
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancements in molecular disassembly of optical probes: a paradigm shift in sensing, bioimaging, and therapeutics
Karolina
Saczuk
 a,
Marta
Dudek
a,
Katarzyna
Matczyszyn
ab and
Marco
Deiana
*a
a,
Marta
Dudek
a,
Katarzyna
Matczyszyn
ab and
Marco
Deiana
*a
aInstitute of Advanced Materials, Faculty of Chemistry, Wrocław University of Science and Technology, 50-370 Wrocław, Poland. E-mail: m.deiana@pwr.edu.pl
bInternational Institute for Sustainability with Knotted Chiral Meta Matter (WPI-SKCM(2)), Hiroshima University, Higashi-Hiroshima, Hiroshima 739-8526, Japan
First published on 4th July 2024
Abstract
The majority of self-assembled fluorescent dyes suffer from aggregation-caused quenching (ACQ), which detrimentally affects their diagnostic and therapeutic effectiveness. While aggregation-induced emission (AIE) active dyes offer a promising solution to overcome this limitation, they may face significant challenges as the intracellular environment often prevents aggregation, leading to disassembly and posing challenges for AIE fluorogens. Recent progress in signal amplification through the disassembly of ACQ dyes has opened new avenues for creating ultrasensitive optical sensors and enhancing phototherapeutic outcomes. These advances are well-aligned with cutting-edge technologies such as single-molecule microscopy and targeted molecular therapies. This work explores the concept of disaggregation-induced emission (DIE), showcasing the revolutionary capabilities of DIE-based dyes from their design to their application in sensing, bioimaging, disease monitoring, and treatment in both cellular and animal models. Our objective is to provide an in-depth comparison of aggregation versus disaggregation mechanisms, aiming to stimulate further advancements in the design and utilization of ACQ fluorescent dyes through DIE technology. This initiative is poised to catalyze scientific progress across a broad spectrum of disciplines.
 Karolina Saczuk | Karolina Saczuk is a PhD student at the Institute of Advanced Materials, Wrocław University of Science and Technology (WUST), Poland. She earned her master's degree in Chemistry and Materials Science, along with two bachelor's degrees in Chemical Technology and Chemical and Process Engineering, both from WUST. Prior to her PhD candidacy, under the guidance of Dr Marco Deiana and Professor Katarzyna Matczyszyn, Karolina accumulated experience in the manufacturing sector, primarily within the food and automotive industries. Currently, she is focusing her research on photodynamic therapy, G-quadruplex biology, and hypoxia. |
 Marta Dudek | Marta Dudek is an Assistant Professor at the Institute of Advanced Materials, Wrocław University of Science and Technology (WUST), Poland. She obtained her PhD degree in 2019 in the field of Chemical Science under the guidance of Prof. Katarzyna Matczyszyn. She gained additional experience during several internships at the University of Strasbourg (France), École Normale Supérieure in Cachan (France), Institute de Sciences Chimiques de Rennes (France), Humboldt University (Germany) and Umeå University (Sweden). Her scientific interests involve the design, synthesis, and characterization of new azobenzene photoswitches as well as their application in biological settings. |
 Katarzyna Matczyszyn | Katarzyna Matczyszyn is a physical chemist and a Professor at the Wrocław University of Science and Technology (WUST), Poland. She is also an affiliated member of the WPI SKMC2 at Hiroshima University, Japan. Previously, she worked at CEA Saclay, École Normale Supérieure de Cachan, and Université Pierre and Marie Curie in France. Her major scientific interest lies in light-matter interactions, particularly with biologically significant materials. She works with photoactive molecules (mostly photochromes), various types of nanoparticles (including plasmonic and carbon nanodots), lyotropic liquid crystals, and in the field of nonlinear optics. She is particularly interested in novel approaches to photodynamic therapy. |
 Marco Deiana | Marco Deiana is an Assistant Professor at the Institute of Advanced Materials, Wrocław University of Science and Technology (WUST), Poland. He completed his PhD in Materials Engineering at WUST in 2018 under the supervision of Prof. Katarzyna Matczyszyn. Subsequently, he joined Umeå University in Sweden as a postdoctoral fellow from 2018 to 2023, working in the research group of Prof. Nasim Sabouri. In 2023, Dr Deiana returned to WUST with a grant from the National Science Centre (NCN) to serve as the Principal Investigator. His research interests include G-quadruplex biology, hypoxia, light-activated therapies, molecular chirality, and self-assembly. |
1 Introduction
The self-assembly of synthetic molecular building blocks represents a powerful approach for fabricating materials with precise properties and functions.1–3 Guided by thermodynamic principles, supramolecular systems generally attain their lowest free-energy state via self-assembly, with monomeric units initially in a higher free-energy state.4 However, alternative systems involving kinetically controlled supramolecular polymerization5–7 or out-of-equilibrium processes8–11 may also be engineered. The dynamic balance between the assembled and unassembled states hinges on their free-energy difference, wherein the formation rate of structured assemblies generally surpasses the rate of disassembly. Such molecular aggregates arise via non-covalent interactions, including π–π stacking, hydrogen bonding, and dipole–dipole forces.12,13 Specific molecular arrangements can guide self-assembly into uniform structures, such as J-type aggregates, while hydrophobic interactions in π-extended systems tend to produce heterogeneous nanoparticles of varying sizes, known as H-type aggregates.12,14–18 These variations significantly influence the optical properties of luminescent materials and, in the context of biomedical research, their translation into clinical applications.19,20Traditional fluorescent molecules are highly effective emitters when isolated, but often experience aggregation-caused quenching (ACQ) at the concentrations commonly used for sensing and imaging.21 This phenomenon, which arises from intermolecular π-stacking, reduces their effectiveness in biological applications, although a handful of ACQ dyes have been reported to be suitable for such uses, particularly in the contexts of drug delivery22–25 and photothermal therapy (PTT).26 Conversely, the phenomenon of aggregation-induced emission (AIE) signifies a paradigm shift, where organic molecules emit light upon aggregation, offering a counterpoint to ACQ by promoting luminescence through the restriction of intramolecular motions.21,27
Despite advancements in understanding and applying AIE, integrating it into biological systems remains challenging.28,29 This difficulty arises, for instance, because cell media typically contain serum supplements, and intracellular environments tend to reduce aggregation, leading to the disassembly of the probes.30 These limitations underscore the necessity for innovative sensing mechanisms that can navigate the constraints of both ACQ and AIE.
Enter disaggregation-induced emission (DIE), a mechanism that harnesses signal amplification through the disassembly of aggregated probes, offering a promising alternative for probe development.31–35 DIE involves chemical compounds that exhibit enhanced emissive properties upon interaction with specific biomolecules or analytes, effectively transforming from a self-quenched state to a highly luminescent form. Despite its potential, DIE remains a relatively underexplored area with much to offer in sensing, bioimaging, and therapeutic applications.
This review aims to summarize the advancements in the field of DIE research, with a specific focus on the development of DIE-active probes that target key macromolecules—namely nucleic acids, proteins, and lipids. These biological targets are specifically chosen as they constitute the primary focus of existing DIE research, supported by well-established and robust data.
While other elements such as macrocyclic cavitands,36–41 signalling molecules (e.g., adenosine-5′-triphosphate,42–45 sulfur dioxide46 or gluthatione47), metal ions,48–51 and various stimuli (e.g., surfactants,52,53 pH54–57 or temperature58) also play significant roles, either directly or indirectly, as disaggregating triggers, they are excluded from this review to maintain a concentrated exposition on the fundamental principles of DIE and their translational potential in advanced clinical research.
This comprehensive overview of DIE mechanisms and current research directions emphasizes the need for precise data interpretation to prevent misconceptions, especially for those new to the field. The discussion will highlight the challenges, limitations, and future prospects of DIE research, emphasizing its transformative potential in advancing the design and optimization of molecular probes across various applications.
2 Disaggregation-induced emission
Compounds exhibiting ACQ characteristics predominate posing significant limitations for sensor and drug development.However, ACQ, despite its common perception as an unfavourable occurrence, can be ingeniously utilized in the design of activatable probes. This approach leverages the fact that introducing a significant alteration in the environment of ACQ-based probes can shift the equilibrium from aggregated states to their monomeric forms. This transition, occurring under specific conditions, can enhance and recover the readout signal, thereby shifting from an “OFF” to an “ON” state.
The process of leveraging ACQ for probe development can occur in at least two simplified scenarios.59 In the simplest scenario, all molecules form molecular aggregates with ACQ character, and the transition from aggregate to monomer enhances the readout signal (Fig. 1(A)). Alternatively, an equilibrium might exist between molecular aggregates with ACQ character and monomeric species (Fig. 1(B)). In this equilibrium, complexation occurs in two steps: initially, the formation of a complex between the monomer and the host molecule at low host concentrations; and following saturation of this binding event, a second binding event at higher host concentrations leads to the disassembly of the molecular aggregates. In both scenarios, the parameters governing the disassembly process require that the association constant of the aggregated molecules (Kaggregation) be lower than or similar to the association constant resulting from the complexation with the host matrix (Kcomplex). In other words, the Gibbs free energy (ΔG) governing the aggregation process should be higher than or similar to the ΔG value resulting from the complexation event (ΔGaggregation > ΔGcomplex).
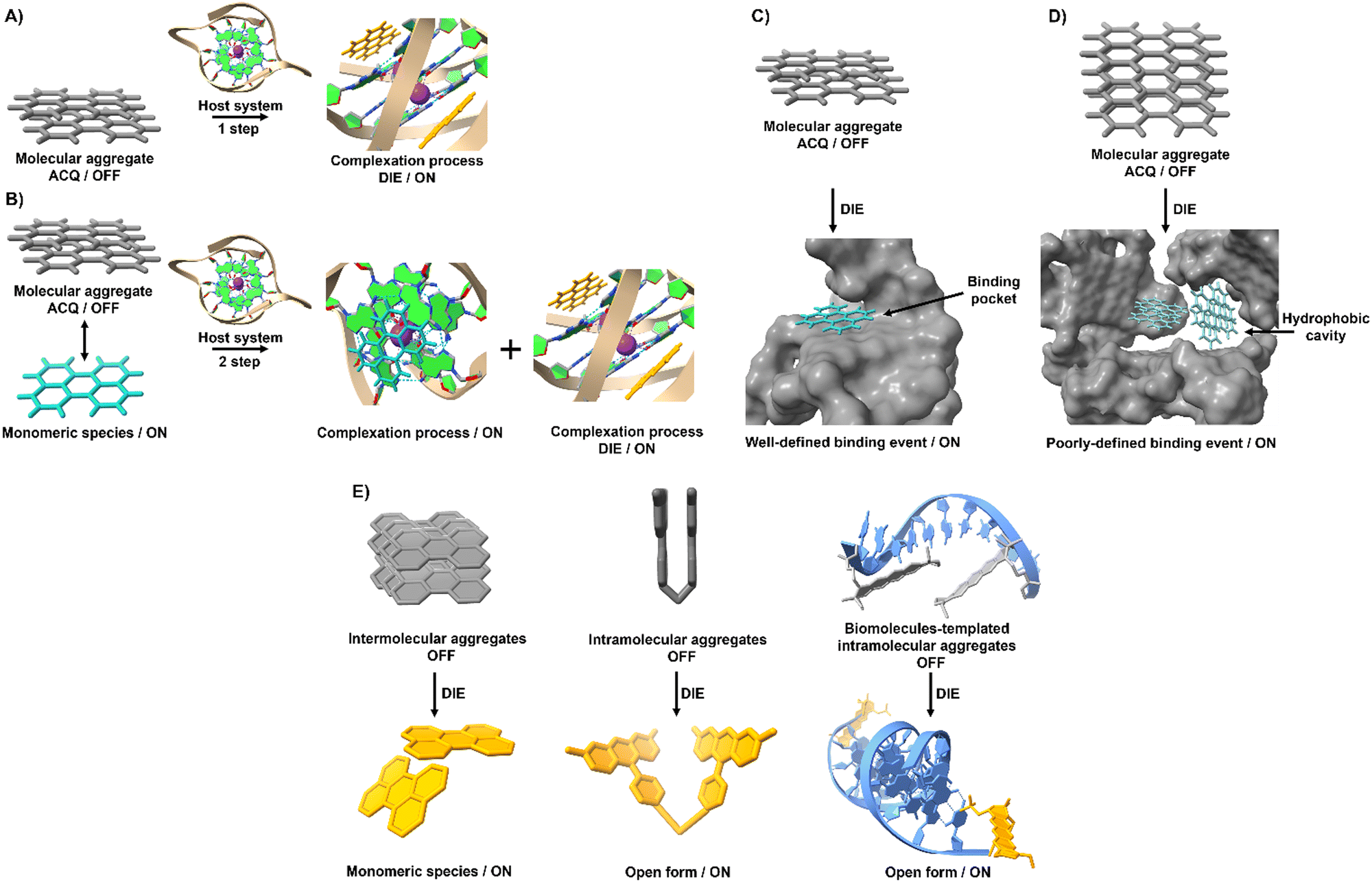 | ||
| Fig. 1 Graphical illustration depicting the simplified mechanisms of molecular recognition mediated by self-assembled probes used in the design of DIE-active dyes. (A) In an aqueous environment, only molecular aggregates are formed by an ACQ-active probe. When a targeted receptor (host) is present, the aggregate state disassembles to form a complex if the free energy of the complex is lower than or similar to that governing the assembly process. (B) An equilibrium is maintained between aggregated and monomeric species. The introduction of a target receptor initiates two binding processes: initially, the monomers in solution bind to the receptor until saturation; subsequently, excess host triggers the disassembly of the aggregates. (C) Molecular recognition mediated by an intramolecularly formed biological structure with a well-defined binding pocket triggers disassembly of the optical probe, resulting in a precise binding event characterized by a specific stoichiometry. (D) Intermolecularly interacting biological matrices can form hydrophobic cavities that coordinate with self-assembled probes, driving their molecular disassembly and resulting in poorly defined complexes that do not adhere to conventional stoichiometry. (E) Various molecular arrangements of ACQ probes demonstrate DIE mechanisms through intermolecular, intramolecular, and biologically templated self-assembly aggregates. | ||
Complexation between aggregated dyes and targeted biomolecules often leads to the formation of well-defined complexes with specific stoichiometry.32,33,60–62 Nonlinear fitting models describing common binding stoichiometries (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, or 2:1) usually represent experimental data accurately. These quantitative binding parameters are often associated with models such as Job's plots or mole ratio methods, which describe the system's stoichiometry. Structural investigations performed by nuclear magnetic resonance (NMR) spectroscopy or molecular dynamics (MD) simulations provide valuable insights into the molecular nature of these systems.
:1, 1:2, or 2:1) usually represent experimental data accurately. These quantitative binding parameters are often associated with models such as Job's plots or mole ratio methods, which describe the system's stoichiometry. Structural investigations performed by nuclear magnetic resonance (NMR) spectroscopy or molecular dynamics (MD) simulations provide valuable insights into the molecular nature of these systems.
However, this scenario typically occurs when inter or intramolecular dye aggregates coordinate with intramolecularly formed biomolecules possessing specific binding pockets (Fig. 1(C)). The situation can become more complicated when intermolecular processes at the biological level influence the complexation process. For instance, intermolecularly interacting biological structures may create hydrophobic domains that do not selectively coordinate with molecular aggregates (Fig. 1(D)). As a result, the disassembly of the probe might not follow the expected aggregation-to-monomer transition, leading to unusual binding events, with association curves that do not exhibit saturation even in the presence of high concentrations of biological templates.
Understanding these complex interactions requires further investigation, as the interplay between dye aggregates and biological structures can significantly impact the efficiency and accuracy of DIE dyes in practical applications.
To harness ACQ effectively for DIE, two main rational design strategies have been proposed, focusing on either intermolecular32,35,61,62 or intramolecular34,63,64 self-assembly (Fig. 1(E)). Intermolecular aggregation typically leads to the formation of supramolecular structures with loosely defined arrangements. In contrast, intramolecular aggregation results in well-defined systems, facilitating precise and controlled self-assembly. Further, incorporating biological templates65,66 into the supramolecular assembly process can optimize ACQ dyes for optical performance in DIE-based sensing applications.
3 Biomolecules-mediated DIE
DIE-active dyes, either used independently or in conjunction with various biomolecules, have led to the creation of ultrasensitive fluorogenic agents boasting exceptional recognition and imaging properties. The subsequent sections delve into the assembly and utilization of this category of materials, focusing on their interaction with different biomolecules: nucleic acids, proteins, and lipids.3.1 DNA-mediated DIE
DNA is essential to the central dogma of molecular biology, functioning not only as a repository for genetic information but also playing a key role in various biological processes with significant clinical implications.67,68 So far, only a limited number of probes that interact directly with double-stranded (ds) DNA have demonstrated DIE character.69,70 They function by directly disassembling upon binding to dsDNA, either through a single-step disassembly mechanism69 or by undergoing a DNA-templated aggregation process first, followed by disassembly at higher duplex concentrations70 (Fig. 2(A) and (B)).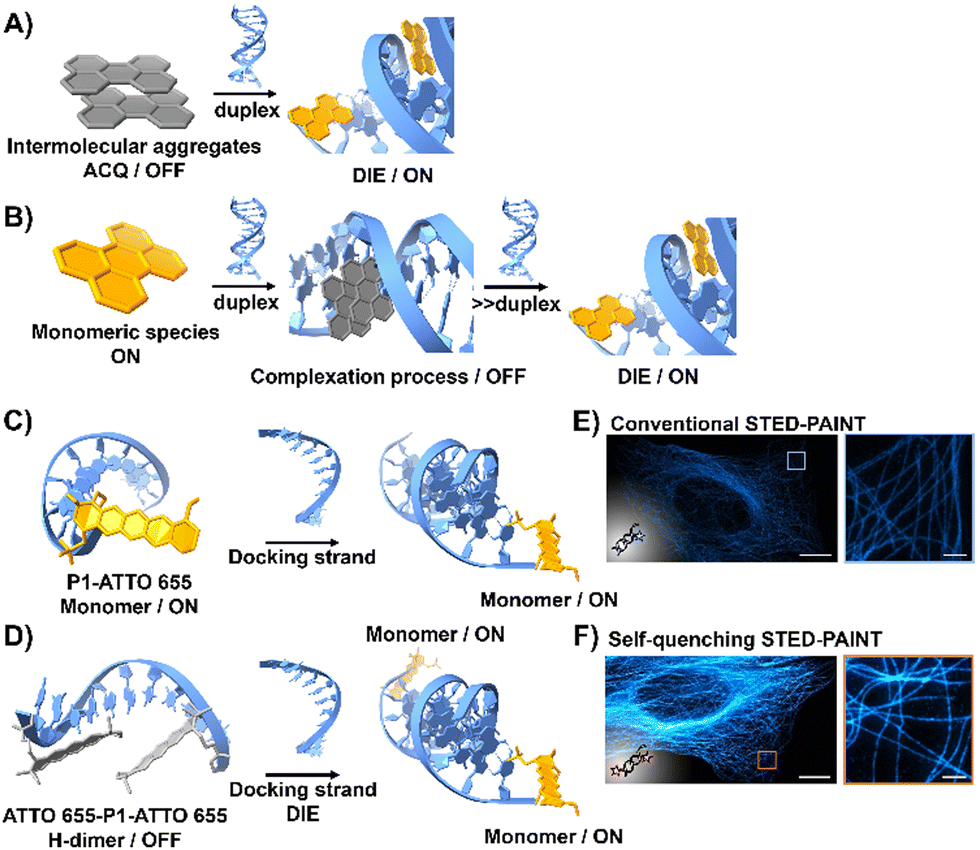 | ||
| Fig. 2 (A) Intermolecular aggregates exhibiting ACQ characteristics can transform into highly emissive monomeric species bound to duplex DNA through the DIE mechanism. (B) In the presence of low concentrations of duplex DNA, the highly emissive monomeric state of a probe self-assembles within the grooves, rather than at intercalation sites. At higher duplex concentrations, a new competitive equilibrium forms between pre-existing DNA-dye aggregates and monomer-like DNA complexes through the DIE process. (C) and (D) Schematic diagrams of optical designs include: (C) the single ATTO 655 dye attached to the 3′-end of the imager strand (P1-ATTO 655), and (D) two ATTO 655 dyes at both the 5′- and 3′-ends of the imager strand, forming a self-quenching H-type dimer (ATTO 655-P1-ATTO 655). Upon introduction of the complementary docking strand, the H-dimer disassembles, leading to a significant increase in fluorescence. (E) and (F) STED microscopy images of U2OS cells immunolabeled for microtubules using either P1-ATTO 655 (E) or the dual-labelled ATTO 655-P1-ATTO 655 (F), demonstrating the effects of dye placement and configuration on fluorescence enhancement. The scale bars represent 10 μm for overviews and 1 μm for detailed zoom-ins. Reproduced from ref. 66 with permission from the Wiley-VCH. | ||
Heilemann and colleagues have proposed an intriguing application of DNA-labelled DIE-active dyes to enhance super-resolution imaging techniques.66 They developed a short-distance H-type self-quenched fluorophore dimer, utilizing the oxazine fluorophore ATTO 655 attached to both the 5′- and 3′-ends of a 9 nucleotide-long DNA imager strand (P1) (ATTO 655-P1-ATTO 655), and assessed its performance against a version labelled with a single ATTO 655 dye at the 3′-end (P1-ATTO 655) (Fig. 2(C) and (D)).66 Spectroscopic analysis revealed that the dual-labelled strand ATTO 655-P1-ATTO 655 shifted from a weakly emissive H-type dimer to a highly fluorescent monomeric state upon forming the docking-imager duplex, markedly enhancing fluorescence and stability. The use of the dual-labelled strand in stimulated emission depletion (STED) imaging71 of immunolabeled α-tubulin in human osteosarcoma U2OS cells resulted in a significant improvement in image quality, achieving a five-fold increase in signal-to-background ratio (SBR) compared to the single-labelled strand (Fig. 2(E) and (F)).66
By applying the principles of hybridization and fluorogenic dimers, the sensitivity of HIV RNA detection in reverse transcription quantitative polymerase chain reaction (RT-qPCR) has been enhanced.65 Beacon probes tagged with dyes such as R6G, ROX, and Cy5 transitioned from dim aggregates to bright monomers upon hybridization or heating. Notably, ROX exhibited exceptional performance, surpassing even the Abbott RealTime HIV-1 kit.65 When tested in spiked human plasma and clinical samples from patients receiving highly active antiretroviral therapy (HAART), this method improved the detection of low-copy HIV RNA, thereby reducing the occurrence of false negatives.65
DNA and RNA can form various non-canonical structures, with the G-quadruplex (G4) being particularly notable (Fig. 3(A)).72 This alternative DNA structure is characterized by its guanine-rich composition, stability, and polymorphism (Fig. 3(B)).73 It plays a significant role in cancer research due to its association with genomic instability.74 Techniques like immunofluorescence, using the G4-specific antibody BG4,75 and chromatin immunoprecipitation followed by sequencing (ChIP-Seq)76,77 have enhanced our capacity to detect these structures within cells and chromatin. However, these methods only identify a portion of G4 sites. Imaging G4s in living cells remains challenging,78,79 partially due to probe aggregation, which decreases the effectiveness of tracking these structures dynamically. These probes often employ hydrophobic chromophores to enhance selectivity for G4 over dsDNA, which may cause ACQ issues.80 However, the interaction of these probes with G4 structures can lead to DIE, offering a solution to some of the challenges in fluorescence imaging and suggesting the possibility for real-time mapping of G4 structures (Fig. 3(C)).
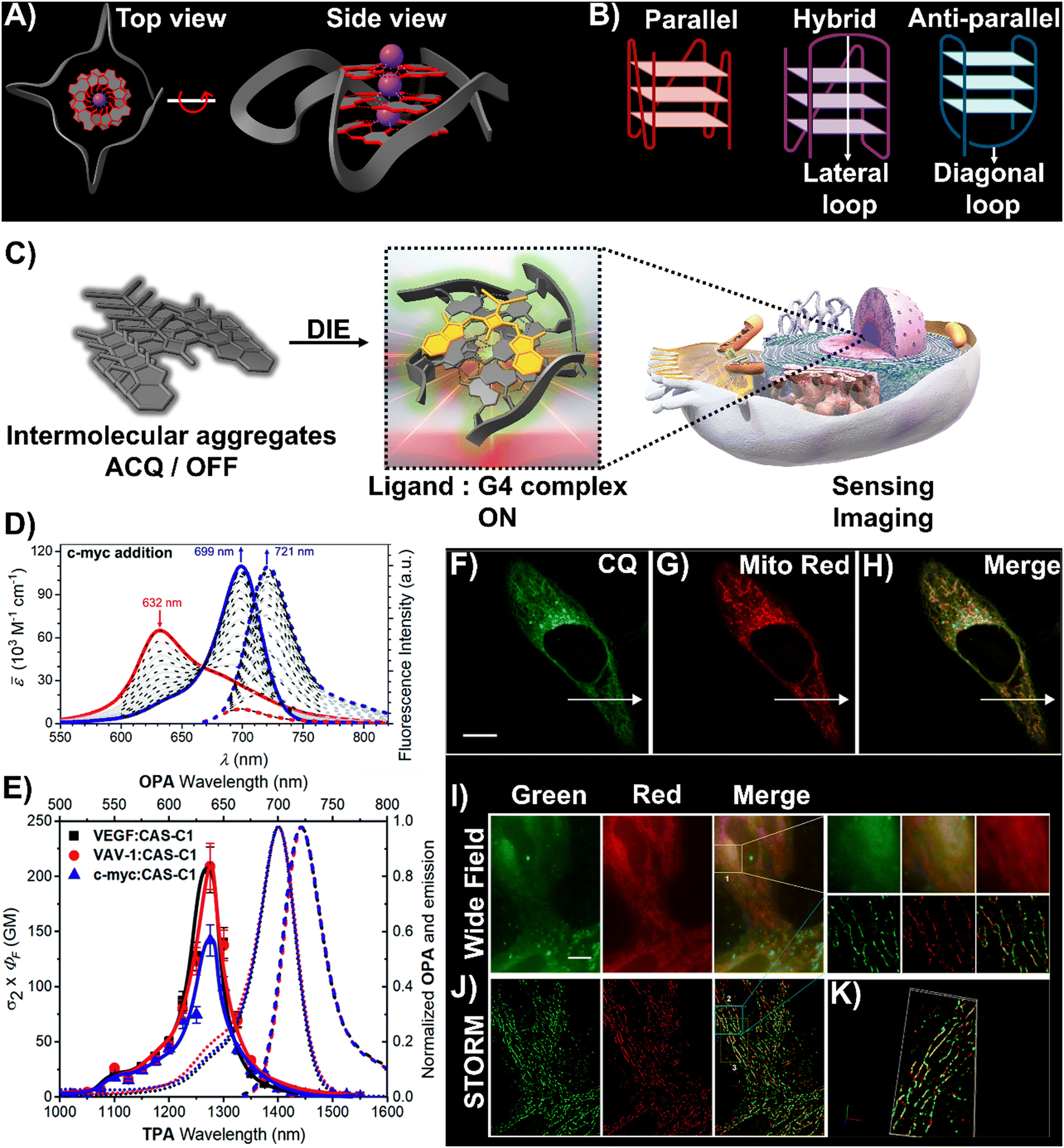 | ||
| Fig. 3 (A) Visualization of G4 structures with top and side views of the X-ray crystallographic structure of parallel human telomeric DNA G-quadruplex (PDB: 1KF1). (B) Depiction of G4 structures forming various topologies: parallel, hybrid, and antiparallel. White arrows indicate lateral and diagonal loops in hybrid and antiparallel G4s, respectively. Created with BioRender.com. (C) Schematic of the DIE process facilitated by G4 structures. (D) Spectrophotometric and fluorometric analyses of the CAS-C1 molecule with incremental additions of the c-MYC G4. (E) Graph showing the two-photon molecular brightness (σ2 × ΦF) of the CAS-C1 complex with G4 structures from VEGF, VAV-1, and c-MYC. Reproduced from ref. 33 with permission from the Royal Society of Chemistry. (F)–(H) Colocalization studies using CQ compound (F), MitoTracker Red CMXRos (G), and their merged imaging (H) in live HepG2 cells. (I) and (J) Widefield (I) and reconstructed STORM images (J) of HepG2 cells stained with CQ and MitoTracker Red CMXRos, including magnified views. Scale bar: 10 μm. (K) Three-dimensional volume representation of STORM-based colocalization between CQ and MitoTracker Red CMXRos. Reprinted with permission from ref. 81. Copyright 2021 American Chemical Society. | ||
A broad array of DIE-active dyes has been identified for detecting G4s in DNA or RNA, both in test tubes and within cancer cells. Specifically, dyes that function through DIE predominantly form intermolecular aggregates and target parallel G4 structures. This preference is due to the more accessible external tetrads found in parallel G4s, in contrast to the less accessible structures of hybrid and antiparallel G4 morphologies that incorporate lateral or diagonal loops (Fig. 3(B)).82 Additionally, parallel G4s may form intermolecular structures that facilitate the disassembly of ACQ-based dyes. These dyes encompass various molecular structures, including squaraines,33,60 core-extended naphthalene diimides,83–85 coumarin32,81,86–88- and cyanine89–91-based dyes, boron-dipyrromethene (BODIPY) analogues,62,92–94 quinazoline-quinazolinone derivatives,61,95 and more96–100 (Scheme 1).
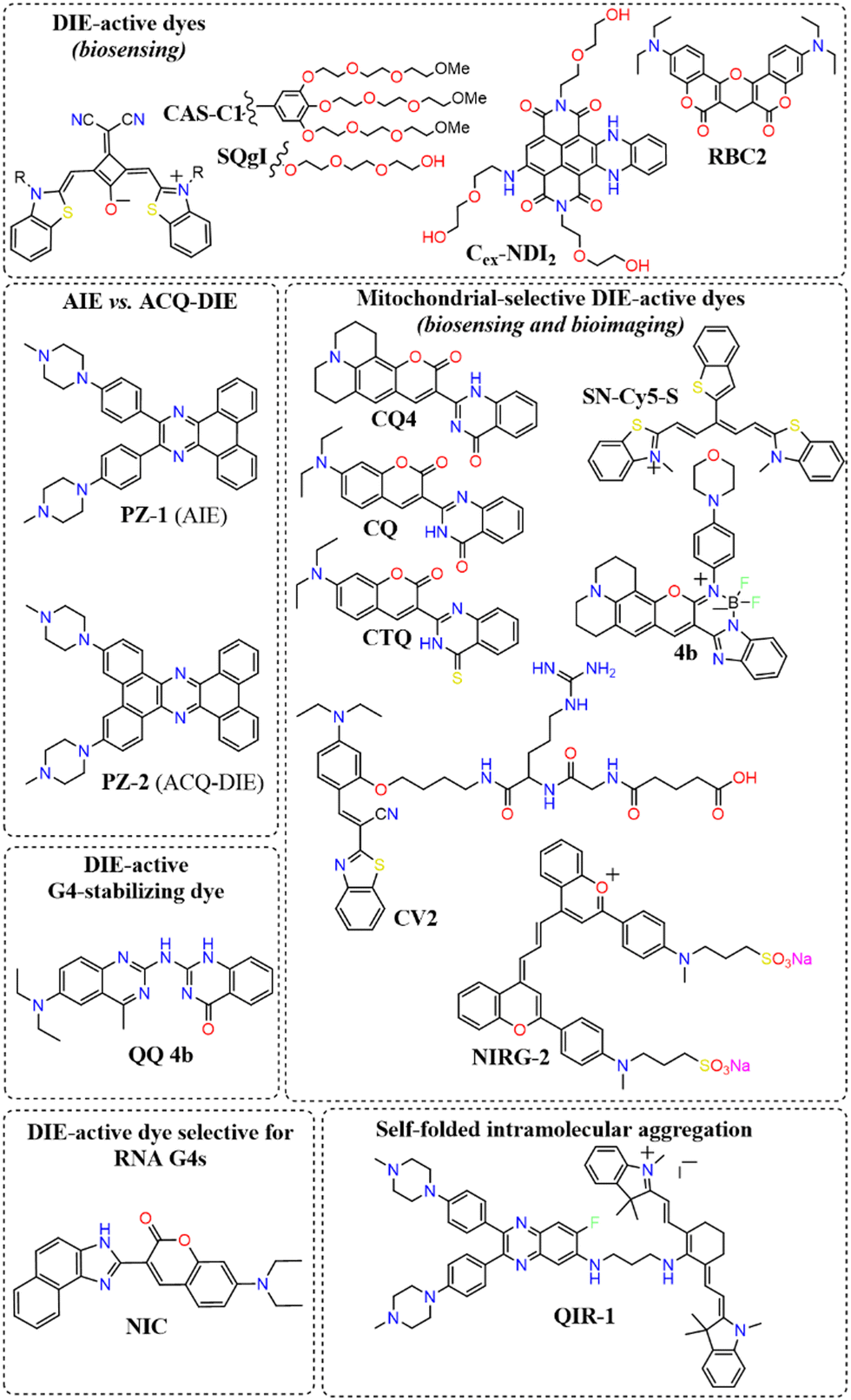 | ||
| Scheme 1 Representative molecular structures of various DIE-active dyes targeting G4 structures. | ||
In this context, Würthner and colleagues developed two near-infrared (NIR) fluorescent probes: amphiphilic dicyanovinyl-substituted squaraine dyes named SQgI60 and CAS-C1.33 These dyes were functionalized with triethylene glycol chains to enhance water solubility without altering the overall charge of the molecules, thereby promoting self-assembly likely driven by hydrophobic effects and/or dipole–dipole interactions among the squaraine scaffolds (Scheme 1).16,101 In an aqueous environment, these probes tended to form non-emissive molecular aggregates with an H-type character (Fig. 3(D)). However, upon binding to biologically relevant parallel G4 structures, there was a notable increase in their photoluminescence quantum yields (ΦF), with values of 0.61 for SQgI and 0.74 for CAS-C1 (Fig. 3(D)).33,60 Additionally, the interaction of CAS-C1 with parallel G4 structures increased the dye's two-photon absorption cross-section (σ2), with values ranging from 273 to 312 GM at 1275 nm, depending on the G4 structure it bound to (Fig. 3(E)).33 This property enabled the CAS-C1:G4 complexes to function in a NIR-to-NIR mode, a highly requested configuration feature in nonlinear microscopy.
Ultrabright coumarin-quinazolinone dyes (CQ,81CQ432 and CTQ,86Scheme 1) designed for DIE have been reported to detect parallel G4 structures with high selectivity in both test-tube experiments and within cancer cells. Some of these dyes have proven to be effectively integrated not only in traditional confocal laser scanning microscopy32,81 (CLSM, Fig. 3(F)–(H)) but also in advanced super-resolution bioimaging methods such as two-photon imaging81 and stochastic optical reconstruction microscopy (STORM)81 (Fig. 3(I)-(K)).
For this category of dyes, a carefully engineered design has also been utilized to minimize the background fluorescence of aggregates, aiming to develop absolute ACQ probes with a higher activation ratio upon binding to G4 structures, thereby achieving lower SBR. This has been demonstrated, for instance, by replacing oxygen atoms with sulfur atoms (CTQ,86Scheme 1), a structural modification also widely used in photodynamic therapy (PDT)102–104 to enhance intersystem crossing.
Research has shown that fluorescence imaging in the NIR-II range (900–1700 nm) offers significant advantages over imaging in the NIR-I range (650–900 nm).105–107 NIR-II imaging exhibits minimal autofluorescence and reduced photon scattering in living tissue, allowing for deeper penetration, higher resolution, and improved SBR. These benefits have garnered considerable interest for in vivo biological studies. In this context, Zhang and colleagues designed a cyanine derivative functionalized with two molecular rotor groups (NIRG-2, Scheme 1), demonstrating the capability to selectively detect DNA G4s, providing a light-up response in the NIR-II window.108 The molecules exhibited self-assembly properties in phosphate buffered saline (PBS), with an absorption band that was hypsochromically shifted. Upon complexation with a G4 structure, a potential disassembly occurred, reverting to a monomer-like absorption band and activating the fluorescence signal by halting the rotational movement of the rotor groups. NIRG-2 had been effectively utilized to distinguish G4 structures in vivo, enabling the differentiation of tumours from normal tissues based on G4 abundance. Additionally, NIRG-2 successfully tracked lymphatic metastasis in tumours through G4 detection.108
Diverging from the conventional intermolecular ACQ systems, DIE-active dyes that form excimer-emissive aggregates, which disassemble into highly emissive monomeric forms in the presence of parallel G4 structures, have also been developed (Fig. 4(A)). Specifically, Kim's group engineered a peptidyl fluorogenic probe named CV2 (Scheme 1), combining a zwitterionic dipeptide receptor with a cyanovinylene dye linked by a tetramethylene linker.98 CLSM and fluorescence lifetime imaging (FLIM) assessments confirmed CV2's specificity for G4 structures, with its green monomeric fluorescence significantly diminished by Deoxyribonuclease I (DNase I) or competitive small-molecule G4 ligands. The probe's potential for monitoring G4 folding and unfolding dynamics was further demonstrated by inhibiting native helicases in HeLa cells, resulting in enhanced fluorescence (Fig. 4(B)–(D)). Furthermore, CV2 proved effective in identifying mitochondrial DNA (mtDNA) damage, as cells exposed to DNA-damaging agents showed reduced CV2 fluorescence, indicative of mtDNA depletion.98
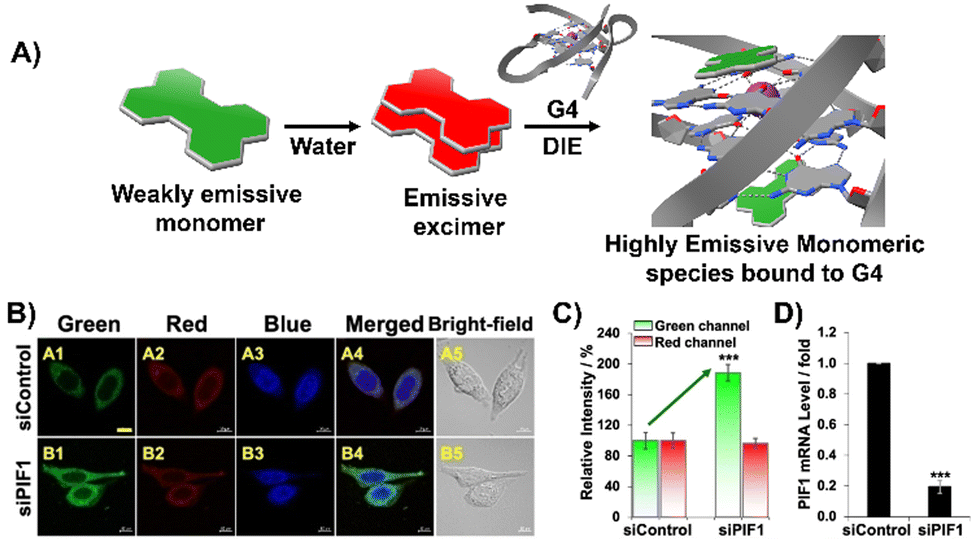 | ||
| Fig. 4 (A) Schematic representation of the DIE operated by CV2. In aqueous environments, CV2 tends to self-assemble into stable aggregates that exhibit excimer emission. Upon interacting with G4, these aggregates disassemble, shifting from red-emissive aggregates to green-emissive monomeric forms. (B) Investigating the impact of endogenous PIF1 knockdown on the fluorescence activation of CV2 by DNA G4s in live HeLa cells, with DAPI used for nuclear staining. (C) Comparison of green and red fluorescence intensities in HeLa cells loaded with CV2, contrasting control with PIF1-silenced cells. (D) Assessment of PIF1 knockdown efficiency via quantification of PIF1 mRNA levels using RT-qPCR in both control and PIF1-silenced HeLa cells. Reprinted with permission from ref. 98. Copyright 2022 Wiley-VCH. | ||
Besides forming intermolecular aggregated structures, dyes active in DIE can also detect G4s through the disassembly of intramolecular aggregates, as demonstrated by compound QIR-1 (Scheme 1).89 However, to date, dyes working in this configuration for G4 detection are still limited,109 and further work is needed to unlock their full potential.
Aiming to enhance the drug-like properties of DIE-active probes, a quinazoline-quinazolinone derivative (QQ 4b, Scheme 1) has been developed.61 This molecule not only displays selective fluorogenic light-up responses towards G4s upon disassembly but also shows sequence specificity. Furthermore, this agent has been demonstrated to inhibit DNA polymerase activity through the stabilization of G4 structures.
To date, the majority of dyes active in DIE are known to bind and sense DNA G4s. An exception is the molecule NIC (Scheme 1), which is capable of detecting and monitoring the dynamics of RNA G4s within cells.87
The DIE principle has been leveraged to create a supramolecular platform for the discovery of new G4-ligands. This technique departs from previous methods that rely on G4-induced dye disaggregation, instead using the self-assembly of the cyanine dye IR786 on the G4 template.110IR786 forms bright monomers, but in the presence of G4 structures, it transitions to dimly emissive aggregates, as illustrated in Fig. 5.110 This change in optical properties upon interaction with G4s has led to the development of a turn-on fluorescence displacement assay, specifically designed to probe interactions between G4s and potential ligands. Introduction of a G4-ligand into the IR786:G4 mixture disrupts this aggregation, resulting in increased fluorescence from IR786. This innovative method offers a novel strategy for ligand screening and the identification of compounds that engage with G4 structures.
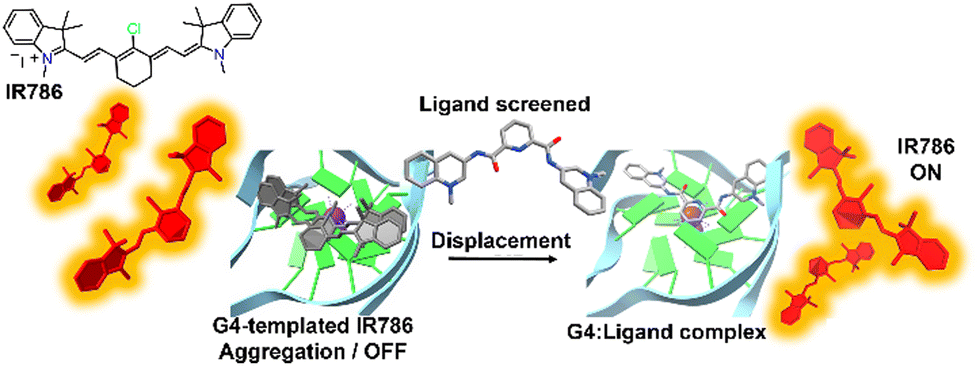 | ||
| Fig. 5 Schematic illustration of the assembly of IR786 to form H-aggregates on DNA G4. The emissive, isolated IR786 binds to G4, forming non-emissive aggregates. Introduction of a high-affinity G4 ligand can displace IR786 from the G4 template, restoring its fluorescence intensity.110 | ||
Recently, a direct comparison of G4 recognition capabilities was conducted between an AIE dye, PZ-1, and a DIE dye, PZ-2 (Scheme 1).97 These two molecules, which differ only in the size of their aromatic regions, demonstrated marked contrasts in performance. The DIE-active probe, PZ-2, significantly outperformed its AIE counterpart, PZ-1, which exhibited minimal changes in emission properties. Specifically, the DIE probe showed approximately 100-fold increase in fluorescence upon binding to G4 structures. This stark difference highlights the superior capability of the DIE-active probe in detecting G4 structures, offering both lower background emission and a more distinct switch-on fluorescence effect compared to its AIE counterpart.
3.2 RNA aptamers-mediated DIE
RNA is essential in numerous biological processes, and to understand its roles, scientists have devised various techniques for visualizing RNA molecules within cells.111 Among these techniques are the fusion of RNA-binding proteins with fluorescent proteins,112,113 and RNA-based fluorogenic modules114 where bulky fluorescent proteins are replaced by smaller fluorogens.115 A cutting-edge approach involves a semi-synthetic strategy that harnesses engineered RNA aptamers.116–119 These aptamers are designed to specifically bind and activate the fluorescence of certain dyes, which are otherwise non-emissive until they interact with the aptamer. RNA aptamers are short, structured nucleotide sequences that can be engineered through a process known as the systematic evolution of ligands by exponential enrichment (SELEX).120,121 This method fine-tunes their ability to recognize specific dyes with remarkable precision, enhancing their affinity for fluorogenic dyes.Despite advancements in this field, the practical application of dye-aptamer systems has been limited by the dyes’ brightness and stability under light.122
In response to this limitation, the Klymchenko group developed a novel dye known as Gemini-561 (G561), which utilizes sulforhodamine B (SRB) linked dyes through an innovative approach based on intramolecular dimerization-caused quenching (DCQ) of fluorescence (Fig. 6(A) and (B)).34 This method allows for the use of lower concentrations of the probe while significantly enhancing the SBR. Through a combination of genetic engineering and selective processes, RNA aptamers were specifically designed to bind this dye, yielding a notable variant named o-Coral.34 This binding interaction, with a dissociation constant of 73 nM, disrupted the aggregated state of G561, triggering a 13-fold increase in fluorescence (Fig. 6(C) and (D)).123o-Coral was capable of forming a bright complex with G561, distinguishing itself by its ability to directly visualize specific RNA molecules in mammalian cells without the need for multiple tagging.34,123 This system represented a significant leap in RNA imaging, offering a powerful tool for observing RNA molecules in their natural cellular context with unprecedented clarity and specificity.
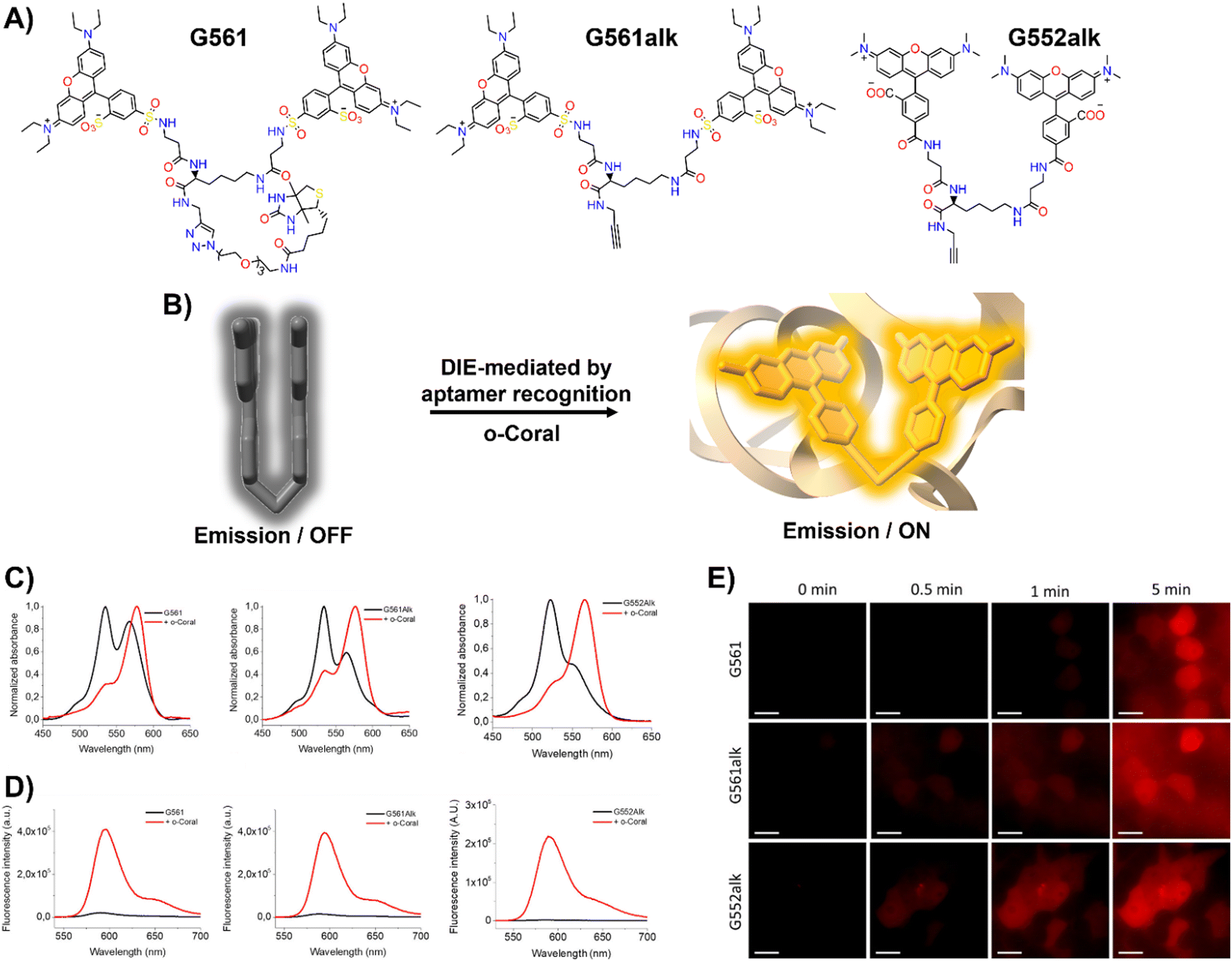 | ||
| Fig. 6 (A) Chemical structures of intramolecularly formed self-assembled probes. (B) Concept of the fluorogenic response of self-quenched dyes upon aptamer binding. (C), (D) Absorption (C) and emission (D) spectra of G561, G561alk and G552alk in the absence and presence of o-Coral. (E) Fast detection of overexpressed o-Coral-tagged U6 RNAs in live cells with the fluorogens. Scale bars are 20 μm. Reprinted with permission from ref. 123. Copyright 2022 American Chemical Society. | ||
Further evaluations of chemical modifications and enhancements to G561 resulted in the development of two dyes: the non-biotinylated Gemini-561-alkyne (G561alk) and its closely related analogue, Gemini-552-alkyne (G552alk) (Fig. 6(A)).123 The latter features a carboxylate group in place of a sulfonate and dimethylamino groups instead of diethylamino. It demonstrated a remarkable 111-fold enhancement in fluorescence and an increased affinity constant of 7.5 nM for o-Coral (Fig. 6(C) and (D)).123 These refinements resulted in even greater sensitivity and brightness in RNA imaging applications, enabling effective detection of o-Coral-tagged RNAs in live cells (Fig. 6(E)).123
Parallel to these developments, Xu and colleagues reported on a naphthalimide-based fluorophore, Nap, which self-assembled into nanoparticles for RNA imaging (Fig. 7(A)).124 By binding with a guanine-rich RNA aptamer, NapRA, Nap's fluorescence was enhanced, enabling the visualization of mCherry mRNA in E. coli. Integrating NapRA into mCherry's 3′-untranslated region did not affect gene expression or protein functionality but allowed for the selective illumination of cells expressing mCherry-NapRA (Fig. 7(B)–(D)).124 The study also utilized structured illumination microscopy (SIM) for super-resolution imaging, pinpointing mRNA locations within bacterial cells, particularly around the nucleoid area (Fig. 7(E)–(H)).124 This aligned with findings on mRNA distribution and suggested that newly transcribed mRNA first appeared near the nucleoid, potentially moving to the periphery later.
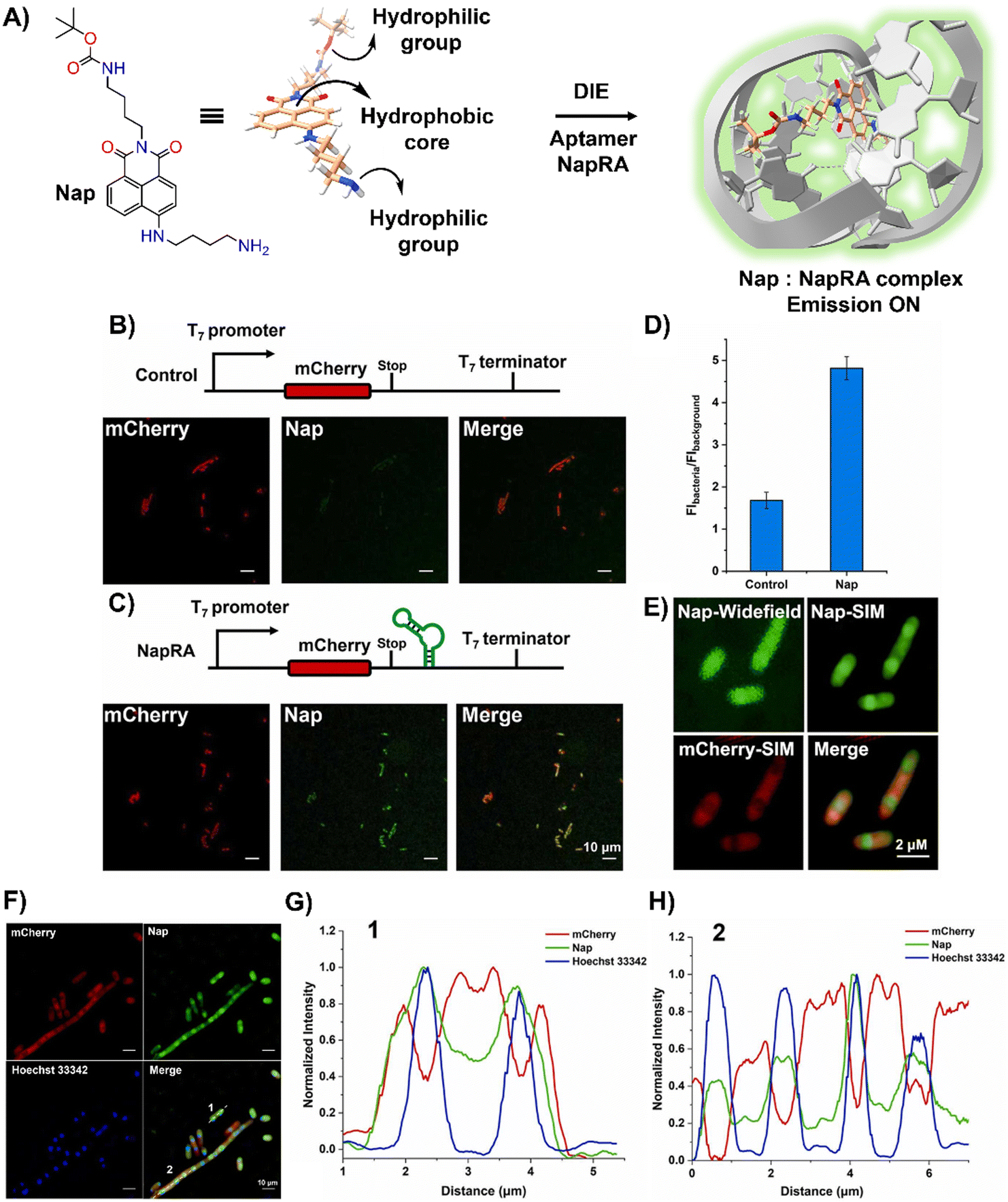 | ||
| Fig. 7 (A) Schematic illustration of the strategy based on the aggregation–disaggregation of Nap for RNA labelling. (B) Expression scheme of control mCherry mRNA and no-wash confocal imaging in bacteria using Nap. (C) Expression scheme of mCherry-NapRA mRNA and no-wash confocal imaging with Nap. (D) Ratio of Nap fluorescence intensity in bacteria expressing mCherry-NapRA mRNA compared to the control. (E) Widefield and SIM imaging of E. coli expressing mCherry-NapRA mRNA treated with Nap. (F) SIM imaging of E. coli expressing mCherry-NapRA mRNA treated with Nap and Hoechst 33342. (G) Normalized intensity profile along dotted line 1 in (F). (H) Normalized intensity profile along dotted line 2 in (F). Scale bar: 10 μm. Reprinted with permission from ref. 124. Copyright 2023 Elsevier. | ||
3.3 DNA-mediated disaggregation in the context of phototherapeutics
DNA-driven molecular disassembly has been utilized to impart stimuli-responsiveness to materials with phototherapeutic and chemotherapeutic properties. Specifically, ACQ effects are widely recognized for their significant limitations on activating dyes, such as in PDT for the generation of reactive oxygen species (ROS).15,125In this field, Yoon and colleagues showcased the spontaneous formation of complexes between the water-soluble photosensitizer (PS), zinc(II) phthalocyanine tetrasubstituted with 6,8-disulfonate-2-naphthyloxy groups (PcS), and the renowned anticancer drug, mitoxantrone (MA), leading to the creation of uniform PcS-MA-based nanoparticles (Fig. 8(A)).126
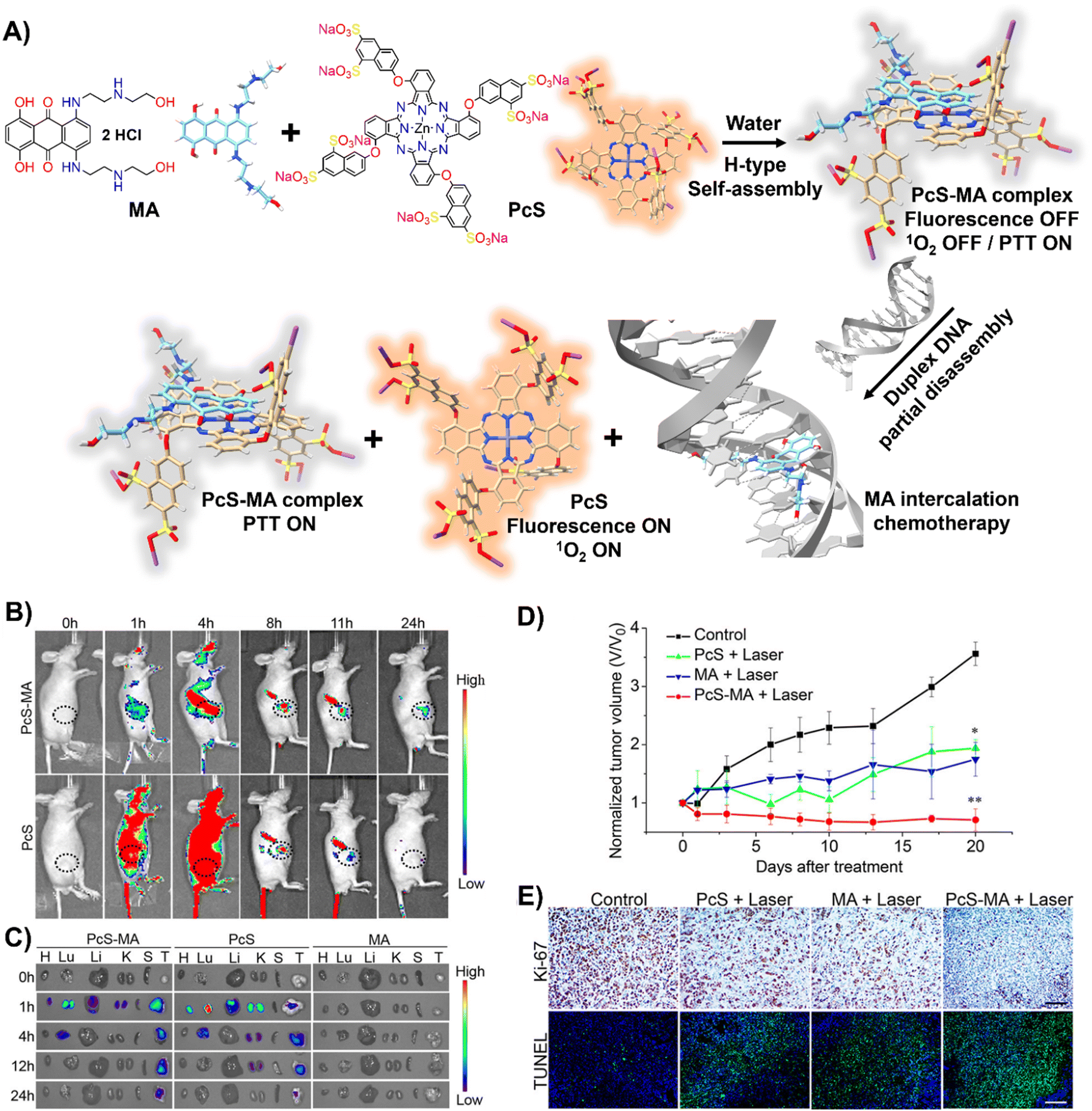 | ||
| Fig. 8 (A) Schematic illustration of a nanotheranostic agent constructed via supramolecular interactions between PcS and MA, featuring nucleic-acid-driven activatable properties for enhanced fluorescent imaging, PDT, PTT, and synergized chemotherapy. (B) and (C) In vivo and ex vivo fluorescence imaging of tumour-bearing mice before and after intravenous injection of PcS-MA, PcS, and MA. (B) MCF7 tumour model: tumour sites are highlighted with dotted circles. (C) SW620 tumour model: labels—H for heart, Lu for lung, Li for liver, K for kidney, S for spleen, T for tumour. (D) Phototherapeutic efficacy of PcS, MA, and PcS-MA in mice with MCF7 tumours. (E) Histological examination of MCF7 tumours on day 21 post-treatment. Proliferation detected with Ki-67 antibody (brown signals), and nuclei counterstained with hematoxylin. Apoptosis was assessed by TUNEL assay (positive signals in green), with nuclei stained by DAPI (blue). Scale bars: 50 μm for Ki-67 images, 100 μm for TUNEL images. Reprinted with permission from ref. 126. Copyright 2018 American Chemical Society. | ||
These nanoparticles formed H-type non-emissive aggregates, which, while unable to generate singlet oxygen (1O2), retained their intrinsic photothermal capabilities (PTT). The introduction of duplex DNA into the binary system facilitated the intercalation of MA, driving the disassembly of the PcS-MA complex and restoring the fluorescence intensity and the photosensitization efficiency of PcS. The absence of interference from metal ions and proteins underscored the system's specificity for DNA recognition. In vivo and ex vivo studies showed that PcS-MA nanoparticles accumulated efficiently in tumour tissues of mice, and laser irradiation significantly reduced tumour growth and induced apoptosis (Fig. 8(B)–(E)).
G4 DNA and RNA structures, characterized by their guanine-rich regions, are particularly susceptible to oxidation due to the electron-rich nature of guanine, which is the most oxidation-prone of the DNA bases (Fig. 9(A)).127–129 This susceptibility is heightened in G4 structures, where the guanine bases are π-stacked, effectively lowering their ionization potential and rendering them more vulnerable to oxidative processes. As a result, G4 structures are acutely responsive to ROS, making them focal points for PSs.130–134 Recent studies have shown that self-assembled inactive PSs, upon binding to G4 structures, can disassemble and catalyse the production of ROS, potentially causing DNA damage (Fig. 9(B)).135,136
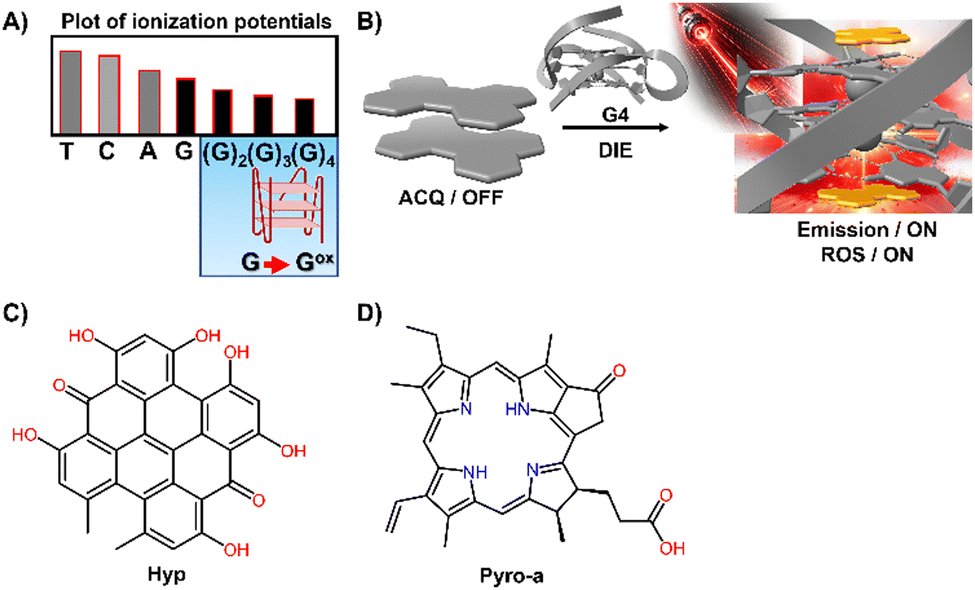 | ||
| Fig. 9 (A) Plot of ionization potentials showing that guanine has the lowest potential, making guanine-rich sequences particularly susceptible to oxidation. (B) Schematic representation showing how a G4 mediates the disassembly of ACQ-active dyes, restoring their fluorescence properties and facilitating the generation of ROS. (C) and (D) Chemical structures of Hyp and Pyro-a. | ||
For example, the interaction of DNA G4s with Hypericin (Hyp), a polycyclic quinone extracted from Hypericum possessing antiviral and anticancer activities, has been shown to amplify its photosensitizing effects upon DIE (Fig. 9(C)).135 The efficiency of 1O2 quantum yield (ΦΔ) in the complex formed with the c-KIT G4 structure in water was notably high at 0.67, underscoring the significance of these interactions in a biologically relevant medium.
Similarly, Pyropheophorbide a (Pyro-a), a derivative of chlorophyll known for its efficacy as a PS, demonstrated increased fluorescence and photosensitizing efficiency upon binding to parallel G4 structures (Fig. 9(D)).136 These observations highlight the critical role of preventing dye aggregation to boost the biosensing capabilities and phototherapeutic applications of ACQ dyes and other inactive materials.
Although this research area is still in its infancy, the strategic targeting of cancer-associated molecules with such innovative approaches heralds promising developments in phototherapy.
3.4 Serum proteins-mediated DIE
Human serum albumin (HSA) is an important thiol-containing protein found in blood and urine, with plasma concentrations typically ranging from 35 to 50 g L−1, and urine levels under 30 mg L−1.137,138 HSA is essential for regulating plasma colloidal osmotic pressure and facilitating the transport of various substances.139,140 Notably, HSA plays a significant role in tumour nutrition, possibly due to the overexpression of albumin receptors and albumin-binding proteins on cancer cells, which promotes albumin accumulation and degradation in tumour tissues.141 The rapid diagnosis of changes in HSA concentrations in body fluids is thus of clinical importance.142,143 Researchers have explored the DIE concept, leveraging albumin's capacity to bind hydrophobic self-assembled molecules and convert them into highly monomeric species. To date, compounds like squaraine,140,142,144 BODIPY,145–149 coumarin,138,150 chalcone,151 and cyanine152,153 have been used for both qualitative and quantitative biosensing of serum albumin proteins (SAP) in vitro and in vivo.Most studies apply the DIE concept directly to target SAP in blood samples,154 using noncovalent interactions between ligand aggregates and albumin (Fig. 10(A)). This approach utilizes molecules with enhanced hydrophobic surfaces to improve aggregate stability in protic solvents through π–π and C–H⋯π interactions, enhancing their interaction with the protein's hydrophobic cavities.153–155 The molecules have been specifically designed to exhibit selectivity towards HSA over other biomolecules156 like DNA, RNA, pepsin, and trypsin, with detection limits typically ranging from 1 to 140 nM.154,157 Furthermore, site-specific binding probes not only detect HSA in bodily fluids but also monitor changes in the protein's conformation and enable fluorescence imaging of SAP in living cells, demonstrating the versatility of HSA probes.154,157
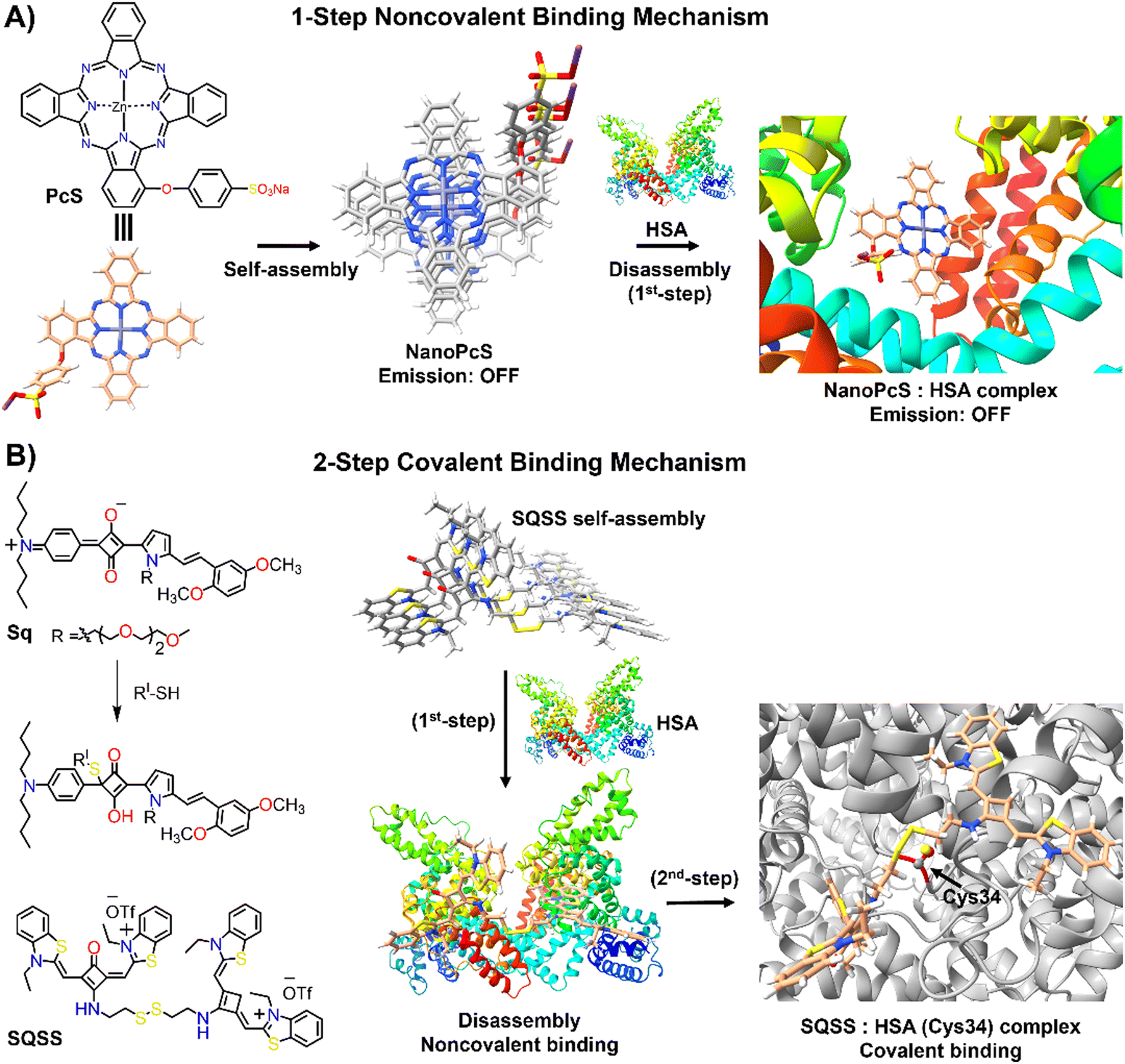 | ||
| Fig. 10 Schematic representation of the molecular recognition mechanisms mediated by DIE-active dyes for detecting HSA. Panel (A) shows the noncovalent binding approach, exemplified by the molecule PcS,158 used to sense HSA. The self-assembled structure of PcS gradually disassembles in the presence of HSA through a one-step mechanism. Panel (B) depicts the covalent binding approach, illustrated by the molecules Sq142 and SQSS.140 In an aqueous solution, SQSS molecules self-assemble. Upon encountering HSA, a two-step binding process begins with an initial noncovalent binding mechanism, which is then followed by a selective covalent binding. This second step is driven by a thiol-sensitive motif and the Cys34 residue of HSA. | ||
Yoon and colleagues demonstrated the specific binding of an exogenous probe, composed of zinc(II) phthalocyanine mono-α-substituted with 4-sulfonatophenoxyl (PcS), to albumin in vivo (Fig. 10(A)).158 This was accomplished using a transgenic mouse system, where albumin was tagged with yellow fluorescent protein (YFP) through intraperitoneal injections of tamoxifen. PcS aggregates were administered intravenously, and plasma analysis one hour post-treatment showed that the aggregates were trapped and disassembled by albumin. Confocal imaging of liver and lung sections from treated mice revealed overlapping fluorescence between PcS and albumin-YFP. This method provides a valuable technique for tracking specific proteins in vivo systems.
Another strategy involves forming a covalent bond between the squaraine-based probes (Sq142 or SQSS140) and the serum protein (Fig. 10(B)). In this method, the probe is engineered with a thiol-sensitive motif, enabling it to react with the cysteine (Cys) residues of SAP. The interaction proceeds through a two-step mechanism: initially, compact probe assemblies loosen via noncovalent interactions, followed by gradual disassembly driven by the presence of Cys34 in SA through covalent binding. This results in a newly formed chromophore that emits a strong fluorescent signal with high selectivity towards SAP over other thiol-containing proteins, achieving detection limits as low as 0.53 nM.140 It is noteworthy that the covalent binding to Cys34 may involve a base-catalysed nucleophilic addition, which restricts its practical use to basic conditions (pH ∼ 8).142
Laboratory testing is generally required to detect HSA. However, there is a critical need for point-of-care (POC) monitoring of HSA, particularly for elderly or chronically ill patients, as low levels of HSA—a condition known as hypoalbuminemia—indicate disease states such as hepatic impairment, chronic kidney disease, protein-losing enteropathy, burns, and sepsis.159,160 In response, Liu and colleagues designed a 2′-hydroxychalcone derivative named 4MC, capable of detecting HSA through the DIE process (Fig. 11(A)).1514MC formed excimer-emissive aggregates that disassembled into highly emissive monomeric forms in the presence of HSA. The concentration of HSA was determined by the emission ratio of aggregated to monomeric probes, resulting in visually distinct red and green colour changes (Fig. 11(B) and (C)). As a proof of concept, a microfluidic paper-based analytical device (μPAD) was integrated with 4MC to create a POC device for HSA detection in whole blood. This μPAD featured an outer layer of plasma separation membrane and a detection pad constructed from wax-patterned filter paper impregnated with 4MC. Demonstrations showed that such a device could be effectively used at home, with HSA levels assessable using a UV lamp for excitation and a smartphone for detection (Fig. 11(D)–(G)). The glowing spots on the paper device could be captured by the smartphone's camera and directly analysed using a dedicated app to provide RGB values.151
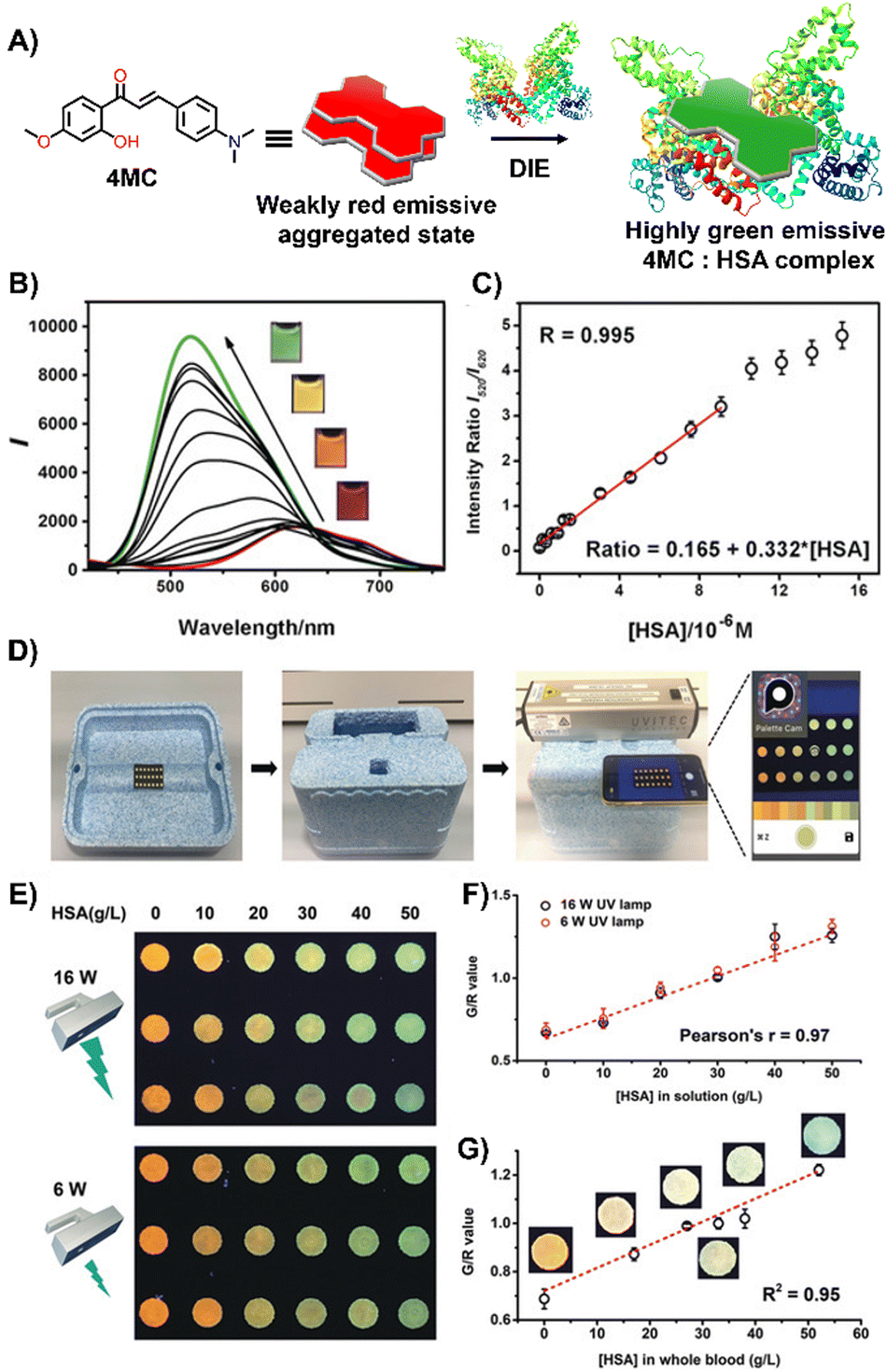 | ||
| Fig. 11 (A) Chemical structure and ratiometric detection method of 4MC for HSA. (B) and (C) Fluorescent spectra (B) and the intensity ratios (I520/I620, monomer/aggregate) (C) of 4MC in response to increasing concentrations of HSA in water. The inset shows photographs under UV irradiation (365 nm) with varying concentrations of HSA. (D) Setup of the mobile-phone-based testing system. (E) Photograph of test paper used to analyse aqueous HSA solutions at increasing concentrations, excited by different handheld UV lamps (16 W and 6 W power, 365 nm). (F) and (G) Calibration curves for aqueous HSA solutions (F) and whole blood samples (G), respectively. Reprinted with permission from ref. 151. Copyright 2020 Wiley-VCH. | ||
3.5 Membrane proteins-mediated DIE
Membrane proteins, which are associated with or attached to cellular membranes or within cell compartments or organelles, play pivotal roles in functions such as molecule transport, cell signalling, cell adhesion, and pathogen recognition.161,162 In this context, the development of protein-targeting probes that enable real-time, highly selective imaging of protein dynamics is of significant importance for diagnosing various diseases, including cancer.163,164A variety of dyes sensitive to protein membranes have been identified.161,165–167 One of the pioneering efforts in this area was led by Hamachi and colleagues, who developed a self-assembled supramolecular ligand-tethered probe (1, Scheme 2) to visualize folate receptors in KB cells, observing robust fluorescence on the cell surface.168
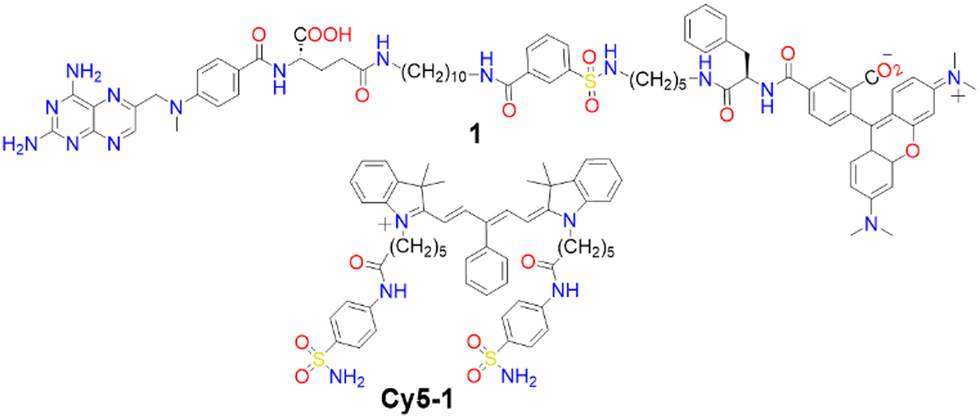 | ||
| Scheme 2 Chemical structures of compound 1 and Cy5-1. | ||
Subsequently, Tan and co-workers designed a near-IR fluorogenic Cy5 probe with a benzenesulfonamide ligand (Cy5-1, Scheme 2) targeting human carbonic anhydrase (hCA).169 In PBS buffer, the probe forms a self-assembled aggregate with a ΦF of 0.019. The presence of hCAII protein triggers disassembly, resulting in an increased ΦF of approximately 0.28, with a LOD of 17 nM. Cy5-1 was used to image HeLa cells known for overexpressing hCAIX on the extracellular surface under hypoxic conditions. Live-cell imaging with CLSM revealed intense surface fluorescence, illustrating Cy5-1's capability to detect changes in the hypoxic environment of cancer cells.
To enhance the drug-like properties of DIE-active probes, Nesterov and colleagues developed phthalocyanine-based sensors specific for the epidermal growth factor receptor (EGFR), with exceptionally low LODs of 3 nM165 and 2 nM.167 These probes selectively label EGFR tyrosine kinase inside cells, making them suitable for selective fluorescent imaging of the protein in both fixed and live cells.167
Stimuli-responsive dyes have also been employed for imaging protein membranes, using the DCQ mechanism.162–164,170 This strategy, pioneered by Klymchenko's research team, was used to image the oxytocin receptor,64,162 the biotin receptor,163 and αvβ3 integrin170 in both in vitro settings and live cells. The successful cellular visualization of membrane proteins with specially designed probes prompted Klymchenko and Bonnet to explore this approach further for visualizing the naturally expressed oxytocin receptor in living mice using a specially designed probe, Cy5.5 decorated with a PEG linker and the peptidic carbetocin (CBT) ligand (dCy5.5-PEG–CBT, Fig. 12(A) and (B)).64 The probe was intravenously administered to lactating Swiss mice, and imaging showed visualization of the mammary glands and liver with specificity superior to that of the monomeric probe mCy5.5-PEG-CBT (Fig. 12(C)–(F)).64
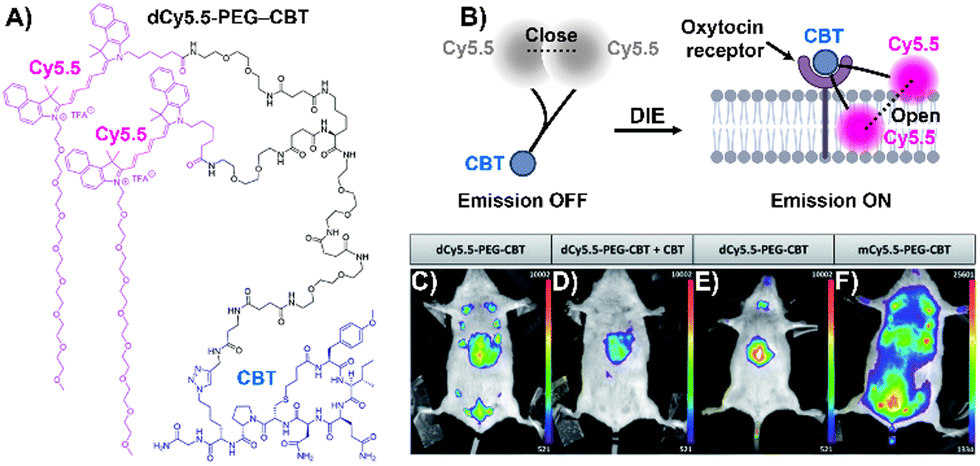 | ||
| Fig. 12 (A) Chemical structure of dCy5.5-PEG-CBT. (B) Recognition mechanism mediated by dCy5.5-PEG-CBT. (C)–(F) In vivo images of lactating mice (C), (D), and (F) and a naïve mouse (E) injected intravenously via the tail with 7.5 nmol of dCy5.5-PEG-CBT (C) and (E), 7.5 nmol of dCy5.5-PEG-CBT along with 450 nmol of CBT (D), or 7.5 nmol of mCy5.5-PEG-CBT (F), taken 30 minutes prior to imaging. Reproduced from ref. 64 with permission from the Royal Society of Chemistry. | ||
In light of these findings, Qian and colleagues implemented the DCQ strategy for real-time imaging of the EGFR receptor with antibody-based fluorogenic probes.171 Cellular studies on A431 (EGFR+) and HepG2 (EGFR−) cells showed intense fluorescence on the former, indicating excellent selectivity. Live-cell imaging demonstrated intracellular fluorescence signifying internalization of the antibody–EGFR complex. Furthermore, fluorescence-activated cell sorting allowed for quantitative analysis of endogenous EGFR expression levels in A431 cells, revealing a linear relationship between fluorescence intensity and probe concentration.171
3.6 Proteins-mediated disaggregation in the context of phototherapeutics
Molecular probes that possess both DIE and photosensitizing properties, and that target overexpressed or cancer-promoting proteins, represent a novel approach in activatable PDT to enhance treatment selectivity.103,104,172,173 This section discusses three distinct strategies that have been employed so far (Fig. 13).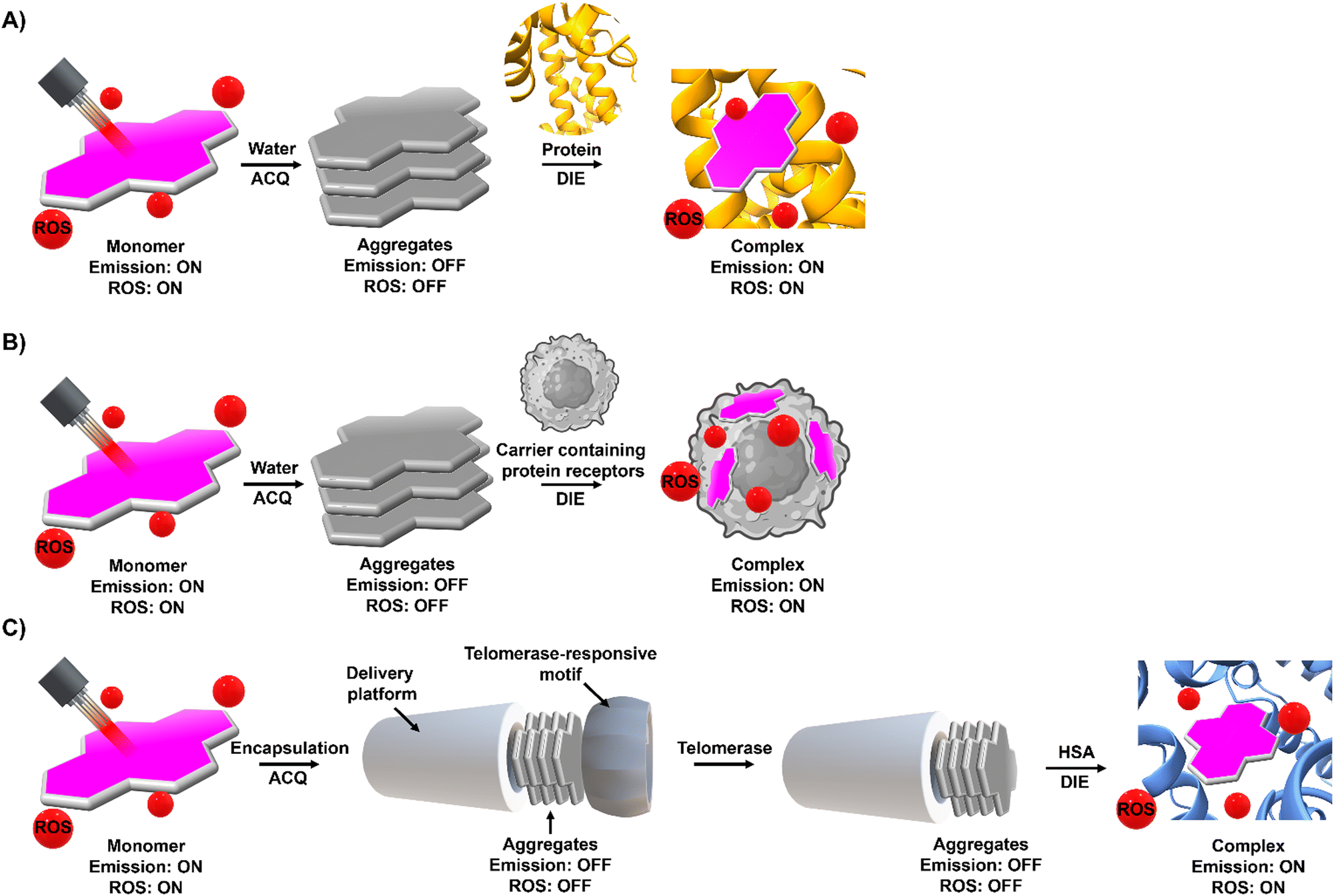 | ||
| Fig. 13 Phototherapeutic strategies used by DIE-activated PSs coordinated with proteins. (A) In its monomeric form, typically found in organic solvents, the PS is photoactive. However, transitioning to an aqueous environment leads to the formation of molecular aggregates, causing a loss of photosensitization capability. Binding to a protein can restore the PS to a monomer-like state, reactivating its ROS production. (B) Similarly to (A), in an aqueous solution, the PS forms molecular aggregates that inhibit its photosensitization properties. Encapsulating the PS in a biological matrix containing proteins disperses the PS into a monomeric form, thereby restoring its ROS production capability. Once encapsulated within the matrix, the PS can be utilized as a delivery system. (C) The PS is encapsulated within an artificial delivery platform that initially quenches its photosensitizing properties, further secured by a protein-responsive motif, such as a DNA sequence. Interaction with specific triggers, in this case telomerase and HSA, leads to the elongation of the DNA sequence and the opening of the delivery platform. This allows direct interaction with HSA, forming a well-defined complex with the PS that restores its monomeric form and ROS production capability. | ||
The first strategy involves the direct disaggregation of the PS in the presence of targeted proteins which leads to increased ΦF and ΦΔ (Fig. 13(A)).102,158,173–176 The structural modification of PSs with specific anchor groups allows targeting of particular proteins. For example, using the folate receptor enhances the cellular uptake of folate-porphysome into cancer cells.174 When combined with laser treatment, this method partially inhibited tumour growth.174 Additionally, developed phthalocyanine derivatives, modified with anchor groups such as biotin (Pc-4TEG-B,173PC-VH175) to target the biotin receptor or sulfonate derivatives (PcS,158PcS-4177) to target albumin, were administered to tumour-bearing mice (Scheme 3). Light irradiation led to approximately 40% tumor growth inhibition with Pc-4TEG-B,173 55% or 85% with PcS-4,177PC-VH175 respectively, and even complete inhibition and reduction in tumour volume with PcS158 treatment.
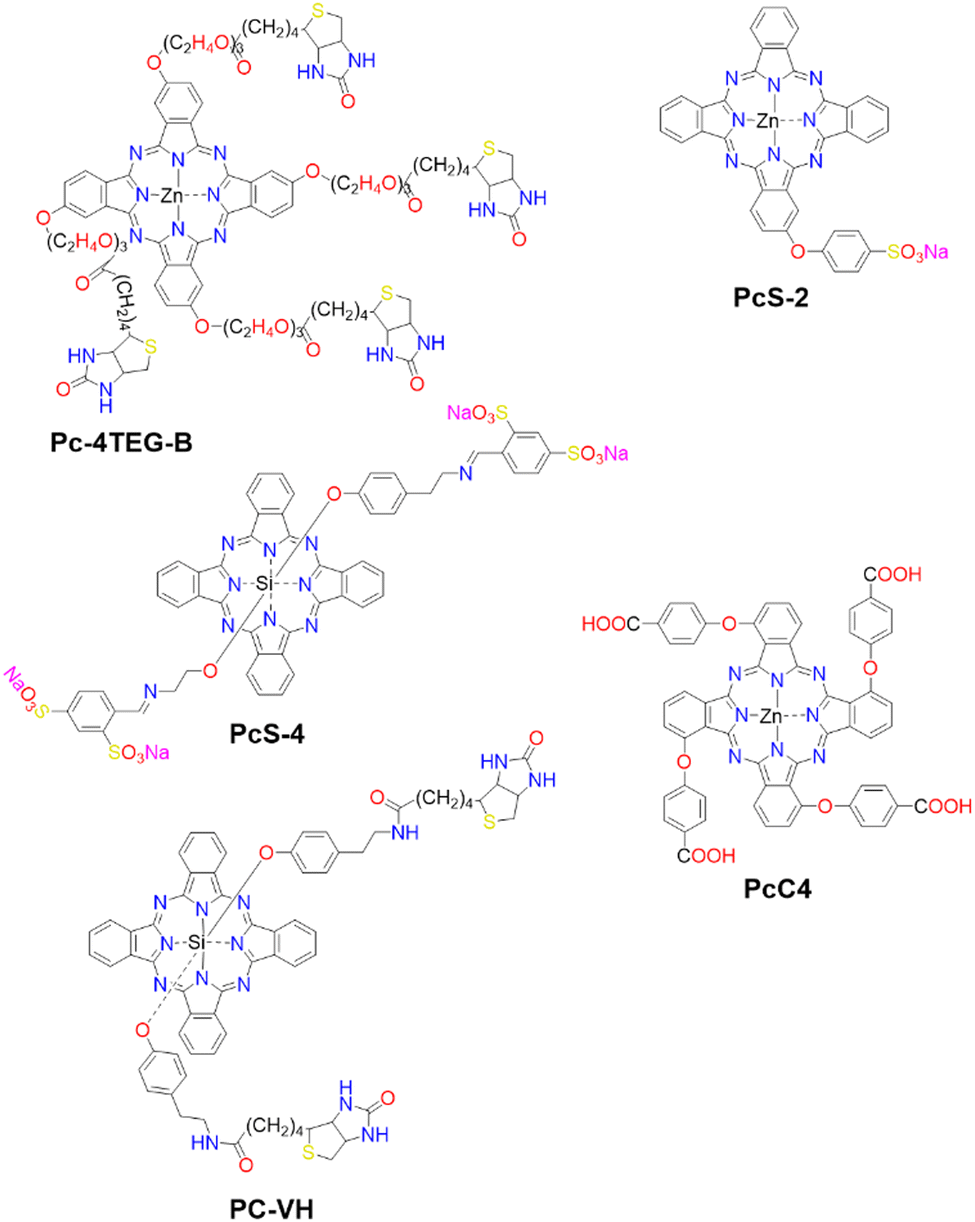 | ||
| Scheme 3 Molecular Structures of PSs employing various mechanisms: Pc-4TEG-B, PcS-4, and PC-VG function through noncovalent interactions (refer to Fig. 13(A)). PcS-2 is embedded within a cell-based drug carrier (see Fig. 13(B)), and PcC4 is encapsulated in a drug delivery platform responsive to two protein triggers (see Fig. 13(C)). | ||
The second strategy utilizes a drug carrier system to deliver the ligand to specific sites (Fig. 13(B)). Yoon and colleagues designed a system using mesenchymal stem cells (MSCs), which naturally possess tumour tropism mediated by growth factors,178 combined with a phthalocyanine derivative (PcS-2,179Scheme 3) that forms H-type aggregates in water. After internalization of PcS-2 by MSCs and subsequent spontaneous disassembly of the aggregates in the presence of albumin, the complex was intraperitoneally administered to HCT116 tumour-bearing mice.179 This resulted in targeted accumulation in the tumour compared to the broader distribution of PcS-2 alone in the abdomen. Irradiation with a 655 nm laser triggered the PDT mechanism, resulting in a 37% inhibition of tumour growth in mice treated with MSC-PcS-2.179
The third strategy aims to enhance tumour specificity and minimize side effects by targeting two cancer-related proteins (Fig. 13(C)).180 This approach involved a sequentially activated, protein-responsive PS, based on a zinc(II) phthalocyanine derivative (PcC4, Scheme 3) entrapped in mesoporous silica nanoparticles (MSNs), with wrapping DNA (O1) serving as a biogate.180 The PcC4-MSN-O1 system exhibited self-quenching photoactivity, which was dramatically activated in sequence by telomerase and then albumin. Cellular studies demonstrated selective phototoxicity towards cancer cells with overexpressed telomerase.180 Furthermore, the administration of PcC4-MSN-O1 with light irradiation to HeLa tumour-bearing mice led to the inhibition of tumour growth, without showing any pathological changes in the mouse tissues, thereby demonstrating both the effectiveness and biocompatibility of this approach.180
3.7 Lipids-mediated DIE
Lipids, encompassing fatty acids, glycerolipids, glycerophospholipids, sphingolipids, and sterols, are vital for various cellular functions and as disease indicators for conditions like tumours and cardiovascular issues.181–187 Fatty acids are essential for energy, cell structure, and signalling;188 sphingolipids and glycerophospholipids for membrane structure;189,190 and steroids, such as progesterone and cortisol, for regulating reproduction191 and immunity.192 Despite their significance, the efficient detection and imaging of lipids and steroids in biological settings continue to be a challenge.193 Up to now, several DIE-active dyes sensitive to lipids have been reported, with some examples highlighted in this and the following sections.29,35,63,194–202Jiang and colleagues developed a molecular probe, IR1061, for in vivo imaging of lipids in fatty liver diseases,203 leveraging the DIE process.194 In organic solvents, IR1061 emitted brightly in the NIR-II region but remained non-fluorescent in aqueous environments due to ACQ (Fig. 14(A)). The probe's fluorescence was activated in PBS with DSPE-PEG2000 liposomes, indicating disaggregation and maintaining stability and specificity under various conditions.194In vivo experiments using atherosclerosis (AS) models (apolipoprotein E knock-out, ApoE−/− mice on a high-fat diet) and healthy C57bl/6 controls showed that only AS mice exhibited a significant, time-dependent increase in liver fluorescence, unlike other organs or controls (Fig. 14(B) and (C)).194 A similar pattern was observed in an obesity (OB) model, where IR1061 specifically highlighted lipid accumulation in the livers of OB mice (Fig. 14(D) and (E)).194 These findings demonstrated IR1061's potential as a DIE-based tool for the specific imaging of fatty liver conditions in AS and OB mouse models.
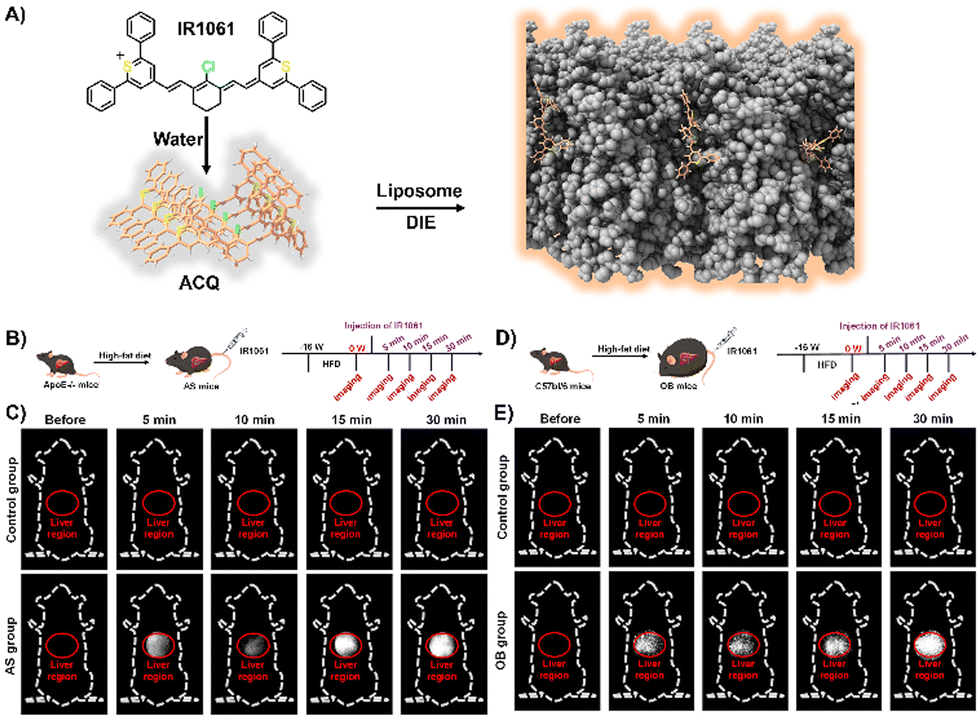 | ||
| Fig. 14 (A) Schematic illustration depicting the use of lipid-activated IR1061 for imaging in AS and OB mouse models. (B) Development of the AS model. (C) Fluorescence imaging comparing the control and AS groups before and after IR1061 injection. (D) Development of the OB model. (E) Fluorescence imaging of the control and OB groups before and after IR1061 injection. Reprinted with permission from ref. 194. Copyright 2024 American Chemical Society. | ||
AS is the leading cause of cardiovascular diseases worldwide,204,205 characterized by the buildup of lipid-rich plaques in arteries that can lead to heart attacks and strokes.206 Cathepsin B (CTB), an enzyme present in macrophages, is crucial in plaque formation and acts as a key biomarker for identifying vulnerable plaques and assessing cardiovascular risk.207–210 However, the use of molecular imaging to track CTB faced challenges in probe penetration and specificity, limiting the identification of high-risk plaques. Given that atherosclerotic plaques largely consist of foam cells derived from macrophages,211 targeting lipid levels could improve the differentiation of atherosclerotic from normal tissues.212–214 This highlighted the need for more accurate and effective methods to measure CTB activity in plaques to better manage cardiovascular risk.
Zhang and colleagues created a CTB-responsive, lipid-sensitive probe, L-CRP, for precise imaging of CTB in atherosclerotic plaques.195L-CRP featured a hydrophilic CTB-responsive dipeptide, a lipophilic alkyl chain for nanoparticle formation, and a caged hemicyanine structure that prevented charge transfer until CTB cleaved the dipeptide. This cleavage triggered monomer formation in lipid-rich environments, enhancing photoacoustic (PA) and fluorescence signals (Fig. 15(A)).195 Unlike the control probe CRP, L-CRP specifically activated in the presence of both CTB and lipids, avoiding false positives from other molecules. Its design enabled L-CRP to target CTB activity within foam cells rich in lipids, distinguishing them from other cell types like M1 macrophages (Fig. 15(B)–(D)). In vivo experiments with atherosclerosis-induced ApoE−/− mice showed L-CRP effectively highlighting atherosclerotic lesions, confirmed by various staining methods, and differentiated them from healthy tissues.195 This specificity, demonstrated in human artery tissues (Fig. 15(E)–(H)), underscored L-CRP's potential for clinical use in identifying atherosclerotic enzymatic activity, offering a new avenue for cardiovascular disease management.195
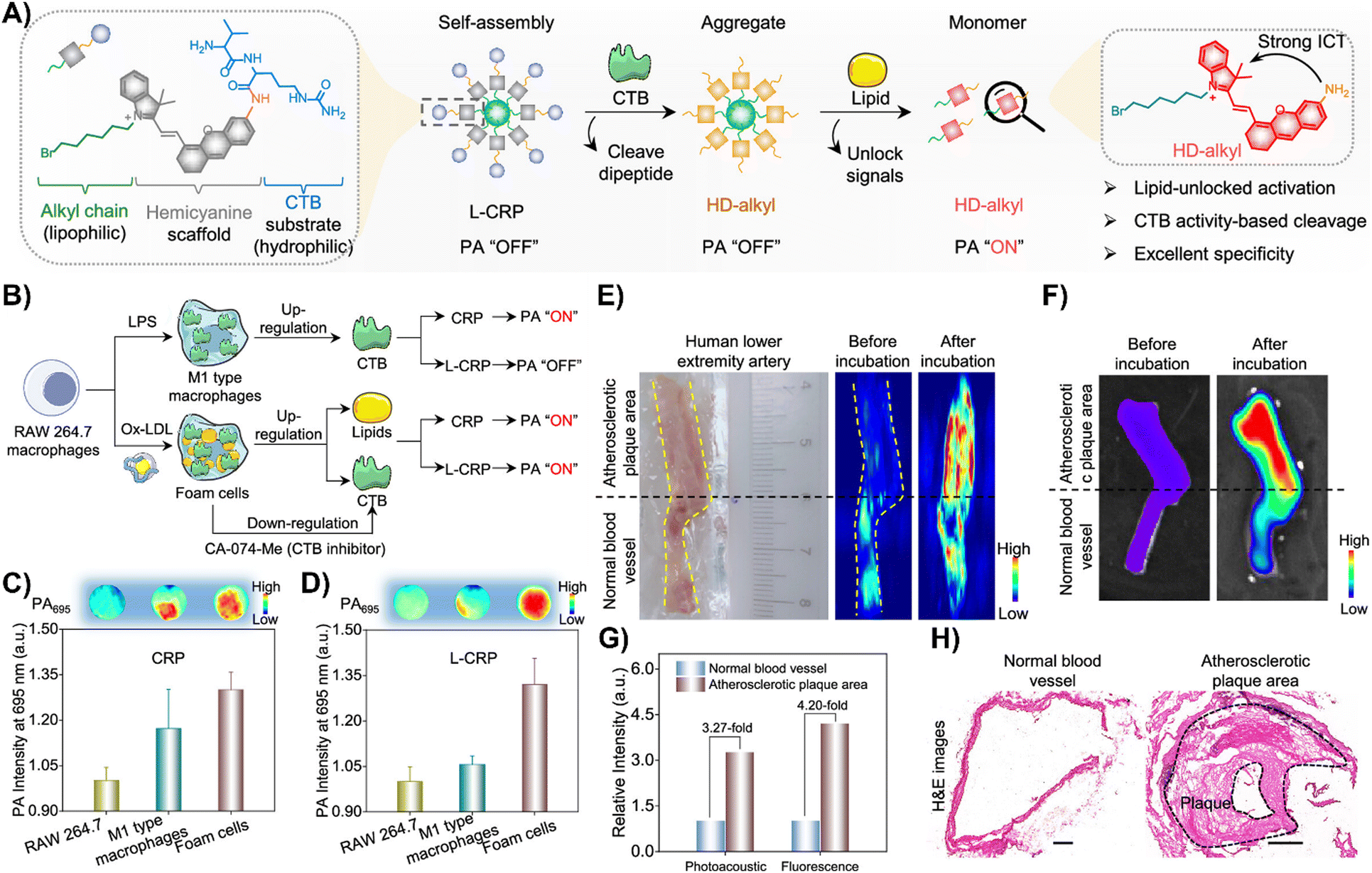 | ||
| Fig. 15 (A) Chemical structure and activation mechanism of the lipid-unlocked CTB-responsive probe (L-CRP): composed of a hydrophilic CTB-responsive dipeptide, a lipophilic alkyl chain, and a caged hemicyanine unit, L-CRP aggregates in hydrophilic environments both before and after CTB incubation. In lipophilic environments, CTB recognition triggers the release of HD-alkyl as a monomer. (B) Construction of M1-type macrophages and foam cells: LPS induces M1-type macrophages with increased CTB expression. Ox-LDL causes foam cells to upregulate lipids and CTB, while the CTB inhibitor CA-074-Me decreases CTB levels in foam cells. (C) and (D) Photoacoustic intensity of CRP and L-CRP after various treatments, with insets showing PA images at 695 nm. (E) Bright field and PA imaging of a human lower extremity artery before and after L-CRP incubation in CTB buffer. The yellow dotted curves outline the blood vessel; the black dotted line marks the boundary between atherosclerotic plaque (above) and normal vessel (below). (F) Fluorescence imaging of the human lower extremity artery pre and post L-CRP incubation. (G) Quantitative analysis of PA and fluorescence intensities from panels (E) and (F). (H) Hematoxylin and eosin (H&E) staining of normal and atherosclerotic areas of the blood vessel. Scale bar = 1000 μm. Reprinted with permission from ref. 195. Copyright 2023 American Chemical Society. | ||
3.8 Plasma membranes-mediated DIE
Effective plasma membrane (PM) staining is essential for scientific studies like cell identification, translocation assays, and exploring PM dynamics, given its vital role in processes like cell division, endocytosis, and signal transduction.215–218 The PM's boundary function enables the development of DIE-active probes that transition from non-fluorescent in cell media to fluorescent upon membrane contact.35,63,196–198,219 This approach minimizes background noise and facilitates real-time, selective imaging of the membrane bilayer.The Klymchenko group had developed a BODIPY-based dye, B-2AZ196 also referred as MemBright-488,196 along with six cyanine-based dyes under the MeMBright family,35 which emitted from green to near-infrared (Fig. 16(A) and (B)). These intermolecularly self-assembled probes transition from non-emissive in phosphate buffer (PBS) to bright upon interaction with DOPC vesicles, mimicking the plasma membrane (PM).35,196 They demonstrated high PM specificity, aligning with PM markers and highlighting intercellular nanotubes. The MeMBright probes also excelled in two-photon microscopy, confirming PM selectivity in live cells and ex vivo tissues (Fig. 16(C)).35MB-Cy3.5, tested for photostability, showed exceptional resistance to photobleaching during extensive 3D liver imaging and enhanced neuron identification in brain sections.35 Super-resolution imaging with MB-Cy3.5 detailed dendritic spine morphologies and visualized “en-passant” synapses, proving the probes’ potential in sophisticated imaging applications.35
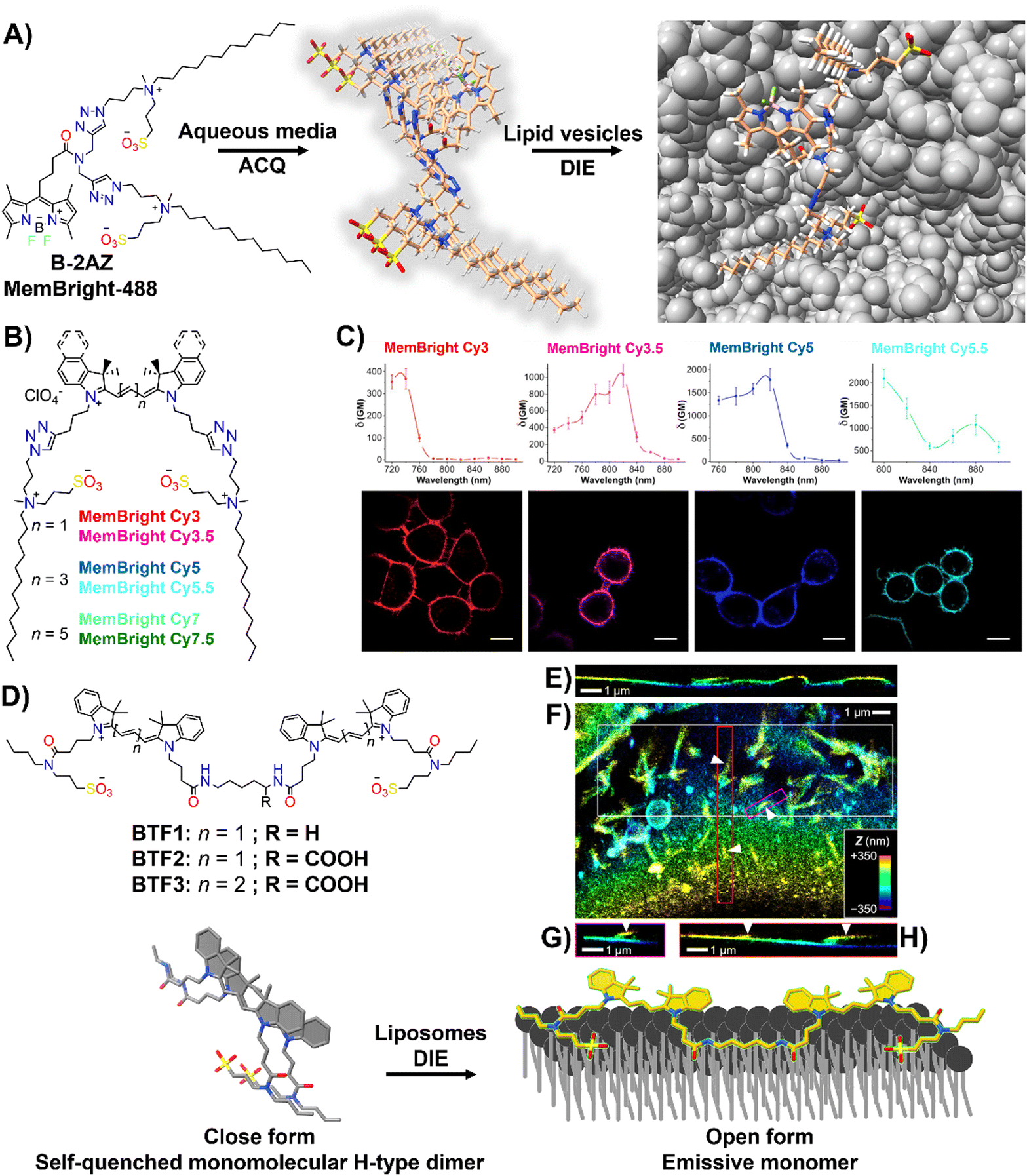 | ||
| Fig. 16 (A) Chemical structure of B-2AZ (MemBright-488), illustrating its propensity for intermolecular aggregation and lipid-vesicle-induced disaggregation. (B) Chemical structures of various MemBright dyes. (C) Graphs of two-photon absorption cross-sections alongside images of KB cells stained with MB-Cy3, MB-Cy3.5, MB-Cy5, and MB-Cy5.5. Reprinted with permission from ref. 35. Copyright 2019 Elsevier. (D) Chemical structures of Cy3-based BTF1 and BTF2, and Cy5-based BTF3 probes, highlighting the concept of a switchable cyanine-based intramolecular dimeric probe that becomes fluorescent upon reversible binding to biomembranes. (E)–(H) 3D-PAINT with BTF2 reveals the intricate 3D structures and dynamics of the plasma membrane in living cells. (E) Presented is a vertical cross-sectional view of the 3D-PAINT data from a COS-7 cell, showcasing varying membrane heights. (F) In-plane view of a different cell, emphasizing numerous nanoscale tubules. (G) and (H) Vertical cross-sectional views through the magenta (G) and red (H) boxes in (F), displaying tubules extending from the cell surface (indicated by arrowheads). Note: in the vertical cross-sectional views in (E), (G), and (H), the vertical dimension indicates depth into the sample. Reprinted with permission from ref. 63. Copyright 2022 American Chemical Society. | ||
The same group also focused on enhancing fluorescent probes for single molecule localization microscopy (SMLM) of cell membranes, addressing challenges in resolution, brightness, and photostability. They introduced intramolecularly formed fluorogenic dimers (BTF1 and BTF2), composed of Cy3 dyes linked by cadaverine or lysine, and featuring membrane anchor sulfonate groups (Fig. 16(D)).63 These dimers were designed to enhance PAINT imaging220 by reducing plasma membrane affinity and facilitating on/off switching (Fig. 16(D)).63 Spectroscopy had shown that these dimers self-quenched in PBS but brightened in organic solvents and with DOPC, indicating effective disassembly for membrane binding. BTF2, with a lower DOPC affinity due to its hydrophilic lysine linker, and the far-red Cy5-based BTF3 variant, were tested in live cell microscopy, demonstrating superior single-molecule fluorescence switching, greater brightness, and enhanced localization precision compared to commercially available PM stains.63BTF2 also allowed 3D imaging of COS-7 fibroblast-like cell membranes, unveiling dynamic membrane reshaping (Fig. 16(E)–(H)). BTF3 further improved super-resolution imaging over monomeric Cy5-based DiD dye, highlighting diverse nanoscopic membrane structures with limited dye movement.63 This advancement introduced dimer-based probes that significantly improved super-resolution imaging of cell membranes by reducing dye diffusion and enhancing imaging resolution and stability.
An alternative method based on DIE to stain the PM was proposed by Xu and colleagues.198 They had developed a series of amide-containing receptors for Zn2+, incorporating a naphthalimide fluorophore and a hydrophobic alkyl chain of varying lengths, designated as ZTRS-alkyl, to anchor to membranes.198 Notably, the dodecyl-substituted probe, ZTRS-C12, formed nanoaggregates in aqueous solution (Fig. 17). In the presence of Zn2+, ZTRS-C12 displayed a 1.5-fold increase in fluorescence. However, when dissolved in a mixed solution of HEPES buffer and CH3CN, serving as a disaggregating solvent, ZTRS-C12 exhibited a significant 13-fold enhancement in fluorescence, demonstrating its ability to sense Zn2+ in its monomeric form.198 In the human colon cancer cell line HT-29, ZTRS-C12, while attached to the exterior of the cell plasma membrane in a monomer-like state, initiated a fluorescence response upon complexing with Zn2+. The fluorescence signal was eliminated upon the addition of EDTA, which displaces zinc from the probe complex. ZTRS-C12 demonstrated exceptional selectivity for Zn2+ over other biologically relevant heavy and transition metal ions.198
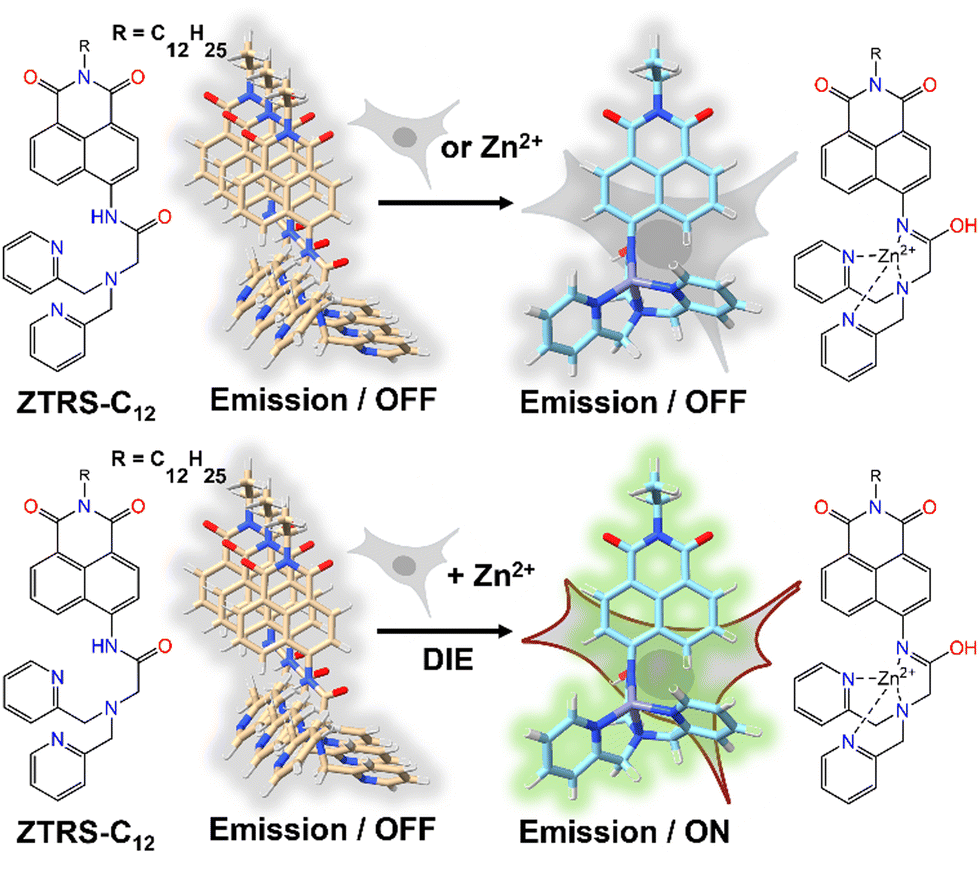 | ||
| Fig. 17 Design of the fluorogenic probe ZTRS-C12 for imaging Zn2+ at the plasma membrane surface of living cells. In its self-assembled state, ZTRS-C12 displayed minimal fluorescence changes when introduced to cells alone or with Zn2+ separately (top panel). However, the simultaneous addition of ZTRS-C12 and Zn2+ to cells triggered its disassembly and activated the fluorescence via complexation with Zn2+ (bottom panel). | ||
3.9 Lipid droplets- and exosomes-mediated DIE
Lipid droplets (LDs), present in organisms from prokaryotes to humans, contain a metabolic lipid core within a phospholipid monolayer membrane.221,222 Beyond serving as lipid storage,223 LDs are integral to processes like membrane trafficking,224 protein degradation,225 inflammation,226 and are implicated in diseases such as obesity and diabetes,186 as well as cancer.227 Given their varied number, size, and composition, imaging and analysing LD dynamics are essential for unravelling their biological significance.The Klymchenko group introduced StatoMerocyanines (SMCy) fluorophores, based on indolenine and dioxaborine barbiturate structures connected by polymethyne chains (Fig. 18(A)).200 These lipophilic dyes, non-emissive in water due to aggregation, exhibited up to 1700-fold fluorescence enhancement in oils (Fig. 18(B)–(D)). When incubated with KB cells, SMCy selectively stained LDs (Fig. 18(E) and (F)). Utilized in multicolor tissue imaging, SMCy dyes effectively highlighted lipid structures in mouse adipose tissue and liver, demonstrating their ability for 3D visualization and tracking of lipid-rich vesicles and LD exchange between cells, proving their suitability for advanced imaging techniques like two-photon microscopy (Fig. 18(G)).200
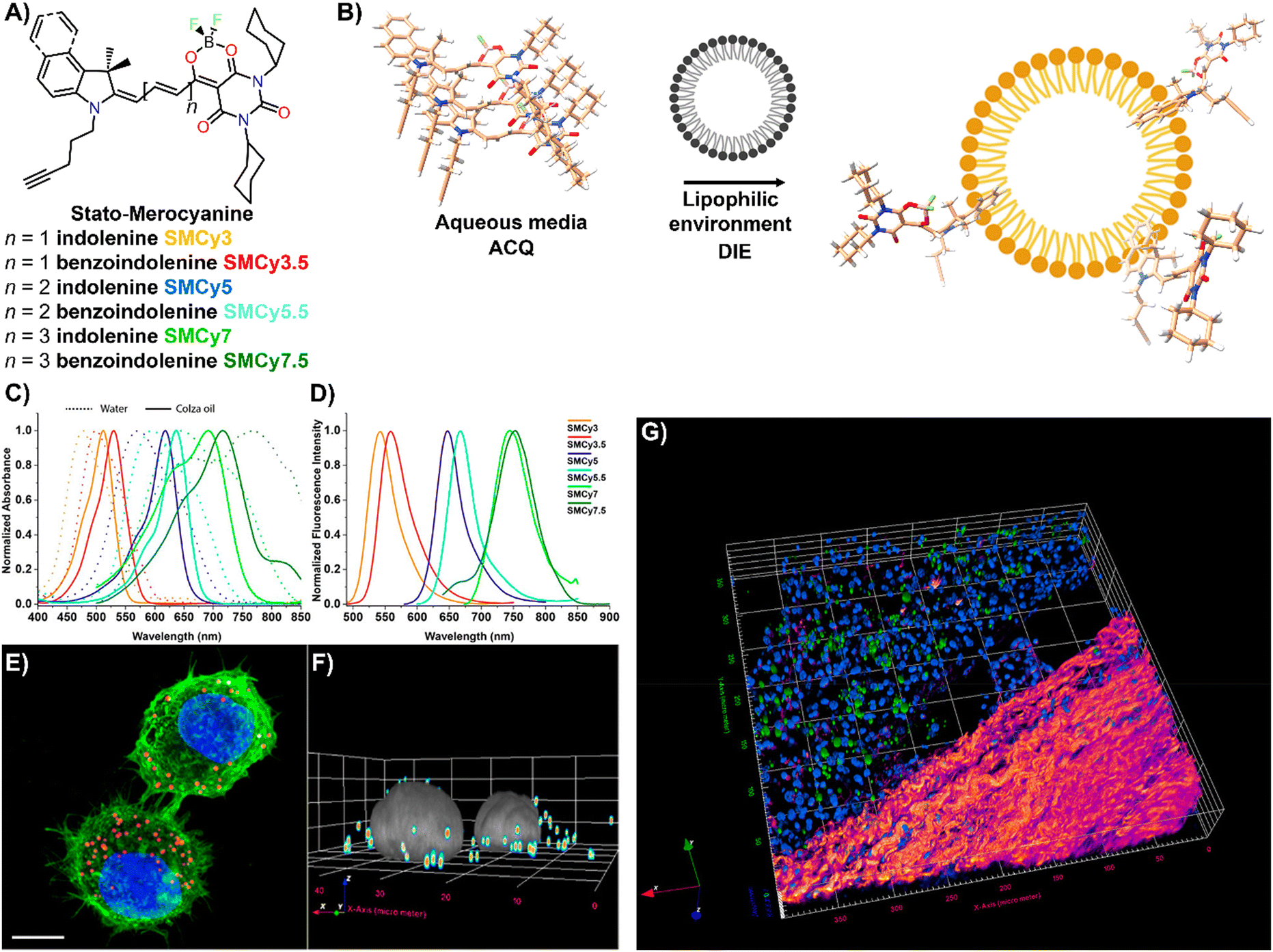 | ||
| Fig. 18 (A) Chemical structures of the six SMCy family members. (B) Operational recognition mechanism of SMCy dyes. (C) and (D) Normalized absorption spectra of SMCy dyes in water (dashed lines) and colza oil (solid lines) (C), along with their normalized emission spectra in oil (D). (E) Maximum intensity projection image of KB cells displaying LDs with SMCy5 (red), nuclei stained with Hoechst (blue), and plasma membranes marked by MemBright-488 (green). Scale bar: 10 μm. (F) 3D image showing the LDs (coloured spots) and nuclei (gray). (G) Two-photon excitation 3D imaging of a mouse liver slice treated with Hoechst and SMCy5.5, depicting nuclei (blue), lipid droplets (green), and collagen fibers (fire-coloured). The excitation wavelength was 810 nm. Reprinted with permission from ref. 200. Copyright 2018 American Chemical Society. | ||
Zhou and team introduced CM2P, a two-photon lipophilic coumarin-based probe for super-resolution imaging and dynamic LD tracking (Fig. 19(A)).29 Exhibiting ACQ in PBS and forming nanoparticles, CM2P's fluorescence intensified in oil/water emulsions via a DIE process (Fig. 19(A)). It selectively stained LDs under both one- and two-photon imaging (Fig. 19(B) and (C)). Employed in STED microscopy, CM2P achieved higher resolution than conventional CLSM, and enabled real-time LD observation with two-photon microscopy (Fig. 19(D)).29
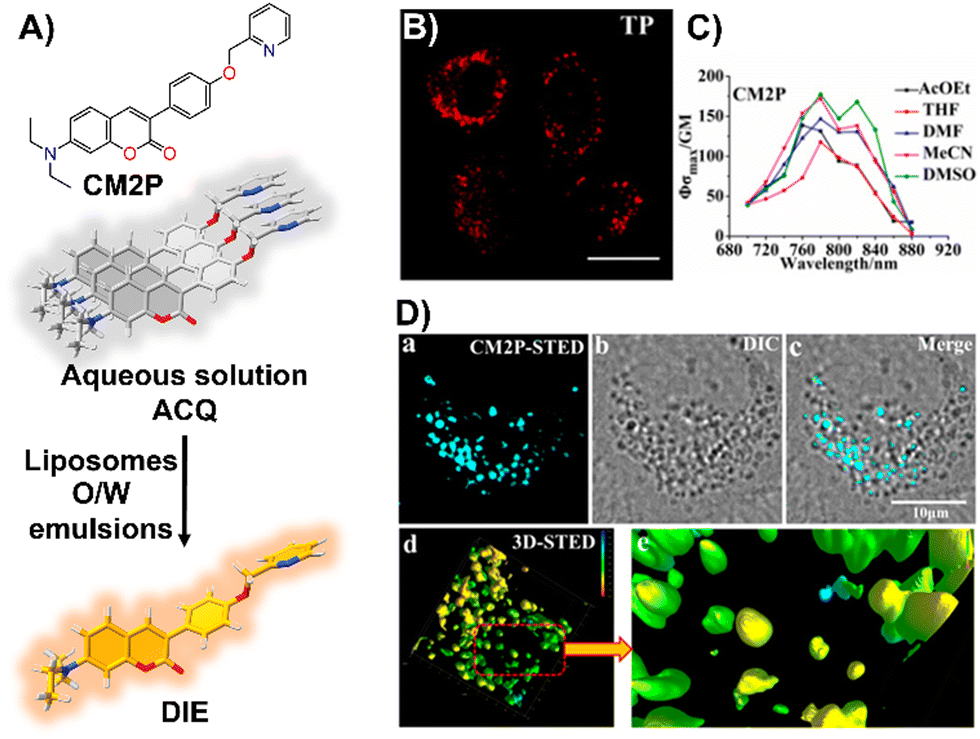 | ||
| Fig. 19 (A) Chemical structure of CM2P and its DIE mechanism mediated by liposomes or oil/water emulsions. (B) and (C) Two-photon fluorescence imaging of HeLa cells stained with CM2P (B) and the two-photon molecular brightness of CM2P across various solvents (C). (D) Morphological characterization of LDs in live HeLa cells stained with CM2P: (a) STED image, (b) bright-field image, (c) merged image of (a) and (b), (d) 3D-STED image, and (e) zoomed-in view of (d). Rainbow calibration bar, Z-scale (0–10 μm); scale bar: 10 μm. Reprinted with permission from ref. 29. Copyright 2019 American Chemical Society. | ||
Exosomes are small vesicles (40–150 nm) released by cells, carrying proteins, mRNA, DNA, and microRNAs crucial for intercellular communication, inflammation, and tissue repair.228–231 Serving as biomarkers, they provide a non-invasive means for disease diagnosis and management, with higher prevalence in tumor cells, influencing cancer development and progression.230–233
Nishizawa's team developed a novel exosome detection method using an asymmetric cyanine dye, ApoC-TRC12, based on Thiazole Red and modified with a hydrophobic unit and a membrane-binding peptide from Apolipoprotein A-I (Fig. 20(A)).202 In PBS, ApoC-TRC12 forms H-type aggregates, showing broad absorption and low fluorescence, which disaggregate in DMSO. Fluorescence tests with synthetic vesicles showed that ApoC-TRC12 lights-up with 130 nm vesicles, suggesting potential selective exosome detection through a DIE process, yet demonstrates minimal change with larger 440 nm vesicles. Tested on exosomes from K562 and A549 cells, ApoC-TRC12 achieved rapid detection with LODs of 3.5 × 103 and 2.1 × 103 particles per μL, respectively, outperforming the commercial MemGlow 640 probe (Fig. 20(B) and (C)).202
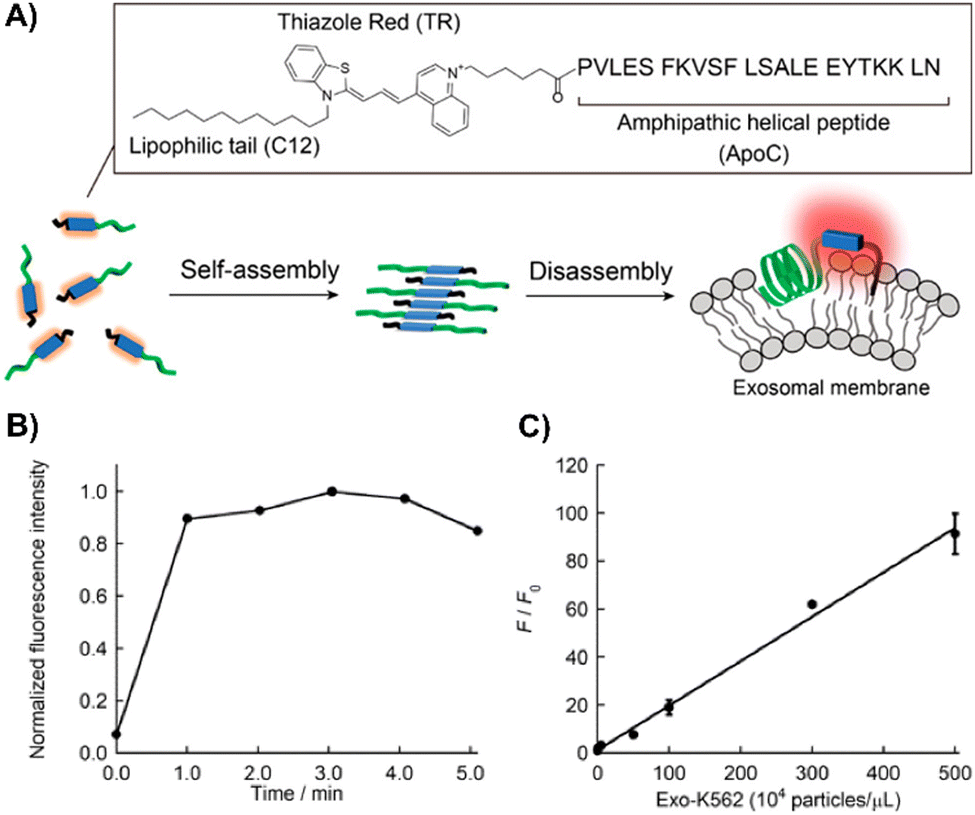 | ||
| Fig. 20 (A) Schematic depiction of the self-assembly/disassembly mechanism for fluorescence sensing by ApoC-TRC12, used in exosome analysis. (B) Time-dependent fluorescence changes at 656 nm for ApoC-TRC12 following the addition of exosomes derived from K562 cells (Exo-K562). (C) Calibration curve for Exo-K562 based on the fluorescence response of ApoC-TRC12. F and F0 represent the fluorescence intensities at 656 nm with and without Exo-K562, respectively. Reprinted with permission from ref. 202. Copyright 2023 American Chemical Society. | ||
4 Self-disassembly and partially unknown processes-mediated DIE
In this section, we aim to highlight how disaggregation can lead to misleading results. DIE is not solely caused by binding to specific receptors that trigger probe disassembly. Instead, DIE may also result from inherent disassembly processes or poorly understood effects (Fig. 21(A)). These inaccuracies are frequently associated with the sample preparation method, which can involve various solvents or their mixtures, the sample's duration of storage and conditions, experimental temperatures, and the presence of impurities in the sample.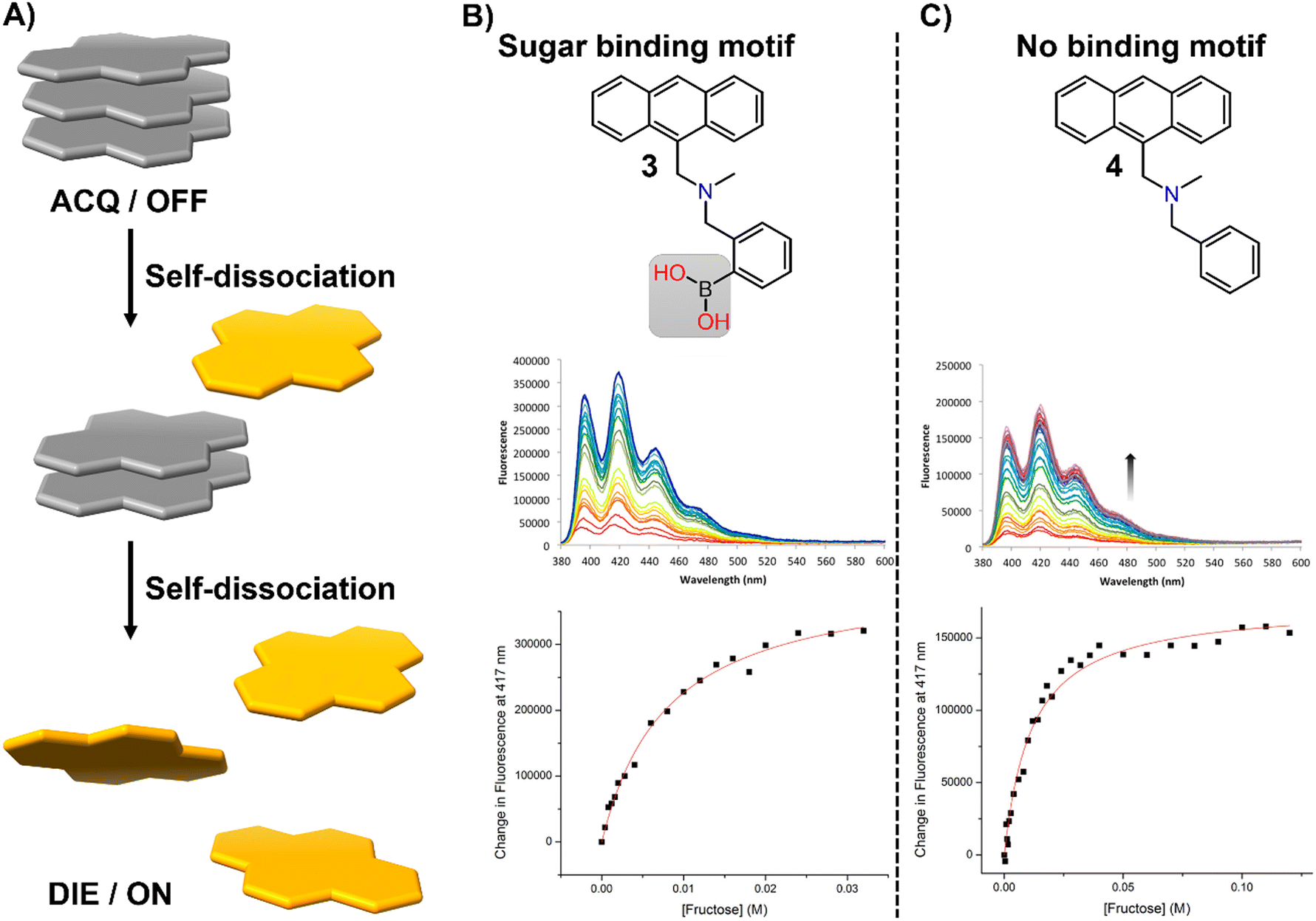 | ||
| Fig. 21 (A) Schematic representation of the self-dissociative mechanism that self-assembling probes may undergo, potentially leading to artifacts and misleading results. (B) Chemical structures of compounds 3 and 4, along with their fluorescence spectral changes associated with the presence of fructose. Reprinted with permission from ref. 234. Copyright 2017 American Chemical Society. | ||
In their research on sugar-binding mechanisms, Anslyn and colleagues explored the activation mechanism of a well-established boronic acid-based saccharide sensor, referred to as compound 3, and compared its fluorescence response to that of a structurally analogous compound 4, which lacks the boronic acid group, thereby inhibiting sugar binding (Fig. 21(B) and (C)).234 Both compounds demonstrated fluorescence enhancements in the absence of fructose, a phenomenon attributed to an autocatalyzed disassembly process. Introducing fructose into a solution pre-equilibrated with compounds 3 and 4 resulted in a modest two-fold increase in the fluorescence intensity of compound 3, while changes in compound 4 were minimal.234 These results indicate that fructose may slightly boost the fluorescence of compound 3, potentially due to a binding interaction or by promoting further disaggregation of the probe, but this does not entirely account for the sensor's activation.234 This highlights the necessity for caution in studies that involve DIE processes with biological molecules, as autocatalyzed disaggregation may produce misleading outcomes.
Similar behaviour has been found in an entirely different sugar-binding study, in which Davis and colleagues discovered that the UV-visible absorption of tetraphenylporphine tetrasulfonate (TPPS), a porphyrin-based host, changed in the presence of glucose.235 These alterations were more accurately linked to variations in the porphyrin's aggregation state rather than to direct glucose binding. The changes in TPPS's UV-visible spectra upon adding glucose appeared to stem from complex, kinetically slow shifts in the porphyrin's aggregation state. Such changes could occur even without any additions to TPPS, and other substances like glycerol could similarly trigger these effects.235 Titration experiments might have suggested 1:1 binding, but this was misleading. While these findings were specific to TPPS, it was probable that other simple porphyrins would exhibit similar behaviour. Consequently, claims of carbohydrate recognition by porphyrins in aqueous solutions should have been approached with caution, especially if changes in aggregation state might explain the observed phenomena.
5 Conclusions and outlook
Initially overlooked, DIE has rapidly evolved into a vibrant and expanding field of research, establishing itself as an indispensable sensing technique through significant advancements over the past decade. The shift from focusing on π–π complex packing and intermolecular interactions32,35,124,158,196 to exploring sophisticated intramolecular dimer models34,63,64 stabilized by covalent bonds has markedly enhanced our comprehension of the fluorescence amplification mechanisms. Discoveries such as enhanced ROS generation102,135,158,173 and ultrasensitive fluorescence assays29,63,81,200 have paved the way for novel applications in bioimaging, sensing, and therapeutic interventions.Thanks to global scientific contributions, DIE research has achieved remarkable progress, addressing clinical diagnosis and pathology,194,195 drug delivery,126,179,180 and precision medicine.61,95,98,108 This review emphasizes the expansion of DIE research from in vitro studies of biologically relevant biomacromolecules and organelles to include tissues and live animals, with DIE-active agents significantly advancing imaging techniques beyond conventional methods.
Despite the global development of numerous DIE-active materials, creating molecular aggregates with precise and consistent optical properties remains a significant challenge. Most DIE dyes exhibit H-type characteristics, leading to poorly defined molecular architectures. In contrast, J-type aggregates typically show well-defined packing behaviour, which can be controlled by incorporating specific hydrogen bonding moieties.236 J-type structures also exhibit unique optical features, such as red-shifted absorption bands and significantly enhanced radiative decay rate (superradiance).19 This may allow their assembly and disassembly to be finely controlled before and after binding events with biological targets, resulting in notable optical changes useful for sensing and imaging applications.
Many DIE-active molecules with superior self-assembly and molecular recognition capabilities are yet to be discovered. Interest in these molecules extends beyond their luminescent properties to their pharmacological potential and applications in diagnostics and therapy. To improve the clarity of bioimaging background signals, there is a push to develop DIE-active dyes for NIR imaging, multiphoton fluorescence microscopy, and super-resolution imaging. This development requires designing fluorescent dyes that completely quench fluorescence when aggregated and fully recover upon monomeric binding to targets, necessitating further optimization of the chemical structures of current DIE-active dyes. Artificial intelligence (AI) presents a promising avenue to revolutionize DIE research by streamlining the discovery process and cutting costs.
Numerous spectroscopic and microscopy methods, coupled with MD simulations, have been employed to decipher the disassembly mechanism of DIE-active dyes triggered by biologically relevant biomolecules. However, there remains a lack of in-depth studies focusing on elucidating the complexation processes at the atomic level using NMR spectroscopy or X-ray crystallography. Obtaining such data would have a tremendous impact on understanding the driving forces behind dye disassembly, providing unprecedented opportunities to design materials with custom-tailored recognition motifs.
Additional potential applications of DIE dyes include the development of smart materials sensitive to specific external stimuli. These dyes can be engineered to act as pH or temperature sensors, disaggregating at specific thresholds. This property is particularly useful in drug delivery systems, where DIE dyes can ensure the targeted and controlled release of drugs within specific cellular organelles in response to pH or temperature changes. Moreover, DIE dyes can be designed to respond to the presence of specific ions or heavy metals, making them valuable in medical diagnostics for monitoring electrolyte balance and in environmental monitoring for detecting pollutants.
In summary, the past decade's research into DIE-active materials has already showcased their immense potential in health and life sciences. Looking ahead, we anticipate that DIE will continue to drive groundbreaking discoveries and introduce innovative methods for understanding biological systems and enhancing therapeutic and diagnostic outcomes.
Author contributions
K. Saczuk and M. Dudek contributed to literature curation and manuscript writing, while K. Matczyszyn took part in manuscript editing. M. Deiana spearheaded the review by conceiving, designing and administering it, supervising the work, leading literature curation, manuscript writing, figure design, review and editing. All authors have read and approved the final manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Deiana would like to acknowledge financial support from project no. 2022/47/P/NZ5/01156, which is co-funded by the National Science Centre and the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 945339. K. Matczyszyn acknowledges funding from the National Science Centre within the Opus UMO-2019/35/B/ST4/03280 project.Notes and references
- A. Schulz and F. Würthner, Angew. Chem., Int. Ed., 2022, 61, e202114667 CrossRef CAS PubMed.
- X. Hu, J. O. Lindner and F. Würthner, J. Am. Chem. Soc., 2020, 142, 3321–3325 CrossRef CAS PubMed.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500–4518 CrossRef CAS PubMed.
- A. P. Deshmukh, A. D. Bailey, L. S. Forte, X. Shen, N. Geue, E. M. Sletten and J. R. Caram, J. Phys. Chem. Lett., 2020, 11, 8026–8033 CrossRef CAS PubMed.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- J. Leira-Iglesias, A. Tassoni, T. Adachi, M. Stich and T. M. Hermans, Nat. Nanotechnol., 2018, 13, 1021–1027 CrossRef CAS PubMed.
- A. Mishra, S. Dhiman and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 2740–2756 CrossRef CAS PubMed.
- S. Dhiman, A. Jain, M. Kumar and S. J. George, J. Am. Chem. Soc., 2017, 139, 16568–16575 CrossRef CAS PubMed.
- M. Hecht and F. Würthner, Acc. Chem. Res., 2021, 54, 642–653 CrossRef CAS PubMed.
- F. Würthner, Acc. Chem. Res., 2016, 49, 868–876 CrossRef PubMed.
- K. Cai, J. Xie, D. Zhang, W. Shi, Q. Yan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 5764–5773 CrossRef CAS PubMed.
- E. Feng, Y. Liu, S. Lv, D. Liu, S. Huang, Z. Li and F. Song, Adv. Funct. Mater., 2022, 32, 2209258 CrossRef CAS.
- V. Grande, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2018, 9, 6904–6911 RSC.
- J. L. Banal, T. Kondo, R. Veneziano, M. Bathe and G. S. Schlau-Cohen, J. Phys. Chem. Lett., 2017, 8, 5827–5833 CrossRef CAS PubMed.
- W. P. Bricker, J. L. Banal, M. B. Stone and M. Bathe, J. Chem. Phys., 2018, 149, 024905 CrossRef PubMed.
- S. Xu, H.-W. Liu, S.-Y. Huan, L. Yuan and X.-B. Zhang, Mater. Chem. Front., 2021, 5, 1076–1089 RSC.
- J. Heo, D. P. Murale, H. Y. Yoon, V. Arun, S. Choi, E. Kim, J.-S. Lee and S. Kim, Aggregate, 2022, 3, e159 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- F. Xia, W. Fan, S. Jiang, Y. Ma, Y. Lu, J. Qi, E. Ahmad, X. Dong, W. Zhao and W. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 21660–21672 CrossRef CAS PubMed.
- X. Hu, W. Fan, Z. Yu, Y. Lu, J. Qi, J. Zhang, X. Dong, W. Zhao and W. Wu, Nanoscale, 2016, 8, 7024–7035 RSC.
- X. Ji, Y. Cai, X. Dong, W. Wu and W. Zhao, Nanoscale, 2023, 15, 9290–9296 RSC.
- H. He, S. Jiang, Y. Xie, Y. Lu, J. Qi, X. Dong, W. Zhao, Z. Yin and W. Wu, Nanoscale Horiz., 2018, 3, 397–407 RSC.
- S. Atchimnaidu, D. Perumal, K. S. Harikrishanan, H. V. P. Thelu and R. Varghese, Nanoscale, 2020, 12, 11858–11862 RSC.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- H. Wang, Q. Li, P. Alam, H. Bai, V. Bhalla, M. R. Bryce, M. Cao, C. Chen, S. Chen, X. Chen, Y. Chen, Z. Chen, D. Dang, D. Ding, S. Ding, Y. Duo, M. Gao, W. He, X. He, X. Hong, Y. Hong, J.-J. Hu, R. Hu, X. Huang, T. D. James, X. Jiang, G.-I. Konishi, R. T. K. Kwok, J. W. Y. Lam, C. Li, H. Li, K. Li, N. Li, W.-J. Li, Y. Li, X.-J. Liang, Y. Liang, B. Liu, G. Liu, X. Liu, X. Lou, X.-Y. Lou, L. Luo, P. R. McGonigal, Z.-W. Mao, G. Niu, T. C. Owyong, A. Pucci, J. Qian, A. Qin, Z. Qiu, A. L. Rogach, B. Situ, K. Tanaka, Y. Tang, B. Wang, D. Wang, J. Wang, W. Wang, W.-X. Wang, W.-J. Wang, X. Wang, Y.-F. Wang, S. Wu, Y. Wu, Y. Xiong, R. Xu, C. Yan, S. Yan, H.-B. Yang, L.-L. Yang, M. Yang, Y.-W. Yang, J. Yoon, S.-Q. Zang, J. Zhang, P. Zhang, T. Zhang, X. Zhang, N. Zhao, Z. Zhao, J. Zheng, L. Zheng, Z. Zheng, M.-Q. Zhu, W.-H. Zhu, H. Zou and B. Z. Tang, ACS Nano, 2023, 17, 14347–14405 CrossRef CAS PubMed.
- H. Xu, H. Zhang, G. Liu, L. Kong, X. Zhu, X. Tian, Z. Zhang, R. Zhang, Z. Wu, Y. Tian and H. Zhou, Anal. Chem., 2019, 91, 977–982 CrossRef CAS PubMed.
- E. G. Kaye, K. Kailass, O. Sadovski and A. A. Beharry, ACS Med. Chem. Lett., 2021, 12, 1295–1301 CrossRef CAS PubMed.
- D. Zhai, W. Xu, L. Zhang and Y.-T. Chang, Chem. Soc. Rev., 2014, 43, 2402–2411 RSC.
- M. Deiana, K. Chand, J. Jamroskovic, I. Obi, E. Chorell and N. Sabouri, Angew. Chem., Int. Ed., 2020, 59, 896–902 CrossRef CAS PubMed.
- V. Grande, C.-A. Shen, M. Deiana, M. Dudek, J. Olesiak-Banska, K. Matczyszyn and F. Würthner, Chem. Sci., 2018, 9, 8375–8381 RSC.
- F. Bouhedda, K. T. Fam, M. Collot, A. Autour, S. Marzi, A. Klymchenko and M. Ryckelynck, Nat. Chem. Biol., 2020, 16, 69–76 CrossRef CAS PubMed.
- M. Collot, P. Ashokkumar, H. Anton, E. Boutant, O. Faklaris, T. Galli, Y. Mély, L. Danglot and A. S. Klymchenko, Cell Chem. Biol., 2019, 26, 600–614 CrossRef CAS PubMed.
- A. Das, S. Das, A. Biswas and N. Chattopadhyay, J. Phys. Chem. B, 2021, 125, 13482–13493 CrossRef CAS PubMed.
- P. Paul, S. Samanta, A. Chatterjee, A. Mallick and T. Majumdar, Phys. Chem. Chem. Phys., 2023, 25, 10166–10174 RSC.
- Q. Bai, S. Zhang, H. Chen, T. Sun, C. Redshaw, J.-X. Zhang, X.-L. Ni, G. Wei and Z. Tao, ChemistrySelect, 2017, 2, 2569–2573 CrossRef CAS.
- M. Sayed, S. Jha and H. Pal, Phys. Chem. Chem. Phys., 2017, 19, 24166–24178 RSC.
- Q. Wang, Q. Zhang, Q.-W. Zhang, X. Li, C.-X. Zhao, T.-Y. Xu, D.-H. Qu and H. Tian, Nat. Commun., 2020, 11, 158 CrossRef PubMed.
- H. Yin, F. Dumur, Y. Niu, M. M. Ayhan, O. Grauby, W. Liu, C. Wang, D. Siri, R. Rosas, A. Tonetto, D. Gigmes, R. Wang, D. Bardelang and O. Ouari, ACS Appl. Mater. Interfaces, 2017, 9, 33220–33228 CrossRef CAS PubMed.
- H.-B. Cheng, Z. Sun, N. Kwon, R. Wang, Y. Cui, C. O. Park and J. Yoon, Chem. – Eur. J., 2019, 25, 3501–3504 CrossRef CAS PubMed.
- P. Zhang, M.-S. Zhu, H. Luo, Q. Zhang, L.-E. Guo, Z. Li and Y.-B. Jiang, Anal. Chem., 2017, 89, 6210–6215 CrossRef CAS PubMed.
- L.-X. Huang, Q. Guo, Y. Chen, P. Verwilst, S. Son, J.-B. Wu, Q.-Y. Cao and J. S. Kim, Chem. Commun., 2019, 55, 14135–14138 RSC.
- B. Muthuraj, S. R. Chowdhury, S. Mukherjee, C. R. Patra and P. K. Iyer, RSC Adv., 2015, 5, 28211–28218 RSC.
- L. Liu, C. Liu, L. Wang, X.-C. Shen and H. Chen, Sens. Actuators, B, 2022, 371, 132542 CrossRef CAS.
- Y. Hu, Y. Wang, X. Wen, Y. Pan, X. Cheng, R. An, G. Gao, H.-Y. Chen and D. Ye, Research, 2020, 4087069 CAS.
- J. Yang, C.-C. Dong, X.-L. Chen, X. Sun, J.-Y. Wei, J.-F. Xiang, J. L. Sessler and H.-Y. Gong, J. Am. Chem. Soc., 2019, 141, 4597–4612 CrossRef CAS PubMed.
- L. K. Kumawat, A. A. Abogunrin, M. Kickham, J. Pardeshi, O. Fenelon, M. Schroeder and R. B. P. Elmes, Front. Chem., 2019, 7, 354 CrossRef CAS PubMed.
- B. Andreiuk, A. Reisch, E. Bernhardt and A. S. Klymchenko, Chem. – Asian J., 2019, 14, 836–846 CrossRef CAS PubMed.
- V. Kshtriya, B. Koshti, D. K. Pandey, S. Kharbanda, C. Kanth P, D. K. Singh, D. Bhatia and N. Gour, Soft Matter, 2021, 17, 4304–4316 RSC.
- M. Qiao, R. Zhang, S. Liu, J. Liu, L. Ding and Y. Fang, ACS Appl. Mater. Interfaces, 2022, 14, 32706–32718 CrossRef CAS PubMed.
- J. Fan, L. Ding and Y. Fang, Langmuir, 2019, 35, 326–341 CrossRef CAS PubMed.
- T. Jing and L. Yan, Talanta, 2017, 170, 185–192 CrossRef CAS PubMed.
- W. Tian, J. Zhang, J. Yu, J. Wu, H. Nawaz, J. Zhang, J. He and F. Wang, Adv. Opt. Mater., 2016, 4, 2044–2050 CrossRef CAS.
- X. Liu, K. Jia, Y. Wang, W. Shao, C. Yao, L. Peng, D. Zhang, X.-Y. Hu and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 4843–4850 CrossRef CAS PubMed.
- Y. Liu, C. Xu, L. Teng, H.-W. Liu, T.-B. Ren, S. Xu, X. Lou, H. Guo, L. Yuan and X.-B. Zhang, Chem. Commun., 2020, 56, 1956–1959 RSC.
- K. Sou, L. Y. Chan, S. Arai and C.-L. K. Lee, Sci. Rep., 2019, 9, 17991 CrossRef PubMed.
- V. G. Panse, P. Vogel, W. E. Trommer and R. Varadarajan, J. Biol. Chem., 2000, 275, 18698–18703 CrossRef CAS PubMed.
- V. Grande, F. Doria, M. Freccero and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 7520–7524 CrossRef CAS PubMed.
- M. Deiana, K. Chand, J. Jamroskovic, R. N. Das, I. Obi, E. Chorell and N. Sabouri, Nanoscale, 2020, 12, 12950–12957 RSC.
- M. Deiana, K. Chand, E. Chorell and N. Sabouri, J. Phys. Chem. Lett., 2023, 14, 1862–1869 CrossRef CAS PubMed.
- I. O. Aparin, R. Yan, R. Pelletier, A. A. Choi, D. I. Danylchuk, K. Xu and A. S. Klymchenko, J. Am. Chem. Soc., 2022, 144, 18043–18053 CrossRef CAS PubMed.
- L. Esteoulle, F. Daubeuf, M. Collot, S. Riché, T. Durroux, D. Brasse, P. Marchand, J. Karpenko, A. S. Klymchenko and D. Bonnet, Chem. Sci., 2020, 11, 6824–6829 RSC.
- V. M. Farzan, M. V. Kvach, I. O. Aparin, D. E. Kireev, T. A. Prikazchikova, A. V. Ustinov, V. V. Shmanai, G. A. Shipulin, V. A. Korshun and T. S. Zatsepin, Talanta, 2019, 194, 226–232 CrossRef CAS PubMed.
- L. F. Kessler, A. Balakrishnan, N. S. Deußner-Helfmann, Y. Li, M. Mantel, M. Glogger, H.-D. Barth, M. S. Dietz and M. Heilemann, Angew. Chem., Int. Ed., 2023, 62, e202307538 CrossRef CAS PubMed.
- A. M. D’Amico and K. M. Vasquez, DNA Repair, 2021, 99, 103049 CrossRef PubMed.
- F. J. Groelly, M. Fawkes, R. A. Dagg, A. N. Blackford and M. Tarsounas, Nat. Rev. Cancer, 2023, 23, 78–94 CrossRef CAS PubMed.
- T. Sakamoto, D. Hasegawa and K. Fujimoto, Org. Biomol. Chem., 2018, 16, 7157–7162 RSC.
- M. Deiana, B. Mettra, K. Matczyszyn, D. Pitrat, J. Olesiak-Banska, C. Monnereau, C. Andraud and M. Samoc, Biomacromolecules, 2016, 17, 3609–3618 CrossRef CAS PubMed.
- H. Blom and J. Widengren, Chem. Rev., 2017, 117, 7377–7427 CrossRef CAS PubMed.
- K. D. Makova and M. H. Weissensteiner, Trends Genet., 2023, 39, 109–124 CrossRef CAS PubMed.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed.
- N. Kosiol, S. Juranek, P. Brossart, A. Heine and K. Paeschke, Mol. Cancer, 2021, 20, 40 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, Nat. Genet., 2016, 48, 1267–1272 CrossRef PubMed.
- R. Hänsel-Hertsch, A. Simeone, A. Shea, W. W. I. Hui, K. G. Zyner, G. Marsico, O. M. Rueda, A. Bruna, A. Martin, X. Zhang, S. Adhikari, D. Tannahill, C. Caldas and S. Balasubramanian, Nat. Genet., 2020, 52, 878–883 CrossRef PubMed.
- M. Deiana, J. Jamroskovic, I. Obi and N. Sabouri, Chem. Commun., 2020, 56, 14251–14254 RSC.
- M. Deiana, M. Mosser, T. Le Bahers, E. Dumont, M. Dudek, S. Denis-Quanquin, N. Sabouri, C. Andraud, K. Matczyszyn, C. Monnereau and L. Guy, Nanoscale, 2021, 13, 13795–13808 RSC.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- S. Liu, L. Bu, Y. Zhang, J. Yan, L. Li, G. Li, Z. Song and J. Huang, Anal. Chem., 2021, 93, 5267–5276 CrossRef CAS PubMed.
- M. Deiana, I. Obi, M. Andreasson, S. Tamilselvi, K. Chand, E. Chorell and N. Sabouri, ACS Chem. Biol., 2021, 16, 1365–1376 CrossRef CAS PubMed.
- M. Zuffo, A. Guédin, E.-D. Leriche, F. Doria, V. Pirota, V. Gabelica, J.-L. Mergny and M. Freccero, Nucleic Acids Res., 2018, 46, e115–e115 CrossRef PubMed.
- F. Doria, M. Nadai, M. Zuffo, R. Perrone, M. Freccero and S. N. Richter, Chem. Commun., 2017, 53, 2268–2271 RSC.
- M. Zuffo, F. Doria, S. Botti, G. Bergamaschi and M. Freccero, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1303–1311 CrossRef CAS PubMed.
- H. Yao, S. Liu, Z. Xing, Y. Miao, Z. Song, G. Li and J. Huang, Anal. Chem., 2022, 94, 15231–15239 CrossRef CAS PubMed.
- Y. Dong and M.-H. Hu, Bioorg. Chem., 2023, 141, 106879 CrossRef CAS PubMed.
- H.-Z. He, K. Li, K.-K. Yu, P.-L. Lu, M.-L. Feng, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2020, 56, 6870–6873 RSC.
- X.-D. Wang, Y.-S. Liu and M.-H. Hu, Bioorg. Chem., 2024, 143, 107006 CrossRef CAS PubMed.
- L.-X. Wang, J.-T. Zhang, X. Sun, D.-W. Yang and Y.-L. Tang, Dyes Pigm., 2021, 185, 108882 CrossRef CAS.
- L. Guan, Y. Zhou, X. Li, Y. Mao, A. Li, Y. Fu, W. Liu, S. Dong, Z. Liang, Y. Zhang, Q. Zhao and L. Zhang, Anal. Chem., 2023, 95, 9288–9296 CrossRef CAS PubMed.
- G.-F. Liu, Y.-S. Chen, Z.-L. Wang, D. Gu and M.-Q. Wang, Dyes Pigm., 2024, 225, 112107 CrossRef CAS.
- H.-Y. Li, H.-W. Cao, X.-X. Lang, Y.-S. Chen and M.-Q. Wang, J. Mater. Chem. B, 2022, 10, 7772–7779 RSC.
- M.-Q. Wang, J.-J. Gao, Q.-Q. Yu and H.-B. Liu, New J. Chem., 2020, 44, 13557–13564 RSC.
- J. Jamroskovic, M. Doimo, K. Chand, I. Obi, R. Kumar, K. Brännström, M. Hedenström, R. Nath Das, A. Akhunzianov, M. Deiana, K. Kasho, S. Sulis Sato, P. L. Pourbozorgi, J. E. Mason, P. Medini, D. Öhlund, S. Wanrooij, E. Chorell and N. Sabouri, J. Am. Chem. Soc., 2020, 142, 2876–2888 CrossRef CAS PubMed.
- X.-D. Wang and M.-H. Hu, Sens. Actuators, B, 2023, 392, 134075 CrossRef CAS.
- M.-H. Hu, Sens. Actuators, B, 2021, 328, 128990 CrossRef CAS.
- A. Pandith, Y. Luo, Y. Jang, J. Bae and Y. Kim, Angew. Chem., Int. Ed., 2023, 62, e202215049 CrossRef CAS PubMed.
- A. Pandith, U. Nagarajachari, R. K. G. Siddappa, S. Lee, C. J. Park, K. Sannathammegowda and Y. J. Seo, Bioorg. Med. Chem., 2021, 35, 116077 CrossRef CAS PubMed.
- L.-M. Zhang, Y.-X. Cui, L.-N. Zhu, J.-Q. Chu and D.-M. Kong, Nucleic Acids Res., 2019, 47, 2727–2738 CrossRef CAS PubMed.
- D. Görl and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 12094–12098 CrossRef PubMed.
- M. Deiana, P. Josse, C. Dalinot, A. Osmolovskyi, P. S. Marqués, J. M. A. Castán, L. Abad Galán, M. Allain, L. Khrouz, O. Maury, T. Le Bahers, P. Blanchard, S. Dabos-Seignon, C. Monnereau, N. Sabouri and C. Cabanetos, Commun. Chem., 2022, 5, 142 CrossRef CAS PubMed.
- Y.-L. Lee, Y.-T. Chou, B.-K. Su, C.-C. Wu, C.-H. Wang, K.-H. Chang, J.-A. A. Ho and P.-T. Chou, J. Am. Chem. Soc., 2022, 144, 17249–17260 CrossRef CAS PubMed.
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS PubMed.
- Y. Chen, S. Wang and F. Zhang, Nat. Rev. Bioeng., 2023, 1, 60–78 CrossRef.
- Z. Qin, T.-B. Ren, H. Zhou, X. Zhang, L. He, Z. Li, X.-B. Zhang and L. Yuan, Angew. Chem., Int. Ed., 2022, 61, e202201541 CrossRef CAS PubMed.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- R.-X. Wang, Y. Ou, Y. Chen, T.-B. Ren, L. Yuan and X.-B. Zhang, J. Am. Chem. Soc., 2024, 146, 11669–11678 CrossRef CAS PubMed.
- L. Rong, J. Cao, Y. Dai, W. Chen and N. Fu, Dyes Pigm., 2024, 227, 112159 CrossRef CAS.
- B.-L. Wang and C. Jiang, Anal. Chem., 2019, 91, 1541–1547 CrossRef CAS PubMed.
- P. Le, N. Ahmed and G. W. Yeo, Nat. Cell Biol., 2022, 24, 815–824 CrossRef CAS PubMed.
- E. Tutucci, M. Vera, J. Biswas, J. Garcia, R. Parker and R. H. Singer, Nat. Methods, 2018, 15, 81–89 CrossRef CAS PubMed.
- A. R. Buxbaum, G. Haimovich and R. H. Singer, Nat. Rev. Mol. Cell Biol., 2015, 16, 95–109 CrossRef CAS PubMed.
- F. Bouhedda, A. Autour and M. Ryckelynck, Int. J. Mol. Sci., 2018, 19, 44 CrossRef PubMed.
- C. Li, A. G. Tebo and A. Gautier, Int. J. Mol. Sci., 2017, 18, 1473 CrossRef PubMed.
- J. R. Babendure, S. R. Adams and R. Y. Tsien, J. Am. Chem. Soc., 2003, 125, 14716–14717 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- R. L. Strack, M. D. Disney and S. R. Jaffrey, Nat. Methods, 2013, 10, 1219–1224 CrossRef CAS PubMed.
- W. Song, R. L. Strack, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 1198–1201 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- K. Y. Han, B. J. Leslie, J. Fei, J. Zhang and T. Ha, J. Am. Chem. Soc., 2013, 135, 19033–19038 CrossRef CAS PubMed.
- K. T. Fam, R. Pelletier, F. Bouhedda, M. Ryckelynck, M. Collot and A. S. Klymchenko, Anal. Chem., 2022, 94, 6657–6664 CrossRef CAS PubMed.
- C. Yan, L. Miao, Y. Zhang, X. Zhou, G. Wang, Y. Li, Q. Qiao and Z. Xu, Sens. Actuators, B, 2023, 386, 133731 CrossRef CAS.
- Y. Li, T. Ma, H. Jiang, W. Li, D. Tian, J. Zhu and Z. A. Li, Angew. Chem., Int. Ed., 2022, 61, e202203093 CrossRef CAS PubMed.
- X. Li, S. Yu, D. Lee, G. Kim, B. Lee, Y. Cho, B.-Y. Zheng, M.-R. Ke, J.-D. Huang, K. T. Nam, X. Chen and J. Yoon, ACS Nano, 2018, 12, 681–688 CrossRef CAS PubMed.
- A. M. Fleming and C. J. Burrows, J. Am. Chem. Soc., 2020, 142, 1115–1136 CrossRef CAS PubMed.
- Y. Ding, A. M. Fleming and C. J. Burrows, J. Am. Chem. Soc., 2017, 139, 2569–2572 CrossRef CAS PubMed.
- A. M. Fleming, B. L. Guerra Castañaza Jenkins, B. A. Buck and C. J. Burrows, J. Am. Chem. Soc., 2024, 146, 11364–11370 CAS.
- W. Chen, Y. Zhang, H.-B. Yi, F. Wang, X. Chu and J.-H. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202300162 CrossRef CAS PubMed.
- M. Deiana, J. M. Andrés Castán, P. Josse, A. Kahsay, D. P. Sánchez, K. Morice, N. Gillet, R. Ravindranath, A. K. Patel, P. Sengupta, I. Obi, E. Rodriguez-Marquez, L. Khrouz, E. Dumont, L. Abad Galán, M. Allain, B. Walker, H. S. Ahn, O. Maury, P. Blanchard, T. Le Bahers, D. Öhlund, J. von Hofsten, C. Monnereau, C. Cabanetos and N. Sabouri, Nucleic Acids Res., 2023, 51, 6264–6285 CrossRef CAS PubMed.
- M. Cheng, Y.-X. Cui, J. Wang, J. Zhang, L.-N. Zhu and D.-M. Kong, ACS Appl. Mater. Interfaces, 2019, 11, 13158–13167 CrossRef CAS PubMed.
- A. Ferino, G. Nicoletto, F. D’Este, S. Zorzet, S. Lago, S. N. Richter, A. Tikhomirov, A. Shchekotikhin and L. E. Xodo, J. Med. Chem., 2020, 63, 1245–1260 CrossRef CAS PubMed.
- Q.-y Ma, X. Li, W. Zhou, X.-f Li, Y.-c Liu, G.-l Feng, H. Tan, Y. Zhang and G.-w Xing, Chem. Commun., 2023, 59, 10287–10290 RSC.
- Z. Hu, D. Wang, Q. Zhou, J. Jie and H. Su, J. Phys. Chem. B, 2024, 128, 576–584 CrossRef CAS PubMed.
- C. Okamoto, A. Momotake and Y. Yamamoto, J. Phys. Chem. B, 2023, 127, 4514–4522 CrossRef CAS PubMed.
- Y. Wang, F. Huo and C. Yin, J. Phys. Chem. B, 2024, 128, 1121–1138 CrossRef CAS PubMed.
- Z.-G. Wang, X.-J. Yan, H.-B. Liu, D.-L. Zhang, W. Liu, C.-Z. Xie, Q.-Z. Li and J.-Y. Xu, J. Mater. Chem. B, 2020, 8, 8346–8355 RSC.
- M. Sasmal, A. S. Musha Islam, D. Moni, D. Maiti, A. Dutta and M. Ali, ACS Appl. Bio Mater., 2022, 5, 5854–5864 CrossRef CAS PubMed.
- Z. Zheng, H. Li, S. Sun and Y. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 44336–44343 CrossRef CAS PubMed.
- E. N. Hoogenboezem and C. L. Duvall, Adv. Drug Delivery Rev., 2018, 130, 73–89 CrossRef CAS PubMed.
- P. Anees, S. Sreejith and A. Ajayaghosh, J. Am. Chem. Soc., 2014, 136, 13233–13239 CrossRef CAS PubMed.
- Y.-R. Wang, L. Feng, L. Xu, Y. Li, D.-D. Wang, J. Hou, K. Zhou, Q. Jin, G.-B. Ge, J.-N. Cui and L. Yang, Chem. Commun., 2016, 52, 6064–6067 RSC.
- J. Qu, W. Meador, P. Cheah, E. E. L. Tanner, J. Delcamp and Y. Zhao, RSC Adv., 2023, 13, 27549–27557 RSC.
- J.-Z. Li, H.-L. Lin, H.-Y. Li, H.-W. Cao, X.-X. Lang, Y.-S. Chen, H.-W. Chen and M.-Q. Wang, Dyes Pigm., 2023, 216, 111357 CrossRef CAS.
- G. B. Guseva, A. A. Ksenofontov, P. S. Bocharov, E. V. Antina and L. E. Nikitina, J. Mol. Liq., 2023, 371, 121078 CrossRef CAS.
- L. P. Jameson, N. W. Smith, O. Annunziata and S. V. Dzyuba, Phys. Chem. Chem. Phys., 2016, 18, 14182–14185 RSC.
- N. Shivran, M. Koli, G. Chakraborty, A. P. Srivastava, S. Chattopadhyay and S. Mula, Org. Biomol. Chem., 2021, 19, 7920–7929 RSC.
- C. Yu, W. Miao, J. Wang, E. Hao and L. Jiao, ACS Omega, 2017, 2, 3551–3561 CrossRef CAS PubMed.
- C. Li, T. Wang, M. Fan, N. Wang, X. Lin, Y. Sun and X. Cui, Nano Lett., 2022, 22, 1954–1962 CrossRef CAS PubMed.
- Z. Luo, T. Lv, K. Zhu, Y. Li, L. Wang, J. J. Gooding, G. Liu and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 3131–3136 CrossRef CAS PubMed.
- L. Long, X. Tan, Z. Liu, Y. Liu, X. Cao and C. Shi, Photochem. Photobiol., 2022, 98, 935–944 CrossRef CAS PubMed.
- S. Samanta, S. Halder and G. Das, Anal. Chem., 2018, 90, 7561–7568 CrossRef CAS PubMed.
- X. Fan, Q. He, S. Sun, H. Li, Y. Pei and Y. Xu, Chem. Commun., 2016, 52, 1178–1181 RSC.
- J. Park and Y. Kim, ChemBioChem, 2019, 20, 350–354 CrossRef CAS PubMed.
- T. Gao, S. Yang, X. Cao, J. Dong, N. Zhao, P. Ge, W. Zeng and Z. Cheng, Anal. Chem., 2017, 89, 10085–10093 CrossRef CAS PubMed.
- Y. Yu, Y. Huang, F. Hu, Y. Jin, G. Zhang, D. Zhang and R. Zhao, Anal. Chem., 2016, 88, 6374–6381 CrossRef CAS PubMed.
- X. Li, S. Yu, Y. Lee, T. Guo, N. Kwon, D. Lee, S. C. Yeom, Y. Cho, G. Kim, J.-D. Huang, S. Choi, K. T. Nam and J. Yoon, J. Am. Chem. Soc., 2019, 141, 1366–1372 CrossRef CAS PubMed.
- F. Wong, Nat. Clin. Pract. Gastroenterol. Hepatol., 2007, 4, 43–51 CrossRef CAS PubMed.
- A. N. Friedman and S. Z. Fadem, J. Am. Soc. Nephrol., 2010, 21, 223–230 CrossRef CAS PubMed.
- K. Mizusawa, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 13386–13395 CrossRef CAS PubMed.
- I. A. Karpenko, M. Collot, L. Richert, C. Valencia, P. Villa, Y. Mély, M. Hibert, D. Bonnet and A. S. Klymchenko, J. Am. Chem. Soc., 2015, 137, 405–412 CrossRef CAS PubMed.
- K. T. Fam, L. Saladin, A. S. Klymchenko and M. Collot, Chem. Commun., 2021, 57, 4807–4810 RSC.
- K. T. Fam, M. Collot and A. S. Klymchenko, Chem. Sci., 2020, 11, 8240–8248 RSC.
- G. T. Ducharme, Z. LaCasse, T. Sheth, I. V. Nesterova and E. E. Nesterov, Angew. Chem., Int. Ed., 2020, 59, 8440–8444 CrossRef CAS PubMed.
- K. Mizusawa, Y. Ishida, Y. Takaoka, M. Miyagawa, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 7291–7293 CrossRef CAS PubMed.
- M. Mohamed, A. K. Klenke, M. V. Anokhin, H. Amadou, P. J. Bothwell, B. Conroy, E. E. Nesterov and I. V. Nesterova, ACS Sens., 2023, 8, 1109–1118 CrossRef CAS PubMed.
- T. Yoshii, K. Mizusawa, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2014, 136, 16635–16642 CrossRef CAS PubMed.
- T.-C. Hou, Y.-Y. Wu, P.-Y. Chiang and K.-T. Tan, Chem. Sci., 2015, 6, 4643–4649 RSC.
- P. Ashokkumar, M. Collot and A. S. Klymchenko, Chem. – Eur. J., 2021, 27, 6795–6803 CrossRef CAS PubMed.
- W. Wang, Y. Zhang, H. Zhao, X. Zhuang, H. Wang, K. He, W. Xu, Y. Kang, S. Chen, S. Zeng and L. Qian, Chem. Sci., 2021, 12, 13477–13482 RSC.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- X. Li, C. y Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880–10886 CrossRef CAS PubMed.
- C. S. Jin, L. Cui, F. Wang, J. Chen and G. Zheng, Adv. Healthcare Mater., 2014, 3, 1240–1249 CrossRef CAS PubMed.
- D. Li, X.-Z. Wang, L.-F. Yang, S.-C. Li, Q.-Y. Hu, X. Li, B.-Y. Zheng, M.-R. Ke and J.-D. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 36435–36443 CrossRef CAS PubMed.
- X. Li, J. S. Oh, Y. Lee, E. C. Lee, M. Yang, N. Kwon, T. W. Ha, D.-Y. Hong, Y. Song, H. K. Kim, B. H. Song, S. Choi, M. R. Lee and J. Yoon, Biomater. Res., 2023, 27, 23 CrossRef CAS PubMed.
- H. Liu, L.-L. Lv, H. Wen, D.-M. Zhao, J. Wu, M.-R. Ke, B.-Y. Zheng, J. Li, X. Li and J.-D. Huang, ACS Appl. Mater. Interfaces, 2022, 14, 28581–28590 CrossRef CAS PubMed.
- B. Cao, M. Yang, Y. Zhu, X. Qu and C. Mao, Adv. Mater., 2014, 26, 4627–4631 CrossRef CAS PubMed.
- X. Li, C. y Kim, J. M. Shin, D. Lee, G. Kim, H.-M. Chung, K.-S. Hong and J. Yoon, Biomaterials, 2018, 187, 18–26 CrossRef CAS PubMed.
- X. Li, H. Fan, T. Guo, H. Bai, N. Kwon, K. H. Kim, S. Yu, Y. Cho, H. Kim, K. T. Nam, J. Yoon, X.-B. Zhang and W. Tan, ACS Nano, 2019, 13, 6702–6710 CrossRef CAS PubMed.
- E. Fahy, S. Subramaniam, R. C. Murphy, M. Nishijima, C. R. H. Raetz, T. Shimizu, F. Spener, G. van Meer, M. J. O. Wakelam and E. A. Dennis, J. Lipid Res., 2009, 50, S9–S14 CrossRef PubMed.
- S. Neugebauer, E. J. Giamarellos-Bourboulis, A. Pelekanou, A. Marioli, F. Baziaka, I. Tsangaris, M. Bauer and M. Kiehntopf, Crit. Care Med., 2016, 44, 1649–1662 CrossRef CAS PubMed.
- L. S. Eberlin, I. Norton, A. L. Dill, A. J. Golby, K. L. Ligon, S. Santagata, R. G. Cooks and N. Y. R. Agar, Cancer Res., 2012, 72, 645–654 CrossRef CAS PubMed.
- B. D. Levy, C. B. Clish, B. Schmidt, K. Gronert and C. N. Serhan, Nat. Immunol., 2001, 2, 612–619 CrossRef CAS PubMed.
- E. J. Delikatny, S. Chawla, D.-J. Leung and H. Poptani, NMR Biomed., 2011, 24, 592–611 CrossRef CAS PubMed.
- N. Krahmer, R. V. Farese and T. C. Walther, EMBO Mol. Med., 2013, 5, 973–983 CrossRef PubMed.
- F. Baenke, B. Peck, H. Miess and A. Schulze, Dis. Models & Mech., 2013, 6, 1353–1363 Search PubMed.
- P. C. Calder, J. Parenter. Enteral Nutr., 2015, 39, 18S–32S Search PubMed.
- M. L. Kraft, Front. Cell Dev. Biol., 2017, 4, 154 Search PubMed.
- D. Hishikawa, T. Hashidate, T. Shimizu and H. Shindou, J. Lipid Res., 2014, 55, 799–807 CrossRef CAS PubMed.
- M. H. Faulds, C. Zhao, K. Dahlman-Wright and J.-Å. Gustafsson, J. Endocrinol., 2012, 212, 3–12 CAS.
- E. Adriaenssens, S. Lottin, T. Dugimont, W. Fauquette, J. Coll, J. P. Dupouy, B. Boilly and J. J. Curgy, Oncogene, 1999, 18, 4460–4473 CrossRef CAS PubMed.
- J. Krämer, R. Kang, L. M. Grimm, L. De Cola, P. Picchetti and F. Biedermann, Chem. Rev., 2022, 122, 3459–3636 CrossRef PubMed.
- K. Wang, X.-L. Wen, X.-Y. Chen, Y. Yue, Y.-S. Yang, H.-L. Zhu, M.-Y. Wang and H.-X. Jiang, Anal. Chem., 2024, 96, 2264–2272 CrossRef CAS PubMed.
- Y. Ma, J. Shang, L. Liu, M. Li, X. Xu, H. Cao, L. Xu, W. Sun, G. Song and X.-B. Zhang, J. Am. Chem. Soc., 2023, 145, 17881–17891 CrossRef CAS PubMed.
- M. Collot, E. Boutant, M. Lehmann and A. S. Klymchenko, Bioconjugate Chem., 2019, 30, 192–199 CrossRef CAS PubMed.
- D. Wu, S. Cheung, G. Sampedro, Z.-L. Chen, R. A. Cahill and D. F. O’Shea, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2272–2280 CrossRef CAS PubMed.
- F. Deng, L. Liu, Q. Qiao, C. Huang, L. Miao and Z. Xu, Chem. Commun., 2019, 55, 15045–15048 RSC.
- N. Curtin, D. Wu, R. Cahill, A. Sarkar, P. M. Aonghusa, S. Zhuk, M. Barberio, M. Al-Taher, J. Marescaux, M. Diana and D. F. O’Shea, Int. J. Med. Sci., 2021, 18, 1541–1553 CrossRef CAS PubMed.
- M. Collot, T. K. Fam, P. Ashokkumar, O. Faklaris, T. Galli, L. Danglot and A. S. Klymchenko, J. Am. Chem. Soc., 2018, 140, 5401–5411 CrossRef CAS PubMed.
- N. Curtin, M. Garre, J.-B. Bodin, N. Solem, R. Méallet-Renault and D. F. O’Shea, RSC Adv., 2022, 12, 35655–35665 RSC.
- K. Ohira, Y. Sato and S. Nishizawa, ACS Sens., 2023, 8, 522–526 CrossRef CAS PubMed.
- L. A. Díaz, J. P. Arab, A. Louvet, R. Bataller and M. Arrese, Nat. Rev. Gastroenterol. Hepatol., 2023, 20, 764–783 CrossRef PubMed.
- V. Mehta and E. Tzima, Nature, 2016, 540, 531–532 CrossRef CAS PubMed.
- H. Zheng, H. Li, Y. Wang, Z. Li, B. Hu, X. Li, L. Fu, H. Hu, Z. Nie, B. Zhao, D. Wei, B. W. Karlson, M. L. Bots, X. Meng, Y. Chen, Y. Wang, S. Cao, Y. Cai, D. Peng, Y. Fu, Z. Yuan, Y. Dong, G. Ma, W. Hong, E. Xu, H. Li, W. Yue, Y. Wang, Z. Zheng, N. Zhang, X. Zhang and W. Fu, Stroke, 2022, 53, 3004–3013 CrossRef CAS PubMed.
- J. L. M. Björkegren and A. J. Lusis, Cell, 2022, 185, 1630–1645 CrossRef PubMed.
- T. Weiss-Sadan, I. Gotsman and G. Blum, FEBS J., 2017, 284, 1455–1472 CrossRef CAS PubMed.
- K. J. Moore, S. Koplev, E. A. Fisher, I. Tabas, J. L. M. Björkegren, A. C. Doran and J. C. Kovacic, J. Am. Coll. Cardiol., 2018, 72, 2181–2197 CrossRef CAS PubMed.
- E. R. H. Walter, S. M. Cooper, J. J. Boyle and N. J. Long, Dalton Trans., 2021, 50, 14486–14497 RSC.
- I. Abd-Elrahman, K. Meir, H. Kosuge, Y. Ben-Nun, T. Weiss Sadan, C. Rubinstein, Y. Samet, M. V. McConnell and G. Blum, Stroke, 2016, 47, 1101–1108 CrossRef CAS PubMed.
- B. Ma, Y. Xiao, Q. Lv, G. Li, Y. Wang and G. Fu, Adv. Mater., 2023, 35, 2206129 CrossRef CAS PubMed.
- M. Sang, Y. Huang, L. Wang, L. Chen, Nawsherwan, G. Li, Y. Wang, X. Yu, C. Dai and J. Zheng, Adv. Sci., 2023, 10, 2207066 CrossRef CAS PubMed.
- Z. Ye, M. Ji, K. Wu, J. Yang, A.-A. Liu, W. Sun, D. Ding and D. Liu, Angew. Chem., Int. Ed., 2022, 61, e202204518 CrossRef CAS PubMed.
- M. Sang, Y. Huang, Z. Liu, G. Li, Y. Wang, Z. Yuan, C. Dai and J. Zheng, ACS Sens., 2023, 8, 893–903 CrossRef CAS PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- E. Sezgin, I. Levental, S. Mayor and C. Eggeling, Nat. Rev. Mol. Cell Biol., 2017, 18, 361–374 CrossRef CAS PubMed.
- S. Rello, J. C. Stockert, V. Moreno, A. Gámez, M. Pacheco, A. Juarranz, M. Cañete and A. Villanueva, Apoptosis, 2005, 10, 201–208 CrossRef CAS PubMed.
- V. V. Shynkar, A. S. Klymchenko, C. Kunzelmann, G. Duportail, C. D. Muller, A. P. Demchenko, J.-M. Freyssinet and Y. Mely, J. Am. Chem. Soc., 2007, 129, 2187–2193 CrossRef CAS PubMed.
- M. Collot, R. Kreder, A. L. Tatarets, L. D. Patsenker, Y. Mely and A. S. Klymchenko, Chem. Commun., 2015, 51, 17136–17139 RSC.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- R. V. Farese and T. C. Walther, Cell, 2009, 139, 855–860 CrossRef CAS PubMed.
- S. Martin and R. G. Parton, Nat. Rev. Mol. Cell Biol., 2006, 7, 373–378 CrossRef CAS PubMed.
- A. R. Thiam, R. V. Farese Jr and T. C. Walther, Nat. Rev. Mol. Cell Biol., 2013, 14, 775–786 CrossRef CAS PubMed.
- J. K. Zehmer, Y. Huang, G. Peng, J. Pu, R. G. W. Anderson and P. Liu, Proteomics, 2009, 9, 914–921 CrossRef CAS PubMed.
- J. A. Olzmann, C. M. Richter and R. R. Kopito, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1345–1350 CrossRef CAS PubMed.
- P. T. Bozza and J. P. B. Viola, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2010, 82, 243–250 CrossRef CAS PubMed.
- Q. Liu, Q. Luo, A. Halim and G. Song, Cancer Lett., 2017, 401, 39–45 CrossRef CAS PubMed.
- L. Balaj, R. Lessard, L. Dai, Y.-J. Cho, S. L. Pomeroy, X. O. Breakefield and J. Skog, Nat. Commun., 2011, 2, 180 CrossRef PubMed.
- B. K. Thakur, H. Zhang, A. Becker, I. Matei, Y. Huang, B. Costa-Silva, Y. Zheng, A. Hoshino, H. Brazier, J. Xiang, C. Williams, R. Rodriguez-Barrueco, J. M. Silva, W. Zhang, S. Hearn, O. Elemento, N. Paknejad, K. Manova-Todorova, K. Welte, J. Bromberg, H. Peinado and D. Lyden, Cell Res., 2014, 24, 766–769 CrossRef CAS PubMed.
- I. Li and B. Y. Nabet, Mol. Cancer, 2019, 18, 32 CrossRef PubMed.
- W. Sun, Y. Ren, Z. Lu and X. Zhao, Mol. Cancer, 2020, 19, 135 CrossRef PubMed.
- M. Boyiadzis and T. L. Whiteside, Leukemia, 2017, 31, 1259–1268 CrossRef CAS PubMed.
- A. Yokoi, A. Villar-Prados, P. A. Oliphint, J. Zhang, X. Song, P. De Hoff, R. Morey, J. Liu, J. Roszik, K. Clise-Dwyer, J. K. Burks, T. J. O’Halloran, L. C. Laurent and A. K. Sood, Sci. Adv., 2019, 5, eaax8849 CrossRef CAS PubMed.
- B. M. Chapin, P. Metola, S. L. Vankayala, H. L. Woodcock, T. J. Mooibroek, V. M. Lynch, J. D. Larkin and E. V. Anslyn, J. Am. Chem. Soc., 2017, 139, 5568–5578 CrossRef CAS PubMed.
- C. M. Renney, G. Fukuhara, Y. Inoue and A. P. Davis, Chem. Commun., 2015, 51, 9551–9554 RSC.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |