
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a dendritic cell-targeted self-assembled polymeric nanoparticle for selective delivery of mRNA vaccines to elicit enhanced immune responses†
Chen-Yo
Fan
a,
Szu-Wen
Wang
a,
Cinya
Chung
a,
Jia-Yan
Chen
a,
Chia-Yen
Chang
a,
Yu-Chen
Chen
a,
Tsui-Ling
Hsu
a,
Ting-Jen R.
Cheng
a and
Chi-Huey
Wong
 *ab
*ab
aGenomics Research Center, Academia Sinica, Taipei 115, Taiwan
bDepartment of Chemistry, The Scripps Research Institute, La Jolla, California 92037, USA. E-mail: wong@scripps.edu
First published on 25th June 2024
Abstract
Recent development of SARS-CoV-2 spike mRNA vaccines to control the pandemic is a breakthrough in the field of vaccine development. mRNA vaccines are generally formulated with lipid nanoparticles (LNPs) which are composed of several lipids with specific ratios; however, they generally lack selective delivery. To develop a selective delivery method for mRNA vaccine formulation, we reported here the synthesis of polymeric nanoparticles (PNPs) composed of a guanidine copolymer containing zwitterionic groups and a dendritic cell (DC)-targeted aryl-trimannoside ligand for encapsulation and selective delivery of an mRNA to dendritic cells. A DC-targeted SARS-CoV-2 spike mRNA–PNP vaccine was shown to elicit a stronger protective immune response in mice compared to the traditional mRNA–LNP vaccine and those without the selective delivery design. It is anticipated that this technology is generally applicable to other mRNA vaccines for DC-targeted delivery with enhanced immune response.
Introduction
The global pandemic caused by the outbreak of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) in 2019 has infected more than 750 million people and caused more than 7 million deaths. Since then, a great deal of global efforts has been made to develop safe, effective, and scalable vaccines to contain the pandemic. There have been more than 400 vaccines investigated to target SARS-CoV-2,1 of which the development of spike mRNA vaccines represents a major breakthrough in the field.2,3 After immunization, the mRNA encoding spike protein is translated to the spike protein in vivo to elicit immune responses, and there is no need to manufacture the spike protein in vitro for administration. This breakthrough in mRNA vaccine development has stimulated new research activities to further improve the technology. The drawbacks of using mRNA to replace protein vaccines include: it is difficult to conduct the post-translational modification of protein at the mRNA level; mRNA is generally unstable and requires low temperatures (−70 °C) for storage; mRNA must be encapsulated in nanomaterials such as lipid nanoparticles (LNPs) for administration;4–7 the lipids used in the mRNA formulation are often mixed in specific ratios to form LNPs and may require microfluidic device to produce LNPs in controlled size;8 and the fate of mRNA after delivery is not well-understood and may cause undesirable side effects.9 One of our interests in the field is to develop an alternative method for encapsulation of mRNA vaccines and selective delivery to antigen presenting cells to elicit enhanced and broadly protective immune responses.Recently, polymers with disulfide linkages have been used as functional materials for a wide range of applications.10–12 Due to their dynamic and reversible properties, these polymers have been used for intracellular delivery of various cargoes.13,14 Disulfide exchange to expose thiols followed by release of cargoes after uptake is achieved by reduction in the cytosol.15,16 The polymers with disulfide linkages are not only biodegradable in the intracellular environment, but also capable of thiol-mediated uptake, making possible efficient protein, siRNA, and mRNA delivery to the cytosol.17 In previous studies, a guanidine-containing polymer was demonstrated to promote the delivery of RNA-based therapeutics.18 However, these polymer–drug complexes showed relatively low efficacy in vivo due to the problem of serum instability caused by nonspecific protein adsorption and insufficient endosomal escape.12,19,20
Despite advances in polymer synthesis,21–23 there is still lack of efficient nanocarriers for selective delivery of mRNA vaccine to antigen presenting cells,24,25 though an siRNA–liposome with a trimeric N-acetyl-galactosamine (GalNAc) targeting the asialo-glycoprotein receptor on hepatocytes has been developed and approved by the FDA for the treatment of homozygous familial hypercholesterolemia.26–28
In this study, we intended to develop a glycan-functionalized polymer as an improved polymer-based mRNA carrier and we started with a thiol-initiator and different propagators to generate a series of polymers for encapsulation of mRNA as an mRNA–polymeric nanoparticle (mRNA–PNP) complex and evaluation of immune responses and antibody protection activity. We finally identify a new type of polymers containing guanidine and zwitterionic groups and a sugar ligand designed to target the DC-SIGN receptor on dendritic cells for encapsulation and selective delivery of SARS-CoV-2 spike mRNA vaccine to dendritic cells to elicit enhanced immune responses.
Results and discussion
We first designed a series of polymers and explored their ability to deliver the mRNA encoding green fluorescent protein (GFP) in transfection study and the mRNA encoding SARS-CoV-2 spike protein for vaccine development. As shown in Fig. 1, the propagator P1 containing a guanidine group and the propagator P2 containing three guanidine groups were designed to facilitate the encapsulation of mRNA by forming strong salt bridges between the guanidinium groups of the polymer and the phosphate groups of mRNA.24 The zwitterions were designed to enhance cell membrane fusion to facilitate uptake. We envisioned that once the polymer-encapsulated mRNA reaches the cytoplasm, the disulfide linkage will be degraded by intracellular glutathione to release mRNA and avoid the accumulation of high molecular weight polymers inside the cell to cause cytotoxicity.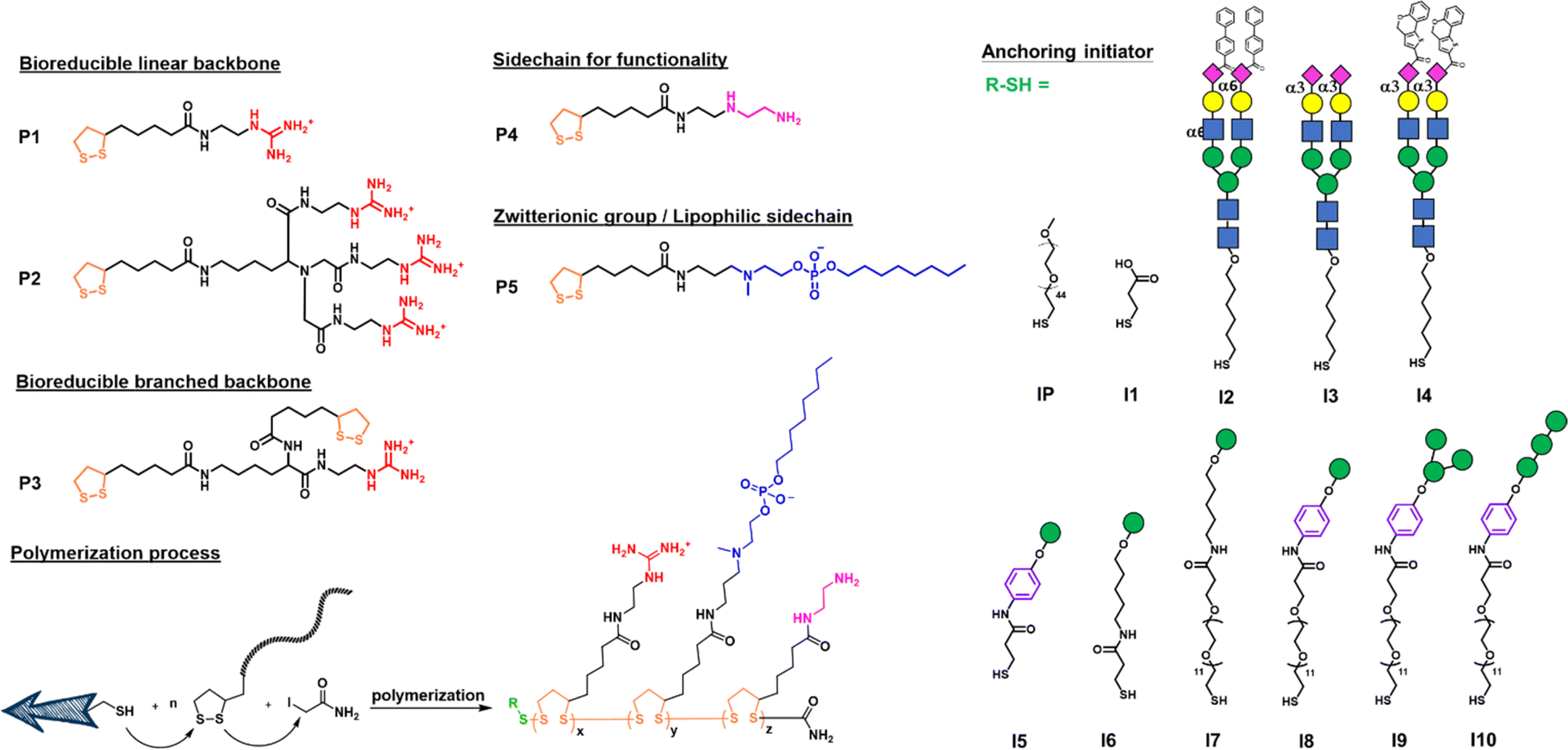 | ||
Fig. 1 Designed structures of bioreducible propagators (P1, P2, P3, P4 and P5) and anchoring initiators (IP, I1, I2, I3, I4, I5, I6, I7, I8, I9 and I10) for polymerization (x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y = 1:1, z = 0.1 or 0). Glycans that are recognized by antigen presenting cells are shown with specified colors and shape symbols: blue square, GlcNAc; green circle, Man; yellow circle, Gal; pink diamond, Neu5Ac. :y = 1:1, z = 0.1 or 0). Glycans that are recognized by antigen presenting cells are shown with specified colors and shape symbols: blue square, GlcNAc; green circle, Man; yellow circle, Gal; pink diamond, Neu5Ac. | ||
The mono-guanidine disulfide (P1) was synthesized according to the procedure reported12 and the tri-valent guanidine disulfide monomer was synthesized from nitrilotriacetic acid to generate the tri-valent guanidine monomer (see more synthetic details in ESI†). Propagator P3 containing two strained disulfides nearby the guanidine group was designed to form a branched structure of the polymer, whereas propagator P4 containing the diethylene-triamine moiety was designed to facilitate the escape of entrapped molecule from degradation and its terminal amine residue was used for functionalization. Propagator P5 containing a zwitterion group was intended to reduce serum protein adsorption, enhance membrane fusion, and enable the delivery of mRNA from the nanocarrier to immune cells.22 The polymerization of P1, P2, P3, P4 or P5 was conducted in degassed water solution at room temperature. A mixture of initiator and propagators in 1 M TEOA buffer, pH 7 was vigorously mixed for 30 min, and the reaction was terminated by adding 0.5 M iodoacetamide.
To identify the optimal polymer for efficient intracellular delivery of GFP–mRNA as a model, homo-polymers and hetero-copolymers were synthesized by co-polymerization of different propagators and their encapsulation ability and transfection efficiency in HEK293T cells were evaluated. As shown in Fig. S1,† all copolymers containing the guanidine group were able to encapsulate GFP–mRNA. Particularly, the P2/P3, P2/P4 and P2/P5 copolymers with tri-valent guanidine moieties exhibited higher capability to encapsulate the mRNA and were comparable with the result with the traditional transfection agent polyethyleneimine (PEI). Next, we evaluated the transfection efficiency of GFP–mRNA in HEK293T cells by using different copolymers. First, we found that the mRNA encapsulated with a hetero polymer is important to achieve lysosomal escape and subsequent translation of the mRNA to protein. The result indicated that P1 polymer was not favorable for intracellular delivery, while the hetero-polymer P1/P4, P2/P4, P1/P5, or P2/P5 was able to transfect HEK293T cells and release the mRNA for translation to GFP. Particularly, the polymer containing the zwitterionic group (Fig. 2A and S2†) was highly effective probably due to membrane fusion as described above. However, we found that the branched polymers P1/P3 and P2/P3 did not show a satisfactory release of mRNA cargoes, although P2/P3 showed a good ability to complex with GFP–mRNA. These results indicated that the propagator is an important component in transfection and the co-polymers generated from P1/P5 and P2/P5 exhibited the highest transfection efficiency, and they did not show any apparent cytotoxicity at the ratio of mRNA–polymer = 1:3 (Fig. S3†).
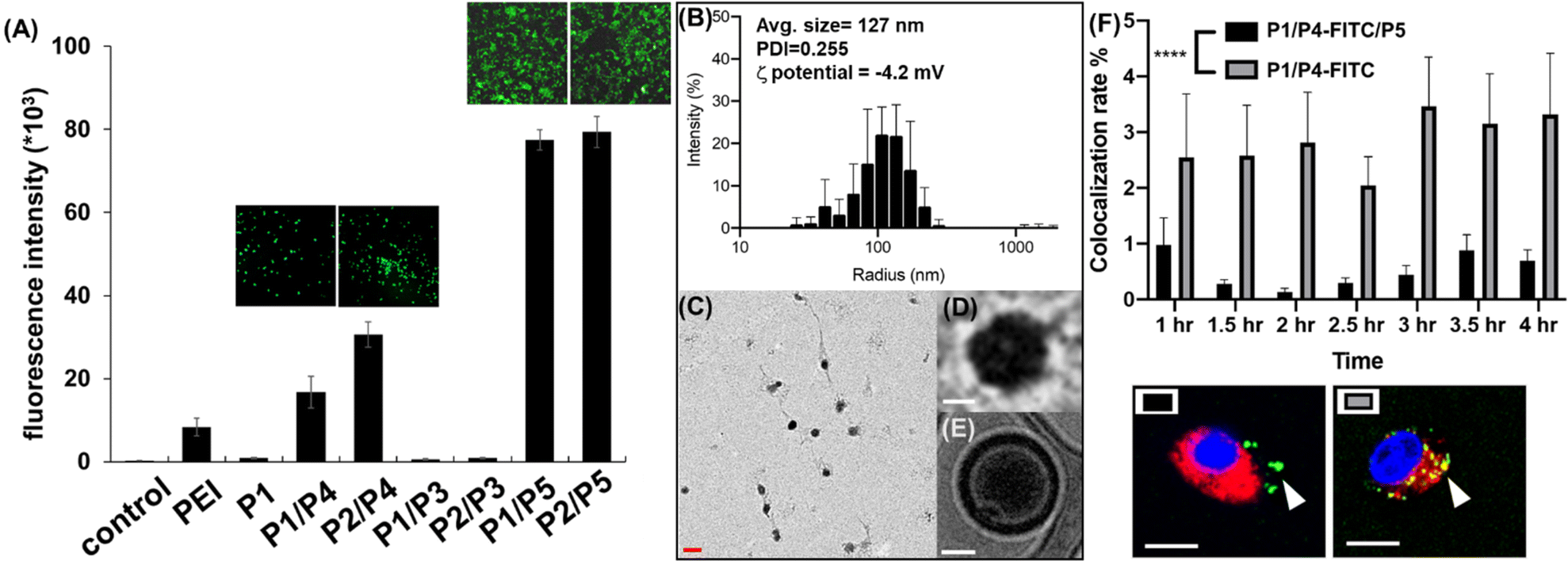 | ||
| Fig. 2 (A) Mean fluorescence intensity of HEK293T cells after transfection with GFP–mRNA–PNP (PNP:PEI (MW = 25k), P1, P1/P4, P2/P4, P1/P3, P2/P3, P1/P5 and P2–P5). (B) Size distribution, (C) TEM image, (D) enlarged TEM image, (E) enlarged CryoEM of mRNA–PNP (I1-P1/P5). White/red scale bar represents 50/300 nm. (F) Colocalization rate of lysosome and mRNA–PNP by confocal fluorescence imaging with P1/P4/P5 or P1/P4 conjugated to FITC. The blue channel is Hoechst, the red channel is LysoTracker, and the green channel is mRNA–PNP–FITC. White scale bar represents 10 μm. | ||
To examine the morphology of mRNA–polymer complex, we first mixed the P1 polymer with mRNA and it was found to form an electron-dense cluster nanoparticle (Fig. S4†), which is in general less efficacious because of serum protein adsorption.29 On the other hand, the complex of P1/P5 co-polymer with mRNA was found to be sphere-like (Fig. 2C and D).29 The polymers are likely to form a layer with positive charge, which can encapsulate the mRNA, and facilitate subsequent cellular uptake through the engagement of zwitterions.
The molecular weight and polymerization index were characterized by gel permeation chromatography (GPC). As shown in Fig. S5,† the polymers displayed a unimodal peak but rather broad molecular weight distribution in the GPC chromatogram and appeared at the early elution time, indicating its polymeric state with high molecular weight. The molecular weight was estimated to be 10.2 kDa (PDI = 1.33) for mRNA–PNP (P1/P5). Based on the result of various copolymers used for GFP–mRNA encapsulation, the P1/P5 co-polymer was selected for further studies, including its ability to complex with mRNA, the physiochemical properties of the resulting nanocomplexes, and the efficiency and selectivity of transfection.
Wild type spike mRNA was prepared and encapsulated by P1/P5 co-polymer at different N/P ratios (Fig. S6†). The result showed that the P1/P5 co-polymer exhibited a good ability to complex with spike mRNA at the N/P ratio of 1, and the average particle size of the resulting mRNA–P1/P5 co-polymer complex was ca. 127 nm, as revealed by CryoEM, TEM and dynamic light scattering (DLS) analysis (Fig. 2B–E). To ensure the fully encapsulation of mRNA, a N/P ratio of 3 was employed and the release and translation of mRNA were observed presumably attributed to GSH-mediated polymer degradation, as it took place in the presence of GSH in a time-dependent manner (Fig. S7†).
We then transfected the spike mRNA–P1/P5 complex (3 μg) in HEK293T cells and performed western blotting. After 48 h post transfection, cells were analyzed for spike-protein translation by western blotting with spike-specific antibody. A significant band at ∼250 kDa corresponding to SARS-CoV-2 spike protein was observed compared to the spike mRNA as negative control (Fig. S8†). This study confirmed that P1/P5 co-polymer is an effective nanocarrier for mRNA transfection in vitro.
Zwitterionic lipids have been reported to improve serum resistance and cell membrane fusion.25 To visualize the location of mRNA–PNP in cells, an FITC-labelled polymer was synthesized from P1/P4/P5, in which FITC was conjugated to the polymer through the amine group on P4. The confocal fluorescence imaging confirmed that mRNA–PNP (I1-P1/P4/P5) exhibited effective mRNA–PNP escape from lysosomes within 2 h; however, the mRNA–PNP (I1-P1/P4) did not exit from lysosomes even in 4 h after internalization (Fig. S9†). As shown in Fig. 2F, the colocalization of lysosome with mRNA–PNP (I1-P1/P4/P5) was lower than the mRNA–PNP (I1-P1/P4), indicating that the alkylated zwitterion residue may significantly improve membrane fusion and lysosomal escape.
In order to selectively deliver the mRNA vaccine to antigen-presenting cells (APCs) especially dendritic cells, we designed the initiator with different glycan heads recognized by the lectin receptors such as Siglec-1, Siglec-2, Siglec-5/E and DC-SIGN which are expressed predominately on DCs and macrophages.30 For Siglec-1 (sialoadhesin), 9-N-(4H-thieno[3,2-c]chromene-2-carbamoy-l)-Neu5Ac-α2,3-Gal-GlcNAc was found to be the most reactive ligand.31 Besides, 9-biphenyl Neu5Ac-α2,6-Gal derivatives showed an increased affinity for Siglec-2,32 and Siglec-5/E prefers the sialosides with the Neu5Ac-α2,3-Gal-GlcNAc structure.33 All ligands were synthesized and used as initiators for incorporation into the polymer which was evaluated for selective delivery of mRNA to dendritic cells.
I2–I5 initiated polymers were prepared and I1 initiated polymer bearing no glycan ligand was used as control. Assuming the polymerization efficiencies of all the initiators are similar, polymers with fixed density of glycan ligands allowed us to examine how these functionalized polymers influence the cellular uptake of mRNA. To assess mRNA–PNP uptake via Siglecs, we compared the binding and internalization of mRNA–PNP into T cells, B cells, and bone marrow-derived dendritic cells (BMDCs). FITC-conjugated mRNA–PNP was incubated with each cell line for 1 h and evaluated by flow cytometry. The I2 mRNA–PNP with 9BPCNeu5Ac conjugated N-glycan intended to target Siglec-2 showed a higher cellular uptake by all APCs compared to I1 mRNA–PNP without glycan modification (Fig. 3A and S11†). However, I2 mRNA–PNP did not show a selective uptake by dendritic cells. Similar results were obtained by using I3 and I4 mRNA–PNP to target Siglec-5/E and Siglec-1, respectively, in which all glycan-decorated mRNA–PNPs exhibited a better uptake by all APCs than the glycan-free mRNA–PNP but there was lack of selectivity toward DCs. With these discouraging results, we decided to evaluate the other C-type lectin DC-SIGN as targeting receptor.
DC-SIGN recognizes high mannose glycans and can route antigens for antigen processing to elicit robust immune responses, especially the CD4+ and CD8+ T cell responses,34–36 making it an attractive receptor for selective mRNA vaccine delivery. It has been used as a target for selective delivery of vaccines and mannose has been used as a ligand for such a delivery system.37 The initiator I5 with aryl mannoside was synthesized as a reference for mRNA–PNP preparation as it was shown to bind preferentially to DC-SIGN.38 The DC-SIGN-mediated cellular uptake by APCs was evaluated by flow cytometry. As shown in Fig. 3A, the I5 mRNA–PNP with the aryl mannose head (I5-P1/P4-FITC/P5) exhibited ∼34% higher cellular uptake by BMDCs compared to the mRNA–PNP (I1-P1/P4-FITC/P5) without the glycan head. On the other hand, B cells and T cells with insignificant DC-SIGN expression showed only a slight increase of fluorescence signal when treated with mRNA–PNP (I1-P1/P4-FITC/P5). These data showed a proof of principle that efficient internalization and selective uptake of mRNA–PNP (I5-P1/P4-FITC/P5) by dendritic cells can be achieved through DC-SIGN receptor targeting.
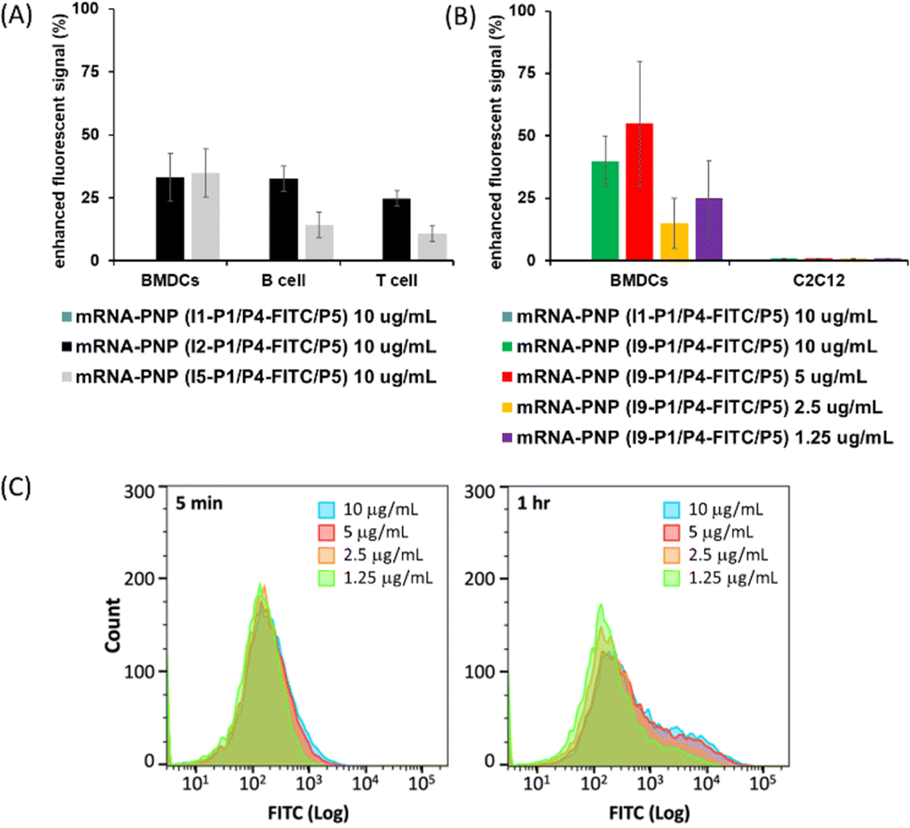 | ||
| Fig. 3 (A) Enhanced FITC signal as uptake measure of targeted mRNA–PNP (I2-P1/P4-FITC/P5) (I5-P1/P4-FITC/P5) by BMDCs, B cells, and T cells from mouse splenocytes compared to nontargeted mRNA–PNP (I1-P1/P4-FITC/P5). (B) Enhanced FITC signal as uptake measure of targeted mRNA–PNP (I9-P1/P4-FITC/P5) by BMDCs and C2C12 cells compared to nontargeted mRNA–PNP (I1-P1/P4-FITC/P5) under different concentrations of mRNA–PNP. Fluorescent signals were analysed by flow cytometry after treatment with mRNA–PNP for 1 h and. (C) Flow cytometry analysis of FITC signals after incubation of mRNA–PNP (I9-P1/P4-FITC/P5) at different concentrations. Incubation time: 5 min, 1 h. | ||
Based on this approach, I6, I7, I8, I9, and I10 with different mannoside structure and spacer were synthesized and the corresponding mRNA–PNPs were prepared for study. I9 with aryl-α-1,3-α-1,6-trimannoside containing a branch type mannose and a hydrophobic presentation was first investigated. Although the mRNA–polymer cluster with aryl mannose head is likely to influence the specificity of receptor binding, our comparative binding study demonstrated that the linear tri-mannose ligand is more specific for mannose receptors (MRs),39 while the branched trimannoside is prone to binding to DC-SIGN. Besides, antigen internalization and signaling rely on DC-SIGN engagement on the cell surface and in the endosome, and the pH of each environment differs. We therefore evaluated binding of DC-SIGN to the mRNA–PNP generated from I5-P1/P5, I6-P1/P5, I7-P1/P5, I8-P1/P5, I9-P1/P5 and I10-P1/P5 under different pH values. We found that the mRNA–PNP from I5-P1/P5, I8-P1/P5, I9-P1/P5 and I10-P1/P5 bind to the DC-SIGN extracellular domain (ECD) at pH 7.4 and 5.0 with nearly the same affinity. However, the mRNA–PNP generated from I6-P1/P5 and I7-P1/P5 lost their binding affinity at lower pH (Fig. S13†) to DC-SIGN.40 This binding results indicated that aryl trimannoside interacts with DC-SIGN in the acidic endosomal compartments. Such binding stability may enhance DC-SIGN-mediated signaling and its synergism with that of endosomal-resident Toll-like receptors such as TLR7. The strong binding of PNP with aryl-mannoside to DC-SIGN may be due to the dense display of the ligand and the aryl group may engage in the CH–π and hydrophobic interactions.41 In addition, for receptor-targeting delivery, ligands are commonly linked with a carrier, and the distance between the carrier and ligand is regulated by the presence of a spacer.42 The mRNA–PNP carrying a longer ligand (I6-P1/P5 with Man-Ar-PEG12, and I7-P1/P5 with Man-PEG12) showed slightly higher affinity toward DC-SIGN. Overall, the mRNA–PNP with aryl-trimannoside (I9-P1/P5) exhibited the highest affinity and lowest KD to DC-SIGN. The increased affinity may be due to a clustering effect and the spatial arrangement of the ligand in the polymer, and the aryl moiety may facilitate its hydrophobic interaction. The aryl-trimannoside ligand and the resulting polymer were also characterized by 1H NMR (Fig. S14–S18†), IR (Fig. S19†) and quantitative glycan analysis (Fig. S20†).
The preferential uptake of mRNA–PNP (I9-P1/P5) by BMDCs and C2C12 muscle cells was also evaluated by flow cytometry. As shown in Fig. 3B, FITC signals increased when BMDCs were incubated with mRNA–PNP (I9-P1/P5) especially at the PNP concentration of 10 and 5 μg mL−1. Fig. 3C showed FITC signals at different incubation intervals and the ratios of I9-P1/P5 versus I1-P1/P5 signals indicated that the mRNA–PNP had a better uptake than the ones without glycans. On the other hand, when C2C12 muscle myoblasts cells, a cell line without mannose receptors were treated with mRNA–PNP (I9-P1/P5) or mRNA–PNP (I1-P1/P5), both samples showed very similar FITC profiles (geometric mean ratio of I9-P1/P5 versus I1-P1/P5 = 100.9%) (Fig. S22†). These data suggested that there is no selectivity towards C2C12 muscle cells.
We further tested whether the uptake of mannosylated mRNA–PNPs are dependent on DC-SIGN. A binding study using ELISA-based measurement including DC-SIGN, macrophage mannose receptor (MMR), MINCLE, Dectin-2, and langerin was conducted. As shown in Fig. S23,† we observed that the branched type of aryl-trimannoside I9 showed a better preference to DC-SIGN compared to the linear type of aryl-trimannoside I10. In addition, I10 was strongly bound to either DC-SIGN, MMR, Dectin-2 or langerin with no significant selectivity. However, I9 presented a more selective binding toward DC-SIGN. These data supported that the efficient uptake of mRNA–I9-P1/P5 by DCs is more likely mediated by DC-SIGN.
To further demonstrate and investigate the efficiency of DC-SIGN-mediated cellular uptake, we compared the uptake of mannosylated I9-initiated PNPs and the I1-initiated PNPs as control by two cell lines: U937 cells and engineered U937 cells with DC-SIGN expression (U937/DC-SIGN),43 using the conjugated FITC to track the uptake of mRNA–PNPs. Based on the flow cytometry analysis, the control mRNA–PNP (I1-P1/P4-FITC/P5) was barely interacting with U937 cells or U937/DC-SIGN cells (Fig. 4A and C). In contrast, the mRNA–PNP (I9-P1/P4-FITC/P5) was intensely bound to the DC-SIGN expressing cell (Fig. 4D) with 10-fold stronger than with U937 cell (Fig. 4C). To further investigate the role of DC-SIGN in receptor-mediated endocytosis, it was found that the endocytosis of mRNA–PNP (I9-P1/P4-FITC/P5) at 4 °C was significantly reduced and the ligand-receptor binding was fully inhibited by addition of EDTA (Fig. 4E), consistent with the role of calcium required in glycan recognition by DC-SIGN. Besides, U937/DC-SIGN cells pre-treated with blocking antibodies against DC-SIGN resulted in a significant decrease in mRNA–PNP (I9-P1/P4-FITC/P5) uptake, supporting that the internalization of mRNA–PNP (I9-P1/P4-FITC/P5) is mediated by DC-SIGN (Fig. 4G and H). Taken together, these data indicated that uptake of mRNA–PNP (I9-P1/P4-FITC/P5) occurred by DC-SIGN-mediated endocytosis, while mRNA–PNPs without mannose ligand showed a poor uptake by cells.
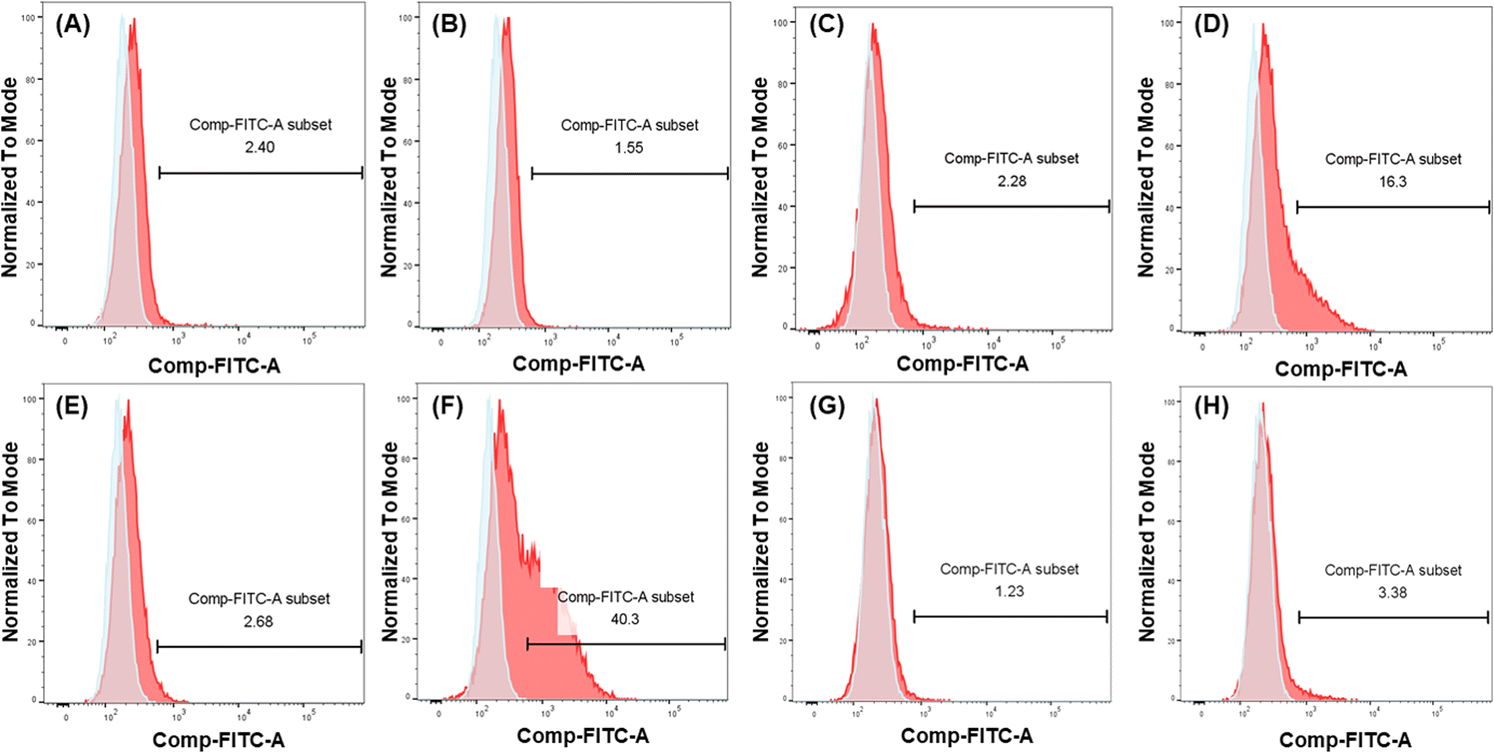 | ||
| Fig. 4 Flow cytometry analysis of mRNA–PNPs uptake by U937 cells and DC-SIGN expressed U937 cells at different conditions. (A) mRNA–PNP (I1-P1/P4-FITC/P5) incubated with U937 cells at 37 °C for 30 min. (B) mRNA–PNP (I9-P1/P4-FITC/P5) incubated with U937 cells at 37 °C for 30 min. (C) mRNA–PNP (I1-P1/P4-FITC/P5) incubated with U937/DC-SIGN cells at 37 °C for 30 min. (D) mRNA–PNP (I9-P1/P4-FITC/P5) incubated with U937/DC-SIGN cells at 37 °C for 30 min. (E) mRNA–PNP (I9-P1/P4-FITC/P5) incubated with U937/DC-SIGN cells in the presence of 10 mM EDTA at 4 °C for 60 min and then 37 °C for 30 min. (F) mRNA–PNP (I9-P1/P4-FITC/P5) incubated with U937/DC-SIGN cells at 4 °C for 60 min and then 37 °C for 30 min. (G) mRNA–PNP (I1-P1/P4-FITC/P5) incubated with U937/DC-SIGN cells (with anti-DC-SIGN antibody pre-blocked at 4 °C for 20 min) at 37 °C for 30 min. (H) mRNA–PNP (I9-P1/P4-FITC/P5) incubated with U937/DC-SIGN cells (with anti-DC-SIGN antibody pre-blocked at 4 °C for 20 min) at 37 °C for 30 min. All of the mRNA–PNPs were washed off after first incubation. | ||
We next evaluated the effect of wild type spike mRNA–PNP with or without the aryl mannoside head on vaccination and immune responses. BALB/c mice were immunized with the vaccines on day 0 and received a boost on day 14 (Fig. 5A). As a positive control, a group treated with spike mRNA–LNP (including ALC-0315, DSPC, ALC-0159, and cholesterol commonly used in current mRNA vaccine formulation) was included. We observed a 10000-fold difference in titer of anti-spike antibodies in the serum of the mRNA–PNP (I1-P1/P5) and the mRNA–LNP groups (Fig. 5B). However, the titer of spike-specific antibody response in mice after second immunizations with the aryl-trimannoside containing mRNA–PNP (I9-P1/P5) was 3.1-fold higher than the two control groups on days 28. The serum was then tested for the ability to neutralize pseudovirus-mediated entry to the ACE2-expressing cells. While the level of antisera from mRNA–PNP (I1-P1/P5) was similar to the mRNA–LNP group, a significantly higher level of neutralizing antibodies in the mRNA–PNP (I9-P1/P5) group was observed (Fig. 5C), and the spike-specific antibody titer and pseudovirus neutralization activity were well correlated.
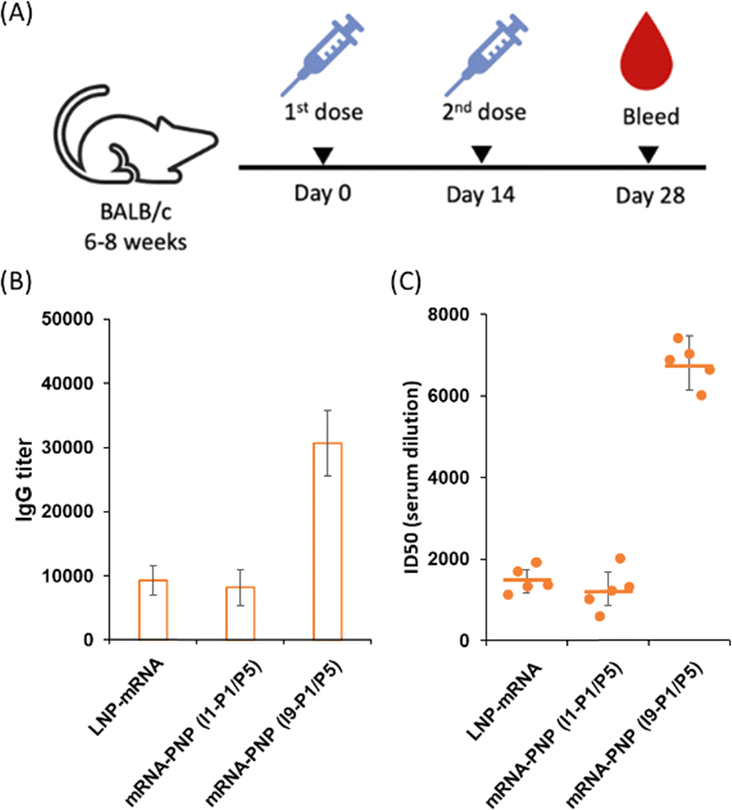 | ||
| Fig. 5 (A) BALB/c mice were immunized with 15 μg of wild type spike, mRNA–LNP, mRNA–PNP (I1-P1/P5), and mRNA–PNP (I9-P1/P5) and boosted on day 14 (n = 5). (B) Serum levels of spike-specific IgGs from spike mRNA–PNP were monitored over 28 days post priming by ELISA. (C) Sera obtained from mice after the last immunization were evaluated for their neutralization activities against SARS-CoV-2 by a pseudovirus-based neutralization assay. Neutralization titers (ID50) were calculated as the reciprocal of the serum dilution that resulted in a 50% reduction in RLUs compared to virus control wells after subtraction of background RLU. The ID50 values are labelled on the plots with standard error of mean. | ||
Conclusions
In conclusion, we have developed and investigated a series of polymers with different glycan ligands for selective mRNA vaccine delivery and demonstrated that the polymer containing guanidine and zwitterionic groups exhibited great efficiency for mRNA vaccine delivery both in vitro and in vivo. In addition, the polymer generated by initiator I9 with an aryl trimannoside head and propagators P1/P5 is an effective carrier for selective delivery of mRNA vaccines to dendritic cells for translation, processing, presentation, and efficient immune responses against the spike antigen. More detailed studies of CD4+ and CD8+ T-cell responses and further evaluation of the mRNA–PNP in animal challenge study are ongoing. We anticipate that this method is generally applicable to other mRNAs for vaccine design and therapeutical development.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
C.-Y. Fan and C.-H. Wong conceived and designed the experiments. C.-Y. Fan, S.-W. Wang, C. Chung, J.-Y. Chen, C.-Y. Chang, Y.-C. Chen performed the experiments and analysed the data. C.-H. Wong directed the project. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National RNAi Core Facility at Academia Sinica for pseudotyped lentivirus neutralization assay and related services. This research was supported by Academia Sinica.References
- J. Zhao, S. Zhao, J. Ou, J. Zhang, W. Lan, W. Guan, X. Wu, Y. Yan, W. Zhao, J. Wu, J. Chodosh and Q. Zhang, Front. Immunol., 2020, 11, 602256 CrossRef CAS PubMed.
- E. J. Anderson, N. G. Rouphael, A. T. Widge, L. A. Jackson, P. C. Roberts, M. Makhene, J. D. Chappell, M. R. Denison, L. J. Stevens, A. J. Pruijssers, A. B. McDermott, B. Flach, B. C. Lin, N. A. Doria-Rose, S. O'Dell, S. D. Schmidt, K. S. Corbett, P. A. Swanson, M. Padilla, K. M. Neuzil, H. Bennett, B. Leav, M. Makowski, J. Albert, K. Cross, V. V. Edara, K. Floyd, M. S. Suthar, D. R. Martinez, R. Baric, W. Buchanan, C. J. Luke, V. K. Phadke, C. A. Rostad, J. E. Ledgerwood, B. S. Graham and J. H. Beigel, N. Engl. J. Med., 2020, 383, 2427–2438 CrossRef CAS PubMed.
- E. E. Walsh, R. W. Frenck, A. R. Falsey, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, K. Neuzil, M. J. Mulligan, R. Bailey, K. A. Swanson, P. Li, K. Koury, W. Kalina, D. Cooper, C. Fontes-Garfias, P.-Y. Shi, Ö. Türeci, K. R. Tompkins, K. E. Lyke, V. Raabe, P. R. Dormitzer, K. U. Jansen, U. Şahin and W. C. Gruber, N. Engl. J. Med., 2020, 383, 2439–2450 CrossRef CAS PubMed.
- K. S. Corbett, B. Flynn, K. E. Foulds, J. R. Francica, S. Boyoglu-Barnum, A. P. Werner, B. Flach, S. O'Connell, K. W. Bock, M. Minai, B. M. Nagata, H. Andersen, D. R. Martinez, A. T. Noe, N. Douek, M. M. Donaldson, N. N. Nji, G. S. Alvarado, D. K. Edwards, D. R. Flebbe, E. Lamb, N. A. Doria-Rose, B. C. Lin, M. K. Louder, S. O'Dell, S. D. Schmidt, E. Phung, L. A. Chang, C. Yap, J.-P. M. Todd, L. Pessaint, A. Van Ry, S. Browne, J. Greenhouse, T. Putman-Taylor, A. Strasbaugh, T.-A. Campbell, A. Cook, A. Dodson, K. Steingrebe, W. Shi, Y. Zhang, O. M. Abiona, L. Wang, A. Pegu, E. S. Yang, K. Leung, T. Zhou, I.-T. Teng, A. Widge, I. Gordon, L. Novik, R. A. Gillespie, R. J. Loomis, J. I. Moliva, G. Stewart-Jones, S. Himansu, W.-P. Kong, M. C. Nason, K. M. Morabito, T. J. Ruckwardt, J. E. Ledgerwood, M. R. Gaudinski, P. D. Kwong, J. R. Mascola, A. Carfi, M. G. Lewis, R. S. Baric, A. McDermott, I. N. Moore, N. J. Sullivan, M. Roederer, R. A. Seder and B. S. Graham, N. Engl. J. Med., 2020, 383, 1544–1555 CrossRef CAS PubMed.
- A. M. Reichmuth, M. A. Oberli, A. Jaklenec, R. Langer and D. Blankschtein, Ther. Delivery, 2016, 7, 319–334 CrossRef CAS PubMed.
- L. Miao, L. Li, Y. Huang, D. Delcassian, J. Chahal, J. Han, Y. Shi, K. Sadtler, W. Gao, J. Lin, J. C. Doloff, R. Langer and D. G. Anderson, Nat. Biotechnol., 2019, 37, 1174–1185 CrossRef CAS PubMed.
- Y. Zhang, C. Sun, C. Wang, K. E. Jankovic and Y. Dong, Chem. Rev., 2021, 121, 12181–12277 CrossRef CAS PubMed.
- D. Chen, K. T. Love, Y. Chen, A. A. Eltoukhy, C. Kastrup, G. Sahay, A. Jeon, Y. Dong, K. A. Whitehead and D. G. Anderson, J. Am. Chem. Soc., 2012, 134, 6948–6951 CrossRef CAS PubMed.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed.
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- Z. Shu, I. Tanaka, A. Ota, D. Fushihara, N. Abe, S. Kawaguchi, K. Nakamoto, F. Tomoike, S. Tada, Y. Ito, Y. Kimura and H. Abe, Angew. Chem., Int. Ed., 2019, 58, 6611–6615 CrossRef CAS PubMed.
- J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed.
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 CrossRef CAS PubMed.
- J. Mosquera, I. García and L. M. Liz-Marzán, Acc. Chem. Res., 2018, 51, 2305–2313 CrossRef CAS PubMed.
- G. Gasparini and S. Matile, Chem. Commun., 2015, 51, 17160–17162 RSC.
- P. Yuan, H. Zhang, L. Qian, X. Mao, S. Du, C. Yu, B. Peng and S. Q. Yao, Angew. Chem., Int. Ed., 2017, 56, 12481–12485 CrossRef CAS PubMed.
- J. Guo, T. Wan, B. Li, Q. Pan, H. Xin, Y. Qiu and Y. Ping, ACS Cent. Sci., 2021, 7, 990–1000 CrossRef CAS PubMed.
- T. Miyazaki, S. Uchida, H. Hatano, Y. Miyahara, A. Matsumoto and H. Cabral, Eur. Polym. J., 2020, 140, 110028 CrossRef CAS.
- P. Zhang, Y. Wang, J. Lian, Q. Shen, C. Wang, B. Ma, Y. Zhang, T. Xu, J. Li, Y. Shao, F. Xu and J.-J. Zhu, Adv. Mater., 2017, 29, 1702311 CrossRef PubMed.
- E. Derivery, E. Bartolami, S. Matile and M. Gonzalez-Gaitan, J. Am. Chem. Soc., 2017, 139, 10172–10175 CrossRef CAS PubMed.
- O. A. W. Haabeth, J. J. K. Lohmeyer, A. Sallets, T. R. Blake, I. Sagiv-Barfi, D. K. Czerwinski, B. McCarthy, A. E. Powell, P. A. Wender, R. M. Waymouth and R. Levy, ACS Cent. Sci., 2021, 7, 1191–1204 CrossRef CAS PubMed.
- S. Liu, X. Wang, X. Yu, Q. Cheng, L. T. Johnson, S. Chatterjee, D. Zhang, S. M. Lee, Y. Sun, T.-C. Lin, J. L. Liu and D. J. Siegwart, J. Am. Chem. Soc., 2021, 143, 21321–21330 CrossRef CAS PubMed.
- H. J. Kim, S. Ogura, T. Otabe, R. Kamegawa, M. Sato, K. Kataoka and K. Miyata, ACS Cent. Sci., 2019, 5, 1866–1875 CrossRef CAS PubMed.
- P. K. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed.
- M. A. Jackson, T. A. Werfel, E. J. Curvino, F. Yu, T. E. Kavanaugh, S. M. Sarett, M. D. Dockery, K. V. Kilchrist, A. N. Jackson, T. D. Giorgio and C. L. Duvall, ACS Nano, 2017, 11, 5680–5696 CrossRef CAS PubMed.
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef CAS PubMed.
- Y. Zhou, P. Teng, N. T. Montgomery, X. Li and W. Tang, ACS Cent. Sci., 2021, 7, 499–506 CrossRef CAS PubMed.
- H. Cui, X. Zhu, S. Li, P. Wang and J. Fang, ACS Omega, 2021, 6, 16259–16265 CrossRef CAS PubMed.
- M. Machtakova, H. Thérien-Aubin and K. Landfester, Chem. Soc. Rev., 2022, 51, 128–152 RSC.
- T. B. H. Geijtenbeek and S. I. Gringhuis, Nat. Rev. Immunol., 2016, 16, 433–448 CrossRef CAS PubMed.
- C. M. Nycholat, C. Rademacher, N. Kawasaki and J. C. Paulson, J. Am. Chem. Soc., 2012, 134, 15696–15699 CrossRef CAS PubMed.
- W. Peng and J. C. Paulson, J. Am. Chem. Soc., 2017, 139, 12450–12458 CrossRef CAS PubMed.
- P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef CAS PubMed.
- S. K. Singh, J. Stephani, M. Schaefer, H. Kalay, J. J. García-Vallejo, J. den Haan, E. Saeland, T. Sparwasser and Y. van Kooyk, Mol. Immunol., 2009, 47, 164–174 CrossRef CAS PubMed.
- P. J. Tacken, I. J. M. de Vries, R. Torensma and C. G. Figdor, Nat. Rev. Immunol., 2007, 7, 790–802 CrossRef CAS PubMed.
- R.-J. E. Li, T. P. Hogervorst, S. Achilli, S. C. Bruijns, T. Arnoldus, C. Vivès, C. C. Wong, M. Thépaut, N. J. Meeuwenoord, H. van den Elst, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov, S. J. van Vliet, F. Fieschi, J. D. C. Codée and Y. van Kooyk, Front. Chem., 2019, 7, 650 CrossRef CAS PubMed.
- A. Singh, Nat. Nanotechnol., 2021, 16, 16–24 CrossRef CAS PubMed.
- M. M. Alam, C. M. Jarvis, R. Hincapie, C. S. McKay, J. Schimer, C. A. Sanhueza, K. Xu, R. C. Diehl, M. G. Finn and L. L. Kiessling, ACS Nano, 2021, 15, 309–321 CrossRef CAS PubMed.
- H. Gao, C. Gonçalves, T. Gallego, M. François-Heude, V. Malard, V. Mateo, F. Lemoine, V. Cendret, F. Djedaini-Pilard, V. Moreau, C. Pichon and P. Midoux, Carbohydr. Res., 2020, 487, 107877 CrossRef CAS PubMed.
- G. Tabarani, M. Thépaut, D. Stroebel, C. Ebel, C. Vivès, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229–21240 CrossRef CAS PubMed.
- K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152–15160 CrossRef CAS PubMed.
- F. Wang, W. Xiao, M. A. Elbahnasawy, X. Bao, Q. Zheng, L. Gong, Y. Zhou, S. Yang, A. Fang, M. M. S. Farag, J. Wu and X. Song, Front. Pharmacol, 2018, 9, 980 CrossRef PubMed.
- J.-H. Wang, A. M. Janas, W. J. Olson, V. N. KewalRamani and L. Wu, J. Virol., 2007, 81, 2497–2507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06575h |
| This journal is © The Royal Society of Chemistry 2024 |