, Processes, 2020, 8, 1–45 CrossRef.
- IEA, District Heating, Paris, 2022 Search PubMed.
- S. Sun, H. Sun, P. T. Williams and C. Wu, Sustainable Energy Fuels, 2021, 5, 4546–4559 RSC.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- C. J. Koch, A. Alagaratnam, A. Goeppert and G. K. S. Prakash, Sustainable Energy Fuels, 2023, 7, 2824–2829 RSC.
- R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed.
- R. Sen, C. J. Koch, A. Goeppert and G. K. S. Prakash, ChemSusChem, 2020, 13, 6318–6322 CrossRef CAS PubMed.
- S. Kar, R. Sen, J. Kothandaraman, A. Goeppert, R. Chowdhury, S. B. Munoz, R. Haiges and G. K. S. Prakash, J. Am. Chem. Soc., 2019, 141, 3160–3170 CrossRef CAS PubMed.
- S. Kar, A. Goeppert and G. K. S. Prakash, ChemSusChem, 2019, 12, 3172–3177 CrossRef CAS PubMed.
- A. Goeppert, H. Zhang, R. Sen, H. Dang and G. K. S. Prakash, ChemSusChem, 2019, 12, 1712–1723 CrossRef CAS PubMed.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed.
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- Y. Meng, J. Jiang, Y. Gao, F. Yan, N. Liu and A. Aihemaiti, J. CO2 Util., 2018, 27, 89–98 CrossRef CAS.
- M. Jahandar Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC.
- F. Vega, A. Sanna, B. Navarrete, M. M. Maroto-Valer and V. J. Cortés, Greenhouse Gases: Sci. Technol., 2014, 4, 707–733 CrossRef CAS.
- Y. Du, Y. Yuan and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2017, 58, 1–9 CrossRef CAS.
- S. B. Fredriksen and K.-J. Jens, Energy Procedia, 2013, 37, 1770–1777 CrossRef CAS.
- R. Sen, A. Goeppert and G. K. S. Prakash, J. Organomet. Chem., 2022, 965–966, 122331 CrossRef CAS.
- C. Chen, S.-T. Yang, W.-S. Ahn and R. Ryoo, Chem. Commun., 2009, 3627–3629 RSC.
- H. Zhang, A. Goeppert, G. A. Olah and G. K. S. Prakash, J. CO2 Util., 2017, 19, 91–99 CrossRef CAS.
- S. Meth, A. Goeppert, G. K. S. Prakash and G. A. Olah, Energy Fuels, 2012, 26, 3082–3090 CrossRef CAS.
- M. Mahmoudkhani and D. W. Keith, Int. J. Greenhouse Gas Control, 2009, 3, 376–384 CrossRef CAS.
- C. J. Koch, V. Galvan, A. Goeppert and G. K. S. Prakash, Green Chem., 2023, 25, 1803–1808 RSC.
- C. J. Koch, A. Alagaratnam, A. Goeppert and G. K. S. Prakash, Isr. J. Chem., 2023, 63, e202200119 CrossRef CAS.
- F. I. Azof, M. Vafeias, D. Panias and J. Safarian, Hydrometallurgy, 2020, 191, 105184 CrossRef CAS.
- R. Sen, A. Goeppert and G. K. S. Prakash, Angew. Chem., Int. Ed., 2022, 61, e202207278 CrossRef CAS PubMed.
- S. Kar, J. Kothandaraman, A. Goeppert and G. K. S. Prakash, J. CO2 Util., 2018, 23, 212–218 CrossRef CAS.
- J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS.
- S. Xie, Z. Li, H. Li and Y. Fang, Catal. Rev.: Sci. Eng., 2023, 1–40 CrossRef.
- A. Shima, M. Sakurai, Y. Sone, M. Ohnishi and T. Abe, in 42nd International Conference on Environmental Systems 2012, ICES 2012, American Institute of Aeronautics and Astronautics Inc., 2012 Search PubMed.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef CAS.
- E. Moioli and A. Züttel, Sustainable Energy Fuels, 2020, 4, 1396–1408 RSC.
- R. Sen, C. J. Koch, V. Galvan, N. Entesari, A. Goeppert and G. K. S. Prakash, J. CO2 Util., 2021, 54, 101762 CrossRef CAS.
- A. Gallo, J. L. Snider, D. Sokaras, D. Nordlund, T. Kroll, H. Ogasawara, L. Kovarik, M. S. Duyar and T. F. Jaramillo, Appl. Catal., B, 2020, 267, 118369 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- J. A. Onrubia-Calvo, A. Bermejo-López, S. Pérez-Vázquez, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Fuel, 2022, 320, 123842 CrossRef CAS.
- G. Wang, Y. Guo, J. Yu, F. Liu, J. Sun, X. Wang, T. Wang and C. Zhao, Chem. Eng. J., 2022, 428, 132110 CrossRef CAS.
- S. J. Park, M. P. Bukhovko and C. W. Jones, Chem. Eng. J., 2021, 420, 130369 CrossRef CAS.
- S. Jo, J. H. Lee, T. Y. Kim, J. H. Woo, H.-J. Ryu, B. Hwang, S. C. Lee, J. C. Kim and K. L. Gilliard-AbdulAziz, Fuel, 2022, 311, 122602 CrossRef CAS.
- H. Sun, Y. Zhang, S. Guan, J. Huang and C. Wu, J. CO2 Util., 2020, 38, 262–272 CrossRef CAS.
- S. Cimino, F. Boccia and L. Lisi, J. CO2 Util., 2020, 37, 195–203 CrossRef CAS.
- M. A. Arellano-Treviño, N. Kanani, C. W. Jeong-Potter and R. J. Farrauto, Chem. Eng. J., 2019, 375, 121953 CrossRef.
- S. J. Park, Y. Kim and C. W. Jones, ChemSusChem, 2020, 13, 2988–2995 CrossRef CAS PubMed.
- S. Sun, Y. Zhang, C. Li, Y. Wang, C. Zhang, X. Zhao, H. Sun and C. Wu, Sep. Purif. Technol., 2023, 323, 124399 CrossRef.
- M. S. Duyar, S. Wang, M. A. Arellano-Treviño and R. J. Farrauto, J. CO2 Util., 2016, 15, 65–71 CrossRef CAS.
- Z. Lv, J. Ruan, W. Tu, X. Hu, D. He, X. Huang and C. Qin, Sep. Purif. Technol., 2023, 309, 123044 CrossRef CAS.
- E. García-Bordejé, A. B. Dongil, J. Moral, J. M. Conesa, A. Guerrero-Ruiz and I. Rodríguez-Ramos, J. CO2 Util., 2023, 68, 102370 CrossRef.
- J. Kothandaraman and D. J. Heldebrant, Green Chem., 2020, 22, 828–834 RSC.
- J. Kothandaraman, J. S. Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, Adv. Energy Mater., 2022, 2202369 CrossRef CAS.
- J. Kothandaraman, J. Saavedra Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, ChemSusChem, 2021, 14, 4812–4819 CrossRef CAS PubMed.
- C. J. Koch, Z. Suhail, A. Goeppert and G. K. S. Prakash, ChemCatChem, 2023, e202300877 CrossRef CAS.
- A. R. Richard and M. Fan, Fuel, 2018, 222, 513–522 CrossRef CAS.
- Z. Liang, Y. Zhang, G. Zhang, J. Liu, Y. Cai, Y. Wang, Y. Zhao, G. Li and K. Bei, Int. J. Hydrogen Energy, 2023, 58(49), 18644–18656 CrossRef.
- G. Owen, C. M. Hawkes, D. Lloyd, J. R. Jennings, R. M. Lambert and R. M. Nix, Appl. Catal., 1987, 33, 405–430 CrossRef CAS.
- J. B. Branco, A. C. Ferreira, A. P. Gonçalves and C. O. Soares, Catal. Commun., 2016, 84, 103–107 CrossRef CAS.
- S.-Q. Yang, J.-P. He, N. Zhang, X.-W. Sui, L. Zhang, Z.-X. Yang and R. H. Xuebao, J. Fuel Chem. Technol., 2018, 46, 179–188 CrossRef CAS.
- Z. Bian, W. Zhong, Y. Yu, Z. Wang, B. Jiang and S. Kawi, Int. J. Hydrogen Energy, 2021, 46, 31041–31053 CrossRef CAS.
- C. Zhang, X. Hu, Z. Zhang, L. Zhang, D. Dong, G. Gao, R. Westerhof and S. S. A. Syed-Hassan, Fuel, 2018, 227, 307–324 CrossRef CAS.
- K. M. Van Allsburg, E. C. D. Tan, J. D. Super, J. A. Schaidle and F. G. Baddour, Nat. Catal., 2022, 5, 342–353 CrossRef CAS.
- F. Bibak and F. Meshkani, Fuel, 2024, 366, 131048 CrossRef CAS.
- J. Gao, C. Jia, J. Li, M. Zhang, F. Gu, G. Xu, Z. Zhong and F. Su, J. Energy Chem., 2013, 22, 919–927 CrossRef CAS.
- K. Stangeland, D. Kalai, H. Li and Z. Yu, Energy Procedia, 2017, 105, 2022–2027 CrossRef CAS.
- Y. Onodera, Y. Takimoto, H. Hijiya, T. Taniguchi, S. Urata, S. Inaba, S. Fujita, I. Obayashi, Y. Hiraoka and S. Kohara, NPG Asia Mater., 2019, 11, 75 CrossRef CAS.
- L. Vidal, A. Gharzouni and S. Rossignol, in Handbook of Sol-Gel Science and Technology, ed. L. Klein, M. Aparicio and A. Jitianu, Springer, 2018 Search PubMed.
- F. Rajabipour, E. Giannini, C. Dunant, J. H. Ideker and M. D. A. Thomas, Cem. Concr. Res., 2024, 53, 293–303 Search PubMed.
- L. Falbo, M. Martinelli, C. G. Visconti, L. Lietti, C. Bassano and P. Deiana, Appl. Catal., B, 2018, 225, 354–363 CrossRef CAS.
- E. Moioli, N. Gallandat and A. Züttel, React. Chem. Eng., 2019, 4, 100–111 RSC.
- H. K. D. Nguyen, T. H. Dang, N. L. T. Nguyen, H. T. Nguyen and N. T. Dinh, Can. J. Chem. Eng., 2018, 96, 832–837 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- M. S. Duyar, A. Gallo, J. L. Snider and T. F. Jaramillo, J. CO2 Util., 2020, 39, 101151 CrossRef CAS.
- O. Omoregbe, H. T. Danh, S. Z. Abidin, H. D. Setiabudi, B. Abdullah, K. B. Vu and D. V. N. Vo, Procedia Energy, 2016, 148, 1388–1395 CrossRef CAS.
- M. H. Amin, S. Putla, S. Bee Abd Hamid and S. K. Bhargava, Appl. Catal., A, 2015, 492, 160–168 CrossRef CAS.
- W. Ahmad, M. N. Younis, R. Shawabkeh and S. Ahmed, Catal. Commun., 2017, 100, 121–126 CrossRef CAS.
- S. C. Bart, Inorg. Chem., 2023, 62, 3713–3714 CrossRef CAS PubMed.
- J. A. Peters, K. Djanashvili, C. F. G. C. Geraldes and C. Platas-Iglesias, Coord. Chem. Rev., 2020, 406, 213146 CrossRef CAS.
- P. Mierczynski, A. Mierczynska, R. Ciesielski, M. Mosinska, M. Nowosielska, A. Czylkowska, W. Maniukiewicz, M. I. Szynkowska and K. Vasilev, Catalysts, 2018, 8, 380 CrossRef.
- F. Schüth, M. D. Ward and J. M. Buriak, Chem. Mater., 2018, 30, 3599–3600 CrossRef.
- S. L. Scott, ACS Catal., 2018, 8, 8597–8599 CrossRef CAS.
- K. A. M. L. Cruz, F. R. P. Rocha and M. C. Hespanhol, ACS Sustain. Chem. Eng., 2024, 12, 6169–6181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00306c |
| This journal is © The Royal Society of Chemistry 2024 |