
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-defined tricobalt tetraoxide's critical morphology effect on the structure–reactivity relationship
Sami Barkaouia,
Noureddine Elboughdiri *bc,
Djamel Ghernaoutcd and
Yacine Benguerbae
*bc,
Djamel Ghernaoutcd and
Yacine Benguerbae
aLaboratoire Matériaux Traitement et Analyse, National Research Institute of Physical and Chemical Analysis, Technological Pole Sidi Thabet, 2020 Sidi Thabet, Tunisia. E-mail: samibarkaoui501@gmail.com
bChemical Engineering Process Department, National School of Engineering Gabes, University of Gabes, Gabes 6011, Tunisia. E-mail: ghilaninouri@yahoo.fr
cChemical Engineering Department, College of Engineering, University of Ha'il, PO Box 2440, Ha'il 81441, Saudi Arabia
dChemical Engineering Department, Faculty of Engineering, University of Blida, PO Box 270, Blida 09000, Algeria. E-mail: djamel_andalus@yahoo.fr
eLaboratoire de Biopharmacie et Pharmacotechnie (LBPT), Université Ferhat ABBAS Sétif-1, Sétif, Algeria. E-mail: benguerbayacine@yahoo.fr
First published on 8th July 2024
Abstract
This review focuses on exploring the intricate relationship between the catalyst particle size and shape on a nanoscale level and how it affects the performance of reactions. Drawing from decades of research, valuable insights have been gained. Intentionally shaping catalyst particles makes exposing a more significant percentage of reactive facets possible, enabling the control of overactive sites. In this study, the effectiveness of Co3O4 nanoparticles (NPs) with nanometric size as a catalyst is examined, with a particular emphasis on the coordination patterns between oxygen and cobalt atoms on the surface of these NPs. Investigating the correlation between the structure and reactivity of the exposed NPs reveals that the form of Co3O4 with nanometric size can be modified to tune its catalytic capabilities finely. Morphology-dependent nanocatalysis is often attributed to the advantageous exposure of reactive crystal facets accumulating numerous active sites. However, experimental evidences highlight the importance of considering the reorganization of NPs throughout their actions and the potential synergistic effects between nearby reactive and less-active aspects. Despite the significant role played by the atomic structure of Co3O4 NPs with nanometric size, limited attention has been given to this aspect due to challenges in high-resolution characterizations. To bridge this gap, this review strongly advocates for a comprehensive understanding of the relationship between the structure and reactivity through real-time observation of individual NPs during the operation. Proposed techniques enable the assessment of dimensions, configuration, and interfacial arrangement, along with the monitoring of structural alterations caused by fluctuating temperature and gaseous conditions. Integrating this live data with spectroscopic methods commonly employed in studying inactive catalysts holds the potential for an enhanced understanding of the fundamental active sites and the dynamic behavior exhibited in catalytic settings.
1. Introduction
A catalyst's principal role is to facilitate the acceleration of a given process without compromising the reaction's integrity. Pt-group metals such as Ir, Ru, Pt, and Pd are acknowledged for their exceptional catalytic activity.1,2 However, their high cost and scarcity on Earth have motivated scholars to investigate cost-effective alternatives with an abundance on the planet.3,4 Transition metals including cobalt (Co), Fe, and Ni have surfaced as viable substitutes for catalysts based on platinum group metals.5–8 First-row transition metals such as Co exhibit diverse properties, acting as electron inks or sources, existing in various oxidation states, and participating in electron exchange.9–12 With its three unoccupied d orbitals, cobalt bonds with surface-bound chemical species, enhancing catalytic activity, especially structural flaws such as vacancies near the crystal lattice surface.13 Cobalt-based catalysts including tricobalt tetraoxide (Co3O4) have gained significant attention in Europe for their extensive use in energy and environmental industries. The catalytic properties of Co, attributed to its partly filled d orbital (3d7), allow for facile composite creation by combining it with other elements or supports. Co and Co-based nanostructures have been investigated to enhance the surface area of catalysts, therefore exposing a more significant number of active sites and allowing the selective exposure of the most active catalytic centers. Co's ability to transition between the Co2+ and Co3+ oxidation states based on redox conditions makes it an ideal reagent complex builder.14Cobalt's dual oxidation state takes advantage of surplus electrons during a reaction, demonstrating its adaptability. The spinel crystal structure of Co3O4 contributes to its multifunctional semiconductor properties, and Co3O4 nanoparticles (NPs) exhibit direct optical band gaps, making them suitable for visible light photocatalysis.15,16 Co3O4's varied spin states, such as high, low, and intermediate spin, make it intriguing from a fundamental and spintronic perspective.16 Cobalt's versatility extends to its environmental impact, as demonstrated by Co3O4's ability to oxidize various compounds, including carbon monoxide (CO), volatile organic compounds, sulfur dioxide (SO2), and hydrocarbons. Co3O4 is employed in processes such as three-way catalytic conversion, phenol oxidation, diesel soot oxidation, and clean energy production, such as hydrogen through steam reforming methanol and ethanol.17–20 Additionally, Co3O4 serves as a commercial catalyst in the oxidation, hydrogenation, and hydrogenolysis of esters.21
This review examines recent advancements in the shape engineering of Co3O4 with nanometric size. It focuses on their catalytic performance, which is influenced by the coordination patterns of oxygen and cobalt atoms on their surface. The analysis encompasses progress in this field, exploring the structure–reactivity relationship concerning exposed NPs. The review concludes with a summary and a perspective on future developments, aiming to inform readers about the potential prospects involving Co3O4-based catalysts.
2. Preparation strategy of the morphological Co3O4
Much effort has been dedicated to preparing Co3O4 with well-controlled shapes, sizes, and crystal structures. Co3O4 has been engineered into zero-dimensional (0D) NPs,22 one-dimensional (1D) structures such as nanorods (NRs),23 nanowires,24,25 two-dimensional (2D) nanodiscs, or nanosheets,26–29 three-dimensional (3D) nanocubes (NCs),30,31 and even hierarchical nanoflowers or more complex structures.32–34 Some of the more well-liked methods to form these nanostructures are coprecipitation, ultrasonic spray pyrolysis, thermal decomposition, microwave-assisted, hydrothermal, and solvothermal methods are examples of physical and chemical processes that have been used for the preparation of Co3O4 with nanometric size.35–40 NPs enclose outstanding features, such as a simple and economical synthesis method, high surface area, good stability, and uncomplicated recovery. These properties put together more approval than other synthesis strategies of the prepared catalysts. Researchers have tried to prepare Co3O4 with nanometric size by different shapes using different methods to obtain a cost-effective, simple procedure, shorter time through an effective manner, and rectify the purity of the synthesized prepared sample. These processes include.2.1. Coprecipitation
The coprecipitation method is a simple, efficient, and economical method for the mass production of ultrafine nano-powders. Homogeneity, purity, and reactivity of the prepared oxide are the other advantages of this method. This method was used to prepare Co3O4 with nanometric size.35 First, Co(NO3)2·6H2O was dissolved in deionized water. Secondly, ammonium oxalate was added to the solution with continuous stirring. The precipitate was then washed with deionized water and dried at room temperature. Finally, it was calcined at 400–500 °C for 3 h. The average size of the obtained NPs was from 40 to 350 nm, and the Co3O4 NPs have an average diameter of 100 nm.2.2. Utrasonic spray pyrolysis
Ultrasonic spray pyrolysis is an efficient, controlled, and versatile synthesis method. It is frequently used to prepare transition metal oxides, particularly Co3O4,36 through high purity and narrow size distribution.Three different precursor solutions were prepared by dissolving cobalt acetate, cobalt chloride, or cobalt nitrate in distilled water with a concentration of cobalt salt as 0.5 mol L−1.36 The starting solution was aerosolized using an ultrasonic nebulizer (Omron, model NB-150U) with a frequency of 1.75 MHz. The spray pyrolysis temperature was kept at 750 °C. The obtained powders were collected at the reactor exit. The prepared Co3O4 samples from cobalt acetate, cobalt chloride, and cobalt nitrate are denoted as A–Co3O4, C–Co3O4, and N–Co3O4. According to the X-ray diffraction (XRD) data of A–Co3O4, C–Co3O4, and N–Co3O4 samples, all the prepared samples adopted a spinel-type cubic structure. The characteristic diffraction peaks are sharp, and no impurities or a second phase were detected, affirming that high-purity Co3O4 was obtained. Scanning electron microscopy (SEM) was used to examine the shapes of the A–Co3O4, C–Co3O4, and N–Co3O4 samples. For the A–Co3O4 powders, the dimple and wrinkle surface can be observed. C–Co3O4 sample has a porous spherical morphology, and microspheres are developed from various closely packed primary particles; moreover, abundant voids are left among adjacent particles. The N–Co3O4 sample has a durian-like shape with a 0.5–3 μm size distribution, suggesting a hollow inner structure.
2.3. Thermal decomposition
The thermal decomposition of metal oxides performed in high boiling point organic solvents and the existence of surfactants are highly relevant. This process is mainly recognized for preparing excellent-quality NPs with small sizes, high crystallinity, and narrow particle size distributions, although the resulting NPs are very stable in organic solvents.41–45 Nonetheless, this approach has some associated drawbacks, e.g., it requires preparation at high reaction temperatures, an inert atmosphere, and long processing times, resulting in increased energy and time consumption.For example, cobalt oxalate was used as a precursor for synthesizing Co3O4NRs by thermal decomposition.37 0.6 g of cobalt oxalate and 5 mL oleylamine (as a surfactant) were placed in a 50 mL two-neck distillation flask and heated up to 140 °C for 1 h. The resulting solution was added to 5 g of triphenylphosphine (as a surfactant) at 240 °C. The black solution was maintained under stirring at 240 °C for 45 min and then cooled to room temperature. The final sample was washed with ethanol several times to remove the excessive surfactant. Transmission electron microscopy (TEM) was used to verify the size and shape of the prepared samples. The TEM images of Co3O4NRs demonstrated that the materials had rod-like shapes. The length of NRs was 400–550 nm, and their diameters were about 20 nm.
2.4. Microwave-assisted methods
Microwave-assisted chemistry is becoming essential in every area of synthetic chemistry since it can boost some competitive advantages over other preparation methods. It could reduce the processing times and enhance the crystallization level of the particles. These advantages of microwave–hydrothermal methods over conventional hydrothermal methods arise from the direct interaction of the microwaves with the ions or molecules in the solution and with the solid phases dispersed in the liquid medium. In effect, it is essential to underline that the efficiency in the conversion capacity of microwave energy into thermal energy is governed by the physics variables: loss tangent, relaxation time, and penetration depth.46 Non-aqueous solvents (glycerol, ethylene glycol (EG), propylene glycol) have been frequently used47,48 to avoid or minimize the agglomeration process between the particles.This method produces high yields, simple to operate, and efficient in terms of being environmentally friendly and energy-consuming. Also, it has been extensively applied to prepare inorganic nanostructured materials49–56 with applications, e.g., electrodes,57 humidity sensors,58 or catalytic devices.52 The method's versatility for synthesizing NPs has been especially reported.59 The microwave-assisted hydrothermal route has been developed to prepare Co3O4 with NRs' shape.40 The method involved two steps: first, NRs of cobalt hydroxide carbonate were prepared by mixing 50 mL of 0.6 M Co(NO3)2·6H2O and 2.4 g of CO(NH2)2 under 500 W microwave irradiated for 3 min. Subsequently, the cobalt hydroxide carbonate NRs were calcined under air at 400 °C for 3 h to fabricate Co3O4NRs. After the thermal decomposition of cobalt hydroxide carbonate precursor under 400 °C for three hours, a single phase of well-crystallized Co3O4 with the cubic structure was obtained, and no peaks of the other phase were detected, indicating that the sample was of high purity. The as-prepared sample was bamboo-like NRs with a diameter varying from 30 to 60 nm and a length of 100 to 1000 nm.
2.5. Hydrothermal and solvothermal methods
The hydrothermal method is one of the best-used processes for preparing nanomaterials. It is essentially a solution reaction-based approach. To control the shape of the prepared materials, either low-pressure or high-pressure conditions can be used depending on the vapor pressure of the main composition in the reaction. It has numerous advantages over the other conventional methods such as energy saving, simplicity, cost-effectiveness, acceleration interaction between solid and species, better nucleation control, higher dispersion, pollution-free (as the reaction is done in a closed system), higher rate of the reaction, and lower temperature of operation in the presence of a suitable solvent. Also, it provides highly crystalline particles with better control over their size and shape.The solvothermal process is similar in its technology to the hydrothermal one, as it is carried out in autoclaves at high temperatures and pressure, through just one difference: instead of water, the synthesis is carried out in organic solvents. Co3O4 nanostructures with different morphologies (NCs, nanowires, nanobundles, nanoplates (NLs), and nanoflowers) have been prepared,38,52 and the experimental details of the preparation of Co3O4 nanostructures with different shapes are summarized in Table 1.
| Shape | Cobalt salt (mM) | Temperature (°C) | Reaction time (h) | Structure-directing agents |
|---|---|---|---|---|
| Nanocubes (NCs) | 2 | 180 | 12 | 15 mL of ammonia (6%) |
| Nanowires | 2 | 150 | 5 | 30 mL ethanol (99.9%) and 3 mmol of urea |
| Nanobundles | 2 | 120 | 12 | 2 mmol urea |
| Nanoplates (NLs) | 2 | 150 | 15 | 3 mL NaOH solution (3.25 mM) with 2 mL ammonia (6%) |
| Nanoflowers | 2 | 180 | 12 | 30 mL ethanol and 15 mL ammonia (6%) |
3. Tuning the morphology of Co3O4 for the catalytic reaction
Cobalt oxide is used mainly as a catalyst. Co3O4 was used as a model in oxidizing CO.60–65 An initial investigation revealed that Co3O4 might facilitate the oxidation of CO at temperatures as low as −54 °C. The activity was significantly decreased, however, when the reaction gas included trace amounts of moisture (3–10 ppm), which obscured the active Co3+ sites.66,67 Cobalt oxide's activity and durability in the CO oxidation process were increased by changing its form from spherical NPs to NRs, demonstrating a solid morphology-dependent impact.64 NR-shaped cobalt hydroxycarbonate was generated by precipitating cobalt acetate with sodium carbonate in EG. As seen in Fig. 1a–c, further calcination at 450 °C in air converted this precursor into rod-shaped Co3O4 NR measuring 200–300 nm in length and 10–20 nm in diameter. The CO oxidation method using spherical NPs yielded an initial CO conversion of 30% at −77 °C. However, as the time-on-stream increased, this conversion decreased to around 10% (Fig. 1h). More active and stable than Co3O4 NP catalysts, NR catalysts demonstrated 100% CO conversion in the first 6 h and maintained an 80% CO conversion for ∼12 h after the reaction. | ||
| Fig. 1 Transmission electron microscope (TEM) and high-resolution transmission electron microscope (HRTEM) images of cobalt-based nanostructures: (a, and b) cobalt hydroxide carbonate; (c–f) Co3O4 nanorods (NRs), (c) low-magnification bright-field view, and (d–f) high-resolution views at {110}, {1–10}, and {100}; (g) NR morphological illustration. Catalytic performance of Co3O4: (h) CO conversion efficiency over time for Co3O4 nanoparticles (NPs) and NRs in a continuous-flow reactor at −77 °C; (i) reaction rate (rCO) vs. CO or O2 concentrations for Co3O4 NRs; (j) Arrhenius plots (based on ref. 64, Copyright 2009, Nature Publishing Group). | ||
In contrast to the spherical NPs, Co3O4 NR demonstrated an approximately one-order-of-magnitude increase in the rate of CO oxidation. At −77 °C, the Co3O4 NR reaction rate was 3.91 × 10−6 molCO g−1 s−1. Conversely, the value of the NPs was just 4.66 × 10−7 molCO g−1 s−1. The high-resolution transmission electron microscope (HRTEM) analysis revealed that the Co3O4 NPs were enclosed by a configuration consisting of eight {111} and six {001} planes. Conversely, the Co3O4 NR preferred to reveal the {110} planes, constituting an estimated 40% of their overall surface area (Fig. 1g). It was found that Co3+ species functioned as active sites for CO oxidation on the {110} plane.
The performance of Co3O4 nanobelts (NBs) and NCs in CO oxidation has been investigated.63 The reaction rate of Co3O4 NC, which mostly exposed the ∼{001} facets, was 0.62 μmol g−1 s−1, as opposed to the 0.85 μmol g−1 s−1 seen on NBs terminated by the {110} plane. The specific conversion rate indicates that at 56 °C, Co3O4 NB exhibits 1.37 times the activity of CO3O4 NCs, demonstrating that the Co3O4 NB are significantly more active than Co3O4 NC. As shown by these studies, the activation of the surface layer lattice oxygen on the {110} planes is more pronounced in the presence of Co3+ species compared to the {001} planes. Furthermore, it was shown that Co3O4 nanowires (NWs) enclosed in {111} planes and measuring around 3 nm in diameter had a notably increased rate of CO oxidation at 248 °C, amounting to 161.75 μmol CO g−1 s−1.61 The enhanced performance resulted from the increased surface area and the profusion of Co3+ cations on the surfaces.
A catalytic study for CO oxidation62 indicates that Co3O4 NR exposed to {111} planes exhibited enhanced activity at an activation energy of 40 kJ mol−1, whereas Co3O4NLs exposed to the same planes had superior activity at a reduced activation energy of 21 kJ mol−1. Significant morphology-dependent effects on CO oxidation have been observed, contradicting prior hypotheses to some degree (maybe due to the porous structures amid cracks and interspaces in the Co3O4 nanostructures). The formation of Co3O4 NS, Co3O4 NB, and Co3O4 NC by hydrothermal synthesis of a cobalt hydroxide precursor followed by direct thermal breakdown was investigated in kinetic experiments for methane (CH4) combustion (Fig. 2a–f).30 The specific rates (rCH4) for Co3O4 NC (343 °C), Co3O4 NB (319 °C), and Co3O4 NS (313 °C) were 1.25, 2.28, and 2.72 μmol g−1, respectively, as shown in Fig. 2g. Additionally, the T50, representing the temperature at which half of the methane conversion occurred, exhibited a decreasing trend in the same sequence. The structural study indicated that the most prevalent planes on Co3O4 NS, Co3O4 NB, and Co3O4 NC were {112}, {110}, and {001}, respectively. Beyond these crystal planes, the methane combustion process persisted in the following order: {112} > {110} ![[right double angle bracket]](https://www.rsc.org/images/entities/char_27eb.gif) {001}. It can be deduced that manipulating the structure of nanostructured cobalt oxides leads to a substantial display of catalytically active sites. This is supported by the enhanced CH4 combustion activity observed in Co3O4 as a nanosheet, which exposes the more reactive {112} planes. The catalytic activity of Co3O4 supported on stainless steel wire mesh, produced by the ammonia evaporation process, was investigated with the preferred oxidation (PROX) of CO.68 The 500 nm-diameter mesoporous Co3O4 nanowires' diameter is 3.4 nm, and they have a Brunauer–Emmett–Teller (BET) surface area of 71 m2 g−1. This structured catalytic system is very stable over the whole temperature range of 100–175 °C due to its low-pressure drop and high heat exchange rate; furthermore, its exceptional catalytic activity is twice that of the highest-performing Co3O4 catalyst previously documented.
{001}. It can be deduced that manipulating the structure of nanostructured cobalt oxides leads to a substantial display of catalytically active sites. This is supported by the enhanced CH4 combustion activity observed in Co3O4 as a nanosheet, which exposes the more reactive {112} planes. The catalytic activity of Co3O4 supported on stainless steel wire mesh, produced by the ammonia evaporation process, was investigated with the preferred oxidation (PROX) of CO.68 The 500 nm-diameter mesoporous Co3O4 nanowires' diameter is 3.4 nm, and they have a Brunauer–Emmett–Teller (BET) surface area of 71 m2 g−1. This structured catalytic system is very stable over the whole temperature range of 100–175 °C due to its low-pressure drop and high heat exchange rate; furthermore, its exceptional catalytic activity is twice that of the highest-performing Co3O4 catalyst previously documented.
 | ||
Fig. 2 Scanning electron microscopy (SEM) and high-resolution transmission electron microscope (HRTEM) analysis with structural models of Co3O4 nanostructures: (a and b) Co3O4NS; (c and d) Co3O4NB; (e and f) Co3O4NC. (g) Methane conversion efficiency vs. temperature for Co3O4 at a GHSV of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 (based on ref. 30, Copyright 2008, American Chemical Society). 000 h−1 (based on ref. 30, Copyright 2008, American Chemical Society). | ||
Although PROX was believed to have an active Co3+ site, its mechanism may have been distinct from the low-temperature oxidation of the CO reaction. Researchers69 stated that the turnover frequency of Co3O4 NC, composed of six 100-facet facets, was 3.5 to 4 times more than that of Co3O4 NS, Co3O4 NB, and Co3O4 NP. Besides reducing Co2+ in hydrogen-rich environments, spectroscopic investigations revealed that Co3O4 NC's bulk and surface Co3+ sites were only modestly stabilized. For selective CO oxidation, the optimum pair Co3+/Co2+ was used. By using a sequence of Co3O4 catalysts, including exposed {111}, {110}, and {100} planes, it was verified that Co3+ functioned as the active site. As shown from the linear relationship between the number of Co3+ surface areas and the quantity of CO2 produced,70 the 100 facets positively impacted the PROX. Analyses of different Co3O4 attributes indicate that the phase and surface characteristics, including shape, surface area, and facets, significantly affect the catalytic activity. As seen in Fig. 3a–l,16 the synthesis of Co3O4, including a variety of Co3O4 NR {110}, Co3O4 NC {100}, and nano-octahedron {111} (NO) facets has been completed. The catalytic reactivity of Co3O4 NR, Co3O4 NC, and Co3O4 NO was the highest for phenol oxidation by the persulfate (PS) process. Fig. 3m and n demonstrated that the Co3O4 NR exhibited the lowest adsorption energy estimated by the density functional theory (DFT). This confirms that PS is more easily activated via a non-radical pathway on the Co3O4 {110} plane.16
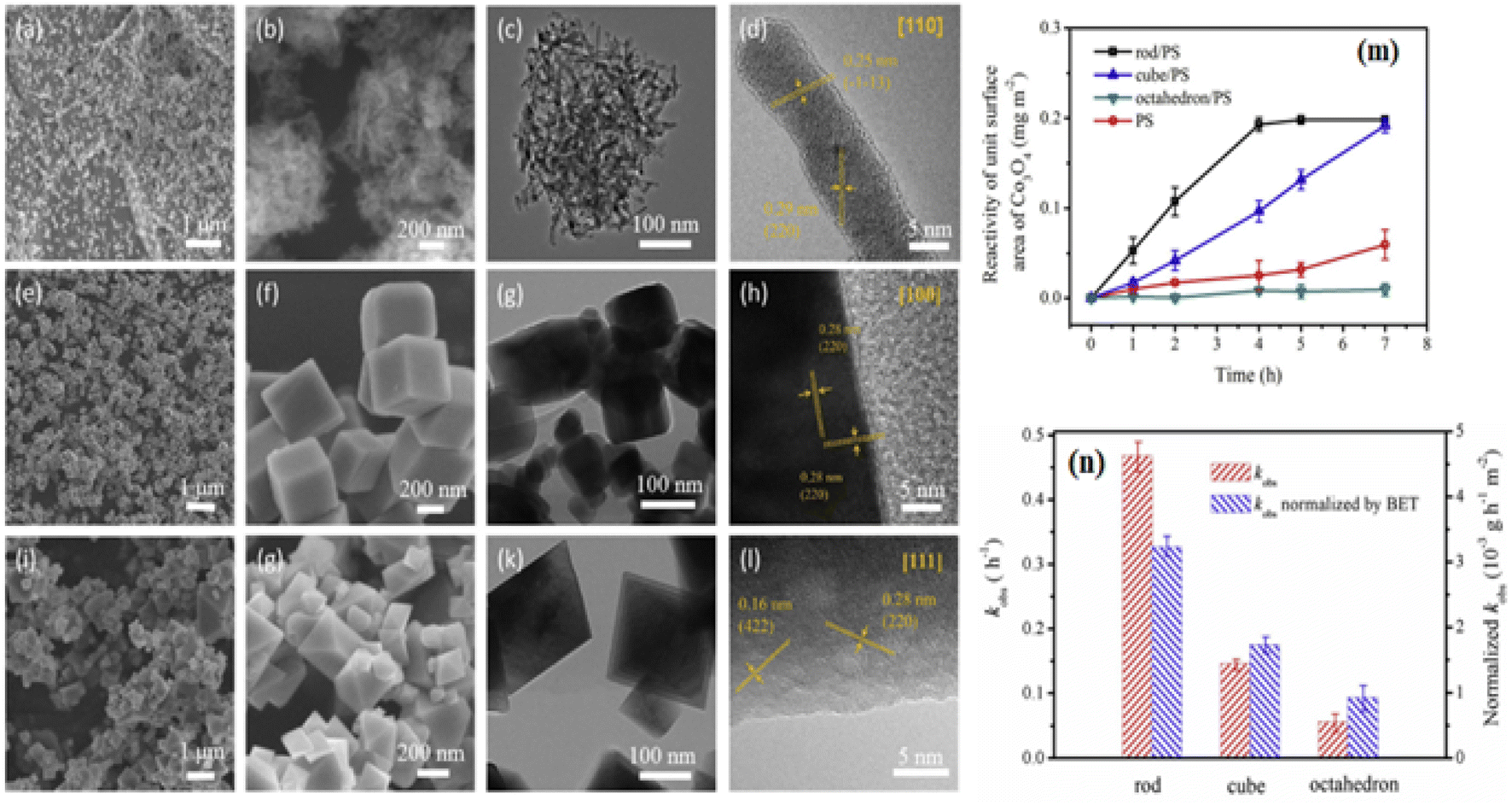 | ||
| Fig. 3 Morphological and catalytic characteristics of Co3O4 nanostructures: scanning electron microscopy (SEM) and high-resolution transmission electron microscope (HRTEM) images of (a–d) Co3O4NR {110}, (e–h) Co3O4NC {100}, and (i–l) Octahedra {111}; (m) phenol oxidation reaction rates using peroxydisulfate and Co3O4 at pH 11; (n) comparison of rate constants and Brunauer–Emmett–Teller (BET)-normalized rate constants for different Co3O4 facets (adapted from ref. 16, © 2020 Elsevier Ltd). | ||
To degrade 5-sulfosalicylic acid, four distinct 3D Co3O4 catalysts were fabricated, each with a unique morphology (Fig. 4): Co3O4 NC {111}, Co3O4 NLs {110}, Co3O4 NNs (nanoneedles, {110}), and Co3O4NFs (nanoflowers, {112}).71 Primarily, Co3O4 NF ({112} facets) is the most beneficial 3D Co3O4 catalyst for the oxidation activation to degrade 5-sulfosalicylic acid71 due to its plentiful Co2+ and more reactive surface, in addition to its most excellent surface area (121.1 m2 g−1). The core–shell contrast ratio of the as-prepared Co3S4@Co3O4 core–shell octahedron catalyst via hydrothermal and post-surface lattice anion exchange is comparatively less than that of the other core–shell structures.72 This is because the concentrations of Co3S4 and Co3O4 are close. The hexagonal shape of the selected area electron diffraction pattern, as seen in Fig. 5E, corresponds to both the {111} facet exposure and the close-packed hexagonal pattern observed in the inset of Fig. 5D in HRTEM. The lattice spacing of the {220} pattern is 0.33 nm. As seen in Fig. 5G, electrochemical CO2 reduction reaction (CRR) and oxygen reduction reaction (ORR) were investigated using a core–shell configuration of Co3O4 NO coated with a Co3S4 surface. A distinctive electronic configuration is bestowed by the heterojunction separating the p-type Co3O4 core and the n-type Co3S4 shell, enabling both catalytic processes.
 | ||
| Fig. 4 Morphological analysis of 3D Co3O4: (A) scanning electron microscopy (SEM) images of (a and b) Co3O4NC, (c and d) Co3O4nanoplates (NLs), (e and f) Co3O4NN, and (g and h) Co3O4NF. (B) Transmission electron microscope (TEM) images with electron diffraction patterns of (a–c) Co3O4NC, (d–f) Co3O4NLs, (g–i) Co3O4NN, and (j–l) Co3O4NF. (C) Comparative analysis of Co3O4 catalysts in oxone activation for 5-sulfosalicylic acid degradation of (a) Co3O4NC, (b) Co3O4NLs, (c) Co3O4NN, and (d) Co3O4NF (adapted from ref. 71, © 2020 Elsevier BV). | ||
 | ||
| Fig. 5 Structural and compositional analysis of Co3O4 and Co3S4 nanostructures: (A) FE-scanning electron microscopy (FE-SEM) image of Co3O4NO; (B) Co3S4@Co3O4NO; (C) Co3S4 nanoneedles. (D) Transmission electron microscope (TEM) and high-resolution transmission electron microscope (HRTEM) images of Co3S4@Co3O4. (E) Selected area electron diffraction pattern of Co3S4@Co3O4. (F) EDX elemental mapping of Co3S4@Co3O4. (G) Schematic of heterojunction-assisted Co3S4@Co3O4 for oxygen reduction reaction (ORR) and CO2 reduction reaction (CRR) (adapted from ref. 72, © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
To solve the recovery issue and make a reusable, eco-friendly “green” catalyst, the optimum catalyst is Co3O4 with nanometric size attached to a particular substrate with solid adhesion. Chemical (sol–gel), physical (pulsed laser deposition, or PLD), and electrochemical (electroless) methods have been used to create coatings that are reconstructed with Co3O4 with nanometric size. Fig. 6a and b shows that the Co3O4 NPs generated using the PLD approach without post-annealing treatment have a mixed amorphous–nanocrystalline phase, a tiny average size of 18 nm, a narrow size distribution of σ = 3 nm, a perfectly spherical form, and allow a degree of accumulation.73,74
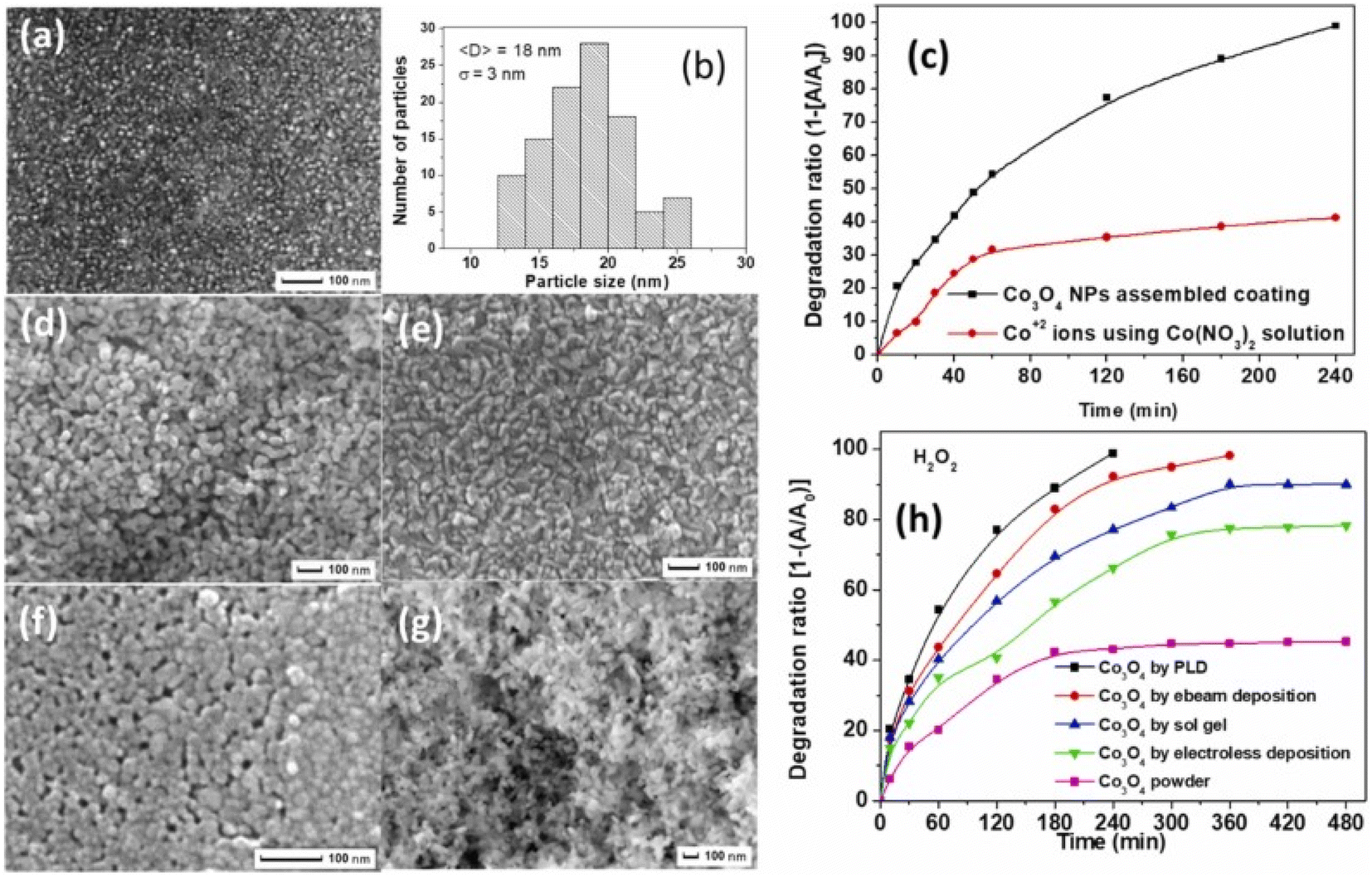 | ||
| Fig. 6 Co3O4 catalysts synthesized via various methods and their photocatalytic performance: (a) SEM images of coatings prepared by PLD, (b) particle size distribution histogram for PLD coatings, (c) time-dependent photocatalytic degradation of MB using Co3O4 NPs assembled coating via PLD and cobalt nitrate and (d–g) SEM images respectively of coatings prepared by (d) electroless, (e) electron beam, (f) sol–gel depositions, and powder form; (h) comparative photocatalytic efficiency of powder Co3O4 and coatings by different methods (adapted from ref. 73 and 74 © 2012 Elsevier BV). | ||
In a methylene blue (MB) solution, the activity of a homogeneous catalyst generating Co2+ ions was compared to that of a thin coating catalyst constructed with heterogeneous Co3O4 with nanometric size. Complete mineralization of MB dye was achieved in 240 min, indicating a far greater degradation rate than the 40% removed by Co2+ ions (Fig. 6c). In the same study, researchers73 found that coatings made of assembled Co3O4 with nanometric size had a slightly lower catalytic activity but still demonstrated good recycling capability. Fig. 6d–g shows that PLD-deposited Co3O4 coatings have the superior photo-degradation rate of MB dye when compared to Co3O4 coatings made using other processes (i.e., electro-beam deposition, sol–gel, and electroless) that have almost equal particle-like morphology (Fig. 6h).
4. Co3O4-supported metal nanoparticles (NPs)
Co3O4 has been considered an active support for heterogeneous catalysis for a very long time and is distinguished by its solid metal support interactions. As stated, Co3O4 with nanometric size has an evident morphological influence on CH4 combustion in the following sequence: Co3O4 NS, Co3O4 NB, and Co3O4 NC.30 Despite applying the same quantity of Pd NPs to these materials, the Pd/Co3O4 NS catalyst continued to produce the most methane combustion. The PdO {111} and Co3O4 NS formed a geometrically advantageous match, particularly on the {112} facet (Fig. 7a–c), which enhanced the solid metal support interactions and subsequently facilitated the activation of C–H bonds.75 The number of missing neighbors of a Co3O4 unit cell on a plane {112} is five for the NS shape. PdO must be sited in the 5-fold center of the surface of Co3O4 NS as a thin discrete film through a matching geometry and strong coordination rather than a top or bridge site.76 | ||
| Fig. 7 Microscopic analysis of 5% Pd-doped Co3O4NS: (a) scanning electron microscopy (SEM), (b) transmission electron microscope (TEM), and (c) high-resolution transmission electron microscope (HRTEM) images highlighting of PdO {002} and 0.466 nm of Co3O4 {111} (reproduced with permission from ref. 75, Copyright 2011, WILEY-VCH Verlag Gmbh& Co). Detailed TEM and scanning transmission electron microscopy (STEM) analysis of Co3O4NR catalysts: (d) TEM, (e) HRTEM, and (f) STEM image of Pt atoms singularly dispersed on Co3O4NR (reproduced with Permission from ref. 77, Copyright; American Chemical Society). | ||
Due to its low activation barrier of 29.6 kJ mol−1, single Pt atoms attached to Co3O4 demonstrate significant catalytic activity in the water gas shift process at 200 °C (turnover frequency = 0.58 molH2 per sitePt per s). The significantly decreased activation energy observed for these individual Pt atoms may indicate that the interaction between the atoms and the 110-faced Co3O4 NR substantially customized the chemical environment of the active sites (Fig. 7d–f).77
Using straightforward hydrothermal and solvothermal techniques, anion adsorption was employed to deposit gold NPs onto Co3O4 materials produced in various forms, including rods, polyhedra, and cubes.78 Au catalysts based on Co3O4 were characterized using TEM and HRTEM. The predicted morphologies of the Co3O4 supports are cube-shaped, rod-shaped, and polyhedron (NH)-shaped (Fig. 8). Research into the exposed planes of various morphological Co3O4 materials has led to the discovery that the morphology of the support plays a crucial role in determining the catalytic activity. Co3O4 NR shows {110} planes most of the time on HRTEM, whereas the {011} and {001} planes are the most prominent on Co3O4 NH and Co3O4 NC structures, respectively. The {110} plane has the most excellent oxygen vacancies, which are very important for the oxidation of ethylene, in comparison to the {011} and {001} planes. Consequently, the ethylene conversion rate of 93.7% was achieved by Au/Co3O4 NR, demonstrating their exceptional catalytic activity. Ethanol conversion was 85.5% for the Au/Co3O4 NH catalyst. At 0 °C, the ethylene conversion on Au/Co3O4 NC was 26.8%, which was the lowest value recorded.
 | ||
| Fig. 8 Transmission electron microscope (TEM) images of Co3O4NR (a), Co3O4NH (d), and Co3O4NC (g). High-resolution transmission electron microscope (HRTEM) images of Au/Co3O4NR (b, and c), Au/Co3O4NH (e, and f), and Au/Co3O4NC (h, and i). Source: reprinted with permission from ref. 78, ©2011 Elsevier BV. | ||
Our prior research79 examined the effect of Co3O4 crystallization on EG oxidation supports in the form of Co3O4 NCs and NLs. As shown in Fig. 9a–c, Au NPs in the Au/Co3O4 NCs samples exhibited a quasi-truncated octahedron structure with Au {111} and {100} faces and had an average size of 2.0 nm. As shown by the interplanar distance of 0.29 nm, corresponding to the {220} crystal plane of cubic Co3O4 oxides, Au NPs are anchored consistently on the Co3O4 {001} facet. Furthermore, the inter-planar spacing of 0.46 nm corresponds to the lattice fringes seen in Au/Co3O4 NL catalysts and is caused by the Co3O4{111} facets of Co3O4 NL oxides. The uniform loading of Au particles onto the Co3O4 {111} facet resulted in the formation of a quasi-truncated octahedron encircled by Au {111} and {100} facets, as seen in Fig. 9d–f. Under these conditions, the Co3O4 NC and Co3O4 NL constituents remained dormant during the aerobic oxidation of EG. With the addition of Au NPs, the catalytic activity of EG oxidation processes was substantially enhanced. Therefore, when subjected to glycol oxidation facilitated by intrinsic defects and surface oxygen vacancies, Au/Co3O4 NL {111} exhibited much greater selectivity and catalytic activity than its Au/Co3O4 NC {001} counterpart (Fig. 9g). One potential catalyst for the oxidation of EG using Au NPs is Co3O4 NL {111}, which facilitates the activation of O2 via the oxygen vacancies on its surface.
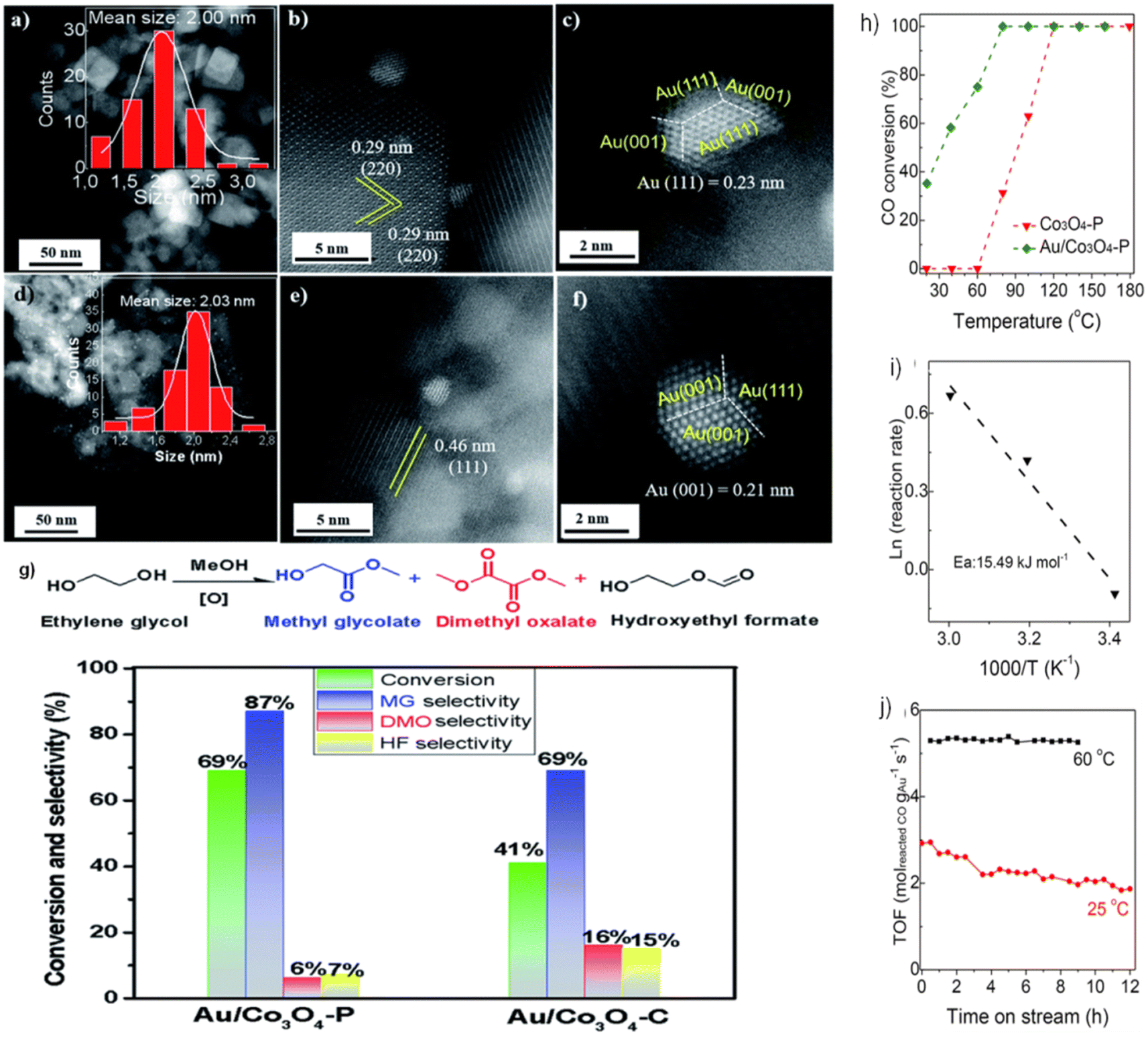 | ||
| Fig. 9 Scanning transmission electron microscopy (STEM) imaging and catalytic performance of Au-doped Co3O4: (a–c) Au particles on Co3O4 {001} in Au/Co3O4NC; (d–f) Au particles on Co3O4 {111} in Au/Co3O4 nanoparticles (NPs); (g) catalytic efficiency of Au/Co3O4NPs and Au/Co3O4NC in ethylene glycol (EG) oxidation. Reprinted with Permission from ref. 79, 2021 Royal Society of Chemistry. Performance analysis of Co3O4 and Au/Co3O4 in CO oxidation: (h) temperature-dependent catalytic activity for CO oxidation; (i) Arrhenius plots showing rate vs. 1/T for CO oxidation over Au/Co3O4; (j) durability tests at 25 °C and 60 °C with CO conversion rates from 25% to 45%. Reprinted with Permission from ref. 80, 2023 Royal Society of Chemistry. | ||
Furthermore, the catalysts Au/Co3O4 P were evaluated in the CO oxidation processes.80 The catalytic activity was substantially enhanced by adding Au NPs, as shown in Fig. 9h. This resulted in a noteworthy CO conversion of 35% at 20 °C and complete at 80 °C. As depicted in Fig. 9i, the activation energy (Ea) for CO oxidation in Au/Co3O4 P is 15.49 kJ mol−1. Therefore, oxygen molecules follow the Langmuir–Hinshelwood mechanism, which catalyzes CO oxidation at low temperatures (20–60 °C) via Au/Co3O4 P {111}. In particular, rather than traversing the surface lattice oxygen sites, CO should be adsorbed onto oxygen vacancies at the surface and activated by Au NPs. The durability of the Au/Co3O4 P catalysts was also evaluated at temperatures of 25 and 60 °C (Fig. 9j). Throughout the twelve-hours CO oxidation process at 25 °C, the Au/Co3O4 P catalyst activity decreased from 2.92 to 1.87 molreactedCO gAu−1 s−1. A minimum activity of 5.26–5.39 molreactedCO gAu−1 s−1 was recorded for 9 h at 60 °C. This phenomenon might be primarily attributed to the surface oxygen vacancies and inherent defects of Co3O4 {111}, which activated O2. Similarly, the presence of Au0, Auδ+, and Au+ species on the surface of Au NPs further enhanced the activation of CO.
5. Chemical nature of the oxide particle morphology
Many people think that certain cobalt cations are abundant at the active sites. Co3O4 NR, rich in Co3+ cations and having mostly exposed {110} surfaces, is very active in low-temperature CO oxidation.64 Moreover, among Co3O4 NR, Co3O4 NC, and Co3O4 NP, Co3O4 NS with mostly exposed {111} planes enriched in Co2+ cations are the most active.38 At low temperatures, a Co3O4 SiO2 nanocomposite devoid of ordered planes but abundant in Co2+ proved an exceptionally active catalyst.81 However, these findings were mainly obtained via catalytic research, and direct spectroscopic evidence of the active surface oxidation state was absent.Contrary to comparable nanostructures, there have been consistent findings on the shape influence of Co3O4 with nanometric size in catalyzing oxidation processes (as shown above). The many reaction routes can contribute, including changing the reaction conditions (primarily the gas and temperature). CO may be oxidized by the Langmuir–Hinshelwood method, which requires surface oxygen species, or the Mars-van Krevelen mechanism, which utilizes lattice oxygen species, according to spectroscopic observations82 and the spectroscopically examined possible reaction pathways/elementary steps of CO oxidation on Co3O4 are configured in Fig. 10, the former exhibited dominance at over 100 °C due to oxygen vacancy formation and the Co3+/Co2+ redox cycle. Conversely, at lower temperatures, the latter demonstrated dominance. One possible reaction mechanism is that CO adsorbs onto Co3+cations and then absorbs oxygen from the surface lattice coordinated to three Co3+ cations. The oxygen vacancy is then filled with oxygen from the gas phase, according to the Mars–van Krevelen mechanism.64
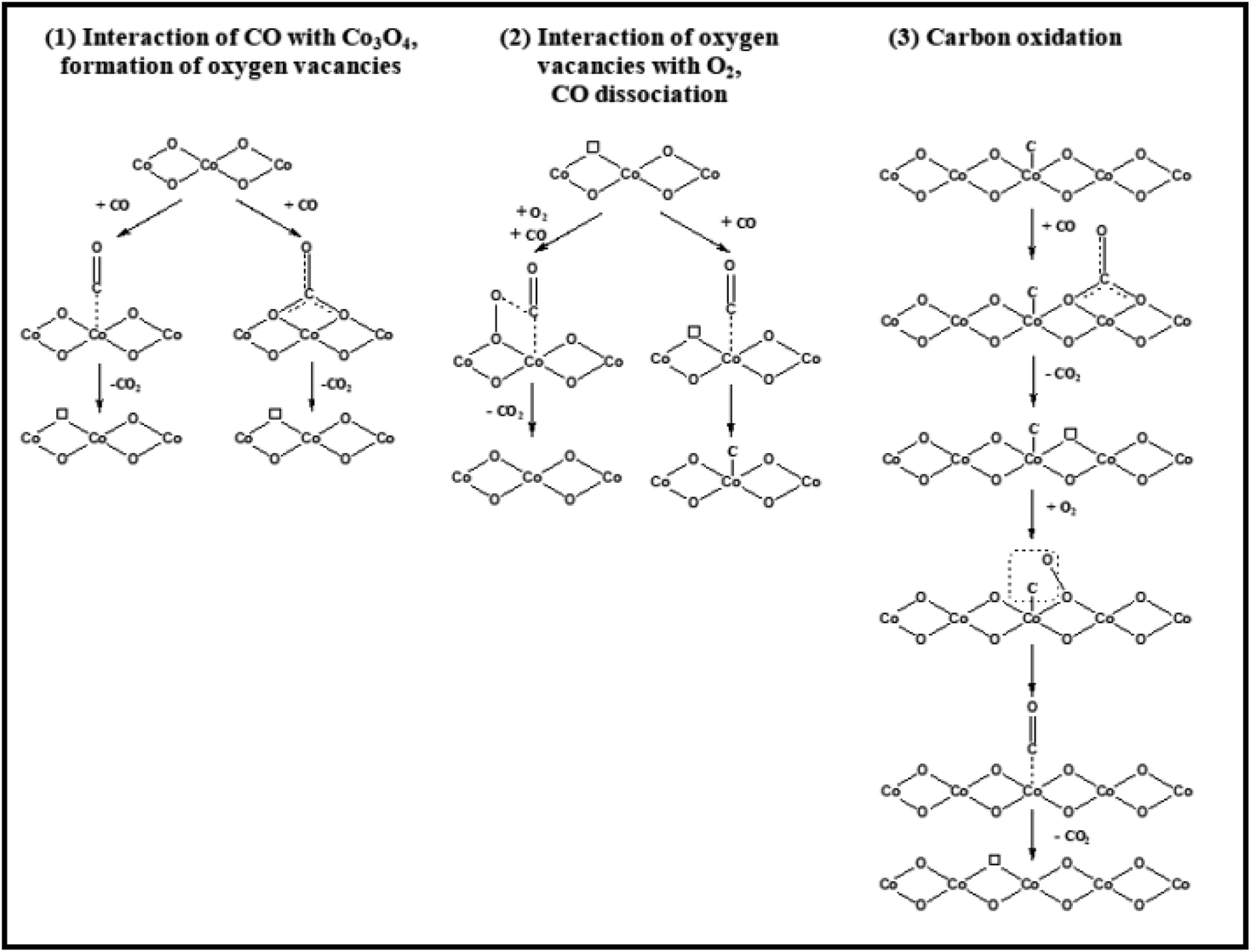 | ||
| Fig. 10 Schematic representation of CO oxidation on Co3O4. Reprinted with Permission from ref. 82, 2018 American Chemical Society. | ||
Spectroscopic evidence is lacking, although an interaction between molecularly adsorbed CO and O–O peroxo species has been postulated by analyzing the impact of pretreatment conditions,65 although no peroxo O–O species were found using in situ Raman spectroscopy.81 According to in situ infrared research, CO adsorbed on Co2+ sites interacted with an oxygen atom bound to a nearby Co3+ cation, and the gas phase oxygen was used to fill the oxygen vacancy.83 Isotopes are vital in the redox Mars–van Krevelen process and are responsible for CO oxidation.84,85
Theoretical investigations into the CO oxidation pathway on Co3O4 have also shown differences.86–89 For instance, a Mars–van Krevelen process involving mostly exposed {110} planes in Co3O4 has been proposed, as shown in Fig. 11.88
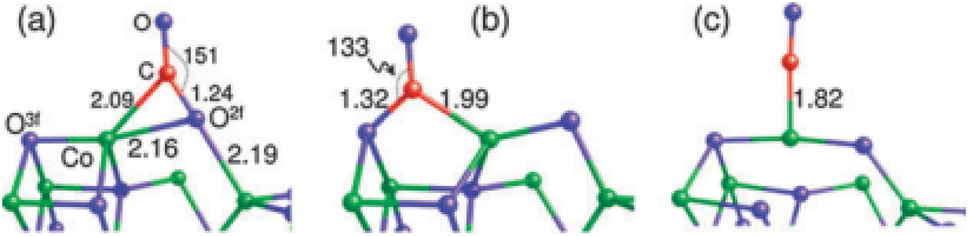 | ||
| Fig. 11 Three adsorption configurations of CO on Co3O4(110): (a) on O2f; (b) on O3f; (c) on Co. Bond lengths are in angstroms; bond angle are in degrees. Co, green, O, blue, and C, red. Reprinted with Permission from ref. 88, 2011 Royal Society of Chemistry. | ||
Theoretically, the octahedrally coordinated Co2+ site in CoO90 would be the most active site for the PROX of CO in the hydrogen-rich stream. According to DFT calculations, the generated carbonates should make the {001} facet of Co3O4 less reactive by blocking the surface sites on that facet but not on CoO {001}, as shown in Fig. 12.
 | ||
| Fig. 12 Potential energy diagrams for (a) the hydrogenation of Co3O4 {001} and (b) the oxidation of CO to CO2 on Co3O4 {001} and CoO {001}. For each transition state (hollow boxes), reaction barriers are given in kJ mol−1. Selected intermediates are shown as a side view along [110], using the following color codes: black (C), blue (Co), red (O), and white (H). Reprinted with Permission from ref. 90, 2019 American Chemical Society. | ||
Surface and lattice oxygen species interact concurrently in the reaction network, making methane oxidation on Co3O4 catalysts more difficult. There were three distinct temperature/conversion phases in the methane oxidation process, identified by the presence or absence of the adsorbed or lattice oxygen and the catalyst's redox state.91 At temperatures between 300 and 450 °C, the dominating superficial Langmuir–Hinshelwood structure produces a stoichiometric {100} surface on Co3O4 NC with a regular size of around 40–60 nm and with the preferential exposure {100}, as previously shown for CH4 combustion on these particles. At temperatures ranging from 450 to 650 °C, where O2 nearly occupies the oxygen vacancies generated by the emission of CO2 and H2O, the imperfect surface area is delineated by the coexistence of the interfacial (Mars–van Krevelen) and suprafacial (Langmuir–Hinshelwood) mechanisms.92,93 At temperatures over 650 °C and with a non-stoichiometric surface area, the completion of oxygen vacancies is only partial, resulting in a substantial reduction in catalyst activity and the combustion of CH4 via the Mars–van Krevelen technique.91
Theoretical computations have led to the notion that the C–H bond in CH4 would be activated by the doubly coordinated lattice oxygen (O2c) across the {110} surface. Therefore, the {110} surface is expected to exhibit more activity than the {100} surface, devoid of any O2c sites.94 Assuming dissociation of CH4 on the Co–O pair; researchers95 indicated that the reactivity of methane combustion increased as follows: {001} < {011} < {112}. Experimental observation of cubic Co3O4 revealed the less active {001} facet, while flower-shaped Co3O4 exhibited the active {111} facet.96 As compared to spherical NPs enclosed in the {001} and {111} facets or Co3O4 NRs exposed to the {110} and {001} facets, Co3O4 NLs encased in the {112} facet showed higher activity in the CH4 combustion process.97 The surface remodeling during reaction circumstances may contribute to the contradicting findings on the reactive facets. It has been shown by molecular modeling of Co3O4 NPs that the form may be maintained; however, when exposed to oxidizing and reducing atmospheres, the relative ratio of {111}/{100}/{110} facets changes dynamically.98 Under conditions rich in hydrogen gas, the faceting {110} plane was preferentially exposed. At the same time, the {111} surface remained untreated due to the development of oxygen surface vacancies and their subsequent diffusion toward the bulk. Nevertheless, the oxygen-rich conditions promoted the {111} termination. Therefore, it was necessary to describe the shape of the active catalysts. Recent breakthroughs in high-resolution microscopic and spectroscopic methods have opened the door to studying the functions of shaped-synchronized NPs in terms of their dynamic performance. Nitric oxide (NO) may be reduced with CO by reshaping Co3O4 NRs with an exposed {110} surface into non-stoichiometric CoO1−x NR (Fig. 13a and b).99 The structure-modified NRs generate nitrogen gas by selectively reducing nitrogen oxides (NOx) with CO at temperatures ranging from 250 to 520 °C. Environmental transmission electron microscopy (ETEM) and ambient pressure X-ray photoelectron spectroscopy showed that the non-stoichiometric CoO1−x NRs had a rock-salt (RS) structure. The 100% selectivity was brought about by the active phase, which included around 25% oxygen vacancies. Electron transport microscopy measurements in environments rich in hydrogen showed that CO3 was reduced to CO, indicating the formation of a boundary contact for particles larger than 15 nm but not for smaller ones, showing that smaller NPs undergo rapid reduction.100 ETEM identified a two-step phase transition during the heating experiment, as shown in Fig. 13c and d. In the low-temperature range of 200 to 280 °C, the wurtzite (WZ) CoO was spontaneously oxidized to spinel (SP) Co3O4 owing to the residual oxygen in the TEM. Secondly, under low oxygen partial pressure conditions, SP Co3O4 was reduced to RS CoO at temperatures reaching 280 °C.101 These visual results show that the as-prepared oxide NPs changed significantly under response conditions.
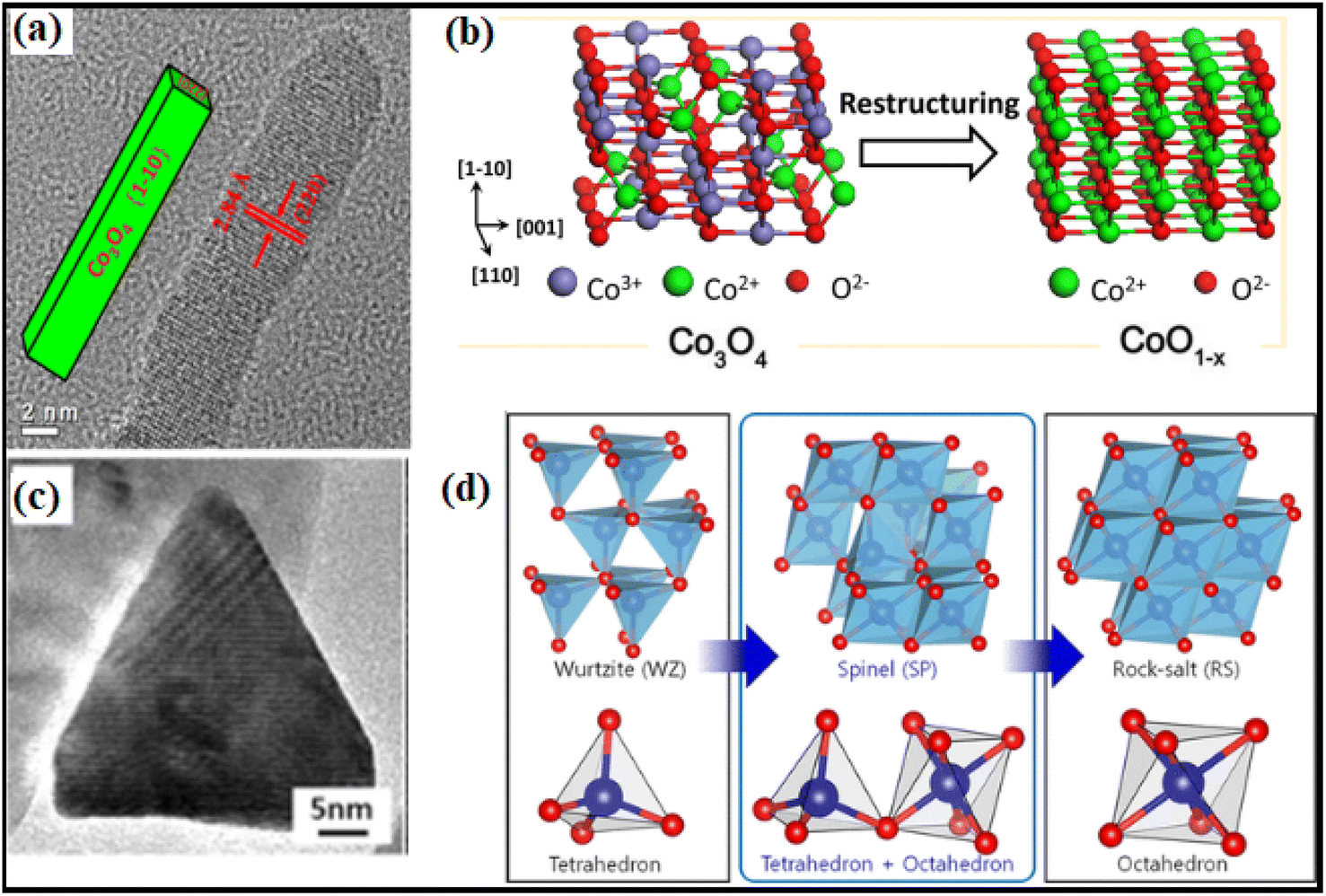 | ||
| Fig. 13 Structural transformation of Co3O4NR: (a) high-resolution transmission electron microscope (HRTEM) image; (b) schematic illustration of Co3O4 to CoO transformation under reaction conditions; (c) HRTEM image of CoO hexagonal pyramid; (d) illustration of the phase transformation from metastable wurtzite (WZ) CoO to stable rock-salt (RS) CoO via the intermediate spinel (SP) Co3O4. Reprinted with Permission from ref. 90 and 101 Copyrights 2013 and 2019, American Chemical Society. | ||
6. Concluding remarks and perspectives
Extensive exploration into the field of nanocatalysis utilizing Co3O4 nanometrics has undeniably demonstrated that the size and shape of the catalyst at the nanoscale level profoundly impact its catalytic effectiveness. A growing body of evidence suggests that the configuration of the nanometric Co3O4 is always critical in achieving optimal levels of selectivity, stability, and catalytic activity. This technology's advancement has been significant due to the incorporation of morphology-dependent nanocatalysts, an innovative tool for finely adjusting catalytically active sites. Both theoretical and experimental investigations have been extensive into the morphology-dependent nanocatalysis of nanometric Co3O4. Specifically, the arrangement of surface Co3+/Co2+ and O sites,102–104 focusing on the oxygen vacancy, has been linked to the catalytic properties of reactive surface facets. However, there are conflicting reports regarding the effectiveness of similar nanostructures in catalyzing different processes or even the same reaction under identical conditions. This suggests that the form-dependency of nanometric Co3O4, as documented, is highly susceptible to variations in reaction parameters and established reaction pathways.The relationship between the catalytic activities of nanometric Co3O4 and the selectively exposed facets induced by shape has been demonstrated through experimental evidence. However, it cannot be ruled out that adjacent facets may work together synergistically. Initially designed nanostructures may undergo structure, morphology, and chemistry changes under actual reaction conditions. The catalytic properties observed in the experiments are determined by the dynamic behavior of the catalyst particles in response to temperature and the reactive environment rather than their state when prepared or recently used. In some instances, the activation of species in a multi-molecule chemical reaction may occur through diffusion on adjacent facets, resulting in a synergistic effect where the species activated by the adsorbed reactant can adsorb and stimulate a different type of reactant. In situ studies, physical and chemical analyses, and dynamic characterization techniques must be employed in operational environments to fully understand functional nanostructures.
To gain a deeper understanding of the relationships within nanostructured catalysts, further exploration is needed to develop improved experimental and theoretical methods.105–107 Variations in temperature and reactive gas fluctuations can impact the well-defined form of Co3O4 nanometric, leading to changes in its electrical and geometric properties. This, in turn, influences the proportion of active surfaces and the coordination environments of oxygen and cobalt atoms on the surface, ultimately affecting the development of active sites. The lack of published studies on the atomic structure of nanometric Co3O4 can be attributed to the limited availability of high-resolution spectroscopic and microscopic characterizations among researchers worldwide. Also, studying active sites' dynamic performance under operational conditions would provide valuable insights into the structure–reactivity relationship. By employing techniques that allow for real-time assessment of size, shape, interfacial structure, and gas-induced structural changes at the active sites of individual nanoparticles, combined with spectroscopic methods, we can significantly enhance our understanding of the inherent active regions and dynamic capabilities of nanostructured catalysts within catalytic environments.
Abbreviation
| BET | Brunauer–Emmett–Teller |
| CRR | CO2 reduction reaction |
| DFT | Density functional theory |
| EG | Ethylene glycol |
| MB | Methylene blue |
| NBs | Nanobelts |
| NCs | Nanocubes |
| NH | Polyhedron |
| NLs | Nanoplates |
| NPs | Nanoparticles |
| NRs | Nanorods |
| ORR | Oxygen reduction reaction |
| PLD | Pulsed laser deposition |
| PS | Persulfate |
| PROX | Preferred oxidation |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| XRD | X-ray diffraction |
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Noble-metal single-atoms in thermocatalysis, electrocatalysis, and photocatalysis, Energy Environ. Sci., 2021, 14, 2954–3009, 10.1039/D1EE00247C.
- J. Lin, X. Wang and T. Zhang, Recent progress in CO oxidation over Pt-group-metal catalysts at low temperatures, Chin. J. Catal., 2016, 37, 1805–1813, DOI:10.1016/S1872-2067(16)62513-5.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Recent progress on multimetal oxide catalysts for the oxygen evolution reaction, Adv. Energy Mater., 2018, 8, 1702774, DOI:10.1002/aenm.201702774.
- J. Kibsgaard and I. Chorkendorff, Considerations for the scaling-up of water splitting catalysts, Nat. Energy, 2019, 4, 430–433, DOI:10.1038/s41560-019-0407-1.
- G. Huang, Z. Xiao, R. Chen and S. Wang, Defect engineering of cobalt-based materials for electrocatalytic water splitting, ACS Sustain. Chem. Eng., 2018, 6, 15954–15969, DOI:10.1021/acssuschemeng.8b04397.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Recent progress in cobalt-based heterogeneous catalysts for electrochemical water splitting, Adv. Mater., 2016, 28, 215–230, DOI:10.1002/adma.201502696.
- P. Hu and M. Long, Cobalt-catalyzed sulfate radical-based advanced oxidation: a review on heterogeneous catalysts and applications, Appl. Catal., B, 2016, 181, 103–117, DOI:10.1016/j.apcatb.2015.07.024.
- S. Li, X. Hao, A. Abudula and G. Guan, Nanostructured Co-based bifunctional electrocatalysts for energy conversion and storage: current status and perspectives, J. Mater. Chem. A, 2019, 7, 18674–18707, 10.1039/C9TA04949E.
- A. L. Smith, K. I. Hardcastle and J. D. Soper, Redox-active ligand-mediated oxidative addition and reductive elimination at square planar cobalt (III): Multielectron reactions for cross-coupling, J. Am. Chem. Soc., 2010, 132, 14358–14360, DOI:10.1021/ja106212w.
- Y. Popat, M. Orlandi, N. Patel, R. Edla, N. Bazzanella, S. Gupta, M. Yadav, S. Pillai, M. K. Patel and A. Miotello, Pulsed laser deposition of CoFe2O4/CoO hierarchical-type nanostructured heterojunction forming a Z-scheme for efficient spatial separation of photoinduced electron-hole pairs and highly active surface area, Appl. Surf. Sci., 2019, 489, 584–594, DOI:10.1016/j.apsusc.2019.05.314.
- R. Edla, A. Tonezzer, M. Orlandi, N. Patel, R. Fernandes, N. Bazzanella, K. Date, D. C. Kothari and A. Miotello, 3D hierarchical nanostructures of iron oxides coatings prepared by pulsed laser deposition for photocatalytic water purification, Appl. Catal., B, 2017, 219, 401–411, DOI:10.1021/am501021e.
- Q. Zhou, Transition-metal catalysis and organocatalysis: where can progress be expected?, Angew. Chem., Int. Ed., 2016, 55, 5352–5353, DOI:10.1002/anie.201509164.
- R. Zhang, Y.-C. Zhang, L. Pan, G.-Q. Shen, N. Mahmood, Y.-H. Ma, Y. Shi, W. Jia, L. Wang and X. Zhang, Engineering cobalt defects in cobalt oxide for highly efficient electrocatalytic oxygen evolution, ACS Catal., 2018, 8, 3803–3811, DOI:10.1021/acscatal.8b01046.
- M. Staniuk, O. Hirsch, N. Kränzlin, R. Böhlen, W. van Beek, P. M. Abdala and D. Koziej, Puzzling mechanism behind a simple synthesis of cobalt and cobalt oxide nanoparticles: in situ synchrotron X-ray absorption and diffraction studies, Chem. Mater., 2014, 26, 2086–2094, DOI:10.1021/cm500090r.
- A. Diallo, A. C. Beye, T. B. Doyle, E. Park and M. Maaza, Green synthesis of Co3O4 nanoparticles via Aspalathus linearis: Physical properties, Green Chem. Lett. Rev., 2015, 8, 30–36, DOI:10.1080/17518253.2015.1082646.
- C. Wang, S. Jia, Y. Zhang, Y. Nian, Y. Wang, Y. Han, Y. Liu, H. Ren, S. Wu and K. Yao, Catalytic reactivity of Co3O4 with different facets in the hydrogen abstraction of phenol by persulfate, Appl. Catal., B, 2020, 270, 118819, DOI:10.1016/j.apcatb.2020.118819.
- Y. Lü, W. Zhan, Y. He, Y. Wang, X. Kong, Q. Kuang, Z. Xie and L. Zheng, MOF-templated synthesis of porous Co3O4 concave nanocubes with high specific surface area and their gas sensing properties, ACS Appl. Mater. Interfaces, 2014, 6, 4186–4195, DOI:10.1021/am405858v.
- K. J. Lee, T.-H. Kim, T. K. Kim, J. H. Lee, H.-K. Song and H. R. Moon, Preparation of Co3O4 electrode materials with different microstructures via pseudomorphic conversion of Co-based metal–organic frameworks, J. Mater. Chem. A, 2014, 2, 14393–14400, 10.1039/C4TA02501F.
- H.-M. Jeong, S.-Y. Jeong, J.-H. Kim, B.-Y. Kim, J.-S. Kim, F. Abdel-Hady, A. A. Wazzan, H. A. Al-Turaif, H. W. Jang and J.-H. Lee, Gas selectivity control in Co3O4 sensor via concurrent tuning of gas reforming and gas filtering using nanoscale hetero-overlayer of catalytic oxides, ACS Appl. Mater. Interfaces, 2017, 9, 41397–41404, DOI:10.1021/acsami.7b13998.
- M. Saeed, M. Ilyas and M. Siddique, Oxidative degradation of phenol in aqueous medium catalyzed by lab prepared cobalt oxide, J. Chem. Soc. Pak., 2012, 34, 626–633, DOI:10.1007/s13369-013-0545-x.
- B. Karasu and S. Turan, Effects of cobalt, copper, manganese and titanium oxide additions on the microstructures of zinc containing soft porcelain glazes, J. Eur. Ceram. Soc., 2002, 22, 1447–1455, DOI:10.1016/S0955-2219(01)00456-3.
- T. He, D. Chen, X. Jiao, Y. Wang and Y. Duan, Solubility-controlled synthesis of high-quality Co3O4 nanocrystals, Chem. Mater., 2005, 17, 4023–4030, DOI:10.1021/cm050727s.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, Thermal formation of mesoporous single-crystal Co3 O4 nano-needles and their lithium storage properties, J. Mater. Chem., 2008, 18, 4397–4401, 10.1039/B810093D.
- K. T. Nam, D.-W. Kim, P. J. Yoo, C.-Y. Chiang, N. Meethong, P. T. Hammond, Y.-M. Chiang and A. M. Belcher, Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes, Science, 2006, 312, 885–888, DOI:10.1126/science.1122716.
- Y. Li, B. Tan and Y. Wu, Mesoporous Co3O4 nanowire arrays for lithium ion batteries with high capacity and rate capability, Nano Lett., 2008, 8, 265–270, DOI:10.1021/nl0725906.
- Y. Hou, H. Kondoh, M. Shimojo, T. Kogure and T. Ohta, High-yield preparation of uniform cobalt hydroxide and oxide nanoplatelets and their characterization, J. Phys. Chem. B, 2005, 109, 19094–19098, DOI:10.1021/jp0521149.
- X. Liu, R. Yi, N. Zhang, R. Shi, X. Li and G. Qiu, Cobalt hydroxide nanosheets and their thermal decomposition to cobalt oxide nanorings, Chem.–Asian J., 2008, 3, 732–738, DOI:10.1002/asia.200700264.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, Selective and controlled synthesis of α-and β-cobalt hydroxides in highly developed hexagonal platelets, J. Am. Chem. Soc., 2005, 127, 13869–13874, DOI:10.1021/ja0523338.
- J. T. Sampanthar and H. C. Zeng, Arresting butterfly-like intermediate nanocrystals of β-Co(OH)2 via ethylenediamine-mediated synthesis, J. Am. Chem. Soc., 2002, 124, 6668–6675, DOI:10.1021/ja012595j.
- L. Hu, Q. Peng and Y. Li, Selective synthesis of Co3O4 nanocrystal with different shape and crystal plane effect on catalytic property for methane combustion, J. Am. Chem. Soc., 2008, 130, 16136–16137, DOI:10.1021/ja806400e.
- J. Feng and H. C. Zeng, Size-controlled growth of Co3O4 nanocubes, Chem. Mater., 2003, 15, 2829–2835, DOI:10.1021/cm020940d.
- L. X. Yang, Y. J. Zhu, L. Li, L. Zhang, H. Tong, W. W. Wang, G. F. Cheng and J. F. Zhu, A facile hydrothermal route to flower-like cobalt hydroxide and oxide, Eur. J. Inorg. Chem., 2006, 4787–4792, DOI:10.1002/ejic.200600553.
- H.-P. Cong and S.-H. Yu, Shape control of cobalt carbonate particles by a hydrothermal process in a mixed solvent: an efficient precursor to nanoporous cobalt oxide architectures and their sensing property, Cryst. Growth Des., 2009, 9, 210–217, DOI:10.1021/cg8003068.
- A.-M. Cao, J.-S. Hu, H.-P. Liang, W.-G. Song, L.-J. Wan, X.-L. He, X.-G. Gao and S.-H. Xia, Hierarchically structured cobalt oxide (Co3O4): the morphology control and its potential in sensors, J. Phys. Chem. B, 2006, 110, 15858–15863, DOI:10.1021/jp0632438.
- R. Lakra, R. Kumar, D. N. Thatoi, P. K. Sahoo and A. Soam, Synthesis and characterization of cobalt oxide (Co3O4) nanoparticles, Mater. Today: Proc., 2021, 41, 269–271, DOI:10.1016/j.matpr.2020.09.099.
- Y. Li, X. Li, Z. Wang, H. Guo and T. Li, Distinct impact of cobalt salt type on the morphology, microstructure, and electrochemical properties of Co3O4 synthesized by ultrasonic spray pyrolysis, J. Alloys Compd., 2017, 696, 836–843, DOI:10.1016/j.jallcom.2016.12.038.
- M. Salavati-Niasari, N. Mir and F. Davar, Synthesis and characterization of Co3O4 nanorods by thermal decomposition of cobalt oxalate, J. Phys. Chem. Solids, 2009, 70, 847–852, DOI:10.1016/j.jpcs.2009.04.006.
- M. M. Shahid, P. Rameshkumar and N. M. Huang, Morphology dependent electrocatalytic properties of hydrothermally synthesized cobalt oxide nanostructures, Ceram. Int., 2015, 41, 13210–13217, DOI:10.1016/j.ceramint.2015.07.098.
- J. Park, X. Shen and G. Wang, Solvothermal synthesis and gas-sensing performance of Co3O4 hollow nanospheres, Sens. Actuators, B, 2009, 136, 494–498, DOI:10.1016/j.snb.2008.11.041.
- T.-L. Lai, Y.-L. Lai, C.-C. Lee, Y.-Y. Shu and C.-B. Wang, Microwave-assisted rapid fabrication of Co3O4 nanorods and application to the degradation of phenol, Catal. Today, 2008, 131, 105–110, DOI:10.1016/j.cattod.2007.10.039.
- A. Demortiere, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Bégin-Colin, Size-dependent properties of magnetic iron oxide nanocrystals, Nanoscale, 2011, 3, 225–232, 10.1039/C0NR00521E.
- S. G. Kwon and T. Hyeon, Colloidal chemical synthesis and formation kinetics of uniformly sized nanocrystals of metals, oxides, and chalcogenides, Acc. Chem. Res., 2008, 41, 1696–1709, DOI:10.1021/ar8000537.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Synthesis of monodisperse spherical nanocrystals, Angew. Chem., Int. Ed., 2007, 46, 4630–4660, DOI:10.1002/anie.200603148.
- E. Kang, J. Park, Y. Hwang, M. Kang, J.-G. Park and T. Hyeon, Direct synthesis of highly crystalline and monodisperse manganese ferrite nanocrystals, J. Phys. Chem. B, 2004, 108, 13932–13935, DOI:10.1021/jp049041y.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, Monodisperse MFe2O4(M = Fe, Co, Mn) nanoparticles, J. Am. Chem. Soc., 2004, 126, 273–279, DOI:10.1021/ja0380852.
- C. O. Kappe, and A. Stadler, Microwave Organic and Medicinal Chemistry, Willey, 2001, vol. 25, pp. 12–20, DOI:10.1002/3527606556.
- T. Thongtem, A. Phuruangrat and S. Thongtem, Preparation and characterization of nanocrystalline SrWO4 using cyclic microwave radiation, Curr. Appl. Phys., 2008, 8, 189–197, DOI:10.1016/j.cap.2007.08.002.
- J. Bi, L. Wu, Z. Li, Z. Ding, X. Wang and X. Fu, A facile microwave solvothermal process to synthesize ZnWO4 nanoparticles, J. Alloys Compd., 2009, 480, 684–688, DOI:10.1016/j.jallcom.2009.02.029.
- A. M. Peiró, C. Domingo, J. Peral, X. Domènech, E. Vigil, M. A. Hernández-Fenollosa, M. Mollar, B. Marí and J. A. Ayllón, Nanostructured zinc oxide films grown from microwave activated aqueous solutions, Thin Solid Films, 2005, 483, 79–83, DOI:10.1016/j.tsf.2004.12.030.
- S. Bhattacharyya and A. Gedanken, Microwave-assisted insertion of silver nanoparticles into 3-D mesoporous zinc oxide nanocomposites and nanorods, J. Phys. Chem. C, 2008, 112, 659–665, DOI:10.1021/jp0760253.
- J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Toward greener nanosynthesis, Chem. Rev., 2007, 107, 2228–2269, DOI:10.1021/cr050943k.
- S.-E. Park and K.-M. Choi, Green catalysis by microwave synthesized nanostructured materials, J. Phys. Chem. Solids, 2008, 69, 1501–1504, DOI:10.1016/j.jpcs.2007.10.119.
- V. Abdelsayed, A. Aljarash, M. S. El-Shall, Z. A. Al Othman and A. H. Alghamdi, Microwave synthesis of bimetallic nanoalloys and CO oxidation on ceria-supported nanoalloys, Chem. Mater., 2009, 21, 2825–2834, DOI:10.1021/cm9004486.
- M. Estruga, C. Domingo and J. A. Ayllón, Microwave radiation as heating method in the synthesis of titanium dioxide nanoparticles from hexafluorotitanate-organic salts, Mater. Res. Bull., 2010, 45, 1224–1229, DOI:10.1016/j.materresbull.2010.05.015.
- S. Yin, Z. Luo, J. Xia and H. Li, Microwave-assisted synthesis of Fe3O4 nanorods and nanowires in an ionic liquid, J. Phys. Chem. Solids, 2010, 71, 1785–1788, DOI:10.1016/j.jpcs.2010.09.016.
- I. Bilecka, M. Kubli, E. Amstad and M. Niederberger, Simultaneous formation of ferrite nanocrystals and deposition of thin films via a microwave-assisted nonaqueous sol–gel process, J. Sol-Gel Sci. Technol., 2011, 57, 313–322, DOI:10.1007/s10971-010-2165-1.
- S. Saremi-Yarahmadi, B. Vaidhyanathan and K. G. U. Wijayantha, Microwave-assisted low temperature fabrication of nanostructured α-Fe2O3 electrodes for solar-driven hydrogen generation, Int. J. Hydrogen Energy, 2010, 35, 10155–10165, DOI:10.1016/j.ijhydene.2010.08.004.
- R. G. Deshmukh, S. S. Badadhe and I. S. Mulla, Microwave-assisted synthesis and humidity sensing of nanostructured α-Fe2O3, Mater. Res. Bull., 2009, 44, 1179–1182, DOI:10.1016/j.materresbull.2008.09.044.
- I. Bilecka and M. Niederberger, Microwave chemistry for inorganic nanomaterials synthesis, Nanoscale, 2010, 2, 1358–1374, 10.1039/B9NR00377K.
- C. Xu, Y. Liu, C. Zhou, L. Wang, H. Geng and Y. Ding, An in situ dealloying and oxidation route to Co3O4nanosheets and their ambient-temperature CO oxidation activity, ChemCatChem, 2011, 3, 399–407, DOI:10.1002/cctc.201000275.
- Y. Sun, P. Lv, J.-Y. Yang, L. He, J.-C. Nie, X. Liu and Y. Li, Ultrathin Co3O4 nanowires with high catalytic oxidation of CO, Chem. Commun., 2011, 47, 11279–11281, 10.1039/C1CC14484G.
- Y. Teng, Y. Kusano, M. Azuma, M. Haruta and Y. Shimakawa, Morphology effects of Co3O4 nanocrystals catalyzing CO oxidation in a dry reactant gas stream, Catal. Sci. Technol., 2011, 1, 920–922, 10.1039/C1CY00113B.
- L. Hu, K. Sun, Q. Peng, B. Xu and Y. Li, Surface active sites on Co3O4 nanobelt and nanocube model catalysts for CO oxidation, Nano Res., 2010, 3, 363–368, DOI:10.1007/s12274-010-1040-2.
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta and W. Shen, Low-temperature oxidation of CO catalysed by Co3O4 nanorods, Nature, 2009, 458, 746–749, DOI:10.1038/nature07877.
- Y. Yu, T. Takei, H. Ohashi, H. He, X. Zhang and M. Haruta, Pretreatments of Co3O4 at moderate temperature for CO oxidation at −80°C, J. Catal., 2009, 267, 121–128, DOI:10.1016/j.jcat.2009.08.003.
- D. Perti and R. L. Kabel, Kinetics of CO oxidation over Co3O4/γ-Al2O3. Part I: Steady state, AIChE J., 1985, 31, 1420–1426, DOI:10.1002/aic.690310903.
- Y.-F. Y. Yao, The oxidation of hydrocarbons and CO over metal oxides: III. Co3O4, J. Catal., 1974, 33, 108–122, DOI:10.1016/0021-9517(74)90250-4.
- G. Marbán, I. López, T. Valdés-Solís and A. B. Fuertes, Highly active structured catalyst made up of mesoporous Co3O4 nanowires supported on a metal wire mesh for the preferential oxidation of CO, Int. J. Hydrogen Energy, 2008, 33, 6687–6695, DOI:10.1016/j.ijhydene.2008.07.067.
- M. Khasu, T. Nyathi, D. J. Morgan, G. J. Hutchings, M. Claeys and N. Fischer, Co3O4 morphology in the preferential oxidation of CO, Catal. Sci. Technol., 2017, 7, 4806–4817, 10.1039/C7CY01194F.
- G. Grzybek, K. Ciura, J. Gryboś, P. Indyka, A. Davó-Quiñonero, D. Lozano-Castelló, A. Bueno-Lopez, A. Kotarba and Z. Sojka, CO-PROX reaction over Co3O4|Al2O3 Catalysts—Impact of the spinel active phase faceting on the catalytic performance, J. Phys. Chem. C, 2019, 123, 20221–20232, DOI:10.1021/acs.jpcc.9b03025.
- M.-H. Li, W. Da Oh, K.-Y. A. Lin, C. Hung, C. Hu and Y. Du, Development of 3-dimensional Co3O4 catalysts with various morphologies for activation of Oxone to degrade 5-sulfosalicylic acid in water, Sci. Total Environ., 2020, 724, 138032, DOI:10.1016/j.scitotenv.2020.138032.
- Y. Yan, K. Li, X. Chen, Y. Yang and J. M. Lee, Heterojunction-assisted Co3S4@Co3O4core–shell octahedrons for supercapacitors and both oxygen and carbon dioxide reduction reactions, Small, 2017, 13, 1701724, DOI:10.1002/smll.201701724.
- T. Warang, N. Patel, R. Fernandes, N. Bazzanella and A. Miotello, Co3O4 nanoparticles assembled coatings synthesized by different techniques for photo-degradation of methylene blue dye, Appl. Catal., B, 2013, 132–133, 204–211, DOI:10.1016/j.apcatb.2012.11.040.
- T. Warang, N. Patel, A. Santini, N. Bazzanella, A. Kale and A. Miotello, Pulsed laser deposition of Co3O4 nanoparticles assembled coating: Role of substrate temperature to tailor disordered to crystalline phase and related photocatalytic activity in degradation of methylene blue, Appl. Catal., A, 2012, 423–424, 21–27, DOI:10.1016/j.apcata.2012.02.037.
- L. Hu, Q. Peng and Y. Li, Low-temperature CH4catalytic combustion over Pd catalyst supported on Co3O4nanocrystals with well-defined crystal planes, ChemCatChem, 2011, 3, 868–874, DOI:10.1002/cctc.201000407.
- I. Chorkendorff, and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, Wiley-VCH, Weinheim, 2003, pp. 172–173, DOI:10.1002/3527602658.
- S. Zhang, J. Shan, Y. Zhu, A. I. Frenkel, A. Patlolla, W. Huang, S. J. Yoon, L. Wang, H. Yoshida, S. Takeda and F. Feng, Tao, WGS catalysis and in situ studies of CoO1–x, PtCon/Co3O4, and PtmCom'/CoO1–xnanorod catalysts, J. Am. Chem. Soc., 2013, 135, 8283–8293, DOI:10.1021/ja401967y.
- W. J. Xue, Y. F. Wang, P. Li, Z.-T. Liu, Z. P. Hao and C. Y. Ma, Morphology effects of Co3O4 on the catalytic activity of Au/Co3O4 catalysts for complete oxidation of trace ethylene, Catal. Commun., 2011, 12, 1265–1268, DOI:10.1016/j.catcom.2011.04.003.
- X. Wei, S. Barkaoui, J. Chen, G. Cao, Z. Wu, F. Wang and G. Li, Investigation of Au/Co3O4 nanocomposites in glycol oxidation by tailoring Co3O4 morphology, Nanoscale Adv., 2021, 3, 1741–1746, 10.1039/D1NA00053E.
- Q. Shi, Z. Li, C. Cao, G. Li and S. Barkaoui, Robust 2 nm-sized gold nanoclusters on Co3O4 for CO oxidation, Nanoscale Adv., 2023, 5, 5385–5389, 10.1039/D3NA00561E.
- C.-J. Jia, M. Schwickardi, C. Weidenthaler, W. Schmidt, S. Korhonen, B. M. Weckhuysen and F. Schüth, Co3O4–SiO2nanocomposite: A very active catalyst for CO oxidation with unusual catalytic behavior, J. Am. Chem. Soc., 2011, 133, 11279–11288, DOI:10.1021/ja2028926.
- L. Lukashuk, N. Yigit, R. Rameshan, E. Kolar, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, K. Föttinger and G. Rupprechter, Operando insights into CO oxidation on cobalt oxide catalysts by NAP-XPS, FTIR, and XRD, ACS Catal., 2018, 8, 8630–8641, DOI:10.1021/acscatal.8b01237.
- M. J. Pollard, B. A. Weinstock, T. E. Bitterwolf, P. R. Griffiths, A. Piers Newbery and J. B. Paine, A mechanistic study of the low-temperature conversion of carbon monoxide to carbon dioxide over a cobalt oxide catalyst, J. Catal., 2008, 254, 218–225, DOI:10.1016/j.jcat.2008.01.001.
- J. Jansson, A. E. C. Palmqvist, E. Fridell, M. Skoglundh, L. Österlund, P. Thormählen and V. Langer, On the catalytic activity of Co3O4 in low-temperature CO oxidation, J. Catal., 2002, 211, 387–397, DOI:10.1006/jcat.2002.3738.
- J. Jansson, Low-temperature CO oxidation over Co3O4/Al2O3, J. Catal., 2000, 194, 55–60, DOI:10.1006/jcat.2000.2924.
- H.-F. Wang, R. Kavanagh, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, Origin of extraordinarily high catalytic activity of Co3O4 and its morphological chemistry for CO oxidation at low temperature, J. Catal., 2012, 296, 110–119, DOI:10.1016/j.jcat.2012.09.005.
- X.-Y. Pang, C. Liu, D.-C. Li, C.-Q. Lv and G.-C. Wang, Structure sensitivity of CO oxidation on Co3O4: A DFT study, ChemPhysChem, 2013, 14, 204–212, DOI:10.1002/cphc.201200807.
- D. Jiang and S. Dai, The role of low-coordinate oxygen on Co3O4(110) in catalyticCO oxidation, Phys. Chem. Chem. Phys., 2011, 13, 978–984, 10.1039/C0CP01138J.
- P. Broqvist, I. Panas and H. Persson, A DFT study on CO oxidation over Co3O4, J. Catal., 2002, 210, 198–206, DOI:10.1006/jcat.2002.3678.
- L. Zhong, T. Kropp, W. Baaziz, O. Ersen, D. Teschner, R. Schlögl, M. Mavrikakis and S. Zafeiratos, Correlation between reactivity and oxidation state of cobalt oxide catalysts for CO preferential oxidation, ACS Catal., 2019, 9, 8325–8336, DOI:10.1021/acscatal.9b02582.
- F. Zasada, J. Janas, W. Piskorz, M. Gorczyńska and Z. Sojka, Total oxidation of lean methane over cobalt spinel nanocubes controlled by the self-adjusted redox state of the catalyst: Experimental and theoretical account for interplay between the Langmuir–Hinshelwood and Mars–Van Krevelen mechanisms, ACS Catal., 2017, 7, 2853–2867, DOI:10.1021/acscatal.6b03139.
- F. Zasada, W. Piskorz, J. Janas, E. Budiyanto and Z. Sojka, Dioxygen activation pathways over cobalt spinel nanocubes—From molecular mechanism into ab initio thermodynamics and 16O2/18O2exchange microkinetics, J. Phys. Chem. C, 2017, 121, 24128–24143, DOI:10.1021/acs.jpcc.7b09597.
- F. Zasada, J. Gryboś, E. Budiyanto, J. Janas and Z. Sojka, Oxygen species stabilized on the cobalt spinel nano-octahedra at various reaction conditions and their role in catalytic CO and CH4 oxidation, N2O decomposition and oxygen isotopic exchange, J. Catal., 2019, 371, 224–235, DOI:10.1016/j.jcat.2019.02.010.
- W. Hu, J. Lan, Y. Guo, X.-M. Cao and P. Hu, Origin of efficient catalytic combustion of methane over Co3O4(110): Active low-coordination lattice oxygen and cooperation of multiple active sites, ACS Catal., 2016, 6, 5508–5519, DOI:10.1021/acscatal.6b01080.
- S. Wang, C. Zhao, S. Li and Y. Sun, First principles prediction of CH4 reactivities with Co3O4 nanocatalysts of different morphologies, Phys. Chem. Chem. Phys., 2017, 19, 30874–30882, 10.1039/C7CP04516F.
- Z. Chen, S. Wang, W. Liu, X. Gao, D. Gao, M. Wang and S. Wang, Morphology-dependent performance of Co3O4 via facile and controllable synthesis for methane combustion, Appl. Catal., A, 2016, 525, 94–102, DOI:10.1016/j.apcata.2016.07.009.
- Y. Sun, J. Liu, J. Song, S. Huang, N. Yang, J. Zhang, Y. Sun and Y. Zhu, Exploring the effect of Co3O4nanocatalysts with different dimensional architectures on methane combustion, ChemCatChem, 2016, 8, 540–545, DOI:10.1002/cctc.201501056.
- F. Zasada, J. Gryboś, W. Piskorz and Z. Sojka, Cobalt spinel (111) facets of various stoichiometry—DFT+U and ab initio thermodynamic investigations, J. Phys. Chem. C, 2018, 122, 2866–2879, DOI:10.1021/acs.jpcc.7b11869.
- S. Zhang, J. Shan, Y. Zhu, L. Nguyen, W. Huang, H. Yoshida, S. Takeda and F. Feng, Tao, Restructuring transition metal oxide nanorods for 100% selectivity in reduction of nitric oxide with carbon monoxide, Nano Lett., 2013, 13, 3310–3314, DOI:10.1021/nl4015292.
- M. R. Ward, E. D. Boyes and P. L. Gai, In situ aberration-corrected environmental TEM: Reduction of model Co3O4 in H2 at the atomic level, ChemCatChem, 2013, 5, 2655–2661, DOI:10.1002/cctc.201300047.
- K. Y. Jang, S. J. Ahn, J.-H. Kwon, K. M. Nam and Y. H. Kim, Novel route from a wurtzite to a rock-salt structure in CoO nanocrystals: In situ transmission electron microscopy study, J. Phys. Chem. C, 2019, 123, 10689–10694, DOI:10.1021/acs.jpcc.9b01548.
- Q. Shi, Y. Zhang, Z. Li, Z.-K. Han, L. Xu, A. Baiker and G. Li, Morphology Effects in MnCeOx solid solution-catalyzed NO reduction with CO: active sites, water tolerance, and reaction pathway, Nano Res., 2023, 16, 6951–6959, DOI:10.1007/s12274-023-5407-6.
- Z. Li, Y. Zhang, Q. Jiang, L. Xu, Z. Han, A. Baiker and G. Li, CuCeOx/CuO catalyst derived from layered double hydroxide precursor: Catalytic performance in NO reduction with CO in the presence of water and oxygen, Langmuir, 2023, 39, 6957–6963, DOI:10.1021/acs.langmuir.2c03258.
- Q. Shi, A. Raza, L. Xu and G. Li, Bismuth oxyhalide quantum dots modified titanate-necklaces with exceptional population of oxygen vacancies and photocatalytic activity, J. Colloid Interface Sci., 2022, 625, 750–760, DOI:10.1016/j.jcis.2022.06.066.
- J. Zhang, Y. Xie, Q. Jiang, S. Guo, J. Huang, L. Xu, Y. Wang and G. Li, Facile Synthesis of Cobalt Clusters-CoNx Composites: Synergistic Effect Boosts up Electrochemical Oxygen Reduction, J. Mater. Chem. A, 2022, 10, 16920–16927, 10.1039/D2TA04413G.
- Z. Li, L. Xu, Z. Babar, A. Raza, Y. Zhang, X. Gu, Y. Miao, Z. Zhao and G. Li, Fabrication of MXene-Bi2WO6 heterojunction by Bi2Ti2O7 hinge for extraordinary LED-light-driven photocatalytic performance, Nano Res., 2024, 17, 4729–4736, DOI:10.1007/s12274-023-6408-1.
- Y. Zhang, Z. Li, J. Zhang, L. Xu, Z. Han, A. Baiker and G. Li, Nanostructured Ni-MoCx: An Efficient Non-Noble Metal Catalyst for the Chemoselective Hydrogenation of Nitroaromatics, Nano Res., 2023, 16, 8919–8928, DOI:10.1007/s12274-023-5598-x.
| This journal is © The Royal Society of Chemistry 2024 |