
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning nanoparticles’ internal structure: fluorinated single-chain nanoparticles (SCNPs) generated by chain collapse of random copolymers†
Marah
Alqaisi
 a,
Justus F.
Thümmler
a,
Florian
Lehmann
b,
Franz-Josef
Schmitt
c,
Leonie
Lentz
c,
Fabian
Rieder
c,
Dariush
Hinderberger
b and
Wolfgang H.
Binder
*a
a,
Justus F.
Thümmler
a,
Florian
Lehmann
b,
Franz-Josef
Schmitt
c,
Leonie
Lentz
c,
Fabian
Rieder
c,
Dariush
Hinderberger
b and
Wolfgang H.
Binder
*a
aMacromolecular Chemistry, Institute of Chemistry, Faculty of Natural Science II (Chemistry, Physics and Mathematics), Martin-Luther-University Halle-Wittenberg, von-Danckelmann-Platz 4, Halle D-06120, Germany. E-mail: wolfgang.binder@chemie.uni-halle.de
bPhysical Chemistry, Institute of Chemistry, Faculty of Natural Science II (Chemistry, Physics and Mathematics), Martin-Luther-University Halle-Wittenberg, von-Danckelmann-Platz 4, Halle D-06120, Germany
cInstitute of Physics, Faculty of Natural Science II (Chemistry, Physics and Mathematics) Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 3, D-06120 Halle, Germany
First published on 13th May 2024
Abstract
The generation of nanosized compartments in single chain nanoparticles (SCNPs) is a promising approach to generate individualized confinement-zones on a small scale for drug-encapsulation or catalysis. We here report the synthesis and characterization of compartmented, fluorinated SCNPs generated by single-chain collapse of amphiphilic copolymers. Polyethylene glycol (PEG) functionalized monomers were utilized as hydrophilic moieties, while hydrophobic residues were introduced using different mole fractions of either aliphatic or fluorinated monomers. Single chain collapse and subsequently crosslinking via copper-catalyzed azide–alkyne click reactions in selective and non-selective solvents yields internally structured SCNPs with hydrodynamic radii of 2.5–5.8 nm. All of the SCNPs exhibited water solubility, but displayed different compartmentations sized <1 nm, depending on the type of hydrophobicity and the monomer ratio. Investigations using continuous wave electron paramagnetic resonance and decay associated fluorescence spectroscopy, specifically targeting the hydrophobic cores of the SCNPs, revealed significant differences between the aliphatic and fluorinated cores of the SCNPs as probed by different specific molecular labels, finally allowing specific embedding of molecules into the specific compartments.
Introduction
In nature a variety of non-covalent interactions, encompassing a spectrum of attractive and repulsive forces, are employed to arrange molecules into complex assemblies, often with a remarkable degree of order.1,2 Thus the self-assembly of polymers in solution generates well-known assemblies such as block-copolymeric micelles, primarily guided by incompatibilities between the polymer blocks and the surrounding solvent molecules, in addition to supramolecular inter/intrachain interactions within the polymer segments.3–6 Collapsing single polymer chains in interaction with surrounding solvent molecules results in single-chain nanoparticles (SCNPs), where the interplay of forces (intermolecular and intramolecular solvent/polymer and polymer/polymer interactions) generates nanoparticles in dimensions of a single polymer chain. If the so-generated assemblies are fixed by crosslinking the collapsed chain, such SCNPs can offer unique advantages for various applications, such as in catalysis7–11 and sensing,12,13 as well as in nanomedicine and drug delivery.14–17 It is exciting to note that the imperfect confinement of such a collapsed polymer chain bears resemblance to intrinsically disordered proteins (IDP),18,19 where a delicate balance between disorder and order gives rise to compartmentalized, sometimes highly organized assemblies (i.e. protein fibers).20 The size and morphology of such SCNPs depend on many factors such as the chosen crosslinking chemistry and the subsequent density and the initial conditions of chain collapse. Thus, intra-chain crosslinking is crucial in preserving the compactness of the polymer chain in addition to the length of the crosslinker to control the final shape of the nanoparticle. It has been shown that short crosslinkers induce predominantly short-range loops along the polymer chain, leading to less compacted ellipsoid-shaped SCNPs.21,22 Crosslinking chemistries for single-chain collapse are manifold, among them irreversible covalent, reversible covalent (dynamic) or noncovalent chemical bonds, all of which affect the final properties of the produced particles as well as the overall structure.23 Next to the crosslinking chemistry, which is used to fix the SCNP in its collapsed state, the design of the initial copolymer is responsible for the SCNPs’ nanostructure.24 The use of amphiphilic copolymers promotes the formation of internal structures, such as Janus-shapes or core–shell structures,25 which can further be used to encapsulate e.g. drugs or catalysts, improving their function and efficiency. We have recently reported the synthesis of water-soluble, amphiphilic SCNPs with defined internal hydrophobic compartments, displaying hydrodynamic radii of 4–5 nm, bearing compartments which are generated during collapse of the polymer chain in a selective solvent.26 Those SCNPs can selectively be loaded with different labels, then situated inside the SCNPs’ internal structure. When collapsed in water as a selective solvent, those labels experience a well-defined, hydrophobic, and dense surrounding inside the core of the SCNPs, whereas THF as a nonselective solvent produces more sparse, less defined structures in which the hydrophobic compartments are positioned in multiple regions distributed across the particle in a sparse structure.27 A similar pathway, dependent on the assembly/collapse process leading to compartmentation was observed by Delledonne et al.28 reporting the formation of unimer micelles serving as carriers for a fluorescent dye for biological applications. A random copolymer, composed of hydrophilic/hydrophobic segments, then generated small unimolecular micelles (<10 nm), featuring a hydrophobic fluorinated core formed through hydrophobic/hydrophilic interactions in water. As no covalent crosslinking was introduced the embedding of a hydrophobic fluorescent dye was accomplished dynamically within the hydrophobic core once dissolved in a nonselective solvent. Ko et al.29 reported the formation of well-defined nanoparticles from folding a single amphiphilic fluorous random copolymer for encapsulating a fluorinated agrochemical. In a similar fashion, Koda et al.30,31 produced biocompatible amphiphilic random copolymers, with PEG chains and per-fluorinated alkane pendants, based on hydrophilic/hydrophobic interactions to generate multi-chain micellar aggregates in addition to single-chain unimer micelles bearing a hydrophilic shell and a fluorinated hydrophobic compartment in water. Matsumoto et al.32 produced double-core or multicompartment micellar morphologies from orthogonal folding and self-assembly of fluorous/amphiphilic random block copolymers in water. Shibata et al.33 reported the investigation of self-folding and self-assembly of amphiphilic random copolymers compromising hydrophilic PEG side chains (4.5 or 8.5 oxyethylene units) in addition to hydrophobic butyl or dodecyl pendants. It was revealed that the pendant design played an important role in inducing intramolecular self-folding or intermolecular self-assembly.We here report on the synthesis and investigation of crosslinked, water-soluble, amphiphilic SCNPs featuring defined hydrophobic/hydrophilic monomer combinations together with different embedded internal compartments depending on the choice of the respective monomers. The hydrophobic entities are incorporated via either purely aliphatic or fluorinated monomer sidechains, representing the first report of core-crosslinked, fluorinated SCNPs, which have previously are known only as non-crosslinked unimolecular micelles. This design allows the SCNPs to adopt different compartments, guided by their hydrophilicity, hydrophobicity and fluorophilicity. The size/volume of both, the aliphatic/fluorinated and hydrophilic parts were controlled by varying the molar ratios of the monomers in the precursor copolymers. Using RAFT polymerization, we varied the content of the hydrophobic monomers ranging from 10 mol% to 30 mol% of the aliphatic or fluorinated monomer ratio in order to modulate compartment formation after chain collapse. To probe the influence of the additional fluorinated monomer on the final shape, morphology, size, and physicochemical properties of the SCNPs, chain collapse was conducted in two different solvents (water and THF). Several methods were used to reveal the nature of the so-formed SCNPs, such as size exclusion chromatography (SEC) and 1H-diffusion-ordered nuclear magnetic resonance (1H-DOSY NMR) spectroscopy, and we also focused on continuous wave electron paramagnetic resonance (CW-EPR) spectroscopy and decay associated fluorescence spectroscopy (DAS) confirming differences between the aliphatic and fluorinated compartments inside the SCNPs (see Scheme 1).
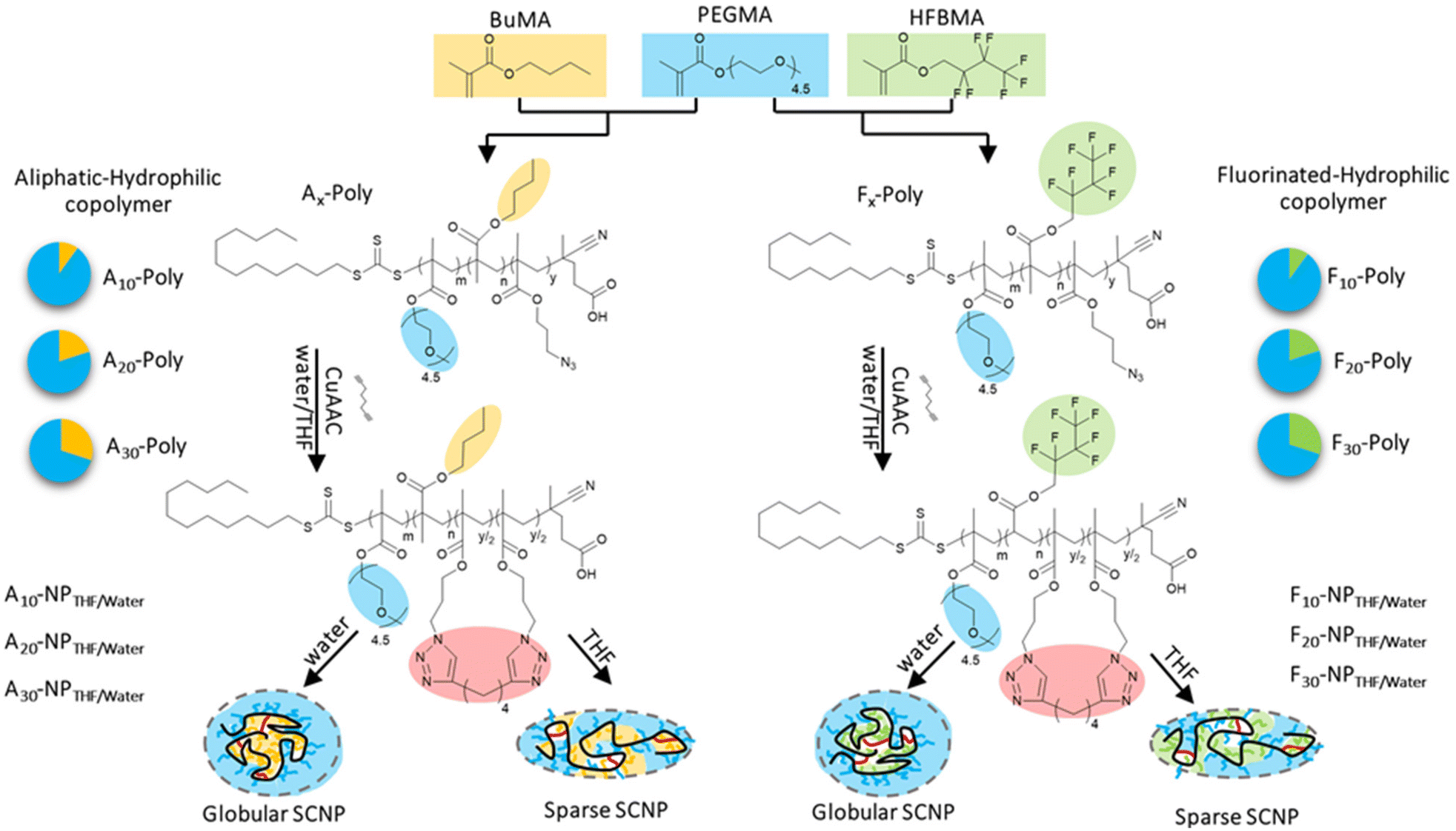 | ||
| Scheme 1 Synthesis of random copolymers from three different methacrylate monomers. Each polymer type displays two different philicities and is produced in three different molar compositions. Crosslinking after single chain collapse generates the desired single chain nanoparticles (SCNPs) with the desired compartments. | ||
Experimental
Materials
2,2′-Azobis(2-methylpropionitrile) AIBN (98%, Sigma-Aldrich), 4-cyano-4-(dodecylsulfanylthiocarbonyl)sulfanylpentanoic acid CDPA (97%, abcr), poly(ethylene glycol)methyl ether methacrylate PEGMA (average Mn 300 Da, containing 300 ppm BHT as an inhibitor and 100 ppm MEHQ as an inhibitor, ≤100%, Sigma-Aldrich), butyl methacrylate BuMA (99%, containing monomethyl ether hydroquinone as an inhibitor, Sigma-Aldrich), 2,2,3,3,4,4,4-heptafluorobutyl methacrylate HFBMA (97%, containing MEHQ as an inhibitor, Sigma-Aldrich), chloroform-d CDCl3 (stabilized with Ag, ≤100%, ARMAR), 1,7-octadiyne (98%, Sigma-Aldrich), copper(II) sulphate pentahydrate (≥99%, VEB Laborchemikalien), copper(I) bromide (98%, Sigma-Aldrich), (+)-sodium L-ascorbate (≥98%, Sigma-Aldrich), N,N,N′,N′′,N′′-pentamethyl diethylenetriamine PMDTA (99%, Sigma-Aldrich), deuterium oxide D2O (99.9%, Sigma-Aldrich), 2,2′-bipyridine (≥99%, Sigma-Aldrich), 5-DOXYL stearic acid, ammonium salt 5-DSA (>99%, Avanti polar lipids). The chemicals were used as received if not mentioned otherwise. All monomers were passed through a column of basic aluminum oxide prior to use to remove the inhibitors and subsequently dried using activated molecular sieves (2 Å). AIBN was re-crystallized from methanol.Characterization methods
1H-, 13C-, 19F-, and DOSY-NMR spectra were measured with an Agilent Technologies 400 MHz VNMRS (400 MHz) in deuterated solvents like CDCl3 and D2O and analyzed with MestReNova 14.2.1-27684 from Mestrelab Research S.L. SEC measurements were performed at 30 °C on a Viscotek GPCmax VE 2001 from Malvern applying a CLM3008 precolumn and a CLM3008 main column with 3.0 mg mL−1 sample concentration. ESI-ToF spectroscopy measurements were performed on an ESI-quadrupole time-of-flight mass spectrometer system (compact LC-MS system) from Bruker Daltonics GmbH & Co. KG with sample concentration 0.1–0.3 mg ml−1 in methanol. Attenuated total reflection-infrared (ATR-FTIR) spectroscopy measurements were done on a Bruker Tensor VERTEX 70 spectrometer equipped with a golden gate unit and the software Opus 8.2 was used to analyze the spectra. AFM imaging was performed on mica surfaces using a Nanosurf CoreAFM with Tap190AI-G cantilevers in the phase-contrast mode. Turbidimetry measurements were done using a JASCO J-1500 with a PTC-510 cell holder at 400 nm with fixed sample concentration of 1.0 mg mL−1. Absorption measurements were conducted on a UV/Vis spectrometer from PerkinElmer at room temperature. Fluorescence measurements were performed using a Cary Eclipse Fluorescence Spectrometer from Agilent using Hellma Analytics quartz 10 mm glass cuvettes at room temperature. CW EPR measurements were conducted using a Miniscope MS 5000 (Magnettech GmbH, Berlin, and Freiberg Instruments, Freiberg, Germany) in water at 25 °C and the simulations were done using Matlab (The Mathworks, Inc.) in combination with the EasySpin program. The fluorescence quantum yields (φf) were determined in aqueous solutions at room temperature (25 μM) with a 405 nm ps laser diode driven at 20 MHz (LDH-405, Picoquant, Berlin). Decay associated spectra (DAS) were determined from time- and wavelength-correlated fluorescence data performed with a setup as described in ref. 34 and 35 (see the ESI†).Synthesis of random copolymers Ax-poly
AIBN (≈1 mmol equivalent), the chain-transfer agent CDPA (≈5 mmol equivalent), PEGMA (1 mol equivalent), BuMA and APMA (see Table S1 in the ESI†) were dissolved in dry DMSO (c = 3.3 M). The reaction mixture was purified of oxygen via 5 freeze–pump–thaw cycles. Subsequently, the reaction was carried out for 3 hours at 70 °C. The reaction was quenched using an ice bath. The polymer solution was transferred for dialysis in water for three days using dialysis tubes with 5 kDa molecular weight cutoff, followed by an additional 3 days of dialysis in THF. The copolymers were characterized using SEC in THF and 1H-NMR in CDCl3 (see Scheme 1, Table 1 and Table S1 in the ESI†).| Sample |
m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n:ya :n:ya |
M
n,appb/kDa |
PDIa | G |
r
hd/nm |
T
cpe/°C |
Sample |
m:n:ya |
M
n,appb/kDa |
PDIa | G |
r
hd/nm |
T
cpe/°C |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Molar ratio of PEGMA:BuMA:APMA in Ax-poly and of PEGMA:HFBMA:APMA in Fx-poly, analyzed from 1H-NMR spectra in CDCl3.
b Measured by SEC in THF.
c Calculated from the (apparent) molecular weights using G = 100%(Mn,polymer − Mn,app,SCNP)/Mn.polymer.
d Calculated from 1H-DOSY NMR measurements in D2O using rh = (kB·T)/(6π·η·D).
e Measured by turbidimetry, Tcp = T at 90% transmission.
|
|||||||||||||
| A10-poly | 79:11:10 |
14.6 | 1.2 | — | 4.0 | 50 | F10-poly | 79:9:12 |
15.5 | 1.3 | — | 4.1 | 48 |
| A10-NPwater | 7.9 | 1.2 | 46% | 5.8 | 55 | F10-NPwater | 9.5 | 1.3 | 40% | 5.6 | 58 | ||
| A10-NPTHF | 11.4 | 1.2 | 22% | 3.3 | 56 | F10-NPTHF | 12.3 | 1.3 | 21% | 3.7 | 62 | ||
| A20-poly | 69:20:10 |
14.6 | 1.3 | — | 3.4 | 43 | F20-poly | 71:19:9 |
17.1 | 1.4 | — | 3.5 | 42 |
| A20-NPwater | 10.8 | 1.5 | 26% | 4.9 | 45 | F20-NPwater | 11.3 | 1.3 | 34% | 5.5 | 44 | ||
| A20-NPTHF | 13.2 | 1.3 | 10% | 2.5 | 53 | F20-NPTHF | 10.8 | 1.5 | 37% | 3.1 | 45 | ||
| A30-poly | 62:27:10 |
17.0 | 1.3 | — | 3.6 | 38 | F30-poly | 61:30:9 |
19.0 | 1.5 | — | 4.2 | 35 |
| A30-NPwater | 9.1 | 1.7 | 46% | 4.0 | 44 | F30-NPwater | 10.0 | 1.6 | 47% | 5.0 | 43 | ||
| A30-NPTHF | 17.0 | 1.5 | 0% | 3.2 | 40 | F30-NPTHF | 17.8 | 2.0 | 11% | 3.7 | 44 | ||
1H NMR (400 MHz, CDCl3, δ in ppm): 0.84–0.99 (–CH3 protons of the backbone, 3H), 1.21–1.85 (–CH2– protons of the backbone, 2H), 3.51–3.62 (–O-CH2-CH2-O– protons of PEGMA, 4H), 3.34 (–O-CH3 protons of PEGMA, 3H), 3.38 (–COO-CH2– protons of APMA, 2H), 3.90 (–COO-CH2– protons of BuMA, 2H) and 4.04 (–COO-CH2– protons of PEGMA, 2H).
Synthesis of random copolymers Fx-poly
AIBN (≈1 mmol equivalent), the chain-transfer agent CDPA (≈5 mmol equivalent), PEGMA (1 mol equivalent), HFBMA and APMA (see Table S1 in the ESI†) were dissolved in dry DMSO (c = 3.3 M). The reaction mixture was purified from oxygen via 5 freeze–pump–thaw cycles. Subsequently, the reaction was carried out for 3 hours at 70 °C. The reaction was quenched using an ice bath. The polymer solution was transferred for dialysis in water for three days using dialysis tubes with 5 kDa molecular weight cutoff, followed by an additional 3 days of dialysis in THF. The copolymers were characterized using SEC in THF and 1H- and 19F-NMR in CDCl3 (see Scheme 1, Table 1 and Table S1 in the ESI†).1H NMR (400 MHz, CDCl3, δ in ppm): 0.82–1.03 (–CH3 protons of the backbone, 3H), 1.21–1.89 (–CH2– protons of the backbone, 2H), 3.50–3.65 (–O-CH2-CH2-O– protons of PEGMA, 4H), 3.33–3.37 (–O-CH3 protons of PEGMA, 3H), 3.38–3.42 (–COO-CH2– protons of APMA, 2H), 4.05–4.08 (–COO-CH2– protons of PEGMA, 2H) and 4.38–4.42 (–COO-CH2– protons of HFBMA, 2H).
Crosslinking reaction of Ax-poly and Fx-poly in water
Crosslinking reactions were conducted in an oxygen-free environment using copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) click reaction with high dilutions (polymer solution concentration ≈10−6 M). The precursor polymer and octadiyne as crosslinker were dissolved in 20–25 mL dry THF. This solution was slowly added to the reaction solution via a syringe pump (1 mL h−1). The reaction solution contained copper(II) sulfate pentahydrate (CuSO4·5H2O, 0.5 mmol), PMDTA (2.0 mmol), and sodium ascorbate (7.6 mmol) in deionized water (250–300 mL). After complete addition of the polymer solution, the product (Ax-NPwater or Fx-NPwater) was extracted with DCM, dissolved in THF and purified by dialysis. The nanoparticles were characterized using SEC in THF and 1H- and 19F-NMR in CDCl3 (see Scheme 1, Table 1 and Table S1 in the ESI†).1H NMR (400 MHz, CDCl3, δ in ppm): 0.85–1.04 (–CH3 protons of the backbone, 3H), 1.24–1.85 (–CH2– protons of the backbone, 2H), 3.33–3.39 (–O-CH3 protons of PEGMA, 3H), 3.54–3.67 (–O-CH2-CH2-O– protons of PEGMA, 4H), 3.94 (–COO-CH2– protons of BuMA, 2H), 4.05–4.08 (–COO-CH2– protons of PEGMA, 2H) and 4.38–4.42 (–COO-CH2– protons of HFBMA, 2H).
Crosslinking reaction of Ax-poly and Fx-poly in THF
Crosslinking reactions were conducted in a water and oxygen-free environment using copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) click reaction with high dilution (polymer solution concentration ≈10−6 M). The precursor polymer and octadiyne as crosslinker were dissolved in 20–25 mL dry THF. This solution was slowly added to the reaction solution via a syringe pump (1 mL h−1). The reaction mixture contained copper(I) bromide (CuBr, 0.4–0.5 mmol) and 2,2′-bipyridine (1.3 mmol) dissolved in dry THF (250–300 mL). After complete addition of the polymer solution, water was added and the product (Ax-NPTHF or Fx-NPTHF) was extracted with DCM, dissolved in THF and purified by dialysis. The nanoparticles were characterized using SEC in THF and 1H- and 19F-NMR in CDCl3 (see Scheme 1, Table 1 and Table S1 in the ESI†).1H NMR (400 MHz, CDCl3, δ in ppm): 0.82–1.05 (–CH3 protons of the backbone, 3H), 1.24–2.17 (–CH2– protons of the backbone, 2H), 3.33–3.39 (–O-CH3 protons of PEGMA, 3H), 3.53–3.66 (–O-CH2-CH2-O– protons of PEGMA, 4H), 3.94 (–COO-CH2– protons of BuMA, 2H), 4.05–4.09 (–COO-CH2– protons of PEGMA, 2H) and 4.42 (–COO-CH2– protons of HFBMA, 2H).
Results and discussion
Design and synthesis strategy
The amphiphilic polymers and resulting SCNPs were designed and synthesized to balance the influence of their solvophilicity on the resulting nanostructure and physicochemical behavior. The use of the aliphatic monomer n-butyl methacrylate (BuMA) in the Ax-poly copolymer and the fluorinated monomer 2,2,3,3,4,4,4-heptafluorobutyl methacrylate (HFBMA) in the Fx-poly copolymer allowed to generate polymers and SCNPs with varying hydrophobic strengths, as depicted in Scheme 1. The design of the two major types of random copolymers (Ax-poly and Fx-poly) was motivated to investigate the segregation between aliphatic and fluorinated side chains in view of compartment formation, as well as their impact on loading/binding to the hydrophobic compartments upon SCNP formation. The precursor polymers were synthesized as random copolymers by reversible-addition–fragmentation chain-transfer (RAFT) polymerization. The monomer feeds used mixtures of the hydrophilic poly(ethylene glycol)methyl ether methacrylate (PEGMA, Mn = 300 g mol−1, DPPEG = 4.5), either BuMA or HFBMA as hydrophobic entities, and azidopropyl methacrylate (APMA) as the crosslinking unit. Six different precursor random copolymers were designed that differ in their hydrophobic/hydrophilic molar composition, namely: 10 mol% aliphatic (A10-poly), 20 mol% aliphatic (A20-poly), 30 mol% aliphatic (A30-poly), 10 mol% fluorinated (F10-poly), 20 mol% fluorinated (F20-poly) and 30 mol% fluorinated (F30-poly) monomers (see Scheme 1). Monomer conversion ranged between 20 and 40% with resulting molecular weights varying from 15 to 19 kg mol−1 and PDI values between 1.2 and 1.5, as indicated by 1H-NMR spectroscopy and SEC in THF (see Table 1). In addition, all polymers carry a third methacrylate monomer (APMA) with an azide group (–N3) as a crosslinking site in amounts of 9–12 mol% in the final chains. The compositions of the final polymers (see Table 1) are in line with the initial feed ratios as proven by 1H-NMR spectroscopy.The copolymers were then probed for the preparation of the SCNPs to study collapse and crosslinking of the copolymer chains in different solvents. As the chain conformation and folding is based on the delicate solvent/polymer interactions, dependent on the ratio of hydrophobicity/hydrophilicity in the copolymer, two different routes were used: (I) crosslinking in a selective solvent (water), which is a good solvent for the PEGMA monomer and a nonsolvent for the hydrophobic monomers (BuMA and HFBMA); and (II) in a nonselective solvent (THF), which is a good solvent for all monomers. Subsequently the produced morphologies were preserved by covalent crosslinking through copper-catalyzed azide–alkyne click-reactions (CuAAC) since this method has proven its efficacy in producing SCNPs.26,27,36,37 All six types of precursor copolymers display on average 5–7 crosslinking sites, provided by the presence of the azide functional group (–N3) from the monomer APMA. The crosslinking process was designed to guarantee intrachain crosslinking and avoid multiple chain interactions by using the continuous addition procedure, in which the precursor polymers together with the external crosslinker (octadiyne) were slowly added to the crosslinking solution via a syringe pump. Thus, the final shape and size of the SCNPs as well as the formation of compartments within the SCNPs are dependent on the folding process of the single precursor polymer chains, which is influenced by polymer/solvent interactions, and on the chemical composition. Twelve different types of SCNPs were produced and reported, namely: A10-NPwater, A20-NPwater and A30-NPwater from Ax-poly crosslinked in water; A10-NPTHF, A20-NPTHF and A30-NPTHF from Ax-poly crosslinked in THF; F10-NPwater, F20-NPwater and F30-NPwater from Fx-poly crosslinked in water; F10-NPTHF, F20-NPTHF and F30-NPTHF from Fx-poly crosslinked in THF (see Scheme 1).
Size determination
After crosslinking the SCNPs their size was investigated using size exclusion chromatography (SEC), which reflected a change in the apparent molecular weights of the precursor copolymers and their corresponding SCNPs. In addition to size changes as determined by changes in the hydrodynamic radius (rh) we used 1H-DOSY-NMR to determine the size of the SCNPs. ATR-IR spectroscopy was used for each polymer before and after crosslinking to identify the vibrational band of the azide functional group (–N3) from the monomer APMA around 2100 cm−1, indicative of the progress of the crosslinking chemistry.38 In all cases after crosslinking no presence of the vibrational band at 2100 cm−1 for the SCNPs was observed. This indicates a complete crosslinking by formation of the 1,2,3-triazole ring and thus stabilization of the SCNPs can be assumed. This observation is supported through ATR-IR spectra of F30-poly and its corresponding SCNPs (F30-NPTHF/water) and A30-poly and its corresponding SCNPs (A30-NPTHF/water) (see Fig. 1a and Fig. S1 in the ESI†). Further, 1H-NMR spectra for the SCNPs were inspected for the presence of the protons of any potentially unreacted alkyne groups (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) at a chemical shift of 1.92 ppm and they showed no presence. In conclusion, the functional groups of azide (–N3) and alkyne (CH) were completely consumed in CuAAC click-reactions to produce 1,2,3-triazole rings, part of the crosslinker, with on average 3 crosslinkers per SCNP. In addition, 1H-NMR spectroscopy shows a broadening of the SCNPs’ spectra due to restricted mobility of the copolymer side chains when compared with the spectra of the corresponding precursor polymers.39,40 Full width at half-maximum (FWHM) analysis at 3.37 ppm, which corresponds to the chemical shift of –O-CH3 of PEG in F20-poly and F20-NPTHF, shows an example with 6.1 mHz and 14 mHz, respectively (see Fig. 1b, and Table S2 and Fig. S2, S6 and S7 in the ESI†).s
CH) at a chemical shift of 1.92 ppm and they showed no presence. In conclusion, the functional groups of azide (–N3) and alkyne (CH) were completely consumed in CuAAC click-reactions to produce 1,2,3-triazole rings, part of the crosslinker, with on average 3 crosslinkers per SCNP. In addition, 1H-NMR spectroscopy shows a broadening of the SCNPs’ spectra due to restricted mobility of the copolymer side chains when compared with the spectra of the corresponding precursor polymers.39,40 Full width at half-maximum (FWHM) analysis at 3.37 ppm, which corresponds to the chemical shift of –O-CH3 of PEG in F20-poly and F20-NPTHF, shows an example with 6.1 mHz and 14 mHz, respectively (see Fig. 1b, and Table S2 and Fig. S2, S6 and S7 in the ESI†).s
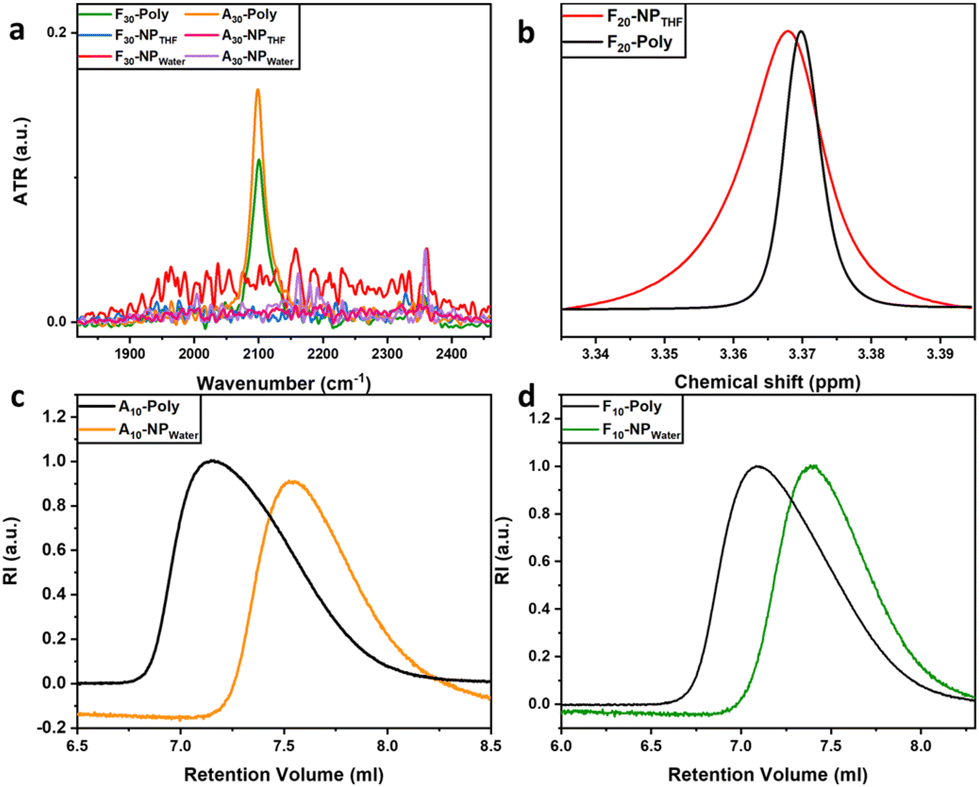 | ||
| Fig. 1 (a) ATR-IR absorption spectra for F30-poly, F30-NPTHF/water, A30-poly and A30-NPTHF/water. (b) 1H-NMR spectra in CDCl3 of F20-poly (black) and F20-NPTHF (red). SEC curves in THF for (c) A10-poly and A10-NPwater and (d) F10-poly and F10-NPwater. | ||
SEC is a commonly used method to investigate successful SCNP formation through reduced hydrodynamic volumes for the SCNPs when compared to the respective precursor polymers.41,42 For all copolymer species, the peaks of the precursor polymers appear at lower retention volumes than the ones corresponding to the SCNPs, indicating the reduction of the hydrodynamic volumes due to successful collapse/crosslinking.43 This behavior is observed in all SCNPs and their respective precursor polymers (see Fig. 1c, d and Fig. S10 in the ESI†). The compaction factor (G) is an indication of the compactness of the SCNP after undergoing intramolecular covalent crosslinking. It's a direct insight into the size change and a relative quantification using the molecular weights of the precursor copolymer chains and the apparent molecular weights of the corresponding SCNPs attained from SEC. All SCNPs show up to 47% compaction in comparison with their corresponding precursor copolymers (see Table 1). Further, the general trend shows that SCNPs crosslinked in water show higher compactions than those crosslinked in THF. This supports the conclusion that the morphology produced in water is more globular and compact and the morphology produced in THF is rather loose and less compact, hence resulting in sparse SCNPs.37 Moreover, size determination measurements were carried out using 1H-DOSY NMR in deuterated water (D2O). The Stokes–Einstein equation (see Table 1) was employed to estimate the hydrodynamic radii, taking into account the temperature and the viscosity of the solvent during the measurement.44 During folding and collapsing, the individual polymer chains undergo rearrangement due to intermolecular interactions (solvent/polymer) and intramolecular interactions (chain segments of the polymer), thus different behavior is expected for the collapse in different solvents. As an example, for the water-collapsed SCNPs, A10-poly displays a rh of 4.0 nm, while its corresponding nanoparticle A10-NPwater exhibits a rh of 5.8 nm. All other sample pairs follow the same behavior (Fig. 2a), with increased rh of the crosslinked SCNPs when compared to the corresponding precursor polymers. This indicates a larger solvation layer around the SCNPs, because of the internal structures which are formed during the single-chain collapse.45 Since the chain has undergone folding and collapsing events this implies that the randomly-dispersed side chains of different philicities along the backbone are induced to rearrange due to intermolecular interactions (solvent–side chain) and intramolecular interactions (side chain–side chain) to end up with most of the hydrophilic side chains facing the solvent phase and hydrophobic side chains folded inside. Additionally, 1H-DOSY NMR spectra were measured for those SCNPs which were crosslinked in THF. In contrast to the crosslinking behavior in water, for both species, Ax-NPTHF and Fx-NPTHF, the size reduces but does not change drastically after crosslinking in THF. We assume that when SCNPs are formed in a nonselective solvent like THF, the arrangement of the monomers and their philicities take place locally and across the chain. Consequently, the resulting SCNPs exhibit a looser structure with separate local domains of different natures, rather than a core–shell globular structure. Instead, they adopt a sparse structure, resulting in a smaller solvation layer around the particle compared to SCNPs formed in water as a selective solvent. This is reflected in the behavior of the hydrodynamic radii which appear smaller in this case, as visible in Fig. 2a. It is worth mentioning that dynamic light scattering (DLS) measurements were conducted as a tool to observe size differences. After inspection of the data (Fig. S16 and Table S4†), it has been concluded that there are differences in rh among the corresponding species when compared with those attained from 1H-DOSY. This is possibly due to particle agglomeration happening during the DLS measurement, in addition to the challenging nature of the measured particles’ size that pushes the DLS method and its accuracy to its limits. Nonetheless, a general trend has been noticed in DLS measurements that shows the precursor copolymers and their corresponding SCNPs are in the same size range. We have chosen to focus on 1H-DOSY data since it offers more precision.42 AFM imaging is used as an additional tool to navigate the conformation and size change due to the collapse and crosslinking events. The images show obvious changes in the measured heights of the detected particles deposited on mica surfaces. The dimensions of the SCNPs appear to be significantly smaller than the dimensions of the precursor polymer. In the case of the precursor polymer, A10-poly, the average height of the scanned particles is 14.2 nm and for its corresponding SCNPs (A10-NPTHF) the average height is 10.3 nm (see Fig. 2b and c, and Fig. S10 and S11 in the ESI†).
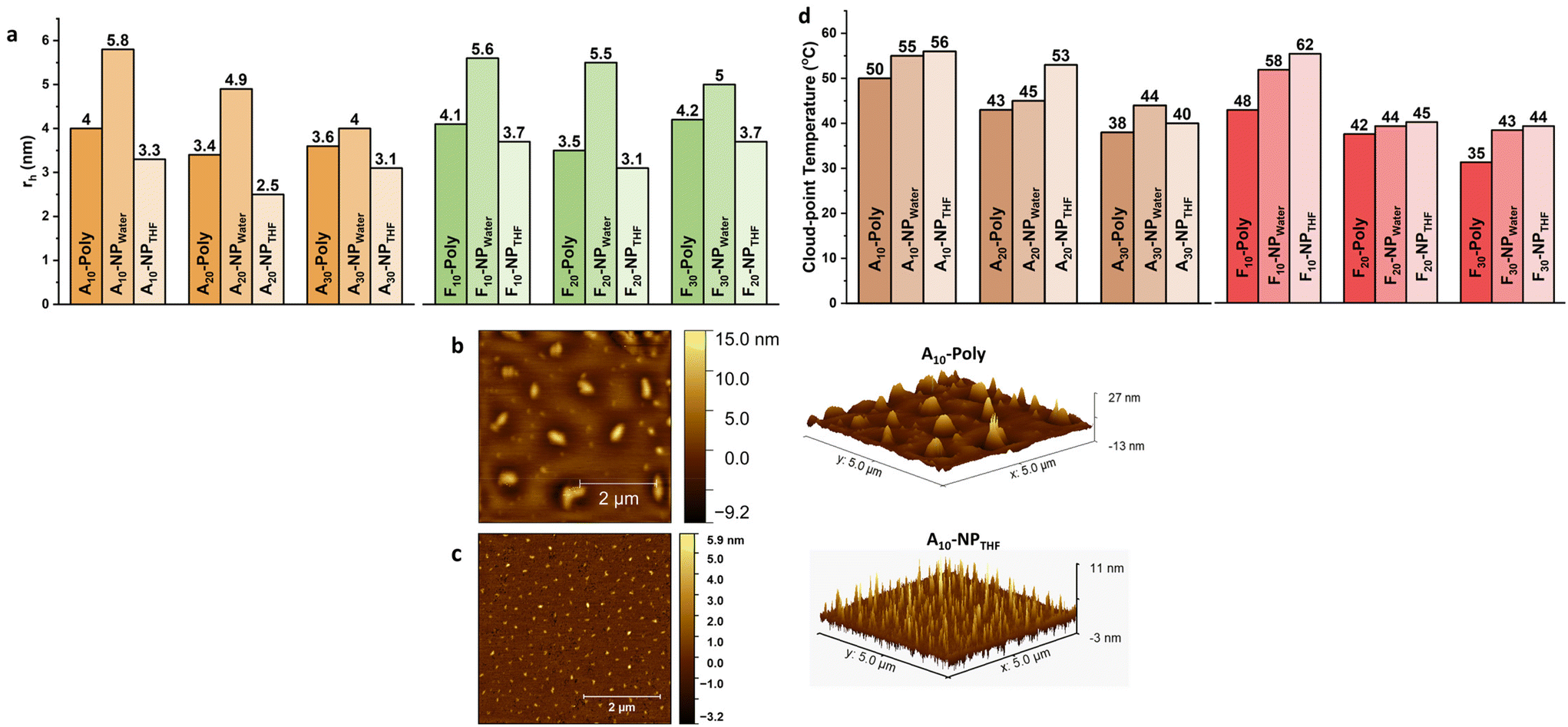 | ||
| Fig. 2 (a) Hydrodynamic radii (nm) as reported from 1H-DOSY NMR spectra in D2O for Ax-poly and the corresponding nanoparticles (Ax-NPwater/THF) and Fx-poly and its corresponding nanoparticles (Fx-NPwater/THF) crosslinked in water and in THF. AFM images of (b) A10-poly and (c) A10-NPTHF. (d) Cloud point temperatures, Tcp as determined from turbidimetry analysis for all copolymer species before and after crosslinking reactions in water and in THF. | ||
Thermal properties and core investigations
Turbidimetry was used to probe changes in solubility as a function of temperature by the thermo-responsive behavior of the poly(ethylene glycol)-side chains of the monomer PEGMA (see Scheme 1), constituting 70 to 90% mole fraction of the copolymers.46 Cloud-point temperatures (Tcp) were observed at 90% transmittance of the aqueous solutions at a fixed concentration of 1.0 mg ml−1 for the precursor polymers as well as the SCNPs. All of the precursor copolymers and the corresponding SCNPs exhibited sharp LCST behavior at various temperatures (Fig. S18†). Precursor copolymer types A10-poly and F10-poly and the corresponding SCNPs exhibited the highest Tcp, due to the higher content of PEG side chains. All in all, the observed elevation of Tcp for all SCNPs in comparison with their precursor copolymer chains suggests enhanced solubility of the SCNPs upon stabilizing the folded structure with internal covalent crosslinking. Hence, the solubility is mainly dominated by the hydrophilic PEG chains, increasing the overall solubility in water. This effect is particularly pronounced in the samples that were crosslinked in THF, whose resulting morphology after the collapse and crosslinking events in THF resembles a sparse structure. The compactness of the SCNPs can be described by the G factor. Hence, SCNPs formed in THF have a larger surface area compared to a sphere, leading to increased exposure to surrounding water molecules. The overall effect is reflected in higher solubility and correspondingly higher Tcp (see Fig. 2d). Additionally, it was noticed that the aliphatic SCNPs showed sharper LCST-type phase separation behavior in comparison with the fluorinated SCNPs (see Fig. S18†).To delve deeper into the nature of the SCNPs derived from both types of copolymers and to probe philicity differences between the aliphatic and fluorophilic cores, decay associated spectra (DAS) were determined following the encapsulation of fluorescent dyes. The dye which was used for emission and absorption measurements is a modified version of Brooker's Merocyanine (BM), known for its unique solvatochromism behavior, which depends on the polarity of the surrounding media. Notably, it exhibits the highest energy absorption in water and the lowest in chloroform, accompanied by a color change from yellow to blue, respectively.47,48 The modification of the dye involves attaching either an aliphatic chain (A-BM) or a fluorinated chain (F-BM) to reach the desired selectivity for each of the compartmeents. Further this modification aims to facilitate the diffusion of the dye molecule into either the aliphatic core or the fluorinated core of the nanoparticle. Overall by this modification the dye molecule will preferentially diffuse into the aliphatic compartment, the same principle applies to the dye with the fluorinated side chain. The technique has been previously employed by Delledonne et al.28 and was now adapted for our system (see the ESI†) to check for the formation of internal compartments inside the SCNPs.
We focussed on the cores of the following SCNPs, A30-NPwater and F30-NPwater. Absorption wavelengths for A-BM and F-BM when dissolved in water are 367 nm and 375 nm, respectively. Both dyes experience a red shift when encapsulated in the core of the SCNPs (A30-NPwater and F30-NPwater). More precisely, A-BM shifts to 388 nm and 390 nm, respectively. F-BM shifts to 400 nm in both cores (see Table 2 and Fig. S15 in the ESI†).
| Species | Parameters | F-BM | A-BM |
|---|---|---|---|
| λ: absorption wavelength. τ: fluorescence life-time of the dominant component (black). φ: fluorescence quantum yield. | |||
| F30-NPwater | λ/nm | 400 | 390 |
| τ (black)/ps | 118 | 77 | |
| φ/% | 2.2 | 1.8 | |
| A30-NPwater | λ/nm | 400 | 388 |
| τ (black)/ps | 100 | 77 | |
| φ/% | 1.8 | 1.7 | |
| Water | λ/nm | 375 | 367 |
| φ/% | 0.7 | 0.7 | |
DAS shows three different fluorescence components (see Fig. 3a–d). The dominating fluorescence component (black) for A-BM in A30-NPwater and F30-NPwater experiences the shortest life-time (77 ps) when compared with F-BM in A30-NPwater (100 ps) and the longest life-time in F30-NPwater (118 ps). This correlates with the highest quantum yield observed for F-BM in F30-NPwater (2.2%) and indicates that F-BM exhibits reduced quenching by water molecules in the fluorinated core. This is reasoned by the higher affinity of F-BM in the F30-NPwater core in comparison to the A30-NPwater core. The second fluorescence component (red) has the longest life-time (412 ps) when A-BM is encapsulated in the aliphatic core of A30-NPwater possibly indicating a stronger affinity of A-BM towards the aliphatic core than the fluorinated core of F30-NPwater. The third fluorescence component (1.1–1.9 ns, blue) is very small in amplitude and therefore not further evaluated. Fluorescence spectra of A-BM and F-BM in both cores were normalized to the corresponding quantum yields at 25 °C in aqueous solutions (see Fig. 3e and Table 2). The quantum yields φA/F-BM of both dyes in water, which in their added amounts are equal to 0.7%,48 were used to calculate the relative quantum yields of the dyes when encapsulated in A30-NPwater and F30-NPwater cores, φA/F-BM, A and φA/F-BM, F, respectively. The quantum yields φA-BM, A, φA-BM, F and φF-BM, A show comparable and higher values compared with φF-BM and φA-BM in water (1.7%–1.8%). The highest quantum yield was recorded for F-BM in the fluorinated core, φF-BM, F = 2.2%, clearly indicating that fluorescence quenching is most strongly suppressed in a fluorinated environment when compared with an aliphatic environment. Further investigations targeting the cores of A30-NPwater and F30-NPwater were carried out using the potential of continuous wave electron paramagnetic resonance (CW-EPR) spectroscopy, relying on a spin-labeled fatty acid, 5-doxyl-stearic acid (5-DSA), expected to specifically probe the hydrophobic cores of the SCNPs. Subsequently, CW-EPR spectra simulations were applied to identify the amount of bound 5-DSA within the SCNPs, as well as the strength of these bonds, by analyzing the inhibited motion of the bound spin probe.49 The CW-EPR spectra represent the sum of all paramagnetic signals inside the SCNP solutions, originating from two components: (I) freely solvated and rotating 5-DSA, and (II) SCNP-bound 5-DSA molecules. It was observed that the aliphatic SCNPs which were crosslinked in water (A30-NPwater) exhibit a higher binding capacity towards hydrophobic molecules like 5-DSA. For a SCNP/5-DSA ratio of 2/1, more than 96% of the 5-DSA molecules are located within the core of A30-NPwater. The core of F30-NPwater merely captures approximately 88% of the spin probe. Hence, the aliphatic probe has a higher affinity to be bound to the aliphatic core of A30-NPwater than to the fluorinated core of F30-NPwater. Furthermore, the rotation correlation time of SCNP-bound 5-DSA amounts to 7.59 ns. This value is identical for both cores of A30-NPwater and F30-NPwater and is higher than that of the free 5-DSA in water (0.10–0.18 ns), caused by the strong encapsulation of 5-DSA in the SCNP cores, decreasing its individual movement (see Fig. 3f and g, and Fig. S13 and Table S3 in the ESI†).
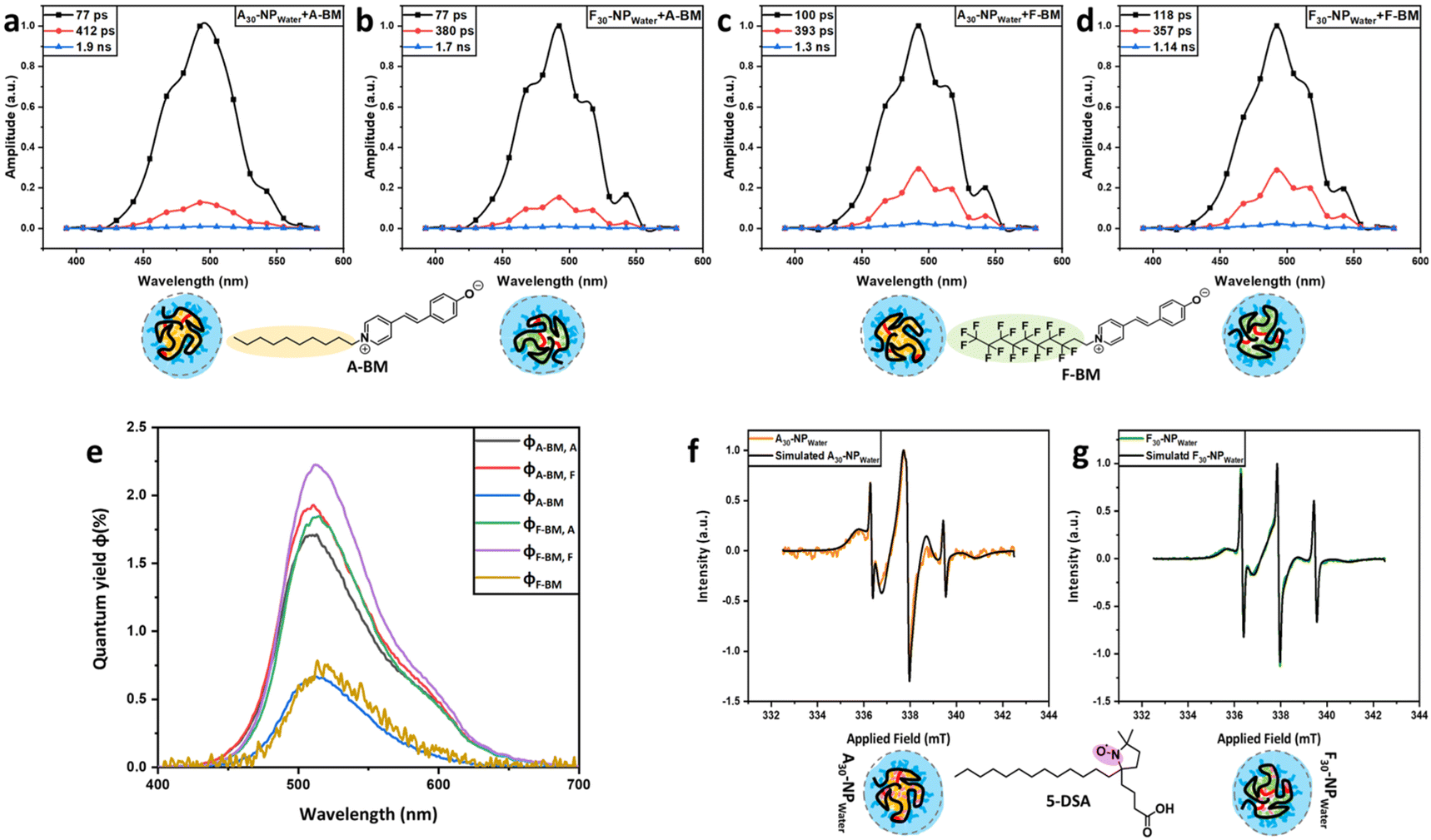 | ||
| Fig. 3 DAS of A-BM in (a) A30-NPwater and (b) F30-NPwater; DAS of F-BM in (c) A30-NPwater and (d) F30-NPwater. (e) Emission spectra of A-BM and F-BM. CW-EPR spectra and simulations of 5-DSA in the core of (f) A30-NPwater and (g) F30-NPwater. | ||
Conclusion
We here investigated the structural and physicochemical differences caused by segregation of hydrophobic and fluorophilic parts inside an amphiphilic single chain nanoparticle, SCNPs to generate internal nanosized compartments therein. Two sets of random amphiphilic copolymers were synthesized as precursor polymers, containing either hydrophilic–aliphatic or hydrophilic–fluorophilic monomers, and subsequently subjected to single-chain collapse, either in water as a selective solvent, or in THF as a nonselective solvent. The hydrophobic/hydrophilic ratio was systematically varied across the two types of the copolymers in three different hydrophobic molar compositions: 30 mol%, 20 mol% and 10 mol% in the final polymers. We explored the impact of changing this ratio on the final shape and morphology of the produced SCNPs and thus could reveal the formation of internal hydrophobic/fluorophilic cores resulting from the collapse and intra-chain crosslinking of copolymer chains in a selective solvent like water. Both EPR spectroscopy and fluorescence spectroscopy (DAS) indicated a clear difference of the formed SCNPs in different collapsing media and indicate the formation of water soluble SCNPs from random polymer chains with different philicity-distributions inside the SCNPs. When an amphiphilic random polymer is collapsed and crosslinked in a selective solvent like water, the resulting morphology is a more globular, core–shell arrangement inside the SCNP. Conversely, when the same random polymer is crosslinked in a nonselective solvent like THF, it forms a sparse structure, with the different philicities distributed locally. The presented methods allow to generate two different types of SCNPs from the same precursor polymer chain based on the choice of the collapsing solvent, and a tuning of the compartments inside the corresponding SCNP, thus forming a novel approach to modulate the SCNPs’ inner and outer structures – an important prerequisite for further exploitation of their function as catalysts and nanoreactors. An investigation aimed at elucidating the nature of the hydrophobic cores (aliphatic or fluorinated) was conducted by using DAS and CW-EPR spectroscopy of specifically labelled reported molecules, selective for either the hydrophobic or the fluorophilic compartment inside the SCNPs. Both methods showed that the choice of the type of hydrophobicity (aliphatic or fluorophilic) has influence on specifically locating small molecules inside parts of the SCNP cores. Hence, fluorinated molecules have a higher affinity to the fluorinated compartments, and aliphatic molecules to aliphatic compartments. This finding is important for the applications of SCNPs, e.g. in terms of drug delivery and selective catalysis, wherein an enzyme-like behavior can be reached via sleective compartimentation behavior. Herein even small differences in binding affinities can have major influences on the efficiency of the release of small substrate- or drug molecules, located inside the specific compartments, and released upon external triggers, such as NIR-activation or photoacoustic responses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DFG project INST 271/444-1 FUGG for financial support; the DFG-project BI1337/16-1; BI 1337/14-1 and the TP B02 within the GRK 2670, W69000789, project nr. 436494874.References
- D. B. Varshey, J. R. G. Sander, T. Friščić and L. R. MacGillivray, in Supramolecular Chemistry, 2012 Search PubMed.
- S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688–714 CrossRef.
- C. L. D. Gibb and B. C. Gibb, in Supramolecular Chemistry, 2012 Search PubMed.
- S. Chen, B.-D. Lechner, A. Meister and W. H. Binder, Nano Lett., 2016, 16, 1491–1496 CrossRef CAS PubMed.
- D. Heinz, E. Amado and J. Kressler, Polymers, 2018, 10, 960 CrossRef PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, A. Arbe, J. Kohlbrecher, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2015, 36, 1592–1597 CrossRef CAS PubMed.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
- J. Rubio-Cervilla, E. Gonzalez and J. A. Pomposo, Nanomaterials, 2017, 7, 341–360 CrossRef PubMed.
- M. Artar, E. R. J. Souren, T. Terashima, E. W. Meijer and A. R. A. Palmans, ACS Macro Lett., 2015, 4(10), 1099–1103 CrossRef CAS PubMed.
- M. A. J. Gillissen, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Polym. Chem., 2012, 3, 3166–3174 RSC.
- A. Latorre-Sanchez and J. A. Pomposo, Chem. Commun., 2015, 51, 15736–15738 RSC.
- A. B. Benito, M. K. Aiertza, M. Marradi, L. Gil-Iceta, T. S. Zahavi, B. Szczupak, M. Jimenez-Gonzalez, T. Reese, E. Scanziani, L. Passoni, M. Matteoli, M. D. Maglie, A. Orenstein, M. Oron-Herman, G. Kostenich, L. Buzhansky, E. Gazit, H. J. Grande, V. Gomez-Vallejo, J. Llop and I. Loinaz, Biomacromolecules, 2016, 17, 3213–3221 CrossRef CAS PubMed.
- A. P. P. Kröger, N. M. Hamelmann, A. Juan, S. Lindhoud and J. M. J. Paulusse, ACS Appl. Mater. Interfaces, 2018, 10, 30946–30951 CrossRef PubMed.
- Y. Yang, Y. Li, Q. Lin, C. Bao and L. Zhu, ACS Macro Lett., 2016, 5, 301–305 CrossRef CAS PubMed.
- A. P. P. Kröger and J. M. J. Paulusse, J. Controlled Release, 2018, 286, 326–347 CrossRef PubMed.
- J. A. Pomposo, I. Perez-Baena, F. Lo Verso, A. J. Moreno, A. Arbe and J. Colmenero, ACS Macro Lett., 2014, 3, 767–772 CrossRef CAS PubMed.
- A. J. Moreno, F. Lo Verso, A. Arbe, J. A. Pomposo and J. Colmenero, J. Phys. Chem. Lett., 2016, 7, 838–844 CrossRef CAS PubMed.
- E. Verde-Sesto, A. Arbe, A. J. Moreno, D. Cangialosi, A. Alegría, J. Colmenero and J. A. Pomposo, Mater. Horiz., 2020, 7, 2292–2313 RSC.
- S. Liao, L. Wei, L. A. Abriata and F. Stellacci, Macromolecules, 2021, 54, 11459–11467 CrossRef CAS.
- R. Upadhya, N. S. Murthy, C. L. Hoop, S. Kosuri, V. Nanda, J. Kohn, J. Baum and A. J. Gormley, Macromolecules, 2019, 52, 8295–8304 CrossRef CAS PubMed.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- G. M. t. Huurne, A. R. A. Palmans and E. W. Meijer, CCS Chem., 2019, 1, 64–82 CrossRef.
- A. Nitti, R. Carfora, G. Assanelli, M. Notari and D. Pasini, ACS Appl. Nano Mater., 2022, 5, 13985–13997 CrossRef CAS.
- J. F. Hoffmann, A. H. Roos, F.-J. Schmitt, D. Hinderberger and W. H. Binder, Angew. Chem., Int. Ed., 2021, 60, 7820–7827 CrossRef CAS PubMed.
- J. F. Thümmler, A. H. Roos, J. Krüger, D. Hinderberger, F.-J. Schmitt, G. Tang, F. G. Golmohamadi, J. Laufer and W. H. Binder, Macromol. Rapid Commun., 2023, 44, 2200618 CrossRef PubMed.
- A. Delledonne, E. Guazzelli, S. Pescina, A. Bianchera, G. Galli, E. Martinelli and C. Sissa, ACS Appl. Nano Mater., 2023, 6(17), 15551–15562 CrossRef CAS PubMed.
- J. H. Ko, A. Bhattacharya, T. Terashima, M. Sawamoto and H. D. Maynard, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 352–359 CrossRef CAS.
- Y. Koda, T. Terashima, M. Sawamoto and H. D. Maynard, Polym. Chem., 2015, 6, 240–247 RSC.
- Y. Koda, T. Terashima and M. Sawamoto, Macromolecules, 2016, 49, 4534–4543 CrossRef CAS.
- M. Matsumoto, M. Sawamoto and T. Terashima, ACS Macro Lett., 2019, 8, 320–325 CrossRef CAS PubMed.
- M. Shibata, M. Matsumoto, Y. Hirai, M. Takenaka, M. Sawamoto and T. Terashima, Macromolecules, 2018, 51, 3738–3745 CrossRef CAS.
- F. J. Schmitt, E. G. Maksimov, P. Hätti, J. Weißenborn, V. Jeyasangar, A. P. Razjivin, V. Z. Paschenko, T. Friedrich and G. Renger, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1461–1470 CrossRef CAS PubMed.
- F. J. Schmitt, E. G. Maksimov, H. Suedmeyer, V. Jeyasangar, C. Theiss, V. Z. Paschenko, H. J. Eichler and G. Renger, Photonics Nanostruct., 2011, 9, 190–195 CrossRef.
- S. Neumann, M. Biewend, S. Rana and W. H. Binder, The CuAAC: Principles, Homogeneous and Heterogeneous Catalysts, and Novel Developments and Applications, report 1022-1336, 2020.
- J. F. Thümmler, R. Maragani, F.-J. Schmitt, G. Tang, S. M. Rahmanlou, J. Laufer, H. Lucas, K. Mäder and W. H. Binder, Chem. Commun., 2023, 59, 11373–11376 RSC.
- X. S. Gai, B. A. Coutifaris, S. H. Brewer and E. E. Fenlon, Phys. Chem. Chem. Phys., 2011, 13, 5926–5930 RSC.
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Macromolecules, 2014, 47, 589–600 CrossRef CAS.
- K. Akiyoshi, S. Deguchi, N. Moriguchi, S. Yamaguchi and J. Sunamoto, Macromolecules, 1993, 26, 3062–3068 CrossRef CAS.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS PubMed.
- E. Blasco, B. T. Tuten, H. Frisch, A. Lederer and C. Barner-Kowollik, Polym. Chem., 2017, 8, 5845–5851 RSC.
- J. Engelke, J. Brandt, C. Barner-Kowollik and A. Lederer, Polym. Chem., 2019, 10, 3410–3425 RSC.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed.
- H. Zhang, L. Zhang, J. You, N. Zhang, L. Yu, H. Zhao, H.-J. Qian and Z.-Y. Lu, CCS Chem., 2021, 3, 2143–2154 CrossRef CAS.
- A. Gandhi, A. Paul, S. O. Sen and K. K. Sen, Asian J. Pharm. Sci., 2015, 10, 99–107 CrossRef.
- V. Z. Shirinian and A. A. Shimkin, in Heterocyclic Polymethine Dyes, ed. L. Strekowski, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, ch. 110, pp. 75–105 Search PubMed.
- V. Cavalli, D. C. da Silva, C. Machado, V. G. Machado and V. Soldi, J. Fluoresc., 2006, 16, 77–86 CrossRef CAS PubMed.
- P. D. Martin, B. Svensson, D. D. Thomas and S. Stoll, J. Phys. Chem. B, 2019, 123, 10131–10141 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00355a |
| This journal is © The Royal Society of Chemistry 2024 |