
Scalable preparation of macroporous collagen microgels by air bubble-induced breakup and ice templating†
Sushant
Singh
 a,
Wing Yan
Chu
b,
Rojin
Ostadsharif Memar
a,
Andrew
De Carlo
a,
Teodor
Veres
ac and
Axel
Günther
*ab
a,
Wing Yan
Chu
b,
Rojin
Ostadsharif Memar
a,
Andrew
De Carlo
a,
Teodor
Veres
ac and
Axel
Günther
*ab
aDepartment of Mechanical and Industrial Engineering, University of Toronto, Toronto, Ontario M5S3G8, Canada. E-mail: axel.guenther@utoronto.ca
bInstitute of Biomedical Engineering, University of Toronto, Toronto, Ontario M5S 3G9, Canada
cLife Sciences Division, National Research Council of Canada, 75 Boulevard de Mortagne, Boucherville, QC, Canada
First published on 2nd July 2024
Abstract
Collagen I, the most abundant protein of the extracellular matrix, has found widespread use in three-dimensional cell culture, and increasingly also in bioprinting and biofabrication applications. However, several limitations remain, such as the capacity to locally recapitulate the multiscale organization of collagen in native tissues. Bioprinting cellular collagen structures with high feature fidelity so far either requires a more rapidly gelling biopolymer to be added or an acellular collagen structure to be defined before the delivery of cells. Here, we report the flow synthesis of macroporous collagen microgels (MCMs) that serve as building blocks for granular bioinks. Obtained bioinks offer excellent printability, provide an avenue to faithfully recapitulate the multiscale collagen organization of native tissues, and overcome the aforementioned limitations. Viscous collagen solutions with concentrations as high as 10 mg ml−1 are consistently converted into droplets using a parallelized microfluidic device via air bubble induced droplet breakup into a continuous oil phase. MCMs are obtained by inducing gelation, oil removal, and washing, and incorporating internal pores of tunable size via ice templating at freezing rates between 0.1 and 10 °C min−1. Independent control over the MCM diameter (175–250 μm) and porosity (58–76%) allows the extracellular matrix structure to be tailored to different tissue engineering applications. The wall structures within MCMs share similarities with the highly compacted and recapitulated collagen porosity in native tissues. This approach in the future can be used to 3D print more complicated biomimetic structures that require cell positioning during printing.
Introduction
Collagen type I is the most abundant constituent of the extracellular matrix (ECM) in many mammalian tissues. The fibril-forming protein exhibits a hierarchical organization1–3 that spans at least four orders of spatial magnitude: from the tropocollagen molecules that self-assemble in a characteristic D-period banding pattern, to tissue specific collagen fiber topologies that modulate tensile and compressive behavior.A wide range of collagen-based bulk (and to a limited extent granular) biomaterials have been presented and shown to promote cell attachment combined with excellent biocompatibility and low immunogenicity. Collagen-based biomaterials are therefore widely used in different cell culture and tissue engineering applications,4 as wound dressings in the clinic,5 and in biofabrication6–9 and bioprinting applications. However, extrusion bioprinting with neutral pH and acidic solutions of collagen I remains a challenge, due in part to the minute-long gelation time and the limited shear-thinning rheological behavior, often requiring dedicated workflows and support baths, and often producing mechanically soft gels.10,11 Structures that are extrusion bioprinted from solutions of collagen I often exhibit poor feature fidelity and sag after only depositing a few layers. In addition, the low concentrations (3–5 mg ml−1) of neutral pH collagen solution that is frequently used for three-dimensional (3D) cell culture leads to significant remodelling and contraction within only a few days of in vitro culture.12 Options to reduce unwanted contraction via in situ chemical cross-linking are limited by the mild conditions required to ensure the viability of cells within the collagen solution. While photo-crosslinking or adding biopolymers with shorter gelation times may be compatible with in vitro assays, they often impose regulatory hurdles concerning limited clinical adoption.
One approach to overcome these limitations and to obtain collagen structures with excellent feature fidelity is to bioprint them into a granular support bath containing gelatin microgels.13,14 However, the extrusion of acidic collagen solutions is incompatible with the one-step formation of cellular structures, and their high concentration (≥35 mg ml−1) results in relatively dense gels.15,16 Granular materials prepared from microgels have become frequently considered bioink candidates, due to their excellent printability.17–19 While collagen microgels have been reported,12,20–23 they are usually prepared from dilute collagen solutions, i.e. 2–3 mg ml−1, without templating, and therefore lack micron-sized pores next to areas of high collagen compaction and cross-linking, making them prone to rapid remodelling.12 Only very few approaches have demonstrated the capacity for collagen microgels to faithfully replicate the multiscale collagen organization found in native tissues that include the characteristic D-periodic banding as well as pores several micrometers in size and regions with densely packed collagen fibers.24,25
Here, we report a facile method for the production of macroporous26 collagen microgels (MCMs) that overcomes several of the remaining limitations. The derived granular bioink provides excellent printability and ensures extrusion bioprinted structures closely match target dimensions. Tailoring the MCM based bioink's porosity allows for fine tuning of the porosity within the printed construct and promotes cell migration.
Compared with previously reported conditions (Table 1), the presented emulsification method is compatible with at least four times higher collagen concentration, translating into an almost 45-fold increase in viscosity: from a concentration of 2 mg ml−1 (viscosity 0.03 Pa s) to a concentration of 10 mg ml−1 (viscosity 1.4 Pa s).27 Air bubble-induced breakup enables the consistent formation of collagen droplets into a continuous oil phase with a viscosity of 0.06 Pa s, significantly lower than the one of the collagen solution. This higher concentration of collagen is expected to reduce compaction and premature degradation during tissue culture12,25 thereby lowering the reliance on crosslinking. To induce porosity we opted for ice templating since the approach is scalable and has previously been used in casted collagen sheets.28 Our custom freezing setup enables subsequent sublimation after ice templating for long-term sample storage. Following ice templating, the microgels undergo changes in their overall morphology and exhibit inter-microgel porosity at certain freezing rates. However, our objective is to induce intra-microgel porosity to facilitate cell migration within these interstitial spaces post jamming.
| Material | Collagen concentration | Number of FDGs | Production rate | Emulsification method | Microgel size; CV | Ref. |
|---|---|---|---|---|---|---|
| Atelocollagen | 9 mg ml−1 | 10 | 160 s−1 (for 200 μm droplets) | Air bubble induced FFG | 175–250 μm; 5% | Present work |
| Atelocollagen | 2 mg ml−1 | 1 | 191 s−1 (for 100 μm droplets) | Axisymmetric FFG | 50–300 μm; 4% | 12 |
| Collagen | 2 mg ml−1 | 1 | 150 s−1 (for 72.4 μm droplets) | FFG & membrane emulsification | 75 μm shrunk to 10 μm; 3.5% | 20 |
| Collagen + alginate | 0.4 mg ml−1 | 1 | NA | Centrifuge DG | 118 μm; 10% | 21 |
| Collagen + alginate | 2 mg ml−1 | 1 | NA | FFG | 440 μm; ∼5% | 22 |
| Collagen + ultra-long single-stranded DNA | 2 mg ml−1 | 1 | NA | T-Junction DG | 500–1000 μm; NA | 23 |
| Atellocollagen + GAG/PEG | 4 mg ml−1; diluted during emulsification | 15 | 278–5800 s−1 | FFG | 25–200 μm; NA | 24, 25 |
Our work primarily centers around four key areas: (1) ensuring the consistent synthesis of collagen microgels, particularly for higher concentrations; (2) automating the washing process of microgels with quantitative testing; (3) achieving porosity values and pore sizes exceeding those typically attainable in microgels; and (4) evaluating printability of bioinks prepared from MCMs for extrusion bioprinting applications.
Materials and methods
Microfluidic device fabrication
Droplet/bubble microfluidic devices were fabricated using multi-layer soft lithography. We prepared two molds, one for flow distribution, and the other for droplet/bubble generation via flow focusing generators (referred to as the FFG layer).29 The mold for the FFG layer was prepared from the negative photoresists SU-8 2025 and 2050 (Kayaku Advanced Materials, Massachusetts, USA) in a two-step process involving spin-coating and UV patterning. Alignment marks on the two photomasks for the FFG layer to be aligned with respect to one another. This process resulted in microchannels with three different depths: 40 μm for the flow resistance channels, 110 μm for the upstream channels, and 300 μm for the downstream channels (Fig. S1†). The mold for the flow distribution layer was prepared from SU-8 2100 spinning two layers of 250 μm, providing a total thickness of 500 μm. We followed manufacturer instructions regarding spinning speed, baking times, and UV exposure rates. To achieve a uniform thickness of SU 8 across the device, hot plates were leveled using a NIST-calibrated digital surface inclinometer (Digi-Pas pioneers Smart 2-AXIS master Machinist Precision Level & Digital Inclinometer, McMaster Carr, Ontario, Canada).After the two molds were prepared, the fabrication process continued with casting the FFG layer. First, premixed and de-gassed polydimethylsiloxane (PDMS, SYLGARD™ 184 Silicone Elastomer Kit, Dow Chemical Company, Michigan, USA) was cast onto the FFG layer mold and baked at 80 °C for one hour. Next, PDMS precursor was poured onto the distribution layer mold and degassed. The cured FFG layer was carefully placed over the PDMS precursor, aligning the respective channels with the FFG microchannels on one and the distribution microchannels on the other layer. The stacked PDMS layers were then placed in an oven for one hour to complete curing. To establish fluid connections, the inlet and outlet channels for collagen, air, and oil, as well as the individual FFGs, were fluidically connected to the distribution channels using a 0.75 mm biopsy punch. An additional layer of pre-cured PDMS was subjected to corona treatment and bonded to the FFG side of the microchannel layer to seal the device on the top. Subsequently, 2 and 3 mm diameter inlet and outlet holes were punched. The distribution side of this layer also underwent corona treatment and was placed over a pre-cured, thick slab (∼10 mm) of PDMS. The entire device was then placed into an oven and cured for one hour at 80 °C. After cooling to room temperature, Aquapel (item number 47100, Aquapel Glass Treatment, Quebec, Canada) to render the microchannel walls hydrophobic.30 Aquapel solution was injected into the channels for 5 min, followed by manual withdrawal using a syringe. The unreacted solution was replaced by injecting and aspirating mineral oil. Finally, the device was baked at 80 °C for 1 h. The oil was passed through 0.22 μm syringe filters, before use. For device characterization, a device with four parallelized droplet generators was designed, while all subsequent experiments were conducted using a device with ten parallelized droplet generators (Fig. S2†).
Preparation of solutions and air-bubble induced droplet formation
For the disperse phase, a 9 mg ml−1 solution of atelocollagen was prepared from FibriCol® type I collagen solution (10 mg ml−1, bovine collagen, Advanced BioMatrix, California, USA). To achieve this, two stock solutions were prepared. The first consisted of 0.05 M acetic acid in 10 × phosphate buffer saline (solution 1). The second consisted of 0.05 M acetic acid in deionized water (solution 2). The two solutions were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio (solution 1:solution 2), affording the flexibility to adjust the collagen concentration as needed. The latter was then mixed with the collagen solution at the same 1:9 ratio and passed through a 10 μm syringe filter. For the continuous oil phase, heavy mineral oil (mineral oil, pure, Thermo Scientific Chemicals, Massachusetts, USA) was combined with 5% vol/vol Span 80 (Sigma Aldrich, Ontario, Canada). Similarly, to create a basic oil phase, 0.2–0.3% vol/vol triethylamine (Sigma Aldrich, Canada) was added along with 5% vol/vol Span 80. A calibration curve for the pH as a function of the triethylamine concentration was prepared (ESI† Fig. S3) according to a previously published protocol.23 To supply the microfluidic device, pressure pumps equipped with flow rate sensors (model PneuWave ECO Pump, CorSolutions, New York, USA) were employed. Flow rate sensors were calibrated according to manufacturer's instructions. The prepared collagen solution was stored at 4 °C to prevent premature gelation and denaturation of collagen. Downstream of the microfluidic device, an external vial was connected to deliver the prepared emulsion through a T-junction where the basic oil containing triethylamine was supplied from a glass syringe to avoid plunger swelling, at a flow rate controlled by a syringe pump (model PhD 2000, Harvard Apparatus, Massachusetts, USA). The droplets of collagen solution and interspersed air bubbles flowed into a collection vial. To initiate the in-flow fibrillogenesis of collagen, the outlet tube was immersed in a water bath at 37 °C with sufficient length for a residence time of approximately 45 minutes.
:9 ratio (solution 1:solution 2), affording the flexibility to adjust the collagen concentration as needed. The latter was then mixed with the collagen solution at the same 1:9 ratio and passed through a 10 μm syringe filter. For the continuous oil phase, heavy mineral oil (mineral oil, pure, Thermo Scientific Chemicals, Massachusetts, USA) was combined with 5% vol/vol Span 80 (Sigma Aldrich, Ontario, Canada). Similarly, to create a basic oil phase, 0.2–0.3% vol/vol triethylamine (Sigma Aldrich, Canada) was added along with 5% vol/vol Span 80. A calibration curve for the pH as a function of the triethylamine concentration was prepared (ESI† Fig. S3) according to a previously published protocol.23 To supply the microfluidic device, pressure pumps equipped with flow rate sensors (model PneuWave ECO Pump, CorSolutions, New York, USA) were employed. Flow rate sensors were calibrated according to manufacturer's instructions. The prepared collagen solution was stored at 4 °C to prevent premature gelation and denaturation of collagen. Downstream of the microfluidic device, an external vial was connected to deliver the prepared emulsion through a T-junction where the basic oil containing triethylamine was supplied from a glass syringe to avoid plunger swelling, at a flow rate controlled by a syringe pump (model PhD 2000, Harvard Apparatus, Massachusetts, USA). The droplets of collagen solution and interspersed air bubbles flowed into a collection vial. To initiate the in-flow fibrillogenesis of collagen, the outlet tube was immersed in a water bath at 37 °C with sufficient length for a residence time of approximately 45 minutes.
Liquid handler setup and sampling
We observed heavy mineral oil to lead to increased stickiness between the microgels, rendering the washing process more difficult. To mitigate the effect, the emulsion was manually washed 2–3 times with hexadecane (Sigma Aldrich, Canada).12 To initiate the washing process, a 10 mL solution containing microgels immersed in hexadecane was transferred into 10 mL deep well plates (catalog number 95040470B, KingFisher™ plastics for 24 deep-well format, Thermo Fisher Scientific, Ontario, Canada) which were subsequently placed inside an automated liquid handler (model JANUS® G3, PerkinElmer, Waltham, Massachusetts, USA). A programmed sequence of steps was executed. First, 4 mL of excess supernatant was removed, and 4 mL of the surfactant Tween 20 (Sigma Aldrich, Canada) solution (0.1% vol/vol) was then added to reduce the interfacial tension and remove pockets of hexadecane remaining within or in between microgels. The solution was thoroughly mixed by aspiration to ensure the homogenization of the suspension. A 30 min wait period allowed the microgels to settle to the bottom. The settling time was determined from a measurement of the sedimentation velocity of the collagen microgels (Fig. S4†). The described washing sequence was carried out five times in a surfactant solution, followed by five times in distilled water. A sample of the supernatant was collected after each washing step and analyzed to determine the concentration of residual Tween 20. Fluorescent dye Vybrant™ DiI (Thermo Fisher Scientific, Canada) was added at a concentration of 1 mM in ethanol. Each supernatant sample was treated with DiI, resulting in a final dye concentration of 720 nM. Prepared samples were thoroughly mixed, incubated for 30 min at 37 °C, filled into 96 well plates and analyzed with a high content imaging system (PerkinElmer Operetta CLS High Content Analysis System, PerkinElmer, USA). Confocal images were captured for each sample at 100% intensity, with a binning factor of 2 and an exposure time of 1 s. The images provided intensity readings for further analysis. A calibration curve was established for Tween 20 concentrations from 0.001% to 0.1% (vol/vol) treated with Dil as described.31 Fluorescence emission intensity of these samples was then determined using the PerkinElmer FL6500 Fluorescence Spectrophotometer (PerkinElmer, USA) for excitation at a wavelength of 550 nm and the emission wavelength of 550 to 620 nm. The bandwidth of the excitation and emission bandpass filters were 5.0 nm and 2.5 nm, respectively, and the gain was set to 1. Similarly, a calibration curve was established using the integrated confocal microscope (Perkin Elmer Operetta) using an excitation wavelength of 586 nm.MCM formation via ice templating
For ice templating of the collagen microgels, a custom setup was prepared (Fig. S5–S7†). Three thermoelectric coolers (HP-127-1.4-2.5-72, TE Technologies, Michigan, USA) were operated in parallel and in closed-loop mode. A thermistor (MP-3176, TE Technologies, USA) read the actual temperature and supplied it to a programmable controller (Temperature Controller TC-720, TE Technologies, USA). At the cold side, a machined aluminium plate, designed as a sample holder, was securely affixed to the thermoelectric cooler according to manufacturer recommendations. On the warm side, a modified heat sink (CP 4012, Wieland Microcool, Alabama, USA) was perfused with temperature-controlled water supplied by an external chiller unit (model 6550T21A130E 6500 Chiller with Turbine Pump, PolyScience, Illinois, USA). Heat conduction was improved by inserting a graphite pad sheet (EYG-S131810, Panasonic Electronic Components, DigiKey, Ontario, Canada) between the aluminium plate and the thermoelectric cooler in lieu of thermal paste. This adjustment improved the uniformity of the temperature across the custom sample holder. The described configuration allowed for the efficient removal of heat and reached a temperature as low as −57 °C within the freezing stage throughout the 24 h testing period (Fig. S5†). To maintain a controlled sublimation environment, the entire aluminium sample holder was surrounded by a custom chamber made from polymethyl methacrylate. The chamber contained gaskets for an airtight seal. The entire assembly was connected to a cold trap (model 7460020 CentriVap® −84 °C Cold Trap, Labconco, Illinois, USA) and a rotary vane vacuum pump (model RV 12, Edwards Vacuum, Burgess Hill, UK) capable of achieving a vacuum pressure of approximately 54 mTorr (7.2 Pa). The described setup allowed us to lyophilize MCM samples in situ with well controlled cooling rates (Fig. S4†).Scanning and transmission electron microscopy of microgels
The primary fixation of the microgels involved immersion in a solution containing 4% paraformaldehyde and 1% glutaraldehyde, both diluted in a 0.1 M phosphate buffer at a pH of 7.2. Fixation was either carried out for a minimum of 1 h or extended overnight for optimal results. Subsequently, the MCMs underwent primary washing with a 0.1 M phosphate buffer at the same pH (7.2), with a requirement of at least three buffer changes conducted within 45 min. The microgels were then subjected to secondary fixation, involving post-fixation with 1% osmium tetraoxide (Sigma Aldrich, Canada) buffered in a 0.1 M phosphate buffer at a pH of 7.2. This secondary fixation process was also carried out for a minimum of 1 h, prior to a secondary washing step using distilled water. The latter involved two changes of distilled water within 30 min. Dehydration was carried out by serial dilution in ethanol, consisting of the following ethanol concentrations: 30% (two changes in 15 min), 50% (two changes in 20 min), 70% (two changes in 30 min), 90% (two changes in 45 min), and 100% (three changes in 1 h). Following dehydration, critical point drying was conducted for at least 10 min in the critical point drying purge phase i.e. in liquid carbon dioxide. Finally, the samples were coated with a 5 to 12 nm gold layer, prior to imaging on an environmental scanning electron microscope (model Prisma E, voltage 30 kV, Thermo Fischer Scientific, Canada).For transmission electron microscopy (TEM), the MCMs underwent a series of processing steps.32 Initially, they were washed three times in a 0.1 M cacodylate buffer (pH 7.4) for 15 min each, fixed for 60 min at room temperature, before being kept overnight at 4 °C in a mixture of 2.5% glutaraldehyde in a 0.1 M cacodylate buffer with the same pH. Following fixation, the samples were again washed three times in 0.1 M cacodylate buffer at pH 7.4, for 15 min each. The next step was en-bloc reduced osmium staining with 1% osmium tetroxide and 1.5% potassium ferrocyanide in a 0.1 M cacodylate buffer at pH 7.4 for 1 h at room temperature. Samples were then washed 5 times with double distilled water for 3 min each. Samples were stained with tannic acid; fresh 1% tannic acid in 0.1 M cacodylate buffer for 2 h at 4 °C, then tannic acid stain was replaced and left for another 2 h at 4 °C. Samples were washed 5 times with double distilled water for 3 min each. Secondary fixation was performed with 2% osmium tetroxide in double distilled water for 40 min at room temperature. Samples were washed 3 times with double distilled water for 5 min each. Staining was performed with 1% uranyl acetate in double distilled water at 4 °C overnight. Samples were washed 3 times with double distilled water for 5 min each. Serial dehydration with ethanol was performed at the following volume concentrations: 30%, 50%, 70%, 90%, 100%, 100%, 100%, 100% for 10 min per step. Samples were infiltrated using dilutions of the EMbed 812 resin kit (Electron Microscopy Sciences, Pennsylvania, USA) in propylene oxide: 100% propylene oxide (Sigma Aldrich, Canada) for 10 min, 30% (v/v) EMbed 812 resin in propylene oxide for 4 h. Followed by 50% (v/v) Embed 812 resin in propylene oxide overnight, 70% (v/v) EMbed 812 resin in propylene oxide for 1 h, 90% (v/v) EMbed 812 resin in propylene oxide for 1 hour, 100% EMbed 812 resin for 1 h, repeat twice more 100% EMbed 812 resin for 1 h. Resin embedded samples were placed into silicone moulds and cured at 60 °C for 48–72 h. Resin-embedded microgel samples were removed from moulds, trimmed, and 60–80 nm thin sections were cut with a microtome (model Reichert-Jung Ultracut E, Leica Mikrosysteme, Vienna, Austria). Precision cut sections were post-stained in 5% uranyl acetate solution for 15 min, washed with deionized water, followed by a 15 min staining with Reynolds lead citrate, and washing with deionized water, prior to TEM imaging (voltage 120 kV, model Talos™ L120C TEM from Thermo Fisher Scientific, USA).
Porosity measurements
To assess porosity, a solution containing 0.1% weight/volume fluorescein isothiocyanate–dextran (FITC–dextran, Sigma Aldrich, Canada) was mixed with the previously prepared collagen solution and subsequently emulsified. The relatively high initial dye concentration was selected to account for the notable dilution of FITC–dextran during the washing steps. The microgels were then subjected to centrifugation at 2000g and loaded into a 1 mm × 1 mm cross section channel that was defined by bonding a soft lithographically patterned PDMS substrate to a glass coverslip. This step was taken to prevent unwanted sample dehydration due to evaporation during imaging. Samples were 3D imaged using a light sheet microscope (model Stellaris 5, Leica Microsystems, Wetzlar, Germany) equipped with a 63× oil immersion objective (NA 1.4), where a 25 μm separation between imaging planes was selected in the depth (z) direction. Acquired z stacks were imported into a 3D reconstruction and image analysis program (Imaris 10.0, Oxford Instruments, Abingdon, UK). Rendering provided information on the particle topology and porosity.33Rheology
For the rheological analysis of the granular bioink, the sample was introduced into a rheometer (model Discovery HR3 Hybrid Rheometer, TA Instruments, Delaware, USA) in a parallel plate configuration. A 60 mm sandblasted stainless steel plate (AR-G2, TA Instruments, USA) was used to reduce slippage.34 A 1000 μm gap was maintained. One plate was set to rotate at an angular velocity of 3 s−1 during the loading phase while the other one was stationary. Prior to initiating the experiment, the axial force was adjusted to 0 N. Once the sample was loaded, the granular bioink was conditioned by applying a shear rate of 500 s−1 for a specified duration in both the clockwise and the anticlockwise directions. Following this conditioning, the bioink was allowed to rest for a period of 3 min before the experiment commenced. During the flow sweep experiments, the bioink was subjected to shear rates that ranged from 500 s−1 to 0.05 s−1. During the oscillation amplitude experiments, bioink samples were strained across a range from 0.1% to 1000%. During the oscillation time sweeps, the bioink samples were subjected to a strain of 300% for 50 s followed by a 1% strain for 50 s. The sequence was repeated twice during the experiment. All experiments are reported as the average of three measurements from three different batches of prepared collagen microgels.Cell culture with microgels and immunostaining
To culture cells, the microgels underwent manual washing. Adult human multipotent stromal cells (RoosterKit™-hUC-1M-XF, RoosterBio, Maryland, USA), were cultured in xeno-free basal media (Rooster Nourish – MSC-XF). All cells were within the first three passages and cultured in accordance with the manufacturer's recommendations. Cells were mixed with microgels at a concentration of 1 × 106 ml−1, prior to cell seeding in 96 well plates. Culture medium was changed every other day, and samples were fixed on the 21st day of the culture period. On this day, the samples were fixed with a 4% paraformaldehyde solution for 20 min, in accordance with manufacturer guidelines for specific staining procedures. Applied stains include phalloidin FITC (ab235137, Abcam, Ontario, Canada) for the cytoskeleton, Hoechst (ab228551, Abcam, Canada) for the cell nuclei, Alexa Fluor® 647 anti-alpha smooth muscle actin antibody (ab202296, Abcam, Canada) for smooth muscle actin, and primary antibody (ab138492, Recombinant Anti-Collagen I antibody, Abcam, Canada) along with a secondary antibody ab72465 (Goat Anti-Rabbit IgG H&L preabsorbed with PE, Abcam, Canada) for collagen staining. Imaging was performed on a Zeiss LSM 880 super-resolution confocal microscope (Zeiss, Baden-Württemberg, Germany).Bioink preparation and bioprinting
Prior to printing, granular bioink was prepared from MCMs in several steps. Initially, MCMs were homogeneously dispersed in phosphate buffer saline by vertexing, followed by centrifugation at 4000g to separate MCMs from the supernatant. The resulting MCM suspension was transferred into two syringe barrels, which were again centrifuged for further compaction. Excess supernatant was removed and a luer lock connector coupled the syringes containing the jammed microgels.17 During 40 cycles, the granular bioink was mixed by periodically transferring it between the two syringe barrels via back-and-forth translation of the syringe plungers. The entire bioink volume was then filled into one of the syringes, capped and stored at 4 °C overnight. For extrusion bioprinting, modifications were made to an original PRUSA i3 3D printer (Prusa Research, Prague, Czech Republic) by connecting it to an external syringe pump (Harvard Apparatus Phd 2000, Harvard Apparatus, USA) for bioink delivery. A tapered dispensing tip with an internal diameter of 0.8 mm (6699A3, McMaster Carr, Canada) at the exit served as the printhead, due to its improved extrusion performance over printheads with a sudden constriction.35 The granular bioink was extrusion bioprinted to define a rectangular grid within one plane, with a flow rate of 6 ml min−1 and a feed rate of 7.6 mm s−1. The optimal printhead-to-printing-surface distance and minimum filament-to-filament distance were determined through filament printing tests. Dimensions, including filament width, spacing and height, were measured using surface profilometry (Keyence VHX7000 microscope system). The printability index indicating shape fidelity and printing accuracy,36 Pr = L2/16A,37,38 was determined using ImageJ, where l and A are the perimeter and area of the square within the bioprinted grid, respectively.Statistical analysis
Quantification data are presented as mean ± one standard deviation. One-way ANOVA followed by Tukey's multiple comparisons test or Brown-Forsythe ANOVA with Dunnett's multiple comparison test for statistical analysis using the GraphPad Prism software version 10.02. A p-value <0.05 was considered significant. Significant differences are indicated by *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.Results and discussion
Our method of obtaining granular bioink from flow synthesized MCMs is summarized in Fig. 1. The first step is the microfluidic emulsification of high-concentration collagen solution.39 For the device, we designed parallelized microfluidic droplet generators for the emulsification of acidic collagen solution at concentrations of up to 10 mg ml−1. We incorporated established ladder geometry design principles.19,40 We made two designs, one with four flow focusing generators (FFGs) and another with ten FFGs. To the best of our knowledge, this is the first report of parallelizing the bubble-induced emulsification technique. Our microfluidic device with ten FFGs reduces the time of producing collagen droplets within a continuous oil phase at volumes relevant to bioprinting purposes. Downstream of the device, an oil and water-soluble base (triethylamine)41 is via a T-connector added to the oil phase, initiating gelation of collagen under low-shear conditions, at 37 °C. The collagen microgels (CMs) and air bubbles and the oil are collected from the exit of the microfluidic device. Due to buoyancy, the air bubbles rise to the top and coalesce. The retained colloidal suspension is then washed using an automated liquid handler, which simultaneously samples the excess supernatant to determine the concentration of the remaining surfactant, Tween 20. Post-washing, collected samples are frozen at different rates to induce porosity within the CMs, producing MCMs. Finally, granular bioinks are prepared for extrusion bioprinting applications. | ||
| Fig. 1 Flow synthesis of macroporous collagen microgels (MCMs), washing, constituting, and extrusion-bioprinting of granular bioink. (A) Parallelized bubble/droplet generator with 10 individual flow-focusing generators (FFGs). Scale bar: 2 cm. (B) Top view of device with three distribution channels and one common collection channel. (C) Air bubble induced emulsification method enables collagen emulsification. (D) Off-chip, the emulsion is mixed with basic oil carrying triethylamine, inducing collagen gelation. (E) Air bubbles rise to top and coalesce. (F) Oil is removed from the emulsion by automated washing using a liquid handler. (G) Freezing provides control over MCM pore size, followed by jamming and (H) extrusion bioprinting the granular bioink. | ||
Air bubble induced collagen emulsification
Enhancing the breakup of the collagen solution by synchronous air bubble formation was essential. Furthermore, when we used previously published protocols12 to form collagen microgels, both with and without air bubble chopping, we observed significant droplet fragmentation when collagen gelation was triggered within microfluidic channels (Fig. S8†). We attribute this phenomenon to the combined effects of the non-Newtonian rheological behavior of the collagen solution and the onset of gelation at the collagen droplet interface. These gelled collagen fragments are assumed to be dislodged by the shear forces. However, a key difference could also be the source of collagen employed previously by Matsunaga et al.12 Air bubbles induced the hydrodynamically confined collagen jet to break up into droplets, as shown in the image sequence in Fig. 2A. In the absence of the air phase, the collagen solution filament that is on both sides hydrodynamically confined by the continuous oil phase elongates, without breaking up. Only when both the collagen solution and air are supplied, consistent emulsification is observed. Once an air bubble forms, the collagen phase begins to create a bulb in the narrow orifice, which grows until it reaches its maximum size, referred to as growth cessation. During this stage, the air phase travels towards the narrow orifice, eventually forming a bubble via a Rayleigh–Plateau instability, causing the neck of the bulb to collapse, forming droplets of collagen solution. Next, we investigated the impact of the air flow rate, Qb, or the air pressure on the droplet generation frequency with the device with 4 FFGs. Similar to findings reported by Zhou et al.,39 we observed a critical air flow rate below which the collagen solution broke up non uniformly sized droplets, as shown on the left side of Fig. 2B. However, once this critical air flow rate was exceeded, stable emulsification with a uniform droplet size was achieved for the selected durations that extended up to 10 h. Surprisingly, increasing Qb did not lead to an increase in the frequencies for generating air bubbles, fb, or droplets of collagen solution, fc. Both frequencies remained constant. Nevertheless, a continued increase in air bubble diameter, Db, was observed. Similarly, we increased the collagen flow rate, Qc, and measured the resulting droplet diameter, Dc, produced by all four FFGs in the common collection channel. This increase in collagen flow rate yielded a linear increase in Qc, as demonstrated in Fig. 2C and D.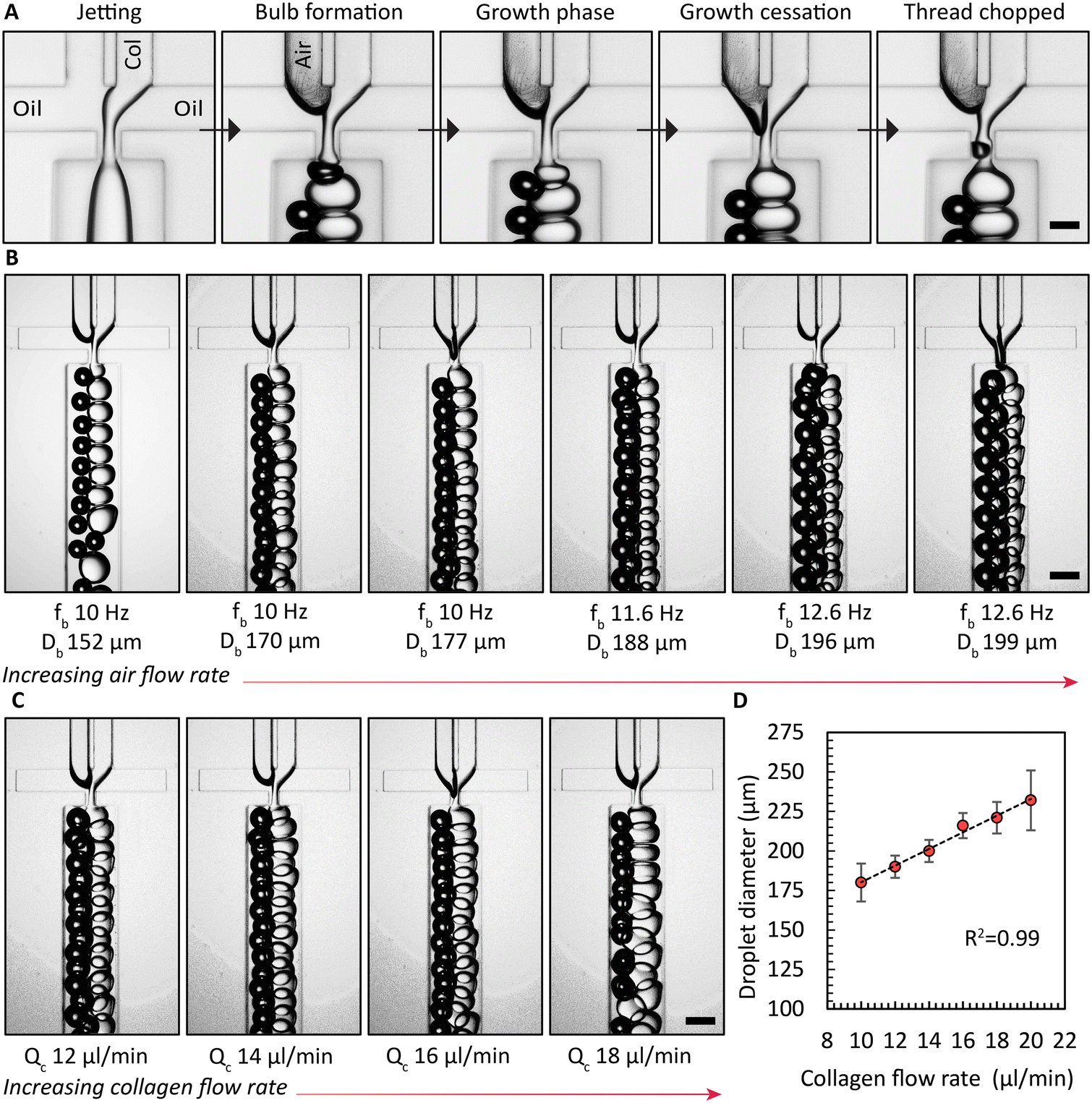 | ||
| Fig. 2 Air-bubble induced breakup of collagen solution in a microfluidic device with four FFGs. (A) Image sequence displaying different stages of collagen droplet formation. Col: collagen. Scale bar: 100 μm. (B) Effect of air pressure or flow rate on bubble/droplet generation frequency. Air pressures, from left to right: 1.4, 1.6, 1.8, 2, 2.2, and 2.4 PSI or 9.6, 11, 12.4, 15.1, and 16.5 kPa. Scale bar: 100 μm. (C) Collagen flow rate varied from 12 to 18 μl min−1. Scale bar: 100 μm. (D) Measured 40 collagen droplet diameters for each condition, showing a linear trend. | ||
It is noteworthy that the design principle we have implemented was previously reported by Yadavali et al.42 to parallelize over 20000 droplet generators. In applications demanding the production of microgels at rates higher than 1 l h−1, the choice of substrate material used in microfluidic device fabrication must be considered to withstand the heightened inlet pressures. In such cases, devices fabricated from thermoplastics or silicon substrates are expected to be beneficial due to improved bonding strength between layers and resistance to biofouling.43 Such approaches that go beyond the scope of the present work are expected to further extend the time for continuous microgel formation beyond the 48 h running time reported in this work.
Automated washing and sampling
An important yet often overlooked aspect of microgel formation is the necessary removal of oil and washing of surfactants. When aiming to scale microgel production to throughputs exceeding 1 l h−1, automating44,45 the laborious washing process becomes inevitable. Ultimately, a combination of in-line washing procedures46 and automated handling for sampling and quality control is not only required for practical reasons but will be required to satisfy the rigorous regulatory requirments47 for ultimate clinical use. Here, we demonstrate surfactant removal during washing and sampling using an automated liquid handler with microgels produced using the device with ten FFGs for sufficient quantities. While the exact binding mechanism of Dil to Tween 20 is not completely understood, it is expected to be similar to its use in tagging cell membranes.48 We were able to improve process flow as the liquid handler is integrated with a confocal microscope enabling intensity measurements within the same platform. The R2 values obtained from the calibration curves generated by the fluorescence spectrophotometer agreed favorably with those obtained from the confocal microscope (4% difference, Fig. 3A–D), suggesting the confocal sensor as a suitable alternative or complement to the protocols used in previous studies. Using this approach, we successfully washed the collagen microgels while retaining the supernatant from each well for later analysis. As shown in Fig. 3E, the intensities of the supernatant decreased with each washing step. Finally, utilizing the calibration curve shown in Fig. 3D, we determined the residual amount of Tween 20 remaining in the sample. Our findings indicated that after five washes, the surfactant concentration of Tween 20 was reduced to only ∼10 ppm from initially 1000 ppm. According to a prior study, the residual surfactant concentration is deemed safe for cell culture and is not expected to cause unwanted lysis or cell wall damage.49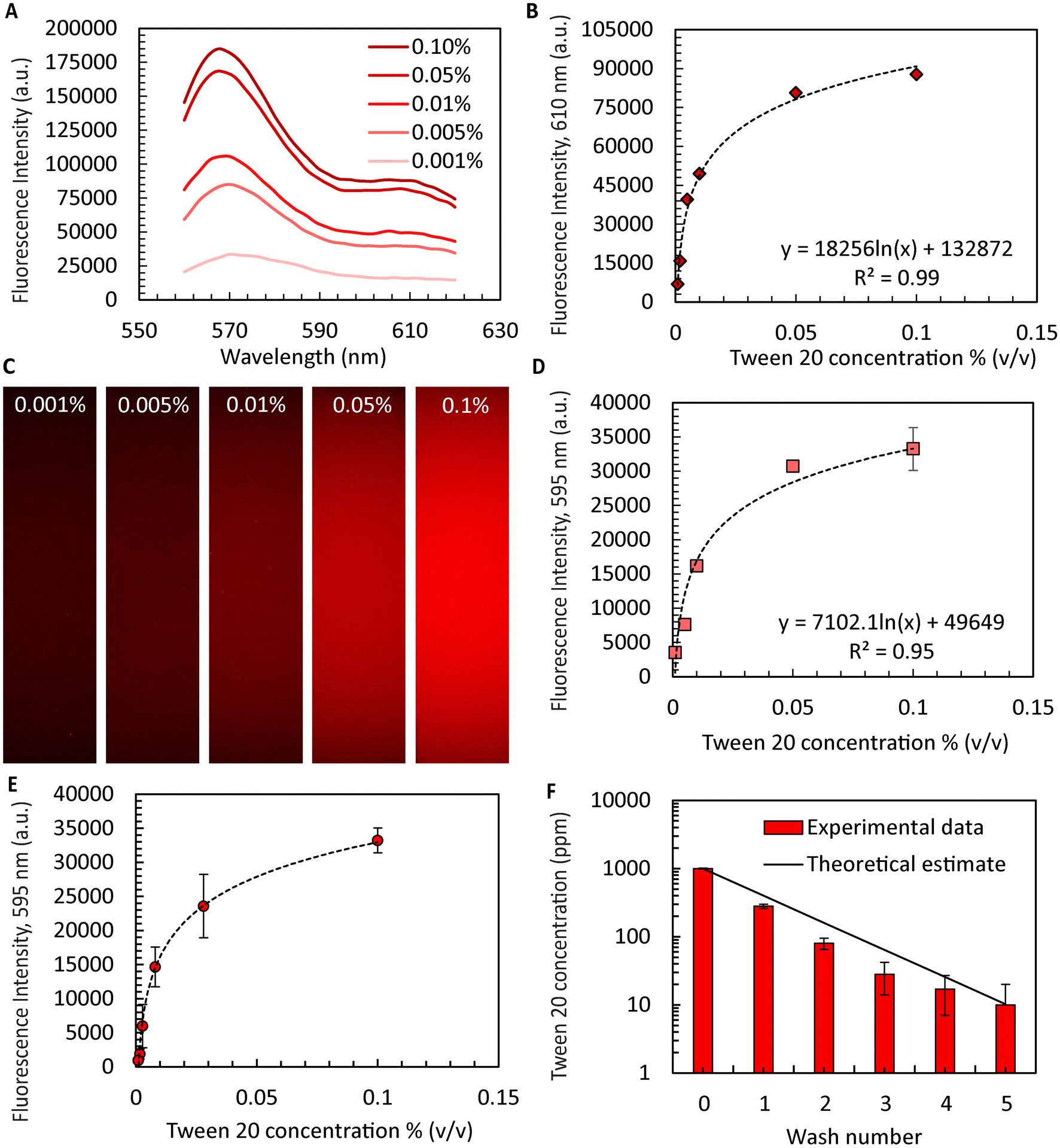 | ||
| Fig. 3 Quantification of automated collagen microgel washing for surfactant removal. (A) Fluorescence emission spectra were generated at an excitation wavelength of 550 nm using fluorescent dye Dil for different concentrations of surfactant Tween 20 (n = 3). (B) Using a fluorescence spectrophotometer, fluorescence intensity was plotted at 610 nm with the curve fit equation displayed (n = 3). (C) Images were generated from a confocal microscope at same surfactant concentrations (Texas red channel). (D) Fluorescence intensity readouts obtained from confocal microscope was plotted at 595 nm with the curve fit equation displayed (n = 3). (E) Measured reduction in Dil intensity after each of the five washing steps (from right to left) (n = 3). (F) Intensity data from (E) used to calculate Tween 20 concentration in ppm using the calibration curve generated in (D) (n = 3). | ||
Macroporous collagen microgels
Fig. 4A shows the CMs obtained after washing. Air bubble induced breakup provided CMs with a narrow size distribution (coefficient of variation 5%, Fig. 4B). We used SEM to evaluate the morphology of the microgels. The images in Fig. 4C and D reveal the well-defined collagen fibers, including the expected fiber bundles. To assess the uniformity of the gelation process, the samples were examined at three distinct locations using TEM. All areas exhibited consistent fiber area coverage (Fig. 4E), and the characteristic D-periodic banding pattern of collagen was consistently observed from the MCM edge to the center (Fig. 4F). The measurements of fiber areas at these locations demonstrated no significant differences (Fig. 4G). We hypothesize that this uniform gelation is attributed to the recirculation within the droplet,50 induced by the viscous drag of the continuous oil phase. Therefore, our approach allows us to robustly form collagen microgels with all the properties expected in a collagen gel. | ||
| Fig. 4 Ultrastructure of CMs. (A) Bright-field micrograph of collagen microgels in deionized water after washing, with size distribution shown in (B). Scale bar: 100 μm. (C) and (D) Scanning electron micrographs illustrating collagen fibers and fiber bundles. Scale bars: 50 μm and 1.5 μm for (C) and (D), respectively. Three locations selected for transmission electron microscopy (TEM) in (C), including the center, middle, and edge, are illustrated. (E) TEM images showing fiber coverage at the three locations. Scale bar: 2 μm. (F) TEM image showing D-periodic banding pattern in collagen across the entire microgel, from the center to the edge. Scale bar: 100 nm. (G) Fiber area data obtained from images in (E) from 5 microgels. Repeated measures one-way ANOVA with Tukey's multiple comparisons test was performed for statistical analysis. | ||
To enhance the porosity within the granular inks prepared from collagen microgels, we conducted ice templating at the freezing rates 0.1, 1, and 10 °C min−1, until the final temperature of −20 °C was reached. Depending on the applied freeing rate, the MCMs exhibited one of two morphologies. At 0.1 °C min−1, the microgels are compressed by the formation of larger ice crystals that are comparable in size with the microgels themselves, forming star-shaped structures with the initially loose collagen fibers being confined within MCM internal walls (Fig. 5A and B). At the two higher freezing rates, the microgels form a core-like structure where the thickness of these walls reduces, revealing intra-microgel porosity (Fig. 5C–F). After thawing, the Feret diameter of the MCMs was measured. As depicted in Fig. 6A–C, the ice crystals induced significant local compaction within the microgels. Another intriguing result was the smaller change in Feret diameter for the freezing rates of 1 and 10 °C min−1 (Fig. 6D). TEM images (Fig. 6E–G) reveal a similar effect on wall thickness post-ice templating. Collagen fibers within the wall seem aligned parallel to each other, similar to reports by Divakar et al.51 We hypothesize that the minimal alteration in microgel attributes under higher freezing rate conditions may be attributed to the ice crystals not exceeding a critical threshold in size, beyond which the desired ice templating effect becomes evident.
 | ||
| Fig. 5 Scanning electron microscopy of macroporous collagen microgels (MCMs). (A) and (B) Representative images of microgels frozen at 0.1 °C min−1 showing the formation of wall like structures. Scale bars: 50 μm and 100 μm for (A) and (B), respectively. (C) Representative images of microgels frozen at 1 and 10 °C min−1 showing reduced depth of walls. Scale bars: 50 μm. (D) Freeze-fractured microgels reveal the internal microstructure of the microgels frozen at 1 and 10 °C min−1. Scale bars: 50 μm. (E) and (F) Show the inter microgel porosity inside the wall structure shown in (F). Scale bars: 10 μm and 1.5 μm for (E) and (F), respectively. | ||
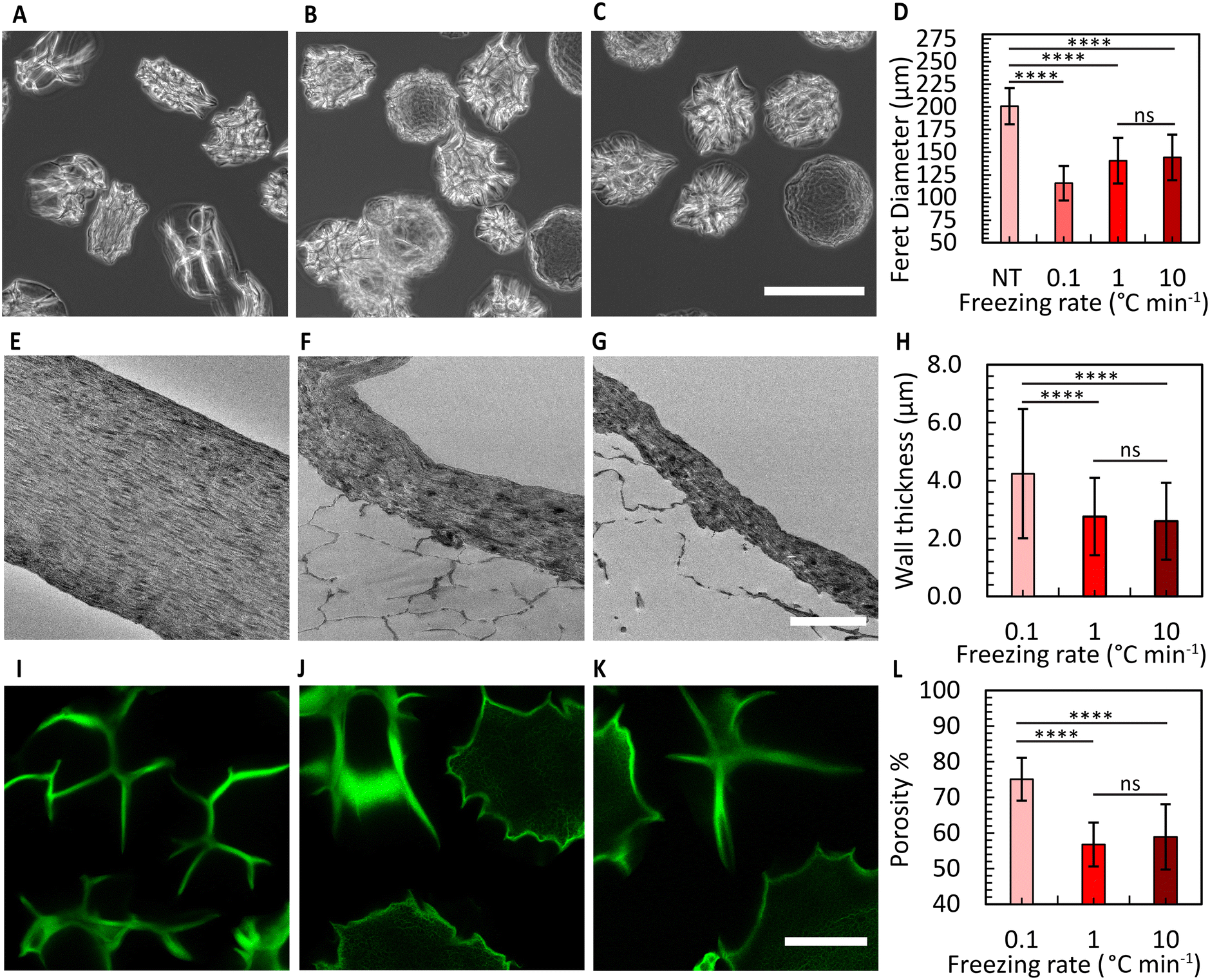 | ||
| Fig. 6 Ultrastructure of MCMs. Freezing conditions, shown from left to right, are 0.1, 1, and 10 °C min−1 for all images. (A)–(C) MCMs in deionized water after ice templating. Scale bar: 200 μm. (D) Feret diameter resulting from these images, where NT stands for non-templated microgels. More than 100 microgels were measured to obtain these results. Brown–Forsythe ANOVA with Dunnett's multiple comparison test for statistical analysis. (E)–(G) Transmission electron microscope (TEM) images illustrating MCM walls after ice templating. Scale bar: 2 μm. (H) Wall thickness extracted from images from (E)–(G). Brown–Forsythe ANOVA with Dunnett's multiple comparison test for statistical analysis. (I)–(K) Fluorescence micrographs of jammed MCMs and (L) corresponding porosity data. Scale bar: 50 μm. Ordinary one-way ANOVA with Tukey's multiple comparisons test was performed for statistical analysis. ns: nonsignificant change. | ||
Brightfield images revealed distinctions between the three freezing rate conditions. In the 0.1 °C min−1 case, the shape of all MCMs appeared irregular, whereas in the other two conditions, some of the MCMs exhibited a more spherical morphology (Fig. S9†). To determine the overall porosity of the granular ink created from these MCMs, we prepared a granular material from jammed MCMs, and conducted 3D imaging using a confocal microscope (Fig. 6I–K). The results indicated the overall porosity to increase to nearly 75% for the freezing rate of 0.1 °C min−1, with a relatively smaller increase in porosity for the other two conditions (Fig. 6I–L). We attribute the difference to reduced compaction that is expected at higher freezing rates, corresponding to a lower overall porosity. The pores within the ice-templated spherical microgels are less than 5 μm wide in size and therefore expected to have a minimal effect on cell migration. In comparison to granular materials prepared from spherical microgels that have not been ice-templated,17 our MCM-based bioink exhibits an approximately threefold higher overall porosity.
Cell culture and bioprinting
To attain bioink volumes sufficient for extrusion bioprinting applications, we increased the total number of FFGs to ten, using the same design principle (Fig. S10†). To evaluate the long-term stability and compatibility of these microgels with cells, we conducted a culture experiment using umbilical cord-derived mesenchymal stromal cells (UC-MSCs) with both CM and MCMs. Culturing UC-MSCs, at a density of 100000 cells per cm2, within the prepared microgels allowed the microarchitecture of the microgels to be maintained for over three weeks, enabling the cells to occupy inter-particle spaces. The ability of the granular bioink to preserve porosity, prevent premature contraction and degradation, holds significant promise for various tissue culture applications requiring extended culture periods. Examples include differentiation of induced pluripotent stem cells,52in vitro wound healing models,53 and endothelial cell-induced vascularization.54 As shown in Fig. 7A and B, the orientation of the cells is influenced by the pores, potentially providing an avenue to mimic the microarchitecture of human tissues. For example, the porosity of decellularized human dermis is 68%55 which falls within the range of porosities obtained in the MCM based granular bioinks. Additionally, in the case of CMs, the cells exhibit a more homogeneous organization compared to the MCMs. However, we predict that the heterogeneous organization of cells in the MCMs more accurately mimics the complexity of native tissues. It is important to note that in this study, a direct and fair comparison between the non-templated and ice-templated conditions cannot be firmly established. This is due to the significant increase in local collagen concentration after ice templating, resulting in a higher collagen content per unit volume in the frozen samples.
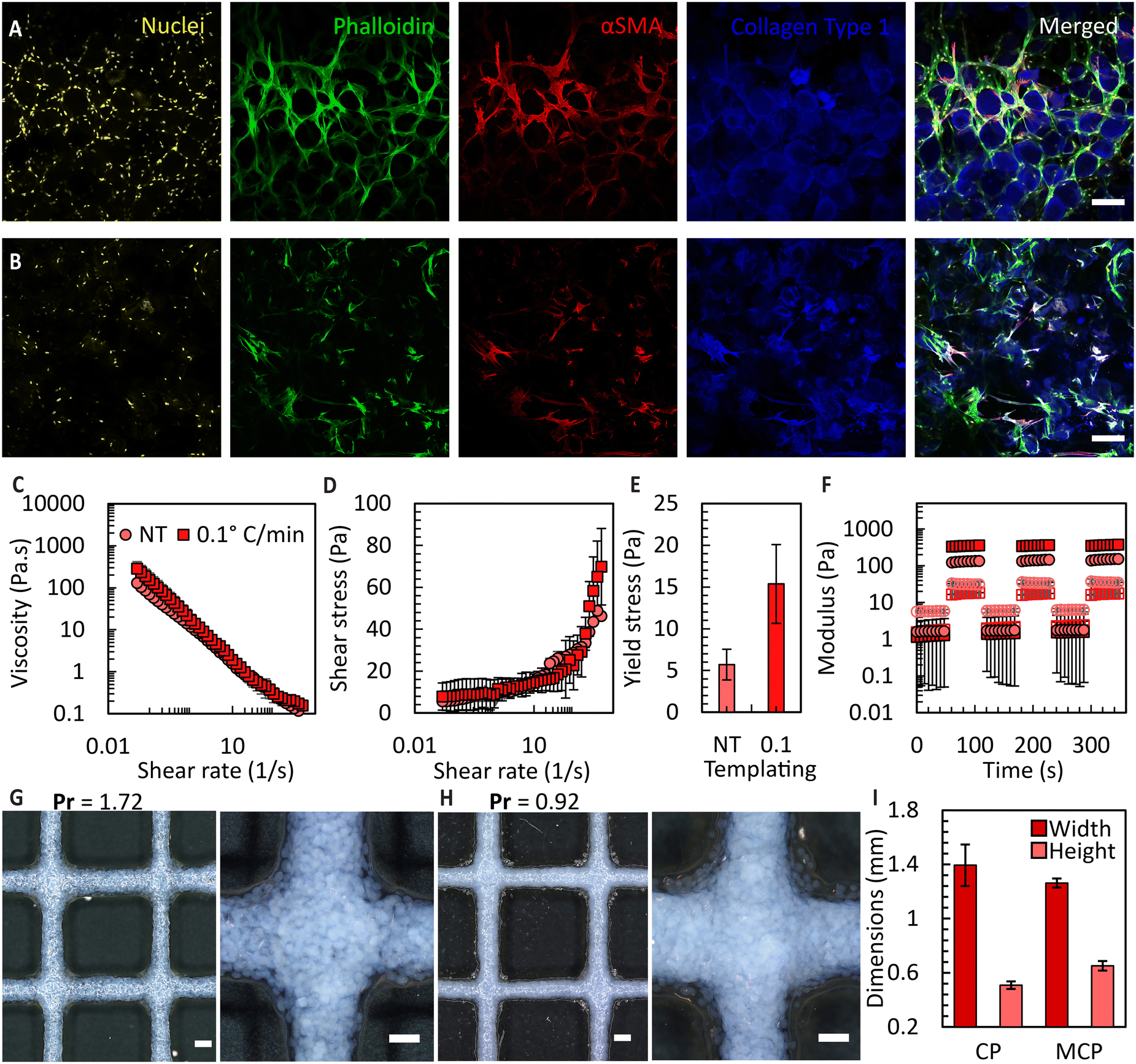 | ||
| Fig. 7 CM and MCM-derived bioink. (A) and (B) Cell morphology at day 21 of culture with collagen microgels (top row) and the 0.1 °C min−1 ice-templated MCMs (bottom row). Scale bar: 200 μm. (C) Viscosity versus shear rate for both the non-templated and MCM-based granular bioinks (n = 3). (D) Shear stress versus shear rate for these formulations, and (E) resulting yield stress values (n = 3). (F) Storage modulus (closed symbols) and loss modulus (open symbols) for same bioinks (n = 3). (G) Square grids printed using the non-templated microgel derived bioink and (H) using MCM-derived bioink. Scale bar: of 1000 μm (left) and 500 μm (right). (I) Dimensions of filaments defining grid patterns (n = 3). | ||
Subsequently, we prepared granular bioinks using these microgels and conducted a rheological assessment. As shown in Fig. 7C, the MCM samples exhibited higher viscosity across all the applied shear rates, suggesting their suitability for bioprinting. By examining the shear stress vs. shear rate responses for these fluids (Fig. 7D), we were able to determine a yield stress value by curve fitting using the Herschel–Bulkley model. The MCM-derived granular bioinks displayed an approximate three times increase in the overall yield stress compared to CM-derived bioinks, as depicted in Fig. 7E. These results, in combination with the porosity data, indicate that ice templating not only enhances the porosity of the microgels threefold but also elevates the yield stress of the fluid. We attribute this viscosity increase to two primary factors: (1) the irregular morphology of the MCM outer surface, which increases interaction points between neighbouring microgels in a dense packing, and (2) the greater number of MCMs per unit volume compared to the CM microgels. Both influences increase the overall interaction within the fluid, consequently requiring higher shear force to disrupt these interactions. Lastly, we conducted oscillatory sweeps to show that the jammed bioink can recover its flow properties after multiple rounds of shearing (Fig. 7F). Amplitude sweep data can be found in Fig. S11.†
Finally, we extrusion-bioprinted the granular bioink into square grid patterns, to determine the printability index (Pr)36 and feature fidelity. The Pr value is a dimensionless parameter that compares the ideal rectangular pore shape with the actual results obtained. The initial filaments were designed to have a diameter of 1 mm, but the flattening of these filaments during printing led to dimensional changes. As expected, the MCM-based bioink exhibited reduced spreading after bioprinting, as indicated by the Pr value of 0.92 in comparison to 1.72 attained for the CM case (Fig. 7G and H), primarily due to the increased yield stress. Moreover, we achieved higher resolution filaments with bioinks prepared from jammed MCMs, with approximately a 10% reduction in filament spreading and a 22% increase in height compared to the CM case.
Conclusions
In conclusion, we introduced a microfluidic approach to address key challenges in the preparation of collagen-based bioinks. We designed and employed a parallelized microfluidic droplet/bubble generator to introduce collagen solutions at concentrations of up to 10 mg ml−1. The air-induced emulsification technique has been used in a parallelized device for the first time, ensuring consistent emulsification of collagen solution and increasing the rate of collagen microgel formation tenfold. By adjusting the air flow rate, we achieved stable emulsification. Furthermore, this approach may be useful for emulsification methods such as step emulsification where a minimum viscosity ratio is needed for droplet formation.56 We monitored surfactant removal during washing using an automated liquid handler with built-in fluorescent readout, ensuring levels sufficiently low for cell culture applications. The presented macroporous collagen microgels showcased uniformity in gelation within the microgel, supported by various microscopy techniques, and a notable increase (>3×) in overall porosity of the granular hydrogel compared to traditionally formed spherical microgels. The increased porosity is attributed to ice templating at different freezing rates, and provides opportunities for improved nutrient diffusion, cell migration, and cell signalling.Our study revealed that MCMs cultured with UC-MSCs maintained the bioink's microarchitecture for up to three weeks. This feature has significant implications for long-term tissue culture applications, particularly in the context of mimicking the porosity of native tissue structures. The rheological assessment showed MCM-based bioinks to exhibit viscosity and yield stress values that make them highly suitable for extrusion-based bioprinting. The ability to recover flow properties after repeated shearing highlights the consistent printability of the granular bioink. Our MCM-derived bioink outperformed non-templated bioinks, as indicated by the reduced filament spreading and improved filament height. Overall, our approach introduces valuable advancements in collagen-based bioink production, offering promising solutions for improving the bioprinting of complex tissue structures with enhanced feature fidelity and long-term stability. We anticipate the use of MCM-based bioinks to find widespread use for applications in tissue engineering, for instance to improve wound healing and promote vascularization.
We consider the presented flow processing approaches to be extendable to other animal-derived collagen sources, including bovine telocollagen, as well as bacteria or plant-derived human recombinant collagen. The use of in-flow liquid–liquid phase separators57,58 may in the future allow some of the remaining batch steps, such oil removal and washing, to be transferred to a flow format in the future. We consider the employed microfluidic device architecture to be scalable and provide future opportunities for preparing MCM-based bioinks at liter per hour throughput.
Data availability
The data supporting this article have been included as part of the ESI.† Data are also available from a repository under link: https://doi.org/10.57760/sciencedb.10115.Author contributions
S. Singh was responsible for conceptualization, investigation, data curation, formal analysis, visualization, writing the original draft, and reviewing and editing. W. Chu performed the investigation and validation of the bioprinting experiments and contributed to the review and editing of the manuscript. R. O. Memar carried out the investigation and validation of the automated washing and measurements related to tween concentrations. A. D. Carlo investigated and validated the bioprinting experiments alongside W. Chu. T. Veres supervised all the students and reviewed and edited the document. A. Günther contributed to conceptualization, investigation, secured funding, supervised the project, assisted with the original draft, and reviewed and edited the document.Conflicts of interest
The authors are not aware of a conflict of interest.Acknowledgements
We thank Dr. Lindsey Fiddes for assistance with sample preparation for transmission electron microscopy and imaging, and Dr. Dan Voicu for support throughout the various stages of microfluidic device preparation. We thank Durgesh Kavishvar from Dr. Arun Ramchandran's laboratory at the University of Toronto for his help with setting up the rheology experiments. We gratefully acknowledge the cold trap as a gift from late Professor Charles A. Ward. We acknowledge support from NSERC (AG, RGPIN-2017-06781, RGPIN-2024-06528, I2IPJ 576571-22) the Barbara and Frank Milligan Graduate Fellowship (SS) and the Undergraduate Student Summer Research Pivot Fellowship (RO). Device fabrication was carried out at the CRAFT Device and Tissue Foundries, open research facilities supported by the University of Toronto, NRC (Disruptive Technology Solutions for Cell and Gene Therapy Challenge program), the Canada Foundation for Innovation and the Ontario Research Fund (Ontario-Québec Center for Organ-on-a-Chip Engineering, Center for Advancing Neurotechnological Innovation to Application).References
- Y. Wang, Z. Wang and Y. Dong, ACS Biomater. Sci. Eng., 2023, 9, 1132–1150 CrossRef CAS PubMed.
- P. Fratzl and R. Weinkamer, Prog. Mater. Sci., 2007, 52, 1263–1334 CrossRef CAS.
- A. Gautieri, S. Vesentini, A. Redaelli and M. J. Buehler, Nano Lett., 2011, 11, 757–766 CrossRef CAS PubMed.
- E. Rezvani Ghomi, N. Nourbakhsh, M. Akbari Kenari, M. Zare and S. Ramakrishna, J. Biomed. Mater. Res., Part B, 2021, 109(12), 1986–1999 CrossRef CAS PubMed.
- L. J. Gould, Adv. Wound Care, 2015, 5, 19–31 CrossRef PubMed.
- C. Haynl, E. Hofmann, K. Pawar, S. Förster and T. Scheibel, Nano Lett., 2016, 16, 5917–5922 CrossRef CAS PubMed.
- L. Huang, K. Nagapudi, R. P. Apkarian and E. L. Chaikof, J. Biomater. Sci., Polym. Ed., 2001, 12, 979–993 CrossRef CAS PubMed.
- S. Rele, Y. Song, R. P. Apkarian, Z. Qu, V. P. Conticello and E. L. Chaikof, J. Am. Chem. Soc., 2007, 129, 14780–14787 CrossRef CAS PubMed.
- S. Malladi, D. Miranda-Nieves, L. Leng, S. J. Grainger, C. Tarabanis, A. P. Nesmith, R. Kosaraju, C. A. Haller, K. K. Parker, E. L. Chaikof and A. Günther, ACS Biomater. Sci. Eng., 2020, 6, 4236–4246 CrossRef CAS PubMed.
- W. Gao, N. Vaezzadeh, K. Chow, H. Chen, P. Lavender, M. D. Jeronimo, A. McAllister, O. Laselva, J. X. Jiang, B. K. Gage, S. Ogawa, A. Ramchandran, C. E. Bear, G. M. Keller and A. Günther, Adv. Healthcare Mater., 2021, 10, e2001746 CrossRef PubMed.
- W. Gao, K. R. Kanagarajah, E. Graham, K. Soon, T. Veres, T. J. Moraes, C. E. Bear, R. A. Veldhuizen, A. P. Wong and A. Günther, Small, 2024, e2309270, DOI:10.1002/smll.202309270.
- Y. T. Matsunaga, Y. Morimoto and S. Takeuchi, Adv. Mater., 2011, 23, H90–H94 CrossRef CAS PubMed.
- T. J. Hinton, Q. Jallerat, R. N. Palchesko, J. H. Park, M. S. Grodzicki, H.-J. Shue, M. H. Ramadan, A. R. Hudson and A. W. Feinberg, Sci. Adv., 2015, 1, e1500758 CrossRef PubMed.
- A. Lee, A. R. Hudson, D. J. Shiwarski, J. W. Tashman, T. J. Hinton, S. Yerneni, J. M. Bliley, P. G. Campbell and A. W. Feinberg, Science, 2019, 365(6452), 482–487 CrossRef CAS PubMed.
- V. L. Cross, Y. Zheng, N. Won Choi, S. S. Verbridge, B. A. Sutermaster, L. J. Bonassar, C. Fischbach and A. D. Stroock, Biomaterials, 2010, 31, 8596–8607 CrossRef CAS PubMed.
- G. Charras and E. Sahai, Nat. Rev. Mol. Cell Biol., 2014, 15, 813–824 CrossRef CAS PubMed.
- C. B. Highley, K. H. Song, A. C. Daly and J. A. Burdick, Adv. Sci., 2019, 6, 1801076 CrossRef PubMed.
- A. Ding, O. Jeon, D. Cleveland, K. L. Gasvoda, D. Wells, S. J. Lee and E. Alsberg, Adv. Mater., 2022, 34(15), e2109394 CrossRef PubMed.
- J. E. Mealy, J. J. Chung, H. H. Jeong, D. Issadore, D. Lee, P. Atluri and J. A. Burdick, Adv. Mater., 2018, 30, 1705912 CrossRef PubMed.
- M. Yamada, A. Hori, S. Sugaya, Y. Yajima, R. Utoh, M. Yamato and M. Seki, Lab Chip, 2015, 15, 3941–3951 RSC.
- S. Yoshida, M. Takinoue and H. Onoe, Adv. Healthcare Mater., 2017, 6, 1601463 CrossRef PubMed.
- L. Hidalgo San Jose, P. Stephens, B. Song and D. Barrow, Tissue Eng., Part C, 2018, 24, 158–170 CrossRef CAS PubMed.
- H. Zhao, Z. Wang, S. Jiang, J. Wang, Z. Hu, P. E. Lobie and S. Ma, Cell Rep. Phys. Sci., 2020, 1, 100047 CrossRef.
- E. Samiei, T. Veres and A. Günther, Proceedings of 25th International Conference on Miniaturized Systems for Chemistry and Life Sciences (MicroTAS), Chemical and Biological Microsystems Society (CBMS), Palm Springs, California, USA and online, Oct 10-14, 2021, pp. 105–106, ISBN 9781713855736 Search PubMed.
- E. Samiei, T. Veres and A. Günther, Microfluidic Synthesis of Collagen-Based Microgels for Tissue Engineering Applications, In review, 2023 Search PubMed.
- L. Wu, Y. Li, Z. Fu and B.-L. Su, Natl. Sci. Rev., 2020, 7, 1667–1701 CrossRef CAS PubMed.
- Advanced BioMatrix, Collagen Viscosity, https://advancedbiomatrix.com/collagen-viscosity.html, (accessed 25 october 2023).
- H. Joukhdar, A. Seifert, T. Jüngst, J. Groll, M. S. Lord and J. Rnjak-Kovacina, Adv. Mater., 2021, 33, 2100091 CrossRef CAS PubMed.
- M. Muluneh and D. Issadore, Lab Chip, 2013, 13, 4750–4754 RSC.
- C. Feng, K. Takahashi and J. Zhu, Front. Bioeng. Biotechnol., 2022, 10, 891213 CrossRef PubMed.
- A. Martos, M. Berger, W. Kranz, A. Spanopoulou, T. Menzen, W. Friess, K. Wuchner and A. Hawe, J. Pharm. Sci., 2020, 109, 646–655 CrossRef CAS PubMed.
- T. Starborg, N. S. Kalson, Y. Lu, A. Mironov, T. F. Cootes, D. F. Holmes and K. E. Kadler, Nat. Protoc., 2013, 8, 1433–1448 CrossRef PubMed.
- T. H. Qazi, V. G. Muir and J. A. Burdick, ACS Biomater. Sci. Eng., 2022, 8, 1427–1442 CrossRef CAS PubMed.
- V. Mansard, L. Bocquet and A. Colin, Soft Matter, 2014, 10, 6984–6989 RSC.
- S. Xin, K. A. Deo, J. Dai, N. K. R. Pandian, D. Chimene, R. M. Moebius, A. Jain, A. Han, A. K. Gaharwar and D. L. Alge, 2021, 7, eabk3087.
- A. Schwab, R. Levato, M. D'Este, S. Piluso, D. Eglin and J. Malda, Chem. Rev., 2020, 120, 11028–11055 CrossRef CAS PubMed.
- L. Ouyang, R. Yao, Y. Zhao and W. Sun, Biofabrication, 2016, 8, 035020 CrossRef PubMed.
- N. Soltan, L. Ning, F. Mohabatpour, P. Papagerakis and X. Chen, ACS Biomater. Sci. Eng., 2019, 5, 2976–2987 CrossRef CAS PubMed.
- C. Zhou, P. Zhu, Y. Tian, X. Tang, R. Shi and L. Wang, Lab Chip, 2017, 17(19), 3310–3317 RSC.
- S. Yadavali, H.-H. Jeong, D. Lee and D. Issadore, Nat. Commun., 2018, 9, 1222 CrossRef PubMed.
- J. M. de Rutte, J. Koh and D. Di Carlo, Adv. Funct. Mater., 2019, 29, 1900071 CrossRef.
- S. Yadavali, D. Lee and D. Issadore, Sci. Rep., 2019, 9, 12213 CrossRef PubMed.
- J. Wu, S. Yadavali, D. Lee and D. A. Issadore, Appl. Phys. Rev., 2021, 8(3), 031304 CAS.
- M. J. Randall, A. Jüngel, M. Rimann and K. Wuertz-Kozak, Front. Bioeng. Biotechnol., 2018, 6, 154 CrossRef PubMed.
- S. C. Fox, D. Siallagan, M. Schmid Daners and M. Meboldt, Curr. Opin. Biomed. Eng., 2019, 10, 165–173 CrossRef.
- M. G. Mohamed, S. Kheiri, S. Islam, H. Kumar, A. Yang and K. Kim, Lab Chip, 2019, 19, 1621–1632 RSC.
- F. J. O'Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- D. Axelrod, Biophys. J., 1979, 26, 557–573 CrossRef CAS PubMed.
- T. Hua, X. Zhang, B. Tang, C. Chang, G. Liu, L. Feng, Y. Yu, D. Zhang and J. Hou, BMC Vet. Res., 2018, 14, 138 CrossRef PubMed.
- H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., Int. Ed., 2003, 42(7), 768–772 CrossRef CAS PubMed.
- P. Divakar, K. Yin and U. G. K. Wegst, J. Mech. Behav. Biomed. Mater., 2019, 90, 350–364 CrossRef CAS PubMed.
- J. W. Lambshead, L. Meagher, J. Goodwin, T. Labonne, E. Ng, A. Elefanty, E. Stanley, C. M. O'Brien and A. L. Laslett, Sci. Rep., 2018, 8, 701 CrossRef PubMed.
- G. Sriram, M. Alberti, Y. Dancik, B. Wu, R. Wu, Z. Feng, S. Ramasamy, P. L. Bigliardi, M. Bigliardi-Qi and Z. Wang, Mater. Today, 2018, 21, 326–340 CrossRef CAS.
- Y. Aghazadeh, S. T. Khan, B. Nkennor and S. S. Nunes, Pharmacol. Ther., 2022, 231, 107976 CrossRef CAS PubMed.
- Y. Wang, R. Xu, W. He, Z. Yao, H. Li, J. Zhou, J. Tan, S. Yang, R. Zhan, G. Luo and J. Wu, Tissue Eng., Part C, 2015, 21, 932–944 CrossRef CAS PubMed.
- C. He, X. Liu, Y. Dong, C. Zhu, Y. Ma and T. Fu, Chem. Eng. Process., 2023, 185, 109309 CrossRef CAS.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. C. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704–5708 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00595j |
| This journal is © The Royal Society of Chemistry 2024 |