
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransformations of carbohydrate derivatives enabled by photocatalysis and visible light photochemistry
Daniel J.
Gorelik†
,
Shrey P.
Desai†
,
Sofia
Jdanova†
,
Julia A.
Turner†
and
Mark S.
Taylor
 *
*
Department of Chemistry, University of Toronto, 80 St. George St., Toronto, ON M5S 3H6, Canada. E-mail: mtaylor@chem.utoronto.ca
First published on 2nd January 2024
Abstract
This review article highlights the diverse ways in which recent developments in the areas of photocatalysis and visible light photochemistry are impacting synthetic carbohydrate chemistry. The major topics covered are photocatalytic glycosylations, generation of radicals at the anomeric position, transformations involving radical formation at non-anomeric positions, additions to glycals, processes initiated by photocatalytic hydrogen atom transfer from sugars, and functional group interconversions at OH and SH groups. Factors influencing stereo- and site-selectivity in these processes, along with mechanistic aspects, are discussed.
 Daniel J. Gorelik | Daniel Gorelik received his BSc and PhD degrees from the University of Toronto. His doctoral research with Prof. Mark S. Taylor was focused on the development of methods for site-selective functionalization of carbohydrates. He is a postdoctoral researcher at the Massachusetts Institute of Technology, Department of Chemistry in the group of Prof. Alison Wendlandt. |
 Shrey P. Desai | Shrey P. Desai received a BSc in Biochemistry from York University where he conducted his Honours Thesis work with Professor Arturo Orellana on pyridine functionalization. He started his PhD studies with Professor Mark S. Taylor in 2019 at the University of Toronto. His doctoral research is focused on applications of organoboron catalysis and visible light photocatalysis for a variety of transformations, including epoxide ring-openings, conjugate additions, glycosylations, and cycloadditions. |
 Sofia Jdanova | Sofia Jdanova received her BSc from the University of Toronto in 2019. She is currently pursuing her PhD at the University of Toronto under the supervision of Prof. Mark Taylor. Her research focuses on the development and mechanistic study of transition metal-catalyzed, site-selective functionalizations of carbohydrate derivatives. |
 Julia A. Turner | Julia A. Turner received a BSc in Biochemistry and Chemistry from Western University in 2019, where she completed a thesis with Prof. Robert H. E. Hudson synthesizing fluorescent nucleobase analogs. She started her PhD at the University of Toronto in 2019 under the supervision of Prof. Mark S. Taylor. Her work is focused on methodology development and computational study of site-selective radical formation on carbohydrates via photoredox and hydrogen atom transfer co-catalysis. |
 Mark S. Taylor | Mark S. Taylor earned his BSc degree from the University of Toronto in 2000, followed by a PhD in Chemistry at Harvard University in 2005, where he conducted research on enantioselective catalysis under the guidance of Prof. Eric Jacobsen. After postdoctoral research with Prof. Timothy Swager at MIT, he returned to the University of Toronto in 2007 to take a faculty position in the Department of Chemistry. Prof. Taylor has mentored more than 45 graduate students conducting research in the areas of catalysis, carbohydrate chemistry and physical organic chemistry. |
1. Introduction
Carbohydrates have their origins in photochemistry: the fixation of CO2 by photoautotrophs drives life on our planet and yields sugars as the most abundant renewable chemical feedstocks. Making use of energy from light to enable transformations that are kinetically and/or thermodynamically unfavorable offers significant potential to advance the synthetic chemistry of carbohydrates; indeed, photochemical transformations of sugars have been explored for decades.1 However, because carbohydrates typically do not absorb light in the visible region, such methods required either the use of high-energy ultraviolet (UV) light or the covalent installation of photoactive groups.Photoredox catalysis provides a general approach for harnessing the energy of incident photons for transformations of substrates that do not possess a chromophore suitable for direct excitation.2 The development and applications of this transformative synthetic technology have been detailed in review articles.3 In brief, absorption of light by a photocatalyst results in an excited state that is capable of acting as either an oxidant or a reductant by participating in a single electron transfer (SET) event with a suitable substrate. Intermediates generated by the redox process (usually radical species) can be exploited in diverse ways to enable new reactivity. A subsequent SET step with an intermediate or reagent takes place to close the photoredox cycle. An alternative mode of activation is energy transfer photocatalysis, wherein an interaction between the substrate and the excited-state photocatalyst results in indirect excitation of the former.4 For the transformations of carbohydrate derivatives described in this review, it is the photoredox catalysis manifold, rather than energy transfer photocatalysis, that generally predominates.
Photocatalysts employed in the transformations described in this review are depicted in Fig. 1, along with the available excited-state redox potentials,5–8 which often serve as important reference data for researchers in the area. For example, catalysts with high excited-state reduction potentials 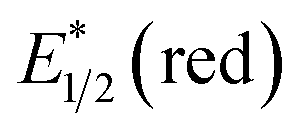 (e.g., Ir-2, Acr-1+, Acr-2+) serve as potent photooxidants, while those with high negative excited-state oxidation potentials
(e.g., Ir-2, Acr-1+, Acr-2+) serve as potent photooxidants, while those with high negative excited-state oxidation potentials 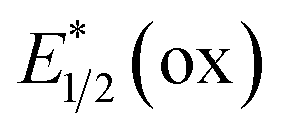 (e.g., fac-Ir(ppy)3) are strong photoreductants.
(e.g., fac-Ir(ppy)3) are strong photoreductants.
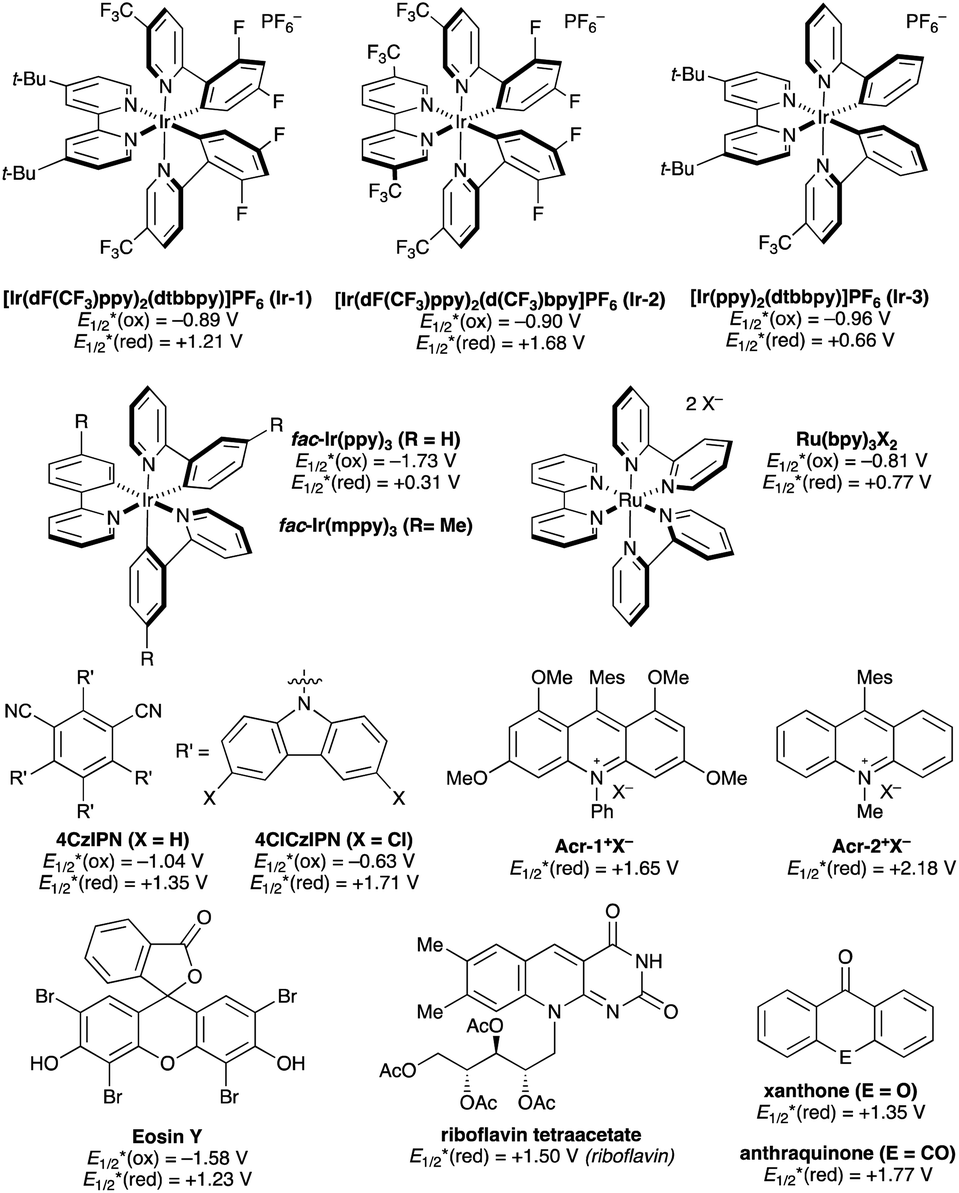 | ||
| Fig. 1 Representative photocatalysts. Reported excited state redox potentials (versus the standard calomel electrode, SCE) are listed. | ||
Advances in visible light photocatalysis have sparked interest in transformations that proceed via direct excitation of substrate-derived species (e.g., electron donor–acceptor (EDA) or charge-transfer complexes9) or transition metal complexes10 by visible light, in the absence of a photosensitizer; applications of such processes in carbohydrate chemistry will also be discussed.
The influence of visible light photocatalysis on the progress of the organic synthesis field has been profound, and carbohydrate chemistry is no exception in this regard. Photocatalysis has been implemented to provide innovative solutions to classic problems in carbohydrate chemistry (e.g., glycosylation, protection of hydroxyl groups), and to enable new types of reactivity for sugar derivatives. Here, we aim to provide an overview of the applications of visible light photocatalysis and photochemistry to carbohydrate-derived substrates. The major topics covered are photocatalytic glycosylations, generation of radicals at the anomeric position, transformations involving radical formation at non-anomeric positions, additions to glycals, processes initiated by photocatalytic hydrogen atom transfer from sugars, and functional group interconversions at OH and SH groups. We anticipate that this relatively broad coverage will provide a distinct type of perspective from more specialized reviews discussing photochemical glycosylations,11 the chemistry of anomeric radicals,12 biomimetic radical reactions of sugars13 and photocatalytic C–H and C–C bond activation of carbohydrate derivatives.14 While a representative carbohydrate derivative may be included among a panel of structurally diverse substrates to explore the scope and limitations of a method, the focus here is on studies that have examined the reactivity of carbohydrate-derived substrates in depth.
2. Activation of glycosyl donors
Glycosylation, the formation of a covalent linkage to the anomeric position of a sugar by nucleophilic substitution, is the key step in the synthesis of oligosaccharides, steroidal glycosides, glycoconjugates and carbohydrate analogs. Synthetic methodology development for glycosylation centers around the identification of leaving groups that can be displaced from the anomeric carbon under appropriate activation conditions. Considerations include stereoselectivity (α- versus β-glycoside formation), ease of preparation and handling of the glycosyl donor (the precursor to the anomeric electrophile), and tuning of its reactivity to enable sequential or one-pot glycosylations (e.g., through ‘arming’ or ‘disarming’ via protective group variation).15Photoredox catalysis and visible light photochemistry have been employed to generate reactive electrophiles through oxidation of a functional group at the anomeric position of a glycosyl donor. Because this topic has been reviewed in depth elsewhere,11 representative examples have been selected to illustrate the approach. Photoacid generation, which takes advantage of the activity of Brønsted acids as catalysts for glycosyl donor activation,16 is also described in the above-mentioned reviews and will not be covered here.
Reactions of water or alcohols with aryl glycosides17 or thioglycosides18 under UV irradiation in the presence of dicyanoarenes (1,4-dicyanobenzene with phenanthroline as photosensitizer, or 1,4-dicyanonaphthalene), were reported more than three decades ago. The proposed mechanisms — photoinduced electron transfer from the aromatic aglycon to generate a radical cation, nucleophilic displacement of a (thio)phenoxy radical, and reduction of the latter by the dicyanoarene radical anion — incorporate elements of what would now be termed a photoredox catalysis process. These studies established a conceptual foundation for activation of glycosyl donors through light-promoted oxidation of an electron-rich anomeric substituent, but were not adopted as preparative methods, likely due to the requirement for UV irradiation and an excess of the glycosyl acceptor (generally water or methanol). Analogous reactivity of aryl selenoglycosides upon UV irradiation was reported by the groups of Furuta and Iwamura,19 and Crich (Scheme 1a),20 using pyrylium and quinolinium photocatalysts, respectively. These contributions included examples of disaccharide synthesis using protected glycoside acceptors.
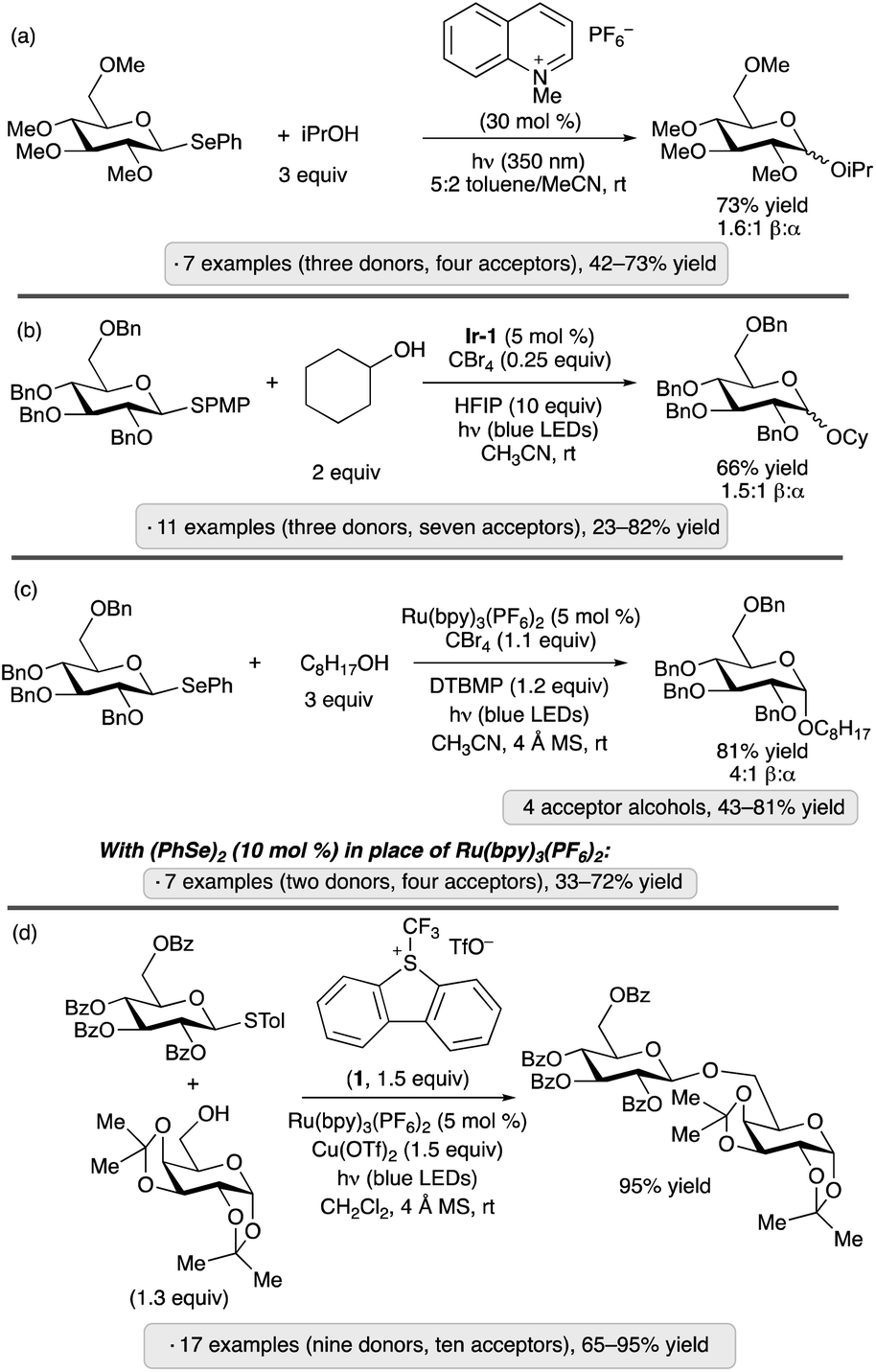 | ||
| Scheme 1 O-Glycosylations via photoredox catalysis reported by: (a) Crich and co-workers;20 (b) Bowers and co-workers;21 Ragains and co-workers;22 (d) Ye and co-workers.23 PMP denotes 4-methoxyphenyl; Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
Applications of modern visible light photocatalysis to glycosylation were reported by the groups of Bowers and Ragains in 2013. Bowers and co-workers found that upon irradiation with a blue light-emitting diode (LED) in the presence of photocatalyst Ir-1, substoichiometric BrCX3 (X = Cl or Br) and hexafluoroisopropanol (HFIP, 10 equiv.) in acetonitrile, para-methoxyphenyl (PMP) thioglycosides underwent glycosidation reactions with various primary, secondary and tertiary alcohols (Scheme 1b).21 A proposed photocatalytic cycle is depicted in Scheme 2. SET from excited-state Ir-1 to BrCX3 results in a strongly oxidizing Ir(IV) species that accepts an electron from the aryl thioglycoside. Expulsion of thiyl radical forms an oxacarbenium ion or related electrophilic species,24 which reacts with the glycosyl acceptor. Because the released thiyl radical can serve as an oxidative quencher for Ir-1 in subsequent turnovers of the photoredox cycle, a stoichiometric quantity of BrCX3 was not needed. β-Configured PMP thioglycosides derived from benzyl or methyl ether-protected glucopyranosose, as well as a 2,6-dideoxypyranose, were employed as donors. The authors proposed that epimerization under the acidic conditions of the reaction was responsible for the formation of mixtures of anomers.
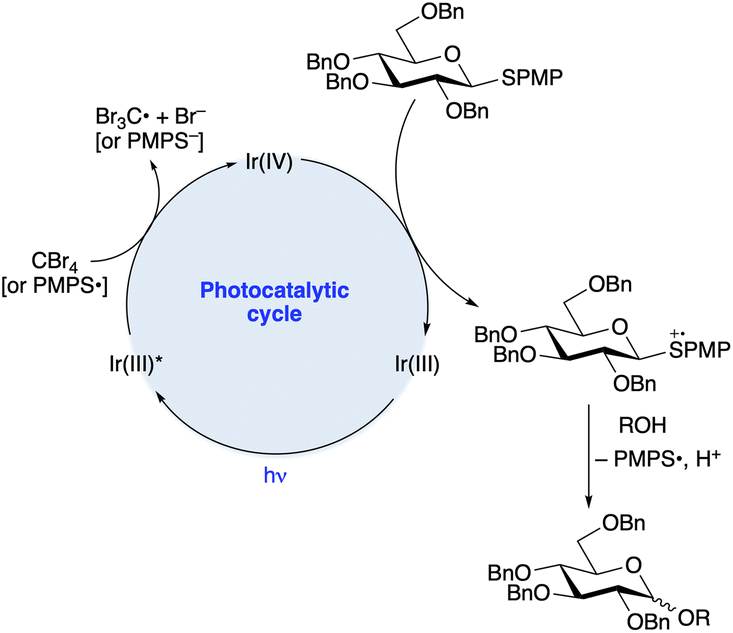 | ||
| Scheme 2 Proposed mechanism for the O-glycosylation shown in Scheme 1b. | ||
The Ragains group's approach to glycosylation via photoredox catalysis employed phenyl selenoglycoside donors, along with Ru(bpy)3(PF6)2 as the photocatalyst, tetrabromomethane and 2,6-di-tert-butyl-4-methylpyridine (DTBMP, Scheme 1c).22 A per-O-benzylated β-glucopyranoside donor was employed, forming α-glycosides as the major products. The mechanism likely involves oxidation of the excited-state photocatalyst by CBr4, followed by SET from the selenoglycoside. Nucleophilic substitution at the anomeric center via loss of the phenylseleno radical results in glycosidic bond formation. A variant of the protocol was developed using (PhSe)2 in place of the Ru(II) photocatalyst (seven examples of gluco or galacto configuration, 33–72% yield).
Ye and co-workers have also explored the combination of a photoredox catalyst, electron-accepting reagent and thioglycoside for O-glycosylation of alcohols.23 The optimized protocol employed catalytic Ru(bpy)3(PF6)2, Umemoto's reagent (1) and copper(II) triflate under visible light irradiation (Scheme 1d). Glycosylations using sialyl donors,25 as well as couplings of aryl glycosides rather than thioglycosides, have been achieved by Ye's group using related protocols.26 In the latter study, syntheses of a variety of O- and N-glycosides were demonstrated, using gluco-, galacto- and manno-configured donors. 1,2-trans-Configured glycosides were obtained selectively with ‘disarmed’ per-O-acylated donors, and tolerance of protective groups that are employed frequently in carbohydrate chemistry (esters, benzyl ethers, acetals and ketals) was demonstrated. The proposed mechanism follows along the general lines of those discussed in the preceding paragraphs – namely, quenching of the excited-state photocatalyst by Umemoto's reagent, followed by oxidation of the anomeric substituent (thioaryl or aryloxy) by the resulting Ru(III) complex, and finally displacement of the thiyl or aryloxy radical to generate the glycoside. Control experiments suggested that the Cu(OTf)2 additive served to suppress a competing reaction of the trifluoromethyl radical with dibenzothiophene.
Ragains and co-workers found that 4-PMP-substituted 3-butenylthioglycosides were activated as glycosyl donors upon irradiation with blue LEDs in the presence of Umemoto's reagent in dichloromethane (Scheme 3).27 Based on spectroscopic evidence and computational modelling, the authors proposed that the thioglycoside and Umemoto's reagent interacted to form an EDA complex capable of being excited by visible light. After SET, cyclization of the radical cation and recombination with trifluoromethyl radical resulted in the formation of a reactive glycosyl donor under conditions that did not cause activation of unfunctionalized thioglycosides. The method was applied to per-O-benzylated glucopyranosyl donors, as well as a tri-O-benzylated derivative having an acetate group at the 2-position. The former resulted in mixtures of anomers (roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α:β), while couplings of the latter were β-selective.
:1 α:β), while couplings of the latter were β-selective.
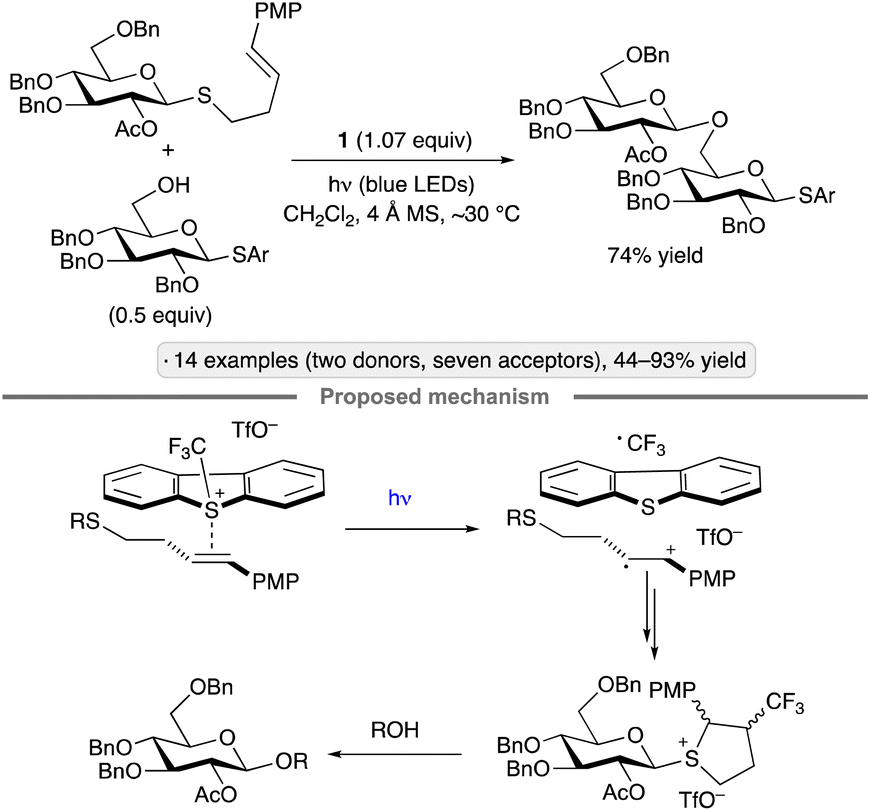 | ||
| Scheme 3 O-Glycosylation via excitation of the EDA complex of a thioglycoside with Umemoto's reagent (1). PMP denotes 4-methoxyphenyl. | ||
Visible light excitation of an EDA complex underlies a protocol for activation of methallyl glycosyl sulfones reported by Niu, Houk and co-workers in 2022.28 The optimized conditions involve irradiation of the glycosyl donor in the presence of 1-iodoperfluorobutane (C4F9I), ammonium hydrogenphosphate and catalytic triphenylphosphine oxide (Scheme 4). The consistent 1,2-cis-stereoselectivity arising from couplings of ‘armed’ per-O-benzylated pyranosyl and furanosyl donors is a noteworthy aspect of this method. The initial stages of the proposed mechanism are reminiscent of that proposed by Ragains and co-workers for the transformation shown in Scheme 3,27 with excitation of the halogen-bonded complex resulting in the formation of a perfluoroalkyl radical that adds to the alkene. Fragmentation of the β-sulfinyl radical, followed by loss of SO2, results in an anomeric radical (see the next section for a discussion of the reactivity of such species). Reaction of the latter with iodine atom generates a glycosyl iodide. The authors proposed that the 1,2-cis-stereoselectivity arises from an associative substitution reaction of the β-configured glycosyl iodide, which is the more reactive of the pair of rapidly interconverting anomers. Computational modelling pointed towards a hydrogen bond between the acceptor OH group and the ether oxygen at C-2 as a stabilizing interaction in the transition state leading to the 1,2-cis-glycoside.
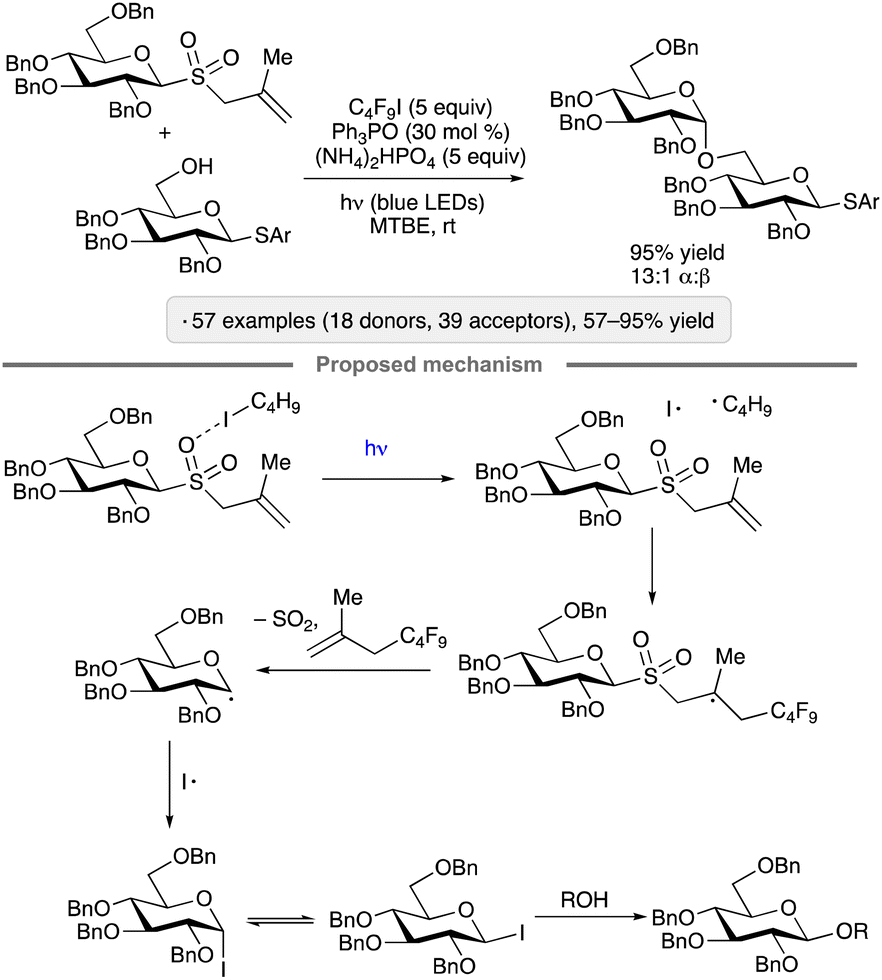 | ||
| Scheme 4 O-Glycosylation via excitation of the halogen-bonded complex between a glycosyl sulfone and 1-iodoperfluorobutane. Ar denotes 4-chlorophenyl. | ||
Wen and Crich employed O-glycosyl 2,2,6,6-tetramethylpiperidinoxides (Tempol glycosides, synthesized from the corresponding glycosyl fluorides) as donors in photocatalytic glycosylations using complex Ir-2 under irradiation by blue LEDs (Scheme 5a).29 Unlike the blue light-mediated processes shown in Scheme 1b–d, this protocol did not require an electrophilic/reducing reagent, since the nitroxyl radical released in the glycosylation event was able to serve as an oxidant to close the photoredox cycle. Armed donors of gluco, manno or galacto configuration were employed. While the stereoselectivity at the anomeric position was generally modest, a dependence of the α:β ratio on the configuration of the starting Tempol glycoside 2 was observed, consistent with an associative pathway for the displacement of the radical leaving group after oxidation.
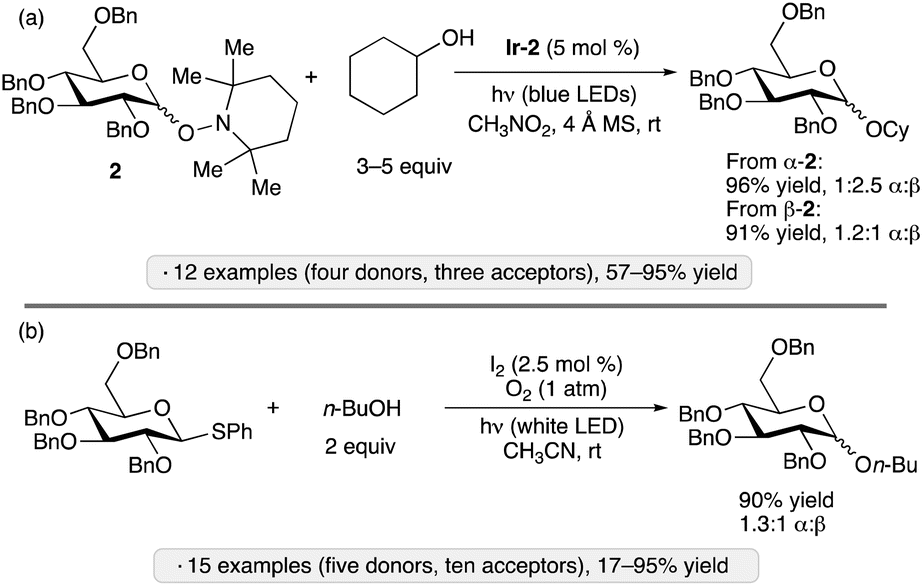 | ||
| Scheme 5 Photocatalytic O-glycosylations reported by the groups of Crich29 (a) and Opatz (b).30Ir-2 denotes [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6. | ||
A distinct reagent combination for photocatalytic glycosylation was disclosed by Opatz and co-workers, who used I2 (2.5 mol%) to activate aryl thioglycosides upon irradiation with a white household LED bulb in acetonitrile under an oxygen atmosphere (Scheme 5b).30 Armed glycosyl donors gave good yields and modest stereoselectivities, while disarmed donors reacted more sluggishly (<40% yield) but with 1,2-trans stereoselectivity. The authors proposed two mechanisms, both of which were supported by experimental observations: an ionic pathway, in which I2 served as the electrophilic activator for the thioglycoside, with the released iodide being reoxidized by photogenerated singlet oxygen; and a SET pathway, wherein the thioglycoside was oxidized by triplet oxygen (generated by photosensitization with I2), followed by expulsion of the thiyl radical in the glycosylation step.
Visible light excitation of catalytic copper complexes has been used to achieve the synthesis of 1,2-cis-configured O-glycosides from glycosyl bromides (Scheme 6).31 Gluco- and galactopyranosyl donors bearing benzyl ether groups at the 2-position, as well as a 2-O-benzylated ribofuranosyl bromide, underwent stereoselective couplings with primary and secondary alcohols in the presence of catalytic copper(I) iodide, Xantphos and 4,7-diphenyl-1,10-phenanthroline (BPhen). Experiments with added TEMPO and a 2-O-allyl-protected substrate capable of 5-exo-trig cyclization did not point to the intermediacy of a free anomeric radical. Subsequent computational studies suggested that a photoexcited Cu(I) complex served to reduce a second equivalent of Cu(I) BPhen, forming an active Cu(0)-like species that engaged in a sequence of oxidative addition, ligand exchange, one-electron oxidation and reductive elimination to generate the O-glycoside in a stereospecific fashion.32 A simplified version of the calculated cycle is depicted in Scheme 6.
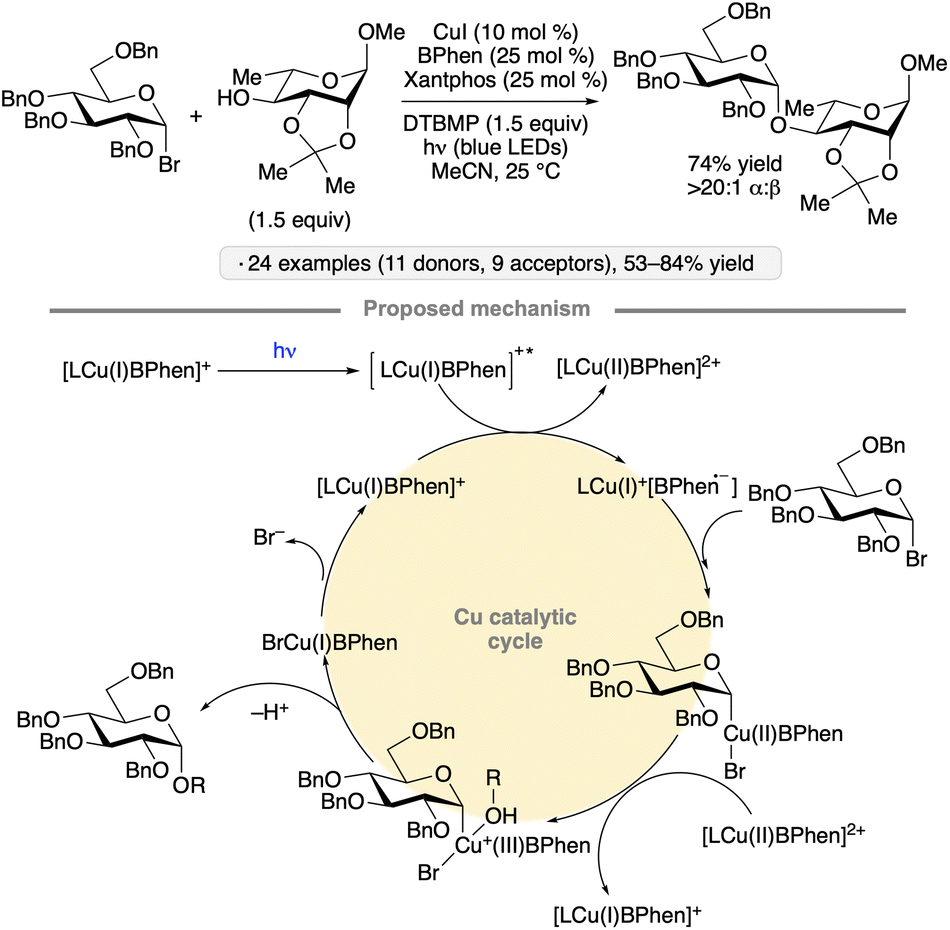 | ||
| Scheme 6 Visible light-induced, copper-catalyzed O-glycosidic bond formation from glycosyl halides. BPhen denotes 4,7-diphenyl-1,10-phenanthroline; DTBMP denotes 2,6-di-tert-butyl-4-methylpyridine. | ||
A mechanistically distinct pathway for photocatalytic substitution at the anomeric position was explored by Nagorny and co-workers, who developed a protocol for the synthesis of glycosyl fluorides from the corresponding hemiacetals.33 The transformation was accomplished using SF6 gas and Hünig's base, along with 4,4′-dimethoxybenzophenone as photocatalyst under irradiation at 365 nm in 1,2-dichloroethane (Scheme 7). The proposed mechanism involves photocatalytic generation of transient amounts of SF4, a reagent capable of promoting deoxyfluorination, from SF6 and iPr2NEt. Tolerance for ester, ether (Bn and PMB) and benzylidene acetal groups, as well as glycosidic linkages, was demonstrated.
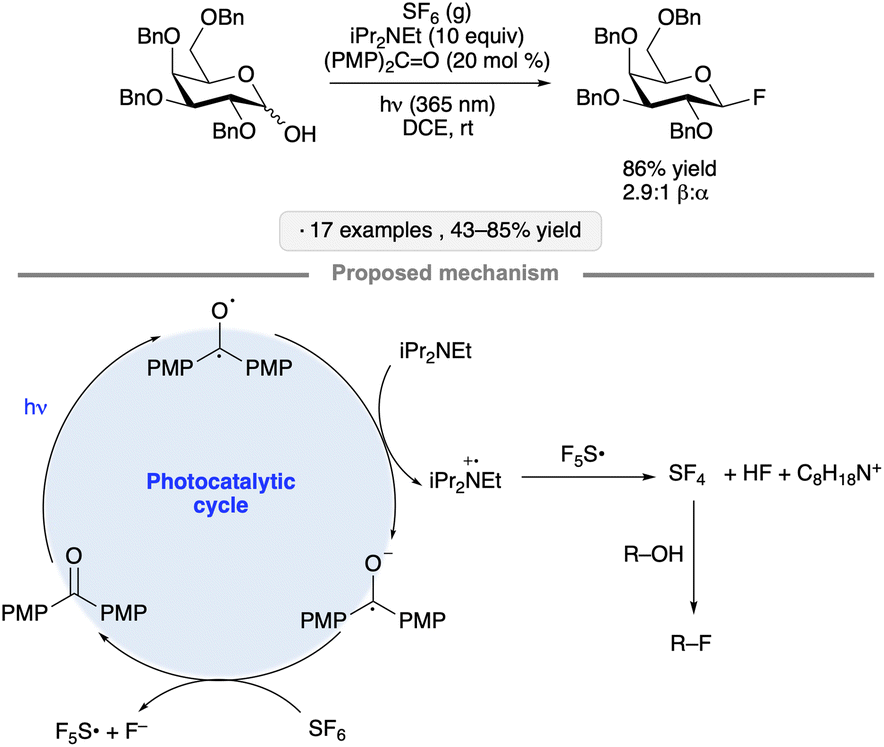 | ||
| Scheme 7 Photocatalytic synthesis of glycosyl fluorides and proposed reaction mechanism. PMP denotes 4-methoxyphenyl. | ||
3. Anomeric radicals
Anomeric radicals serve as the basis for alternative approaches for glycosidic bond construction, offering distinct and potentially complementary features in comparison to the reactions of glycosyl electrophiles.34 Foundational contributions from the group of Giese in the 1980s provided insight into the structure, bonding and reactivity of radicals arising from homolytic cleavage of an anomeric C–X bond.35 By enabling access to anomeric radicals from a variety of precursors under mild conditions, photocatalysis has created new opportunities to develop synthetic methods that exploit the reactivity of these versatile intermediates.3.1. Glycosidations via radical substitutions of glycosyl halides
The utility of photocatalysis for glycosyl radical formation was established by Gagné and co-workers in 2010, with the demonstration of C-glycoside formation from glycosyl bromides and electrophilic alkenes upon irradiation with blue LEDs in the presence of [Ru(bpy)3](BF4)2, iPr2NEt and Hantzsch ester 3 (Scheme 8).36 High α-stereoselectivities were observed for reactions of per-O-acylated gluco-, galacto- and mannopyranosyl halides, consistent with previously documented stereochemical outcomes of additions of anomeric radicals to alkenes.35 In comparison to the protocol initially reported by Giese and Dupuis (UV photolysis in the presence of tributyltin hydride),35a the photocatalytic procedure offers several practical advantages, which is notable given the prevalence of C-glycosides as natural product substructures and metabolically stable analogs of O-glycosides.37 The authors' proposed mechanism involves SET from iPr2NEt to the excited-state photocatalyst, priming the latter for reduction of the glycosyl halide. After addition of the anomeric radical to the alkene, the C-glycoside product is formed by hydrogen atom transfer to the resulting radical from either 3 or a trialkylamine-derived species (denoted R–H in Scheme 8). The Gagné group went on to develop a flow photoreactor for couplings of glycosyl bromides with acrolein on scales as high as 18 mmol, and employed the products in the synthesis of C-linked glycopeptide and glycolipid analogs.38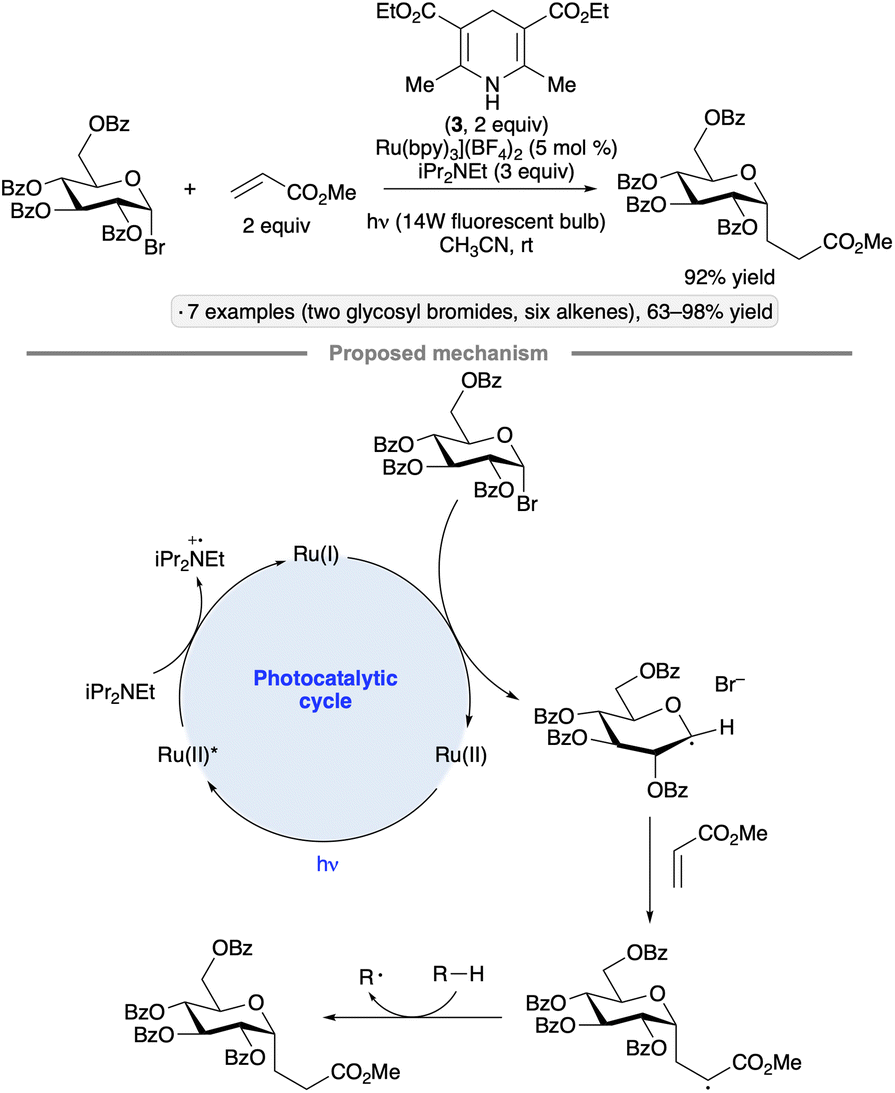 | ||
| Scheme 8 Photocatalytic synthesis of C-glycosides from glycosyl halides and proposed reaction mechanism. | ||
Photocatalytic activation of glycosyl halides has proved to be a versatile method for the synthesis of diverse types of C-glycosides. The use of Ir-1 along with tris(trimethylsilyl)silane as a silyl radical precursor enabled alkenylations of α-acetobromoglucose with 2,2′-diaryl-substituted vinyl sulfones (Scheme 9a).39 By using α-trifluoromethylstyrenes as radical trapping reagents, Hu and co-workers accomplished the photocatalytic synthesis of gem-difluoroalkene-substituted C-glycosides (Scheme 9b).40 In addition to α-selective couplings of per-O-acylated pyranosyl bromides, the authors demonstrated 1,2-trans-selective C-glycosylations of isopropylidene-protected furanosyl bromides. Nitrogen heterocycles have also been used as trapping agents for photocatalytically generated anomeric radicals, enabling the preparation of C-nucleoside analogs and other heterocyclic C-glycosides (e.g., Scheme 9c).41 Triethylamine served as reductive quencher for the excited-state photocatalyst (see Scheme 8 for a related mechanism) and as base for rearomatization of the heterocycle after radical addition and single-electron reduction. Ester-protected furanosyl and pyranosyl bromides gave rise to C-glycosides with 1,2-trans-stereoselectivity, while the stereochemical outcomes of reactions of isopropylidene ketal-protected congeners were variable: ribo- and mannofuranosides gave 1,2-cis-selectivity, as shown in Scheme 9c, while lyxofuranosides and mannopyranosides favored the 1,2-trans-configured products. Trapping of anomeric radicals with N-sulfonyl methacrylamide derivatives results in intramolecular aryl transfer (Truce–Smiles-type rearrangement), followed by desulfonylation, generating functionalized C-glycosides (Scheme 9d).42 For the mechanism of glycosyl radical formation from (TMS)3SiOH and base in the presence of a photoredox catalyst, see Scheme 15 below.
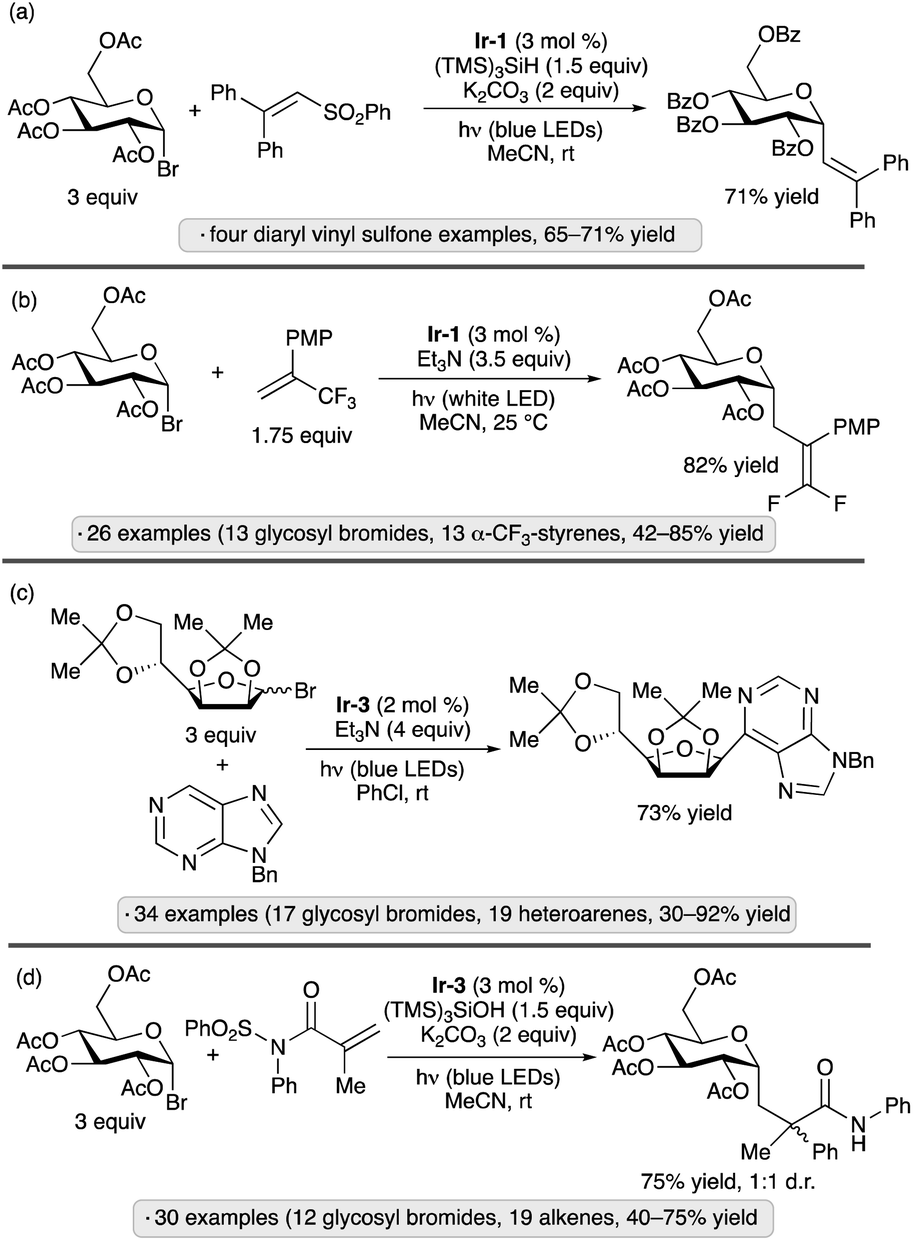 | ||
| Scheme 9 Photocatalytic reactions of glycosyl bromides with (a) vinyl sulfones;39 (b) trifluoromethylstyrenes;40 (c) nitrogen heterocycles;41 and (d) N-sulfonyl methacrylamides.42Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; Ir-3 denotes [Ir(ppy)2(dtbbpy)]PF6. PMP denotes 4-methoxyphenyl. | ||
Reductive couplings with haloalkynes enable the synthesis of alkynyl C-glycosides from glycosyl halides. The protocol developed by Yu and co-workers employs the organic photocatalyst Eosin Y along with iPr2NEt as the reductant, and is tolerant of ester and isopropylidene ketal protective groups (Scheme 10).43 The authors proposed that the glycosyl radical was generated by halogen atom transfer (XAT) to the radical arising from photooxidation and deprotonation of the trialkylamine base. Addition to the alkynyl bromide and expulsion of bromine atom forms the C-glycoside product, with photocatalyst turnover being accomplished by reduction of Br˙ to Br−.
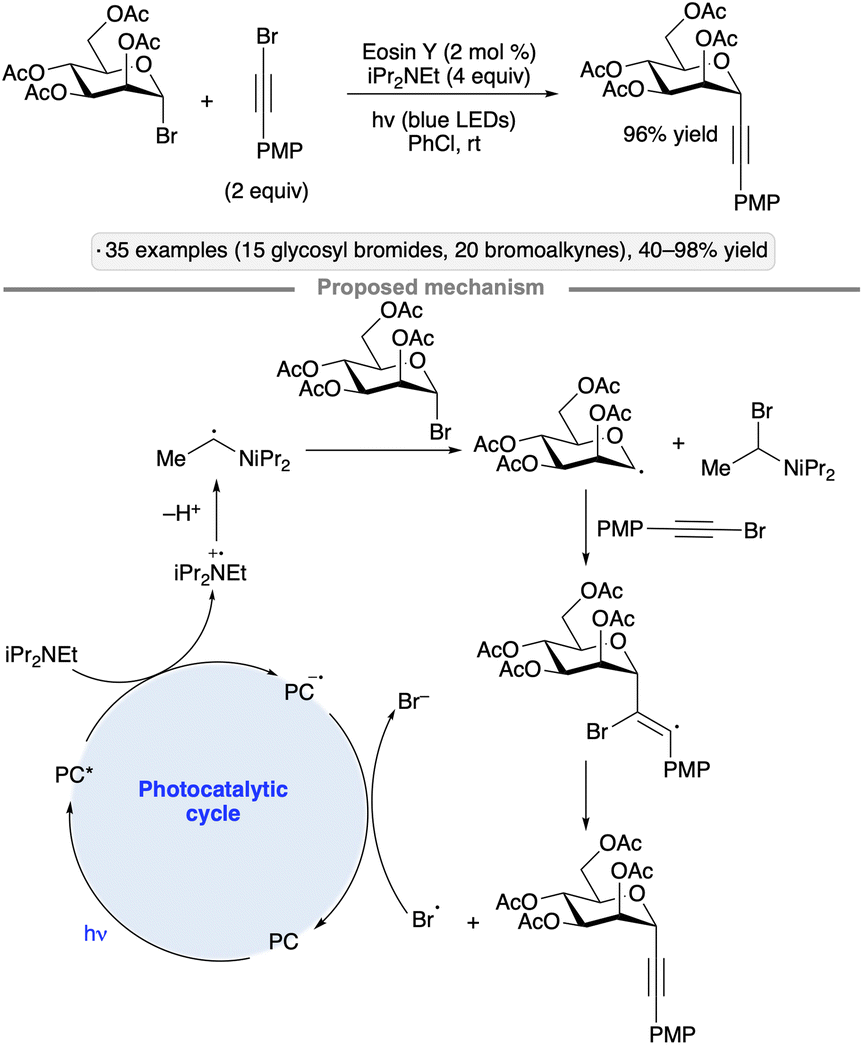 | ||
| Scheme 10 Reductive couplings of bromoalkynes with glycosyl bromides. | ||
Reductive couplings of aryl bromides and glycosyl chlorides have been used to synthesize aryl C-glycosides. The protocol developed by Zhang, Niu and co-workers employs a Ni(II) complex and Ir-1 photocatalyst,44 with Hantzsch ester 3 as the stoichiometric reductant (Scheme 11).45 Per-O-acetylated pyranosyl chlorides were coupled with para-bromoanisole, giving access to 1,2-trans-configured C-aryl glycosides in yields ranging from 34% to 53%. Various bromoarenes and bromoheteroarenes were coupled with the glucopyranosyl chloride partner to furnish the corresponding β-C-glycosides. Alkoxy, alkyl and halo (F, Cl) substituents were tolerated at the meta and para positions, whereas examples bearing strong electron-withdrawing groups or ortho substituents were not reported. The proposed mechanism involves SET from the Hantzsch ester to photoexcited Ir-1. Deprotonation of the Hantzsch ester-derived radical cation results in the generation of a reductant capable of forming the glycosyl radical from the glycosyl chloride. The Ni(II) complex arising from oxidative addition to the bromoarene interacts with the glycosyl radical, enabling reductive elimination to form the C-glycoside. SET from reduced Ir-1 to the resulting Ni(I) intermediate closes the coupled catalytic cycles.
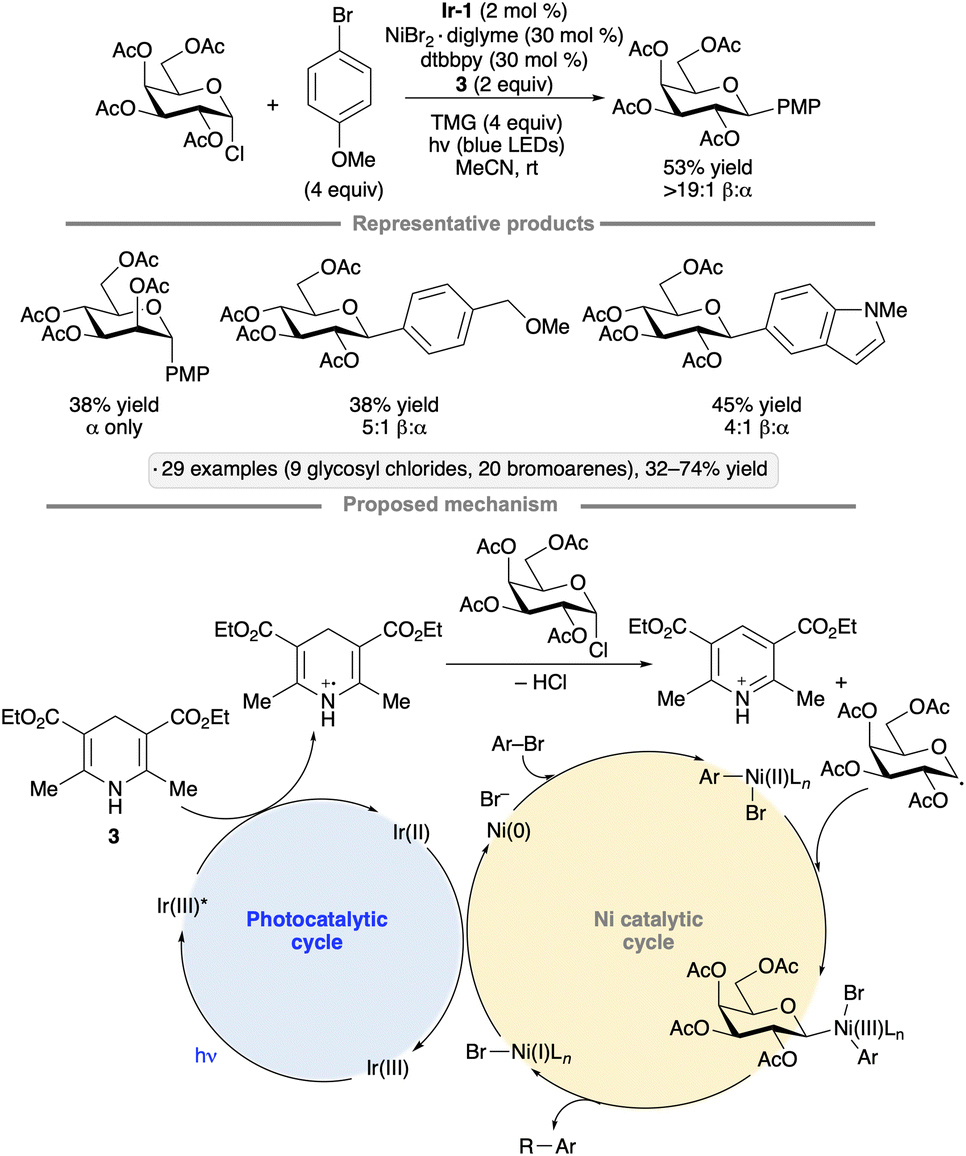 | ||
| Scheme 11 Synergistic nickel/photoredox catalysis for reductive couplings of glycosyl chlorides with aryl bromides. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine; TMG denotes 1,1,3,3-tetramethylguanidine; PMP denotes 4-methoxyphenyl. | ||
Considering the relevance of 2-(α-mannopyranosyl)tryptophan as a naturally occurring C-glycosylpeptide moiety, the group of Goddard-Borger aimed to develop a method that would enable late-stage incorporation of this motif into peptide substrates.46 Evaluation of reaction conditions for the reductive coupling of acetobromomannose with a protected 2-bromotryptophan revealed that the transformation could be accomplished in 78% yield upon irradiation with violet LEDs (390 nm) in the presence of a nickel(II) complex, Hantzsch ester, iPr2NEt and MgCl2 in acetonitrile at room temperature (Scheme 12). Other aryl and heteroaryl bromides, including 2-bromotryptophan-containing dipeptides, underwent α-C-mannosylation under these conditions. The Hantzsch ester likely serves as a photoreductant for the glycosyl bromide, generating an anomeric radical that engages an arylnickel complex prior to C–C bond-forming reductive elimination. An alternative protocol was developed for C-mannosylations of more complex, 2-bromotryptophan-containing peptides; the 4CzIPN photocatalyst was employed in place of the Hantszsch ester, and a mannosyl chloride was used as the glycosylating reagent. These conditions were used to incorporate a 2-(α-mannopyranosyl)tryptophan moiety into a protected decapeptide in 30% yield.
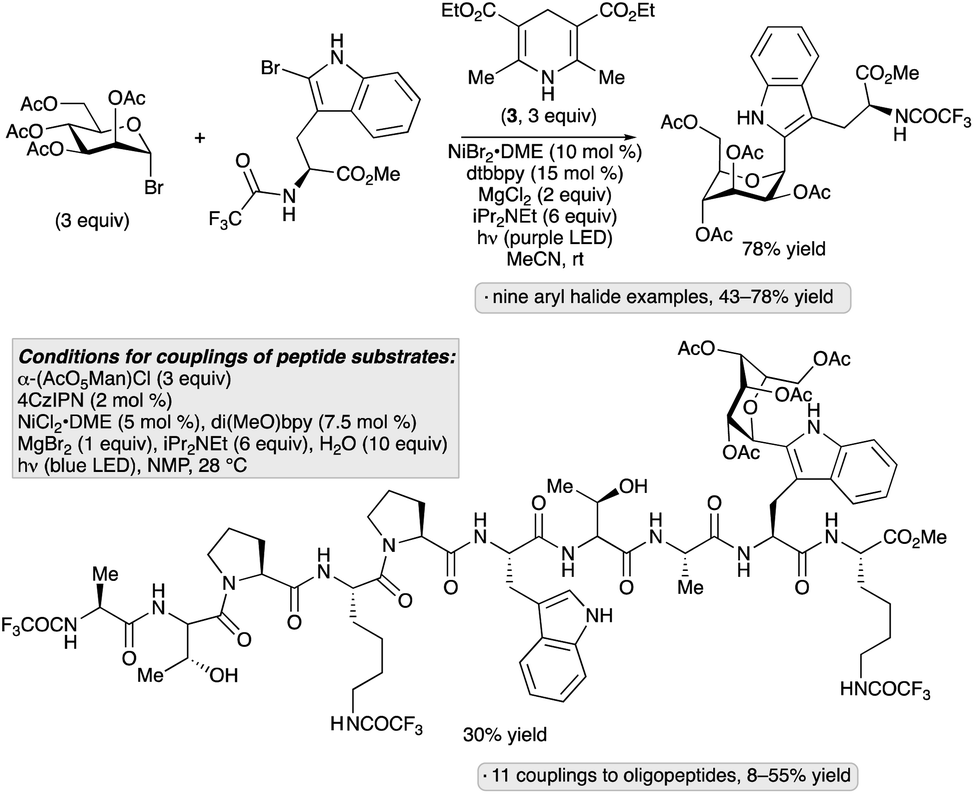 | ||
| Scheme 12 Photocatalytic coupling of 2-bromotryptophan derivatives with per-O-acetylated mannosyl halides. 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine; di(MeO)bpy denotes 4,4′-dimethoxy-2,2′-bipyridine. | ||
Nickel catalysis has been used to synthesize C-alkyl glycosides from glycosyl bromides via reductive couplings with N-hydroxyphthalimide esters or N-alkylpyridinium salts (Scheme 13).47 The procedure was applied to a range of primary and secondary redox-active electrophiles, including amino acid and peptide derivatives. The scope of glycosyl donors employed includes armed furanosyl and pyranosyl bromides or chlorides. The authors proposed that EDA complexes were formed from the redox-active electrophile, Hantzsch ester 3 and a Lewis basic additive (lithium iodide and triethylamine for the NHP esters and N-alkylpyridinium salts, respectively). Visible light excitation resulted in electron transfer from 3 to the redox-active electrophile, leading to fragmentation and alkyl radical formation. The radical cation derived from 3 served to reduce Ni(I) to Ni(0), which underwent oxidation to the glycosyl bromide. Reaction of the alkyl radical with the resulting glycosylnickel(II) complex, followed by reductive elimination, generated the alkyl C-glycoside product.
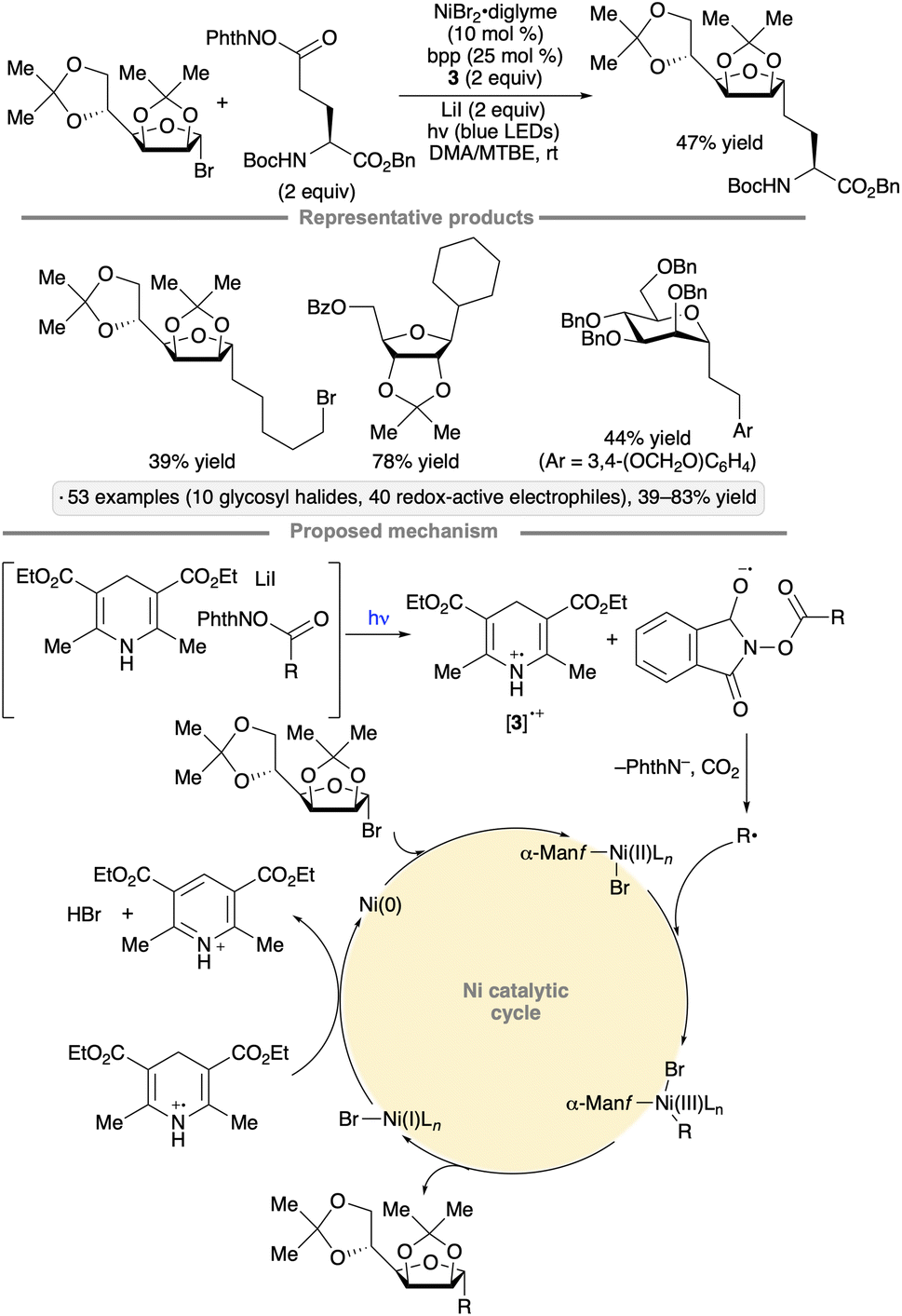 | ||
| Scheme 13 C-Glycosylation via couplings of glycosyl halides with redox-active electrophiles. bpp denotes 2,6-bis(pyrazol-1-yl)pyridine. | ||
Visible light-induced, Pd-catalyzed Heck-type reactions have been employed as an alternative approach for the synthesis of C-glycosides.48 Using a Pd(Xantphos) complex as catalyst under blue LED irradiation, ester-protected pyranosyl bromides underwent 1,2-trans-selective couplings with styrene derivatives to yield C-vinyl glycosides (Scheme 14). Isopropylidene-protected mannopyranosyl and mannofuranosyl donors also underwent stereoselective coupling. The authors proposed that oxidative addition of a photoexcited Pd(0) species provides access to the anomeric radical intermediate that engages the alkene to form the carbon–carbon bond.10
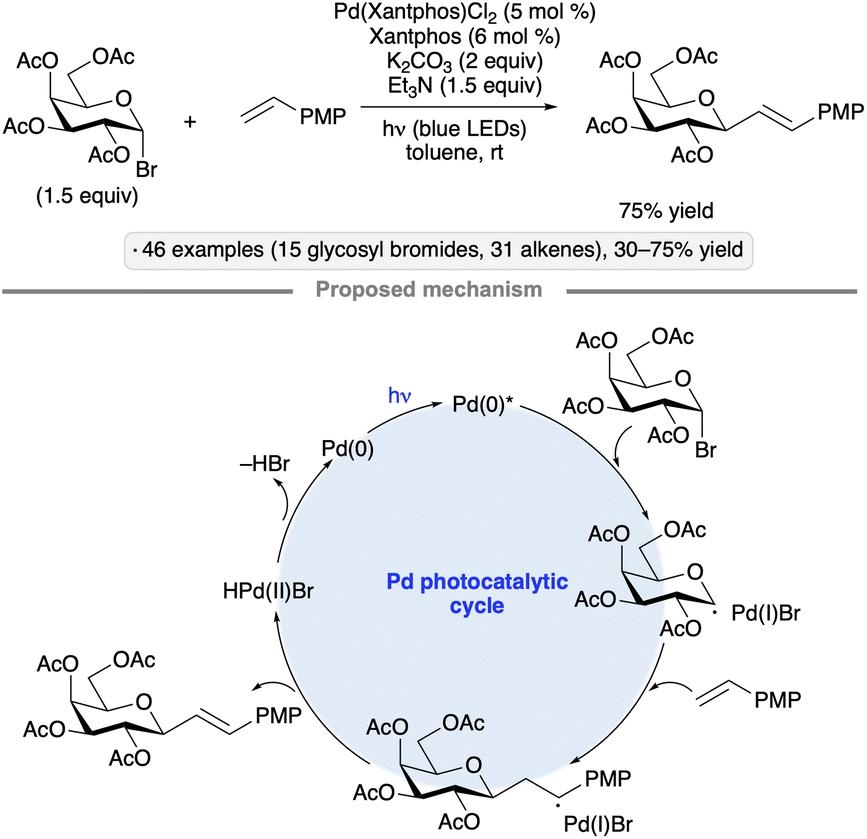 | ||
| Scheme 14 Visible light-induced, Pd-catalyzed coupling of glycosyl halides with styrene derivatives. | ||
Wang and co-workers used thiosulfonates as trapping agents for anomeric radicals, enabling a photocatalytic synthesis of thioglycosides from glycosyl bromides (Scheme 15).49 The authors found that tris(trimethylsilyl)silanol, in combination with K3PO4 and the 4ClCzIPN photocatalyst, a potent photoreductant, resulted in efficient S-glycosylation upon blue light irradiation. The hydrodehalogenation product was obtained using other potential reductants such as iPr2NEt, Hantzsch ester or ascorbic acid. The protocol yielded α-1,2-cis-configured thioglycosides from ester-protected gluco- and galactopyranosyl bromides, whereas manno-configured bromides gave 1,2-trans stereoselectivity. An exception to this pattern was per-O-acetylated xylosyl bromide; the 1,2-trans-selectivity obtained for this substrate likely arises from axial attack on an anomeric radical in the 1C4 conformation.50 Tolerance for benzyl, isopropylidene ketal, silyl ether protective groups and functionalized thiosulfonates, along with applications to the synthesis of S-furanosides and selenoglycosides, were demonstrated. The proposed mechanism for radical generation involves reduction of photoexcited 4ClCzIPN by the silanoate, followed by a Brook-type rearrangement to generate the silyl radical that engages in halogen atom transfer with the glycosyl halide. Trapping of the anomeric radical with the thiosulfonate releases a sulfonyl radical that serves as an oxidant to close the photocatalytic cycle.
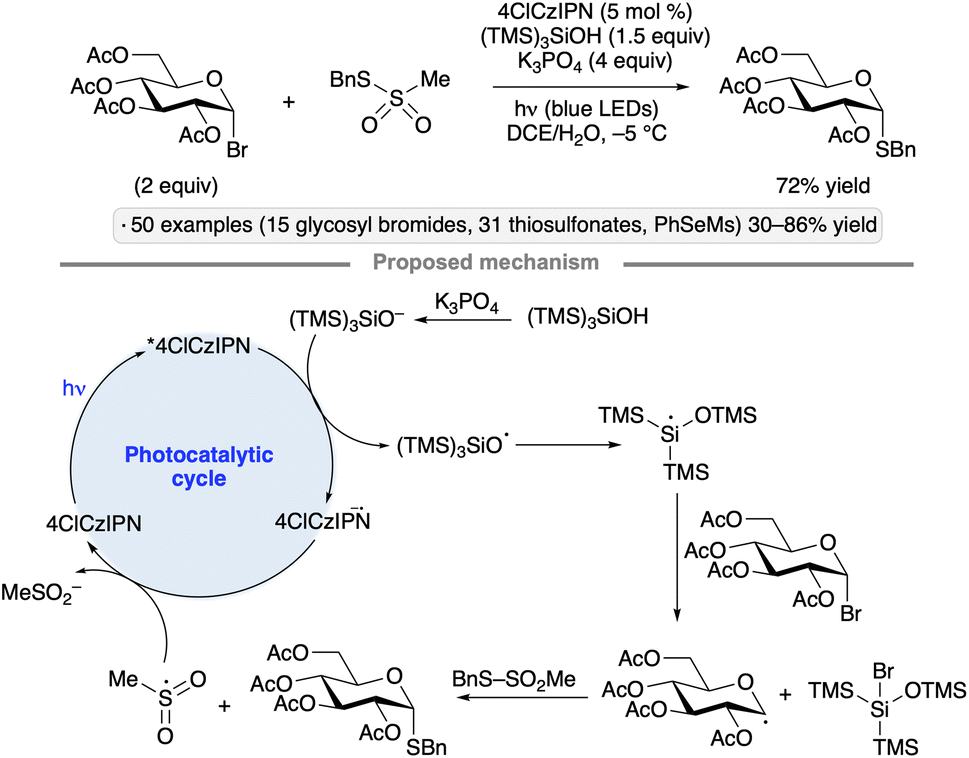 | ||
| Scheme 15 Photocatalytic synthesis of thioglycosides from glycosyl bromides. 4ClCzIPN denotes 1,2,3,5-tetrakis(3,6-dichlorocarbazol-9-yl)-4,6-dicyanobenzene. | ||
3.2. Other classes of glycosyl radical precursors
While glycosyl bromides show versatile reactivity as precursors to anomeric radicals, they are prone to hydrolysis and their synthesis often requires harsh reagents. Efforts have been made to identify alternative anomeric substituents that can be activated towards homolytic bond cleavage under photocatalytic conditions. In 2019, Wang, Zhang and co-workers disclosed a photocatalytic, decarboxylative reaction for the synthesis of furanosyl C-glycosides (Scheme 16a).51 In the presence of the organic photocatalyst 4CzIPN, a nickel salt and 2,2′-bipyridine (bpy), per-O-benzylated ribofuranose-1-carboxylic acid was coupled with a range of aryl, heteroaryl and vinyl halides. The requisite glycosyl-1-carboxylic acid starting material was synthesized in 30% overall yield from the per-O-benzylated glycosyl 1-acetate by C-glycosidation with cyanide followed by hydrolysis. The resulting β-C-glycoside products are of potential interest as nucleoside analogs. While most of the synthesized compounds were per-O-benzylated ribofuranosyl C-glycosides, the method was also used to synthesize a 2-deoxyfuranoside and a ‘reversed’ glycoside (see Section 4.2). The depicted photocatalytic cycle draws on mechanistic proposals for related synergistic nickel/photocatalytic decarboxylative couplings.44,52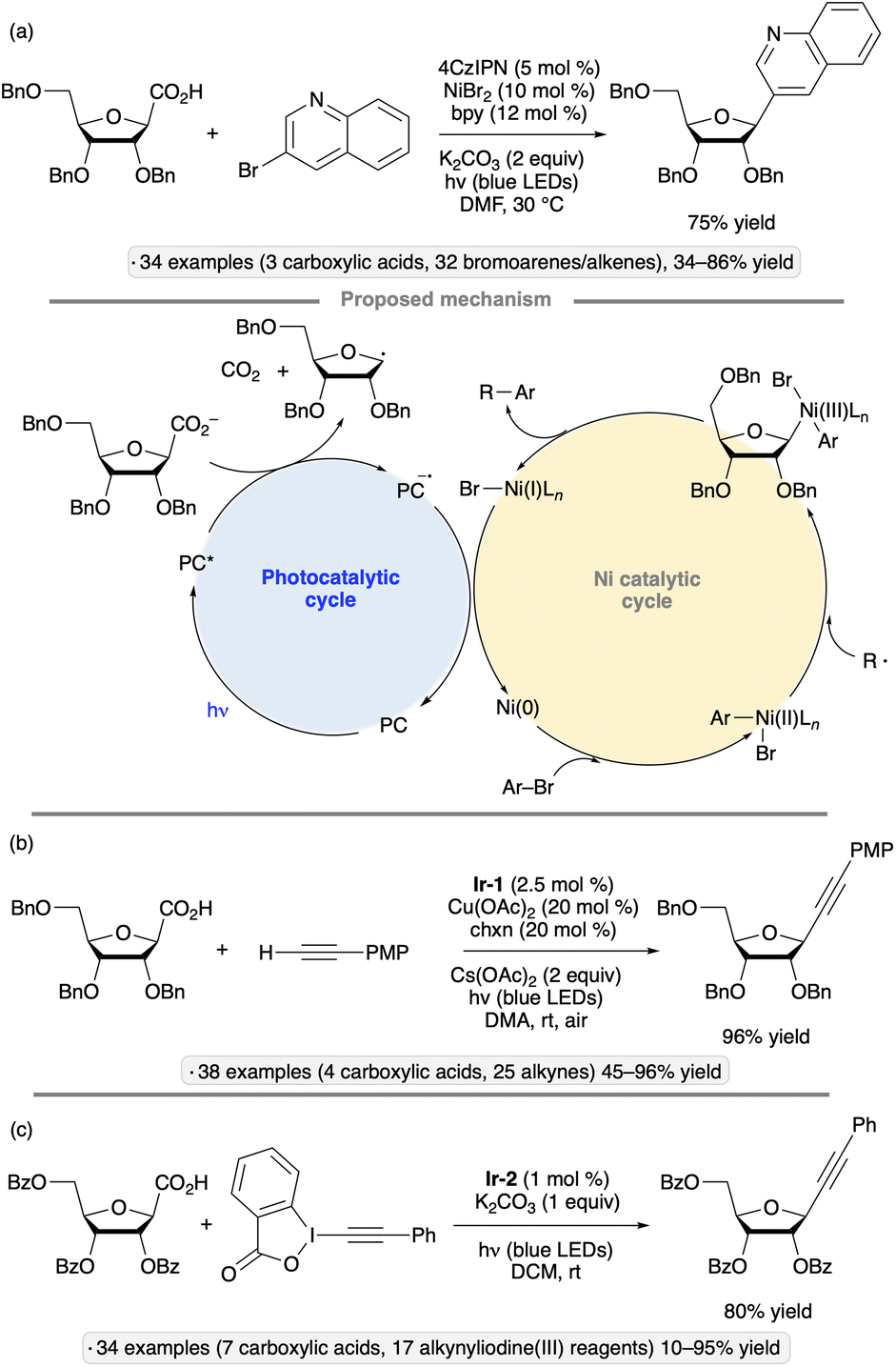 | ||
| Scheme 16 Photocatalytic, decarboxylative couplings of glycosyl-1-carboxylates with (a) bromoarenes;51 (b) alkynes;53 and (c) alkynyliodine(III) reagents.54 bpy denotes 2,2′-bipyridine; Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; chxn denotes trans-1,2-diaminocyclohexane; Ir-2 denotes [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6. | ||
C-Alkynyl glycosides have also been synthesized from glycosyl-1-carboxylates via decarboxylative couplings with terminal alkynes. Zhu and Messaoudi employed a copper(I) co-catalyst along with Ir-1, while irradiating with blue LEDs under an air atmosphere, to accomplish this transformation (Scheme 16b).53 Similarly to the reaction shown in Scheme 16a, the method was primarily used to access per-O-benzylated β-C-ribofuranosides, but tolerance of alternative substitution patterns (2,3-O-isopropylidene ketal protection, deoxygenation at C2) was demonstrated, along with an example of reversed glycoside synthesis. The proposed mechanism for carbon–carbon bond formation involves reductive elimination from a Cu(III) species generated from the anomeric radical (produced via SET from photoexcited Ir-1 and the carboxylate as described in the preceding paragraph) and a Cu(II) acetylide. Oxidation by O2 was invoked to close the photocatalytic cycle (Ir(II) to Ir(III)) and to generate the reactive copper acetylide (Cu(I) to Cu(II)).
An alternative approach to the synthesis of alkynyl C-glycosides from glycosyl-1-carboxylates was disclosed by the group of Zhang, who used alkynyliodine(III) reagents as coupling agents under irradiation with blue LEDs in the presence of Ir-2 (Scheme 16c).54 Per-O-benzoylated ribofuranosyl-1-carboxylates were coupled with phenylacetylene derivatives to give β-C-alkynyl glycoside products. Stereoselective couplings of per-O-benzylated furanosyl and pyranosyl donors were also demonstrated. The authors proposed that after addition of the glycosyl radical to the ethynylbenziodoxolone, the alkyne product was generated by elimination of an iodanyl radical, which underwent reduction with concomitant regeneration of the Ir-2 photocatalyst.
Diao and co-workers have explored dihydropyridine-(DHP)-derived glycosyl esters as precursors to anomeric radicals under photocatalytic conditions. The key preparative advantage is the ability to access the requisite substrates by esterification of readily available glycosyl hemiacetals. The group's initial report involved C-aryl glycoside synthesis from aryl halides in the presence of a Ni(II) complex (Scheme 17).55 The proposed pathway for formation of the anomeric radical is depicted. Oxidation of the dihydropyridyl group by the excited photocatalyst, followed by deprotonation, results in a radical that can fragment with loss of a substituted pyridine. Further decarboxylative fragmentation results in the anomeric radical, which engages the Ni(II) aryl halide complex as shown previously (Schemes 11 and 16a). The reduced photocatalyst is responsible for reduction of Ni(I) to Ni(0). Manno- and ribofuranosides bearing isopropylidene ketal or ether protective groups at the 2- and 3-positions were coupled effectively to give β-configured products (1,2-trans-stereoselectivity). Reactions of pyranoside-derived glycosyl esters were also conducted, with good yields being obtained for 2,3-O-isopropylidene-bearing, manno-configured substrates and 2-deoxy congeners, versus lower yields (>30%) for other protected gluco-, galacto- and mannopyranosides. Various aryl and heteroaryl halide partners could be employed, with the exception of readily oxidizable heterocycles (e.g., furan and pyrrole derivatives).
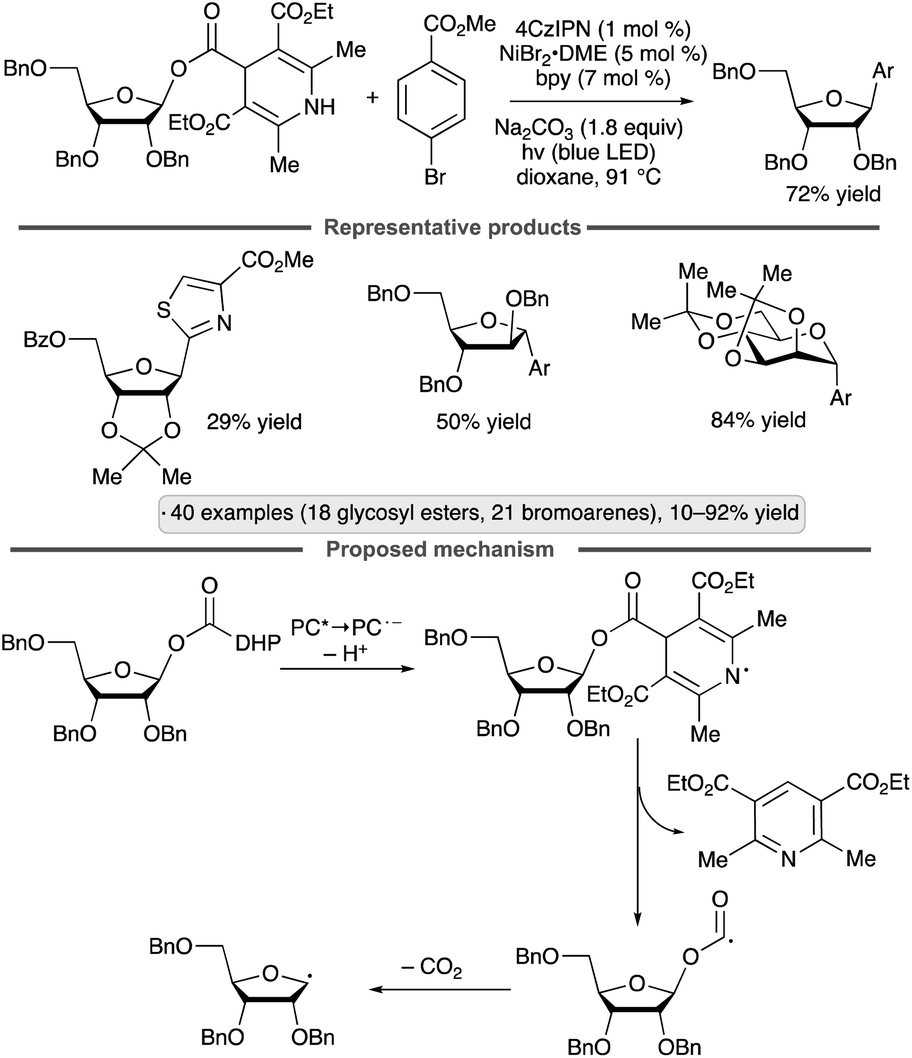 | ||
| Scheme 17 Photocatalytic C-arylations of dihydropyridine-derived glycosyl esters. 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; bpy denotes 2,2′-bipyridine; Ar denotes 4-(MeO2C)C6H4. | ||
A related method for the synthesis of C-acyl glycosides was reported by the Diao group in 2021 (Scheme 18a).56 In place of the aryl bromide coupling partner, a carboxylic acid was used to acylate the anomeric radical in the presence of diethyl dicarbonate as activating reagent.57 1,2-Trans stereoselectivity was obtained with a variety of pyranosyl and furanosyl DHP esters, although strongly disarmed systems gave lower yields (e.g., 23% for a 2,3,4,6-tetra-O-acetylated mannopyranoside DHP ester). The scope of carboxylic acid partners was likewise broad, including primary, secondary and tertiary aliphatic acids as well as benzoic acid derivatives.
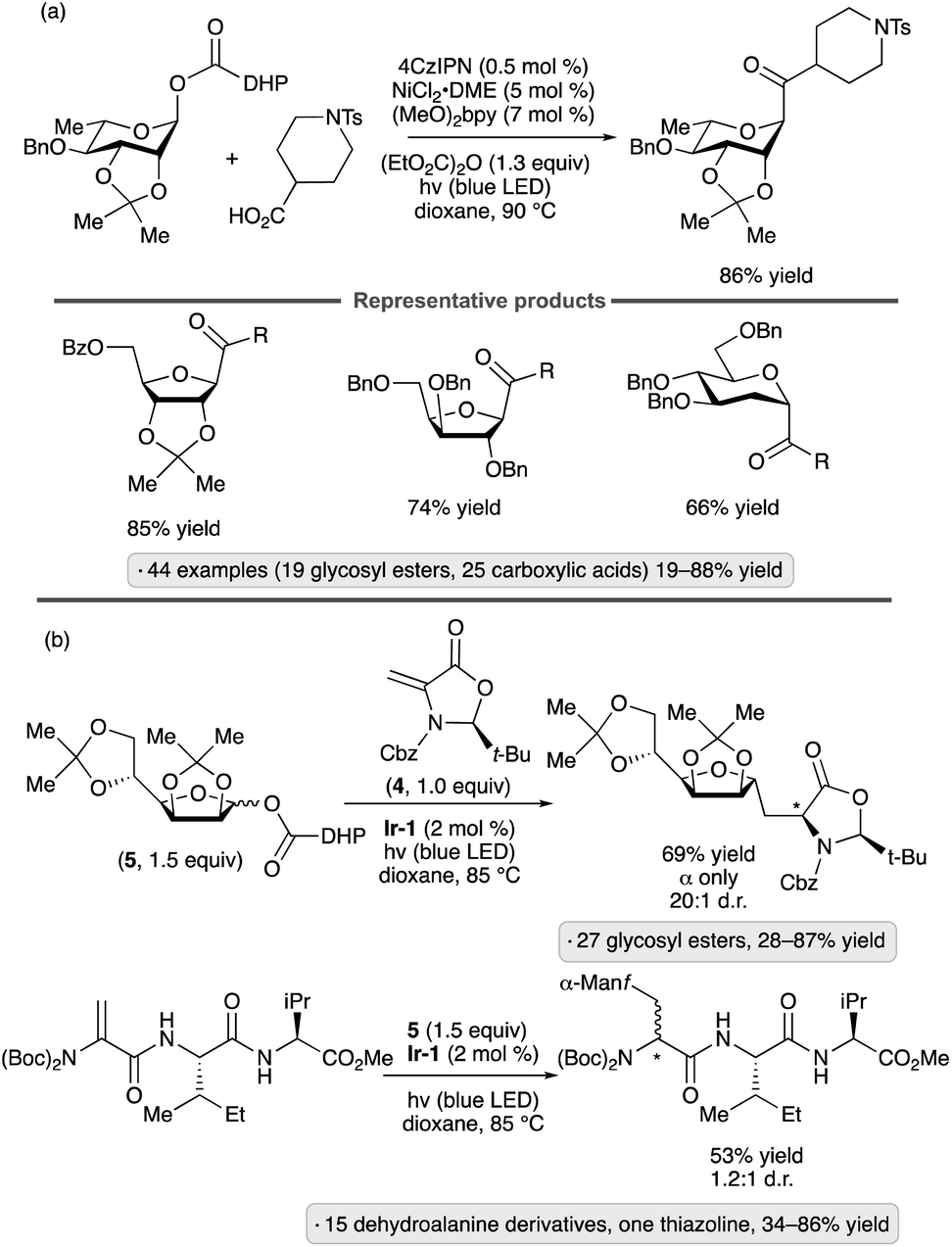 | ||
| Scheme 18 Photocatalytic C-glycosidations of dihydropyridine-derived glycosyl esters. (a) C-Acylation by coupling with esters. (b) Addition to dehydroalanine derivatives. 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; (MeO)2bpy denotes 4,4′-dimethoxy-2,2′-bipyridine; Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
DHP esters have also been employed as precursors to C-glycosyl amino acids and C-glycosyl peptides via radical additions to dehydroalanine derivatives (Scheme 18b).58 Couplings with chiral oxazolidine 4 took place with high levels of diastereoselectivity at the newly formed α-amino acid chirality center. Both furanosyl and pyranosyl DHP esters participated in the reaction, with variable levels of 1,2-trans stereoselectivity being obtained depending on the sugar configuration and the choice of protective groups. Consistent with the work of Diao and co-workers,55 the transformation was most efficient when employing substrates derived from armed sugars. Mannofuranosylations of protected dehydroalanine moieties in di-, tri- and tetrapeptide substrates were demonstrated, giving rise to roughly 1:1 mixtures of diastereomers at the α-amino acid chirality center.
Activation of alcohols by addition to azolium salts, followed by photocatalytic oxidation, has been employed to achieve deoxygenative arylation reactions in the presence of a Ni(II) co-catalyst (Scheme 19).59C-Aryl glycosides were generated from glycosyl hemiacetal substrates, and the protocol was applied to substrates having free OH groups at positions other than the anomeric center (e.g., C2 of a ribofuranoside, C1 of a fructopyranose).
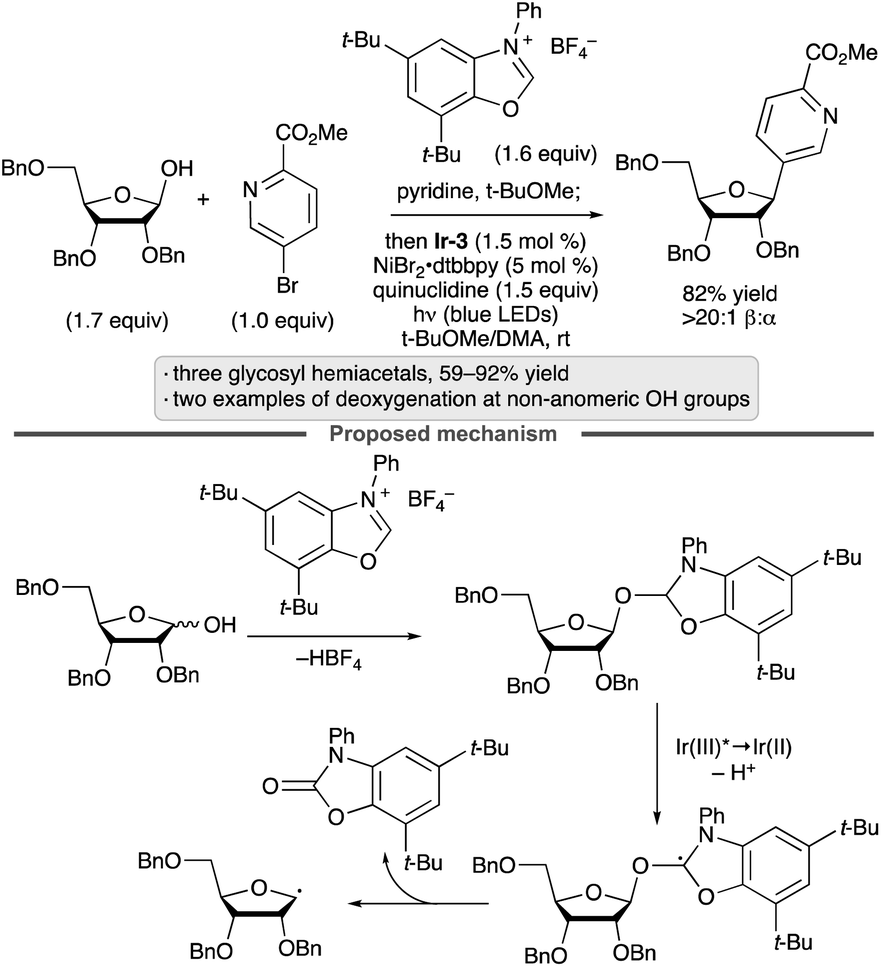 | ||
| Scheme 19 Photocatalytic, deoxygenative arylations of alcohols mediated by an azolium salt. Ir-3 denotes [Ir(ppy)2(dtbbpy)]PF6; dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine. | ||
Walczak and co-workers achieved the synthesis of thioglycosides from glycosyl stannanes in the presence of a copper(I) catalyst with irradiation by blue LEDs (Scheme 20).60 The authors proposed that the absorption of light served to promote homolysis of the disulfide reagent to generate thiyl radicals, which were then trapped by a glycosylcopper(I) complex. Equatorially configured gluco- and 2-deoxypyranosyl stannanes gave rise to axial thioglycosides as the major products, an observation consistent with an outer-sphere mechanism involving an anomeric radical intermediate.
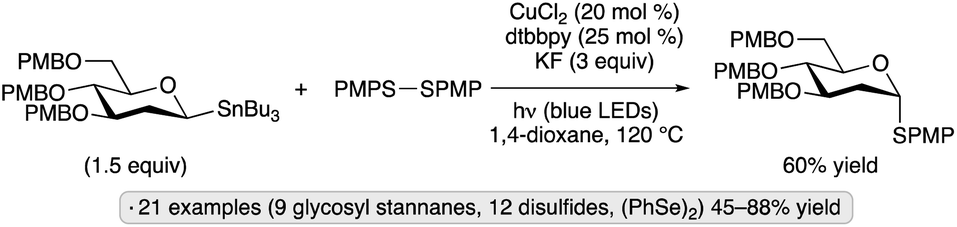 | ||
| Scheme 20 Visible light-induced, copper-catalyzed couplings of a glycosyl stannane with a disulfide. dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine; PMP denotes 4-methoxyphenyl. | ||
Glycosyl trifluoroborates have emerged as another class of useful precursors to anomeric radicals. The group of Hirai synthesized β-configured 2-deoxypyranosyl trifluoroborates from the corresponding glycal-derived N-methyliminodiacetic acid (MIDA) boronate esters.61 Congeners bearing silyl ether and cyclic silylene protective groups were prepared by this route. Upon irradiation with blue light in the presence of Ir-1 and a nickel(II) complex,62 the anomeric trifluoroborates underwent couplings with aryl, heteroaryl and vinyl bromides to generate α-configured 2-deoxy-C-glycosides (Scheme 21a). An alternative approach to the synthesis of glycosyl trifluoroborates was reported soon after by the group of Walczak, who subjected glycosyl chlorides to a sequence of lithium–halogen exchange, borate quenching and transesterification to the MIDA boronate prior to trifluoroborate formation.63 The method provided access to α-stereoisomers, and was conducted on 2-deoxypyranosyl chlorides as well as derivatives having a free 2-OH group. Tolerance for benzyl and silyl ether groups was demonstrated, and trifluoroborates bearing free OH groups could be further elaborated by glycosylation reactions with trichloroacetimidate donors. C-Arylations using Ir-1 and a nickel co-catalyst resulted in α-configured products from 2-deoxypyranosyl starting materials, and mixtures of anomers (∼2:1 to 4:1 α:β) from gluco- and galacto-configured variants having free 2-OH groups (Scheme 21b). Trapping of the glycosyl radicals by C–S and C–Se bond formation was also demonstrated. The methods reported by the two groups likely proceed by similar mechanisms, with SET from the trifluoroborate to the excited photocatalyst serving as the key step to generate the anomeric radical. Conformational and stereoelectronic effects in the radical intermediate control the stereoselectivity of C-glycoside formation, regardless of the initial configuration of the trifluoroborate starting material.
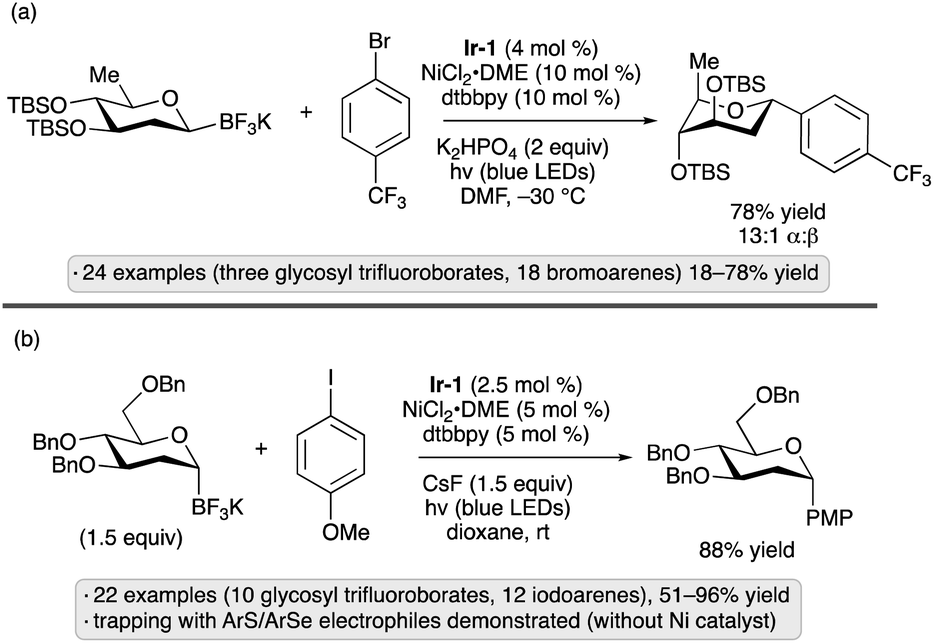 | ||
| Scheme 21 C-Glycoside formation from glycosyl trifluoroborates via photocatalysis reported by (a) Hirai and co-workers;61 (b) Walczak and co-workers.63Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine. | ||
Niu and co-workers have pioneered applications of glycosyl sulfones as precursors to anomeric radicals. Couplings of methallyl glycosyl sulfones with disulfides were achieved upon irradiation in the presence of catalytic Eosin Y, generating products with an axially oriented S-glycosidic linkage (Scheme 22).64 The ability to employ unprotected glycosyl sulfones is a key advantage of this class of precursors. A first-generation variant of the protocol employed pre-synthesized mixed disulfides as reagents, whereas the second-generation protocol relied on in situ disulfide formation using an isothiazolone reagent. Direct conjugations of unprotected carbohydrates to complex peptides, as well as a Cys-containing mutant of an affibody, were demonstrated. Computational modelling supported a proposed mechanism involving addition of a thiyl radical (likely formed by energy transfer from electronically excited Eosin Y) to the methyallyl group, followed by fragmentation with loss of SO2 to form the glycosyl radical. The latter engages disulfide to form the S-glycoside with the release of thiyl radical, which can propagate a chain process.
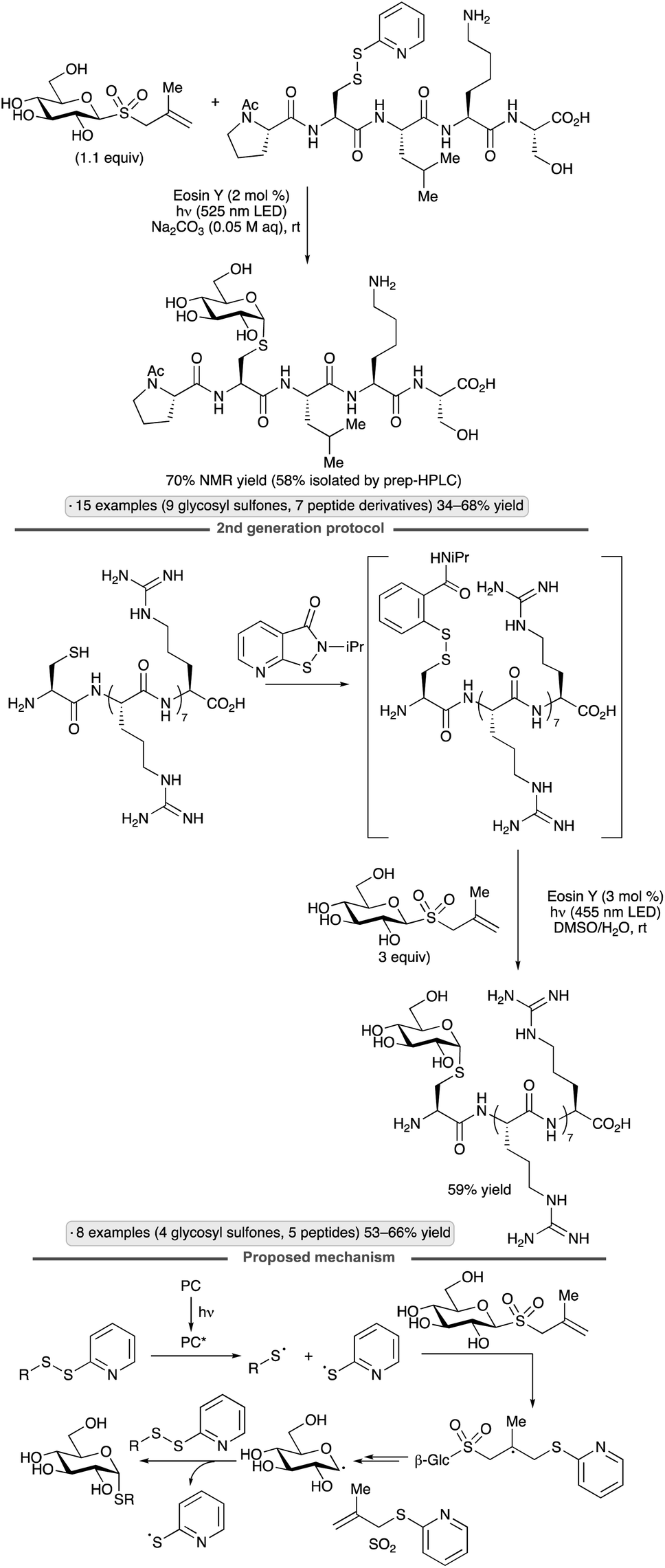 | ||
| Scheme 22 Synthesis of S-glycosides from methallyl glycosyl sulfones. | ||
The Niu group developed conditions for couplings of glycosyl methallyl sulfones with iodoarenes to form C-aryl glycosides.65 Ru(bpy)3Cl2·6H2O was employed as photocatalyst, along with a nickel(II) co-catalyst and sodium toluenesulfinate (Scheme 23). The latter was proposed to undergo oxidation to form a toluenesulfonyl radical, which adds to the methallyl group, ultimately leading to the formation of glycosyl radical in a manner similar to that shown in Scheme 22. Binding of the glycosyl radical to an arylnickel(II) species, followed by reductive elimination, generates the C-aryl glycoside. 1,2-Trans-selective couplings of gluco-, galacto- and manno-configured pyranosyl sulfones, as well as a ribofuranosyl derivative, were achieved, generally without protection of the sugar OH groups; indeed, a direct comparison between the free glucopyranosyl sulfone and the corresponding per-O-benzylated species showed that the former gave a higher yield (81% versus 68%). In addition to iodoarenes, a variety of iodinated heterocyclic partners were successfully subjected to the protocol, including derivatives of benzothiophene, pyridine, quinoline and indole. Computational modelling suggested that the use of unprotected glycosyl radical precursors accelerated the addition to the arylnickel(II) complex.
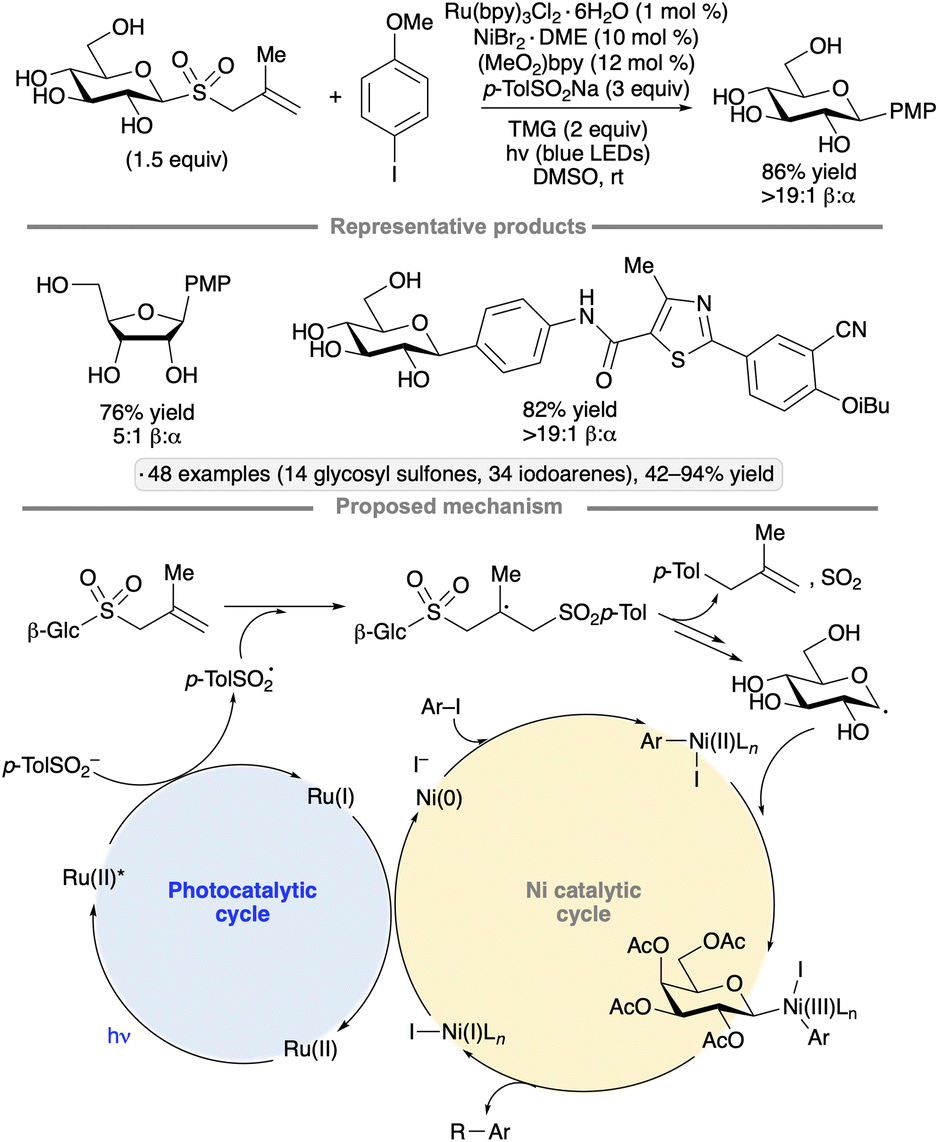 | ||
| Scheme 23 Photoredox/nickel co-catalysis for synthesis of C-aryl glycosides from methallyl glycosyl sulfones. (MeO)2bpy denotes 4,4′-dimethoxy-2,2′-bipyridine; TMG denotes 1,1,3,3-tetramethylguanidine; PMP denotes 4-methoxyphenyl. | ||
The group of Koh has achieved couplings of glycosyl heteroaryl sulfones with unsaturated reaction partners upon irradiation in the presence of CsOAc and Hantzsch ester 3 (Scheme 24).66 The authors proposed that a ternary EDA complex derived from the sulfone, 3 and CsOAc underwent photoexcitation and radical formation via electron transfer. Loss of the sulfinate anion generates the glycosyl radical, which can be trapped by additions to alkenes or alkynes, or by cross-couplings with haloalkene, heteroaryl sulfonate or disulfide reagents. The protocol was applied to unprotected heteroaryl glycosyl sulfones, as well as derivatives bearing ester, ether, silyl ether and ketal protective groups. α-Selectivity was obtained using a range of pyranosyl radical precursors, including derivatives of glucose, mannose, galactose and N-acetylneuraminic acid, as well as 2-deoxy congeners, while 2-deoxyribofuranosyl sulfones gave rise to mixtures of anomers.
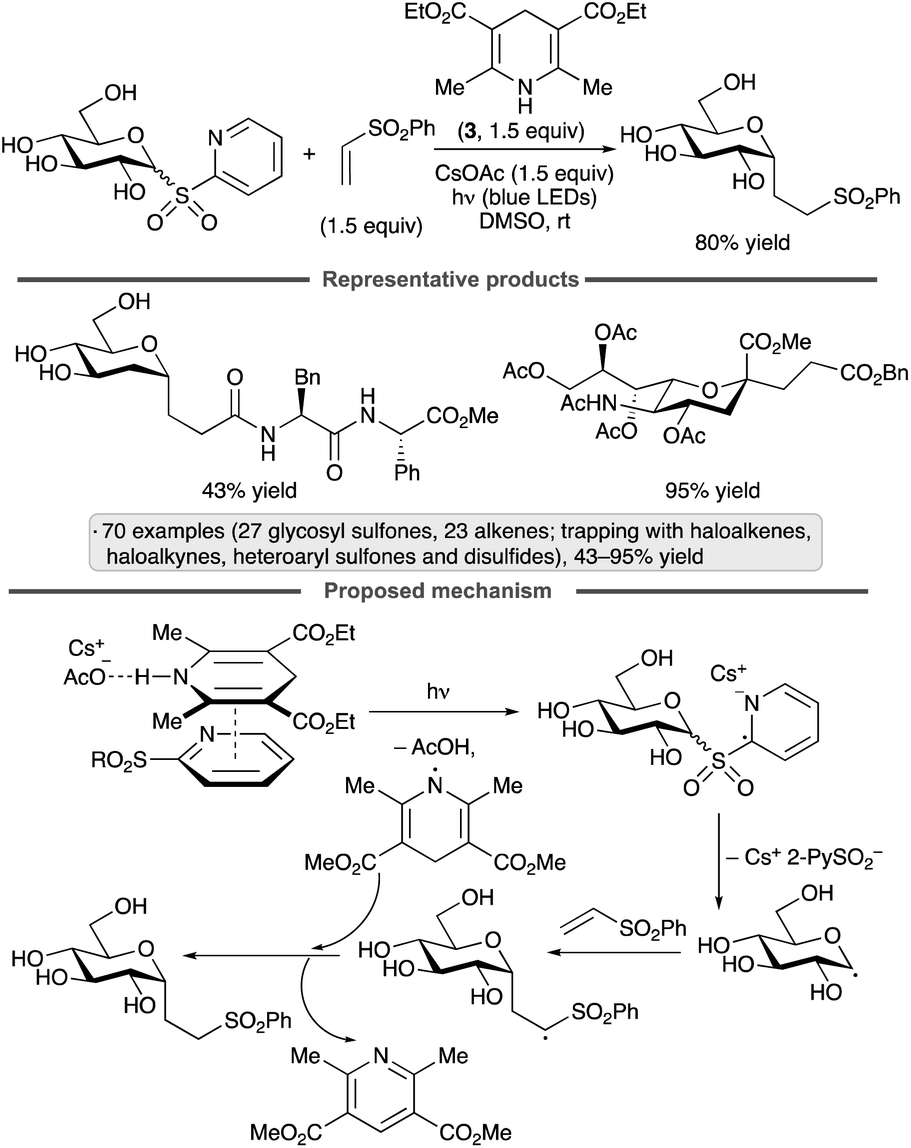 | ||
| Scheme 24 Heteroaryl glycosyl sulfones as precursors to anomeric radicals. | ||
Sodium glycosyl sulfinates have been explored as precursors to anomeric radicals by the groups of Niu67 and Hirai.68 Niu's group generated the sulfinate salts from carboxyethyl sulfone precursors via β-elimination, while Hirai and co-workers used a base-mediated cleavage of 2-pyrimidyl sulfones. The protocol initially developed by Niu and co-workers employed trifluoroacetic acid and tert-butyl hydroperoxide to activate the sulfinate towards anomeric radical formation in the absence of light, likely by hydrogen atom transfer from the sulfinic acid. The group went on to develop a photocatalytic variant, using Ru(bpy)3Cl2·6H2O in DMSO, to achieve Giese additions to a variety of electron-deficient alkenes (Scheme 25a). The transformation was employed to synthesize DNA–carbohydrate conjugates. The method developed by the group of Hirai employed an acridinium photocatalyst in a 4:1 methanol/water mixture (Scheme 25b). In both cases, the formation of the glycosyl radical likely takes place by SET from the sulfinate to the photoexcited catalyst, followed by loss of SO2. It is noteworthy that the transformations can be accomplished without additional reagents, and are tolerant of polar, protic solvents, including water.
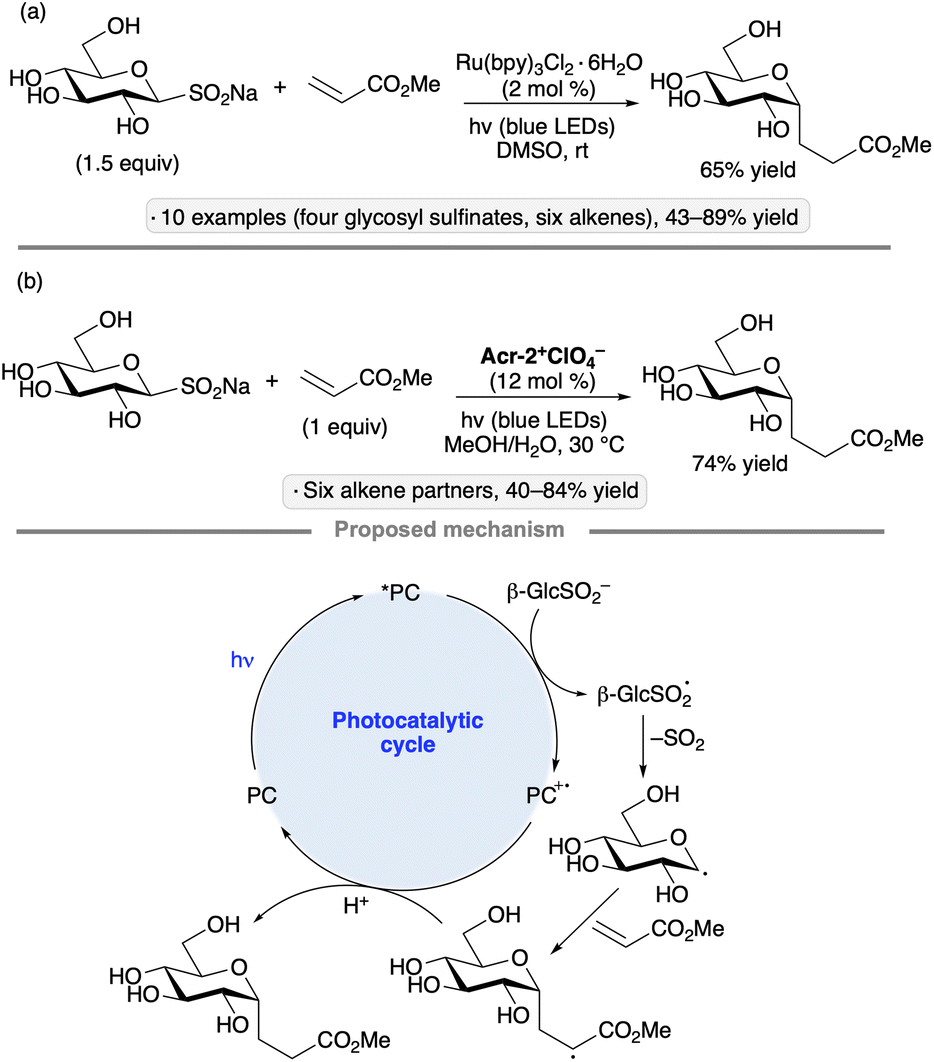 | ||
| Scheme 25 Photocatalytic C-glycosidations of sodium pyranosyl sulfinates developed by (a) Niu and co-workers67b and (b) Hirai and co-workers.68 See Fig. 1 for the structure of Acr-2+. | ||
A method for the synthesis of pyridyl C-glycosides from methyl glycosyl sulfoxides was developed by Niu, Zhang and co-workers (Scheme 26).69 The mechanism proposed by the authors is a radical chain, initiated by the formation of methoxy radical from a N-methoxypyridinium salt-derived EDA complex. Radical substitution of the glycosyl sulfoxide by MeO˙ generates a glycosyl radical. Addition of the latter to the N-methoxypyridinium salt, followed by elimination of methoxy radical, resulted in the α-configured, heterocyclic C-glycoside product while propagating the chain.
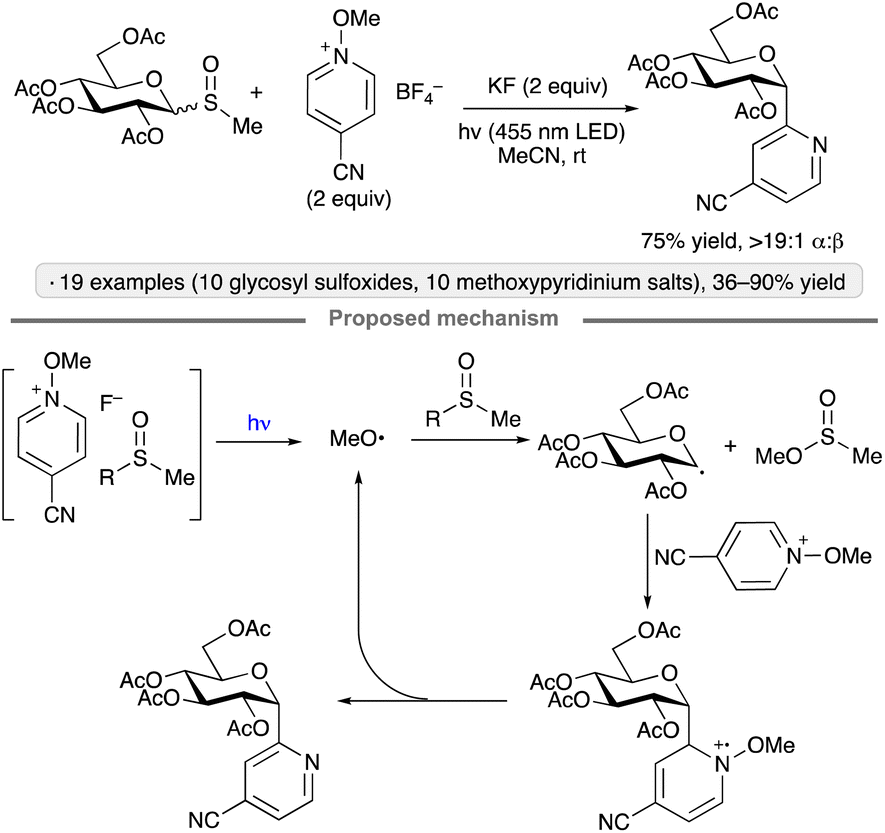 | ||
| Scheme 26 C-Glycoside synthesis via photoinitiated couplings of glycosyl methyl sulfoxides with N-methoxypyridinium salts. | ||
3.3. Rearrangements of anomeric radicals
1,2-Acyloxy migrations (Surzur–Tanner rearrangements) of anomeric radicals provide methods for the synthesis of C2-modified derivatives, as exemplified by Giese and co-workers’ synthesis of 2-deoxysugars from per-O-acylated glycosyl bromides upon treatment with Bu3SnH and AIBN.70 Recent work by the group of Ngai has added a new dimension to this mode of reactivity – an example of a class of transformations known as ‘spin-center shifts’71 – using transition metal complexes to trigger the 1,2-acyloxy migrations and to enable subsequent functionalization of the resulting radicals. The group's initial report described the palladium-catalyzed reactivity of 2-O-acylated pyranosyl halides upon irradiation with blue LEDs; conditions were identified to generate the corresponding 2-deoxyglycosyl esters, their 2-deuterated isotopologs, or the 2-iodo-2-deoxyglycosyl esters arising from an isomerization reaction (Scheme 27).72 Photoexcitation of the Pd(0) complex provided access to organometallic intermediates having radical character, thus enabling the acyloxy migration to take place. A concerted mechanism was proposed for the latter based on the results of radical clock, crossover and substituent effect experiments. Atom transfer or reductive elimination steps deliver the products of reduction, deuteration or isomerization with regeneration of the Pd(0) catalyst.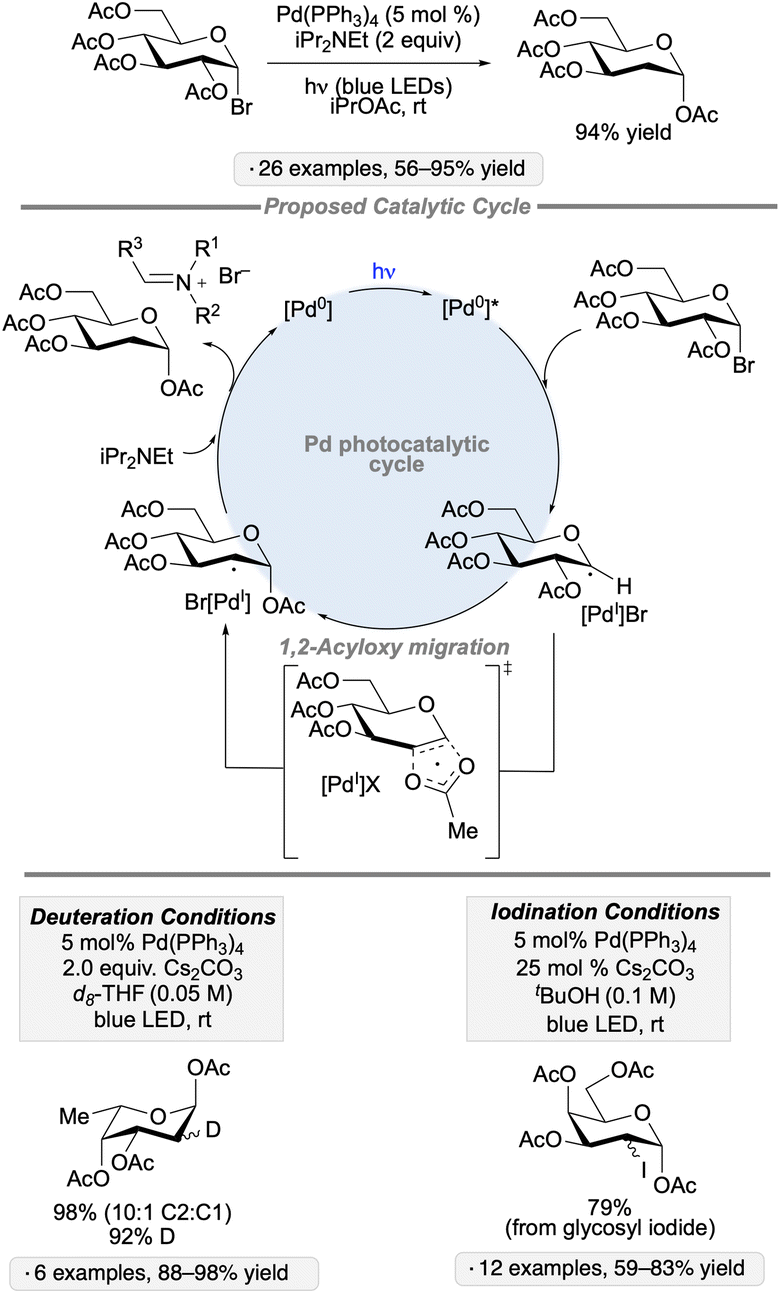 | ||
| Scheme 27 Visible light-induced, Pd-catalyzed reductions and isomerizations of glycosyl halides via 1,2-acyloxy migration. | ||
Building on this result, the Ngai group explored alternative reagents to trap the C2 radical intermediates, thus allowing access to new product classes. Using silyl enol ethers in combination with a Pd(Xantphos) catalyst, 2-oxoethyl substituents were installed at the 2-position,73 while visible light-induced, migratory Mizoroki–Heck-type reactions with styrene derivatives resulted in 2-alkenylated pyranoside products (Scheme 28).74 For both transformations, products having an axially oriented substituent at the 2-position were obtained from gluco- and galacto-configured glycosyl halides. The former gave higher diastereoselectivities than the latter, likely due to steric shielding of the β-face by the axial group at C4 when using galactopyranosides. A subsequent report from the group showed that C2-arylation could be achieved by nickel catalyzed couplings with boronic acids (without the need for visible light excitation).75
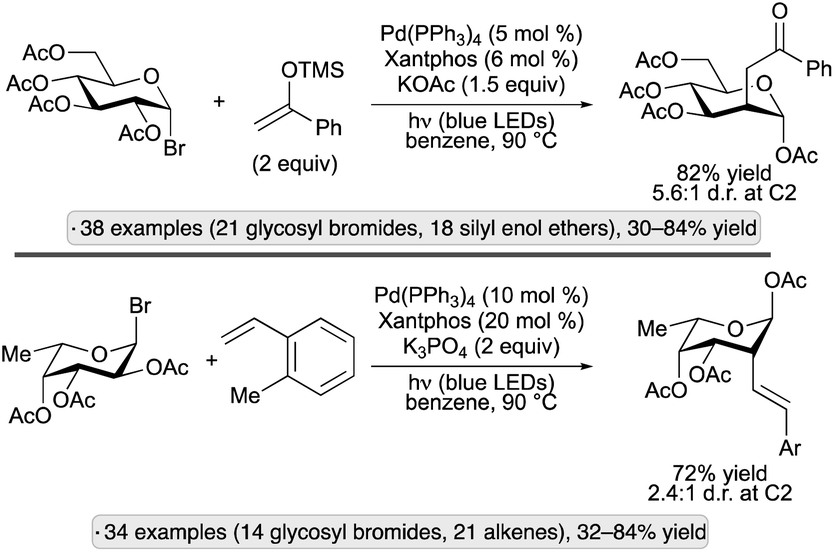 | ||
| Scheme 28 Visible light-induced, Pd-catalyzed couplings of acetylated glycosyl bromides with silyl enol ethers and styrene derivatives. | ||
4. Radicals at non-anomeric positions via C–C, C–O or C–X bond homolysis
4.1. Synthesis of 2-deoxyglycosides via radical substitution at C2
The stereoselective synthesis of 2-deoxyglycosides is challenging; strategies that rely on anchimeric assistance or steric shielding by a protected OH group at the 2-position are not available in such cases.76 Glycosidation of a donor bearing a temporary substituent at the 2-position, followed by reductive removal of the latter, provides a useful and general solution to this problem.77 Accordingly, mild and selective methods to achieve reductive cleavage of, for example, carbon–halogen or carbon–sulfur bonds at the 2-position are needed. In recent years, photocatalytic and photochemical methods have been developed to address this problem.Building on photocatalytic, reductive deiodinations reported by the groups of Stephenson and Lee,78 Wan and co-workers developed a procedure for the reduction of axial 2-iodo substituents in α-pyranosides (Scheme 29). The precise role of the arenethiol additive, which was used in place of the Hantzsch ester component from the Stephenson protocol, was not established. The yields of deiodination were generally greater than 90%, and an impressive 87% yield was obtained in the fourfold deiodination of a tetrasaccharide derivative. An evaluation of nine alternative protocols for the preparation of an α-2-deoxyglucopyranoside from the 2-iodo precursor revealed that the photocatalytic procedure provided the highest yield.
 | ||
| Scheme 29 Synthesis of an α-2-deoxyglucopyranoside by photocatalytic deiodination. | ||
A photocatalytic protocol for reductive desulfurization of thiosugars has been developed by Dong and co-workers.79 After selective S-deacetylation of a thioester precursor, irradiation with blue light in the presence of Ru(bpy)3Cl2 and tris(carboxyethyl)phosphine hydrochloride (TCEP·HCl) furnished the corresponding deoxysugar (Scheme 30). In combination with a stereoselective glycosidation of manno-configured 2-(thioacetyl)pyranoside donors, the method enabled the selective preparation of α-2-deoxyglycosides. Dethiolations at the anomeric position, as well as C4 and C6, were also demonstrated. In comparison to a method reported earlier by the same group involving direct excitation with UV light from a mercury lamp,80 the photocatalytic method displayed lower levels of side reactivity (e.g., for substrates bearing free OH groups or benzyl ether protective groups). The proposed mechanism involves oxidation of the thiol by photoexcited Ru(bpy)3Cl2, followed by reduction of the radical cation by TCEP.
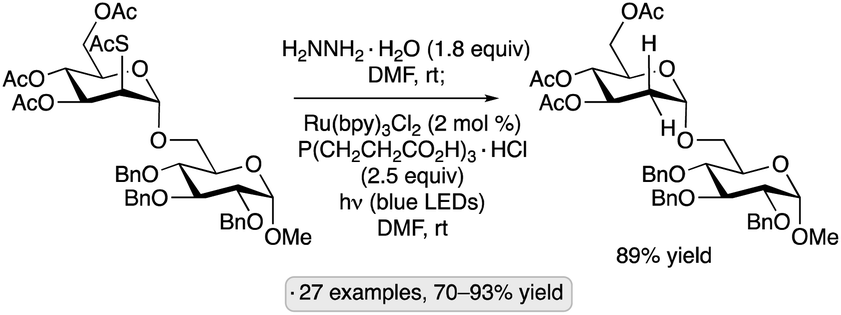 | ||
| Scheme 30 A one-pot procedure for selective S-deacetylation and photocatalytic dethiolation. | ||
4.2. Synthesis of ‘reversed’ glycosides
Methods for radical formation have been developed to enable the synthesis of ‘reversed’ glycosides by incorporation of a substituent at the 5-position of a pyranoside or the 4-position of a furanoside (that is, the other position adjacent to the ring oxygen). Radical fragmentations feature prominently in this class of transformations. Hari and co-workers devised an approach for the preparation of 5′-carba analogues of nucleoside 5′-phosphates by couplings of 5′-O-phthalimidonucleoside derivatives with vinyl phosphonates, using Hantzsch ester 3 as a reductant and fac-Ir(ppy)3 as photocatalyst (Scheme 31).81 Methyl acrylate, acrylonitrile and a vinyl sulfonamide were also employed as coupling partners. The authors proposed that, after oxidation of 3 by photoexcited Ir(ppy)3, SET from the reduced photocatalyst to the N-alkoxyphthalimide group followed by fragmentation of the radical anion led to the formation of an alkoxy radical. β-Fragmentation of the latter with expulsion of formaldehyde led to the carbon-centered radical at the 4′-position, which then added to the alkene in the C–C bond-forming step.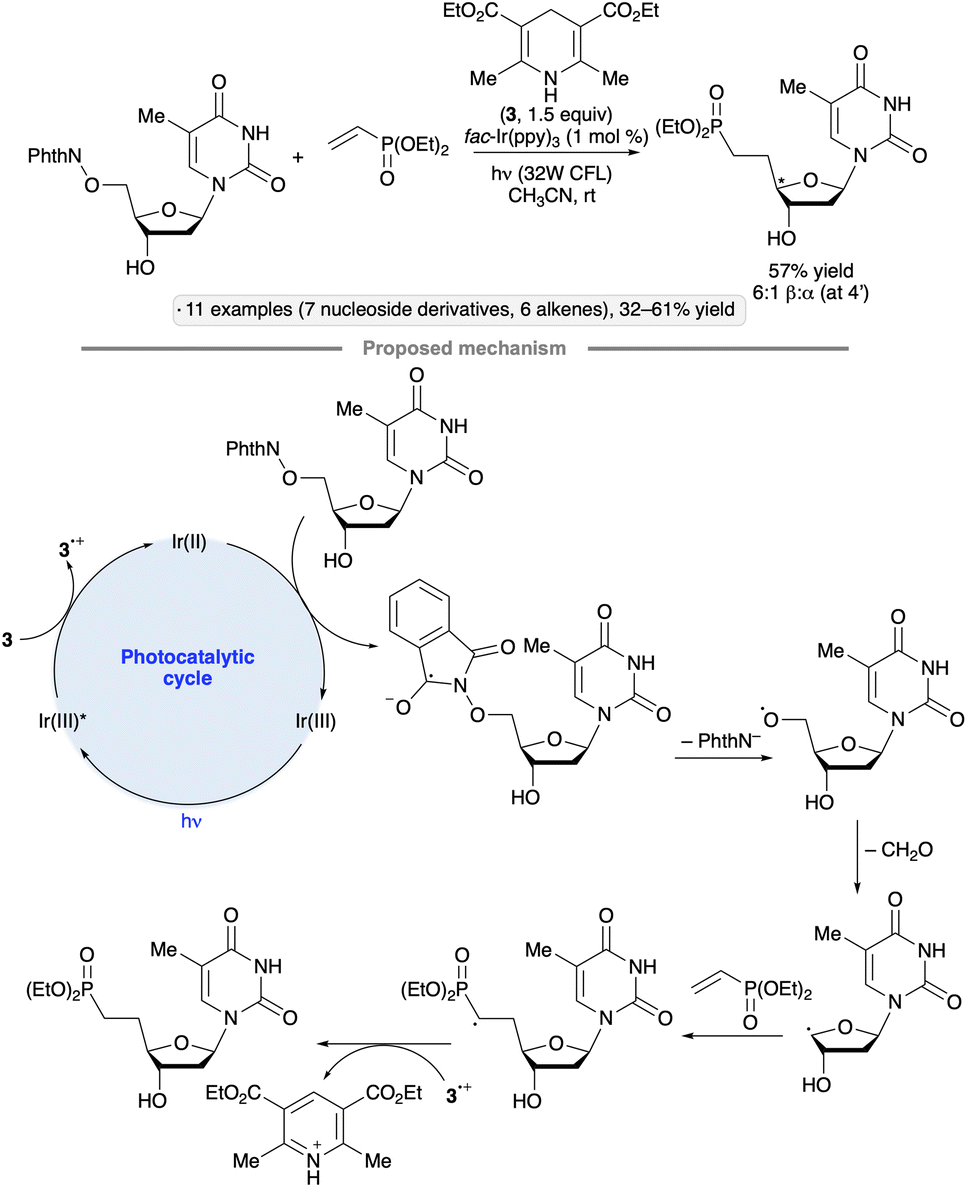 | ||
| Scheme 31 Photocatalytic synthesis of a 5′-carbanucleoside-5′-phosphate by fragmentation of an alkoxy radical. | ||
Elimination of formaldehyde from alkoxy radicals, generated directly from alcohols via cerium photocatalysis, provides another pathway for the synthesis of reversed glycosides.82 Transformations of nucleoside derivatives similar to that shown in Scheme 31 were accomplished directly from substrates having a free 5′-OH group upon irradiation at 365 nm in the presence of CeBr3 and Bu4N+Br− (Scheme 32). Isopropylidene-protected ribonucleosides underwent dehydroxymethylation–alkylation with a trisubstituted alkene to give a mixture of diastereomers at the C4′ and C5′ chirality centers. The authors proposed that the key alkoxy radical was generated by ligand-to-metal charge transfer (LMCT) excitation of a Ce(IV) alkoxide complex generated in situ.
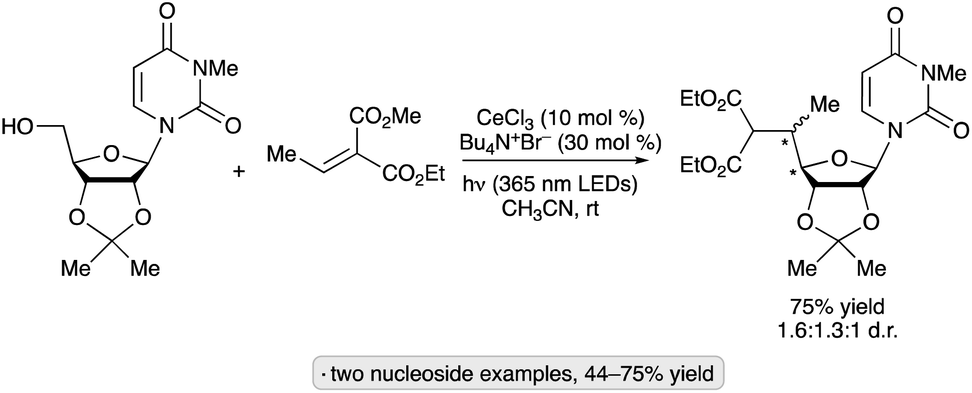 | ||
| Scheme 32 Cerium-photocatalyzed dehydroxymethylation–alkylation of a protected ribonucleoside. | ||
The Molander group employed carbohydrate-substituted DHPs as precursors to reversed C-acyl and C-aryl glycosides through dual photoredox/nickel catalysis.83 The DHP group was constructed via condensation from the formyl-substituted carbohydrate precursor, which in turn arose from oxidation of the primary OH group (Scheme 33a). C-Acylation was accomplished using dimethyl dicarbonate as promoter, and usually resulted in mixtures of stereoisomers – generally less than 5:1 d.r. – at the newly formed chirality center (Scheme 33b). Pyranoside and furanoside substrates were employed, including derivatives with isopropylidene ketal, ether, silyl ether, and free OH substituents. Using aryl or heteroaryl bromides as coupling partners, C-arylation was achieved (e.g., Scheme 33c). The mechanism for radical formation is reminiscent of that depicted in Scheme 17; after loss of the substituted pyridine, the formyl radical undergoes decarbonylation. Light-induced reactions of DHP derivatives of carbohydrates have also been employed to achieve allylations,84 trifluoromethylthiolations85 and couplings to form pyridine-derived C-glycosides.86
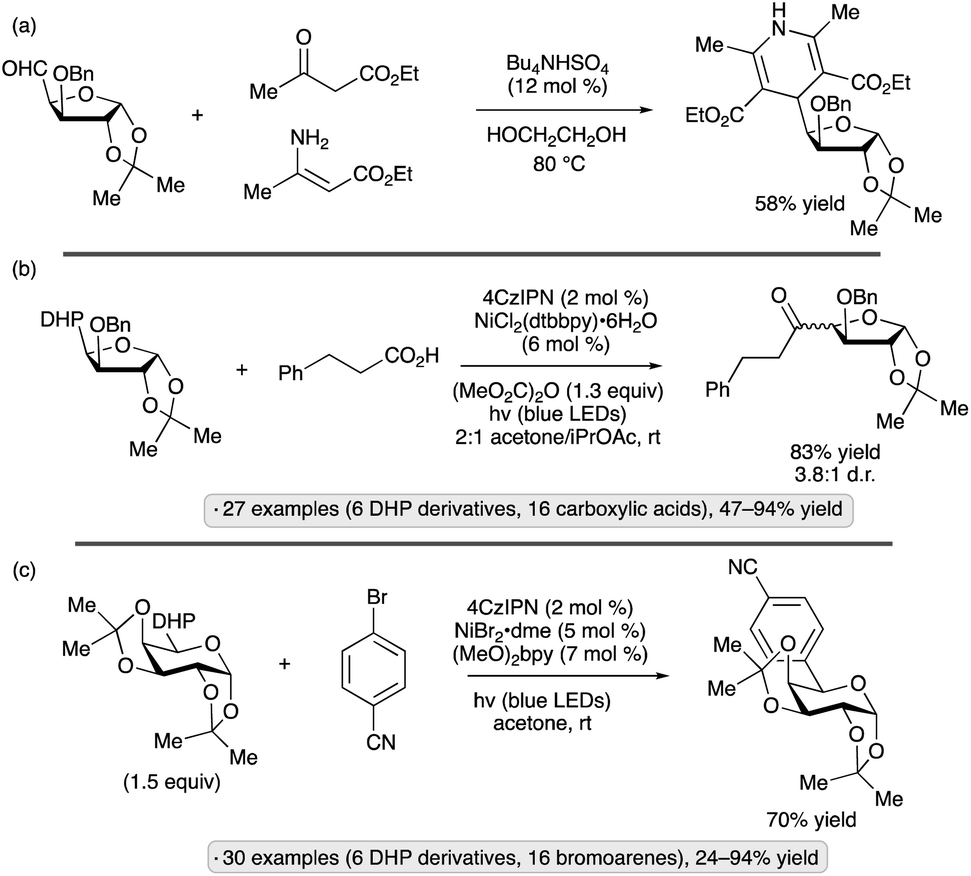 | ||
| Scheme 33 Synthesis of ‘reversed’ C-glycosides from dihydropyridyl (DHP) derivatives. (a) Preparation of substrates by condensation reactions. (b) C-Acylation with carboxylic acids. (c) C-Arylation with bromoarenes. 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine; (MeO)2bpy denotes 4,4′-dimethoxy-2,2′-bipyridine. | ||
Olofsson and co-workers achieved a synthesis of reversed C-vinyl glycosides by couplings of DHP-substituted sugar derivatives with vinylbenziodoxolones (e.g., 8, Scheme 34).87 The use of hypervalent iodine reagents obviates the need for a transition metal co-catalyst, and the process is stereospecific with respect to the configuration of the vinyl group. The protocol was applied to derivatives of galactopyranose, mannopyranose, arabinofuranose and ribofuranose, giving varying degrees of diastereoselectivity, along with various styrene-derived di- and trisubstituted vinylbenziodoxolones. The proposed mechanism involves oxidation of the DHP group by the excited photocatalyst, followed by a concerted radical substitution on the vinylbenziodoxolone. The displaced benziodoxolone radical is reduced by the photocatalyst radical anion, closing the photocatalytic cycle and generating iodobenzoate.
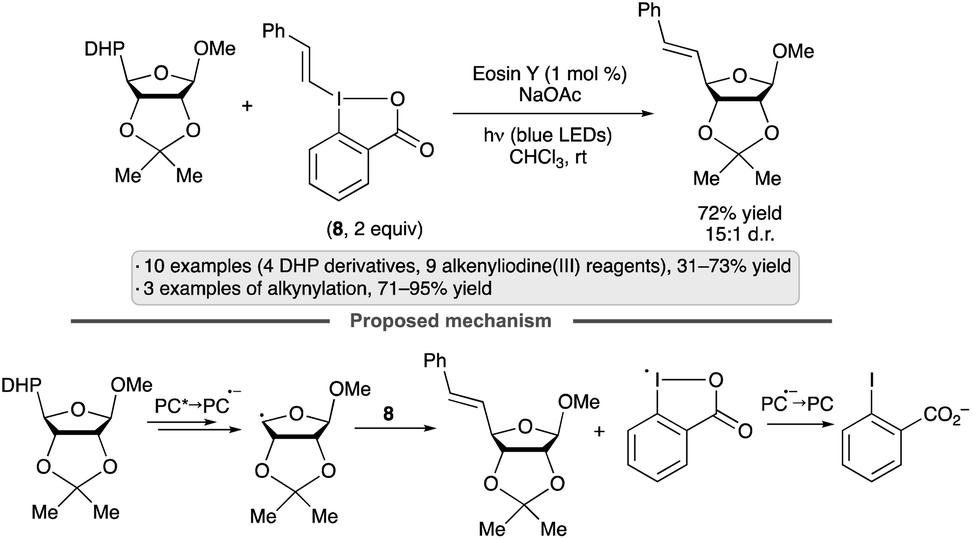 | ||
| Scheme 34 Synthesis of reversed vinyl glycosides by couplings of dihydropyridyl (DHP) derivatives with vinylbenziodoxolones. | ||
Uronic acid-derived N-hydroxytetrachlorophthalimide esters have been used as precursors to reversed C-glycosyl amino acid derivatives using Hantzsch ester 3 as photoreductant and hydrogen atom donor, and glyoxylate-derived imines as electrophilic radical traps, in the absence of a photocatalyst (Scheme 35).88 The anomeric radical intermediate is generated by fragmentation of the N-hydroxyphthalimide-derived radical anion with loss of CO2, while the ammonium salt additive serves to activate the imine by Brønsted acid catalysis. α-Hexo-pyranoside substrates resulted in 1,5-trans-selectivity, while β-pentofuranose derivatives were 1,4-trans-selective. All products were formed as mixtures of diastereomers (∼1:1 d.r.) at the α-amino ester chirality center.
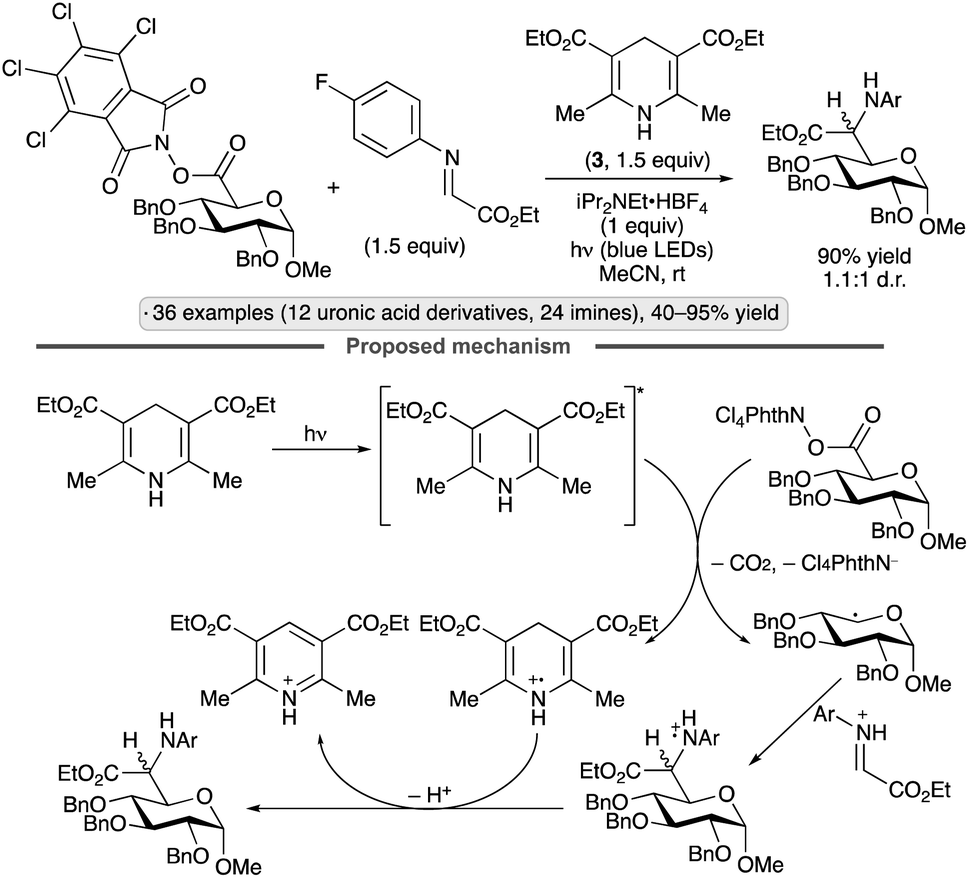 | ||
| Scheme 35 Synthesis of reversed C-glycosyl amino acids from tetrachlorophthaloyl esters. | ||
Photodecarboxylative Giese-type reactions of uronic acid-derived N-hydroxyphthalimide (NHP) esters have been used to synthesize L-configured pyranoside derivatives from D-sugar congeners (Scheme 36).89 The highest levels of L:D stereoselectivity were achieved using bis(ketal)-protected bicyclic substrates. Computational modelling supported the authors' proposal of a 4C1 conformation for the key radical intermediate, which underwent axial-selective alkylation to deliver the L-configured product. A sequence of decarboxylative alkenylation, bis(ketal) deprotection and ozonolysis was used to synthesize methyl β-L-guloside from an α-configured mannuronic acid starting material.
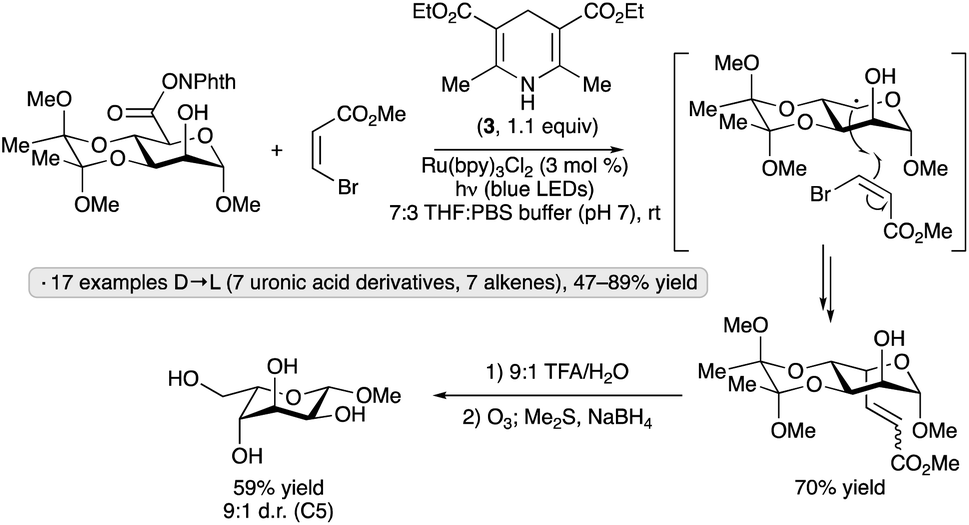 | ||
| Scheme 36 Synthesis of methyl β-L-guloside from a mannuronic acid derivative via photocatalytic, decarboxylative Giese-type reaction. | ||
An N-hydroxyphthalimide ester was employed as the precursor to a 2′-O,4′-C-ethylene-bridged 5-methyluridine derivative via cyclization of a 4′-radical intermediate (Scheme 37).90 The bridged nucleotide analog structure can confer increased affinity for nucleic acid binding partners along with reduced rates of degradation by nuclease enzymes, advantageous features in the context of developing oligonucleotide-based therapeutic agents. Irradiation of the 5-methyluridine-derived NHP ester with a 32 W compact fluorescent light (CFL) bulb in the presence of fac-Ir(ppy)3 and Hantzsch ester 3 resulted in the ethylene-bridged analog with the (S)-configuration at the 6′-position. Minimization of steric strain between the alkene and the 3′-OBn group was proposed as a rationale for the diastereoselectivity of the radical cyclization.
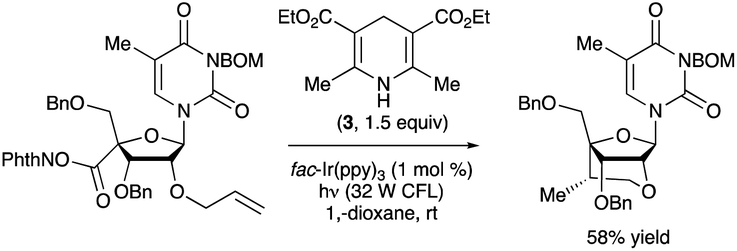 | ||
| Scheme 37 Synthesis of a bridged nucleoside by photocatalytic, decarboxylative radical cyclization. BOM denotes benzyloxymethyl. | ||
Diastereoselective couplings of uronic acid-derived NHP esters with N-(8-quinolinyl)glycine derivatives have been achieved by irradiation with blue LEDs in the presence of Cu(OTf)2 and PHANEPHOS, a chiral phosphine ligand (Scheme 38).91 The stereochemistry at the reversed glycoside chirality center was dependent on the configuration and substitution pattern of the glycoside substrate, whereas the outcome at the α-glycosyl amino acid chirality center was controlled by the chiral phosphine ligand. Armed furanoside and pyranoside substrates bearing benzyl or isopropylidene ketal groups were found to participate effectively. Oligopeptides up to six amino acids in length bearing an N-terminal quinolinylglycine moiety were successfully coupled with the carbohydrate-derived NHP esters. The authors proposed that SET from a photoexcited Cu(I) – quinoline complex to the NHP ester served to initiate the formation of the reversed glycosyl radical. After capture of the radical to form an alkylcopper(II) species, LMCT of the latter and deprotonation generate the glycine-derived radical, ultimately leading to C–C bond-forming reductive elimination. In a closely related, contemporaneous study, Liang and co-workers found that Xyl-BINAP also served as an effective chiral ligand for such coupling reactions.92
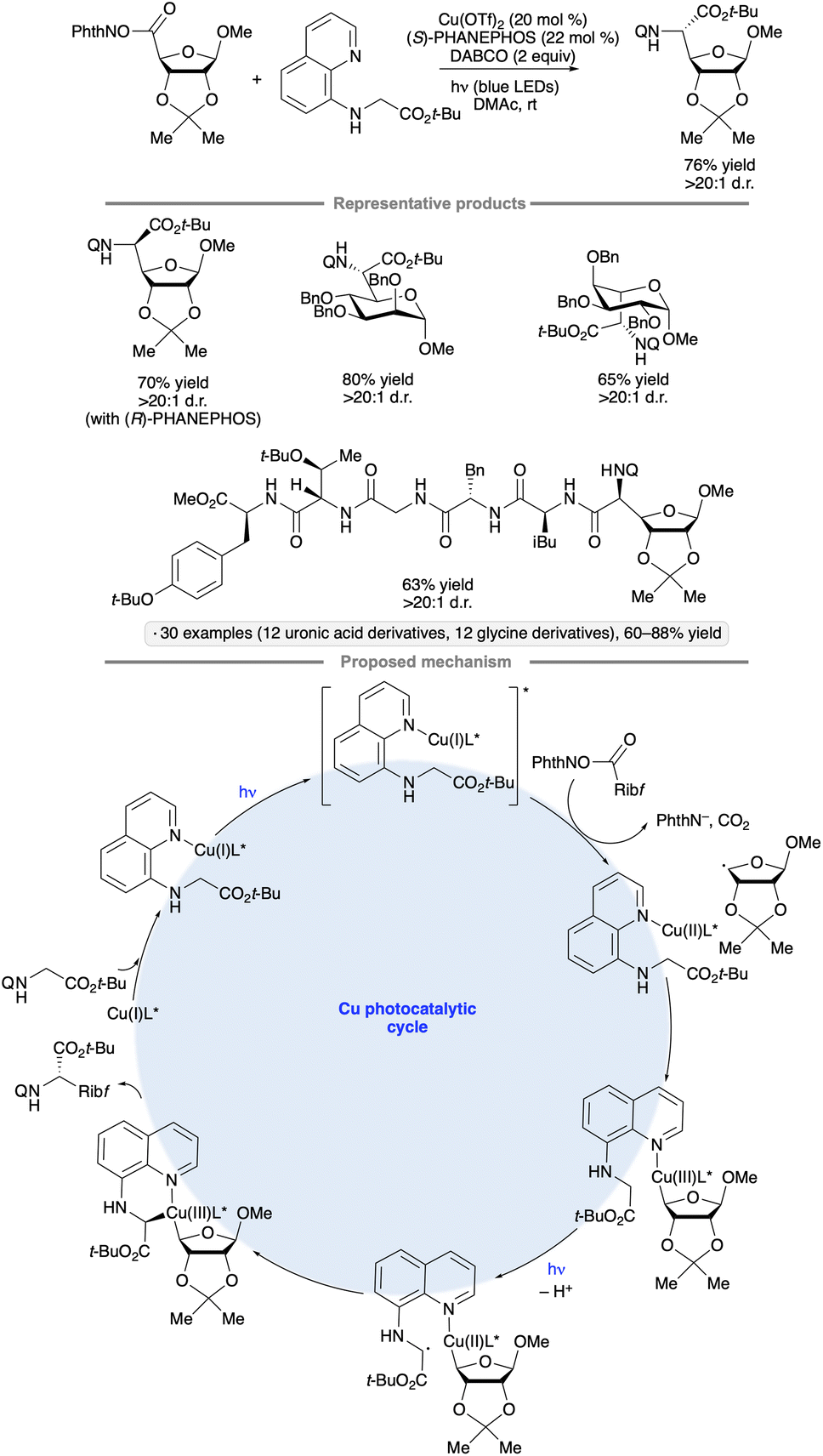 | ||
| Scheme 38 Ligand-controlled, diastereoselective couplings of N-(8-quinolinyl)glycine tert-butyl ester with uronic acid-derived NHP esters. Q denotes 8-quinolinyl. | ||
An alternative approach to the synthesis of reversed C-acyl glycosides was reported by the Molander group (Scheme 39).93 In contrast to the transformation shown in Scheme 33b, which employed DHP derivatives as precursors to reversed glycosyl radicals in the presence of the 4CzIPN photocatalyst, the method relied upon radical formation from the non-sugar component, a NHP ester. The formation of a donor–acceptor complex of the latter with Hantzsch ester 3 enables alkyl radical formation without the need for a photocatalyst. Generation of a mixed dicarbonate from the uronic acid and dimethyl dicarbonate activated the sugar component towards oxidative addition to the nickel complex. The sugar partners employed include uronic acid derivatives of galactopyranose, arabinofuranose, ribofuranose and uridine, as well as 2-ketogluconic acid. Primary, secondary and tertiary NHP esters underwent the coupling, with the latter class giving <50% yields.
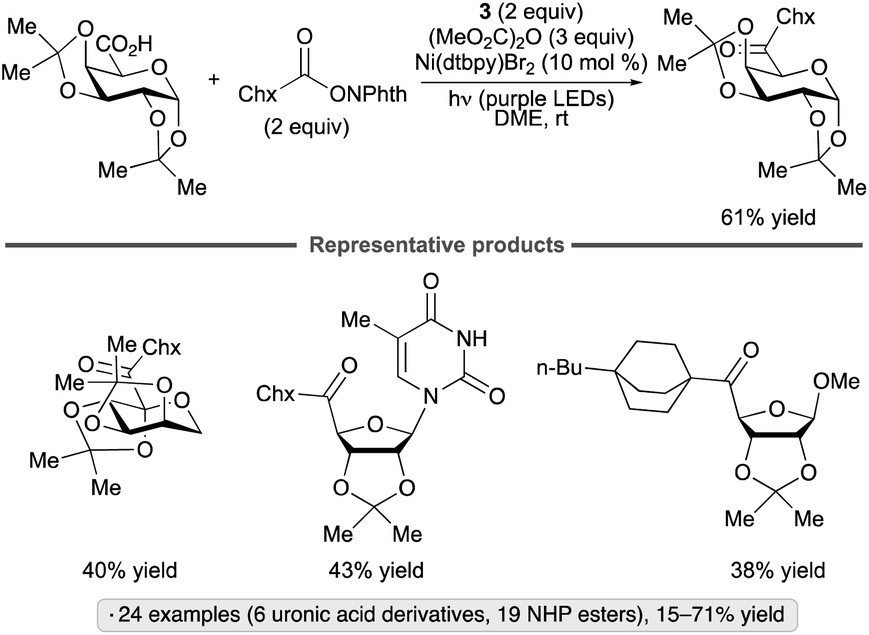 | ||
| Scheme 39 Synthesis of reversed C-acyl glycosides. Chx denotes cyclohexyl. | ||
Reversed glycosyl fluorides have been synthesized by decarboxylative fluorinations of uronic acid derivatives in the presence of an acridinium photocatalyst (Scheme 40).94 Li and co-workers found that disarmed (benzoyl-protected) substrates tended to give lower yields of the reversed glycosyl fluorides than their armed, benzyl-protected counterparts. Nucleoside analogs arising from fluorination at C4′ were also accessed using this approach.
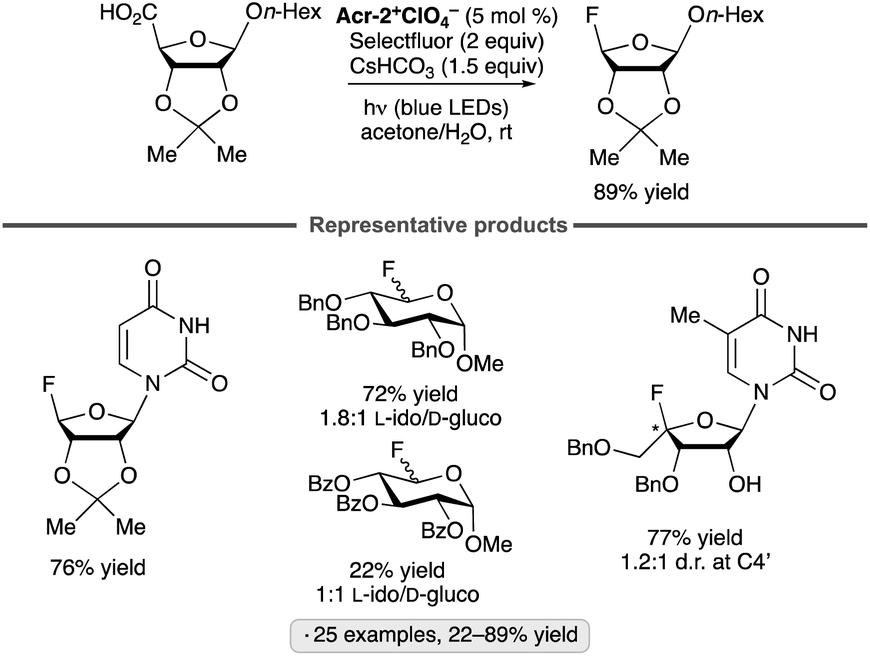 | ||
| Scheme 40 Synthesis of reversed glycosyl fluorides from uronic acid derivatives. See Fig. 1 for the structure of Acr-2+. | ||
4.3. Acyclic products via radical fragmentations
β-Fragmentation of sugar-derived alkoxy radicals provides a pathway for accessing functionalized acyclic products from pyranoside or furanoside precursors. Photocatalysis has been employed to generate the alkoxy radicals needed to accomplish such transformations. The Knowles group developed a three-catalyst system consisting of an Ir(III) photocatalyst, a phosphate salt and a thiol hydrogen atom transfer agent to accomplish the endergonic chain-scission/dehomologation of anomeric hemiacetals (Scheme 41).95 Optimization of the hydrogen bond-accepting phosphate salt proved to be important; the authors speculated that the rate of proton-coupled electron transfer (PCET) to generate the key alkoxy radical was sensitive to structural variation of this component. The protocol was applied effectively to tetra-O-benzylated gluco- and galactopyranoses, as well as a tri-O-benzylated ribofuranose.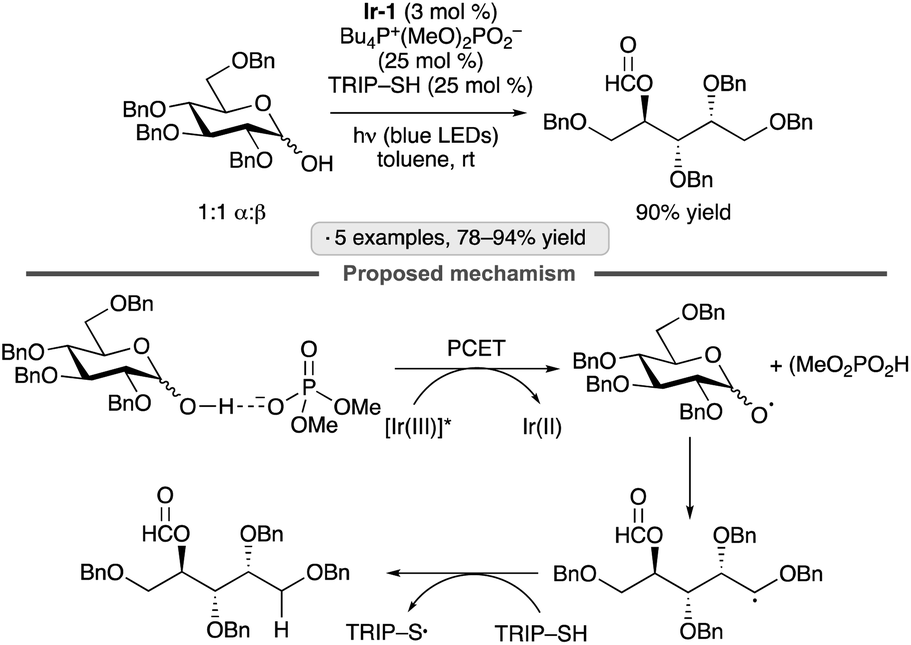 | ||
| Scheme 41 Photocatalytic chain-scission/dehomologation of an anomeric hemiacetal. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; TRIP denotes 2,4,6-triisopropylphenyl. | ||
A sequence of alkoxy radical fragmentation, 1,5-HAT, radical oxidation and intramolecular acetal formation has been used as a key step in the synthesis of polyoxamic acid, a trihydroxylated α-amino acid (Scheme 42).96 A substrate having a free 6-OH group capable of cyclization onto the benzylic oxacarbenium ion was employed to avoid the formation of product mixtures arising from formyl group migration.
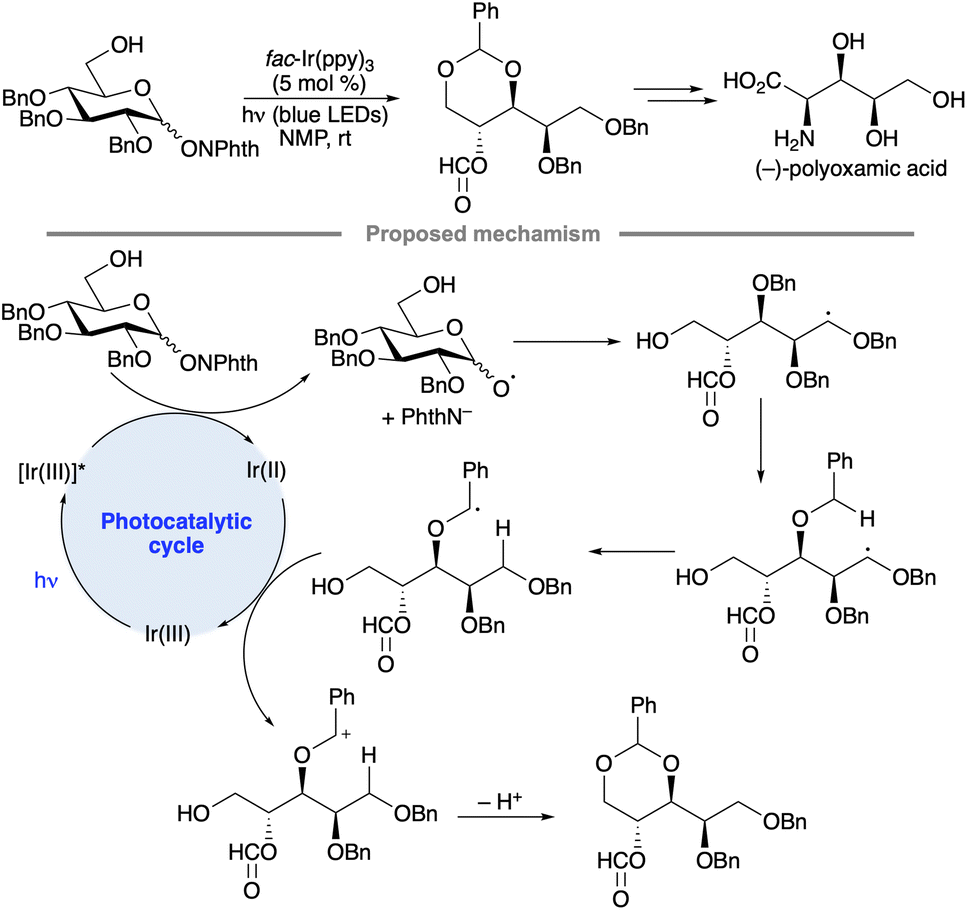 | ||
| Scheme 42 Photocatalytic alkoxy radical fragmentation/1,5-HAT sequence. | ||
Ring fragmentation has been combined with radical borylation to achieve the synthesis of an aldose-derived organoboron compound, a transformation reported by Noble, Aggarwal and co-workers (Scheme 43).97 A radical chain mechanism was proposed, wherein excitation of the N,N-dimethylacetamide (DMAc) complex B2Cat2·DMAc or the N-alkoxyphthalimide by blue light ultimately leads to the formation of a DMAc-stabilized boryl radical. Addition of the latter to the N-alkoxyphthalimide, followed by alkoxy radical formation and ring-opening fragmentation, results in the functionalized alkyl radical that is trapped by B2Cat2 to generate the borylated product.
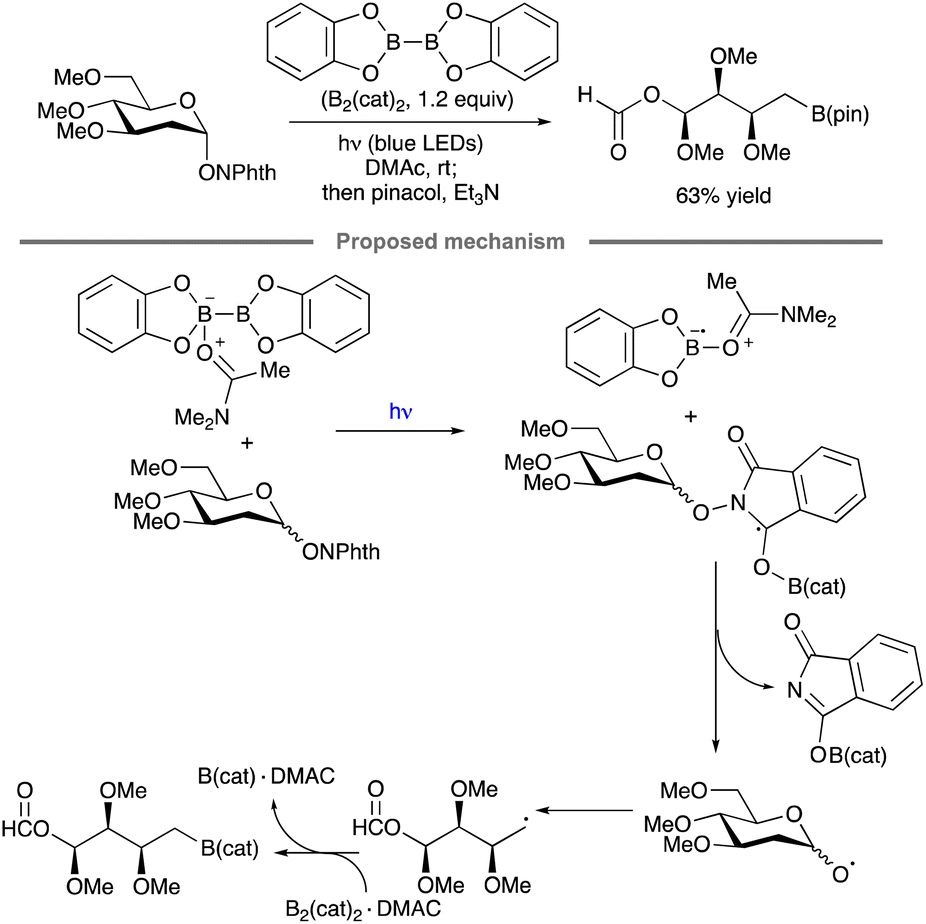 | ||
| Scheme 43 Blue light-initiated synthesis of an aldose-derived boronic ester. | ||
5. Additions to glycals
Glycals, cyclic enol ethers derived from carbohydrates, are versatile reagents that are employed extensively in synthesis.98 Additions of radicals to glycals give rise to sugar-derived radicals at either the anomeric position or at C2; these can be trapped in a variety of ways to achieve further functionalizations.99 In addition to the photocatalytic methods discussed below, photoinitiated protocols for additions to glycals via radical mechanisms have been developed, including thiol–ene reactions that can yield either thioglycosides or 2-thiosugars.100The groups of Xiong and Ye developed a photocatalytic C–H trifluoromethylation of glycals using Umemoto's reagent 1 (Scheme 44).101 Compatibility with benzyl, 4-methoxybenzyl (PMB) and methyl ether groups, as well as esters and a primary arenesulfonate group, was demonstrated. The method was also applied to an unsaturated N-acetylneuraminic acid derivative. The authors' proposed mechanism involves photoreduction of 1 to generate trifluoromethyl radical, which adds to the 2-position of the glycal. Oxidation of the anomeric radical forms an oxacarbenium ion, which undergoes elimination to form the functionalized glycal product.
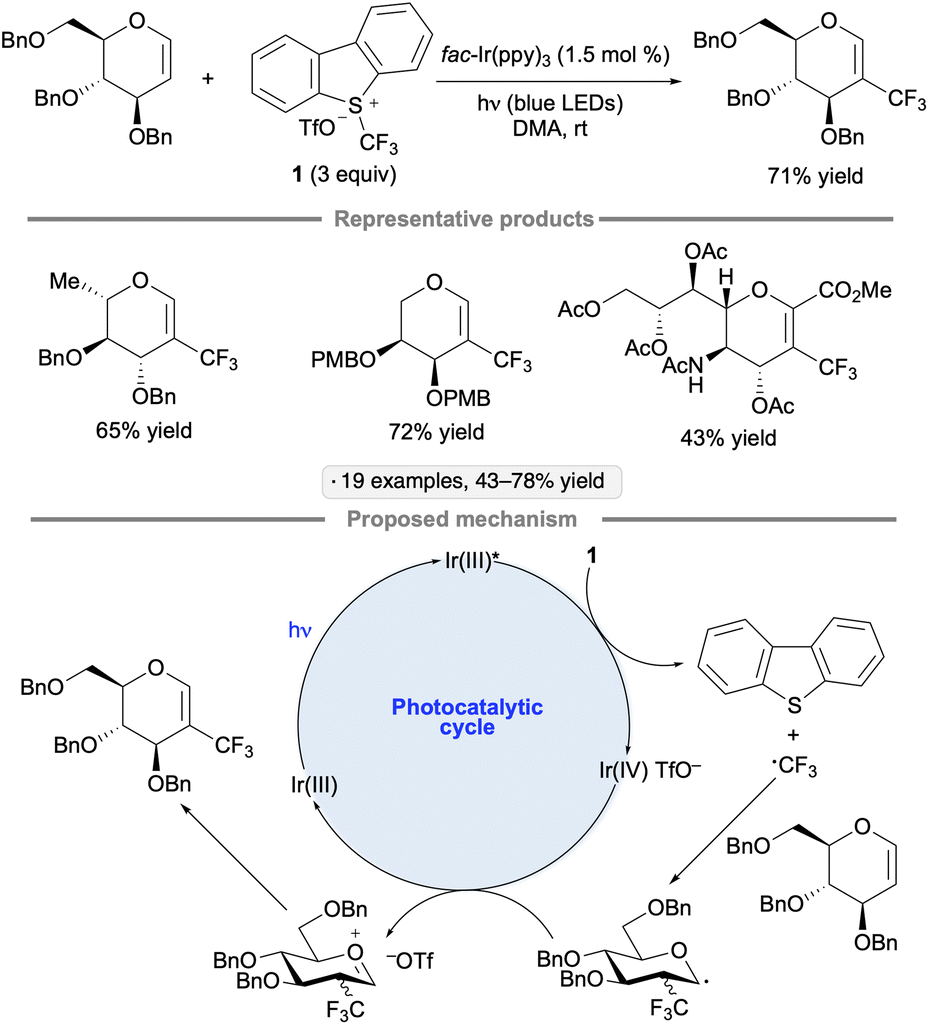 | ||
| Scheme 44 Photocatalytic C–H trifluoromethylation of glycals with Umemoto's reagent. | ||
Exo-glycals, isomers having an exocyclic alkene group,102 have also been subjected to photocatalytic C–H trifluoromethylations with the 2,8-difluorinated Umemoto reagent 6.103Z-Configured products were obtained selectively from pyranose- and furanose-derived exo-glycal substrates bearing benzyl ether, silyl ether and ester protective groups (Scheme 45). Computational modelling suggested that both thermodynamic and kinetic factors favor the Z-over the E-configured products. The proposed reaction pathway – trifluoromethyl radical addition to the enol ether, oxidation of the anomeric radical and elimination to generate the functionalized alkene – is analogous to that shown in Scheme 44.
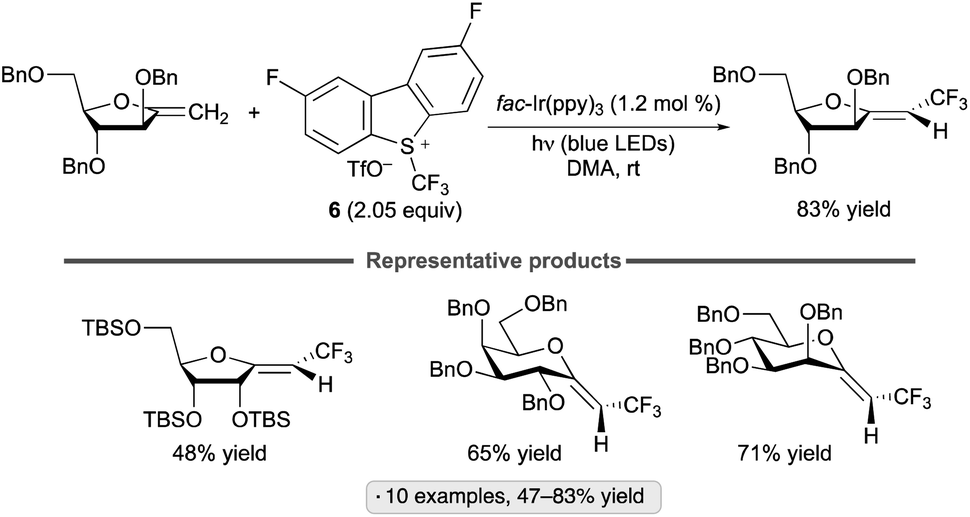 | ||
| Scheme 45 Photocatalytic C–H trifluoromethylation of exo-glycals. | ||
Hu and co-workers explored cyclizations of glycal-derived imidate radicals as a way to achieve C-glycoside synthesis with concomitant C–N bond construction at the 2-position.104 Irradiation of the N-phenylhydroxylamine-derived imidate 7 (ref. 105) in the presence of Ir-3, iPr2NEt and alkene partner (either a styrene derivative or an acrylic acid derivative) resulted in a dihydrooxazole-bearing C-glycoside product (Scheme 46). In the proposed mechanism, the N-centered radical arising from reduction of the imidate engages the glycal in a 5-exo-tet cyclization, forming an anomeric radical that is trapped by the alkene. Hydrogen atom transfer to the resulting stabilized radical generates the C-glycoside product. 1,2-Trans stereoselectivity was observed across the several glycal configurations examined, likely a consequence of steric shielding of the concave face of the fused bicyclic anomeric radical intermediates.
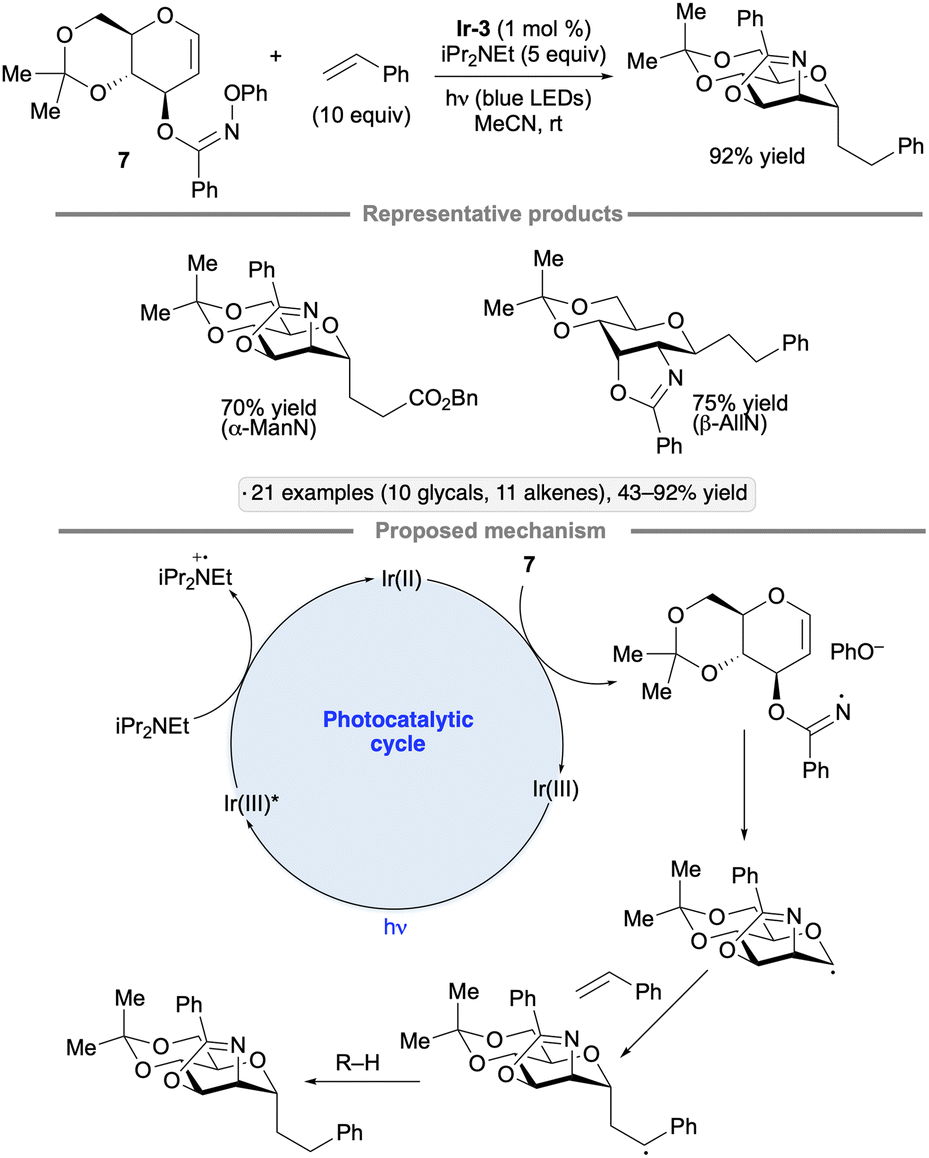 | ||
| Scheme 46 Photocatalytic process for net aminoalkylation of glycals via anomeric radicals. Ir-3 denotes [Ir(ppy)2(dtbbpy)]PF6. | ||
Selective additions of secondary phosphine oxides to the 2-position of glycals has been achieved under blue light irradiation with the 4CzIPN photocatalyst and dilauroyl peroxide (LPO) as an additive (Scheme 47).106 The reaction gave rise to 1-deoxypyranoside products, with the stereochemistry of the newly formed C–P bond at C2 being trans to the substituent at the 3-position. Given that C-alkyl glycosides were not generated in the presence of alkenes as potential trapping agents, and that deuteration at the 1-position resulted when the reaction was conducted with D2O as an additive, the authors proposed that a glycosyl anion was formed by reduction of the glycosyl radical intermediate. The role of dilauroyl peroxide, which was not essential for reactivity but was found to improve the yield of the transformation, is unclear.
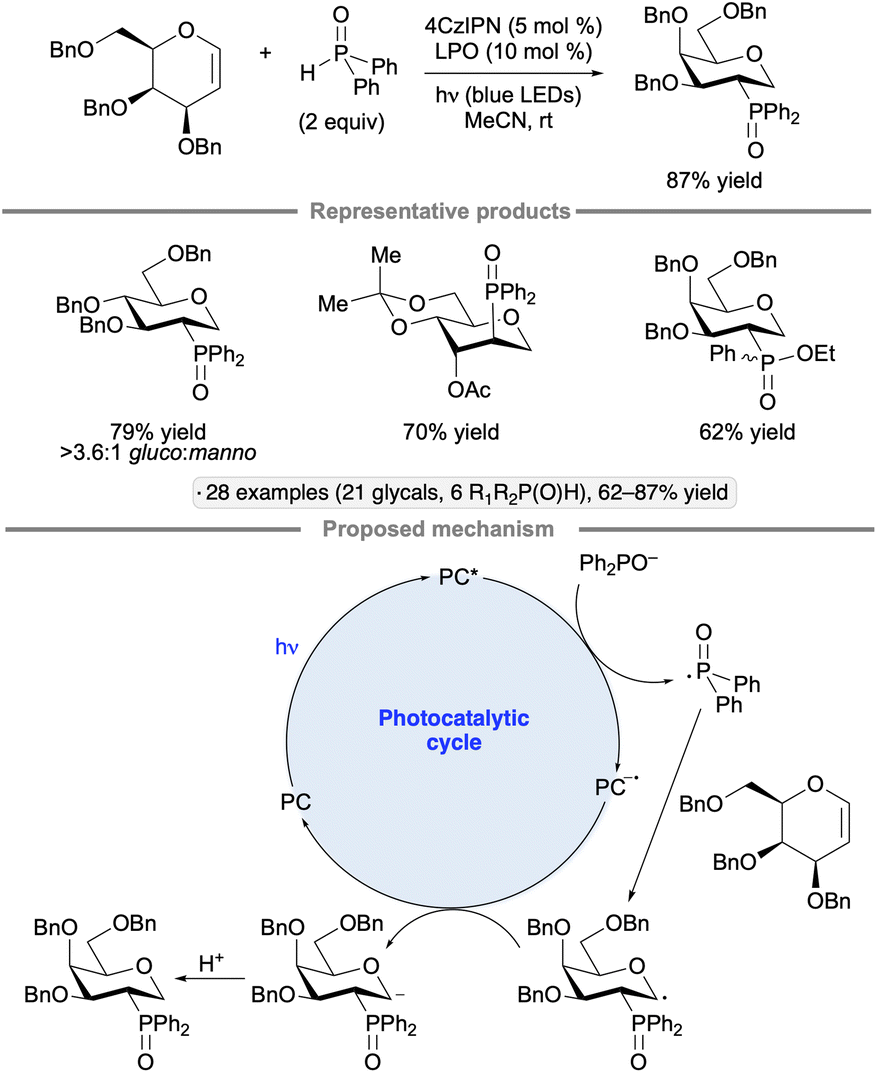 | ||
| Scheme 47 Photocatalytic addition of secondary phosphine oxides to glycals. 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene. LPO denotes dilauroyl peroxide. | ||
Additions of alcohols to glycals, generating 2-deoxyglycoside products, have been achieved by blue light irradiation in the presence of fac-Ir(ppy)3 and BrCH2CN (Scheme 48).107 Glucal and galactal derivatives resulted in α-2-deoxyglycosides, while glycals having an axially oriented substituent at the 3-position (allo stereochemistry) were converted to β-deoxyglycosides. The method was employed in the iterative synthesis of an α-2-deoxygalactopyranoside 20-mer and in the preparation of the cardiac glycoside digoxin. The results of light on/off experiments, along with a quantum yield exceeding 1, were consistent with a chain process initiated by the formation of bromine radical from BrCH2CN and the excited photocatalyst.
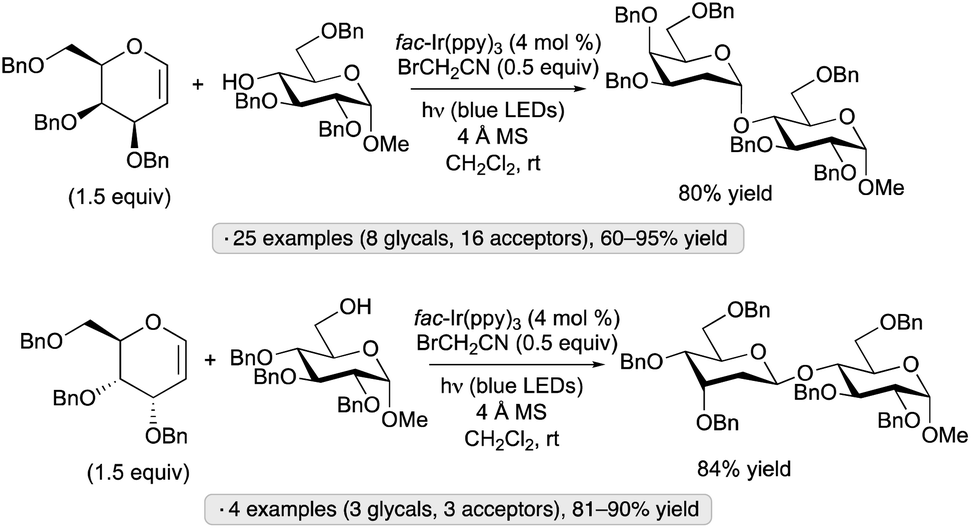 | ||
| Scheme 48 Photoinitiated additions of alcohols to glycals. | ||
2-Acetoxyglycals have been used as substrates for photocatalytic couplings with iodoperfluoroalkanes to furnish perfluoroalkyl C-glycoside products.108 Unlike the transformation shown in Scheme 44, this reaction does not involve an anomeric radical; rather, the regioselectivity of addition to the acetoxy glycal results in the formation of a radical at the 2-position. Under the optimized conditions of 0.5 mol% Ir-1, 4 equivalents of iodoperfluoroalkane, 1 equivalent of Cs2CO3, in acetonitrile under blue LED irradiation, peracetylated glucals and galactals reacted to give the α-configured products in 32% to 59% yields (Scheme 49); the mass balance consisted primarily of unreacted glycal starting material. A mixture of α-lyxo and β-xylo-configured C-glycosides was obtained from a pentose-derived acetoxyglycal, consistent with the higher conformational flexibility of the latter in comparison to the hexose derivatives. In the proposed photocatalytic cycle, carbonate acts as a reductant towards Ir-1*, generating a reducing species that reacts with the iodoperfluoroalkane to form a perfluoroalkyl radical. After addition to the glycal, the radical at C2 accepts a hydrogen atom – likely from the solvent – to form the glycoside product. Mukherjee and co-workers showed that 2-deoxy-O-glycosides can be synthesized from glycals by a related approach involving the addition an alkoxy radical to the 1-position, followed by hydrogen atom transfer to the C2-centered radical.109 The transformation was achieved using tert-butyl hydroperoxide as the alkoxy radical precursor and Eosin Y as photocatalyst.
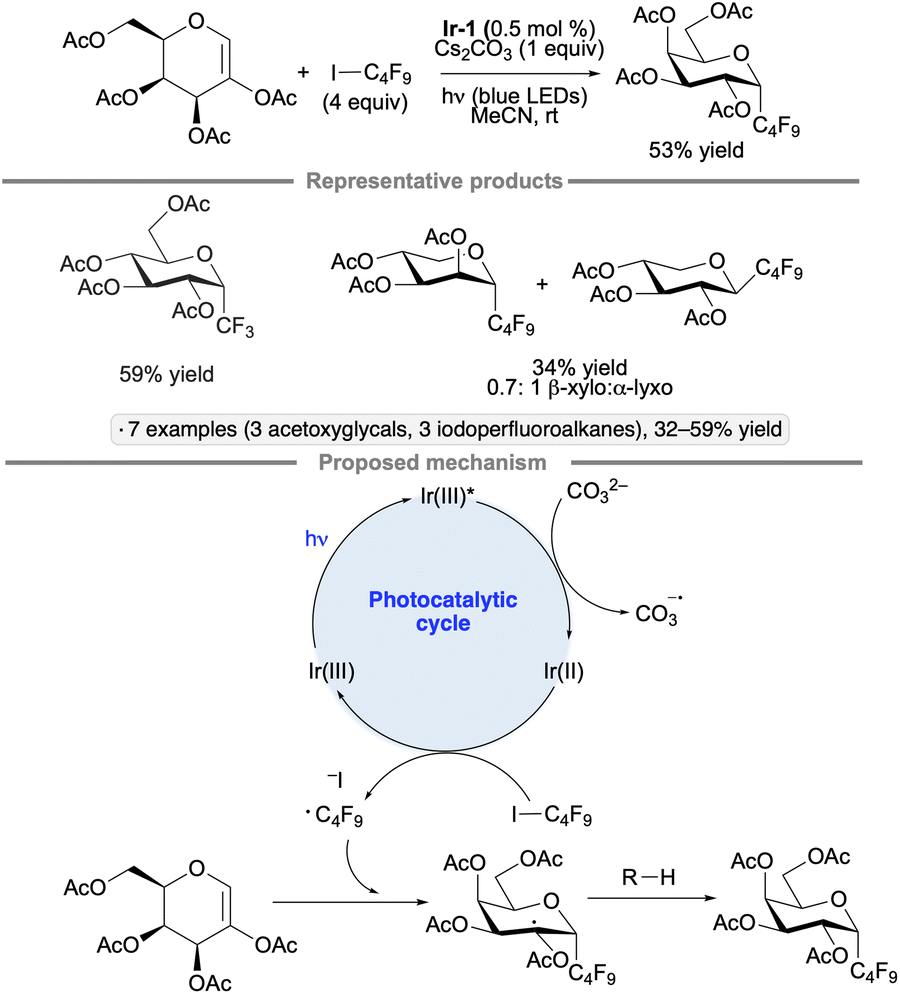 | ||
| Scheme 49 Synthesis of perfluorinated C-glycosides by photocatalytic radical addition to 2-acetoxyglycals. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
6. Hydrogen atom transfer from carbohydrate derivatives
Selective hydrogen atom transfer (HAT) from a C–H bond in a sugar-derived substrate serves as the key step in the mechanism of catalysis for several important classes of enzymes, including the ribonucleotide reductases and S-adenosylmethionine (SAM)-dependent enzymes.110 The discovery and development of processes that combine a photocatalyst with an HAT-mediating co-catalyst111 have created new opportunities to devise laboratory methods that mimic such enzymatic reactions.13 Key precedent was reported by MacMillan and co-workers, who invented a protocol for selective C–H functionalization at the α-position of alcohols using Ir-1, quinuclidine as HAT co-catalyst and tetra-n-butylammonium dihydrogenphosphate (Bu4N+H2PO4−, TBAP) under blue light irradiation.112 Hydrogen bond donation from the hydroxy (OH) group to the H2PO4− anion accelerates HAT to the quinuclidinium radical cation, an electrophilic reagent that reacts preferentially with hydridic C–H bonds due to polarity matching.113 For example, bis(isopropylidene)glucopyranose underwent selective C-alkylation at the 6-position upon coupling with methyl acrylate (Scheme 50). In the proposed photocatalytic cycle, the quinuclidinium radical cation is generated by SET to the excited-state Ir complex. After HAT and addition to the alkene, reduction of the resulting radical, protonation and cyclization result in the lactone product.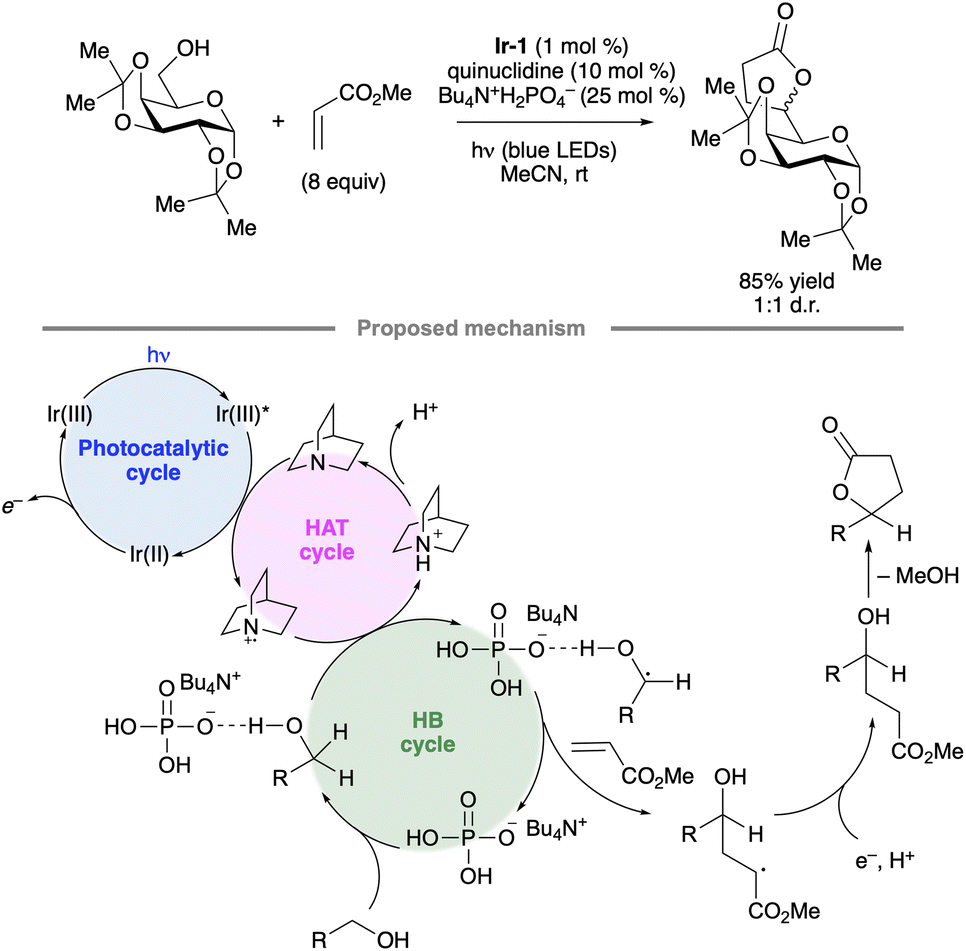 | ||
| Scheme 50 Selective C-alkylation of a protected galactopyranose by photoredox/HAT/hydrogen bonding co-catalysis. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
6.1. C-Alkylations of carbohydrate derivatives
Applications of the Ir-1/quinuclidine/TBAP co-catalyst system to unprotected pyranosides were investigated in a study led by Witte and Minnaard.114 Methyl α-glucopyranoside underwent site-selective C-alkylation at the 3-position using vinyl sulfone, vinyl phosphonate and enone coupling partners (Scheme 51a). Alkylation at C3 was also achieved for 6-O-protected derivatives, methyl α-xylopyranoside and methyl α-allopyranoside. Mixtures of products were obtained from methyl α-mannopyranoside, methyl β-xylopyranoside and methyl β-glucopyranoside, whereas methyl β-galactopyranoside did not react under the conditions employed. Considering that C3 of an α-glucopyranoside is neither the site of the weakest C–H bond115 nor the most sterically accessible position, the selectivity of the HAT step is noteworthy. A subsequent computational study using density functional theory (DFT) indicates that the transition state for HAT from the 3-position is stabilized by C–H⋯O hydrogen bonding interactions between the electron-deficient α-hydrogens of the quinuclidinium cation and the nonbonding electrons of the axially oriented anomeric oxygen.116,117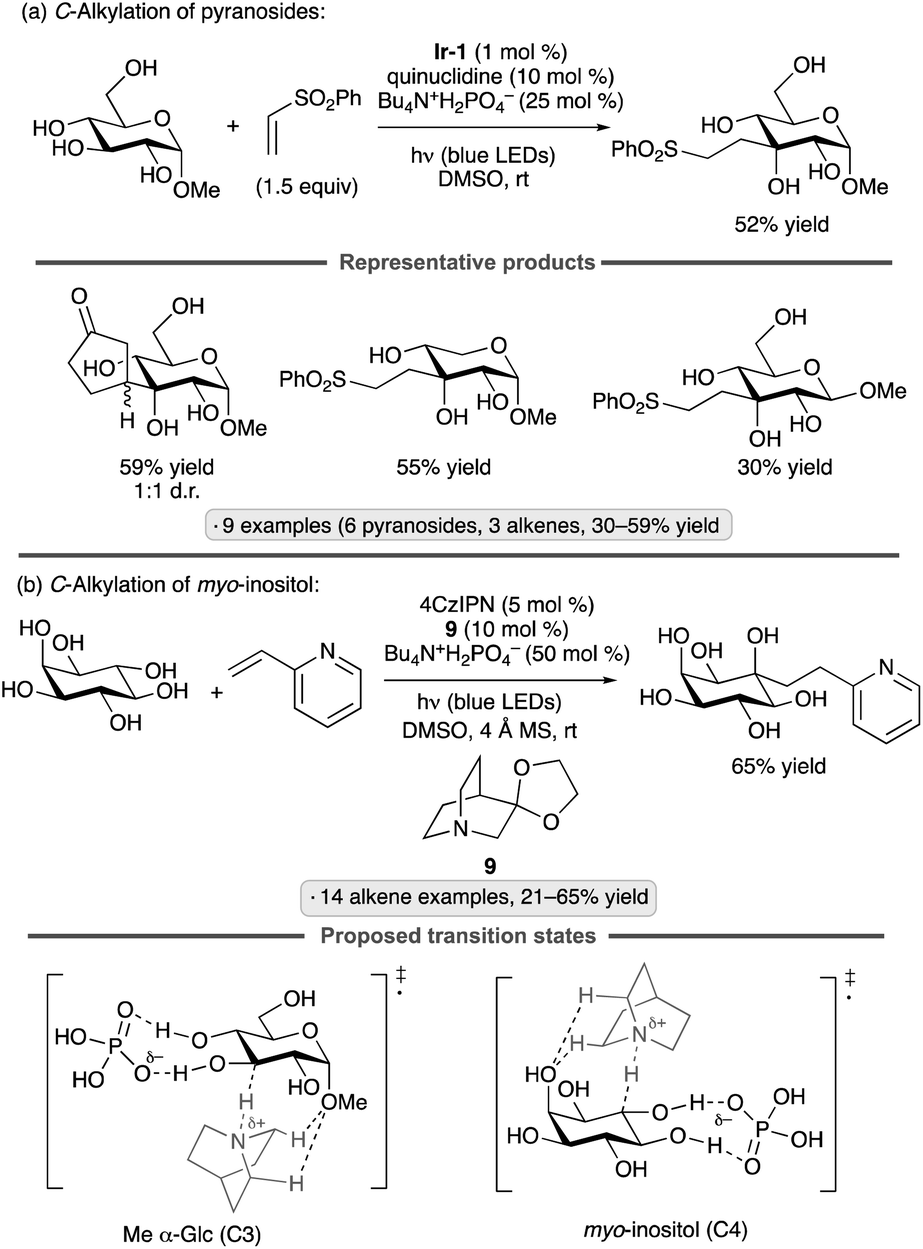 | ||
| Scheme 51 Site-selective C-alkylations of pyranosides and myo-inositol, and proposed transition state structures for selective HAT. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene. | ||
Liu, Wang and co-workers found that 3-quinuclidinone-derived ketal 9, in combination with 4CzIPN and TBAP, constituted an effective catalyst system for couplings of myo-inositol with alkenes, including vinylpyridines, electron-deficient styrene derivatives and phenyl vinyl sulfone (Scheme 51b).118 Products arising from selective HAT from the 4-position, followed by equatorial approach of the alkene to the resulting radical, were obtained. Although a detailed rationale for the observed site-selectivity was not provided, this is another instance of a 1,3-diaxial relationship between the reactive hydrogen and a hydroxy/alkoxy group. It is likely that C–H⋯O hydrogen bonds of the type described in the preceding paragraph operate to stabilize this transition state.
The Taylor group has shown that diphenylborinic acid (Ph2BOH) can act as a co-catalyst in place of TBAP to achieve selective, photocatalytic HAT from cis-diol-containing substrates (Scheme 52).119Galacto- and manno-configured substrates, which did not undergo selective C-alkylation under the prototypical MacMillan-type conditions with co-catalytic TBAP, were coupled with methyl acrylate to yield butyrolactone products. After abstraction of the equatorial hydrogen from the cis-1,2-diol group, C–C bond formation took place with retention of configuration. Lower yields, along with altered diastereoselectivity or site-selectivity in some instances, were observed when the organoboron catalyst was omitted. It was proposed that the formation of a tetracoordinate, anionic borinic ester from the cis-1,2-diol120 accelerated the HAT step through a combination of electrostatic effects (Coulombic attraction to the quinuclidinium radical cation) and polarity matching (stabilization of partial positive charge at carbon in the transition state).113 DFT modelling indicated that borinic ester formation caused a reduction in bond dissociation enthalpies while enhancing an intrinsic kinetic preference for HAT from the 3-position of an α-rhamnopyranoside. Addition of the alkene to the convex face of the bicyclic, borinic ester-derived radical would account for the observed diastereoselectivity.
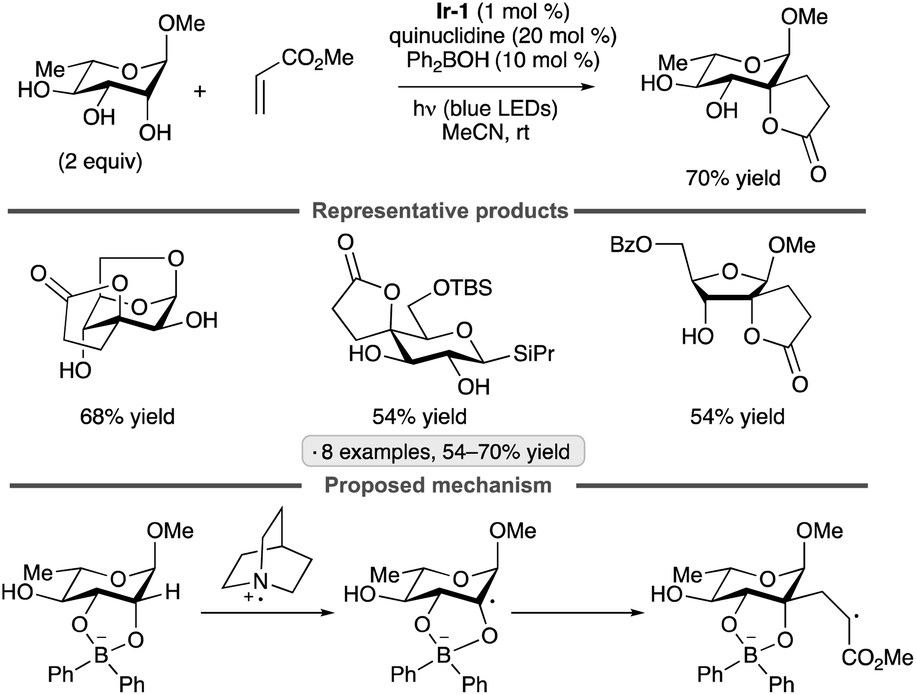 | ||
| Scheme 52 Selective C-alkylation of cis-1,2-diol groups in carbohydrates using diphenylborinic acid (Ph2BOH) as a co-catalyst. Key steps of the proposed mechanism are depicted. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
Evaluation of a range of organotin co-catalysts revealed that Ph2SnCl2 also serves to accelerate HAT from diol groups in carbohydrate-derived substrates (Scheme 53).121 The Ir-1/quinuclidine/PhSnCl2 co-catalyst system gave higher yields than Ir-1/quinuclidine/Ph2BOH for couplings of cis-diols with phenyl vinyl sulfone, acrylonitrile and a vinyl phosphonate, reaction partners for which lactonization of the alkylation product (which was apparently needed for efficient turnover when using organoboron catalysts) cannot take place. Activation of the 1,2-trans diol group in α-glucopyranosides was also possible using Ph2SnCl2, in keeping with established reactivity patterns for organotin-catalyzed O-functionalizations.122
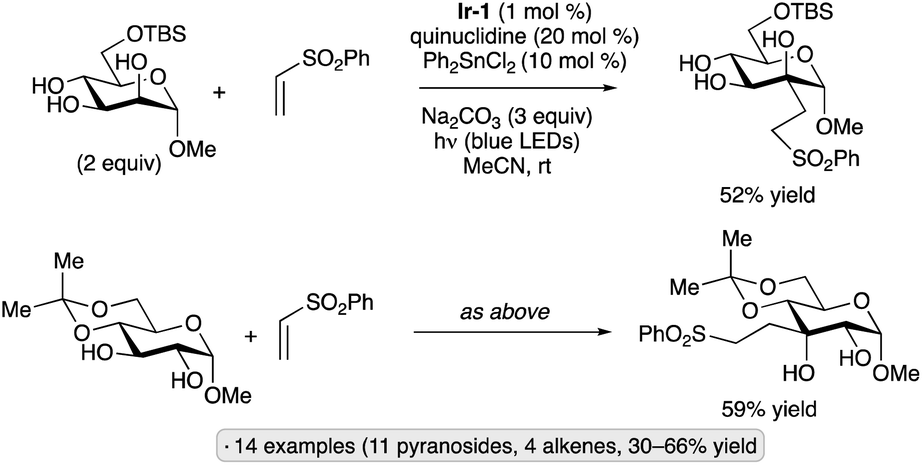 | ||
| Scheme 53 Ph2SnCl2 as a co-catalyst for selective C-alkylations of diol groups in pyranosides. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
A report from Li and Kubinobu documents examples of switching the site of C-alkylation of ketal-protected pyranosides by changing the structure of the hydrogen atom-abstracting photocatalyst (Scheme 54).123 For example, 6-O-acetylated bis(isopropylidene)galactopyranose underwent C3-selective alkylation upon excitation with UV light (365 nm) in the presence of anthraquinone as photocatalyst, versus C4-selective alkylation in the presence of tetrabutylammonium decatungstate (TBADT). The authors proposed that relative bond dissociation enthalpies play a role in the site-selectivity of HAT to the semiquinone radical, whereas photoexcited decatungstate selectively abstracts the hydrogen atom from the most sterically accessible position.
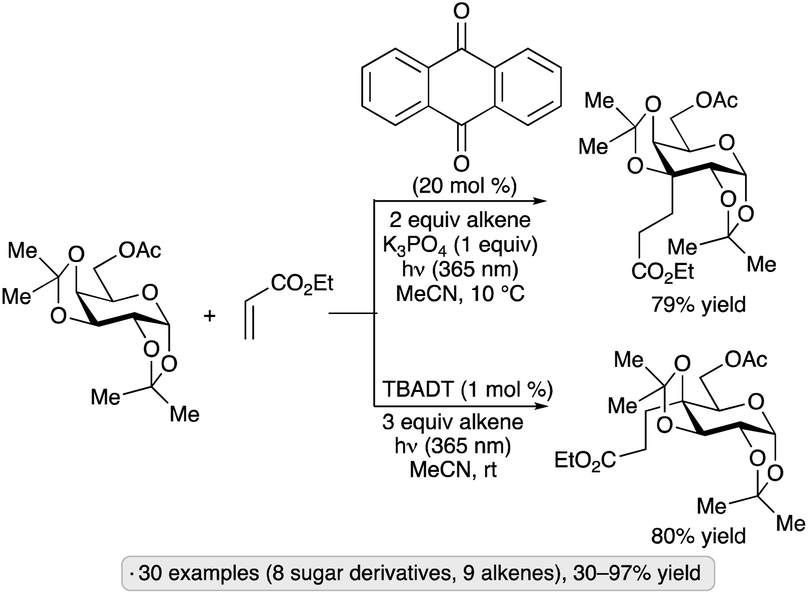 | ||
| Scheme 54 C3- versus C4-selective alkylation of a galactopyranose derivative depending on the photocatalyst employed. TBADT denotes [Bu4N+]4[W10O32]4−. | ||
6.2. Synthesis of ketodeoxysugars by redox isomerization of diol groups
1,2-Radical migrations accompanied by elimination of a leaving group from the position adjacent to the initial radical, also termed ‘spin-center shifts’,71 have been implicated in the reaction pathways for several important enzymes, including the ribonucleotide reductases and SAM-dependent radical lyases.110 Laboratory mimicry of such processes, using photoredox/HAT catalysis to generate the initial sugar-derived radical, has been a productive approach for the development of synthetic methods that provide access to building blocks for the preparation of nucleoside analogs and rare sugars.The Taylor group explored redox isomerizations of furanosides to ketodeoxysugars, taking inspiration from the mode of catalysis of the ribonucleotide reductases. The hypothesis was that an organoboron catalyst could accelerate both the HAT step (see Scheme 52) as well as the subsequent dehydration via spin-center shift.124 Evaluation of diarylborinic and arylboronic acid co-catalysts revealed that only the latter class was effective in promoting the transformation of protected β-ribofuranoside 10 to 3-deoxy-2-ketosugar 11, with pentafluorophenylboronic acid (C6F5B(OH)2) providing the highest yield (Scheme 55). Furanosides having a cis-stereochemical relationship between the 2-OH and 3-OH group (β-ribo- or α-lyxo-configuration) and an ester protective group at the 5-position were appropriate substrates for this transformation; other protective groups at C5 gave rise to lower levels of regioselectivity (3-deoxy versus 2-deoxyketosugar). Stereoselective reduction and N-glycosylation reactions were used to synthesize 3-deoxy analogs of uridine from the ketodeoxysugar products.
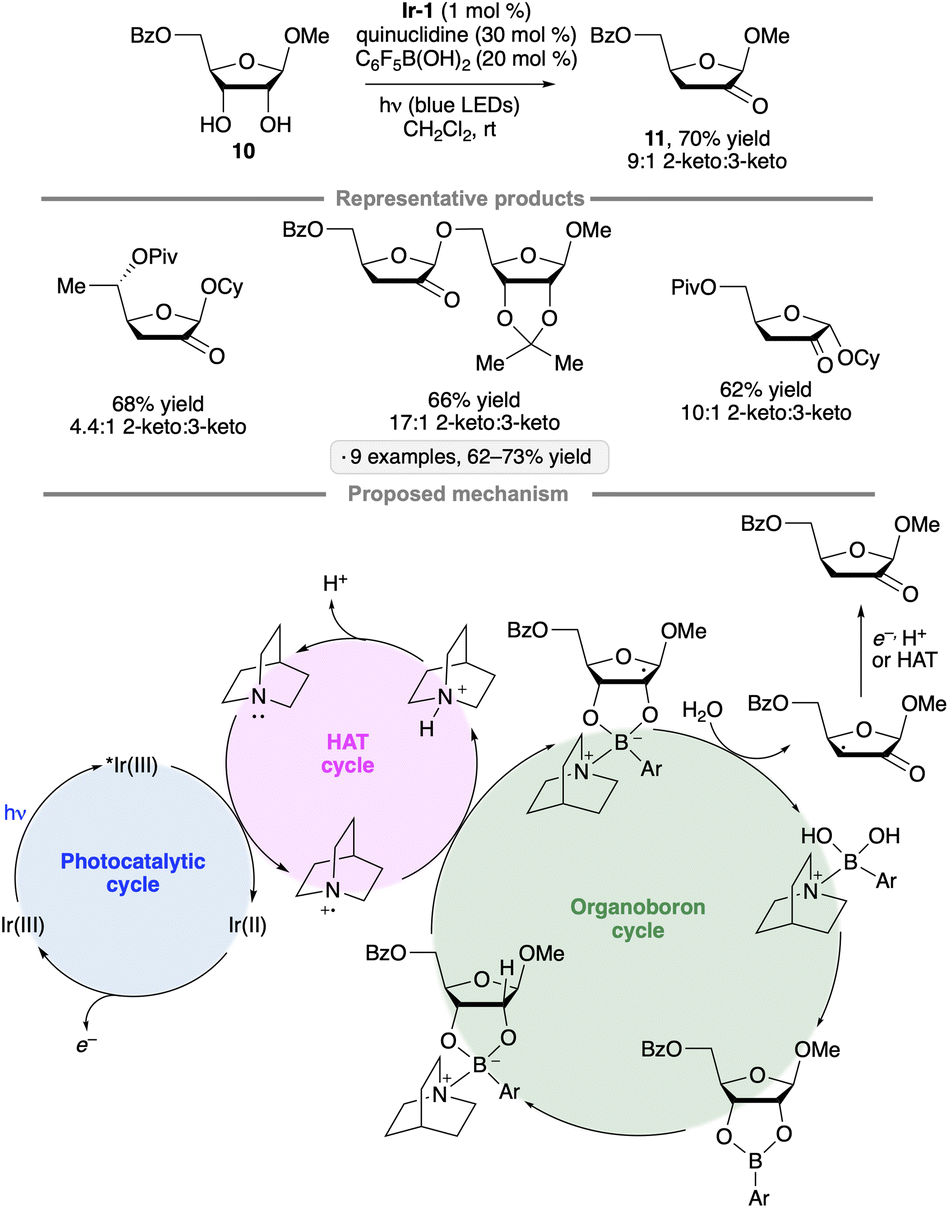 | ||
| Scheme 55 Redox isomerizations of furanosides to 3-deoxy-2-ketosugars. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; Cy denotes cyclohexyl; Ar denotes pentafluorophenyl. | ||
Photocatalytic HAT followed by spin-center shift serves as the basis for a synthesis of 2-deoxyaldonates from free aldoses, reported by Murakami and co-workers (Scheme 56).125 Irradiation of an aqueous solution of the aldose with 365 nm UV light in the presence of sodium hydroxide and a benzophenone-derived bis-sulfonate (BPSS) resulted in the formation of the redox isomerization product. Cyclization to the 2-deoxy-aldonolactones was accomplished by treatment with Me3SiCl (TMSCl) in water, facilitating product isolation. In the proposed reaction mechanism, deprotonation of the hemiacetal OH group activates the C1 position towards HAT to the ketyl radical resulting from photoexcitation of BPSS. Dehydration/radical migration is followed by SET, which closes the photocatalytic cycle.
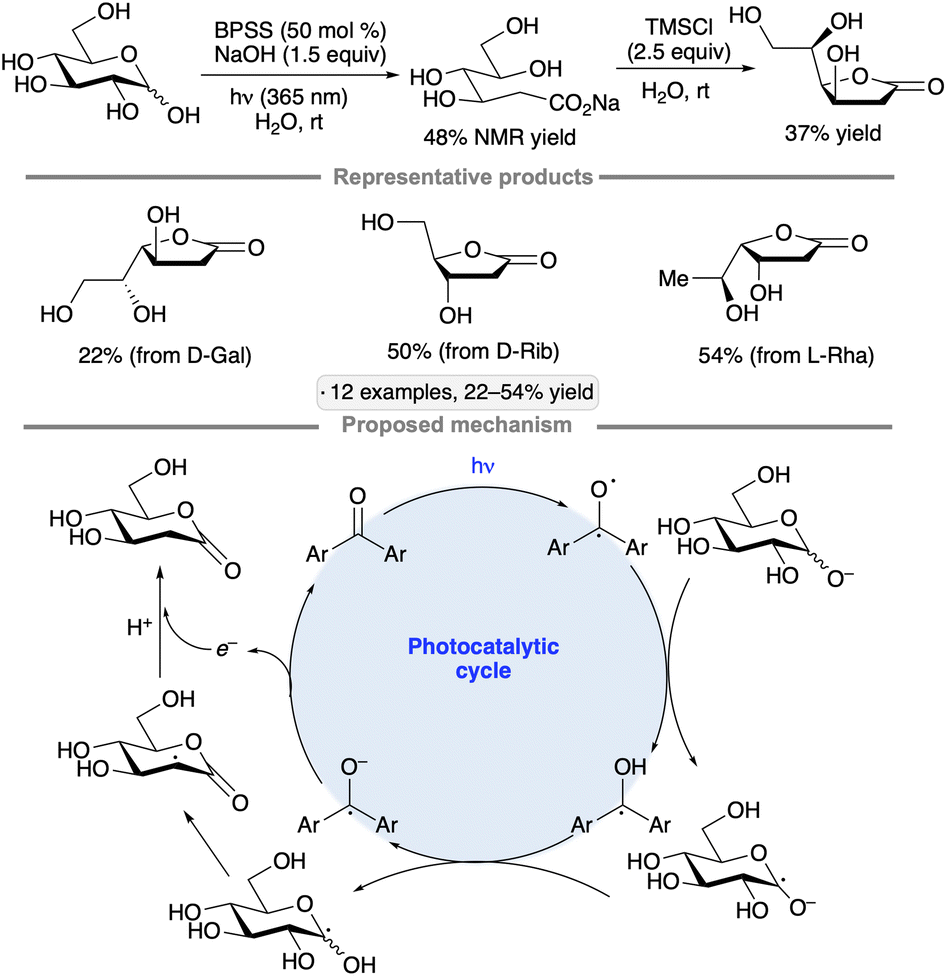 | ||
| Scheme 56 Synthesis of 2-deoxyaldonates from aldoses via photocatalytic redox isomerization. BPSS denotes benzophenone disodium sulfonate; Ar denotes a sulfonated aryl group (roughly 2:1 meta/para). | ||
A method for the synthesis of 2-deoxy- and 4-deoxy-2-ketosugars by redox isomerizations of pyranoside derivatives was developed by Wendlandt and co-workers.126 The authors found that manganese(II) acetate accelerated the formation of ketodeoxysugar from the radical generated by C3-selective HAT from the sugar to the quinuclidinium radical cation (Scheme 57). An axially oriented OH group adjacent to the site of radical formation was needed for the formation of the ketodeoxysugar products. Whether the redox isomerization is accompanied by oxidation of Mn(II) to Mn(III) (path A) or proceeds via a more conventional spin-center shift, accelerated by Mn(II) (path B) is not clear at this stage. The L-rhamnopyranoside-derived 3-keto-2-deoxysugar was employed to achieve expeditious syntheses of methyl glycosides of the rare L-sugars digotoxose, olivose, ristosamine and mycarose.
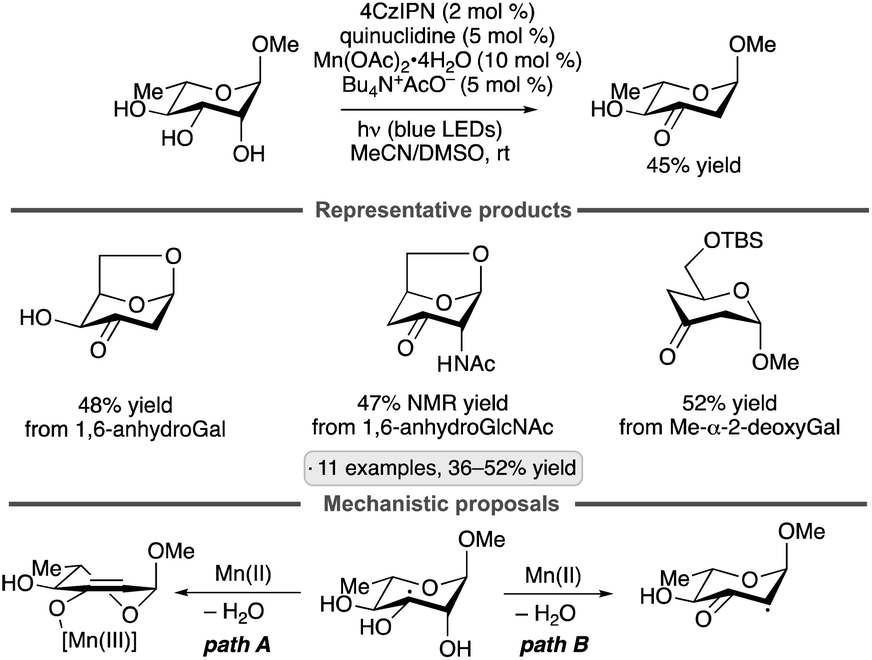 | ||
| Scheme 57 Redox isomerizations of pyranosides using a photoredox/HAT/Mn(II) co-catalyst system. 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene. | ||
The Taylor group described an alternative approach to redox isomerizations of pyranosides,127 taking advantage of the accelerating effect of O-acylation on the rate of the 1,2-migration/elimination step.128 By employing monoacylated pyranosides as substrates, deoxygenation via cleavage of an equatorial C–O bond could be accomplished (e.g., 12 → 13, Scheme 58), in contrast to the strict requirement for an axial OH group when using Mn(II) co-catalysis. The site of O-acylation dictated the position of deoxygenation, thus enabling the synthesis of 16 and 17 from rhamnopyranoside derivatives 14 and 15. As a consequence of C3-selective HAT from the α-configured pyranoside substrates to the quinuclidinium radical cation, 3-ketosugar products were accessed using this method.
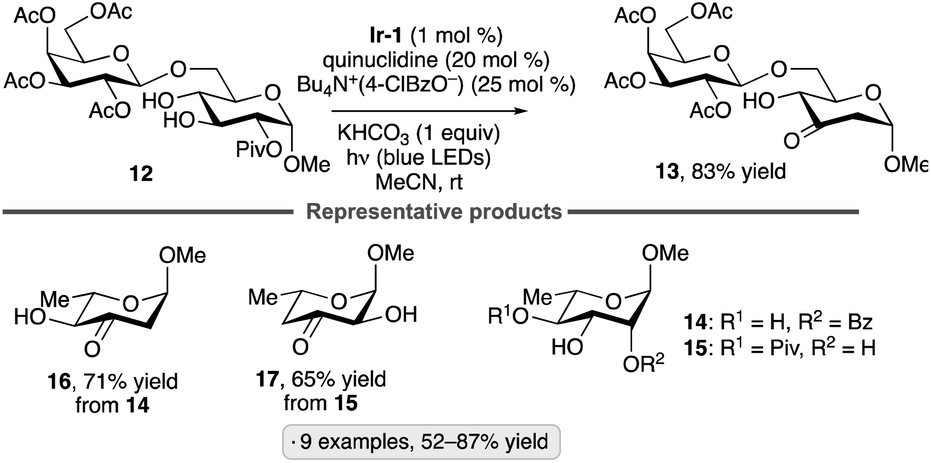 | ||
| Scheme 58 Redox isomerizations of pyranoside-derived monoesters. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
6.3. Epimerization of carbohydrates
Sequential hydrogen atom transfers provide a pathway for epimerization of carbohydrates, provided that the delivery of the hydrogen atom to the sugar-derived radical results in a net inversion of configuration. Radical SAM epimerases operate by such mechanisms.13 Wendlandt and co-workers reported that the combined use of quinuclidine and adamantanethiol as HAT-mediating co-catalysts, along with 4CzIPN and tetrabutylammonium 4-chlorobenzoate (Bu4N+(4-ClBzO−)), enabled the light-mediated epimerization of methyl α-glucopyranoside to its C3 epimer (allo stereochemistry, Scheme 59).129 Selective epimerization at C3 was achieved for other α-gluco-configured substrates, and with 1,6-anhydroglucose, galactose and mannose, while β-galacto-configured pyranosides underwent epimerization at C2 to give products of talo configuration. The method was also applied to unprotected monosaccharides, which present an additional level of complexity due to the potential for mutarotation and pyranose/furanose interconversion. In the proposed catalytic cycle, photogenerated quinuclidinium radical cation abstracts the hydrogen atom from the 3-position of the glucopyranoside, followed by hydrogen atom donation to the less hindered face of the radical by adamantanethiol. The photocatalytic cycle is closed by reduction and protonation of the thiyl radical.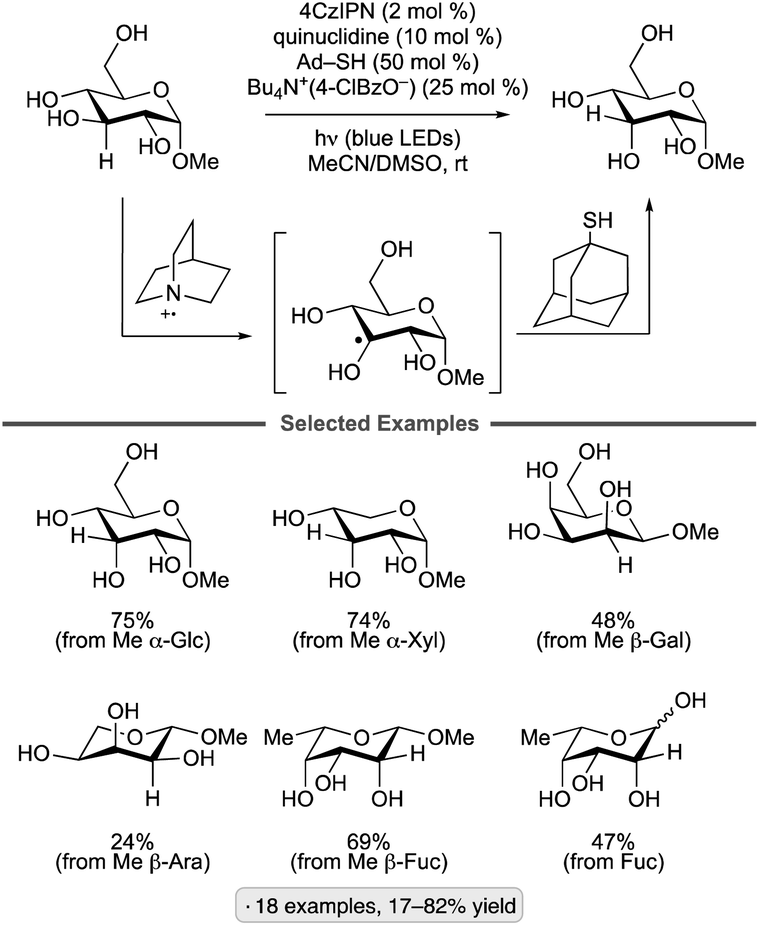 | ||
| Scheme 59 Site-selective epimerization of glycosides and free sugars. 4CzIPN denotes 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; Ad–SH denotes 1-adamantanethiol. | ||
The preference for quinuclidine radical cation to engage in HAT with C–H bonds having a 1,3-diaxial relationship with an oxygen substituent (see Section 4.1) is evident in the examples shown in Scheme 59. The Wendlandt group demonstrated that a complementary mode of site-selectivity could be achieved by using photoactivated decatungstate in place of quinuclidinium radical cation to achieve the initial HAT step.130 Under the optimized conditions depicted in Scheme 60, selective equatorial-to-axial epimerizations of OH groups were achieved for several pyranoside substrates. A related protocol employing sodium decatungstate (NaDT) and an amine-substituted disulfide pre-catalyst with an aqueous co-solvent enabled epimerizations of OH groups in unprotected sugars and cyclitols. Deuterium exchange experiments implicated the difference in rate of HAT from equatorial versus axial C–H bonds to photoexcited decatungstate anion, a consequence of steric effects in the transition state, as the determinant of selectivity. Efficient syntheses of rare sugars and cyclitols, including L-quinovose, D-idose and scyllo-inositol, were achieved using this approach.
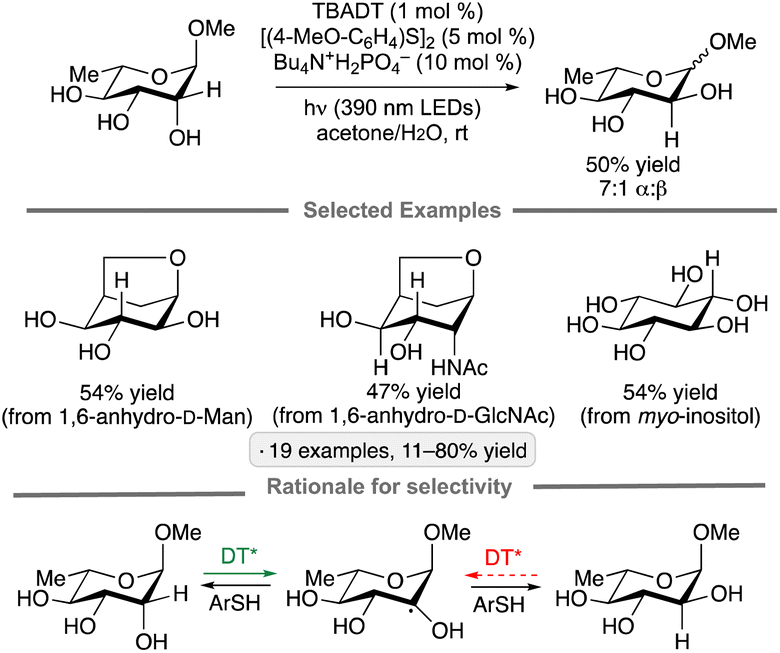 | ||
| Scheme 60 Selective equatorial-to-axial epimerizations of OH groups. TBA denotes Bu4N+; DT denotes [Bu4N+]4[W10O32]4−. | ||
Taking advantage of selective boronic ester formation to create a thermodynamic preference for cis- versus trans-1,2-diol motifs, Oswood and MacMillan accomplished selective epimerizations of OH groups in various diol-containing substrates, including carbohydrate derivatives (Scheme 61).131 The optimized protocol involves the use of tetrabutylphosphonium decatungstatate (TBPDT) and diphenyl disulfide to accomplish epimerization via photocatalytic HAT, along with methylboronic acid as the diol-complexing agent. The methylboronic ester product was converted to the corresponding free diol by treatment with pinanediol. Whereas methyl α-glucopyranoside underwent epimerization at C2 to yield the α-mannopyranoside, a C3-selective epimerization (gluco to allo stereochemistry) was achieved using the β-anomer. The results are consistent with a sterically controlled H-atom abstraction by the photoexcited decatungstate anion.
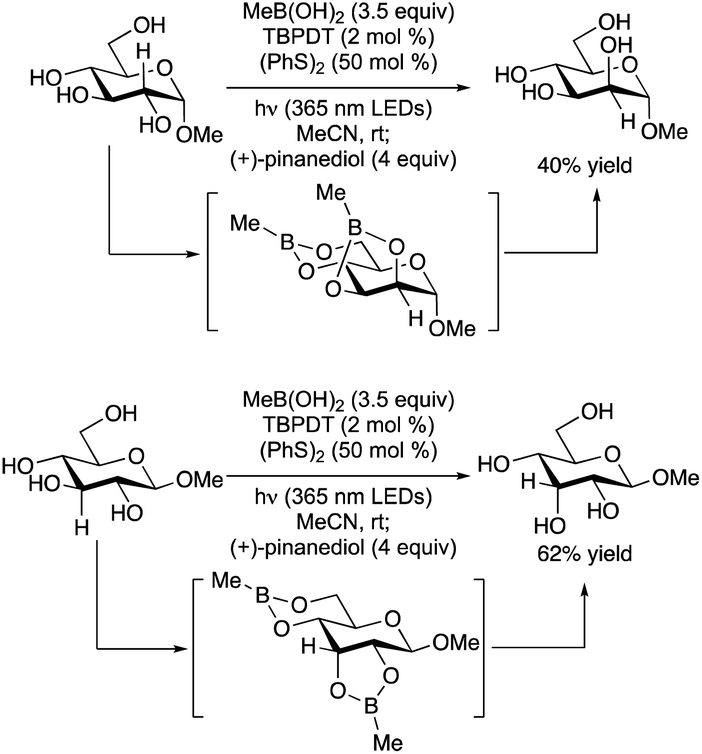 | ||
| Scheme 61 Methylboronic acid-mediated photocatalytic trans-to-cis diol epimerization of glucopyranosides. DBPDT denotes tetrabutylphosphonium decatungstate, [Bu4P+]4[W10O32]4−. | ||
6.4. Site-selective oxidations
Site-selective, aerobic oxidations of carbohydrate derivatives to ketosugars have been achieved by subjecting pyranosides to the conditions of photocatalytic HAT.132 α-Glucopyranosides underwent oxidation at C3 under an air atmosphere in acetonitrile using an acridinium photocatalyst and quinuclidine as HAT co-catalyst (Scheme 62). The superoxide anion O2−, generated by SET from the reduced photocatalyst to O2, was proposed to act as the trapping agent for the sugar-derived radical. Molecular sieves (4 Å pore size) were found to have a beneficial effect on yield, likely by preventing the buildup of hydrogen peroxide, which is a byproduct of the aerobic oxidation process. Selective oxidations of cis-diol groups were also conducted, using the oxidation-resistant 2-carboxyphenylboronic acid (2-CBPA) as a bond-weakening co-catalyst. The authors proposed that manno-configured substrates underwent initial oxidation at the 2-position, in keeping with previous observations on organoboron/HAT co-catalysis (e.g., Scheme 52), followed by a base-mediated isomerization to the more stable 3-ketosugar. The oxidation/isomerization was accompanied by epimerization at the 2-position, giving rise to the α-gluco-configured product. Selective, blue-light-mediated aerobic oxidations of α-glucopyranosides at the 3-position were also achieved by Liu, Wang and co-workers, using riboflavin tetraacetate and quinuclidine as the photoredox/HAT co-catalyst system, along with TBAP and MnO2 as additives.133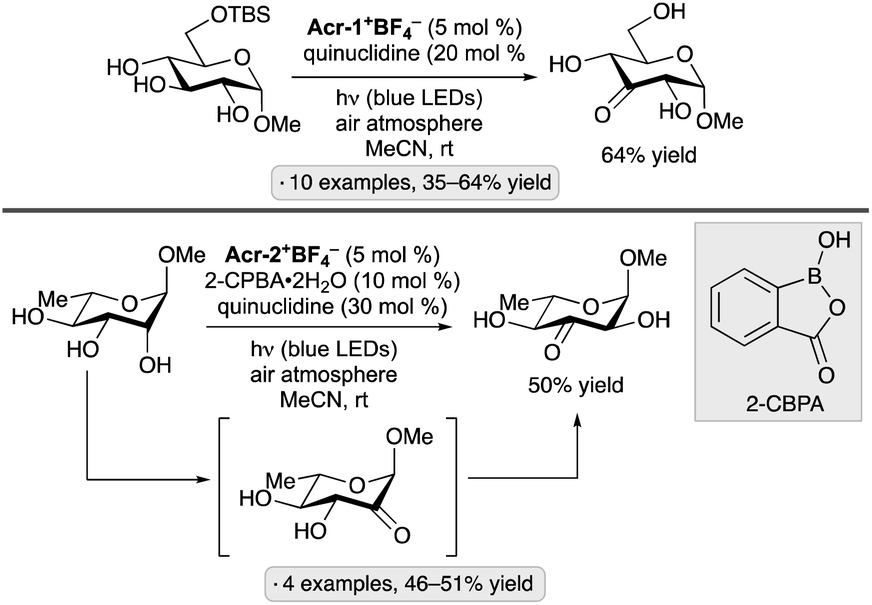 | ||
| Scheme 62 Photocatalytic aerobic oxidations of carbohydrate derivatives. See Fig. 1 for the structure of Acr-1+ and Acr-2+. 2-CBPA denotes 2-carboxyphenylboronic acid. | ||
Lectka, Dudding and co-workers have investigated the photochemical reactivity of sugar-derived ketals with Selectfluor in the presence of xanthone as a photoinitiator (Scheme 63).134 α-Fluoroketals were generated site-selectively from various bis(isopropylidene)furanose and pyranose substrates (the transformation is included in this section because the gem-oxyfluoride products, which were stable to isolation when bearing the ketal protective group, are at the ketosugar oxidation level). Noting that the 2-position of bis(isopropylidene)galactopyranose is not the site of the weakest C–H bond, the authors used DFT calculations (IEFPCM(MeCN)UB3LYP/6-311++G(2d,2p)//UB3LYP/6-31G(d)) to model the transition state for HAT to the Selectfluor-derived radical dication. As depicted in Scheme 63, the transition state is stabilized by noncovalent interactions, including C–H⋯O hydrogen bonds between the quinuclidinium α-hydrogens and a ketal oxygen.
 | ||
| Scheme 63 Photocatalytic method for the preparation of carbohydrate-derived α-fluoroketals. | ||
6.5. Intramolecular HAT
Intramolecular hydrogen atom transfer provides a strategy for generating radicals at positions that might otherwise be difficult. A pioneering example was achieved by the Rovis group, who showed that an anomeric radical could be generated by intramolecular HAT to a pendant amidyl radical in a 2-deoxy-C-glycoside substrate.135 Kuninobu and co-workers took advantage of intramolecular HAT to achieve C2-selective functionalizations of a sulfamate-bearing fructopyranoside derivative (Scheme 64).136 The N-centered radical generated by oxidation and deprotonation of the sulfamate group underwent 1,6-HAT to form the radical at the 2-position. Hydrolysis of the sulfamate to the corresponding alcohol was accomplished treatment with sodium azide.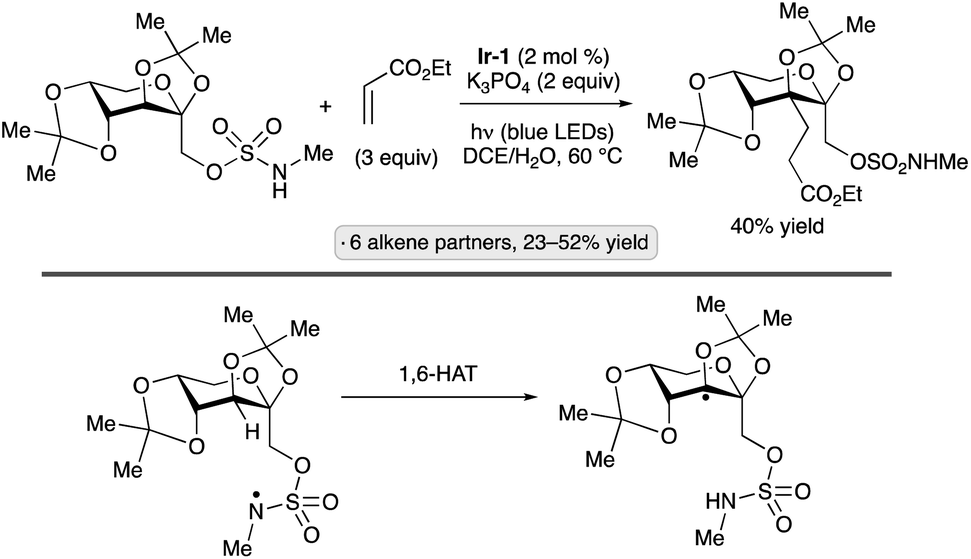 | ||
| Scheme 64 Selective C2-alkylation of a fructopyranose-derived sulfamate by intramolecular HAT. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
Blue light-mediated cyclizations of carbohydrate-derived trichloroacetimidates to oxazolines were developed by Shaw and Kumar, with 1,5-HAT to a nitrogen-centered radical serving as a key step in the process (Scheme 65).137N-Iodination of the trichloroacetimidate in situ, followed by photochemical homolysis of the N–I bond, resulted in the formation of an N-centered radical. After 1,5-HAT, the generated carbon-centered radical underwent oxidation and cyclization to form the oxazoline. α-Manno-configured trichloroacetimidates gave rise to cis-fused oxazoline products, along with isomeric oxazolines arising from an O-to-N rearrangement of the imidate as side products in some cases. Spiro-fused oxazolines were obtained from gluco- and mannopyranosides having a trichloroacetimidate group at the 6-position.
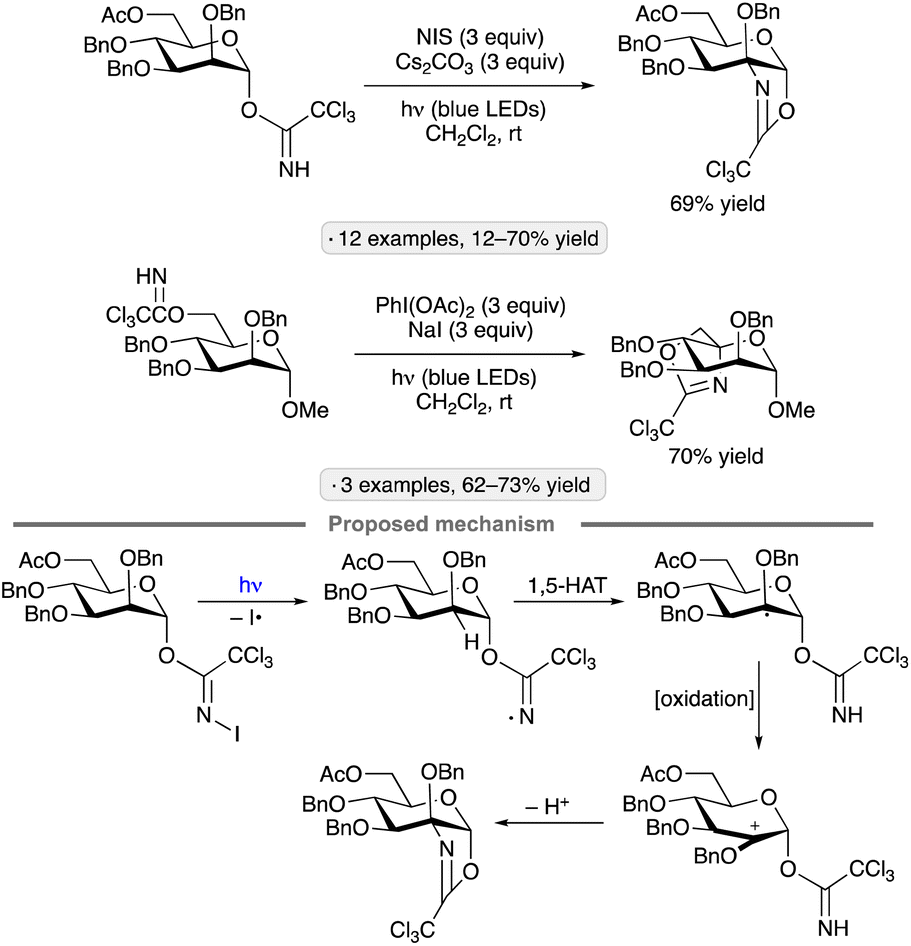 | ||
| Scheme 65 Synthesis of fused oxazoline derivatives from sugar-derived trichloroacetimidates. | ||
Pérez-Martín, Suárez and co-workers have devised processes for cyclization and epimerization reactions of sugar derivatives initiated by intramolecular HAT to alkoxy radicals generated from N-alkoxyphthalimides.138 Both transformations depicted in Scheme 66 involve intramolecular 1,5-HAT to an alkoxy radical; the spiroketal is formed by subsequent 1,2-phosphatoxy migration and cyclization, whereas the gluco-to-ido epimerization involves a stereoselective hydrogen atom transfer to the radical at the 5-position. In general, the use of photocatalytic conditions gave rise to lower yields of the desired products than were obtained using azobis(isobutyronitrile) (AIBN) as an initiator in the absence of light.
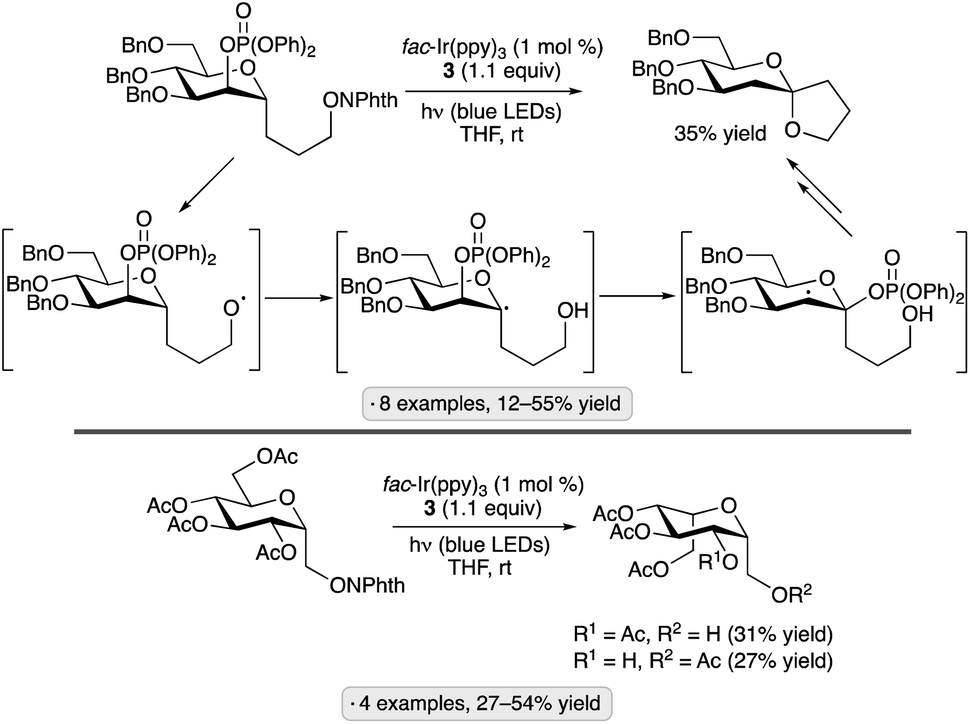 | ||
| Scheme 66 Photocatalytic processes involving 1,5-HAT to alkoxy radicals. | ||
7. Functional group interconversions at carbohydrate OH and SH groups
7.1. Functionalization of OH groups
A method for the synthesis of O-aryl glycosides from anomeric hemiacetals and bromoarenes was reported by the groups of Wang and Xiao (Scheme 67).139 Adapting a protocol developed by the MacMillan group,140 the authors employed the Ir-1 photocatalyst along with a Ni(II)/dtbbpy co-catalyst to achieve the C–O couplings. Benzyl-protected pyranose derivatives underwent O-arylation, generally resulting in axially configured glycosides as the major products. Electron-deficient bromoarenes or bromopyridines were the most suitable coupling partners; the anomeric stereoselectivity was found to vary depending on the phenol partner employed. The protocol complements conventional methods for O-aryl glycoside synthesis, which generally involve displacements of leaving groups from the anomeric center by phenoxide nucleophiles. According to the mechanistic proposal advanced by the MacMillan group, oxidation of an arylnickel(II) alkoxide to the corresponding Ni(III) species by photoexcited Ir-1 accelerates C–O bond-forming reductive elimination, generating a Ni(I) complex that undergoes SET with the reduced photocatalyst to close both cycles.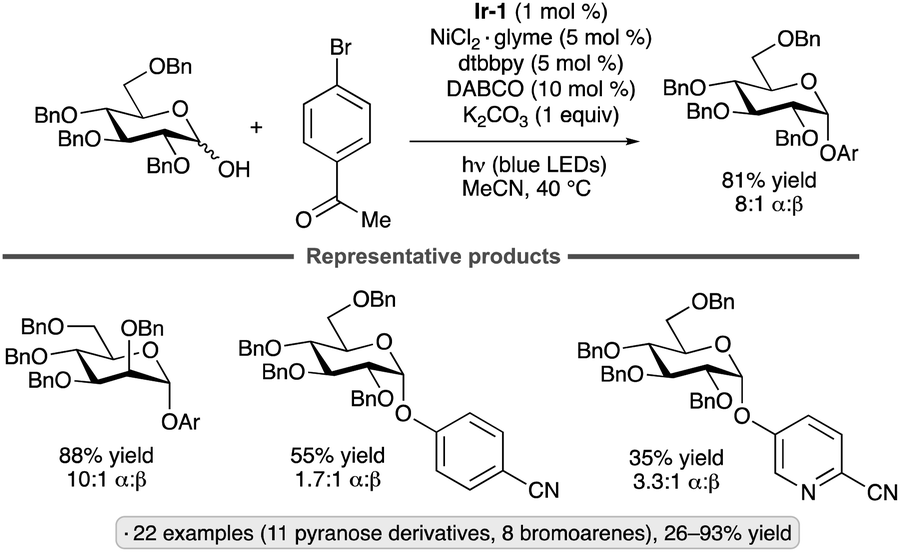 | ||
| Scheme 67 Synthesis of aryl glycosides by couplings of anomeric hemiacetals with bromoarenes. Ir-1 denotes [Ir(dF(CF3)ppy)2(dtbbpy)]PF6; dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine; Ar denotes 4-acetylphenyl. | ||
7.2. Functionalization of SH groups
Thiol–ene reactions of glycosyl thiols are a powerful tool for the conjugation of sugars to complex partners, as the conditions employed are mild and functional-group tolerant, and the resulting S-glycosidic linkages are often resistant to enzyme-mediated hydrolysis.141 Building on a UV-initiated conjugation of glycosyl thiols reported by the Davis group,142 Wang and co-workers developed a blue light-initiated coupling of thiols, including glycosyl congeners, with alkenes, in the presence of the acridinium photocatalyst Acr-2+BF4− (Scheme 68).143 Mixed organic/aqueous solvent (1:1 H2O/MeCN by volume) was also a suitable solvent for the transformation.144 Oxidation of the thiol by photoexcited Acr-2+, followed by deprotonation, was proposed to generate the reactive thiyl radical, thus initiating the radical chain process for the thiol–ene reaction. The authors proposed that adventitious oxygen is responsible for re-oxidation of [Acr-2]˙. A red-light-induced protocol has been developed for thiol–ene and thiol–yne reactions of per-O-acetylated β-glucopyranosyl thiol, using non-metallated tetraphenylporphyrin as photocatalyst.145
 | ||
| Scheme 68 Blue light-initiated thiol–ene reaction of a glycosyl thiol. See Fig. 1 for the structure of Acr-1+. | ||
Synergistic nickel/photocatalysis has been employed to achieve functionalizations of the SH group in glycosyl thiols. The group of Messaoudi showed that per-O-acylated 1-thiosugars could be coupled with aryl, heteroaryl, vinyl and alkynyl iodides or bromides to generate the corresponding β-S-glycosides (Scheme 69).146 Ammonium silicate salt 18 serves as a precursor to an alkyl radical, which generates the reactive thiyl radical via HAT.147
 | ||
| Scheme 69 Couplings of 1-glycosylthiols with haloarenes and haloalkenes. dtbbpy denotes 4,4′-di-tert-butyl-2,2′-bipyridine; Ar denotes 4-cyanophenyl; PMP denotes 4-methoxyphenyl. | ||
7.3. Protective group manipulations
Protocols for visible light-mediated oxidative deprotection of benzyl ethers in carbohydrate derivatives were reported by Seeberger, Pieber and co-workers (Scheme 70).148 Irradiation of sugar-derived benzyl ethers with green light (525 nm) in the presence of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) resulted in the corresponding free alcohols, including examples of substrates bearing functional groups that would be incompatible with conventional conditions for debenzylation (hydrogenolysis or Birch reduction). An alternative protocol employing catalytic DDQ under an air atmosphere, along with tert-butyl nitrite as an additive, was also described. The key benzylic oxacarbenium ion leading to C–O bond cleavage could arise via a combination of SET and HAT steps involving the triplet excited state of DDQ. The nitrite likely serves as a precursor to nitrous oxide, which acts as a redox mediator for the regeneration of DDQ by aerobic oxidation. A variant of the protocol using 440 nm blue light with relatively short irradiation times (3–10 min) was demonstrated in a flow reactor. | ||
| Scheme 70 Green light-mediated, selective deprotection of pyranoside-derived benzyl ethers. | ||
Another approach for visible-light-mediated deprotections of benzyl ethers in carbohydrate derivatives was reported by Xia and co-workers.149 Using the subsitituted phenol pre-catalyst DBPP, along with co-catalytic n-butanethiol and cesium formate as stoichiometric reductant, debenzylations of alcohols, esters, amides and anilines were achieved, as well as deprotections of other moieties having benzylic C–X bonds, including PMB, BOM, CBz and benzylidene acetal groups (Scheme 71). In the authors' proposed photocatalytic cycle, single-electron reduction of the substituted aryl group is accomplished by the photoexcited phenoxide catalyst. Fragmentation of the substrate-derived radical anion generates the alkoxide along with tolyl radical, which is trapped via HAT from the thiol co-catalyst. Regeneration of the photocatalyst and thiol is accompanied by oxidation of formate to CO2.
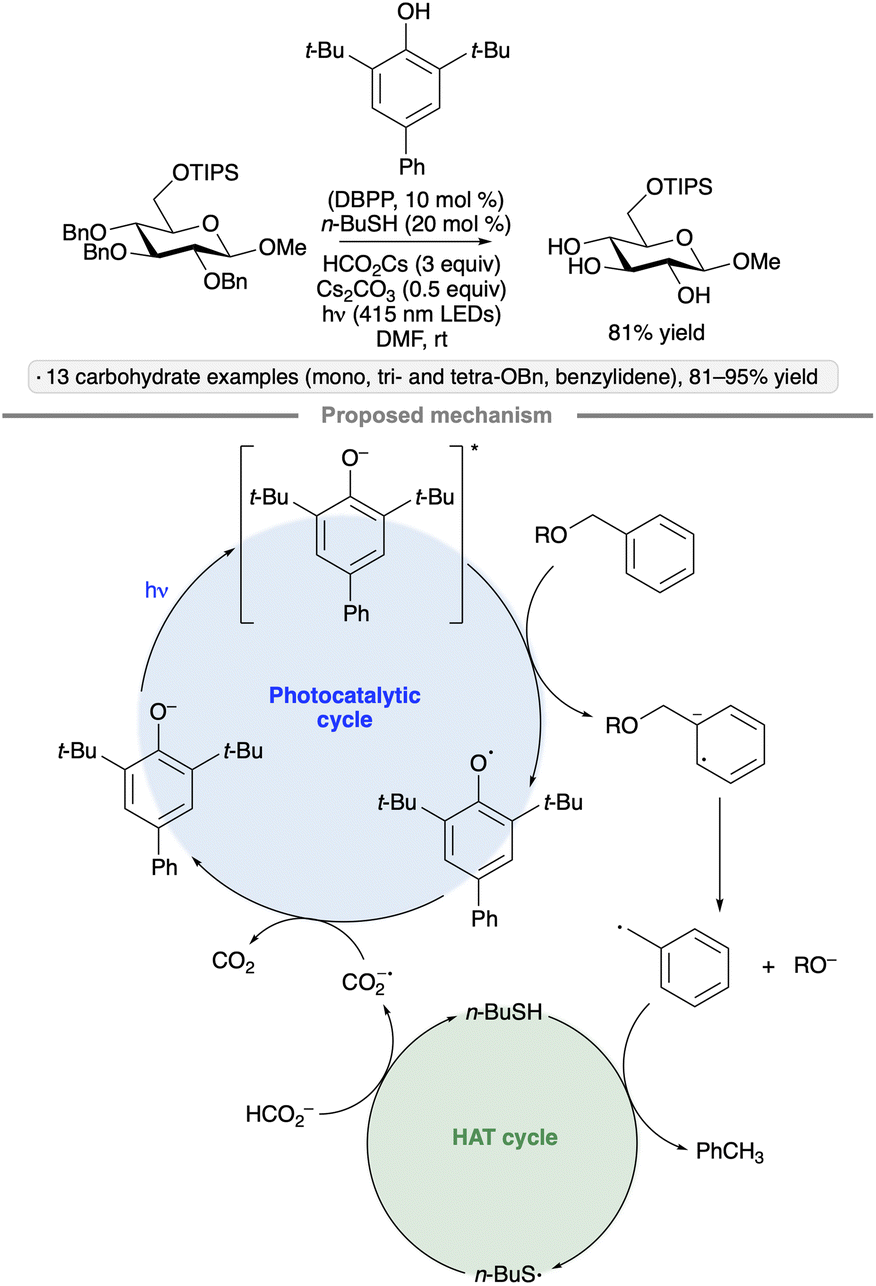 | ||
| Scheme 71 Visible light-mediated debenzylation using a phenoxide photocatalyst and co-catalytic thiol. | ||
Carbonates bearing substituted 4-bora-3a,4a-diaza-s-indacene (BODIPY) moieties have been explored as visible light-labile protective groups for OH groups in carbohydrates. Identifying a protective group that was stable to the conditions of thioglycoside activation with N-iodosuccinimide and triflic acid, while also undergoing visible light-induced deprotection at a reasonable rate, proved to be a key consideration in the study conducted by the groups of Seeberger and Winter.150 The F-BODIPY-derivative was found to possess an appropriate balance of these characteristics, enabling the glycosylation–deprotection–glycosylation sequence depicted in Scheme 72. The proposed mechanism for the deprotection step involves photoinduced heterolytic cleavage of the C–O bond, generating an ion pair.
 | ||
| Scheme 72 Sequential glycosylations enabled by a visible light-sensitive, BODIPY-based protective group. | ||
8. Conclusions
We hope that it will be clear from the preceding discussion that the application of photocatalysis and photochemistry to carbohydrate synthesis is a vibrant and active area of research that is evolving rapidly along several fronts. Radical chemistry makes it possible to conduct key bond constructions and functional group manipulations on derivatives having free OH groups, a significant advantage in the context of carbohydrate substrates. A strong argument can be made that photocatalytic protocols already constitute the state-of-the-art methods for addressing important challenges in carbohydrate chemistry, including C-glycoside synthesis and the preparation of rare sugar building blocks. The future of this field is full of promise. Given the rapid pace at which light-induced reactions of organic compounds are being discovered and developed, it is a certainty that new transformations of sugars will be identified, providing access to unexplored regions of chemical space. Exciting opportunities exist to build understanding of the factors that influence stereoselectivity and site-selectivity in processes involving sugar-derived radical intermediates, and to develop new selective processes that rely on reagent or catalyst control. Much of the chemistry described in this review has been conducted using monosaccharide derivatives, the simplest sugars. Applications to oligosaccharides, polysaccharides, nucleic acid polymers and other complex glycans present challenges, but could provide new tools to interrogate and perturb the functions of these important classes of biomolecules.Author contributions
All authors contributed to writing the original draft and reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC and the Province of Ontario.Notes and references
- (a) R. W. Binkley, Adv. Carbohydr. Chem. Biochem., 1981, 38, 105–193 CrossRef; (b) E. R. Binkley and R. W. Binkley, Carbohydrate Photochemistry, American Chemical Society, Washington DC, 1998 Search PubMed.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (b) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (e) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS; (f) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (g) J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- G. C. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L.-C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn'ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883–5889 RSC.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed.
- (a) J. R. Ragains, in Comprehensive Glycoscience, ed. J. Barchi Jr, Elsevier, Oxford, 2nd edn, 2021, ch. 11, vol. 2, pp. 328–364 Search PubMed; (b) Z. Azeem and P. K. Mandal, Adv. Synth. Catal., 2023, 365, 2818–2849 CrossRef CAS.
- (a) L.-Y. Xu, N.-L. Fan and X.-G. Hu, Org. Biomol. Chem., 2020, 18, 5095–5109 RSC; (b) A. Chen, L. Xu, Z. Zhou, S. Zhao, T. Yang and F. Zhu, J. Carbohydr. Chem., 2022, 40, 361–400 CrossRef; (c) T. Ghosh and T. Nokami, Carbohydr. Res., 2022, 522, 108677 CrossRef CAS PubMed; (d) W. Shang and D. Niu, Acc. Chem. Res., 2023, 56, 2473–2488 CrossRef CAS PubMed; (e) Y. Jiang, Y. Zhang, B. C. Lee and M. J. Koh, Angew. Chem. Int. Ed., 2023, 62, e2023051238 Search PubMed.
- C. E. Suh, H. M. Carder and A. E. Wendlandt, ACS Chem. Biol., 2021, 16, 1814–1828 CrossRef CAS PubMed.
- A. Shatskiy, E. V. Stepanova and M. D. Kärkäs, Nat. Rev. Chem., 2022, 6, 782–805 CrossRef CAS.
- Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, ed. A. V. Demchenko, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) R. Iwata, K. Uda, D. Takahashi and K. Toshima, Chem. Commun., 2014, 50, 10695–10698 RSC; (b) K. Kimura, T. Eto, D. Takahashi and K. Toshima, Org. Lett., 2016, 18, 3190–3193 CrossRef; (c) G. Zhao and T. Wang, Angew. Chem., Int. Ed., 2018, 57, 6120–6124 CrossRef CAS; (d) J. Liu, S. Yin, H. Wang, H. Li and G. Ni, Carbohydr. Res., 2020, 490, 107963 CrossRef CAS PubMed; (e) J. Li, G. Zhao and T. Wang, Synlett, 2020, 31, 823–828 CrossRef CAS; (f) G. Zhao, J. Li and T. Wang, Chem. Commun., 2021, 57, 12659–12662 RSC.
- (a) J. D. Timpa, M. G. Legendre, G. W. Griffin and P. K. Das, Carbohydr. Res., 1983, 117, 69–80 CrossRef CAS; (b) J. D. Timpa and G. W. Griffin, Carbohydr. Res., 1984, 131, 185–196 CrossRef CAS; (c) S. Hashimoto, I. Kurimoto, Y. Fujii and R. Noyori, J. Am. Chem. Soc., 1985, 107, 1427–1429 CrossRef CAS.
- G. W. Griffin, N. C. Bandara, M. A. Clarke, W.-S. Tsang, P. J. Garegg, S. Oscarson and B. A. Silwanis, Heterocycles, 1990, 30, 939–947 CrossRef CAS.
- T. Furuta, K. Takeuchii and M. Iwamura, Chem. Commun., 1996, 157–158 RSC.
- I. Cumptsey and D. Crich, J. Carbohydr. Chem., 2011, 30, 469–485 CrossRef.
- W. J. Wever, M. A. Cinelli and A. A. Bowers, Org. Lett., 2013, 15, 30–33 CrossRef CAS PubMed.
- M. Spell, X. Wang, A. E. Wahba, E. Conner and J. Ragains, Carbohydr. Res., 2013, 369, 42–47 CrossRef CAS PubMed.
- R.-Z. Mao, F. Guo, D.-C. Xiong, Q. Li, J. Duan and X.-S. Ye, Org. Chem. Front., 2016, 3, 737–743 RSC.
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 17, 8242–8284 CrossRef PubMed.
- Y. Yu, D.-C. Xiong, R.-Z. Mao and X.-S. Ye, J. Org. Chem., 2016, 81, 7134–7138 CrossRef CAS PubMed.
- Y. Cao, M. Zhou, R.-Z. Mao, Y. Zou, F. Xia, D.-K. Liu, J. Liu, Q. Li, D.-C. Xiong and X.-S. Ye, Chem. Commun., 2021, 10899–10902 RSC.
- M. L. Spell, K. Deveaux, C. G. Bresnahan, B. L. Bernard, W. Sheffield, R. Kumar and J. R. Ragains, Angew. Chem., Int. Ed., 2016, 55, 6515–6519 CrossRef CAS PubMed.
- C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q.-D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686–694 CrossRef CAS PubMed.
- P. Wen and D. Crich, Org. Lett., 2017, 19, 2402–2405 CrossRef CAS.
- M. Krumb, T. Lucas and T. Opatz, Eur. J. Org Chem., 2019, 4517–4521 CrossRef CAS.
- Y. Fu, J. L. Dickson, R. S. Loka, H. Xu, R. N. Scaugaard, H. B. Schlegel, L. Luo and H. M. Nguyen, ACS Catal., 2020, 10, 5990–6001 CrossRef.
- R. N. Schaugaard, H. M. Nguyen and H. B. Schlegel, Inorg. Chem., 2021, 60, 12801–12812 CrossRef CAS.
- S. Kim, Y. Khomutnyk, A. Bannykh and P. Nagorny, Org. Lett., 2021, 23, 190–194 CrossRef CAS PubMed.
- L.-Y. Xu, N.-L. Fan and X.-G. Hu, Org. Biomol. Chem., 2020, 18, 5095–5109 RSC.
- (a) B. Giese and J. Dupuis, Angew. Chem., Int. Ed., 1983, 22, 622–623 CrossRef; (b) J. Dupuis, B. Giese, D. Rüegge, H. Fischer, H.-G. Korth and R. Sustmann, Angew. Chem., Int. Ed., 1984, 23, 896–898 CrossRef; (c) B. Giese and J. Dupuis, Tetrahedron Lett., 1984, 25, 1349–1352 CrossRef CAS.
- R. S. Andrews, J. J. Becker and M. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274–7276 CrossRef CAS PubMed.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS.
- R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 4140–4143 CrossRef CAS PubMed.
- Q.-Q. Zhou, S. J. S. Düsel, L.-Q. Lu, B. König and W.-J. Xiao, Chem. Commun., 2019, 55, 107–110 RSC.
- C.-Y. Li, Y. Ma, Z.-W. Lei and X.-G. Hu, Org. Lett., 2021, 23, 8899–8904 CrossRef CAS PubMed.
- L. Xia, W. Fan, X.-A. Yuan and S. Yu, ACS Catal., 2021, 11, 9397–9406 CrossRef CAS.
- R.-Q. Jiao, Y.-N. Ding, M. Li, W.-Y. Shi, X. Chen, Z. Zhang, W.-X. Wei, X.-S. Li, X.-P. Gong, Y.-Y. Luan, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2023, 25, 6099–6104 CrossRef CAS PubMed.
- L. Xia, M. Jin, Y. Jiao and S. Yu, Org. Lett., 2022, 24, 364–368 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- Z.-D. Mou, J.-X. Wang, X. Zhang and D. Niu, Adv. Synth. Catal., 2021, 363, 3025–3029 CrossRef CAS.
- R. Mao, S. Xi, S. Shah, M. J. Roy, A. John, J. P. Lingford, G. Gäde, N. E. Scott and E. D. Goddard-Borger, J. Am. Chem. Soc., 2021, 143, 12699–12707 CrossRef CAS PubMed.
- Y. Wei, Q. Wang and M. J. Koh, Angew. Chem., Int. Ed., 2023, 62, e202214247 CrossRef CAS.
- M. Li, Y.-F. Qiu, C.-T. Wang, X.-S. Li, W.-X. Wei, Y.-Z. Wang, Q.-F. Bao, Y.-N. Ding, W.-Y. Shi and Y.-M. Liang, Org. Lett., 2020, 22, 6288–6293 CrossRef CAS.
- P. Ji, Y. Zhang, F. Gao, F. Bi and W. Wang, Chem. Sci., 2020, 11, 13079–13084 RSC.
- H. Abe, S. Shuto and A. Matsuda, J. Am. Chem. Soc., 2001, 123, 11870–11882 CrossRef CAS PubMed.
- Y. Ma, S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang and Y. Zhang, Chem. Commun., 2019, 55, 14657–14660 RSC.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- M. Zhu and S. Messaoudi, ACS Catal., 2021, 11, 6334–6342 CrossRef CAS.
- K. Lu, Y. Ma, S. Liu, S. Guo and Y. Zhang, Chin. J. Chem., 2022, 40, 681–686 CrossRef CAS.
- Y. Wei, B. Ben-zvi and T. Diao, Angew. Chem., Int. Ed., 2021, 60, 9433–9438 CrossRef CAS.
- Y. Wei, J. Lam and T. Diao, Chem. Sci., 2021, 12, 11414–11419 RSC.
- C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645–17651 CrossRef CAS PubMed.
- A. Chen, S. Zhao, Y. Han, Z. Zhou, B. Yang, L.-G. Xie, M. A. Walczak and F. Zhu, Chem. Sci., 2023, 14, 7569–7580 RSC.
- Z. Dong and D. W. C. MacMillan, Nature, 2021, 598, 451–456 CrossRef CAS PubMed.
- F. Zhu, S.-q. Zhang, Z. Chen, J. Rui, X. Hong and M. A. Walczak, J. Am. Chem. Soc., 2020, 142, 11102–11113 CrossRef CAS.
- D. Takeda, M. Yoritate, H. Yasutomi, S. Chiba, T. Moriyama, A. Yokoo, K. Usui and G. Hirai, Org. Lett., 2021, 23, 1940–1944 CrossRef CAS PubMed.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS.
- E. M. Miller and M. A. Walczak, Org. Lett., 2021, 23, 4289–4293 CrossRef CAS PubMed.
- L.-Q. Wan, X. Zhang, Y. Zou, R. Shi, J.-G. Cao, S.-Y. Xu, L.-D. Deng, L. Zhou, Y. Gong, X. Shu, G. Y. Lee, H. Ren, L. Dai, S. Qi, K. N. Houk and D. Niu, J. Am. Chem. Soc., 2021, 143, 11919–11926 CrossRef CAS.
- C. Zhang, S.-Y. Xu, H. Zuo, X. Zhang, Q.-D. Dang and D. Niu, Nat. Synth., 2023, 2, 251–260 Search PubMed.
- Q. Wang, B. C. Lee, T. J. Tan, Y. Jiang, W. H. Ser and M. J. Koh, Nat. Synth., 2022, 1, 967–974 CrossRef.
- (a) S. Xu, W. Zhang, C. Li, Y. Li, H. Zeng, Y. Wang, Y. Zhang and D. Niu, Angew. Chem., Int. Ed., 2023, 62, e202218303 CrossRef CAS PubMed; (b) H. Zeng, Y. Li, R. Wu, D. Li, Y. Zhang, S. Xu and D. Niu, Org. Lett., 2023 DOI:10.1021/acs.orglett.3c00833 , in press doi: .
- T. Miura, M. Yoritate and G. Hirai, Chem. Commun., 2023, 59, 8564–8567 RSC.
- D. Xie, Y. Wang, X. Zhang, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2022, 61, e202204922 CrossRef CAS.
- B. Giese, K. S. Gröninger, T. Witzel, H.-G. Korth and R. Sustmann, Angew. Chem., Int. Ed., 1987, 26, 233–234 CrossRef.
- (a) P. Wessig and O. Muehling, Eur. J. Org Chem., 2007, 14, 2219–2232 CrossRef; (b) B. Matsuo, A. Granados, J. Majhi, M. Sharique, G. Levitre and G. A. Molander, ACS Org. Inorg. Au, 2022, 2, 435–454 CrossRef CAS PubMed; (c) F.-L. Zhang, B. Li, K. N. Houk and Y.-F. Wang, JACS Au, 2022, 2, 1032–1042 CrossRef CAS PubMed.
- G. Zhao, W. Yao, J. N. Mauro and M.-Y. Ngai, J. Am. Chem. Soc., 2021, 143, 1728–1734 CrossRef CAS PubMed.
- G. Zhao, U. Mukherjee, L. Zhou, Y. Wu, W. Yao, J. N. Mauro, P. Liu and M.-Y. Ngai, Chem. Sci., 2022, 13, 6276–6282 RSC.
- W. Yao, G. Zhao, Y. Wu, L. Zhou, U. Mikherjee, P. Liu and M.-Y. Ngai, J. Am. Chem. Soc., 2022, 144, 3353–3359 CrossRef CAS PubMed.
- G. Zhao, W. Yao, I. Kevlishvili, J. N. Mauro, P. Liu and M.-Y. Ngai, J. Am. Chem. Soc., 2021, 143, 8590–8596 CrossRef CAS PubMed.
- C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, T. Ladduwahetty, J. L. Randall and A. Chucholowski, J. Am. Chem. Soc., 1986, 108, 2466–2467 CrossRef CAS PubMed; (b) W. R. Roush and C. E. Bennett, J. Am. Chem. Soc., 1999, 121, 3541–3542 Search PubMed.
- (a) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed; (b) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS.
- T. Luo, Y.-F. Guo, T.-T. Xu and H. Dong, J. Org. Chem., 2023, 88, 8024–8033 CrossRef CAS PubMed.
- J.-T. Ge, L. Zhou, T. Luo, J. Lv and H. Dong, Org. Lett., 2019, 21, 5903–5906 CrossRef CAS PubMed.
- Y. Ito, A. Kimura, T. Osawa and Y. Hari, J. Org. Chem., 2018, 83, 10701–10708 CrossRef CAS PubMed.
- K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef CAS PubMed.
- (a) S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6610–6613 CrossRef CAS; (b) A. Dumoulin, J. K. Matsui, A. Gutiérrez-Bonet and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6614–6618 CrossRef CAS PubMed; (c) J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed.
- (a) J. K. Matsui, A. Gutiérrez-Bonet, M. Rotella, R. Alam, O. Gutierrez and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 15847–15851 CrossRef CAS PubMed; (b) Z.-J. Wang, S. Zheng, E. Romero, J. K. Matsui and G. A. Molander, Org. Lett., 2019, 21, 6543–6547 CrossRef CAS PubMed.
- A. Lipp, S. O. Badir, R. Dykstra, O. Gutierrez and G. A. Molander, Adv. Synth. Catal., 2021, 363, 3507–3520 CrossRef CAS PubMed.
- I. Kim, S. Park and S. Hong, Org. Lett., 2020, 22, 8730–8734 CrossRef CAS PubMed.
- K. B. Pal, E. M. Di Tomasso, A. K. Inge and B. Olofsson, Angew. Chem., Int. Ed., 2023, e202301368 Search PubMed.
- P. Ji, Y. Zhang, Y. Wei, H. Huang, W. Hu, P. A. Mariano and W. Wang, Org. Lett., 2019, 21, 3086–3092 CrossRef CAS.
- I. C. S. Wan, M. D. Witte and A. J. Minnaard, Org. Lett., 2019, 21, 7669–7673 CrossRef CAS PubMed.
- Y. Ito, N. Tsutsui, T. Osawa and Y. Hari, J. Org. Chem., 2019, 84, 9093–9100 CrossRef CAS PubMed.
- R. Qi, C. Wang, Z. Ma, H. Wang, Q. Chen, L. Liu, D. Pan, X. Ren, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2022, 61, e202200822 CrossRef CAS PubMed.
- Y.-N. Ding, N. Li, Y.-C. Huang, Y. An and Y.-M. Liang, Org. Lett., 2022, 24, 4519–4523 CrossRef CAS PubMed.
- M. Escolano, M. J. Cabrera-Afonso, M. Ribagorda, S. O. Badir and G. A. Molander, J. Org. Chem., 2022, 87, 4981–4990 CrossRef CAS PubMed.
- H. Ding, N. Yan, P. Wang, N. Song, Q. Sun, T. Li and M. Li, Org. Chem. Front., 2022, 9, 2808–2814 RSC.
- E. Ota, H. Wang, N. L. Frye and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 1457–1462 CrossRef CAS PubMed.
- T. Matsuoka, S. Inuki, T. Miyagawa, S. Oishi and H. Ohno, J. Org. Chem., 2020, 85, 8271–8278 CrossRef CAS PubMed.
- C. Su, R. Madhavachary, A. Noble and V. K. Aggarwal, Org. Lett., 2020, 22, 7213–7218 CrossRef PubMed.
- (a) S. J. Danishefsky and M. T. Bilodeau, Angew. Chem., Int. Ed., 1996, 35, 1380–1419 CrossRef CAS; (b) H. H. Kinfe, Org. Biomol. Chem., 2019, 17, 4153–4182 RSC.
- G. Goti, ChemCatChem, 2022, 14, e202200290 CrossRef CAS.
- A. Borbás, Chem. – Eur. J., 2020, 26, 6090–6101 CrossRef PubMed.
- B. Wang, D.-C. Xiong and X.-S. Ye, Org. Lett., 2015, 17, 5698–5701 CrossRef CAS PubMed.
- C. Taillefumier and Y. Chapleur, Chem. Rev., 2004, 104, 263–292 CrossRef CAS PubMed.
- C. J.-M. Frédéric, J. Cornil, M. Vandamme, L. Dumitrescu, A. Tikad, R. Robiette and S. P. Vincent, Org. Lett., 2018, 20, 6769–6773 CrossRef PubMed.
- W.-Z. Shi, H. Li, G.-C. Mu, J.-L. Lu, Y.-H. Tu and X.-G. Hu, Org. Lett., 2021, 23, 2659–2663 CrossRef CAS PubMed.
- K. M. Nakafuku, S. C. Fosu and D. A. Nagib, J. Am. Chem. Soc., 2018, 140, 11202–11205 CrossRef CAS PubMed.
- H. Li, K.-C. Yu, J.-K. Su, W. Ouyang, N.-L. Fan and X.-G. Hu, Green Chem., 2022, 24, 8280–8291 RSC.
- K.-M. Liu, P.-Y. Wang, Z.-Y. Guo, D.-C. Xiong, X.-J. Qin, M. Liu, M. Liu, W.-Y. Xue and X.-S. Ye, Angew. Chem., Int. Ed., 2022, 61, e202114726 CrossRef CAS PubMed.
- E. W. M. Flores, M. L. Uhrig and A. Postigo, Org. Biomol. Chem., 2020, 18, 8724–8734 RSC.
- F. Rasool, A. H. Bhat, N. Hussain and D. Mukherjee, ChemistrySelect, 2016, 1, 6553–6557 CrossRef.
- (a) J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98(143), 13705–13762 Search PubMed; (b) M. W. Ruszczycky, Y. Ogasawara and H.-W. Liu, Biochim. Biophys. Acta, 2012, 1824, 1231–1244 CrossRef CAS PubMed.
- (a) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (b) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (c) J. Zhang and M. Rueping, Chem. Soc. Rev., 2023, 52, 4099–4120 RSC.
- J. A. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- I. C. Wan, M. D. Witte and A. J. Minnaard, Chem. Commun., 2017, 53, 4926–4929 RSC.
- J. A. Turner, T. Adrianov, M. A. Zakaria and M. S. Taylor, J. Org. Chem., 2022, 87, 1421–1433 CrossRef CAS PubMed.
- J. A. Turner, T. Adrianov and M. S. Taylor, J. Org. Chem., 2023, 88, 5713–5730 CrossRef CAS PubMed.
- F. Ghorbani, S. A. Harry, J. N. Capilato, C. R. Pitts, J. Joram, G. N. Peters, J. D. Tovar, I. Smajlagic, M. A. Siegler, T. Dudding and T. Lectka, J. Am. Chem. Soc., 2020, 142, 14710–14724 CrossRef CAS PubMed.
- H. Cao, T. Guo, X. Deng, X. Huo, S. Tang, J. Liu and X. Wang, Chem. Commun., 2022, 58, 9934–9937 RSC.
- V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed.
- M. S. Taylor, Acc. Chem. Res., 2015, 48, 295–305 CrossRef CAS PubMed.
- D. J. Gorelik, J. A. Turner, T. S. Virk, D. A. Foucher and M. S. Taylor, Org. Lett., 2021, 23, 5180–5185 CrossRef CAS PubMed.
- V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed.
- Y. Li and Y. Kuninobu, Adv. Synth. Catal., 2023, 365, 2577–2587 CrossRef CAS.
- V. Dimakos, D. Gorelik, H. Y. Su, G. E. Garrett, G. Hughes, H. Shibayama and M. S. Taylor, Chem. Sci., 2020, 11, 1531–1537 RSC.
- Y. Masuda, H. Tsuda and M. Murakami, Angew. Chem., Int. Ed., 2020, 59, 2755–2759 CrossRef CAS PubMed.
- H. M. Carder, C. E. Suh and A. E. Wendlandt, J. Am. Chem. Soc., 2021, 143, 13798–13805 CrossRef CAS PubMed.
- J. A. Turner, N. Rosano, D. J. Gorelik and M. S. Taylor, ACS Catal., 2021, 11, 11171–11179 CrossRef CAS.
- R. Lenz and B. Giese, J. Am. Chem. Soc., 1997, 11, 2784–2794 CrossRef.
- Y. Wang, H. M. Carder and A. E. Wendlandt, Nature, 2020, 578, 403–408 CrossRef CAS PubMed.
- H. M. Carder, Y. Wang and A. E. Wendlandt, J. Am. Chem. Soc., 2022, 144, 11870–11877 CrossRef CAS PubMed.
- C. J. Oswood and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 93–98 CrossRef CAS PubMed.
- D. J. Gorelik, V. Dimakos, T. Adrianov and M. S. Taylor, Chem. Commun., 2021, 57, 12135–12138 RSC.
- M. Wu, Q. Jiang, Q. Tian, T. Guo, F. Cai, S. Tang, J. Liu and X. Wang, CCS Chem., 2022, 4, 3599–3608 CrossRef CAS.
- J. N. Capilato, C. R. Pitts, R. Rowshanpour, T. Dudding and T. Leckta, J. Org. Chem., 2020, 85, 2855–2864 CrossRef CAS PubMed.
- D.-F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897–14900 CrossRef CAS PubMed.
- Y. Li, S. Miyamoto, T. Torigoe and Y. Kuninobu, Org. Biomol. Chem., 2021, 19, 3124–3127 RSC.
- M. Shaw and A. Kumar, Org. Lett., 2019, 21, 3108–3113 CrossRef CAS PubMed.
- (a) E. I. León, A. Martín, A. S. Montes, I. Pérez-Martín, M. del Sol Rodríguez and E. Suárez, J. Org. Chem., 2021, 86, 14508–14552 CrossRef PubMed; (b) A. S. Montes, E. I. León, A. Martín, I. Pérez-Martín and E. Suárez, Eur. J. Org Chem., 2022, e202101391 CrossRef CAS.
- H. Ye, C. Xiao, Q.-Q. Zhou, P. G. Wang and W.-J. Xiao, J. Org. Chem., 2018, 83, 13325–13334 CrossRef CAS PubMed.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330–334 CrossRef CAS PubMed.
- L. McSweeney, F. Dénès and E. M. Scanlan, Eur. J. Org Chem., 2016, 2080–2095 CrossRef CAS.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 CrossRef CAS PubMed.
- G. Zhao, S. Kaur and T. Wang, Org. Lett., 2017, 19, 3291–3294 CrossRef CAS PubMed.
- S. Kaur, D. P. Luciano, X. Fan, G. Zhao, S. Messier, M. M. Walker, Q. Zhang and T. Wang, Tetrahedron Lett., 2021, 86, 153499 CrossRef CAS.
- K. Rybicka-Jasínska, T. Wdowik, K. Łuczak, A. J. Wierzba, O. Drapała and D. Gryko, ACS Org. Inorg. Au, 2022, 2, 422–426 CrossRef PubMed.
- M. Zhu, G. Dagousset, M. Alami, E. Magnier and S. Messaoudi, Org. Lett., 2019, 21, 5132–5137 CrossRef CAS PubMed.
- M. Jouffroy, C. B. Kelly and G. A. Molander, Org. Lett., 2016, 18, 876–879 CrossRef CAS PubMed.
- C. Cavedon, E. T. Sletten, A. Madani, O. Niemeyer, P. H. Seeberger and B. Pieber, Org. Lett., 2012, 23, 514–518 CrossRef PubMed.
- K. Liang, X. Li, D. Wei, C. Jin, C. Liu and C. Xia, Chem, 2023, 9, 511–522 CAS.
- S. Leichnitz, K. C. Dissanayake, A. H. Winter and P. H. Seeberger, Chem. Commun., 2022, 58, 10556–10559 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |