
Photopolymerization activated by photobase generators and applications: from photolithography to high-quality photoresists†
Han-Wen
Pei
 ab,
Kai
Ye
c,
Yizhi
Shao
c,
Dan
Chen
c,
Zhao-Yan
Sun
ab,
Tao
Gong
*c,
Dandan
Liu
*d and
Ke
Sun
*ce
ab,
Kai
Ye
c,
Yizhi
Shao
c,
Dan
Chen
c,
Zhao-Yan
Sun
ab,
Tao
Gong
*c,
Dandan
Liu
*d and
Ke
Sun
*ce
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei, 230026, China
cInstitute of Intelligent Manufacturing Technology, Shenzhen Polytechnic University, 4089 Shahe West Road, Shenzhen, 518055, China. E-mail: gongtao@szpt.edu.cn; sunke0101@szpt.edu.cn
dFAW-Volkswagen Automotive Co. Ltd., Changchun, 130021, China. E-mail: dandan.liu@faw-vw.com
eShenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China
First published on 3rd January 2024
Abstract
Light-induced polymerization has become a very attractive technology for advanced manufacturing of polymers. In fact, the photopolymerization processes, and even the final properties of the polymerized materials, are determined by the photoinitiator (PI), which is the key component that generates the active species during irradiation. Among the many photoactive compounds utilized as PIs, photoacid generators (PAGs) and photobase generators (PBGs) efficiently generate the bases or acids required to activate polymerization. Despite the obvious advantages of PAGs, PBGs exhibit wavelength specificity, better photostability, excellent compatibility with a wider range of monomers, etc. Furthermore, these organic compounds promote photopolymerizations followed by various photochemical processes encompassing ring-opening polymerization, the thiol–epoxy click reaction and thiol-Michael addition. With the aim of expanding the number of available PBGs, intensive research has been devoted in the last decade to designing and synthesising PBGs that promote photopolymerization with higher initiation capacities. In this review, we systematically discuss the recent advances made with organic PBGs during 2019–2023 to provide deeper insight into these photoactive compounds and broaden their application.
1 Introduction
Photopolymerization is an energy-saving and environmentally friendly technology that is triggered by photosensitive species, namely, photoinitiators (PIs), that are activated by UV or visible light.1–4 Many photoinitiators (PIs) have been discovered, and organic initiators have opened new avenues to photopolymerization by providing fast kinetics and access to a broader range of monomers.5–9 As conventional organic initiators, photoacid generators (PAGs) and photobase generators (PBGs) efficiently generate active species for photopolymerization.10–14 In most circumstances, the photopolymerization process is strongly inhibited by atmospheric oxygen, which affects the properties of the final material.15,16 However, PAGs and PBGs avoid these drawbacks, including oxygen inhibition, small molecule migration and yellowing.17 Hence, the utilization of PAGs and PBGs broadens the range of photopolymerization techniques; however, the choice between a PAG or a PBG depends on their specific advantages and disadvantages in the application.10,13,18–20PAGs are compounds that generate strong acids upon exposure to light, and these acids are the active species that activate polymerization reactions.21–23 Indeed, PAGs have specific characteristics, e.g., high sensitivity and fast reactions, and they are critical components that control the initiation and progression of photopolymerization reactions.11,12 For this reason, intensive research had been performed on photoacid generators to determine the photochemical mechanisms of cationic polymerization, radical polymerization, ring-opening polymerization, and even one- and two-photon sensitive polymerization.24–28 For instance, coumarin scaffold iodonium salts were utilized as efficient PAGs to construct interpenetrating polymer network (IPN) polymeric materials following cationic/free radical mechanisms;29 Also, new coumarin based iodonium salts reported recently were performed in cationic polymerization, even induce the low reactive monomers like oxetanes and glycidyl ethers.30 Moreover, the progress was also made in environmental and industrial aspects: PAGs were utilized to produce degradable polyester by inducing the ROP of cyclic esters under 365 nm LED light irradiation;31 On the other hand, PAGs also participate in the preparation of high-performance photoresists during the fabrication of MEMS devices.32–34 However, photoacid generators also have specific defects, such as sensitivity to light and temperature, which can impact the stability and repeatability of the photopolymerization process.16,35 As ideal substitutes for PAGs, PBGs activate photopolymerization by generating strong bases, and PBGs exhibit better stability in air, lower costs, poor reactivity with metals and a lower susceptibilities to the degradation or corrosion processes resulting from the strong acidities of PAGs after processing.36–38 With the rapid development of artificial light sources, PBGs that are sensitive to long-wavelength light have been used in photopolymerizations because of their specific advantages at visible light wavelengths, including safety and the high energy processes seen with light emitting diodes (LED).37,39,40 However, fewer PBGs have been reported in past decades compared with PAGs, thus hindering development for practical application.
For instance, the PBGs described in prior publications only generated primary or secondary amines, such as O-acryloximes, carbamates, and formamide derivatives or served to control the acidity of the polymerization system.13,41–44 Among these, only a few PBGs were found with high photobase-generating efficiencies and thermostabilities. In recent years, more efficient PBGs have been developed to release amidines that serve as reactive nucleophiles to trigger anionic ring-opening polymerizations and thiol-Michael, thiol-isocyanate, epoxy polymerization, and thiol–thiol reactions under irradiation, while the range of bases released by the PBGs now includes tertiary amines, sulfonamides, guanidines, phosphazenes, and even carbanions.45–49 In this review, newly developed PBGs described in the literature published in 2019–2023 are discussed systematically and classified in terms of their efficacies in photopolymerization processes. Overall, the use of PBGs in photopolymerization has been a powerful stimulus in the development of lithography with high-quality and high-resolution patterns. This emerging technology even enables the production of high-performance photoresists with improved reliability in microelectronic devices relative to those from PAGs.50,51
2 State of the art
As mentioned above, intense effort has been devoted to the development of suitable monomers and photoinitiators for photopolymerization. However, most of the substantial achievements involved photoinitiation systems with photosensitive organic compounds used for radical photopolymerizations.52–57 Indeed, these photopolymerizations are sensitive to atmospheric oxygen, result in volume shrinkage during irradiation, and are applicable with only a limited range of monomers. Fortunately, PBGs are less prone to these limitations that affect the sensitivity and speed of photopolymerization reactions, and they often exhibit monomer conversion rates and are efficient photoinitiators or precursors.58,59 For instance, Asemani et al.60 used a PBG initiatior and organosilane precursors and fabricated organic–inorganic hybrid (OIH) thin films for metal-corrosion protection via efficient photoinitiated sol–gel reactions activated with UV irradiation. Here, both the superbase and acid generated from the PBG and PAG induced dark polymerization via sol–gel condensation reactions with very high initiation capacities even after the UV radiation was stopped. A range of photoinitiators and precursors, UV light intensities, curing conditions, and even film thicknesses enhanced the anti-corrosion performance. Indeed, this work used PBGs (or PAGs) and provided new sustainable photoinitiated sol–gel processes providing high-performance prototype primer-less coating systems.60 In fact, PBGs are compatible with a wide range of monomers to prepare new photopolymers following photochemical mechanisms with the liberation of bases: ring-opening polymerization of cyclic esters; thiol-Michael addition; cationic polymerization of epoxy groups and thiol–epoxy polymerization, etc. Based on their specific characteristics, comprising inexpensive and environmentally friendly properties, greater photostabilities under irradiation, PBGs should undoubtedly broaden the versatility of photopolymerization, and corresponding applications.50,612.1 Mechanism of photopolymerization initiated by photobase generators
In the past decade, PBGs were mainly molecules that released primary and secondary amines. The development of different PBGs that efficiently release strong bases was important for photopolymerization and cross-linking reactions; the base generation capacities of photocaging salts were found to depend on the position of the chromophore, which is still under development. PBGs have been widely used in photodecarboxylations, thiol–epoxy, thiol-isocyanate or thiol-Michael reactions, in which they release photocaged bases to initiate alternating propagation and chain transfer steps with radicals.62–66 Here, the photocaged bases included strong organic bases showing promise for PBG development, such as 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) or 1,1,3,3-tetramethylguanidine (TMG).67–70 For instance, DBU abstracted protons from selected thiols to generate thiolates serving as nucleophiles in desirable reactions with electrophiles such as epoxide rings. In 2020, Chen et al.71 fabricated thick composites from a thiol–epoxy formulation containing the newly developed PBG TBD·HBPh4 (the chemical structure and photochemical mechanism are illustrated in Table 1), which generated facile reactions when irradiated at 385 nm. It was noted that the resulting composites exhibited excellent uniformity, which was confirmed with functional group conversions, Tg values, and thermal decompositions observed at different radii and depths. It was well documented that the photopolymerization process was affected by the concentration of PBG, the irradiation time, and the curing temperature. As expected, the mechanical properties of the cured materials were enhanced by the addition of 1 wt% glass fibres into the initial formulation, and 2 wt% carbon nanotubes were also used in the same process for the fabrication of dark composites. In 2022, Yu et al.72 used a dual-curing process for digital light processing (DLP 3D printing), which included both orthogonal thiol–ene and thiol–epoxy reactions initiated with TBD-HBPh4 as the PBG. The photocured formulation was composed of the glycidyl methacrylate (abbrev. GMA) cross-linker, diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (abbrev. TPO) and TBD-HBPh4/isopropyl-9H-thioxanthen (ITX). Photocuring of the isotropic tensile photosensitive resin was followed by a step polymerization process, as the bases released from TBD-HBPh4 initiated a thiol–epoxy reaction to increase interfacial adhesion between the layers. Remarkably, the 3D product exhibited low anisotropy with each printed orientation (<5%) but extremely high tensile strength and toughness, which were approximately 5 times greater than those seen after heating for 5 hours at 120 °C.| Photobase generators | Photobase generation | Irradiation conditions | Conversion (or Mna) | λ max/εmax | Ref. |
