
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of SiOx based anode materials for next generation lithium-ion batteries
Yuanteng
Yang
a,
Yanxia
Liu
a,
Xiaoli
Jiang
a,
Lin
Zhao
a,
Penglei
Wang
 *b and
Yagang
Zhang
*a
*b and
Yagang
Zhang
*a
aSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu, 611731, Sichuan, China. E-mail: ygzhang@uestc.edu.cn; plwang@phy.cuhk.edu.hk
bDepartment of Physics, the Chinese University of Hong Kong, Shatin, Hong Kong SAR 999077, China
First published on 12th January 2024
Abstract
Lithium-ion batteries (LIBs) have gained significant importance in various fields, as they play a vital role in energy storage and conversion. The design and development of anode materials are of the utmost significance in enhancing the electrochemical performance of LIBs. SiOx has emerged as a promising candidate for the next generation of anode materials, primarily due to its high energy density, which has garnered widespread attention. However, the commercialization of SiOx has been hindered by challenges such as low conductivity and significant volume change during cycling. The design of SiOx/C composites offers a potential solution to circumvent most of the inherent drawbacks of SiOx, as carbon materials possess excellent electrochemical and physical properties. In this review, we aim to provide an in-depth analysis of SiOx with different dimensions, with a specific focus on the influence of the oxidation degree on the electrochemical performance. We classify the recent representative progress in SiOx/C composites based on their structural characteristics and highlight the structure–activity relationship of different materials. Additionally, strategies for improving the performance of SiOx based anode materials are discussed, including the use of carbon sources, doping, surface modification, prelithiation and the use of binders. Finally, the challenges and research prospects are also described, aiming to provide insights for the rational design of advanced SiOx/C composites.
1. Introduction
In light of the depletion of fossil fuels and the challenges posed by climate change, there is a pressing need to develop green energy solutions to alleviate the energy crisis. Lithium-ion batteries (LIBs) have been comprehensively improved and are widely used in various fields due to their higher specific capacity and longer cycle life compared to traditional batteries.1,2 The widespread interest and utilization of LIBs in diverse electronic devices, along with their integration with renewable energy sources like wind and tidal power, have led to a surge in practical applications.3,4 The specific capacity is one of the most important indicators for evaluating LIBs. Research indicates that the capacity of LIBs is consistently advancing at a rate of 7% to 10% per year. This highlights the crucial importance of continuous innovation in battery technology to meet the ever-increasing demand for energy storage solutions.5 LIBs primarily comprise an anode, a cathode, separators, and electrolytes (Fig. 1a). The anode material holds significant importance in the overall system, playing a pivotal role in determining the specific capacity of the battery (Fig. 1b).6 In the early days of LIB development, coke was commonly used as the anode material. Most anode materials currently used in commercially available LIBs still consist of carbon materials. This choice is primarily driven by the excellent cycle performance and enhanced safety characteristics offered by carbon-based anodes.7,8 However, the low specific capacity of graphite (372 mA h g−1) has been unable to meet the demand of LIBs. In order to break through the problem of low specific capacity, it is necessary to seek the next generation of LIB anode materials.8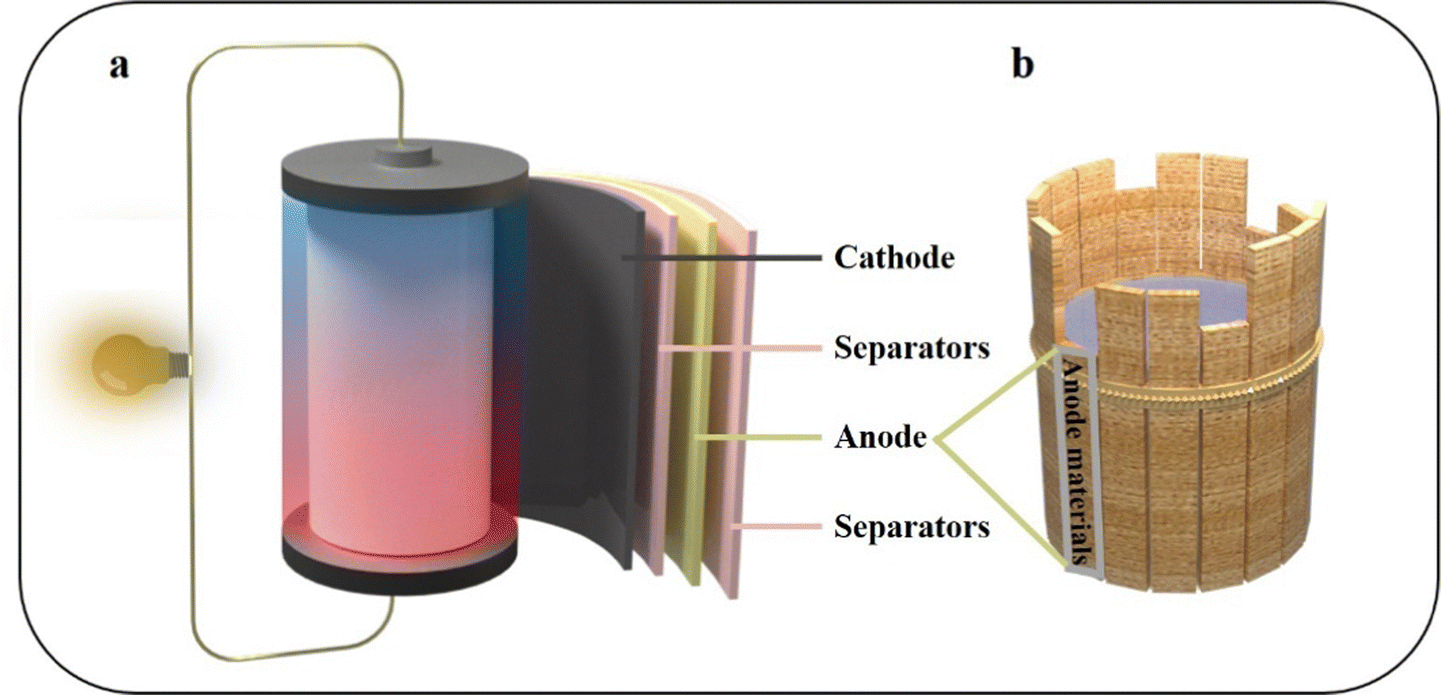 | ||
| Fig. 1 Schematic diagram of (a) the LIBs structure and (b) anode materials, which play a decisive role in LIBs. | ||
Si is considered one of the most promising next-generation anode materials for LIBs due to its high specific capacity of approximately 3400 mA h g−1, which is much higher than those of carbon materials. This makes it highly attractive for use in high performance batteries that require a high energy density. In addition to its high specific capacity, Si is also abundant in nature and easy to obtain, making it an attractive option for large-scale battery production.9 Nevertheless, one major challenge encountered with silicon as a negative electrode is its significant volume expansion (300%) during the charging/discharging process. The volume expansion makes the electrode deform and the electrode material structure collapse. At the same time, the solid–electrolyte interphase (SEI) film will also break, causing the Si to be in contact with the electrolyte, constantly generating the SEI, consuming the electrolyte, and seriously affecting the electrochemical performance. In addition, the electronic transmission capacity of Si based materials is also far less than that of graphite, which severely limits their application in LIBs.10,11
It is found that SiOx is a mixture of Si and SiO2 and some metastable silicon oxides, so the theoretical specific capacity is also between those of SiO2 (1600 mA h g−1) and Si (4200 mA h g−1).12 However, SiOx exhibits significantly reduced volume expansion compared to pure Si, making it a promising candidate for high-performance batteries. The lithium intercalation mechanism is different due to the change of Si species.13 An inert layer of LiO2 and lithium silicate will be formed in the outer layer of SiOx, which will significantly improve the stability of the SEI and ultimately improve its cycling performance.14 Unfortunately, SiOx still has a volume expansion of more than 160%, and the addition of oxides has significantly reduced the conductivity, which poses a challenge for its commercialization.15 The electrochemical performance of SiOx based anode materials can be effectively improved through reasonable material design.16,17 The design of hollow structures offers a viable solution to address the issue of volume expansion, making it possible to mitigate this change. Additionally, the use of SiOx/C composite anode materials is widely recognized as the most effective method to overcome this limitation. The second challenge associated with SiOx is low conductivity.18 Combining carbon with SiOx in a composite material can not only mitigate volume changes, but also take advantage of the excellent chemical properties and superior conductivity of carbon, which can facilitate the transfer of both electrons and ions.19 While there have been many reviews summarizing the research progress of Si based anode materials for LIBs, research focused on SiOx is relatively limited.20,21 This review aims to provide a comprehensive summary and analysis of notable research advancements in the field of SiOx anode structural design. Specifically, it emphasizes the distinguishing features of SiOx core structures and their profound impact on electrochemical performance. The importance of oxygen content is analysed, and the value of x should be reasonably controlled during the synthesis of SiOx. According to the different structures of SiOx/C composites, the synthesis methods and the structure–activity relationship are summarized. Furthermore, it highlights several optimization strategies aimed at enhancing the performance of SiOx/C, with particular emphasis on the hybridization of diverse carbon sources with SiOx. Lastly, this review discusses the current challenges that SiOx/C composites still encounter, aiming to stimulate discussions and generate ideas that can contribute to the progress of SiOx/C composites. It is hoped that this review will inspire further advancements in the field.
2. Crystal structures of SiOx and carbon materials
SiOx based anode materials are considered one of the most attractive alternatives to graphite because of their high specific capacity and stable voltage platform. To make better use of SiOx, we must have a more comprehensive understanding of SiOx.22 In the past decades, many efforts have been made to analyse the crystal structure and energy storage mechanism of SiOx.23 Due to the limited characterization instruments, the early understanding of SiOx was controversial and incomplete. Some researchers argued that SiOx, as reported by Philipp, exists in a single-phase state, where continuous Si–Si and Si–O bonds randomly distribute within the SiO random network, forming SiOx materials.24,25 On the other hand, another group of researchers supported the notion that SiOx is a two-phase mixture, with SiO2 and Si randomly mixed together.26 By 2003, Hohl et al.27 and Fuglein et al.28 characterized SiOx by more advanced characterization methods (XPS, HRTEM, NMR, ESR, etc.) and proposed an interface cluster model. In Fig. 2a, the white regions may signify clusters of SiO2, while the black regions may represent clusters of Si. Additionally, the grey regions could denote either transitional silicon oxide (SiO) or a blend of SiO2 and Si. In the past decade, researchers have also proposed an intermediate model, according to which amorphous SiO2 and amorphous Si exist in amorphous SiO, and the transition phase SiOx is wrapped by the two phases (Fig. 2b).29 The energy storage mechanism of SiOx is similar to that of Si in the sense that both materials will generate a lithium–Si alloy (such as Li13Si4, Li7Si3, and Li12Si7) during the charge/discharge process in a lithium-ion battery. However, in the case of SiOx, additional reactions occur, resulting in the formation of Li silicates (such as Li4SiO4, Li2SiO3, and Li2SiO5) and Li2O. Li2O can act as a Li+ diffusion medium, facilitating Li+ transport and improving the rate performance of the battery. Moreover, Li2O and Li silicate can also buffer the volume expansion of SiOx during cycling.30 According to research findings, a higher oxygen content in SiOx can lead to the formation of additional by-products such as Li silicate (Li2SiO3) and Li2O during the initial charge/discharge cycles of the battery. The reaction that produces these by-products is irreversible, which will consume a large amount of Li+ and greatly reduce the initial coulomb efficiency (ICE). The Si in SiOx has a regular tetrahedral structure with good crystallinity, so it has strong rigidity, but weak elasticity and conductivity. With the insertion of Li+, the change of volume will produce large stress in the electrode. If the stress exceeds the elastic range of the material, it will lead to cracking or even pulverization of the electrode material, which will seriously affect the life of the battery.31 After 20 years of research and development, it is indeed apparent that utilizing high-capacity SiOx-based anode materials in lithium-ion batteries (LIBs) remains a challenging task.32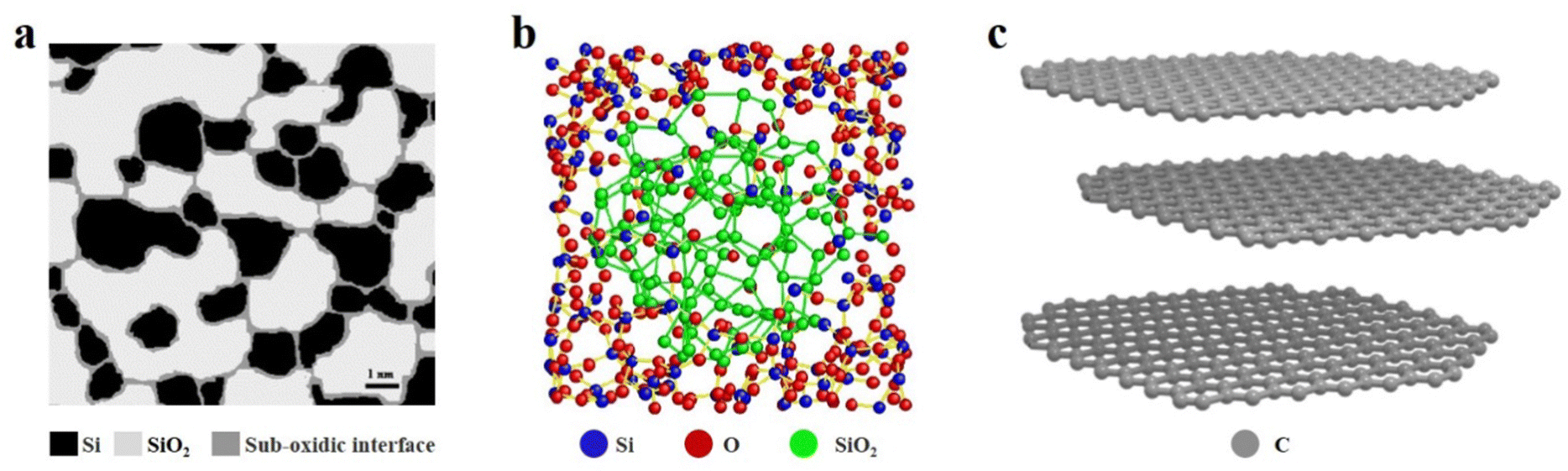 | ||
| Fig. 2 (a) Schematic illustration of the interface cluster mixture structure of a-SiO. Reproduced with permission.27 Reproduced from ref. 27 with permission from Elsevier, Copyright 2003. (b) Reconstructed heterostructure model of amorphous SiO.29 Reproduced from ref. 29 with permission from Springer Nature, Copyright 2016. (c) Crystal structure model of graphite. | ||
The application of carbon materials in the anode of LIBs is well-explored, with graphite being the most commonly used and mature material. The layered structure is the unique characteristic of graphite, and the interlaced graphene layers are connected by van der Waals forces and metal bonds (Fig. 2c).33 The force between graphene layers in graphite is weak, allowing the layers to easily slip past each other, which contributes to the excellent mechanical properties of graphite. This property is beneficial in the context of LIBs as graphite can provide a stress buffer to accommodate the volume changes that occur in the SiOx material during cycling. By serving as a mechanical support, graphite can help to prevent the degradation and fracture of the SiOx material, thereby improving the overall cycling stability and performance of the battery.34 The carbon of graphite is sp2 hybridized, and six carbon atoms form a planar hexagonal structure on the same plane. In addition, each carbon atom has a delocalized valence electron to form a big π key, and the electrons can move freely between the graphene layers. The delocalized Π electrons endow graphite with excellent electronic conductivity. Alongside graphite, the carbon materials in SiOx/C composites include graphene, carbon nanotubes, multiwalled carbon nanotubes (MWCNTs), fullerene, etc.35 These materials have desirable mechanical and electrochemical properties. For instance, the inclusion of carbon nanotubes in SiOx can enhance conductivity by providing additional pathways for ion and electron transport.36 Nevertheless, the practical utilization of these materials is often hindered by high production costs or complex manufacturing processes, thereby limiting their industrial-scale production.37
The lower specific capacity of carbon materials limits their application as anode materials when used alone. Research has shown that the combination of SiOx and carbon materials can achieve the superposition of their advantages.38
3. SiOx design
3.1. SiOx structure design
In order to realize the potential of SiOx/C composites as anode materials with excellent electrochemical performance, it is essential to carefully design the SiOx precursors.39 The size and morphology of the SiOx core can significantly impact the electrochemical performance of the composites. It is essential for a valuable process to not only prioritize cost considerations but also place significant emphasis on environmental friendliness.40 Some of the most common nanostructures used in SiOx precursors include nanoparticles, porous nanoparticles, hollow nanoparticles, nanowires, and nanofilms. These structures can significantly improve the cycle stability and coulomb efficiency of SiOx based anode materials. By optimizing the synthesis and design of SiOx precursors, it is possible to achieve SiOx/C composites with excellent electrochemical performance and long-term cycling stability.41,423.1.1.1. 0D SiOx particles. SiOx particles are one of the most fundamental designs and have been intensively researched due to their manufacturing compatibility and commercial availability.43 Miraculously, it has been shown that reducing the diameter of the SiOx core can attenuate the volume expansion. In the process of material exploration, various experimental routes have been developed to synthesize homogeneous SiOx nanoparticles.44 Tao et al.45 prepared a mixture of tetraethyl orthosilicate (TEOS) and citric acid under intense stirring. The mixture was polymerized with ethylene glycol through polyesterification reaction, and then the polymer gel was obtained by ultrasonic atomization. Finally, SiOx/C was obtained by spray drying in an Ar/H2 mixture atmosphere. Xi et al.46 chose the Si particles from a chemical company and then modified the surface oxide by HCl pickling. Si particles were mixed with formaldehyde–water solution and resorcinol to form the precursors of phenolic resin coated Si/SiO2. Subsequently, Si/SiOx/C was obtained after high-temperature carbonization of the precursor. Similarly, Cao et al.47 reported a simple and less costly method to obtain SiOx by ball-milling Si powder in air, which can achieve simple control of oxygen content.
3.1.1.2. 3D SiOx particles. The secondary structure adjustment of SiOx particles can enhance their cycle performance. Porous SiOx particles were etched on the surface to leave many channels, which can shorten the transmission distance of ions and electrons and achieve high-rate charging/discharging.48 Kin et al.49 reported an efficient strategy for preparing porous SiOx. It involved mechanical milling of SiO2 with zinc, and then Zn/SiO2 was calcined at high temperature. Because the melting point and boiling point of Zn are relatively low, the redundant Zn will evaporate more easily to remove after reducing SiO2. SiOx with many mesopores can be obtained by this process. Zeng et al.50 synthesized cauliflower like Si/SiOx particles by incomplete magnesium thermal reduction of SiO2. SiO2 nanoparticles were prepared by hydrothermal reaction of TEOS, urea, pentanol and cetylpyridinium bromide. Then SiO2 particles, Mg and NaCl were subjected to high temperature solid phase reaction at 700 °C to obtain porous CF-Si/SiOx-700. Finally, the redundant Mg and MgO were washed away by HCl. CF-Si/SiOx-700 was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), which revealed the uniform diameter of the sample (300–500 nm). In addition, it can be clearly seen that there are many pores connected to the internal space which enhance the electronic conductivity and shorten the ion transport channel. Owing to this unique structure, it shows excellent rate performance at various current densities. CF-Si/SiOx-700 delivered a reversible capacity of 600 mA h g−1 and a capacity retention rate of 99% after 400 cycles, which represents its outstanding structural stability. In a similar manner, Zeng et al.51 conducted a study where they utilized an Al/Si alloy as the raw material and employed selective etching with HCl to produce Al-doped porous Si/SiOx composites (Fig. 3a). Multiple experiments were conducted at different HCl concentrations and durations to achieve the desired results. From SEM images, it can be seen that a uniform three-dimensional porous structure is formed, which can enhance the electronic and ionic conductivity (Fig. 3b). As the etching depth increased, there was a reduction in Si4+ and the outer layer acted as a protective barrier, thereby improving the cycling stability. The presence of residual Al plays a crucial role in enhancing the conductivity and preserving the porous structure. Furthermore, during the initial charge/discharge, the SiOx outer layer undergoes various side reactions, leading to a significant decrease in the resistance of the battery system during subsequent cycles (Fig. 3c). In addition, the composite material exhibits a promising specific capacity of 2914 mA h g−1 at 0.1 A g−1 (Fig. 3d). Raza et al.52 obtained porous SiOx/Mg2SiO4/SiOx composites through high-temperature reduction of SiO by Mg. The introduction of silicate into this multilayer structure significantly improves the electrochemical reversibility and almost completely suppresses the volume change of SiOx during electrochemical reaction. The porous structure of the composite not only ensures enough electrolyte and electron transport pathways, but also enriches the active sites on the surface.
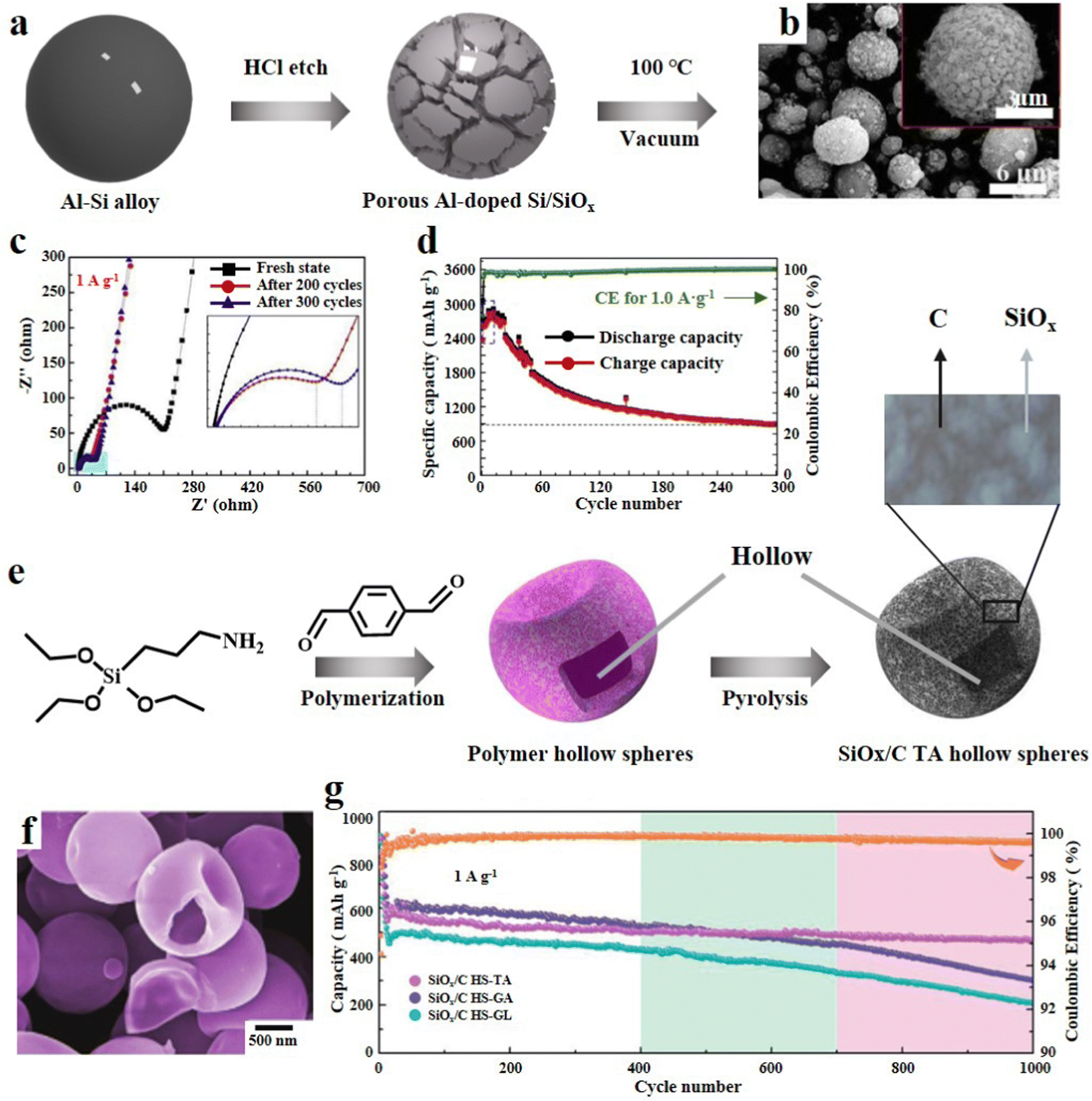 | ||
| Fig. 3 (a) Schematic diagram of the synthesis of Al-doped porous Si/SiOx, (b) SEM images, (c) EIS results and (d) cycling performance at 0.1 A g−1.51 Reproduced from ref. 51 with permission from Elsevier, Copyright 2020. (e) Schematic illustration of the synthetic procedure of SiOx/C HS-TA, (f) SEM image and (g) long-term cycling performance at 1.0 A g−1.54 Reproduced from ref. 54 with permission from John Wiley and Sons, Copyright 2021. | ||
Hollow SiOx particles can fundamentally solve the intrinsic volume effect of SiOx. Sufficient internal reserved space has been proved to be very effective in mitigating the dramatic volume change during delithiation.53 Zhou et al.54 judiciously used dialdehyde molecules, triaminopropyltriethoxysilane (APTES) and terephthalaldehyde (TA), as the precursor of carbon and Si, and synthesized polymer hollow spheres (PHSs) in one step through aldehyde amine condensation reaction (SiOx/C HS-TA). Furthermore, the PHSs were obtained by using a crosslinking agent, and the size, morphology and carbon content could be precisely tuned (Fig. 3e). In addition, organic carbon and organosilicon in PHSs generate a carbon matrix and a SiOx unit after high-temperature pyrolysis, resulting in the formation of an inherent ultra-fine hollow architecture (Fig. 3f). This SiOx/C composite can not only alleviate the stress of cycling, but also result in excellent electrochemical performance in terms of reversible capacity, rate performance and coulomb efficiency (Fig. 3g). Xu et al.55 proposed an environmentally friendly, efficient and simple route for preparing hollow SiOx composites encapsulated by carbon conductive networks. With polyacrylic acid (PAA) as the soft template, TEOS was hydrolysed and condensed to form a SiO2 layer. When the PAA template was immersed in an aqueous solution, it dissolved to form hollow SiO2 spheres. Then, SiO2 particles were mixed with phenolic resin, and a layer of carbon shell was formed in the carbothermal reduction process, while SiO2 was reduced to the SiOx matrix. Li et al.56 reported a composite of self-assembled, metal organic framework (MOF) template induced hollow SiOx nanoparticles coated with N-doped graphene aerogels. This nanocomposite possesses a 3D porous structure with abundant pores and a high specific surface area, and exhibits rapid high Li+ transport and electron transfer.
3.2. Oxygen content of SiOx
Compared with Si based anodes, SiOx based anodes have more stable cycle performance, which can be attributed to the addition of oxygen element, and the oxygen content has a critical influence on the performance of the anode.66,67 Having a deep understanding of the effect of the oxygen content in SiOx is indeed crucial for advancing the application of SiOx-based anode materials in LIBs. Yang et al.68 studied scientifically the effect of oxygen content in SiOx (0.8, 1 and 1.1) as the anode material on the electrochemical cycle of LIBs. Through XRD results, crystalline Si, amorphous SiO, and amorphous SiO2 phases were found in SiOx, and it was proved that they all participated in the energy storage reaction with Li+. The increase in oxygen content in SiOx will diminish the insertion of lithium-ions, thus reducing the specific capacity of the material. However, the sacrifice of specific capacity alleviates the volume expansion of the electrode, thus significantly improving the cycle stability. Moon et al.69 have provided valuable insights into the influence of oxygen content on the electrochemical performance of SiOx through extensive theoretical calculations. Employing density functional theory (DFT) calculations, the Li–Si–O ternary phase diagram is obtained, which has practical significance to understand the reaction products between SiOx and lithium (Fig. 4a). The increase in oxygen content will significantly affect the conductivity. According to the energy band theory, the oxygen content is proportional to the band gap (Fig. 4b). After further mathematical analysis, they speculated that, with the increase of oxygen content, there will be a higher voltage platform, which is more favourable for Li embedding. The voltage values of Si and SiOx were compared in the experiment, confirming the accuracy of the theoretical calculation. By employing similar mathematical approaches, it is conjectured that as the oxygen content decreases, the propensity for SiOx to undergo elastic softening increases, potentially leading to material disintegration.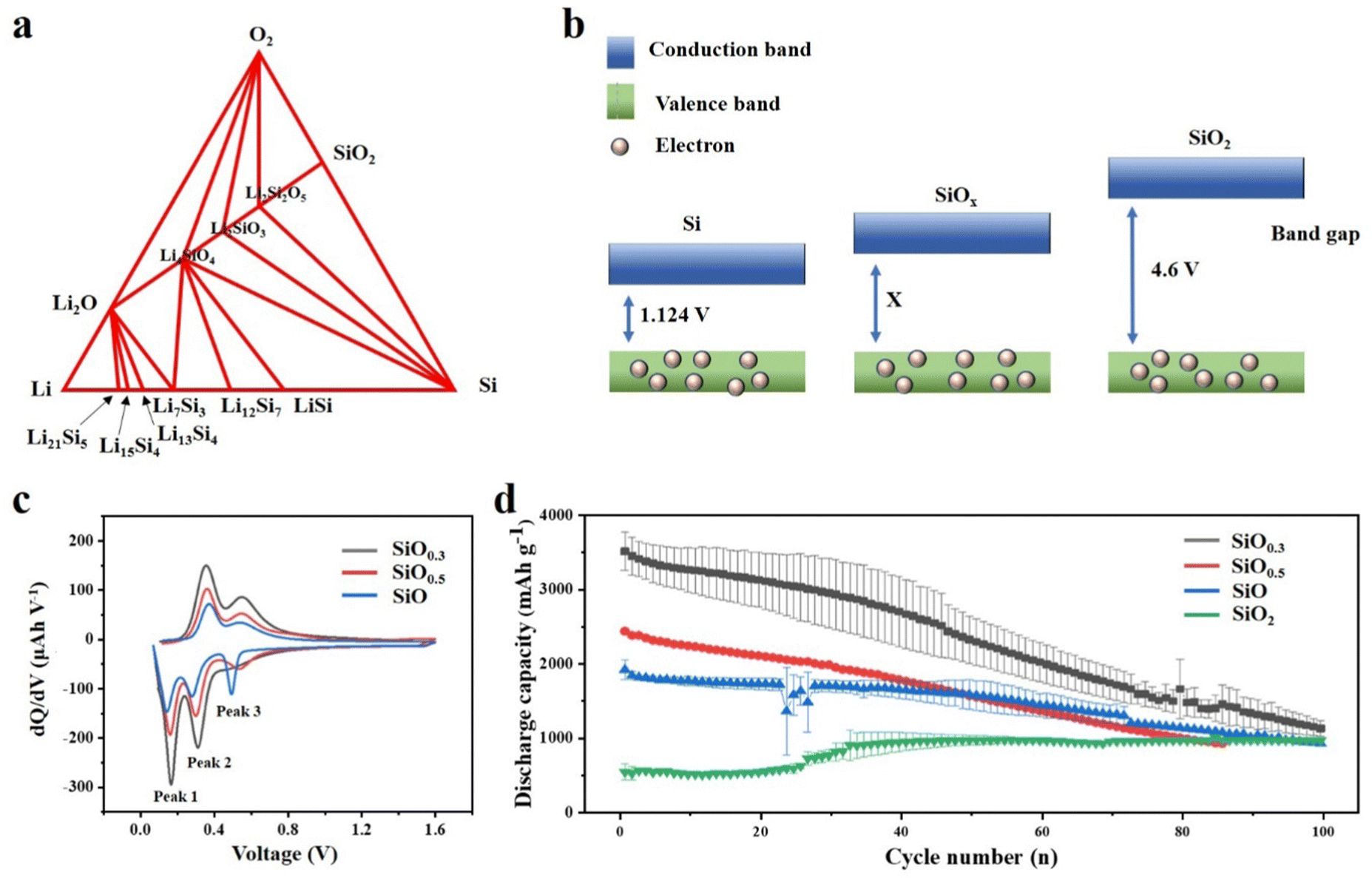 | ||
| Fig. 4 (a) Ternary phase diagram of Si, O, and Li using density functional theory calculations.69 Reproduced from ref. 69 with permission from John Wiley and Sons, Copyright 2020. (b) Band gap of Si, SiOx and SiO2. (c) dQ/dV curves of the 10th cycle data for three SiOx films and (d) discharge capacity vs. cycle number of various SiOx thin film electrodes over 100 cycles.70 Reproduced from ref. 70 with permission from American Chemical Society, Copyright 2022. | ||
Recently, Cho et al.70 reported the effect of oxygen content on the structural evolution of SiOx during electrochemical cycling. Thin films (50 nm) with different oxidation degrees were prepared by magnetron sputtering, and the changes of the SiOx surface were investigated. The flow rate of oxygen/argon entering the chamber determines the oxygen content in the chamber. Thus, SiOx films with different oxygen contents can be obtained. The films were also analysed by electrochemical analysis and in situ X-ray photoelectron spectroscopy (XPS). As shown in Fig. 4c, different oxygen contents will cause a voltage shift of peak 3, which is due to the oxygen related reaction inside the battery and is irreversible in the first few cycles (Fig. 4c). Peak 3 can be stable over about 30 cycles. However, with the increase of oxygen content, a higher voltage appears at peak 3. It is speculated that this is due to the inertness of Si–O, and the Si–Si after lithiation can be recycled reversibly, which increases the conductivity and capacity. When the SiOx film is assembled into a half cell for testing, it can be clearly observed that, with the increase of oxygen content, the capacity retention rate increases, but the reversible capacity decreases as expected (Fig. 4d). Moreover, through the electrochemical impedance test of SiO0.3, SiO and SiO2, it is shown that the total resistance is significant, increasing with the increase of oxygen content, which is consistent with the analysis of the energy band theory. This is also due to the inherent insulation of the Si–O bond, and the difficulty of Si–O bond breaking hinders the diffusion of Li+. Moreover, it can be seen from the EIS images that the overall resistance tends to decrease from the 25th cycle, which is a result of the activation of electrode materials. Similarly, the design of this nano-scale thin film makes it possible to observe the macroscopic changes of the electrode surface more clearly. By comparing the electrode materials with different oxygen contents after cycling, it can be seen that the high oxygen content makes the electrode material more difficult to crack. These advantages of high oxygen content can be attributed to the formation of a stable SEI, which has a certain buffering effect on the change of electrode volume.
Ge et al.71 obtained SiOx/C with different oxygen contents through a simple and scalable process. SiO2 microspheres were prepared by hydrolysis and condensation of TEOS. Using hexadecyltrimethylammonium bromide (CTAB) as the template, porous SiO2 was obtained after the soft template was removed by annealing in air. Subsequently, SiO2 and Mg were mixed at different mass ratios and reacted at high temperature. HCl solution was used to remove the template MgO. Finally, SiOx/C with different oxygen contents were obtained by polymerization of formaldehyde and resorcinol on the surface of SiOx followed by carbonization. An appropriate amount of Mg can achieve good electrochemical performance. After 300 cycles, the best SiOx/C composite possessed a capacity of 1010.4 mA h g−1 at 1 A g−1. Cao et al.47 reported a simple and cheap synthesis route to obtain SiOx. Controlling the exposure time of Si powder to air during the milling process enables the manipulation of the oxygen content in SiOx. Fan et al.72 studied the diffusion and distribution of oxygen in SiOx during heat treatment. After heat treatment of commercial SiOx particles, it is found that oxygen will diffuse to the particle surface. During the process, an amorphous oxygen-rich layer forms around the SiOx, while a crystalline Si core gradually develops within it. It is worth noting that the diffusion of oxygen in SiOx can also be influenced by temperature. At higher temperatures, the diffusion of oxygen is enhanced, which can result in a more complete outward shift of oxygen from the SiOx material. This means that the oxygen content of SiOx can be adjusted by controlling the temperature during synthesis or processing. Furthermore, with higher temperature treatment, a larger pore structure will be formed after HF etching. Different etching times can also generate SiOx particles with various pore sizes. This report serves as a theoretical foundation for the research and development of porous SiOx materials.
4. Structural design of SiOx/C composites
SiOx has a high specific capacity, but its severe volume expansion and poor conductivity limit its further development and also lead to unrestricted growth of the SEI and electrode material pulverization (Fig. 5a).73 The exceptional physical properties of carbon materials can be effectively combined with SiOx to mitigate its volume expansion and enhance electrical conductivity. Moreover, the experimental results show that the combination of SiOx and carbon materials is one of the most effective ways to improve the cycle performance of SiOx.74 The most widely adopted method for compounding SiOx particles is by employing graphite as a coating material. Graphite has good ductility and thus it can be physically coated with a layer of carbon on the surface of Si particles.75 It is very important to reasonably design the structure of the SiOx/C composites because a small change in the 3D structure of the SiOx/C composites will have a significant impact on the electrochemical performance.76 The main structures are the core–shell structure, yolk–shell structure, and some other novel structures, such as the SiOx embedded graphite layer structure, nanotube structure, etc. The synthesis method and the characteristics of the structure–activity relationship are also described in the following.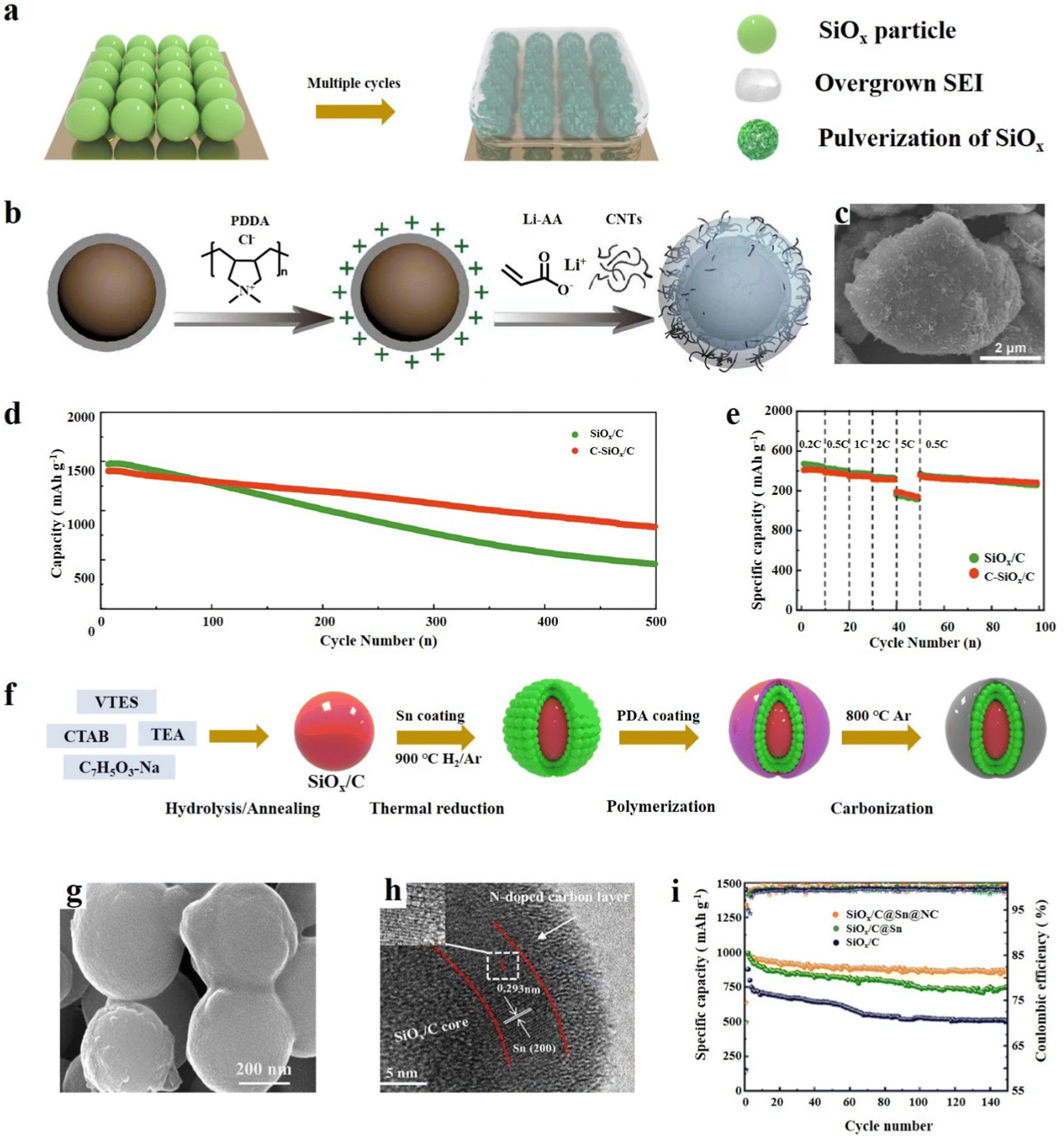 | ||
| Fig. 5 (a) Schematic diagram of the failure mechanism of SiOx based anode materials during cycling. (b) Schematic illustration of the preparation of C-SiOx/C, (c) SEM image, and (d) cycling performance of Li‖SiOx/C and (e) Li‖C-SiOx/C at 0.2C for the first three cycles and at 0.5C for the subsequent cycles and rate capabilities.15 Reproduced from ref. 15 with permission from Elsevier, Copyright 2020. (f) Schematic diagram of the preparative route for SiOx/C@Sn@NC, (g, h) SEM and TEM images and (i) cycle performance at 0.2 A g−1.86 Reproduced from ref. 86 with permission from Elsevier, Copyright 2023. | ||
4.1. Core–shell SiOx/C
The SiOx/C core–shell structure involves utilizing SiOx as the core material and completely enveloping SiOx with graphite. This approach enables the integration of the advantageous properties of both SiOx and graphite, offering a synergistic strategy.77 The carbon layer can not only efficiently buffer the volume change during cycling, but also significantly increase the electrical conductivity. Overgrowth and suppressed cracking of the SEI layer are considered crucial factors that significantly impact performance, and the tight coating of SiOx with the carbon layer avoids contact between SiOx and the electrolyte, thus forming a stable SEI film to enhance the cycle performance.Gu et al.78 demonstrated a simple way to coat SiOx with polydopamine (PDA) with rich hydroxyl groups; PDA is able to form a stable carbon layer on the surface of SiOx (SiOx@PDA@GNH). The functional groups on dopamine can bond with the graphene nanotube hybrid (GNH) to form a hybrid layer, and the coating layer is evenly distributed and has good electrolyte wetting properties. Compared with SiOx and SiOx@PDA, the lithiation/delithiation voltage of SiOx@PDA@GNH is obviously higher, resulting in a higher discharge power. It was found that SiOx@PDA@GNH shows a significantly better cycling performance and has a capacity of 1269 mA h g−1 at 0.05C. After subjecting the SiOx and SiOx@PDA@GNH electrodes to 50 cycles of testing in half cells, significant differences were observed in their electrode structures as revealed by SEM analysis. The SiOx@PDA@GNH electrode exhibited no noticeable signs of peel off or cracks, indicating its enhanced stability and structural integrity. Transition growth of the SEI and particle pulverization can lead to decreased coulombic efficiency (ICE), and storage performance may continue to degrade during repeated Li uptake/release cycles.79 Li et al.15 dispersed SiOx/C and polydimethylammonium chloride (PDDA) in distilled water to coat SiOx/C. Lithium polyacrylate (Li-PAA) was then formed on the surface under the initiation of ammonium persulfate, while simultaneously embedding MWCNTs to obtain C-SiOx/C particles (Fig. 5b). Electron microscopy characterization revealed that the surface of the material particles is rough and porous, which may be a result of the embedding of polypropylene lithium and carbon nanotubes (Fig. 5c). The rate performance test showed that C-SiOx/C has better electrochemical reversibility (Fig. 5d). Owing to the flexible Li–PAA interface, the material can maintain structural integrity during long cycling and continuously deliver high reversible capacity (Fig. 5e). Zhang et al.80 developed an efficient and mild method to synthesise a core–shell SiOx/C composite for LIBs. They mixed citric acid with SiOx through ball milling and obtained a uniform core–shell structure material after carbonization. The SiOx/C shows superb electrochemical performance with a high specific capacity of 1296.3 mA h g−1 and has a capacity retention of 65.1% after 200 cycles. The mild approach and excellent properties of SiOx/C make its commercial large-scale production possible. Recently, Lee et al.81 reported a method of physically combining SiOx with graphene to form a durable and high-energy hybrid anode. Graphene shows strong ductility and good adhesion on the SiOx surface, which can greatly reduce the volume expansion during multiple cycles. Thus, this synthesis method provides an efficient and simple method for the production of high-performance SiOx anode materials. Although physical strategies for synthesizing SiOx are simple and easy to operate, they can also result in the formation of many large defects on the surface of SiOx. These defects can lead to direct contact between the SiOx material and the electrolyte, which can cause the electrolyte to be consumed and result in the overgrowth of the SEI.74 SiOx can generate a uniform and dense coating on the surface by electrostatic interaction or chemical binding. Zhang et al.82 designed a sol–gel method to synthesize SiOx/C core–shell structure anode materials. They employed a wet-milling method to reduce the particle size of SiOx, which can help to mitigate the volume expansion of the electrode material during the charge and discharge cycles. After 170 cycles, it still had 97.3% capacity retention. SiOx particles were synthesized by Shi et al.83 with equimolar amounts of Si and SiOx by thermal evaporation. SiOx was mixed with dopamine solution to obtain a SiOx precursor coated with polydopamine. The polydopamine coating served as a carbon source during subsequent high-temperature carbonization, resulting in the formation of a SiOx@C composite material. The carbon coating stabilized the structure of the material and contributed to the formation of a stable SEI, and the rate performance was significantly improved. At a current density of 0.1 A g−1, the core–shell structure material exhibited a reversible capacity of 1514 mA h g−1. Similarly, Lv et al.84 reported a method to synthesize core–shell SiOx/C nanostructured materials through sol–gel synthesis, which can achieve high yield and provide experimental guidance for industrial production. This SiOx/C carbon layer was relatively thin and evenly distributed, and a specific capacity of 820 mA h g−1 could still be delivered after 100 cycles.
It is indeed worth noting that the research achievements primarily focused on single-layer carbon coatings. However, it has been observed that multi-layer coatings are more effective in mitigating the volume change of SiOx, leading to improved cycle performance.85 Li et al.86 have developed a highly efficient anode material, SiOx/C@Sn@NC, with a double-layer coating structure. This material exhibits outstanding electrochemical performance, and its preparation involves the hydrolysis of vinyltriethoxysilane (VTES) using the TEA (C6H15N) catalyst in a solution of sodium salicylate and CTAB to obtain a precursor. The precursor is then carbonized at high temperature, yielding SiOx/C nanoparticles which are then coated with Sn and N-doped carbon (Fig. 5f). The SEM analysis confirms that the material is uniformly layered and coated, while TEM shows clear lattice stripes of Sn and amorphous N-doped carbon on the outermost layer (Fig. 5g and h). The combination of multiple active components and the double-layer coating structure significantly enhances ion diffusion and charge transfer, and promotes the formation of a stable SEI capable of absorbing stress during electrode cycling. Comparison studies reveal that both Sn and N-doped carbon can improve the specific capacity and cycling performance of the material (Fig. 5i). Wang et al.87 designed a method to synthesize SiOx/C@C through solvent-free ball milling and the sol–gel method. The whole synthesis process has no solvent participation, which reduces the risk of waste and potential pollution. Carbon encapsulation and doping jointly improve the electrochemical performance of SiOx, showing a reversible capacity of 546 mA h g−1 after 1000 cycles at 1 A g−1. Similarly, Guo et al.88 designed a bilayer structure in which the Si–SiOx particles were coated with carbon and a polymer. Each layer of coating has a specific role, and the main effect of the inner layer of carbon coating is to improve the conductivity. The outer layer of the composite material can take advantage of the elasticity of the polymer, can well limit the volume expansion of Si–SiOx and enhance structural integrity during cycling. In the half cell test, the materials exhibited a stable capacity of 794 mA h g−1 at 2C after 900 cycles and a capacity retention of 83%. Moreover, in the full battery test, the capacity loss was only 20% after 1100 cycles. In addition to bilayers of carbon, SiOx itself can also be used as a shell. Zhou et al.89 designed a double-layer structure with an inner layer of SiOx and an outer layer of diatomic co-doped carbon layer. The inner layer SiOx here also acts as a buffer layer to control the volume change of the Si core, and the outer carbon layer rich in defects acts as a conductive layer. Together, the two layers can significantly improve the electrochemical performance of the material. Zhu et al.90 reported a homogeneous multilayer core–shell structure Si@SiOx@C material, in which Si was coated with SiOx and soft carbon. The thickness of SiOx can be adjusted to 2–15 nm through adjusting the calcination temperature. The multilayer structure protects the Si particles during cycling, thus enhancing the structural integrity, and significantly improves the conductivity.
As a novel and efficient SiOx/C composite structure, the porous carbon layer coating structure has attracted extensive attention. The porous carbon layer acts as a conductive matrix and can be used as a buffer layer to reduce the volume expansion of SiOx to enhance the cycle stability. The structure can reduce the length of the diffusion path of Li-ions and enhance the conductivity between electrolyte and electrode materials. Zhang et al.91 reported a porous P@C/SiOx core–shell structure material; the activated material showed a reversible specific capacity of 1151.8 mA g−1 at a current density of 0.1 A g−1 and an excellent ICE of 79.2%. However, the coulomb efficiency of carbon coated rice husk without treatment was only 45.7%. Similarly, Cui et al.92 also used rice husk as both Si and carbon sources to obtain mesoporous SiOx/C core–shell composites by carbothermic reduction of precursors. In the half battery test, the capacity attenuation of the material was less than 0.5% after 200 cycles, making it a good substitute for the graphite material. Despite the notable enhancement in electrochemical performance achieved with the use of porous coatings, the design of the coating structure for a porous carbon layer remains a challenging task. Currently, there is no well-established method for creating the desired porous structure and regulating the pore size, necessitating further experimental investigations by researchers.93
4.2. Yolk@shell SiOx/C
The yolk–shell structure is a distinctive structure based on the core–shell structure and leaves a certain space between the carbon layer and the SiOx core.94 The conventional tightly wrapped structure is limited in its ability to fully control the expansion of SiOx, which can potentially result in the cracking of the carbon layer and even structural collapse.95 The design of reserved buffer space can allow the SiOx core to shrink more freely, and the SEI film and even the entire electrode structure can maintain good integrity, thus enhancing the cycle stability (Fig. 6a).96 The yolk–shell structure is usually synthesized by the sacrificial template method. Cui et al.97 first reported the Si anode material with yolk shell structure. They synthesized a kind of amorphous carbon as the shell, which was coated on the Si nanoparticles by the sol–gel method. This yolk–shell structure benefits from this layer of SiO2 as a sacrificial layer. This layer of SiO2 is finally etched with HF to form the gap, so the thickness of SiO2 can determine the size of the final void space. Tactfully, it can be adjusted through experimental methods to achieve controllable SiO2 thickness. It was found that the sacrificial coating thickness should be larger than 30 nm to reserve enough space, so that the outer carbon shell will not be damaged when Si expands. However, this layer of void space cannot be too large, which will reduce the bulk density. The material exhibits a specific capacity of 2800 mA h g−1 at a C/10 current density.97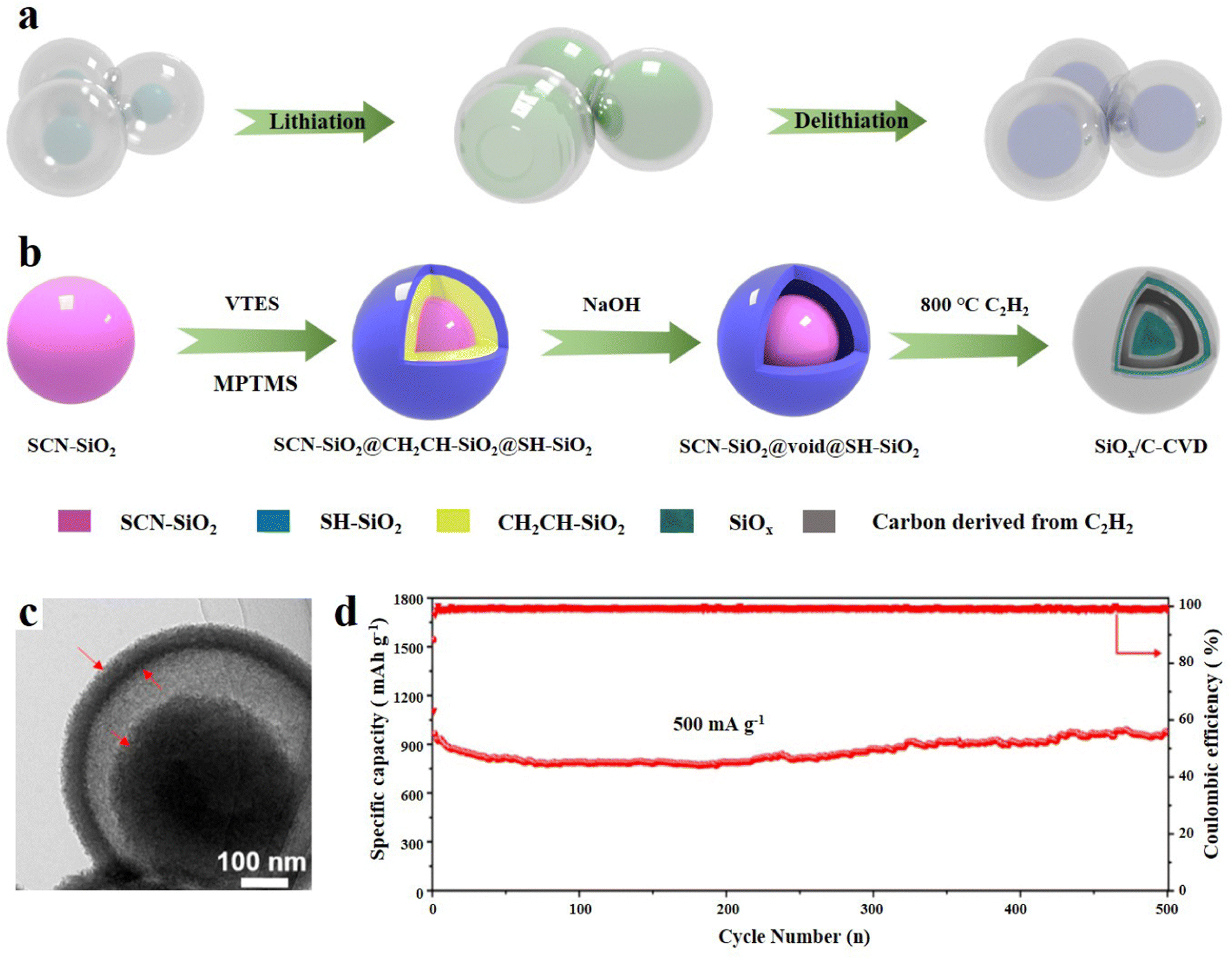 | ||
| Fig. 6 (a) The evolution of the yolk@shell structure during lithiation/delithiation. (b) Schematic illustration of the preparation of SiOx/C-CVD, (c) TEM image and (d) long-term cycling performance at 500 mA g−1.98 Reproduced from ref. 98 with permission from Elsevier, Copyright 2019. | ||
Liu et al.98 synthesized SiOx/C composites with a yolk@shell structure through the sol–gel process and selective etching. Three kinds of SiO2 with different functional groups (–SCN, –CHCH2 and –SC) were formed by hydrolysis and condensation of different silanes and coated layer by layer. First, cyano containing silane was hydrolysed to synthesize SCN-SiO2 microspheres, which were then coated. The middle layer is CH2CH–SiO2 and the outer layer is SC-SiO2. The ingenious thing is that the –CHCH2 of the intermediate layer shows weak alkalinity, which can be removed by the selective reaction of hot sodium hydroxide, thus forming a precursor with a yolk@shell structure (SiO2@void@SH-SiO2). After high temperature annealing in an Ar atmosphere, the organic phase converted into amorphous carbon, and finally SiOx@void@C was obtained. The chemical vapor deposition (CVD) technology is used for annealing in flowing acetylene and argon to form a uniform semi-graphitized carbon layer in the outer layer of SiOx (Fig. 6b). The void space can be clearly seen from the TEM images (Fig. 6c). The incorporation of a semi-graphitized carbon layer serves a dual purpose: it enhances the electron diffusion rate and addresses the issue of traditional carbon layers being prone to cracking. This unique yolk@shell structure contributes to the exceptional electrochemical performance of the material, particularly its impressive rate performance. In the first 150 cycles, the capacity attenuation is only 3%, and under a high current density of 1 A g−1 the discharge capacity is 725 mA h g−1 (Fig. 6d). Interestingly, the capacity increases after 200 cycles, which is mainly due to the reversible side reaction between the electrolyte and electrode materials. Moreover, NaOH solution is used to remove the silica layer, which is safer and more environmentally friendly than traditional HF etching.99 In addition, the design of a semi-graphitized carbon coating can also provide some experimental reference for other anode materials with an obvious volume effect.100
The incorporation of a double-layer carbon shell remains an exceptionally efficient structural design concept within the yolk@shell structure.101 Recently, Luo et al.102 synthesized a new type of bilayer yolk@shell SiOx@C@C anode material by a hydrothermal method. Firstly, Si nanospheres were obtained by hydrolysis and condensation of vinyl triethoxysilane, which were then annealed in an argon atmosphere to form SiOx/C composites. A surface modifier (PDDA) was used to treat the surface of SiOx balls, so that a layer of methyl methacrylate (PMMA) can be coated on their surface. The formation of the yolk@shell structure is because the PDDA in the middle layer will self-react and release gas during high temperature treatment, resulting in a cavity in the overall structure. The conductivity of SiOx can be significantly enhanced by incorporating additional internal carbon shells in its structure. Moreover, the design of a double layer carbon shell aids in the formation of a more stable SEI film. The uniform coating of the inner carbon shell on the core effectively prevents undesired side reactions between the electrolyte and SiOx, thereby minimizing electrolyte consumption. Based on the above findings, the yolk@shell structure shows superior capacity and cycle performance to the core–shell SiOx/C@C structure. After 500 cycles at 0.5 A g−1, the material exhibits a discharge capacity of 770 mA h g−1, with an ICE of 99%. Wang et al.103 synthesized yolk@shell N,S co-doped SiOx/C@void@C by a one-step method. Co-doping based on the double-layer carbon shell further improves the overall conductivity, and the material still has 464 mA h g−1 energy density under a high current density of 5 A g−1. The combination of a simple and efficient synthesis method and the demonstrated excellent electrochemical performance firmly establishes this material as a highly promising candidate for anode materials in LIBs.
Environmentally friendly synthesis routes hold greater practical value.104 Zhang et al.105 developed a HF-free method to synthesize a yolk@shell structure SiOx/C@C composite. Two layers of the polymer shell are generated on the Si core, and the middle layer is polydopamine, which can be selectively etched by NaOH to form voids. The outer layer of polypyrrole can form a N-doped carbon shell after carbonization at high temperature, which greatly improves the conductivity. This composite can still provide 804 mA h g−1 capacity at 0.1 A g−1 after 1000 cycles. Recently, Chen et al.106 also designed a HF-free strategy to synthesize a new type of SiOx/C@void@Si/C yolk@shell structure. SiO2 containing vinyl is synthesized through a hydrothermal reaction, and upon vinyl carbonization, amorphous carbon is formed. This amorphous carbon can create a core–shell structure, with SiOx embedded within it. Through the use of molten AlCl3, metal Al is then able to reduce SiO2 at low temperatures, resulting in the generation of Si. As Ostwald ripening occurs, the newly formed Si diffuses outward and merges, ultimately forming a perfectly spherical structure. With a sufficient reaction time, a void space is created between layers, forming the yolk@shell structure. In addition, the gap size of the void space can be effectively adjusted by the time of aluminothermic reduction. The synergistic effect of the two layers of carbon not only enhances the conductivity, but also mitigates the effect of the volume change of SiOx and Si in maintaining structural integrity. SiOx/C@void@Si/C shows excellent cycle stability. After 550 cycles, there is an initial reversible specific capacity of 1093 mA h g−1 at 0.2 A g−1. Similarly, Gong et al.107 reported a unique SiOx@TiO2@C negative electrode material. Tetrabutyl titanate was hydrolysed to coat TiO2 on the surface of SiOx by the sol–gel method, and phenolic resin was formed on the surface of SiOx by the reaction between resorcinol and formaldehyde, and a carbon shell was formed after calcination. TiO2 is a typical amphoteric oxide that can be etched by NaOH to avoid the environmental harm caused by HF. Furthermore, the size of the void space can be adjusted by varying the etching time. By adapting well to the volume expansion of SiOx, TiO2 can effectively enhance the electrochemical performance. A specific capacity of 1290 mA h g−1 is delivered under a current density of 0.1 A g−1.
4.3. Novel structures of SiOx/C
The SiOx/C composite exhibits a distinctive and adaptable structure–activity relationship.50,108 The addition of carbon not only improves the conductivity of SiOx, but also maintains the micro-integrity of the electrode material.109 The continuous exploration of the SiOx/C structure has led to the steady optimization of the physical and electrochemical properties of SiOx based anode materials.110 The core–shell structure and yolk@shell structure of SiOx/C have been previously discussed, highlighting their structural characteristics and performance characteristics. Furthermore, SiOx/C composites showcase a wide array of structures that go beyond the aforementioned types, and demonstrate exceptional performance.111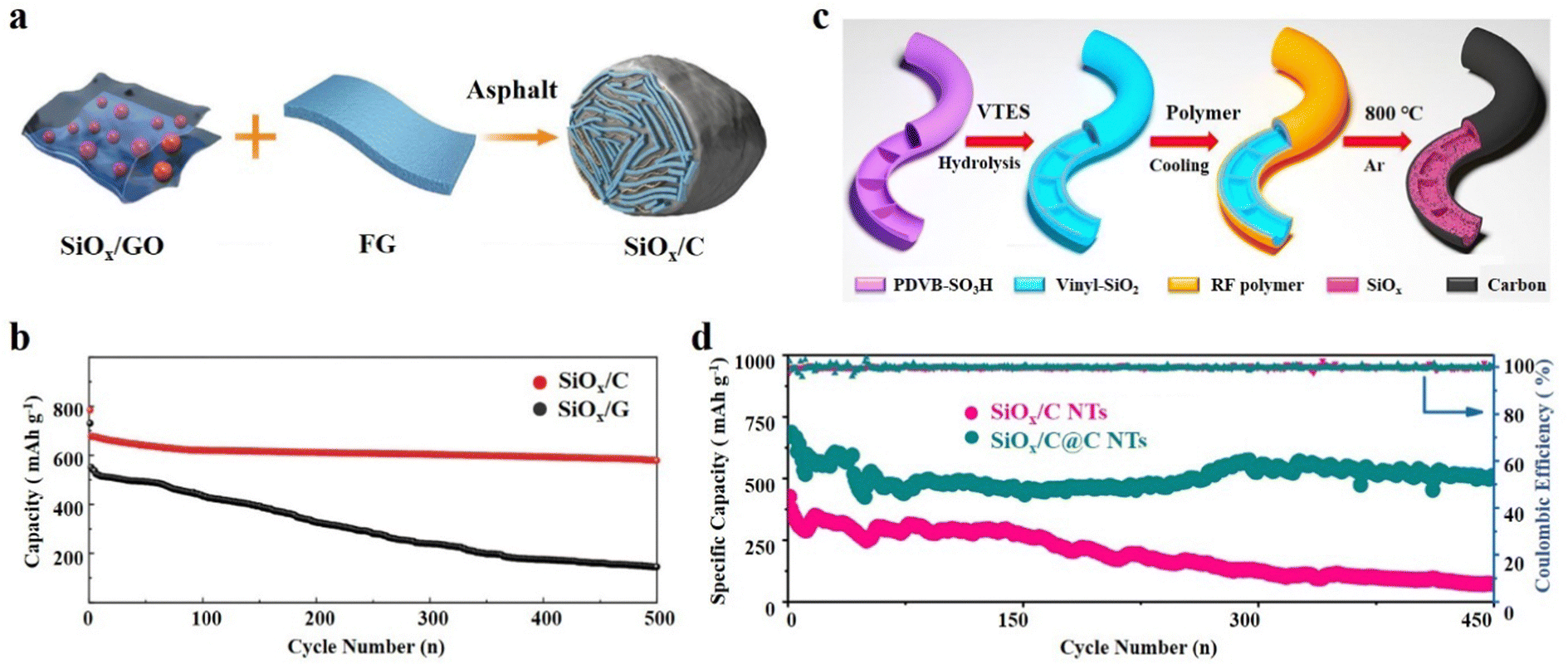 | ||
| Fig. 7 (a) Schematic illustration of the synthetic process of the SiOx/C hydrogel and (b) cycling performance.117 Reproduced from ref. 117 with permission from John Wiley and Sons, Copyright 2017. (c) Schematic illustration of the preparation process of SiOx/C@C NTs and (d) the long-term cycling performance at 500 mA g−1 of SiOx/C@C NTs.120 Reproduced from ref. 120 with permission from Elsevier, Copyright 2022. | ||
5. Rational design strategy for SiOx/C
5.1. Carbon types in SiOx/C
During charging/discharging, volume expansion and contraction can lead to mechanical damage and a decrease in the electrochemical performance of the electrode material.121,122 Carbon materials have good ductility and conductivity, and their composites with SiOx effectively alleviate the inherent shortcomings of SiOx, greatly promoting the development of SiOx/C composite materials.123 Due to the excellent performance of carbon materials and their abundant reserves on the Earth, researchers have developed various allotropes of carbon, such as graphene, carbon nanotubes, and fullerenes, by utilizing biomass, polymer carbonization, etc. (Fig. 8a and b). Different carbon types in SiOx/C composites have unique physical and chemical properties, and there is desirable method to composite each carbon materials with SiOx.124 For example, graphene and carbon nanotubes are suitable for mechanical mixing and co-precipitation, while polymer carbonization is more suitable for the sol–gel method. The currently commercialized SiOx/C material is a composite of SiOx and carbon materials added to graphite or graphene through a specific process.125 However, the amount of SiOx added is still relatively low, resulting in a lower specific capacity of the composite material.126,127 | ||
| Fig. 8 (a) Commercial carbon materials for SiOx/C composites. (b) Several carbon material precursors. | ||
The development of high value SiOx/C composites using inexpensive biomass has gained attention due to renewable and rich reserves of carbon in nature. Biomass is often used as both a Si source and a carbon source, and biomass can be easily carbonized at high temperatures to obtain graphite like materials.134 Yu's team utilized sugarcane leaves as raw materials and employed a molten salt-assisted low-temperature aluminothermic reaction to prepare a porous Si–SiOx@Cu composite material.135 They mixed sugarcane leaf powder with CuCl2 and ZnCl2 and calcined the mixture under an argon atmosphere. After thoroughly washing away the chloride salt with HCl and drying, they obtained a porous SiOx/C precursor. The precursor was reacted with the mixture of AlCl3 and Al powder in an argon atmosphere, and Al and Al-containing composites were washed away to obtain Si–SiOx@C. The oxygen content of Si can be adjusted by varying the amount of aluminium powder added. After 400 cycles, a reversible capacity of 1562.8 mA h g−1 was observed at 0.2 A g−1. Similarly, Chen and colleagues prepared layered porous carbon-coated SiOx/C composites using bamboo shoot shells as raw materials.136 Guo et al.137 employed dried bamboo leaves to directly calcine and generate the SiOx/NC precursor which was then calcined to produce N-doped carbon. The precursor was then subjected to a hydrothermal reaction with Ni(OAc)2·4H2O to obtain Ni3Si2O5(OH)4/NC, which was further used to prepare a series of Ni/SiOx/NC composite anodes by calcination at varying temperatures. It is worth noting that animal protein can also produce doped carbon during the calcination process, which significantly enhances the overall conductivity of the material. On the other hand, Meng and team surface-treated commercial Si powder to obtain Si@SiOx, which was mixed with egg white and transferred to polytetrafluoroethylene hydrothermal kettle for molecular cooking. Finally, the product Si@SiOx@C was obtained by calcination in a tube furnace. The outer layer of N-doped carbon not only enhances the conductivity but also maintains the interface stability of the electrode material. This double-layer structure effectively acts as a buffer, alleviating the internal strain experienced by the Si core during cycling.138
| Anode | Cycling stability (mA h g−1) | Rate capability (mA h g−1) | Carbon types | Ref. |
|---|---|---|---|---|
| SiOx/G | 624 after 1000 cycles at 1 A g−1 | 661 at 10 A g−1 | Graphene | 146 |
| SiOx/TiO2@MLG | 966 after 400 cycles at 2 A g−1 | 251 at 10 A g−1 | Graphene | 141 |
| SiOx@CNTs/C | 1080 after 400 cycles at 0.5 A g−1 | 284 at 10 A g−1 | Carbon nanotubes and graphite | 18 |
| C-SiOx/C | 234 after 200 cycles at 0.5 C | 1191 at 5 C | Carbon nanotubes and graphite | 15 |
| SiOx/MWCNTs/N-doped C | 621 after 450 cycles at 0.1 A g−1 | 388 at 800 mA g−1 | MWCNTs and graphite | 145 |
| NC@SiOx@m-C | 583 after 500 cycles at 1 A g−1 | 495 at 2 A g−1 | Graphite | 130 |
5.2. Doped carbon materials
Doping carbon materials, such as graphite, in SiOx/C composites offers several benefits. Firstly, it increases the density of defects in graphite, which reduces the energy barrier and band gap, thereby improving the electronic conductivity of the SiOx/C composite. Additionally, carbon doping increases the electronegativity and the presence of surface-active sites on graphite, enhancing its interaction with SiOx (Fig. 9a).147 Han et al.148 employed a high-temperature heating process to decompose organosilicon compounds within a sealed vessel. This decomposition resulted in the formation of SiOCN (SiO/N-doped carbon) microspheres. Through CVD technology, a carbon layer is grown on the surface of SiOCN in the mixed atmosphere of CH4 and H2. In this continuous high-temperature CVD process, with the conversion of C–H into C–C bonds through dehydrogenation reaction, the SiOCN is reduced to VG@SiOx/NC and the N atoms come from the amino group of the organosilicon. As observed from the SEM images, VG@SiOx/NC maintains the spherical morphology of highly round, smooth, and well-dispersed SiOCN microspheres. Moreover, the porous structure form on the surface, which not only plays a critical role of the Li+ transport route during cycling but also improves the contact of the electrolyte with active materials. The N doping of the carbon layer can also cause the formation of large defects. These defects can increase the lithium-ion accommodation and be favourable for buffering the volume expansion of SiOx. More importantly, the VG@SiOx/NC fixed three-dimensional structure shows stable electrochemical performance during the charging/discharging process, which delivered a high initial reversible capacity of 1323.8 mA h g−1. Hu et al.149 formed a resin coating layer on the surface of SiOx through m-phenylenediamine and formaldehyde by amine aldehyde condensation reaction. After high temperature annealing, SiOx@NC with a core–shell structure is obtained. This core–shell enveloping structure can well buffer the volume change issue of SiOx in lithium storage because the carbon layer is highly mechanically stable and highly electrically conductive. As a result, SiOx@NC manifests a high reversible capacity with outstanding durability.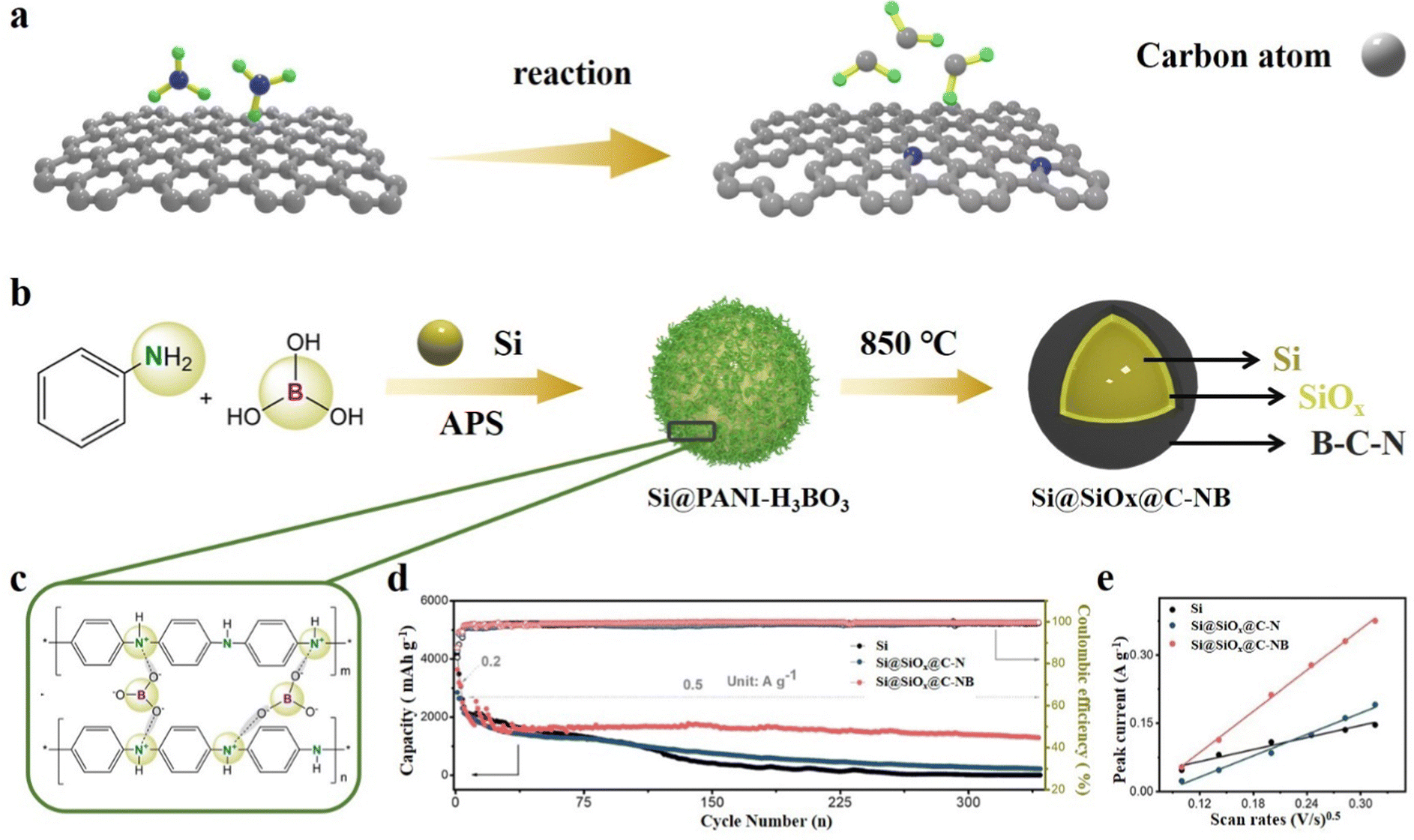 | ||
| Fig. 9 (a) Schematic diagram of doping manufacturing defects. (b) Synthetic approach of Si@SiOx@C-NB, (c) polymer molecular structure, (d) cycling stability for 340 cycles at 0.5 A g−1 and (e) linear fits for varying sweeping rate CV profiles.89 Reproduced from ref. 89 with permission from John Wiley and Sons, Copyright 2022. | ||
The co-doping of non-metallic atoms can significantly improve the mechanical and electrochemical properties of the carbon layer. Zhou et al.89 reported a double-layer coated structure of Si particles with a B,N co-doped carbon outer layer and a SiOx inner layer through the polymer-H3BO3 copyrolysis process. This structure benefits from the incorporation of polyaniline as both a carbon-network provider and a N source. Polyaniline contributes to the formation of a conductive network, enhancing the overall electrical conductivity of the composite. Additionally, the presence of boric acid enables electrostatic interactions with polyaniline, leading to the formation of a polymer network on the surface of Si particles (Fig. 9b and c). At high temperatures, with the precipitation of CO2, H2O and NH3, Si@SiOx@C-NB is obtained. Because of the synergistic effects of N and B atoms, the flexible carbon layer of Si@SiOx@C-NB contributes to better LIB performance. The Si@SiOx@C-NB composite revealed a charge capacity of 690 mA h g−1 at 5 A g−1 and it is obviously better than that of the single atom N-doped composite (Fig. 9d). The cyclic voltammetry peak current of different materials confirm the optimization of electronic conductivity by doping (Fig. 9e). Shi et al.150 synthesized composite materials by embedding SiOx in N,S co-doped graphene, with thiourea (CN2H4S) as the source of both N and S. Graphene oxide, SiOx and thiourea are ultrasonically dispersed and annealed at high temperature to obtain SiOx/N,S-rGO. The wrinkled N,S co-doped graphene can increase the specific surface area and enhance the electronic conductivity between SiOx microparticles. Meanwhile, it may also relieve the stress and accommodate the volume changes during cycling. Zhang et al.151 used rice husk as the source of both C and Si, NH4HB4O7 as the heteroatom source and a porogen reagent to synthesize a B,N co-doped porous C/SiOx composite by a one-step calcination strategy. With the homogeneous distribution of B and N in the porous C framework, the electronic and Li+ conductivity can be substantially enhanced and produce abundant adsorption sites for lithium-ion storage. Due to the unique porous structure and B,N co-doping, the composite material achieves large coulomb efficiency and specific capacity. Similarly, Guo et al.152 also used rice husk as the Si/C source and synthesized an Fe and N co-doped SiOx/Fe–N–C composite by an electrospray carbonization process. Fe single atoms encapsulated in N-doped graphite frameworks improve the dispersion of SiOx, so that the SiOx/Fe–N–C composite shows excellent electrochemical stability. Metals can also be embedded in the SiOx matrix, which can greatly improve the reaction kinetics of SiOx.
5.3. Surface modification
The carbon coating improves the conductivity of the material, buffers the volume change of SiOx, and greatly improves the cycle stability.153 Despite its potential, the diffusion of the carbon layer to Li+ is limited, making it challenging to form a stable SEI. This issue impedes the overall performance improvement of SiOx/C composites. Therefore, it is a promising surface modification strategy to combine hybrid coatings with other high-performance materials.138 Coating with metal oxides has been proved to be efficient and feasible, such as Al2O3,154 CeO2,155 MgO,156 ZrO2,157,158 and TiO2.141,159 The surface modification of electrodes with metal oxides can reduce the direct contact between the SiOx core and the electrolyte, and reduce the consumption of Li+ to form a stable SEI.160,161 Xiao et al.162 synthesized SiOx@TiO2@C composite materials with a double-layer structure via an expandable experimental route. The process involves mixing SiOx particles with titanium butoxide, followed by heating and stirring to produce SiOx@TiO2 precursors. CVD technology is then utilized to initiate a carbon coating reaction using propylene as the carbon source, resulting in the final product SiOx@TiO2@C. TiO2 has strong mechanical stability, and the electrode material has small volume changes after lithium insertion, which can maintain the stability of the SEI. High electrochemical reversibility will not cause too many side reactions and capacity attenuation. Furthermore, the inclusion of a TiO2 layer in the composite contributes to enhancing the overall conductivity of the material. TiO2 possesses favourable electronic conductivity and ionic conductivity properties, allowing it to facilitate efficient charge transport within the material. This improved conductivity enables the composite to adapt to high currents, making it suitable for applications that require high-power performance. It is hybridized with a highly graphitized carbon layer and uniformly coated on the surface of SiOx particles, forming an efficient conductive network, greatly reducing side reactions, and significantly improving the ICE. The thermal effect of the reaction between LixSi and the electrolyte affects the safety of LIBs, which can lead to combustion and even explosion. The experimental results demonstrate that direct carbon coating on the surface of SiOx@C has limited suppression of thermal effects. However, the use of SiOx@TiO2 significantly inhibits the generation of heat. This more stable thermal performance can be attributed to the excellent thermal stability of the lithium titanate phase. In the traditional magnesium thermal reduction reaction to generate SiOx based negative electrode materials, HCl solution is usually used to wash away the remaining Mg and the reaction by-product MgO. Then a porous structure will be formed to alleviate the volume expansion problem. However, recent studies have found that MgO can dehydrate the electrolyte and eliminate the conditions causing HF generation, thus avoiding the corrosion of the collector and SiOx. In a study conducted by Xu et al.,155 SiOx was obtained from SiO2 through a process called magnesium thermal reduction. During this process, in situ MgO particles were incorporated into the SiOx matrix, resulting in the formation of a SiOx/MgO composite. Subsequently, a polymer shell was formed through phenolic condensation, encapsulating the SiOx/MgO composite. After annealing, the composite transformed into SiOx/MgO/Mg2SiO4/C. In the final composite structure, both MgO and Mg2SiO4 phases were uniformly distributed within the SiOx matrix. This distribution plays a crucial role in buffering volume changes that occur during charge and discharge cycles. By buffering volume changes from all directions, the composite effectively inhibits material cracking and maintains structural integrity during cycling. In addition, the formation of Mg2SiO4 inhibits the formation of lithium silicate, greatly improving the initial coulomb efficiency. Cui et al.163 used rice husk as a raw material to obtain the porous SiO2/C precursor after carbonization. Then the precursor was ground with Al powder and annealed at high temperature, obtained the Al-doped porous C/SiO2. Al2O3 with a suitable band gap and superior ionic conductivity can improve the rate performance of the material. Similarly, Al2O3, as an amphoteric oxide, can inhibit the side reaction of LiPF6 based electrolyte to generate HF. Although Al2O3 has a negative impact on the electronic conductivity, the remaining Al nanoparticles can alleviate the influence of Al2O3 on conductivity. LIBs exhibit high ICE and excellent cycle stability. The metal oxide layer is relatively inert, which is not conducive to electronic conductivity. In recent years, SiOx has also been coated with lithium salts. Kang et al.164 reported a single layer LiF coated SiOx anode material. LiF has a high Young's modulus and interface energy, which allow it to adhere well to SiOx. During battery cycling, an inorganic-rich SEI layer is formed, which is distinct from the traditional SEI and offers superior conductivity and structural stability. Furthermore, the use of LiF in the SEI layer can enhance the thermal stability, reduce electrolyte decomposition, and improve overall battery performance. Gu et al.165 designed a Li3PO4 coating, which can reduce the energy barrier of Li+ transmission, and combined it with the carbon conductive framework, which greatly improved the transmission capacity of electrons and ions. In addition, Li3PO4 contributes to the formation of a stable SEI and the ICE is also improved.5.4. Prelithiation
The ion conductivity and cycling performance of SiOx based electrode materials exhibit a positive correlation with the oxygen content.166 Pan et al.167 conducted electrochemical impedance testing and constant current intermittent titration experiments and found that the diffusion rate of Li+ was higher in SiOx than in Si, which enabled SiOx to charge/discharge more rapidly. After oxygen enrichment on the surface of SiOx, a large amount of LixSiOy and Li2O will be generated during the first charge and discharge to form an SEI, resulting in irreversible consumption of Li+ and thus reducing the ICE. In order to compensate for the loss of active lithium after the battery is assembled, prelithiation is an efficient strategy.168 The fundamental concept behind prelithiation is to utilize an external lithium source to compensate for the lithium consumption that occurs during the initial cycle of the battery. Various prelithiation strategies exist, each requiring a distinct approach and set of materials.169 In general, all prelithiation strategies can be divided into chemical prelithiation and electrochemical prelithiation from the reaction mechanism. Cathode prelithiation additives can be used easily, and electrochemical prelithiation is the most common for anode materials.170Chemical prelithiation is realized by a chemical reaction between lithium metal or other lithium rich materials and active materials.171 Lithium metal is the most typical anode prelithiation material because of its lowest electrochemical potential, and it can be lithiated through direct contact with electrode materials.172 When lithium metal contacts with active substances, internal short circuit or self-discharge will occur, which will cause Li+ to embed into the anode material and react to lithiate the anode material. In addition, this lithiation process can also form a stable SEI on the surface of the anode material, reducing the side reaction of SiOx. Although the strategy of directly adding lithium directly improves the ICE, the addition amount of lithium metal also needs to be controlled because the excess lithium will produce dendrites during cycling, which will puncture the separators and affect the safety of the battery. Meng et al.173 pressed SiOx, the resistance buffer layer (RBL) and lithium foil together. The RBL is carbon nanotubes coated with PVB, which has high electronic and ionic conductivity. By adjusting the coating parameters of the RBL, the resistance value can be changed, thereby altering the electrode current during lithiation. Lower resistance values lead to higher currents, resulting in faster lithiation rates and greater degrees of lithiation. This simple method realizes the adjustable rate and degree of lithiation and can also make the lithiation more uniform. The treated SiOx anode significantly improved the ICE (68% to 87%) in the full battery and could stably circulate for more than 200 cycles. In addition to direct contact with lithium metal, some lithium rich compounds can also be added to the anode materials to achieve prelithiation, such as Li2.6Co0.4N, LixSi, etc. Zhao et al.174 used SiOx as the raw material to obtain the LixSi/Li2O composite through low-cost synthesis. The LixSi nanodomain is uniformly embedded in the Li2O crystal, and the ICE of this lithiated composite material is increased from 52.6% to 93.8%; even when mixed with other anode materials, the ICE can exceed 100%. In addition, irreversible Li2O can be used as a protective layer on the surface of materials, which can maintain stable electrochemical performance even in an environment with 40% relative humidity. Although the LixSi alloy has low oxidation–reduction potential and high specific capacity, it can be used for prelithiation of almost all kinds of anode materials. However, LixSi is unstable and easily deteriorates when exposed to air, so its industrial application still faces great challenges. Solution chemical prelithiation is also an efficient and safe prelithiation strategy. The initial coulomb efficiency of Li+ is improved by inserting the electrode material into a lithium rich organic solution for redox reaction. The reaction in solution can make the lithiation process uniform and maintain the integrity of the electrode.175 In the research conducted by Yan et al.,175 they employed a method of prelithiation by dissolving lithium tablets in a biphenyl solution of tetrahydrofuran. The SiOx/C material was then exposed to the lithium biphenyl complex present in the composite solution. As a result of this interaction, a reaction occurred between SiOx/C and the lithium species. This reaction led to the formation of a lithium–silicon oxide compound, denoted as LixSiOy, on the surface of the SiOx/C material. It can obviously cause lithium consumption during the first cycle and greatly improve the coulomb efficiency.
Electrochemical prelithiation is considered a viable and user-friendly approach compared to chemical prelithiation.176 Prior to assembling the electrode material into the battery, a half cell is created using lithium metal or other lithium-containing electrode materials as the counter electrode.177 This half cell is then charged to compensate for the lithium depletion that occurs in the negative electrode material.178 The speed and degree of prelithiation can be precisely controlled by adjusting various parameters. Electrochemical prelithiation can form a uniform and dense SEI, but the need to assemble and disassemble the battery system increases the difficulty of large-scale application.177 Additionally, specific charging voltage parameters for prelithiation are associated with different electrode materials. When the voltage is excessively high, full lithiation of lithium may not occur, while excessively low levels can cause the formation of observable lithium dendrites. In conclusion, multiple prelithiation methods exhibit unique characteristics. However, research on prelithiation technology remains relatively underdeveloped and immature at present.176
5.5. Binders
The inherent drawback of SiOx based negative electrode materials is the expansion of volume during the cycling process. Although the use of carbon materials in composites has effectively addressed this issue, there is still potential for further improvement.179 In order to overcome these shortcomings, a lot of research has been done to promote the industrialization of SiOx.180 The design of the binder also shows great potential for improving the performance of SiOx based materials.181 The adhesive combines the active material and the conductive agent, so the first consideration standard of the adhesive is strong interface adhesion and strong Li+ conductivity.182 The volume expansion of SiOx will cause the material to crack or even pulverize, so an efficient binder should have good mechanical elasticity and strength. Elasticity can absord the stress in the process of circulation and keeps particles in contact.183 The development of adhesives should mainly focus on designing polymer adhesives rich in –COOH, –OH, –NH2 and other polar groups.184 The traditionally used adhesive is polyvinylidene fluoride (PVDF), but its application in SiOx based materials cannot provide good adhesion and mitigate buffer volume change. Sodium carboxymethyl cellulose (CMC) is a commonly used adhesive due to containing a large number of hydroxyl and carboxyl groups, which enable strong adhesion to the SiOx surface. However, during significant volume changes of SiOx, CMC can slide and cause the material to detach or fall off. In view of this, Xue et al.185 used CMC as the binder to add a small amount of Si particles as the nano-comb when making the slurry. The polar groups in the polymer can interact with the surface of Si nanoparticles to open the molecular chain of CMC. Stretched molecular chains can provide maximum adhesion and can also increase the surface contact with SiOx to generate a uniform conductive network. Liao et al.186 obtained a three-dimensional cross-linked adhesive, c-CMC-IDA150, by in situ thermal shrinkage and reaction between iminodiacetic acid (IDA) and CMC. The crosslinking degree can be well adjusted by adjusting the mass fraction of IDA and heat treatment temperature. c-CMC-IDA150 has many contact sites interacting with SiOx, which improves the surface adhesion. In addition, c-CMC-IDA150 can also adapt to the displacement and volume change of SiOx particles during circulation.The 3D network structure serves as an excellent stress buffer for SiOx volume changes, thereby preserving the integrity of the electrode structure.187 Furthermore, the 3D network can enhance the overall electronic conductivity of materials and facilitate the formation of stable SEI layers. The majority of binders exhibit favourable elasticity, helping to maintain the overall structure of the electrode.188 Nevertheless, the volume of SiOx may undergo significant changes if the elasticity of the binder is not suitable. Volume expansion extends the diffusion path of Li+, which seriously affects the rate performance of the material. Compared with van der Waals forces and covalent bonds, hydrogen bonds have a dynamic self-healing ability and strong mechanical properties. Tang et al.189 introduced tannic acid into linear polyacrylic acid, which formed a stable and reversible 3D cross-linking network structure under the action of a large number of hydrogen bonds. Hydrogen bonding can also provide SiOx based materials with strong interfacial adhesion and cohesiveness. Compared with the CMC binder, a large number of hydrogen bonds can be dynamically broken and recombined at room temperature, resulting in excellent electrode stability. After 50 cycles, no significant electrode cracking was observed, and 1025 mA h g−1 was still achievable after 250 cycles under 500 mA g−1. Similarly, Weng et al.190 utilized free radical polymerization of acrylamide and acrylic acid to produce covalent–noncovalent networks with exceptional elasticity. The presence of carboxyl and amide groups within the network of a material enables the formation of hydrogen bonds. These hydrogen bonds play a crucial role in enhancing the structural stability of the electrode. Furthermore, materials that are rich in hydroxyl groups (–OH) exhibit strong interactions with other components, such as binders. This strong interaction helps to mitigate binder agglomeration, which refers to the clustering or agglomeration of the binder material within the electrode.
As binders usually exhibit relatively weak conductivity, it is crucial to control the proportion of the binder. In recent years, increasing the conductivity of adhesives has also attracted extensive attention. Song et al.184 designed a cross-linked conductive adhesive (CCB) with strong mechanical properties. Starting from the molecular level, it not only enhances the intrinsic electronic conductivity, but also shows good mechanical properties during electrochemical cycling through the covalent bond connection between the binders.
6. Conclusion
SiOx (silicon oxide) is considered one of the most promising anode materials for lithium-ion batteries due to its high specific capacity. However, it is important to note that there are still challenges and issues that need to be well addressed in order to realize its full potential. The huge volume change and low electron transmission are the two key issues of SiOx during cycling. In this review, we systematically summarize the design of SiOx/C composite materials, focusing on their three-dimensional structure from internal and external aspects. Firstly, based on the design of the SiOx core, the characteristics of spherical structures, nanowires and nanofilms in electrochemical performance are discussed. The hollow structure of SiOx based anode materials offers plenty of space to accommodate the volume expansion during cycling, resulting in exceptional stress-buffering capabilities. Nanowires and nanofilms exhibit higher volume energy density, as well as an enhanced electron and ion transport ability. The oxidation degree of SiOx is controlled by adjusting the amount of the reducing agent. A suitable oxygen content generates a more stable SEI, and excessive lithium will not be consumed during the initial cycle. The recombination with carbon has been proved through many studies to greatly improve the conductivity of SiOx and provide a stable electrode/electrolyte interface. In addition, carbon materials have good elasticity and can buffer the volume change of SiOx during charging. The different SiOx/C composite structures of SiOx/C are distinguished. The yolk@shell structure can adapt to the volume change of SiOx well without affecting the specific mass capacity. The coating of the carbon layer can also reduce the direct contact between the electrolyte and SiOx, and improve the stability of the SEI. On the basis of SiOx/C, the carbon source can also be the development direction of advanced SiOx/C composites, and the influence of carbon content on their specific capacity and cycle stability is emphasized. Furthermore, this review also summarizes various non-metallic atom doping methods that can enhance the intrinsic conductivity of the carbon coating layer. During the initial cycles, SiOx electrodes may undergo significant side reactions, which can reduce the coulombic efficiency. To address this issue, a promising strategy is prelithiation of the electrode before cycling, thereby compensating for the lithium consumption that occurs during the first cycle. As SiOx/C composites have better structural stability and conductivity during cycling, the choice of the binder will also determine their application. Three-dimensional crosslinked adhesives have a strong influence on mechanical and conductive properties and could be crucial for high performance Si/C Li ion batteries.Despite ongoing research efforts to improve the electrochemical properties of high performance SiOx based anode materials, challenges remain. For instance, to address the serious volume expansion and poor conductivity of SiOx electrodes, researchers have developed hollow or porous SiOx particles by the sacrificial template approach. However, this remains a challenge for industrial scale production due to the high production costs and complex processes involved. While improving the performance parameters of SiOx in energy storage is crucial, attention must also be given to its scalability for mass production. The controlled, predictable, economical, and systematic combination of SiOx and carbon can guide the development of advanced and practical SiOx/C composites and electrode materials, while also providing valuable insights into other high capacity energy storage systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Key Research and Development Projects of Sichuan Province (2023YFG0222), the “Tianfu Emei” Science and Technology Innovation Leader Program in Sichuan Province (2021), the University of Electronic Science and Technology of China Talent Start-up Funds (A1098 5310 2360 1208), and the National Natural Science Foundation of China (21464015 and 21472235).References
- Z. Li, W. Tang, Y. Yang, G. Lai, Z. Lin, H. Xiao, J. Qiu, X. Wei, S. Wu and Z. Lin, Adv. Funct. Mater., 2022, 32, 220615 Search PubMed.
- K. Andrew, O. Seok Hyeon, A. Arindam, R. S. Bhaskar, K. Sandeep and P. Rajkumar, J. Mater. Chem. A, 2023, 15, 7833–7866 Search PubMed.
- Z. Song, T. Zhang, L. Wang, Y. Zhao, Z. Li, M. Zhang, K. Wang, S. Xue, J. Fang, Y. Ji, F. Pan and L. Yang, Small Methods, 2022, 6, 2101591 CrossRef CAS PubMed.
- Q. Li, R. Yi, Y. Xu, X. Cao, C. Wang, W. Xu and J.-G. Zhang, J. Power Sources, 2022, 548, 232063 CrossRef CAS.
- R. Li, C. Fu, B. Cui, C. Cui, X. Mu, Y. Gao, G. Yin and P. Zuo, J. Power Sources, 2023, 554, 232337 CrossRef CAS.
- F. Lin, J. Luo, Y. Zhang, J. Zhu, H. A. Malik, Z. Wan and C. Jia, J. Mater. Chem. A, 2023, 11, 2544–2567 RSC.
- Y. T. Malik, S.-Y. Shin, J. I. Jang, H. M. Kim, S. Cho, Y. R. Do and J.-W. Jeon, Small, 2022, 19, 2206141 CrossRef PubMed.
- D. Wang, C. Zhou, B. Cao, Y. Xu, D. Zhang, A. Li, J. Zhou, Z. Ma, X. Chen and H. Song, Energy Storage Mater., 2020, 24, 312–318 CrossRef.
- S. E. Wang, M. J. Kim, J. W. Lee, J. Chun, J. Choi, K. C. Roh, Y. C. Kang and D. S. Jung, Small Methods, 2022, 6, 2200430 CrossRef CAS PubMed.
- Q. Zhang, C. Zhang, W. Luo, L. Cui, Y.-J. Wang, T. Jian, X. Li, Q. Yan, H. Liu, C. Ouyang, Y. Chen, C.-L. Chen and J. Zhang, Adv. Sci., 2020, 7, 2200749 Search PubMed.
- W. Zhang, S.-B. Son, H. Guthrey and C. Ban, Energy Storage Mater., 2022, 49, 111–121 CrossRef.
- W. Chen, R. V. Salvatierra, M. Ren, J. Chen, M. G. Stanford and J. M. Tour, Adv. Mater., 2020, 32, 2002850 CrossRef CAS PubMed.
- G. Ge, G. Li, X. Wang, X. Chen, L. Fu, X. Liu, E. Mao, J. Liu, X. Yang, C. Qian and Y. Sun, Nano Lett., 2021, 21, 3127–3133 CrossRef CAS PubMed.
- J. Im, J.-D. Kwon, D.-H. Kim, S. Yoon and K. Y. Cho, Small Methods, 2022, 6, 2101052 CrossRef CAS PubMed.
- G. Li, L.-B. Huang, M.-Y. Yan, J.-Y. Li, K.-C. Jiang, Y.-X. Yin, S. Xin, Q. Xu and Y.-G. Guo, Nano Energy, 2020, 74, 104890 CrossRef CAS.
- H. Li, H. Li, Z. Yang, L. Yang, J. Gong, Y. Liu, G. Wang, Z. Zheng, B. Zhong, Y. Song, Y. Zhong, Z. Wu and X. Guo, Small, 2021, 17, 2102641 CrossRef CAS.
- Y. Li, Y. Qian, J. Zhou, N. Lin and Y. Qian, Nano Res., 2022, 15, 230–237 CrossRef CAS.
- H. Tian, H. Tian, W. Yang, F. Zhang, W. Yang, Q. Zhang, Y. Wang, J. Liu, S. R. P. Silva, H. Liu and G. Wang, Adv. Funct. Mater., 2021, 31, 2101796 CrossRef CAS.
- Y.-F. Tian, G. Li, D.-X. Xu, Z.-Y. Lu, M.-Y. Yan, J. Wan, J.-Y. Li, Q. Xu, S. Xin, R. Wen and Y.-G. Guo, Adv. Mater., 2022, 34, 2200672 CrossRef CAS.
- Y. Weng, G. Chen, F. Dou, X. Zhuang, Q. Wang, M. Lu, L. Shi and D. Zhang, J. Energy Storage, 2020, 32, 101856 CrossRef.
- W. Zhang, Y. Weng, W. Shen, R. Lv, F. Kang and Z.-H. Huang, Carbon, 2020, 158, 163–171 CrossRef CAS.
- M. Ashuri, Q. He and L. L. Shaw, J. Power Sources, 2023, 559, 232660 CrossRef CAS.
- H. Li, S. Zhu, S. Li, Q. Zhang, J. Zhao and L. Zhang, Chem. J. Chin. Univ., 2021, 42, 2342–2358 CAS.
- H. R. Philipp, J. Phys. Chem. Solids, 1971, 32, 1935–1945 CrossRef CAS.
- H. R. Philipp, J. Non-Cryst. Solids, 1972, 8–10, 627–632 CrossRef CAS.
- R. J. Temkin, J. Non-Cryst. Solids, 1975, 17, 215–230 CrossRef CAS.
- A. Hohl, T. Wieder, P. A. van Aken, T. E. Weirich, G. Denninger, M. Vidal, S. Oswald, C. Deneke, J. Mayer and H. Fuess, J. Non-Cryst. Solids, 2003, 320, 255–280 CrossRef CAS.
- E. Fuglein and U. Schubert, Chem. Mater., 1999, 11, 865–866 CrossRef.
- A. Hirata, S. Kohara, T. Asada, M. Arao, C. Yogi, H. Imai, Y. Tan, T. Fujita and M. Chen, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- R. Wang, H. Li, Y. Wu, H. Li, B. Zhong, Y. Sun, Z. Wu and X. Guo, Adv. Energy Mater., 2022, 12, 2202342 CrossRef CAS.
- A. Su, J. Li, J. Dong, D. Yang, G. Chen and Y. Wei, Small, 2020, 16, 2001714 CrossRef CAS.
- S. Bazlen, P. Heugel, O. von Kessel, W. Commerell and J. Tuebke, J. Energy Storage, 2022, 49, 104044 CrossRef.
- H. P. Boehm, R. Setton and E. Stumpp, Pure Appl. Chem., 1994, 66, 1893–1901 CrossRef CAS.
- T. Yao, N. Ozawa, T. Aikawa and S. Yoshinaga, Solid State Ionics, 2004, 175, 199–202 CrossRef CAS.
- H. Wu, L. Zheng, J. Zhan, N. Du, W. Liu, J. Ma, L. Su and L. Wang, J. Power Sources, 2020, 449, 227513 CrossRef CAS.
- Z. Ma, A. Song, Z. Liu, Y. Guo, X. Yang, Q. Li, Y. Fan, L. Dai, H. Tian, X. Qin, H. Liu, G. Shao and G. Wang, Adv. Funct. Mater., 2023, 33, 2301112 CrossRef CAS.
- H. Wu, L. Zheng, N. Du, B. Sun, J. Ma, Y. Jiang, J. Gong, H. Chen and L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 22323–22331 CrossRef CAS.
- N. Yao, Y. Zhang, X. Rao, Z. Yang, K. Zheng, K. Swierczek and H. Zhao, Int. J. Miner., Metall. Mater., 2022, 29, 876–895 CrossRef CAS.
- X. Zhu, B. Liu, J. Shao, Q. Zhang, Y. Wan, C. Zhong and J. Lu, Adv. Funct. Mater., 2023, 33, 2213363 CrossRef CAS.
- S. Zhu, H. Li, Z. Hu, Q. Zhang, J. Zhao and L. Zhang, Acta Phys.-Chim. Sin., 2022, 38, 628233 Search PubMed.
- X. Zhou, Z. Qi, Q. Liu, J. Tian, M. Liu, K. Dong and Z. Lei, Front. Mater., 2021, 7, 628233 CrossRef.
- G. Zhu, D. Chao, W. Xu, M. Wu and H. Zhang, ACS Nano, 2021, 15, 15567–15593 CrossRef CAS PubMed.
- X. Zhang, H. Shi, P. Lv, J. Liu and H. Zhang, Energy Technol., 2021, 9, 2100400 CrossRef CAS.
- M. Li, C. Ye, Z. Li, Q. Lin, J. Cao, F. Liu, G. Song and S. Kawi, J. Mater. Chem. A, 2022, 10, 6330–6350 RSC.
- J. Tao, Z. Yan, J. Yang, J. Li, Y. Lin and Z. Huang, Carbon Energy, 2022, 4, 129–141 CrossRef CAS.
- Q. Hua, D. Dai, C. Zhang, F. Han, T. Lv, X. Li, S. Wang, R. Zhu, H. Liao and S. Zhang, Nanoscale Res. Lett., 2018, 13, 134–139 CrossRef PubMed.
- Y. Cao, J. C. Bennett, R. A. Dunlap and M. N. Obrovac, Chem. Mater., 2018, 30, 7418–7422 CrossRef CAS.
- P. Zhang, L. Wang, J. Xie, L. Su and C.-A. Ma, J. Mater. Chem. A, 2014, 2, 3776–3782 RSC.
- H.-S. Kim, W. Cho, D. Park, K. Kim, W.-S. Jung, H. Choi and J.-H. Kim, J. Alloys Compd., 2019, 803, 325–331 CrossRef CAS.
- K. Zeng, T. Li, X. Qin, G. Liang, L. Zhang, Q. Liu, B. Li and F. Kang, Nano Res., 2020, 13, 2987–2993 CrossRef.
- K. Wang, Y. Tan, P. Li and J. Sun, Electrochim. Acta, 2020, 353, 136538 CrossRef CAS.
- A. Raza, J. Y. Jung, C.-H. Lee, B. G. Kim, J.-H. Choi, M.-S. Park and S.-M. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 7161–7170 CrossRef CAS PubMed.
- T. Wu, Q. Ke, M. Lu, P. Pan, Y. Zhou, Z. Gu, G. Cui and H. Lu, Catalysts, 2022, 12, 2–28 Search PubMed.
- X. Zhou, Y. Liu, Y. Ren, T. Mu, X. Yin, C. Du, H. Huo, X. Cheng, P. Zuo and G. Yin, Adv. Funct. Mater., 2021, 31, 2101145 CrossRef CAS.
- T. Xu, Q. Wang, J. Zhang, X. Xie and B. Xia, ACS Appl. Mater. Interfaces, 2019, 11, 19959–19967 CrossRef CAS.
- C. Guo, Y. Xie, K. Pan and L. Li, Nanoscale, 2020, 12, 13017–13027 RSC.
- O. J. Sanumi, P. G. Ndungu and B. O. Oboirien, J. Power Sources, 2022, 543, 231849 CrossRef.
- A. Staerz, H. G. Seo, T. Defferriere and H. L. Tuller, J. Mater. Chem. A, 2022, 10, 2618–2636 RSC.
- V. Pavlenko, S. Khosravi, S. Zoltowska, A. B. Haruna, M. Zahid, Z. Mansurov, Z. Supiyeva, A. Galal, K. I. Ozoemena, Q. Abbas and T. Jesionowski, Mater. Sci. Eng., R, 2022, 149, 100682 CrossRef.
- Z. Li, Q. He, L. He, P. Hu, W. Li, H. Yan, X. Peng, C. Huang and L. Mai, J. Mater. Chem. A, 2017, 5, 4183–4189 RSC.
- A. Kim, S. Lim, D.-H. Peck, S.-K. Kim, B. Lee and D. Jung, Nanomaterials, 2012, 2, 206–216 CrossRef CAS PubMed.
- Z. Wang, L. Kong, Z. Guo, X. Zhang, X. Wang and X. Zhang, Chem. Eng. J., 2022, 428, 131060 CrossRef CAS.
- Q. Liu, Z. Cui, R. Zou, J. Zhang, K. Xu and J. Hu, Small, 2017, 13, 1603754 CrossRef PubMed.
- M. Chen, X. Ma, B. Chen, R. Arsenault, P. Karlson, N. Simon and Y. Wang, Joule, 2019, 3, 2622–2646 CrossRef CAS.
- H. Takezawa, S. Ito, H. Yoshizawa and T. Abe, J. Power Sources, 2016, 324, 45–51 CrossRef CAS.
- L. F. Castaneda, J. F. Rodriguez and J. L. Nava, Chem. Eng. J., 2021, 413, 127529 CrossRef CAS.
- L. Su, J. Xie, Y. Xu, L. Wang, Y. Wang and M. Ren, J. Alloys Compd., 2016, 663, 524–530 CrossRef CAS.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 152–153, 125–129 CrossRef CAS.
- J. Moon, Int. J. Energy Res., 2021, 45, 7315–7325 CrossRef CAS.
- J. H. Cho, X. Xiao, M. W. Verbrugge and B. W. Sheldon, ACS Appl. Energy Mater., 2022, 5, 13293–13306 CrossRef CAS.
- J. Ge, H. Shen, F. Zhou, Y. Li, N. Yuan, W. Yang, H. Zhou, B. Xu, R. Guo and P. Xu, J. Mater. Chem. A, 2022, 10, 1928–1939 RSC.
- S. Fan, X. Zhou, J. Tang, Y. Ma and J. Yang, Appl. Surf. Sci., 2022, 579, 152179 CrossRef CAS.
- J. Chai, N. Han, S. Feng, X. Huang, B. Tang and W. Zhang, Chem. Eng. J., 2023, 453, 139768 CrossRef CAS.
- S. Chae, S.-H. Choi, N. Kim, J. Sung and J. Cho, Angew. Chem., Int. Ed., 2020, 59, 110–135 CrossRef CAS PubMed.
- P. Li, H. Kim, S.-T. Myung and Y.-K. Sun, Energy Storage Mater., 2021, 35, 550–576 CrossRef.
- J. Gao, C. Chen, Q. Dong, J. Dai, Y. Yao, T. Li, A. Rundlett, R. Wang, C. Wang and L. Hu, Adv. Mater., 2021, 33, 2005305 CrossRef CAS PubMed.
- J. Sung, J. Ma, S. H. Choi, J. Hong, N. Kim, S. Chae, Y. Son, S. Y. Kim and J. Cho, Adv. Mater., 2019, 33, 1900970 CrossRef PubMed.
- H. Gu, Y. Wang, Y. Zeng, M. Yu, T. Liu, J. Chen, K. Wang, J. Xie and L. Li, ACS Appl. Mater. Interfaces, 2022, 14, 17388–17395 CrossRef CAS.
- Z. Cao, X. Zheng, Q. Qu, Y. Huang and H. Zheng, Adv. Mater., 2021, 33, 2103178 CrossRef CAS PubMed.
- J. Zhang, X. Zhang, C. Zhang, Z. Liu, J. Zheng, Y. Zuo, C. Xue, C. Li and B. Cheng, Energy Fuels, 2017, 31, 8758–8763 CrossRef CAS.
- L. Lee, W. To, A. Ran, J.-H. Lee, S. M. Hwang and Y.-J. Kim, Chem. Eng. J., 2022, 442, 136166 CrossRef CAS.
- J. Zhang, P. Ma, X. Zhang, Z. Liu, J. Zheng, Y. Zuo, C. Xue, B. Cheng and C. Li, Energy Technol., 2019, 7, 1800800 CrossRef.
- L. Shi, W. Wang, A. Wang, K. Yuan, Z. Jin and Y. Yang, J. Power Sources, 2016, 318, 184–191 CrossRef CAS.
- P. Lv, H. Zhao, C. Gao, T. Zhang and X. Liu, Electrochim. Acta, 2015, 152, 345–351 CrossRef CAS.
- B. Liang, Y. Liu and Y. Xu, J. Power Sources, 2014, 267, 469–490 CrossRef CAS.
- H. Li, J. Peng, Z. Wu, X. Liu, P. Liu, B. Chang and X. Wang, Chem. Eng. J., 2023, 462, 142172 CrossRef CAS.
- Z. Wang, H. Zhang, X. Zhang, X. Wang and X. Zhang, Composites, Part B, 2022, 247, 110308 CrossRef CAS.
- J. Guo, G. Zhao, T. Xie, D. Dong, C. Ma, L. Su, L. Gong, X. Lou, X. Guo, J. Wang and Y. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 19023–19032 CrossRef CAS PubMed.
- J. Zhou, Y. Lu, L. Yang, W. Zhu, W. Liu, Y. Yang and K. Liu, Carbon Energy, 2022, 4, 399–410 CrossRef CAS.
- G. Zhu, Y. Gu, S. Heng, Y. Wang, Q. Qu and H. Zheng, Electrochim. Acta, 2019, 323, 134840 CrossRef CAS.
- H. Zhang, K. Liu, Y. Liu, Z. Lang, W. He, L. Ma, J. Man, G. Jia, J. Cui and J. Sun, J. Power Sources, 2020, 447, 227400 CrossRef CAS.
- J. Cui, Y. Cui, S. Li, H. Sun, Z. Wen and J. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 30239–30247 CrossRef CAS PubMed.
- Y.-F. Tian, G. Li, D.-X. Xu, Z.-Y. Lu, M.-Y. Yan, J. Wan, J.-Y. Li, Q. Xu, S. Xin, R. Wen and Y.-G. Guo, Adv. Mater., 2022, 34, 2200672 CrossRef CAS PubMed.
- P. Li, G. Zhao, X. Zheng, X. Xu, C. Yao, W. Sun and S. X. Dou, Energy Storage Mater., 2018, 15, 422–446 CrossRef.
- H. Tian, F. Huang, Y. Zhu, S. Liu, Y. Han, M. Jaroniec, Q. Yang, H. Liu, G. Q. M. Lu and J. Liu, Adv. Funct. Mater., 2018, 28, 1801737 CrossRef.
- Q. Shi, J. Zhou, S. Ullah, X. Yang, K. Tokarska, B. Trzebicka, H. Q. Ta and M. H. Ruemmeli, Energy Storage Mater., 2021, 34, 735–754 CrossRef.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed.
- Z. Liu, Y. Zhao, R. He, W. Luo, J. Meng, Q. Yu, D. Zhao, L. Zhou and L. Mai, Energy Storage Mater., 2019, 19, 299–305 CrossRef.
- Q. Wang, M. Zhu, G. Chen, N. Dudko, Y. Li, H. Liu, L. Shi, G. Wu and D. Zhang, Adv. Mater., 2022, 34, 2109658 CrossRef CAS.
- Q. Man, Y. An, C. Liu, H. Shen, S. Xiong and J. Feng, J. Energy Chem., 2023, 76, 576–600 CrossRef CAS.
- P. Ares and K. S. Novoselov, Nano Mater. Sci., 2022, 4, 3–9 CrossRef CAS.
- H. Luo, X. Zhang, C. Xu, W. He, Z. Wang, W. Cai and Y. Zhang, ACS Appl. Energy Mater., 2022, 5, 8982–8989 CrossRef CAS.
- H. Wang, X. Que, Y. Liu, X. Wu, Q. Yuan, J. Lu and W. Gan, J. Alloys Compd., 2021, 874, 159913 CrossRef CAS.
- M. Ku, D. Park, M. Kim, M. Choi and W. Choi, J. Mater. Chem. A, 2023, 11, 15277–15285 RSC.
- Y. Zhang, G. Hu, Q. Yu, Z. Liu, C. Yu, L. Wu, L. Zhou and L. Mai, Mater. Chem. Front., 2020, 4, 1656–1663 RSC.
- W. Chen, S. Kuang, H. Wei, P. Wu, T. Tang, H. Li, Y. Liang, X. Yu and J. Yu, J. Colloid Interface Sci., 2022, 610, 583–591 CrossRef CAS PubMed.
- Q. Gong, H. Wang, W. Song, B. Sun, P. Cao, S. Gu, X. Sun and G. Zhou, Chem. – Eur. J., 2021, 27, 2654–2661 CrossRef CAS PubMed.
- H. Tian, H. Tian, S. Wang, S. Chen, F. Zhang, L. Song, H. Liu, J. Liu and G. Wang, Nat. Commun., 2020, 11, 5025 CrossRef CAS.
- D. Wang, T. Wang, M. He, T. Wang and H. Wang, Small, 2021, 17, 2103673 CrossRef PubMed.
- T. Liu, Y. Qu, J. Liu, L. Zhang, B. Cheng and J. Yu, Small, 2021, 17, 14903–14909 Search PubMed.
- H. Wang, C.-J. Yao, H.-J. Nie, K.-Z. Wang, Y.-W. Zhong, P. Chen, S. Mei and Q. Zhang, J. Mater. Chem. A, 2020, 8, 11906–11922 RSC.
- Q. Yu, P. Ge, Z. Liu, M. Xu, W. Yang, L. Zhou, D. Zhao and L. Mai, J. Mater. Chem. A, 2018, 6, 14903–14909 RSC.
- S. Sim, P. Oh, S. Park and J. Cho, Adv. Mater., 2013, 25, 4498–4503 CrossRef CAS PubMed.
- K. Xiao, Q. Yin, X. Wu and C. Huang, Nano Mater. Sci., 2022, 4, 383–392 CrossRef CAS.
- C. Duan, K. Liang, Z. Zhang, J. Li, T. Chen, D. Lv, L. Li, L. Kang, K. Wang, H. Hu and H. Xi, Nano Mater. Sci., 2022, 4, 351–365 CrossRef CAS.
- L. Zhang, J. Deng, L. Liu, W. Si, S. Oswald, L. Xi, M. Kundu, G. Ma, T. Gemming, S. Baunack, F. Ding, C. Yan and O. G. Schmidt, Adv. Mater., 2014, 26, 4527–4532 CrossRef CAS PubMed.
- Q. Xu, J.-K. Sun, Y.-X. Yin and Y.-G. Guo, Adv. Funct. Mater., 2018, 28, 1705235 CrossRef.
- S. Park, J. Sung, S. Chae, J. Hong, T. Lee, Y. Lee, H. Cha, S. Y. Kim and J. Cho, ACS Nano, 2020, 14, 11548–11557 CrossRef CAS PubMed.
- H. Xue, Y. Cheng, Q. Gu, Z. Wang, Y. Shen, D. Yin, L. Wang and G. Huang, Nanoscale, 2021, 13, 3808–3816 RSC.
- Z. Wang, L. Kong, Z. Guo, X. Zhang, X. Wang and X. Zhang, Chem. Eng. J., 2022, 428, 131060 CrossRef CAS.
- S. Li, K. Wang, G. Zhang, S. Li, Y. Xu, X. Zhang, X. Zhang, S. Zheng, X. Sun and Y. Ma, Adv. Funct. Mater., 2022, 32, 2200796 CrossRef CAS.
- W.-H. Li, Y.-M. Li, X.-F. Liu, Z.-Y. Gu, H.-J. Liang, X.-X. Zhao, J.-Z. Guo and X.-L. Wu, Adv. Funct. Mater., 2022, 32, 2201038 CrossRef CAS.
- H.-J. Liang, Z.-Y. Gu, X.-X. Zhao, J.-Z. Guo, J.-L. Yang, W.-H. Li, B. Li, Z.-M. Liu, Z.-H. Sun, J.-P. Zhang and X.-L. Wu, Sci. Bull., 2022, 67, 1581–1588 CrossRef CAS PubMed.
- Q. Liu, R. Xu, D. Mu, G. Tan, H. Gao, N. Li, R. Chen and F. Wu, Carbon Energy, 2022, 4, 458–479 CrossRef CAS.
- H. Tian, J. Liang and J. Liu, Adv. Mater., 2019, 31, 1903886 CrossRef CAS PubMed.
- X. Min, G. Xu, B. Xie, P. Guan, M. Sun and G. Cui, Energy Storage Mater., 2022, 47, 297–318 CrossRef.
- M. Peng, K. Shin, L. Jiang, Y. Jin, K. Zeng, X. Zhou and Y. Tang, Angew. Chem., Int. Ed., 2022, 61, e202206770 CrossRef CAS.
- J. Scharf, M. Chouchane, D. P. Finegan, B. Lu, C. Redquest, M.-C. Kim, W. Yao, A. A. Franco, D. Gostovic, Z. Liu, M. Riccio, F. Zelenka, J.-M. Doux and Y. S. Meng, Nat. Nanotechnol., 2022, 17, 446–459 CrossRef CAS.
- H. Li, H. Li, Y. Lai, Z. Yang, Q. Yang, Y. Liu, Z. Zheng, Y. Liu, Y. Sun, B. Zhong, Z. Wu and X. Guo, Adv. Energy Mater., 2022, 12, 2102181 CrossRef CAS.
- Q. Ouyang, G. Li, X. Zhang, X. Zhao, Y. Wang, Q. Wang, Z. Fan, J. Wang and L. Li, Chem. Eng. J., 2023, 460, 141762 CrossRef CAS.
- G. Hu, R. Yu, Z. Liu, Q. Yu, Y. Zhang, Q. Chen, J. Wu, L. Zhou and L. Mai, ACS Appl. Mater. Interfaces, 2021, 13, 3991–3998 CrossRef CAS PubMed.
- R. F. H. Hernandha, P. C. Rath, B. Umesh, J. Patra, C.-Y. Huang, W.-W. Wu, Q.-F. Dong, J. Li and J.-K. Chang, Adv. Funct. Mater., 2021, 31, 2104135 CrossRef CAS.
- Q. Sun, J. Li, M. Yang, S. Wang, G. Zeng, H. Liu, J. Cheng, D. Li, Y. Wei, P. Si, Y. Tian and L. Ci, Small, 2023, 19, 2300759 CrossRef CAS PubMed.
- L. Sun, Y. Liu, R. Shao, J. Wu, R. Jiang and Z. Jin, Energy Storage Mater., 2022, 46, 482–502 CrossRef.
- W. Chen, H. Liu, S. Kuang, H. Huang, T. Tang, M. Zheng, Y. Fang and X. Yu, Carbon, 2021, 179, 377–386 CrossRef CAS.
- W. Chen, D. Xu, S. Kuang, Z. Wu, H. Hu, M. Zheng and X. Yu, J. Power Sources, 2021, 489, 229459 CrossRef CAS.
- X. Guo, Y.-Z. Zhang, F. Zhang, Q. Li, D. H. Anjum, H. Liang, Y. Liu, C.-S. Liu, H. N. Alshareef and H. Pang, J. Mater. Chem. A, 2019, 7, 15969–15974 RSC.
- T. Meng, B. Li, C. Liu, Q. Wang, D. Shu, S. Ou, M. S. Balogun, H. Su and Y. Tong, Energy Storage Mater., 2022, 46, 344–351 CrossRef.
- J. A. Buledi, N. Mahar, A. Mallah, A. R. Solangi, I. M. Palabiyik, N. Qambrani, F. Karimi, Y. Vasseghian and H. Karimi-Maleh, Food Chem. Toxicol., 2022, 161, 112843 CrossRef CAS PubMed.
- Y. Mu, M. Han, B. Wu, Y. Wang, Z. Li, J. Li, Z. Li, S. Wang, J. Wan and L. Zeng, Adv. Sci., 2022, 9, 2104685 CrossRef CAS PubMed.
- H. Xue, Y. Wu, Y. Zou, Y. Shen, G. Liu, Q. Li, D. Yin, L. Wang and J. Ming, Adv. Funct. Mater., 2020, 30, 1910657 CrossRef CAS.
- L. Lee, W. T. A. Ran, J.-H. Lee, S. M. Hwang and Y. J. Kim, Chem. Eng. J., 2022, 442, 136166 CrossRef CAS.
- J. Dai, H. Feng, K. Shi, X. Ma, Y. Yan, L. Ye and Y. Xia, Chemosphere, 2022, 307, 135833 CrossRef CAS PubMed.
- T. H. Nguyen, D. Yang, B. Zhu, H. Lin, T. Ma and B. Jia, J. Mater. Chem. A, 2021, 9, 7366–7395 RSC.
- Y. Ren, X. Wu and M. Li, Electrochim. Acta, 2016, 206, 328–336 CrossRef CAS.
- Q. Xu, J.-K. Sun, Z.-L. Yu, Y.-X. Yin, S. Xin, S.-H. Yu and Y.-G. Guo, Adv. Mater., 2018, 30, 1707430 CrossRef PubMed.
- X. Yue, S. Huang, J. Cai, Y. Jin and P. K. Shen, J. Mater. Chem. A, 2017, 5, 7784–7790 RSC.
- M. Han, Y. Mu, F. Yuan, J. Liang, T. Jiang, X. Bai and J. Yu, J. Mater. Chem. A, 2020, 8, 3822–3833 RSC.
- G. Hu, K. Zhong, R. Yu, Z. Liu, Y. Zhang, J. Wu, L. Zhou and L. Mai, J. Mater. Chem. A, 2020, 8, 13285–13291 RSC.
- L. Shi, Y. Li, Y. Xing, R. Lin, G. Cheng, J. Ding and K. H. Lam, Electrochim. Acta, 2021, 390, 138841 CrossRef CAS.
- H. Zhang, Y. Du, N. Zhang, P. Lin, W. Liu, Z. Wen, J. Cui and J. Sun, Microporous Mesoporous Mater., 2022, 335, 111794 CrossRef CAS.
- X. Guo, H. Xu, W. Li, Y. Liu, Y. Shi, Q. Li and H. Pang, Adv. Sci., 2022, 10, 2206084 CrossRef PubMed.
- D. Cao, X. Sun, Y. Li, A. Anderson, W. Lu and H. Zhu, Adv. Mater., 2022, 34, 2200401 CrossRef CAS PubMed.
- Q.-H. Wu, B. Qu, J. Tang, C. Wang, D. Wang, Y. Li and J.-G. Ren, Electrochim. Acta, 2015, 156, 147–153 CrossRef CAS.
- H.-W. Ha, K. H. Jeong, N. J. Yun, M. Z. Hong and K. Kim, Electrochim. Acta, 2005, 50, 3764–3769 CrossRef CAS.
- Z. Wang, C. Wu, L. Liu, F. Wu, L. Chen and X. Huang, J. Electrochem. Soc., 2002, 149, A466–A471 CrossRef CAS.
- J. Cho, H. Kim and B. Park, J. Electrochem. Soc., 2004, 151, A1707–A1711 CrossRef CAS.
- Y.-M. Lin, H.-C. Wu, Y.-C. Yen, Z.-Z. Guo, M.-H. Yang, H.-M. Chen, H.-S. Sheu and N.-L. Wu, J. Electrochem. Soc., 2005, 152, A1526–A1532 CrossRef CAS.
- Y. Jiang, S. Liu, Y. Ding, J. Jiang, W. Li, S. Huang, Z. Chen, B. Zhao and J. Zhang, J. Power Sources, 2020, 467, 228301 CrossRef CAS.
- T. Tao, S. Lu, Y. Fan, W. Lei, S. Huang and Y. Chen, Adv. Mater., 2017, 29, 1700542 CrossRef PubMed.
- H. Tian, A. Song, H. Tian, J. Liu, G. Shao, H. Liu and G. Wang, Chem. Sci., 2021, 12, 7656–7676 RSC.
- Z. Xiao, C. Yu, X. Lin, X. Chen, C. Zhang, H. Jiang, R. Zhang and F. Wei, Nano Energy, 2020, 77, 104890 Search PubMed.
- J. Cui, J. Yang, J. Man, S. Li, J. Yin, L. Ma, W. He, J. Sun and J. Hu, Electrochim. Acta, 2019, 300, 470–481 CrossRef CAS.
- T. Kang, J. Tan, X. Li, J. Liang, H. Wang, D. Shen, Y. Wu, Z. Huang, Y. Lu, Z. Tong and C.-S. Lee, J. Mater. Chem. A, 2021, 9, 7807–7816 RSC.
- L. Gu, J. Han, M. Chen, W. Zhou, X. Wang, M. Xu, H. Lin, H. Liu, H. Chen, J. Chen, Q. Zhang and X. Han, Energy Storage Mater., 2022, 52, 547–561 CrossRef.
- X.-Y. Yue, Y.-X. Yao, J. Zhang, S.-Y. Yang, Z. Li, C. Yan and Q. Zhang, Adv. Mater., 2022, 34, 2110337 CrossRef CAS PubMed.
- S. Pan, L. Yang, P. Su, H. Zhang and S. Zhang, Small, 2022, 18, 2202137 Search PubMed.
- B. H. Park, G.-W. Lee, S. B. Choi, Y.-H. Kim and K. B. Kim, Nanoscale, 2020, 12, 22140–22149 RSC.
- X. Song, Y. Lu, C. Li, F. Wang and X. Zhao, J. Energy Storage, 2022, 51, 104390 CrossRef.
- Y. Shen, X. Shen, M. Yang, J. Qian, Y. Cao, H. Yang, Y. Luo and X. Ai, Adv. Funct. Mater., 2021, 31, 2101181 CrossRef CAS.
- Y. Huang, C. Liu, F. Wei, G. Wang, L. Xiao, J. Lu and L. Zhuang, J. Mater. Chem. A, 2020, 8, 18715–18720 RSC.
- Y. Xue, J. Deng, C. Wang, R. G. Mendes, L. Chen, Y. Xiao, Q. Zhang, T. Zhang, X. Hu, X. Li, M. H. Rümmeli and L. Fu, J. Mater. Chem. A, 2016, 4, 5395–5401 RSC.
- Q. Meng, G. Li, J. Yue, Q. Xu, Y.-X. Yin and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 32062–32068 CrossRef CAS PubMed.
- J. Zhao, H.-W. Lee, J. Sun, K. Yan, Y. Liu, W. Liu, Z. Lu, D. Lin, G. Zhou and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7408–7413 CrossRef CAS PubMed.
- M.-Y. Yan, G. Li, J. Zhang, Y.-F. Tian, Y.-X. Yin, C.-J. Zhang, K.-C. Jiang, Q. Xu, H.-L. Li and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 27202–27209 CrossRef CAS.
- K. Zou, W. Deng, P. Cai, X. Deng, B. Wang, C. Liu, J. Li, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2020, 31, 2005581 CrossRef.
- M. Fan, Q. Meng, X. Chang, C. F. Gu, X. H. Meng, Y. X. Yin, H. Li, L. J. Wan and Y. G. Guo, Adv. Energy Mater., 2022, 12, 2103630 CrossRef CAS.
- Z. Jingxi, C. Xi, S. Gang, W. Hailong, D. Yanhao and W. Chang-An, J. Mater. Chem. A, 2023, 11, 8757–8765 RSC.
- W. Wu, Y. Kang, M. Wang, D. Xu, J. Wang, Y. Cao, C. Wang and Y. Deng, J. Power Sources, 2020, 464, 228224 CrossRef.
- W. Kunther, S. Ferreiro and J. Skibsted, J. Mater. Chem. A, 2017, 11, 8757–8765 Search PubMed.
- T. Meng, B. Li, Q. Wang, J. Hao, B. Huang, F. L. Gu, H. Xu, P. Liu and Y. Tong, ACS Nano, 2020, 14, 7066–7076 CrossRef CAS PubMed.
- Z. Wang, X. Xu, C. Chen, T. Huang and A. Yu, J. Power Sources, 2022, 539, 231604 CrossRef CAS.
- X. Sun, L. Geng, S. Yi, C. Li, Y. An, X. Zhang, X. Zhang, K. Wang and Y. Ma, J. Power Sources, 2021, 499, 229936 CrossRef CAS.
- Z. Song, S. Chen, Y. Zhao, S. Xue, G. Qian, J. Fang, T. Zhang, C. Long, L. Yang and F. Pan, Small, 2021, 17, 2102256 CrossRef CAS PubMed.
- S. Xue, Y. Fu, Z. Song, S. Chen, Y. Ji, Y. Zhao, H. Wang, G. Qian, L. Yang and F. Pan, Energy Environ. Mater., 2022, 5, 1310–1316 CrossRef CAS.
- H. Liao, W. He, N. Liu, D. Luo, H. Dou and X. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 49313–49321 CrossRef CAS PubMed.
- A. N. Preman, H. Lee, J. Yoo, I. T. Kim, T. Saito and S.-K. Ahn, J. Mater. Chem. A, 2020, 8, 25548–25570 RSC.
- Z. Shi, Q. Liu, Z. Yang, L. A. Robertson, S. R. Bheemireddy, Y. Zhao, Z. Zhang and L. Zhang, J. Mater. Chem. A, 2021, 10, 1380–1389 RSC.
- W. Tang, L. Feng, X. Wei, G. Lai, H. Chen, Z. Li, X. Huang, S. Wu and Z. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 56910–56918 CrossRef CAS PubMed.
- Z. Weng, S. Di, L. Chen, G. Wu, Y. Zhang, C. Jia, N. Zhang, X. Liu and G. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 42494–42503 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |