
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halo-perfluoroalkoxylation of gem-difluoroalkenes with short-lived alkali metal perfluoroalkoxides in triglyme†‡
Koki
Kawai
a,
Yoshimitsu
Kato
b,
Taichi
Araki
b,
Sota
Ikawa
b,
Mai
Usui
b,
Naoyuki
Hoshiya
c,
Yosuke
Kishikawa
c,
Jorge
Escorihuela
 d and
Norio
Shibata
*ab
d and
Norio
Shibata
*ab
aDepartment of Nanopharmaceutical Sciences, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan. E-mail: nozshiba@nitech.ac.jp
bDepartment of Life Science and Applied Chemistry, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan
cTechnology Innovation Center, DAIKIN Industries, Ltd, 1-1 Nishi-Hitotsuya, Settsu, Osaka 566-8585, Japan
dDepartamento de Química Orgánica, Universitat de València, Avda. Vicente Andrés Estellés S/N, Burjassot 46100, Valencia, Spain
First published on 22nd May 2024
Abstract
Alkali metal alkoxides play a pivotal role in nucleophilic alkoxylation reactions, offering pathways for the synthesis of ethers, including the increasingly sought-after trifluoromethyl ethers. However, the synthesis of long-chain perfluoroalkyl ethers remains a substantial challenge in this field. Through the innovative use of triglyme to encapsulate potassium ions, we enhanced the stability of short-lived, longer-chain perfluoroalkoxy anions, thereby facilitating efficient nucleophilic perfluoroalkoxylation reactions. This method provides a new precedent for the halo-perfluoroalkoxylation of gem-difluoroalkenes and offers a versatile tool for the design of perfluoroalkyl ethers, including those containing complex moieties of heterocycles and drug molecules. We also demonstrated the utility of the resulting halo-perfluoroalkoxyl adducts through various chemical transformations to valuable diverse perfluoroalkyl ethers.
Introduction
Alkali metal alkoxides are recognized as key reagents in organic synthesis, especially for nucleophilic alkoxylation reactions that establish carbon–oxygen (C–O) bonds and produce ethers.1 Recent breakthroughs have thrusted this area into new territories, especially in the synthesis of trifluoromethyl ethers (R-OCF3),2 promising exciting developments in agrochemicals and pharmaceuticals.3 Traditionally, the production of alkali metal trifluoromethoxides [M][OCF3] requires fluorophosgene (carbonyl fluoride, F2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and alkali metal fluorides (M–F, Fig. 1a),4 and the recent introduction of safer shelf-stable reagents for nucleophilic trifluoromethylation has advanced significantly (Fig. 1b).5 Perfluoroalkyl ethers are prized for their high lipophilicity, improved metabolic stability, and unparalleled thermal and chemical resilience, and they can be used in various industries.6–8 Most of the unique properties are due to the ether oxygens inserted between the perfluorinated carbon backbones.7b For example, compared to perfluoroalkyl carboxylic acids, perfluoropolyether carboxylic acids allow easy formation of non-covalent hydrogen bonds with water, resulting in increased hydrophilicity.8a The presence of ether linkages in perfluoroalkyl ether carboxylic acids results in increased bond dissociation energies for the adjacent carbon-fluorine bonds, which affects their reactivity and degradation results.8b This structural feature generally promotes more effective degradation under reductive conditions than non-oxygenated variants by facilitating the cleavage of carbon–oxygen bonds and enhancing defluorination. There are also reports that perfluoroalkylether sulfonates degrade more effectively than perfluoroalkyl sulfonates in subcritical water in the presence of oxygen8c due to their ether linkages. In addition, perfluoroalkyl compounds with long perfluorinated chains consisting of more than 10 perfluorinated carbons exhibit a crystalline nature,9 which would also be an additional advantage of perfluoroalkyl ethers.10 Despite rapid progress in nucleophilic trifluoromethoxylation, the synthesis of longer perfluoroalkyl ethers remains a significant challenge,11,12 particularly for ethers with perfluoroalkyl moieties on both sides, represented by RCF2–O–CF2R′.12 To understand the difficulty of nucleophilic longer-chain perfluoroalkoxylation reactions, our investigation began with density functional theory (DFT) calculations to assess the feasibility of generating perfluoroalkoxides from their precursors, represented by two critical reactions: the traditional generation of [K][OCF3] from OCF2 and potassium fluoride (KF) and the formation of potassium perfluorohexanolate [K][OCF2C5F11] from perfluorohexanoyl fluoride (OCF(C5F11)) and KF (Fig. 1c). Our results reveal a stark contrast in the reaction Gibbs free energy (ΔGR) between these processes (see ESI‡ for details). The formation of [K][OCF3] had a ΔGR of −10.9 kcal mol−1, indicating a favorable reaction pathway. In contrast, the generation of [K][OCF2C5F11] had a ΔGR of −1.8 kcal mol−1, suggesting a less favorable process. To overcome this discrepancy and enhance the synthetic feasibility of long-chain perfluoroalkyl ethers, we introduced a novel method that involves encapsulating K+ ions with two molecules of triglyme (G3). This approach significantly increases the stability of the OCF2C5F11 anion, facilitating the efficient synthesis of this complex molecule, [K(G3)2][OCF2C5F11], with a ΔGR of −33.9 kcal mol−1, indicating a highly favorable reaction process. This result not only deepens our understanding of the stability of alkali metal perfluoroalkoxides but also realizes the halo-perfluoroalkoxylation of gem-difluoroalkenes 1 using [K(G3)2][OCF2Rf] generated in situ from perfluoroalkanoyl fluorides 2 (OCFRf) with KF, yielding valuable difluoroalkyl perfluoroalkyl ethers 3 (X = I), 4 (X = Br) or 6 (X = Cl) with chain lengths from C3 to C6 (Fig. 1d). This halo-perfluoroalkoxylation reaction was achieved with regioselectivity, and various perfluoroalkyl ethers with functional groups relevant to materials and medicines were obtained in high yields. Finally, we explored the various chemical transformations of the resulting halo-perfluoroalkoxy products to demonstrate their potential as building blocks.
O) and alkali metal fluorides (M–F, Fig. 1a),4 and the recent introduction of safer shelf-stable reagents for nucleophilic trifluoromethylation has advanced significantly (Fig. 1b).5 Perfluoroalkyl ethers are prized for their high lipophilicity, improved metabolic stability, and unparalleled thermal and chemical resilience, and they can be used in various industries.6–8 Most of the unique properties are due to the ether oxygens inserted between the perfluorinated carbon backbones.7b For example, compared to perfluoroalkyl carboxylic acids, perfluoropolyether carboxylic acids allow easy formation of non-covalent hydrogen bonds with water, resulting in increased hydrophilicity.8a The presence of ether linkages in perfluoroalkyl ether carboxylic acids results in increased bond dissociation energies for the adjacent carbon-fluorine bonds, which affects their reactivity and degradation results.8b This structural feature generally promotes more effective degradation under reductive conditions than non-oxygenated variants by facilitating the cleavage of carbon–oxygen bonds and enhancing defluorination. There are also reports that perfluoroalkylether sulfonates degrade more effectively than perfluoroalkyl sulfonates in subcritical water in the presence of oxygen8c due to their ether linkages. In addition, perfluoroalkyl compounds with long perfluorinated chains consisting of more than 10 perfluorinated carbons exhibit a crystalline nature,9 which would also be an additional advantage of perfluoroalkyl ethers.10 Despite rapid progress in nucleophilic trifluoromethoxylation, the synthesis of longer perfluoroalkyl ethers remains a significant challenge,11,12 particularly for ethers with perfluoroalkyl moieties on both sides, represented by RCF2–O–CF2R′.12 To understand the difficulty of nucleophilic longer-chain perfluoroalkoxylation reactions, our investigation began with density functional theory (DFT) calculations to assess the feasibility of generating perfluoroalkoxides from their precursors, represented by two critical reactions: the traditional generation of [K][OCF3] from OCF2 and potassium fluoride (KF) and the formation of potassium perfluorohexanolate [K][OCF2C5F11] from perfluorohexanoyl fluoride (OCF(C5F11)) and KF (Fig. 1c). Our results reveal a stark contrast in the reaction Gibbs free energy (ΔGR) between these processes (see ESI‡ for details). The formation of [K][OCF3] had a ΔGR of −10.9 kcal mol−1, indicating a favorable reaction pathway. In contrast, the generation of [K][OCF2C5F11] had a ΔGR of −1.8 kcal mol−1, suggesting a less favorable process. To overcome this discrepancy and enhance the synthetic feasibility of long-chain perfluoroalkyl ethers, we introduced a novel method that involves encapsulating K+ ions with two molecules of triglyme (G3). This approach significantly increases the stability of the OCF2C5F11 anion, facilitating the efficient synthesis of this complex molecule, [K(G3)2][OCF2C5F11], with a ΔGR of −33.9 kcal mol−1, indicating a highly favorable reaction process. This result not only deepens our understanding of the stability of alkali metal perfluoroalkoxides but also realizes the halo-perfluoroalkoxylation of gem-difluoroalkenes 1 using [K(G3)2][OCF2Rf] generated in situ from perfluoroalkanoyl fluorides 2 (OCFRf) with KF, yielding valuable difluoroalkyl perfluoroalkyl ethers 3 (X = I), 4 (X = Br) or 6 (X = Cl) with chain lengths from C3 to C6 (Fig. 1d). This halo-perfluoroalkoxylation reaction was achieved with regioselectivity, and various perfluoroalkyl ethers with functional groups relevant to materials and medicines were obtained in high yields. Finally, we explored the various chemical transformations of the resulting halo-perfluoroalkoxy products to demonstrate their potential as building blocks.
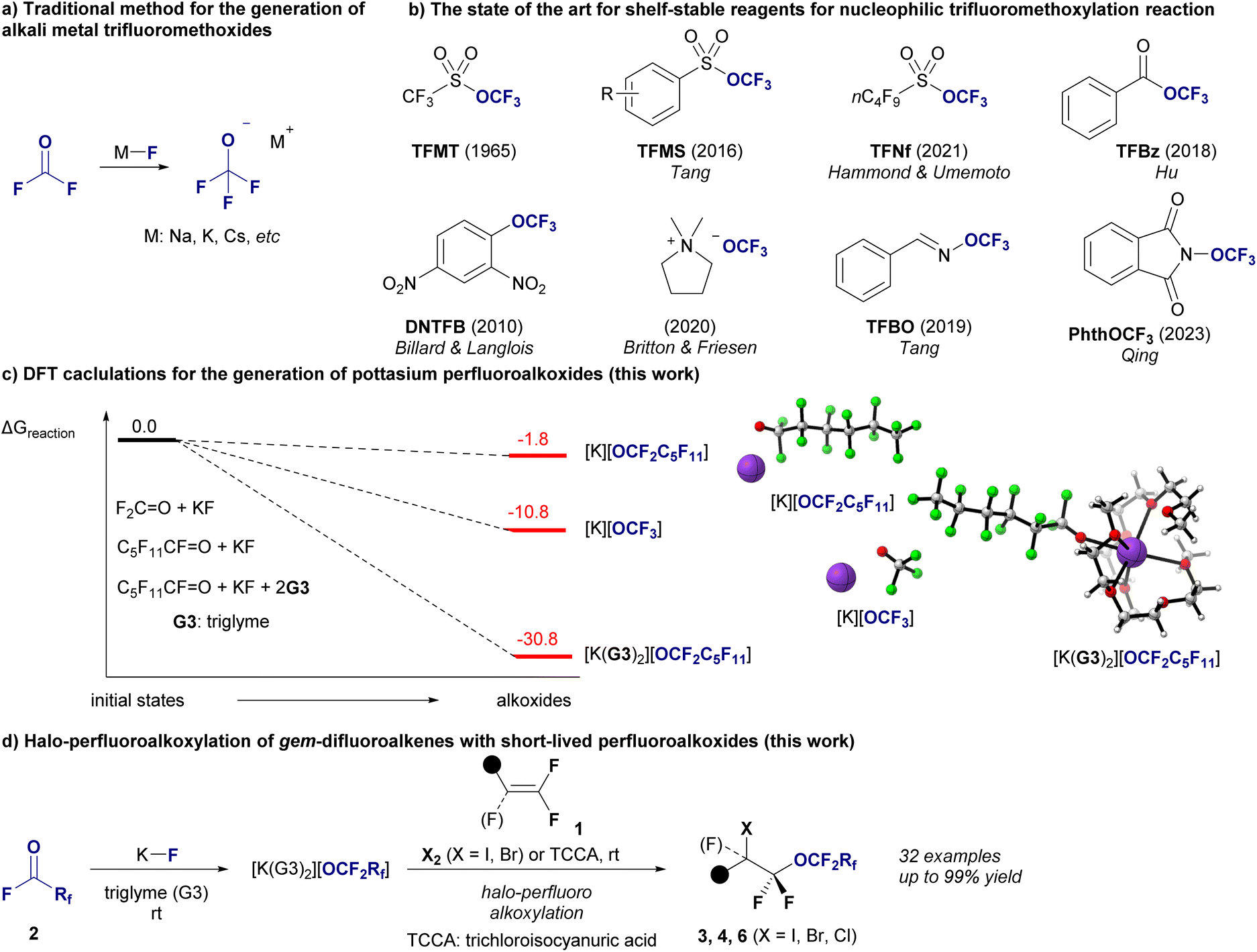 | ||
| Fig. 1 Background and strategy of nucleophilic perfluoroalkoxylations. (a) Traditional method for the preparation of alkali metal trifluoromethoxides. (b) Shelf-stable reagents for nucleophilic trifluoromethoxylation. (c) DFT-calculated reaction Gibbs free energy B3LYP-D3/6-311+G(d,p) in ether (SMD), in kcal mol−1 for the reactions of KF with F2CO (−10.8 kcal mol−1), C5F11CFO (−1.8 kcal mol−1) and C5F11CFO with triglyme (−30.8 kcal mol−1) (this work). (d) Halo-perfluoroalkoxylation of olefins (this work). | ||
Results and discussion
The halo-perfluoroalkoxylation reaction was optimized using gem-difluoroalkene 1a and alkali metal perfluoroalkoxide generated in situ from undecafluorohexanoyl fluoride 2a and inorganic fluoride as model substrates in the presence of halogen sources (Table 1). Glymes were used as the solvent to stabilize the labile perfluoroalkoxide by encapsulating metal cations, based on our previous research on trifluoromethylation reactions involving the labile trifluoromethyl anion.13 Initially, 2a was treated with sodium fluoride (NaF), KF, or cesium fluoride (CsF) in triglyme for 15 min at room temperature to generate metal perfluoroalkoxides. After fluoroalkene 1a was added to the perfluoroalkoxide solution, I2 was added to the reaction mixture. NaF treatment did not yield the desired product (entry 1), KF treatment produced 3aa in 38% yield (entry 2), and CsF treatment resulted in 25% yield (entry 3). A lower yield (4%) was obtained when N-iodosuccinimide (NIS) was used instead of I2 (entry 4). Switching the solvent to MeCN, DMF, Et2O, or THF using KF did not increase the yield (entries 5–8), even in the presence of 18-crown-6-ether (18-C-6, entry 9). Subsequently, we varied the equivalents of acyl fluorides 2a, KF, and I2. A yield of 38% was obtained when using 2.0 equiv. of I2 (entry 10). Using triglyme/THF solved (entry 11) and increasing the number of equivalents of 2a and KF to 2.0 equiv. (entry 12) led to decrease in yields. Remarkably, employing 2.0 equivalents of 2a, KF, and I2 substantially improved the yield to 70% (entry 13), with further enhancement to an outstanding 89% yield achieved by increasing the equivalents to 3.0 (entry 14). Encouraged by the success of iodo-perfluoroalkoxylation and subsequent bromo-perfluoroalkoxylation of 1a with 2a using bromine in the triglyme, we noted the significant impact of reactant quantities (entries 15–17). Notably, a high yield of the desired compound 4aa (88%) was obtained by employing 3.0 equivalents of reagents (entry 17). The addition of reagents is critical for successful transformation. When the generated alkoxide and Br2 were first mixed, followed by the addition of alkene 1a, the yield of the desired fluoroalkyl ether 4aa decreased to 56%, and 1a was recovered in 36% yield (entry 18).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2a (Y equiv.) | MF | Halogen source (Z equiv.) | Solvent (0.1 M) | Yieldb3aa (%) | Yieldb4aa (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a, MF, halogen source, solvent (1.0 mL), room temperature for 18 h. b 19F NMR yields were determined using C6F6 as an internal standard. c 18-C-6 (1.0 equiv.) was added. d 1a (0.3 mmol) was used for 6 h e Isolated yield. f 1a was added after stirring Br2 with the perfluoroalkoxide generated for 15 min. | ||||||
| 1 | 1.0 | NaF | I2 (1.0) | Triglyme | 0 | |
| 2 | 1.0 | KF | I2 (1.0) | Triglyme | 38 | |
| 3 | 1.0 | CsF | I2 (1.0) | Triglyme | 25 | |
| 4 | 1.0 | KF | NIS (1.0) | Triglyme | 4 | |
| 5 | 1.0 | KF | I2 (1.0) | MeCN | 18 | |
| 6 | 1.0 | KF | I2 (1.0) | DMF | 9 | |
| 7 | 1.0 | KF | I2 (1.0) | Et2O | 0 | |
| 8 | 1.0 | KF | I2 (1.0) | THF | 0 | |
| 9c | 1.0 | KF | I2 (1.0) | THF | 7 | |
| 10 | 1.0 | KF | I2 (2.0) | Triglyme | 38 | |
| 11 | 1.0 | KF | I2 (2.0) | Triglyme/THF (1/2) | 22 | |
| 12 | 2.0 | KF | I2 (1.0) | Triglyme | 31 | |
| 13 | 2.0 | KF | I2 (2.0) | Triglyme | 70 | |
| 14 | 3.0 | KF | I2 (3.0) | Triglyme | 89 | |
| 15 | 1.0 | KF | Br2 (1.0) | Triglyme | 48 | |
| 16 | 2.0 | KF | Br2 (2.0) | Triglyme | 70 | |
| 17d | 3.0 | KF | Br2 (3.0) | Triglyme | 88e | |
| 18f | 3.0 | KF | Br2 (3.0) | Triglyme | 56 | |
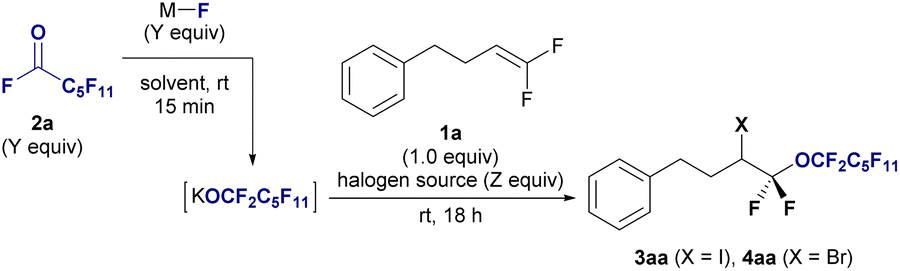
Next, we explored the substrate scope of haloperfluoroalkoxylation under optimal reaction conditions (Fig. 2). To demonstrate the functional tolerance of this method, various gem-difluoroalkenes with diverse reactive functionalities, including benzyl ether (1b), ether (1c), ester (1d), tosylate (1e), ketone, and amide groups (1f), were selected as substrates for iodo-perfluoro-hexyloxylation. All the reactions proceeded smoothly, furnishing desired α-perfluorohexyl α,α-difluoroalkyl ethers 3 in high yields (82–96%) and regioselectivity, irrespective of the functional group. Notably, chemoselectivity was observed in the iodo-perfluoro-hexyloxylation of perfluoroallylbenzene (1g). The desired perfluorinated ether 3ga was obtained in 81% yield without SNAr reactions at the perfluorophenyl moiety. A gram-scale reaction was also conducted using perfluoroalkoxide on 6.0 mmol (1.01 g) of 1a under optimized conditions, resulting in the isolation of iodoperfluoroalkyl ether 3aa in 92% yield (Fig. 2, 3aa: 3.47 g isolated).
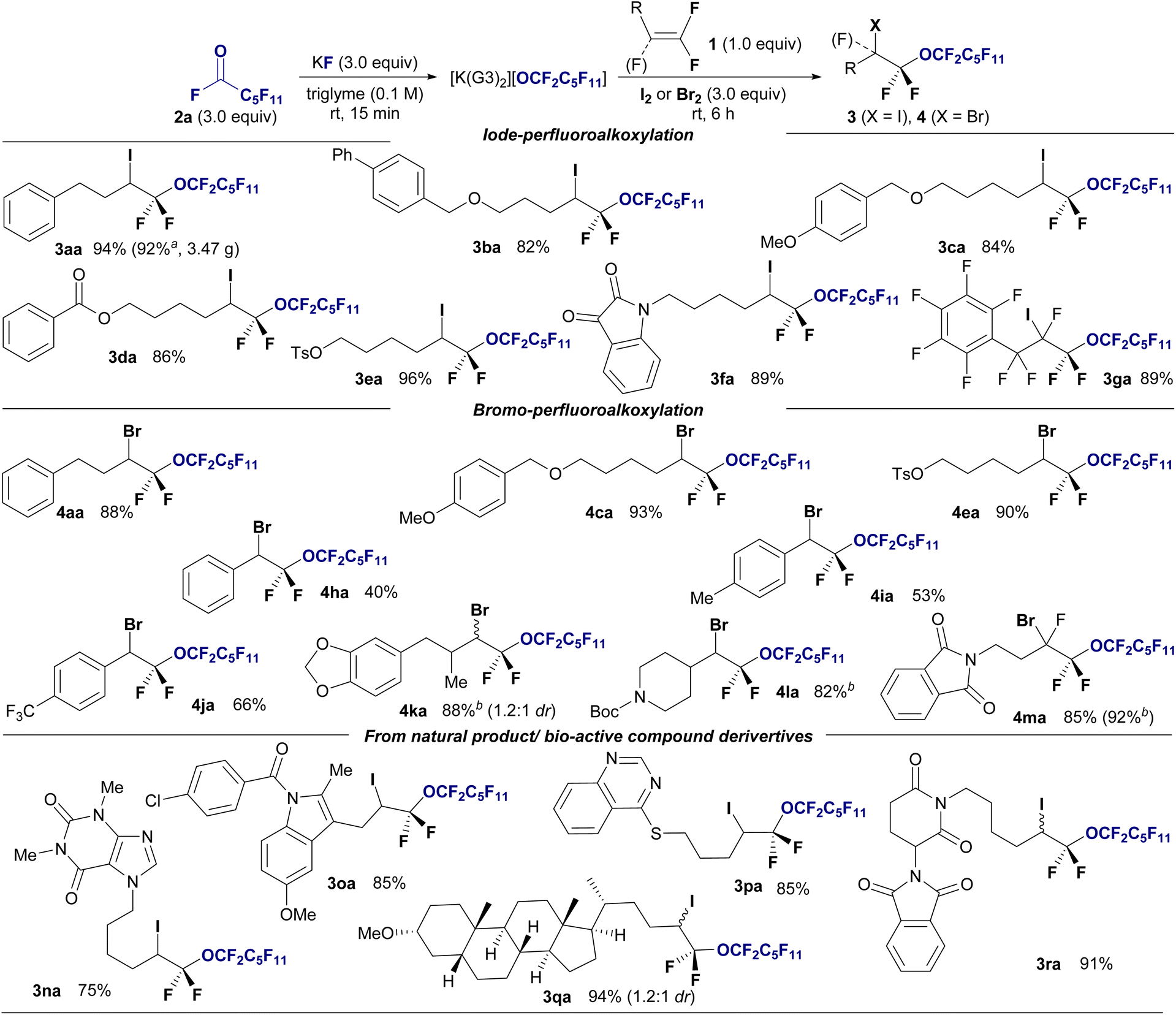 | ||
| Fig. 2 Substrate scope of the halo-perfluoroalkoxylation of gem-difluoroalkenes 1 using undecafluorohexanoyl fluoride 2a. Isolated yields are shown. Reaction conditions: 1 (0.3 mmol), 2a (0.9 mmol), KF (0.9 mmol), I2 or Br2 (0.9 mmol) in triglyme (3.0 mL), stirring under atmosphere of N2 at room temperature. (a) Gram scale reaction. 1 (6.0 mmol, 1.01 g) was used. (b) 1 (0.3 mmol), 2a (1.5 mmol), KF (1.5 mmol), Br2 (1.5 mmol) in triglyme (6.0 mL). | ||
We expanded the substrate scope of bromoperfluoroalkoxylation using Br2. Gem-difluoroalkenes bearing benzyloxy and methoxy moieties (1c) and tosylate (1e) reacted effectively with potassium perfluorohexyloxide 2a, which was generated in situ in the presence of Br2, providing the corresponding perfluorohexyloxyl difluoroalkyl ethers in excellent yields (4aa, 88%; 4ca, 93%; 4ea, 90%). Gem-difluorostyrene derivatives (1h–1j) with nonsubstituted (H), electron-donating (Me), and electron-withdrawing (CF3) groups also interacted with perfluoroalkoxides under bromination conditions, affording the corresponding bromoperfluoroalkyl ethers in moderate to good yields (4ha: 40%, 4ia: 53%, 4ja: 66%). The bromo-perfluoroalkylation of gem-difluoroalkenes with a secondary alkyl γ-position furnished the corresponding ethers (4ka: 88% (1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), 4la: 82%). Significantly, the trifluorinated alkene 1m efficiently reacted with 2a, KF, and bromine, yielding bis(α,α-difluoro)ether in a high yield (4ma: 92%). Subsequently, reactions involving various gem-difluoroalkenes bearing natural products or biologically relevant moieties were explored. Gem-difluoroalkenes 1, containing theophylline (1n), indomethacin (1o), an insecticidal agent (1p), lithocholic acid (1q), and thalidomide (1r), underwent iodoperfluoroalkoxylation using 1a, KF, and I2 in the triglyme, resulting in the synthesis of drug-conjugated perfluoroalkyl diethyl ether in high yields and with excellent regioselectivity (3na: 75%; 3oa: 85%; 3pa: 85%; 3qa: 75%; 3ra: 91%). Biologically attractive molecules with perfluoroalkyl chains are expected to be used as decoy molecules in drug discovery.14
:1 dr), 4la: 82%). Significantly, the trifluorinated alkene 1m efficiently reacted with 2a, KF, and bromine, yielding bis(α,α-difluoro)ether in a high yield (4ma: 92%). Subsequently, reactions involving various gem-difluoroalkenes bearing natural products or biologically relevant moieties were explored. Gem-difluoroalkenes 1, containing theophylline (1n), indomethacin (1o), an insecticidal agent (1p), lithocholic acid (1q), and thalidomide (1r), underwent iodoperfluoroalkoxylation using 1a, KF, and I2 in the triglyme, resulting in the synthesis of drug-conjugated perfluoroalkyl diethyl ether in high yields and with excellent regioselectivity (3na: 75%; 3oa: 85%; 3pa: 85%; 3qa: 75%; 3ra: 91%). Biologically attractive molecules with perfluoroalkyl chains are expected to be used as decoy molecules in drug discovery.14
This methodology was applied to halo-perfluoroalkoxylation reactions with shorter perfluoroalkoxy moieties ranging from C3 to C5 to demonstrate a modular approach (Fig. 3a). Perfluoroacyl fluorides (2b–2d) were generated ex situ from their corresponding perfluorocarboxylic acids (5) via deoxyfluorination using Ishikawa's reagent and NaF. The perfluoropropoxylation, perfluorobutyloxylation, and perfluoropentoxylation of fluoroolefines proceeded smoothly, affording the corresponding perfluoroalkoxyethers in good to high yields (4ab, 76%; 4ac, 65%; 4ad, 99%). Furthermore, perfluoro-isopropoxylation was performed using hexafluoroacetone and KF in triglyme with difluoroalkene 1a in the presence of Br2 to afford the desired branched perfluorinated alkyl ether 4ae in 83% yield.
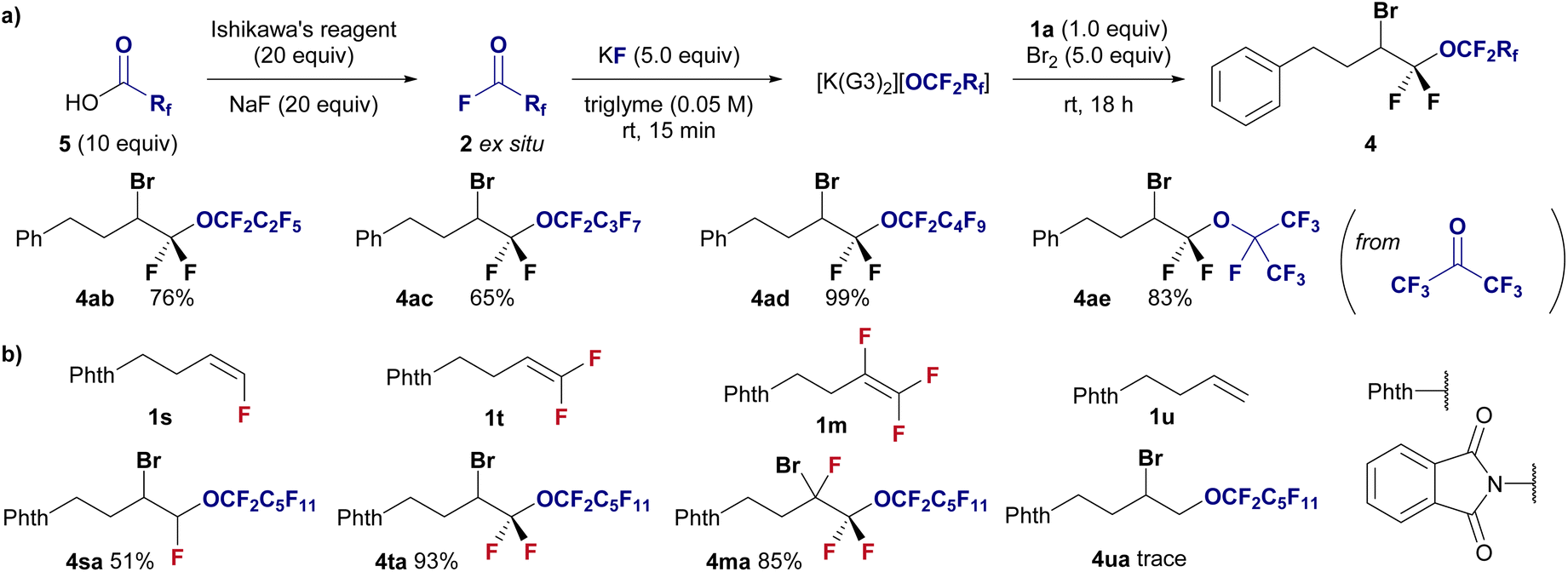 | ||
| Fig. 3 (a) Substrate scope of the halo-perfluoroalkoxylation of gem-difluoroalkene 1a using various perfluoroacylfluorides 2. Isolated yields are shown. Reaction conditions: 1a (0.3 mmol), 2 (excess), KF (1.5 mmol), Br2 (1.5 mmol) in triglyme (6.0 mL), stirring under atmosphere of N2 at room temperature. (b) Investigation of fluoroalkene selective reaction using undecafluorohexanoyl fluoride 2a under the standard conditions in Fig. 2. | ||
We further investigated the effect of fluorine on the reactivity of alkenes in halo-perfluoroalkoxylation reactions under standard conditions (Fig. 3b). Notably, the effect of fluorine substitution is obvious, and mono- (1s), di- (1t), and tri-fluorinated (1m) alkenes are well accepted as substrates (4sa, 51%; 4ta, 93%; 4ma, 85%), whereas the nonfluorinated alkene 1u remains unreactive (4ua, trace). This highly fluoroalkene-selective transformation enhances the advantages of this methodology.
Although I2 and Br2 are effective for this conversion, we did not investigate the use of chlorine due to its gasueous, highly oxidative nature, which makes it difficult to optimize the reaction conditions. Upon further investigation, trichloroisocyanuric acid (TCCA) was found to be a suitable chlorine source for chloro-perfluoro alkoxylation, allowing the production of bis(α,α)difluoro ethers 6 from 2a and 1 in yields ranging from to 40–58%. Chloro-perfluoroalkoxylation was also selective to fluorinated alkenes 1, with nonfluorinated alkene 1u remaining unreactive (6ua, 0%) (Fig. 4).
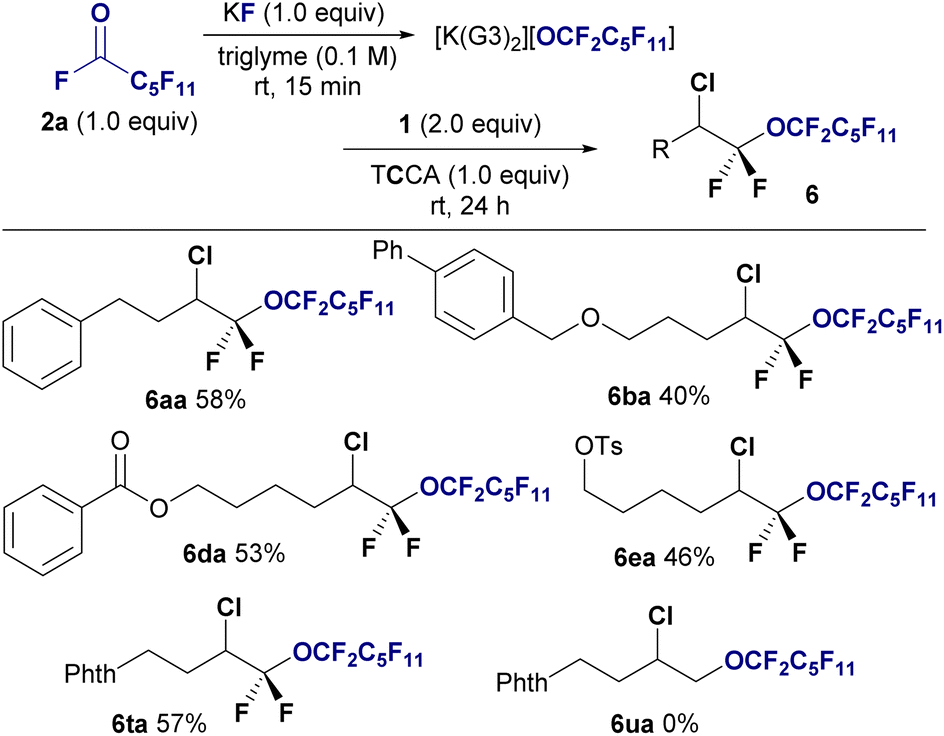 | ||
| Fig. 4 Substrate scope of the chloro-perfluoroalkoxylation of gem-difluoroalkenes 1 using undecafluorohexanoyl fluoride 2a. Isolated yields are shown. Reaction conditions: 1 (0.6 mmol), 2a (0.3 mmol), KF (0.3 mmol), TCCA (0.3 mmol) in triglyme (3.0 mL), stirring under atmosphere of N2 at room temperature. | ||
Furthermore, several chemical transformations were performed to demonstrate the synthetic utility of the obtained halo-perfluoroalkyl ether products (Fig. 5). First, radical coupling of 3aa with TEMPO in the presence of TMS3SiH at room temperature resulted in the formation of the TEMPO adduct 7 in 71% yield. Subsequently, the piperidinyl protecting group of 7 was reductively removed using zinc and acetic acid, yielding oxygenated compound 8 in quantitative yield. Employing tributyl stannane facilitated the reduction of iodine in 3aa using AIBN, leading to hydrogenated product 9 in 93% yield. Additionally, the radical allylation of 3aa using triethylborane provided corresponding allylated product 10 in 77% yield. The Giese radical addition of 3aa yielded the desired olefin 11 in 58% yield. Finally, HI elimination from 3aa using m-CPBA afforded corresponding E-alkene 12 as a single isomer in 67% yield.
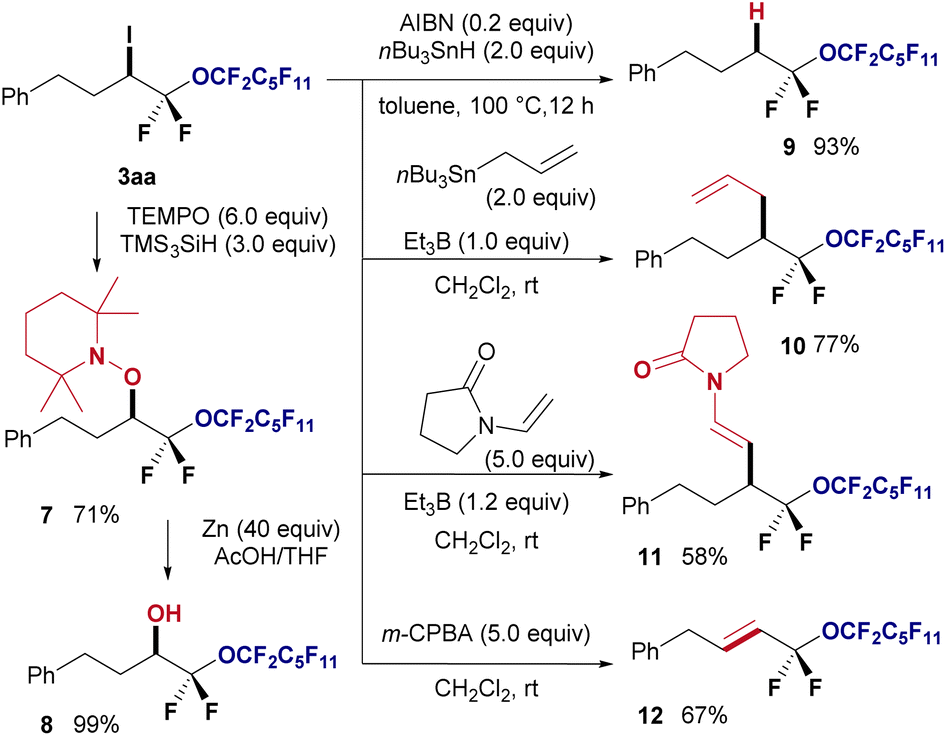 | ||
| Fig. 5 Synthetic applications. | ||
DFT calculations were conducted to assess the stabilization of perfluoroalkoxides in the presence of triglymes. The calculations were performed at the B3LYP-D3/6-311+G(d,p) (SDD for K and Cs) level of theory using Gaussian 16 software.15 As indicated in Fig. 1c. Initially, the formation of perfluoroalkoxides from KF and acylfluoride 2a in ether was calculated to be exothermic (Fig. 6a, −1.8 kcal mol−1). Complex formation involving two triglyme molecules (G3) coordinated to alkali metal cations resulted in complexes [Na(G3)2][OCF2C5F11], [K(G3)2][OCF2C5F11], and [Cs(G3)2][OCF2C5F11], indicating that the potassium cation had the greatest stabilizing effect (Fig. 6b, 33.9 kcal mol−1). The M+–O(G3) distances in the tetra-dentated G3 structures, that is, [Na(G3)2][OCF2C5F11], [K(G3)2][OCF2C5F11], and [Cs(G3)2][OCF2C5F11], ranged from 2.80–3.00, 2.90–3.10, and 3.20–3.40 Å, respectively. This implies that the deformation energy of G3 molecules, which is the energy increase due to the deformation of G3 geometries when G3 molecules are coordinated to the M+ cation, is less destabilizing in [K(G3)2][OCF2C5F11].16 Subsequently, a control experiment was conducted (Fig. 6c). Alkali metal fluoride salts were compared under optimal reaction conditions using triglyme: KF resulted in 89% yield, CsF yielded a slightly lower yield, and NaF did not react. The effects of solvents were further investigated. The yield decreased to 51% when the reaction was conducted in MeCN with KF, and no reaction occurred in Et2O. The yield of the CsF/MeCN system was greater than that of the KF/MeCN system but significantly lower than that of the CsF/triglyme system. Based on these calculations and experimental observations, the yields of the larger cation, cesium salt, were greater in MeCN, and potassium salt exhibited a greater stabilizing effect than the cesium salt in the presence of triglyme.
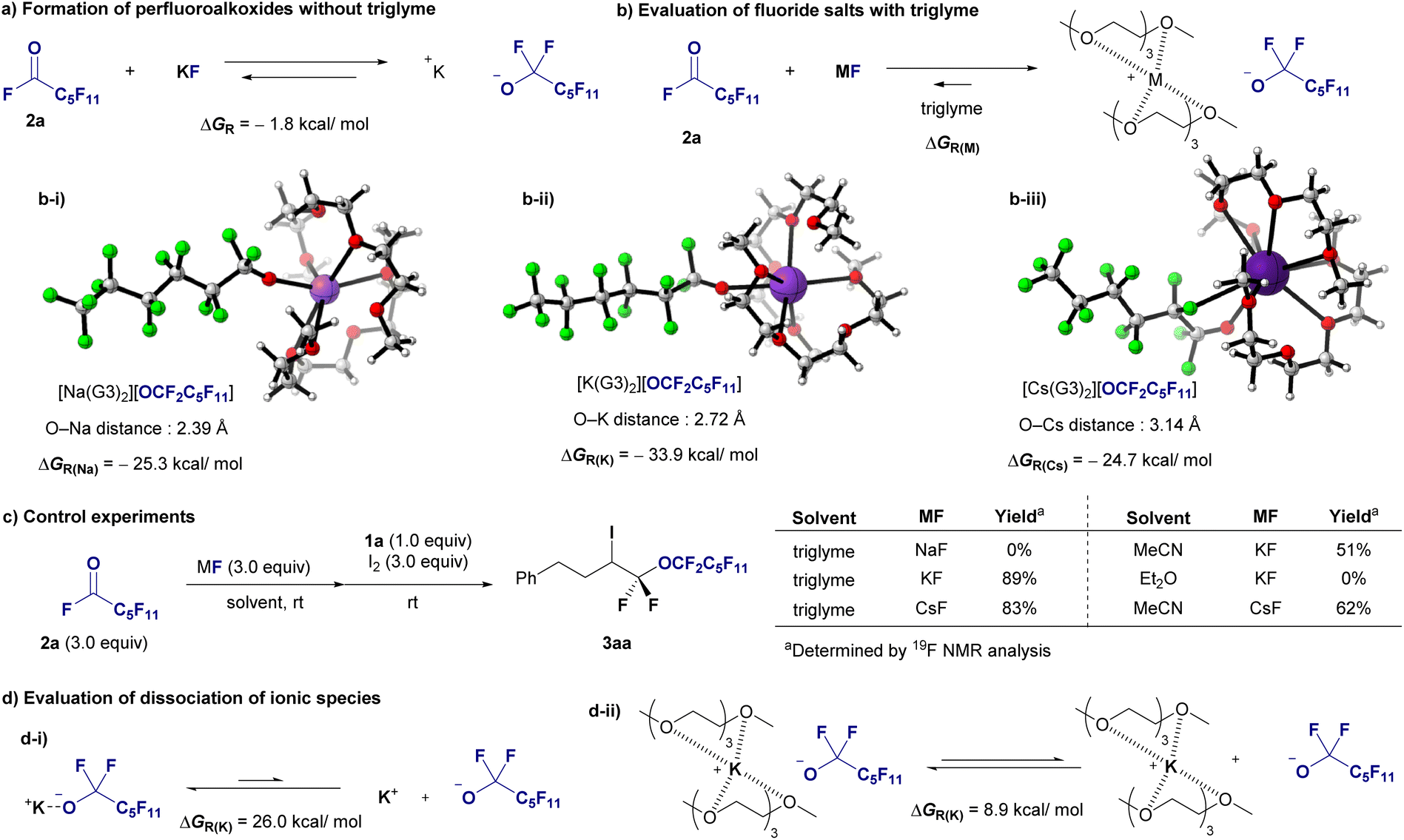 | ||
| Fig. 6 Mechanistic studies. (a) Formation of perfluoroalkoxides without triglyme. (b) Evaluation of the fluoride salt with triglyme. Optimized structures of fluoride salt with two triglyme molecules. (c) Control experiments. (d) Evaluation of dissociation of ionic species. | ||
In addition, these outcomes might also be attributed to the enhanced reactivity of perfluoroalkoxide anions because of the better dissociation of the ionic species. We thus performed additional DFT calculations to evaluate the dissociation of ionic species (Fig. 6d). The dissociation of ionic pair [K][OCF2C5F11] had a ΔGR of 26.0 kcal mol−1, indicating a not favourable process; whereas the process involving the encapsulation of K+ ions with two molecules of triglyme facilitates the dissociation of the ionic pair [K(G3)2][OCF2C5F11], with a ΔGR of 8.9 kcal mol−1. As shown by the ΔGR values, the process is more favourable in the presence of triglyme. Based on the results of Fig. 6a, b-ii, and d, both factors, stabilization and reactivity of the anion, resulted in diglyme playing important roles for the successful transformation. These two factors were more favourable in the presence of triglyme as inferred from the computed Gibbs energies, despite the dissociation is still an endergonic process. Also, the difference in Gibbs energies stabilization in the presence and the absence of triglyime (ΔΔGR = −33.9 − (−1.8) = −32.1 kcal mol−1) is more significant than that for the dissociation (ΔΔGR = 26.0–8.9 = 17.1 kcal mol−1).
Conclusion
We designed a novel approach to enhance the stability of short-lived, longer-chain perfluoroalkoxy anions to facilitate efficient nucleophilic perfluoroalkoxylation reactions. This method involves regioselective halo-perfluoroalkoxylation of gem-difluoroalkenes using potassium perfluoroalkoxides generated in situ from KF and perfluoroacylfluorides in a triglyme with bromine, iodine, or TCCA at room temperature. This methodology is broadly applicable to substrates containing analogs derived from bioactive compounds, thus enabling the synthesis of difluoroalkyl perfluoroalkyl ethers with C3–C6 perfluoroalkyl chains. The reaction is performed under mild conditions at rt in an environmentally friendly solvent, triglyme.17 Moreover, it encompasses a wide range of gem-difluoroalkenes as substrates, producing halo-perfluoroalkoxyl adducts that are suitable for diverse chemical transformations. This methodology is highly chemoselective toward fluorinated alkenes, whereas nonfluorinated alkenes remain intact. The extension of this methodology to the synthesis of fluorinated polymers consisting of bis(α,α-difluoro)ethers (–CF2–O–CF2–) is currently under investigation.Data availability
The data that support the findings of this study are available within the article and the ESI.‡ Details about materials and methods, experimental procedures, characterization data, and NMR spectral are included.Author contributions
KK optimized the reaction conditions. KK, YK, TA, MU and SI surveyed the substrate scope, analyzed the data, and then discussed the results with NH, YK and NS. JE performed DFT calculation and analyzed the data. KK and NS wrote the manuscript. NS supervised the project. All authors contributed to the manuscript and have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the CREST program of the Japan Science and Technology Agency, entitled “Precise Material Science for Degradation and Stability” (grant number: JPMJCR21L1). The computational resources from the Servei d’Informàtica de la Universidad de Valencia (SIUV) are gratefully acknowledged for providing access to the supercomputing resources. Profs Tsutomu Konno and Shigeyuki Yamada (Kyoto Institute of Technology) helped with mass spectrometry.Notes and references
- A. W. Williamson, J. Chem. Soc., 1852, 4, 229–239 Search PubMed.
- (a) F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827–856 CrossRef CAS PubMed; (b) A. Tlili, F. Toulgoat and T. Billard, Angew. Chem., Int. Ed., 2016, 55, 11726–11735 CrossRef CAS PubMed; (c) B.-Y. Hao, Y.-P. Han, Y. Zhang and Y.-M. Liang, Org. Biomol. Chem., 2023, 21, 4926–4954 RSC.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallowb and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed; (d) B. M. Johnson, Y.-Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed; (e) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (f) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- M. E. Redwood and C. J. Willis, Can. J. Chem., 1965, 43, 1893–1898 CrossRef CAS.
- (a) A. A. Kolomeitsev, M. Vorobyev and H. Gillandt, Tetrahedron Lett., 2008, 49, 449–454 CrossRef CAS; (b) O. Marrec, T. Billard, J.-P. Vors, S. Pazenok and B. R. Langlois, J. Fluorine Chem., 2010, 131, 200–207 CrossRef CAS; (c) O. Marrec, T. Billard, J.-P. Vors, S. Pazenok and B. R. Langlois, Adv. Synth. Catal., 2010, 352, 2831–2837 CrossRef CAS; (d) S. Guo, F. Cong, R. Guo, L. Wang and P. Tang, Nat. Chem., 2017, 9, 546–551 CrossRef CAS PubMed; (e) M. Zhou, C. Ni, Y. Zeng and J. Hu, J. Am. Chem. Soc., 2018, 140, 6801–6805 CrossRef CAS PubMed; (f) Y. Li, Y. Yang, J. Xin and P. Tang, Nat. Commun., 2020, 11, 755 CrossRef PubMed; (g) J. J. Newton, B. J. Jelier, M. Meanwell, R. E. Martin, R. Britton and C. M. Friesen, Org. Lett., 2020, 22, 1785–1790 CrossRef CAS PubMed; (h) Z. Lu, T. Kumon, B. Hammond and T. Umemoto, Angew. Chem., Int. Ed., 2021, 60, 16171–16177 CrossRef CAS PubMed; (i) W.-J. Yuan, C.-L. Tong, X.-H. Xu and F.-L. Qing, J. Org. Chem., 2023, 88, 4434–4441 CrossRef CAS PubMed.
- (a) G. Caporiccio, C. Corti, S. Saldini and G. Carniselli, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 515–519 CrossRef CAS; (b) W. Morales, W. R. Jones Jr and D. H. Buckley, Analysis of a Spacecraft Instrument Ball Bearing Assembly Lubricated by a Perfluoroalkylether Nasa Technical Memorandum, TM-87260, 1986 Search PubMed; (c) K. J. L. Paciorek and R. H. Kratzer, J. Fluor. Chem., 1994, 67, 167–175 CrossRef; (d) T. Imae, Curr. Opin. Colloid Interface Sci., 2003, 8, 307–314 CrossRef CAS; (e) M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC; (f) P. Kirsch, Moden Fluoroorganic Chemistry, Wiley-VCH, 2013 CrossRef; (g) P. Lipovská, L. Rathouská, O. Šimůnek, J. Hošek, V. Kolaříková, M. Rybáčková, J. Cvačka, M. Svoboda, J. Kvíčala and J. Fluor, Chem, 2016, 191, 14–22 Search PubMed.
- (a) A. S. W. Lo and I. T. Horváth, Green Chem., 2015, 17, 4701–4714 RSC; (b) R. Zhou, Y. Jin, Y. Shen, P. Zhao, Y. Zhou and J. Leather, Sci. Eng., 2021, 3, 6 CAS; (c) A. Zaggia and B. Ameduri, Curr. Opin. Colloid Interface Sci., 2012, 17, 188–195 CrossRef CAS.
- (a) M. Strynar, S. Dagnino, R. McMahen, S. Liang, A. Lindstrom, E. Andersen, L. McMillan, M. Thurman, I. Ferrer and C. Ball, Environ. Sci. Technol., 2015, 49, 11622–11630 CrossRef CAS PubMed; (b) M. J. Bentel, Y. Yu, L. Xu, H. Kwon, Z. Li, B. M. Wong, Y. Men and J. Liu, Environ. Sci. Technol., 2020, 54, 2489–2499 CrossRef CAS PubMed; (c) H. Hori, M. Murayama and S. Kutsuna, Chemosphere, 2009, 77, 1400–1405 CrossRef CAS PubMed.
- (a) H. W. Starkweather Jr, Macromolecules, 1986, 19, 1131–1134 CrossRef; (b) T. Hasegawa, Chem. Rec., 2017, 17, 903–917 CrossRef CAS PubMed; (c) J. Kvíčala, The Curious World of Fluorinated Molecules, ed. K. Seppelt, Elsevier, 2021, pp. 319–342 Search PubMed.
- The perfluoroalkyl ether derivative, hexafluoropropylene oxide dimer acid (HFPO-DA, GenX) was developed as an industrial replacement for straight-chain perfluoroalkyl substances; however, its use has been discussed in recent studies. Further research is needed to determine its environmental impact, according to the following references (a) What is HFPO-Dimer Acid? Chemours website, https://www.chemours.com/en/about-chemours/genx (accessed in May 2024); (b) EU court again rules against Chemours on GenX ‘forever chemicals’ CHEM Trust website, https://chemtrust.org/genx_court_appeal/(accessed in May 2024); (c) G. Munoza, J. Liub, S. V. Duya and S. Sauvé, Trends Environ. Anal. Chem., 2019, 23, e00066 CrossRef; (d) W. Sun, X. Zhang, Y. Qiao, N. Griffin, H. Zhang, L. Wang and H. Liu, Ecotoxicol. Environ. Saf., 2023, 259, 115020 CrossRef CAS PubMed.
- (a) B. J. Jelier, J. L. Howell, C. D. Montgomery, D. B. Leznoff and C. M. Friesen, Angew. Chem., Int. Ed., 2015, 54, 2945–2949 CrossRef CAS PubMed; (b) S. Fujioka, K. Hirano, N. Hoshiya, A. Yamauchi, Y. Kishikawa and M. Uchiyama, Chem. Commun., 2023, 59, 8290–8293 RSC.
- (a) F. W. Evans, M. H. Litt, A. M. Weidler-Kubanek and F. P. Avonda, J. Org. Chem., 1968, 33, 1839–1844 CrossRef CAS; (b) K. K. Johri and D. D. DesMarteau, J. Org. Chem., 1983, 48, 242–250 CrossRef CAS; (c) W. A. Kamil, F. Haspel-Hentrich and J. M. Shreeve, Inorg. Chem., 1986, 25, 376–380 CrossRef CAS; (d) I. Wlassics, V. Tortelli, S. Carella, C. Monzani and G. Marchionni, Molecules, 2011, 16, 6512–6540 CrossRef CAS PubMed.
- (a) T. Saito, J. Wang, E. Tokunaga, S. Tsuzuki and N. Shibata, Sci. Rep., 2018, 8, 11501 CrossRef PubMed; (b) Y. Fujihira, Y. Liang, M. Ono, K. Hirano, T. Kagawa and N. Shibata, Beilstein J. Org. Chem., 2021, 17, 431–438 CrossRef CAS PubMed; (c) Y. Fujihira, K. Hirano, M. Ono, H. Mimura, T. Kagawa, D. M. Sedgwick, S. Fustero and N. Shibata, J. Org. Chem., 2021, 86, 5883–5893 CrossRef CAS PubMed.
- (a) N. Kawakami, O. Shoji and Y. Watanabe, Angew. Chem., Int. Ed., 2011, 50, 5315–5318 CrossRef CAS PubMed; (b) F. E. Zilly, J. P. Acevedo, W. Augustyniak, A. Deege, U. W. Hausig and M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 2720–2724 CrossRef CAS PubMed; (c) N. Kawakami, O. Shoji and Y. Watanabe, Chem. Sci., 2013, 4, 2344–2348 RSC; (d) O. Shoji, T. Kunimatsu, N. Kawakami and Y. Watanabe, Angew. Chem., Int. Ed., 2013, 52, 6606–6610 CrossRef CAS PubMed; (e) Z. Cong, O. Shoji, C. Kasai, N. Kawakami, H. Sugimoto, Y. Shiro and Y. Watanabe, ACS Catal., 2015, 5, 150–156 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc, Wallingford CT, 2016 Search PubMed.
- T. Kimura, K. Fujii, Y. Sato, M. Morita and N. Yoshimoto, J. Phys. Chem. C, 2015, 119, 18911–18917 CrossRef CAS.
- (a) S. Tang and H. Zhao, RSC Adv., 2014, 4, 11251 RSC; (b) D. Di Lecce, V. Marangon, H.-G. Jung, Y. Tominaga, S. Greenbaum and J. Hassoun, Green Chem., 2022, 24, 1021–1048 RSC.
Footnotes |
| † Dedicated to Professor Santos Fustero and Professor Günter Haufe on the Occasion of their 75th Birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02084g |
| This journal is © The Royal Society of Chemistry 2024 |