
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBaeyer–Villiger oxidation: a promising tool for the synthesis of natural products: a review
Summaya Fatimaa,
Ameer Fawad Zahoor *a,
Samreen Gul Khana,
Syed Ali Raza Naqvia,
Syed Makhdoom Hussainb,
Usman Nazeerc,
Asim Manshaa,
Hamad Ahmadd,
Aijaz Rasool Chaudhrye and
Ahmad Irfanf
*a,
Samreen Gul Khana,
Syed Ali Raza Naqvia,
Syed Makhdoom Hussainb,
Usman Nazeerc,
Asim Manshaa,
Hamad Ahmadd,
Aijaz Rasool Chaudhrye and
Ahmad Irfanf
aDepartment of Chemistry, Government College University Faisalabad, 38000 Faisalabad, Pakistan. E-mail: fawad.zahoor@gcuf.edu.pk
bDepartment of Zoology, Government College University Faisalabad, 38000 Faisalabad, Pakistan
cDepartment of Chemistry, University of Houston, 3585 Cullen Boulevard, Texas 77204-5003, USA
dDepartment of Chemistry, University of Management and Technology, Lahore 54000, Pakistan
eDepartment of Physics, College of Science, University of Bisha, PO Box 551, Bisha 61922, Saudi Arabia
fDepartment of Chemistry, College of Science, King Khalid University, PO Box 9004, Abha 61413, Saudi Arabia
First published on 25th July 2024
Abstract
Baeyer–Villiger oxidation is a well-known reaction utilized for the synthesis of lactones and ester functionalities from ketones. Chiral lactones can be synthesized from chiral or racemic ketones by employing asymmetric Baeyer–Villiger oxidation. These lactones act as key intermediates in the synthesis of most of the biologically active natural products, their analogues, and derivatives. Various monooxygenases and oxidizing agents facilitate BV oxidation, providing a broad range of synthetic applications in organic chemistry. The variety of enzymatic and chemoselective Baeyer–Villiger oxidations and their substantial role in the synthesis of natural products i.e., alkaloids, polyketides, fatty acids, terpenoids, etc. (reported since 2018) have been summarized in this review article.
1. Introduction
The Baeyer–Villiger oxidation reaction is a primitive conversion of aliphatic or cyclic ketones to their corresponding esters and lactones.1 The conversion is carried out in the presence of peracids i.e., per sulfuric acid, perchloric acid, meta-chloroperoxybenzoic acid (m-CPBA), and hydrogen peroxide under acidic conditions.2 In 1899, Adolf Von Baeyer3 with his student, Victor Villiger accomplished the renowned BVO reaction for the first time by using Caro's acid (potassium mono persulfate: KHSO5) as a novel oxidant to convert cyclic ketones into respective lactones with 50% yield.4 Its conversion into a catalytic process was achieved in 1993 by the use of Pt(II) complexes.5In 1950, three different possible intermediates related to Baeyer–Villiger oxidation reaction were distinguished by Speers and Doering.6 The first intermediate (dioxirane) was proposed by Baeyer and Villiger while the second was carbonyl oxide7 and the third one was “Criegee intermediate”.8,9 Doering and Dorfman10 in 1953, elucidated the mechanism of Baeyer–Villiger oxidation. They illustrated that the reaction occurs by the insertion of an oxygen atom between a carbonyl carbon and an adjacent carbon atom in ketone. As a result, an ester or lactone is generated through the formation of a “Criegee intermediate” as shown in Scheme 1.
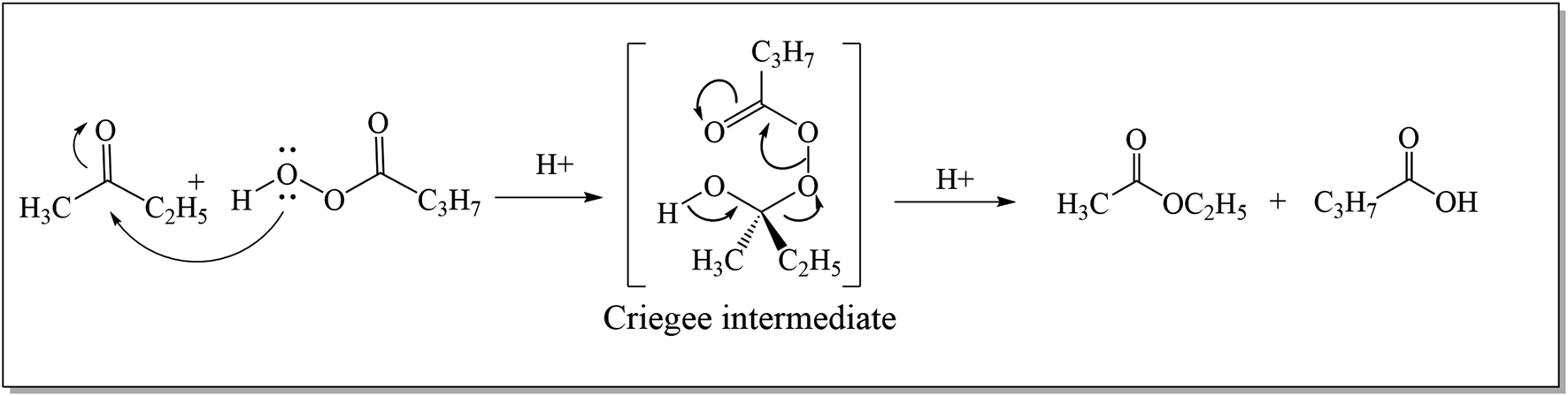 | ||
| Scheme 1 General mechanism of Baeyer–Villiger oxidation. | ||
In 1994, Bolm11 and Strukul12 individually demonstrated that there is a probability to obtain specific stereoselectivity of Criegee intermediate by applying the combination of two different solvents and catalysts (giving 92% and 58% ee values).1
Reagents used for the Baeyer–Villiger oxidation are m-CPBA, peracetic acid, perfluoro acetic acids, H2O2/protic acid, H2O2/Lewis acid, and H2O2/base systems.7 It has also been reported by Murahashi in 1992 that when an aldehyde and molecular oxygen are added, peroxides are produced in situ. This drawback was eliminated by Kaneda in 1994 by utilizing Fe-based catalysts.13 Fe catalysts are favorable because they are less harmful and easily available. Nowadays, efficient transformations can be carried out with high enantioselectivities by using chiral metal complexes14–16 (transition metals17 i.e., Co,18 Zr,19–21 Pt,12,22 Hf23 or Cu24 and non-transition metals i.e., Mg,25 Al,26–29 etc.) and optically active enantiopure organo-catalysts.15 Besides this, Baeyer–Villiger oxidation, carried out in the presence of hydrogen peroxide is more convenient due to low cost of H2O2, high content of oxygen, easy handling and production of water as a byproduct.30 Enzymatic BVOs are also environmentally safe in which molecular oxygen is used as an atom-efficient oxidant and water as a solvent for biosynthesis.1 Many BVMOs are also used as biocatalysts, thus contributing to green chemistry approaches.31
Baeyer–Villiger oxidation reaction offers powerful methodologies and synthetic tools for industrial and natural product syntheses. In recent years, an enormous number of natural products have been isolated from insects, plants, and marine sources (invertebrates and sponges) with a wide range of pharmaceutical activities.32–34 BVO reaction is of great interest in organic chemistry due to its diverse implementations in the synthesis of antibiotics, monomers for polymerization,35 pheromones, and steroids3 etc. In the biosynthetic pathway, MtmOIV [a Baeyer Villiger monooxygenase (BVMO)] is also responsible for the synthesis of mithramycin36 (anticancer antibiotic). Potential BVMOs have also a major role in the biosynthesis of aurafurones37 and aflatoxins38 which belong to the class of polyketides.
Taking into consideration the deadly aspects of cancer, several research groups are striving to synthesize and develop effective anti-cancer agents.39,40 Prostaglandins 4 are biologically active hormones (chemical messengers) that have a role in pharmaceuticals synthesis,41 such as latanoprost is a valuable prostaglandin-related drug. Similarly, steroidal lactones are valuable compounds due to their antiandrogenic and anti-cancerous action.42 These suppress the 5α-reductase43 enzyme and therefore are used to treat androgen-dependent ailments i.e., steroidal testololactone is used to treat breast cancer.44 BVO reaction can also be employed within the synthetic approach leading to steroidal lactones. For example, dehydroepiandrosterone (DHEA) is converted to testololactone by using BVMO.45 Similarly, zoapatanol (diterpenoid) 5 is a biologically active molecule obtained by BVO. It naturally occurs in the zoapatle plant (Montanoa tomentosa)46 and is used to treat labor pain.47 Fig. 1 elaborates the structures of some biologically active compounds synthesized via involving BVO reaction.
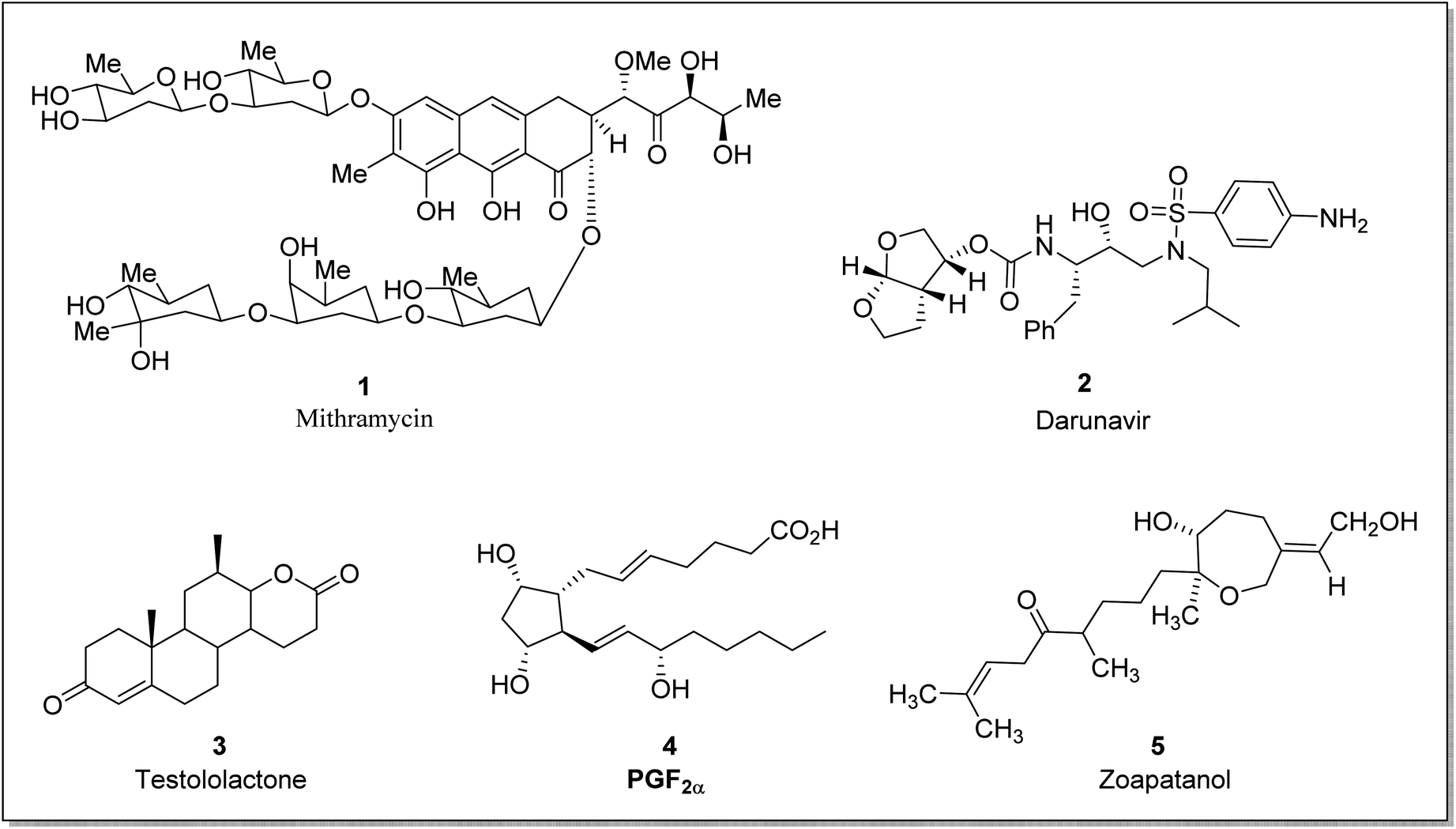 | ||
| Fig. 1 Structures of a few biologically active natural products synthesized by BVO. | ||
Brink et al. in 2004, provided an overview of reaction features, mechanisms and catalysts of BVO reaction by utilizing green reaction conditions.48 Gonzalo et al. in 2021, presented a review on BVMOs-assisted biosynthesis of a variety of compounds.49 In 2016, Bucko et al. also reported different bio-technological approaches towards novel monooxygenase-promoted Baeyer–Villiger oxidation.50 In 2018, Perkel et al. published a review to provide organized information on configuration of esters and lactones synthesized by the Baeyer–Villiger oxidation reaction.51 The current review provides a critical update on the role of Baeyer–Villiger oxidation reaction towards the synthesis of natural products (alkaloids, terpenoids, polyketides, and fatty acids) with high enantio- and stereoselectivity, and efficient yields, reported since 2018.
2. Review of literature
2.1. Synthesis of alkaloid-based natural products via Baeyer–Villiger oxidation
Several natural alkaloids are isolated from herbs and these exhibit antiproliferative, antiviral, antibacterial, insecticidal, and antimetastatic properties against cancer cells. Baeyer–Villiger oxidation reaction plays a key role in the synthesis of alkaloid-based natural compounds i.e., synthesis of homoproaporphine alkaloids, indole-based alkaloids, and other marine alkaloids etc. (Fig. 2.)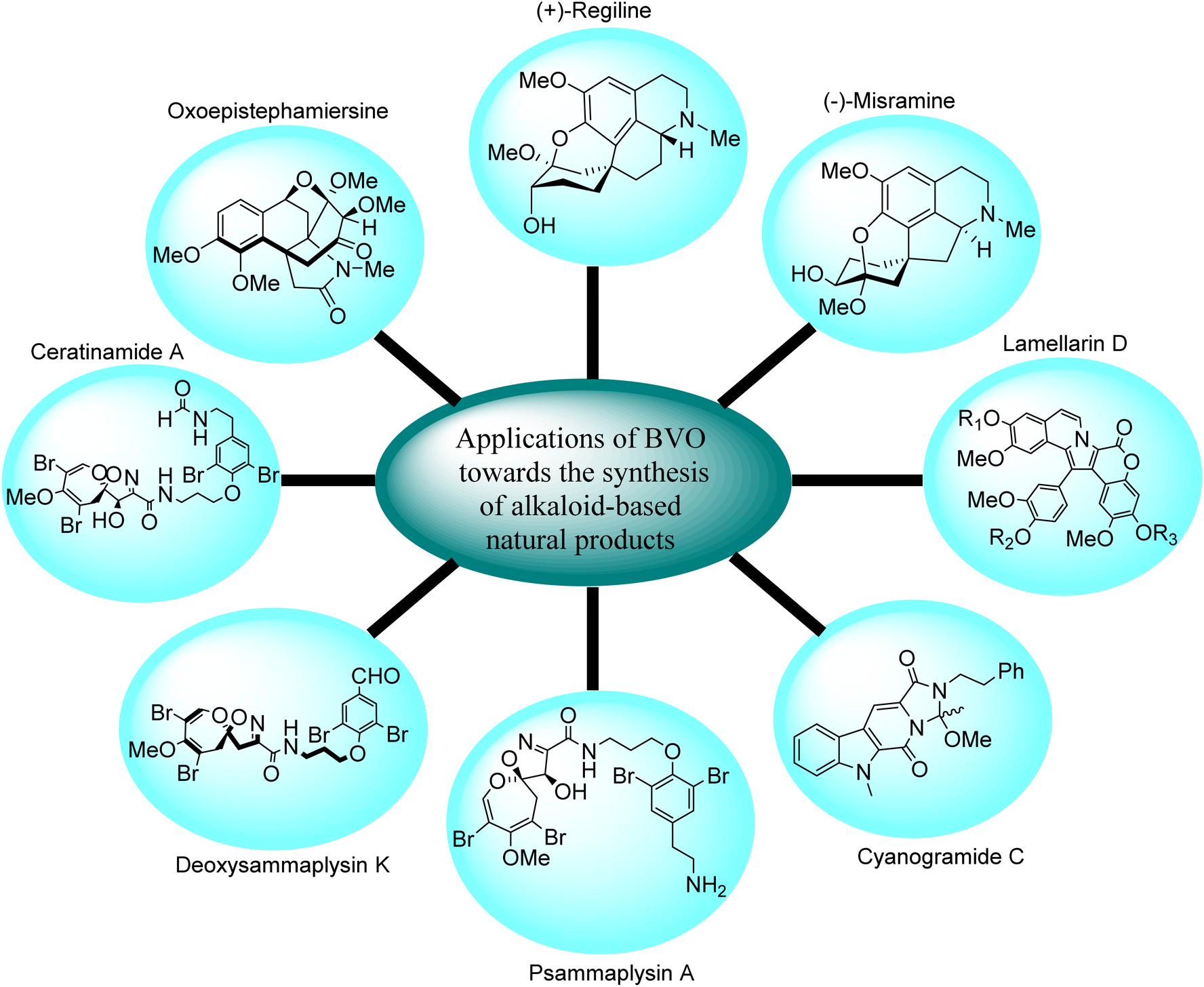 | ||
| Fig. 2 Pictorial representation of alkaloid-based natural products synthesized by Baeyer–Villiger oxidation reaction. | ||
Five-membered heterocyclic compounds are fundamental in medicinal chemistry due to their antifungal, antibacterial, and antiviral properties.52 Homoproaporphine alkaloids (i.e., (+)-regeline, (+)-jolantidine, and (+)-regilinine, etc.) are naturally occurring five-membered heterocyclic compounds that belong to biologically active tetrahydroisoquinones.53 This class consists of over 10 members, some of them exhibit anticholinesterase activity54 and are also used as drugs to prevent the demolition of the neurotransmitter acetylcholine in the nervous system. The synthesis of stereogenic carbons of these alkaloids has always been a challenging task for chemists.55–57 In 1975, the only diastereoselective synthesis (d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :r = 1:1) of regeline was presented by Kametani group in which they employed the oxidative coupling of the phenethylisoquinoline to obtain ring system.58 However, to achieve the first enantioselective total synthesis of homoproaporphine alkaloids in 5.3 to 9.6% overall yield (13 to 16 steps), Pu et al. in 2020, presented the total synthesis by utilizing many fundamental pathways including Baeyer–Villiger oxidation.59 In the first step of synthesis, the chiral intermediate 6 was treated with N-bromo succinimide (NBS) in an acidic medium to afford aryl bromide 7 in 94% yield. In the next step, compound 7 was converted to enol ether 8 over a few steps. Then, compound 8 upon treatment with trifluoroacetic acid (TFA) followed by acylation, generated a chiral ketone 9 in 90% yield. Consequently, ketone 9 underwent m-CPBA-mediated Baeyer–Villiger oxidation in the presence of lithium carbonate in dichloromethane (DCM) to give compound 10 in 92% yield (d:r ∼5.7:1), which upon further reduction with diisobutylaluminium hydride (DIBAL-H) in DCM and consecutive oxidation with pyridinium chlorochromate (PCC) in the presence of DCM yielded aldehyde-lactone 11 in 65% yield. Subsequently, Pinacol-type cyclization of compound 11 with samarium(II)iodide and removal of the N-benzyloxycarbonyl group from compound 12 resulted in the synthesis of (+)-jolantidine 13 in 73% yield. Afterwards, the insertion of a methyl group to hemiketal 12 with CH(OMe)3 and camphorsulfonic acid (CSA) in methanol furnished compound 14 in 85% yield. Compound 14 underwent Swern oxidation followed by LiAlH4-mediated reduction in THF to afford the desired natural compound, regilinine 15 in 66% yield. In another approach, compound (+)-15 on reduction with LiAlH4 in THF furnished (+)-regiline 16 in 69% yield which upon further treatment with boron tribromide (BBr3) in dichloromethane synthesized (+)-kesselridine 17 in 78% yield. To achieve the synthesis of natural product 18 in 86% yield, compound 17 was treated with CH(OMe)3 and CSA in methanol (Scheme 2).
:r = 1:1) of regeline was presented by Kametani group in which they employed the oxidative coupling of the phenethylisoquinoline to obtain ring system.58 However, to achieve the first enantioselective total synthesis of homoproaporphine alkaloids in 5.3 to 9.6% overall yield (13 to 16 steps), Pu et al. in 2020, presented the total synthesis by utilizing many fundamental pathways including Baeyer–Villiger oxidation.59 In the first step of synthesis, the chiral intermediate 6 was treated with N-bromo succinimide (NBS) in an acidic medium to afford aryl bromide 7 in 94% yield. In the next step, compound 7 was converted to enol ether 8 over a few steps. Then, compound 8 upon treatment with trifluoroacetic acid (TFA) followed by acylation, generated a chiral ketone 9 in 90% yield. Consequently, ketone 9 underwent m-CPBA-mediated Baeyer–Villiger oxidation in the presence of lithium carbonate in dichloromethane (DCM) to give compound 10 in 92% yield (d:r ∼5.7:1), which upon further reduction with diisobutylaluminium hydride (DIBAL-H) in DCM and consecutive oxidation with pyridinium chlorochromate (PCC) in the presence of DCM yielded aldehyde-lactone 11 in 65% yield. Subsequently, Pinacol-type cyclization of compound 11 with samarium(II)iodide and removal of the N-benzyloxycarbonyl group from compound 12 resulted in the synthesis of (+)-jolantidine 13 in 73% yield. Afterwards, the insertion of a methyl group to hemiketal 12 with CH(OMe)3 and camphorsulfonic acid (CSA) in methanol furnished compound 14 in 85% yield. Compound 14 underwent Swern oxidation followed by LiAlH4-mediated reduction in THF to afford the desired natural compound, regilinine 15 in 66% yield. In another approach, compound (+)-15 on reduction with LiAlH4 in THF furnished (+)-regiline 16 in 69% yield which upon further treatment with boron tribromide (BBr3) in dichloromethane synthesized (+)-kesselridine 17 in 78% yield. To achieve the synthesis of natural product 18 in 86% yield, compound 17 was treated with CH(OMe)3 and CSA in methanol (Scheme 2).
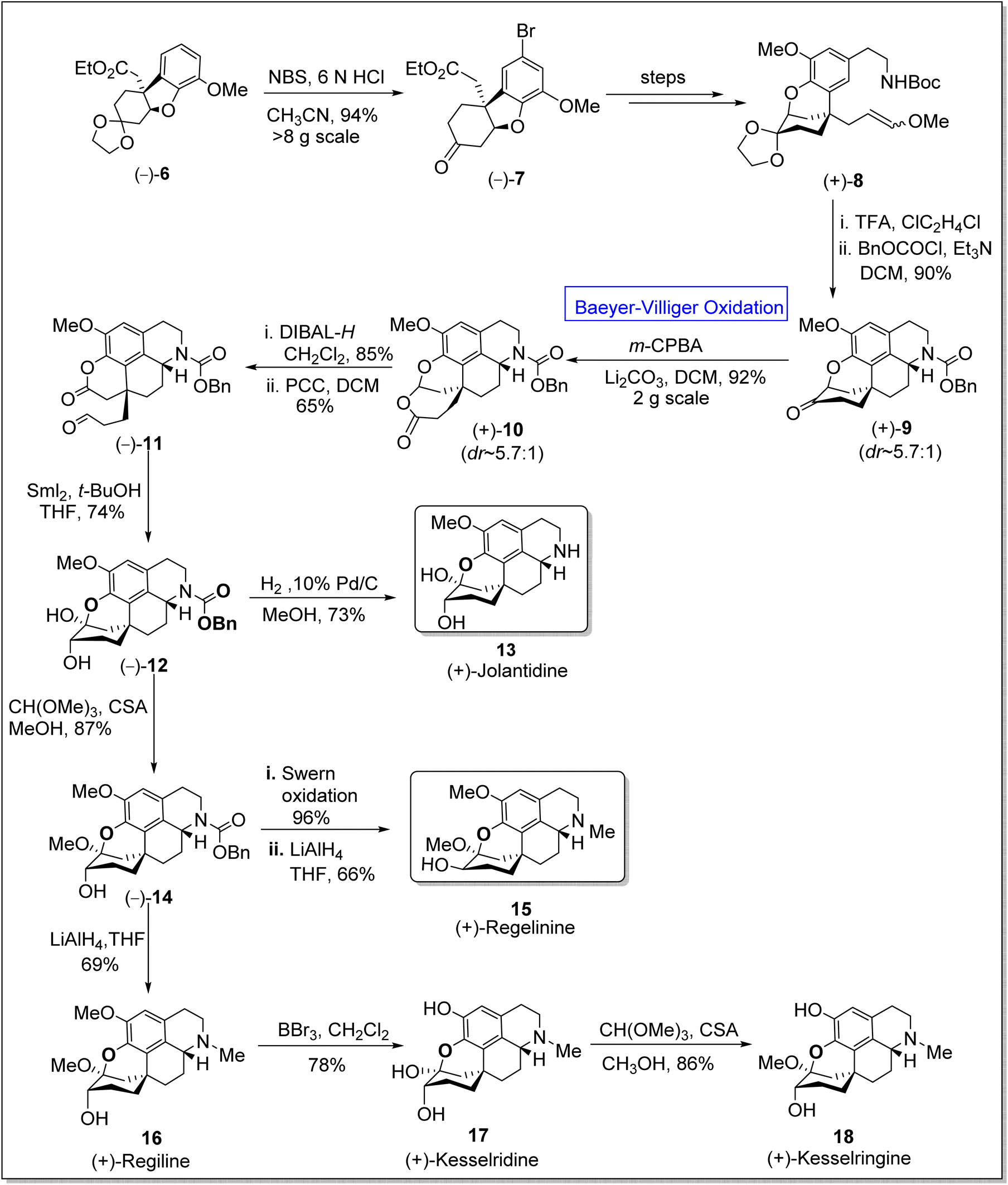 | ||
| Scheme 2 Synthesis of homoproaporphine alkaloids; jolantidine 13, regelinine 15, and kesselringine 18 via Baeyer–Villiger oxidation. | ||
(−)-Misramine 26 is a pentacyclic proaporphine alkaloid,52 a member of isoquinoline alkaloids.60 The former is a biologically active compound and was found in Roemeria hybrida and R. dodecandra.61 In 2018, Yoshida and Takao et al. synthesized the (−)-misramine with overall 2.0% yield (in twenty four steps) from 2,4,6-tribromoanisole by employing asymmetric Friedel–Crafts 1,4-addition. However, Pu et al. in 2021 presented the enantioselective total synthesis of the natural product, (−)-misramine 26 in 11.4% overall yield (in nine steps) by utilizing Baeyer–Villiger oxidation as a significant step.60 The synthesis was commenced with the reaction of chiral intermediate (−)-19 with TFA and methyl chloroformate in DCM to afford a pentacyclic intermediate 20 in 66% yield. Compound 20 underwent Baeyer–Villiger oxidation with m-CPBA in DCM to generate a lactone 21 in 80% yield. An aldehyde-lactone 22 was synthesized from lactone 21 in a sequence of a few steps. Then, the aldehyde-lactone 22 experienced pinacol-type cyclization in the presence of THF to furnish hydroxy ketone 24 in 72% yield instead of compound 23. Methylation of compound 24 with CH(OMe)3 in methanol followed by Swern oxidation with (COCl)2, DMSO, and triethyl amine (NEt3) in DCM produced a ketone 25 in 92% yield. Reduction of compound 25 with LiAlH4 in THF furnished the desired product (−)-26 in 89% yield (Scheme 3).
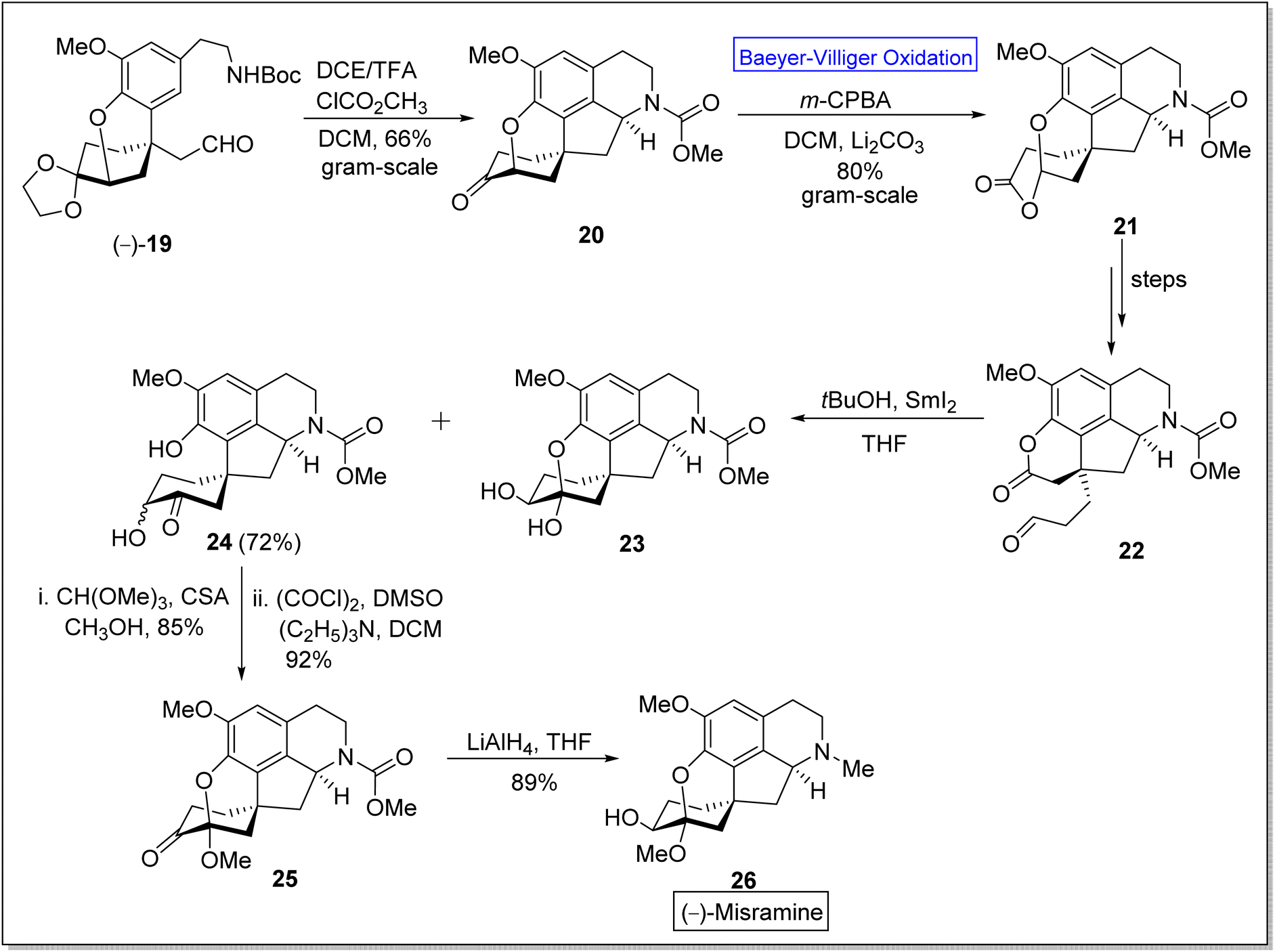 | ||
| Scheme 3 Synthesis of (−)-misramine 26 via Baeyer–Villiger oxidation. | ||
Lamellarins are marine alkaloids isolated from mollusk Lamellaria species.62 Lamellarins show broad range anticancer,63,64 antibacterial,65 antioxidant, and multidrug resistant activities.66,67 The typical derivatives of lamellarin D 31 show different IC50 values i.e., some exhibit 3 nM, 10 nM and 15 nM against fine lines of human lung, colon, and hepatocellular cancer cells respectively. Lamellarin D is the most notable compound owing to its biological potential. Despite the potential activities of lamellarin D 31, its further advancement was restricted due to its poor solubility,68 which is due to the pentacyclic framework of Lamellarin. To improve the solubility of target compound 31, Zheng and colleagues in 2021, reported the synthesis of glycosidic lamellarin derivatives in multiple steps with overall 0.6–2.8% yield by utilizing the Baeyer–Villiger oxidation reaction as a key step.69 The synthesis was accomplished with Baeyer–Villiger oxidation of commercialized isovaniline 27 with H2O2 in the presence of selenium dioxide and DCM to afford an ester 28. In the next step, ester 28 was converted into compound 29 over a few steps. Subsequently, compound 29 was reacted with di-cyclohexyl carbodiimide (DCC) in DCM and then with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in DCM to synthesize the compound 30 which further furnished the derivatives of lamellarin D 31 over a few steps (Scheme 4).
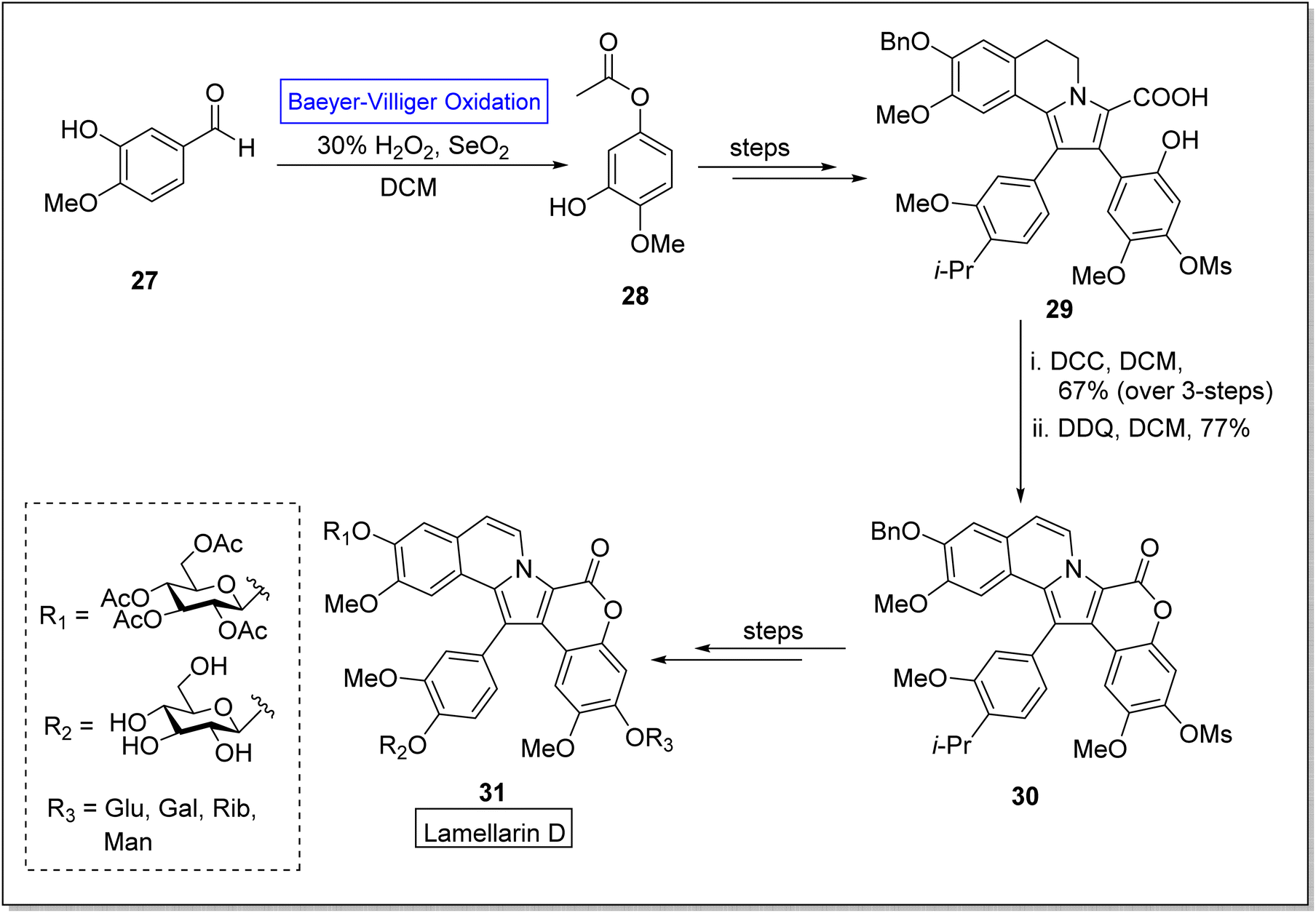 | ||
| Scheme 4 Synthesis of glycosylated derivatives of Lamellarin D 31 via Baeyer–Villiger oxidation. | ||
Cyanogramides (B 38 and C 39) are marine-derived indole-based alkaloids70 which possess pyrrolo[1,2-c] imidazole system. These were isolated by Lian, Zhang, and associates independently.71 These show reverse resistance in the cancer cells.72 In 2018, Sarnes and coworkers attempted to synthesize cyanogramide by employing enantioselective thiourea-mediated spiro-annulation which consisted hydantoin rather than 2-methoxy-2-methylimidazolidinone. This route resulted in failure of ring closure. Later, in 2020, this drawback was addressed by Zhu, Zhang and colleagues by elucidating the biosynthetic process.73 Marinacarboline E 35 is the precursor for the synthesis of cyanogramide B 38 and C 39. Sarnes and companions in 2022, presented the synthesis of marinacarboline E 35, cyanogramides B 38 and C 39 by employing Baeyer–Villiger oxidation as a basic step.70 In the first step of synthesis, L-tryptophan methyl ester 32 was converted into the natural product stellarine C 33 in 61% yield via Pictet–Spengler reaction with an aldehyde in the presence of iodine, p-toluene sulfonic acid and methanol. In the next step, methylation of β-carboline 33 in the presence of NaH, MeI in DMF followed by saponification with LiOH in THF to generate the compound 34 in 90% yield. Then, HATU (hexafluorophosphate azabenzotriazole tetramethyl uranium)-mediated coupling of compound 34 with 2-phenyl ethylamine furnished marinacarboline E 35 (for the first time in 4-steps) in 85% yield. Thereafter, the marinacarboline E 35 was subjected to Baeyer–Villiger oxidation with m-CPBA in the presence of magnesium monoperoxyphthalate (MMPA) to synthesize tricyclic pyridone 36. Subsequently, compound 36 was converted into ketene aminal 37 over a sequence of a few steps. Furthermore, the ketene aminal 37 was treated with p-TsOH and chloroform to afford the cyanogramide B 38 in 80% yield. In the last step, the treatment of natural product 38 with NaH and MeI in DMF furnished the cyanogramide C 39 in 84% yield (Scheme 5).
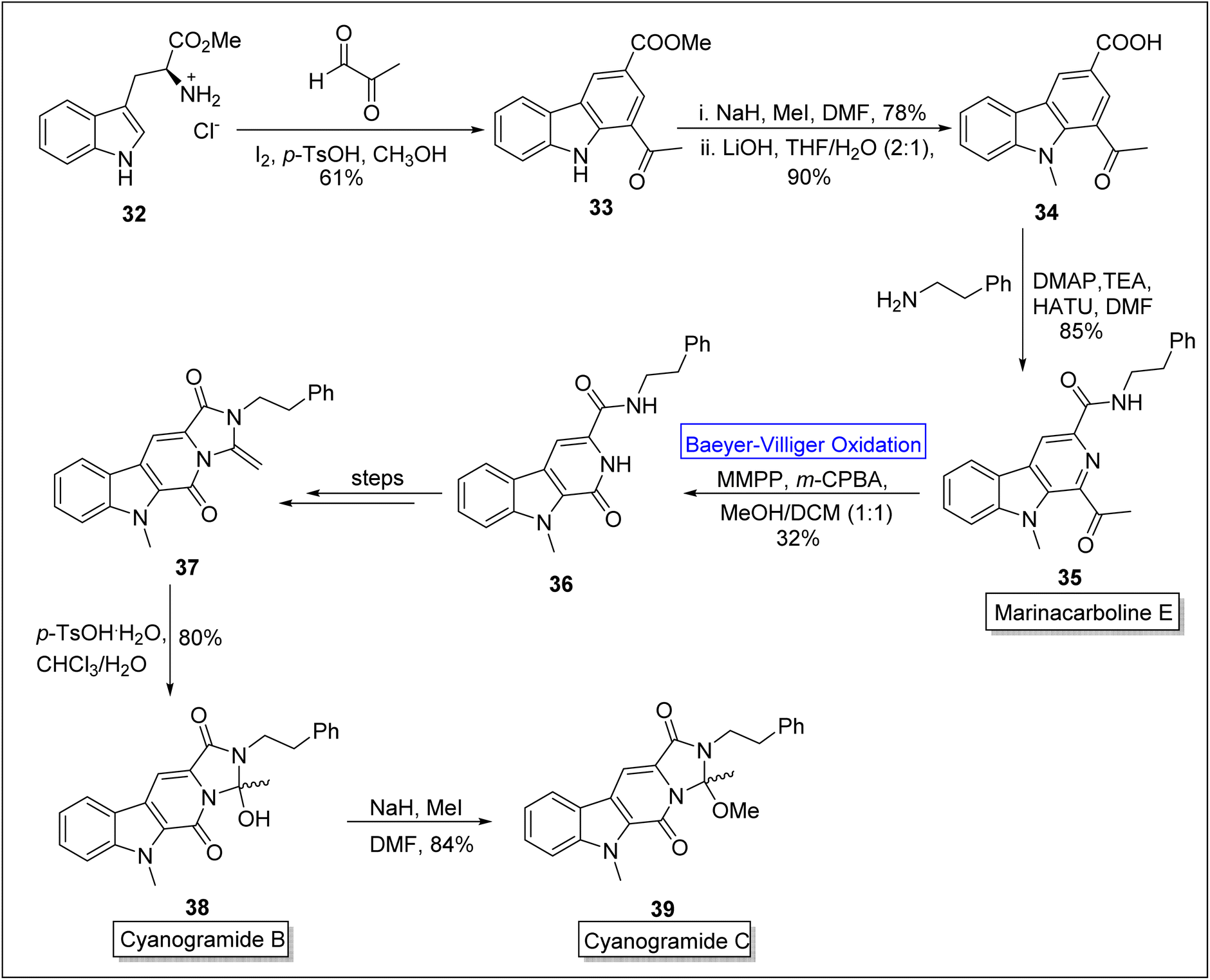 | ||
| Scheme 5 Synthesis of cyanogramides B 38 and C 39 via Baeyer–Villiger oxidation. | ||
Psammaplysins are marine alkaloids that belong to natural dihydrooxepine-spiroisoxazoline based-products.74 Initially, psammaplysin A 44 was extracted by Kashman from Psammaplysilla purpura75,76 in 1982 and it exhibited anti-malarial, anti-biotic, anti-cancer, and anti-HIV activities.77–80 In 2022, Paciorek et al. reported the first total synthesis of psammaplysin A 44 in thirteen steps by using Baeyer–Villiger oxidation as an essential step.74 In the beginning, compound 40 was reacted with NBS, NEt3, ethyl nitroacetate in MeCN, and then treated with triethyl phosphite (P(OEt)3) and 1,4-dioxane to synthesize a ketone 41 in 99% yield. In the next step, ketone 41 on treatment with TBSCl, imidazole (imH) in DCM followed by m-CPBA induced Baeyer–Villiger oxidation in the presence of Na2HPO4 and DCM generated a lactone 42 in 87% yield (over 2-steps). Then, lactone 42 was further reacted with potassium hexamethyldisilazide (KHMDS), diphenyl chlorophosphate (PhO)2POCl, and hexamethylphosphoramide (HMPA) in tetrahydrofuran (THF) to afford the compound 43 in 86% yield. Subsequently, compound 43 delivered the targeted natural product 44 over a few steps (Scheme 6).
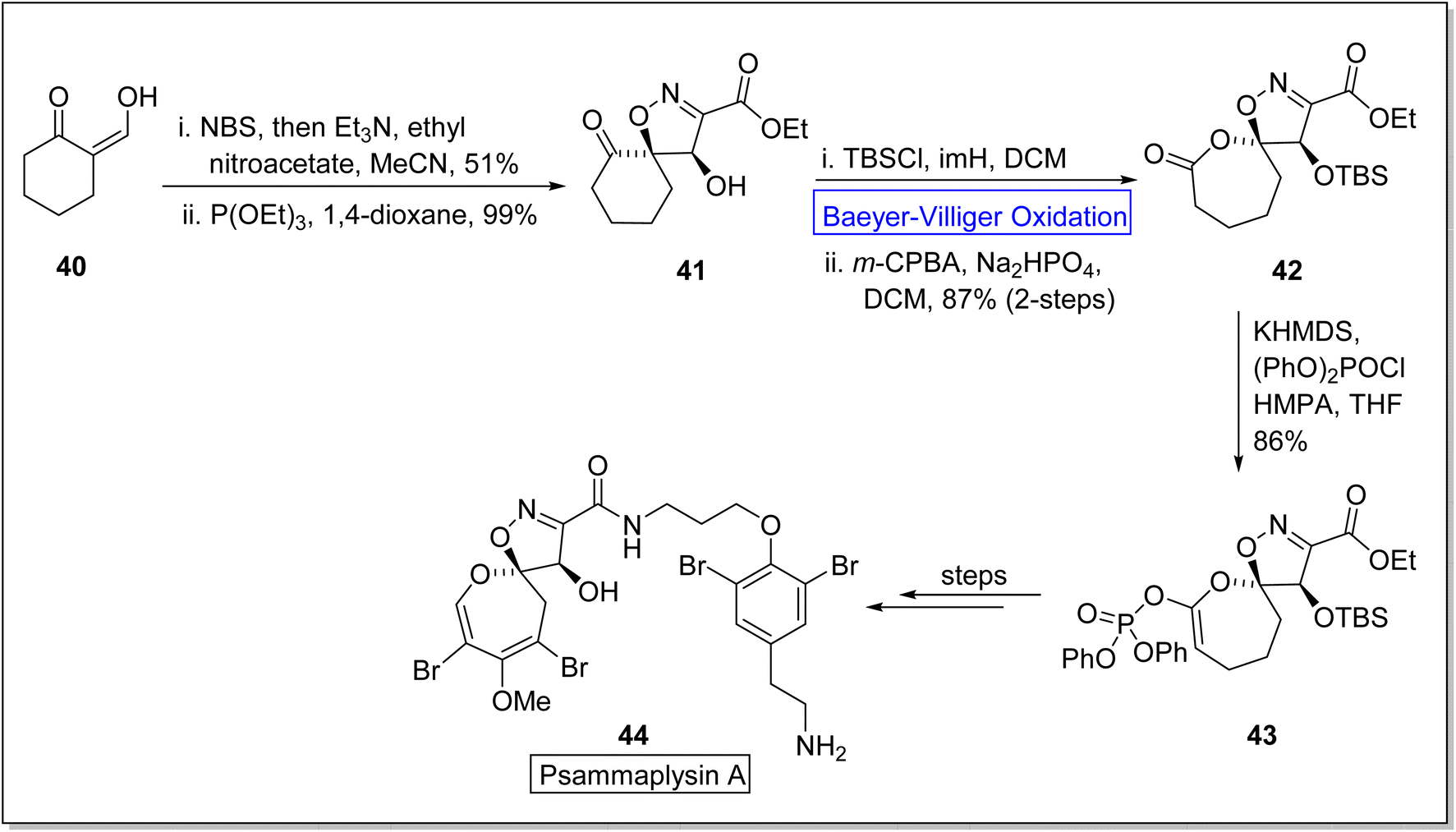 | ||
| Scheme 6 Synthesis of psammaplysin A 44 via Baeyer–Villiger oxidation. | ||
Pyrazoles are remarkable five-membered heterocyclic compounds that exhibit broad-range spectrum of pharmaceutical applications and play a vital role in natural product syntheses. For example, these are exploited in the synthesis of psammaplysins by using different synthetic routes. Psammaplysin K, O, and ceratinamides are novel bromotyrosine derivatives obtained from sea sponge81–84 Pseudoceratina purpurea.85 These exhibit anti-fouling, anti-cancer, and anti-bacterial activities.86–88 Despite all these excellent activities, no synthetic approach for theses natural spirooxepin isoxazolineswas reported till 2022. However, in 2022, Zhang et al. presented the synthesis of 7-deoxypsammaplysins K 155, O 154b, and 7-deoxyceratinamide A 154a by leveraging Baeyer–Villiger oxidation as a key step.89 The synthesis was initiated with the reduction of 2-bromo-2-cyclohexanone 45 with NaBH4, CeCl3·7H2O to produce a compound which was further reacted with tert-butyldimethylsilyl triflouromethane sulfonate (TBSOTf) in NEt3 and then underwent oxidation with chromium trioxide (CrO3) in 3,5-dimethylpyrazole (3,5-DMP) to synthesize silyl ether 46 in 57% yield. Ether 46 experienced the Baeyer–Villiger oxidation with urea hydrogen peroxide (UHP) in the presence of Na2HPO4 and trifluoroacetic anhydride (TFAA) to furnish a lactone 47 in 52% yield. Furthermore, the reaction of lactone 47 with Petasis reagent (Cp2TiMe2) and tetra-n-butyl ammonium fluoride (TBAF) in the presence of DMP, NaHMDS, and TfOMe (3-steps) generated oxepine 48 in 50% yield. The compound 48 was further treated with hydroxyamino acetate chloride 49 by using THF, NaHCO3, NaHPO4, N,N-diisopropylethylamine (DIPEA), DCM, and then with NBS as a bromide agent, tetrabutylammonium tribromide (TBATB), PTB and 2,6-DPBP in DCM to afford an ester 50. Subsequently, ester 50 was reacted with LiOH·H2O solution to synthesize an acid 51 which further underwent coupling with 52a & 52b and 53 individually in the presence of N,N,N′,N′-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and N-methylimidazole (NMI) respectively. These coupling reactions resulted in the construction of corresponding natural products 54a and 54b (in 58% and 60% yields respectively) and 55 in 81% yield (Scheme 7).
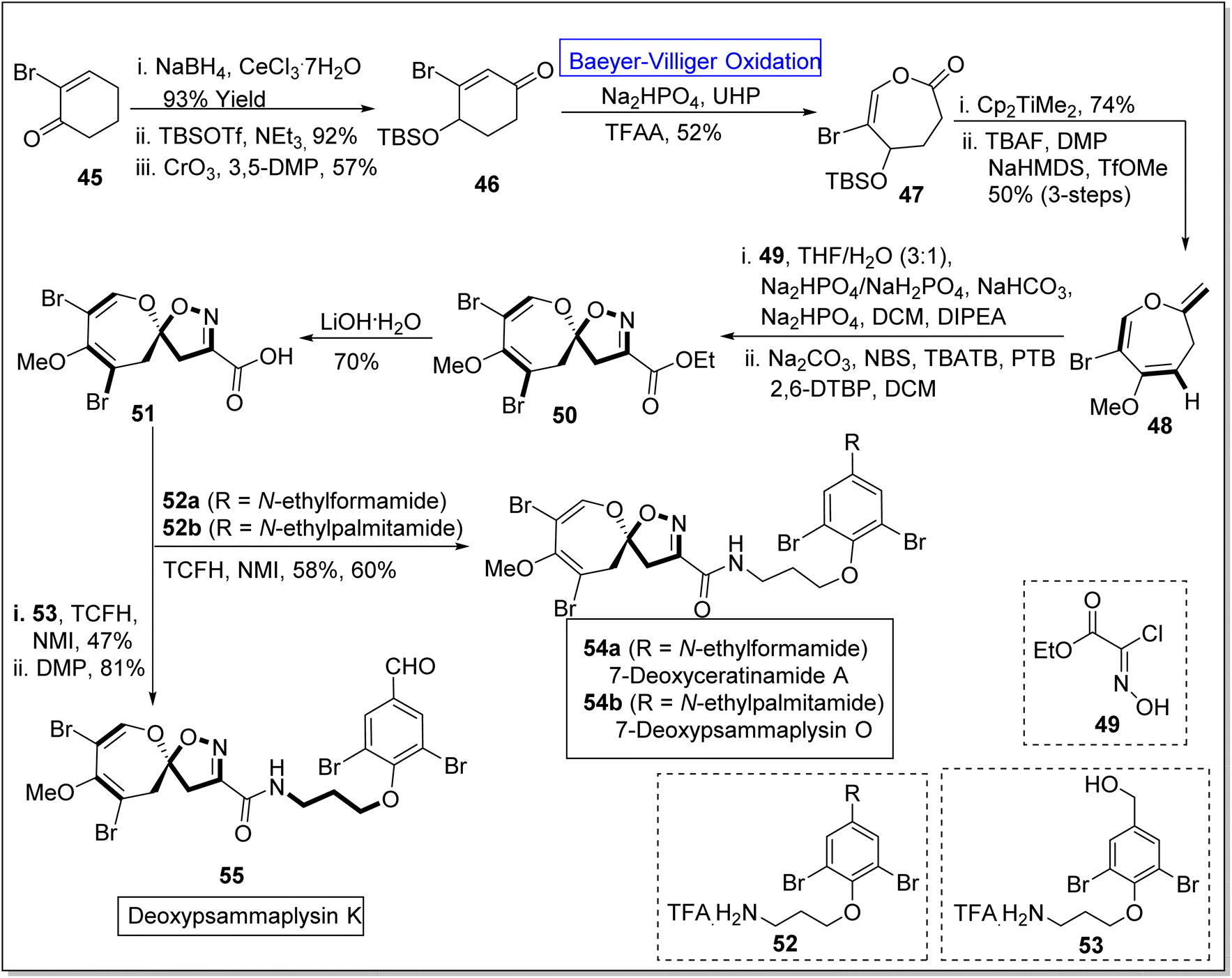 | ||
| Scheme 7 Synthesis of deoxypsammaplysins O 54b, K 55 and deoxyceratinamide A 55 via Baeyer–Villiger oxidation. | ||
Psammaplysins 65 and 67 are a distinctive class of bromotyrosine-based spiroisoxazolines, which are marine alkaloids. These consist of more than 300 members.90 Psammaplysins A, M, Q, and O contain more than 35 members linked with amide bond. These compounds exhibit antiviral, antimicrobial, and anticancer activities74 with sub-micromolar IC50 values. Despite the known biological activities, the total synthesis of these compounds had not been achieved for 40 years. For the first time, in 2022, Magauer group synthesized the racemic Psammaplysin A (in 13 steps) by employing Henry reaction but they faced the difficulty installing carbon 7 hydroxyl group. This drawback was eliminated in 2023 by Morrow and Smith by the use of enediol ether dipolarophile. In 2023, Morrow & Smith presented the total synthesis of psammaplysins A 65 and first total synthesis of psammaplysin O, Q, M, (65 and 67) and ceratinamide A 67 by utilizing Baeyer Villiger oxidation as a fundamental step to convert 6-membered ketone 56 into 7-membered lactone 57.91 The synthesis was started with the preparation of lactone 57 in 99% yield from ketone 56 via subsequent Baeyer–Villiger oxidation with m-CPBA. Benzyloxymethylenation of lactone 57 afforded enol ether 58 in 53% yield (E/Z = 1:1), (E)-58 was further treated with compound 59 under optimized conditions (i-Pr2NEt, MTBE) to furnish a single diastereomer spiroisoxazoline 60 in 44% yield. Ketal deprotection of this compound 60 with acetone and THF in acidic media yielded ketone 61 which underwent many steps to synthesize a spirocycle 62. Aminolysis of this compound 62 furnished the phthalimide-protected moiety 64 in 49% yield on treatment with compound 63, zirconium tert-butoxide (Zr(OtBu)4), 1-hydroxy-7-azabenzotriazole (HOAT), THF, and PhMe. Consequently, compound 64 further delivered the desired natural psammaplysin A 65 (in 11 steps) in 68% yield by reacting with hydrazine and pyridine in DCM and methanol. In another approach, compound 64 on treatment with compound 66, amine, Zr(OtBu)4, and HOAT in THF generated the respective natural products 67 (Scheme 8).
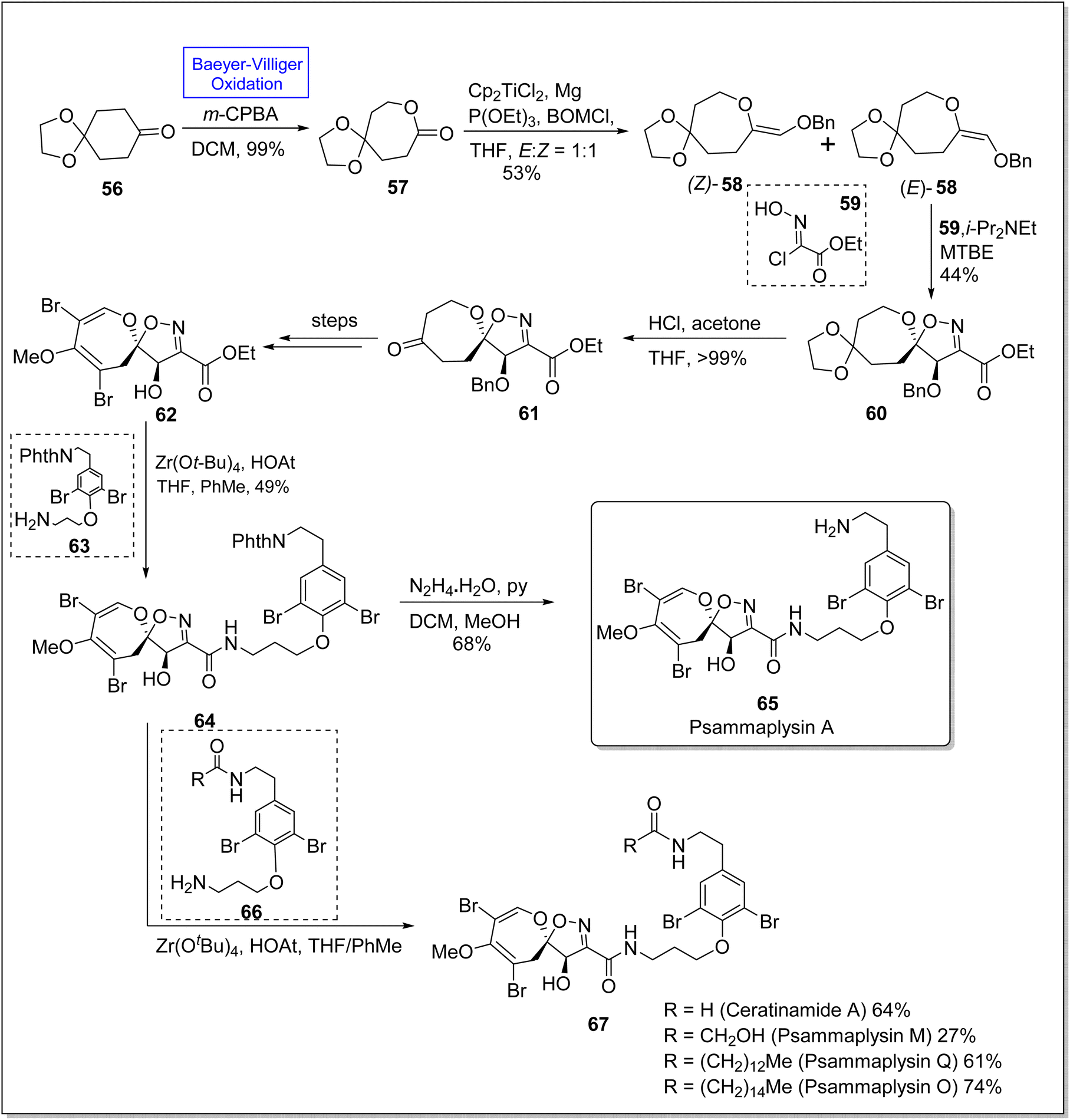 | ||
| Scheme 8 Synthesis of natural marine alkaloids; psammaplysins 65 and 67 via Baeyer–Villiger oxidation. | ||
Metaphanine 75 and oxoepistiphemiersine 77 are hasubanan alkaloids which are isolated from genus Stephania.92 Such alkaloids are widely used in medicines due to their anti-microbial, cytotoxic, and anti-viral activities.93–96 These share benzannulated aza [4.4.3] propellane skeleton and vary due to different patterns on arene ring in 2023, Sun et al. reported the total synthetic divergent strategy of hasubanan alkaloids (in 12–13 steps) by utilizing Baeyer–Villiger oxidation reaction as a fundamental step.97 Synthetic scheme was initialized with the condensation of cyclohexanedione monoethylene acetal 68 with cyclohexylamine, deprotection with LDA, and alkylation with iodide followed by treatment with 69, Ph3PMeBr, tBuOK, THF, and HCl to generate a compound 70 in 87% yield. Then, the compound 70 gave a tetracyclic diketone 71 in 72% yield under optimized conditions. Compound 71 was further subjected to the Baeyer–Villiger oxidation with m-CPBA in the presence of NaHCO3 and it resulted in the synthesis of a lactone 72 in 75% yield with outstanding regioselectivity. Thereafter, lactone 72 yielded a tetracyclic oxo-metaphanine 73 over a few steps, which further delivered a sulphamide 74 in 80% yield by reacting with lithium bis(trimethylsilyl)amide (LHMDS), MOMBr in THF and Lawesson's reagent in DCM. Finally, compound 74 afforded the desired compound, Metaphanine 75 in 82% yield on reduction with Raney Ni in DCM. In another approach, compound 73 generated a methyl enol ether 76 in 90% yield via methylation with LHMDS. Finally, Brown's hydroboration-oxidation of enol ether 76 followed by Dess-Martin oxidation furnished the oxoepistiphemiersine 77 in 90% yield (Scheme 9).
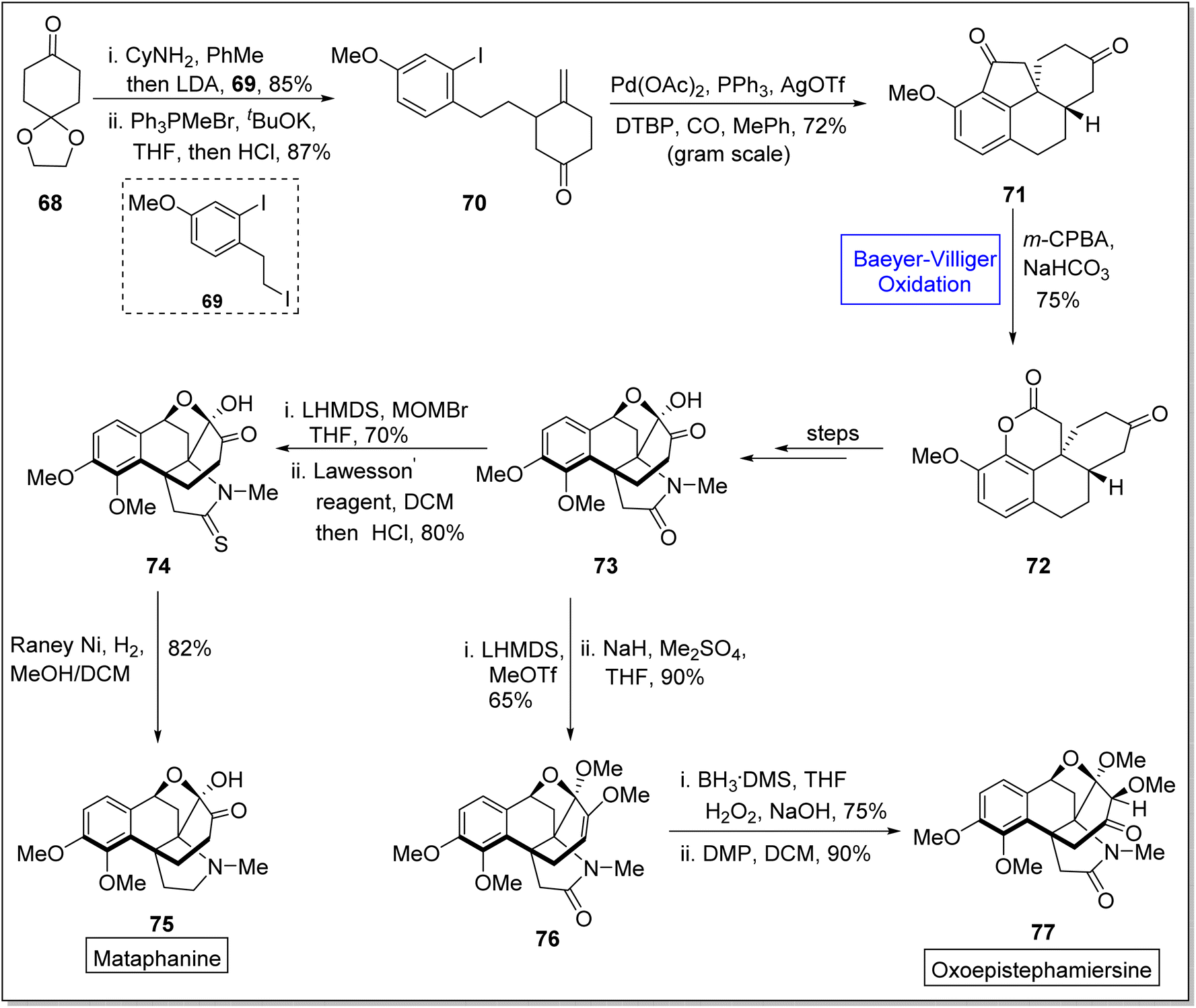 | ||
| Scheme 9 Synthesis of metaphanine 76 and oxoepistiphemiersine 77 via Baeyer–Villiger oxidation. | ||
2.2. Synthesis of polyketide-based natural products via Baeyer–Villiger oxidation
In the field of natural product-based medicines, polyketides are of pivotal significance due to their anti-cancer and anti-bacterial activities.98 Owing to these activities, many researchers have synthesized different naturally occurring polyketides by employing Baeyer–Villiger oxidation reaction. Fig. 3 shows the pictorial representation of structures of some of naturally occurring polyketides synthesized via BVO reaction.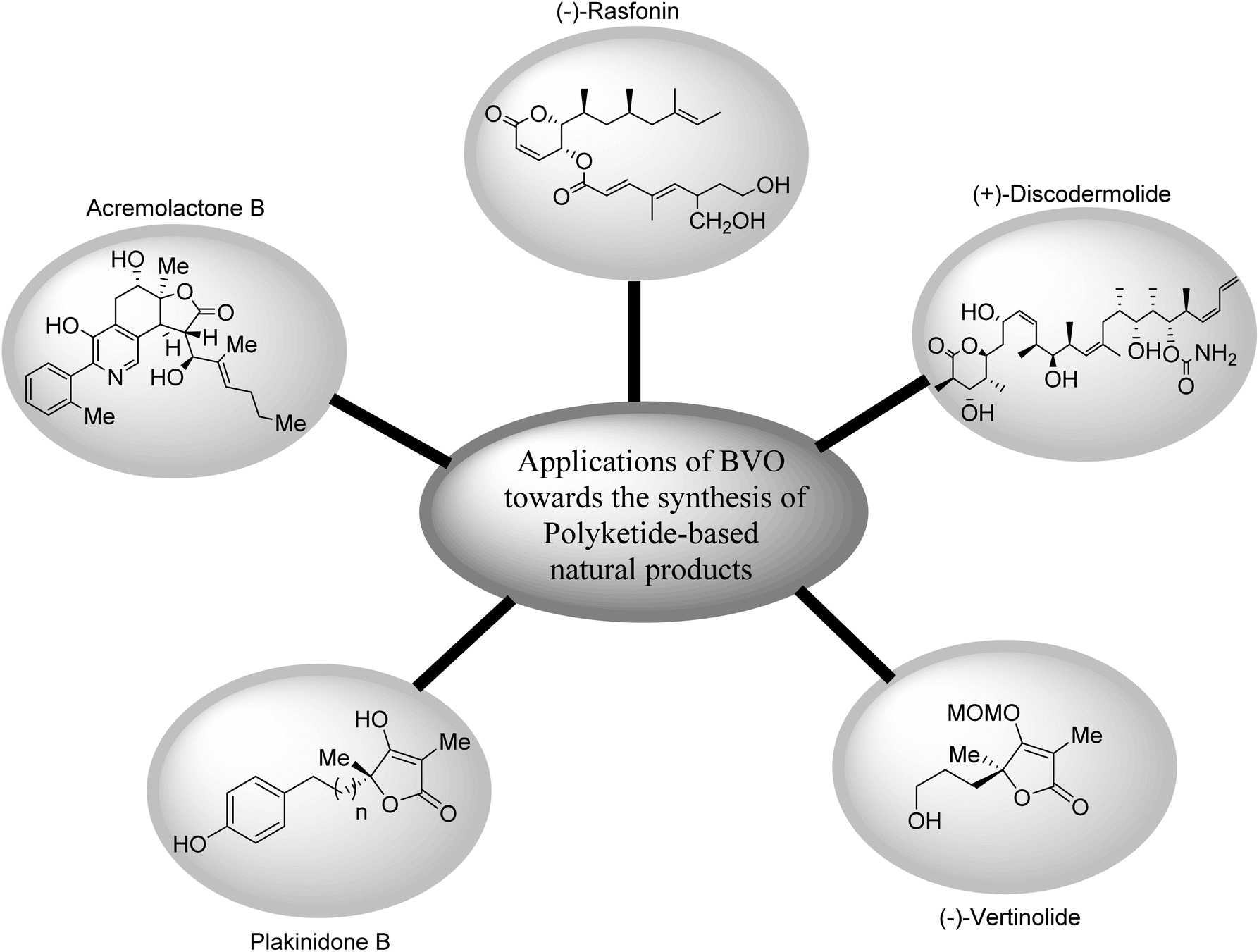 | ||
| Fig. 3 Structures of some naturally occurring polyketides synthesized via Baeyer–Villiger oxidation. | ||
(−)-Rasfonin 82 is a natural polyketide which is obtained from a fungal strain, Cephalotrichum gorgonifer.99 It is a pharmaceutically significant bioactive compound that display antitumor activity and induces necroptosis, apoptosis, and autophagy in human cells.100–103 Wu and co-workers in 2019, reported the total synthesis of (−)-rasfonin by utilizing regioselective asymmetric Baeyer–Villiger oxidation.104 The synthesis was commenced with the Baeyer Villiger oxidation of 3-substituted cyclic ketone 78 with m-CPBA in the presence of scandium(III) triflouromethanesulfonate Sc(OTf)3 and EtOAc to afford a lactone 79 in 96–99% yield with 93–97% ee. Then, the lactone 79 was converted to compound 80 in 97% yield with 90% ee on treatment with EtSH and AIMe3 in THF. Afterwards, compound 80 was made to react with tert-butyl(chloro)diphenylsilane (TBDPSCl), NaH, and EtOAc to generate 1,3-dimethyl thioester 81 in 91% yield with 90% ee. Subsequently, thioester 81 further underwent a series of reactions to furnish the (−)-rasfonin 82 (Scheme 10).
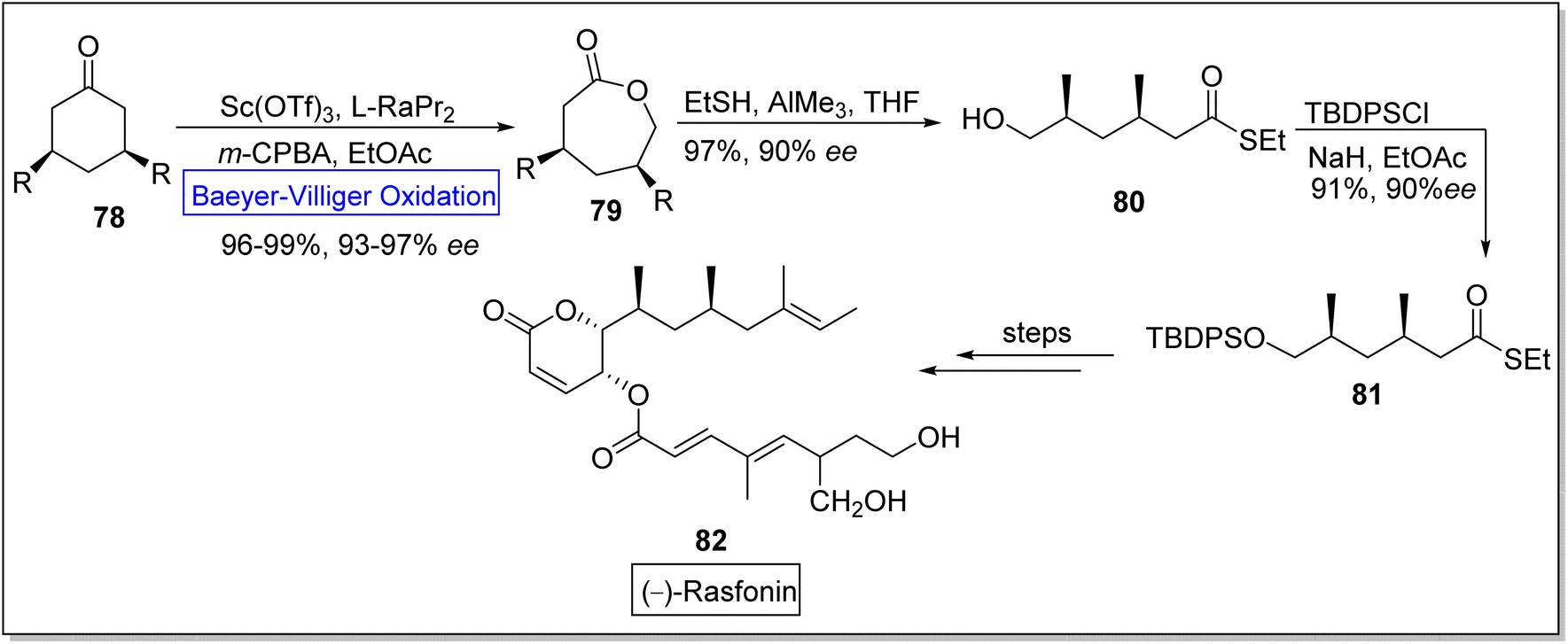 | ||
| Scheme 10 Synthesis of (−)-rasfonin 82 via Baeyer–Villiger oxidation. | ||
(+)-Discodermolide is a recently discovered cytotoxic polyketide. It is a very efficient antimitotic105,106 natural product which is extracted from a sea sponge Discodermia dissolute.105,107 Gunasekara and coworkers, in 1990, discovered this novel polyketide.107 This product consists of thirteen chiral centers. Schreiber and associates synthesized the antipods of discodermolide but absolute configuration of this natural compound remained unclear.108 To achieve the anticipated synthesis of (+)-discodermolide, Dubasi and Verala in 2022, reported the total synthesis of key fragments of the natural product by utilizing Baeyer–Villiger oxidation as one of the key reactions.109 The synthesis was initiated with the DIBAL-H mediated reduction of bicyclic ketone 83 in DCM followed by reaction with NaH, BnBr, and THF to afford compound 84. Then, the compound 84 experienced asymmetrical hydroboration with Ipc2BH in the presence of PCC and DCM to furnish ketone 85 in 87% yield. Ketone 85 underwent Baeyer–Villiger oxidation with m-CPBA and NaOH in DCM to synthesize lactone 86 in 90% yield. Furthermore, compound 87 was obtained in 80% yield by the treatment of lactone 86 with TBDPS followed by reduction with LiAlH4 which further generated a keto compound 88 in 92% yield via Dess-Martin periodonane-mediated (DMP) oxidation in DCM. Keto compound 88 was converted into two isomers; β-isomer 89 as minor isomer in 9% yield and α-isomer 90 as major isomer in 83% yield. α-Isomer 90 on reaction with TBAF in THF gave a triol 91 in 88% yield which further afforded the subunit of discodermolide 92 over a few steps. In another approach, methylation of lactone 86 followed by reduction with LiAlH4 generated a triol 93 in 85% yield. In the final step, triol 93 yielded the subunit 94 over a few steps (Scheme 11).
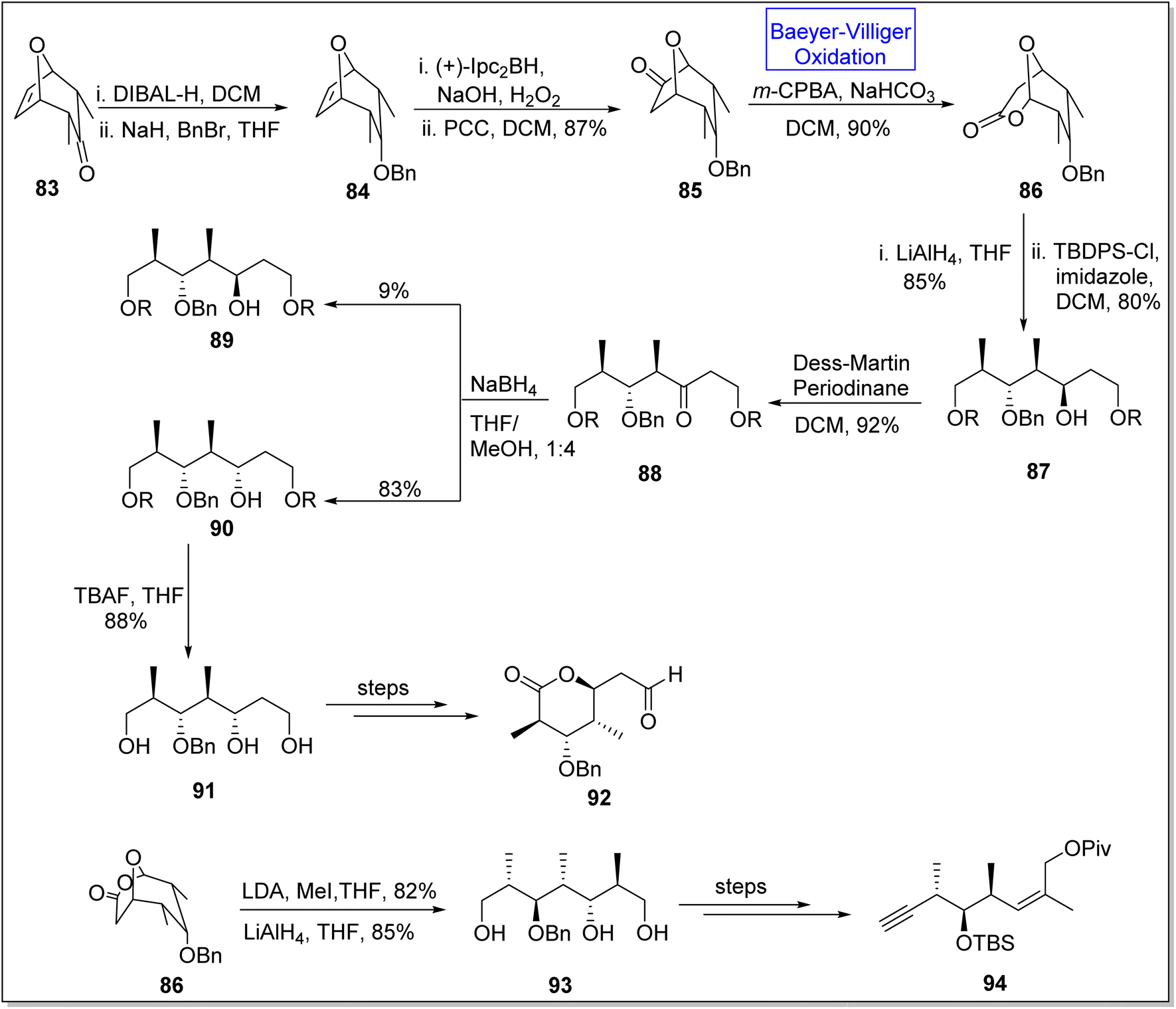 | ||
| Scheme 11 Synthesis of C1–7 92 and C8–15 94 subunits of (+)-discodermolide via Baeyer–Villiger oxidation. | ||
Epoxides are three-membered heterocyclic compounds and experience epoxide ring opening reactions due to ring strain. Therefore, these are the substantial intermediates used in natural products synthesis110 such as synthesis of (+)-discodermolide subunits. (+)-Discodermolide is a natural marine polyketide107 and a very efficient anticancer agent.111 In 1990, it was extracted from a sea sponge Discodermia dissolute and it possess 13 stereogenic centers. Since several synthetic approaches for this natural compound and its subunits have been reported but in 2020, Si and Kaliappan established the novel non-aldol synthetic strategy for the synthesis of two subunits of (+)-discodermolide by utilizing Baeyer–Villiger oxidation as a key reaction.112 The synthesis was accomplished with the homologation of alcohol 95 via oxidation in the presence of (COCl)2, DMSO, Et3N in DCM followed by Wittig reaction and reduction to afford an allylic alcohol 96 in 88%. In the next step, the alcohol 96 underwent Baeyer–Villiger oxidation with m-CPBA to generate an epoxy alcohol 97 in 14:1 diastereomeric ratio with 82% yield. Furthermore, this epoxy alcohol 97 was subjected to oxidation and homologation under Wittig olefination conditions to synthesize epoxy ester 98 in 85% yield (over 2-steps). Subsequently, Shimizu reaction of compound 98 under optimized conditions provided a hydroxyester 99 in diastereomer ratio >10:1 (81% yield). Then, compound 99 synthesized the 1,3-diol 100 in 47% yield over a few steps. Primary alcohol 100 was mesylated with MsCl and underwent Finkelstein reaction to furnish the subunit of discodermolide 101 in 46% yield (over 2-steps). In another route, compound 97 afforded a primary alcohol 102 in 75% yield via many steps which further underwent oxidation to yield the C9–C13 subunit 103 of discodermolide in 64% yield over 2 steps (Scheme 12).
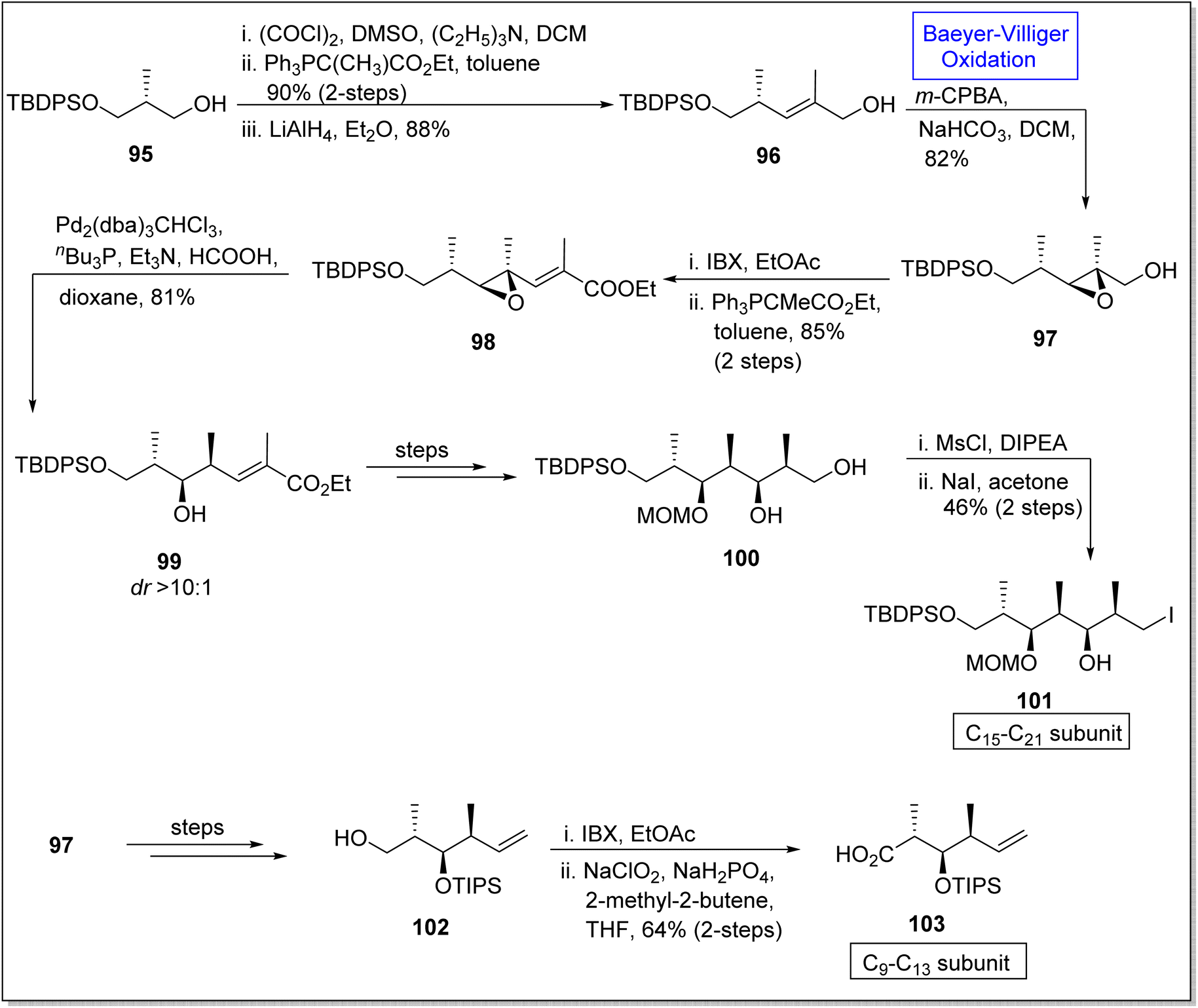 | ||
| Scheme 12 Synthesis of C15–21 101 and C9–13 103 subunits of (+)-discodermolide via Baeyer–Villiger oxidation. | ||
Spirocyclic moieties captivate the researchers due to their ring system and effective biological activities. Many natural products contain spirocyclic ring system and are used in pharmaceutical industry.113 Spirocyclic ligands are also used in the natural product synthesis. Such as vertinolide is a natural product which is synthesized in the presence of spirocyclic chiral phosphoric acid ligand 106. Vertinolide 108 is a derivative of β-tetronic acid and it is extracted from Verticillium intertextum. It belongs to fungal-metabolites, myotoxins.114,115 Vertinolide 108 inhibits the root and shoot growth in Lattuce seedlings. Plakinidone B 111 is a five-membered perlactone which is isolated from Angulospiculatus and it exhibits anti-viral and anti-microbial properties. In 2023, Liu et al. reported an enentioselective synthesis of vertinolide and 1st total synthesis of the plakinidone by utilizing the highly enantioselective Baeyer–Villiger oxidation as a significant step.116 The synthesis was begun with the treatment of cyclobutane-1,3-dione 104 with cumene hydroperoxide 105 (Baeyer–Villiger oxidation) and spirocyclic chiral phosphoric acid ligand 106 in the presence of (C2H5)2Cl2 and then protected with methoxy methyl chloride (MOMCl) and NaH in THF to give an enolate ether 107 in 82% yield (86% ee). Then, enolate ether 107 afforded the vertinolide 108 in 81% yield (86% ee) by reacting with LDA, CH3I, in THF followed by Pd/C-catalyzed reduction in EtOAc. Furthermore, cyclobutene-1,3-dione 109 was reacted with 105 and 106 in DCE and then with BnBr and NaH in THF to synthesize compound 110 in 78% yield (>99% ee). Methylation of compound 110 with MeI and LDA in THF followed by debenzylation furnished the plakinidone B 111 in 72% yield over 2-steps (Scheme 13).
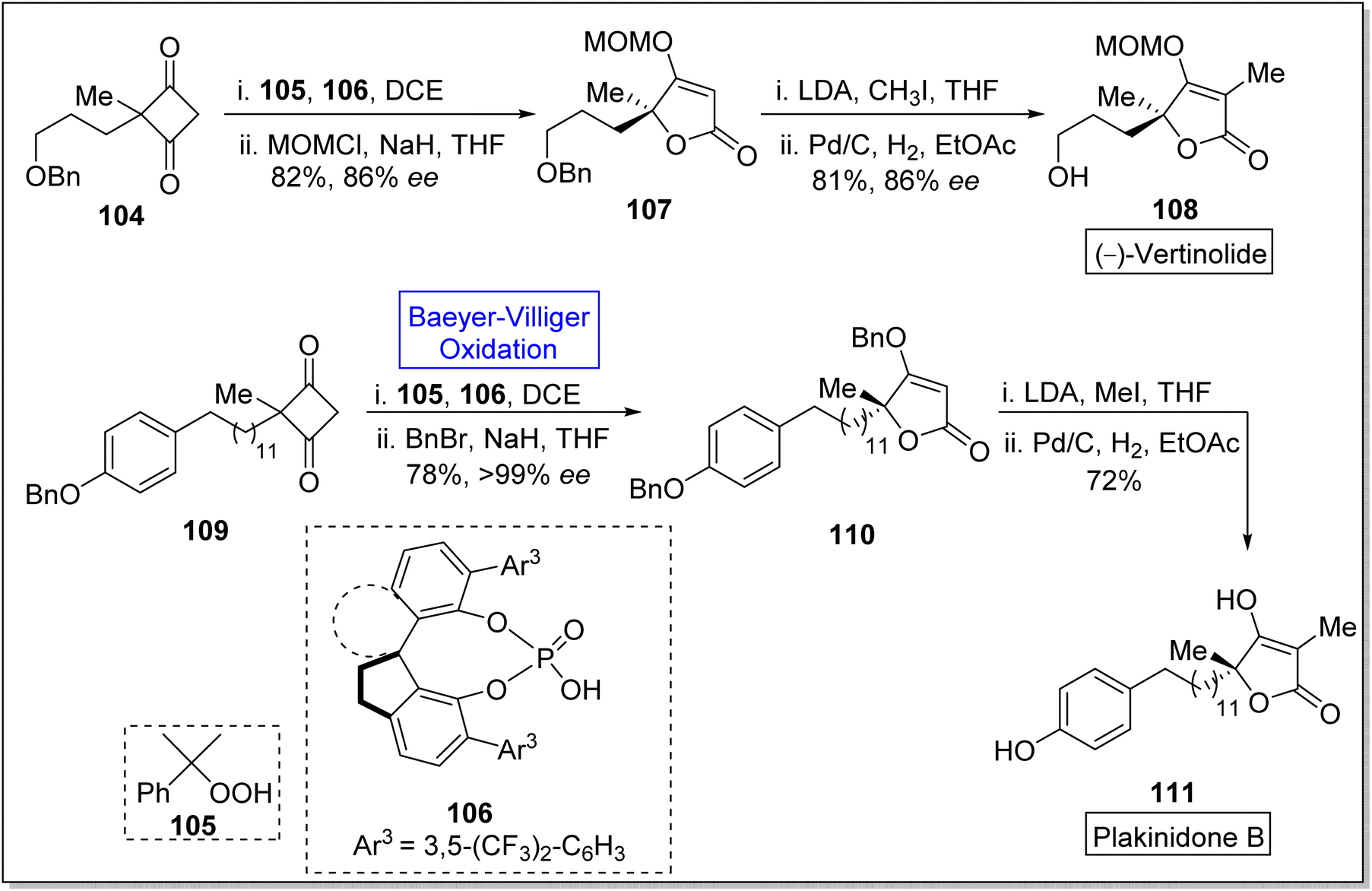 | ||
| Scheme 13 Synthesis of vertinolide 108 and Plakinidone B 111 via Baeyer–Villiger oxidation. | ||
Acremolactone B is an azaphilone-type fungal polyketide. Acrolactones were isolated by Sussa and group from the fungus, Acremonium roseum.117,118 Such natural products have gained significant importance due to vast range of biological activities such as herbicidal, anti-cancer, anti-viral, anti-inflammatory and anti-microbial activities.119 In 2023, Ba et al. reported the first total asymmetric synthesis of acremolactone B on gram scale by utilizing Baeyer–Villiger oxidation as a key step.120 The synthesis was inaugurated with [2 + 2] photocycloaddition of compound 112 with 1,1-diethoxyethene in the presence of NaOMe and H2SO4 to afford the mixture of cis and trans products 113a and 113b (19% and 41% respectively) in 2-steps. In the next step, m-CPBA-induced Baeyer–Villiger oxidation of compound 113b generated highly regio and chemo-selective γ-lactone 114 in 97% yield. Then, the reaction of compound 114 with Me3Ph3N+Br− followed by triflation with Tf2O in Et3N generated the compound 115 in 78% yield (over 2 steps). The compound 115 was subjected to Stille-Magita coupling with Pd2dba3, Ph3As, LiCl (Farina Protocol), and tributyl(vinyl)stannane to furnish bromo diene 116 in 85% yield. Subsequently, treatment of compound 116 with K2OsO2(OH)4, NMO, and NaIO4 afforded the corresponding bicyclic intermediate 117 with overall 86% yield. Afterwards, a 4-step sequence from compound 117 to 118 resulted in the formation of a pyridine ring. Deprotonation of compound 118 with LDA and acylation with acyl cyanide 119 synthesized the compound 120 in 92% yield which upon further reduction with DIBAL-H in HF synthesized the natural product 121 in 90% yield (Scheme 14).
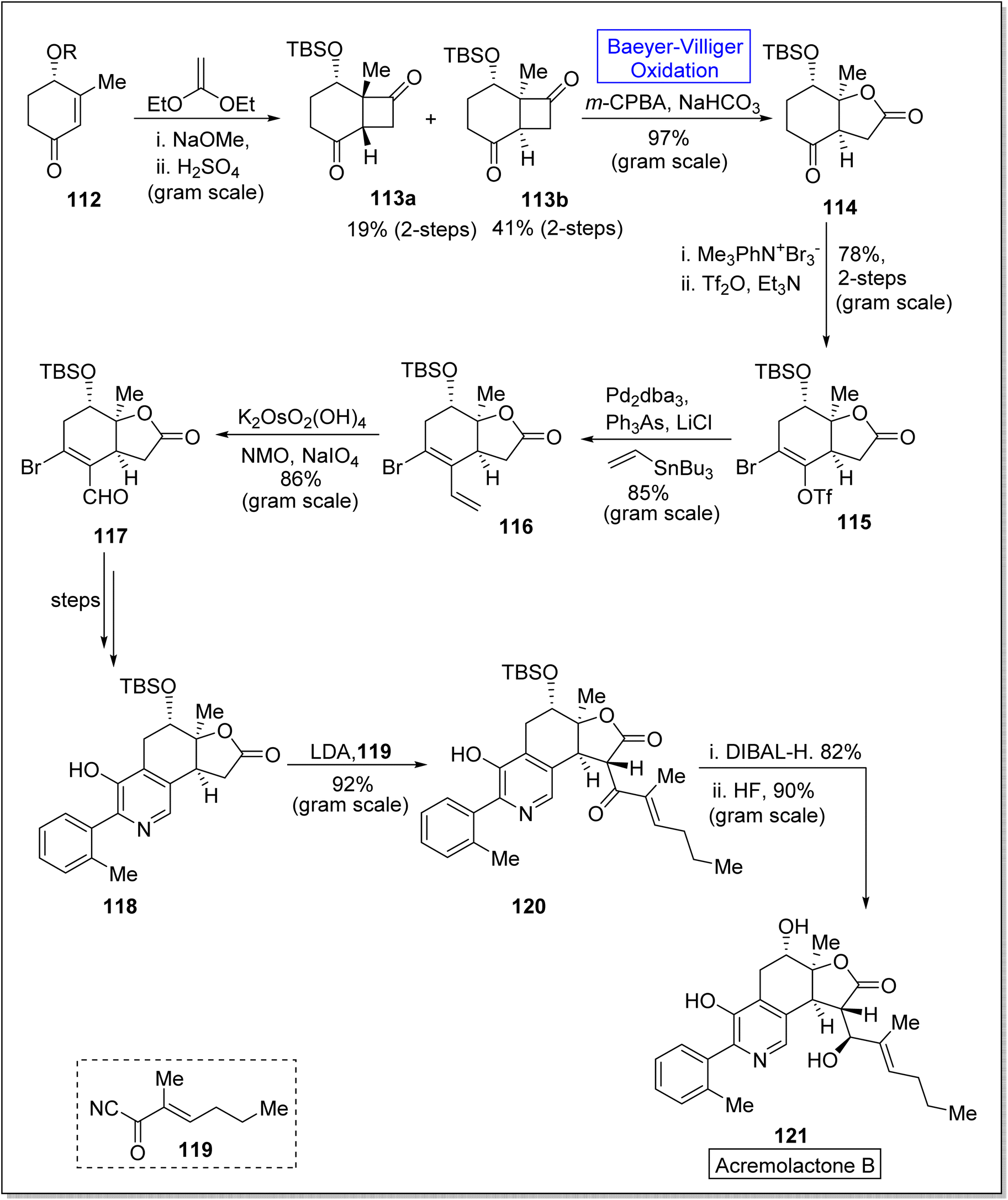 | ||
| Scheme 14 Synthesis of acremolactone B 121 via Baeyer–Villiger oxidation. | ||
2.3. Synthesis of terpene-based natural products via Baeyer–Villiger oxidation
Terpenes are a diverse class of natural compounds. They are isolated from plants, animals fungi, pathogens, insects, and endophytes. They exhibit anti-microbial, anti-inflammatory, anti-cancer, anti-oxidant and anti-diabetic activities.121,122 Baeyer–Villiger oxidation is one of those appealing reactions used for the synthesis of naturally occurring terpenes and terpenoids (Fig. 4). | ||
| Fig. 4 Pictorial framework elaborating the structures of some terpene-based natural products obtained by involving BVO as a key step. | ||
Friedelanes are natural triterpenoids obtained from leaves and branches of Maytenus robusta Reissek.123 These exhibit biological potential against breast and ovarian cancer cells124 with a decrease in IC50 value (IC50 < 100 μM). In 2020, Aguilar et al. presented the synthesis of friedelane derivatives by utilizing Baeyer–Villiger oxidation as a significant step.123 In the synthetic route, compound 122 was converted into a lactone i.e., friedelane derivative 123 via m-CPBA catalyzed Baeyer–Villiger oxidation in chloroform. Similarly, the same compound 122 afforded the other friedelane derivative 124 on treatment with NH2OH, HCl, and pyridine in methanol (Scheme 15).
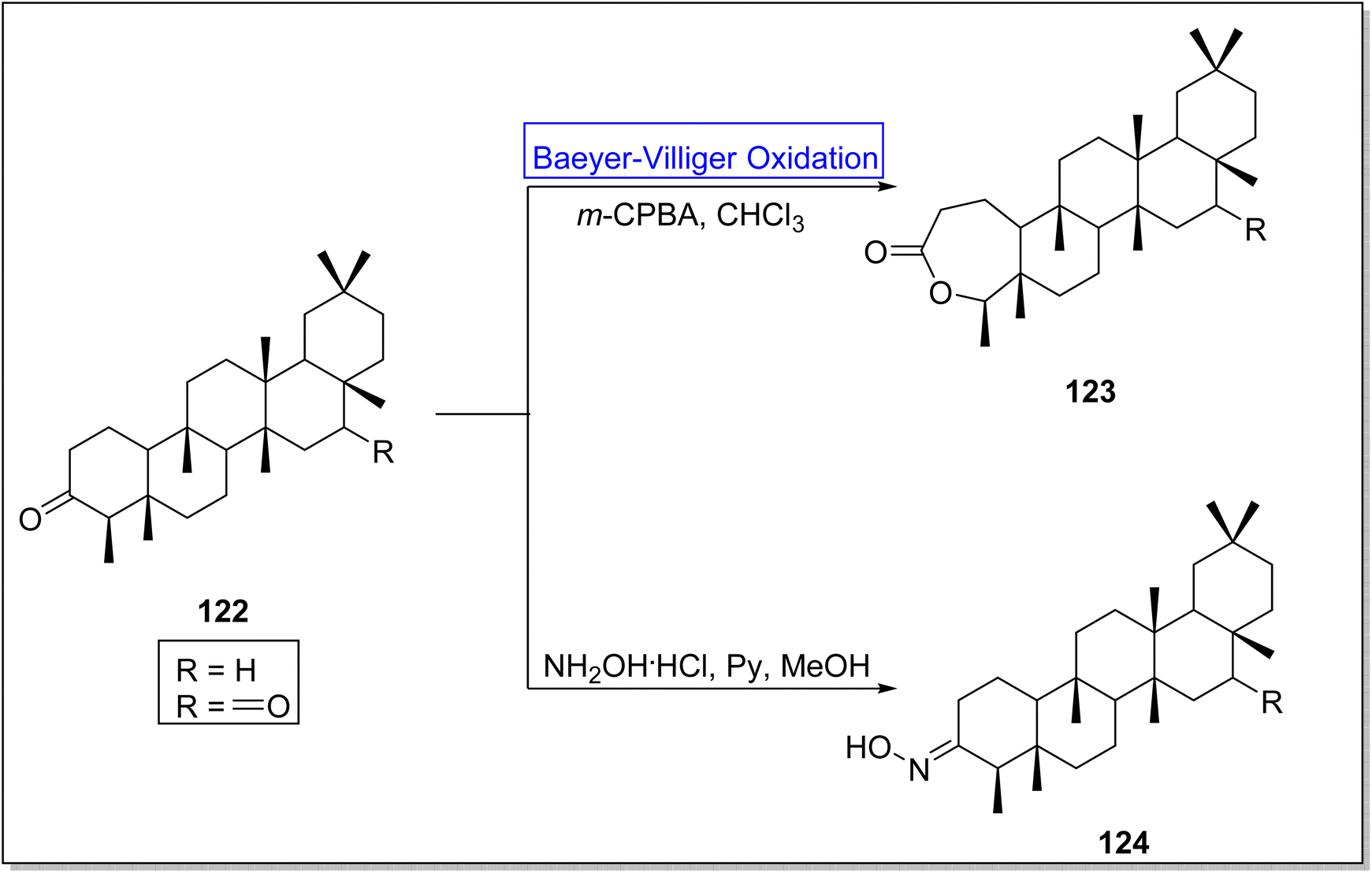 | ||
| Scheme 15 Synthesis of friedelanes 123 and 124 via Baeyer–Villiger oxidation. | ||
Cross-coupling reactions are remarkable synthetic tools that are used for the synthesis of naturally occurring biologically active molecules and their analogues i.e., anhydro ryanodol 131. Anhydro ryanodol 131 is a diterpenoid125 and a hydrolysis product of natural compound ryanodine which is isolated from a shrub; Ryana speciosa. It exhibits insecticidal properties. Initially, ryanodol was synthesized by Deslongchamps' by harnessing Diels–Alder reaction and oxidative cleavage.126,127 Prior to 2020, Reisman achieved the synthesis of ryanodol by employing Pauson–Khand reaction and SeO2-catalyzed polyoxidation.125 However, in 2020, Du et al. reported the unique approach of formal and total synthesis of anhydroryanodol 131 by utilizing the BV oxidation as a key step through the formation of ryanoids (which further led to successful synthesis of anhydroryanodol) and carbocyle formation.128 For this purpose, an alkyne 125 was treated with Cu-mediated i-PrMgCl, iodine, added with MnO2 in DCM (67%), C4H3liO in THF and NBS followed by the reaction with TBSOTf, benzylamine, DCM, and then with ClRh(PPh3)3, PhH to give a hemiacetal 26 (α and β with 3:1 ratio). Acylation of hemiacetal 126 with acyl cyanide 127 followed by methylation with KHMDS and methyl iodide synthesized the 1,3-diketone 128 in 59% yield (d:r ≥ 20). Then compound 128 underwent Sonogashira coupling and Stille coupling to generate the subsequent enyne 129 in 58% yield. Then, this enyne 142 was converted into compound 130 over a few steps. Finally, compound 130 afforded anhydroryanodol 131 in 38% yield via Baeyer–Villiger oxidation with m-CPBA, hydration and desilylation followed by the reaction with trissulfonium difluorotrimethylsilicate (TASF). Thereafter, compound 131 generated ryanodol 132 in 38% yield via epoxidation and reductive cyclization (2-step sequence) in dichloro ethane (DCE) and THF (Scheme 16).
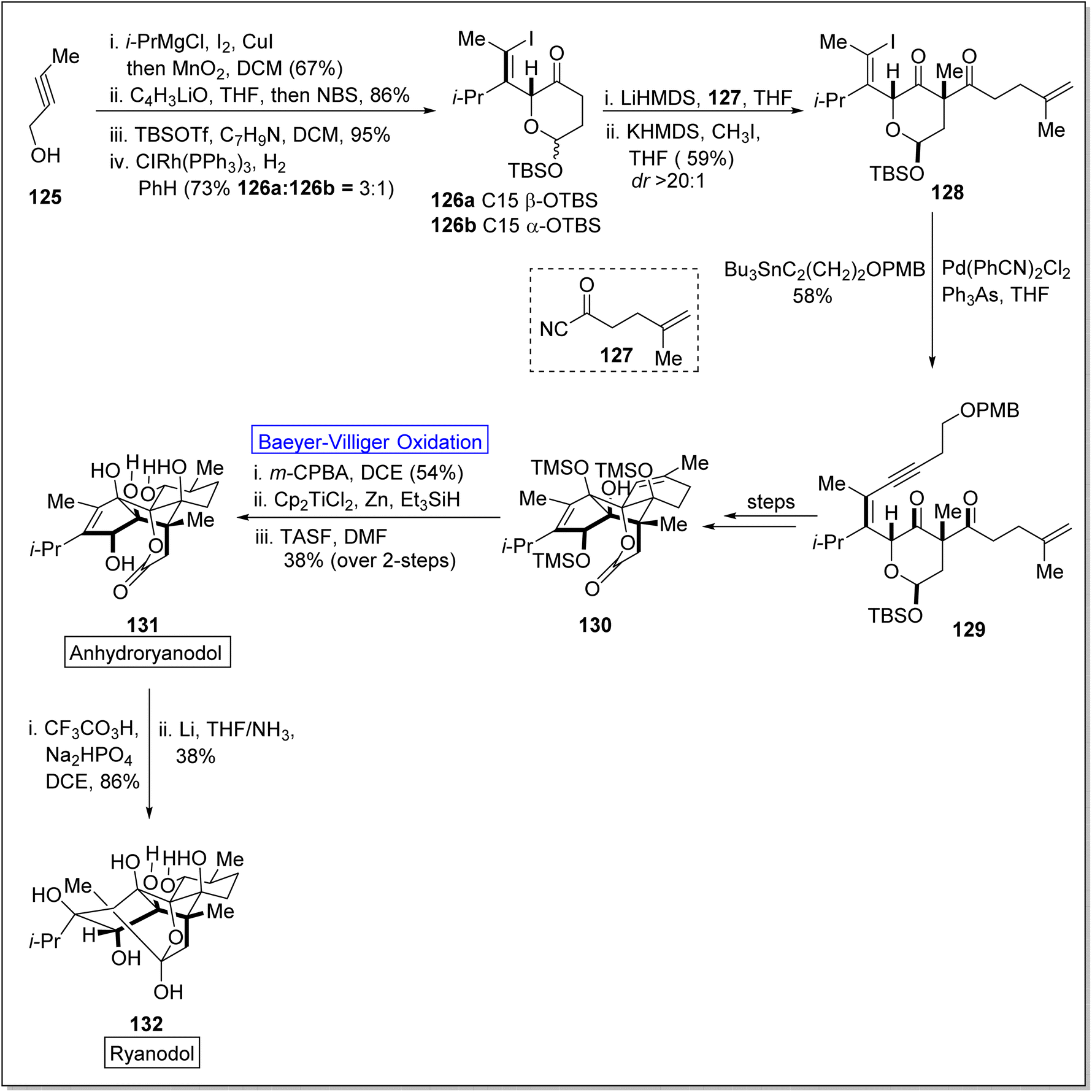 | ||
| Scheme 16 Synthesis of anhydroryanodol 132 via Baeyer–Villiger oxidation. | ||
Strigolactones are naturally occurring vital hormones which play a crucial role in agriculture for plant development. Zealactone 137 is an abundant strigolactone, isolated from roots of corn (in eight steps).129,130 Its naturally occurring diastereomers have IC50 value of 0.59 μM against ZmD14-dependent YLG hydrolysis. In 2020, Yoshimura and co-workers established the total synthesis of zealactone 1a/2 in 8 steps with overall 21% yield via Baeyer–Villiger oxidation reaction.131 In the first step of synthesis, cyclobutanone 133 underwent the Baeyer–Villiger oxidation with m-CPBA in the presence of Bu4NOH in DCM to produce a γ-butyrolactone 134 in 61% yield. In the next step, lactone 134 was reacted with TBAF and then it was hydrostannylated with HSnBu3 and azobisisobutyronitrile (AIBN) in toluene to yield the respective stannane 135 in 97% yield. Then, compound 135 was treated with vinyliodide 136, Pd2dba3, p(furyl)3, and 1,4-dioxane to furnish the zealactone 137 in 61% yield (Scheme 17).
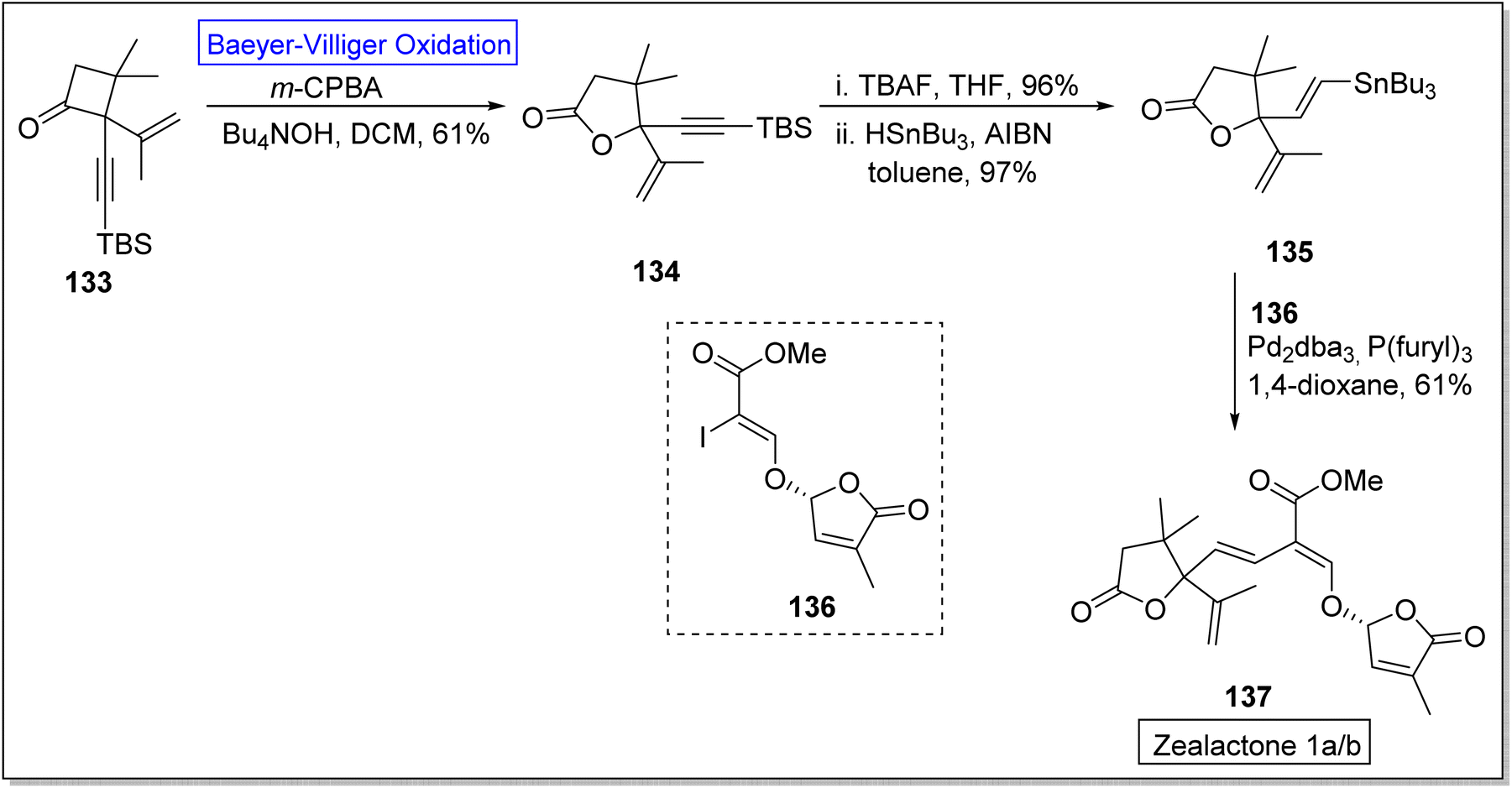 | ||
| Scheme 17 Synthesis of zealactone 137 via Baeyer–Villiger oxidation. | ||
Propindilactone G 146 belongs to a large family of polycyclic Schisandra nortriterpenoids, which were isolated from Schizandraceae family of plants.132,133 These reveal herbal medicinal properties and are used to treat insomnia, hepatitis and asthenia.134 In 2015, Yang and colleagues presented the synthesis of the propindilactone G by employing the Diels–Alder and Pauson–Khand reaction to achieve ring system.135 However, in 2020, Wang et al. reported the biosynthesis of propindilactone G in 58% yield from steroidal lactone by utilizing significant reactions including Baeyer–Villiger oxidation reaction.134 The synthesis was commenced with the reaction of steroidal lactone 138 with 3-iodobenzoic acid (IBX) followed by selective chlorination and E2 reaction to afford olefin 139. Then, compound 139 underwent a series of reactions to generate a ketone 140 in 82% yield. Ketone 140 was further subjected to three steps sequence including demethylation with CH3I and tBuOK, Baeyer–Villiger oxidation with m-CPBA and Mukaiyama dehydrogenation with lithium bis(trimethylsilyl)amide (LiHMDS) to synthesize an unsaturated lactone 142 in 62% yield. Compound 142 was converted to a diol 143 in 82% yield by addition of Zn/HOAc and p-toluene sulfonic acid in methanol. Swern oxidation of compound 143 followed by reaction with 2-ethoxyvinylbromide 144 and HCl gave an unsaturated aldehyde 145 in 59% yield which further produced the targeted compound 146 over a few steps. Stereochemistry of target product 146 (at C-17) was opposite to that of steroidal lactone 138 (Scheme 18).
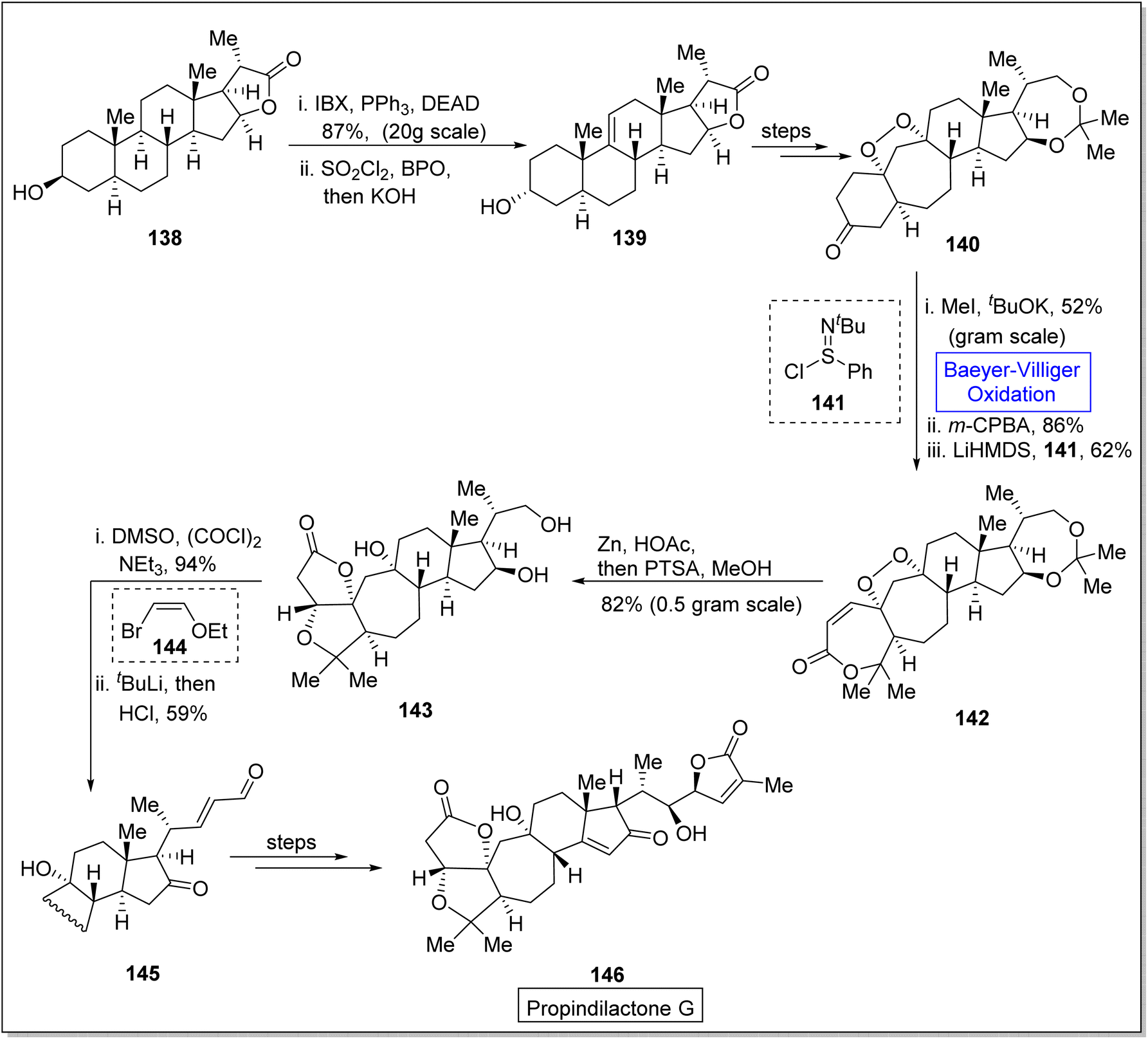 | ||
| Scheme 18 Synthesis of propindilactone G 146 via Baeyer–Villiger oxidation. | ||
Euonyminol 152 is a sesquiterpene136,137 with dihydro-β-agrofuran nucleus and is pharmaceutically significant due to anti-proliferative, anti-oxidant, and anti-microbial activities.137 In 1995, White and coworkers reported the synthesis of less oxygenated euonyminol. In 2021, Tomanik and coworkers presented the first enantioselective total synthesis of highly oxidized euonyminol by utilizing Baeyer–Villiger oxidation reaction as a key step.138 The synthesis embarked with the conversion of diene 147 into a diastereomer 148 in 70% yield via oxidation with Shi ketone and oxone in the presence of K2CO3. Then, compound 148 afforded an α-keto lactone 149 over a few steps which further underwent Baeyer–Villiger oxidation using magnesium monoperoxyphthalate (MMPP) and diazomethane to give a methyl ester 150 in 78% yield. Treatment of methyl ester 150 with HF.TEA, Hg(OTf)2, and tetramethyl urea (TMU) followed by Dess–Martin periodinane (DMP) furnishing a neopentyl aldehyde 151 in 72% yield (3-step sequence). Compound 151 generated the natural product, euonyminol 152 in a sequential process (Scheme 19).
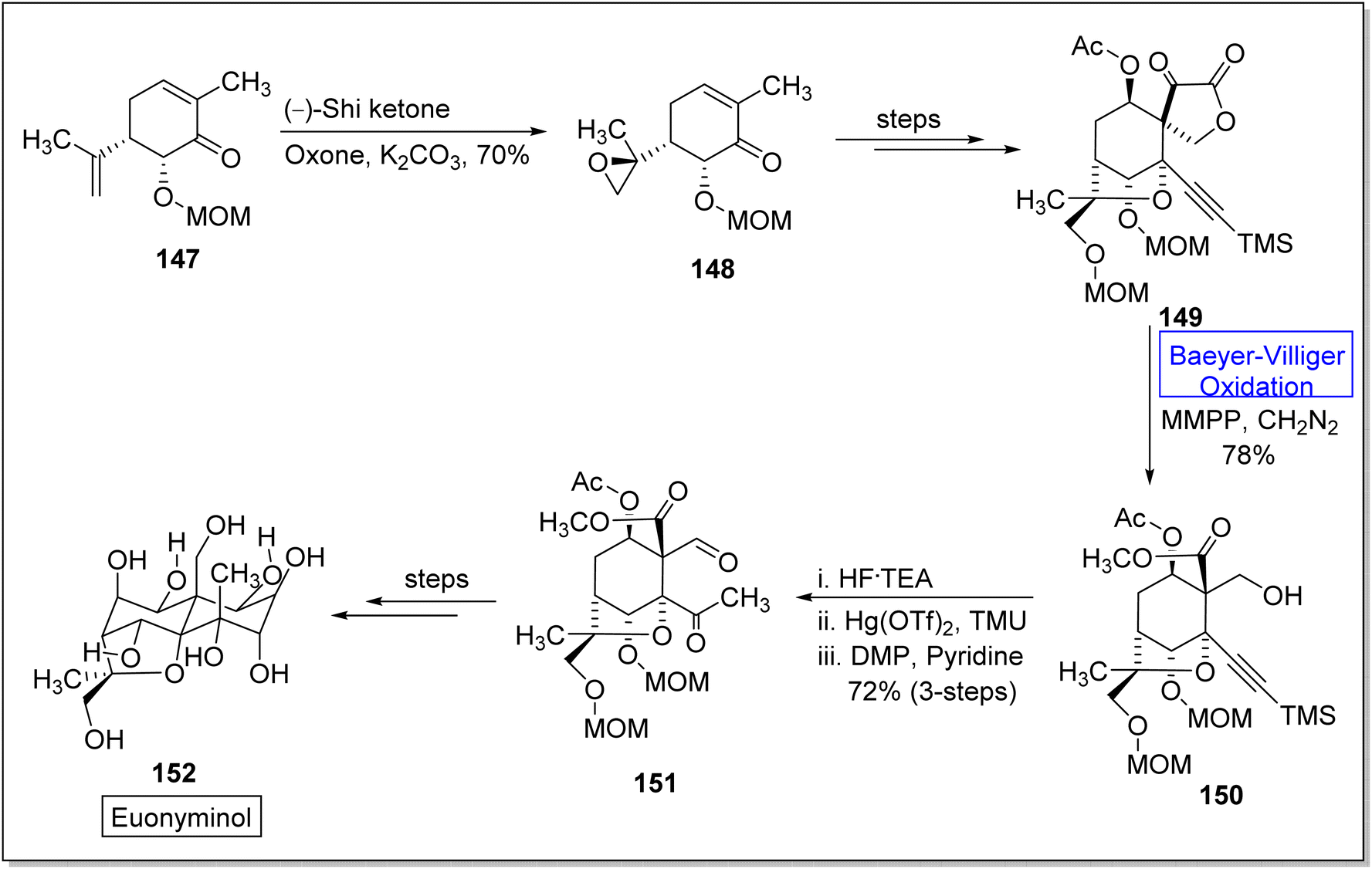 | ||
| Scheme 19 Synthesis of euonyminol 152 via Baeyer–Villiger oxidation. | ||
Asnovolin A (158) and B (159) are DMOA (3,5-dimethylorsellinic acid)-derived natural spiromeroterpenoids,139 which are pharmaceutically pivotal due to anti-bacterial activity.140,141 Yang et al. in 2022, reported the asymmetric total synthesis of asnovolins in 9–11 steps for the first time, by employing many key reactions including regioselective BV oxidation.142 The synthesis was inaugurated with the conversion of hydroxy enone 153 into neopentyl iodide 154 in the presence of Tf2O, 2,6-lutidine, DCM, NaI, acetone and NaBH4, in methanol. Neopentyl iodide 154 over a few steps afforded the compound 155 which upon further treatment with IBX, Na2HPO4, and DMSO furnishing an enone 156 (72%) with regiocontrol. Compound 156 generated an asnovolin J 157 over a few steps. Subsequently, the product 157 underwent Baeyer–Villiger oxidation with m-CPBA in DCM to synthesize an asnovolin A 158 in 76% yield which upon further reaction with p-TsOH in methanol afforded the targeted product, asnovolin E 159 in 41% yield (Scheme 20).
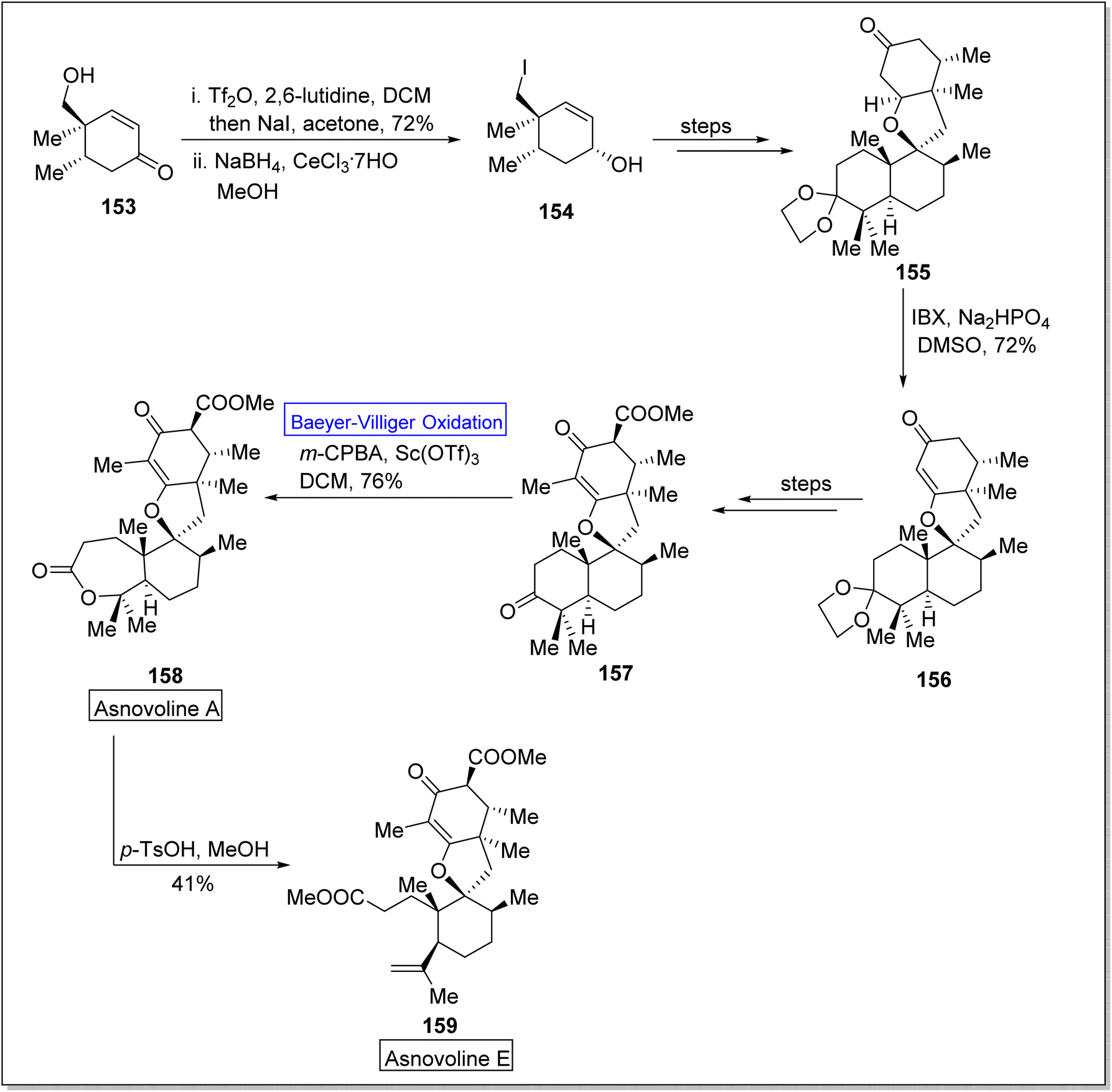 | ||
| Scheme 20 Synthesis of asnovoline A 158 and E 159 via Baeyer–Villiger oxidation. | ||
2.4. Synthesis of fatty acid-based natural products via Baeyer–Villiger oxidation
Baeyer–Villiger oxidation reaction also play a fundamental role in the synthesis of naturally occurring fatty acids which are important in medicinal field due to their biological activities (Fig. 5).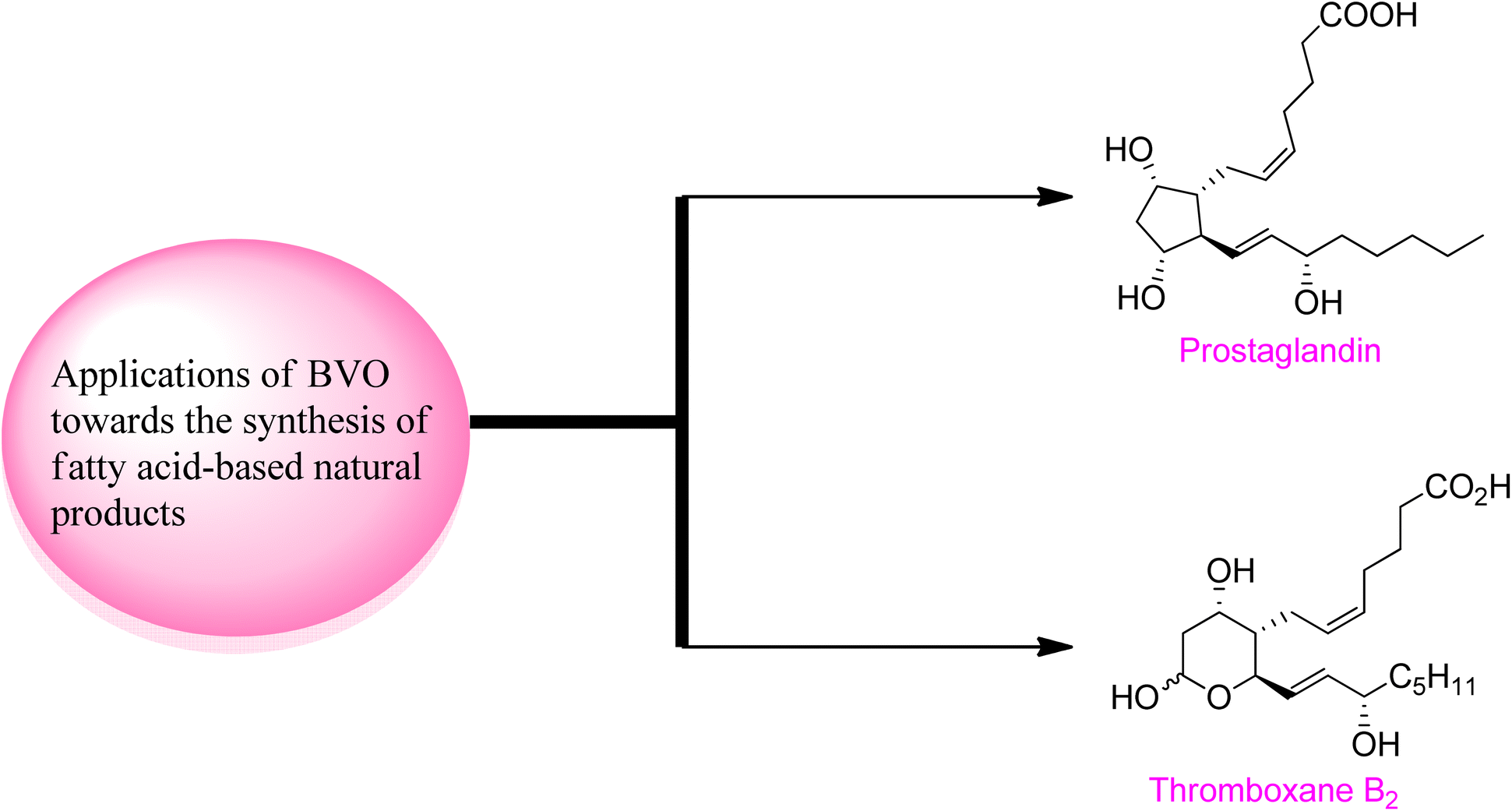 | ||
| Fig. 5 Fatty acid-based natural products synthesized by involving BVO as a key step. | ||
Prostaglandins (PGF2α) are significant chemical messengers which exhibit biological activities and have broad range applications in medicinal field.143,144 These exhibit wide range of pharmaceutical activities in contraction and dilation of smooth muscles, control of hormonal release, cell growth, suppression of acid in stomach and anti-glaucoma drugs.143 Prior to 2019, Aggarwal and Hayashi achieved the synthesis of different prostaglandins by harnessing bond disconnection methodology.145 However, these syntheses comprised lengthy schemes and complex protection/deprotection protocols. Zhu and colleagues in 2019, presented a novel protocol of enantioselective (90–99% ee) Baeyer–Villiger oxidation for the synthesis of prostaglandins in 8 steps with overall 20% yield from a lactone (−)-162.146 In the first step of synthesis, cyclobutanone 160 was subjected to Baeyer–Villiger oxidation with H2O2 in the presence of a catalyst 161 in chloroform to afford the corresponding lactone (−)-162 in 45% yield with 95% ee. In the next step, reductive dechlorination of lactone 162 with Zn/NH4Cl followed by Prins reaction with paraformaldehyde and deformylation gave diol 163 in 79% over 2-steps which further furnished an allylic alcohol 164 over a few steps. Reduction of compound 164 with DIBAL-H followed by Wittig olefination synthesized the targeted compound 4 with an overall 20% yield (Scheme 21).
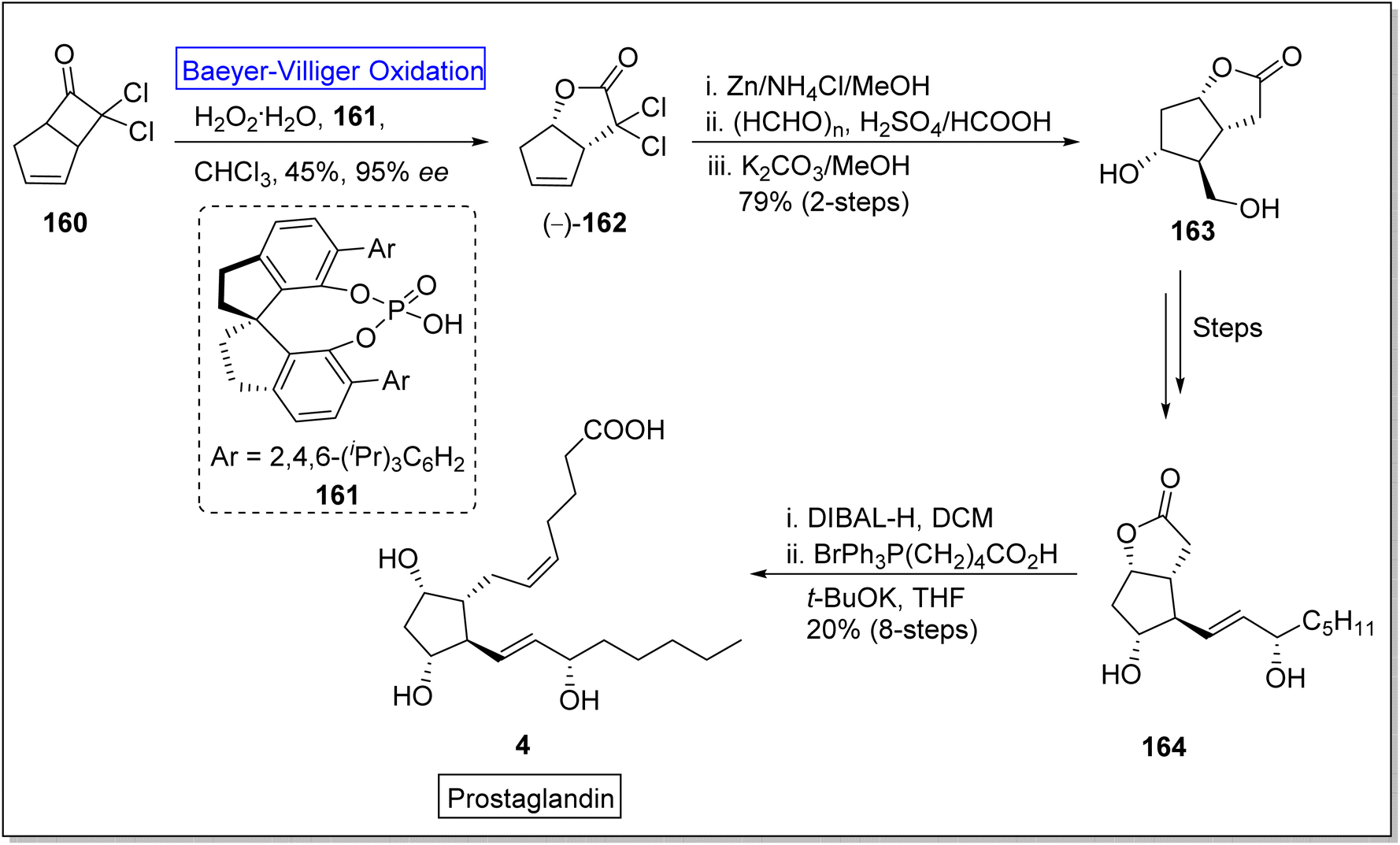 | ||
| Scheme 21 Synthesis of prostaglandin 4 via Baeyer–Villiger oxidation. | ||
Thromboxane B2 (TxB2) and thromboxane A2 (txA2) belong to lipid family known as prostanoids, which are subclass of eicosanoids containing prostaglandins. TxB2 causes thrombosis147 and effect immune system148 and it is considered as a fundamental scaffold to study prostanoid-associated biochemical phenomena. Previously reported synthetic strategies of TxB2 were lengthy, time consuming, and suffer from poor atom economy. In 2020, Jing et al. reported a twelve-step synthesis of natural prostanoid (TxB2) in overall 5% yield from 2,5-dimethoxytetrahydrofuran.149 Baeyer–Villiger oxidation is one of the key reactions involved in this asymmetric synthetic strategy. The synthesis was initialized with the formation of enal intermediate 166 in 29% yield (99% ee) from the aldol reaction of succinaldehyde 165 with L-proline. In the next step, treatment of compound 166 with para methoxy benzyl acetal (PMBOH) resulted in the synthesis of two diastereomers α-167 and β-167 (β/α 1.7:1). Then, α-isomer of PMBOH 167 was treated with vinyl cuprate using trimethylsilyl chloride (TMSCl), Et3N, and triphenyl phosphene (PPh3) to synthesize compound 168. The compound 168 was subjected to Baeyer–Villiger oxidation with m-CPBA to afford a lactone β-169 in 64% yield and α-169 in 63% yield (over three steps). Reduction, oxy methylation, and deprotection of PMB of lactone β-169 furnished a diastereomer β-170 (in 80% yield) over a few steps. Furthermore, olefination of the diastereomer β-170 using phosphonium salt with tBuOK resulted in the corresponding alkene 171 in 97% yield. Consequently, alkene 171 upon further treatment with TBAF in THF followed by hydrolysis generated the final product TxB2 172 in 90% yield (Scheme 22).
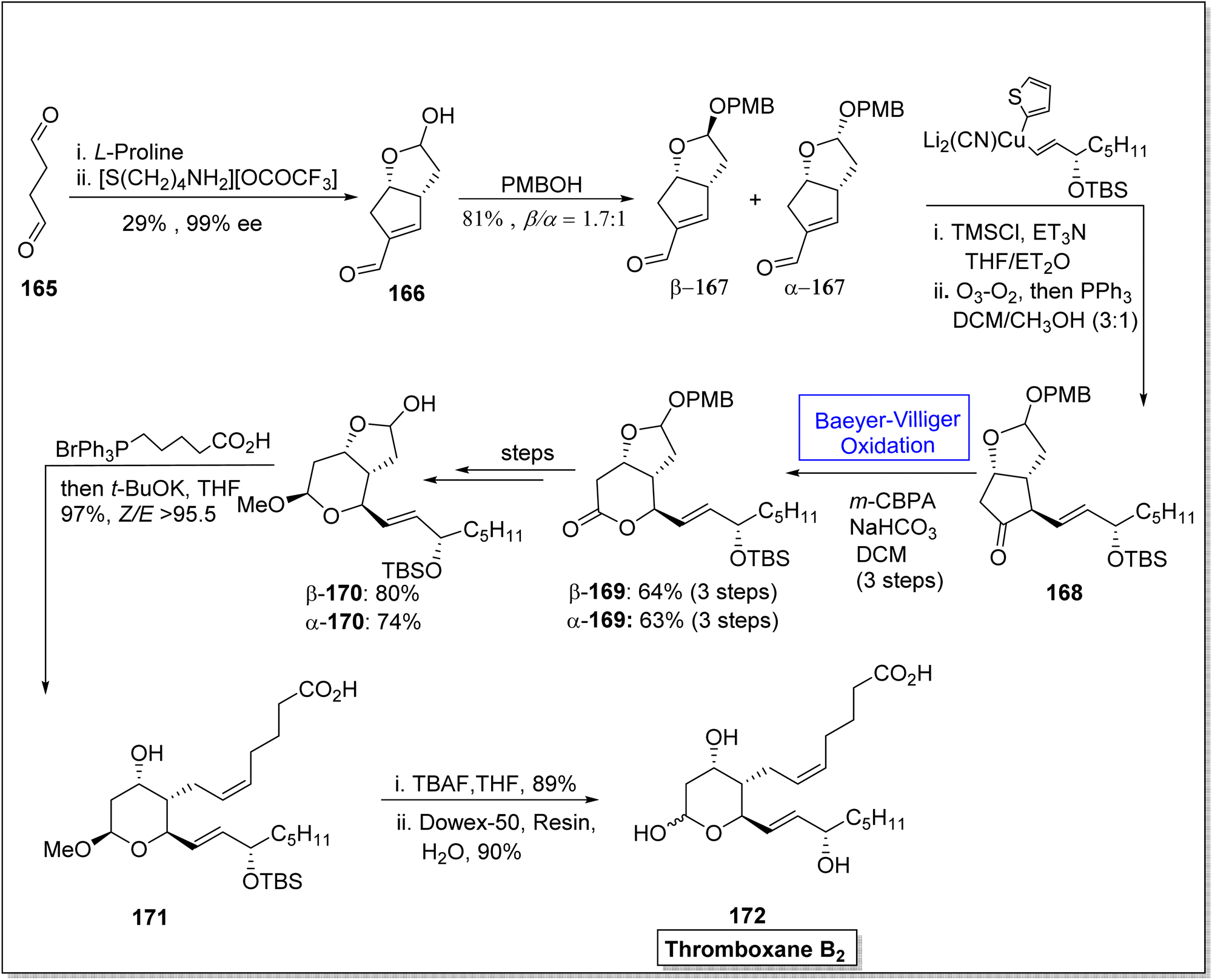 | ||
| Scheme 22 Synthesis of thromboxane B2 172 via Baeyer–Villiger oxidation. | ||
2.5. Synthesis of miscellaneous natural compounds via Baeyer–Villiger oxidation
Many other natural products can also be synthesized through Baeyer–Villiger oxidation reaction. Such as nepetoidin B, protoanemonin, terfestatins, darunavir; a protease inhibitor, cinnamic acid dimers and some hydroquinones (Fig. 6).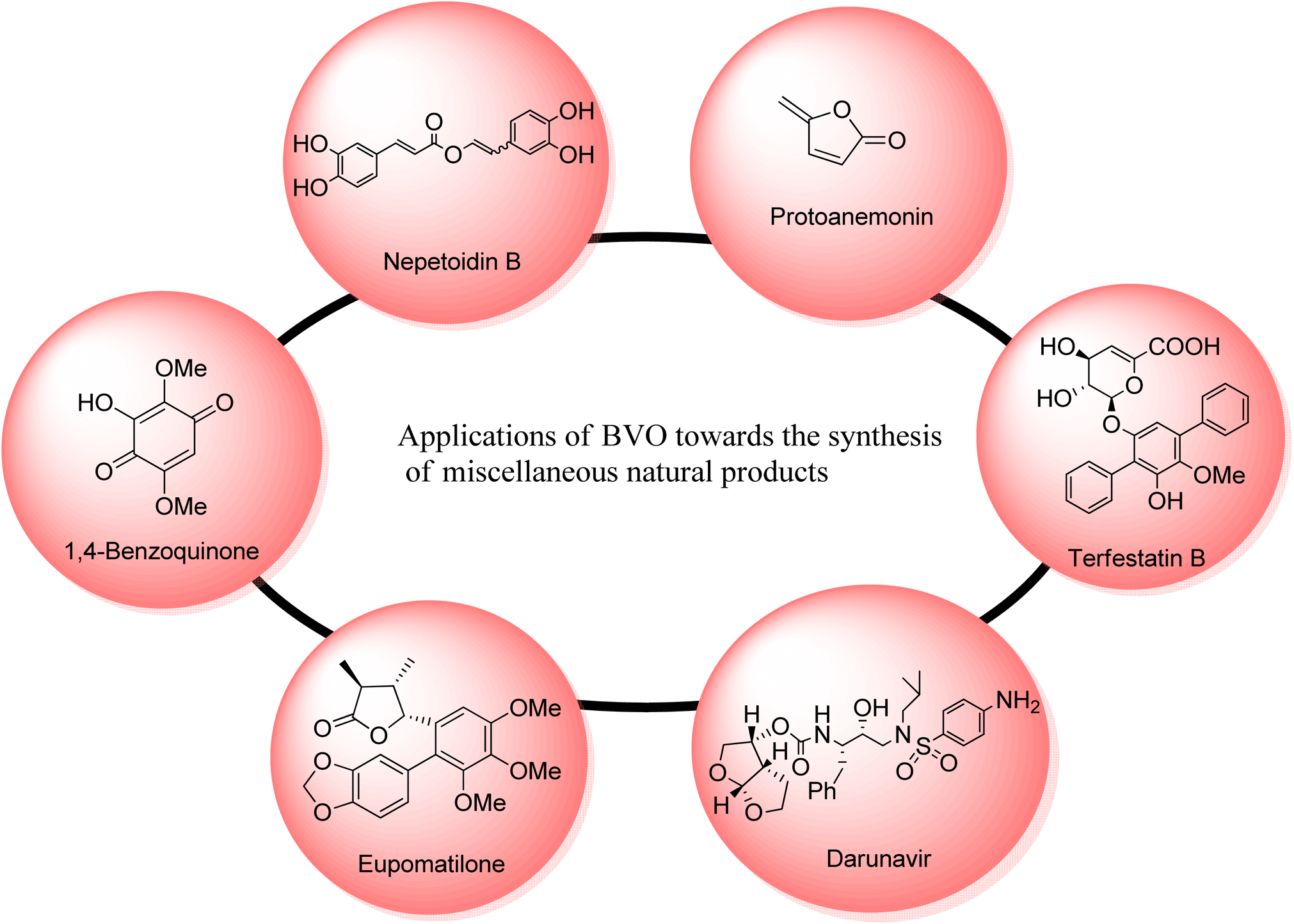 | ||
| Fig. 6 Miscellaneous natural products synthesized by involving BVO as a key step. | ||
Nepetoidin B 175 belongs to a class of natural products and initially it was extracted from Plecranthus caninus, but now it is obtained from various plants and herbs.150 It reveals many biological activities i.e., anti-bacterial, anti-cancer, and anti-fungal activities.151,152 Despite of these medicinal features, no synthetic strategies of compound 175 have been reported yet. In 2018, Timokhin et al. reported a two-step first synthesis of nepetoidin B (94% ee) in overall 17% yield by employing Baeyer Villiger oxidation as a significant step.153 In the first step of schematic route, 1,5-bis(3,4-dimethoxyphenyl)-1,4-pentadiene-3-one 173 was converted into tetramethylated nepetoidin B 174 in 40% yield via Baeyer–Villiger oxidation with oxone in DMF. In the second step, compound 174 delivered the target product, nepetoidin 175 in overall 17% yield via demethylation using BBr3 (Scheme 23).
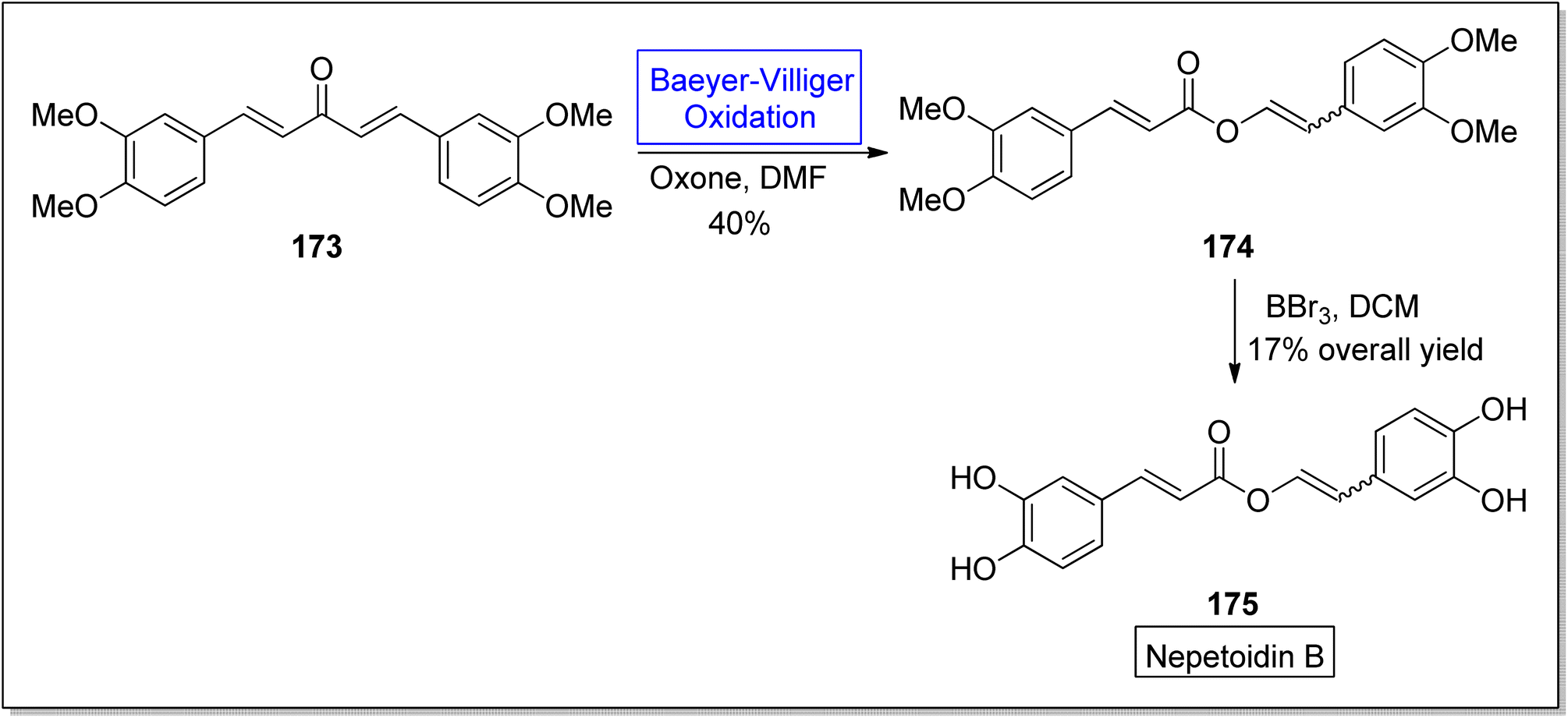 | ||
| Scheme 23 Synthesis of nepetoidin B 175 via Baeyer–Villiger oxidation. | ||
Various efforts to synthesize anti-microbial agents have been carried out to overcome several microbes causing diseases.154 Protoanemonin 179 is an antimicrobial155 γ-lactone which is obtained from members of Ranunculaceae family.156 Protoanemonin is famous for its pervasive and irritant properties, hence, may cause dermatitis.156–159 Alibes and group synthesized protoanemonin in overall 25% yield via photo-oxidation process.160 Similarly, Mliki and Trabelsi attempted to synthesize the targeted compound using CH3COOH and sodium perborate but did not detect the product.161 All previously reported methods involved high cost, scarcely available and contaminated reagents. In 2020, Martinez and companions reported the 1st one-pot selective synthesis of γ-alkylidenebutenolide (protoanemonin) in overall 25% yield from D-fructose 176 through 5-(hydroxy methyl) furfural 177 by employing Baeyer–Villiger oxidation.162 In the beginning, D-fructose 176 was converted into 5-(hydroxy methyl) furfural 177 in 98% yield in the presence of hypophosphorous acid (HPA). Homogenous autocatalysis and H2O2-mediated-Baeyer–Villiger oxidation of compound 177 in chlorinated solvent afforded the protoanemonin 179 in 94% yield from 177 and 28% yield from 176 (Scheme 24).
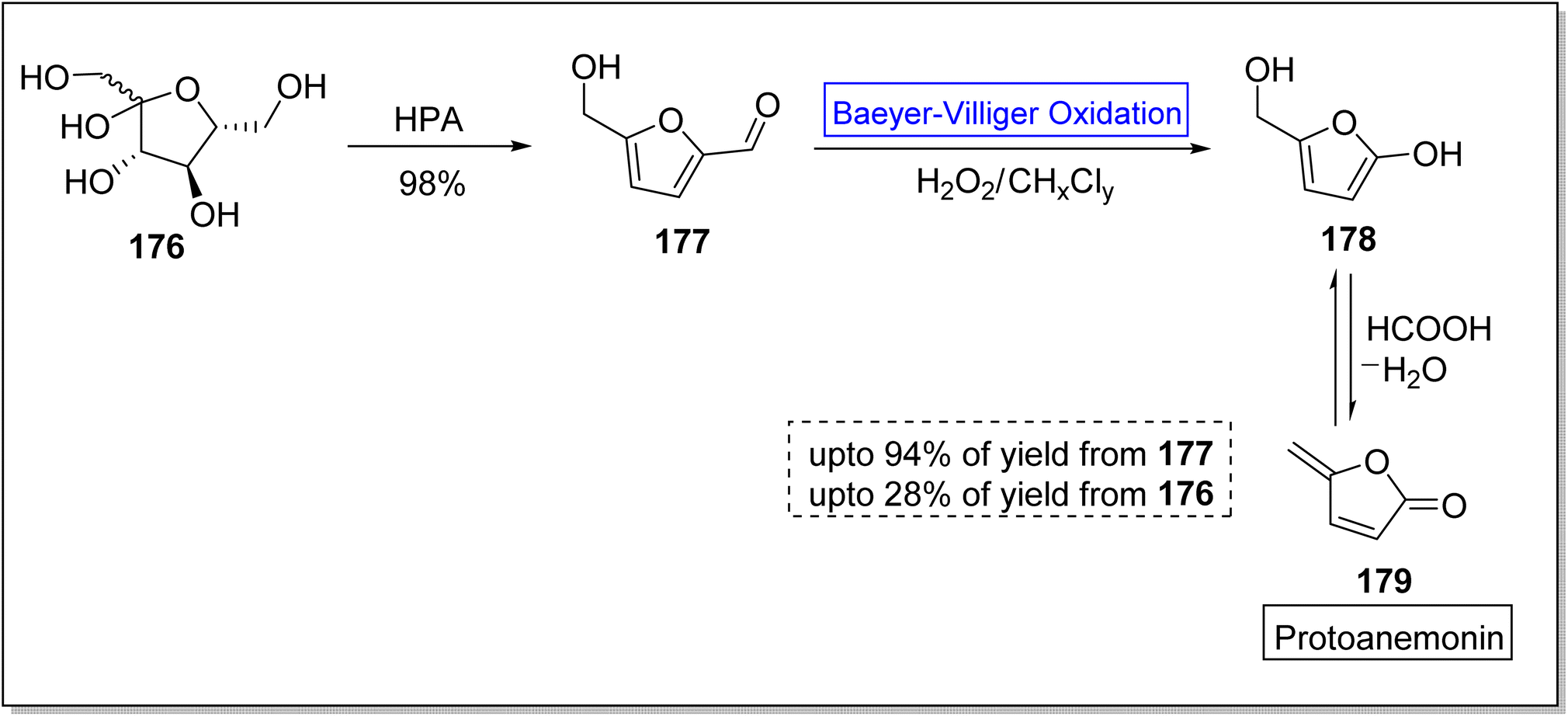 | ||
| Scheme 24 Synthesis of protoanemonin 179 via Baeyer–Villiger oxidation. | ||
Terfestatin A 186 and B 187 are natural products which belong to terfestatin family and both are isolated from Streptomyces species. Terfestatin A 186 and B 189 reveal potent inhibitory and neuroprotective activities, respectively.163 In 2020, Sugawara and colleagues presented the synthesis of terfestatin A with 21% overall yield (in 5-steps) and 1st total synthesis of the terfestatin B with 30% overall yield (in 8-steps) from aromatic aldehyde by engaging Baeyer–Villiger oxidation reaction as a key step.164 For this purpose, 2,5-dibromohydroquinone 180 was converted into an aromatic aldehyde 181 in 78% yield by reacting with hexamethylene tetramine (HMTA) and TFA. Then, aldehyde 181 under optimized conditions afforded a terphenyl derivative 182 in 91% yield. Glucosylation of compound 182 with glycosyl bromide 183 was carried out to give a monomer 184 in 66% yield. Subsequently, the monomer 184 underwent the Baeyer–Villiger oxidation with m-CPBA to generate a compound 185. Furthermore, compound 185 was reacted with TBAF and LiOH, in THF to synthesize terfestatin A 186 in 39% yield. In another approach, O-methylation of compound 182 with MeI in the presence of K2CO3 in DMF afforded compound 187. Compound 187 underwent Baeyer–Villiger oxidation with m-CPBA to furnish compound 188 in 90% yield which further synthesized terfestatin B 189 over a few steps (Scheme 25).
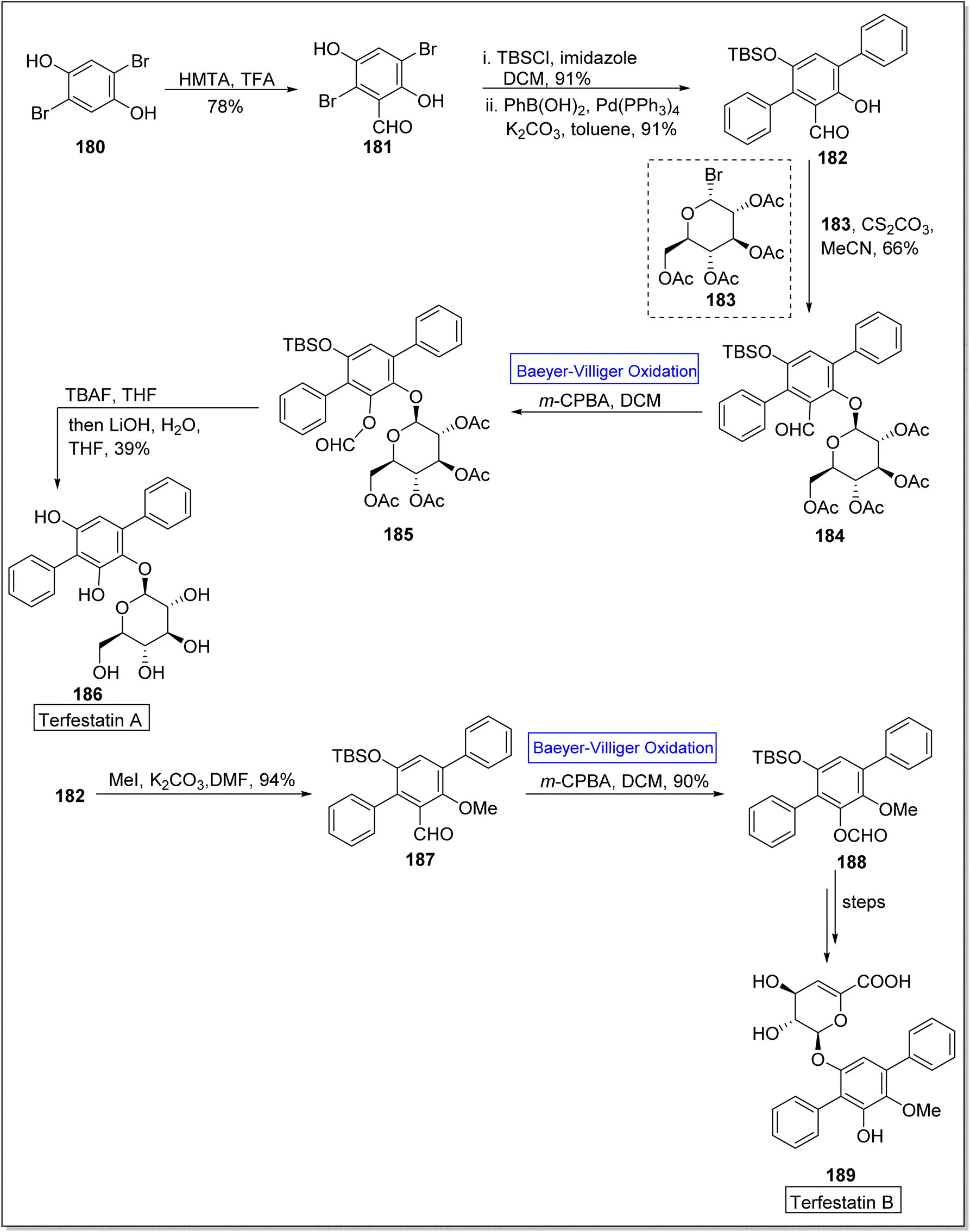 | ||
| Scheme 25 Synthesis of terfestatin A 186 and terfestatin B 189 via Baeyer–Villiger oxidation. | ||
Darunavir 2 is a potent natural drug which belongs to protease inhibitor therapeutics, an important part of cART (combination antiretroviral therapies) procedure.165 It is widely used to treat HIV type-1 disease and AIDS.166,167 The key element of darunavir 2 is bis-THF [bicyclic (3R,3aS,6aR)-bis-tetrahydrofuran] ligand alcohol. Initially, Ghosh and co-workers readily synthesized bis-THF alcohol via lipase-promoted enzymatic resolution of employing (3R)-diethyl malate as precursor. However, they obtained the target molecule in 92–96% ee. Quaedflieg and colleagues synthesized bis-THF by employing diastereoselective Micheal addition as key step, Black et al. reported the synthesis by employing Mukaiyama aldol reaction, and Xie group carried out the Lewis acid-mediated synthesis. In 2020, Ghosh et al. extended their work to achieve more enantioselective synthesis of optically pure bis-THF ligand (99% ee) from xylose.168 The synthesis was carried out by utilizing many named reactions including Baeyer–Villiger oxidation as significant step. The synthesis was commenced with the selective protection of alcohol as benzoate derivative of benzoyl chloride in presence of DMAP and pyridine to generate a compound 191 in 95% yield. Then, Swern oxidation of compound 191 followed by Wittig olefination with (carboethoxy methylene) triphenyl phosphorane in DCM afforded an α,β-unsaturated ester 192 in 85% yield with E/Z = 1:8 (over 2-steps). Afterwards, catalytic hydrogenation of Z-isomer 192 with 10% Pd/C in ethanol followed by reaction with BF3·OEt2 and Et3SiH in the presence of K2CO3 and methanol produced a bicyclic alcohol 193 in 87% yield. In the next step, compound 193 was converted into lactone 194 in 41% yield over 3-steps including Dess-Martin oxidation, Baeyer–Villiger oxidation with m-CPBA and acidification in MeOH. Reduction of methyl acetal 194 with LiAlH4 in THF gave bis-THF alcohol 195 in 63% yield. When ligand 195 was reacted with active N,N′-disuccinimidyl carbonate 196 in MeCN, it yielded carbonate 197 in 65% yield. In the last step, compound 197 upon further reaction with Cbz (benzyl chlorocarbonate) derivative 198, furnished the darunavir 2 in 53% yield (Scheme 26).
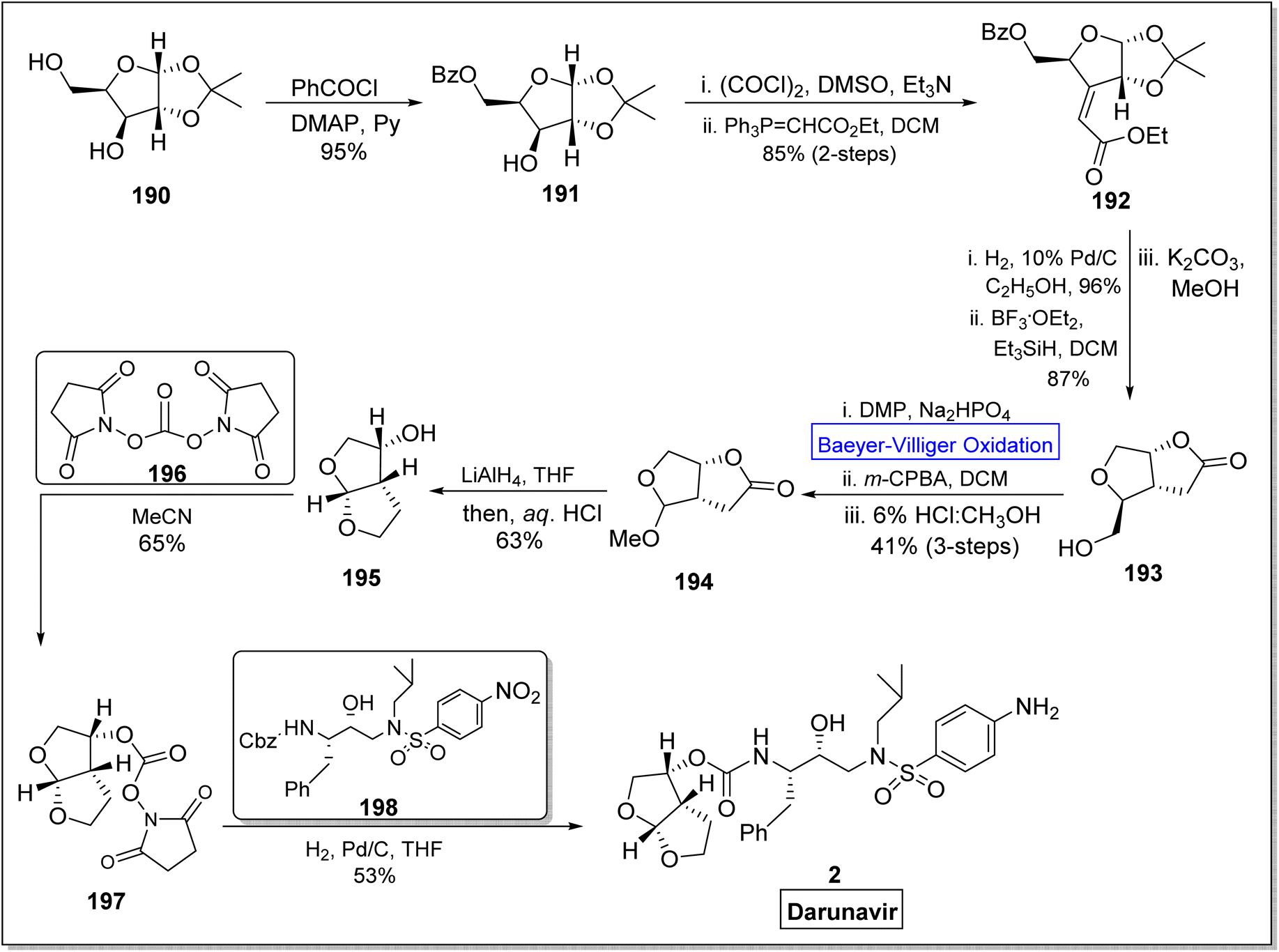 | ||
| Scheme 26 Synthesis of protease inhibitor; darunavir 2 via Baeyer–Villiger oxidation. | ||
Eupomatilones are natural products (cinnamic acid dimers)169 related to lignan family and these were isolated by Carroll and Taylor (in 1991) from a shrub Eupomatia bennettii.170 These are dispersed throughout the plant stems, leaves, roots, seeds and fruits, and exhibit anti-fungal, anti-HIV and anti-cancer activities. In 2022, Zhang et al. achieved the total synthesis of natural eupomatilones 204 and 205 from a cyclic ketone 199 by employing Cu(II)complex-catalyzed asymmetric BV oxidation.171 The synthesis was initiated with the Pd-regulated arylation of 3-methylcyclobutanne-1-one 199 with 1-bromo-3,4,5-trimethyloxybenzene 200 to prepare a racemic precursor 201 in 65% yield (d:r = 5.6:1). To improve the diastereomeric ratio, the precursor 201 was treated with p-toluenesulfonic acid in chloroform. Then, compound 201 underwent Baeyer–Villiger oxidation with m-CPBA followed by reaction with Cu(NTf2)2 to furnish a chiral lactone 202 in 48% yield with 92% ee. Furthermore, lactone 202 afforded a γ-butyrolactone 203 over a few steps. Finally, compound 203 afforded the eupomatilone 204 in 67% yield with 94% ee via reacting with Eschenmoser's salt in THF followed by Baeyer–Villiger oxidation with m-CPBA. In another route, compound 203 produced eupomatilone 205 in 70% yield with 95% ee via methylation with LiHMDS and MeI in THF (Scheme 27).
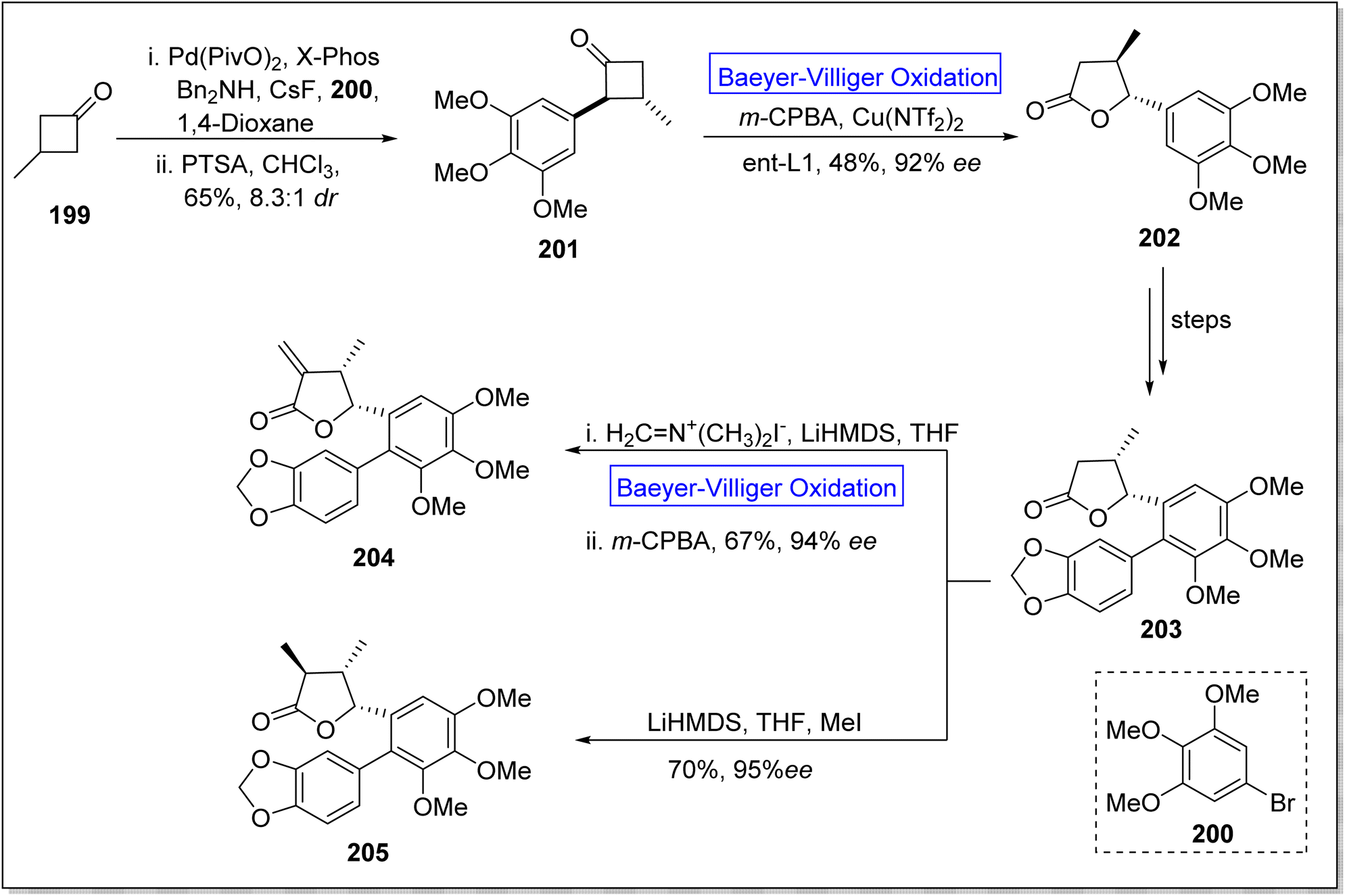 | ||
| Scheme 27 Synthesis of eupomatilones 204 and 205 via Baeyer–Villiger oxidation. | ||
Hydroquinones and 1,4-benzoquinones are natural metabolites and these are widespread in marine organisms,172,173 plants, and animals.174,175 These compounds exhibit anti-oxidant and anti-cancer activities in animals. In 2023, Tsyganov et al. reported the synthesis of 3-hydroxy-2,5-dimethoxybenzo-1,4-quinone1,4-benzoquinone 209 by using Baeyer–Villiger oxidation reaction as a key step.176 The synthesis was accomplished with the formation of apiol aldehyde 207 from apiol alkene 206 in the presence of KOH, chloroform, methanol, and pyridine. Apiol aldehyde 207 underwent Baeyer–Villiger oxidation with H2O2 in the presence of H2SO4 and methanol to furnish a phenol 208 in 78% yield. Then, oxidation of phenol 208 followed by ring opening of dioxolane resulted in the synthesis of target compound 209 in overall 73% yield (Scheme 28).
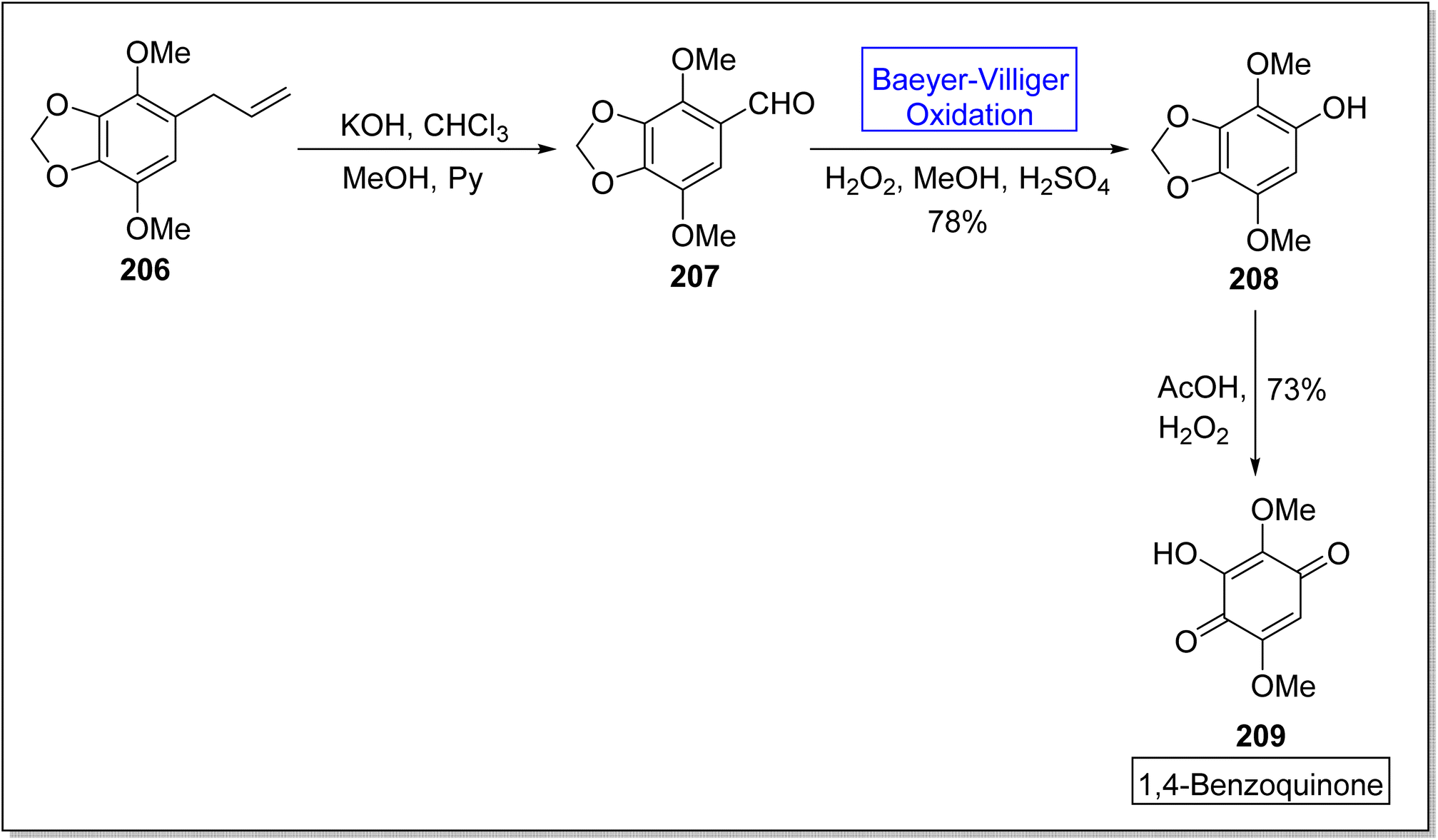 | ||
| Scheme 28 Biosynthesis of benzoquinone 209 via Baeyer–Villiger oxidation. | ||
2.6. Baeyer–Villiger oxidation in the biosynthesis of natural products
There is a diversity of enzymes which can be used in selective oxidation reactions in nature. These may include oxidases,177 peroxidases,178 monooxygenases,179 dioxygenases,180 and dehydrogenases.177 Baeyer–Villiger monooxygenases play a key role in the biosynthesis of many naturally occurring compounds (Fig. 7).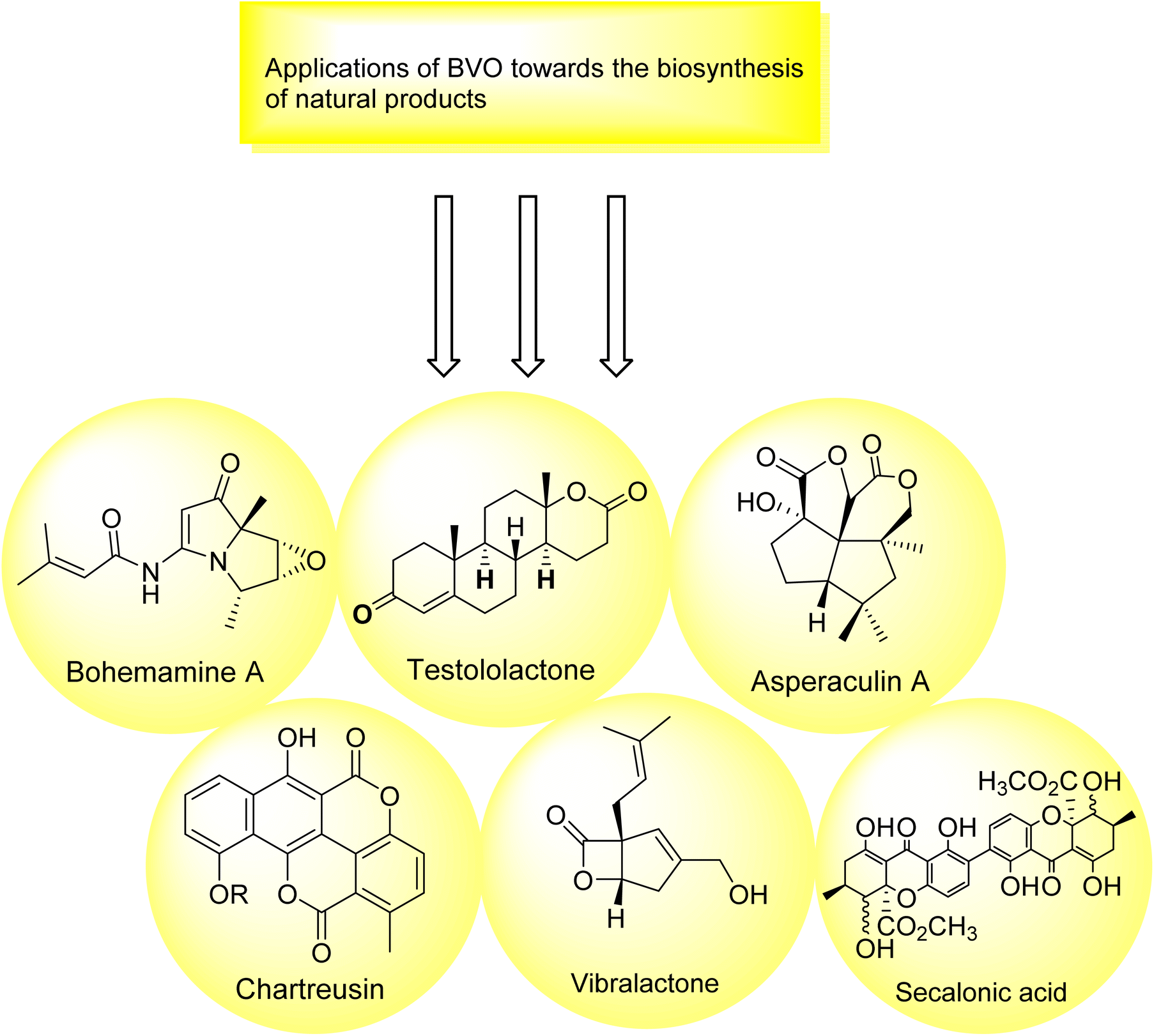 | ||
| Fig. 7 Pictorial representation of role of BVO in biosynthesis. | ||
Bohemamines (BHMs) 214 are pyrrolizidine-based bacterial alkaloids. These are obtained from Streptomyces species and homospermidine. These are biologically active natural compounds. These are used for self-defense against amoebae. Baeyer–Villiger monooxygenases BhmK/BhmJ and BhmG has a key role in the economical biosynthesis of BHMs. In 2020, Liu et al. presented the biosynthesis of BHMs from genes by utilizing BVMOs (BhmK and BhmJ) as biocatalysts.181 The synthetic scheme of BHMs was demonstrated into two routes. In the first route, compound BhmJ was treated with a co-enzyme to afford BHM 210 which was further reacted with BhmK to generate 211. Resulted compound 211 by reacting with Baeyer–Villiger monooxygenase afforded BHM D 212 while in another route, compound 211 synthesized compound 213 which further generated BHMs 214a, 214b & 214c on treatment with BhmA (Scheme 29).
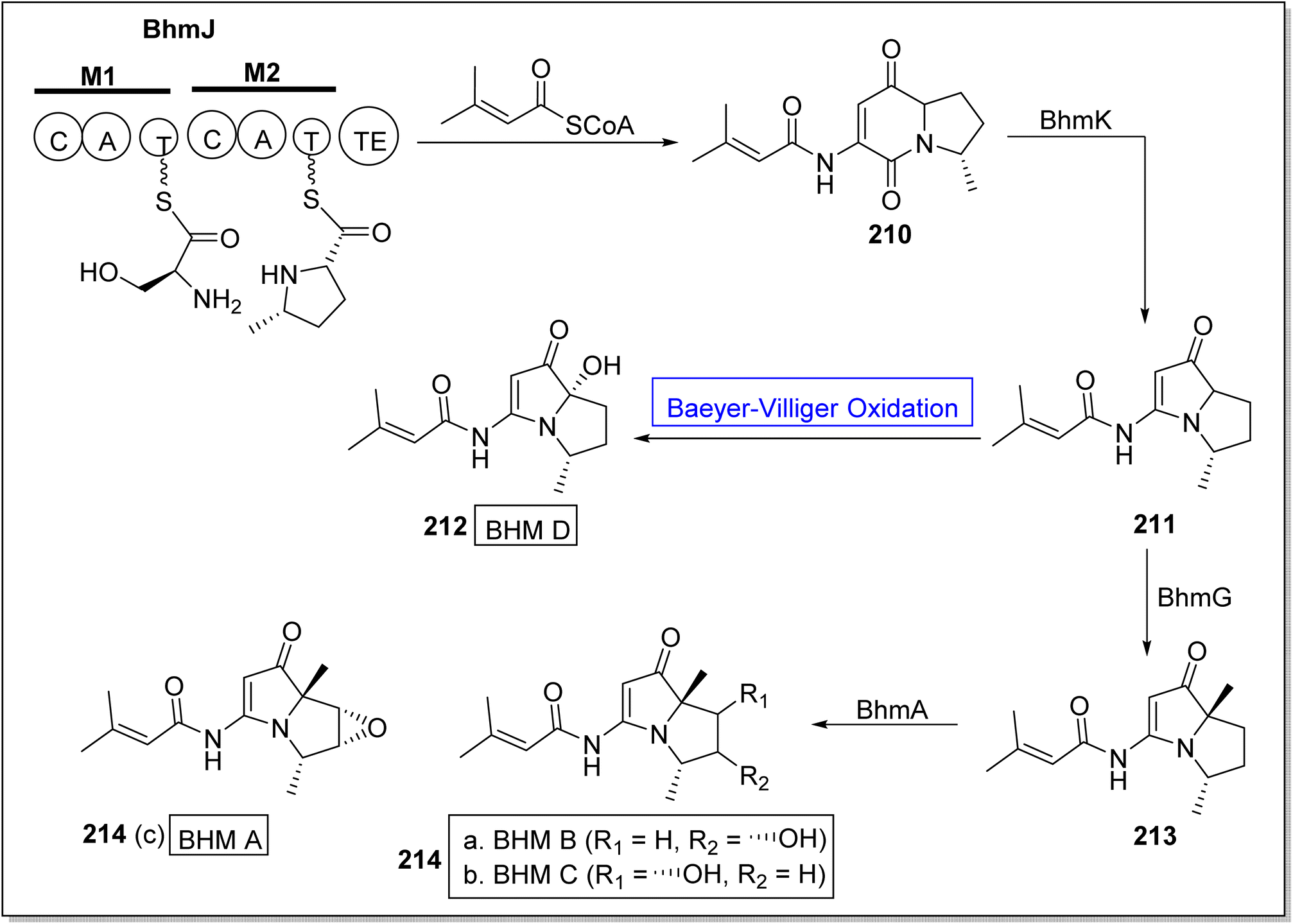 | ||
| Scheme 29 Synthesis of bohemamines 212 and 214 (a–c) via Baeyer–Villiger oxidation. | ||
Testosterone 216 is a significant male hormone responsible for the male characteristics. Testololactone is also a steroidal compound used to treat breast cancer, prostate cancer and prostatic hyperplasia.182 In 2020, Paula et al. reported the biosynthesis of testosterone and testololactone by utilizing Baeyer–Villiger oxidation as a key reaction.182 The conversion was proceeded with the formation of testosterone 216 from progesterone 215 by using Baeyer–Villiger monooxygenase followed by hydrolysis. Then, compound 216 further delivered testololactone 3 via dehydrogenation and Baeyer–Villiger oxidation using BVMO (Scheme 30).
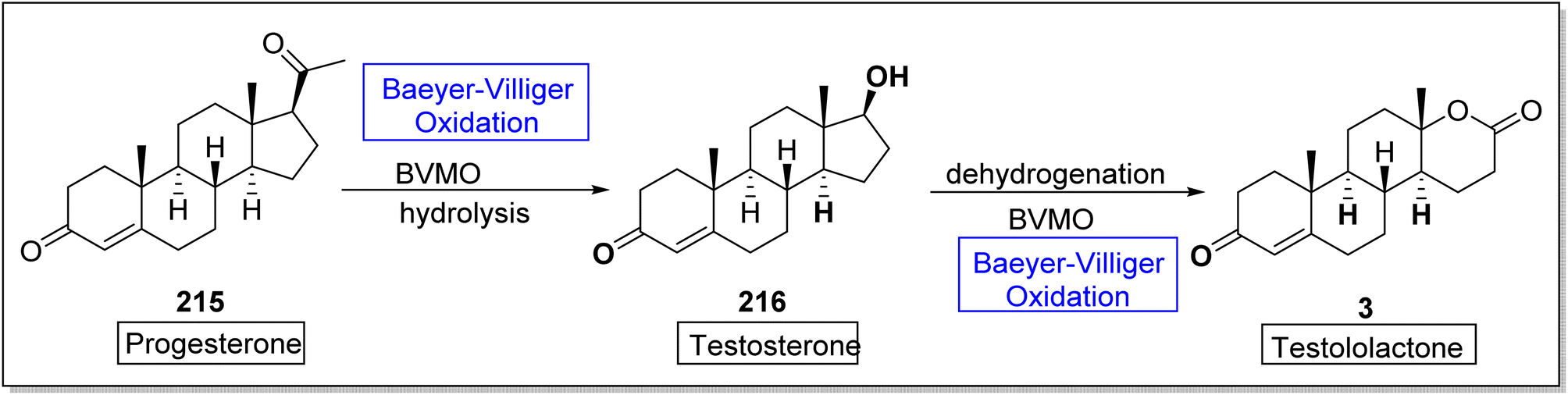 | ||
| Scheme 30 Synthesis of testosterone 216 and testololactone 3 via Baeyer–Villiger oxidation. | ||
Asperculin A 220 and penifulvin D 222 are fungal sesquiterpenes which consist of [5.5.5.6] dioxafenestrane ring.183–185 These are biologically active natural products. Primarily, asperculin A 220 and penifulvin D 222 were extracted from a terrestrial fungus; Penicillium griseofulvum and marine fungus; Aspergillus aculeatus respectively. In their previous work, Wei and colleagues reported that a 3-gene cluster (peni gene) was responsible for the biosynthesis of penifulvin A. Although the pathway was fully established but stereo and regiochemical hydroxylation on sp3 carbon remained unclear. In 2021, Wei and co-workers presented the biosynthesis of asperculin A and penifulvin D by using Baeyer–Villiger monooxygenases PeniC and AspeB which provided valuable biocatalysts and wide range of strategies for nonactivated C-oxidation modification.186 The synthesis was began with the formation of a scaffold silphinene 218 from a precursor FPP (farnesyl pyrophosphate) 217 in presence of sesquiterpene cyclase (PeniA) and AspeG. The scaffold 218 generated penifulvin A 219 over a few steps. Compound 219 in one approach, afforded asperculin A 220 via AspeB-catalyzed Beyer–Villiger oxidation in the presence of PeniF and AspeC. In another approach, it furnished penifulvin D 222 on reaction with PeniF (BVMO) after synthesizing penifulvin A 221 in the presence of PeniC (Scheme 31).
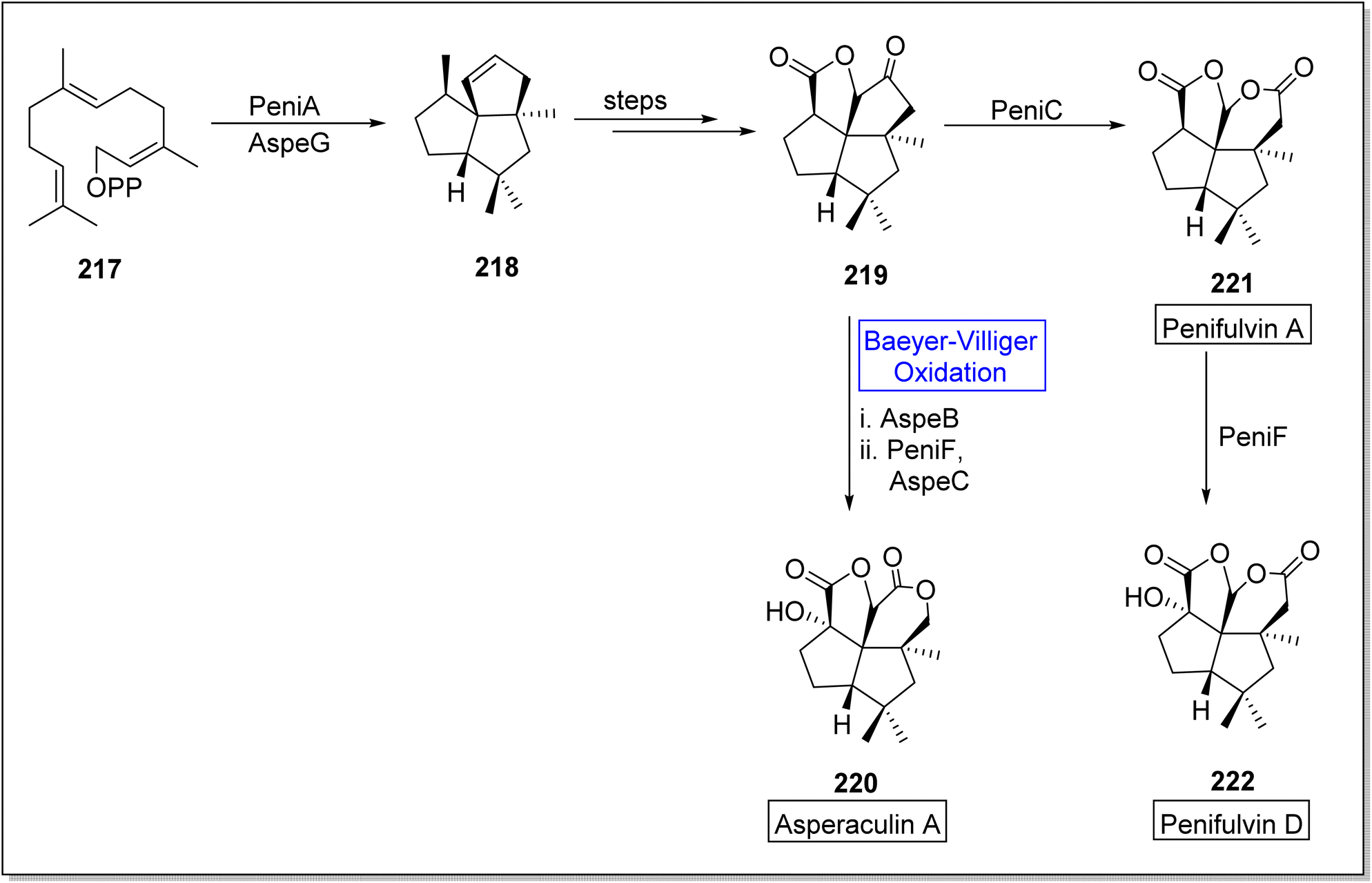 | ||
| Scheme 31 Synthesis of asperaculin A 220 and penifulvin D 222 via Baeyer–Villiger oxidation. | ||
Chartreusin 227 is a glycosidic aromatic polyketide which is extracted from Streptomyces chartreusis. It is well-known for its potent biological activity against tumor cells.187 Initially, Xu and colleagues investigated the role of flavin-dependent ChaZ in the gene cluster of cha but chaZ gene suppressed the synthesis of compound 227. For better understanding, Jiao et al. in 2021, presented the biosynthesis of natural compound chartreusin 227 by using flavin-dependent BVMO (ChaZ), redox enzymes, and NADPH-dependent ketoreductase ChaE.187 Schematic route demonstrated the synthesis of compound 224 from a cofactor-scaffold (acetyl-CoA + MeI-CoA) 223 in the presence of a framework of enzymes which was further converted into decaketide scaffold of tetracyclic intermediate 225 over a few steps. Consequently, scaffold, resomycin C 225 underwent ChaZ-catalyzed Baeyer–Villiger oxidation to furnish a pentacyclic intermediate 226. Compound 226 generated the desired pentacyclic natural product; chartreusin 227 in the presence of ketoreductase ChaE, dioxygenase ChaP, ChaGT1 and, ChaGT2 (Scheme 32).
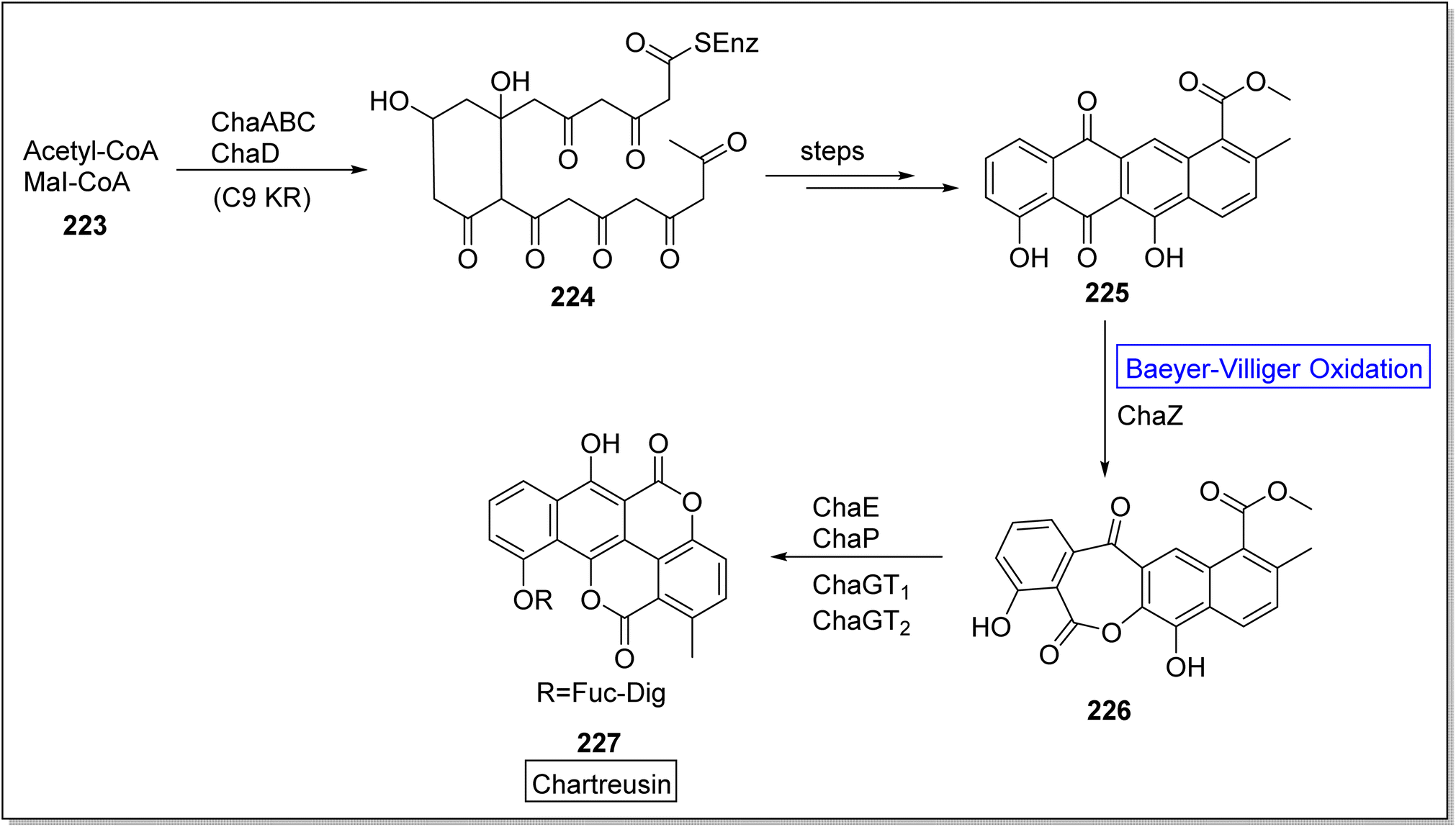 | ||
| Scheme 32 Synthesis of chartreusin 227 via Baeyer–Villiger oxidation. | ||
Secalonic acid D 231 is a fungal xanthone-based homodimer of blennolide B. It is biologically active natural product which encourages cancerous cells apoptosis,188 suppresses DNA topoisomerase I,189 and represses tumor angiogenesis.190 Broad range specificity of enzyme AacuE (used for the synthesis of dimeric molecules) make it an efficient biocatalyst. Initially, Wei group attempted to achieve the total bioinspired synthesis of compound 231 but they obtained four isomers of targeted compound 231 owing to activity of A.oryzae (an endogenous enzyme). In 2021, Wei et al. successfully achieved the biosynthesis of secalonic acid D by overexpressing the BVMO (AacuH) that competed with endogenous enzyme.191 In the beginning, crysophanol 228 underwent AacuH-catalyzed Baeyer–Villiger oxidation followed by hydrolysis to generate monodictyphenone 229. Then, methyl esterification of compound 229 with methyltransferase AacuQ in the presence of NsrG synthesized the compound 230. Compound 230 afforded the xanthone diamers (secalonic acid B, D, and F having specific stereochemistry) 231 over a few steps (Scheme 33).
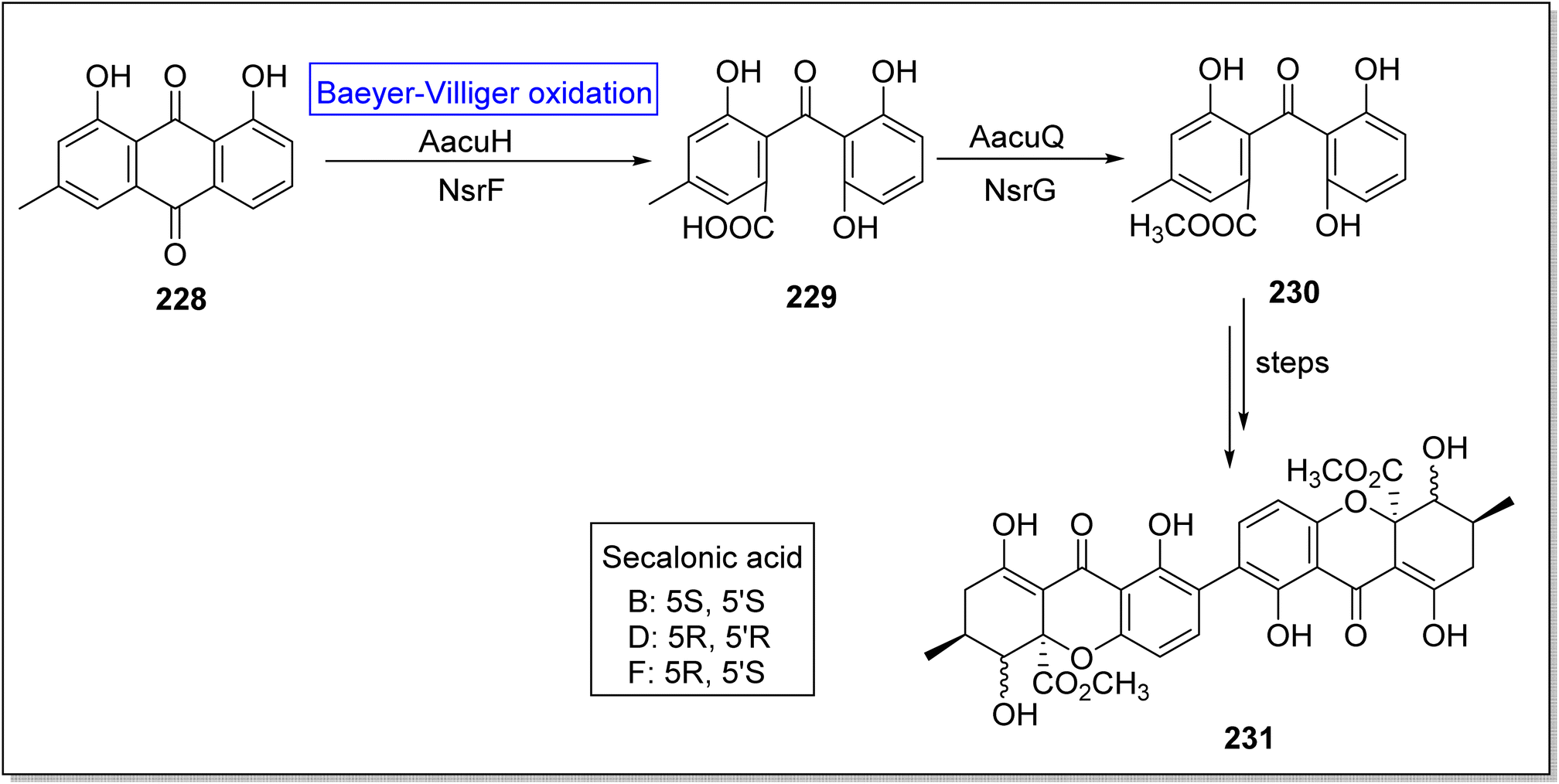 | ||
| Scheme 33 Synthesis of secalonic acid (B, D and F) 231 via Baeyer–Villiger oxidation. | ||
Vibralactone 236 is a rare and potent natural product containing oxepine-2(3H)-one ring. It is isolated from a basidiomycete fungus Boreostereum vibrans; Sterum vibrans and Stereum mushrooms, together with 1,5-seco-vibralactone. Vibralactone 236 has the potential to inhibit pancreatic lipase with 0.4 μg mL−1 value of IC50. Feng et al. in 2023, reported the total biosynthesis of vibralactone 236 by using Baeyer–Villiger monooxygenases.192 For this purpose, 4-hydroxybenzoate 232 was catalyzed by UbiA prenyltransferases (VibP1/VibP2) in the presence of isoprenoid precursor, dimethylallyl pyrophosphate (DMAPP), a carboxylic acid reductase BvCAR, NADPH and ATP to generate an aldehyde 233. Then, aldehyde 233 was catalyzed by reductases BvARs and NADPH to furnish a benzylic alcohol 234. Thereafter, alcohol 234 underwent a VibO-interceded enzymatic Baeyer–Villiger oxidation in the presence of NADPH and O2 to afford a 1,5-seco-vibralactone 235. Subsequently, compound 235 afforded the targeted product 236 through VibC action (Scheme 34).
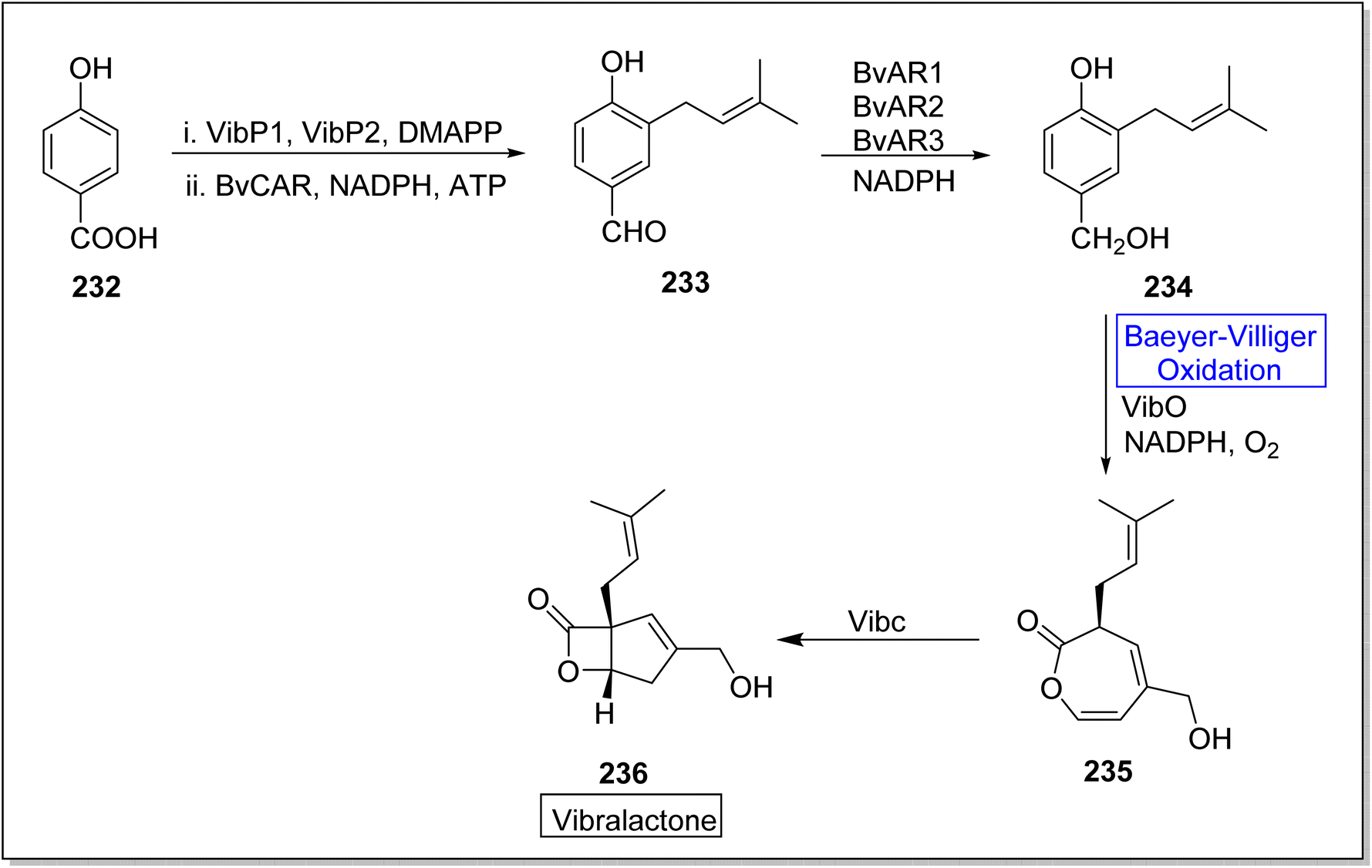 | ||
| Scheme 34 Synthesis of vibralactone 236 via Baeyer–Villiger oxidation. | ||
3. Conclusion
This review highlights the synthesis of natural products and their derivatives in which Baeyer–Villiger oxidation has been used as a key reaction. Baeyer–Villiger oxidation reaction offers the chiral lactones as valuable intermediates which can be used for consecutive reactions. Such conversions can be carried out in the presence of eco-friendly reagents, novel Baeyer–Villiger monooxygenases and mild reaction conditions to achieve high enantioselectivity, regioselectivity, and desired amount of yield with no or less side products. The utility of BV oxidation in the synthesis of various natural products i.e., polyketides, terpenoids, quinones, fatty acids, and alkaloids which are pharmaceutically important due to their potent biological activities have been demonstrated. The efficiency and superiority of this reaction is expected to motivate organic chemists to focus on Baeyer–Villiger oxidation reaction to develop efficient pathways for further advancements towards the synthesis of natural products in future.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Irfan extends his appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Groups Research Project under grant number (RGP2/156/45). A. R. Chaudhry is thankful to the Deanship of Graduate Studies and Scientific Research at the University of Bisha, for supporting this work through the Fast-Track Research Support Program.References
- A. Cavarzan, G. Bianchini, P. Sgarbossa, L. Lefort, S. Gladiali, A. Scarso and G. Strukul, Chem.–Eur. J., 2009, 15, 7930–7939 CrossRef CAS PubMed.
- I. A. Yaremenko, V. A. Vil, D. V. Demchuk and A. O. Terent'ev, Beilstein J. Org. Chem., 2016, 12, 1647–1748 CrossRef CAS PubMed.
- H. Leisch, K. Morley and P. C. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed.
- C. Jiménez-Sanchidrián and J. R. Ruiz, Tetrahedron, 2008, 64, 2011–2026 CrossRef.
- M. D. T. Frisone, F. Pinna and G. Strukul, Organometallics, 1993, 12, 148–156 CrossRef.
- W. v. E. Doering and L. Speers, J. Am. Chem. Soc., 1950, 72, 5515–5518 CrossRef CAS.
- M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 1999, 737–750 CrossRef.
- R. Criegee, Justus Liebigs Ann. Chem., 1948, 560, 127–135 CrossRef CAS.
- W. Adam, Acc. Chem. Res., 1989, 22, 13 CrossRef.
- W. v. E. Doering and E. Dorfman, J. Am. Chem. Soc., 1953, 75, 5595–5598 CrossRef CAS.
- C. Bolm, G. Schlingloff and K. Weickhardt, Angew Chem. Int. Ed. Engl., 1994, 33, 1848–1849 CrossRef.
- A. Gusso, C. Baccin, F. Pinna and G. Strukul, Organometallics, 1994, 13, 3442–3451 CrossRef CAS.
- L. Belaroui and A. Bengueddach, Clay Miner., 2012, 47, 275–284 CrossRef CAS.
- S.-I. Murahashi, S. Ono and Y. Imada, Angew. Chem., Int. Ed. Engl., 2002, 41, 2366–2368 CrossRef CAS PubMed.
- S. Xu, Z. Wang, X. Zhang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2008, 47, 2840–2843 CrossRef CAS PubMed.
- B. Wang, Y.-M. Shen and Y. Shi, J. Org. Chem., 2006, 71, 9519–9521 CrossRef CAS PubMed.
- R. Ashraf, A. F. Zahoor, K. G. Ali, U. Nazeer, M. J. Saif, A. Mansha, A. R. Chaudhry and A. Irfan, RSC Adv., 2024, 14, 14539–14581 RSC.
- T. Uchida and T. Katsuki, Tetrahedron Lett., 2001, 42, 6911–6914 CrossRef CAS.
- A. Watanabe, T. Uchida, R. Irie and T. Katsuki, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5737–5742 CrossRef CAS PubMed.
- A. Watanabe, T. Uchida, K. Ito and T. Katsuki, Tetrahedron Lett., 2002, 43, 4481–4485 CrossRef CAS.
- C. Bolm and O. Beckmann, Chirality: The Pharmacological, Biological, and Chemical Consequences of Molecular Asymmetry, 2000, vol. 12, pp. 523–525 Search PubMed.
- C. Paneghetti, R. Gavagnin, F. Pinna and G. Strukul, Organometallics, 1999, 18, 5057–5065 CrossRef CAS.
- K. Matsumoto, A. Watanabe, T. Uchida, K. Ogi and T. Katsuki, Tetrahedron Lett., 2004, 45, 2385–2388 CrossRef CAS.
- C. Bolm, T. K. K. Luong and G. Schlingloff, Synlett, 1997, 1997, 1151–1152 CrossRef.
- C. Bolm, O. Beckmann, A. Cosp and C. Palazzi, Synlett, 2001, 2001, 1461–1463 CrossRef.
- C. Bolm, J.-C. Frison, Y. Zhang and W. D. Wulff, Synlett, 2004, 2004, 1619–1621 CrossRef.
- J.-C. Frison, C. Palazzi and C. Bolm, Tetrahedron, 2006, 62, 6700–6706 CrossRef CAS.
- C. Bolm, O. Beckmann and C. Palazzi, Can. J. Chem., 2001, 79, 1593–1597 CrossRef CAS.
- C. Bolm, O. Beckmann, T. Kühn, C. Palazzi, W. Adam, P. B. Rao and C. R. Saha-Möller, Tetrahedron: Asymmetry, 2001, 12, 2441–2446 CrossRef CAS.
- T. D. Bradley, A. Dragan and N. C. Tomkinson, Tetrahedron, 2015, 71, 8155–8161 CrossRef CAS.
- G. de Gonzalo, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2010, 11, 2208–2231 CrossRef CAS PubMed.
- S. Pandit, P. Sharma, A. Prakash, B. Lal, R. Bhuyan, I. Ahmad and A. Kuila, Ind. Crops Prod., 2024, 211, 118262 CrossRef CAS.
- K. Kumari, T. Syed, A. Krishna, S. Muvvala, A. Nowduri, C. Sridhar and A. Saxena, Nat. Prod. Res., 2023, 37, 3402–3408 CrossRef CAS PubMed.
- K. Nicolaou, C. R. Hale, C. Nilewski and H. A. Ioannidou, Chem. Soc. Rev., 2012, 41, 5185–5238 RSC.
- F. Yan, C. Li, X. Liang, S. Guo, Y. Fu and L. Chen, Recent Pat. Chem. Eng., 2013, 6, 43–56 CrossRef CAS.
- M. Gibson, M. Nur-e-alam, F. Lipata, M. A. Oliveira and J. Rohr, J. Am. Chem. Soc., 2005, 127, 17594–17595 CrossRef CAS PubMed.
- B. Frank, S. C. Wenzel, H. B. Bode, M. Scharfe, H. Blöcker and R. Müller, J. Mol. Biol., 2007, 374, 24–38 CrossRef CAS PubMed.
- Y. Wen, H. Hatabayashi and H. Arai, Appl. Environ. Microbiol., 2005, 71, 3192–3198 CrossRef CAS PubMed.
- R. Akhtar, A. F. Zahoor, A. Rasul, M. Ahmad, M. N. Anjum, M. Ajmal and Z. Raza, Pak. J. Pharm. Sci., 2019, 32, 2215–2222 Search PubMed.
- I. Shahzadi, A. F. Zahoor, B. Tüzün, A. Mansha, M. N. Anjum, A. Rasul, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, PLoS One, 2022, 17, e0278027 CrossRef CAS PubMed.
- K. Zhu, S. Hu, M. Liu, H. Peng and F. E. Chen, Angew. Chem., 2019, 131, 10028–10032 CrossRef.
- A. Świzdor, Molecules, 2013, 18, 13812–13822 CrossRef PubMed.
- M. Garrido, E. Bratoeff, D. Bonilla, J. Soriano, Y. Heuze and M. Cabeza, J. Steroid Biochem. Mol. Biol., 2011, 127, 367–373 CrossRef CAS PubMed.
- M. J. Balunas, B. Su, R. W. Brueggemeier and A. D. Kinghorn, Anti-Cancer Agents Med. Chem., 2008, 8, 646–682 CrossRef CAS PubMed.
- H.-M. Liu, H. Li, L. Shan and J. Wu, Steroids, 2006, 71, 931–934 CrossRef CAS PubMed.
- M. L. Cotter, Org. Magn. Reson., 1981, 17, 14–17 CrossRef CAS.
- V. V. Kane and D. L. Doyle, Tetrahedron Lett., 1981, 22, 3027–3030 CrossRef CAS.
- G.-J. Ten Brink, I. Arends and R. Sheldon, Chem. Rev., 2004, 104, 4105–4124 Search PubMed.
- G. de Gonzalo and A. R. Alcántara, Catalysts, 2021, 11, 605 CrossRef CAS.
- M. Bučko, P. Gemeiner, A. Schenkmayerová, T. Krajčovič, F. Rudroff and M. D. Mihovilovič, Appl. Microbiol. Biotechnol., 2016, 100, 6585–6599 CrossRef PubMed.
- A. Perkel, S. Voronina and G. Borkina, Russ. Chem. Bull., 2018, 67, 779–786 CrossRef CAS.
- U. Nazeer, A. Mushtaq, A. F. Zahoor, F. Hafeez, I. Shahzadi and R. Akhtar, RSC Adv., 2023, 13, 35695–35732 RSC.
- M. Shamma, J. L. Moniot, M. Shamma and J. L. Moniot, Isoquinoline Alkaloids Research 1972–1977, 1978, pp. 271–292 Search PubMed.
- E. Rozengart, N. Basova and A. Suvorov, J. Evol. Biochem. Physiol., 2006, 42, 408–416 CrossRef CAS.
- R. Rios, Chem. Soc. Rev., 2012, 41, 1060–1074 RSC.
- L. K. Smith and I. R. Baxendale, Org. Biomol. Chem., 2015, 13, 9907–9933 RSC.
- A. Ding, M. Meazza, H. Guo, J. W. Yang and R. Rios, Chem. Soc. Rev., 2018, 47, 5946–5996 RSC.
- T. Kametani, K. Fukumoto, F. Satoh, K. Kigasawa and H. Sugi, Chem. Informationsdienst, 1976, 7, 921–925 Search PubMed.
- L.-Y. Pu, F. Yang, J.-Q. Chen, Y. Xiong, H.-Y. Bin, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2020, 22, 7526–7530 CrossRef CAS PubMed.
- L. Y. Pu, F. Yang, J. Q. Chen, Y. Xiong, J. H. Xie and Q. L. Zhou, Adv. Synth. Catal., 2021, 363, 785–790 CrossRef CAS.
- S. El-Masry, Z. Mahmoud, M. Amer, A. J. Freyer, E. Valencia, A. Patra and M. Shamma, J. Org. Chem., 1985, 50, 729–730 CrossRef CAS.
- R. J. Andersen, D. J. Faulkner, C. H. He, G. D. Van Duyne and J. Clardy, J. Am. Chem. Soc., 1985, 107, 5492–5495 CrossRef CAS.
- C. Bailly, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 363–378 CrossRef CAS PubMed.
- K. Tangdenpaisal, R. Worayuthakarn, S. Karnkla, P. Ploypradith, P. Intachote, S. Sengsai, B. Saimanee, S. Ruchirawat and M. Chittchang, Chem.–Asian J., 2015, 10, 925–937 CrossRef CAS PubMed.
- H. Zhang, M. M. Conte, X.-C. Huang, Z. Khalil and R. J. Capon, Org. Biomol. Chem., 2012, 10, 2656–2663 RSC.
- A. Quesada, G. Gravalos and J. Fernandez Puentes, Br. J. Cancer, 1996, 74, 677–682 CrossRef CAS PubMed.
- F. Plisson, X. C. Huang, H. Zhang, Z. Khalil and R. J. Capon, Chem.–Asian J., 2012, 7, 1616–1623 CrossRef CAS PubMed.
- D. Pla, A. Francesch, P. Calvo, C. Cuevas, R. Aligué, F. Albericio and M. Alvarez, Bioconjugate Chem., 2009, 20, 1100–1111 CrossRef CAS PubMed.
- L. Zheng, T. Gao, Z. Ge, Z. Ma, J. Xu, W. Ding and L. Shen, Eur. J. Med. Chem., 2021, 214, 113226 CrossRef CAS PubMed.
- D. M. Sarnes, P. G. Jones and T. Lindel, Org. Lett., 2022, 24, 2479–2482 CrossRef CAS PubMed.
- L. Qin, W. Yi, X.-Y. Lian and Z. Zhang, J. Nat. Prod., 2020, 83, 2686–2695 CrossRef CAS PubMed.
- P. Fu, F. Kong, X. Li, Y. Wang and W. Zhu, Org. Lett., 2014, 16, 3708–3711 CrossRef CAS PubMed.
- Y. Zhu, Q. Zhang, C. Fang, Y. Zhang, L. Ma, Z. Liu, S. Zhai, J. Peng, L. Zhang and W. Zhu, Angew. Chem., Int. Ed., 2020, 59, 14065–14069 CrossRef CAS PubMed.
- J. Paciorek, D. Höfler, K. R. Sokol, K. Wurst and T. Magauer, J. Am. Chem. Soc., 2022, 144, 19704–19708 CrossRef CAS PubMed.
- Y. Kashman, A. Groweiss, S. Carmely, Z. Kinamoni, D. Czarkie and M. Rotem, Pure Appl. Chem., 1982, 54, 1995–2010 CrossRef CAS.
- M. Rotem, S. Carmely, Y. Kashman and Y. Loya, Tetrahedron, 1983, 39, 667–676 CrossRef CAS.
- B. R. Copp, C. M. Ireland and L. R. Barrows, J. Nat. Prod., 1992, 55, 822–823 CrossRef CAS PubMed.
- I. W. Mudianta, T. Skinner-Adams, K. T. Andrews, R. A. Davis, T. A. Hadi, P. Y. Hayes and M. J. Garson, J. Nat. Prod., 2012, 75, 2132–2143 CrossRef CAS PubMed.
- T. Ichiba, P. J. Scheuer and M. Kelly-Borges, J. Org. Chem., 1993, 58, 4149–4150 CrossRef CAS.
- D. M. Ramsey, M. A. Islam, L. Turnbull, R. A. Davis, C. B. Whitchurch and S. R. McAlpine, Bioorg. Med. Chem. Lett., 2013, 23, 4862–4866 CrossRef CAS PubMed.
- P. P. Principe and W. S. Fisher, J. Nat. Prod., 2018, 81, 2307–2320 CrossRef CAS PubMed.
- S.-i. Kurimoto, T. Ohno, R. Hokari, A. Ishiyama, M. Iwatsuki, S. Ōmura, J. i. Kobayashi and T. Kubota, Mar. Drugs, 2018, 16, 463 CrossRef CAS PubMed.
- D. T. Youssef, H. Z. Asfour and L. A. Shaala, Mar. Drugs, 2021, 19, 433 CrossRef CAS PubMed.
- R. Kumar, C. L. Bidgood, C. Levrier, J. H. Gunter, C. C. Nelson, M. C. Sadowski and R. A. Davis, J. Nat. Prod., 2020, 83, 2357–2366 CrossRef CAS PubMed.
- S. Tsukamoto, H. Kato, H. Hirota and N. Fusetani, Tetrahedron, 1996, 52, 8181–8186 CrossRef CAS.
- R. J. Andersen and D. J. Faulkner, Tetrahedron Lett., 1973, 14, 1175–1178 CrossRef.
- A. I. de Medeiros, R. C. Gandolfi, A. Secatto, R. M. Falcucci, L. H. Faccioli, E. Hajdu, S. Peixinho and R. G. Berlinck, Immunopharmacol. Immunotoxicol., 2012, 34, 919–924 CrossRef PubMed.
- L. A. Shaala, D. T. Youssef, J. M. Badr, M. Sulaiman and A. Khedr, Mar. Drugs, 2015, 13, 1621–1631 CrossRef CAS PubMed.
- L. Zhang, R. Wang, C. Wang, B. Liu, J. Yang, Z. Zhang, J. Huang and Z. Yang, Org. Lett., 2022, 24, 3786–3791 CrossRef CAS PubMed.
- M. N. Salib, M. T. Jamison and T. F. Molinski, J. Nat. Prod., 2020, 83, 1532–1540 CrossRef CAS PubMed.
- A. P. Morrow and M. W. Smith, J. Am. Chem. Soc., 2024, 146, 2913–2918 CrossRef CAS PubMed.
- S. M. King and S. B. Herzon, J. Org. Chem., 2014, 79, 8937–8947 CrossRef CAS PubMed.
- D. K. Semwal, U. Rawat, R. Semwal, R. Singh, P. Krishan, M. Singh and G. J. P. Singh, J. Asian Nat. Prod. Res., 2009, 11, 1045–1055 CrossRef CAS PubMed.
- A. R. Carroll, T. Arumugan, J. Redburn, A. Ngo, G. P. Guymer, P. I. Forster and R. J. Quinn, J. Nat. Prod., 2010, 73, 988–991 CrossRef CAS PubMed.
- M. Tomita, T. Ibuka, Y. Inubuahi and K. Takeda, Tetrahedron Lett., 1964, 5, 3605–3616 CrossRef.
- M.-H. Yan, P. Cheng, Z.-Y. Jiang, Y.-B. Ma, X.-M. Zhang, F.-X. Zhang, L.-M. Yang, Y.-T. Zheng and J.-J. Chen, J. Nat. Prod., 2008, 71, 760–763 CrossRef CAS PubMed.
- Y. K. Sun, J. B. Qiao, Y. M. Xin, Q. Zhou, Z. H. Ma, H. Shao and Y. M. Zhao, Angew. Chem., 2023, 135, e202310917 CrossRef.
- K. J. Weissman, Methods Enzymol., 2009, 459, 3–16 CAS.
- A. Schüller, L. Studt-Reinhold, H. Berger, L. Silvestrini, R. Labuda, U. Güldener, M. Gorfer, M. Bacher, M. Doppler and E. Gasparotto, Fungal Biol. Biotechnol., 2023, 10, 1–19 CrossRef PubMed.
- B. Hou, S. Liu, E. Li and X. Jiang, Chem. Biodiversity, 2020, 17, e2000743 CrossRef CAS PubMed.
- Q. Lu, S. Yan, H. Sun, W. Wang, Y. Li, X. Yang, X. Jiang, Y. Che and Z. Xi, Cell Death Dis., 2015, 6, e2005 CrossRef CAS PubMed.
- Z. Xiao, L. Li, Y. Li, W. Zhou, J. Cheng, F. Liu, P. Zheng, Y. Zhang and Y. Che, Cell Death Dis., 2014, 5, e1241 CrossRef CAS PubMed.
- F. Zhang, T. Yan and W. Guo, J. Peking Univ. (Health Sci.), 2019, 51, 234–238 CAS.
- W. Wu, W. Cao, L. Hu, Z. Su, X. Liu and X. Feng, Chem. Sci., 2019, 10, 7003–7008 RSC.
- S. Gunasekera, G. Paul and R. Longley, J. Nat. Prod., 2002, 65, 1643–1648 CrossRef CAS PubMed.
- S. P. Gunasekera, S. J. Mickel, R. Daeffler, D. Niederer, A. E. Wright, P. Linley and T. Pitts, J. Nat. Prod., 2004, 67, 749–756 CrossRef CAS PubMed.
- S. P. Gunasekera, M. Gunasekera, R. E. Longley and G. K. Schulte, J. Org. Chem., 1990, 55, 4912–4915 CrossRef CAS.
- D. T. Hung, J. B. Nerenberg and S. L. Schreiber, J. Am. Chem. Soc., 1996, 118, 11054–11080 CrossRef CAS.
- N. Dubasi and R. Varala, Caribbean Journal of Sciences and Technology, 2022, 10, 28–35 CrossRef.
- R. Munir, A. F. Zahoor, U. Nazeer, M. A. Saeed, A. Mansha, A. Irfan and M. U. Tariq, RSC Adv., 2023, 13, 35172–35208 RSC.
- E. ter Haar, R. J. Kowalski, E. Hamel, C. M. Lin, R. E. Longley, S. P. Gunasekera, H. S. Rosenkranz and B. W. Day, Biochemistry, 1996, 35, 243–250 CrossRef CAS PubMed.
- D. Si and K. P. Kaliappan, Asian J. Org. Chem., 2020, 9, 1205–1212 CrossRef CAS.
- K. Babar, A. F. Zahoor, S. Ahmad and R. Akhtar, Mol. Diversity, 2021, 25, 2487–2532 CrossRef CAS PubMed.
- K. Matsuo and Y. Sakaguchi, Chem. Pharm. Bull., 1997, 45, 1620–1625 CrossRef CAS.
- L. Trifonov, H. Hilpert, P. Floersheim, A. Dreiding, D. Rast, R. Skrivanova and L. Hoesch, Tetrahedron, 1986, 42, 3157–3179 CrossRef CAS.
- C. Liu, F.-L. Zou, K.-G. Wen, Y.-Y. Peng, Q.-P. Ding and X.-P. Zeng, Org. Lett., 2023, 25, 5719–5723 CrossRef CAS PubMed.
- J.-M. Gao, S.-X. Yang and J.-C. Qin, Chem. Rev., 2013, 113, 4755–4811 CrossRef CAS PubMed.
- C. Chen, H. Tao, W. Chen, B. Yang, X. Zhou, X. Luo and Y. Liu, RSC Adv., 2020, 10, 10197–10220 RSC.
- N. Osmanova, W. Schultze and N. Ayoub, Phytochem. Rev., 2010, 9, 315–342 CrossRef CAS.
- M. Ba, F. He, L. Ren, W. Whittingham, P. Yang and A. Li, Angew. Chem., Int. Ed., 2023, e202314800 Search PubMed.
- S. Perveen and A. Al-Taweel, Terpenes and Terpenoids, BoD–Books on Demand, 2018 Search PubMed.
- A. Ullah, S. Munir, Y. Mabkhot and S. L. Badshah, Molecules, 2019, 24, 678 CrossRef CAS PubMed.
- M. G. Aguilar, G. F. Sousa, F. C. Evangelista, A. P. Sabino, S. A. Vieira Filho and L. P. Duarte, Nat. Prod. Res., 2020, 34, 810–815 CrossRef CAS PubMed.
- A. Petronelli, G. Pannitteri and U. Testa, Anti-Cancer Drugs, 2009, 20, 880–892 CrossRef CAS PubMed.
- K. V. Chuang, C. Xu and S. E. Reisman, Science, 2016, 353, 912–915 CrossRef CAS PubMed.
- P. Deslongchamps, A. Bélanger, D. J. Berney, H.-J. Borschberg, R. Brousseau, A. Doutheau, R. Durand, H. Katayama, R. Lapalme and D. M. Leturc, Can. J. Chem., 1990, 68, 115–126 CrossRef CAS.
- P. Deslongchamps, A. Bélanger, D. J. Berney, H.-J. Borschberg, R. Brousseau, A. Doutheau, R. Durand, H. Katayama, R. Lapalme and D. M. Leturc, Can. J. Chem., 1990, 68, 127–152 CrossRef CAS.
- K. Du, M. J. Kier, Z. D. Stempel, V. Jeso, A. L. Rheingold and G. C. Micalizio, J. Am. Chem. Soc., 2020, 142, 12937–12941 CrossRef CAS PubMed.
- S. Al-Babili and H. J. Bouwmeester, Annu. Rev. Plant Biol., 2015, 66, 161–186 CrossRef CAS PubMed.
- B. Zwanenburg and D. Blanco-Ania, J. Exp. Bot., 2018, 69, 2205–2218 CrossRef CAS PubMed.
- M. Yoshimura, M. Dieckmann, P. Y. Dakas, R. Fonné-Pfister, C. Screpanti, K. Hermann, S. Rendine, P. Quinodoz, B. Horoz and S. Catak, Helv. Chim. Acta, 2020, 103, e2000017 CrossRef CAS.
- W.-L. Xiao, R.-T. Li, S.-X. Huang, J.-X. Pu and H.-D. Sun, Nat. Prod. Rep., 2008, 25, 871–891 RSC.
- Y.-M. Shi, W.-L. Xiao, J.-X. Pu and H.-D. Sun, Nat. Prod. Rep., 2015, 32, 367–410 RSC.
- Y. Wang, B. Chen, X. He and J. Gui, J. Am. Chem. Soc., 2020, 142, 5007–5012 CrossRef CAS PubMed.
- L. You, X.-T. Liang, L.-M. Xu, Y.-F. Wang, J.-J. Zhang, Q. Su, Y.-H. Li, B. Zhang, S.-L. Yang and J.-H. Chen, J. Am. Chem. Soc., 2015, 137, 10120–10123 CrossRef CAS PubMed.
- M. Getasetegn, Phytochem. Rev., 2016, 15, 907–920 CrossRef CAS.
- O. Wolfes, Arch. Pharm., 1930, 268, 81 CrossRef CAS.
- M. Tomanik, Z. Xu and S. B. Herzon, J. Am. Chem. Soc., 2021, 143, 699–704 CrossRef CAS PubMed.
- E. Okuyama, M. Yamazaki and Y. Katsube, Tetrahedron Lett., 1984, 25, 3233–3234 CrossRef CAS.
- R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC.
- M. Jiang, Z. Wu, L. Liu and S. Chen, Org. Biomol. Chem., 2021, 19, 1644–1704 RSC.
- F. Yang and J. A. Porco Jr., J. Am. Chem. Soc., 2022, 144, 12970–12978 CrossRef CAS PubMed.
- I. Dams, J. Wasyluk, M. Prost and A. Kutner, Prostaglandins Other Lipid Mediators, 2013, 104, 109–121 CrossRef PubMed.
- C. D. Funk, Science, 2001, 294, 1871–1875 CrossRef CAS PubMed.
- A. Pelšs, N. Gandhamsetty, J. R. Smith, D. Mailhol, M. Silvi, A. J. Watson, I. Perez-Powell, S. Prévost, N. Schützenmeister and P. R. Moore, Chem.–Eur. J., 2018, 24, 9542–9545 CrossRef PubMed.
- K. Zhu, S. Hu, M. Liu, H. Peng and F. Chen, Angew. Chem., Int. Ed., 2019, 131, 10028–10032 CrossRef.
- M. Hamberg, J. Svensson and B. Samuelsson, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2994–2998 CrossRef CAS PubMed.
- R. Morchón, E. Carretón, R. García, T. Zueva, V. Kartashev and F. Simón, J. Helminthol., 2020, 94, e67 CrossRef PubMed.
- C. Jing and V. K. Aggarwal, Org. Lett., 2020, 22, 6505–6509 CrossRef CAS PubMed.
- S. Arihara, P. Rüedi and C. H. Eugster, Helv. Chim. Acta, 1975, 58, 447–453 CrossRef CAS.
- W. Zhou, H. Xie, X. Xu, Y. Liang and X. Wei, J. Funct. Foods, 2014, 6, 492–498 CrossRef CAS.
- R. J. Grayer, M. R. Eckert, N. C. Veitch, G. C. Kite, P. D. Marin, T. Kokubun, M. S. Simmonds and A. J. Paton, Phytochemistry, 2003, 64, 519–528 CrossRef CAS PubMed.
- V. Timokhin, M. Regner, Y. Tsuji, J. Grabber and J. Ralph, Synlett, 2018, 29, 1229 CrossRef CAS.
- A. F. Zahoor, M. Yousaf, R. Siddique, S. Ahmad, S. A. R. Naqvi and S. M. A. Rizvi, Synth. Commun., 2017, 47, 1021–1039 CrossRef CAS.
- D. Mares, A. Bonora, G. Sacchetti, M. Rubini and C. Romagnoli, Cell Biol. Int., 1997, 21, 397–404 CrossRef CAS PubMed.
- M. B. Müller, J. Bertrams and F. C. Stintzing, J. Pharm. Biomed. Anal., 2020, 188, 113370 CrossRef PubMed.
- E. Teuscher and U. Lindequist, Natural Poisons and Venoms: Plant Toxins: Terpenes and Steroids, Walter de Gruyter GmbH & Co KG, 2023 Search PubMed.
- A. Bonora, G. Dall'Olio and A. Bruni, Planta Med., 1985, 51, 364–367 CrossRef CAS PubMed.
- F. Stintzing, E. Selinger, U. Lindequist, K. Wende, H. Wegele and U. Meyer, J. Anthropol. Med., 2010, 63, 567–573 Search PubMed.
- R. Alibés, J. Font, A. Mulá and R. M. Ortuño, Synth. Commun., 1990, 20, 2607–2615 CrossRef.
- K. Mliki and M. Trabelsi, Res. Chem. Intermed., 2016, 42, 8253–8260 CrossRef CAS.
- J. J. Martínez, L. A. Páez, L. F. Gutiérrez, O. H. Pardo Cuervo, H. A. Rojas, G. P. Romanelli, J. Portilla, J. C. Castillo and D. Becerra, Asian J. Org. Chem., 2020, 9, 2184–2190 CrossRef.
- L. Wang, M. Li, J. Tang, Y. Lin, K. Sidthipong, N. Sumida, N. Kushida and K. Umezawa, J. Antibiot., 2017, 70, 987–990 CrossRef CAS PubMed.
- S. Sugawara, Y. Meguro, S. Sato, M. Enomoto, Y. Ogura and S. Kuwahara, Tetrahedron Lett., 2020, 61, 151891 CrossRef CAS.
- A. K. Ghosh, H. L. Osswald and G. Prato, J. Med. Chem., 2016, 59, 5172–5208 CrossRef CAS PubMed.
- J. Robertson and J. Feinberg, Expert Opin. Pharmacother., 2012, 13, 1363–1375 CrossRef CAS PubMed.
- J. M. Llibre, A. Imaz and B. Clotet, AIDS Rev., 2013, 15, 112–121 Search PubMed.
- A. K. Ghosh, S. B. Markad and W. L. Robinson, J. Org. Chem., 2020, 86, 1216–1222 CrossRef PubMed.
- S. Mitra, S. R. Gurrala and R. S. Coleman, J. Org. Chem., 2007, 72, 8724–8736 CrossRef CAS PubMed.
- R. S. Coleman and S. R. Gurrala, Org. Lett., 2004, 6, 4025–4028 CrossRef CAS PubMed.
- C.-S. Zhang, Y.-P. Shao, F.-M. Zhang, X. Han, X.-M. Zhang, K. Zhang and Y.-Q. Tu, Chem. Sci., 2022, 13, 8429–8435 RSC.
- M. Ochi, H. Kotsuki, S. Inoue, M. Taniguchi and T. Tokoroyama, Chem. Lett., 1979, 8, 831–832 CrossRef.
- R. J. Capon, E. L. Ghisalberti and P. R. Jefferies, Phytochemistry, 1983, 22, 1465–1467 CrossRef CAS.
- R. Thomson, Naturally Occurring Quinones, Elsevier, 2012 Search PubMed.
- J. Pennock, Terpenoids in Plants, ed. J. B. Pridham, Academic Press, London, 1967, pp. 129–146 Search PubMed.
- D. V. Tsyganov, D. V. Demchuk, O. I. Adaeva, L. D. Konyushkin, M. E. Minyaev, V. N. Khrustalev and V. V. Semenov, Mendeleev Commun., 2023, 33, 539–542 CrossRef CAS.
- R. D. Schmid and V. Urlacher, Modern Biooxidation: Enzymes, Reactions and Applications, John Wiley & Sons, 2007 Search PubMed.
- D. W. Wong, Appl. Biochem. Biotechnol., 2009, 157, 174–209 CrossRef CAS PubMed.
- V. B. Urlacher and R. D. Schmid, Curr. Opin. Chem. Biol., 2006, 10, 156–161 CrossRef CAS PubMed.
- T. D. Bugg and S. Ramaswamy, Curr. Opin. Chem. Biol., 2008, 12, 134–140 CrossRef CAS PubMed.
- L. Liu, S. Li, R. Sun, X. Qin, J. Ju, C. Zhang, Y. Duan and Y. Huang, Org. Lett., 2020, 22, 4614–4619 CrossRef CAS PubMed.
- S. F. C. de Paula and A. L. M. Porto, Biocatal. Agric. Biotechnol., 2020, 25, 101546 CrossRef.
- S. H. Shim, D. C. Swenson, J. B. Gloer, P. F. Dowd and D. T. Wicklow, Org. Lett., 2006, 8, 1225–1228 CrossRef CAS PubMed.
- S. H. Shim, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2006, 69, 1601–1605 CrossRef CAS PubMed.
- N. Ingavat, C. Mahidol, S. Ruchirawat and P. Kittakoop, J. Nat. Prod., 2011, 74, 1650–1652 CrossRef CAS PubMed.
- Q. Wei, H.-C. Zeng and Y. Zou, ACS Catal., 2021, 11, 948–957 CrossRef CAS.
- F. W. Jiao, Y. S. Wang, X. T. You, W. Wei, Y. Chen, C. L. Yang, Z. K. Guo, B. Zhang, Y. Liang and R. X. Tan, Angew. Chem., 2021, 133, 26582–26588 CrossRef.
- J.-y. Zhang, L.-y. Tao, Y.-j. Liang, Y.-y. Yan, C.-l. Dai, X.-k. Xia, Z.-g. She, Y.-c. Lin and L.-w. Fu, Cell Cycle, 2009, 8, 2444–2450 CrossRef CAS PubMed.
- R. Hong, Pharm. Biol., 2011, 49, 796–799 CrossRef CAS PubMed.
- S. K. Guru, A. S. Pathania, S. Kumar, D. Ramesh, M. Kumar, S. Rana, A. Kumar, F. Malik, P. Sharma and B. Chandan, Cancer Res., 2015, 75, 2886–2896 CrossRef CAS PubMed.
- X. Wei, X. Chen, L. Chen, D. Yan, W.-G. Wang and Y. Matsuda, J. Nat. Prod., 2021, 84, 1544–1549 CrossRef CAS PubMed.
- K.-N. Feng, Y. Zhang, M. Zhang, Y.-L. Yang, J.-K. Liu, L. Pan and Y. Zeng, Nat. Commun., 2023, 14, 3436 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |