
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddition of allyl Grignard to nitriles in air and at room temperature: experimental and computational mechanistic insights in pH-switchable synthesis†‡
Blanca
Parra-Cadenas
 a,
Israel
Fernández
*b,
Fernando
Carrillo-Hermosilla
*a,
Joaquín
García-Álvarez
*c and
David
Elorriaga
*d
a,
Israel
Fernández
*b,
Fernando
Carrillo-Hermosilla
*a,
Joaquín
García-Álvarez
*c and
David
Elorriaga
*d
aDepartamento de Química Inorgánica, Orgánica y Bioquímica-Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Ciencias y Tecnologías Químicas, Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain
bDepartamento de Química Orgánica I and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain
cLaboratorio de Química Sintética Sostenible (QuimSinSos), Departamento de Química Orgánica e Inorgánica, (IUQOEM), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Química, Universidad de Oviedo, E33071 Oviedo, Spain. E-mail: garciajoaquin@uniovi.es
dGroup of Bioorganometallic Chemistry and Catalysis (BIOMCAT), Departamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, E33071 Oviedo, Spain
First published on 4th March 2024
Abstract
A straightforward and selective conversion of nitriles into highly substituted tetrahydropyridines, aminoketones or enamines by using allylmagnesium bromide as an addition partner (under neat conditions) and subsequent treatment with different aqueous-based hydrolysis protocols is reported. Refuting the conventional wisdom of the incompatibility of Grignard reagents with air and moisture, we herein report that the presence of water allows us to promote the chemoselective formation of the target tetrahydropyridines over other competing products (even in the case of highly challenging aliphatic nitriles). Moreover, the careful tuning of both the reaction media employed (acid or basic aqueous solutions for the hydrolysis protocol) and the electronic properties of the starting nitriles allowed us to design a multi-task system capable of producing either β-aminoketones or enamines in a totally selective manner. Importantly, and for the first time in the chemistry of main-group polar organometallic reagents in non-conventional protic solvents (e.g., water), both experimental and computational studies showed that the excellent efficiency and selectivity observed in aqueous media cannot be replicated by using standard dry volatile organic solvents (VOCs) under inert atmosphere conditions.
Introduction
The introduction of organolithium (RLi) and Grignard (RMgX) reagents in organic synthesis constituted one of the greatest achievements in synthetic chemistry, opening the door to the selective and efficient formation of new C–C and C–X bonds.1 The importance of these synthetic organometallic tools is that it is estimated that around 95% of drug synthesis in the pharmaceutical industry employs these reagents in at least one reaction step.2 However, their utilization typically relies on the use of: (i) rigorously dried, aprotic, toxic and volatile organic solvents (VOCs); (ii) an inert atmosphere (usually N2 or Ar); and (iii) low temperatures (ranging from 0 to −78 °C),1 these being conditions directly related to the reactivity of these highly polar organometallic reagents, which could lead to undesired side reactions in the presence of moisture and oxygen or when working at room temperature.1 However, recent innovations in organometallic synthesis have demonstrated that at variance with conventional wisdom, polar organolithium and Grignard reagents can be used: (i) in air; (ii) in sustainable, protic, non-toxic and non-dried solvents [such as water or deep eutectic solvents (DESs)]; and (iii) at room temperature, thus reducing costs, waste and consumption of energy and fossil resources.3–5Importantly, and aside from the sustainable point of view, the use of the aforementioned protic and polar solvents (water6 and DESs) in polar organometallic chemistry permitted either the discovery of new reactions/mechanistic pathways or the improvement of selectivity, yields or reaction times (in already known reactions) that cannot be replicated using traditional, dry and toxic VOC solvents.3,4 This has allowed the extension of the use of aerobic/moisture/ambient temperature compatible with RLi/RMgX synthetic protocols to a wide variety of different areas of interest for general chemistry, including (among others) lithium halogen exchange processes, metalation reactions, Pd-catalyzed C–C coupling protocols or anionic polymerizations.3,4 In this vein, Hevia, Capriati and some of us have previously reported the possibility of promoting the efficient and selective addition of RLi/RMgX reagents to nitriles in water,4d glycerol,7a and biomass-derived ethereal solvents (cyclopentyl methyl ether, CPME)7b or even under neat conditions7c (see Scheme 1). Building on these previous studies but going significantly beyond by trying to have (for the first time in this chemistry) a rational and mechanistically well-supported theory for aerobic/moisture-compatible polar organometallic chemistry, we present herein a combined experimental/computational study that reveals the crucial role of water in the addition of allyl Grignard reagents to aromatic nitriles for the chemoselective formation of tetrahydropyridines, aminoketones or enamines, which cannot be mimicked using traditional, dry and toxic organic solvents.
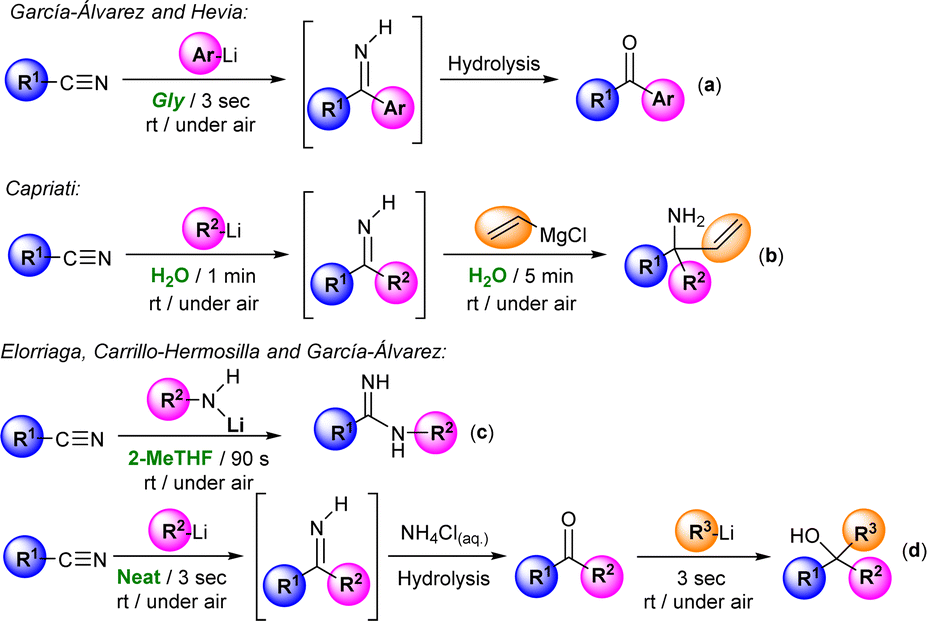 | ||
| Scheme 1 Previous experimental work on the addition of organolithium/organomagnesium reagents to nitriles working at room temperature, in air and using glycerol (Gly) or water as solvent and under neat conditions. | ||
As depicted in Scheme 1d, we have recently described a modular double addition of RLi/RMgX to nitriles to produce tertiary alcohols under bench-type reaction conditions (room temperature and in air/moisture) without any halfway purification step.7c Worth mentioning is that in the first step of this one-pot tandem methodology, Grignard reagents only showed poor to moderate conversion into the desired intermediate ketone. In the same line, only a few examples in the literature reported that Grignard reagents are capable of triggering their addition reaction into aromatic nitriles in conventional organic solvents, with most of them having in common the employment of allyl Grignard as a nucleophile.8–10 For instance, and as paradigmatic cases, we can mention the formation of: (i) tertiary carbinamines, by double addition of this Grignard reagent to α-alkoxy nitriles;8 or (ii) Z/E isomers of the stable primary enamine 1-aminobutadiene, when mixing one equivalent of aromatic nitriles with equimolar quantities of allyl Grignard (controlled hydrolysis with saturated ammonia solution is mandatory).9
Previously, Grassberger et al. also reported the reaction between one equivalent of allyl Grignard and different aromatic nitriles,10 forming the corresponding tetrahydropyridines11 in moderate yields after hydrolysis with water. The proposed mechanism suggests a metal-assisted aza-Diels–Alder reaction (aza-DA),12,13 thus involving the cycloaddition of the two intermediates formed by the nucleophilic addition of the allyl Grignard reagents to the nitrile (Scheme 2). Although the mechanism is quite plausible, no experimental or theoretical support for this proposal was presented.
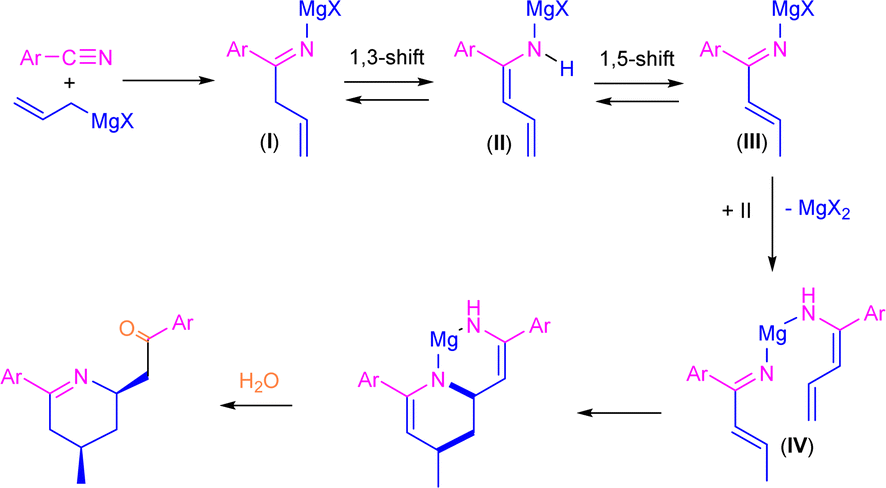 | ||
| Scheme 2 Previously proposed mechanism for the synthesis of tetrahydropyridine derivatives through addition of allylmagnesium bromide to aromatic nitriles. | ||
Thus, and expanding the scope of aqueous/air compatible polar s-block organometallic chemistry to uncharted territories, herein we report the design of the first straightforward, selective and multi-task system capable of converting nitriles into highly substituted tetrahydropyridines, aminoketones or enamines by using allylmagnesium bromide as an addition partner and subsequent fine selection of the aqueous hydrolytic conditions. Combining theoretical calculations with reactivity studies, here we provide informative mechanistic insights on how cooperative effects between RMgX reagents and water can be maximised when designing new magnesium-promoted organic transformations.
Results and discussion
Reaction of allylmagnesium bromide with benzonitrile in the absence of external solvents in air/moisture: experimental studies
Bearing in mind the success of our previous methodologies in the design of the modular double addition of RLi/RMgX reagents to nitriles in air (Scheme 1d),7c we decided to export our previous experience to the addition reaction of allyl Grignard to aromatic nitriles in the absence of external solvents. First, we explored, as a model reaction, the addition of commercially available allylmagnesium bromide to benzonitrile (PhCN, 1a) in the presence of air/moisture, at room temperature and without any additional organic solvent. It is well known that the addition of highly polar organometallic reagents (RLi/RMgX) to nitriles usually gives rise to the formation of the corresponding imine, which evolves upon hydrolysis into the concomitant ketone.1 However, when we carried out the addition protocol under the aforementioned bench-type reaction conditions followed by a standard hydrolytic quenching at room temperature (by using saturated aqueous solution of NH4Cl), we observed the formation of a mixture containing the tetrahydropyridine 2a and one of the isomers of the primary enamine 1-amino-1-phenyl-1,3-butadiene (3a) as a by-product in a 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 ratio (Scheme 3), instead of either the expected imine [PhC(
:85 ratio (Scheme 3), instead of either the expected imine [PhC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)CH2CHCH2] or ketone [PhC(O)CH2CHCH2]. Although the formation of 2a was already reported,10 the concomitant synthesis of enamine 3a should not be expected under our reaction conditions taking into account the previous results reported by Grassberger and co-workers, as they described the formation of mixtures of the tetrahydropyridine 2a with the corresponding carbinamine 4a.10 In this sense, the presence of enamine 3a in our reaction mixture can be explained by tautomerisation of the corresponding imine compound, formed upon hydrolysis of I as shown in Scheme 3. This tautomerisation equilibrium is shifted towards enamines by the conjugative effect of the butadiene skeleton as reported by Erker.9 These types of substituted 1,3-butadienes have E/Z isomers around the double bond between C1–C2, but in our case only one of the isomers was present. Taking into account this experimental observation, which is not consistent with the mechanism previously proposed by Grassberger and co-workers (Scheme 2), we decided to focus our attention on the proposal of a new version of the mechanism for the synthesis of 2a.
NH)CH2CHCH2] or ketone [PhC(O)CH2CHCH2]. Although the formation of 2a was already reported,10 the concomitant synthesis of enamine 3a should not be expected under our reaction conditions taking into account the previous results reported by Grassberger and co-workers, as they described the formation of mixtures of the tetrahydropyridine 2a with the corresponding carbinamine 4a.10 In this sense, the presence of enamine 3a in our reaction mixture can be explained by tautomerisation of the corresponding imine compound, formed upon hydrolysis of I as shown in Scheme 3. This tautomerisation equilibrium is shifted towards enamines by the conjugative effect of the butadiene skeleton as reported by Erker.9 These types of substituted 1,3-butadienes have E/Z isomers around the double bond between C1–C2, but in our case only one of the isomers was present. Taking into account this experimental observation, which is not consistent with the mechanism previously proposed by Grassberger and co-workers (Scheme 2), we decided to focus our attention on the proposal of a new version of the mechanism for the synthesis of 2a.
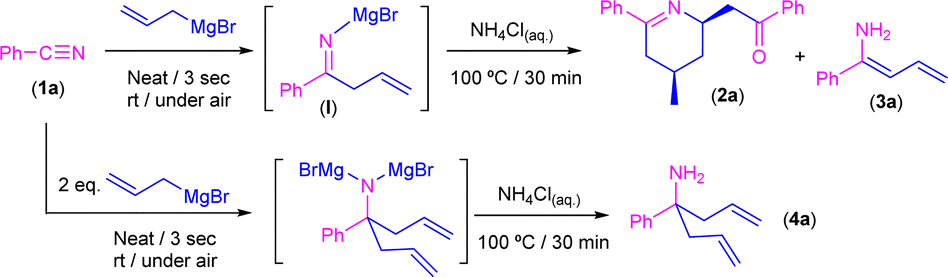 | ||
| Scheme 3 Addition of commercially available allyl magnesium bromide to benzonitrile (PhCN, 1a) in the presence of air/moisture, at room temperature and without any additional organic solvent. | ||
Thus, we first directed our efforts to try to rationalise which of the E/Z isomers of enamine 3a were obtained under our experimental conditions. In this sense, from the two possible enamine isomers Z/E formed, only the isomer Z is able to place the “dienophile” tail in the right position to undergo the aza-DA reaction, which thus implies that the unreacted isomer must be E-3a. Important to mention here is the fact that it has already been established that the interconversion between both Z/E isomers requires high levels of energy.14 Therefore, to force the deliberated transformation between Z/E-3a isomers, we decided to implement a new set of experimental conditions, which imply the heating of the reaction media at 100 °C for 30 minutes after hydrolysis with saturated ammonium chloride solution. Satisfactorily, under these new conditions, tetrahydropyridine 2a was obtained as the unique reaction product in 99% yield. On the other hand, when two equivalents of the Grignard reagent were added, the main product detected was the carbinamine 4a formed by a double addition reaction.15
In order to monitor the quantitative transformation of benzonitrile (1a) into tetrahydropyridine 2a, we carried out, in a Young NMR tube and under an inert atmosphere, the reaction between nitrile 1a and allyl Grignard, giving rise in the first term to the corresponding iminic-type intermediate I. After 24 hours, we observed that compound I is totally stable under the inert atmosphere, at room temperature and in the absence of protic sources and thus did not evolve into the aforementioned possible reaction intermediates II and III (Scheme 2) under these Schlenk-type conditions. All our aforementioned experimental observations suggest that: (i) the initial isomerization reactions and the subsequent aza-Diels–Alder cycloaddition only take place during the hydrolysis step (thus the presence of water in the reaction media is mandatory) with NH4Cl at 100 °C; and (ii) the magnesium metallic fragment is not involved in the formation of the observed tetrahydropyridine 2a.
Reaction of allylmagnesium bromide with benzonitrile in the absence of external solvents in air/moisture: computational studies
Tacking all these experimental pieces of evidence into account, we decide to complete our mechanistic studies by performing density functional theory (DFT) calculations, at the dispersion corrected PCM(diethylether)-B3LYP-D3/def2-TZVPP//PCM(diethylether)-B3LYP-D3/def2-SVP level. As shown in Fig. 1, the final aza-Diels–Alder reaction involving species IV (see Scheme 2), despite being clearly exergonic (ΔGR = −8.2 kcal mol−1), requires a relatively high free activation barrier (ΔG≠ = 28.8 kcal mol−1), which is actually in the limit to be compatible with a process occurring at room temperature. Similar high barriers were obtained either by using other functionals (M06-2X/def2-SVP or BP86-D3/def2-SVP) or by including a solvent molecule attached to the magnesium atom (Me2O as a model of an ethereal solvent which is present in the commercially available solution of the allyl Grignard, see Fig. 1b). These results therefore suggest that the key aza-Diels–Alder reaction is unlikely to take place during the first step of the transformation.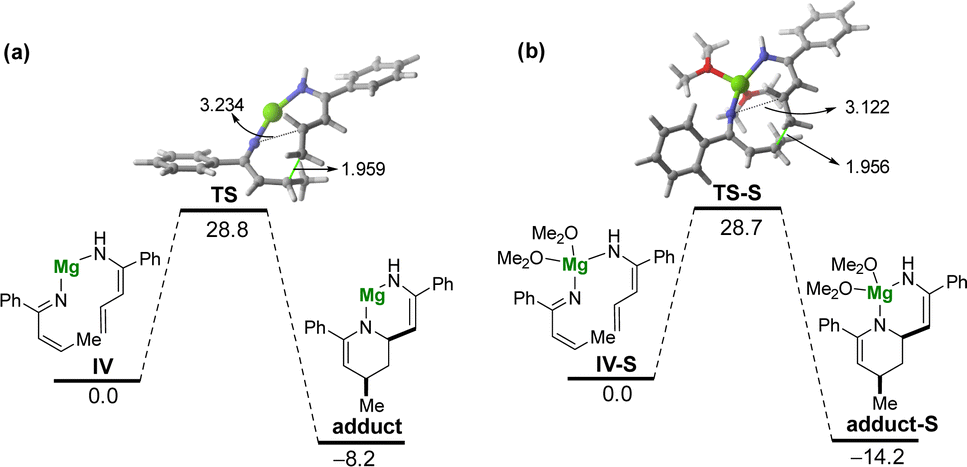 | ||
| Fig. 1 Computed reaction profile for the magnesium-assisted Diels–Alder reaction involving: (a) species IV; and (b) species IV-S where two molecules of solvent are coordinated to the metal atom. Relative free energies (ΔG, at 298 K) and bond distances are given in kcal mol−1 and angstroms, respectively. All data have been computed at the PCM(Et2O)/B3LYP-D3/def2-TZVPP//PCM(Et2O)/B3LYP-D3/def2-SVP level. | ||
Having all these results in mind and taking into account that we have probed experimentally that magnesium compound I does not evolve into the corresponding tetrahydropyridine 2a under traditional Schlenk-type conditions (which indicates that formation of 2a takes place only after hydrolysis of this organomagnesium compound), we decided to study computationally the process without the assistance of magnesium. To this end, our calculations (Fig. 2) begin from the imine A formed upon protonation of magnesium-imine I in Scheme 3 due to the addition of an aqueous solution of NH4Cl. Imine A tautomerises to its more stable Z-enamine B, which may serve as a dienophile in the subsequent Diels–Alder cycloaddition. Then, the expected 1,5-H migration leading to imine C takes place either directly viaTS1 (ΔG≠ = 28.8 kcal mol−1) or assisted by water (as a water dimer, ΔG≠ = 31.6 kcal mol−1). Once the α,β-unsaturated imine C is formed, the aza-Diels–Alder cycloaddition reaction may occur through two alternative pathways. On the one hand, the α,β-unsaturated imine C may act as a diene and react with B acting as an activated dienophile leading to tetrahydropyridine D in a highly exergonic transformation (ΔGR = −14.7 kcal mol−1) via the concerted and highly asynchronous transition state TS2. Alternatively, C may also react with the non-conjugated imine A, leading to the formation of the isomeric tetrahydropyridine E again in a highly exergonic transformation (ΔGR = −11.5 kcal mol−1) via the analogous concerted transition state. In both cases, the computed free activation barriers are relatively high (>30 kcal mol−1) which are compatible with the high temperature (100 °C) required in our experimental studies. From the data in Fig. 2, the former pathway (involving C + B) seems favoured over the latter (involving C + A) from both kinetic (ΔΔG≠ = 3.3 kcal mol−1) and thermodynamic (ΔGR = 3.2 kcal mol−1) points of view. Final hydrolysis of D leads to the experimentally observed tetrahydropyridine 2a, where the enamine group in D has been transformed into the corresponding ketone.
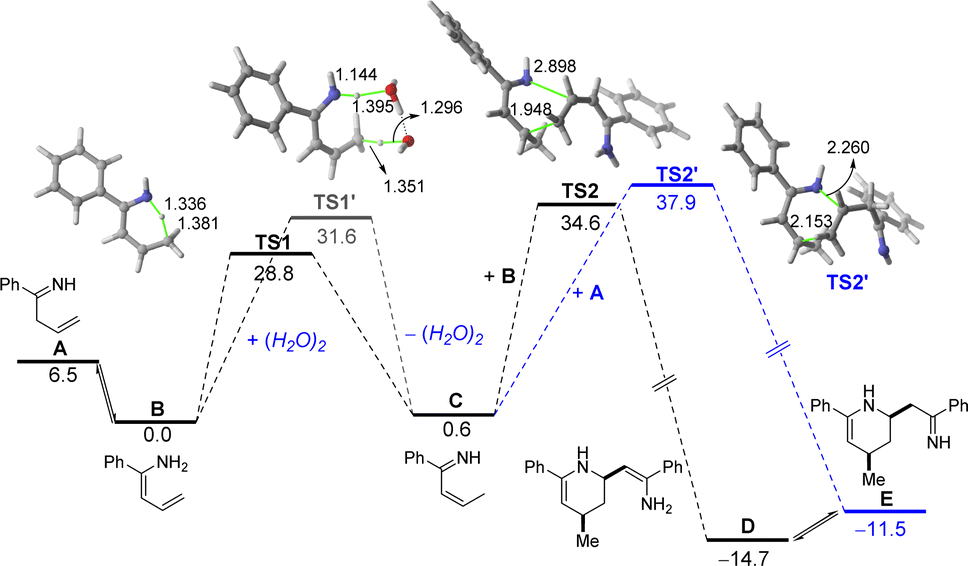 | ||
| Fig. 2 Computed reaction profile for the transformation of imine A into the two possible tautomeric tetrahydropyridines D and E. Relative free energies (ΔG, at 298 K) and bond distances are given in kcal mol−1 and angstroms, respectively. All data have been computed at the PCM(water)/B3LYP-D3/def2-TZVPP//PCM(water)/B3LYP-D3/def2-SVP level. | ||
Moreover, in our experimental protocol, the saturated solutions of ammonium chloride used in the hydrolysis step present a pH range of 5–6. Thus, we decided to extend our DFT studies to this more realistic situation starting with the protonated enamine B–H (Fig. 3). Our calculations indicate that the corresponding aza-Diels–Alder cycloaddition involving the protonated α,β-unsaturated imine C–H proceeds similarly to that involving its neutral counterpart C. Indeed, no substantial differences in the computed barrier (ΔG≠ = 34.9 kcal mol−1) or reaction energy (ΔGR = −13.6 kcal mol−1) were found.
