
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyester biodegradability: importance and potential for optimisation†
Yue
Wang
 ab,
Robert-Jan
van Putten
c,
Albert
Tietema
b,
John R.
Parsons
b and
Gert-Jan M.
Gruter
*ac
ab,
Robert-Jan
van Putten
c,
Albert
Tietema
b,
John R.
Parsons
b and
Gert-Jan M.
Gruter
*ac
avan ‘t Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: g.j.m.gruter@uva.nl
bInstitute for Biodiversity and Ecosystem Dynamics (IBED), University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
cAvantium Support BV, Zekeringstraat 29, 1014 BV Amsterdam, The Netherlands. E-mail: robert-jan.vanputten@avantium.com
First published on 5th March 2024
Abstract
To reduce global CO2 emissions in line with EU targets, it is essential that we replace fossil-derived plastics with renewable alternatives. This provides an opportunity to develop novel plastics with improved design features, such as better reusability, recyclability, and environmental biodegradability. Although recycling and reuse of plastics is favoured, this relies heavily on the infrastructure of waste management, which is not consistently advanced on a worldwide scale. Furthermore, today's bulk polyolefin plastics are inherently unsuitable for closed-loop recycling, but the introduction of plastics with enhanced biodegradability could help to combat issues with plastic accumulation, especially for packaging applications. It is also important to recognise that plastics enter the environment through littering, even where the best waste-collection infrastructure is in place. This causes endless environmental accumulation when the plastics are non-(bio)degradable. Biodegradability depends heavily on circumstances; some biodegradable polymers degrade rapidly under tropical conditions in soil, but they may not also degrade at the bottom of the sea. Biodegradable polyesters are theoretically recyclable, and even if mechanical recycling is difficult, they can be broken down to their monomers by hydrolysis for subsequent purification and re-polymerisation. Additionally, both the physical properties and the biodegradability of polyesters are tuneable by varying their building blocks. The relationship between the (chemical) structures/compositions (aromatic, branched, linear, polar/apolar monomers; monomer chain length) and biodegradation/hydrolysis of polyesters is discussed here in the context of the design of biodegradable polyesters.
1. The problems caused by plastics
Plastics are used for many applications and play a vital role in industry, (food) packaging, textiles, transportation and other uses in daily life. It was reported that the annual production of plastics worldwide was 390 million tons in 2021.1 At approximately 40% of the end-use market, packaging represents the largest volume demand for virgin plastics in Europe.2 The annual global plastic production is expected to reach 1 billion tons by the end of 2050, with an associated annual CO2 footprint predicted at 2.8 billion tons (2.8 Gt).3 The increasing production of plastics from fossil-based resources does not conform with targets to reduce global CO2 emission. Thus, more sustainable feedstocks, such as biomass, CO2 (using carbon capture and utilisation (CCU)) and recycled waste materials, will be required for the next generations of plastics. By changing the feedstock, a substantial portion of the 1 Gt CO2 emissions (2023) related to plastics can be avoided and when using renewable energy for the plastics production and a significant amount of CO2 for feedstock, it is possible to achieve net-zero emissions.3–6In addition to the impact of plastic manufacturing on climate change, plastic waste is also a severe problem. It is estimated that 6–17 million tons of plastic waste accumulates in the environment annually.7 Incorrectly disposed plastic waste leads to environmental pollution and threatens ecosystems (Fig. 1).8,9 An illustrative example was presented in the 2019 United Nations Environment Programme report that a pregnant sperm whale washed up dead on a Mediterranean beach with close to 25 kg of plastic waste in her stomach.10 Unrecyclable single-use plastic packaging has no post-use value, which is the main cause for plastic pollution, because there is a high risk that these materials will end up in the environment following improper disposal. As conventional plastics are resistant to (bio)degradation, they remain in the environment, and eventually disintegrate into micro- and nanoplastics (MNPs). Prarat and Hongsawat found that microplastics in samples collected from the shore areas in Thailand are mostly polyethylene (PE), polypropylene (PP) and polystyrene (PS).11 Additionally, MNPs interact with (hydrophobic) contaminants (as carrier). This raises additional concerns about MNPs as they could contribute to the migration and accumulation of pollutants in the food chain worldwide.12–17 For example, in a simple artificial food chain experiment, zebrafish were fed with Artemia nauplii (crustaceans) in which very small (1–20 μm) nanoplastics were accumulated with and without benzo[a]pyrene as a persistent organic model pollutant (POP).18 Microplastics passed the intestinal tracts of zebrafish without significant accumulation and without signs of severe disease even after chronic dietary exposure. The potential of microplastics to act as a vector for POPs was tracked and uptake and distribution within intestinal tracts of zebrafish in histological cryosections was shown. Although problems caused by MNPs in the human body have not been reported yet, the observation of microplastics in the human body raises serious public concern. Recently, plastic particles, including PS, PE and poly(ethylene terephthalate) (PET), were detected and quantified in human blood by Leslie et al.19 To avoid risks to human health and ecosystems, it is necessary to change our attitude towards plastics and their use and waste-management, and to improve relevant infrastructure.
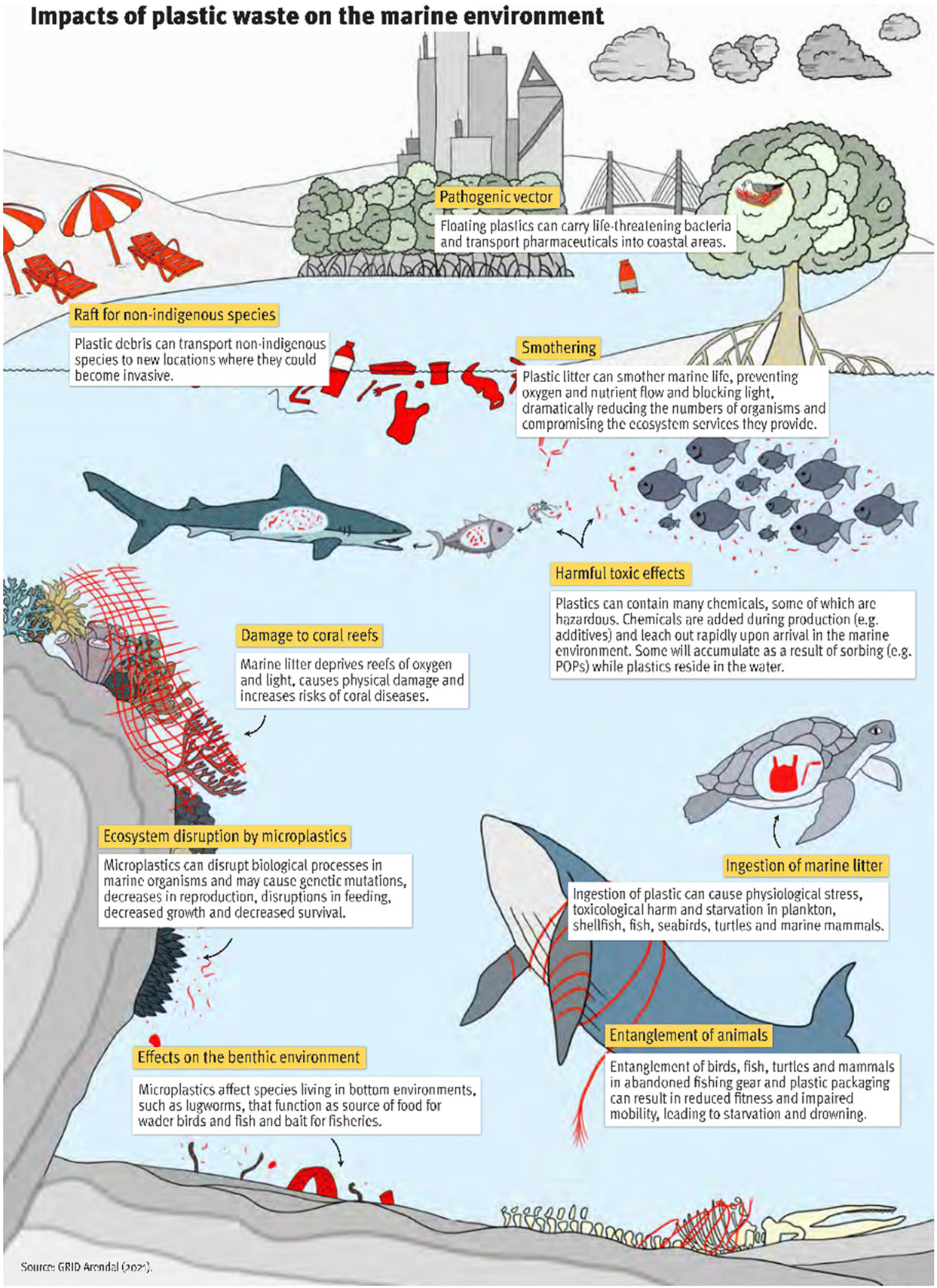 | ||
| Fig. 1 Impacts of plastic waste on the marine environment. Reprinted from GRID-Arendal.20 | ||
2. Biodegradable plastic: a pollution mitigation supplement to reduce, reuse and recycle
Due to the convenience, low cost and light weight of plastics, it is neither feasible nor sustainable to replace them with other materials for certain applications.20,21 For instance, in developing countries, the use of plastic bottled water remains essential for public health with regard to sanitation.22 Particularly, the outbreak of COVID-19 raised our reliance on plastic products with the increased production of personal protective equipment, disposable tableware, and packaging waste.20,23 Furthermore, plastic packaging improves the shelf-life for food, as well as the efficiency of transportation and distribution, which subsequently decreases food waste, provides consumers with a wider variety of food, and contributes to worldwide food security.20 Consequently, plastics are widely considered to be essential for sanitation and food safety.Recycling of plastic waste is a popular method for addressing the carbon footprint and pollution contributions from plastics.24 However, this relies heavily on the infrastructure of solid waste collection systems. In 2020, only 34.6% of Europe's plastic waste was recycled, and this represents the highest level of waste management globally.2 Moreover, this value was calculated based on the amount of collection instead of circulation. This demonstrates that even for what is widely considered as high-quality infrastructure for waste management, recycling levels are low, meaning that efficient recycling in the absence of this infrastructure is impossible in less fortunate areas across the globe.
It is not cost-effective to separate complex and contaminated plastic waste with laminated flexible structures (such as food packaging) for conventional mechanical recycling.25 Alternatively, chemical recycling can be implemented to convert plastics into smaller molecules, but this is not adopted as commonly as mechanical recycling.22,25,26 Currently, only 0.2% of the 34.6% recycled post-consumed plastic in Europe has been chemically recycled.2 For chemical recycling, it is important to realise that the circularity potential varies vastly for different plastic materials. While nylons, polycarbonates and polyesters such as PET can be depolymerised to their monomers at very high yields, this is not the case for polyolefins. It is not possible to selectively depolymerise PE and PP respectively back to ethylene and propylene, and a maximum monomer yield of 40–50% is expected from pyrolysis, followed by hydrotreating of polyolefin waste.
Particularly for the packaging segment, which typically holds the highest percentage of recycled plastics, it is estimated that only 14% of plastic packaging is recycled globally, while most packaging plastics are leaked into the environment (32%) or end up in landfills (40%).27,28 For applications where single-use plastics are necessary, and collection for recycling is not possible, environmental biodegradability of the plastic should become a required design feature.
The three R's: reduce, reuse and recycle, are a well-known framework to minimise the footprint of plastics. However, as discussed there are limitations, especially in the field of packaging. Therefore, next-generation plastics are expected to be either (1) fully closed-loop recyclable when the relevant infrastructure is available or (2) designed to degrade completely (without the generation of harmful residuals, e.g. mineralisation to CO2) in the environment over time. Even if a plastic's degradation takes years rather than months, an endless accumulation of plastics into the environment can be prevented, and biodegradable plastics could be used as a supplemental and transitional technology to the three R's framework. (Bio)degradable characteristics may be particularly beneficial for applications with an expected short lifetime and/or when collection would be challenging, for example with (food) packaging, mulch films, fishery materials and disposable medical items.
3. Shedding light on biodegradable polyesters
3.1 Understanding biodegradation: definitions, mechanisms, and influencing factors
The International Union of Pure and Applied Chemistry (IUPAC) defines a biodegradable polymer as “a polymer susceptible to degradation by biological activity, with the degradation accompanied by a lowering of its molar mass”.29 This definition emphasises the deterioration of the polymer.Various studies provide slightly different descriptions of biodegradation mechanisms. According to our interpretation, the biodegradation of plastics generally follows these main steps (Fig. 2).30–37
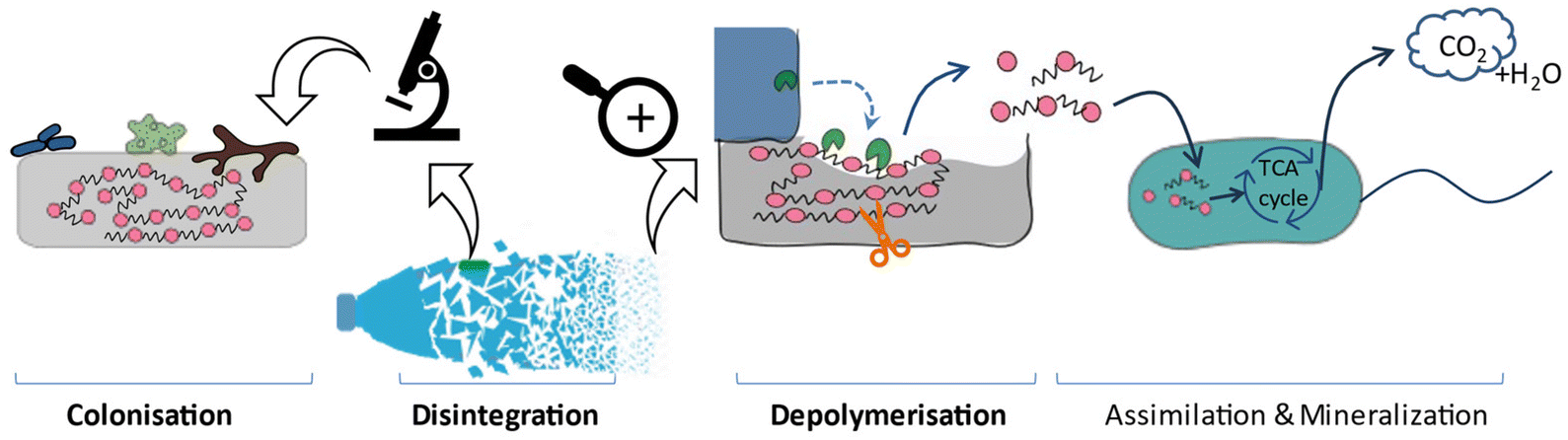 | ||
| Fig. 2 Plastic biodegradation under aerobic conditions. Solid arrows represent carbon flow.33,38 | ||
• Colonisation/film formation of microorganisms: microorganisms can excrete extracellular polymeric substances (EPS, e.g. polysaccharides, proteins) to allow them to adhere to the plastic surface.39 While some studies consider this to be a crucial initial step in biodegradation, the formation of the film doesn't necessarily indicate that erosion has occurred on the polymeric surface.40,41 However, it could have changed the buoyancy of the plastics and enhanced the interactions between hydrophobic and hydrophilic phases, which could affect the following steps indirectly.33,37,42,43
• Disintegration (macroscopic scale): polymers fragment into smaller size particles due to a combination of independent and interdependent driving forces, including mechanical stress, light, heat, oxygen, water and microorganisms. This leads to alterations in both the physical (e.g. morphology, weight loss) and mechanical properties (e.g. ductility and tensile strength) of the polymers, and can further-accelerate the formation of MNPs. In addition to these physical processes, such as shearing44 and interaction with animals,36 disintegration is generally associated with depolymerisation, which can be considered to be the macroscopic result of depolymerisation.
• Depolymerisation (molecular scale, e.g. hydrolysis for polyesters): the scission of linkages within the polymer chains decreases the molecular weight of the polymer and releases small molecules, including oligomers and monomers, to the environment. Since polymers are too large (high molecular weights) to be taken up by microorganisms, depolymerisation is a prerequisite for the subsequent assimilation stage. The energy required for this cleavage can originate from various sources, such as thermal energy, light energy, mechanical energy, chemical energy, and/or biological energy.37 Therefore, certain literature classifies distinct degradation pathways according to the energy source, as extensively discussed in the literature.36,37,45 The role of microorganisms is of particular interest, and comprehensive reviews33,36,46,47 have covered various types of microorganisms, including bacteria,48 fungi,49 algae, and their combined interactions,36 as well as various enzymes.50
• Assimilation and mineralisation: microorganisms can take up small molecules released from the previous stage and utilise them as substrate for metabolism and biomass growth.38 In this phase, polymer carbon is converted into CO2 and biomass under aerobic conditions (or to CO2 and CH4 under anaerobic conditions), typically via the tricarboxylic acid cycle (TCA).33,49,51 Generally, these end products resulting from complete biodegradation do not pose an ecological toxicity risk. On the contrary, if depolymerized products cannot be fully assimilated or mineralised, they may present ecotoxic hazard under specific conditions. Microorganisms in this stage may differ from those involved in depolymerisation.37
Clearly, disintegration of a polymer is only the beginning of this process and therefore not a good definition for biodegradability. Thus, we recommend the published scientific opinion of the European Union, where biodegradation of plastics is considered as “the microbial conversion of all its organic constituents to carbon dioxide (CO2) (or carbon dioxide and methane in conditions where oxygen is not present), new microbial biomass and mineral salts, within a timescale short enough not to lead to lasting harm or accumulation in the open environment”.52 This emphasises that the ‘results’ from biodegradation of (biodegradable) plastics should have relatively less environmental impact on nature. The “timescale short enough not to lead to lasting harm or accumulation” is powerful, as this will be very different for each material and for all local circumstances. “Lasting harm and accumulation” are criteria that can be objectively assessed.
Consumers expect that the so-called biodegradable plastics can biodegrade fast in nature without resulting in any negative impact on the environment.53 However, some of those commercial plastics claimed to be biodegradable were tested under industrial composting conditions, as described in standard methods ISO 14855,54 ASTM D6400,55 EN 13432.56 The fact that industrial composting provides more favourable conditions (i.e. higher temperature, higher total concentration of microorganisms, a higher moisture content for soil) can easily explain that polylactic acid (PLA), a well-known bio-based biodegradable polyester, completely degrades within several months under these conditions, yet only very slow degradation has been observed at ambient temperature.57–60 Particularly, the temperature of industrial composting (55–60 °C) could be equal to or above the Tg of poly(lactic acid) (PLA) (55–62 °C), which leads to the glassy and crystalline polymer becoming softer. This change of state is believed to accelerate the biodegradation rate.57 Overall, testing biodegradation at ambient temperature instead of under industrial composting conditions is more representative to evaluate fate-in-nature for biodegradable plastics.
In 2019, Napper and Thompson studied the biodegradation of “compostable” carrier bags. One type completely disappeared within 3 months in a marine environment (note: breakdown into MNPs is not considered to be biodegradation), but minimal loss was observed after 27 months in soil.61 Therefore, it is necessary to define the conditions when describing or claiming the biodegradability of plastics. Both internal and external factors, specifically polymer properties and environmental conditions, could significantly influence the primary degradation mechanisms and (bio)degradation rates.
The composition of polymers, i.e. structural units and their (sequence) distribution,62 plays a decisive role in the properties of the polymer, including the biodegradability (this will be discussed in next section). Separately, other polymer characteristics,33 such as hydrophilicity and hydrophobicity,63 crystallinity64 (versus amorphousness), chain flexibility65 and orientation, molecular weight and distribution, stereochemical structure, Tg, Tm,66 end groups,67 free volume,68 density, and the presence of residuals, and material properties including morphology, shape, specific surface area, colour69 and additives (e.g. antioxidants or photosensitizers), could also directly or indirectly affect the (bio)degradation of plastics in the environment. For instance, increased chain flexibility can enhance enzyme accessibility, therefore facilitating hydrolysis.62,65
External environmental factors include oxygen availability, UV exposure44/weathering, water/moisture/humidity, pH, temperature, salinity, the total amount of microorganisms, and microbial community composition. These factors could directly influence the biodegradation mechanism and rate. They also have indirect effects by shaping microbial communities. For example, salinity has a significant impact on microbial species selection and their metabolic activities.70 Other environmental factors like nutrients (C/N ratio) and additional carbon source71 could primarily influence this process through their effects on microorganisms.
These factors could interact and combine to influence plastic biodegradation. For instance,44 photooxidation can promote the formation of biofilms on the surface of plastics, which, in turn, acts as a shield against UV exposure for the plastic. Furthermore, the biofilm may reduce the buoyancy of the debris in seawater. Subsequently, this accelerates the sinking of debris into unfavourable degradation environments, such as areas with a lower temperature and an absence of light, which may lead to a reduced bioactivity.
3.2 Polyesters hydrolysis: mechanisms and advantages
Polyesters, including polybutylene adipate terephthalate (PBAT), PLA, polybutylene succinate (PBS) and polyhydroxyalkanoates (PHA), are the largest group (>67%) of biodegradable thermoplastics produced on industrial scale, followed by starch blends (26%).4 PHA is produced by bacteria (natural polyesters) and other synthetic polyesters are typically manufactured via esterification polymerisation.The search for more sustainable feedstocks for next generation plastics can generate a very wide range of building blocks (monomers) for polymer synthesis that are not necessarily available from fossil resources. In particular, biomass and CO2 (via CCU) can provide a range of non-fossil building blocks (monomers) for polyesters with unique properties; the monomer's chemical structure (composition) largely determines the thermal and physio-mechanical properties of the resulting polymer. It is possible to tune the properties of the resulting polyesters by selecting appropriate building blocks for the required application.72 For example, Wang and Gruter reported that they were able to tune the Tg of a series of fully renewable (co)polyesters poly(isosorbide oxalate-diols) from 60 to 167 °C by varying the isosorbide/diol ratio.73,74 Kasmi et al. recently reported a potential to tune the Tg of a series of fully biobased (co)polyesters poly(isosorbide furanoate-co-azelate) from −3 to 91 °C by varying the content of 2,5-furandicarboxylic acid (FDCA).75
Polyesters can potentially be recycled by both mechanical and chemical recycling processes.76–79 Compared to polyolefins, the esterification is a chemically reversible process, and can be reversed via hydrolysis or alcoholysis. This means polyesters can be broken down to monomers and these monomers (optionally after purification) can be repolymerised. In this way, the recycling of polyesters is a closed loop process, and downcycling (e.g. by mechanical recycling) can be avoided.
The mechanism of hydrolytic biodegradation for polyesters is described in some studies as a competition between bulk versus surface erosion (Fig. 3).32,80 The predominant mechanism ultimately depends on the diffusivity of the key reactant (e.g., water or catalyst), the rate of polymer bond cleavage, and the matrix dimensions.80 If the diffusivity of the chemical substance throughout the material is faster than the rate of polymer bond cleavage, bulk erosion occurs. This can be observed in the case of abiotic (non-enzymatic) hydrolysis of polyesters. However, when extracellular enzymes are involved in depolymerisation, surface erosion becomes predominant, because the enzymes cannot penetrate the polymer matrix due to their large size. As a result, surface biodeterioration is often mentioned and considered to be an important step in polymer biodegradation. It is important to note that in cases of surface erosion, the material undergoes weight loss, but there is no significant change in the molar mass of the main part of the material. These two mechanisms can also occur simultaneously.
 | ||
| Fig. 3 Schematic illustration of the changes a polymer matrix undergoes during surface erosion and bulk erosion. Reproduced from ref. 80 with permission from Elsevier, copyright 2002. | ||
It is also interesting to mention that faster internal degradation (heterogeneous degradation) in PLA and poly(lactic-co-glycolic acid) (PLGA). Li provided a detailed explanation of this phenomenon. In brief, it can be explained by the concept of autocatalysis in polyesters.63 Autocatalysis refers to the products of a reaction (i.e., hydrolysed carboxylic acids/end-groups) serving as catalysts for the same reaction, thus accelerating the hydrolysis rate of ester bonds.63 The hydrolysis products, i.e. acids, are trapped inside the polymer matrix rather than being released into solution, subsequently accelerating the hydrolysis rate internally compared to the surface.63 The detailed mechanism for acid-catalysed hydrolysis of polyesters is illustrated in Fig. S1. However, Tsuji and Nakahara also reported that adding additional lactic acid (LA) to the solution did not accelerate the hydrolysis of PLA polymer.81 Authors attribute this to differences in the availability of free LA in aquatic solutions compared to the polymer matrix.81
At the molecular scale, the hydrolysis of polyesters can be categorized into two types of scissions: endo- and exo-scission (Fig. 4). endo-Scission refers to the random cleavage of ester bonds along the polyester backbone, resulting in a significant decrease in molecular weight. exo-Scission, on the other hand, involves the cleavage of ester bonds at the chain ends, leading to a rather smaller decrease in molecular weight. Different structures and enzymes can lead to different hydrolysis mechanisms. For example, both poly(1,4-butylene 2,5-furandicarboxylate) (PBF, Tg = 39 °C) and poly(1,4-butylene 2,5-thiophenedicarboxylate) (PBTF, Tg = 25 °C) can be hydrolysed by cutinase enzymes, but they exhibit endo- and exo-wise cleavage, respectively. The authors attributed the difference to the lower flexibility/mobility (i.e., higher Tg) of PBF, which limits the access of cutinases and hinders the random scission of ester bonds along the polymer chains.82
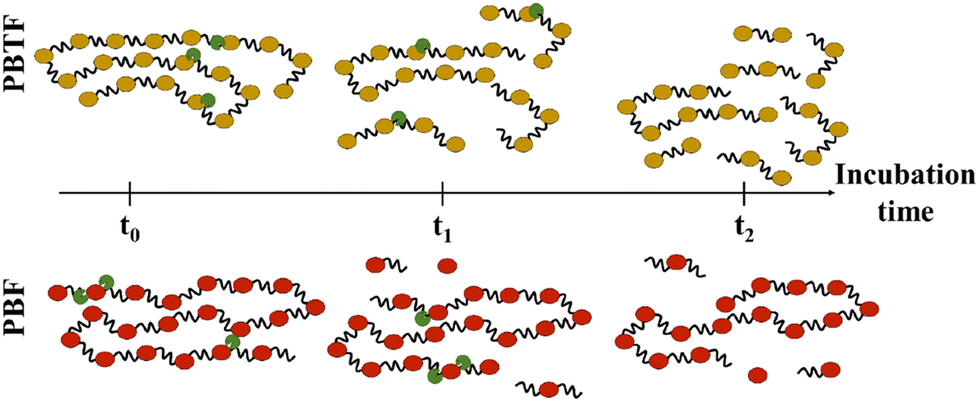 | ||
| Fig. 4 Enzymatic hydrolysis mechanisms. The endo-wise hydrolysis mechanism for PBTF (top) leads to the release of oligomers, which are subsequently hydrolysed to monomers. In contrast, the exo-wise hydrolysis mechanism for PBF (bottom) leads to the immediate release of monomers. Reproduced from ref. 82 with permission from Elsevier, copyright 2019. | ||
For the complete biodegradation of a polyester to take place, it first needs to be hydrolysed to small molecules that can be taken up into the bacterial or fungal cell, and in theory, this hydrolysis can occur enzymatically or non-enzymatically. In principle, specific hydrolases (secreted by fungi and bacteria) are required because the rate of non-enzymatic hydrolysis of polyesters is expected to be much slower under mild conditions, such as ambient temperature and/or neutral pH in nature. The occurrence of non-enzymatic hydrolysis is also significantly dependent on the type of ester bonds present in the polyester. Esters from strong carboxylic acids are more easily formed and also are more easily hydrolysed, especially in aquatic environments. When environmental degradability is important, we should opt for polyesters that undergo non-enzymatic hydrolysis as a mechanism for complete (bio)degradation within a reasonable time frame, and especially in aquatic environments. This means that under conditions where biodegradation is disfavoured but where water is present, for example in the deep sea (darkness, cold and low biological activity), non-enzymatic hydrolysable polyesters are suitable to minimise the accumulation of plastics in the environment.
For hydrolysable polyesters, the MNPs could theoretically be hydrolysed more easily than macroplastics due to relatively larger surface areas. The breaking down of oligomers and monomers will continue in the presence of moisture. If the hydrolysis products are known to be biodegradable and not harmful, then full environmental biodegradation is assured. It is also important to realise that breakdown to monomers and small oligomers is slow and will be diluted in nature, which will mitigate risks of monomers that have some toxicity (at elevated concentration) for bacteria and fungi.
Compared to current packaging material (mainly PP, PE, PET),1 hydrolysable plastics can reduce the lifetime of microplastics, which in-turn reduces the likelihood that they absorb and spread contaminants into the food chain. Therefore, hydrolysable polyesters may also contribute to minimising the negative effects of MNPs.
4. Structural effects on the hydrolysis and biodegradation rate of polyesters
Both chemical structures and physical properties, including molecular weight, glass transition and melting temperature (Tg and Tm), crystallinity, rigidity and hydrophilicity, impact the potential for polyesters to biodegrade within a reasonable time frame in various environments. The chemical structure is a major factor in determining thermomechanical and most physical properties of polyesters. As a consequence, (co)polymerisation of different monomers leading to varying compositions is a commonly employed strategy to tune the final properties of polyesters and make them suitable for certain applications. Biodegradability of polyesters is also expected to be tuneable via the same strategy. Therefore, by making copolyesters, it is possible to design biodegradable/hydrolysable polyesters with favourable high thermal- and good mechanical properties.Understanding how the structure of polyesters affects their biodegradability is important for the development of novel plastics. Therefore, in this section, the impacts of the chemical structure on biodegradation or hydrolysis of polyesters are discussed by presenting specific selected examples (a summary table is provided in the ESI, Table S1†). For a detailed discussion on how thermal and mechanical properties are effected by the chemical structure of polyesters, see Larrañaga and Lizundia72 and references mentioned below.
The conventional polyesters, such as PET, exhibit decent thermal and mechanical properties with their high content of the terephthalic acid (TPA) aromatic building block. However, the TPA polyesters are resistant to hydrolysis and biodegradation, as discussed in detail in the TPA section below. Consequently, strategies aimed at enhancing the biodegradability of conventional polyesters involve incorporating monomers that form ester bonds that more easily hydrolyse, such as aliphatic esters, monomers with more acidic carboxylic acids such as oxalic acid or blending them with biodegradable polyesters. It's important to note that the latter approach is not recommended due to the risk of non-biodegradable polyesters persisting and forming MNPs.
Most commercially available biodegradable polyesters, including PLA, PBS and polyhydroxybutyrate (PHB), are aliphatic polyesters with linear structures. However, they generally possess unfavourable thermal- and/or mechanical properties for many applications. The incorporation of rigid aromatic and (bi)cyclic monomers, like terephthalate, furan dicarboxylate, isosorbide, cyclobutene diol or cyclohexane dimethanol into polyesters typically improves their thermal and mechanical properties. Therefore, copolyesters with rigid (bi)cyclic co-monomers attract a lot of attention, but their synthesis is often challenging.
4.1 Aromatic diacid monomers
It as has been reported widely that higher terephthalate content may reduce the enzymatic degradation rate of copolyesters. When the terephthalate content is above 60 mol% (relative to the total diacid), copolyesters are considered non-biodegradable, similar to purely aromatic polyesters (Fig. 5).62,83 This reduced biodegradability can be attributed to the limited accessibility of the enzyme to catalyse the cleavage of ester bonds, likely caused by the aromatic groups.
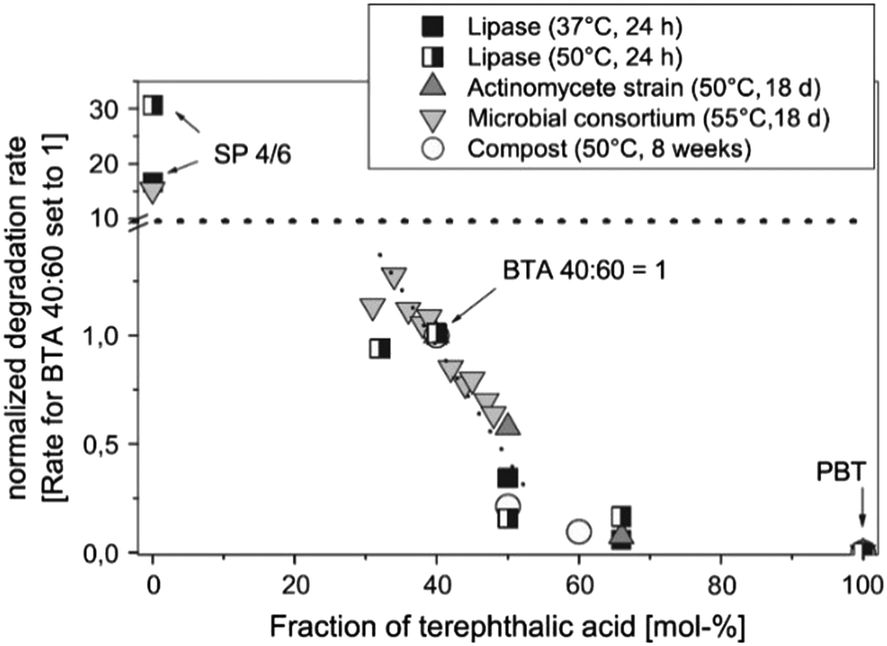 | ||
Fig. 5 Dependence of the degradation rate of aliphatic–aromatic copolyesters (BTA-copolyesters, PBAT) on the content of terephthalic acid in the polymer in different degradation environments. The absolute degradation rates are normalised to the rate for the random copolyesters BTArandom 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60. B: 1,4-Butanediol unit; T: terephthalic acid unit; A: adipic acid unit; SP 4/6: poly(butylene adipate) (SP 4/6). Reproduced from ref. 62 with permission from Elsevier, copyright 2005. :60. B: 1,4-Butanediol unit; T: terephthalic acid unit; A: adipic acid unit; SP 4/6: poly(butylene adipate) (SP 4/6). Reproduced from ref. 62 with permission from Elsevier, copyright 2005. | ||
Interestingly, Marten et al. studied the enzymatic hydrolysis of PBAT50/50 with different micro-structures, including random and strictly alternating, at 37 and 50 °C respectively (Fig. 6).62 It is found that neither of them were significantly hydrolysed at 37 °C, while the alternating one hydrolysed at 50 °C. Even though the molecular weight of the alternating PBAT was only about one third of the other (18 and 51 kg mol−1), this difference was attributed to their different Tm (random, 132 °C; alternating, 85 °C), which also gave an indication of the mobility of the chains. The authors suggested that the chain mobility of polymers increased with an increase in the temperature of the environment, which allows the access by lipase (with a deep active site) for the alternating PBAT50/50 (BTAaltern. 50:50) at 50 °C but not for the random copolyesters (BTAstatist. 50:50). Therefore, they concluded that it may not be possible to design polyesters with high Tm combined with high degradation rate at ambient temperature, unless polyesters could degrade via mechanisms other than enzymatic hydrolysis by lipase (deep active site). This means that polyesters with high Tm could be hydrolysed either non-enzymatically or by other enzymes, for instance enzyme with shallow active sites as shown in the next paragraph.
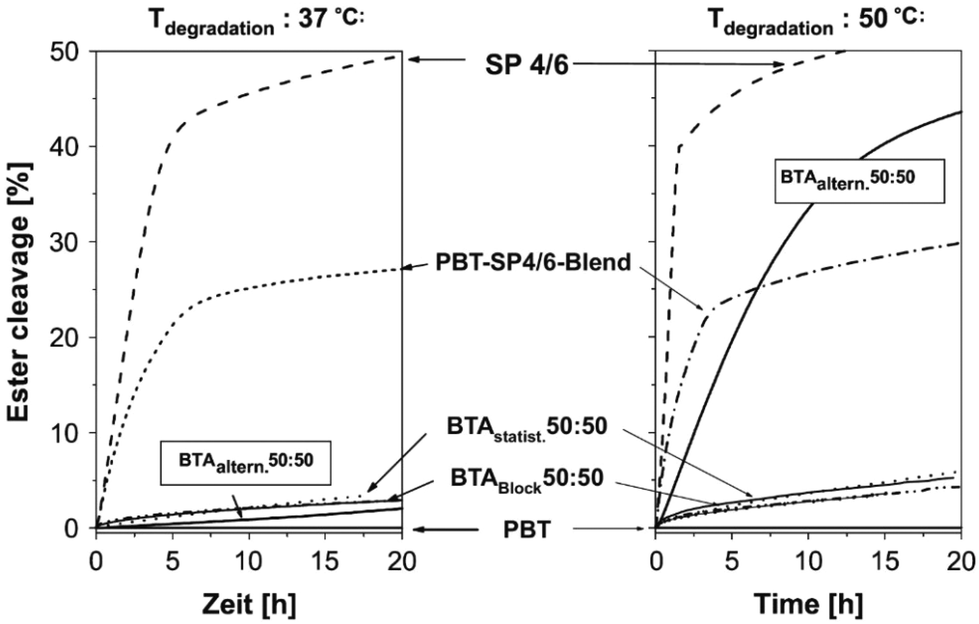 | ||
| Fig. 6 Degradation of polyesters of different micro-structure by a lipase from Pseudomonas sp. (PsL) at 37 °C and 50 °C in water. Degradation was monitored by titrating the acid groups formed during ester cleavage. B: 1,4-Butanediol unit; T: terephthalic acid unit; A: adipic acid unit; SP 4/6: poly(butylene adipate) (SP 4/6). Reproduced from ref. 62 with permission from Elsevier, copyright 2005. | ||
Moreover, Zumstein et al. investigated the relationship between chain flexibility of PBAT (dependent on the ratio of aromatic and aliphatic monomers) and the active site of the enzyme.65 Enzymes with a deep active site (e.g. Rhizopus oryzae lipase (RoL)) require higher chain flexibility to hydrolyse PBAT, while Tm (43–126 °C) has little effect on enzymes with shallow active sites (Fusarium solani cutinase (FsC)) (Fig. 7).
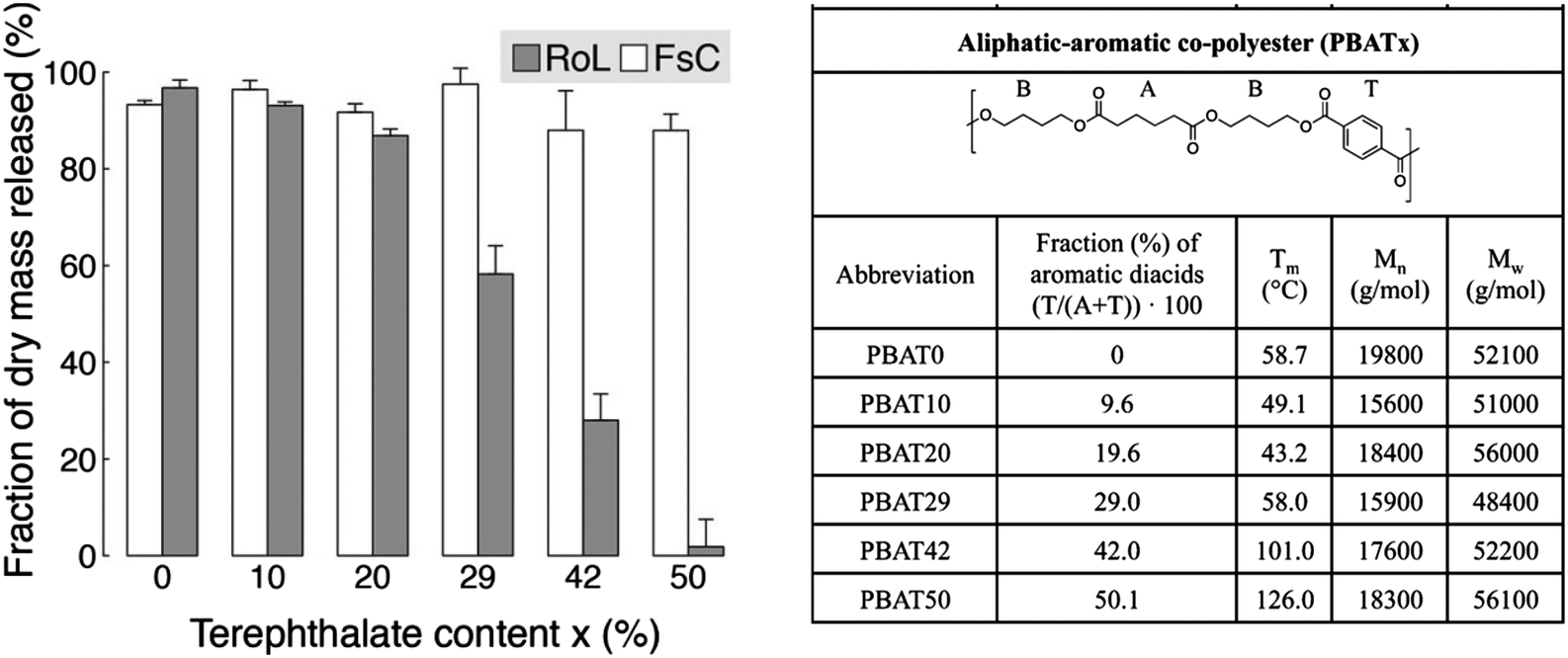 | ||
| Fig. 7 Hydrolysis of poly(butylene adipate-co-terephthalate) (PBATx) thin films by Fusarium solani cutinase (FsC) and Rhizopus oryzae lipase (RoL) at pH 6 and 20 °C, as measured with a quartz-crystal microbalance with dissipation monitoring (QCM-D). The different PBATx polyesters varied in the molar fraction of the aromatic diacid terephthalate (T) to the aliphatic diacid adipate (A) (i.e., x = T/(A + T) × 100). Fraction (%) of initially coated dry polyester mass that was released during the hydrolysis experiments. Error bars represent deviations of duplicate measurements from their mean. Reproduced from ref. 65 with permission from American Chemical Society, copyright 2017. | ||
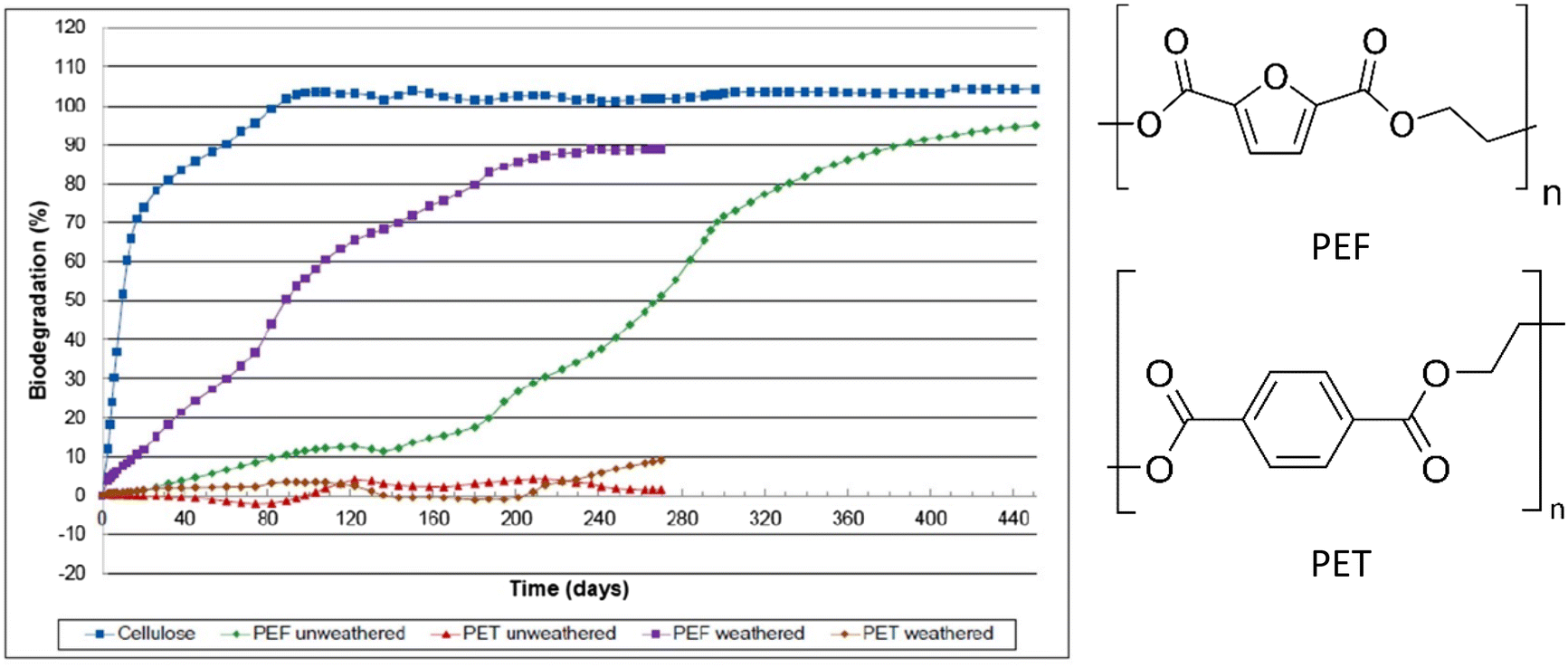 | ||
| Fig. 8 Biodegradation profiles of weathered and un-weathered PEF, as well as weathered and un-weathered PET and cellulose as a reference material. Biodegradation (%) = amount of polymer converted to CO2 (up to 450 days) with air/oxygen @ 58 °C in soil. All curves are the averages of three samples.85 | ||
However, much like TPA, aromatic-aliphatic copolyesters with higher FDCA content were reported to hydrolyse relatively slowly at low temperature (non-industrial composting). For instance, Hu et al. synthesised a series of poly(butylene furandicarboxylate-co-glycolate, PBFGA) copolyesters with different molar ratios of glycolate (GA) and furanoate to conduct hydrolysis experiments with and without porcine pancreas lipase in phosphate-buffered saline at 37 °C.86 Weight loss was observed for PBFGAs with more than 30 mol% GA units (with fully random micro-sequential structures) after 35 days, independent of the presence of enzyme. For PBFGA20, slight weight loss, with visible holes in the surface, was observed for lipase only after 70 days. Higher contents of FDCA further decreased the hydrolysis rate of PBFGA copolyesters (Fig. 9). The authors suggested that the steric hindrance of the furan ring restrains hydrolysis. The threshold of FDCA content for biodegradability is likely higher than for TPA.
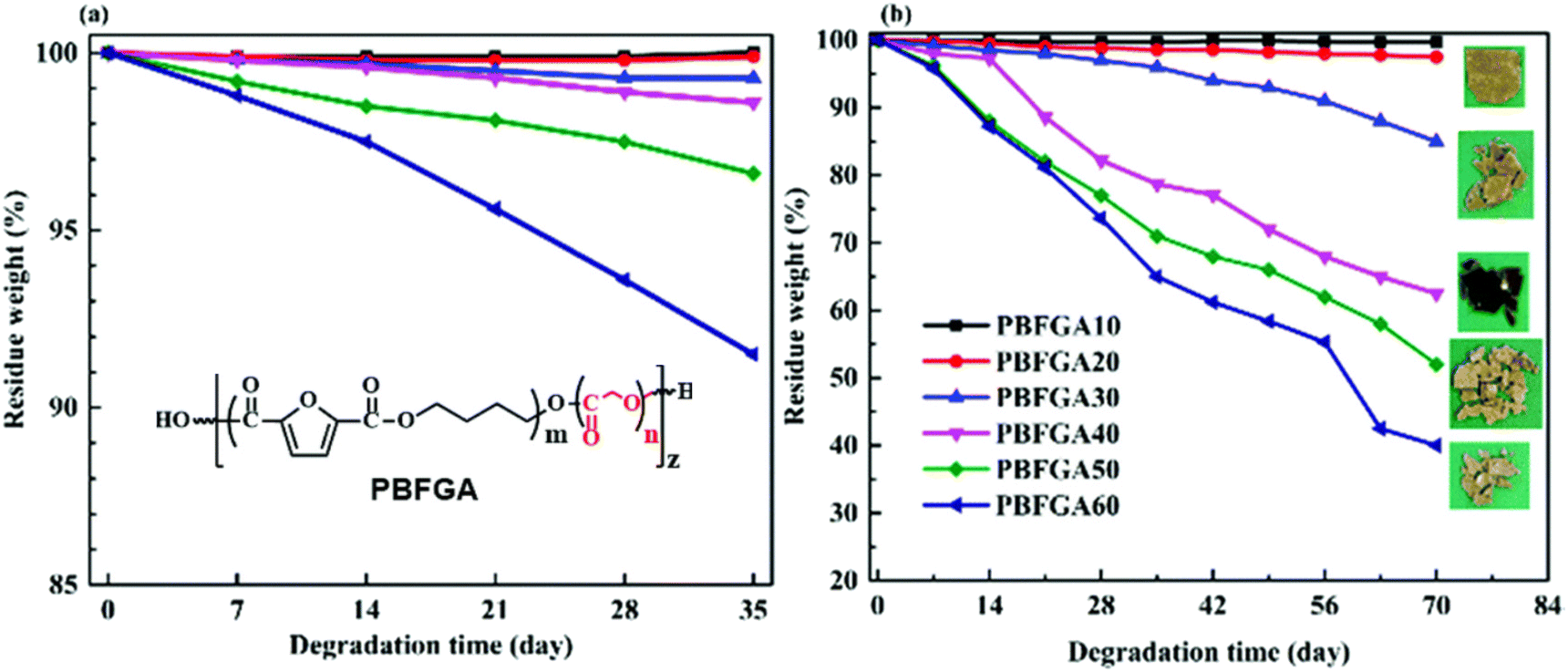 | ||
| Fig. 9 Weight loss curves of PBFGAs during hydrolysis (a), enzymatic degradation (b) and visual observations of degraded films after 70 d at 37 °C. Note: Scales are not equal for (a) and (b). Reproduced from ref. 86 with permission from the Royal Society of Chemistry, copyright 2019. | ||
It is important to note that Tg's of all PBFGAs are around 37 °C (Table 1), and the experiments were performed at 37 °C. The low Tg's indicate the polymer chains have a more flexible structure than for instance PLA (Tg 55–62 °C) or PET (Tg > 70 °C). The faster degrading compositions (40% GA content and higher) were all amorphous, whereas those with less GA exhibited increasing levels of crystallinity with decreasing amounts of GA, resulting in reduced degradation. This suggests that a low Tg (i.e. close to the temperature of degradation) combined with an amorphous structure likely makes the ester bonds more accessible for both water and enzymes.
| 1st heating scan | 1st heating scan after annealing | 2nd heating scan | |||||
|---|---|---|---|---|---|---|---|
| T m (°C) | ΔHm (J g−1) | T m (°C) | ΔHm (J g−1) | T g (°C) | T m (°C) | ΔHm (J g−1) | |
| PBFGA60 | nd | nd | nd | nd | 37.7 | nd | nd |
| PBFGA50 | nd | nd | nd | nd | 37.6 | nd | nd |
| PBFGA40 | nd | nd | 84.5 | 2.7 | 37.3 | nd | nd |
| PBFGA30 | 110 | 0.6 | 112.8 | 17 | 36.3 | nd | nd |
| PBFGA20 | 138.7 | 14.1 | 141.7 | 31 | 38.2 | nd | nd |
| PBFGA10 | 158.7 | 24.1 | 156 | 33.2 | 37.2 | 155.6 | 26.7 |
| PBF | 168.3 | 34.8 | 167.8 | 37.5 | 36.9 | 170.3 | 30.8 |
4.2 Cyclic aliphatic diol monomers
000 ton per year by Roquette in France.
Qi et al. incorporated isosorbide into PBS and obtained a series of random copolymers (PBIS) with a wide range (0–100%) of IS content.87 It was found that the increase in isosorbide almost linearly improves the Tg but reduces the crystallinity and Tm of the resulting copolyesters (Fig. 10a). This was attributed to the molecular structure of isosorbide, which is rigid and asymmetric. Similar effects on Tg were also observed by van der Maas.88 Modification of these properties (i.e. Tg, Tm, crystallinity) are also expected to affect the biodegradation behaviour of PBIS copolyesters.
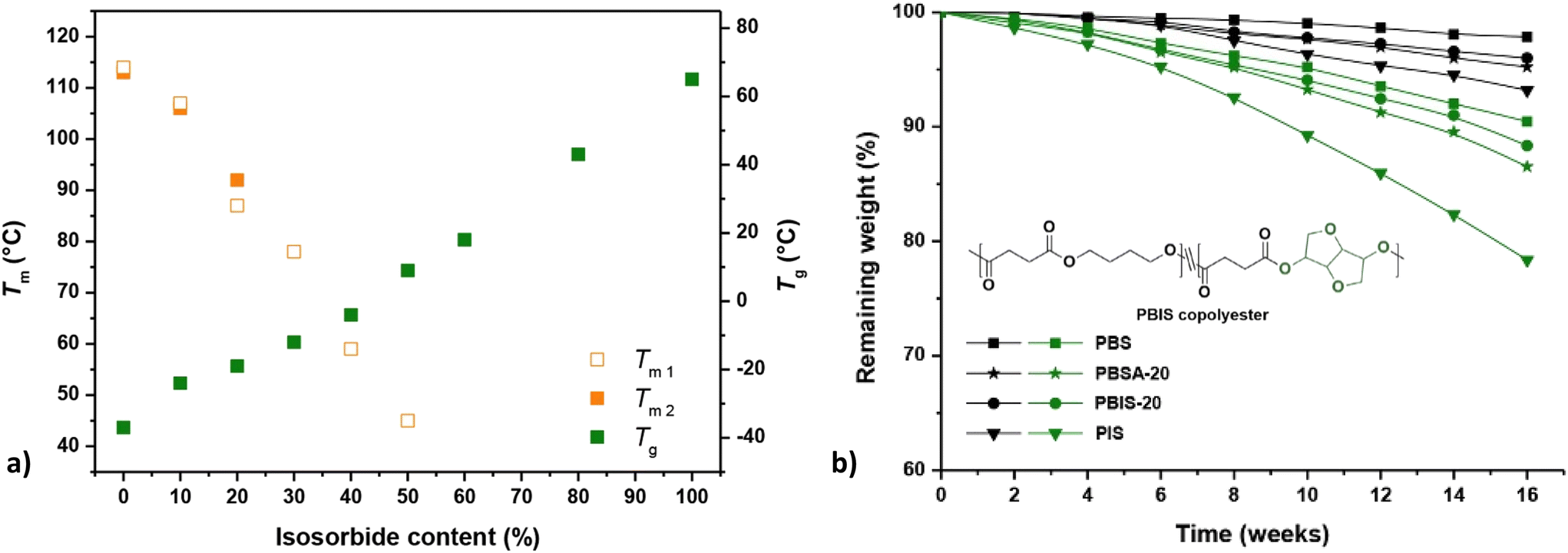 | ||
| Fig. 10 Melting and glass transition temperatures of PBIS copolyesters as a function of isosorbide contents (a). Melting temperatures were obtained during the first and the second heating scans using differential scanning calorimetry (DSC) (Tm1, Tm2). Enzymatic degradation of polyesters (b). Remaining weight of samples tested with (green) or without (black) the presence of porcine pancreas lipase at 37 °C (b). Note: PBSA-20 (A = adipate): Tm1 = 98 °C, Tm2 = 97 °C, Tg = −42 °C. Reproduced from ref. 87 with permission from Elsevier, copyright 2017. | ||
In the same study, Qi et al. compared hydrolysis of homopolymers (PBS and PIS) and copolymers PBIS-20 (20 mol% isosorbide) and PBSA-20 (20 mol% adipate).87 The incorporation of isosorbide was found to facilitate the hydrolysis, especially for enzymatic hydrolysis with porcine pancreas lipase and for the homopolymer PIS (higher content of isosorbide, Fig. 10b). This was attributed to the increased hydrophilicity/hygroscopicity and reduced crystallinity resulting from isosorbide. However, the Tg's of PBS and PBIS-20 were lower than the experiment temperature (37 °C), while the Tg of PIS was rather higher. Together with steric hindrance caused by isosorbide, these factors are actually expected to impede enzymatic hydrolysis of polyesters containing isosorbide.
In addition, we reported the (bio)degradation of poly(isosorbide-co-diol oxalate) (PISOX), a new class of high Tg (tuneable Tg's of sub-zero to 167 °C) renewable (co)polyesters at ambient temperature (25 °C) in soil as well as their non-enzymatic hydrolysis (Table 2 and Fig. 11a).89 This novel material was designed to combine high Tg (>100 °C) with good biodegradation. A representative of copolyesters poly(isosorbide-co-1,6-hexanediol) oxalate with 75/25 mole ratio isosorbide/1,6-hexanediol demonstrated relatively fast biodegradation in soil as well as in the marine environment and the relationship between biodegradation and the non-enzymatic hydrolysis was discussed in our earlier study (Fig. 11b and c).90
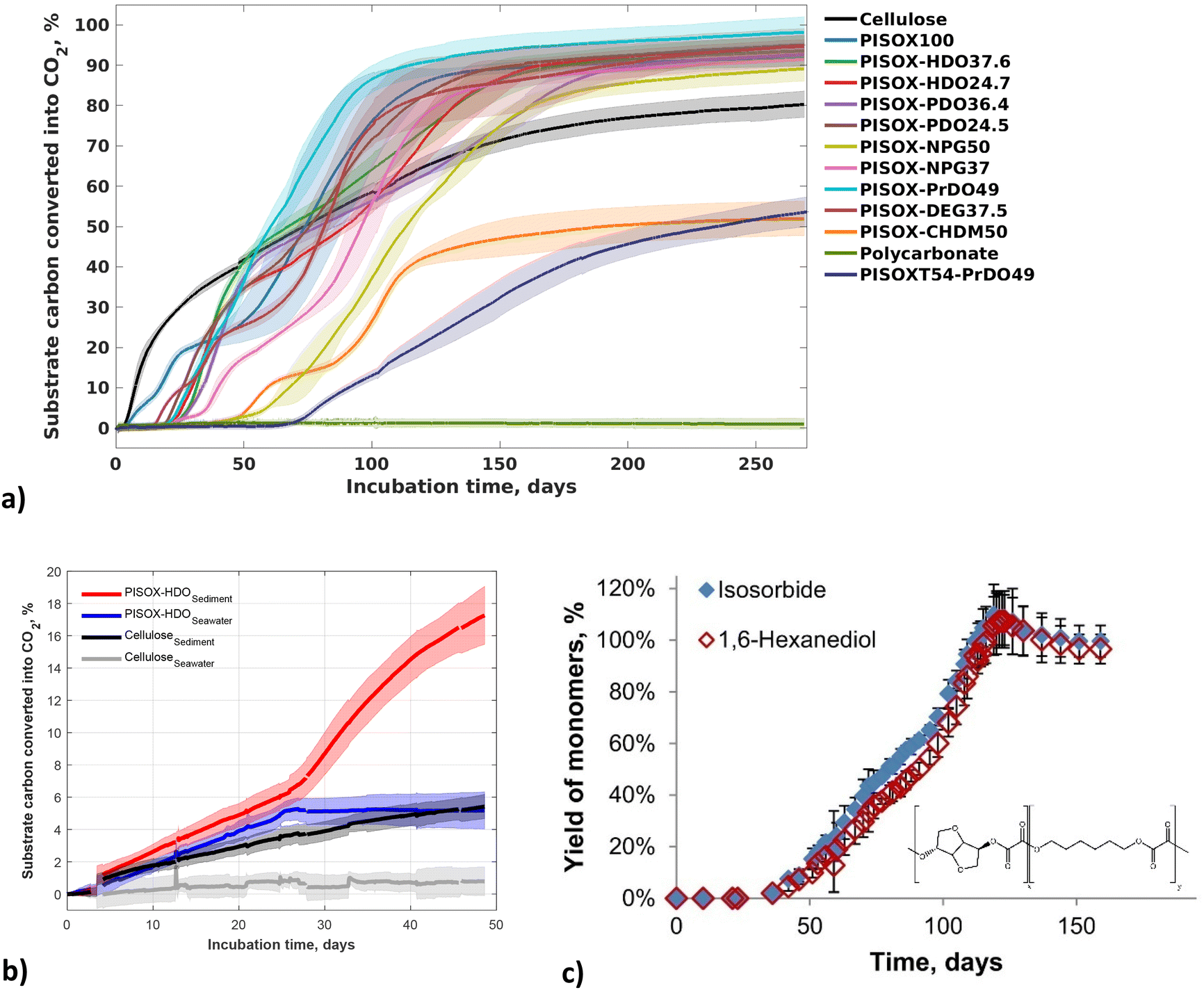 | ||
| Fig. 11 Biodegradation curves of PISOX, PISOX copolyesters, cellulose and polycarbonate (positive and negative reference) with approximately 5 mg (substrate) carbon per gram of dry soil at 25 °C (a). Biodegradation curves of PISOX-HDO25 and cellulose in seawater and on the interface of seawater and sediment (b). Mean biodegradation percentages (lines) were plotted. The shaded area represents the standard deviation of at least three replicates. Hydrolysis of PSIXO-HDO25 (individual yield monomers: isosorbide and 1,6-hexanediol) during 6-month hydrolysis at 25 °C in D2O as percentage of their theoretical maximum release (c). Error bars represent standard deviation of triplicate hydrolysis experiments.60,89 | ||
| Polymer | % IS relative to total diol | Co-monomer | Co-monomer structure | % co-diol relative to total diol | T g, °C |
|---|---|---|---|---|---|
| a % oxalic acid relative to total diacid. b % TPA relative to total diacid. | |||||
| PISOX100 | 100.0% | — | — | — | 167 |
| PISOX-DEG37.5 | 62.5% | Diethylene glycol, DEG |
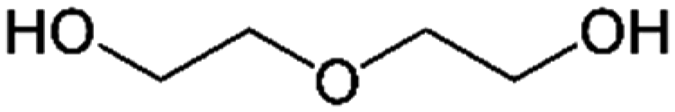
|
37.5% | 88 |
| PISOX-PrDO49 | 51.0% | 1,3-Propanediol, PrDO |
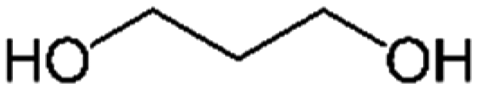
|
49.0% | 85 |
| PISOX-PDO36.4 | 63.6% | 1,5-Pentanediol, PDO |
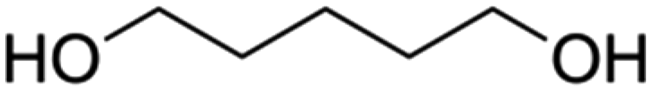
|
36.4% | 85 |
| PISOX-PDO24.5 | 75.5% | 1,5-Pentanediol, PDO | 24.5% | 110 | |
| PISOX-HDO37.6 | 62.4% | 1,6-Hexanediol, HDO |
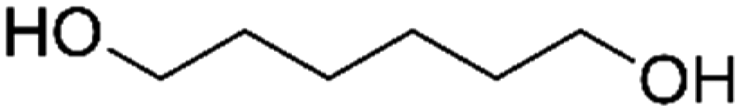
|
37.6% | 76 |
| PISOX-HDO24.7 | 75.3% | 1,6-Hexanediol, HDO | 24.7% | 107 | |
| PISOX-NPG50 | 50.0% | Neopentyl glycol, NPG |
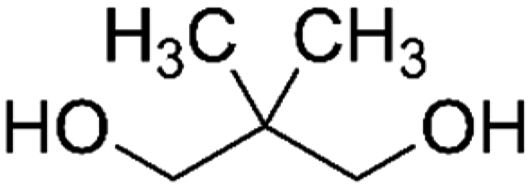
|
50.0% | 83 |
| PISOX-NPG37 | 63.0% | Neopentyl glycol, NPG | 37.0% | 102 | |
| PISOX-CHDM50 | 50.0% | 1,4-Cyclohexanedimethanol, CHDM |
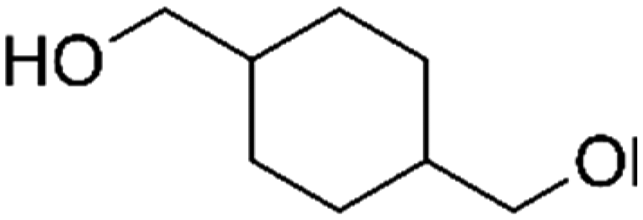
|
50.0% | 101 |
| PISOXT54-PrDO49 | 51.0% | 1,3-Propanediol |
|
49.0% | 102 |
| 46%a | Terephthalic acid | 54%b | |||
In this study,89 all (co)polyesters with non-cyclic co-monomers biodegraded completely within 6 months in soil (Fig. 11a). They show biodegradability comparable to (or even better than) that of cellulose under the same conditions. The PISOX copolymers with 1,4-cyclohexanedimethanol and terephthalic acid as third and fourth comonomer did not exhibit fast biodegradation. However, the PISOX homopolymer, which has a similar content of cyclic rigid monomers, degraded the fastest (Fig. 11a). This indicates that isosorbide, as a biobased rigid cyclic monomer, can provide good thermal (i.e. Tg) and mechanical properties of polymers without sacrificing biodegradability when combined with oxalic acid.
Resistance to heat (i.e. high Tg) for amorphous polymers is generally considered an unfavourable factor for facile environmental biodegradability. An increased difference between Tg and the temperature of the environment negatively affects the mobility of the polymer (like the Tm), and has been attributed to limiting the access to the enzyme, especially for those enzymes with deep active sites. This appears to contrast the biodegradation of PISOX copolyesters, but we saw that non-enzymatic hydrolysis of the oxalate building blocks allowed environmental biodegradability despite the materials having very high Tg's.90
4.3 Linear aliphatic monomers
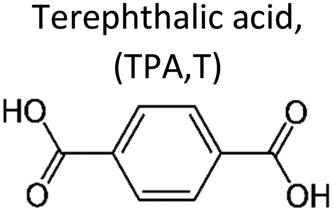
In general, the chain length, or the number of methylene units, of linear aliphatic building blocks is expected to affect thermal–mechanical properties of polyesters as well as biodegradability. To systematically investigate the effect of the number of methylene units on biodegradability of linear polyesters, Baba et al. conducted polycondensations of 1,4-butanediol with nine aliphatic α,ω-dicarboxylic acids (C4–C16) to obtain PBADs (Fig. 12).91 Their environmental biodegradability was determined by biochemical oxygen demand (BOD, i.e. oxygen consumptions) in water (inoculum mixture prepared with 4 soils) at 25 °C. After 30 days, it was found that PBDd (C12) barely biodegraded, and biodegradation rates of PBUd and PBS(u) (C11, C4) were slower than the others (Fig. 12). Despite the long lag phase of PBS(u) and limited incubation time, biodegradability of PBS(u) (during a longer period) could be considered comparable with others, taking into account the steep upward trend at the end of incubation. These results suggested that medium chain length (C4–C8) linear aliphatic building blocks had a similar effect on the biodegradability of polyesters, which is in line with our observations for PISOX copolyesters (Fig. 11a),89 while long chain length diols had a negative effect.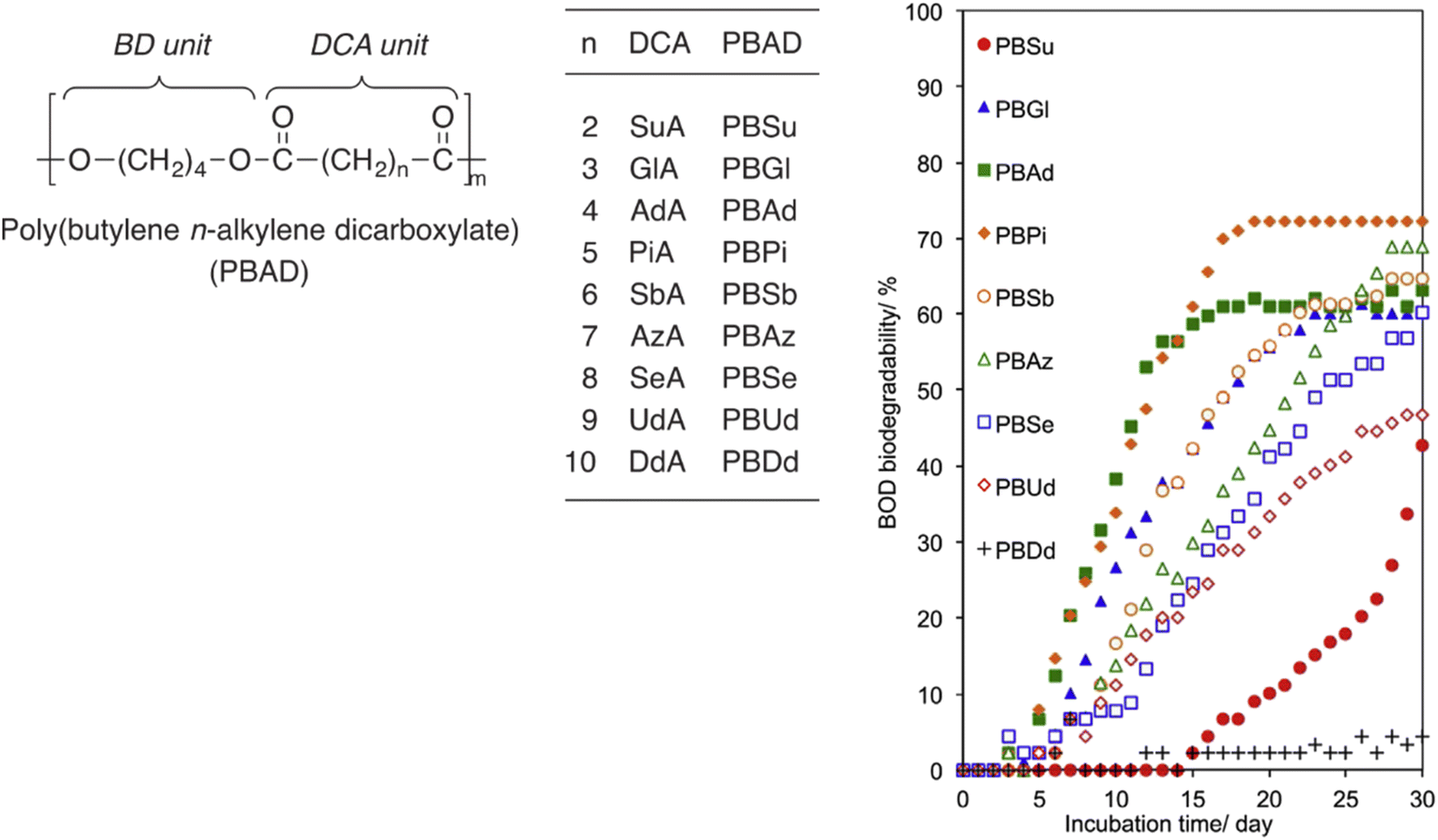 | ||
| Fig. 12 BOD biodegradation curves of PBADs by inoculum mixture of four soils at 25 °C for 30 days. Reproduced from ref. 91 with permission from Elsevier, copyright 2017. | ||
Tachibana et al. also conducted the BOD-test for PBADs with longer chain dicarboxylic acids (C13–C16, Fig. 13).92 As expected, quite limited biodegradation (0–5%) was reported after 45 days.
 | ||
| Fig. 13 BOD biodegradation curves of 1,4-butanediol (BD), n-alkylene dicarboxylic acid (DCAs), including tridecanoic diacid (n = 11; TrdA), tetradecanoic diacid (n = 12; TedA), pentadecanoic diacid (n = 13; PdA), and hexadecanoic diacid (n = 14; HdA), and PBADs at 25 °C for 45 days. Reproduced from ref. 92 with permission from Elsevier, copyright 2021. | ||
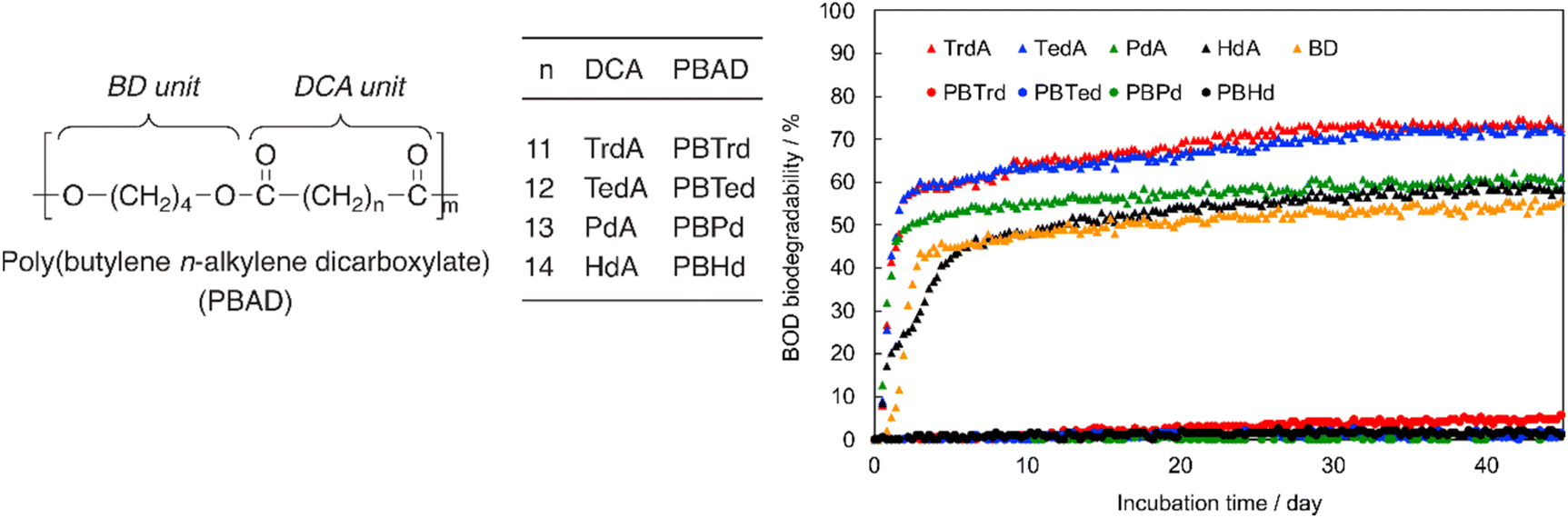
The even–odd effect on Tm was demonstrated clearly with these PBADs (Fig. 14).92 However, this effect did not appear to apply to their biodegradability. Additionally, Tg of PBADs (<−16 °C) are far lower than the incubation temperature.
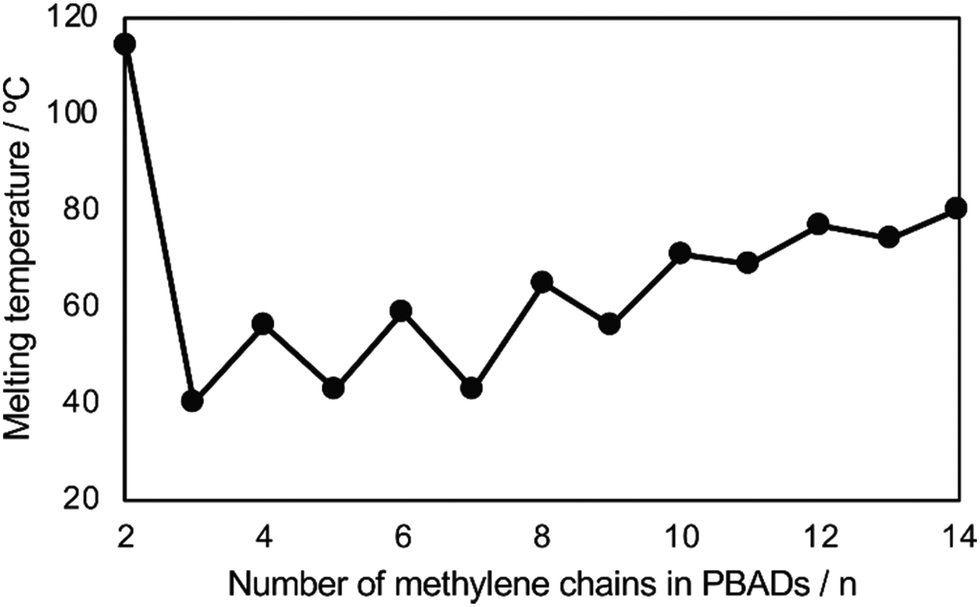 | ||
| Fig. 14 Relationships of Tm of PBAD and number of methylene (n) in the alkylene dicarboxylate of PBADs. Reproduced from ref. 92 with permission from Elsevier, copyright 2021. | ||
Tachibana et al. polymerised oxabicyclodicarboxylic anhydride (OBCA, derived from furfural) and six linear diols of varying chain length units (Fig. 15).93 The same method from the previously-mentioned study was used to assess biodegradability. Generally, no clear effect of the chain length of the diol units on biodegradability could be observed. Three compositions, i.e. diols with chain lengths of 3, 4 and 10 carbon atoms in polyoxabicyclates (POBC), showed high biodegradability in the BOD tests, while POBCs with C2 and C6 showed much lower biodegradability after 90 days. There is therefore no direct relationship between the chain length of the diol and the biodegradability of the polyester. Considering the rapid mineralisation (under the same conditions) of all the diol monomers, the rate-limiting step should be the hydrolysis of the polymers by enzymes produced by the microorganisms in the inoculum. Additionally, in comparison with PBADs, this study showed that incorporation of cyclic building blocks delayed biodegradation significantly.
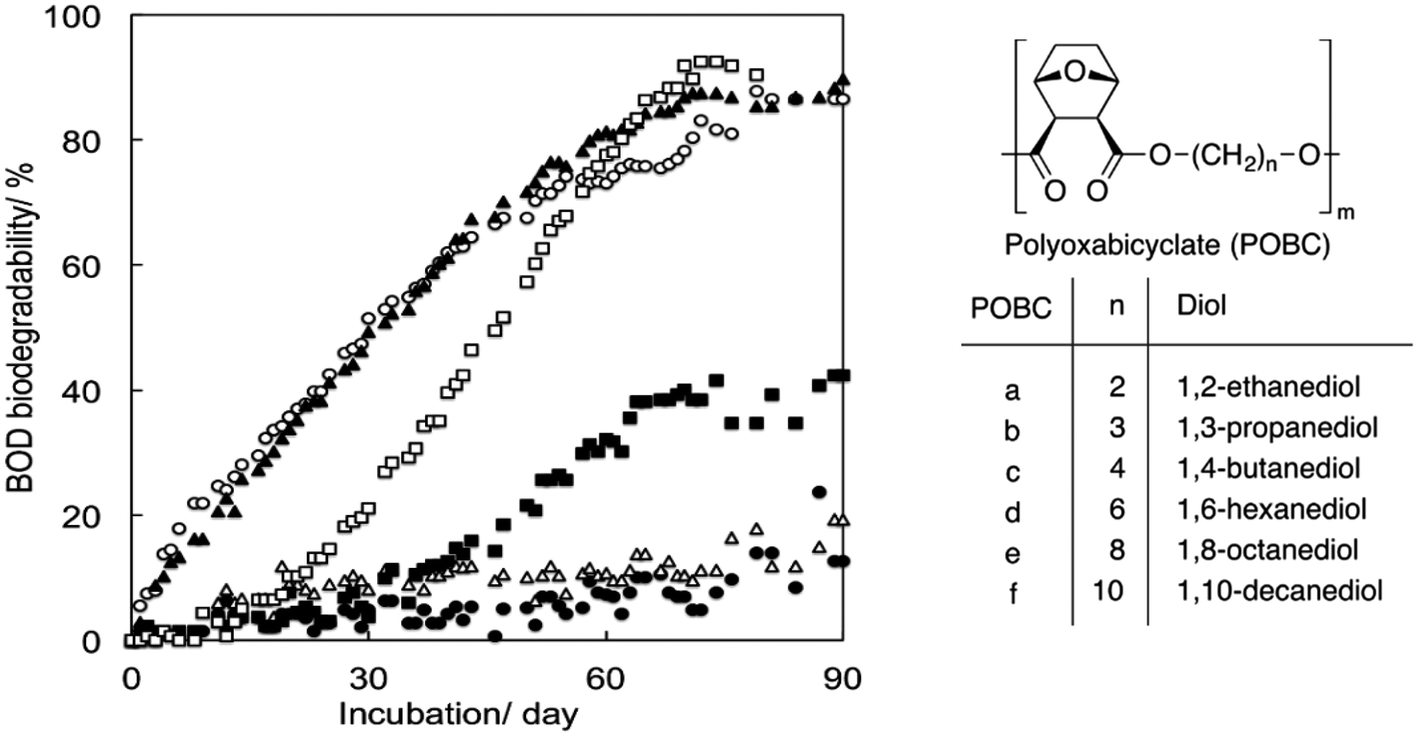 | ||
| Fig. 15 BOD biodegradation curves of POBCs (●: POBC-a, ○: POBC-b, ▲: POBC-c, △: POBC-d, ■: POBC-e, and □: POBC-f) by the mixture inoculum at 25 °C. Reproduced from ref. 93 with permission from Elsevier, copyright 2017. | ||
Therefore, polyoxalates are expected to lead to fast biodegradation in various environments. Combined with isosorbide, poly(isosorbide-co-diol oxalate) polyesters (PISOX-diol) were developed, which demonstrate good mechanical properties, good water vapour- and oxygen barriers, and good thermal properties. Their biodegradation is discussed in Section 4.2 (Fig. 11). Additionally, non-enzymatic hydrolysis of these polyesters was performed at 25 °C in the same study, copolyesters with linear co-diol were non-enzymatically hydrolysed completely within 180 days.89,90 This indicates that facile hydrolysis of oxalate esters is essential for the fast biodegradability of PISOX (co)polyesters at ambient temperature in soil.
Furthermore, the presence of oxalate in polyesters was shown to favour relatively fast biodegradation, independent of the type of the non-cyclic co-diol, such as 1,3-propanediol, diethylene glycol, 1,5-pentanediol, 1,6-hexanediol and neopentyl glycol. Similar to the study of Baba et al., no obvious trend was observed for the chain length of the diol (C3–C6) on the biodegradability of PISOX copolyesters.89,91
4.4 Branched monomers
Specifically, varying the ratio between LA and GA allows for tuning the sensitivity to hydrolysis for PLGA. For example, Zhou et al. presented the non-enzymatic hydrolysis profile of a series of PLGAs with 50% to 100% molar content of LA in phosphate-buffered saline at 37 °C (Fig. 16).98 The losses of microsphere (containing human serum albumin) weight and polymer intrinsic viscosity for PLGAs in a 7-week hydrolytic incubation were presented. Both rates decreased with the increase in LA molar ratios in PLGA in spite of slight increase in Tg.
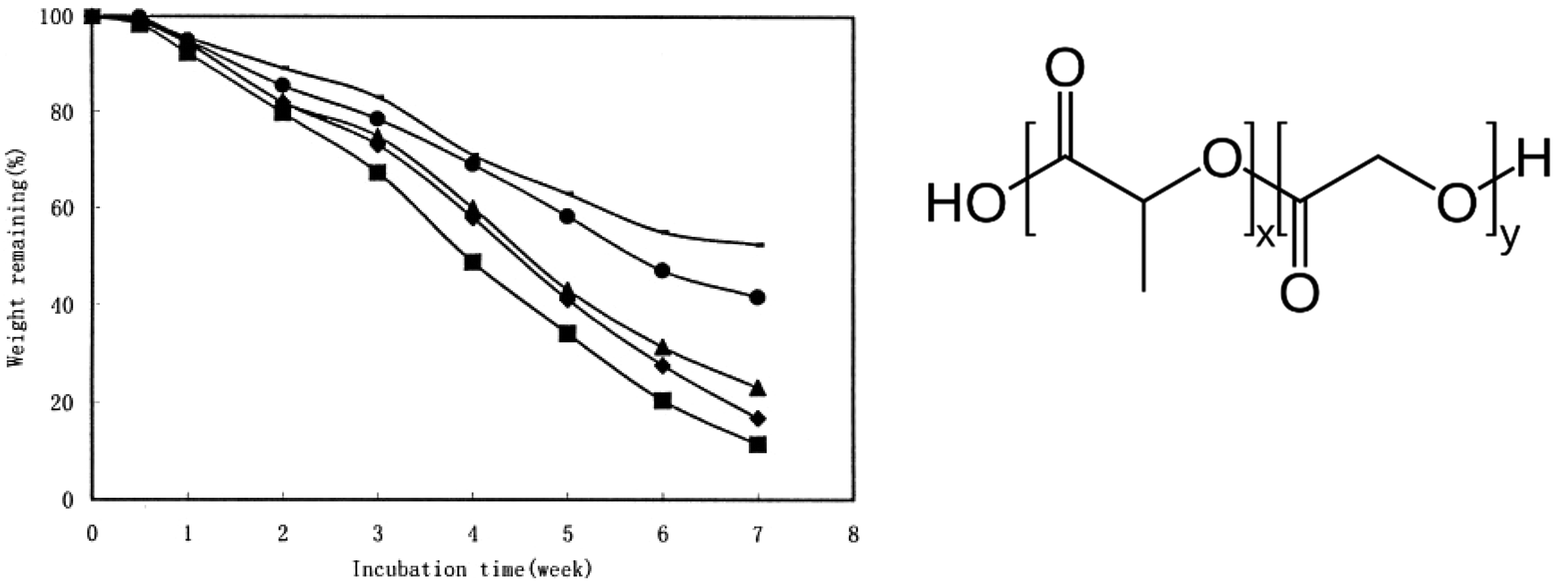 | ||
| Fig. 16 Weight remaining percentage of (—) PLA, (●) PLGA (85/15), (▲) PLGA (75/25), (●) PLGA (65/35), and (■) PLGA (50/50) microspheres containing human serum albumin incubated in phosphate-buffer saline (154 mM, pH 7.4) at 37 °C. Each point represents the mean of three individual samples of microspheres. Reproduced from ref. 98 with permission from John Wiley & Sons Inc., copyright 2003. | ||
The negative impact of LA monomers on hydrolysis for PLGA (LA > 50%) was also reported in other studies.67,97,99 This was attributed to the lower hydrophilicity of LA versus GA. The methyl branch/side-chain makes the lactate ester groups more sterically hindered than the glycolate ester groups, which could reduce the accessibility to water.
The biodegradation and hydrolysis of relatively novel PLGA with high GA content (PLGA12/88 and PLGA6/94) was studied (Fig. 17).60 These polymers biodegraded at a rate similar to cellulose (the reference), which reached approximately 40% mineralisation within 53-day incubation in soil (Fig. 17a).60 However, the non-enzymatic hydrolysis was not that fast (incomplete over 800 days, Fig. 17b).60 The comparison of the hydrolysis profile of these two compositions suggested the competition of two factors that determine the relative hydrolysis rate of these PLGA copolymers: on the one hand, higher LA content results in less hydrophilicity, primarily affecting the early stages of hydrolysis; on the other hand, the presence of crystalline areas with higher GA content appears to slow down the hydrolysis in the later stages.97
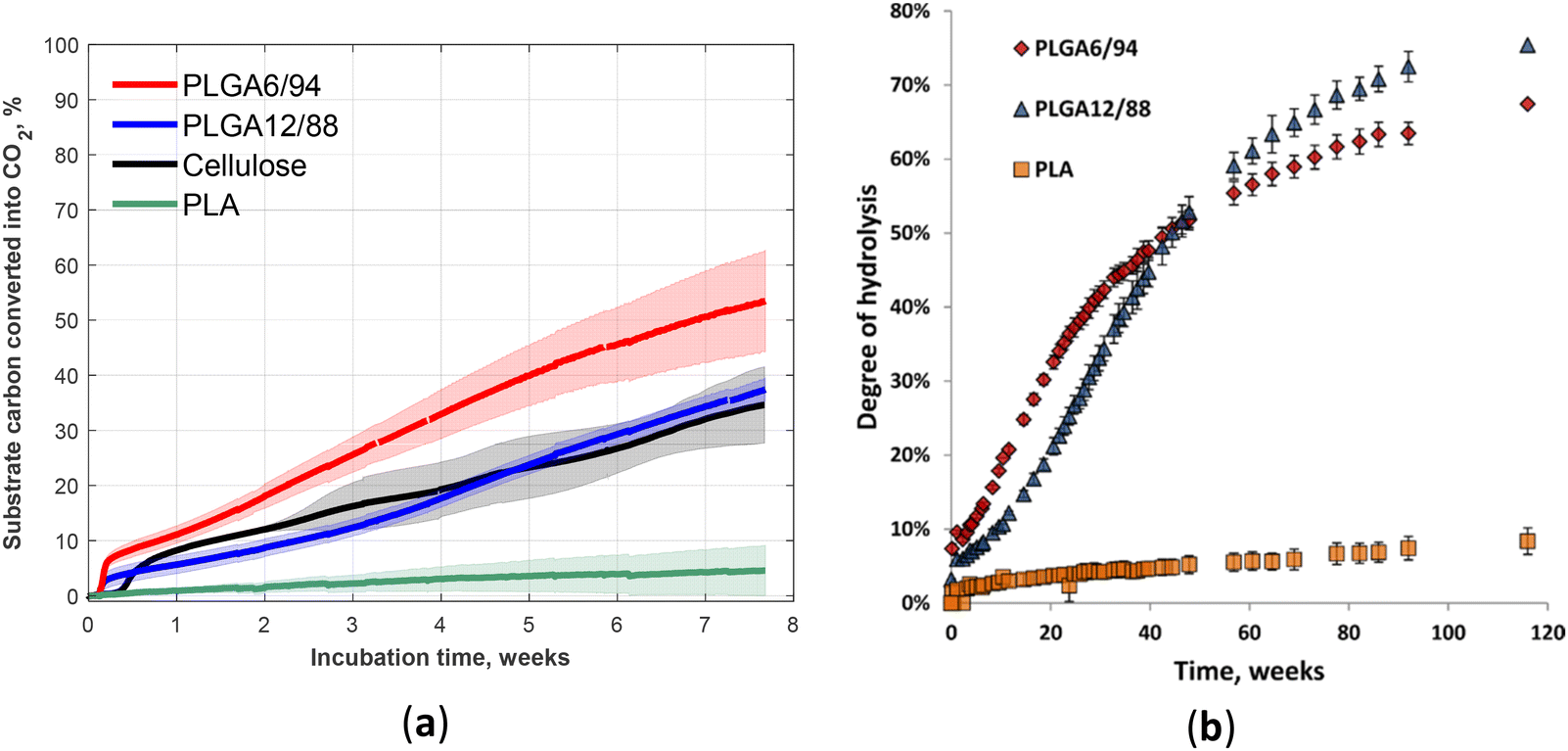 | ||
| Fig. 17 Fifty-three-day biodegradation curves of PLA, PLGA12/88, PLGA6/94 and cellulose (reference) at 25 °C in soil (a). Mean biodegradation (lines) were plotted. The shaded area represents the standard deviation of replicates. Degree of hydrolysis for PLGA6/94, PLGA12/88 and PLA versus time over 116 weeks at 25 °C in D2O (b). The points represent the averages of triplicate experiments, with the error bars representing the standard deviation. | ||
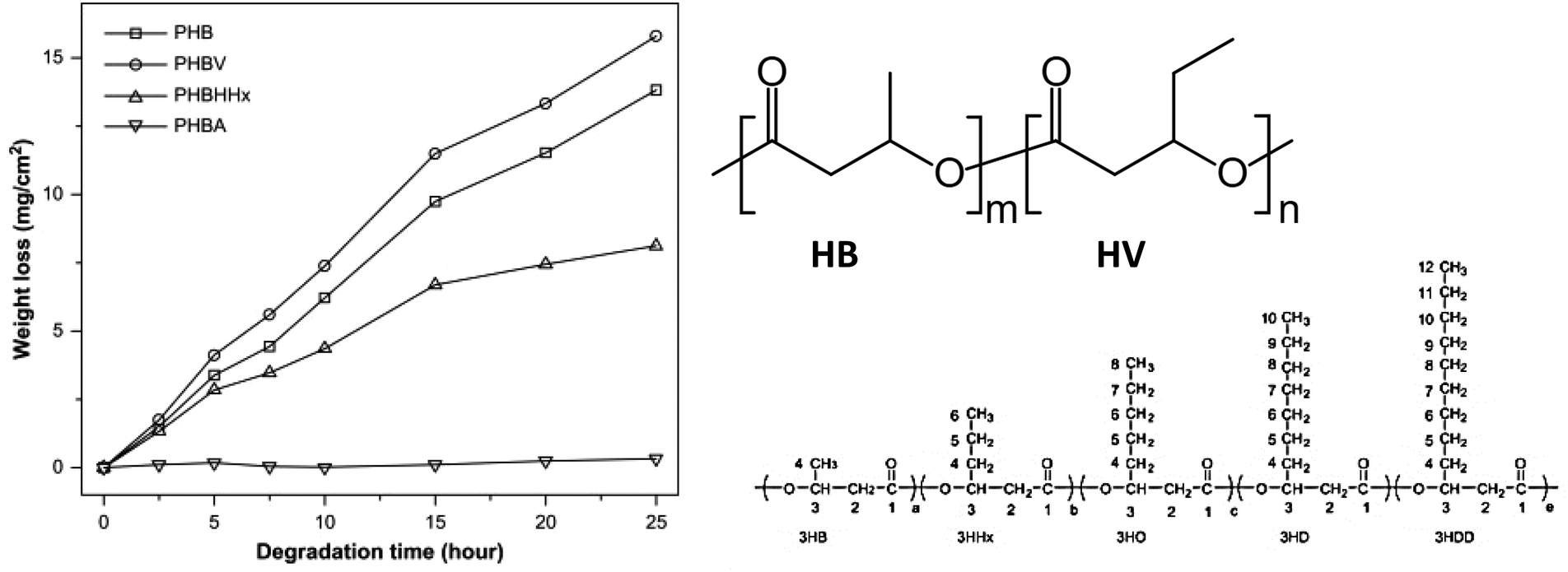 | ||
| Fig. 18 Enzymatic degradation of PHBV (94%), PHB (75%), PHBHHx (39%) and PHBA (0%) by PHA depolymerase produced by Ralstonia pickettii T1 at 37 °C. Structures of monomers and PHBV (m-HB, n-HV, as an example for copolymers). Reproduced from ref. 100 with permission from Elsevier, copyright 2007. | ||
| Sample | P(3HA) compositions (mol%) | Molecular weight | Thermal properties | |||||
|---|---|---|---|---|---|---|---|---|
| HB | Other monomer | HD, HDD | M n (×105) | M w/Mn | T g (°C) | T m (°C) | ΔHm (J g−1) | |
| PHB | 100 | — | — | 1.84 | 2.2 | 3 | 175.9 | 91.5 |
| PHBV | 80.9 | 19.1 (HV) | — | 1.53 | 2.1 | −0.4 | 160.5 | 78.4 |
| PHBHHx | 81.2 | 18.8 (HHx) | — | 5.68 | 1.7 | −0.6 | 103.1 | 75.4 |
| PHBA | 84.8 | 9.4 (HO) | 5.8 | 1.15 | 3.4 | −5.9 | 133.4 | 67.1 |
The biodegradability of these polyesters was evaluated by weight loss (mg cm−2), considering typical surface erosion resulting from enzymatic hydrolysis. It was found that the order of biodegradability (poly(3-hydroxybutyrate – 19 mol% 3-hydroxyvalerate) (PHBV) > poly-3-hydroxybutyrate (PHB) > poly(3-hydroxybutyrate – 19 mol% 3-hydroxyhexanoate) (PHBHHx) > poly(3-hydroxybutyrate – 15 mol% 3-hydroxyalkanoates) (PHBA)) did not correlate with the Tm, Tg, crystallinity and molecular weight but with the side-chain length of comonomers (PHB > PHBV > PHBHHx > PHBA). In general, the biodegradation rate increased with decreasing side-chain length (Fig. 18), except for PHB (the homopolymer), which was an outlier from this trend. Therefore, the authors proposed that the structural effect, i.e. length of side-chain in this case, was the predominant factor determining enzymatic hydrolysis of PHA with intermediate chain length. The side-chain length was considered to affect the hydrolysis in two ways: the depolymerase used in this study only show affinity to PHA with short side-chain, and the higher hydrophobicity of the medium side-chain prevents the access of the enzyme to the crystalline region of the PHA with short side-chains.
5. Concluding remarks and outlook
Considering the predicted growth in plastic demand and the global targets to reduce CO2 emission, it is essential that we initiate a transition to substitute fossil-based plastic feedstock for renewable alternatives. Polyesters allow for a broad selection of feedstock and building blocks (monomers) from biobased and other renewable resources. Furthermore, polyesters can be tuned to provide excellent physico-mechanical, thermal, barrier and biodegradation properties for a wide range of applications.Currently, many plastics utilised on an industrial level accumulate in the environment, and some of these plastics proceed to release microplastic particles; an improvement in waste management is necessary. The three R's (reduce, reuse and recycle) are a well-known framework to deal with these issues. However, (single-use) plastics are important for sanitation and food safety. Recycling heavily relies on collection and recycling infrastructures, which are lacking in many countries, especially in the most populated parts of the world. Therefore, biodegradability should be combined with the three R's.
As a result, next-generation plastics should be either closed-loop recyclable when the infrastructure is available, or designed to degrade completely (e.g. mineralised) over time when ending up in the environment. This necessitates the incorporation of biodegradability as a design feature for applications with an expected short lifetime or collection challenges, such as (food) packaging, mulch films, fishery material and disposable medical items. Furthermore, we should ensure that biodegradable plastics are not advertised in a way that could encourage littering.
Polyesters have the potential to be recycled by both mechanical and chemical processes (Table S1). Therefore, using biodegradable polyesters could help to smooth the transition from the current scenario, where plastics are leaked to the environment, to a future circular economy, where plastics are largely recycled.
The hydrolysable ester bonds offer potential for biodegradation in the environment, especially for polyesters that are susceptible to non-enzymatic hydrolysis at a moderate rate at ambient temperature. Their hydrolysis does not rely on enzymes, so degradation is likely to still take place under unfavourable conditions for biodegradation, such as in the deep sea. Therefore, non-enzymatically hydrolysable polyesters contribute to reducing plastic accumulation in the environment, especially for marine environments. Commercialisation of such polyesters may also reduce the effects of MNPs.
Large quantities of biodegradable polyesters entering the environment can be considered as an input of carbon, which might affect the carbon and nitrogen dynamics.101–103 Some microbial species could be enriched by biodegradation and result in the change of microbial communities, which might decrease the biodiversity.102 For instance, lactic acid could facilitate the growth of lactic acid bacteria, which will generate toxic substance for other microbes and fungi.104 Therefore, qualitative and quantitative investigation into the biodegradation products and their interactions with (micro)organisms and the environment is necessary.
Although the sensitivity to non-enzymatic hydrolysis will reduce the service life of the polymers/plastics (“plastics with an expiry date”), the benefits of environmental biodegradation not requiring specific hydrolase enzymes should outweigh the disadvantages. For many applications, we must consider the trade-off between convenience and reduction in environmental risk. Similar to the variety of thermal and mechanical properties, polyesters also show great potential for designing their biodegradability by selection of building blocks, even though this does require the development of novel synthetic approaches and copolymerisation. Generally, incorporation of cyclic monomers as rigid building blocks, including aromatic and aliphatic compounds, improves certain thermomechanical properties of polyesters. For hydrocarbon-based monomers, this typically leads to a regression in the biodegradation and/or non-enzymatic hydrolysis rate, which becomes worse with higher molar ratio of these cyclic monomers. This could be the result of (1) the decrease in the flexibility of the polymer chain, which decreases the probability of hydrolysis via enzymes with deep active-sites; and/or (2) the steric hindrance/hydrophobicity of the cyclic structure. Biobased isosorbide could be an alternative where the cyclic structure can provide good thermal (i.e. Tg) and mechanical properties to polymers, while also leading to a high level of (bio)degradability.75,87,89
On the other hand, increasing the chain length, i.e. amount of methylene units within aliphatic building blocks for polyesters, could tune the polyester towards being more flexible (and elastomeric), while long chain (>C11) polyesters show significantly less biodegradability than polyesters with medium chain lengths. This may be attributed to the decrease in hydrophilicity. There is no clear trend in biodegradability for short- and middle-length chains. The shortest diacid, oxalic acid, shows sensitivity to hydrolysis. Additionally, (longer) side-chains can result in a raised hinderance to the hydrolysis of polyesters.
The development of novel polyesters from sustainable sources leads to a wide variety of potential compositions. The specific balance between thermal properties, mechanical properties, and biodegradability requires comprehensive investigation on the structure–biodegradability relationship.
Biodegradation is a complex process that may occur via multiple mechanisms at varying rates under different conditions. Given the influence of multiple factors on biodegradation, the use of machine learning could be beneficial for predictive modelling to offer a partially-quantitative approach. These models must consider both internal factors (such as polymer composition) and external factors (e.g. marine environment). This approach can also help identify the significance of factors affecting the biodegradability of a specific type of polyester under defined conditions.105 Consequently, it allows for preliminary screening to select promising polymer compositions suitable for specific applications (computational simulations) and provides control over these internal factors during the polymer design phase.
Moreover, ideally, with the advancement of artificial intelligence (AI) and the development of predictive models, users are allowed to set the requirements for biodegradability and conditions for usage and/or end-of-life, considering specific applications. AI can then assist in the design of (co-)polyester compositions.
However, it's important to emphasis that introducing a product to market entails many factors, and chemical design represents only one aspect within a multidisciplinary process. Biodegradability is just one factor in overall the design.106
To achieve this, we need a comprehensive database of well-characterized polyester biodegradation results under a specified environment. In addition to systematically recoding data, AI could be trained to extract information from published research articles. However, physically conducting experiments will always be required to validate the AI model's output, and to expand the database, which will therefore improve the model.
As biodegradation tests are time-consuming, it is important to employ high-throughput platforms for efficient research into the biodegradability of a polymer.60 This allows for study of the biodegradability of novel polymers with various compositions in a time-efficient way. Therefore, expanding the scope of research into novel materials already in the early stage at a limited cost.
Abbreviations
| CCU | Carbon capture and utilisation |
| FDCA | 2,5-Furandicarboxylic acid |
| BD | 1,4-Butanediol |
| BOD | Biochemical oxygen demand |
| DCA | Dicarboxylic acid |
| DSC | Differential scanning calorimetry |
| GA | Glycolate/glycolic acid |
| GA | Glycolic acid |
| HB | 3-Hydroxybutyrate |
| HDO | 1,6-Hexanediol |
| IS | Isosorbide |
| IUPAC | International Union of Pure and Applied Chemistry |
| LA | Lactic acid |
| M n | Number average molecular weights |
| MNPs | Micro- and nano plastics |
| PBADs | Poly(butylene n-alkylene dicarboxylate) |
| PBAT | Poly(butylene adipate-co-terephthalate) |
| PBDd | Poly(butylene dodecanedioate) |
| PBF | Poly(butylene furandicarboxylate) |
| PBFGA | Poly(butylene furandicarboxylate-co-glycolate) |
| PBIS | Poly(butylene-co-isosorbide) succinate |
| PBS/PBS(u) | Polybutylene succinate |
| PBSA | Poly(butylene succinate-co-butylene adipate) |
| PBTF | Poly(1,4-butylene 2,5-thiophenedicarboxylate) |
| PBUd | Poly(butylene undecanedioate) |
| PC | Polycarbonate |
| PE | Polyethylene |
| PEF | Poly(ethylene 2,5-furandicarboxylate) |
| PET | Poly(ethylene terephthalate) |
| PHA | Polyhydroxyalkanoates |
| PHB | Polyhydroxybutyrate |
| PHBA | Poly(3-hydroxybutyrate – 15 mol% 3-hydroxyalkanoates) |
| PHBHHx | Poly(3-hydroxybutyrate – 19 mol% 3-hydroxyhexanoate) |
| PHBV | Polyhydroxybutyrate-co-hydroxyvalerate |
| PIS | Poly(isosorbide succinate) |
| PISOX/PISOX100 | Poly(isosorbide oxalate) |
| PLA | Poly(lactic acid) |
| PLGA | Poly(lactic-co-glycolic acid) |
| POBC | Polyoxabicyclates |
| POP | Persistent organic pollutant |
| PP | Polypropylene |
| PS | Polystyrene |
| T g | Glass transition temperature |
| Three R's | Reduce, reuse and recycle |
| T m | Melting temperature |
| TPA/T | Terephthalic acid |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded by Netherlands Organisation for Scientific Research (NWO), (grant number 731.017.203). The authors would like to thank LEGO SYSTEM A/S and Avantium for co-funding this project. We thank Bing Wang, Bruno Bottega Pergher, Kevin van der Maas, Maria A. Murcia Valderrama and Daniel H. Weinland for the valuable discussions around polymer properties and synthesis, and Angus McLuskie for refining the language.References
- Plastics – the Facts 2022 – Plastics Europe, https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022/, accessed December 13, 2022.
- PlasticsEurope, Plastics – the Facts 2021, https://plasticseurope.org/knowledge-hub/plastics-the-facts-2021/, accessed April 11, 2022.
- L. A. Hamilton, S. Feit, C. Muffett, M. Kelso, S. M. Rubright, C. Bernhardt, E. Schaeffer, D. Moon, J. Morris and R. Labbé-Bellas, Plastic and Climate: The Hidden Costs of a Plastic Planet, https://www.ciel.org/plasticandclimate/, accessed December 21, 2021.
- European Bioplastics, nova-Institute, Global production capacities of bioplastics 2021, https://www.european-bioplastics.org/bioplastics/materials/, accessed July 13, 2022.
- R. Meys, A. Kätelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Science, 2021, 374, 71–76 CrossRef CAS PubMed.
- S. Spierling, E. Knüpffer, H. Behnsen, M. Mudersbach, H. Krieg, S. Springer, S. Albrecht, C. Herrmann and H.-J. Endres, J. Cleaner Prod., 2018, 185, 476–491 CrossRef.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- Y. Chae and Y.-J. An, Environ. Pollut., 2018, 240, 387–395 CrossRef CAS.
- W. C. Li, H. F. Tse and L. Fok, Sci. Total Environ., 2016, 566–567, 333–349 CrossRef CAS.
- A. Borunda, National Geographic, 2019.
- P. Prarat and P. Hongsawat, Mar. Pollut. Bull., 2022, 174, 113200 CrossRef CAS.
- L. G. A. Barboza, L. R. Vieira, V. Branco, C. Carvalho and L. Guilhermino, Sci. Rep., 2018, 8, 15655 CrossRef.
- P. D. Dissanayake, S. Kim, B. Sarkar, P. Oleszczuk, M. K. Sang, M. N. Haque, J. H. Ahn, M. S. Bank and Y. S. Ok, Environ. Res., 2022, 209, 112734 CrossRef CAS PubMed.
- J.-J. Guo, X.-P. Huang, L. Xiang, Y.-Z. Wang, Y.-W. Li, H. Li, Q.-Y. Cai, C.-H. Mo and M.-H. Wong, Environ. Int., 2020, 137, 105263 CrossRef CAS.
- P. Li, X. Wang, M. Su, X. Zou, L. Duan and H. Zhang, Bull. Environ. Contam. Toxicol., 2021, 107, 577 CrossRef CAS.
- P. Pannetier, B. Morin, F. Le Bihanic, L. Dubreil, C. Clérandeau, F. Chouvellon, K. Van Arkel, M. Danion and J. Cachot, Environ. Int., 2020, 134, 105047 CrossRef CAS.
- H. Qu, R. Ma, H. Barrett, B. Wang, J. Han, F. Wang, P. Chen, W. Wang, G. Peng and G. Yu, Environ. Int., 2020, 136, 105480 CrossRef CAS.
- A. Batel, F. Linti, M. Scherer, L. Erdinger and T. Braunbeck, Environ. Toxicol. Chem., 2016, 35, 1656–1666 CrossRef CAS.
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Environ. Int., 2022, 107199 CrossRef CAS.
- United Nations Environment Programme, Drowning in Plastics – Marine Litter and Plastic Waste Vital Graphics, https://www.unep.org/resources/report/drowning-plastics-marine-litter-and-plastic-waste-vital-graphics, accessed April 7, 2022.
- J. J. Klemeš, Y. V. Fan and P. Jiang, Energy Sources, Part A, 2021, 43, 1549 CrossRef.
- J. Nikiema and Z. Asiedu, Environ. Sci. Pollut. Res., 2022, 29, 24547 CrossRef.
- M. Maissan, COVID-19, https://www.waste2wear.com/covid-19-increased-plastic-waste/, accessed April 15, 2022.
- J. Jiang, K. Shi, X. Zhang, K. Yu, H. Zhang, J. He, Y. Ju and J. Liu, J. Environ. Chem. Eng., 2022, 10, 106867 CrossRef CAS.
- F. A. Cruz Sanchez, H. Boudaoud, M. Camargo and J. M. Pearce, J. Cleaner Prod., 2020, 264, 121602 CrossRef.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- PlasticsEurope, Plastics – the Facts 2013 – Plastics Europe, https://plasticseurope.org/knowledge-hub/plastics-the-facts-2013/, accessed April 13, 2022.
- Ellen MacArthur Foundation, The New Plastics Economy: Rethinking the future of plastics, 2016.
- T. I. U. of P. and A. Chemistry (IUPAC), IUPAC – biodegradable polymer (BT07169), https://goldbook.iupac.org/terms/view/BT07169, accessed August 5, 2022.
- A. Krzan, S. Hemjinda, S. Miertus, A. Corti and E. Chiellini, Polym. Degrad. Stab., 2006, 91, 2819–2833 CrossRef CAS.
- M. C. Krueger, H. Harms and D. Schlosser, Appl. Microbiol. Biotechnol., 2015, 99, 8857–8874 CrossRef CAS.
- J. D. Badia, O. Gil-Castell and A. Ribes-Greus, Polym. Degrad. Stab., 2017, 137, 35–57 CrossRef CAS.
- A.-U.-R. Bacha, I. Nabi, M. Zaheer, W. Jin and L. Yang, Sci. Total Environ., 2023, 858, 160108 CrossRef CAS PubMed.
- S. S. Ali, T. Elsamahy, R. Al-Tohamy, D. Zhu, Y. A.-G. Mahmoud, E. Koutra, M. A. Metwally, M. Kornaros and J. Sun, Sci. Total Environ., 2021, 780, 146590 CrossRef CAS PubMed.
- D. N. Bikiaris, Polym. Degrad. Stab., 2013, 98, 1908–1928 CrossRef CAS.
- L. Liu, M. Xu, Y. Ye and B. Zhang, Sci. Total Environ., 2022, 806, 151312 CrossRef CAS PubMed.
- N. Lucas, C. Bienaime, C. Belloy, M. Queneudec, F. Silvestre and J.-E. Nava-Saucedo, Chemosphere, 2008, 73, 429–442 CrossRef CAS.
- M. Sander, Environ. Sci. Technol., 2019, 53, 2304–2315 CrossRef CAS PubMed.
- F. Cappitelli, P. Principi and C. Sorlini, Trends Biotechnol., 2006, 24, 350–354 CrossRef CAS PubMed.
- I. V. Kirstein, A. Wichels, E. Gullans, G. Krohne and G. Gerdts, PLoS One, 2019, 14, e0215859 CrossRef CAS PubMed.
- S. Wallström, E. Strömberg and S. Karlsson, Polym. Test., 2005, 24, 557–563 CrossRef.
- J. Gerritse, H. A. Leslie, C. A. de Tender, L. I. Devriese and A. D. Vethaak, Sci. Rep., 2020, 10, 10945 CrossRef CAS.
- D. Lobelle and M. Cunliffe, Mar. Pollut. Bull., 2011, 62, 197–200 CrossRef CAS.
- A. L. Andrady, P. W. Barnes, J. F. Bornman, T. Gouin, S. Madronich, C. C. White, R. G. Zepp and M. A. K. Jansen, Sci. Total Environ., 2022, 851, 158022 CrossRef CAS.
- K. Zhang, A. H. Hamidian, A. Tubić, Y. Zhang, J. K. H. Fang, C. Wu and P. K. S. Lam, Environ. Pollut., 2021, 274, 116554 CrossRef CAS.
- J. Yuan, J. Ma, Y. Sun, T. Zhou, Y. Zhao and F. Yu, Sci. Total Environ., 2020, 715, 136968 CrossRef CAS.
- S. S. Ali, T. Elsamahy, E. Koutra, M. Kornaros, M. El-Sheekh, E. A. Abdelkarim, D. Zhu and J. Sun, Sci. Total Environ., 2021, 771, 144719 CrossRef CAS.
- Degradation of plastics and plastic-degrading bacteria in cold marine habitats|SpringerLink, https://link.springer.com/article/10.1007/s00253-018-9195-y, accessed June 30, 2021.
- E. J. Okal, G. Heng, E. A. Magige, S. Khan, S. Wu, Z. Ge, T. Zhang, P. E. Mortimer and J. Xu, Ecotoxicol. Environ. Saf., 2023, 262, 115202 CrossRef CAS PubMed.
- J. Lai, H. Huang, M. Lin, Y. Xu, X. Li and B. Sun, Front. Microbiol., 2022, 13, 1113705 CrossRef PubMed.
- A.-U.-R. Bacha, I. Nabi and L. Zhang, ACS ES&T Engg, 2021, 1, 1481–1501 Search PubMed.
- Science Advice for Policy by European Academies (SAPEA), Biodegradability of plastics in the open environment, Science Advice for Policy by European Academies (SAPEA), DE, 2020.
- M. Nazareth, M. R. C. Marques, M. C. A. Leite and Í. B. Castro, J. Hazard. Mater., 2019, 366, 714–722 CrossRef CAS PubMed.
- The International Organization for Standardization, ISO 14855-1:2012(en), Determination of the ultimate aerobic biodegradability of plastic materials under controlled composting conditions – Method by analysis of evolved carbon dioxide – Part 1: General method, the International Organization for Standardization, 2012.
- ASTM International, ASTM D6400-21, Standard Specification for Labeling of Plastics Designed to be Aerobically Composted in Municipal or Industrial Facilities, ASTM International, ASTM International, West Conshohocken, PA, 2021, vol. 08.003, https://www.astm.org.
- EN13432, CE de NCEN, EN 13432, Packaging – Requirements for Packaging Recoverable Through Composting and Biodegradation – Test Scheme and Evaluation criteria for the Final Acceptance of Packaging, 2000.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- M. Karamanlioglu, R. Preziosi and G. D. Robson, Polym. Degrad. Stab., 2017, 137, 122–130 CrossRef CAS.
- M. Kunioka, F. Ninomiya and M. Funabashi, Polym. Degrad. Stab., 2006, 91, 1919–1928 CrossRef CAS.
- Y. Wang, M. A. Murcia Valderrama, R.-J. van Putten, C. J. E. Davey, A. Tietema, J. R. Parsons, B. Wang and G.-J. M. Gruter, Polymers, 2022, 14, 15 CrossRef CAS PubMed.
- I. E. Napper and R. C. Thompson, Environ. Sci. Technol., 2019, 53, 4775–4783 CrossRef CAS PubMed.
- E. Marten, R.-J. Müller and W.-D. Deckwer, Polym. Degrad. Stab., 2005, 88, 371–381 CrossRef CAS.
- S. Li, J. Biomed. Mater. Res., 1999, 48, 342–353 CrossRef CAS PubMed.
- L. N. Woodard and M. A. Grunlan, ACS Macro Lett., 2018, 7, 976–982 CrossRef CAS PubMed.
- M. T. Zumstein, D. Rechsteiner, N. Roduner, V. Perz, D. Ribitsch, G. M. Guebitz, H.-P. E. Kohler, K. McNeill and M. Sander, Environ. Sci. Technol., 2017, 51, 7476–7485 CrossRef CAS PubMed.
- Y. Tokiwa, B. P. Calabia, C. U. Ugwu and S. Aiba, Int. J. Mol. Sci., 2009, 10, 3722–3742 CrossRef CAS PubMed.
- M. A. Tracy, K. L. Ward, L. Firouzabadian, Y. Wang, N. Dong, R. Qian and Y. Zhang, Biomaterials, 1999, 20, 1057–1062 CrossRef CAS PubMed.
- A. Togo and H. Hagihara, Polym. Degrad. Stab., 2023, 216, 110488 CrossRef CAS.
- R. Jiang, G. Lu, Z. Yan, J. Liu, D. Wu and Y. Wang, J. Hazard. Mater., 2021, 405, 124247 CrossRef CAS PubMed.
- J. S. C. Viera, M. R. C. Marques, M. C. Nazareth, P. C. Jimenez, C. Sanz-Lázaro and Í. B. Castro, J. Hazard. Mater., 2021, 416, 125957 CrossRef CAS PubMed.
- S. Shabbir, M. Faheem, N. Ali, P. G. Kerr, L.-F. Wang, S. Kuppusamy and Y. Li, Sci. Total Environ., 2020, 717, 137064 CrossRef CAS PubMed.
- A. Larrañaga and E. Lizundia, Eur. Polym. J., 2019, 121, 109296 CrossRef.
- B. Wang and G.-J. M. Gruter, PCT patent application by Avantium, WO2018211132A1, 2018 Search PubMed.
- B. Wang and G.-J. M. Gruter, PCT patent application by Avantium, WO2018211133A1, 2018 Search PubMed.
- N. Kasmi, Z. Terzopoulou, Y. Chebbi, R. Dieden, Y. Habibi and D. N. Bikiaris, Polym. Degrad. Stab., 2022, 195, 109804 CrossRef CAS.
- R. Kumar, K. Sadeghi, J. Jang and J. Seo, Sci. Total Environ., 2023, 882, 163446 CrossRef CAS PubMed.
- F. M. Lamberti, L. A. Román-Ramírez and J. Wood, J. Polym. Environ., 2020, 28, 2551–2571 CrossRef CAS.
- S. Thiyagarajan, E. Maaskant-Reilink, T. A. Ewing, M. K. Julsing and J. van Haveren, RSC Adv., 2022, 12, 947–970 RSC.
- A. L. Merchan, T. Fischöder, J. Hee, M. S. Lehnertz, O. Osterthun, S. Pielsticker, J. Schleier, T. Tiso, L. M. Blank, J. Klankermayer, R. Kneer, P. Quicker, G. Walther and R. Palkovits, Green Chem., 2022, 24, 9428–9449 RSC.
- F. von Burkersroda, L. Schedl and A. Göpferich, Biomaterials, 2002, 23, 4221–4231 CrossRef CAS PubMed.
- H. Tsuji and K. Nakahara, J. Appl. Polym. Sci., 2002, 86, 186–194 CrossRef CAS.
- M. Gigli, F. Quartinello, M. Soccio, A. Pellis, N. Lotti, G. M. Guebitz, S. Licoccia and A. Munari, Environ. Int., 2019, 130, 104852 CrossRef CAS PubMed.
- R.-J. Müller, I. Kleeberg and W.-D. Deckwer, J. Biotechnol., 2001, 86, 87–95 CrossRef PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas. United States, 2004 DOI:10.2172/15008859.
- E. de Jong, H. A. Visser, A. S. Dias, C. Harvey and G.-J. M. Gruter, Polymers, 2022, 14, 943 CrossRef CAS PubMed.
- H. Hu, R. Zhang, J. Wang, W. B. Ying, L. Shi, C. Yao, Z. Kong, K. Wang and J. Zhu, Green Chem., 2019, 21, 3013–3022 RSC.
- J. Qi, J. Wu, J. Chen and H. Wang, Polym. Degrad. Stab., 2019, 160, 229–241 CrossRef CAS.
- K. van der Maas, PhD thesis, University of Amsterdam, 2024.
- Y. Wang, K. van der Maas, D. H. Weinland, D. Trijnes, R.-J. van Putten, A. Tietema, J. R. Parsons, E. de Rijke and G.-J. M. Gruter, Environ. Sci. Technol., 2024, 12 DOI:10.1021/acs.est.2c09699.
- Y. Wang, C. J. E. Davey, K. van der Maas, R.-J. van Putten, A. Tietema, J. R. Parsons and G.-J. M. Gruter, Sci. Total Environ., 2022, 815, 152781 CrossRef CAS PubMed.
- T. Baba, Y. Tachibana, S. Suda and K. Kasuya, Polym. Degrad. Stab., 2017, 138, 18–26 CrossRef CAS.
- Y. Tachibana, T. Tsutsuba, K. Kageyama and K. Kasuya, Polym. Degrad. Stab., 2021, 190, 109650 CrossRef CAS.
- Y. Tachibana, M. Yamahata, H. Ichihara and K. Kasuya, Polym. Degrad. Stab., 2017, 146, 121–125 CrossRef CAS.
- E. Schuler, P. A. Ermolich, N. R. Shiju and G.-J. M. Gruter, ChemSusChem, 2021, 14, 1517–1523 CrossRef CAS PubMed.
- European Bioplastics, Institute for Bioplastics and Biocomposites: Berlin, 2020, 2021.
- M. A. Murcia Valderrama, R.-J. van Putten and G.-J. M. Gruter, ACS Appl. Polym. Mater., 2020, 2, 2706–2718 CrossRef CAS PubMed.
- Encyclopedia of Biomaterials and Biomedical Engineering, ed. G. Wnek and G. Bowlin, CRC Press, 2008, pp. 2291–2301 Search PubMed.
- S. Zhou, X. Deng, X. Li, W. Jia and L. Liu, J. Appl. Polym. Sci., 2004, 91, 1848–1856 CrossRef CAS.
- S. Li, H. Garreau and M. Vert, J. Mater. Sci. Mater. Med., 1990, 1, 198–206 CrossRef CAS.
- Z. Li, H. Lin, N. Ishii, G.-Q. Chen and Y. Inoue, Polym. Degrad. Stab., 2007, 92, 1708–1714 CrossRef CAS.
- F. Meng, X. Yang, M. Riksen and V. Geissen, Environ. Res., 2022, 204, 111938 CrossRef CAS PubMed.
- W. Courtene-Jones, A. Martínez Rodríguez and R. D. Handy, Emerging Top. Life Sci., 2022, 6, 423–433 CrossRef PubMed.
- S. Bandopadhyay, L. Martin-Closas, A. M. Pelacho and J. M. DeBruyn, Front. Microbiol., 2018, 9, 819 CrossRef PubMed.
- S. Siedler, R. Balti and A. R. Neves, Curr. Opin. Biotechnol., 2019, 56, 138–146 CrossRef CAS PubMed.
- K. Min, J. D. Cuiffi and R. T. Mathers, Nat. Commun., 2020, 11, 727 CrossRef CAS PubMed.
- R. S. Boethling, E. Sommer and D. DiFiore, Chem. Rev., 2007, 107, 2207–2227 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04489k |
| This journal is © The Royal Society of Chemistry 2024 |