|---|---|---|---|---|---|
| a Determined by GPC. b Ethyl 2-cyanoacrylate. c Butyl 2- cyanoacrylate. d 2-Octyl 2-cyanoacrylate. e (Dimethylamino) benzoate. f 2-Isopropylthioxanthone. g Camphorquinone. | |||||
| TBD-HBPh4 |

|
385 nm; irradiation: ∼20 min; | 80%–90%(epoxy) | <300 nm/— | 71 |
| 90 °C postbaking temperature; | 80%–90%(thiol) | Solvent: acetonitrile | |||
| Thick cylindric thiol–epoxy samples (r = 1.5 cm, h = 4 cm) | |||||
| BDAE-DBU |

|
312 nm; >470 nm | — | — | 74 |
| Ac-C-TBD(or TMG) |

|
455 nm; irradiation time: ∼30 min | <6%(epoxy) | Ac-C-TBD: | 75 |
| <6%(thiol) | 442 nm/14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 458 M−1 cm−1; 458 M−1 cm−1; |
||||
| >50%(+TEMPO, epoxy) | Ac-C-TMG: | ||||
| >50%(+TEMPO, thiol); | 451 nm/11612 M−1 cm−1 |
||||
| Dark curing: <6% | Solvent: acetonitrile | ||||
| Tx-DBU |
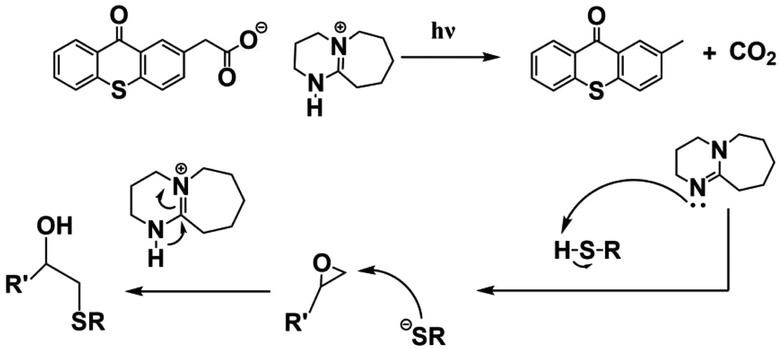
|
Copolymer: PS and PMMA-co-PGMA; | Final polymer: PMMA-co-PGMA-g-PS, Mn = 9600 g mol−1 ([Copolymer]:[PS-SH]:[PBG] = 1:4:1) |
— | 76 |
| DMF as solvent, irradiation time: 6 h at 365 nm; | |||||
| BTIPA-DEA |

|
405 nm; irradiation time: | 415 nm/3845 M−1 cm−1 | 77 | |
| 18 s for ECA; | M n = 4.98 × 104 gmol−1 (ECAb) | Solvent: acetonitrile | |||
| 42 s for BCA; | M n = 5.08 × 104 gmol−1 (BCAc) | ||||
| 76 s for OCA. | M n = 2.25 × 104 gmol−1 (OCAd) | ||||
| NPPOC-DEA |
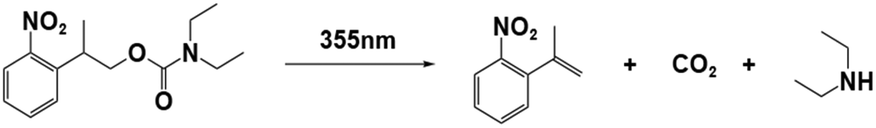
|
355 nm; irradiation time: | 254 nm/4990 M−1 cm−1 | 77 | |
| 28 s for ECA; | M n = 5.59 × 104 gmol−1 (ECA) | Solvent: acetonitrile | |||
| 84 s for BCA; | M n = 8.57 × 104 gmol−1 (BCA) | ||||
| 94 s for OCA. | M n = 2.04 × 104 gmol−1 (OCA) | ||||
| BODIPY-DEA |

|
532 nm; irradiation time: | 550 nm/65180 M−1 cm−1 |
77 | |
| 108 s for ECA; | M n = 7.24 × 104 gmol−1 (ECA); | Solvent: acetonitrile | |||
| 126 s for BCA; | M n = 4.19 × 104 gmol−1 (BCA); | ||||
| 168 s for OCA. | M n = 2.49 × 104 gmol−1 (OCA) | ||||
| BODIPY-I-TMG |
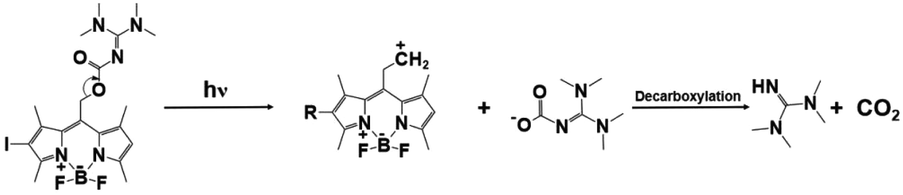
|
505 nm, irradiation time: ∼20 min; | Thiol conversion under 505 nm: ∼98%(butyl thioglycolate + ethyl acrylate); | 535 nm/∼31300 M−1 cm−1 |
78 |
| Dark curing: possible | ∼95%(butyl 3-mercaptopropionate + divinyl sulfone); | Solvent: dichloromethane | |||
| ∼76%(butyl thioglycolate + divinyl sulfone); | |||||
| ∼96%(butyl 3-mercaptopropionate + ethyl acrylate); | |||||
| Dark curing: | |||||
| 10%(butyl thioglycolate + ethyl acrylate) | |||||
| XnAA-TBD |
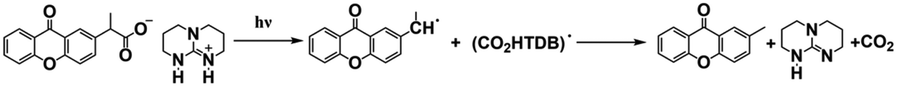
|
— | — | 346 nm/5926 M−1 cm−1 | 79 |
| XnPA-TBD |

|
— | — | 346 nm/5870 M−1 cm−1 | 79 |
| TX-DBN |
|
405 nm | In thiol–ene (PETMP/TTT) system: | 382 nm/25000 M−1 cm−1 |
80 |
| Irradiation: ∼600 s | Double bond conversion: >60%, | Solvent: acetonitrile | |||
| Thiol conversion: >50%; | |||||
| Thiol–epoxy (PETMP/BADGE) system: | |||||
| Both double bond and thiol conversion < 15% | |||||
| NB-TMG |

|
[DCPD]/[PBG]/[HM] = 5000:10:1; |
∼10% at 405 nm with 0.2 wt% EDABe and 0.1% ITXf | — | 81 |
| Monomer: dicyclopendatiene (DPCD); | ∼10% at 405 nm with 1 wt% EDAB and 0.5 wt% benzil at 405 nm; | ||||
| Irradiation: ∼180 s | ∼20% at 475 nm with 0.5 wt% CQg. | ||||
| DMNB-TMG |

|
[DCPD]/[PBG]/[HM] = 5000:10:1; |
<10% at 405 nm with 0.2 wt% EDAB and 0.1% ITX; | — | 81 |
| Monomer: dicyclopendatiene (DPCD); | <10% at 405 nm with 1 wt% EDAB and 0.5 wt% benzil at 405 nm; | ||||
| Irradiation: ∼180 s | <10% at 475 nm with 0.5 wt% CQ. | ||||
| NPPOC-TMG |

|
[DCPD]/[NPPOC-TMG]/[HM] = 5000:10:1 |
∼30% for 475 nm; | — | 81 |
| Monomer: dicyclopendatiene (DPCD) | <10% for 365 nm; | ||||
| Irradiation: ∼180 s | <10% for 475 nm + 365 nm with 0.5 wt% CQ and 1 wt% EDAB; | ||||
| 30%–40% at 405 nm with 0.2 wt% EDAB and 0.1% ITX; | |||||
| 20% at 405 nm with 1 wt% EDAB and 0.5 wt% benzil at 405 nm; | |||||
| 30%–40% at 475 nm with 0.5 wt% CQ. | |||||
| TX-QF-BPh4 |

|
395 nm | Conversion of TMPTA: | 4280 M−1 cm−1@365 nm; | 58 |
| 385 nm | 52.25%@395 nm | 4600 M−1 cm−1 ≈ εmax@385 nm; | |||
| 365 nm | 60.03%@385 nm | 1800 M−1 cm−1@395 nm | |||
| Irradiation: ∼200 s | 58.32%@365 nm | Solvent: acetonitrile | |||
Similar to PAGs, coumarin scaffold PBGs were also designed to induce thiol–ene polymerization under violet/blue light irradiation. In Kiker's work,73 five coumarinylmethyl PBGs with photocaged TMG were synthesized and associated structure–reactivity relationships were fully investigated: Halogenation at the 3-position leads enhancements both in the disparity between maximum C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and S–H conversion and radical contributions to the polymerization mechanism, in contrast, the authors used a simple method by adding tetramethylpiperidinyloxy (TEMPO) to bias photopolymerizations towards a step-growth mechanism. In 2020, Ikuta et al.74 developed a PBG via complexation of a diarylethene derivative (BDAE), which contained a central 5-membered boronate ring combined with DBU serving as a strong base. Basicity control by the BDAE-DBU solution was investigated with both UV-visible and 1H-NMR measurements, which revealed that decomposition of the BDAE generated phenylboronic acid. Then, complexes were formed between the acid and DBU, which reduced the basicity after UV/visible irradiation. Notably, the basicity control in this work was achieved by the BDAE-DBU complex formed via UV or visible irradiation, as shown in Table 1, and it constituted a cyclic process with the addition/release of super amines. However, BDAE complexation did not occur with weak bases such as trimethylamine or triethanolamine. Similarly, Huang et al.82 performed NIR-induced anionic thiol–epoxy photopolymerizations catalysed by lanthanide-based upconversion nanoparticles (UCNPs) during irradiation at 980 nm. Here, PBGs based on thioxanthone derivatives were utilized in the photodecarboxylation process, and they released superbases after absorbing the fluorescence emitted from the UCNPs (see Fig. 1). Markedly, the crosslinked luminescent polymers contained nearly 100% gel content and emitted fluorescence during continuous irradiation at 365 or 980 nm. Moreover, good network uniformity and a curing depth of 11.8 cm were observed after intermittent photo- and thermal activation via postbaking, with excellent photostability and shape memory arising from the thermal or photo stimuli. Hence, this work revealed that thiol–epoxy photopolymerization may enable the fabrication of smart materials for a range of applications, e.g., smart medical devices and robots, with NIR irradiation.
C and S–H conversion and radical contributions to the polymerization mechanism, in contrast, the authors used a simple method by adding tetramethylpiperidinyloxy (TEMPO) to bias photopolymerizations towards a step-growth mechanism. In 2020, Ikuta et al.74 developed a PBG via complexation of a diarylethene derivative (BDAE), which contained a central 5-membered boronate ring combined with DBU serving as a strong base. Basicity control by the BDAE-DBU solution was investigated with both UV-visible and 1H-NMR measurements, which revealed that decomposition of the BDAE generated phenylboronic acid. Then, complexes were formed between the acid and DBU, which reduced the basicity after UV/visible irradiation. Notably, the basicity control in this work was achieved by the BDAE-DBU complex formed via UV or visible irradiation, as shown in Table 1, and it constituted a cyclic process with the addition/release of super amines. However, BDAE complexation did not occur with weak bases such as trimethylamine or triethanolamine. Similarly, Huang et al.82 performed NIR-induced anionic thiol–epoxy photopolymerizations catalysed by lanthanide-based upconversion nanoparticles (UCNPs) during irradiation at 980 nm. Here, PBGs based on thioxanthone derivatives were utilized in the photodecarboxylation process, and they released superbases after absorbing the fluorescence emitted from the UCNPs (see Fig. 1). Markedly, the crosslinked luminescent polymers contained nearly 100% gel content and emitted fluorescence during continuous irradiation at 365 or 980 nm. Moreover, good network uniformity and a curing depth of 11.8 cm were observed after intermittent photo- and thermal activation via postbaking, with excellent photostability and shape memory arising from the thermal or photo stimuli. Hence, this work revealed that thiol–epoxy photopolymerization may enable the fabrication of smart materials for a range of applications, e.g., smart medical devices and robots, with NIR irradiation.
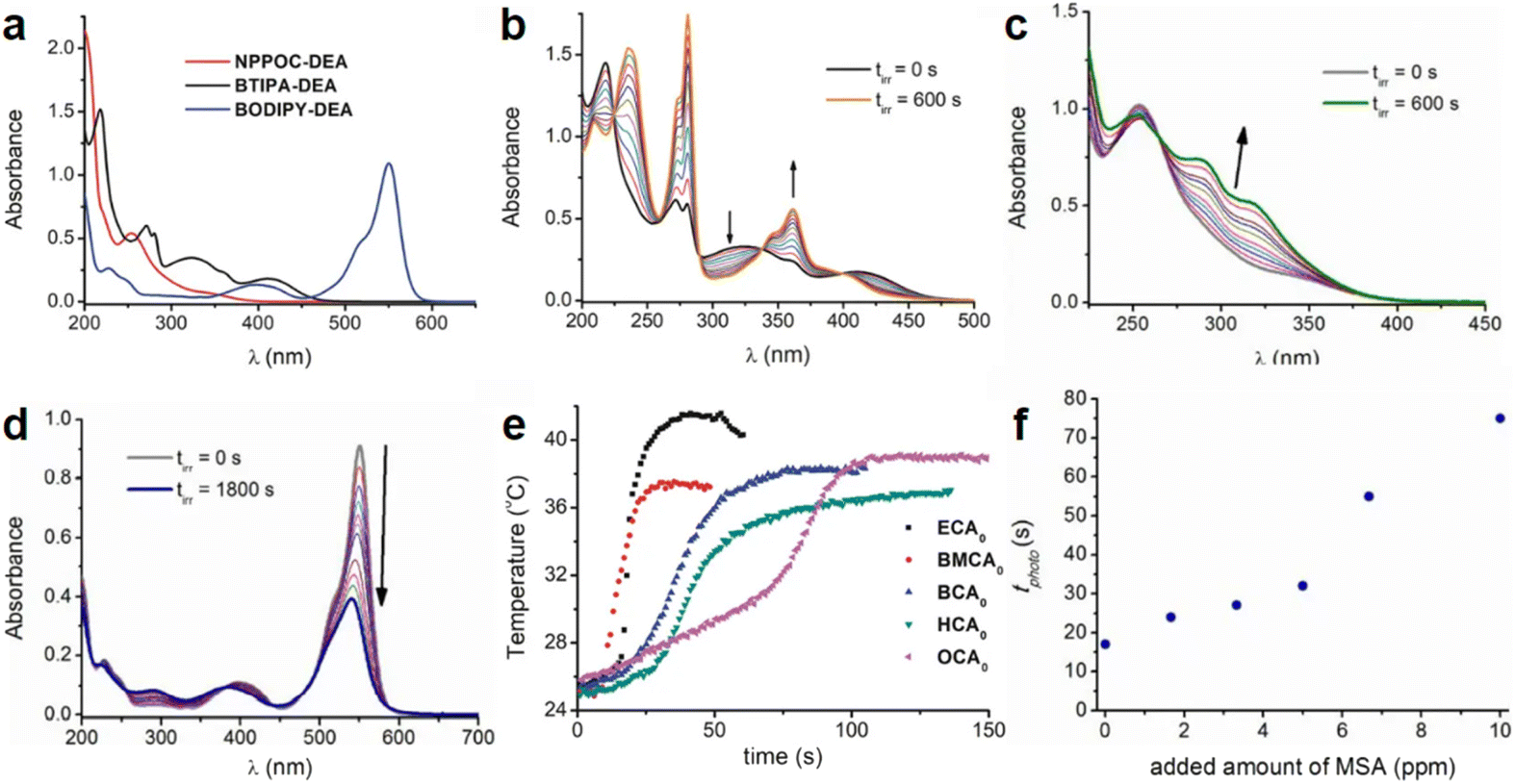 | ||
| Fig. 1 (a) Absorption spectra of NPPOC-DEA (c = 1.60 × 10−4 M), BTIPA-DEA (c = 4.76 × 10−5 M), and BODIPY-DEA (c = 1.68 × 10−5 M) in acetonitrile. (b) Variation in the absorption spectra of BTIPA-DEA (c = 4.76 × 10−5 M) in acetonitrile upon continuous irradiation at λexc = 405 nm (power = 2.8 mW). Spectra were recorded every 60 s for a total irradiation time (tirr) of 600 s. (c) Variation in the absorption spectra of NPPOC-DEA (c = 3.17 × 10−4 M) in acetonitrile upon continuous irradiation at λexc = 355 nm (power = 3.5 mW). Spectra were recorded at different times (0–600 s). The arrow shows the spectral change over time. (d) Variation in the absorption spectra of BODIPY-DEA (c = 1.48 × 10−5 M) in acetonitrile upon continuous irradiation at λexc = 532 nm (power = 28.2 mW). Spectra were recorded at different times (0–1800 s). The arrow shows the spectral change over time. (e) Photopolymerization exotherms of mixtures containing BTIPA-DEA and commercial CA monomers (λexc = 405 nm, power = 280 mW). (f) Variation in the polymerization time (tphoto) with the added quantity of methanesulfonic acid. Reproduced from ref. 77 with permission from the American Chemical Society, copyright 2019. | ||
Later, two unimolecular chromophores, Ac-C-TBD and Ac-C-TMG with azocarbazole moiety were designed by Xu et al. in 2021,75 which successfully used for thiol–epoxy photopolymerization as PBGs without post-baking process. Here, the photopolymerization kinetics were also established by real-time infrared spectroscopy, where both Ac-C-TBD and Ac-C-TMG exhibited better initiation capacity on thiol-polymerization rather than epoxy. For instance, the final conversions for thiol and epoxy achieved 80% and 70% for Ac-C-TBD, respectively. A mechanism was proposed following photodecarboxylation of a thiol by an alkoxide anion and the formation of the disulfide bond, to release strong organic base via the photolysis and ESR experiments (see Table 1). On the other hand, Ac-C-TBD was also proved to be a better photoinitiator than Ac-C-TMG which was summarized from the corresponding thiol and epoxy conversions, owing to stronger alkalinity of the former one.
In 2023, Xie et al.83 developed a series of PBGs based on triphenylamine (TPA) and tetraphenylethelene (TPE) with photocaged amine, including DBU, DBN and TBD. Here, the PBGs could induce a fast thiol–epoxy photopolymerization and suffer a photodecarboxylation process, resulting a high conversion of monomer under near-UV/visible light irradiation. Remarkably, these newly synthesized PBGs not only exhibit excellent light absorption, but also trigger Aggregation-induced emission (AIE) phenomenon with increasing viscosity during the photopolymerization process. Moreover, a linear correlation equation between AIE intensity with monomer conversion was established, giving a new route to trace the monomer conversion quantitatively. Recently, Kiliclar et al.76 modified polymers via side-chain functionalization, grafting and chain extension by applying photoinduced thiol–epoxy click reactions with irradiation at 365 nm; these were initiated with the synthesized PBG DBU-acetate (Tx-DBU). In this work, the authors provided a simple pathway by generating nucleophilic thiolates for addition to epoxide rings, and the Tx-DBU provided both decarboxylation and abstraction of protons from the aliphatic and aromatic thiols (see Table 1). Indeed, this research demonstrated a new role for PBGs in the design of polymeric structures with a variety of topologies. Although all of the studies of thiol–epoxy click photopolymerization mentioned above broadened the application scope of photochemistry, further molecular design for efficient release of the weak bases needed to initiate photopolymerization is necessary for the development of photobase generators.
PBGs for the thiol-Michael addition reaction have exhibited efficient photolysis and base release upon UV irradiation. Indeed, the bases released from the PBGs should be strong initiators of the subsequent photopolymerization process upon light exposure. In 2019, Faggi et al.77 synthesized three carbamate derivatives serving as PBGs for photoinduced polymerization of cyanoacrylate monomers by diethylamine (DEA). They examined the photolyses of three photoinitiators in acetonitrile and used UV-vis absorption spectroscopy to verify the mechanisms of the photobase generation processes. Under irradiation, photolysis of the carbamate groups and the generation of DEA were observed for NPPOC-DEA and BTIPA-DEA (Fig. 1b and c), which was confirmed by nuclear magnetic resonance. BTIPA-DEA underwent photolysis to generate a compound with a nucleophilic pyridine group, which initiated polymerization of the monomer with the generated DEA.
In the case of BODIPY-DEA, no free DEA was observed, indicating the formation of CO2 and a benzylic BODIPY cation, which reacted with DEA to form a new tertiary amine, as shown in Table 1. The photoreaction quantum yields (Φph) were also determined for the PBGs. Large Φph values were measured for NPPOC-DEA and BTIPA-DEA, but the Φph for BODIPY-DEA was very low. With its larger molar extinction coefficient (εmax = 65180 M−1 cm−1) and redshifted absorption, BODIPY-DEA was a reliable PI with higher amine photorelease efficiency than NPPOC-DEA and BTIPA-DEA. Moreover, the highly reactive monomers ethyl 2-cyanoacrylate (ECA), methoxyethyl 2-cyanoacrylate (BMCA), butyl 2-cyanoacrylate (BCA), hexyl 2-cyanoacrylate (HCA), and 2-octyl-2-cyanoacrylate (OCA) were used to evaluate the photoinitiation capacities of the photoinitiators. Interestingly, a series of commercial cyanoacrylate monomers were prepared with the same acid stabilizer (methanesulfonic acid, MSA) at different concentrations, and the viscosities of the systems (ECAst, BMCAst, BCAst, HCAst, and OCAst) were kept constant to investigate the intrinsic response of each monomer to the synthesized photobase generators. All cyanoacrylate photopolymerizations were investigated with calorimetry to record the temperature changes of the monomer–photoinitiator pairs during irradiation (see Fig. 1e). The time required to reach half of the overall thermal change for each sample during photopolymerization (tphoto) was used to quantify the efficiency of CA photopolymerization, and no polymerization was observed in the dark. During irradiation, BTIPA-DEA and NPPOC-DEA exhibited shorter photoinitiation times for cyanoacrylate polymerization, while BODIPY-DEA underwent a larger tphoto because of its lower photoreaction quantum yield and considerable steric effects. Notably, tphoto was increased by the acid stabilizer methanesulfonic acid (see Fig. 1f), and then amine photodeprotection and the intrinsic monomer reactivities for some cyanoacrylates, e.g., BCA, HCA, OCA, led to shorter tphoto. Even though the long alkyl chain CAs had longer polymerization periods under the same irradiation conditions because their low reactivities inhibited the nucleophile activation and propagation steps, shorter photopolymerization times (<190 s) were observed for all monomer–photoinitiator pairs involved in this work compared to the thermal polymerizations of the cyanoacrylates, which had times of several hours (ECA and BMECA) and even several days (BCA, HCA, and OCA). In summary, the PBGs developed in this work dramatically shortened the polymerization processes of cyanoacrylate monomers, especially for the less reactive monomers and even for long alkyl chain cyanoacrylates, providing new perspectives for high-performance applications in the future.
Despite the truth that BODIPY derivatives possess excellent light-emitting properties and flexible structures, novel BODIPY-based PBGs have been also initiated long wavelength-light-induced thiol–ene Michael addition photopolymerization. For instance, two TMG-caged PBGs, BD-TMG and BD-I-TMG, acting as very effective photoinitiators with spatial and temporal control capacity on the release of TMG upon the irradiation of green visible light at 505 nm. Moreover, the BODIPY derivative with heavy atom effect, BD-I-TMG, exhibited higher photoreleasing efficiency (ΦTMG = 0.029) and photolytic efficiency (εΦTMG = 907.7) rather than that of BD-TMG in the absence of iodine atom (ΦTMG = 0.009, εΦTMG = 512.1). Markedly, a cross-linked polymer with relatively homogeneous network was also attained from BD-I-TMG characterized by mechanical properties. As perspective, to achieve more red/infrared light induced polymerization could promote wider applications in for biomedical aspect and wavelength control.78
As mentioned above, the thiol-Michael addition has been widely utilized for mild polymerization of polymeric materials with high monomer conversions and reaction rates. Interestingly, a base amplifier was also utilized in the thiol-Michael addition reaction, which easily underwent autocatalytic decomposition to release additional base. In Sinha's work, the authors systematically investigated the photoinitiation capacities of 2-(2-nitrophenyl)-propyloxycarbonyl (NPPOC)-protected amines (NPPOC-HEX or NPPOC-DEA) photobase generators in the presence of base amplifiers, 9-fluorenylmethyl carbamate (Fmoc) derivatives, to enhance the photoinitiation efficiency of the base-induced thiol-Michael reaction upon irradiation at 365 nm. Here, 1-hexyl acrylate (HA) or ethyl vinyl sulfone (EVS) were used as the Michael acceptors and mixed with butyl 3-mercaptopropionate (BMP) as the thiol source. In the control reaction, only a low thiol conversion (∼35%) was achieved after 60 min for the BMP/HA combination with a 1:1 thiol/vinyl stoichiometric ratio and without any catalyst, while a lower conversion (∼26%) was obtained when NPPOC-Hex (2.5 mol%) was used along with BMP and HA. As expected, higher thiol conversions (∼49%) were achieved after the addition of Fmoc-Hex (5 mol%) as a base amplifier. Furthermore, the same tendency was observed in replacing NPPOC-Hex and Fmoc-Hex with NPPOC-DEA and Fmoc-DEA, respectively. Surprisingly, an extremely high conversion rate of the functional groups (∼97%) for the BMP/EVS combination was found by using NPPOC-DEA and Fmoc-DEA, confirming the promotion of Fmoc derivatives as base amplifiers for reaction kinetics. Interestingly, this system continued to function in the dark after 30 s of irradiation, furnishing 76% conversion after 60 min. Remarkably, the conversion profile for the TMPTA/PETMP monomer combination initiated with a specific ratio of NPPOC-DEA and Fmoc-TMG (20 mol% and 1 mol%, respectively) was established by using confocal Raman spectroscopy to investigate polymerization in the dark. As a result, the conversions of the TMPTA/PETMP system decreased with increases in the diffusion distance of the base from the scanned position to the edge of the illuminated region (marked as ‘x’). On the other hand, the conversion was increased at higher temperatures because of the greater mobility of the base; remarkably, the conversion rate at a distance x of 16.5 mm was also high, >90% at 70 °C. Finally, the mechanical properties of the resulting polymer were evaluated with dynamic mechanical analyses, which indicated higher glass transition temperatures (Tg) than the system without Fmoc derivatives. However, a lower Tg was detected with a lower concentration of catalyst for DVS/PETMP because of the lower conversion rate achieved in the short irradiation time. In conclusion, Fmoc/PBG systems are emerging tools with which to enhance the photosensitivity of photopolymerization, thus broadening the scale of photopolymerization applications, especially in relatively dark environments with lower energy consumption.84
In the same year, Zivic et al.85 reported three newly synthesized thioxanthone chromophores (TX) and used them as PBGs in photopolymerization of polyurethanes (PUs); the release and catalytic activity of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) as a latent base was enhanced by introducing an isopropyl group at the α position of the carboxylate, including a hydrogen for TX-O-DBU, an isopropyl group in iPr-TX-O-DBU, and a second carboxylic functional group in TX-O-(DBU)2. During irradiation at 365 nm, the decarboxylation and base-releasing capacities of the newly developed PBGs were examined by monitoring the loss of the proton α to the carboxylate group via1H NMR spectroscopy. For PBG with a fast decarboxylation rate, the peak for iPr-TX-O-DBU rapidly disappeared within 2 min, and the spectra taken before 30 s and 5 min of irradiation are illustrated in Fig. 2a. Compared to iPr-TX-O-DBU, the decarboxylation rate of TX-O-DBU was only 48% after 2 min, proving that the decarboxylation process was highly dependent on the isopropyl substituent. As illustrated in Fig. 2b, TX-O-(DBU)2 exhibited faster decarboxylation than TX-O-DBU because two equivalents of DBU could theoretically be released from the former, although decarboxylation of the second carboxylate group is much slower than that of the first. Then, the decarboxylation mechanisms were systematically investigated for the isopropyl thioxanthone chromophores via DFT calculations. As a common process, the decarboxylation mechanism involves the formation of a triplet excited state (T1) from the singlet ground state (S0) as the first photochemical step. Two potential mechanisms were proposed by the authors for the subsequent steps: (1) An intramolecular mechanism caused the thioxanthone molecule to generate a second triplet species (T2) from T1 after an electronic transition and a hydrogen transfer. Then, the decarboxylation process occurred and formed a triplet diradical (T3), as shown in Fig. 2c. This mechanism would release free DBU with minimal energy needed, which was proven by calculations of the enthalpy differences for each step in the reaction (see Fig. 2e). (2) An intermolecular charge transfer occurred between the thioxanthone molecules and another molecule. Here, a H+ transfer could occur between two radical ions, one could release DBU with a doublet at the sulfur atom, marked as DA+, and the other one holds an unpaired electron at the carbonylic carbon, marked as DB−. In the next step, the species produced from DA+ after liberation of the DBU (marked as DA) could produce a new radical Dcvia decarboxylation (see Fig. 2d). However, this route is less favourable than the intramolecular mechanism because the former requires 80 kcal mol−1 for charge transfer from the T1 state of one molecule to a second molecule, which is much more endothermic than the intramolecular mechanism, which only requires approximately 15 kcal mol−1 for the hydrogen transfer. In fact, decarboxylation and base generation only slightly endothermic or even exothermic. Furthermore, photopolymerization of polyurethane in the presence of iPr-TX-ODBU and a 365 nm LED was investigated with a rheological study (see Fig. 2f). Based on FT-IR results, the authors concluded that both the conversion and its moduli crossover time were strongly dependent on the light intensity. Then, the photopolymerized system with iPr-TX-ODBU and trifunctional monomer was used to produce coatings and enable 3D printing because the trifunctional monomer exhibited higher conversion than the difunctional isocyanates and alcohols upon irradiation with the same light intensity. After photopolymerization, a tack-free coating was obtained on a glass plate with 100% conversion, as shown in the FTIR analysis (Fig. 2g). Finally, the ultimate 3D product was obtained by irradiation with a 365 nm LED after depositing the first layer (see Fig. 2h).
 | ||
| Fig. 2 (a) 1H NMR spectra of a solution containing iPr-TX-O-DBU (c = 10−2 mol L−1) irradiated at 365 nm for different times with an LED source (spectra were recorded before, after 30 s, and 5 min of irradiation). (b) Percentage of decarboxylation as a function of irradiation time. (c) Proposed intramolecular photodecomposition mechanism. (d) Proposed intermolecular photodecomposition mechanism. (e) Energetics of the intramolecular photodecomposition mechanism for TXO-DBU (red line) and iPr-TX-O-DBU (purple line). (f) Evolution of the storage modulus G′ and the loss modulus G′′ as a function of irradiation time using a 250 mW cm−2 irradiation intensity in the presence of 3.75 mol% iPr-TX-O-DBU. (g) IR spectra of the HDI-Biuret/PCL-900/glycerol mixture before and after 40 s of irradiation with the UV curing conveyor system in the presence of 3.75 mol% iPr-TX-O-DBU. (h) Photograph of a printed specimen. Reproduced from ref. 85 with permission from the American Chemical Society, copyright 2020. | ||
In 2022, Zivic et al.86 used biodegradable monomers and multifunctional trimethylene carbonate (TMC) into photoinduced ring-opening polymerization for the first time. Herein, TBD salts were utilized as superbase photogenerators based on thioxanthone acetic acid to fabricate biodegradable crosslinked poly(ester/carbonate) patterns via stereolithography. Specifically, the liquid formulation, DOD-BisTMC, was irradiated at 385 nm with the poly(ε-caprolactone) triol serving as a photocatalyst and in the presence of the aforementioned PBGs. It was obvious that the PBGs and temperature significantly affected the polymerization kinetics, and various parameters and gel times were fully investigated via photorheometry. After selection of the best experimental conditions, the dark polymerization process was successfully performed by stereolithography, and the thickness of the coating film was dependent on the reaction time in the absence of irradiation; this indicated that dark polymerization had occurred and was unrelated to the irradiation time. Indeed, this work provided a feasible route to biodegradable coatings and objects, even for solvent-free polymerized systems via stereolithography. In fact, further design following this methodology on PBG could be applied for more precise 3DP of various polyesters and polycarbonates, especially the products which are fully degradable.
As discussed above, even though various PBGs were used in photodecarboxylation reactions with near-UV LEDs, those PBGs have rarely been viewed as promising candidates. In 2023, Ley et al.79 synthesized two carboxylic acid-substituted xanthones (Xn) with long T1 lifetimes and presented new mechanisms for photobase generation. Specifically, the investigated PBGs comprised the superbase TBD with acetic acid-substituted Xn (named XnAAH) and propionic acid-substituted Xn (named XnPAH) (see Table 1). The authors provided full photophysical and photochemical characterizations of PBGs by steady-state and time-resolved spectroscopy. The molecules exhibited similar molar absorption coefficients and no evidence of singlet excited states. However, both XnAA-TBD and XnPA-TBD showed lower triplet quantum yields than other derivatives comprising the carboxylic acid precursors XnAAH and XnPAH. Here, it was considered that efficient photodecarboxylation occurred from the second excited triplet state (T2) rather than T1 for the two photobase generators; specifically, the rate constants for intersystem crossing and internal conversion were measured in acetonitrile via femtosecond pump–probe spectroscopy.
Specifically, the authors suggested a mechanism in which the photodecarboxylation process occurred before population of T1, and ethyl and propyl radicals, as short-lived but important intermediates, were generated from homolytic cleavage and underwent base release and the formation of CO2 through rearrangement reactions of the photobase (Table 1). Significantly, the quantum yields of the photobases (φB) were obtained both in argon-saturated solutions and in air-saturated solutions, where lower φB values were detected in the presence of oxygen as a triplet state scavenger than in the absence of oxygen. Hence, it is rational to conclude that the photodecarboxylation process occurred from a triplet state. As a hypothesis, photodecarboxylation with two photobase generators proceeded via ground state → second singlet excited state (S2) → second triplet excited state (T2) upon irradiation. Here, the ultrafast conversions from S2 to T2 were achieved within approximately 2 ps by intersystem crossing (the rate constants are marked as kISC). Afterwards, the φB values obtained were all higher than the quantum yields of the triplet states (φT), and the sum of φB and φT approached 1, indicating complementary approaches from T2, as suggested: (1) the bases were not released from the T1 state, where decarboxylation and release of TBD occurred after hydrogen transfer from intermediate radicals that were generated from the dissociation process (the rate constants are marked as kdiss); and (2) formation of the T1 state with a slow internal conversion rate is marked as kT IC. As a result, the photodecarboxylation of XnAA-TBD investigated in this work was described as shown in Fig. 3, with the addition of rate constants on the femtosecond time scale: kISC ∼4.76 × 1011 s−1, kTIC ∼1.53 × 109 s−1, and kdiss ∼2.20 × 109 s−1. Indeed, the strategy used in this work provided a powerful tool with which to understand and design photorelease mechanisms for other photobase systems according to their molecular structures and the quantum yields for the photobases and excited triplet state. Indeed, this work reveals that a more accurate photophysical analysis is required for precise identification of the photochemical pathways occurring in polymerization systems, giving a deeper understanding of the base generation process to design more efficient systems with the assistance of computational chemistry.
 | ||
| Fig. 3 (a) Perrin-Jablonski diagram of Xn photophysics. The thick arrows show the effect of increasing the solvent polarity on the relative positions of the four excited states. (b) First electronic transition of Xn and the Xn derivatives in acetonitrile: PBGs (XnAA-TBD and XnPA-TBD), the corresponding carboxylic acids (XNAAH and XnPAH), and the corresponding photoproducts of decarboxylation (XnMe and XnEt). (c) Femtosecond pump–probe time-resolved spectroscopy of PBG XnAA-TBD in acetonitrile. (a) Transient spectra at different delays, (b) DADS from the global analysis of the spectrotemporal data, and (c) SADS corresponding to the S2!T2! T1 pathway. (d) Mechanism of TBD release from (a) XnAA-TBD and (b) XnPA-TBD during photodecarboxylation. Copyright 2020, the image was reproduced from ref. 79 with permission from Wiley-VCH. Reproduced from ref. 79 with permission from Wiley-VCH, copyright 2020. | ||
As discussed above, these efficient PBGs mainly underwent photodecarboxylation to release bases and active species; however, several defects cannot be neglected, such as the CO2 released during the process, which may yield bubbles/cracks in the photochemical products, or the poor solubility of the PBGs in polymerization systems. In 2020, Zheng, Y.80 synthesized a PBG complex composed of a nonionic amidine chromophore with thioxanthone and DBN as the superbase (abbrev. TX-DBN). TX-DBN exhibited strong absorption at 380 nm, with excellent solubility in commercial monomers, promoting efficient activation of the dual thiol–epoxy and thiol–ene polymerization to form a hybrid network. In this work, base release was also achieved although bond cleavage and fast hydrogen transfer under 405 nm light; however, there was no CO2 formed during the photochemical reaction (see Table 1).
Interestingly, Foster et al.81 utilized photolysis of PBGs to deactivate polymerization because the latent olefin metathesis catalyst (CQ, ITX or benzil for photosensitize Ru-based metathesis) could potentially be decomposed by deprotonation of the amine-promoted metallacyclobutane. First, they synthesized three PBGs comprising 1,1,3,3-tetramethyl guanidine (TMG) as the base, 2-nitrobenzyl TMG carbamate (NB-TMG), 4,5-dimethoxy-2-nitrobenzyl TMG carbamate (NVOC-TMG), and 2-(2-nitrophenyl)propyl TMG carbamate (NPPOC-TMG) and then combined them with various photosensitizers (HeatMet, HM) for photoinitiation of monomeric dicyclopentadiene under 405/475 nm irradiation. Remarkably, dual-wavelength photoactivation was identified in this work, and the 365 nm irradiation was also used to promote catalyst decomposition and polymerization deactivation at separate irradiation starting times. As a result, all PBGs had poor effects on the polymerization rates under 405 or 475 nm irradiation. In contrast, all polymerizations were deactivated under 365 nm light owing to partial sensitization of the PBG by the photosensitizers (Fig. 4a and b). Furthermore, the CQ + EDAB + NPPOC-TMG resin system was constructed for optimization in the next step because (1) camphorquinone (CQ) exhibited a higher absorption maximum than the other photosensitisers; (2) NPPOC-TMG had the worst effect on monomer conversion among the three investigated PBGs; and (3) the shortest incubation times before gelation were obtained for the resin in the presence of NPPOC-TMG. The optimal ratio of the polymerizable system was [CQ]/[EDAB]/[NPPOC-TMG]/[HM] = 10:20:15:1, which gave the best performance in initiating polymerization (t = 0 s) under 475 nm light and deactivating polymerization under 365 nm light at various time points to decompose the catalyst for systematic investigation (see Fig. 4c–e). For instance, monomer conversion was almost instantaneously inhibited at the onset of 365 nm irradiation in the presence of 15 equiv. of NPPOC-TMG, indicating that the deactivation effects were much more obvious than those seen with 5 equiv. (inhibited obviously from 20 s after irradiation) and 10 equiv. (inhibited obviously from ∼10 s after irradiation) of NPPOC-TMG under the same conditions (see Fig. 4f–i). In summary, the final monomer conversions were significantly decreased by the long irradiation times with a 365 nm light source or high NPPOC-TMG contents in the system.
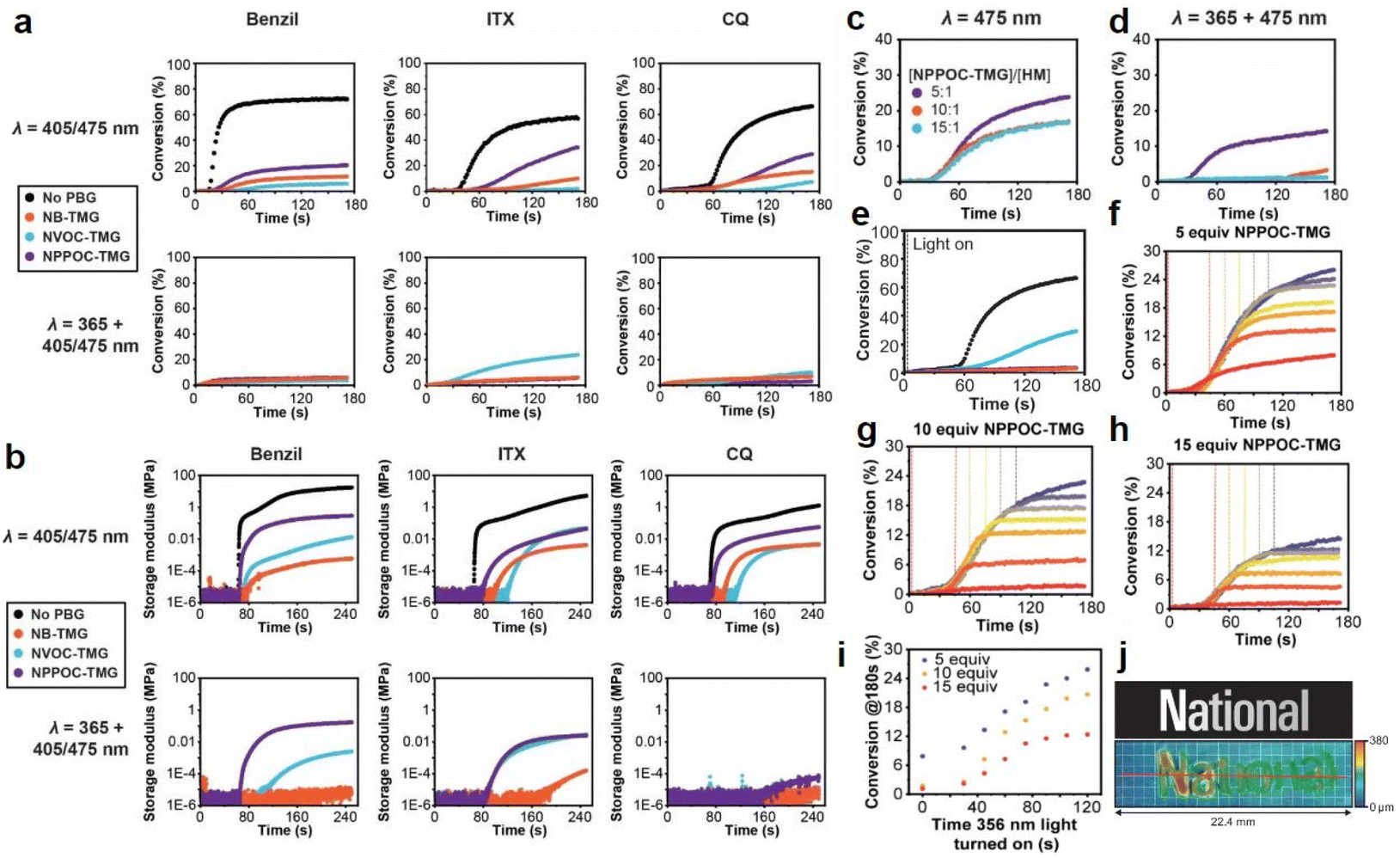 | ||
| Fig. 4 (a) Kinetics of DCPD polymerization measured by FT-IR spectroscopy at 1573 cm−1 for resins formulated with different PSs and PBGs. Samples were irradiated at either 405 nm for benzil and ITX or 475 for CQ (top row) to study the initiation behaviour or at 365 nm + 405/475 nm to study polymerization deactivation (bottom row). The black curves correspond to control polymerizations carried out with the various PSs in the absence of PBG. For each plot, the appropriate PS was mixed with DCPD, HM, and either NB-TMG (blue circles), NVOC-TMG (orange circles), or NPPOC-TMG (purple circles). [DCPD]/[PBG]/[HM] = 5000/10/1 was used for these experiments, with 0.2 wt% EDAB and 0.1% ITX or 1 wt% EDAB and 0.5 wt% benzil or 0.5 wt% CQ as appropriate. (b) Storage modulus vs. time for polymerizations using resins formulated with different PSs and PBGs. Samples were irradiated at either 405 nm for benzil and ITX or 475 for CQ (top row) to study initiation behaviour or 365 nm + 405/475 nm to study polymerization deactivation (bottom row). The black curves correspond to control polymerizations carried out with the various PSs in the absence of PBG. For each plot, the appropriate PS was mixed with DCPD, HM, and either NB-TMG (blue circles), NVOC-TMG (orange circles), or NPPOC-TMG (purple circles). [DCPD]/[PBG]/[HM] = 5000/10/1 was used for these experiments, with 0.2 wt% EDAB and 0.1% ITX or 1 wt% EDAB and 0.5 wt% benzil or 0.5 wt% CQ as appropriate. Kinetics of polymerization as measured by FT-IR spectroscopy at 1573 cm−1 for polymerization of DCPD by HM in the presence CQ, EDAB, and different amounts of NPPOC-TMG and irradiated at either (c) 475 nm or (d) both 365 nm and 475 nm; (e) polymerization kinetics as measured by FT-IR spectroscopy at 1573 cm−1 for optimized photoresin irradiated with 475 nm light in the absence of PBG (black circles) and with PBG at 475 nm (blue circles), 365 nm (purple circles), and both wavelengths (orange circles). Kinetics of DCPD polymerization as measured by FT-IR spectroscopy at 1573 cm−1 for resins formulated using (f) 5, (g) 10, or (h) 15 equiv. of NPPOC-TMG relative to HM. For these experiments, the 475 nm light was turned on at t = 0 to initiate polymerization, and then the 365 nm light was turned on at different time points (t = 0, 30, 45, 60, 75, 90, 105, or 120 s) to decompose the catalyst and thus deactivate polymerization. Monomer conversion significantly slowed or stopped altogether shortly after the 365 nm light was turned on. (i) Plateau (ultimate) conversion as a function of the time that the 365 nm light was turned on during the experiments. (j) Multilayer greyscale image and corresponding topographical image of the tapered height “National” text. The grid represents 1 mm × 1 mm squares. Reproduced from ref. 81 with permission from John Wiley and Sons, copyright 2022. | ||
As indicated by the results described above, selective dual-wavelength olefin metathesis polymerizations (SWOMP) were instantaneously stopped with 15 equiv. of NPPOC-TMG, and the authors adapted this to continuous stereolithographic (SLA) printing of complex objects because the components used in this work, e.g., photosensitizers and PBGs, had no harmful influence on the photoproducts. Remarkably, patterns with varying heights were obtained by SLA printing with this innovative approach (see Fig. 4j). Indeed, the development of PBGs for deactivation enabled the use of wavelength-selective photopolymerization in additive manufacturing. In terms of proper materials selection, continuous SWOMP with the dual-wavelength approach constructed more complex objects with very fast printing rates up to 36 mm h−1 for patterned light and 180 mm h−1 for unpatterned and high-intensity light; this is expected to be a powerful alternative for existing printing methods based on free radical polymerization (FRP) and traditional SLA.
2.2 Photomediated nucleophile-catalysed photopolymerization initiated by photobase generators
Along with the traditional pathway for thiol-Michael reactions on photocaged bases, catalysis of the thiol-Michael addition reaction by strong nucleophiles was also reported. In 2010, Chan et al.87 performed nucleophile-mediated thiol-Michael reactions with methyldiphenylphosphine and dimethylphenylphosphine, and rapid reactions were observed on the subsecond scale. In 2021, Zhang et al.88 fabricated phosphonium tetraphenylborate salts (MDPP·HBPh4) for use as phosphine catalysts in a three-component photoinitiating system with isopropylthioxanthone (ITX) and a radical inhibitor, 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO). Specifically, the authors utilized ITX to release the phosphine from DMPP·HBPh4 and use it as a nucleophile sensitizer to trigger the thiol-Michael addition reaction while simultaneously generating radicals via proton abstraction, and the chemical structures are shown in Fig. 5a. In addition, the radical scavenger TEMPO was added to increase the rate of the addition reaction and form a homogeneous polymer network, which was proven by monitoring the photopolymerization kinetics of thiol and vinyl moieties with real-time FT-IR spectroscopy. As shown in Fig. 5b, the photopolymerization reaction of the PETMP/DVS mixture activated by ITX/MDPP·HBPh4/TEMPO (1/2/2 wt%) were more rapid under irradiation with a 405 nm LED than those of ITX/MDPP·HBPh4 (1/2 wt%) run in the absence of TEMPO. However, when the photopolymerization kinetics of the PETMP/PEGDA mixture were investigated with the same conditions used for the TEMPO reaction, the conversions of the thiol and acrylate monomers exhibited similar tendencies, as shown by the solid lines in Fig. 5c. However, the acrylate monomers seemed more reactive (conversion ∼99%) after 5 min of irradiation (dashed lines) in the thiol-Michael reaction than the thiol monomers (conversion ∼54%) without TEMPO, indicating that the thiol–acrylate and acrylate–acrylate interactions were the dominant reactions in the process. | ||
| Fig. 5 (a) Chemical structures of monomers and initiators used in this study; (b) conversions of thiol and vinyl moieties as a function of reaction time for a stoichiometric PETMP and DVS mixture; (c) conversions of thiol and vinyl moieties as a function of reaction time for a stoichiometric PETMP and PEGDA (Mn = 575) mixture. All photopolymerization products were photocured with either ITX/DMPP·HBPh4/TEMPO (1/2/2 wt%) or ITX/MDPP·HBPh4 (1/2 wt%) and initiated with 40 mW cm−2, 405 nm LED light. (d) Proposed mechanism for the photogeneration of MDPP from MDPP·HBPh4. (e) Conversions of thiol and vinyl moieties as a function of reaction time in TEA ITX/TEA·HBPh4/TEMPO (1/2/2 wt%) catalysed thiol-acrylate reaction in PETMP and PEGDA (Mn = 575). Relatively slow polymerization kinetics and an off-stoichiometric thiol-acrylate reaction were observed, indicating that the photocaged weak nucleophiles were less reactive in the thiol-Michael photopolymerization. (f) Conversions of thiol moieties as a function of reaction time in the ITX/DMPP·HBPh4/TEMPO (1/2/2 wt%) catalysed reaction in PETMP/DVS in 2 min and with continuous irradiation, respectively. The samples were all irradiated with 405 nm, 40 mW cm−2 LED light. Reproduced from ref. 88 with permission from the American Chemical Society, copyright 2021. | ||
As shown in Fig. 5d, the authors proposed a photochemical mechanism in which MDPP·HBPh4 underwent photodeprotection with triplet energy transfer from ITX. Then, a phosphine was released from MDPP·HBPh4 after intramolecular transfer of a proton from MDPP·H+ to the anion BPh4−, which decomposed to aromatic fragments after rearrangement. Then, the nucleophile capacity in the initiation step was also investigated for the thiol-Michael polymerization by examining a photocaged compound (TEA·HBPh4) consisting of tetraphenylborate and triethylamine in the PETMP/PEGDA system. The reaction exhibited lower conversion rates (53% for the thiol group and 97% for the vinyl group after 20 min of irradiation) under initiation of the ITX/TEA·HBPh4/TEMPO combination (1/2/2 wt%) compared to the DMPP· HBPh4 system (see Fig. 5e), revealing the importance of a strong nucleophile in the thiol-Michael polymerization process. Remarkably, anionic reactive intermediates still effected propagation when irradiation was stopped (see Fig. 5f). The radicals had short lifetimes in the dark; thus, the thiol-vinyl reactions were activated by the nucleophile initiation pathway. Finally, the thermomechanical properties of the polymerized PETMP/DVS mixture were damaged by radical-mediated side reactions under 405 nm LED light.
In conclude, many PBGs proved to be able to activate polymerization with different types of photochemical mechanisms. Looking forward, the development of molecular design for efficient PBGs requires the balance among several well-defined properties, such as base-release efficiency, especially for the liberation of strong bases, the catalytic species generated (radicals, cations, or anions) and the photoinitiation capacities, which could give a better mechanistic understanding on photobase generation and photoinitiation processes. Markedly, designing more efficient systems by novel synthetic approaches is a must, which could optimize base generation, the photoinitiated species generated during the photopolymerization. Furthermore, the design on efficient PBGs with strong fluorescence/phosphorescence could also be regarded as targeted direction, due to its potential utilization in targeted applications.
2.3 Advances in PBG-induced photopolymerization: towards elimination of oxygen/water effects
It is very challenging to avoid the negative effects of oxygen/water, even material shrinkage from the photopolymerization process. In 2020, Huang et al.58 designed thioxanthone derivatives as PBGs with quaternary ammonium ions and long fluorinated carbon chains (TX-QF-BPh4), and these exhibited excellent thermostability to over 238 °C under UV irradiation (365–405 nm). Here, the system comprising the PBG furnished higher monomer conversion under 395 nm LED irradiation than ITX, confirming the excellent photoinitiation capacity of the TX-QF-BPh4. As shown in Table 1, the polymerization followed a mechanism with two single-electron transfers (SETs): (1) the first SET process occurred between the borate anion of PBG and the cation to form intermediate 1; (2) the second SET process produced a radical and tertiary amine from 2 to 3 after rapid homolytic cleavage of the CeN bond of the intermediate. Remarkably, TX-QF-BPh4 enabled aggregation on the surface layers of coatings, which was confirmed by using XPS to investigate the F and S contents in different layers and with release of the perfluoroalkyl tert-amine in the absence of coinitiators or additives; this exhibited efficient oxygen inhibition, especially on the surface, during the photopolymerization process. Indeed, the ring-opening reaction also exhibited relatively little material shrinkage and was widely investigated and used in emerging applications. In 2020, Yeo et al.89 examined oxirane ring-opening reactions catalysed by complexation of an acid and a superbase, e.g., benzoylphenylpropionic acid and DBU, respectively (see Fig. 6a). Here, the supramolecule underwent a photodecarboxylation reaction after deprotonation by the ketoprofen under 365 nm irradiation. Then, the base (DBU) was released through an efficient proton-transfer mechanism, which was confirmed by the colour changes of the phenolsulfonphthalein indicator. After irradiation, the PBG solution with chromogenic agent turned pink upon photoirradiation, indicating the release of base at higher pH. In addition, a new absorption signal was detected at ∼575 nm by UV-vis spectroscopy. Interestingly, the authors developed a new 1H NMR strategy named small-molecule-based NMR tube experiments to examine the capacity of the released base to react with thiol and epoxide. Notably, formation of the β-hydroxythioether linkage during the ring-opening reaction was proven to proceed quantitatively by area integration analysis, and it was also confirmed by changing the thiol precursor. Moreover, no thiol–epoxy reaction was observed in the dark or in the absence of PBG. The chemical and mechanical properties of the bulk polymerized products were also investigated for epoxide monomers with different specific properties, encompassing biocompatible polyethylene glycol derivatives, a trifunctional molecule and two compounds on the siloxane motif. The results showed that the elastic modulus was tuned between low and high values by using thiol and epoxy precursors with different chemical properties and surface chemistries to achieve the target materials with different hardnesses. The densitometric results indicated volume shrinkage of ∼5%, while a lower shrinkage of 3.8% was found for the specific system, which was much lower than that seen for free radical polymerization of trifunctional acrylate (∼17%). Compared to the use of the superbase diazabicyclononene (DBN) in bulk polymerizations, the DBU-based PBG required longer times for full gelation under the same photochemical conditions. | ||
| Fig. 6 (a) Synthesis of a photolatent catalyst 1 and its use in proton-transfer reactions using thiol and epoxide functionalities; (b) functional group reactivity considerations in a base-catalysed oxirane ring opening reactions; (c) scanning electron micrographs (SEM) of line (a, width of ca. 70 μm, height ca. 60 μm), star (b, height ca. 1 μm), and grid (c, line width of ca. 10 μm, height ca. 1 μm) patterns. The bottom images (d and e) show the freestanding filaments obtained from patterns b and c, respectively; (d) SEM image showing (a) a silicon master with cylindrical cavities (diameter: ca. 200 nm, height: ca. 200 nm) and replication using precursor combinations of 2 and 7 (b), 3 and 5 (c), and 3 and 7 (d); (e) and (f) fluorescence microscopy images (top = bright field, bottom = λexcitation = 330–385 nm) of the pattern after modification with dye. Reproduced from ref. 89 with permission from the American Chemical Society, copyright 2020. | ||
Finally, this efficient polymerization process was used with soft lithography to fabricate micro- and nanosized objects, even in the absence of a postbaking process, which is commonly used in cationic polymerizations of epoxy systems. Surprisingly, the patterns were separated from the mould through etching as a silicon wafer after the lithography process (see Fig. 6c and d). In this work, photochemical proton-transfer polymerization provided a facile and economic method for sulfonium/carboxylate-based zwitterionic design by selecting proper polymers from among many thiol and epoxide precursors and then achieved a highly uniform network with enhanced mechanical and antibiofouling performance after fabrication, which will likely be widely utilized for applying biofunctional and reactive patterned surfaces in the future. Because they are biodegradable and biocompatible, aliphatic polycarbonates have been widely used in photopolymerizations. However, the design of carbonate-containing monomers has mainly been focused on modifications of carbonate-containing monomers with main- or side-chain functional groups or oligomeric precursors, rather than fabrication of pure polycarbonate networks. Some PBGs have shown the potential to serve in drug delivery, chemotherapeutics and tissue engineering owing to their excellent biocompatibilities and biodegradabilities. Previously, cyclic carbonates were used as PAGs to conduct ring-opening polymerization (ROP) or ring-closing reactions by generating free acids.90 In the recent work of Schandl et al.,91 sustainable, biodegradable and inherently biocompatible aliphatic polycarbonates with network structures were fabricated from cyclic carbonates at elevated temperatures. Here, methyl 4-((hexahydropyrrolo[1,2-a]pyrimidin-1(2H)-yl) methyl)benzoate was synthesized as a PBG, and the tensile tests and DMTA measurements of the targeted materials showed they were highly tunable from elastomers to tough materials based on the ratio of reactive diluent to crosslinker. Indeed, this allowed use of the investigated systems as biocompatible and biodegradable materials in medical applications. In 2020, Gritzapis et al.92 successfully synthesized p-pyridyl oxime carbamate derivatives as “photobase generating DNA-photocleavers” in the presence of plasmid DNA. Interestingly, the amines were released N–O bond homolysis and retained the radical-generation capacity in vivo under hypoxic and anaerobic conditions. Even though the generation of amines in vivo may imbalance the pHs of the tissues, these DNA-photocleavers were not affected by oxygen or pH during DNA photocleavage, and the oxime carbamate derivatives were identified as a new family of photocleavers.
2.4 Photopatterning of nanocarbon material by using photobase generators
Previously, nanocarbon materials have emerged as promising photoinitiators/photocatalysts in photopolymerzation owing to their excellent photochemical/photophysical properties, high extinct coefficients, and eco-friendly, even notable electrical conductivity.93 Previously, bases 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) has been reported as an efficient n-type dopant for the single-walled carbon nanotubes (SWCNT) due to its excellent electron-donating properties.94 In 2023, Tanaka et al.95 successfully fabricated the SWCNT sheets with p–n patterns by utilizing PBG, 2-(9-oxoxanthen-2-yl)propionic acid 1,5,7-triazabicyclo[4.4.0]dec-5-ene salt, as a photoinduced electron dopant. Here, half of PBG-doped SWCNT sheet (p-type) was covered by an aluminum foil photomask and converted to n-type through a 1 min UV irradiation treatment (see Fig. 7a and b). Both the Seebeck coefficients of exposed and masked sections of the SWCNT sheets were examined and a pair of results were obtained, −32.7 mV K−1 and 38.8 mV K−1, respectively (see Fig. 7c). A patterning resolution less than 10 mm at the p–n interface in the photodoped SWCNT sheet was confirmed by using Kelvin force microscopy (KFM). The height and surface profile at the p–n junction interface were illustrated in Fig. 7d and e, respectively. After irradiation, the potential convert to negative and blue for the exposed part, where a clear potential boundary between exposed and masked part could be observed in the surface profile. Remarkably, such high patterning resolution is close to that of the plasma treatment system (∼5 mm, see Fig. 5f). Markedly, owing to the stable cations formed by PBG after doping, the n-type SWCNT sheets achieved extremely long air stability above 100 days. | ||
| Fig. 7 (a) p–n patterning of SWCNT sheets by photoirradiation using an aluminum foil mask. (b) Photographs of masked SWCNT sheets and the UV irradiation process. (c) Seebeck coefficient of pristine (black), non-masked (red), and masked (blue) SWCNT sheets, as measured at room temperature in air. (d) Height profile and (e) surface potential of the p–n interface in a 125 × 125 mm region (red: positive potential, blue: negative potential). (f) Cross-sectional surface potential profiles of the p–n junction interface in regions A–B, C–D, E–F, and G–H. Reproduced from ref. 95 with permission from the Royal Society of Chemistry, copyright 2023. | ||
In another work, Qian et al.96 suggested that the PBG, (E)-1-piperidino-3-(2-hydroxyphenyl)-2-propen-1-one, could be regarded as an efficient base source to fabricate highly homogeneous films epitaxially fused quantum dot (QD) superlattices (epi-SLs) from assembled oleate-capped superlattices, which can be further performed in photopatterning for optoelectronic devices. In photobase illumination method, the PBG releases piperidine under UV light irradiation, then reacts with ethylene glycol to produce glycoxide to trigger by glycoxide–oleate ligand exchange (see Fig. 8b). To evaluate the chemical and structural uniformity of high-quality epi-SLs QD films attained by this method, the films were also approached with base addition by amine point injection (Fig. 8a). In amine point injection method, piperidine was carefully injected into the edge of the floating oleatecapped film (red arrow, see Fig. 8c) and the surrounding region (black rectangle) of injection point was analyzed by FTIR after 1 h diffusion. As shown in Fig. 8d and e, highly inhomogeneous films could be observed from 2D, 3D maps which exhibit gradient decreasing tendency of percent oleate removed in the region, from closest to farthest site of the injection point. Moreover, Fig. 8f illustrates a floating oleate-capped SL film with PBG in ethylene glycol subphase before illumination. After 1 h illumination of a mercury lamp (1.5 mW cm−2) with 254 nm light, oleate loss in specific area of the film (the black rectangle in Fig. 8f) with 4 h exchange in the dark shows in narrow scope of 62.5 ± 5.3% as shown in the map (see Fig. 8g and h). In contrast to films made by amine point injection method, epi-SLs films fabricated by photobase illumination method are not only more homogeneous oleate removal, but also contain less tears and cracks, due to the contactless characteristics of photobase illumination, which avoids the mechanical damaging occurred in point injection process. In fact, we believe that, these works give potential chances to utilized PBGs as a powerful tool to make nanocarbon materials – no matter photodoped SWCNT sheets or photobase-leading candidates for achieving photopatterning, or provide a way to scale up the fabrication of for optoelectronic devices, even wearable thermoelectric applications in the future.
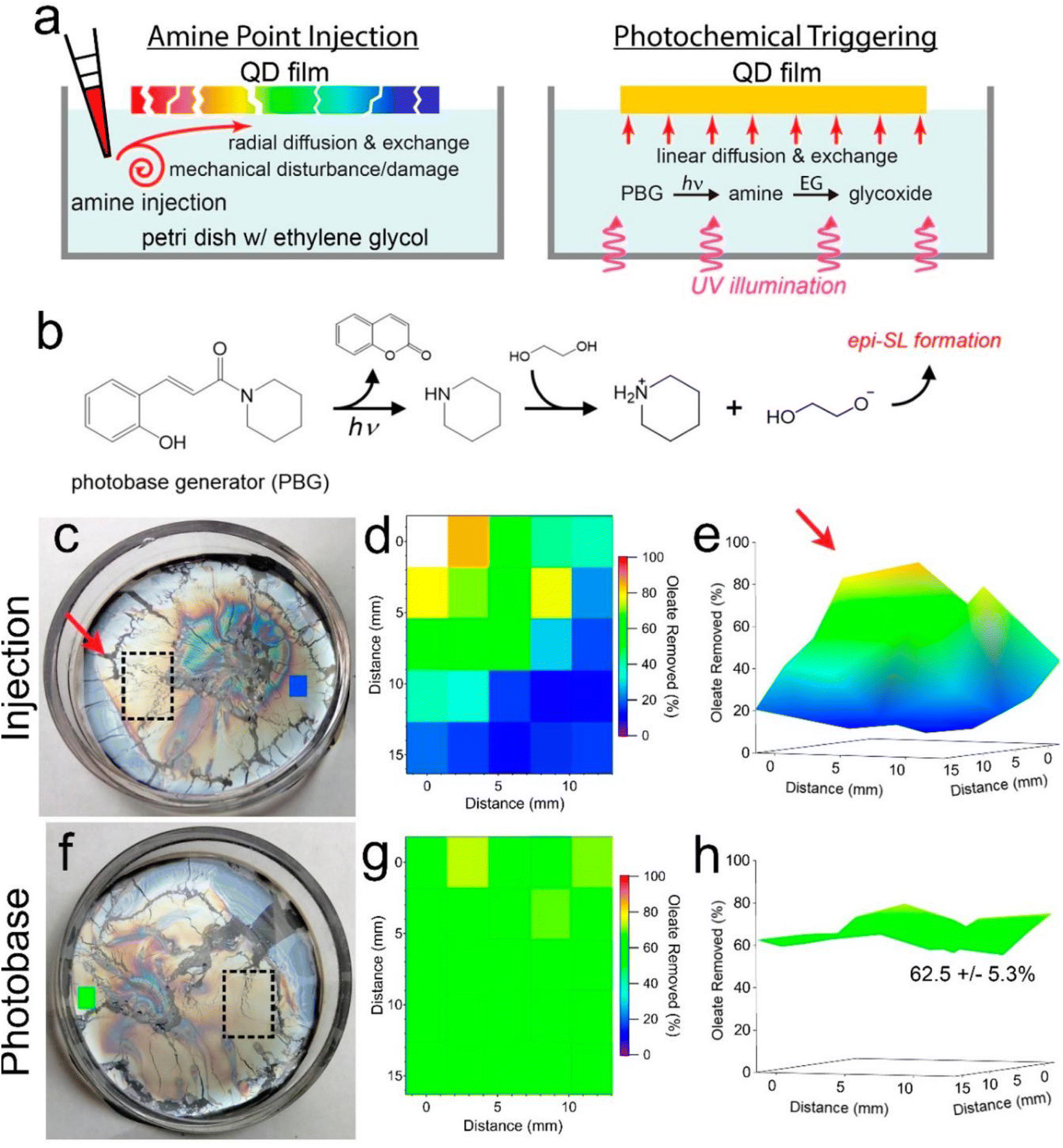 | ||
| Fig. 8 Photochemically triggered epi-SL conversion and uniformity of the resulting films. (a) Schematic depictions of the conventional point injection method (left) compared to our photobase method of making epi-SLs (right). Contactless illumination of a photobase generator (PBG) molecule predissolved in the EG yields a laterally uniform glycoxide concentration and glycoxide–oleate ligand exchange while avoiding the damaging mechanical disturbances of point injection. The color of the QD films denotes the degree of local oleate removal. (b) Proposed reaction cascade for photochemically triggered epi-SL conversion. Photodissociation of the PBG WPBG-027 produces piperidine, which deprotonates EG to form glycoxide, triggering glycoxide–oleate exchange and conversion of the oleate-capped SL to the epi-SL. (c–e) Results for piperidine point injection. The photograph in (c) shows a floating oleate-capped SL film prior to piperidine injection. The red arrow, dashed rectangle, and small blue rectangle denote the location of piperidine injection, the area mapped by FTIR, and an additional point on the far side of the film measured by FTIR, respectively. The diameter of the Petri dish is 5 cm. (d) 2D map (5 × 5 array) of the percent oleate removed from the film as determined by FTIR measurements of the stamp-transferred film. The mapped area is indicated by the dashed rectangle in (c). The injection point is to the left of the upper left corner of the map. One data point is missing from the map due to delamination of the QD film at that location. All quantification corrections were applied as per Fig. 1. As expected, there is a marked oleate concentration gradient in the injection-made film. The blue color of the rectangle on the far side of the film in (c) is consistent with this gradient. Film treatment conditions: 0.5 mL of 11 mM piperidine in EG slowly injected into the 5 mL EG subphase, exchanged for 1 h (1 mM piperidine overall, equivalent to 360 μM glycoxide). (e) 3D view of the map. The red arrow denotes the injection point. (f–h) Results for photobase illumination. (f) Photograph of the floating film before illumination. The dashed rectangle and small green rectangle denote the area mapped by FTIR and an additional point on the far side of the film measured by FTIR, respectively. (g) 2D map (5 × 5 array) of the percent oleate removed, as per (d). The percent oleate removed is constant within experimental error. The green color of the rectangle on the far side of the film in (f) shows that the entire film has uniform ligand exchange. Film treatment conditions: 1 mM PBG in the subphase, 1 h illumination with 254 nm light@1.5 mW cm−2 followed by 4 h of additional exchange in the dark. (h) 3D view of the map. Oleate removal in the mapped region is 62.5 ± 5.3%. Both films are 60 ± 10 nm thick. Reproduced from ref. 96 with permission from the American Chemical Society, copyright 2022. | ||
3 Photobase generators: towards high-quality photoresists
In fact, efficient PBGs for photopolymerization are accessible by balancing the base-release capacity and photoinitiation ability of the superbase through proper structural modifications of the original chromophores via grafting of specific moieties on the photocaging compounds. Furthermore, innovation of the photochemical mechanism is needed, with oxygen or shrinkage resistance that does not negatively affect the photopolymerization process. As mentioned above, photomediated nucleophile-catalysed reactions limit the generation of radicals during polymerization reactions run with visible light irradiation. Hence, chromophores should be designed to afford mechanistic processes that improve the catalytic performance and optimize light-induced polymerization with the PBGs involved. Finally, interest in the development of PBGs is growing since these exhibit remarkable potential to replace PAGs in photonic, electronic, pharmaceutical, and biological applications, which could minimize the damage done to cells or other sensitive materials, such as photoresists.97 As far as we know, photoresists are used in the semiconductor industry to pattern thin films on substrates, and the choice of a photoinitiator is a crucial factor. Most of the photoinitiators utilized for photopatterning materials are PAGs, and examples of PBGs fabricating photoresists are still rare. PBGs efficiently avoid the metal corrosion caused by the acidic species from PAGs during the photopatterning process. For instance, Terada et al.98 reported highly photosensitive PBGs, such as 1,3-bis[(2-nitro-4,5-dimethoxybenzyl)oxycarbonyl-4-piperidyl]propane, to produce various epoxy resins with carboxylic acid groups and excellent solubility. As shown by the photochemical mechanism in Table 1, the PBGs also underwent photodecarboxylation and then a base proliferation reaction in the presence of a base amplifier, 1,3-bis[3-((9-fluorenylmethoxy)carbonyl)aminopropyl]tetramethyldisiloxane. Remarkably, the base-catalysed reaction between the epoxy and carboxylic acid described in this work achieved extremely thin patterned films (∼10 μm) with high photosensitivity (up to 900 mJ cm−2) and resolution (10 μm line-and-space), and metal corrosion was successfully avoided.As alkaline-developable photoresists, Tseng et al.99 developed a negative photosensitive formulation based on PBG, (E)-3-(2-hydroxy-4-methoxyphenyl)-1-(piperidin-1-yl)prop-2-en-1-one (HMPP) and photosensitive polyimides (PSPIs) derived from poly(amic acids) (PAAs) by using a chemical amplification method (Fig. 9a). Notably, they initially utilized a rigid and linear backbone with high thermomechanical stability of the PI, which was composed of p-phenylenediamine (PDA) and 4,4′-biphenyl dianhydride (BPDA), and provided a very clear pattern with an approximately 0.5 μm-thick film due to molecular weight control with PAA(PDA-BPDA) to optimize the high photosensitivity (Fig. 9c–h). In the same year, they fabricated a similar negative-type PSPI composed of PAA(PDA-BPDA/6-FDA) and HMPP. As a result, the resulting PI thin film also exhibited high thermal stability, specifically with excellent ductility, high dimensional stability and even low dielectric properties.100 Nowadays, negative-type PSPIs based on PAA have been widely developed with PBGs.101 For instance, Chang et al. found that the high optical transparency and low rigidity of the polyimide backbone give a significant promotion on the performance of PSPI, especially for the photosensitivity, owing to the designed wide optical bandgap and low rigidity are beneficial for the photochemical reaction and chemical imidization.102 In 2019, Noda et al. fabricated nega-type patterns efficiently attained by polysilane solution with optimum ratio of OXTA-P2tBu (1.8:1) as a PBG under UV irradiation, where the ratio determined by the observations on pH change, ESR and UV absorption spectra, combining with quantum chemical calculation.17 These work re-confirmed the important role of designs on molecular structures, compositions of the PBGs and monomers, even for the design of fluorescent species, no matter these species are derived from organic molecules or nanomaterials, which could become the mainstream method to expand the family of highly efficient PBGs, further promoting their microelectronic applications, particularly in the photoresist aspect in the future.103,104
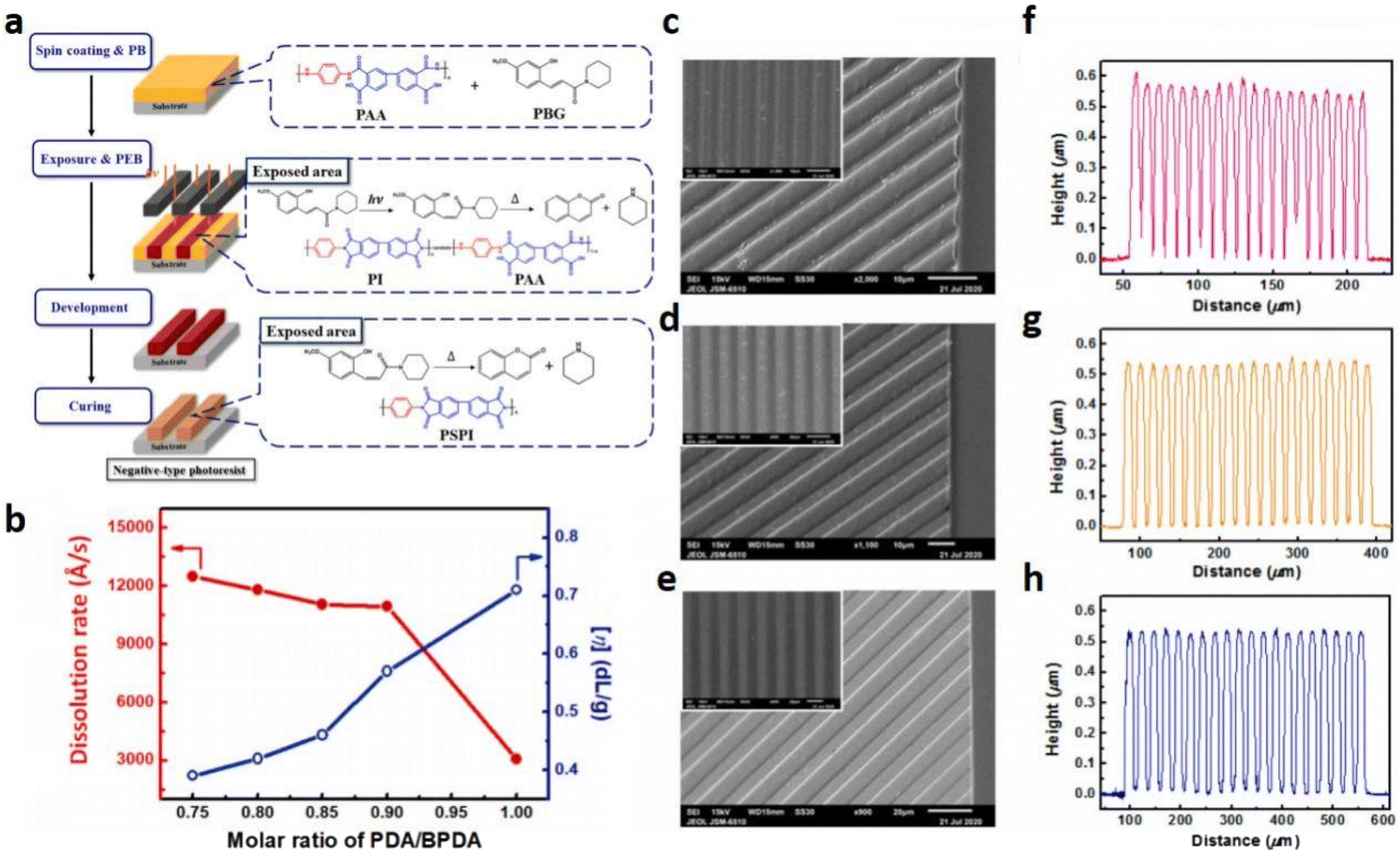 | ||
| Fig. 9 (a) Photolithographic process with the low-temperature curable PSPI with PBG; (b) effect of molecular weight on the dissolution rate of PAA (PDABPDA). The PB condition was fixed at 100 °C for 5 min. SEM images in (c)–(e) cross-sectional view for the PI patterns cured at 250 °C for 1 h with line-and-space patterns featuring 4, 8, and 12 μm, respectively (the inset image is in the front view). (f), (g) and (h) Surface profiles of line and space patterns featuring 4, 8 and 12 μm obtained from the surface profilometer. Reproduced from ref. 99 with permission from Elsevier, copyright 2020. | ||
4 Conclusions and outlook
In this review, recent developments seen with organic photogenerators (PBGs) used in visible light-sensitive photopolymerizations reported from 2019 to early 2023 were discussed and their outstanding characteristics reviewed, e.g., specific range of wavelengths, remarkable photostability, and excellent photoinitiation capacity. The discussion was mainly focused on efficient base release and photoactivation in different photochemical mechanisms, including ring-opening polymerization, the thiol–epoxy click reaction and the thiol-Michael addition reaction. Indeed, most current PBGs undergo decarboxylation or proton-transfer mechanisms, and the majority of the compounds used as photoinitiators were designed and fabricated for radical-mediated polymerization processes carried out under light of different wavelengths; however, a nucleophile initiation pathway seems to be a more efficient strategy with which to broaden development in the future. On the other hand, extensive work on oxygen/water and material shrinkage was also widely investigated and should be continued, wherein the use of PBGs is an efficient way to avoid these negative effects in photopolymerization processes. In conclusion, the design of low-cost and nontoxic PBGs with high base-release and photoinitiation capacities are needed to increase the efficiencies of photopolymerization reactions and enhance the thermal or mechanical properties of the fabricated polymers. As perspective, novel synthesis approaches enable to promote the design of high-performance organic photoinitiators with specific structures and functions, even for fluorescent molecules, and improves the reliability of PBGs. Moreover, PBGs could be imagined to combine with nanocarbon materials for performing photopatterning, further providing a way for emerging other applications, e.g., pharmaceuticals, biological technologies, stereolithography, soft lithography, and high-quality photoresists in microelectronic devices.Author contributions
Conceptualization (T. G., D. L., K. S.); methodology (H.-W. P., K. S.); investigation (H.-W. P., D. L., K. S.); visualization (H.-W. P., K. S.); funding acquisition (Y. S., D. C., T. G., K. S.); project administration (Y. S., T. G.); supervision (T. G., D. L., K. S.); writing – original draft (H.-W. P, K. S.); writing – review & editing (H.-W. P., K. Y., Y. S, D. C., D. L., K. S.).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by the general project of the stable support project of Shenzhen Colleges and Universities (20220811224429001), the National Science Foundation of Guangdong Province (2022A1515011724), the Featured Innovation Project of Guangdong Provincial Department of Education (2021KTSCX275), the Shenzhen Polytechnic Scientific Research Start-up Project (6022312028K), the Postdoctoral Foundation Project of Shenzhen Polytechnic (6022331002K), the Foundation of State Key Laboratory of Digital Manufacturing Equipment and Technology (DMETKF2022027), Special Projects in Key Areas of the Guangdong Provincial Department of Education (2023ZDZX1080) and Shenzhen Science and Technology Program (20231123154444001).References
- P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- M. A. Tasdelen, J. Lalevée and Y. Yagci, Polym. Chem., 2020, 11, 1111–1121 RSC.
- K. Sun, P. Xiao, F. Dumur and J. Lalevée, J. Polym. Sci., 2021, 59, 1338–1389 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- S. Liu, N. Giacoletto, B. Graff, F. Morlet-Savary, M. Nechab, P. Xiao, F. Dumur and J. Lalevée, Mater. Today Chem., 2022, 26, 101137 CrossRef CAS.
- M. Mitterbauer, P. Knaack, S. Naumov, M. Markovic, A. Ovsianikov, N. Moszner and R. Liska, Angew. Chem., Int. Ed. Engl., 2018, 57, 12146–12150 CrossRef CAS PubMed.
- J. Yang, R. Tang, S. Shi and J. Nie, Photochem. Photobiol. Sci., 2013, 12, 923–929 CrossRef CAS PubMed.
- K. Sun, X. Peng, Z. Gan, W. Chen, X. Li, T. Gong and P. Xiao, Catalysts, 2022, 12, 1272 CrossRef CAS.
- B. Corakci, S. O. Hacioglu, L. Toppare and U. Bulut, Polymer, 2013, 54, 3182–3187 CrossRef CAS.
- M. Shirai and M. Tsunooka, Bull. Chem. Soc. Jpn., 1998, 71, 2483–2507 CrossRef CAS.
- C. J. Martin, G. Rapenne, T. Nakashima and T. Kawai, J. Photochem. Photobiol., C, 2018, 34, 41–51 CrossRef CAS.
- J. V. Crivello and E. Reichmanis, Chem. Mater., 2014, 26, 533–548 CrossRef CAS.
- K. Suyama and M. Shirai, Prog. Polym. Sci., 2009, 34, 194–209 CrossRef CAS.
- N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2019, 58, 10410–10422 CrossRef CAS PubMed.
- J. P. Fouassier and J. Lalevée, Photoinitiators for Polymer Synthesis: Scope, Reactivity, and Efficiency, John Wiley & Sons, Weinheim, 2012 Search PubMed.
- A. K. O'Brien and C. N. Bowman, Macromolecules, 2006, 39, 2501–2506 CrossRef.
- K. Noda, S. Kikuchi, N. Ikuma, D. Shiota, M. Furutani and K. Arimitsu, J. Photopolym. Sci. Technol., 2019, 32, 265–270 CrossRef CAS.
- M. Tsunooka, K. Suyama, H. Okumura and M. Shirai, J. Photopolym. Sci. Technol., 2006, 19, 65–71 CrossRef CAS.
- H. Lai, J. Zhang, F. Xing and P. Xiao, Chem. Soc. Rev., 2020, 49, 1867–1886 RSC.
- R. M. Uda and D. Takenaka, Mater. Lett., 2021, 303, 130541 CrossRef CAS.
- S. I. Schlesinger, Polym. Eng. Sci., 1974, 14, 513–515 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, Macromolecules, 1977, 10, 1307–1315 CrossRef CAS.
- M. Ikbal, R. Banerjee, S. Atta, D. Dhara, A. Anoop and N. D. Singh, J. Org. Chem., 2012, 77, 10557–10567 CrossRef CAS PubMed.
- C. Decker and K. Moussa, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 3429–3443 CrossRef CAS.
- N. Zivic, M. Bouzrati-Zerrelli, S. Villotte, F. Morlet-Savary, C. Dietlin, F. Dumur, D. Gigmes, J. P. Fouassier and J. Lalevée, Polym. Chem., 2016, 7, 5873–5879 RSC.
- M. Jin, X. Wu, J. Xie, J. P. Malval and D. Wan, RSC Adv., 2015, 5, 55340–55347 RSC.
- C. Fu, J. Xu and C. Boyer, Chem. Commun., 2016, 52, 7126–7129 RSC.
- X. Sun, M. Jin, X. Wu, H. Pan, D. Wan and H. Pu, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 776–782 CrossRef CAS.
- M. Topa, E. Hola, M. Galek, F. Petko, M. Pilch, R. Popielarz, F. Morlet-Savary, B. Graff, J. Lalevée and J. Ortyl, Polym. Chem., 2020, 11, 5261–5278 RSC.
- F. Petko, A. Świeży, M. Jankowska, P. Stalmach and J. Ortyl, Polym. Chem., 2023, 14, 3018–3034 RSC.
- X. De Pariza, E. Jara, N. Zivic, F. Ruipérez, T. Long and H. Sardon, Polym. Chem., 2021, 12, 4035–4042 RSC.
- G. M. Kim, B. Kim, M. Liebau, J. Huskens, D. N. Reinhoudt and J. Brugger, J. Microelectromech. Syst., 2002, 11, 175–181 CrossRef CAS.
- S. Yoshihisa, S. Atsushi, K. Yoshiyuki, M. Satoshi, H. Nao and D. W. William, J. Photopolym. Sci. Technol., 2005, 18, 125–132 CrossRef.
- A. Nakajima, P. Kang, N. Honda, K. Hikichi, M. Esashi and S. Tanaka, J. Micromech. Microeng., 2007, 17, S230 CrossRef CAS.
- J. G. Kloosterboer, in Electronic Applications, Springer, Berlin, Heidelberg, 1988, pp. 1–61 Search PubMed.
- A. Shiraishi and T. Yamashita, ChemistrySelect, 2020, 5, 2858–2863 CrossRef CAS.
- K. Arimitsu and R. Endo, Chem. Mater., 2013, 25, 4461–4463 CrossRef CAS.
- T. Ohba, D. Nakai, K. Suyama and M. Shirai, Chem. Lett., 2005, 34, 818–819 CrossRef CAS.
- X. Zhang, W. Xi, C. Wang, M. Podgórski and C. N. Bowman, ACS Macro Lett., 2016, 5, 229–233 CrossRef CAS PubMed.
- M. Bouzrati-Zerelli, M. Frigoli, F. Dumur, B. Graff, J. P. Fouassier and J. Lalevée, Polymer, 2017, 124, 151–156 CrossRef CAS.
- H. Salmi, X. Allonas, C. Ley, A. Defoin and A. Ak, Polym. Chem., 2014, 5, 6577–6583 RSC.
- J. Lalevée, X. Allonas, J. P. Fouassier, H. Tachi, A. Izumitani, M. Shirai and M. Tsunooka, J. Photochem. Photobiol., A, 2002, 151, 27–37 CrossRef.
- G. Bucher, J. C. Scaiano, R. Sinta, G. Barclay and J. Cameron, J. Am. Chem. Soc., 1995, 117, 3848–3855 CrossRef CAS.
- J. F. Cameron and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4303–4313 CrossRef CAS.
- J. Sinha, A. Dobson, O. Bankhar, M. Podgórski, P. K. Shah, S. L. W. Zajdowicz, A. Alotaibi, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2020, 36, 249–256 CrossRef CAS PubMed.
- X. Dong, P. Hu, W. Shen, Z. Li, R. Liu and X. Liu, Polymers, 2017, 9, 400 CrossRef PubMed.
- X. Sun, J. P. Gao and Z. Y. Wang, J. Am. Chem. Soc., 2008, 130, 8130–8131 CrossRef CAS PubMed.
- J. Shin, J. Lee and H. M. Jeong, J. Appl. Polym. Sci., 2018, 135, 46070 CrossRef.
- B. El Fouhaili, A. Ibrahim, C. Dietlin, A. Chemtob, X. Allonas and C. Croutxé-Barghorn, Prog. Org. Coat., 2019, 137, 105293 CrossRef CAS.
- M. Sangermano, A. Vitale and K. Dietliker, Polymer, 2014, 55, 1628–1635 CrossRef CAS.
- L. Tseng, Y. Lin, C. Kuo, C. Kuo, M. Ueda and W. Chen, React. Funct. Polym., 2020, 157, 104760 CrossRef CAS.
- K. Sun, Y. Xu, F. Dumur, F. Morlet-Savary, H. Chen, C. Dietlin, B. Graff, J. Lalevée and P. Xiao, Polym. Chem., 2020, 11, 2230–2242 RSC.
- A. Al Mousawi, A. Kermagoret, D. L. Versace, J. Toufaily, T. Hamieh, B. Graff, F. Dumur, D. Gigmes, J. P. Fouassier and J. Lalevée, Polym. Chem., 2017, 8, 568–580 RSC.
- K. Sun, S. Liu, H. Chen, F. Morlet-Savary, B. Graff, C. Pigot, M. Nechab, P. Xiao, F. Dumur and J. Lalevée, Eur. Polym. J., 2021, 147, 110331 CrossRef CAS.
- H. Chen, G. Noirbent, Y. Zhang, D. Brunel, D. Gigmes, F. Morlet-Savary, B. Graff, P. Xiao, F. Dumur and J. Lalevée, Polym. Chem., 2020, 11, 6512–6528 RSC.
- K. Sun, H. Chen, Y. Zhang, F. Morlet-Savary, B. Graff, P. Xiao, F. Dumur and J. Lalevée, Eur. Polym. J., 2021, 151, 110410 CrossRef CAS.
- P. Xiao, F. Dumur, M. Frigoli, M. A. Tehfe, B. Graff, J. P. Fouassier, D. Gigmes and J. Lalevée, Polym. Chem., 2013, 4, 5440–5448 RSC.
- L. Huang, G. Xie and J. Yang, Prog. Org. Coat., 2020, 143, 105604 CrossRef CAS.
- Y. Shirai, A. Sasaki, S. Sato, D. Aoki and K. Arimitsu, ACS Appl. Mater. Interfaces, 2023, 15, 28563–28569 CrossRef CAS PubMed.
- H. R. Asemani, L. Luo and V. Mannari, Prog. Org. Coat., 2020, 147, 105878 CrossRef CAS.
- J. Holländer, R. Hakala, J. Suominen, N. Moritz, J. Yliruusi and N. Sandler, Int. J. Pharm., 2018, 544, 433–442 CrossRef PubMed.
- H. Manchanda and V. Mannari, Prog. Org. Coat., 2019, 127, 222–230 CrossRef CAS.
- A. Gigot, M. Sangermano, L. C. Capozzi and K. Dietliker, Polymer, 2015, 68, 195–201 CrossRef CAS.
- X. Zhang, W. Xi, G. Gao, X. Wang, J. W. Stansbury and C. N. Bowman, ACS Macro Lett., 2018, 7, 852–857 CrossRef CAS PubMed.
- S. Chatani, T. Gong, B. A. Earle, M. Podgórski and C. N. Bowman, ACS Macro Lett., 2014, 3, 315–318 CrossRef CAS PubMed.
- Y. Jian, Y. He, Y. Sun, H. Yang, W. Yang and J. Nie, J. Mater. Chem. C, 2013, 1, 4481–4489 RSC.
- K. Dietliker, R. Hüsler, J. L. Birbaum, S. Ilg, S. Villeneuve, K. Studer, T. Jung, J. Benkhoff, H. Kura, A. Matsumoto and H. Oka, Prog. Org. Coat., 2007, 58, 146–157 CrossRef CAS.
- H. Salmi, X. Allonas and C. Ley, Prog. Org. Coat., 2016, 100, 81–85 CrossRef CAS.
- J. Hwang, D. G. Lee, H. Yeo, J. Rao, Z. Zhu, J. Shin, K. Jeong, S. Kim, H. W. Jung and A. Khan, J. Am. Chem. Soc., 2018, 140, 6700–6709 CrossRef CAS PubMed.
- M. Eckert-Maksić, Z. Glasovac, P. Trošelj, A. Kütt, T. Rodima, I. Koppel and I. A. Koppel, Eur. J. Org. Chem., 2008, 5176–5184 CrossRef.
- L. Chen, Y. Zheng, X. Meng, G. Wei, K. Dietliker and Z. Li, ACS Omega, 2020, 5, 15192–15201 CrossRef CAS PubMed.
- B. Yu, J. Zheng, J. Wu, H. Ma, X. Zhou, Y. Hui, F. Liu and J. He, Polym. Test., 2022, 116, 107767 CrossRef CAS.
- M. Kiker, A. Uddin, L. Stevens, K. Chung, P. Lu and Z. Page, Polym. Chem., 2023, 14, 3843–3850 RSC.
- K. Ikuta, K. Iritani, A. Shiraishi and T. Yamashita, J. Photopolym. Sci. Technol., 2020, 33, 85–90 CrossRef CAS.
- C. Xu, X. Wu, Y. Xiong, Z. Li and H. Tang, J. Polym. Sci., 2021, 59, 3020–3028 CrossRef CAS.
- H. C. Kiliclar, G. Yilmaz, Z. Li, Y. Zheng and Y. Yagci, Macromol. Chem. Phys., 2023, 224, 2200343 CrossRef CAS.
- E. Faggi, J. Aguilera, R. Sáez, F. Pujol, J. Marquet, J. Hernando and R. M. Sebastián, Macromolecules, 2019, 52, 2329–2339 CrossRef CAS.
- S. Yu, O. Reddy, A. Abaci, Y. Ai, Y. Li, H. Chen, M. Guveendiren, K. Belfield and Y. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 45281–45289 CrossRef CAS PubMed.
- C. Ley, A. Siedel, T. Bertaux, C. Croutxé-Barghorn and X. Allonas, Angew. Chem., Int. Ed., 2023, 62, e202214784 CrossRef CAS PubMed.
- Y. Zheng, Y. Yang, H. Yang, F. Han and Z. Li, Prog. Org. Coat., 2020, 148, 105842 CrossRef CAS.
- J. C. Foster, A. W. Cook, N. T. Monk, B. H. Jones, L. N. Appelhans, E. M. Redline and S. C. Leguizamon, Adv. Sci., 2022, 9, 2200770 CrossRef CAS PubMed.
- Y. Huang, L. Li, X. Liu and Z. Li, Polym. Chem., 2022, 13, 3048–3052 RSC.
- G. Xie, Y. Huang, H. Hu and J. Yang, Appl. Res., 2023, e202300052 CrossRef.
- J. Sinha, M. Podgórski, A. Tomaschke, V. L. Ferguson and C. N. Bowman, Macromolecules, 2020, 53, 6331–6340 CrossRef CAS.
- N. Zivic, N. Sadaba, N. Almandoz, F. Ruipérez, D. Mecerreyes and H. Sardon, Macromolecules, 2020, 53, 2069–2076 CrossRef CAS.
- N. Zivic, T. Brossier, F. Crestey, S. Catrouillet, A. Chemtob, V. Héroguez, P. Lacroix-Desmazes, C. Joly-Duhamel, S. Blanquer and J. Pinaud, Prog. Org. Coat., 2022, 172, 107128 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- X. Zhang, X. Wang, S. Chatani and C. N. Bowman, ACS Macro Lett., 2021, 10, 84–89 CrossRef CAS PubMed.
- H. Yeo and A. Khan, J. Am. Chem. Soc., 2020, 142, 3479–3488 CrossRef CAS PubMed.
- I. A. Barker and A. P. Dove, Chem. Commun., 2013, 49, 1205–1207 RSC.
- S. Schandl, T. Koch, J. Stampfl, K. Ehrmann and R. Liska, React. Funct. Polym., 2023, 182, 105460 CrossRef CAS.
- P. S. Gritzapis, P. C. Varras, N. P. Andreou, K. R. Katsani, K. Dafnopoulos, G. Psomas, Z. V. Peitsinis, A. E. Koumbis and K. C. Fylaktakidou, Beilstein J. Org. Chem., 2020, 16, 337–350 CrossRef CAS PubMed.
- K. Sun, Y. Zhang, D. Zhu, X. Peng, J. Zhang, T. Gong, M. Ma and P. Xiao, J. Photochem. Photobiol., C, 2023, 57, 100637 CrossRef CAS.
- S. Horike, Q. Wei, K. Akaike, K. Kirihara, M. Mukaida, Y. Koshiba and K. Ishida, Nat. Commun., 2022, 13, 3517 CrossRef CAS PubMed.
- N. Tanaka, M. Yamamoto, I. Yamaguchi, A. Hamasuna, E. Honjo and T. Fujigaya, J. Mater. Chem. A, 2023, 11, 23278–23287 RSC.
- C. Qian, A. Abelson, A. Miller-Casas, R. Capp, I. Vinogradov, N. Udagawa, N. Ge and M. Law, ACS Nano, 2022, 16, 3239–3250 CrossRef CAS PubMed.
- L. Steidl, S. J. Jhaveri, R. Ayothi, J. Sha, J. D. McMullen, S. Y. C. Ng, W. R. Zipfel, R. Zentel and C. K. Ober, J. Mater. Chem., 2009, 19, 505–513 RSC.
- K. Terada, M. Furutani and K. Arimitsu, Polym. Adv. Technol., 2019, 30, 304–311 CrossRef CAS.
- L. Y. Tseng, Y. C. Lin, C. C. Kuo, C. C. Kuo, M. Ueda and W. C. Chen, React. Funct. Polym., 2020, 157, 104760 CrossRef CAS.
- L. Y. Tseng, Y. C. Lin, C. C. Kuo, C. K. Chen, C. E. Wang, C. C. Kuo, M. Ueda and W. C. Chen, J. Polym. Sci., 2020, 58, 2366–2375 CrossRef CAS.
- L. Liu, C. Song, B. Xue, J. Li, J. Wang and J. Li, J. Semicond., 2018, 39, 026001 CrossRef.
- E. C. Chang, L. Y. Tseng, Y. Liu, C. K. Chen, C. C. Kuo, M. Ueda, Y. C. Lin and W. C. Chen, J. Polym. Sci., 2023, 61, 2122–2132 CrossRef CAS.
- M. C. Fu, T. Higashihara and M. Ueda, Polym. J., 2018, 50, 57–76 CrossRef CAS.
- S. Honda, Commun. Chem., 2023, 6, 170 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00992k |
| This journal is © The Royal Society of Chemistry 2024 |