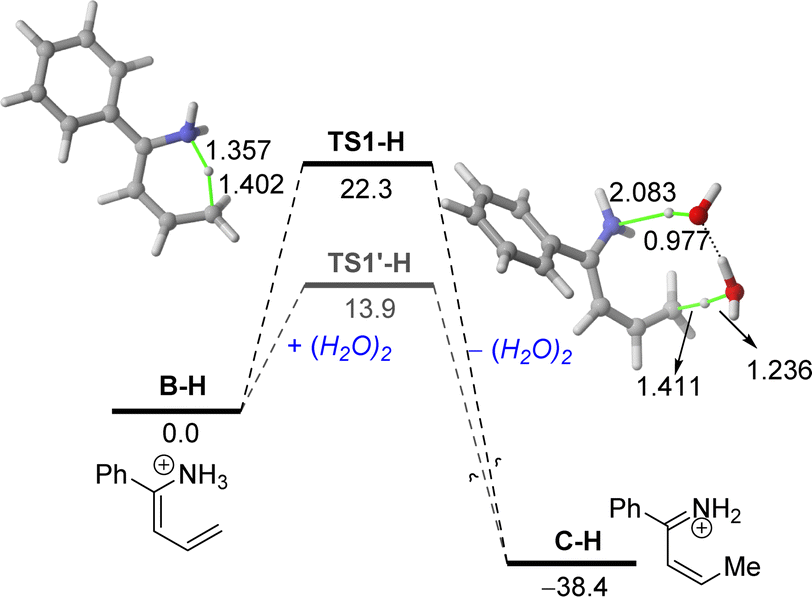 | ||
| Fig. 3 Computed reaction profile for the initial 1,5-H migration from the protonated enamine B–H. Relative free energies (ΔG, at 298 K) and bond distances are given in kcal mol−1 and angstroms, respectively. All data have been computed at the PCM(water)/B3LYP-D3/def2-TZVPP//PCM(water)/B3LYP-D3/def2-SVP level. | ||
However, data in Fig. 3 clearly show that protonation significantly favours the initial 1,5-H migration in the direct pathway and particularly, in the water-assisted reaction. In both cases, the computed barrier is much lower (ΔΔG≠ = 6.5 kcal mol−1 and 17.7 kcal mol−1, for the direct and water-assisted processes, respectively) and the reaction becomes clearly much more exergonic (ΔGR = −38.4 kcal mol−1), which illustrates the beneficial role of protonation in the initial 1,5-H migration reaction. The markedly lower barriers computed in the water-assisted reaction can be mainly ascribed to the easy abstraction of the proton attached to the nitrogen atom by water, as confirmed by the long N⋯H distance in TS1′-H of 2.083 Å. This computational finding also highlights the importance of the presence of water in the reaction media, thus explaining why the quantitative formation of tetrahydropyridine 2a cannot be replicated in traditional and dry organic solvents.
Bearing all these results in mind, we could establish the best conditions for the synthesis of tetrahydropyridine-type compounds as follows: (i) addition of one equivalent of the allyl Grignard reagent to the corresponding nitrile; (ii) under neat conditions; (iii) at room temperature; (iv) in air; and (v) hydrolysis after 3 seconds of reaction (which involves first the addition of 2 mL of a saturated solution of ammonium chloride and secondly heating at 100 °C for 20 min).
Synthesis of tetrahydropyridines 2a–j through addition of allyl magnesium bromide to aromatic/aliphatic nitriles in the presence of air/moisture, at room temperature, and without any additional organic solvent: acidic hydrolysis (NH4Cl)
Once the best conditions were set up, we decided to expand the scope of this transformation to a family of both aromatic and aliphatic nitriles 1a–j (Scheme 4). Thus, by following this procedure, benzonitrile (1a) gave rise to the corresponding tetrahydropyridine 2a in almost quantitative yield (98%). Similar reaction yields were found in the presence of electron-donating groups such as Me (2b) and MeO (2c) in the para-position (97% and 98%, respectively). On the other hand, when the aromatic ring is substituted in the para-position by an electron-withdrawing group such as Br (2d) the yield decreases to 68%, although this can be related to the low solubility of the starting nitrile in the ethereal solvent which is present in the commercially available solution of the allyl Grignard rather than to the electronic effect of the substituent. In good agreement with this experimentally observed solubility effect, we found that the presence of a different electron-withdrawing group (CF3) in the para-position of the starting nitrile 1e is well-tolerated by our system, and almost quantitative formation of CF3-containing tetrahydropyridine 2e (93%) was observed. Similarly, our protocol is also compatible with naphthyl-substituted nitriles (such as 1f) affording the corresponding tetrahydropyridine 2f in a remarkable 88% yield. Regarding steric effects, we should mention that the presence of substituents in the ortho-position of the aromatic ring of the nitrile (e.g., 2-tolylnitrile) completely blocks the reaction, thus recovering the unreacted starting nitrile. Conversely, our system is capable of converting, in almost quantitative yields (89–93%), the corresponding para-substituted aromatic nitriles 1g–h into the desired tetrahydropyridines 2g–h. Finally, and trying to find the limits of our procedure, we decided to focus our attention on the most challenging aliphatic-based nitriles 1i–j.16 Satisfactorily, and for the first time in this chemistry, we were able to obtain the corresponding aliphatic-based tetrahydropyridines 2i–j in quantitative (2i; 99%) to moderate yields (2j, 68%). Finally, we would like to mention that all the obtained tetrahydropyridines (2a–j) presented the expected syn-stereoisomeric conformation, as corroborated by the corresponding NOE NMR experiments.10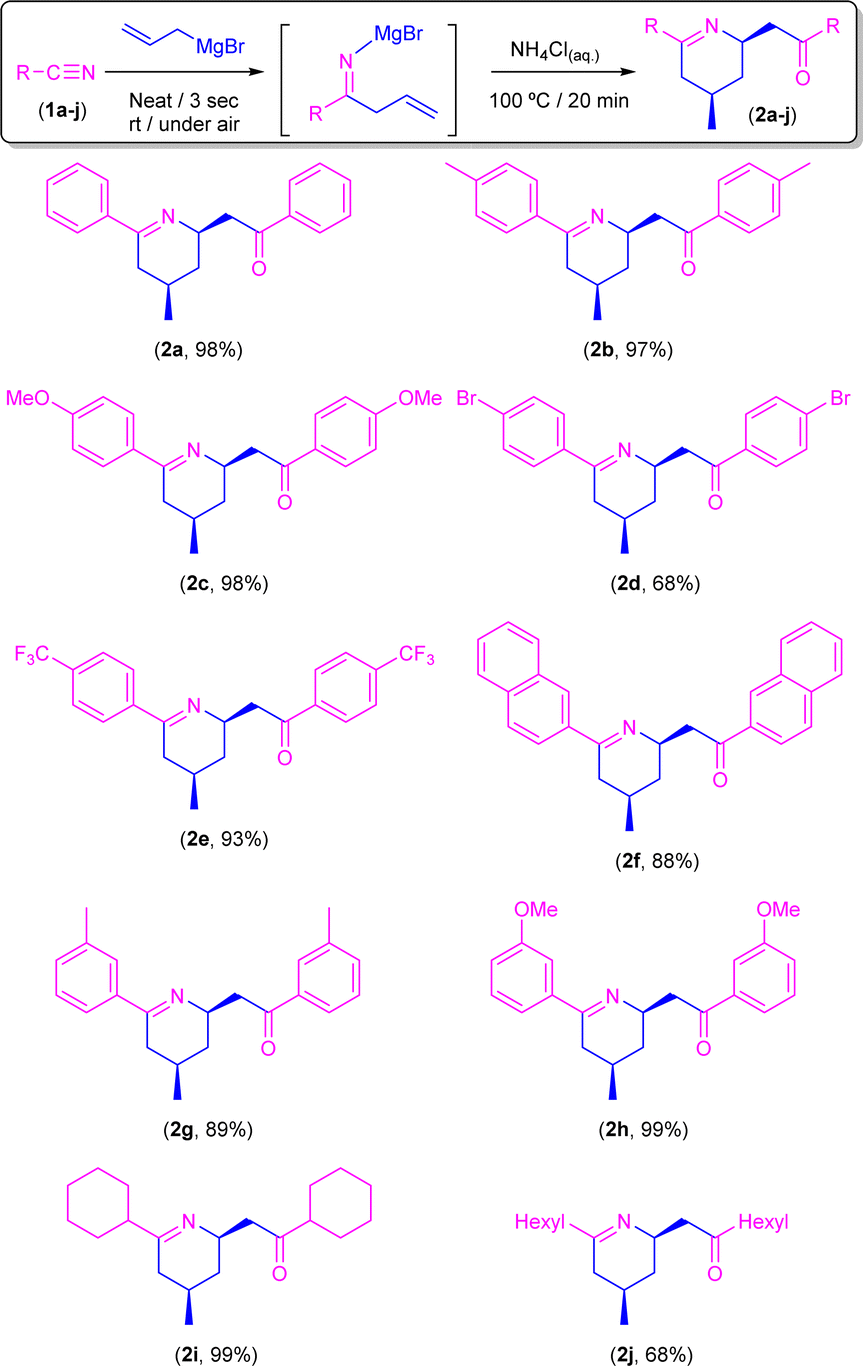 | ||
| Scheme 4 Synthesis of tetrahydropyridines 2a–j through addition of allyl magnesium bromide (0.5 mmol) to different aromatic/aliphatic nitriles (R–CN, 1a–j; 0.5 mmol) in the presence of air/moisture, at room temperature, without any additional organic solvent and with vigorous stirring. After 3 seconds of the addition, the concomitant hydrolysis was promoted by adding 2 mL of a saturated aqueous solution of NH4Cl and heating the obtained reaction crude up to 100 °C for 20 minutes. Yields of the reaction crudes were determined by 1H-NMR using 1,3,5-trimethoxybenzene as an internal standard (0.5 mmol). | ||
Synthesis of β-aminoketones 5a–c,g,i,j or enamines 3d–f through addition of allyl magnesium bromide to nitriles in the presence of air/moisture, at room temperature and without any additional organic solvent: basic hydrolysis (aqueous NH3)
Keeping in mind the astonishing selectivity and efficiency shown by our system in the synthesis of a variety of tetrahydropyridines 2a–j and the previous results reported in the field,8–10 we decided to revisit the hydrolysis step again but changing the proton concentration of the aqueous media employed, thus trying to find a multi-task synthetic procedure just by simple pH-switching. In this sense, it was previously reported that the controlled hydrolysis of the addition reaction under study but using saturated ammonia solution (instead of aqueous NH4Cl) gave a mixture of the two possible enamine isomers (Z/E).9 Thus, we decided to investigate how basic hydrolysis affects the reaction finding that our experimental results totally agreed with the findings previously reported by Erker.9 When compound I was hydrolysed with a saturated solution of ammonia, the two isomers of 3a were obtained as unique products. However, once the mixture of Z/E-3a compounds was isolated and no basic media was present anymore, it was completely transformed into 2a after a week. Remarkably, the implementation of our hydrolytic methodology (heating up the aqueous medium at 100 °C for 20 min) allows the selective and quantitative conversion of the mixture of enamines Z/E-3a into the corresponding β-aminoketone 5a (Scheme 5). This surprising transformation has been studied previously by Tezer and Ozkan17 where the proposed mechanism involves an initial 1,5-sigmatropic H-shift, followed by the hydrolysis of the imine group to form an α,β-unsaturated ketone. This species then undergoes a Michael addition with ammonia as a nucleophile leading to the observed β-aminoketone 5a.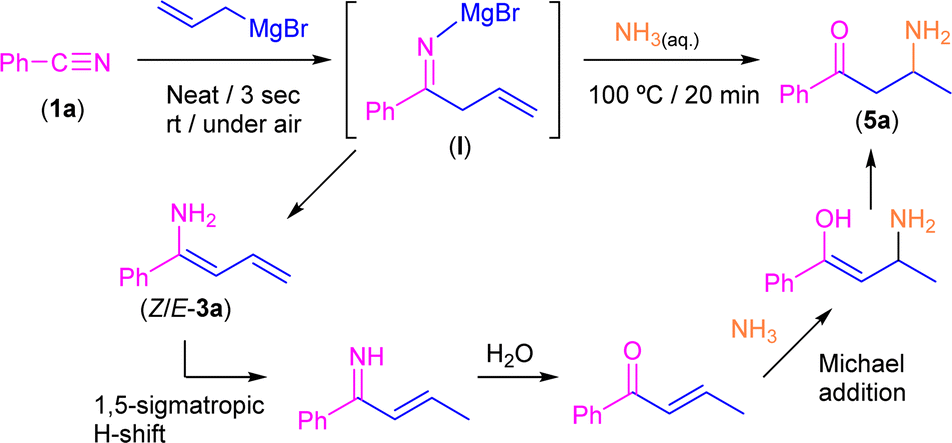 | ||
| Scheme 5 Synthesis of β-aminoketone 5a through addition of allyl magnesium bromide to benzonitrile (1a) in the presence of air/moisture, at room temperature and without any additional organic solvent followed by concomitant hydrolysis with an aqueous NH3 solution. | ||
Taking into account the pivotal role of β-aminoketones in organic synthesis, as they are considered one of the most important scaffolds in the preparation of natural products, drugs and bioactive molecules,18 and once we have set the best reaction conditions that allow the selective conversion of benzonitrile (1a) into β-aminoketone 5a (Scheme 5), we finally decided to focus our attention on the study of the role of the nature of the starting nitriles in this synthetic protocol (Scheme 6). In this case, we found that the electronic properties of the substituents present in the starting aromatic nitriles under study indeed played a pivotal role. Thus, we observed that when the selected aromatic nitriles contain electron-donating groups in either ortho- (1b–c) or para-positions (1g),19 the corresponding β-aminoketones 5b,c,g were obtained with good quantitate yields (Scheme 6). In the same line, the challenging aliphatic nitriles 1i–j follow the same reaction pathway producing the corresponding β-aminoketones 5i–j in moderate to quantitative yields (59–97%).
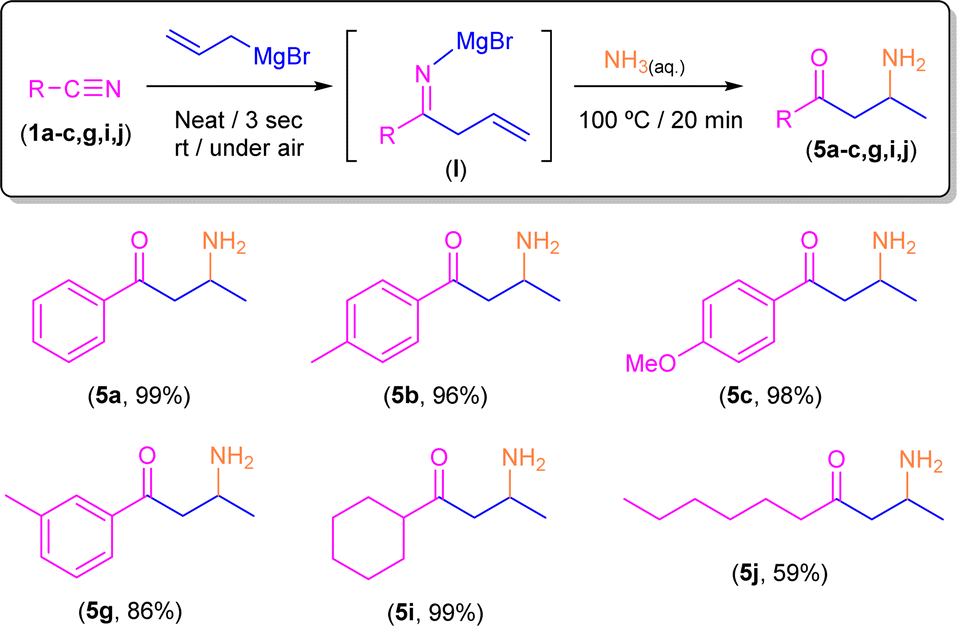 | ||
| Scheme 6 Synthesis of β-aminoketones 5a–c,g,i,j through addition of allyl magnesium bromide to nitriles 1a–c,g,i,j in the presence of air/moisture, at room temperature and without any additional organic solvent followed by concomitant hydrolysis with an aqueous NH3 solution. | ||
Conversely, when aromatic nitriles containing electron-withdrawing (Br, 1d; CF31e) or naphthyl (1f) groups were employed under the same reaction conditions, selective formation of enamines 3d–f in very good Z/E ratios was observed (Scheme 7). In combination, both experimental sets of observations suggest that the corresponding 1,5-sigmatropic H-shift, required for the formation of the corresponding unsaturated imine intermediate (Scheme 5), is particularly favoured when electron-rich aromatic nitriles are employed. In line with this, we computationally found that the 1,5-sigmatropic H-shift involving species B equipped with the strong electronic donor p-C6H4–NH2 substituent proceeds with a lower barrier (ΔΔG≠ = 2.1 kcal mol−1) than the analogous reaction involving the electron-withdrawing p-C6H4–NO2 group. Despite this, reasons for the lack of reactivity observed in substrates 1d–f remain unclear at the moment. At this point, we would like to highlight the different nature of the highly substituted final products that we are able to obtain, ranging from tetrahydropyridines (Scheme 4) or β-aminoketones (Scheme 6) to enamines (Scheme 7) just by fine-tuning the reaction conditions and the electronic nature of the starting nitrile.
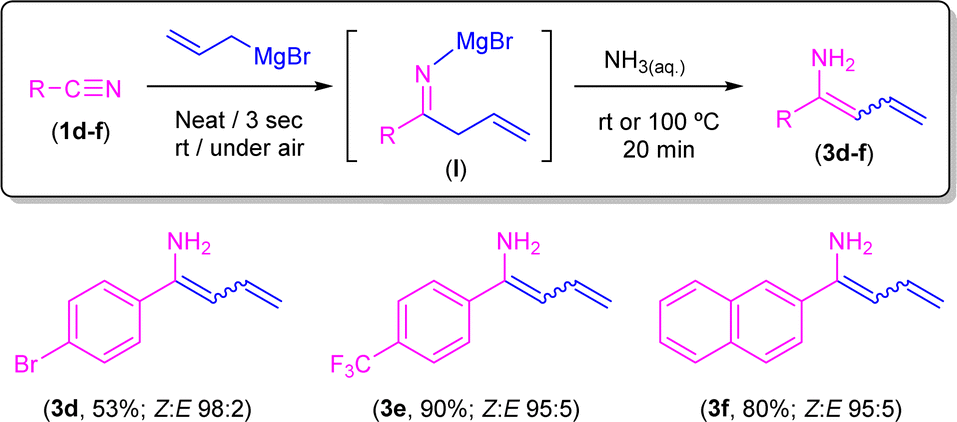 | ||
| Scheme 7 Synthesis of enamines 3d–f through addition of allyl magnesium bromide to nitriles 1d–f in the presence of air/moisture, at room temperature and without any additional organic solvent followed by concomitant hydrolysis with an aqueous NH3 solution. | ||
Conclusions
In summary, in this work we have demonstrated that the use of bench-type reaction conditions (room temperature and the presence of air/moisture) in the addition of a highly polar allyl Grignard reagent to nitriles under neat conditions, together with fine-tuning of the chemical (acidic or basic conditions) and physical (heating at 100 °C) characteristics of the hydrolysis protocol are key factors in the design of a multi-task synthetic protocol capable of selectively producing three different organic scaffolds, namely highly substituted tetrahydropyridines, aminoketones and enamines. Importantly, we have been able to implement the use of both aromatic or aliphatic nitriles in our synthetic protocols, these aliphatic nitriles being traditionally incapable of undergoing addition reactions with polar organometallic reagents (RLi/RMgX) under non-conventional conditions. Moreover, and for the first time in this chemistry, both experimental and computational studies corroborate the benefits associated with the employment of non-conventional bench-type reaction conditions (room temperature in the presence of air/moisture) in the chemistry of polar organometallic reagents, which cannot be replicated when using traditional Schlenk-type synthetic techniques.20 Thus, our synthetic protocol establishes a new route to obtain “a la carte products” by means of a simple and unique reaction.Experimental
General methods and materials
Allylmagnesium bromide (1 M in diethyl ether) was purchased from Sigma Aldrich and its concentration was established by titrating against iodine.21 Infrared spectra were recorded on a Bruker Tensor 27 spectrometer, using an ATR accessory. HRMS were measured in ESI mode, with a TOF mass analyser (Bruker model Impact II). NMR spectra were recorded on a Bruker Avance Neo 400 spectrometer operating at 400.13 MHz for 1H, 100.62 MHz for 13C and 376 MHz for 19F. All 13C and 19F spectra were proton decoupled. 1H and 13C NMR spectra were referenced against an appropriate solvent signal. 19F NMR spectra were referenced against CFCl3. Characterisation details, including 1H, 19F, 13C{1H} NMR spectra and HRMS data, for compounds 2a–j, 3a,d,e,f, 4a and 5a–c,g,i,j are included in the ESI.‡General procedure for the synthesis of tetrahydropyridines 2a–j, in the presence of air and without additional solvent
Synthesis was performed in air and at room temperature. A glass tube was charged with the appropriate nitrile (1a–j, 0.5 mmol) and allylmagnesium bromide (0.5 mmol) was added with vigorous stirring. After 3 seconds of stirring, the reaction was quenched with 2 mL of a saturated solution of NH4Cl and then heated to 100 °C for 20 minutes. After reaching room temperature, 5 mL of distilled water was added, and the mixture was extracted with 2-MeTHF (3 × 5 mL). The combined organic phases were dried over anhydrous MgSO4 and the solvent was concentrated in vacuo. Yields of the reaction crudes were determined by 1H-NMR using 1,3,5-trimethoxybenzene as an internal standard (0.5 mmol). Separation and purification of every compound were carried out using TLC glass plate silica (employing hexane:ethyl acetate mixtures). All reactions were performed in triplicate to ensure good reproducibility of the obtained yields.Procedure for the synthesis of enamines Z/E-3a,d,e,f, in the presence of air and without additional solvent
Synthesis was performed in air and at room temperature. A glass tube was charged with the desired nitrile (3a,d,e,f, 0.5 mmol) and allylmagnesium bromide (0.5 mmol) was added with vigorous stirring. After 3 seconds of stirring, the reaction was quenched with 2 mL of a saturated aqueous solution of NH3 (hydrolysis can be maintained at 100 °C for 20 minutes, without any noticeable change for enamines 3d–f). The reaction mixture was extracted with 2-MeTHF (3 × 5 mL). The combined organic phases were dried over anhydrous MgSO4 and the solvent was concentrated in vacuo. Yield of the reaction crude was determined by 1H-NMR using 1,3,5-trimethoxybenzene as an internal standard (0.5 mmol). Separation and purification of mixtures of Z/E-3a,d,e,f were carried out using TLC glass plate silica (employing hexane:ethyl acetate mixtures). The reaction was performed in triplicate to ensure good reproducibility of the obtained yield.
Procedure for the synthesis of 4a, in the presence of air and without additional solvent
Synthesis was performed in air and at room temperature. A glass tube was charged with benzonitrile (1a, 0.5 mmol) and allylmagnesium bromide (1 mmol) was added with vigorous stirring. After 5 seconds of stirring, the reaction was quenched with 2 mL of a saturated solution of NH4Cl and then 5 mL of distilled water was added, and the reaction mixture was extracted with 2-MeTHF (3 × 5 mL). The combined organic phases were dried over anhydrous MgSO4 and the solvent was concentrated in vacuo. The yield of the reaction crude was determined by 1H-NMR using 1,3,5-trimethoxybenzene as an internal standard (0.5 mmol). Separation and purification of 4a were carried out using TLC glass plate silica (employing hexane:ethyl acetate mixtures). The reaction was performed in triplicate to ensure good reproducibility of the obtained yield.
Procedure for the synthesis of 5a–c,g,i,j, in the presence of air and without additional solvent
Synthesis was performed in air and at room temperature. A glass tube was charged with the desired nitrile (1a–c,g,i,j, 0.5 mmol) and allylmagnesium bromide (0.5 mmol) was added with vigorous stirring. After 5 seconds of stirring, the reaction was quenched with 2 mL of a saturated solution of NH3 and then heated to 100 °C for 20 minutes. After reaching room temperature, 5 mL of distilled water was added, and the reaction was extracted with 2-MeTHF (3 × 5 mL). The combined organic phases were dried over anhydrous MgSO4 and the solvent was concentrated in vacuo. The yield of the reaction crude was determined by 1H-NMR using 1,3,5-trimethoxybenzene as an internal standard (0.5 mmol). Separation and purification of 5a–c,g,i,j were carried out using TLC glass plate silica (employing hexane:ethyl acetate mixtures). The reaction was performed in triplicate to ensure good reproducibility of the obtained yield.
Procedure for the synthesis of intermediate I
Synthesis was performed under an inert atmosphere and at room temperature inside a glovebox. A glass tube was charged with allylmagnesium bromide (0.5 mmol), and the solvent was removed under vacuum. Then, the white solid was dissolved in deuterated THF and transferred into a young NMR tube, and benzonitrile (1a, 0.5 mmol) was added. The reaction under study was monitored by regular 1H NMR measurements.Computational details
All the calculations reported in this paper were obtained with the Gaussian 09 suite of programs.22 All species were optimised using the B3LYP functional23 in conjunction with the D3 dispersion correction suggested by Grimme et al.24 using the standard double-ζ quality def2-SVP25 basis sets for all atoms. Solvent effects were taken into account during the geometry optimisations using the polarisable continuum model (PCM).26 This level is denoted as PCM(solvent)-B3LYP-D3/def2-SVP. All stationary points were characterised using frequency calculations.27 Reactants and products have positive definite Hessian matrices, whereas transition structures show only one negative eigenvalue in their diagonalised force constant matrices, and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration using the intrinsic reaction coordinate (IRC) method.28 In addition, the vibrational calculation provides the thermal Gibbs energy corrections by using the gas ideal-rigid-rotor-harmonic-oscillator approximation. Energy refinements were carried out by means of single-point calculations at the same DFT level using the much larger triple-ζ basis set def2-TZVPP. This level is denoted as PCM(solvent)/B3LYP-D3/def2-TZVPP//PCM(solvent)/B3LYP-D3/def2-SVP.Data availability
All the experimental and computational data supporting this study are included in the main text and the ESI.‡Author contributions
DE formulated the project. All experiments were performed by BPC and DE. The design, analysis and interpretation of all theoretical studies were performed by IF. FCH, JGA and DE conceptualised the work and secured the funding. JGA wrote the first draft of the article. All authors discussed the results and revised the manuscript. All authors have given approval to the final version of the present manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. G. A. thanks MCIN/AEI/10.13039/501100011033 (project numbers CTQ2016-75986-P, RED2018-102387-T and PID2020-113473GB-I00) for financial support. D. E., F. C.-H., and B. P-C. acknowledge financial support from MCIN/AEI/10.13039/501100011033 (project numbers PID2020-117353GB-I00 and RED2018-102387-T) and the Universidad de Castilla-La Mancha (project number 2022-GRIN-34031). D. E. thanks the Junta de Comunidades de Castilla-La Mancha and EU for financial support through the European Regional Development Fund (ERDF; project SBPLY/19/180501/000137). I. F. thanks the Spanish MCIN/AEI/10.13039/501100011033 (grants PID2019-106184GB-I00 and PID2022-139318NB-I00).Notes and references
- For selected reviews/books dealing with organolithium chemistry, see: (a) Z. Rappoport and I. Marek, The chemistry of organolithium compounds, Wiley, Chichester, 2005 CrossRef; (b) V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204 RSC . For selected reviews/books dealing with organomagnesium chemistry, see:; (c) J. F. Garst and M. P. Soriaga, Chem. Soc. Rev., 2004, 248, 623 CAS; (d) The Chemistry of Organomagnesium Compounds, ed. Z. Rappoport and I. Marek, Patai Series, Wiley, Chichester, 2008 Search PubMed; (e) D. Seyferth, Organometallics, 2009, 28, 1598 CrossRef CAS.
- (a) G. Wu and M. Huang, Chem. Rev., 2006, 106, 2596 CrossRef CAS PubMed; (b) S. K. Nair, B. N. Rocke and S. Sutton, Chapter 11: Lithium, Magnesium, and Copper: Contemporary Applications of Organometallic Chemistry in the Pharmaceutical Industry, in Synthetic Methods in Drug Discovery, 2016, vol. 2, pp. 1–74 Search PubMed; (c) L.-C. Campeau and D. E. Fogg, Organometallics, 2019, 38, 1 CrossRef CAS.
- For reviews in the fields, see: (a) J. García-Álvarez, Eur. J. Inorg. Chem., 2015, 5147 CrossRef; (b) J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org Chem., 2015, 6779 CrossRef; (c) J. García-Álvarez, E. Hevia and V. Capriati, Chem.–An Euro. J., 2018, 24, 14854 CrossRef PubMed; (d) E. Hevia, Chimia, 2020, 74, 681 CrossRef CAS PubMed; (e) T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247 CrossRef CAS PubMed; (f) F. M. Perna, P. Vitale and V. Capriati, Curr. Opin. Green Sustain. Chem., 2021, 30, 100487 CrossRef CAS; (g) S. E. García-Garrido, A. Presa Soto, E. Hevia and J. García-Álvarez, Eur. J. Inorg. Chem., 2021, 3116 CrossRef.
- For a selection of studies in this field of research that have made an impact on multidisciplinary general chemistry, see: (a) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969 CrossRef CAS PubMed; (b) L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192 RSC; (c) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145 CrossRef CAS PubMed; (d) G. Dilauro, M. Dell'Aera, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2017, 56, 10200 CrossRef CAS PubMed; (e) G. Dilauro, A. F. Quivelli, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2019, 58, 1813 CrossRef; (f) F. F. Mulks, L. J. Bole, L. Davin, A. Hernán-Gómez, A. Kennedy, J. García-Alvarez and E. Hevia, Angew. Chem., Int. Ed., 2020, 59, 19021 CrossRef CAS PubMed; (g) M. Fairley, L. J. Bole, F. F. Mulks, L. Main, A. R. Kennedy, C. T. O’Hara, J. García-Álvarez and E. Hevia, Chem. Sci., 2020, 11, 6500 RSC; (h) G. Dilauro, C. S. Azzollini, P. Vitale, A. Salomone, F. M. Perna and V. Capriati, Angew. Chem., Int. Ed., 2021, 60, 10632 CrossRef CAS PubMed; (i) F. F. Mulks, B. Pinho, A. W. J. Platten, M. R. Andalibi, A. J. Expósito, K. J. Edler, E. Hevia and L. Torrente-Murciano, Chem, 2022, 8, 3382 CrossRef CAS; (j) G. Dilauro, C. Luccarelli, A. F. Quivelli, P. Vitale, F. M. Perna and V. Capriati, Angew. Chem., Int. Ed., 2023, 62, e2023047 CrossRef PubMed.
- The use of aerobic and non-dried reaction conditions for RLi/RMgX has also been reported in flow processing. For recent revisions in this field, see: (a) A. Nagaki, Tetrahedron Lett., 2019, 60, 150923 CrossRef CAS; (b) M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814 CrossRef CAS; (c) M. Colella, A. Nagaki and R. Luisi, Chem.–Eur. J., 2020, 26, 19 CrossRef CAS PubMed.
- For reviews/books related to the general use of water as non-conventional solvent in organic synthesis, see: (a) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed; (b) Y. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 8103 CrossRef CAS PubMed; (c) U. M. Lindström, Organic Reactions in Water: Principles, Strategies and Applications, Blackwell, Oxford, 1st edn, 2007 CrossRef; (d) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed; (e) Y. J. Zuo and J. Qu, J. Org. Chem., 2014, 79, 6832 Search PubMed.
- (a) M. J. Rodríguez-Álvarez, J. García-Álvarez, M. Uzelac, M. Fairley, C. T. O'Hara and E. Hevia, Chem.–Eur. J., 2018, 24, 1720 CrossRef PubMed; (b) D. Elorriaga, B. Parra-Cadenas, A. Antiñolo, F. Carrillo-Hermosilla and J. García-Álvarez, Green Chem., 2022, 24, 800 RSC; (c) D. Elorriaga, B. Parra-Cadenas, A. Antiñolo, F. Carrillo-Hermosilla and J. García-Álvarez, ChemSusChem, 2022, 15, e202201348 CrossRef CAS PubMed.
- (a) M. Chastrette and G. P. Axiotis, Synthesis, 1980, 889 CrossRef CAS; (b) A. B. Charette, A. Gagnon, M. Janes and C. Mellon, Tetrahedron Lett., 1998, 39, 5147 CrossRef CAS.
- G. Erker, M. Riedel, S. Koch, T. Jödicke and E.-U. Würthwein, J. Org. Chem., 1995, 60, 5284 CrossRef CAS.
- M. A. Grassberger, A. Horvath and G. Schulz, Tetrahedron Lett., 1991, 32, 7393 CrossRef CAS.
- Tetrahydropyridines and piperidines are well-established structural motifs with relevant applications in pharmacology and production of natural products: (a) G. M. Strunz and J. A. Findlay, in The Alkaloids: Chemistry and Pharmacology, ed. A. Brossi, Academic Press, New York, 1985, vol. 26 Search PubMed; (b) P. D. Bailey, P. A. Millwood and P. D. Smith, Chem. Commun., 1998, 633 RSC; (c) Y. Nishimura, T. Satoh, H. Adachi, S. Kondo, T. Takeuchi, M. Azetaka, H. Fukuyasu and Y. Iizuka, J. Med. Chem., 1997, 40, 2626 CrossRef CAS PubMed; (d) N. Zitzmann, A. S. Mehta, S. Carrouee, T. D. Butters, F. M. Platt, J. McCauley, B. S. Blumberg, R. A. Dwek and T. M. Block, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11878 CrossRef CAS PubMed.
- Aza-Diels-Alder reaction (aza-DA) is one of the most powerful tools for the formation of N-containing heterocycles: (a) R. T. Bailey, R. S. Garigipati, J. A. Morton and S. M. Weinreb, J. Am. Chem. Soc., 1984, 106, 3240 CrossRef; (b) R. Lock and H. Waldmann, Liebigs Ann., 1994, 511 CrossRef CAS; (c) A. B. Holmes, A. Kee, T. Ladduwahetty and D. F. Smith, J. Chem. Soc., Chem. Commun., 1990, 1412 RSC.
- In contrast to the standard all-carbon Diels-Alder reaction, the aza-DA reaction has been studied far less. The main feature of this reaction is the presence of a nitrogen atom, usually as an imine group acting as a part of either the dienophile or diene, leading to different N-heterocycles and, usually requiring the presence of a Lewis acid (e.g., ZnCl2, BF3, TiCl4, etc.) to take place: P. Buonora, J.-C. Olsen and T. Oh, Tetrahedron, 2001, 57, 6099 CrossRef CAS.
- M. C. Lin and K. J. Laider, Can. J. Chem., 1968, 46, 973 CrossRef CAS.
- Formation of carbinamines by double addition of RLi/RMgX reagents to nitriles in water was previously reported by Capriati and co-workers (see ref. 4d).
- Aliphatic nitriles have been previously reported as the most recalcitrant reagents to undergo the addition of highly polarised organometallic reagents under bench-type reaction conditions (see refs. 4d and 7).
- N. Tezer and R. Ozkan, J. Mol. Struct.: THEOCHEM, 2001, 564, 79 CrossRef.
- For recent reviews in this field, see: (a) Y. Du, Q. Li, B. Xiong, D. Zhang and M. Wang, Bioorg. Med. Chem., 2010, 18, 4255 CrossRef CAS PubMed; (b) M. Altmeyer, E. Amtmann, C. Heyl, A. J. Scheidig and C. D. Klein, Bioorg. Med. Chem., 2014, 24, 5310 CrossRef CAS PubMed; (c) N. H. Nguyen, A. B. Hughes and B. E. Sleebs, Curr. Org. Chem., 2014, 18, 260 CrossRef CAS; (d) A. Ravn, M. Vilstrup, P. Noerby, K. Daasbjerg and T. Daasbjerg, J. Am. Chem. Soc., 2019, 141, 11821 CrossRef CAS PubMed; (e) M. M. Hammouda and K. M. Elattar, RSC Adv., 2022, 12, 24681 RSC.
- As previously observed in the synthesis of tetrahydropyridines 2a–j (Scheme 4), the presence of ortho-substituents in the starting aromatic nitrile (e.g., 2-tolylnitrile) completely blocks the synthetic procedure.
- For previous examples on computational and experimental studies involving organolithium reagents and aqueous environments (i.e., cooperation between n-Buli and water or coordination of water to the lithium cation within lithium amide mixed aggregates), see: (a) V. Juste-Navarro, I. Delso, T. Tejero and P. Merino, Chem.–Eur. J., 2016, 22, 11527 CrossRef CAS PubMed; (b) Y. Gimbert, D. Lesage, C. Fressigné and J. Maddaluno, J. Org. Chem., 2017, 82, 8141 CrossRef CAS PubMed.
- A. Krasovskiy and P. Knochel, Synthesis, 2006, 5, 890 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. J. Becke, Chem. Phys., 1993, 98, 5648 CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1998, 37, 785 CrossRef PubMed; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. J. Krieg, Chem. Phys., 2010, 132, 154104 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef; (b) J. L. Pascual-Ahuir, E. Silla and I. Tuñón, J. Comput. Chem., 1994, 15, 1127 CrossRef CAS; (c) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- J. W. McIver and A. K. Komornicki, J. Am. Chem. Soc., 1972, 94, 2625 CrossRef CAS.
- C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef.
Footnotes |
| † Dedicated to Professor Jose (Pepe) Gimeno on the occasion of his 75th birthday, a visionary organometallic chemist and an inspiring mentor of Joaquín García-Álvarez. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details, NMR spectra, Cartesian coordinates (in Å) and free energies (in a.u.) of all the stationary points discussed in the text. See DOI: https://doi.org/10.1039/d3sc06403d |
| This journal is © The Royal Society of Chemistry 2024 |