
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in active targeting of ligand-directed polymeric nanomicelles via exploiting overexpressed cellular receptors for precise nanomedicine
Mona M. Agwa *a,
Rehab Elsayed Marzoukb and
Sally A. Sabra*c
*a,
Rehab Elsayed Marzoukb and
Sally A. Sabra*c
aDepartment of Chemistry of Natural and Microbial Products, Pharmaceutical and Drug Industries Research Institute, National Research Centre, 33 El-Behooth St, Dokki, Giza, 12622, Egypt. E-mail: mona.m.agwa@alexu.edu.eg; mm.agwa@nrc.sci.eg; magwa79@gmail.com; Fax: +202 33370931; Tel: +202 33371635
bMedical Biochemistry Department, Faculty of Medicine, Helwan University, Helwan, Cairo, Egypt
cDepartment of Biotechnology, Institute of Graduate Studies and Research, Alexandria University, Alexandria, 21526, Egypt. E-mail: ssabra@uwo.ca; ssabra@alexu.edu.eg
First published on 26th July 2024
Abstract
Many of the utilized drugs which already exist in the pharmaceutical sector are hydrophobic in nature. These drugs are characterized by being poorly absorbed and difficult to formulate in aqueous environments with low bioavailability, which could result in consuming high and frequent doses in order to fulfil the required therapeutic effect. As a result, there is a decisive demand to find modern alternatives to overcome all these drawbacks. Self-assembling polymeric nanomicelles (PMs) with their unique structure appear to be a fascinating choice as a pharmaceutical carrier system for improving the solubility & bioavailability of many drugs. PMs as drug carriers have many advantages including suitable size, high stability, prolonged circulation time, elevated cargo capacity and controlled therapeutic release. Otherwise, the pathological features of some diseased cells, like cancer, allow PMs with particle size <200 nm to be passively uptaken via enhanced permeability and retention phenomenon (EPR). However, the passive targeting approach was proven to be insufficient in many cases. Consequently, the therapeutic efficiency of these PMs can be further reinforced by enhancing their cellular internalization via incorporating targeting ligands. These targeting ligands can enhance the assemblage of loaded cargos in the intended tissues via receptor-mediated endocytosis through exploiting receptors robustly expressed on the exterior of the intended tissue while minimizing their toxic effects. In this review, the up-to-date approaches of harnessing active targeting ligands to exploit certain overexpressed receptors will be summarized concerning the functionalization of the exterior of PMs for ameliorating their targeting potential in the scope of nanomedicine.
1 Introduction
Polymeric micelles (PMs) are nanocarriers comprising a core/shell architecture which are formed by self-assembly of amphiphilic copolymers in an aqueous medium.1,2 Below their critical micellar concentration (CMC), the micellar copolymers exist individually in the solution. Upon increasing the micellar concentration above the CMC, the amphiphilic copolymers can then associate to form nano-size micelles with their hydrophobic segment directed inside to form the core and the hydrophilic segment directed outside towards the outer aqueous medium.3,4 Micelles with relatively low CMC are supposed to be more stable when injected into the circulation because small amounts of individual copolymers will participate in the formation of the self-assembled micelle, and hence when greatly diluted, they will resist this dilution and can preserve their micellar architecture. Moreover, the outer shell of PMs is responsible for their in vivo pharmacokinetic behavior,5,6 besides its ability to minimize the undesired drug interactions with the surrounding environment and to reduce opsonization and uptake by the reticuloendothelial system. PMs, with their unique structure, appear to be a fascinating choice as a drug delivery owing to their unique features including; improved stability, especially upon dilution (for micelles with relatively low CMC), biocompatibility, and improved drug bioavailability.7,8Although PMs can be used for many diseases, cancer therapy is indeed of great interest due to important abnormalities associated with tumor including heterogeneity in the endothelial lining of blood vessels in the tumor in comparison to healthy blood vessels having a permeability cutoff of nearly 7 nm.9 Moreover, the tumor vasculatures usually lack well-defined morphology with increased spacing between endothelial cells, resulting in increased permeability and accumulation of relatively large molecules between these interstitial spaces. Tumors also lack a working drainage system, so accumulation of large molecules at the tumor site will last for a longer time when compared to healthy cells.10,11 Nearly, all the approved nanomedicine-based formulations are based on enhanced permeability and retention effect (EPR) or alternatively named; passive tumor targeting. The main regulator affecting the efficiency of passive targeting is the particle size. Particles with hydrodynamic diameter less than 6–8 nm could be rapidly cleared by renal filtration,12 whereas particles larger than 200 nm might be cleared by liver and spleen.13 However, if EPR effect works well in a certain tumor, it is only responsible for increasing drug concentration in the tumor's vicinity, not in the tumor cells (Fig. 1).
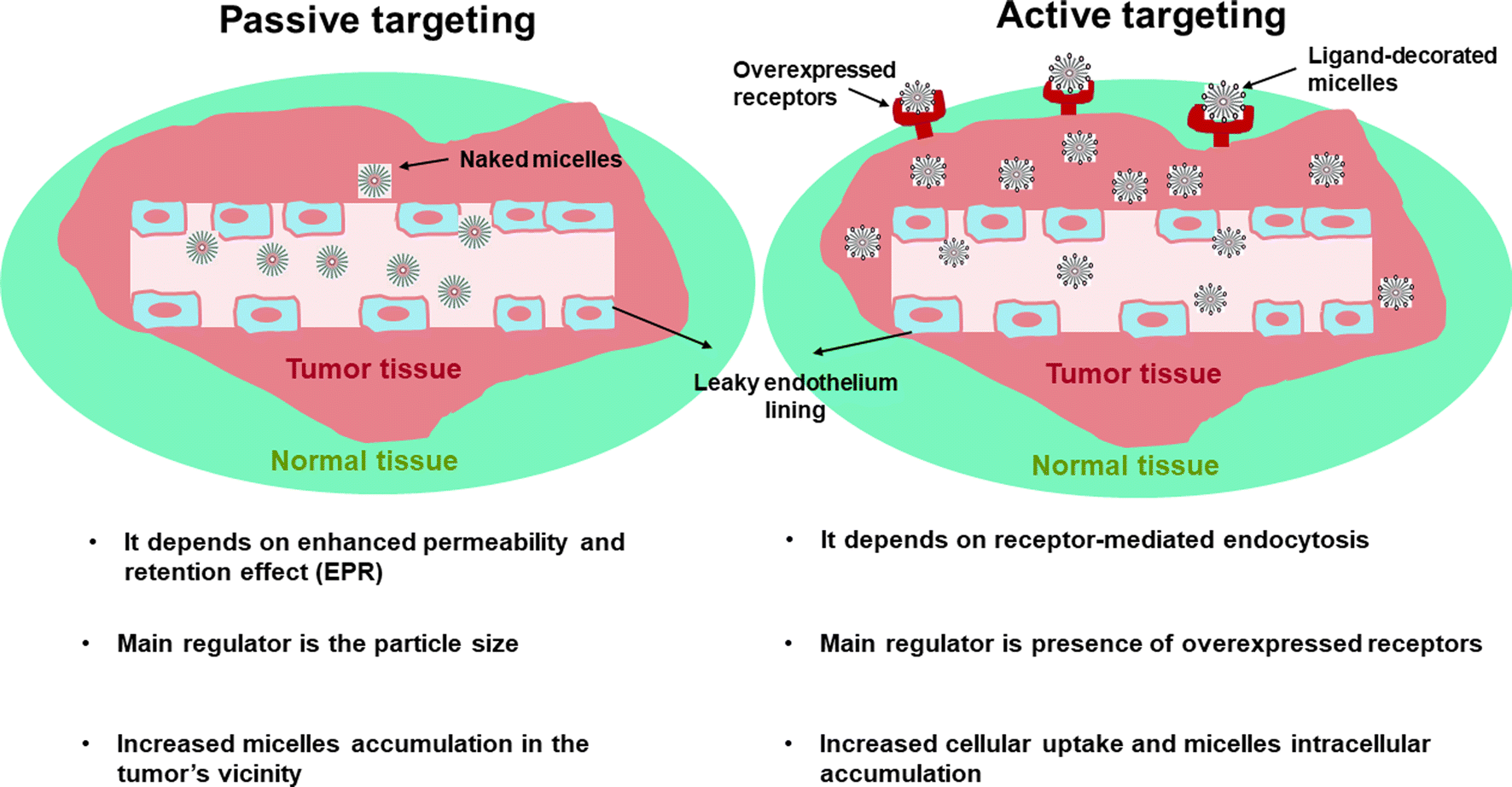 | ||
| Fig. 1 Schematic illustration showing difference between passive and active targeting. | ||
As a result and in order to increase targetabiliy and efficiency of the PMs, targeting ligands including; antibodies, peptides, proteins, sugar moieties and small molecules can be introduced onto the surface of the micelles, which can then recognize target cells or even intracellular organelles inside the diseased organs.14,15 This approach of targeting is named “active targeting” and it works mainly on improving cellular recognition and uptake, and hence increasing the intracellular concentration of drugs causing a significant reduction in the frequency of dosing.16 This is greatly useful especially if the drug is needed to work inside tumor cells or any other diseased cells as this mode of targeting is not limited to tumors only.17 In this review, the most recent approaches in active targeting of PMs via exploiting specific receptors will be highlighted including; human epidermal growth factor receptor-2 (HER-2), integrin receptors, Epidermal growth factor receptors (EGFR), ephrin type-A receptor 2 (EphA2), nucleolin receptors, transferrin receptors (TFR), asialoglycoprotein receptors (ASGPR), glycyrrhetinic acid receptors (GRs), Glucose transporter 1 (GLUT1) receptor, folate receptors (FRs), cell surface adhesion (CD44) receptors, biotin receptors, Mucin 16 (MUC16) receptor, fibronectin (FN) receptor, mannose receptors (MRs), vitamin D3 receptor (vit.D3R), and protease/activated receptors-2 (PARs-2) (Fig. 2). The active targeting delivery of PMs can be achieved through the amalgamation with an active targeting moiety into their exteriors which can recognize and link these receptors to mend their cellular targeting potential.
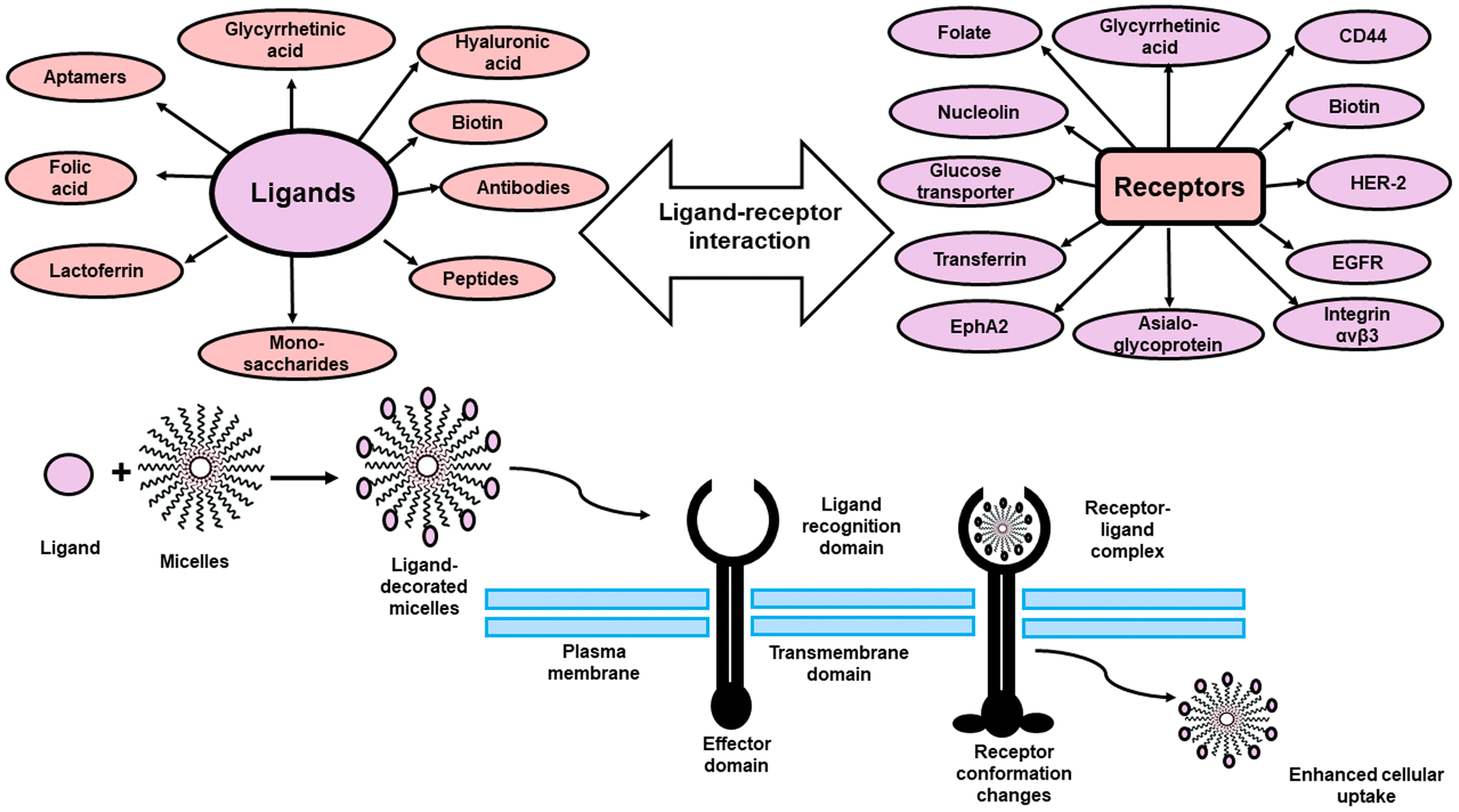 | ||
| Fig. 2 Schematic illustration of different active targeted ligands and their cellular receptors. | ||
2 Polymeric micelles for targeting different receptors
2.1. Human epidermal growth factor receptor-2
Human epidermal growth factor receptor-2 (HER-2) is a transmembrane glycoprotein receptor massively expressed on the exterior of many malignancies. It is 185 kDa with an intracellular tyrosine kinase domain.18 Targeting tumor cells via HER-2 utilizing anti-HER-2 humanized antibody (AB) has manifested therapeutic promise as it can successfully bypass chemotherapeutic resistance.19 In this scope, Peng and his co-workers have constructed targeted nanocrystal micelles against HER-2 expressing breast tumors.20 The amphiphilic micelles comprised from PCL2000/MPEG2000 and PCL5000-/PEG2000-CHO and they were fabricated via emulsification method, followed by loading with cytotoxic paclitaxel (PTX) via solvent evaporation procedure to produce PCL-PEG/PTX nanomicelles. The outer shell of the nanomicelles was grafted with herceptin AB through Schiff base formation between the aldehyde groups of PCL-PEG/PTX and amine groups of herceptin. The targeted PMs exhibited a pH-responsive and controlled release pattern for PTX, assuring its eligibility for targeted tumor delivery. In vitro uptake results showed maximum internalization for coumarin-6 labelled HER/PCL-PEG/PTX nanomicelles following one hour (1 h) incubation with SKBR-3 cells overexpressing HER-2 receptor compared to PCL-PEG/PTX nanomicelles or HER/PCL-PEG/PTX nanomicelles incubated with MDA-MB-231 cells (negative HER-2). Moreover, targeted nanomicelles exhibited significant in vitro cytotoxic effect against SKBR-3 cells compared to PTX, blank micelles, drug-loaded untargeted micelles or targeted nanomicelles against MDA-MB-231 cells. Furthermore, in vivo bio-distribution results revealed a powerful tumor homing assemblage after 1 h of i.v. administration of HER/PCL-PEG/PTX into tumor bearing-female BALB/c nude mice that remained unchangeable for 24 h. The fabricated targeted nanomicelles also displayed an extended plasma residence time with improved anti-malignant potency in terms of reduced tumor volume and prolonged survival time. Moreover, no marks of toxicity were observed during the entire study, confirming their high safety burden.Targeted PMs utilizing biodegradable polyester dendrons (G4OX) as hydrophobic core and linear PEG [poly(ethylene glycol)] polymers as the hydrophilic shell were fabricated by Bolu and his co-workers using dendron–polymer conjugates to form G4OX-PEG.21 Fabricated PMs were loaded with the cytotoxic drug docetaxel (DTX) using solvent evaporation technique, followed by surface modification with trastuzumab (T) monoclonal AB via amidation reaction to form T/G4OX-PEG-DTX for targeting of cells overexpressing HER-2 receptors.22 The targeted PMs exhibited a pH-responsive release pattern for DTX, confirming its suitability for targeted tumor delivery. Moreover, in vitro uptake studies elucidated magnificent cellular assemblage for fluorescently labelled targeted PMs (T/G4OX-PEG) when incubated with MCF-7 and SK-OV-3 cancer cell lines for 4 h and remained for 24 h unlike fluorescently labelled AB-free PMs (G4OX-PEG), confirming the important role exerted by trastuzumab in enhancing cellular uptake via HER-2 receptor-mediated targeting. Also, the fluorescent signals were highly centralized across cell periphery at 4 h, which then extended widely at 24 h due to augmented internalization and fusing with the acidic lysosomes. The quantitative internalization performed by flow cytometry also confirmed the superior uptake for the targeted PMs. In addition, the fabricated targeted PMs loaded with DTX showed powerful in vitro anticancer activity accompanied by low toxicity. Moreover, the powerful anticancer activity for the drug loaded targeted PMs was emphasized via western blot technique and revealed elevated expression of the apoptotic markers; caspase 8 and Bax.
Kim and his colleagues developed targeted self-assembled PMs for enhancing photodynamic therapy (PDT) against HER-2 positive breast cancer cells.23 The PMs consist of pheophorbide a (Pheo a), functionalized methoxy poly[ethylene glycol], block, poly[L-lysine hydrochloride] [PEG-PLL] to form [PEG-PLL-Pheo a] via amide bond formation using DCC conjugation reaction. The HER2 targeting peptide (HLTV) was conjugated to hyaluronic acid (HA) through EDC conjugation procedure to form (HLTV-HA).22 The targeted self-assembled PMs were fabricated via dissolving of PEG-PLL-Pheo a in DMSO, followed by dropwise addition to HLTV-HA aqueous solution under stirring to prepare HLTV-HA/PEG-PLL-Pheo a PMs to target HER-2 receptors overexpressed on the exterior of breast cancer cells. Targeted PMs exhibited higher cellular uptake when incubated with SK-BR-3 cells in comparison to free Pheo a owing to the premium binding strength of HLTV to HER-2 receptors. In vitro anticancer activity elucidated a higher efficiency for HLTV-HA/PEG-PLL-Pheo a PMs following laser irradiation against SK-BR-3 cells when compared to PMs without laser irradiation, confirming the potent photodynamic efficiency of the peptide conjugated PMs. The improved PDT efficacy for the peptide conjugated PMs following laser irradiation was confirmed via a dual fluorescent staining procedure using calcein-AM/ethidium homodimer-1 (EthD-1) for specifying live (green) and dead (red) cells. When a photosensitizing agent is exposed to laser irradiation, ROS are produced causing subsequent cellular death and the amount of the produced ROS can be detected by using 2′,7′-dichlorofluorescein (DCF, a fluorescence indicator) using flow cytometry (FC). Results revealed an enhanced ROS production in case of peptide-conjugated PMs incubated with SK-BR-3 cells under laser irradiation compared to free Pheo a., associated with higher degree of necrosis and apoptosis. Moreover, the superiority of the PDT performance of the targeted PMs against SK-BR-3 cells was proved by employing three dimensional (3D) spheroid cancer cells model and results demonstrated that targeted PMs were efficacious PDT against HER-2 positive breast cancer due to efficient cellular internalization.
In another study, the antitumor potential of (LTVSPWY) peptide and herceptin antibody functionalized PMs were examined against breast cancer cells overexpressing HER2 receptors.24 PMs were synthesized from [(N-3-sulfopropyl-N,N-dimethylammonium)ethyl methacrylate] as the hydrophilic shell and DEGMA (di(ethylene glycol) methyl ether methacrylate)/AEMA (2-aminoethyl methacrylamide)/a vinyl-functionalized, acid-sensitive crosslinker as the hydrophobic core via RAFT polymerization technique to form core cross linked PMs (CCPMs).25 The LTVSPWY peptide and herceptin antibody were grafted to the fabricated CCPMs through EDC conjugation reaction to form LTVSPWY/CCPMs and Her/CCPMs. DOX was then encapsulated into the targeted CCPMs to give LTVSPWY/CCPMs-DOX and Her/CCPMs-DOX. The peptide and antibody targeted PMs exhibited a pH and acid-responsive release pattern for DOX, confirming its suitability for targeted tumor delivery. The targeting potentials of peptide and antibody-conjugated PMs were examined on SKBR3 breast cancer cells (positive HER-2) and MCF-10A normal breast cells (negative HER-2). Results revealed that LTVSPWY/CCPMs-DOX were more effective against SKBR3 cells than Her/CCPMs-DOX, indicating greater targeting efficiency for the peptide than the AB. Moreover, the decoration of the CCPMs with peptide and AB didn't cause any change in the selectivity toward MCF-10A cells with identical cytotoxic effect, confirming minimal uptake by normal MCF-10A cells. In addition, in vitro uptake results revealed a higher uptake for peptide-conjugated PMs than antibody-conjugated PMs. The intensity of the fluorescent signals was quantified and showed a 4-fold and 2-fold increase for the LTVSPWY/CCPMs-DOX and Her/CCPMs-DOX respectively compared to CCPMs-DOX. The apoptotic effect of the fabricated PMs was estimated via annexin V/PI apoptotic assay and results showed that LTVSPWY/CCPMs-DOX were more efficient than Her/CCPMs-DOX and CCPMs-DOX against SKBR3 cells showing a higher apoptotic and necrotic cellular architectures. There was also a massive decline in the expression of Bcl-2 associated with Bax increase in case of peptide-conjugated PMs compared to antibody-conjugated PMs, suggesting that the targeted CCPMs induced the anticancer activity via restraining of Bcl-2 (anti-apoptotic marker protein) and triggering of Bax (apoptotic marker protein). Moreover, in vitro genotoxicity effect evaluated by comet assay suggested that there was massive DNA fragmentation in SKBR3 cells treated with LTVSPWY/CCPMs-DOX compared to Her/CCPMs-DOX or CCPMs-DOX, indicating that LTVSPWY/CCPMs-DOX exhibited more potent apoptotic and cytostatic effects due to their selective targeting and minimal toxicity (Table 1).
| PMs | Drugs | Targeting ligand | Grafting mode | Target organ | Receptor | Key consequences | Ref. |
|---|---|---|---|---|---|---|---|
| PCL-PEG | PTX | Herceptin antibody | Schiff base reaction | Breast cancer | HER-2 | Powerful in vivo antitumor potential via preferable internalization | 20 |
| G4OX-PEG | DTX | Trastuzumab antibody | Amidation reaction | — | HER-2 | Superior in vitro activity due to enhanced cellular uptake via HER-2 receptors | 21 |
| HA/PEG-PLL-Pheo a | HLTV peptide | Simple mixing | — | HER-2 | Efficient in vitro activity due to enhanced cellular internalization of the PDT | 23 | |
| CCPMs | DOX | LTVSPWY peptide & herceptin antibody | EDC conjugation reaction | Breast cancer | HER-2 | Potent in vitro anticancer activity for the peptide-targeted micelles | 24 |
| mPEG-b-PCL & Mal-PEG-PCL | IR-780 | Cetuximab antibody | Thiole maleimide coupling reaction | Colorectal cancer | EGFR | Eminent in vivo antitumor phototherapeutic effect | 26 |
| Mal-PEG/PTMC | DOX | GE11 peptide | Schiff's base reaction & click reaction | Breast cancer | EGFR | Powerful in vivo antitumor efficiency for targeted dual drug loaded micelles | 27 |
| CEL | |||||||
| PEO/b/PCL | A83B4C63 | GE11 peptide | Simple mixing | Colorectal cancer | EGFR | Superior in vivo antitumor performance and prolonged survival duration for GE11 modified micelles | 28 |
| PEO/b/PBCL | Cy5.5 | ||||||
| PIC | Anti-EphA2 (1C1) antibody | Thiole maleimide coupling reaction | Prostate cancer | EphA2 | Efficient in vitro cellular uptake via receptor mediated endocytosis | 29 | |
| N3/PEG/PBLA & N3/PEG/PLL(TFA) | SN-38 | Anti-EphA2 (1C1) antibody | Thiole maleimide coupling reaction | Prostate cancer | EphA2 | Excellent in vitro activity due to enhanced cellular internalization for targeted micelles | 30 |
2.2. Epidermal growth factor receptors
Epidermal growth factor receptor (EGFR) is a tyrosine kinase transmembrane glycoprotein receptor with molecular weight of about 170 KDa. It is massively expressed in multiple tumors and its upregulation is usually associated with low survival rate and bad therapeutic response.18,31 These receptors can internalize into the inner of cancer cells via receptor-directed endocytosis, making them a perfect target for ligand directed antitumor therapy. Various available (EGFR) targeting ligands have been reported such as proteins (mainly ABs) and peptides.32 Targeting EGFR overexpressed on the exterior of colorectal malignant cells (CRC) was realized by Shih and his co-workers via developing EGFR tumor targeted PMs loaded with a photothermal agent.26 Nanomicelles were developed from the amphiphilic block copolymers mPEG-b-PCL and maleimide/terminated PEG-PCL (Mal-PEG-PCL) via ring-opening polymerization technique. These multifunctional micelles were loaded with IR-780 photothermal dye via co-solvent evaporation technique, then IR-780-loaded micelles were further decorated with thiolated Cetuximab (Cet) AB via thiol maleimide coupling reaction to form Cet/mPEG-b-PCL/IR-780 and Cet/Mal-PEG-PCL/IR-780 nanomicelles for selective binding to EGFR on the exterior of CRC cells.22 In vitro uptake study assessed via fluorescent microscope manifested lower uptake for FITC-labeled targeted micelles when incubated with SW-620 cells (low EGFR expression) while, HCT-116 cells (high EGFR expression) exhibited higher uptake. In vivo biodistribution scanning assessed by near-infrared fluorescence imaging demonstrated deeper and higher tumor aggregation for the prepared targeted micelles in comparison to other regions after 96 h post-i.v. administration into HCT-116 tumor-bearing mice. Additionally, the targeted PMs exhibited significantly higher in vivo uptake (0.48 ± 0.06%) than the non-targeted PMs (0.23 ± 0.04%). Moreover, in vivo antitumor efficacy study revealed an outstanding tumor growth repression for Cetuximab decorated IR-780-loaded nanomicelles following laser irradiation when compared to treatment without laser, which might be linked to the elevated accumulation of targeted IR-780 loaded nanomicelles in tumor. Moreover, H&E and NADPH stained tumors from multiple treated groups revealed tremendous necrotic areas in the HCT-116 tumors group treated with Cetuximab decorated IR-780 loaded-nanomicelles exposed to laser irradiation in comparison to all the other treated groups.To achieve rapid and effective drug accumulation into deep tumor tissues, Guo and his colleagues developed multifunctional targeted nanomicelles for enhanced delivery of combinatorial therapy against metastatic breast cancer via exploiting EGFR.27 Nanomicelles were fabricated from Mal-PEG-PCL and trimethylene carbonate (TMC) via ring-opening polymerization, then they were co-loaded with cytotoxic DOX and celecoxib (CEL) using esterification reaction and macromolecular self-assembly, respectively to form Mal-PEG/PTMC-DOX-CEL nanomicelles. The exterior of the dual loaded nanomicelles was grafted with GE11 peptide via Schiff's base reaction and click reaction to form GE11/Mal-PEG/PTMC-DOX-CEL nanomicelles.33 The targeted PMs displayed a pH and acid-triggered release pattern for both DOX and CEL, implying the potential of the targeted PMs for targeted delivery to the acidic tumor environment. In vitro cellular uptake study revealed an efficient uptake evidenced by numerous fluorescent signals in the nuclei for GE11 decorated nanomicelles following 6 h incubation with 4T1 tumor cells (highly expressing EGFR) compared to GE11 free nanomicelles owing to the superior EGFR binding affinity mediated via GE11 peptide. Moreover, in vitro cytotoxicity evaluated via CCK-8 assay showed remarkably augmented antitumor activity for GE11/Mal-PEG/PTMC-DOX-CEL nanomicelles against 4T1 cells when compared to other treated groups. More importantly, in vivo biodistribution experiment examined using 4T1 tumor-bearing BALB/c female mice depicted higher intratumoral accumulation after i.v. injection of GE11/Mal-PEG/PTMC-DOX-CEL nanomicelles (1.6 fold increase) and GE11/Mal-PEG/PTMC-DOX nanomicelles (1.7 fold increase) compared to free DOX or nanomicelles without peptide targeting. More importantly, the visual 3D fluorescence image of the tumor cryosection depicted the appearance of numerous DOX fluorescence signals in the nuclear zone, suggesting the successful nuclear accumulation. In vivo antitumor therapeutic efficiency resulted in superior antitumor performance evidenced by elevated tumor growth repression accompanied by minimized lung metastasis. In addition, there weren't any signs of systemic toxicity in all the nanomicelles-treated groups, confirming their therapeutic antitumor efficiency and biocompatibility of GE11-decorated dual drug loaded nanomicelles.
In another study, the outer shell of the nanomicelles was grafted with dodecapeptide GE11 to exploit EGFR so as to ameliorate deep tumor tissue internalization and retention.28 Fabricated nanomicelles comprising-polyethylene oxide-block-polycaprolactone (PEO/b/PCL) amphiphilic copolymer and PEO/block/(α-benzyl carboxylate-ε-caprolactone)(PEO/b/PBCL) amphiphilic copolymer were prepared via ring-opening polymerization. GE11-grafted nanomicelles were then conjugated with NIR dye Cy5.5 via azide–alkyne click chemistry34 to form GE11-PEO/b/PCL-Cy5.5 and GE11-PEO/b/PBCL-Cy5.5 nanomicelles. Afterwards, the polynucleotide kinase/phosphatase inhibitor A83B4C63 was physically loaded into the GE11 modified nanomicelles/Cy5.5 conjugate. The targeted PMs displayed a controlled release pattern for A83B4C63 at neutral pH. In vitro cellular uptake results disclosed an augmented internalization of GE11-PEO/b/PCL-Cy5.5 and GE11-PEO/b/PBCL-Cy5.5 nanomicelles after incubation with HCT116 cells (high EGFR expression) compared to SW620 cells (weak EGFR expression). In vitro antitumor screening performed against HCT116 tumor cells revealed potent cytotoxicity of GE11-modified nanomicelles loaded with A83B4C63 compared to GE11 free nanomicelles. More importantly, in vivo tumor localization assessed via luminescence imaging revealed higher accumulation for GE11-modified nanomicelles at the tumor region compared to other regions after i.v. injection into HCT116 tumor-bearing nude mice with superior in vivo antitumor effect and prolonged survival rate, suggesting the beneficial effect of GE11 grafting onto nanomicelles surface (Table 1).
2.3. Ephrin type-A receptor 2 (EphA2)
Ephrin type-A receptor 2 (EphA2) is a 130 KDa protein constructed from 967 amino acids and it is a tyrosine kinase receptor.35 It is weakly expressed in proliferating normal epithelial cells, while, it is strongly expressed in many malignancies including; lung, prostate, ovarian and breast, suggesting its potential as a committed therapeutic target for tumour management. As a result, targeting the EphA2 receptor via monoclonal antibodies (mABs) could stimulate the internalization of decorated micelles, which in turn can suppress cancer proliferation.36 In this regard, crosslinked polyion complex nanomicelles decorated with Fabs (antibody fragments) for binding EphA2 receptors were developed by Florinas et al.29 The fabricated nanomicelles comprised of azide decorated cationic PEG-b-poly(amino acids) [azido-PEG-b-poly/(L-lysine)[N3-PEG-b/PLL] and anionic azido-PEG-b-poly/(aspartic acid) [N3-PEG-b/PAsp]] were crosslinked with EDC coupling reaction to form self-assembled electrostatic crosslinked polyion complex nanomicelles termed as (PIC).37,38 The cysteine engineered Anti-EphA2 (1C1) monoclonal AB fragments (Fabs) were produced using Dimasi e. al. method39 to act as a binding site for azide decorated PEG nanomicelles. Fabricated PIC nanomicelles were then decorated with cysteine functionalized Fabs via thiol maleimide coupling reaction to form Fabs-decorated PIC nanomicelles for site specific targeting of the overexpressed EphA2 receptors. The fabricated micelles exhibited high cellular receptor binding affinity (more than 90%) when incubated with PC3 human prostate cancer cells (positive EphA2 receptor) compared with untargeted micelles. In vitro cellular uptake experiment assessed via a real-time fluorescence microscope imager revealed higher cellular uptake for Fabs-decorated PIC nanomicelles when incubated with PC3 cancer cells for 12 h, confirming the utility of these Fab-decorated PIC nanomicelles in efficient targeting via exploiting EphA2 receptors.In a similar study, nanomicelles decorated with Fabs were developed to enhance targeting via recognition of overexpressed EphA2 receptors on the exterior of malignant PC3 prostate cells.30 Nano-micelles were constructed from (N3/PEG/b/poly(benzyl-L-aspartate) (N3/PEG/PBLA) and N3/PEG/b/pLL(TFA)) copolymers via ring-opening polymerization reaction, then the surface of the fabricated nanomicelles was functionalized with Fab antibody through thiol maleimide coupling reaction, followed by loading with the anti-cancer agent; SN-38 via Diels–Alder conjugation reaction. The constructed targeted nanomicelles exhibited higher cellular receptor binding affinity than the untargeted nanomicelles following 10 min incubation with PC3 cancer cells. Existence of Fab antibodies on the exterior of the fabricated micelles was found to enhance their in vitro uptake by PC3 cancer cells after 6 h incubation through ligand-mediated endocytosis compared to untargeted micelles. In vitro cytotoxicity of SN-38 loaded targeted nanomicelles revealed higher cytotoxic effect against cancer cells compared to the untargeted SN-38 loaded nanomicelles (Table 1).
2.4. Integrin receptors
The term “integrin” arises from its role as the integral membrane protein complex connecting the extracellular matrix (ECM) to the cytoskeleton and transducing mechanical and biochemical signals among cells and the surrounding environment in both healthy and diseased conditions.40 It is also the main cell adhesion transmembrane receptor comprising of 18 α-subunits and 8 β-subunits in a non-covalent complex generating 24 functionally prominent heterodimeric transmembrane adhesion receptors. The RGD peptide (Arg-Gly-Asp) is commonly known to target integrin.41 In this regard, Lei et al. successfully fabricated a new dual-targeted nanomicelles decorated with Ang-2 (Angiopep-2) and cRGD peptide to exploit numerous receptors massively expressed on the exterior of glioma cells.42 The outer shell of the nanomicelles was constructed from two types of hydrophilic copolymers; [PEG] and β-cyclodextrin (β-CD) attached together via condensation reaction, followed by decoration with Ang-2 and cRGD via click reaction for targeting LRP-1 (low-density lipoprotein receptor-related protein-1) and αvβ3 integrin receptors, respectively to form Ang-2-PEG-β-CD and cRGD-PEG-β-CD. The inner hydrophobic core was composed of adamantly and PCL (Ad-PCL) conjugated via ring-opening polymerization reaction. The dual targeted micelles (cRGD/Ang-2-PEG-β-CD-PCL) were manufactured via self-assembly between Ang2-PEG-β-CD and cRGD-PEG-β-CD aqueous solutions with Ad-PCL dissolved in DMSO was achieved by dialysis. After that, DOX was encapsulated within the targeted micelles via solvent evaporation method. The constructed targeted PMs displayed a sustained DOX release at a pH 7.4. In vitro cellular internalization study demonstrated that the prepared labeled cRGD/Ang-2-PEG-β-CD-PCL nanomicelles exhibited higher intracellular uptake when incubated with glioma cells (C6 cells) and microvascular endothelial cells (bEnd.3 cells) compared to untargeted nanomicelles. Additionally, the delivery capacity of DOX in the targeted PMs was higher than that of the untargeted PMs by 9.5 times. Moreover, the fabricated dual targeted nanomicelles exhibited augmented in vivo antitumor efficiency after i.v. administration into C6 tumor-bearing mice in comparison to all the examined groups, indicating the potential therapeutic effect of dual targeted nanomicelles for effective glioma drug delivery via facilitated BBB (blood brain barrier) permeation through cRGD and Ang-2 triggered intratumoral drug accumulation.In another study, integrin-targeted self-assembled crosslinked core/shell nanomicelles were constructed from thermo-sensitive mPEG-b-poly[(N-(2-hydroxypropyl) methacrylamide lactate) (mPEG-b-pHPMAmLacn)] block copolymers.43 During self-assembly in an aqueous solution into core/shell framework, the micellar core was crosslinked via free radical polymerization reaction. DTX; as a hydrophobic drug was then co-crosslinked in the micellar internal core through covalent attachment to allow drug release at the site of interest. Fabricated nanomicelles were then decorated with three different densities of cRGD peptide (1, 3.6, and 5 mol%) via BCN (bicyclononyne) conjugation to examine the influence of peptide decoration densities on the internalization efficiency. In vitro cellular uptake study was conducted using four different cell lines having different patterns of integrin expression: A431 cells (low integrin), HUVEC− cells (intermediate integrin), HUVEC+ and 4T1 cells (high integrin). Results revealed an augmented cellular internalization efficiency centralized very adjacent to cell nuclei for cRGD decorated nanomicelles by the target cells (13.2 AF% for HUVEC+ cells, and 15.7 AF% for 4T1 cells) compared to untargeted nanomicelles and cells with low integrin expression (7.9 AF% for both A431 and HUVEC− cells). In addition, internalization was found to be temperature and time-dependent, with the best conditions at 37 °C and 24 h incubation time. Moreover, the uptake potential was also dependent on the type of the cells, with the best results obtained in case of high integrin expressing cells. Interestingly, lower cRGD decoration density (1 mol%) was found to be the most appropriate density for potent targeting and cellular uptake in vivo unlike higher decoration density that displayed lower tumor homing, suggesting the suitability of low cRGD density for tumor targeting, whereas high peptide functionalization density can negatively influence the biodistribution and internalization into solid tumors, stimulate steric hindrance and unfavorable protein corona formation which can trigger blood opsonization and phagocytosis by liver and spleen.
In another recent study, a novel stimuli-responsive nanocarrier was designed against breast cancer by merging double therapeutic carriers comprising mesoporous silica nanoparticles (MSNPs) and mixed micelles.44 MSNPs were loaded with DOX via the formation of Schiff base between MSNPs and DOX as the first carrier. The second carrier was constructed from mixed micelles fabricated via a thin film hydration technique using Pluronic F 127 (PF127–CHO) and RGD-Pluronic P123 (PP123). Mixed micelles (MM) were then loaded with paclitaxel (PTX) via hydrophobic interaction (MM/PTX). The RGD peptide was conjugated to PP123 via EDC conjugation reaction to target the αvβ3 integrin receptors. The whole double drug carrier was prepared via dispersing [AMSN/DOX] in the water dispersions of [MM/PTX] under stirring to obtain targeted MSNPs/DOX-MM/PTX micelles. The fabricated mixed targeted micelles exhibited better therapeutic release at the acidic pH. In vitro cytotoxicity study conducted on MCF-7 cancer cells disclosed premium activity for the double drug carrier [MSNPs/DOX-MM/PTX] with lower toxicity compared to free drugs mixture. In vitro dual staining results for assaying live (green) and dead (red) cells showed that cells treated with MSNPs/DOX-MM/PTX exhibited massive nuclei fragmentation which could be attributed to the presence of RGD peptide as a targeting moiety causing efficient tumor accumulation (Table 2).
| PMs | Drugs | Targeting ligand | Grafting mode | Target organ | Receptor | Key consequences | Ref. |
|---|---|---|---|---|---|---|---|
| PEG-β-CD-PCL | DOX | Ang-2 and cRGD peptide | Click reaction | Brain | LRP-1 | Potent in vivo therapeutic effect owing to superior brain targeting | 42 |
| αvβ3 integrin | |||||||
| mPEG-b-pHPMAmLacn | DTX | cRGD peptide | BCN conjugation | αvβ3 integrin | Enhanced uptake for the lower density cRGD decorated micelles | 43 | |
| MSNPs-MM | PTX | RGD peptide | EDC conjugation reaction | — | αvβ3 integrin | Superior in vitro cellular uptake and antitumor effect | 44 |
| DOX | |||||||
| PEG-PCL | PTX | F3 peptide | Thiole–maleimide coupling reaction | Breast | Nucleolin | Prominent in vivo performance due to better accumulation and pharmacokinetics | 45 |
| D/PLA-PEI | CPT sur-shRNA | AS1411 aptamer | EDC conjugation reaction | Colon | Nucleolin | Eminent in vivo synergistic action owing to preferable tumor homing | 46 |
| β-CD/(PCL-PAEMA)21 | CPT | AS1411 aptamer | Sulfo-SMCC crosslinker | Breast | Nucleolin | Powerful in vivo performance owing to stronger selectivity | 47 |
| PEG3400-PE | TRQ | Transferrin | — | Transferrin | Enhanced in vitro antitumor activity via TF-mediated uptake | 48 | |
| PTX | |||||||
| VPM | CUR | Transferrin | Post insertion method | — | Transferrin | Powerful in vitro antitumor activity for targeted PMs | 49 |
| PEG-PLA | PTX | TF-T12 | EDC conjugation reaction | Brain | Transferrin | Improved in vivo anti-tumor effect due to higher tumor accumulation | 50 |
2.5. Nucleolin receptors
Nucleolin is a multifunctional protein and nucleic acids-binding protein localized in the nucleus and cytoplasm and it is highly expressed in eukaryotic cells. Likewise, it is colonized on the exterior of multiple cells with a vital role in controlling cell proliferation, cell cycle and apoptosis.51 Nucleolin is highly expressed on the exterior of variable malignant cells, and its overexpression is usually associated with weak prognosis. AS1411 aptamer is a 26-base DNA with superior binding capacity towards nucleolin and it was the initial clinically examined aptamer for targeted tumor therapy purposes.52 Exploiting nucleolin receptors for active cellular targeting is a promising therapeutic strategy for cancer. Cai and his co-workers fabricated polymeric micelles from maleimide-terminated PEG-PLA synthesized via ring-opening polymerization method to form HOOC-PEG-PLA followed by an esterification interaction in the presence of HEMI (1H-pyrrole-2,5-dione) to form mal-PEG-PLA.45 Fabricated nanomicelles were further modified with a tumor-homing peptide (F3) through thiol–maleimide coupling reaction to selectively bind overexpressed nucleolin receptors, then they were loaded with PTX via dialysis method. The targeted nanomicelles displayed a pH and acid-triggered release pattern for PTX, implying their potential for targeted delivery to the acidic tumor environment. In vitro cellular studies disclosed an augmented uptake for coumarin-6 labeled F3 decorated nanomicelles compared to F3 free nanomicelles when incubated with MCF-7 cancer cells. The Quantitative findings of flow cytometry further displayed a powerful fluorescence intensity in the cytoplasm after 2 h incubation. Moreover, cellular internalization of coumarin-6-labeled F3 decorated nanomicelles was massively inhibited when cells were pretreated with F3 peptide, confirming that the cellular internalization of the nanomicelles is mainly via F3 peptide–nucleolin receptor interaction. In addition, fabricated F3 modified nanomicelles exhibited 2 fold higher intratumoral accumulation than F3 free nanomicelles and better tumor suppression following i.v. administration into tumor-bearing mice associated with enhanced PTX pharmacokinetic profile and low systemic toxicity.The synergistic anticancer effect for co-delivery of dual small molecule chemotherapy and macromolecular gene therapy loaded PMs against CRC was reported by Sanati and his colleagues.46 In this study, PMs were constructed from poly (DL-lactic acid) (PLA) and polyethyleneimine (PEI) di-block copolymer via EDC conjugation reaction followed by loading of camptothecin (CPT) into the inner hydrophobic micellar core to form PLA-PEI/CPT micelles. On the other hand, sur-shRNA as survivin inhibitor gene therapy was adsorbed on the micellar shell followed by coating with PCAD (poly carboxylic acid dextran) via mixing at room temperature to form D/PLA-PEI/CPT-sur micelles. Targeted PMs were obtained via surface modification of the dually loaded dextran coated micelles with nucleolin targeting aptamer through EDC conjugation reaction to form AS1411-D/PLA-PEI/CPT-sur micelles. The targeted nanomicelles displayed a biphasic release behavior at pH 7.4 that showed fast release during the initial 24 h for CPT adsorbed on the surface followed by slow and constant release for CPT loaded within the nanomicelles. In vitro cellular uptake studies conducted on C26 cells revealed higher uptake in case of AS1411-D/PLA-PEI/CPT-sur micelles compared to D/PLA-PEI/CPT-sur micelles and cells pretreated with AS1411 aptamer. In addition, fabricated aptamer modified PMs exhibited powerful in vivo intratumoral accumulation following intravenous administration into tumor-bearing mice compared to aptamer free PMs with no signs of serious systemic toxicity except for the group treated with free CPT, suggesting the synergistic effect of both CPT as chemotherapeutic drug and sur-shRNA as survivin inhibitor.
In another approach, PMs were developed from a star like polymer comprising 21-arm of β-CD/[PCL/poly(2-aminoethyl methacrylate)] (β-CD/(PCL-PAEMA)21) through cationic ring opening polymerization reaction, followed by loading with CPT using co-lyophilization method to form β-CD/(PCL-PAEMA)21/CPT.47 Modifying the PMs surface with AS1411 aptamer was achieved using sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) crosslinking agent to obtain targeted AS1411/β-CD/(PCL-PAEMA)21/CPT.22 The targeted nanomicelles displayed a pH and acid-triggered release pattern for CPT, implying their potential for targeted delivery to the acidic tumor environment. In vitro cellular uptake and cytotoxicity studies revealed preferable cellular uptake and anticancer effect for the targeted micelles upon incubation with MCF-7 and 4T1 cells compared to L929 cells. In vivo biodistribution screening showed powerful intratumoral targeting by aptamer functionalized PMs 5 h after i.v. administration into tumor-bearing mice, resulting in a significant contraction in the tumor volume and prolonged survival rate compared to all other treated groups. The quantified mean fluorescence intensities were 111, 112, 116, 132, 136, and 147 in the lung, spleen, heart, kidney, liver, and tumor, respectively following 5 h injection (Table 2).
2.6. Transferrin receptors
Transferrin receptor (TfR) is a transmembrane glycoprotein responsible for regulating iron balance and metabolism. In general, the cellular expression level of TfR is weak, but both brain cancer cells and vascular endothelial cells are overexpressing TfRs.53,54 Also, TfRs are highly expressed in tissues demanding plenty of iron for heme composition such as brain capillary endothelial cells to maintain iron balance.22,55 Overexpression of TfR is reported in many malignant cells, making this vital receptor a potential target for cancer therapeutics. Various ligands including transferrin (Tf), lactoferrin, and even peptides can be utilized for decorating the surface of the PMs.56,57In attempts to antagonize MDR (multidrug resistance) in cell lines overexpressing P-glycoprotein (P-gp) via employing targeted drug delivery NPs, Zou and his co-workers developed transferrin-targeted PMs for co-delivery of TRQ (tariquidar); a potent P-gp suppressor and chemotherapeutic paclitaxel (PTX) against MDR ovarian carcinoma cells.48 The PMs were fabricated from p-nitrophenylcarbonyl-PEG-nitrophenyl-carbonate (pNP-PEG3400-pNP) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rh–PE) to develop [pNP-PEG3400-PE] nanomicelles according to previously established method by Dabholkar and his colleagues.58 Targeted PMs were synthesized via the correlation between TF, actively targeted moiety and the distal side of the PEG3400-PE PMs through pNP moiety to form TF/PEG3400-PE micelles for targeting TfRs. Finally, the targeted micelles were co-loaded with PTX and TRQ to form TF/PEG3400-PE/PTX-TRQ PMs. Rhodamine-labeled TF-modified PMs [TF/PEG3400-PE PMs] exhibited higher cellular internalization when incubated with MDR ovarian carcinoma A2780-Adr and SKOV-3TR cell lines overexpressing TFRs on their surfaces. Furthermore, in vitro cytotoxicity study depicted superior cell killing activity for TF/PEG3400-PE/PTX-TRQ PMs against the examined cell lines in comparison to all other-treated groups owing to the effectiveness of TF-directed endocytosis. Moreover, the anticancer results against 3D spheroids SKOV-3TR cells showed higher penetration ability of TF-targeted nanomicelles into the deeper spheroid layers, demonstrating an improved antitumor activity via micellar co-encapsulation of PTX and TRQ, besides TF-targeting against MDR human ovarian cancer.
Another TF functionalized micelles were developed by Muddineti and his colleagues to alter the aqueous solubility of curcumin and improve its targeting potential to solid tumors via exploiting overexpressed TfRs.49 Micelles were comprised from poly(ethylene glycol)(PEG)-ylated vitamin-E/lipid (PE) (VPM) synthesized via acid–amine coupling reactions.59 Targeted nanomicelles were synthesized by functionalizing the prepared VPM micelles with TF via post insertion method60 to develop TF/VPM nanomicelles. In vitro uptake study revealed an augmented intracellular accumulation of TF/VPM-CUR nanomicelles in the middle stacks when incubated with HeLa and HepG2 cells for 4 h compared to untargeted nanomicelles and targeted nanomicelles incubated with TF-pretreated cells. Moreover, higher CUR uptake in the spheroid cells was observed for TF/VPM-CUR nanomicelles (mean fluorescence intensity is 1651) after 4 h incubation compared to TF free nanomicelles (mean fluorescence intensity is 1125), confirming the micellar ability to penetrate the deep tumor mass.
Achieving both safe and efficient targeted therapeutic delivery of PMs through BBB against glioma was successfully conducted by Sun and his co-authors.50 PMs were fabricated from HOOC-PEG-PLA di-block copolymer whose surface was decorated with TF-T12 peptide through EDC conjugation procedure to form targeted TF-T12/PEG-PLA micelles to overcome BBB and efficiently target glioma cells. Targeted PMs were then loaded with PTX via dialysis method. The constructed targeted nanomicelles showed slow release behavior for the loaded PTX in pH 7.4. In vitro cellular homing & cytotoxicity screenings revealed a significant uptake in case of DiR labeled TF-T12/PEG-PLA nanomicelles upon their incubation with U87MG cells with potent anticancer effect compared to unlabeled micelles, which could be related to the presence of TF on the exterior of the micelles that can selectively be attracted to overexpressed TfRs on the surface of U87MG cells. In vivo studies revealed good biodistribution and potent anticancer effect in tumor-bearing mice after i.v. administration of TF-T12/PEG-PLA nanomicelles in terms of reduced proliferation, angiogenesis, and induced apoptosis compared to PEG-PLA-PTX nanomicelles due to their superior potential to cross the BBB barrier. Furthermore, no distinct signals of systemic toxicity were detected following treatment with the prepared micelles, while groups treated with free PTX displayed lung injury and liver fibrosis, suggesting the potential anti-glioma therapeutic efficiency of TF-T12/PEG-PLA-PTX nanomicelles mediated via binding TfRs and overcoming BBB to guarantee higher tumoral PTX accumulation (Table 2).
2.7. Asialoglycoprotein receptors
Asialoglycoprotein receptor (ASGPR); a transmembrane C-type lectin, realizes a broad diversity of ligands comprising terminal galactose or N-acetylgalactosamine residues. The expression of ASGPR was found to be maximum on the surface of hepatocytes.61 Sugar-based ligands usually display a superior recognition and binding affinity towards ASGPR. Consequently, ASGPR is a fascinating target for precise therapeutic and diagnostic liver targeting.62 In this regard, Xiang, et.al., exploited a dual targeting strategy with two different targeting ligands that can accurately bind to two varied overexpressed receptors on the exterior of liver tissues.63 PMs were constructed from N/galactosylated/chitosan/5-fluorouracil acetic acid linked [LA–CHI–FUA] via an ionic crosslinking method.64 First of all, N/galactosylated/chitosan (LA–CHI) was developed via amidation conjugation reaction between lactobionic acid (LA) and chitosan (CHI) using EDC conjugation reaction, followed by subsequent conjugation with 5-fluorouracil acetic acid (FUA) using EDC amidation conjugation reaction. LA–CHI–FUA nanomicelles were then produced using an ionic crosslinking method. Afterward, fabricated LA–CHI–FUA were modified with folic acid (FA), to target the massively expressed folate receptors (FRs) in numerous malignant tissues through EDC conjugation procedure to develop FA/LA–CHI–FUA nanomicelles. The targeted nanomicelles displayed controlled in vitro release manner for FUA at three pH of 5.0, 6.8, and 7.4. In vitro cellular study demonstrated an enhanced uptake within the cytoplasm in case of FITC-labeled FA/LA–CHI–FUA nanomicelles (59-fold greater) when incubated with SMMC-7721 cells (positive FRs and ASGPR) for 4 h compared with A549 cells (negative FRs and ASGPR) and SMMC-7721 cells pretreated with FA and galactose, emphasizing the combined targeting ligands-reinforced cellular uptake. Moreover, FA/LA–CHI–FUA nanomicelles exhibited high cellular viabilities towards HUVEC (human umbilical vein endothelial) cells and L02 (normal hepatocytes) in comparison to free 5-FUA. In addition, in vitro antitumor efficacy demonstrated greater cell killing potency for FA/LA–CHI–FUA nanomicelles compared to both LA–CHI–FUA nanomicelles and free 5-FUA, which could be related to the dual synergistic targeting effect. Moreover, in vivo antitumor study revealed an eminent activity for FA/LA–CHI–FUA nanomicelles after i.v. administration into SMMC-7721 tumor-bearing mice accompanied by lower toxicity compared to free 5-FUA, which emphasized an improved therapeutic effect of dual targeted FA/LA–CHI–FUA nanomicelles via exploiting the massively exposed ASGPRs and folate receptors (FRs) on the exterior of HCC cells.In another study, galactose (Gal)-modified nanomicelles against HCC were fabricated by Mazumder and his co-authors for cancer detection and therapy.65 Fabricated nanomicelles were comprised of poly(ethylene glycol)-b-poly(lactide) (PEG-P-PLA) loaded with superparamagnetic iron oxide (SPIO) NPs and the anticancer molecule; 19-O-triphenylmethylandrographolide (RSPP050) via solvent evaporation method to form PEG-P-PLA/SPIO-RSPP050 nanomicelles. Targeted nanomicelles were fabricated via surface modification of the PEG-P-PLA/SPIO-RSPP050 with Gal via simple mixing and sonication to form Gal/PEG-P-PLA/SPIO-RSPP050 nanomicelles. In vitro intracellular homing depicted an augmented cellular internalization, particularly in the cytoplasm and nuclei for Gal/PEG-P-PLA/SPIO nanomicelles loaded with Nile red when incubated with HepG2 cells (positive ASGPR) for 3 h compared with untargeted nanomicelles or targeted nanomicelles incubated with L929 cells (Negative ASGPR). Also, the constructed Gal/PEG-P-PLA/SPIO-RSPP050 nanomicelles exhibited an eminent killing effect against HepG2 cells compared to free RSPP050, PEG-P-PLA/SPIO-RSPP050 nanomicelles and Gal/PEG-P-PLA/SPIO nanomicelles. In addition, in vitro intracellular iron uptake was estimated via Prussian blue staining and results showed that HepG2 cells exhibited higher iron uptake confirmed by intense blue deposition when incubated with Gal/PEG-P-PLA/SPIO-RSPP050 nanomicelles compared to untargeted nanomicelles, suggesting the potential of this nanoplatform to function in magnetic resonance imaging (MRI) applications due to presence of galactose which could mediate efficient cellular internalization by means of receptor recognition and internalization.
Another notable example of exploiting LA-functionalized nanomicelles for targeting liver tumors with dual imaging purposes involving; MRI and single/photon emission computed tomography (SPECT) was conducted by Assawapanumat and his colleagues.66 Nanomicelles were fabricated from allyl-PEG-b-PLA di-block copolymer followed by conjugation with cysteamine HCl using sequential anionic ring opening polymerization method to form NH2-PEG-b-PLA.67 Fabricated NH2-PEG-b-PLA was then conjugated with the carboxylic end of diethylenetriaminepentaacetic acid-functionalized (DTPA) via its amino terminus using DCC–NHS conjugation reaction to form DTPA-PEG-b-PLA diblock copolymer.68 Targeting human hepatic carcinoma via exploiting ASGPR was achieved via surface decoration of the developed nanomicelles with LA via DCC-NHS conjugation procedure to form targeted LA/DTPA-PEG-b-PLA nanomicelles. The SPIO NPs were incorporated into the targeted blank nanomicelles as a contrast agent for MRI via solvent evaporation method to form LA/DTPA-PEG-b-PLA-SPIO nanomicelles. Labeling of the fabricated nanomicelles with radiolabeled Technetium-99 m [99mTc] was attained via complexation between 99mTc and DTPA on the surface of the micelles using stannous chloride (SnCl2) under acidic conditions to form radiolabeled LA/99mTc-DTPA-PEG-b-PLA-SPIO nanomicelles. 99mTc has good sensitivity in spatial resolution owing to its ability to emit gamma radiation using a gamma camera device. In vitro hemecompatibility of the developed nanomicelles showed low hemolytic effect (<2%) confirming the possibility for further in vivo applications. In vitro cellular uptake study examined via Prussian blue staining revealed a higher accumulation of SPIO inside the HepG2 cells when incubated with targeted LA/99mTc-DTPA-PEG-b-PLA-SPIO nanomicelles compared to the non-targeted ones. In addition, in vitro SPECT imaging elucidated that LA/99mTc-DTPA-PEG-b-PLA-SPIO nanomicelles exhibited strong bright signals (2.5 fold increment) of radionuclide in HepG2 cells after 2 h compared to 9mTc-DTPA-PEG-b-PLA-SPIO nanomicelles or free 9mTc. Moreover, LA-targeted nanomicelles exhibited enhanced in vitro MRI signal (3 fold increment) when incubated for 2 h with HepG2 cells compared to untargeted nanomicelles, suggesting that integrating two different imaging approaches such as SPIO and 9mTc along with active targeting by LA can offer a unique nanosystem that could internalize into HepG2 cells to act as a multimodal contrast agent for liver cancer diagnosis (Table 3).
| PMs | Drugs | Targeting ligand | Grafting mode | Target organ | Receptor | Key consequences | Ref. |
|---|---|---|---|---|---|---|---|
| LA–CHI–FUA | FUA | LA | EDC conjugation reaction | Liver | ASGPR | Enhanced in vivo antitumor effect for dual targeted PMs | 63 |
| FA | FRs | ||||||
| PEG-P-PLA | SPIO | Gal | Simple mixing | — | ASGPR | Improved in vitro anticancer effect and imaging due to high tumor accumulation | 65 |
| RSPP050 | |||||||
| PEG-P-PLA | SPIO | LA | DCC conjugation reaction | — | ASGPR | Stimulated in vitro multimodal contrast effect due to SPECT/MRI signals | 66 |
| 9mTc | |||||||
| PEG-P(Glu)/Gluc-PEG-P(Glu) | Cisplatin | Glu | C6 linkage using ether linkage | Brain | GLUT 1 | Potent in vivo therapeutic effect due to precise intratumoral accumulation | 69 |
| PEO-PPO | CUR | Glu | Microwave-assisted ring opening reaction | Breast | GLUT1 | Superior in vivo intratumoral accumulation via GLUT1 receptor | 70 |
| Soluplus-TPGS | PTX | Glu | Microwave-assisted ring opening reaction | Brain | GLUT1 | Enhanced brain delivery via GLUT 1 receptors associated with improved PTX effect | 71 |
2.8. Glucose transporter 1 receptor
Glucose transporter 1 (GLUT1) is a transporter responsible for cellular uptake of monosaccharide such as galactose and glucose.72 It is highly expressed in many tumors including colon, ovary, lung, kidney, prostate, breast, and brain, whereas its expression is relatively limited in normal tissues due to the higher demand for aerobic glycolysis rate of cancer cells that needs more glucose than the normal cells.73 Also, GLUT1 is strongly located onto BCECs (brain capillary endothelial cells) to facilitate glucose uptake. As a result, GLUT1 receptors could facilitate the delivery of glucose-decorated carriers across the BBB. Based on these findings, GLUT1 receptors could be exploited as a fascinating targeting ligand for early tumor diagnosis and treatment.74 In this light, Suzuki and his co-authors utilized the privilege of cancer permanent glycolysis to design glucose-decorated nanomicelles to bind to the externally expressed GLUT1 on the malignant cells and to overcome the vascular endothelial barrier so as to improve intratumoral delivery therapeutic potency.69 Nanomicelles were composed of poly(ethylene glycol)-poly(L-glutamic acid) (PEG-P(Glu)) and α-glucopyranos-6-O-yl-poly(ethylene glycol)-poly(L-glutamic acid) block copolymer (Gluc-PEG-P(Glu)). Cisplatin was then loaded into the fabricated PMs using Mochida, et.al., method75 to develop (PMs-Pt). Finally, Glu as a tumor targeting moiety was linked to the exterior of cisplatin-loaded PMs via carbon 6 into the α-terminus of the PEG block using ether linkage76 to form Glu/PMs-Pt. The targeted nanomicelles displayed a controlled in vitro release manner for Pt at pH of 7.4. In vitro anticancer efficiency indicated a potent activity for Glu/PMs-Pt against human squamous cell carcinoma (OSC-19) 3D multicellular spheroids cells (high GLUT 1 expression) compared to PMs-Pt, free Pt or Glu/PMs-Pt in case of human glioblastoma-astrocytoma (U87MG) multicellular spheroids cells (low GLUT 1 expression). On the other hand, knocking down GLUT1 receptor expression on OCS19 cells was found to reduce the anticancer efficacy of Glu/PMs-Pt, which proposes that the anticancer activity of Glu-modified cisplatin loaded PMs was mediated via targeting the externally overexpressed GLUT1 on the malignant cells. Furthermore, in vivo biodistribution study assessed by quantifying Pt concentration via ICP-MS elucidated a higher intratumoral accumulation for Glu/PMs-Pt than other organs subsequent to i.v. injection in OCS19 tumor-bearing BALB/c mice. Additionally, the targeted nanomicelles displayed rapid and 2 fold higher intratumoral accumulation within 4 h following injection combared to the untargeted micelles. Moreover, in vivo antitumor study depicted an augmented activity and massive % tumor growth inhibition for Glu/PMs-Pt without any detectable toxicity, unlike the group treated with the free drug which exhibited some signs of toxicity.In another approach conducted by Lecot and his coworkers, a novel targeted glycosylated PMs were developed for directed delivery of CUR to breast cancer cells overexpressing GLUT1 receptor.70 PMs were comprised of poly(ethylene oxide)–poly(propylene oxide) (PEO–PPO) block copolymers fabricated according to a previously reported method.77 Glycosylation of the PMs was achieved via decoration with Glu using microwave-assisted ring opening reaction of gluconolactone in the presence of Sn(Oct)2 to form Glu/PEO-PPO.78 Targeted glycosylated PMs were then loaded with CUR via solvent evaporation method to form Glu/PEO-PPO-CUR. Fabricated glycosylated PMs loaded with CUR exhibited higher cellular internalization in comparison to untargeted CUR-loaded PMs or free CUR when incubated with murine mammary tumor cells (4T1) overexpressing GLUT 1 receptor. In vitro cytotoxic study demonstrated better cytotoxicity for Glu/PEO-PPO-CUR nanomicelles against 4T1 cells in comparison to all the examined groups. Moreover, in vivo biodistribution screening revealed an augmented intratumoral accumulation of Glu/PEO-PPO-CUR nanomicelles that remained for 48 h after their i.v. injection into 4T1 tumor-bearing BALB/c mice compared to PEO-PPO-CUR nanomicelles, suggesting the longer intratumoral residence time of the targeted nanomicelles.
In another attempt, glycosylated mixed nanomicelles were fabricated by Riedel and his co-authors for directed delivery of PTX to glioblastoma cells.71 Mixed nanomicelles were constructed from polyvinyl caprolactam–polyvinylacetate/polyethylene glycol graft copolymer (Soluplus) and D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) through polymer dispersion in water under stirring to form Soluplus-TPGS, then dispersed nanomicelles were loaded with PTX via acetone diffusion technique.79 Glycosylation of the mixed nanomicelles was achieved via decoration with Glu using a microwave-assisted ring opening reaction to form Glu/Soluplus-TPGS-PTX. The in vitro release manner of PTX from the targeted nanomicelles manifested a controlled release behavior through time with no detectable burst effect. In vitro anticancer study showed superior efficiency for Glu/Soluplus-TPGS-PTX against human glioblastoma (U251) cell lines which overexpress GLUT 1 receptor in comparison to Soluplus-TPGS-PTX and free PTX. Moreover, results of cell death via dual staining assay for quantification of live (green) and dead (red) cells were in accordance with in vitro cytotoxicity results manifesting that Glu/Soluplus-TPGS-PTX exhibited superior cell death potential compared to other groups. Meanwhile, this high cellular internalization was not observed in LN229 cells, confirming that the uptake mainly depends on the overexpressed GLUT1 receptors. More importantly, in vivo biodistribution experiment revealed higher brain accumulation (more than 8-fold) for Glu/Soluplus-TPGS-PTX after 0.5 h of i.v. injection into Wistar rats compared to Soluplus-TPGS-PTX or free PTX, suggesting the capability of these targeted mixed nanomicelles to overcome the BBB via binding to GLUT 1 receptors and inhibition of P-glycoprotein (Table 3).
2.9. Folate receptors (FRs)
Functionalization of PMs with folate is an already existing trend by which we can target the massively expressed FRs located externally on the cancerous cells encompassing breast, ovarian, and uterine cancers.80–82 Furthermore, a subtype of FR namely, FR alpha (FRα) was found to have higher affinity towards folic acid and some other analogue molecules.83 This receptor is located within the cellular plasma membranes attached to glycosylphosphatidylinositol molecule which can facilitate the entrance of FA or FA-decorated micelles via receptor-mediated endocytosis.84 FA as a targeting ligand has many advantages over other molecules including; being cheap, stable, with low immunogenicity and ease of conjugation.84 In addition, when it binds to FRα, it begins an endocytotic cascade track, which usually causes accelerated relocation of FRα to the external region of the cell and easy disassembly of functionalized nanosystem with fast endosomal drug release.85 Moreover, the small size of FA usually does not cause a significant increase in the nanocarrier’s size and hence, it does not affect its cellular penetration.86In an interesting study, pluronic F68 triblock co-polymer was conjugated to FA using DCC/NHS coupling reaction, then DTX was incorporated inside the hydrophobic core of the PMs through thin film hydration technique.87 Pluronic F68 is a synthetic and amphiphilic polymer approved by the FDA.82 It is well known to possess a sandwich-like structure comprising two hydrophilic poly(ethylene oxide) (PEO) blocks with a poly(propylene oxide) (PPO) block located within them. Pluronic F68 was selected to be the drug carrier owing to its potential to suppress P-gp (P-glycoprotein), and hence reducing drug efflux, while FA was selected to be the targeting ligand due to its ability to join FR and enhance cellular uptake via receptor-mediated endocytosis. Results revealed that the prepared targeted PMs exhibited higher encapsulation efficiency of about 94.75%, associated with sustained DTX release of about 94 ± 3.9% and 85 ± 4.6% from DTX-loaded pluronic micelles and DTX-loaded FA-decorated pluronic micelles, respectively after 48 h. This sustained drug release in case of FA-decorated micelles might be the coating effect of the conjugation moiety on the surface of the micelles. Furthermore, the prepared FA-coated-DTX-loaded micelles demonstrated amended in vitro anticancer impact against human MDA-MB-231 breast cancer cells when compared to FA-free micelles or free DTX. In addition, when FA-coated micelles were administrated intraperitoneally in healthy rats, they showed no toxicity on lung, liver and kidneys.
In another study, a dual-responsive micelles were fabricated from FA and mPEG grafted polyurethane (FA-PUSS-gimi-mPEG).88 First, mPEG and FA were successively grafted to the polyurethane side chain to form disulfide bond through acid-sensitive benzoic-imine bond and amido bond. The rationale beyond selecting this platform relies on taking the privilege of the long hydrophilic mPEG chain, which will stabilize the micelles in the blood circulation and also protect short FA segment in the polymer side chain. When reaching the tumor site with its acidic environment, mPEG will start to shad due to cleavage of benzoic-imine bond, giving a chance for FA to be exposed to potentiate active targeting. At the same time, charge reversal will occur due to protonation of amino moieties in the acidic environment, which is supposed to reinforce the cellular homing of the micelles. In addition, disulfide bonds found in polyurethane will be cleaved in abundant GSH levels found in the tumor tissue, which in turn will speed up the release of the drug leading to improved drug efficiency. The micelles exhibited negative charge, but the charge turned positive after reaching the tumor acidic environment as mentioned above. Doxorubicin was integrated into the hydrophobic core of the PMs via dialysis technique with faster release at pH 5 and 10 mM GSH when compared to release at physiological pH (pH 7.4), suggesting that the design of the micelles guaranteed no burst release effect under normal physiological condition with low drug release in tumor extracellular environment (pH 6.5) and more rapid drug release in tumor intracellular environment (pH 5). Moreover, the structure changes that occur in the micellar architecture in response to acid redox potentials were found to enhance cellular uptake and drug release in HGC-27 human gastric cancer cells, which were confirmed by more DOX accumulation in the nucleus under acidic condition due to micellar structure instability as a result of multiple synergistic effects.
In hepatic fibrosis, the most important cellular populations that are the major contributors in disease progression are myofibroblastic hepatic stellate cells (MF-HSCs) which are formed as a result of the conversion of quiescent HSCs to MF-HSCs.89 Most antifibrotic drugs work on these cells either by inhibiting their proliferation or stimulating their apoptosis.90 Recognition of ligand-decorated nanocarriers is based on the massively expressed receptors on the external layer of MF-HSCs such as platelet-derived growth factor receptors (PGDFRβ), insulin-like growth factor receptors, and CD44 receptors.91–94 Furthermore, it was found that HSCs selectively overexpress folate receptor alpha (FRα), which is strictly not expressed in other hepatic cells, making it an excellent selective target candidate for antifibrotic nanodrugs.95 Another privilege is that the small size of FA will help in sufficient delivery of nanomedicines via going to the lumen of HSC as large molecules will face difficulties in delivery in case of fibrosis due to loss of LSECs (liver sinusoidal endothelial cells) fenestration and accumulation of ECMs.96
Based on the above information, FA-linked PEG-PCL copolymeric micelles were fabricated via EDC/DMAP conjugation technique, then FA was further attached to the hydrophilic shell of the micelles through EDC/NHS technique.97 CPT was then integrated into the hydrophobic core of the micelles via dialysis method. Morphological analysis disclosed that the copolymers can self-assemble in aqueous medium into spherical micelles with good cytocompatibility. Interestingly, when nile red (NR); a lipophilic dye was encapsulated into FA-linked micelles, it exhibited two fold more release at pH 5 in comparison to pH7.4, indicating a pH-sensitive release profile for NR-loaded micelles, which could be beneficial as HSCs cells become more glycolytic and lactate secretory acidic microenvironment. This acidic environment can afford more antifibrotic drugs at the fibrosis area. In vivo findings in CCl4-hepatic fibrosis rat model revealed that intravenous injection of FA-decorated NR-loaded micelles exhibited the highest accumulation level in liver unlike to non-decorated micelles which in turn might cause an improvement in suppressing fibrogenesis and better recovery of liver function and structure. In addition, much less NR was detected in the spleen and heart, whereas, lung and kidney included the least NR amount.
In another study, thermosensitive and biocompatible poly(2-hydroxyethyl acrylate) (PHEA) and poly(N-isopropylacrylamide) (PNIPAAm) copolymers were conjugated to form polymeric micellar structure employing reversible addition–fragmentation chain transfer polymerization reaction to encapsulate 5-FUA within their core via dialysis method, then these micelles were further functionalized with FA via DCC/DMAP conjugation reaction.98 In vitro drug release study in PBS at physiological conditions revealed release of less than 5% of the drug, confirming permanent attachement of the drug to the polymeric micelles structure. Moreover, in vitro cellular studies showed that fabricated micelles revealed good compatibility with normal host cells including; human skin fibroblasts (CRL-1475), human colorectal fibroblasts (CCD-112CoN) and human monocytic cell line (THP-1), whereas, FA-conjugated micelles exhibited potent cytotoxicity against DLD-1, CaCo-2 and HT-29 human colorectal adenocarcinoma cell lines with induced apoptosis and necrosis potentials.
In another interesting approach, methotrexate–polyethylene glycol (MTX–PEG) covalent conjugate was utilized to functionalize chitosan/2,3-dimethylmaleic anhydride (DMMA) polymeric micelles.99 Firstly, chitosan was attached to DMMA via an amidation reaction to give an anionic polymer, then DOX was integrated into this platform by electrostatic interaction. Afterwards, the external layer of these micelles was linked with MTX–PEG by carbodiimide reaction to improve their targeting potential towards cancer cells as PEG will minimize protein adsorption, while MTX can specifically attach to the externally overexpressed FA receptors on cancer cells. In vitro drug release study conducted at pH 7.4 and 5.4 with and without proteases revealed that the release pattern of DOX was greatly affected with the change in the pH not the presence or absence of protease with only 25.7% released at pH 7.4 and 85.6% at pH 5.4 after 48 h, which might be related to a decrease in the degree of ionization in the drug carboxyl group causing a destruction in the electrostatic interaction between the drug and DMMA. On the other hand, the release profile of MTX was greatly affected by the presence or absence of protease not the pH. At pH 5.4, the release of MTX was about 12%, while it raised up to 48.4% in the presence of protease after 48 h, which might be due to stronger protease activity in acidic pH. In vitro cellular uptake study revealed more DOX accumulation in the nucleus of MDA-MB-231 cancer cells than HK-2 cells normal cells were treated with micelles. In case of free drug, there wasn't any significant difference between its accumulation in both cell types as the free drug can pass across cell membrane via diffusion without any selectivity, whereas in case of drug-loaded micelles, it can enter the cell via receptor-mediated endocytosis. Furthermore, in vitro and in vivo findings showed that the prepared micelles induced generation of autophagosomes causing autophagic death of cancer cells because of lysosomal destruction that might happen due to the presence of DMMA which can hold protons from the outside of the lysosome and allow the entrance of water and chloride ions instead leading at the end to lysosomal rapture (proton sponge effect) (Table 4).
| Micelles composition | Drug | Targeting ligand | Grafting mode | Target organ | Receptor | Key consequences | References |
|---|---|---|---|---|---|---|---|
| Pluronic F68 | DTX | FA | DCC/NHS | — | FR | Enhanced in vitro anticancer effect with no in vivo toxicity in rats upon intraperitoneal administration | 87 |
| (FA-PUSS-gimi-mPEG) | DOX | FA | DCC/NHS | — | FR | Acid/redox-targeting resulted in better cellular uptake in vitro in HGC-27 cells | 88 |
| PEG-PCL | CPT | FA | EDC/NHS | Liver | FRα | Increased recovery from liver fibrosis in vivo in CCl4-induced rats | 97 |
| poly(2-hydroxyethyl acrylate) (PHEA)-poly(N-isopropylacrylamide) (PNIPAAm) | 5-FU | FA | DCC/DMAP | — | FR | In vitro compatibility with normal human cell lines with improved cytotoxicity against human colorectal cell lines | 98 |
| Chitosan/2,3-dimethylmaleic anhydride (DMMA) | DOX | MTX–PEG | EDC/NHS | Breast cancer | FR | Improved in vivo antitumor effect due to accumulation of autophagosomes | 99 |
| Carboxymethyl chitosan/rhein with thioketal linker | Celastrol | GA | EDC/NHS | Liver | GR | ROS-responsive micelles with good accumulation and antitumor efficacy in H22 tumor cells-bearing mice | 100 |
| sHA-DOX, HA-GA | DOX | GA | Amide bond formation in presence of DMT-MM | Liver | GR | Superior antitumor efficacy in H22 tumor-bearing mice | 101 |
| mPEG-HZ-PLA, GA-PEG-PLA | Cou6 | GA | EDC/NHS | Liver | GR | Prolonged circulation in the blood for mixed micelles with better tumor accumulation | 102 |
2.10. Glycyrrhetinic acid receptors (GRs)
Glycyrrhetinic acid (GA) is very common ligand that can target liver cancer cells as there are numerous GARs on the exterior of hepatic parenchymal cells where HCC usually occurs.103 Moreover, GARs are found on the exterior of normal hepatic cells, but at lower expression level.104 Taking this privilege, carboxymethyl chitosan and anthraquinone rhein as the hydrophilic and hydrophobic segment, respectively were conjugated with a thioketal linker to form ROS-sensitive micelles for delivery of the anti-hepatoma drug; celastrol.100 Afterwards, GA was conjugated to the external layer of the assembled micelles by EDC/NHS coupling technique, then the drug was integrated into the hydrophobic core of the micelles employing a dialysis method.In vitro drug release study demonstrated that free celastrol exhibited more than 70% release after 12 h and almost 100% release after 24 h in pH 7.4, whereas drug release from GA coated drug-loaded micelles was about 29% after 12 h and 37% after 24 h. The obtained micelles exhibited their maximum release when they were incubated in pH 5 and 10 mmol mL−1 H2O2 reaching about 58% after 12 h and 81% after 24 h, confirming their sensitivity to elevated ROS level inside tumor cells.
In vitro cellular cytotoxicity study demonstrated that assembled drug-loaded micelles showed an inhibitory effect on BEL-7402 and HepG2 malignant cells with good ROS-responsive properties, associated with better cellular uptake due to overexpressed GA receptors. Furthermore, when these micelles were intravenously administrated in ICR mice impregnated with H22 tumor cells, they displayed longer residence in the circulation leading to enhanced antitumor effect and reduced systemic toxicity compared to free drug. This improved effect might be due to better accumulation of the targeted micelles in the liver and tumor with 1.2 and 1.8 fold increase in comparison to untargeted micelles and very low accumulation in kidney spleen, heart and lung, suggesting superior targetabiliy and selectivity.
In another delightful approach, mixed micelles for treatment of hepatoma were fabricated, in which, sulfated hyaluronic acid (sHA) was linked to DOX via hydrazone linker to develop pH-sensitive micelles and then HA was conjugated to GA to improve cellular uptake of these micelles by cancer cells via GR receptors.101 Sulfated HA was utilized in this nanoplatform instead of high molecular weight HA as they can be simply dissociated by hyaluronidase to yield low-molecular-weight fractions, which were found to enhance tumor proliferation and migration,105 while hyaluronidase cannot degrade sHA, besides, some studies reported that sHA itself can inhibit angiogenesis, so it could be included in managing some solid tumors.106 sHA-DOX was prepared by a two-step reaction. First, sHA was conjugated to adipic acid dihydrazide (ADH) via amide bond. Second, DOX and sHA-ADH were conjugated by an acid-liable hydrazone bond.107 For HA-GA conjugate, it was fabricated via conjugation of HA to aminated GA (GA-N) in the presence of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMT-MM) to form an amide bond.108 Morphological examination indicated the formation of spherical mixed micelles with pH-dependent release of DOX. Moreover, these mixed micelles exhibited cytotoxic effect against HepG2 and HeLa liver carcinoma cells in a dose-dependent manner with more pronounced cellular uptake in case of HeLa cells. More significantly, in vivo studies in H22 hepatic tumor-bearing mice showed superior anti-tumor efficacy with minimal systemic toxicity in case of mixed micelles compared to all the other treated groups.
In a similar approach, actively targeted and pH-sensitive mixed micelles were prepared. They were comprised of 2 co-polymers including; polyethylene glycol methyl ether–hydrazone–polylactic acid (mPEG–HZ–PLA) copolymer which was fabricated by ring-opening polymerization reaction and (GA–PEG–PLA) which was synthesized by carbodiimide coupling technique among amino groups of PEG and carboxylic groups present in GA and PLA. Coumarin-6 (Cou6) was incorporated within the hydrophobic core of the fabricated micelles by thin film hydration technique.102 In vitro drug release study showed that at pH 5, the release was faster in micelles containing hydrazone bond owing to their pH sensitivity and breaking down in the slightly acidic tumor microenvironment to release their payload. In vivo pharmacokinetic results showed that the prepared Cou6-loaded mixed micelles could extend the drug circulation in the blood with better accumulation in the liver and the tumor site after i.v. injection in H22-bearing mice compared to other organs and other treated groups. Micelles revealed prolonged active time in the circulation as they were slowly removed from the circulation due to the possible increased viscosity of the copolymers at the body temperature, besides presence of PEG in the micellar structure which could minimize interaction between micelles and opsonins serum proteins, and hence reduce clearance by the reticuloendothelial system. Interestingly, it was noticed that the accumulation of Cou6 in tumor (198.07 ng mL−1) was higher than that in liver (131.17 ng mL−1) in case of GA-decorated micelles, with greater Cou6 concentration ratio in tumor (1.51) for GA-decorated micelles with hydrazone bond compared to micelles without GA decoration (1.15) and micelles without both GA and hydrazone bond, suggesting that combining both active targeting and a stimulus-responsive effect might give the most pronounced antitumor effect. In addition, when Re (relative intake ratio) values were calculated for tumor and liver, GA-decorated micelles with hydrazone bond exhibited significant higher values in comparison to micelles without GA decoration, indicating that these micelles can efficiently minimize non-target organ distribution (Table 4).
2.11. Multifunctional cell surface adhesion (CD44) receptors
It is a transmembrane glycoprotein with tri domains; the extracellular, the transmembrane, and the intracellular domain.109 CD44 receptors are known to be greatly expressed in multiple malignancies, and they exert a vital role in enhancing tumor invasion, migration, and metastasis. This receptor was first identified as a receptor for HA, and then it was found to bind to several other ligands.110 The privilege of overexpressing CD44 receptors on the external layer of cancer cells was extensively used in developing targeted nanoplatforms for enhanced therapy.22,111In a recent study, HA–SS–PLGA copolymer was fabricated using cysteine as a linker because it includes disulfide bond. HA served as the hydrophilic fraction, while PLGA was the hydrophobic fraction.112 Fabricated self-assembled micelles were shown to be redox-responsive as the –SS– bond was reduced under tumor's glutathione elevated environment. The hydrophobic CUR was then integrated into the micellar core using a dialysis technique. In vitro drug release results indicated that at pH 5.4, 40% of CUR was released after 12 h and 48% after 48 h. Conversely, when GSH was found in the release medium, about 83% of CUR was released after 48 h due to fast disulfide bond breakage causing a disruption in the micellar structure and diffusion of the drug. Interestingly, it was observed that the release of CUR in the acidic medium with GSH was higher than its release in neutral pH with GSH. This could be due to physical entrapment of CUR in the micellar hydrophobic core, which upon exposure to acidic conditions can cause protonation to the carboxyl groups which can in turn affect charge interaction between the drug and the micelle. This interesting finding suggests that these micelles might improve drug release in tumor redox and pH-sensitive environment. Moreover, in vitro cytotoxicity findings disclosed that prepared drug-integrated micelles demonstrated good cellular uptake and tumor inhibition due to the overexpressed CD44 receptors on the surface of MCF-7 cells and tumor cell entrance via receptor-mediated endocytosis, leading to better tumor inhibition. On the other hand, drug-loaded micelles were less toxic on normal cells, which confirms their ability to target drug release.
In another study, podophyllotoxin (PPT) was linked to HA via an ester and disulfide bonding to develop a pH- and redox-sensitive prodrug micelles (HA-SS-PPT).113 PPT is a lignan compound extracted from natural plants of the genus podophyllum with potential antitumor activity mainly via inhibition of microtubule assembly in tumor cells.114 In vitro release study revealed that 21.7% of PPT was released from HA-SS-PPT at pH 7.4, while at pH 5%, it increased to 66.2% after 72 h. When 20 mM GSH was added to PBS at pH 5, the release was elevated to more than 85% due to cleavage of ester bond at low pH, besides cleavage of disulfide bond by GSH, suggesting that these micelles could minimize drug release in the circulation, and hence reducing side effects of PPT, besides improving efficient drug release in tumor microenvironment with low pH and elevated GSH level that produce pore channels in the micelles, causing degradation and erosion in the polymeric micelles. In vitro cellular uptake studies elucidated that the fabricated micelles could accumulate in the tumor site efficiently due to CD44-receptors-mediated endocytosis. Interestingly, when the in vitro release profile of HA-SS-PPT micelles was examined and compared to HA-NH-CO-PPT micelles (pH-sensitive only), they displayed 33% higher release due to their dual-responsiveness nature. Moreover, in vivo study results suggested that these micelles were able to inhibit tumor progression with more than 92% inhibition compared to only 65% in case of HA-NH-CO-PPT micelles, with minimal systemic toxicity.
In another approach, a graft amphiphilic copolymer was developed based on modification of HA with mPEG, deoxycholic acid (DCA) and N-acetyl-L-cysteine (NAC) to give (mPEG–HA(DCA)–NAC) based on successive coupling reaction with EDC/NHS carbodiimide bonding.115 Deoxycholic acid was included in the synthesis to balance the amphiphilicity of the grafted copolymer to facilitate its self-assembly in the aqueous medium, while the thiol groups in NAC can form disulfide bonds in the polymer, and hence can induce the formation of a redox-sensitive nanocarrier. PTX as an anticancer therapy was further integrated into the hydrophobic core of the micelles via ultra-sonication technique. Results indicated that drug-loaded micelles exhibited high encapsulation efficiency (73.8%) and redox-sensitivity towards PTX release in the tumor microenvironment, with 80.4% and 95.1% of PTX were released after 26 h and GSH concentrations 10 mM and 20 mM, respectively, confirming that the drug release from these micelles is triggered by high levels of GSH due to rapid cleavage of disulfide bond. In addition, in vitro findings conducted on MCF-7 cells (overexpressing CD44 receptors) demonstrated a more potent cytotoxic effect when compared to normal peripheral blood mononuclear cells (PBMC) due to increased cellular uptake in case of cancerous cells via receptor-mediated endocytosis. Furthermore, in vivo antitumor efficacy study in H22 tumor-bearing mice proved an augmented effect in case of PTX-loaded mPEG–HA(DCA)–NAC micelles with minimal toxicities to the main organs in comparison to free drug. This improved antitumor effect could be related to better tumor internalization via EPR effect and CD44-receptor endocytosis, in addition to GSH-triggered drug release inside the tumor cells (Table 5).
| Micelles composition | Drug | Targeting ligand | Grafting mode | Target organ | Receptor | Key consequences | References |
|---|---|---|---|---|---|---|---|
| HA–SS–PLGA | CUR | HA | EDC/NHS | — | CD44 | Improved cellular uptake in MCF-7 cell line with decreased proliferation | 112 |
| HA-s-s-PPT | PPT | HA | EDC/NHS | Breast | CD44 | In vivo tumor inhibition (92%) due to better cellular uptake and internalization | 113 |
| mPEG–HA(DCA)–NAC | PTX | HA | EDC/NHS | Liver | CD44 | Superior antitumor efficacy study in H22-tumor bearing mice | 115 |
| Pluronic F68-PCL | Erlotinib | Biotin | DMAP/DCC | — | Biotin receptors | Augmented cellular homing and cytotoxicity on A549 cancer cells | 116 |
| 3s-PCL-SeSe-PEG | DOX | Biotin | DCC/NHS | — | Biotin receptors | Pronounced cytotoxic effect on HeLa and MDA-MB-231 cells with low killing effect on HaCaT cells | 117 |
| CHI-BT–HBS-CB | PTX | Biotin | EDC/NHS | Breast | Biotin receptors | Improved in vivo efficacy with good imaging properties | 118 |
2.12. Biotin receptors
Biotin or (vit. H) receptors are known to be greatly expressed on the external layer of multiple cancer cell types, which can enable the internalization of biotin-decorated PMs into cancer cells.119 In order to maximize the bioavailability of erlotinib; an epidermal growth factor inhibitor, a micellar system was fabricated to encapsulate this drug comprising from biotin-functionalized pluronic F68-PCL copolymer.116 In vitro drug release study showed that the release was slow and pH-dependent with about 84.45% and 89.45% of the drug released after 96 h at pH 7.7 and 5.5, respectively. In vitro cellular uptake and cytotoxicity studies showed that biotin-conjugated micelles offered improved cellular uptake on A549 lung cancer cell line and higher cytotoxicity after 72 h owing to better internalization via massively expressed biotin receptors on the surface of cancer cells.In another study, star-shaped redox-sensitive polymeric micelles were assembled from 3s-PCL-SeSe-PEG amphiphilic copolymer using ring opening reaction.117 Biotin was further attached via its carboxylic group to the amino moiety of 3s-PCL-SeSe-PEG via DCC/NHS conjugation technique, then DOX was integrated into the targeted micellar core using dialysis or solvent exchange reaction. Physicochemical studies showed that the prepared nanomicelles were stable upon high dilution with redox-sensitive drug release profile in the high cancer redox environment. 21% of DOX was released after 72 h in 0.0067 M PBS, whereas, 57%, 65%, and 61% of DOX was released in the first 6 h in 5 mM GSH, 10 mM GSH and 0.1% H2O2, respectively. Interestingly, in vitro cellular uptake study revealed that biotin-coated DOX-loaded micelles exhibited the highest cellular uptake after 6 h with translocation in the nuclei in case of HeLa cells in comparison to HaCaT cells because HeLa cells express higher levels of sodium dependent multivitamin transporter (SMVT) responsible for biotin translocation, besides a higher redox responsiveness responsible for intratumoral DOX release. Moreover, in vitro cellular studies revealed that biotin-decorated blank micelles did not exhibit any toxic effect on HaCaT, HeLa and MDA-MB-231 cell lines, whereas, targeted drug-loaded micelles blocked the proliferation of HeLa and MDA-MB-231 cancer cell lines with 77 and 62% proliferation inhibition, respectively, while the growth of normal HaCaT cells was blocked by only 12% at the same DOX concentration (5 μg mL−1), suggesting that these multi-functional polymeric micelles can act as a promising delivery system if examined in vivo.
Another interesting study functionalized the backbone of chitosan with cetyl 4-formylbenzoate alkyl, 4-(2-hydroxyethoxy) benzophenonesalicylaldazide and biotin to from an amphiphilic copolymer (CHI-BT–HBS-CB) that can self-assemble in the aqueous media to form micelles with active targeting ability and pH-responsiveness.118 Cetyl 4-formylbenzoate alkyl and 4-(2-hydroxyethoxy) benzophenonesalicylaldazide were included in the assembly as the hydrophobic micellar core with aggregation-induced emission (AIE) properties and they were attached to chitosan backbone via benzoic imine bonds and a Schiff-base derivative formation, respectively. PTX was then loaded into the micellar core using a probe-type ultrasonic method.120 Results showed that the prepared micelles displayed a superior AIE effect with triggered drug release in the acidic tumor microenvironment, achieving PTX release after 24 h of about 33.3%, 69.4% and 80.8% at pHs 7.4, 6 and 5, respectively. This increased PTX release in the acidic condition might be attributed to easier cleavage of imine bond in acidic medium, suggesting a selective drug release inside tumor cells, while maintaining their encapsulated payload in the circulation. In addition, there was a selective uptake by MCF-7 cancer cells due to biotin functionalization when compared to MCF-10A cells after 4 h as MCF-7 cells are overexpressing biotin receptors. More importantly, in vivo findings showed that PTX-loaded CHI-BT–HBS-CB micelles demonstrated improved antitumor efficacy with minimal toxicity due to selective tumor accumulation via biotin receptors, followed by PTX release in the cytosol as a result of imine bond cleavage in the acidic tumor microenvironment, suggesting that it could be a good multifunctional nanoplatform for cancer therapy and imaging (Table 5).
2.13. Other miscellaneous receptors
Mucin 16 (MUC16) is a large glycoprotein type I transmembrane commonly known as CA125. It is characterized by limited expression in normal cells, with expression confined to the exposed epithelial membranes of many organs such as female reproductive system, abdominal lining cavity, tracheal surface and ocular surface to serve as a barrier against foreign bodies, while aiding in lubricating of the mucosal layer.121 Overexpression of MUC16 was observed in multiple tumors including; pancreatic, breast, lung, and ovarian cancers. Moreover, its overexpression is usually associated with metastasis and bad prognosis.122 Consequently, MUC16 has been exploited as a fascinating target receptor for malignant tumor therapy via interacting with MUC16 monoclonal antibodies functionalized therapeutics.123 In this regard, Pantshwa and his co-authors developed a novel anti-MUC 16 antibodies functionalized nanomicelles incorporated within hydrogel for directed delivery of MTX against ovarian carcinoma.124 Nanomicelles were synthesized from an aspartic (ASP) inner core and poly(N-isopropylacrylamide) (PNIPAAm) outer shell according to the previously reported method,125 followed by loading with MTX via solvent evaporation method to form PASP-b-PNIPAAm-MTX nanomicelles. Afterwards, drug-loaded nanomicelles were decorated with anti-MUC 16 antibody using EDC conjugation reaction to form MUC/PASP-b-PNIPAAm-MTX nanomicelles for selective binding to the overexpressed MUC 16 receptors. Targeted nanomicelles were then encapsulated within the bioresponsive C–P–N [chitosan–poly(vinylpyrrolidone)–poly(N-isopropylacrylamide)] hydrogel fabricated via free-radical polymerization mechanism to form MUC/PASP-b-PNIPAAm-MTX/C–P–N nanomicelles. Targeted nanomicelles displayed pH- and temperature-responsiveness towards malignant cells after i.p. implantation. Furthermore, in vivo studies performed on ovarian carcinoma-bearing mice after i.p. implantation of the manufactured nanomicelles manifested a perfect therapeutic efficiency for MUC/PASP-b-PNIPAAm-MTX nanomicelles in terms of diminished tumor volume associated with higher survival rate compared to MUC 16-free nanomicelles (PASP-b-PNIPAAm-MTX). Moreover, immunohistochemical analysis manifested a remarkable diminishing in mucin 16 expression for i.p implanted MUC/PASP-b-PNIPAAm-MTX nanomicelles compared to all other groups, which suggests the potential to prevent metastasis relying on MUC16-induced mucoadhesion to other organs within the peritoneum.Fibronectin (FN) is an integral protein found in normal and malignant ECM and it is involved in regulating cellular adhesion, growth, migration and malignant transformation.126 It is overexpressed in many tumor tissues including; breast, colorectal, thyroid, brain, lung and ovary and it is usually accompanied by poor prognosis, suggesting the feasibility of FN as a therapeutic target for cancer management.127 Based on these facts, a dual acting therapy employing PMs decorated with FN-targeting peptides was developed.128 Targeted PMs were synthesized from DSPE-PEG [1,2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol)], followed by decoration with CREKA peptide via simple mixing (C/DSPE-PEG) to specifically bind to overexpressed FN receptors. The targeted PMs were then co-loaded with DOX and vinorelbine (VIN) to prevent breast cancer metastasis (C/DSPE-PEG/D-V). The targeted nanomicelles displayed a pH and acid-triggered release pattern for DOX and VIN, implying their potential for targeted delivery to the acidic tumor environment. In vitro cellular uptake and cytotoxicity studies demonstrated a favorable internalization for C/DSPE-PEG/D-V nanomicelles within the cytoplasm when incubated with 4T1 cancer cells at 3 h unlike the untargeted nanomicelles or free drugs combination. Higher cellular uptake for the C/DSPE-PEG/D-V nanomicelles was associated with disrupting the microtubule cytoarchitecture triggered via VIN due to downregulation of tubulin expression. In vivo pharmacokinetic examination executed in healthy BALB/C mice depicted a prolonged blood residence time for both drugs in case of i.v. administration of targeted dual loaded-nanomicelles when compared to free drugs. Additionally, the targeting potential of the prepared targeted-nanomicelles was examined in vivo by quantifying DOX fluorescence distribution in various body organs in breast cancer lung metastatic BALB/C mice. Results revealed good accumulation in metastatic site in case of C/DSPE-PEG/D-V nanomicelles when compared to all other groups.
Mannose receptors (MRs) are multifunctional endocytic receptors related to C-type lectin family. C-type lectins are the immune receptors responsible for the discrimination of glycan architectures related to pathogens and mutated cells.129 C-type lectins are important owing to their integration in cell proliferation, immune response and programmed cell death. These receptors have exterior domains allowing recognition of multiple glycoconjugate moieties. They can trigger both endocytosis and phagocytosis as well as assisting clearance of soluble and particulate ligands. Upon internalization, ligands are detached from the receptor and the receptor returns to the cell exterior.130 This lectin/carbohydrate binding can be exploited via the development of PMs functionalized with carbohydrate ligands that can bind to overexpressed lectin receptors on the exterior of the target cell, so their targeting can serve as a strategy to manage multiple infections and cancers.131 Based on these premises, Negrete and his co-workers constructed a dual sugar functionalized nanomicelles for directed delivery of Sorafenib (SFB) against HCC.132 Fabricated nanomicelles were designed to target both MRs and ASGPRs via surface functionalization with mannose and galactose, respectively. The delivery system was comprised form polydiacetylene nanomicelles modified with mannose and galactose via simple mixing to form Man/P and Gal/P nanomicelles. Targeted nanomicelles were loaded with SFB under stirring to form Man/P-SFB and Gal/P-SFB nanomicelles. The targeted nanomicelles displayed a controlled in vitro release manner for SFB at pH of 7.4. The pro-apoptotic and anti-proliferative activity of the prepared nanomicelles were measured and results revealed high caspase-3 production and lower cell proliferation in HepG2 cells when treated with Man/P-SFB nanomicelles in comparison to Gal/P-SFB nanomicelles, which could be attributed to the fact that the MR expression by HepG2 cells is relatively higher than ASGPR expression. In vitro cytotoxicity study emphasized that both delivery systems are eligible to internalize into HepG2 cells via endocytosis, liberating their encapsulated payload in the cytoplasm and initiating cellular apoptotic responses with better results in case of nanomicelles targeting MRs.
Vitamin D receptor (vit.DR) is a member of ligand activated nuclear receptor family, which is activated after binding with vitamin D3 (active form).133 It was recognized as one of the remarkable transcription factors employed in P-gp regulation.134 Overexpression of vit.DR on the external layer of malignant cells was previously reported.135,136 Exploiting vit.DR as a therapeutic target for cancer therapy is not very common, unlike other highly exploited receptors involving AR (androgen receptors), ER (estrogen receptors), PR (progesterone receptors), HER-2 receptors, and EGFRs.137 In order to further explore targeting this receptor in cancer therapy, our research team developed targeted nanomicelles to co-deliver combinatorial tumor therapy against breast cancer via exploiting overexpressed vit-DR.138 Nanomicelles were synthesized via solubilizing sodium caseinate (Na CAS) protein in aqueous medium at a concentration above its critical micelle concentration, followed by co-loading with etoposide (ETP) and Vit.D3 using solvent evaporation method to form CAS-ETP-D3. Vit-D3/phosphatidylcholine complex was then used to form an outer coat on the micellar surface via thin film hydration method to give Vit.D3/CAS-ETP-Vit.D3 nanomicelles. Both ETP and vit.D3 displayed controlled release manner from the constructed targeted nanomicelles at pH of 7.4 without any notable initial burst release. Fabricated targeted nanomicelles depicted very low hemolytic % on contrary to free combinational drugs. In vitro anticancer study elucidated that D3/CAS-ETP-D3 nanomicelles exhibited better activity against MDA MB-231 and MCF-7 cells than free drugs. Moreover, the sensitivity of MCF-7 cells towards the targeted nanomicelles was higher than that of MDA MB-231 cancer cells owing to higher expression of vit.DR on MCF-7 cancer cells. Investigation of in vivo biodistrbution study demonstrated 4.3 fold higher tumor accumulation of Vit.D3/CAS-rhodamine B nanomicelles following i.v. administration into tumor-bearing mice in comparison to other organs. Moreover, in vivo anticancer efficacy study demonstrated the best antitumor effect in case of Vit.D3/CAS-ETP-D3 nanomicelles in terms of reduced tumor invasion, angiogenesis, autophagy and augmented apoptosis, besides downregulation of miR-21 and miR-192.
Protease/activated receptors-2 (PARs-2) are members of G protein-coupled receptors that are stimulated via proteolytic splitting of the amino terminus and hence, they can work as sensors for extracellular proteases.139 Elevated expression of PARs-2 has been previously reported in advanced stages of many malignancies such as ovarian, gastric, breast and prostate cancers. Moreover, its overexpression is usually linked to the stage of invasiveness displayed by primary and metastatic cancers.140 Previous studies showed that PARs-2 receptors are also greatly expressed on the exterior of fibroblast cell membranes.141–143 Considering these advantages, targeting fibroblast cells via exploiting PARs-2 can enable specific therapeutic delivery and localization for better wound healing. Lee and his co-workers developed a smart fibroblast targeting PMs via binding overexpressed PARs-2 for better wound healing performance.144 Targeted PMs comprised amphiphilic PEO-b-PCL followed by functionalization with KTTKS; a collagen-derived peptide via thiol–maleimide conjugation technique22 to form KTTKS/PEO-b-PCL nanomicelles. The inner core of the targeted micelles was loaded with CUR through hydrophobic interaction to give KTTKS/PEO-b-PCL-CUR nanomicelles. The loaded CUR displayed controlled release manner from the constructed targeted nanomicelles at pH of 7.4. In vitro cellular uptake studies demonstrated two fold higher accumulation for the peptide-decorated nanomicelles via receptor-mediated endocytosis when incubated with HDF (fibroblast) cells in comparison to SK-MEL-28 (melanocyte) cells and HaCaT (keratinocyte) cells, which resulted in reinforced CUR effect. Moreover, HDF cells exhibited enhanced collagen manufacturing, and lower expression of MMP-2 and 9 when incubated with KTTKS/PEO-b-PCL-CUR nanomicelles. In vitro scratch assay revealed a higher migration rate to the scratched region for HDF cells treated with KTTKS/PEO-b-PCL-CUR nanomicelles, followed by complete closure to the scratched region unlike free CUR and PEO-b-PCL-CUR nanomicelles, which showed lower migration rate. In vivo wound healing study showed faster and complete wound closure for mice topically applying KTTKS/PEO-b-PCL-CUR nanomicelles, associated with efficient tissue reepithelization and high mature collagen deposition at the healed skin area (Table 6).
| PMs | Drugs | Targeting ligand | Grafting mode | Target organ | Receptor | Key consequences | Ref. |
|---|---|---|---|---|---|---|---|
| PASP-b-PNIPAAm | MTX | anti-MUC 16 antibody | EDC conjugation reaction | Ovary | MUC 16 | Improved in vivo activity associated with reduced metastasis | 124 |
| DSPE-PEG | DOX, | CREKA peptide | Simple mixing | Breast | FN | Superior in vivo therapeutic potential in eradicating metastatic tumor | 128 |
| VIN | |||||||
| Polydiacetylene | SFB | Mannose & galactose | Simple mixing | — | MRs, ASGPR | Better in vitro results for nanomicelles targeting MRs | 132 |
| Na CAS | ETP | Vit-D3 | Simple mixing | Breast | Vit.DR | Outstanding in vivo activity without toxicity via exploiting Vit.DR | 138 |
| Vit-D3 | |||||||
| PEO-b-PCL | CUR | KTTKS peptide | Thiol–maleimide conjugation reaction | Skin | PARs-2 | Excellent in vivo wound healing via targeting PAR-2 receptors | 144 |
3 Conclusion and future perspectives
Fabrication of novel delivery systems has been vastly investigated for managing various diseases with special emphasis on actively targeted nanocarriers. As a result, researchers are usually seeking a delivery nanoplatform with high efficiency and minimal toxicity to fulfill an optimal therapeutic potential suitable for human use. Diverse delivery systems have shown great success in the delivery of therapeutics to the diseased sites. Compared to other delivery systems, self-assembled polymeric micelles have gained special concern owing to their unique properties such as ease of construction, small size, high loading capacity, prolonged blood circulation, enhanced pharmacokinetics for the loaded therapeutics, biocompatibility and low toxicity. They are usually composed of two segments, hydrophilic and hydrophobic segments that can form an amphiphilic copolymer, which can easily self-assemble in aqueous environment. The hydrophilic segment composes the exterior shell and the hydrophobic segment forms the interior core, in which hydrophobic molecules can be physically entrapped or chemically conjugated to the micellar core. Another privilege of polymeric micelles is the ease of surface functionalization with varied ligands to actively bind various diseased cells. Despite the high potential for polymeric micelles as delivery systems, there are some issues that need to be handled. Minimizing the toxicity of some utilized copolymers and overcoming the off-target drug release from the micellar system are still a huge challenge to achieve superior therapeutic potential, so attempts are put forward to develop smart targeted micellar systems. The utility of receptor-mediated therapeutics can overcome obstacles linked to drug resistance, low drug bioavailability, fast renal clearance and non-selective distribution. Recently, numerous receptor-targeted micelles have been constructed for drug delivery via attaching one or more ligand to the nanomicellar surface so as to direct the encapsulated drugs into the target site of interest.Preclinical studies in different disease models have shown great promise for ligands-functionalized polymeric micelles in enhancing the therapeutic potential via improvingdrug accumulation, facilitating cellular internalization and minimizing toxicity. Notably, two active targeting polymeric nanomicellar formulations have been transferred to the clinical appraisal of patients with prostate and liver tumors named BIND-014 and PK2, respectively. We are expecting that in the following years, more active targeting polymeric micelles will enter various phases of clinical trials for managing many diseases. However, more efforts should be oriented toward the construction of smart targeting polymeric micelles decorated with multiple ligands to exploit multiple receptors on the cellular surface for more precise targeting.
List of abbreviations
| AB | Antibodies |
| Fabs | Antibody fragments |
| ASGPR | Asialoglycoprotein receptors |
| Ang-2 | Angiopep-2 |
| BBB | Blood brain barrier |
| β-CD | β-Cyclodextrin |
| CD44 | Cell surface adhesion |
| Cet | Cetuximab |
| CEL | Celecoxib |
| CCK-8 | Cell counting kit-8 |
| CLSM | Confocal laser scanning microscope |
| Calcein-AM | Calcein-acetoxymethyl |
| CHI | Chitosan |
| CPT | Camptothecin |
| CRC | Colorectal malignant cells |
| CUR | Curcumin |
| DCC | N,N′-dicyclohexane carbodiimide |
| DOX | Doxorubicin |
| DTX | Docetaxel |
| DCFH | 2′-7′Dichlorofluorescin |
| DCFH-DA | 2′-7′Dichlorofluorescin diacetate |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EPR | Enhanced permeability and retention |
| EGFR | Epidermal growth factor receptors |
| EphA2 | Ephrin type-A receptor 2 |
| ECM | Extracellular matrix |
| FA | Folic acid |
| FC | Flow cytometry |
| FITC | Fluorescein isothiocyanate |
| FN | Fibronectin |
| FRS | Folate receptors |
| GA | Glycyrrhetinic acid |
| GRs | Glycyrrhetinic acid receptors |
| GLUT1 | Glucose transporter-1 receptors |
| 5-FUA | 5-Fluorouracil |
| GSH | Glutathione |
| Gal | Galactose |
| GLU | Glucose |
| HA | Hyaluronic acid |
| HER-2 | Human epidermal growth factor receptors |
| H&E | Hematoxylin and eosin |
| HCC | Hepatocellular carcinoma |
| I.V | Intra venous |
| ICP-MS | Inductively coupled plasma mass spectroscopy |
| kDa | Kilo Dalton |
| LA | Lactobionic acid |
| LSECs | Liver sinusoidal endothelial cells |
| MDR | Multidrug resistance |
| MRI | Magnetic resonance imaging |
| MPEG | Methoxy poly(ethylene glycol) |
| MTX | Methotrexate |
| MSNs | Mesoporous silica nanoparticles |
| MR | Mannose receptor |
| MUC16 | Mucin 16 |
| MF-HSCs | Myofibroblastic hepatic stellate cells |
| NHS | N-hydroxysuccinimide |
| NPs | Nanoparticles |
| NIR | Near infra red |
| PMs | Polymeric nanomicelles |
| PDT | Photodynamic therapy |
| Pheo a | Pheophorbide a |
| PTX | Paclitaxel |
| PI | Propidium iodide |
| PCL | Poly(ε-caprolactone) |
| PAsp | Poly/aspartic acid |
| PCAD | Poly carboxylic acid dextran |
| PLL | Poly-L-lysine |
| PLA | Poly(DL-lactic acid) |
| PEI | Polyethylenimine |
| PEO | Polyethylene oxide |
| RAFT | Reversible addition–fragmentation chain transfer |
| PARs-2 | Protease/activated receptors-2 |
| ROS | Reactive oxygen species |
| SPIONs | Superparamagnetic iron oxide nanoparticles |
| TRQ | Tariquidar |
| TFR | Transferrin receptors |
| T | Trastuzumab |
| TF | Transferrin |
| TMC | Trimethylene carbonate |
| vit.D3R | Vitamin D3 receptor |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Mona M. Agwa: conceptualization; investigation; supervision; validation; visualization; writing – original draft; writing – review & editing. Rehab Elsayed Marzouk: conceptualization; investigation; supervision; validation; visualization; writing – original draft; writing – review & editing. Sally A. Sabra: conceptualization; investigation; supervision; validation; visualization; writing – original draft; writing – review & editing.Conflicts of interest
The authors declare that they have no competing interests.References
- E. I. Amer, S. R. Allam, A. Y. Hassan, E. M. El-Fakharany, M. M. Agwa, S. N. Khattab, E. Sheta and M. H. El-Faham, PLoS Neglected Trop. Dis., 2023, 17, e0011776 CrossRef CAS PubMed.
- V. Junnuthula, P. Kolimi, D. Nyavanandi, S. Sampathi, L. K. Vora and S. Dyawanapelly, Pharmaceutics, 2022, 14, 1860 CrossRef CAS PubMed.
- J. Ma, C. Guo, Y. Tang, J. Xiang, S. Chen, J. Wang and H. Liu, J. Colloid Interface Sci., 2007, 312, 390–396 CrossRef CAS PubMed.
- F. Andrade, D. Rafael, M. Vilar-Hernandez, S. Montero, F. Martinez-Trucharte, J. Seras-Franzoso, Z. V. Díaz-Riascos, A. Boullosa, N. Garcia-Aranda and P. Camara-Sanchez, J. Controlled Release, 2021, 331, 198–212 CrossRef CAS PubMed.
- J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed.
- M. M. Agwa and S. A. Sabra, in Polymeric Micelles for Drug Delivery, Elsevier, 2022, pp. 285–314 Search PubMed.
- A. Varela-Moreira, Y. Shi, M. H. Fens, T. Lammers, W. E. Hennink and R. M. Schiffelers, Mater. Chem. Front., 2017, 1, 1485–1501 RSC.
- A. Jabbari, E. Yaghoobi, H. Azizollahi, S. Shojaee, M. Ramezani, M. Alibolandi, K. Abnous and S. M. Taghdisi, Int. J. Pharm., 2022, 611, 121346 CrossRef CAS.
- P. Carmeliet, Nat. Med., 2003, 9, 653–660 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Contr. Release, 2000, 65, 271–284 CrossRef CAS.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug discovery today, 2006, 11, 812–818 CrossRef CAS.
- H. S. Choi, W. Liu, F. Liu, K. Nasr, P. Misra, M. G. Bawendi and J. V. Frangioni, Nat. Nanotechnol., 2010, 5, 42–47 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS.
- M. M. Agwa, H. Elmotasem, R. I. Moustafa, A. S. Abdelsattar, M. S. Mohy-Eldin and M. M. Fouda, Int. J. Biol. Macromol., 2023, 253, 127460 CrossRef CAS PubMed.
- Y. Zhong, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2014, 15, 1955–1969 CrossRef CAS PubMed.
- C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Nano Today, 2012, 7, 467–480 CrossRef CAS.
- M. M. Agwa, H. Elmotasem, H. Elsayed, A. S. Abdelsattar, A. M. Omer, D. T. Gebreel, M. S. Mohy-Eldin and M. M. Fouda, Int. J. Biol. Macromol., 2023, 239, 124294 CrossRef CAS PubMed.
- D. E. Large, J. R. Soucy, J. Hebert and D. T. Auguste, Adv. Ther., 2019, 2, 1800091 CrossRef PubMed.
- P. Ringhieri, S. Mannucci, G. Conti, E. Nicolato, G. Fracasso, P. Marzola, G. Morelli and A. Accardo, Int. J. Nanomed., 2017, 501–514 CrossRef CAS PubMed.
- J. Peng, J. Chen, F. Xie, W. Bao, H. Xu, H. Wang, Y. Xu and Z. Du, Biomaterials, 2019, 222, 119420 CrossRef CAS PubMed.
- B. S. Bolu, B. Golba, A. Sanyal and R. Sanyal, Biomater. Sci., 2020, 8, 2600–2610 RSC.
- M. M. Agwa and S. Sabra, Int. J. Biol. Macromol., 2021, 167, 1527–1543 CrossRef CAS PubMed.
- Y.-J. Kim, J.-H. Ha and Y.-J. Kim, Nanotechnology, 2021, 32, 275101 CrossRef CAS.
- N. N. Bayram, G. T. Ulu, N. A. Abdulhadi, S. Gürdap, İ. A. İşoğlu, Y. Baran and S. D. İşoğlu, Pharmaceutics, 2023, 15, 733 CrossRef CAS PubMed.
- S. Gurdap, N. N. Bayram, I. A. Isoglu and S. Dinçer İşoğlu, ACS Appl. Polym. Mater., 2022, 4, 6303–6311 CrossRef CAS.
- Y.-H. Shih, T.-Y. Luo, P.-F. Chiang, C.-J. Yao, W.-J. Lin, C.-L. Peng and M.-J. Shieh, J. Contr. Release, 2017, 258, 196–207 CrossRef CAS.
- Z. Guo, J. Sui, Y. Li, Q. Wei, C. Wei, L. Xiu, R. Zhu, Y. Sun, J. Hu and J.-L. Li, J. Mater. Chem. B, 2022, 10, 9266–9279 RSC.
- I. M. de Paiva, M. R. Vakili, A. H. Soleimani, S. A. Tabatabaei Dakhili, S. Munira, M. Paladino, G. Martin, F. R. Jirik, D. G. Hall and M. Weinfeld, Mol. Pharm., 2022, 19, 1825–1838 CrossRef CAS PubMed.
- S. Florinas, M. Liu, R. Fleming, L. Van Vlerken-Ysla, J. Ayriss, R. Gilbreth, N. Dimasi, C. Gao, H. Wu and Z.-Q. Xu, Biomacromolecules, 2016, 17, 1818–1833 CrossRef CAS.
- S. Chen, S. Florinas, A. Teitgen, Z.-Q. Xu, C. Gao, H. Wu, K. Kataoka, H. Cabral and R. J. Christie, Sci. Technol. Adv. Mater., 2017, 18, 666–680 CrossRef CAS.
- F. Masood, Mater. Sci. Eng. C, 2016, 60, 569–578 CrossRef CAS PubMed.
- A. M. Master and A. Sen Gupta, Nanomedicine, 2012, 7, 1895–1906 CrossRef CAS PubMed.
- J. Xu, Y. Liu and S.-h. Hsu, Molecules, 2019, 24, 3005 CrossRef CAS.
- S. M. Garg, I. M. Paiva, M. R. Vakili, R. Soudy, K. Agopsowicz, A. H. Soleimani, M. Hitt, K. Kaur and A. Lavasanifar, Biomaterials, 2017, 144, 17–29 CrossRef CAS PubMed.
- T. Xiao, Y. Xiao, W. Wang, Y. Y. Tang, Z. Xiao and M. Su, J. Hematol. Oncol., 2020, 13, 1–17 CrossRef PubMed.
- P. Zhao, D. Jiang, Y. Huang and C. Chen, J. Genet. Genomics, 2021, 48, 261–267 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294–5299 CrossRef CAS.
- A. Harada and K. Kataoka, Soft Matter, 2008, 4, 162–167 RSC.
- N. Dimasi, C. Gao, R. Fleming, R. M. Woods, X.-T. Yao, L. Shirinian, P. A. Kiener and H. Wu, J. Mol. Biol., 2009, 393, 672–692 CrossRef CAS PubMed.
- X. Pang, X. He, Z. Qiu, H. Zhang, R. Xie, Z. Liu, Y. Gu, N. Zhao, Q. Xiang and Y. Cui, Signal Transduct. Targeted Ther., 2023, 8, 1–42 CrossRef CAS PubMed.
- E. Andreucci, K. Bugatti, S. Peppicelli, J. Ruzzolini, M. Lulli, L. Calorini, L. Battistini, F. Zanardi, A. Sartori and F. Bianchini, Int. J. Mol. Sci., 2023, 24, 1475 CrossRef CAS PubMed.
- Y. Lei, S. Chen, X. Zeng, Y. Meng, C. Chang and G. Zheng, J. Appl. Polym. Sci., 2022, 139, 52358 CrossRef CAS.
- F. De Lorenzi, L. Y. Rizzo, R. Daware, A. Motta, M. Baues, M. Bartneck, M. Vogt, M. van Zandvoort, L. Kaps and Q. Hu, Drug Delivery Transl. Res., 2023, 13, 1195–1211 CrossRef CAS PubMed.
- V. Manjusha, L. Reshma and T. Anirudhan, J. Drug Deliv. Sci. Technol., 2023, 79, 104032 CrossRef CAS.
- Y. Cai, Z. Xu, Q. Shuai, F. Zhu, J. Xu, X. Gao and X. Sun, Biomater. Sci., 2020, 8, 2274–2282 RSC.
- S. Sanati, S. Taghavi, K. Abnous, S. M. Taghdisi, M. Babaei, M. Ramezani and M. Alibolandi, Gene Ther., 2022, 29, 55–68 CrossRef CAS PubMed.
- A. Jabbari, E. Yaghoobi, H. Azizollahi, S. Shojaee, M. Ramezani, M. Alibolandi, K. Abnous and S. M. Taghdisi, Int. J. Pharm., 2022, 611, 121346 CrossRef CAS PubMed.
- W. Zou, C. Sarisozen and V. P. Torchilin, J. Drug Target., 2017, 25, 225–234 CrossRef CAS PubMed.
- O. S. Muddineti, P. Kumari, B. Ghosh and S. Biswas, Pharm. Res., 2018, 35, 1–14 CrossRef CAS PubMed.
- P. Sun, Y. Xiao, Q. Di, W. Ma, X. Ma, Q. Wang and W. Chen, Int. J. Nanomed., 2020, 15, 6673–6688 CrossRef CAS PubMed.
- S. Kitagawa, T. Matsuda, A. Washizaki, H. Murakami, T. Yamamoto and Y. Yoshioka, npj Vaccines, 2022, 7, 115 CrossRef CAS PubMed.
- F. Li, J. Lu, J. Liu, C. Liang, M. Wang, L. Wang, D. Li, H. Yao, Q. Zhang and J. Wen, Nat. Commun., 2017, 8, 1390 CrossRef PubMed.
- H. Kawabata, Free Radic. Biol. Med., 2019, 133, 46–54 CrossRef CAS PubMed.
- S. Sabra and M. M. Agwa, Int. J. Biol. Macromol., 2020, 164, 1046–1060 CrossRef CAS PubMed.
- T. R. Daniels, T. Delgado, J. A. Rodriguez, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 144–158 CrossRef CAS PubMed.
- J. Li, Z. Zhang, B. Zhang, X. Yan and K. Fan, Biomater. Sci., 2023, 1–20 Search PubMed.
- M. M. Agwa, D. A. Abdelmonsif, S. N. Khattab and S. Sabra, Int. J. Biol. Macromol., 2020, 162, 246–261 CrossRef CAS PubMed.
- R. D. Dabholkar, R. M. Sawant, D. A. Mongayt, P. V. Devarajan and V. P. Torchilin, Int. J. Pharm., 2006, 315, 148–157 CrossRef CAS PubMed.
- O. S. Muddineti, P. Kumari, B. Ghosh, V. P. Torchilin and S. Biswas, ACS Appl. Mater. Interfaces, 2017, 9, 16778–16792 CrossRef CAS PubMed.
- C. Sarisozen, A. H. Abouzeid and V. P. Torchilin, Eur. J. Pharm. Biopharm., 2014, 88, 539–550 CrossRef CAS PubMed.
- A. A. D'souza and P. V. Devarajan, J. Contr. Release, 2015, 203, 126–139 CrossRef PubMed.
- C. A. Sanhueza, M. M. Baksh, B. Thuma, M. D. Roy, S. Dutta, C. Préville, B. A. Chrunyk, K. Beaumont, R. Dullea and M. Ammirati, J. Am. Chem. Soc., 2017, 139, 3528–3536 CrossRef CAS PubMed.
- Y. Xiang, W. Huang, C. Huang, J. Long, Y. Zhou, Y. Liu, S. Tang, D.-x. He, X.-w. Tan and H. Wei, Mol. Pharm., 2020, 17, 3223–3235 CrossRef CAS PubMed.
- C. Y. Yu, N. M. Li, S. Yang, Q. Ning, C. Huang, W. Huang, Z. N. He, D. X. He, X. W. Tan and L. C. Sun, J. Appl. Polym. Sci., 2015, 132, 1–7 Search PubMed.
- A. Mazumder, A. Dwivedi, W. Assawapanumat, R. Saeeng, W. Sungkarat and N. Nasongkla, Pharm. Dev. Technol., 2022, 27, 379–388 CrossRef CAS PubMed.
- W. Assawapanumat, S. Udomphon, A. Kampaengtip, S. Yaset, X. Han, P. Nittayacharn, M. T. Nieman, C. Chotipanich, W. Sungkarat and P. Sunintaboon, J. Drug Deliv. Sci. Technol., 2023, 79, 104060 CrossRef CAS.
- X. Yang, W. Deng, L. Fu, E. Blanco, J. Gao, D. Quan and X. Shuai, J. Biomed. Mater. Res., Part A, 2008, 86, 48–60 CrossRef PubMed.
- H. Fonge, H. Lee, R. M. Reilly and C. Allen, Mol. Pharm., 2010, 7, 177–186 CrossRef CAS PubMed.
- K. Suzuki, Y. Miura, Y. Mochida, T. Miyazaki, K. Toh, Y. Anraku, V. Melo, X. Liu, T. Ishii and O. Nagano, J. Contr. Release, 2019, 301, 28–41 CrossRef CAS PubMed.
- N. Lecot, R. Glisoni, N. Oddone, J. Benech, M. Fernández, J. P. Gambini, P. Cabral and A. Sosnik, Adv. Ther., 2021, 4, 2000010 CrossRef CAS.
- J. Riedel, M. Pibuel, E. Bernabeu, D. Poodts, M. Díaz, M. Allo, L. Parola, S. Hajos, J. M. Lázaro-Martínez and M. J. Salgueiro, J. Drug Deliv. Sci. Technol., 2022, 68, 103046 CrossRef CAS.
- B. Cao, R. Zhao, H. Li, X. Xu, J. Gao, L. Chen and B. Wei, Int. J. Biol. Sci., 2023, 19, 104 CrossRef CAS PubMed.
- S. Almahmoud, X. Wang, J. L. Vennerstrom and H. A. Zhong, Molecules, 2019, 24, 2159 CrossRef CAS PubMed.
- L. Su, Y. Feng, K. Wei, X. Xu, R. Liu and G. Chen, Chem. Rev., 2021, 121, 10950–11029 CrossRef CAS PubMed.
- Y. Mochida, H. Cabral, Y. Miura, F. Albertini, S. Fukushima, K. Osada, N. Nishiyama and K. Kataoka, ACS Nano, 2014, 8, 6724–6738 CrossRef CAS PubMed.
- E. C. Calvaresi and P. J. Hergenrother, Chem. Sci., 2013, 4, 2319–2333 RSC.
- M. L. Cuestas, R. J. Glisoni, V. L. Mathet and A. Sosnik, J. Nanopart. Res., 2013, 15, 1–21 CrossRef.
- R. J. Glisoni and A. Sosnik, Macromol. Biosci., 2014, 14, 1639–1651 CrossRef CAS PubMed.
- M. A. Moretton, E. Bernabeu, E. Grotz, L. Gonzalez, M. Zubillaga and D. A. Chiappetta, Eur. J. Pharm. Biopharm., 2017, 114, 305–316 CrossRef CAS PubMed.
- K. Byagari, A. Shanavas, A. Rengan, G. Kundu and R. Srivastava, J. Biomed. Nanotechnol., 2014, 10, 109–119 CrossRef CAS PubMed.
- J. Zhang, L. Wang, H. Fai Chan, W. Xie, S. Chen, C. He, Y. Wang and M. Chen, Sci. Rep., 2017, 7, 46057 CrossRef CAS PubMed.
- W.-C. Ma, Q. Zhang, H. Li, C. A. Larregieu, N. Zhang, T. Chu, H. Jin and S.-J. Mao, Int. J. Pharm., 2013, 450, 21–30 CrossRef CAS PubMed.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Contr. Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- N. Kang, M.-È. Perron, R. E. Prud'Homme, Y. Zhang, G. Gaucher and J.-C. Leroux, Nano Lett., 2005, 5, 315–319 CrossRef CAS PubMed.
- L. E. Kelemen, Int. J. Cancer Res., 2006, 119, 243–250 CrossRef CAS PubMed.
- C. Yue, P. Liu, M. Zheng, P. Zhao, Y. Wang, Y. Ma and L. Cai, Biomaterials, 2013, 34, 6853–6861 CrossRef CAS PubMed.
- N. Rarokar, R. Agrawal, S. Yadav, P. Khedekar, C. Ravikumar, D. Telange and S. Gurav, J. Mol. Liq., 2023, 369, 120842 CrossRef CAS.
- W. Tao, J. Wang, Y. Zhou, Z. Liu, H. Chen, Z. Zhao, H. Yan and X. Liao, Colloids Surf., B, 2023, 222, 113084 CrossRef CAS PubMed.
- O. Khomich, A. V. Ivanov and B. Bartosch, Cells, 2019, 9, 24 CrossRef PubMed.
- D. Ezhilararasan, T. Lakshmi and B. Raut, J. Nanomater., 2021, 2021, 1–9 CrossRef.
- S.-L. Du, H. Pan, W.-Y. Lu, J. Wang, J. Wu and J.-Y. Wang, J. Pharmacol. Exp. Ther., 2007, 322, 560–568 CrossRef CAS PubMed.
- K. S. Kim, W. Hur, S.-J. Park, S. W. Hong, J. E. Choi, E. J. Goh, S. K. Yoon and S. K. Hahn, ACS Nano, 2010, 4, 3005–3014 CrossRef CAS PubMed.
- Q.-Q. Fan, C.-L. Zhang, J.-B. Qiao, P.-F. Cui, L. Xing, Y.-K. Oh and H.-L. Jiang, Biomaterials, 2020, 230, 119616 CrossRef CAS PubMed.
- Z. Chen, A. Jain, H. Liu, Z. Zhao and K. Cheng, J. Pharmacol. Exp. Ther., 2019, 370, 695–702 CrossRef CAS PubMed.
- L. Xu, Q. Bai, X. Zhang and H. Yang, J. Contr. Release, 2017, 252, 73–82 CrossRef CAS PubMed.
- L. D. DeLeve, Hepatology, 2015, 61, 1740–1746 CrossRef CAS PubMed.
- L. Xiang, X. Wang, Y. Shao, Q. Jiao, J. Cheng, X. Zheng, S. Zhou and Y. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 2030–2042 CrossRef CAS PubMed.
- S. Milewska, G. Siemiaszko, A. Z. Wilczewska, I. Misztalewska-Turkowicz, K. H. Markiewicz, D. Szymczuk, D. Sawicka, H. Car, R. Lazny and K. Niemirowicz-Laskowska, Int. J. Mol. Sci., 2023, 24, 1364 CrossRef CAS PubMed.
- Z. Cao, R. Liu, Y. Li, X. Luo, Z. Hua, X. Wang, Z. Xue, Z. Zhang, C. Lu and A. Lu, Breast Cancer Res., 2023, 25, 1–15 CrossRef PubMed.
- X. Zhang, X. Xu, X. Wang, Y. Lin, Y. Zheng, W. Xu, J. Liu and W. Xu, Carbohydr. Polym., 2023, 303, 120439 CrossRef CAS PubMed.
- Z.-p. Li, G.-x. Tian, H. Jiang, R.-y. Pan, B. Lian, M. Wang, Z.-q. Gao, B. Zhang and J.-l. Wu, Int. J. Nanomed., 2019, 14, 9437–9452 CrossRef CAS PubMed.
- Y. Zheng, S. Shi, Y. Liu, Y. Zhao and Y. Sun, J. Appl. Biomater., 2019, 34, 141–151 CrossRef CAS PubMed.
- J. Prakash, L. Beljaars, A. K. Harapanahalli, M. Zeinstra-Smith, A. de Jager-Krikken, M. Hessing, H. Steen and K. Poelstra, Int. J. Cancer, 2010, 126, 1966–1981 CrossRef CAS PubMed.
- T.-H. Ying, J.-H. Tsai, T.-T. Wu, M.-T. Tsai, W.-W. Su, Y.-S. Hsieh and J.-Y. Liu, Chin. J. Physiol., 2008, 51, 269–274 CAS.
- V. B. Lokeshwar, S. Mirza and A. Jordan, Adv. Cancer Res., 2014, 123, 35–65 CrossRef PubMed.
- D.-K. Lim, R. G. Wylie, R. Langer and D. S. Kohane, Biomaterials, 2016, 77, 130–138 CrossRef CAS PubMed.
- C. Fu, H. Li, N. Li, X. Miao, M. Xie, W. Du and L.-M. Zhang, Carbohydr. Polym., 2015, 128, 163–170 CrossRef CAS PubMed.
- G. Tian, R. Pan, B. Zhang, M. Qu, B. Lian, H. Jiang, Z. Gao and J. Wu, Front. Pharmacol, 2019, 10, 1–13 CrossRef PubMed.
- K. A. Iczkowski, Am. J. Transl. Res., 2011, 3, 1–7 CAS.
- L. T. Senbanjo and M. A. Chellaiah, Front. Cell Dev. Biol., 2017, 5, 18 Search PubMed.
- M. M. Agwa, H. Elmotasem, H. Elsayed, A. S. Abdelsattar, A. M. Omer, D. T. Gebreel, M. S. Mohy-Eldin and M. M. Fouda, Int. J. Biol. Macromol., 2023, 239, 124294 CrossRef CAS PubMed.
- H. Wang, Y. Zhang, Y. Liu, Y. Ren, J. Wang, B. Niu and W. Li, Eur. Polym. J., 2022, 177, 111450 CrossRef CAS.
- M. Li, Y. Zhao, J. Sun, H. Chen, Z. Liu, K. Lin, P. Ma, W. Zhang, Y. Zhen and S. Zhang, Carbohydr. Polym., 2022, 288, 119402 CrossRef CAS PubMed.
- J. Wen, F. Liu, B. Tao and S. Sun, Bioorg. Med. Chem. Lett., 2019, 29, 1019–1022 CrossRef CAS PubMed.
- S. Li, W. Zhao, N. Liang, Y. Xu, Y. Kawashima and S. Sun, React. Funct. Polym., 2020, 152, 104608 CrossRef CAS.
- S. Patel, V. Patel, M. Yadav, D. Panjwani, P. Ahlawat, A. Dharamsi and A. Patel, J. Polym. Res., 2023, 30, 14 CrossRef CAS.
- Y. S. Birhan, E. Y. Hanurry, T. W. Mekonnen, H. F. Darge, Y. H. Lin, M. C. Yang and H. C. Tsai, J. Appl. Polym. Sci., 2022, 139, 52327 CrossRef CAS.
- Y. Xu, N. Liang, J. Liu, X. Gong, P. Yan and S. Sun, Carbohydr. Polym., 2022, 290, 119509 CrossRef CAS PubMed.
- Y. Patil, T. Sadhukha, L. Ma and J. Panyam, J. Contr. Release, 2009, 136, 21–29 CrossRef CAS PubMed.
- X. Zhang, N. Liang, X. Gong, Y. Kawashima, F. Cui and S. Sun, Colloids Surf., B, 2019, 177, 11–18 CrossRef CAS PubMed.
- A. Aithal, S. Rauth, P. Kshirsagar, A. Shah, I. Lakshmanan, W. M. Junker, M. Jain, M. P. Ponnusamy and S. K. Batra, Expert Opin. Ther. Targets, 2018, 22, 675–686 CrossRef CAS PubMed.
- C. Liang, Y. Qin, B. Zhang, S. Ji, S. Shi, W. Xu, J. Liu, J. Xiang, D. Liang and Q. Hu, Mol. Cancer Res., 2017, 15, 201–212 CrossRef CAS PubMed.
- S. Das and S. K. Batra, Cancer Res., 2015, 75, 4669–4674 CrossRef CAS PubMed.
- J. M. Pantshwa, K. Rhoda, S. J. Clift, P. Pradeep, Y. E. Choonara, P. Kumar, L. C. Du Toit, C. Penny and V. Pillay, Int. J. Mol. Sci., 2018, 19, 3030 CrossRef PubMed.
- Y. Tamada, H. Takeuchi, N. Suzuki, N. Susumu, D. Aoki and T. Irimura, Cancer Sci., 2007, 98, 1586–1591 CrossRef CAS PubMed.
- M. Jeon, J. Lee, S. J. Nam, I. Shin, J. Lee and S. Kim, Exp. Cell Res., 2015, 333, 116–126 CrossRef CAS PubMed.
- A. Vaidya, H. Wang, V. Qian, H. Gilmore and Z.-R. Lu, Cells, 2020, 9, 1826 CrossRef CAS PubMed.
- Z. Gong, M. Chen, Q. Ren, X. Yue and Z. Dai, Signal Transduct. Targeted Ther., 2020, 5, 1–11 CrossRef PubMed.
- J. M. Galdopórpora, C. Martinena, E. Bernabeu, J. Riedel, L. Palmas, I. Castangia, M. L. Manca, M. Garcés, J. Lázaro-Martinez and M. J. Salgueiro, Pharmaceutics, 2022, 14, 959 CrossRef PubMed.
- S. A. Torres-Pérez, C. E. Torres-Pérez, M. Pedraza-Escalona, S. M. Pérez-Tapia and E. Ramón-Gallegos, Front. Oncol., 2020, 10, 605037 CrossRef PubMed.
- J. M. Irache, H. H. Salman, C. Gamazo and S. Espuelas, Expert Opin. Drug Delivery, 2008, 5, 703–724 CrossRef CAS PubMed.
- M. Negrete, E. Romero-Ben, A. Gutiérrez-Valencia, C. Rosales-Barrios, E. Alés, T. Mena-Barragán, J. A. Flores, M. C. Castillejos, P. de la Cruz-Ojeda and E. Navarro-Villarán, ACS Appl. Bio Mater., 2021, 4, 4789–4799 CrossRef CAS PubMed.
- B. P. Kota, J. D. Allen and B. D. Roufogalis, Basic Clin. Pharmacol. Toxicol., 2011, 109, 97–102 CrossRef CAS PubMed.
- S. Tachibana, K. Yoshinari, T. Chikada, T. Toriyabe, K. Nagata and Y. Yamazoe, Drug Metab. Dispos., 2009, 37, 1604–1610 CrossRef CAS PubMed.
- M. J. Duffy, A. Murray, N. C. Synnott, N. O'Donovan and J. Crown, Crit. Rev. Oncol. Hematol., 2017, 112, 190–197 CrossRef PubMed.
- J. Thorne and M. J. Campbell, Proc. Nutr. Soc., 2008, 67, 115–127 CrossRef CAS PubMed.
- A. Murray, S. F. Madden, N. C. Synnott, R. Klinger, D. O'Connor, N. O'Donovan, W. Gallagher, J. Crown and M. J. Duffy, Endocr. Relat. Cancer, 2017, 24, 181–195 CAS.
- M. M. Agwa, M. M. Abu-Serie, D. A. Abdelmonsif, N. Moussa, H. Elsayed, S. N. Khattab and S. Sabra, Int. J. Pharm., 2021, 607, 120965 CrossRef CAS PubMed.
- A. S. Rothmeier and W. Ruf, Semin. Immunopathol., 2012, 34, 133–149 CrossRef CAS PubMed.
- N. R. Pawar, M. S. Buzza and T. M. Antalis, Cancer Res., 2019, 79, 301–310 CrossRef CAS PubMed.
- Y. Lee, M. Kim, I. D. Kong, J. Ryu, M. Jang, C. Lee and E. H. Choi, Korean J. Dermatol., 2010, 48, 966–974 Search PubMed.
- N. Atrux-Tallau, T. Delmas, S. H. Han, J. W. Kim and J. Bibette, Int. J. Cosmet. Sci., 2013, 35, 310–318 CrossRef CAS PubMed.
- J. H. Cho, J. Y. Kang, S. Kim, H. R. Baek, J. Kim, K.-S. Jang and J. W. Kim, J. Mater. Chem. B, 2021, 9, 4956–4962 RSC.
- Y. Lee, S. Kim, J. Seo, H. K. Kim, Y. P. Han, E. J. Park, J. O. Park, C.-S. Yang and J. W. Kim, Biomater. Sci., 2023, 11, 450–460 RSC.
| This journal is © The Royal Society of Chemistry 2024 |