 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the effect of fusion partners on the enzymatic activity and thermodynamic stability of poly(ethylene terephthalate) degrading enzymes†
Liliana
Oliveira
 ,
Alex
Cahill
,
Len
Wuscher
,
Kerry R.
Green
,
Victoria
Bemmer
and
Bruce R.
Lichtenstein
*
,
Alex
Cahill
,
Len
Wuscher
,
Kerry R.
Green
,
Victoria
Bemmer
and
Bruce R.
Lichtenstein
*
Centre for Enzyme Innovation, School of Biological Sciences, University of Portsmouth, UK. E-mail: bruce.lichtenstein@port.ac.uk
First published on 17th May 2024
Abstract
Plastics are a cornerstone of the modern world, yet the durable material properties that we have come to depend upon have made them recalcitrant environmental pollutants. Biological solutions in the form of engineered enzymes offer low energy and sustainable approaches to recycle and upcycle plastic waste, uncoupling their production and end of life from fossil fuels and greenhouse gases. These enzymes however, encounter immense challenges acting on plastics: facing hydrophobic surfaces, molecular crowding, and high levels of substrate heterogeneity. There have been mixed reports about the benefits of fusing partner domains to polyethylene terephthalate (PET) degrading enzymes, with moderate improvements identified under specific conditions, but no clarity into the factors that underlie the mechanisms. Here, we use the SpyCatcher003:SpyTag003 technology, which demonstrates a profound 47 °C shift in Tm upon irreversible complex formation, to investigate the influence of the thermal stability of the fusion partner on a range of PETases selected for their optimal reaction temperatures. We find that the thermal stability of the fusion partner does not have a positive correlation on the activity of the enzymes or their evident kinetic and thermal stabilities. Instead, it appears that the fusion to less stable SpyCatcher003 tends to increase the measured activation energy of unfolding compared to the more stable complex and wildtype enzymes. Despite this, the fusions to SpyCatcher003 do not show significantly better catalytic activity on PET films, with or without SpyTag003, and were found to be sometimes disruptive. The approach we highlight here, in using a fusion partner with controllable melting temperature, allowed us to dissect the impact of the stability of a fusion partner on enzyme properties. Although fusion stability did not appear to be coupled with identifiable trends in enzymatic activities, careful analysis of the unfolding pathways, and solid and solution activities of a wider range of enzymes may yield a more detailed understanding.
Introduction
Plastic-degrading enzymes encounter unique challenges when tackling their substrates, which generally are hydrophobic solids with relatively immobile crystalline and amorphous domains. The surface of the plastic acts as a potential site for denaturation and aggregation of the enzymes through non-specific adsorption. This, in turn, is anticipated to slow reaction kinetics owing to both reduction in functional enzyme concentrations, and reduced access to substrate due to surface fouling. The issue is more acute at the elevated temperatures that enzymes digesting plastics like polyethylene terephthalate (PET) commonly operate, where the need for a flexible active site works in opposition to the requirement for thermal stability.1,2 Enhanced stability through coupling of enzymes to suitable protein partners has been observed across a variety of enzyme classes acting on soluble substrates.3–5 Despite these successes, the benefits of fusions to enzymes acting at a polymer surface is less clear cut.Fusions to PETases have been reported that enhance, diminish, or leave unperturbed the catalytic turnover of plastic substrates.6,7 The most significant improvement to PET-degrading enzymes realised with a fusion protein appears to be between IsPETase and IsMHETase, which together catalyse the degradation of PET to terephthalic acid (TPA) and ethylene glycol (EG), without substantial accumulation of the mono-glycol MHET, possibly driven by improvement of substrate flux and alleviation of product inhibition.8 Other such dual enzyme fusions show comparatively moderate levels of reported enhancement in the degradation rate of PET under specific conditions.9 Slight improvements were also observed in the activity of PETase fusions with non-catalytic carbohydrate binding domains,10,11 α-synuclein,12 and hydrophobins,13 which are thought to target the enzymes to the polymer surface, effectively decreasing their Km. Most surprising, however, is that fusions to what are likely bystander protein domains, like thioredoxin,14 also appear to have the ability to enhance the enzymatic turnover of PET, suggesting that the assumed mechanisms underlying successful fusions may rather be a direct consequence of the enzyme being bound to a sufficiently stabilising protein partner.
Gross changes in the effective stability of enzymes upon fusion with another domain are not guaranteed, and the extent of these effects depends upon a range of factors including interactions between domains, individual thermal stabilities, as well as their folding pathways and kinetics. Furthermore, enhanced thermodynamic and kinetic stability does not necessarily translate to more efficient enzymes as there are competing considerations that can advantage enzymes with higher dynamics, for instance supporting active site rearrangements and flexibility, at a cost of enzyme longevity and durability.
Most of our understanding of how fusions affect protein properties comes from observations that well folded partners, like maltose binding protein (MBP) or SUMO, help to increase soluble expression yields.15,16 These protein partners are largely thought to serve as molecular chaperones, preventing partially unfolded structures of fused proteins from aggregating in solution.17 This is similar in concept, but not molecular detail, as predicted volume exclusion effects,18 where the high local concentration of the fusion partner effectively increases the energy of unfolding by either stabilising the native state or destabilising the higher volume unfolded ensemble through soft interactions and steric effects,19 respectively. Despite established detailed molecular models, clearly defining the exact role of a fusion partner on an enzyme’s stability and activity remains challenging, as specific factors that may be at play are obscured by competing and often counteracting effects.
The high variation observed in the effects of fusions to PETases leaves unresolved whether the approach, as applied to soluble enzymes, is viable as a strategy to support biocatalytic reactions at solid surfaces. Systematic studies on the costs and benefits of enzyme fusions have been limited by the lack of fusion partners for which biophysical parameters can be adequately controlled with minimal perturbation of their structure and sequence. It is thus difficult to divorce the effects on enzyme fusions from the changing structural properties of the fusion partner. Owing to this, we sought to establish an approach where we could introduce a fusion partner that, through minimal sequence and structural changes, has profoundly varied thermodynamic properties. To accomplish this, we adapted the Spycatcher:SpyTag (SC:ST) system.20
The SpyCatcher:SpyTag technology allows the irreversible assembly of protein domains under a range of conditions via the formation of an isopeptide bond in solution between the separately produced Spy components. The system is similar in size to SUMO tags and has proven amenable to engineering, and the Howarth group has produced increasingly sophisticated and functional variants since their initial report.21,22 Given the nature of the system, a large number of teams have taken advantage of this protein ‘superglue,’ focusing principally on functional assemblies of enzymes,23,24 new materials,25 vaccines,26 but also in applications in cyclising proteins for improved stability.7 However, few if any of these reports make use of the incredible increase in stability of SpyCatcher upon binding to the SpyTag. When in complex, Spycatcher003’s melting temperature (Tm) increases by approximately 47 °C, going from a Tm of 48.3 °C to 95.2 °C.22 This enhanced structural stability upon complex formation is associated with minimal structural perturbation (Fig. 1) as the binding of SpyCatcher to SpyTag depends upon a pre-assembled native β-strand structure.
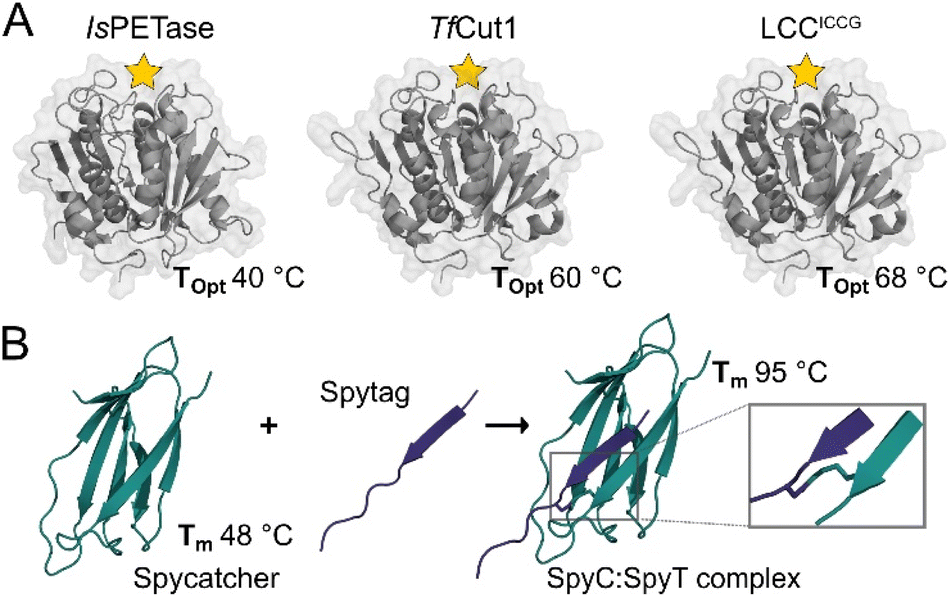 | ||
| Fig. 1 Crystal structures of selected PET hydrolases and the SpyCatcher:SpyTag complex. (A) With active sites indicated by a yellow star, the selected catalytic domains, IsPETase (PDB: 6EQE), TfCut1 (PDB: 7QJR), and LCCICCG (PDB: 8JMO), show a range of optimal reaction temperatures (Topt) when digesting solid PET substrates. (B) SpyCatcher and SpyTag form a covalent complex (PDB: 4MLS) via a spontaneous peptide bond between Lys31 and Asp117, increasing its Tm by 48 °C. | ||
In this study, we report on the effect of SpyCatcher003 as a fusion partner to three established PET-degrading enzymes: IsPETase,27TfCut1 (ref. 28) and LCCICCG,29 selected for their activity optima ranging from 40 °C to 70 °C, allowing us to examine the effect of stability of a fusion partner on the catalytic activity, as well as the thermodynamic and kinetic stability of associated enzymes uncoupled from significant structural changes.
Results and discussion
We designed an approach that allows for the production of the PETase-SpyCatcher003 fusions separate to SpyTag003, which was expressed as a C-terminal fusion to MBP with an installed TEV cut site and internal His-tag (Fig. 2). This allowed the isolation of the expressed fusions with minimal variation from established protocols, as well as providing a unique MBP affinity tag for resolving the bound SpyCatcher003:SpyTag003 assemblies from any unreacted enzyme-fusions. TEV cleavage at the SpyTag003 and chromatography of these assemblies yielded our final Enzyme-SpyCatcher003:SpyTag003 fusions.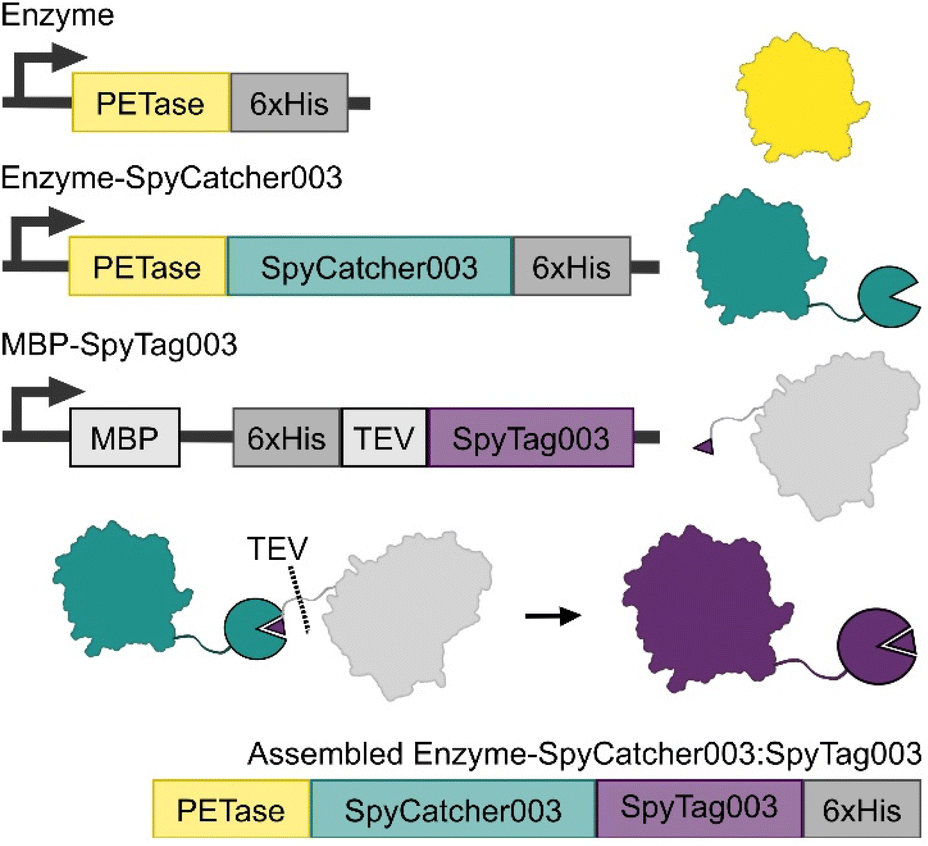 | ||
| Fig. 2 Expression and enzyme constructs used in this study. Wildtype PET hydrolases were expressed as a control (yellow), and in fusion with SpyCatcher003 (teal). SpyTag003 was expressed as a C-terminal fusion to MBP (grey). Once mixed, the Spycatcher003 and SpyTag003 form a covalent complex, after which TEV cleavage upstream of the fused SpyTag003 yields the final assembled enzyme-SpyCatcher003:SpyTag003 complex (purple). | ||
The three PETases used in this study were selected on the basis of their reported Tm as well as their reported optimal temperature for activity Topt. IsPETase was selected as a mesophilic enzyme with optimal activity at 40 °C; TfCut1 was selected as a moderately thermostable enzyme with an optimal enzymatic activity at 60 °C, and lastly engineered LCCICCG was selected for its elevated thermostability and optimal temperature at 68 °C. Finally, all PETase activity tests were carried out at 100 nM enzyme concentration, as this is the concentration where most of the known PET degrading enzymes generally have achieved maximal activity on films prior to any observed concentration dependent inhibition.
Effect of fusion thermostability on PET-degrading activity
Our initial designs for the enzyme fusions used the full length SpyCatcher003, however we observed substantially reduced protein yields and enzymatic activity on PET film with these constructs. Structural modelling via ColabFold30,31 suggested that the N-terminus of the full length SpyCatcher003 is capable of unfolding and wrapping around the globular fold of PETases, blocking the active site groove (ESI. 3†). Removing this N-terminal loop to create the ΔN1 variant restored both expressions for yields and activity. For ease, in the remainder of the report we refer to the ΔN1 variant as Spycatcher003.Activities of the PETases varied only slightly upon fusion to either SpyCatcher003 or SpyCatcher003:SpyTag003 (Fig. 3). IsPETase showed slightly diminished activity at 100 nM protein concentration at 30 °C, and at 40 °C the PET degrading activity was lost (ESI. 4†); some recovery of activity was observed after assembly of the SpyCatcher003:SpyTag003 complex in both cases. While the negative effect of the SpyCatcher003 fusion in this case is somewhat unexpected from a thermal stability perspective, it is in line with our prior observation that the reported IsPETase–IsMHETase fusion shows substantial concentration-dependent inhibition above approximately 75 nM,32 and may reflect the fact that additional crowding at the plastic surface due to an additional domain can force the enzymes into plastic-bound, but unproductive conformations.
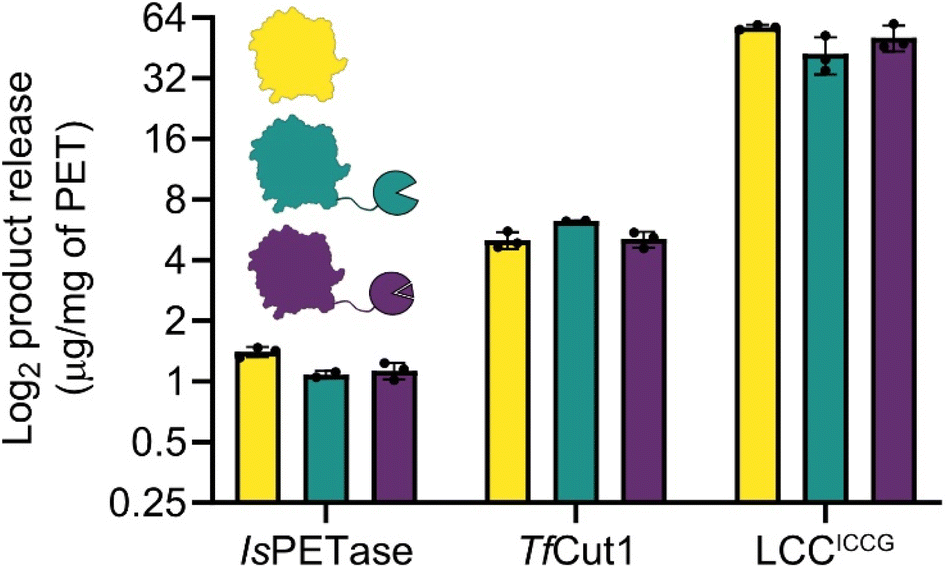 | ||
| Fig. 3 Total product release from amorphous PET film digestions. Despite different fusion stabilities, the activities of PET hydrolases are not disrupted, with the exception of IsPETase, which shows a slight diminishment in activity. TfCut1 has a slight elevation in activity when fused to SpyCatcher003, however this is eliminated in the SpyCatcher003:SpyTag003 fusion complex. The observed activities in LCCICCG are not significantly different. The plot is in log2 scale so that differences between enzymes can be more easily compared, linear plots are in ESI. 4.† | ||
With TfCut1, a small 20% increase in activity was observed with the SpyCatcher003 fusion without the tag, but this improvement was lost upon complex formation with the SpyTag003. This is somewhat surprising as without the tag, the Tm of the Spycatcher003 is lower than the temperature of optimal activity (60 °C), and the SpyCatcher003 is expected to be substantially unfolded under the assay conditions; in contrast, after formation of the SpyCatcher003:SpyTag003 complex, the temperature of the reaction is substantially below the Tm of the complex, and yet there are no apparent benefits in activity.
A similar pattern was observed with LCCICCG, where the unfolded SpyCatcher003 fusion was well tolerated by the enzyme. Even though a slight decrease in total product release was observed; when comparing the SpyCatcher003 fusion to the wildtype enzyme, this difference was not significant. Moreover, the SpyTag003 complex also did not affect PET-degrading activity negatively and is within error of both the SpyCatcher003 fusion and the wildtype.
Effect of fusion thermostability on the thermodynamic and kinetic stabilities of enzymes
We sought to understand whether these subtle changes in enzymatic activity were matched by changes in thermodynamic and kinetic stabilities, therefore we used differential scanning calorimetry to derive stability parameters, using variable scan rates ranging from 24 °C to 192 °C per hour (Table 1). Here, we found Spycatcher003 and Spycatcher003:SpyTag003 to be fully reversible and in equilibrium between the native and denatured state (N = D) with a Tm of 52 °C without SpyTag003 and 98 °C with SpyTag003, slightly higher than previous reports.22 In modelling the irreversible unfolding kinetics of IsPETase, TfCut1 and LCCICCG using the CalFitter server,33,34 all were found to go directly from their native state to a denatured state, with no reversibility (N → D); the Tm was measured to be 53 °C, 78 °C and 95 °C, respectively.| Protein | Model | T m/Tact | E act |
|---|---|---|---|
| a Data could not be resolved between the catalytic domain and the fusion. | |||
| SpyCatcher003 | N = D (Van’t Hoff’s) | 52.11 ± 0.02 | N/A |
| SpyCatcher003:SpyTag003 | N = D (Van’t Hoff’s) | 97.74 ± 0.03 | N/A |
| IsPETase | N → D | 52.67 ± 0.04 | 189.45 ± 2.21 |
| IsPETase-SCa | N → I1 → I2 → D | 44.54 ± 0.01 | 353.54 ± 5.39 |
| 46.58 ± 0.05 | 338.61 ± 2.28 | ||
| 47.45 ± 0.02 | 129.63 ± 7.84 | ||
| IsPETase-SC:ST | N → D | 49.82 ± 0.21 | 149.13 ± 16.14 |
| TfCut1 | N → D | 77.79 ± 0.07 | 210.81 ± 4.01 |
| TfCut1-SC | N → I1 → I2 → D | 75.84 ± 0.16 | 417.10 ± 25.82 |
| 73.15 ± 0.17 | 382.81 ± 7.27 | ||
| 79.07 ± 0.35 | 242.05 ± 10.61 | ||
| TfCut1-SC:ST | N → D | 78.46 ± 0.11 | 195.97 ± 5.13 |
| LCCICCG | N → D | 95.40 ± 0.01 | 516.35 ± 3.56 |
| LCCICCG-SC | N → D | 97.2 ± 0.02 | 561.15 ± 5.16 |
| LCCICCG-SC:STa | N → I → D | 93.47 ± 0.05 | 494.76 ± 7.62 |
| 97.08 ± 0.01 | 524.91 ± 1.24 | ||
Overall, the thermodynamic and kinetic stability data show no clear pattern that applies globally to the three enzymes. The fusions to SpyCatcher003 generally cause an increase of the activation energy (Eact) of unfolding steps, with only a couple of exceptions; however, this does not translate to changes in the temperature at which irreversible unfolding steps occur (Tact). Moreover, the results did not follow a priori predictions, that increased fusion partner stability would be beneficial or less negatively impactful on enzyme stability: here, the SpyTag003 complex did not always benefit the less thermostable PETases, and the SpyCatcher003 on its own did not always disrupt the more thermostable ones.
IsPETase showed a decrease in Tact when bound to both SpyCatcher003 and SpyCatcher003:SpyTag003, although this effect is less substantial when the SpyTag003 is present. This result correlates with the effects seen on the PET activity data, which suggests that there may be an interplay between surface crowding and reduced thermal stability manifested in the activity data. Despite a general increase in Eact when bound to Spycatcher003 alone, this did not positively affect IsPETase’s activity. The data also suggests that, when bound to Spycatcher003, IsPETase passes through two partially unfolded intermediates (I1/I2) before fully denaturing. This was not observed in the data with the SpyCatcher003:SpyTag003 complex. It is worth noting that it was not possible to resolve the melting transitions of IsPETase and SpyCatcher003, and therefore the observed changes in the DSC thermograms may be arising from contributions of both domains.
In the case of TfCut1, the fusion to SpyCatcher003 increased the Eact and changed the unfolding dynamics of the enzyme. Similarly to IsPETase, we observed a transition through two intermediate states prior to denaturation. In this case, the DSC data was fully resolved from that of SpyCatcher003, suggesting that the unfolded fusion domain interacts with the catalytic domain and stabilises these intermediate conformations. Upon complex with the SpyTag003, however, the melting transition of the catalytic domain match that of the wildtype enzyme, correlating with the activity data on PET.
In the case of LCCICCG, the thermodynamic and kinetic parameters seemed rather unchanged by the fusions tested. The enzyme in complex with the SpyTag003 is best fit to a model indicating the formation of a single intermediate prior to denaturation, however similarly to IsPETase, the data could not be resolved between the catalytic domain and the fusion and therefore it is possibly an artefact.
Overall, there seems to be good agreement between the thermodynamic and kinetic stability data, and the ability of the enzymes to degrade PET. Even though that the variations in activity are small, the changes in Tm and Eact are reflected in the activity assays. Surprisingly, increases on Eact were not sufficient to prevent IsPETase from being impacted by SpyCatcher003 at 30 °C, and the TfCut1/LCCICCG catalytic domains were not impaired by an unfolded fusion domain. We may be observing hints that for enzymes acting on solid surfaces, where the interaction with the plastic can be stabilising during the catalytic cycle, the impact of fusions depends upon the intrinsic stability and dynamics of the enzyme domain. Both of the thermotolerant domains showed little changes in activity on the plastics, despite significant but small changes in their melting transitions and mechanisms upon fusing with the SpyCatcher003 domains; while IsPETase proved intolerant to fusions regardless of their stability.
IsPETase is remarkably efficient at digesting PET at lower temperatures because it is dynamic under these reaction conditions and capable of adapting to the rigid and heterogeneous plastic substrate. In contrast, thermotolerant PET hydrolases can sacrifice some amount of this flexibility to become more stable and retain or improve activity because the plastic substrate itself becomes increasingly mobile as temperature increases. Functional protein dynamics in IsPETase may have mixed effects, including underlying its established sensitivity to crowding,32,35 but also, perhaps, to fusions. This highlights the importance of considering functional dynamics when engineering fusions of enzymes working at solid polymer surfaces.
Conclusions
We examined the effects of the thermodynamic and kinetic stability of fusion partners on the activity of PET degrading enzymes. Using SpyCatcher003, we were able to take advantage of its irreversible shift in melting temperature upon covalent association to SpyTag003, allowing us to resolve the role of fusion stability with minimal structural variation. Across the three enzymes tested, with a Topt of 40–70 °C, only small variations in enzymatic activities were observed. Despite evaluation of IsPETase fusion activities well below the Tm of SpyCatcher003, the enzyme proved sensitive to fusions with or without SpyTag003. However, both TfCut1 and LCCICCG demonstrated less substantial changes in activity, with some enhancement observed in TfCut1 and a limited, but insignificant, reduction in the activities of LCCICCG. Both the isolated SpyCatcher003 and SpyCatcher003:SpyTag003 showed fully reversible temperature melt thermograms under VSR-DSC, while all fusion constructs unfolded irreversibly, aligning with their native enzymes.When examining the effect of the fusions on the thermal stabilities of the enzymes, only IsPETase demonstrated a significant perturbation of its melting transitions, becoming significantly destabilised when fused to SpyCatcher003. This was only partially resolved by the SpyCatcher003:SpyTag003 fusion, suggesting that IsPETase may have a general intolerance to fusions that may affect its activities beyond the influence of crowding at surfaces. Otherwise, the fusions, despite their distinct stabilities, did not affect the PETases significantly. Although no direct benefits were observed from the fusions, the high tolerance of the more active PETases to both denatured and folded fusion domains, suggests that as a strategy to introduce additional activities (as opposed to enhanced stability), protein fusions to thermotolerant PET hydrolases may prove viable if selected appropriately.
Materials and methods
Plasmid construction
His6-TEV-SpyTag003 was installed at the end of MBP in the pMal-p4x vector using Gibson assembly.36 All other genes and gene fragments were synthesised by Twist Bioscience, optimised for expression in E. coli. The respective enzymes were cloned into pET21b(+) or pET29a(+) between NdeI and XhoI sites via Gibson assembly. DNA was amplified by PCR using Q5 polymerase (New England Bioscience, NEB) and purified via gel extraction (Monarch Gel Extraction Kit, NEB). Assembly was accomplished following the protocol in ESI. 1,† after which assemblies were transformed into NEB 5-alpha and DNA extracted by miniprep (Qiagen). DNA sequences were confirmed by Sanger sequencing (Eurofins). Sequences and vector information for all constructs can be found in ESI. 2.†Protein expression and purification
All constructs were expressed in BL21 (DE3) or T7Express (NEB), and grown in Terrific Broth (Melford) with selection antibiotic at 37 °C until an OD600 = 1.2. Protein expression was induced using 1 mM IPTG, for 16 hours at 18 °C. Cells were harvested by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g, resuspended in IMAC binding buffer (20 mM Tris–HCl pH 8.0, 300 mM sodium chloride, 40 mM imidazole), homogenised and sonicated at an amplitude of 40%, 50% duty cycle, for a total processing time of 6 minutes. Lysate was then clarified by centrifugation at 55000×g, and filtered through a 0.45 μm MCE filter (Fisher Scientific) before being purified via IMAC on a HisTrap FF 5 mL column (Cytiva) using a gradient elution. Constructs were then further purified through size exclusion chromatography on a HiLoad 16/600 Superdex 75 pg (Cytiva).
000×g, resuspended in IMAC binding buffer (20 mM Tris–HCl pH 8.0, 300 mM sodium chloride, 40 mM imidazole), homogenised and sonicated at an amplitude of 40%, 50% duty cycle, for a total processing time of 6 minutes. Lysate was then clarified by centrifugation at 55000×g, and filtered through a 0.45 μm MCE filter (Fisher Scientific) before being purified via IMAC on a HisTrap FF 5 mL column (Cytiva) using a gradient elution. Constructs were then further purified through size exclusion chromatography on a HiLoad 16/600 Superdex 75 pg (Cytiva).
SpyCatcher003:SpyTag003 assembly and purification
SpyCatcher003, IsPETase-SC, TfCut1-SC and LCCICCG-SC were separately incubated with MBP-SpyTag at room temperature for 5 minutes. The assembled constructs were then isolated using an MBP-Trap column (Cytiva) and eluted with maltose. The protein eluant was desalted using a PD-10 into TEV cleavage buffer (50 mM Tris–HCl pH 8.0, 0.5 mM EDTA, 1 mM DTT) and TEV protease was added at a 1:50 ratio of absorbance at 280 nm (TEV:protein). The cleavage was performed at 30 °C for 2 hours, after which the assemblies were isolated via ion exchange chromatography (AIEX for TfCut1 and LCCICCG, CIEX for IsPETase), followed by a final size exclusion chromatography step for buffer exchange and polishing. As both IsPETase and TfCut1 are most active when bound to calcium, 10 mM CaCl2 was added to both enzymes prior to this final step.
Activity assays on PET
Enzymes were incubated with 10.5 mg of amorphous PET film (Goodfellow ES30-FM-000145), in 500 μl reactions with 100 nM enzyme, 100 mM sodium chloride and 50 mM buffer at the reported optimal pH (glycine pH 9.0 for IsPETase, and sodium phosphate pH 7.5 for TfCut1 and LCCICCG). The reactions were performed in triplicate and incubated in thermomixers at 300 rpm, at reported optimal temperatures for each enzyme (30 °C and 40 °C for IsPETase, 60 °C for TfCut1 and 70 °C for LCCICCG). Reactions were quenched at 24 hours by the addition of an equal volume of methanol.Analysis of product release by HPLC
The analysis method was adapted from a previous reported UPLC method37 to allow for analysis at HPLC pressures (∼300 bar). Remaining plastic was removed, and samples were spun at 10000×g for 10 min to remove particles. The absorbance of samples at 240 nm was measured and diluted to an Abs240 ∼ 1 using 13% acetonitrile/0.1% formic acid in water mixture ahead of analyte analysis by HPLC. A C18 Kinetex LC column (00B-4605-AN) was equilibrated before analysis, and samples were run at 1.1 mL min−1. 0.1% formic acid in water was used as the stationary phase, with HPLC-grade acetonitrile as the mobile phase. 10 μl of sample was loaded onto the column, with an isocratic elution at 13% mobile phase for 0.87 minutes, followed by a step to 95% mobile phase for 1.12 minutes and an equilibration step at 13% mobile phase for a total time of 3.6 minutes. Analytes were identified and integrated using OpenLab software, based on calibration curves.
Differential scanning calorimetry (DSC)
Prior to DSC, all proteins were buffer exchanged into 50 mM sodium phosphate pH 7.5, 100 mM sodium chloride and assessed for homogeneity using size exclusion chromatography. 325 μl of sample were used for the DSC measurements, with the chromatography buffer used as the baseline control. Melting curves were measured at variable scan rates from 24 °C per hour to 192 °C per hour, with low feedback using the MicroCal PEAQ-DSC automated system. Data was integrated using the Malvern software, and fitted to thermal-denaturation models using Calfitter33,34 to derive all the Tm, Tact and Eact.Author contributions
LO carried out all data collection and data analysis. AC, LW, and KG assisted in protein production and purification. The methodology was developed by BRL and LO. BRL and VB acquired funds. BRL conceptualised and supervised the project. The manuscript was written by BRL and LO, and reviewed by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Research England for E3 funding. In addition to E3 funding, this work has been supported by funding from the Royal Society [Grant RGS\R2\212336] to BRL, and the UKRI Engineering Biology Mission Hub [Grant BBSRC/Y007972/1 to BRL and VB].Notes and references
- T. Fecker, P. Galaz-Davison, F. Engelberger, Y. Narui, M. Sotomayor, L. P. Parra and C. A. Ramírez-Sarmiento, Biophys. J., 2018, 114, 1302–1312 CrossRef CAS PubMed.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. Lee Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357 CrossRef CAS PubMed.
- F. S. Aalbers and M. W. Fraaije, ChemBioChem, 2019, 20, 20 CrossRef CAS PubMed.
- T. N. Vigil, M. J. C. Rowson, A. J. Frost and B. W. Berger, Mater. Adv., 2023, 4, 662–668 RSC.
- L. Mestrom, S. R. Marsden, M. Dieters, P. Achterberg, L. Stolk, I. Bento, U. Hanefeld and P. L. Hagedoorn, Appl. Environ. Microbiol., 2019, 85, e03084 CAS.
- L. Dai, Y. Qu, J. W. Huang, Y. Hu, H. Hu, S. Li, C. C. Chen and R. T. Guo, J. Biotechnol., 2021, 334, 47–50 CrossRef CAS PubMed.
- B. Sana, K. Ding, J. W. Siau, R. R. Pasula, S. Chee, S. Kharel, J. B. H. Lena, E. Goh, L. Rajamani, Y. M. Lam, S. Lim and J. F. Ghadessy, Biotechnol. Bioeng., 2023, 120, 3200–3209 CrossRef CAS PubMed.
- B. C. Knott, E. Erickson, M. D. Allen, J. E. Gado, R. Graham, F. L. Kearns, I. Pardo, E. Topuzlu, J. J. Anderson, H. P. Austin, G. Dominick, C. W. Johnson, N. A. Rorrer, C. J. Szostkiewicz, V. Copié, C. M. Payne, H. L. Woodcock, B. S. Donohoe, G. T. Beckham and J. E. McGeehan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25476–25485 CrossRef CAS PubMed.
- J. Zhang, H. Wang, Z. Luo, Z. Yang, Z. Zhang, P. Wang, M. Li, Y. Zhang, Y. Feng, D. Lu and Y. Zhu, Commun. Biol., 2023, 6, 1–18 CrossRef PubMed.
- R. Graham, E. Erickson, R. K. Brizendine, D. Salvachúa, W. E. Michener, Y. Li, Z. Tan, G. T. Beckham, J. E. McGeehan and A. R. Pickford, Chem Catal., 2022, 2, 2644–2657 CrossRef CAS.
- D. Ribitsch, A. O. Yebra, S. Zitzenbacher, J. Wu, S. Nowitsch, G. Steinkellner, K. Greimel, A. Doliska, G. Oberdorfer, C. C. Gruber, K. Gruber, H. Schwab, K. Stana-Kleinschek, E. H. Acero and G. M. Guebitz, Biomacromolecules, 2013, 14, 1769–1776 CrossRef CAS PubMed.
- L. Su, K. Chen, S. Bai, L. Yu and Y. Sun, Biochem. Eng. J., 2022, 188, 108709 CrossRef CAS.
- N. Puspitasari, S. L. Tsai and C. K. Lee, Appl. Biochem. Biotechnol., 2021, 193, 1284–1295 CrossRef CAS PubMed.
- P. Wagner-Egea, V. Tosi, P. Wang, C. Grey, B. Zhang and J. A. Linares-Pastén, Appl. Sci., 2021, 11, 8315 CrossRef CAS.
- P. Sun, J. E. Tropea and D. S. Waugh, Methods Mol. Biol., 2011, 705, 259–274 CrossRef CAS PubMed.
- T. R. Butt, S. C. Edavettal, J. P. Hall and M. R. Mattern, Protein Expression Purif., 2005, 43, 1 CrossRef CAS PubMed.
- R. B. Kapust and D. S. Waugh, Protein Sci., 1999, 8, 1668–1674 CrossRef CAS PubMed.
- A. C. Miklos, C. Li, N. G. Sharaf and G. J. Pielak, Biochemistry, 2010, 49, 6984–6991 CrossRef CAS PubMed.
- M. Sarkar, C. Li and G. J. Pielak, Biophys. Rev., 2013, 5, 187 CrossRef PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- A. H. Keeble, A. Banerjee, M. P. Ferla, S. C. Reddington, I. N. A. K. Anuar and M. Howarth, Angew. Chem., Int. Ed., 2017, 56, 16521–16525 CrossRef CAS PubMed.
- A. H. Keeble, P. Turkki, S. Stokes, I. N. A. K. Anuar, R. Rahikainen, V. P. Hytönen and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26523–26533 CrossRef CAS PubMed.
- A. R. Sutherland, M. K. Alam and C. R. Geyer, ChemBioChem, 2019, 20, 319–328 CrossRef CAS PubMed.
- X. Zhong, Y. Ma, X. Zhang, J. Zhang, B. Wu, F. Hollmann and Y. Wang, Mol. Catal., 2022, 521, 112188 CrossRef CAS.
- X. Gao, J. Fang, B. Xue, L. Fu and H. Li, Biomacromolecules, 2016, 17, 2812–2819 CrossRef CAS PubMed.
- T. K. Tan, P. Rijal, R. Rahikainen, A. H. Keeble, L. Schimanski, S. Hussain, R. Harvey, J. W. P. Hayes, J. C. Edwards, R. K. McLean, V. Martini, M. Pedrera, N. Thakur, C. Conceicao, I. Dietrich, H. Shelton, A. Ludi, G. Wilsden, C. Browning, A. K. Zagrajek, D. Bialy, S. Bhat, P. Stevenson-Leggett, P. Hollinghurst, M. Tully, K. Moffat, C. Chiu, R. Waters, A. Gray, M. Azhar, V. Mioulet, J. Newman, A. S. Asfor, A. Burman, S. Crossley, J. A. Hammond, E. Tchilian, B. Charleston, D. Bailey, T. J. Tuthill, S. P. Graham, H. M. E. Duyvesteyn, T. Malinauskas, J. Huo, J. A. Tree, K. R. Buttigieg, R. J. Owens, M. W. Carroll, R. S. Daniels, J. W. McCauley, D. I. Stuart, K. Y. A. Huang, M. Howarth and A. R. Townsend, Nat. Commun., 2021, 12, 1–16 CrossRef PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- S. Chen, X. Tong, R. W. Woodard, G. Du, J. Wu and J. Chen, J. Biol. Chem., 2008, 283, 25854–25862 CrossRef CAS PubMed.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- L. Avilan, B. R. Lichtenstein, G. König, M. Zahn, M. D. Allen, L. Oliveira, M. Clark, V. Bemmer, R. Graham, H. P. Austin, G. Dominick, C. W. Johnson, G. T. Beckham, J. E. McGeehan and A. R. Pickford, ChemSusChem, 2023, 16, e202202277 CrossRef CAS PubMed.
- S. Mazurenko, J. Stourac, A. Kunka, S. Nedeljković, D. Bednar, Z. Prokop and J. Damborsky, Nucleic Acids Res., 2018, 46, W344–W349 CrossRef CAS PubMed.
- A. Kunka, D. Lacko, J. Stourac, J. Damborsky, Z. Prokop and S. Mazurenko, Nucleic Acids Res., 2022, 50, W145 CrossRef CAS PubMed.
- E. Z. Linda Zhong-Johnson, Z. Dong, C. T. Canova, F. Destro, M. Cañellas, M. C. Hoffman, J. Maréchal, T. M. Johnson, G. S. Schlau-Cohen, M. F. Lucas, R. D. Braatz, K. G. Sprenger, C. A. Voigt and A. J. Sinskey, J. Biol. Chem., 2024, 300, 105783 CrossRef PubMed.
- D. G. Gibson, L. Young, R. Y. Chuang, J. C. Venter, C. A. Hutchison and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS PubMed.
- E. L. Bell, R. Smithson, S. Kilbride, J. Foster, F. J. Hardy, S. Ramachandran, A. A. Tedstone, S. J. Haigh, A. A. Garforth, P. J. R. Day, C. Levy, M. P. Shaver and A. P. Green, Nat. Catal., 2022, 5, 673–681 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further methodologies, protein sequences, structural models and extended data. See DOI: https://doi.org/10.1039/d4fd00067f |
| This journal is © The Royal Society of Chemistry 2024 |