
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining experiment and energy landscapes to explore anaerobic heme breakdown in multifunctional hemoproteins†
Alasdair D.
Keith‡
 *a,
Elizabeth B.
Sawyer
a,
Desmond C. Y.
Choy
a,
Yuhang
Xie
a,
George S.
Biggs
a,
Oskar James
Klein
a,
Paul D.
Brear
b,
David J.
Wales
*a and
Paul D.
Barker
*a
*a,
Elizabeth B.
Sawyer
a,
Desmond C. Y.
Choy
a,
Yuhang
Xie
a,
George S.
Biggs
a,
Oskar James
Klein
a,
Paul D.
Brear
b,
David J.
Wales
*a and
Paul D.
Barker
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: adk44@cam.ac.uk; dw34@cam.ac.uk; pdb30@cam.ac.uk
bDepartment of Biochemistry, University of Cambridge, Sanger Building, Cambridge CB2 1GA, UK
First published on 29th November 2023
Abstract
To survive, many pathogens extract heme from their host organism and break down the porphyrin scaffold to sequester the Fe2+ ion via a heme oxygenase. Recent studies have revealed that certain pathogens can anaerobically degrade heme. Our own research has shown that one such pathway proceeds via NADH-dependent heme degradation, which has been identified in a family of hemoproteins from a range of bacteria. HemS, from Yersinia enterocolitica, is the main focus of this work, along with HmuS (Yersinia pestis), ChuS (Escherichia coli) and ShuS (Shigella dysenteriae). We combine experiments, Energy Landscape Theory, and a bioinformatic investigation to place these homologues within a wider phylogenetic context. A subset of these hemoproteins are known to bind certain DNA promoter regions, suggesting not only that they can catalytically degrade heme, but that they are also involved in transcriptional modulation responding to heme flux. Many of the bacterial species responsible for these hemoproteins (including those that produce HemS, ChuS and ShuS) are known to specifically target oxygen-depleted regions of the gastrointestinal tract. A deeper understanding of anaerobic heme breakdown processes exploited by these pathogens could therefore prove useful in the development of future strategies for disease prevention.
1 Introduction
1.1 Bacterial heme breakdown
Heme is the most abundant iron-containing molecule in vertebrates.1 Many pathogens, which either cannot synthesize heme2 or expend significant energy doing so,3 have developed effective strategies to sequester heme from their hosts. Once internalized by the pathogen, heme can either be transported within the cell for direct utilization, or broken down to extract the iron ion.Current research suggests that the vast majority of heme breakdown enzymes follow a specific mechanism,4–6 referred to in this paper as the ‘canonical heme oxygenase (HO)’ pathway. This mechanism, which is shown in Fig. S1 (ESI†), is aerobic (consuming three O2 molecules) and ultimately yields biliverdin and carbon monoxide (CO).7,8 This canonical HO process is not confined to bacteria, but is also ubiquitous in the eukaryotic domain. Within mammals, the production of biliverdin from HO activity accounts for the greenish-blue color typical of bruising.9 In addition to the useful extraction of iron, this mechanism has further biological benefits, as the co-products can be utilized in a number of downstream processes. For example, within higher order species such as humans, the released CO is an essential neuronal cell signalling molecule10–12 and a potent antioxidant,13,14 and viral replication is inhibited by biliverdin.15,16 Furthermore, biliverdin can be converted to bilirubin via biliverdin reductase (BVR), thus becoming a potent antimutagen, which scavenges hydroperoxyl radicals,17,18 also making it an effective antioxidant.19,20
The prevalence, and consistency, of this canonical HO reaction across a wide range of species, and the product utility, suggest that this is the typical means of heme breakdown in pathogenic bacteria. However, identification of possible alternative heme degradation enzymes, which are structurally distinct from typical HOs and tend to give alternative products, has become a topic of interest. Two important families5 are the IsdG/I (iron surface determinant) proteins discovered in Staphylococcus aureus and Bacillus anthracis,21,22 and the MhuD (Mycobacterium haem utilization) protein from Mycobacterium tuberculosis.23
A family of operons, identified across several pathogenic bacteria, has been identified as important for heme regulation, and include genes encoding putative heme degradative enzymes (Table S1, ESI†). These operons are denoted hem, hmu, chu, shu and phu. The first heme sequestering proteins to be discovered were those of the hem operon of Yersinia enterocolitica.24 The operon consists of 9 genes, hemWXY, hemPR and hemSTUV, separated by intergenic regions (Fig. 1).
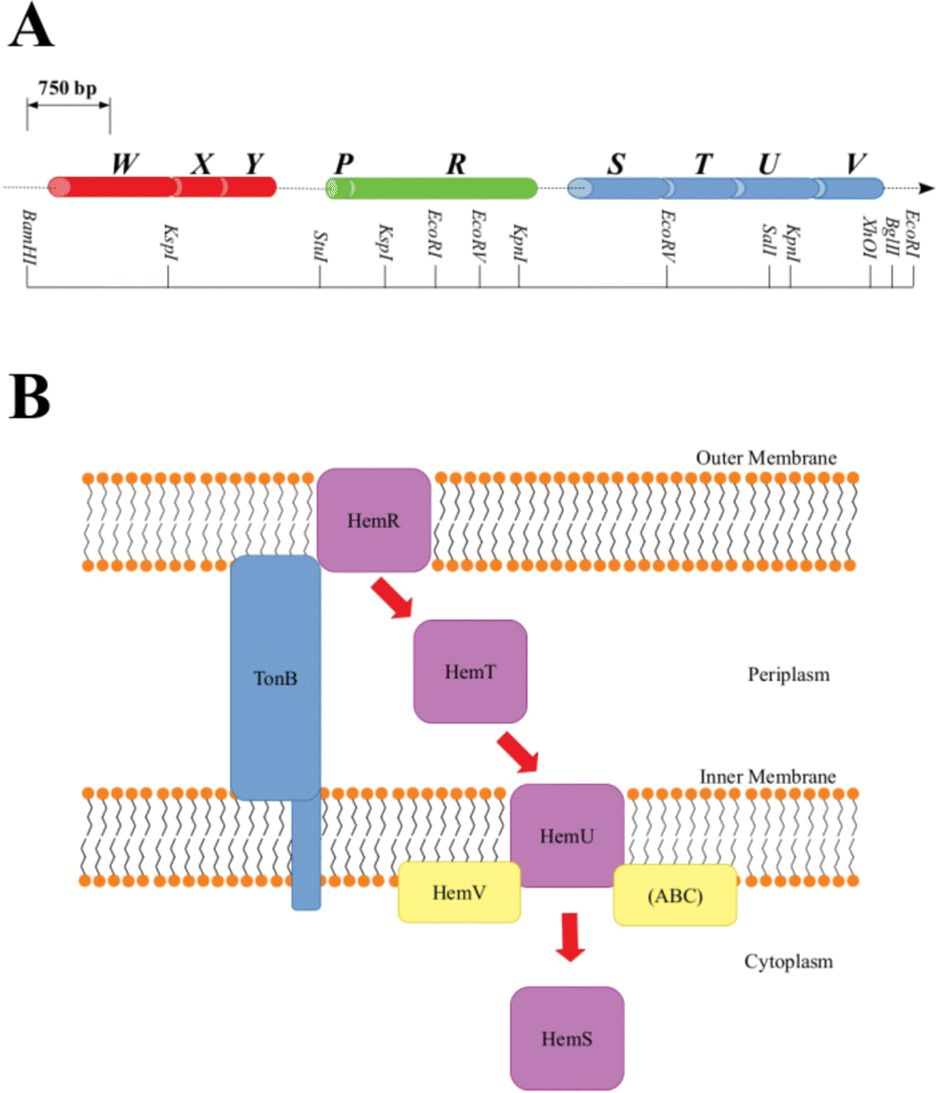 | ||
| Fig. 1 A: Y. enterocolitica hem operon restriction map. Genes are color-coded and separated by intergenic regions, where cylinder width corresponds to the number of base pairs in the gene. B: Representation of the heme-uptake system coded by hem. A purple box indicates a hemoprotein, whereas the other proteins are auxiliaries. Red arrows chart heme progress through the cell. ABC is the ATP-binding cassette. TonB spans the periplasm, allowing signals to be transmitted between the outer membrane and the cytoplasm. | ||
The functions of HemW, HemX and HemY have not been extensively studied. In the chu operon, where the chuWXY genes are more clearly integrated within their respective operons, certain heme transport/breakdown functions have been proposed. LaMattina et al. showed that ChuW, a member of the radical S-adenosylmethionine methyltransferase (RSMT) family, engages in characteristic methyl transfer to otherwise unreactive carbon atoms in heme.25 Such transfer is typically accompanied by appreciable chemical rearrangements,26,27 and it was shown in the case of ChuW that this transfer included the cleavage of the heme porphyrin ring. LaMattina et al. found that the porphyrin would break down to a compound with a consistent but previously unseen mass, but could not determine its exact structure. They designated this compound ‘anaerobilin’, since this novel route of heme catabolism could be carried out anaerobically. Using a ‘deuteroanaerobilin’ (derived from deuteroheme as opposed to heme) surrogate to investigate processes downstream from ChuW catalysis, LaMattina et al. found the product could be further degraded once introduced to ChuY. Indeed, increasing ChuY concentration gave a linear increase in activity, suggesting an enzymatic reaction was taking place. They therefore concluded that the function of ChuY is to reduce a potentially toxic heme breakdown product.25 ChuX is capable of binding a molar equivalent of heme, has a high KD value and has no obvious heme breakdown capabilities, suggesting a role in heme transport.28 Thus, ChuW, ChuX and ChuY work in tandem, with ChuX shuttling anaerobilin from ChuW to ChuY for further breakdown. This heme degradation strategy encoded within the chu operon (and therefore, by implication, in the other operons discussed in this paper too, although this aspect would require a dedicated study) centers around the WXY proteins. This Perspective will discuss an alternative, but closely related, heme breakdown strategy encoded by these operons, centered around the respective S proteins (HemS, HmuS, ChuS, ShuS and PhuS).
As shown in Fig. 1, within the context of the hem operon, the R protein (HemR) acts as a heme acquisition protein.24 HemR consists of a characteristic transmembrane β-barrel, with extracellular loops, which act as receptors. Exogenous heme is detected at the cell surface, bound, and absorbed into the periplasm by HemR or its homologues. These proteins are TonB-dependent,24,29 allowing for the transmission of signals between the outer membrane and the cytoplasm, which allows for the concentration of extracellular heme to be closely monitored, forming part of an effective feedback loop to prevent the cell from becoming overloaded with potentially toxic heme. The feedback loop is sophisticated and beyond the scope of this Perspectives article. However, as we have some new ideas concerning the operation of this loop, and in particular the role of the P proteins (found in some of the operons discussed in this paper, but not in others), a discussion is provided in Section D of the ESI.†
Once in the periplasm, it seems that heme can then either be picked up by the XYZ set of proteins (as discussed above) or by the STUV proteins. Within all of the operons we consider, there is a clear intergenic region (in hem this includes an inverted repeat24 and a Shine–Dalgarno sequence30) between the PR and STUV genes, highlighting that the upstream genes are concerned with heme acquisition, whereas the downstream genes constitute a heme-specific periplasmic binding-protein-dependent transport (PBT) system. PBTs contain one of each of these three classes of protein: a periplasmic binding protein; a hydrophobic inner membrane-spanning protein; and a hydrophilic ATP-binding protein. Ordinarily, they also contain a cytoplasmic protein, implicated either in heme transport or breakdown. The Y. enterocolitica hemSTUV proteins were the first PBT to be characterized from Gram-negative bacteria.31
The T proteins, which transport heme through the periplasm to the inner membrane, have a large central cleft containing a heme-binding tyrosine residue. Some homologues, such as HemT and HmuT, have a further histidine residue inside the cleft, allowing for binding of heme dimer.32
Upon delivery to the inner membrane, heme is taken up by the U protein. These proteins are permeases rich in α-helices to assist in the formation of a membrane-spanning funnel. Such proteins have a conserved EAAX3GX9LLLL sequence, which allows for close interaction with the ATPase of the PBT (i.e. the V protein).33 The V proteins all contain the motifs necessary for ATP hydrolysis: the GX4GK Walker A motif important for phosphoryl-binding;34 and the hhhhDE (where h is a hydrophobic residue) Walker B motif required to bind Mg2+ and effect the hydrolysis.35 ATP hydrolysis provides the energy for the U protein to pump heme across the inner membrane, where it is picked up by the cytosolic S protein.
1.2 The cytosolic S protein
The precise function of the S family of proteins has long been debated, reflected by inconsistencies in annotations (usually as heme transfer proteins or heme-degrading enzymes) in Uniprot36 and throughout the literature. The crystal structure of HemS in both its apo- (PDB: 2J0R)37 and its holo- (PDB: 2J0P)38 forms, as shown in Fig. 2, reveal that HemS consists of two topologically homologous domains joined by an unstructured loop, yielding a pair of large, stacked β-sheets. These stacked central β-sheets are twisted and capped by α-helices, yielding two distinct pockets. Heme binds within the deeper pocket, with one of the free iron axial ligands anchored by histidine (H196),38 and the other by arginine (R102), via an H2O molecule or OH− ion.38 As well as these bonds to the iron, it was apparent that many contacts were being made between selected residues of HemS and the propionate groups of the heme. Binding of heme within the HemS pocket results in an induced fit conformational change.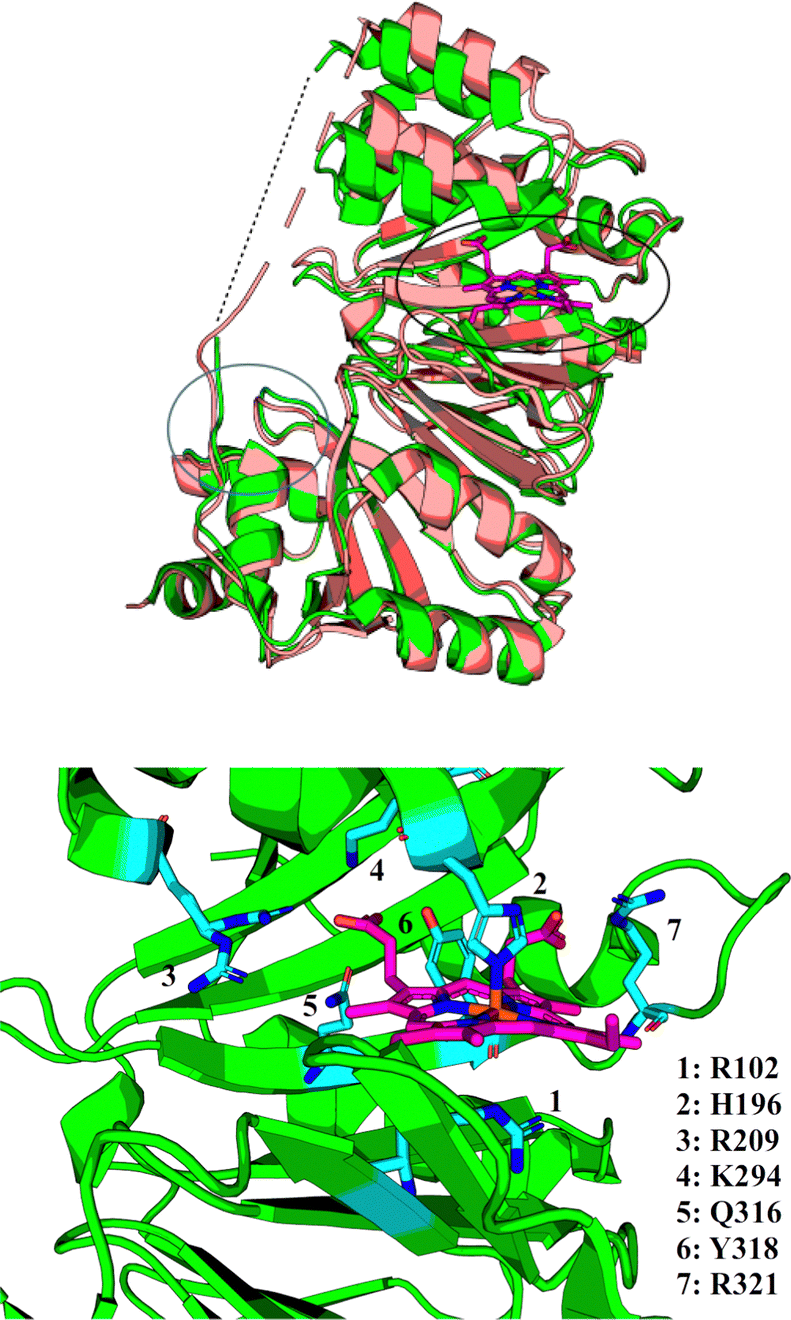 | ||
| Fig. 2 Top: holo-HemS (PDB: 2J0P, green) superimposed on apo-HemS (PDB 2J0R, salmon), with the heme in 2J0P highlighted in magenta. The black circle corresponds to the large cavity, and the blue circle to the small cavity. A dashed line is provided to show the missing loop region from the holo-structure. The apo-loop is also incomplete. The structural overlay clearly shows the large cavity ‘clamping down’ upon heme-binding when compared against the apo-form. Bottom: Magnified representation of the holo-HemS heme-binding pocket from 2J0P. The main residues implicated in heme-binding are shown in cyan. R102 (mediated by H2O/OH−) and H196 interact at the free axial sites of the Fe ion, and the other residues highlighted interact with the propionate groups of HemS. R321 is the only residue which coordinates with the exposed propionate. | ||
Crystal structures for ChuS have also been solved, in both apo- (PDB: 1U9T)39 and holo- (PDB: 4CDP)40 forms, and are very similar to their HemS equivalents. An important difference is that ChuS in its apo-form has a narrower cavity than HemS, whereas its holo-form has a wider cavity, suggesting that HemS contracts more around heme, excluding more solvent, thus resulting in a greater entropic drive to bind heme than ChuS does.
Suits et al., noting that the N- and C-terminal halves of ChuS represented a structural duplication (as is also the case for the other homologues) despite there being only 19% sequence identity, concluded that ChuS was carrying out a similar function to ChuX, which can dimerise to give structurally related pockets to those in ChuS.39 This observation lends support to the hypothesis that the XYZ and STUV sets of proteins are engaged in related – whether complementary or competing – heme breakdown strategies.
The S proteins can act as heme-degrading enzymes, first demonstrated in the aerobic reaction between ascorbic acid and holo-ChuS.39 The reaction is H2O2-dependent and yields a novel tripyrrole product, with a characteristic UV-Visible signal at 560 nm and mass spectrometry (MS) m/z peaks at 514.32 and 437.19. We show in this Perspectives article that this product is distinct from the NADH-dependent breakdown of heme.
Based on its ability to bind, but not degrade, heme in the absence of molecular oxygen, ChuS has also been proposed to fulfil a heme-chaperoning role under anaerobic, iron-replete conditions. In this role, ChuS acts as a storage molecule to mitigate toxicity caused by high cytosolic heme levels; and heme chaperone to ChuW for this latter enzyme to catalyze S-adenosylmethionine-mediated anaerobic breakdown.41
The only other S protein homologue whose crystal structure has been solved is PhuS, available in both its apo- (PDB codes 4IMH and 4MGF)42,43 and holo- (PDB code 4MF9)43 forms. Wilks et al. showed that this protein could break down heme using ascorbate or CPR-NADPH but concluded this was due to coupled oxidation.44 Further, they showed that PhuS acts as a heme chaperone and can deliver heme unidirectionally to the bona fide heme oxygenase, pa-HO or HemO. The arrangement of histidine residues within PhuS that are not present in the homologous S proteins, facilitates this transfer: H210 and H212 lie in close proximity but are more exposed than H209.45,46 Other than the proposed transfer of heme from ChuS to ChuW (which is not an ordinary HO), the homologues of PhuS, which lack these extra histidines, have not been implicated in heme transfer to HOs.
PhuS can also bind DNA, although this function is mutually exclusive with respect to heme-binding.47 Specifically, apo-PhuS was shown to target the prrF1 promoter, an iron-responsive region directly downstream of the phu operon. Under conditions of iron starvation, PrrF bacterial small RNAs (sRNAs) cause degradation of nonessential iron-containing proteins,48–50 thus diverting iron to those proteins essential for cell survival. Additionally, a read-through of the prrF1 terminator, which depends on heme concentration, can yield a longer PrrH transcript, with this sRNA being of suspected importance for infectivity.47 From these results, it was proposed that PhuS fulfils a dual function, delivering heme to HemO and in transcriptional modulation of PrrF/PrrH coincident with heme itself.
HmuS is a close homologue of HemS (89.6% sequence identity) and it is thought that these proteins will behave similarly. ShuS has 98.5% sequence identity with ChuS but did not show heme degradation activity in initial NADPH or ascorbate assays. Apo-ShuS exhibits non-sequence-specific DNA binding.51 Thus, roles for the S proteins in DNA-dependent self-regulation as well as heme breakdown have been proposed. Summaries of the properties and reactivities of the S proteins are given in Table 1 and Fig. 3.
| Species | Protein | Chaperone | DNA-binding | Heme breakdown | holo/apo crystal form |
|---|---|---|---|---|---|
| a HemS perhaps acts as a heme chaperone when the heme concentration is high, as discussed in the results. b Demonstrated to transport heme to ChuW. c Reaction with CPR-NADH system demonstrated. Requires aerobic conditions. d Standard NADPH and ascorbate based assays attempted but no HO activity identified. e Demonstrated to transport heme to HemO. f Reaction with CPR-NADH system demonstrated, although it was suggested the observations may be due to a coupled oxidation process instead. | |||||
| Yersinia enterocolitica | HemS | −a | − | + | Monomer/monomer |
| Yersinia pestis | HmuS | − | − | − | N/A |
| Escherichia coli O157:H7 | ChuS | +41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
− | +39,52c |
Dimer/monomer |
| Shigella dysenteriae | ShuS | − | +51,53 | −d | N/A |
| Pseudomonas aeruginosa | PhuS | +44e |
+47 | +44f |
Dimer/dimer |
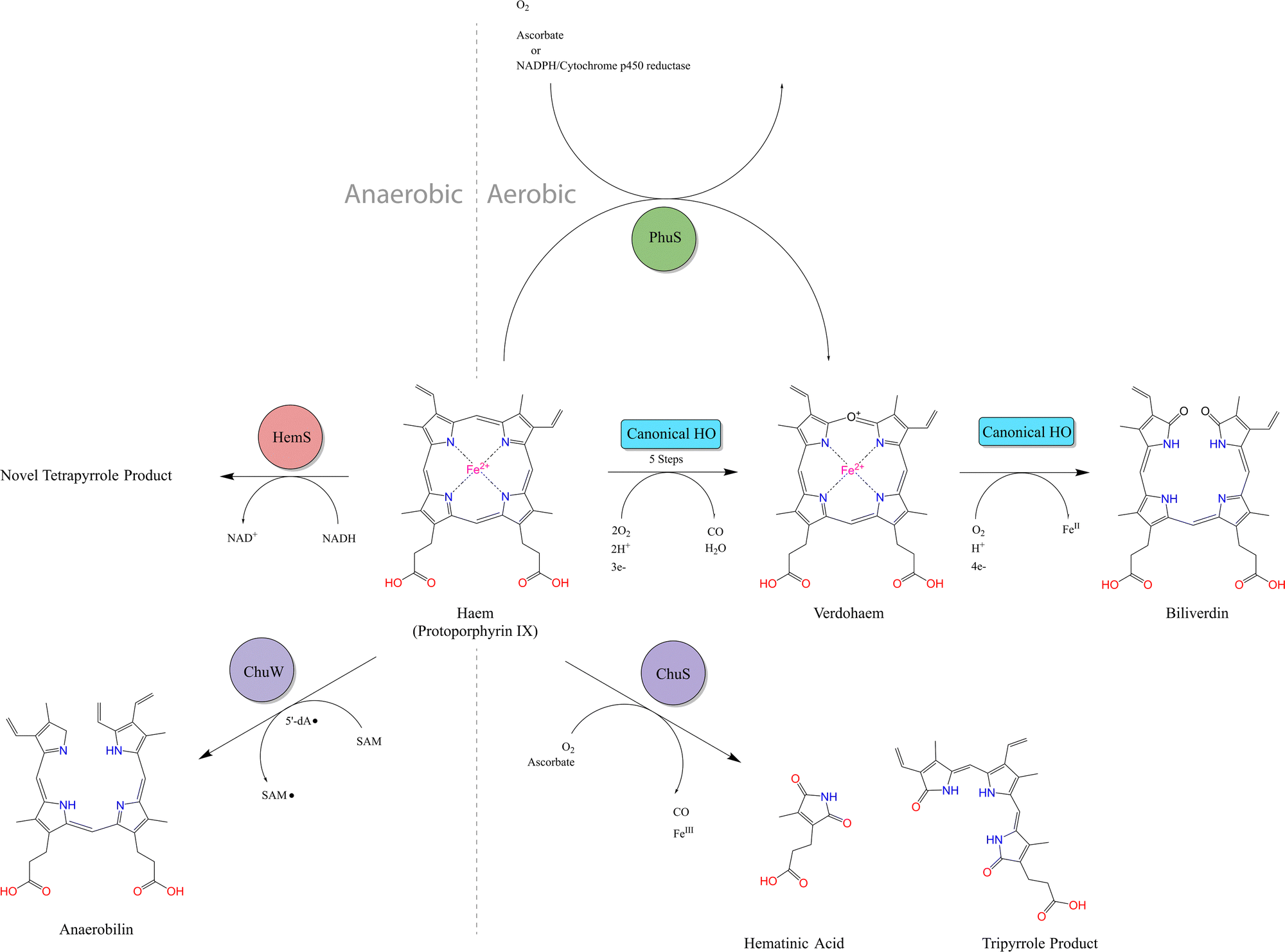 | ||
| Fig. 3 Summary of known heme breakdown pathways. Canonical HO: Fe2+ is extracted from heme using oxygen and reductants such as NADPH/Cytochrome P450 reductase. Biliverdin and CO are produced with ferrous verdoheme as the final intermediate. ChuS: non-canonical HO reaction with ascorbate in oxygen demonstrated to release CO, Fe3+, hematinic acid and a tripyrrole product. Ferric verdoheme is the final intermediate. PhuS: oxidative heme degradation reaction which can use ascorbate or CPR-NADPH as reductants. Products of this reaction have not been characterized, but verdoheme is one of the intermediates and biliverdin is not produced. ChuW: anaerobic heme degradation using radical SAM mechanism to produce anaerobilin. HemS: anaerobic reductive heme degradation using NAD(P)H and producing a novel uncharacterized tetrapyrrole product, as discussed in Results. | ||
1.3 Energy landscape theory
After discovery of the novel anaerobic heme breakdown process discussed in this paper, analysis of the presumed NADH-binding pocket proved difficult by conventional experimental methods or by bioinformatics. Due to transient and weak binding properties, classical titrations did not provide definitive information. Structural and sequence-based analyses were difficult, given this was a protein fold with little homology to known NADH binding sites. Therefore, Energy Landscape Theory (ELT)54–58 and associated computational methodology was employed.59–62ELT recognizes that the potential energy surface (PES) encodes all of the information necessary to determine the structural, thermodynamic and kinetic properties of any molecule.55 Here we focus on local minima, and the connections between them via transition states. We visualize the landscape using disconnectivity graphs,63,64 which afford key insight into qualitative thermodynamic and kinetic properties,55,65–74 and quantitative results from analysis of the underlying kinetic transition network.75–78 Free energies can be derived from the PES through the superposition approach79 where, at a given temperature, T, the canonical partition function, Z(T), is expressed as a sum of the contributions from the basins of attraction of the local minima, with the harmonic approximation typically used to estimate the vibrational partition function of each of these local minima.55,62,65,79–83 This approach provides a means for computations to be directly compared against or predict a suite of experimental outcomes, such as rate constants and heat capacities. It has been used to tackle a wide range of biochemical problems, from protein folding84–87 and coarse-grained viral capsid assembly88–91 to DNA base-pairing92,93 and directed evolution/understanding of natural selection.94,95 Here we use the energy landscape to investigate protein–ligand interactions.
2 Research outline
The original aim of our research was to examine the precise heme-binding capabilities of HemS. However, a small-molecule screen revealed that HemS is capable of degrading heme using NADH via a novel, anaerobic, hydride transfer mechanism. The reaction has proved challenging to characterize due to the weak, transient nature of NADH binding, and the instability of the heme breakdown product. To supplement conventional biophysical techniques, bioinformatics was used to identify possible NADH binding sites, and energy landscape calculations employed to demonstrate a pathway for NADH approach to heme in the main HemS pocket.Having discovered this heme breakdown reaction in HemS, we conducted a phylogenetic investigation, culminating in the selection of appropriate homologues for parallel experimental/energy landscape studies.
A long-standing aim of our research was to determine the structure of the heme breakdown product, which we know to be novel. We have had partial success: although important characteristics of this structure are now known, the precise structure is still elusive.
Our research has generated a wealth of data, which was enough to fill three doctoral theses.96–98 In this Perspectives article, we present our data in order to provide focus and to address three important questions facing current hemoprotein research:
1. Are cytosolic S hemoproteins capable of breaking heme down as an anaerobic, hydride-dependent alternative to canonical heme oxygenase activity?
2. Do these cytosolic S hemoproteins have functions aside from heme breakdown?
3. How can energy landscape theory and simulations provide novel insight for cutting-edge protein–ligand research?
3 Results and discussion
3.1 Heme-binding in HemS and selected homologues
The wavelengths and the extinction coefficients of the three characteristic absorbance bands, α (∼580 nm), β (∼545 nm) and γ, or Soret, (∼410 nm), are very similar for HemS, HmuS, ChuS and ShuS, suggesting that heme is buckled and distorted similarly in each. These UV-Visible spectra are given in Section M of the ESI.† These spectra also showed that the wavelength and intensity of the Soret band of each homologue increase as the pH is increased. The red shift in wavelength is proposed to be due to a change in spin-state of the iron due to the deprotonation of an iron-coordinated water molecule. In the structure, there is a water molecule mediating the bond between R102 and the iron, causing the latter to be high-spin. At higher pH, a OH− ion mediates instead, causing the iron ion to become low spin, resulting in a higher wavelength Soret maximum.With HemS, this study was extended to consider a wider range of pH values. Fig. S11A (ESI†) shows that the trend is valid at least between pH values 4–9. Also, as the pH is decreased, the α and β bands become negligible and the Soret band decreases in absorbance, suggesting that heme is not binding as strongly to the protein. Fig. S11B (ESI†) plots the Soret absorbance against the pH, and gives a best fit to a simple Henderson–Hasselbach equation describing a single titratable group. The fit suggests that the pKa of the axial heme ligand is around 5.5.
Non-denaturing mass spectrometry (ndMS) revealed a range of HemS : heme binding stoichiometries, even in a 1:1 protein:heme mixture. At pH 7, three separate species, apo-HemS, holo-1-HemS and holo-2-HemS, were all detected, as shown at the 11+ charge state in Fig. S12A (ESI†). The 12+, 10+, and 9+ charged species were also detected in the spectrum. Parent ions were quadrupole selected and subjected to 20 V collision energies in order to strip heme from HemS. The MS2 spectra, as shown in Fig. S12B–E (ESI†), reveals that heme can bind to HemS as a dimer and not just as a monomer. Further justification for believing that heme can bind to HemS as a dimer, from ELT-based calculations, is given in Section N of the ESI.† Further ndMS studies, using a 1:8 protein:heme mixture, identified stoichiometries up to holo-9-HemS, as shown in Section O of the ESI.† The significance of these higher order stoichiometries is not currently clear, although it does suggest that heme is capable of surface-associating, or ‘sticking’, to HemS at these higher stoichiometries.
3.2 Reaction with NADH – biophysical investigations
We systematically screened a variety of small molecules to determine their influence on the heme-binding properties of HemS (details in Table S8, ESI†). We found that only NADH and NADPH induce activity. In all experiments where NADH and NADPH were both tested, they behaved identically and we therefore assume that results for NADH alone also apply to NADPH. Further analysis of NADPH is provided in Section T of the ESI,† including its possible biological role in comparison to NADH.Our primary, reproducible observation was that incubation of NADH with apo-HemS and heme showed a large increase in absorbance at 591 nm and a corresponding decrease in the heme Soret peak at 408 nm, although this is partially obscured by the shoulder of the NADH absorbance at 340 nm. Spectra are given in Fig. 4. This reaction is characterized by a dramatic color change from pale green/brown to deep purple.
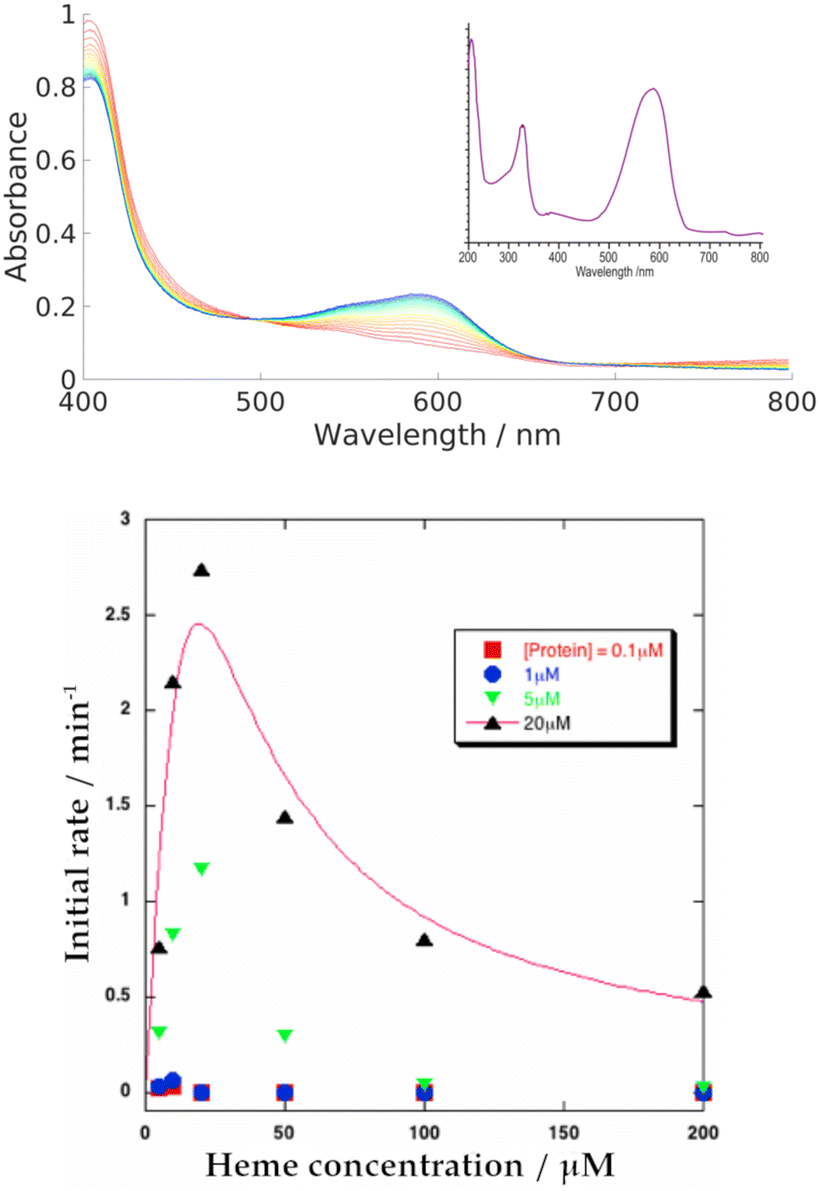 | ||
| Fig. 4 Top: Representative UV-Vis spectra showing heme breakdown by NADH in HemS, where the buffer alone is used as a baseline. Evolution is charted in 1 minute intervals, from 1 min (red) to 20 min (blue), and reveals the formation of a purple heme breakdown product (591 nm). 5 μM HemS and 20 μM heme were pre-equilibrated, and the reaction tracked upon injection of 2000 μM NADH. The inset shows the spectrum of the heme breakdown product following HPLC. Bottom: Plot of the initial rates of the NADH-dependent holo-HemS reaction vs. heme concentration. This experiment was conducted at four different protein concentrations (red: 0.1 μM; blue: 1 μM; green: 5 μM; black: 20 μM). At low heme concentrations, the initial rate of reaction increases. However, a maximum is reached, after which the rate decreases with increasing heme concentration. The solid line describes conventional Michaelis–Menten kinetics but with single site substrate inhibition present. | ||
Under the same conditions at room temperature, holo-HemS alone is stable for two months and showed no heme breakdown in this period. Additionally, the reaction did not occur when free heme and NADH were incubated together. Tests with oxidised, holo-cytochrome b562 (a six-coordinate low-spin cytochrome) showed that NADH could partially reduce heme, but no enzymatic activity was observed. Incubation of free heme and NADH does result in conventional, non-enzymatic ‘coupled oxidation’ and the production of biliverdin. Tests with free-base PPIX, Co(III)-PPIX and Zn(II)-PPIX all showed no activity, suggesting the reaction required iron in the porphyrin.
Increasing the concentration of the protein or NADH both led to increases in the rate of reaction, as tracked by the formation of the 591 nm peak. These trends are illustrated in a series of spectra in Fig. S16 and S17 (ESI†). The trend for heme concentration dependence is also shown in Fig. S15 (ESI†), and is summarized in Fig. 4. This heme-dependent behaviour is more complicated, with the rate of reaction directly dependent on heme concentration only when that heme concentration is low. At higher concentrations, heme begins to inhibit its own breakdown.
Stopped-flow spectroscopy revealed the reaction proceeded via an intermediate with a characteristic peak in the near-IR, which suggests that initial heme breakdown involves a disruption of aromaticity and/or change in the iron oxidation state. Given that the porphyrin is eventually cleaved open to produce the heme breakdown product (HBP), it may be that some sort of sigmatropic rearrangement then occurs. Further analysis of the stopped-flow data will be presented in a follow-up study.
Conventionally, NADH acts as a hydride donor but it can also act as a reducing agent on Fe3+ porphyrins. As discussed in the introduction, NAD(P)H can constitute one part of a group of reactants that, in the presence of dioxygen, produces ferric peroxy species, and thus initiates coupled oxidation, which leads to the breakdown of heme and formation of biliverdin. Direct transfer of hydride from NADH to heme has not been demonstrated in HemS or any of its homologues, to the best of our knowledge. To demonstrate whether this novel reaction involved hydride transfer, deuterium labelling experiments were employed. (R)-NADD and (S)-NADD were synthesized (structures and NMR spectra are provided in Fig. S30, ESI†) and used individually to induce heme breakdown. The resulting HBPs were compared by mass spectrometry, and the rates of reaction tracked by UV-Visible spectroscopy, all of which are shown in Fig. 5. The most abundant signature HBP fragmentation peaks shifted from m/z 569.3 and 462.2 to 570.3 and 463.2 when either (R)-NADD or (S)-NADD were used in place of NADH. Accounting for naturally occurring isotopes, it can be seen that there is a near-50:50 mix of deuterated and non-deuterated HBPs when either (R)-NADD and (S)-NADD were used. This result reveals two features of the heme breakdown reaction. Firstly, that deuteride (and therefore, by implication, hydride) is transferred from NADD/NADH to the porphyrin. Secondly, that such transfer is non-stereospecific in that it can occur from either face of the nicotinamide head.
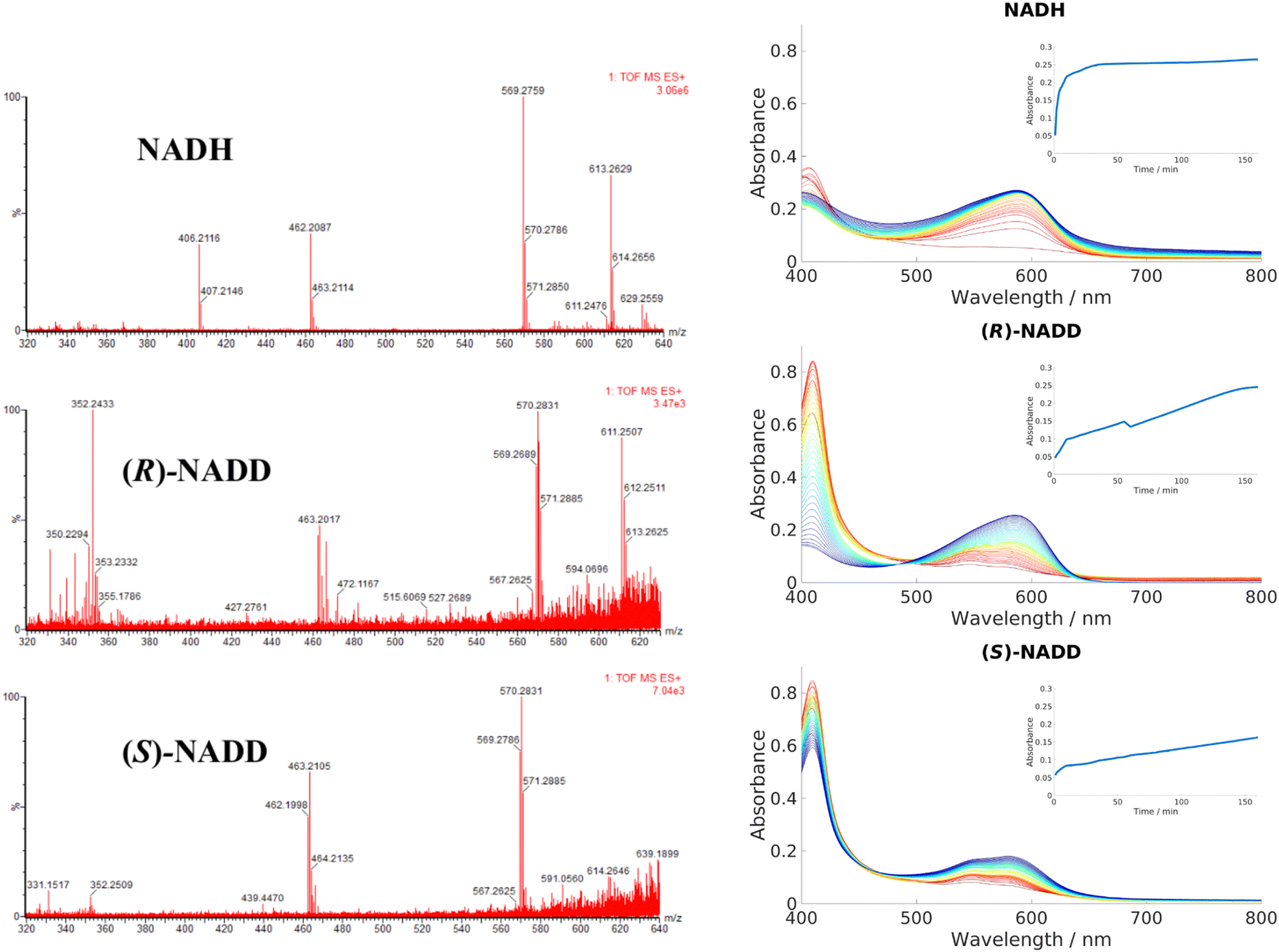 | ||
| Fig. 5 Left: LCMS data for the product mixtures following reactions with NADH, (R)-NADD and (S)-NADD, respectively. Data for the molecular ion (m/z of 613.3 in the NADH-based sample) is inconclusive, but the peaks based at 569.3 and 462.2 have different isotope patterns in the (R)-NADD and (S)-NADD spectra compared to NADH. Right: UV-Visible spectra comparing the rates of reaction between NADH, (R)-NADD and (S)-NADD. Reactants were mixed in the ratio 10 μM wild type (WT) HemS: 10 μM heme: 1000 μM NADH/NADD. Standard procedure in this work was to take a baseline with protein, heme and buffer, and the spectra upon addition of NADH; however, for these experiments, baselines were taken with NADH/NADD, and the data collected upon addition of heme (see Section A of the ESI† for details). Spectra were collected every minute for 10 minutes, then every 5 minutes for 150 minutes, indicated by the color change from red (1 minute) to blue/purple (160 minutes). Insets chart the changing absorbance at 591 nm over time. | ||
UV-Vis data, however, reveals that there is a kinetic differentiation depending on the stereoisomer of NADD used to carry out the reaction, as indicated in the different evolutions of the 408 nm Soret peak. These Soret peaks grew significantly up to absorbances of around 0.8 in the two deuterated experiments, whereas in the non-deuterated case it did not even reach half that value. We conclude that in all three cases, heme was entering the pocket and coordinating to HemS quickly. However, in the case of NADH, conversion to the HBP was fast (thus preventing a build-up of HemS coordinated to heme, as indicated by the peak at 408 nm) whereas for (R)-NADD and (S)-NADD it was slow (where such a build-up did occur). This effect is reflected by the growth of absorbance at 591 nm, indicating increased concentration of the HBP. In the NADH case, this increase occurs quickly (with an initial rate of 306 μA U s−1) so that it has effectively reached a maximum at 0.25 after 40 minutes, as shown by the top inset in Fig. 5. For (R)-NADD, the initial rate is reduced to 97 μA U s−1, and it appears that the absorbance is beginning to level off after 160 minutes, again at an absorbance of around 0.25. (S)-NADD is slower still: its initial rate is 50 μA U s−1. Furthermore, the absorbance is still rising, and has only reached a value of 0.2, after 160 minutes. These relative rates between the two deuterated forms of NADD are reflected in the Soret peak. For (R)-NADD, this peak has decreased almost to zero after 160 minutes, suggesting that the heme added is almost used up. However, after the same amount of time in the (S)-NADD case, the Soret peak is still far from zero, indicating that a significant amount of heme is still to be converted.98
It is therefore apparent that both deuterated forms of NADH reduce the rate of reaction compared to the non-deuterated form, with the (S)-form giving the greatest decrease. Given the LCMS data, which show that the mix of deuterated and non-deuterated products is approximately 50:50 irrespective of the deuterated stereoisomer used, it is not immediately apparent why the (S)-form should be slower than the (R)-form. A possible explanation is that there are, broadly speaking, two different conformations NADH/NADD can adopt when transferring hydride/deuteride over to heme, and that these different conformations are each exclusive in terms of which hydride (either (R)- or (S)-) it can transfer, assuming hydride transfer is the rate determining step. Furthermore, we surmise that both conformations are almost equally likely, but one of them results in a slower rate of transfer of the hydride to heme (perhaps because of the greater distance between the molecules). Since hydrides transfer faster than deuterides, hydride transfer will govern the overall rate. In (S)-NADD, the deuteride is at the (S)-position and the hydride is therefore at the (R)-position. As the rate of reaction is so much slower when the hydride is in this position (as opposed to (R)-NADD, when the hydride is at the (S)-position) we conclude that the rate of transfer is more difficult from the (R)-position. In other words, transfer of a hydride from the (S)-position is faster. The deuterides do not fall into this scenario because of their relatively slow rate of transfer with respect to hydrides. Because these different rates result in no noticeable difference between the proportion of the deuterated and non-deuterated forms in the LCMS spectra for either the (R)-NADD or (S)-NADD cases, we further propose that NADH/NADD must ‘commit’ to a conformation before transferring over one of its hydrides; even though a hydride at the (R)-position transfers more slowly to heme than it does from the (S)-position, this process is still faster than the conformational changes that would be required for the nicotinamide head to switch round and present its (S)-hydride instead.98 The Kinetic Isotope Effects for (R)-NADD and (S)-NADD are, respectively, 3.2 and 6.1, which would suggest these KIEs are primary, C–H/D-breaking, in nature.
3.3 Anaerobic reaction
The reaction has been shown to proceed via hydride transfer, without an apparent role for oxygen. Experiments were therefore conducted to investigate whether the complete reaction could indeed proceed in an anaerobic atmosphere. As described in the introduction, such a feature would be of great interest, since anaerobic heme breakdown strategies within bacterial strains are relatively unknown.Parallel free air/nitrogen atmosphere experiments over a range of HemS:heme:NADH ratios showed not only that the reaction proceeded unhindered under anaerobic conditions, but that it was actually more effective than under aerobic conditions. Illustrative results are given in Fig. 6, which show that a competing side-product is formed under aerobic conditions but not under anaerobic conditions. This competing side-reaction explains why the Soret peak in the aerobic case is depleted faster than in the anaerobic case, yet the 591 nm peak representing the intended HBP grows at approximately the same rate. The ∼700 nm peak is indicative of biliverdin, which was confirmed by mass spectrometry, as shown in Fig. S26 (ESI†). In addition to being the end product of the ‘canonical’ heme oxygenase process described in the introduction, biliverdin can also be produced non-enzymatically and non-regiospecifically via coupled oxidation.5,99,100 In coupled oxidation, NADH acts as a mild reductant, just like ascorbate, and is capable of reducing the heme iron which can then react with oxygen to generate iron superoxy and peroxy species, thus initiating the non-enzymatic heme breakdown. Being a non-enzymatic process, it is typically out-performed in the presence of a heme-breakdown protein by other processes, unless the concentrations of heme and NADH are appreciably higher than that of the enzyme, as is the case in our artificial conditions. Clearly, the heme breakdown reaction in anaerobic conditions by the holo-HemS reaction with NADH differentiates it from the usual, ‘canonical’ method of bacterial heme uptake and breakdown, which requires oxygen to proceed, and hence may go some way to explaining why certain strains of bacteria can survive in oxygen-starved regions of their hosts. Furthermore, the fact that non-enzymatic coupled oxidation competes under aerobic conditions, and that there are no equivalent competing side-reactions under anaerobic conditions, strongly suggests that this heme breakdown pathway has arisen to allow certain pathogens to extract heme in a controlled manner specifically when oxygen levels are low.
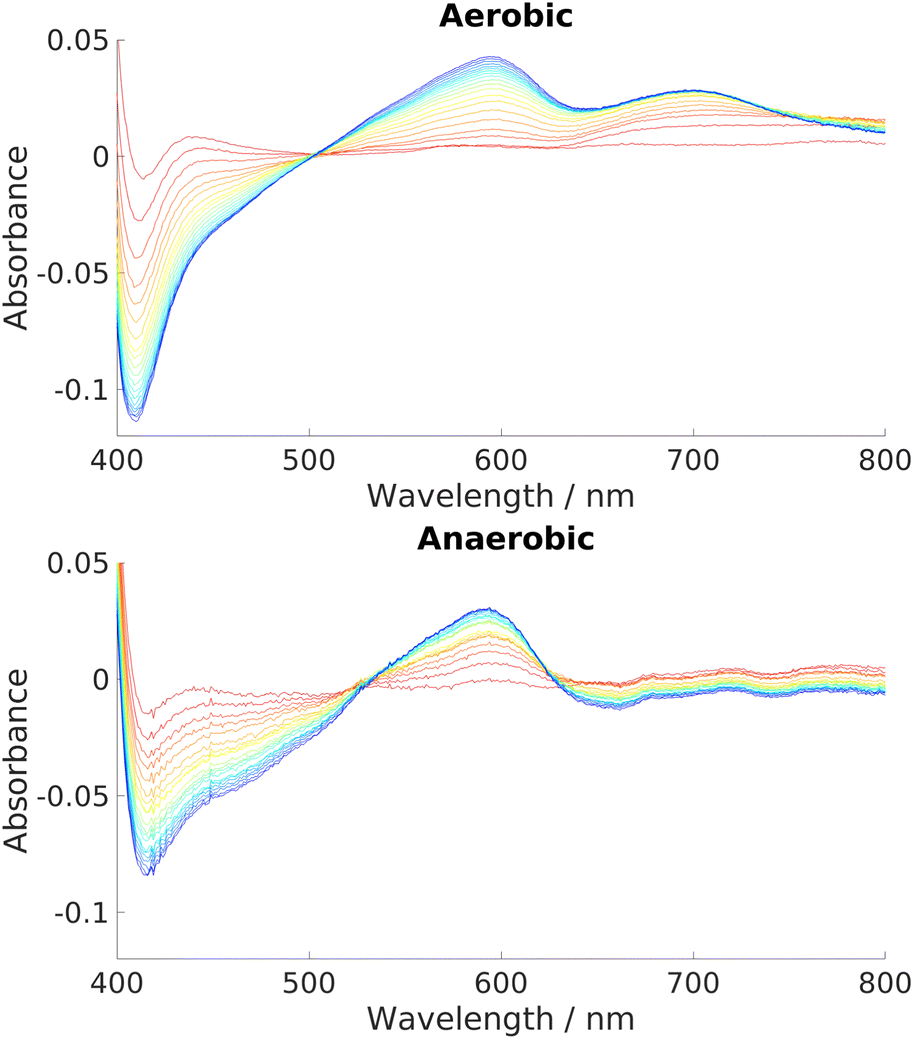 | ||
| Fig. 6 UV-Vis spectra charting the progress of NADH-dependent breakdown of heme in HemS over time. Spectra were recorded for 20 minutes in 1 minute increments, indicated by the color scheme which runs from red (1 minute) to blue/purple (20 minutes). Exact stoichiometries were 1 μM HemS: 20 μM heme: 2000 μM NADH. Top: Aerobic conditions. A peak at around 700 nm develops over time. Bottom: Anaerobic conditions. No such peak develops.98 | ||
3.4 Structure of heme breakdown product
The UV-Visible and mass spectra gathered for this HBP showed that it was a novel compound. Significant analyses to gain insight into its exact structure have been attempted but only with partial success. Complete mass spectra indicate that the parent ion (and so presumably the HBP itself) has a m/z ratio of 613.3, but the abundance of this peak is typically lower than fragments at 569.3 (the base peak) and 462.2, suggesting the HBP is unstable when subjected to ionization. Accurate mass analysis revealed that the 569.3 peak arises because of decarboxylation of the HBP. MS2 analyses of the 613.3 and 569.3 compounds showed that each could be fragmented to give the 462.2 peak, as shown in Fig. S21 (ESI†). This type of fragmentation is not found in the mass spectrometry of heme.101–103 Accurate mass analysis further showed that the 462.2 fragment still contains iron. Two plausible schemes were developed to show how this 462.2 species could arise.Propionate-containing pyrrole:
| 613.2643 − 462.2010 = 151.0633 = C8H9NO2 |
Vinyl-containing pyrrole:
| 569.2739 − 462.2010 = 107.0729 = C7H9N |
This breakdown presented a problem for analysis, as heme contains two copies of each of these potential fragments. To determine whether one of these fragments is consistently lost, or whether it is a mixture, comparative studies with deuteroheme and mesoheme were made. These molecules are chemically very similar to heme and were found to react in the same way with NADH in HemS. Their structures are given in Section R of the ESI,† plus their respective HPLC chromatograms and mass spectra. Table 2 summarizes these data and shows that the difference in masses that deuteroheme and mesoheme have with heme were replicated with those for the higher order products (i.e. those with m/z 613.3 and m/z 569.3), but that this difference was halved with m/z 462.2. These data reveal that it is a vinyl-containing pyrrole that is consistently lost, which in turn implies that the 462.2 species is a tripyrrole which retains the two propionate-bearing pyrroles of the heme molecule, plus one vinyl-containing pyrrole. This analysis further reveals that the α-meso-carbon and either the β-meso-carbon or the δ-meso-carbon of heme (see Fig. S9 for labeling, ESI†) must be broken to produce the 462.2 species. There are no other ions of significant abundance, suggesting that the 613.3 and 569.3 species are broken down consistently, and that the ring of the HBP is already cleaved at one of the meso-carbon positions before ionization in the spectrometer. Were this not the case and the cyclic tetrapyrrole was still intact, there would be no obvious significant preference for which meso-carbon bond was broken first, resulting in a greater variety of fragments.
| LCMS masses | |||||
|---|---|---|---|---|---|
| Heme | Deuteroheme | Mesoheme | |||
| Mass | Mass | Δ vs. heme | Mass | Δ vs. heme | |
| Pre reaction | 616.2 | 564.2 | −52 | 620.2 | +4 |
| Fragments | 613.3 | 561.6 | −52 | 617.2 | +4 |
| 569.3 | 517.2 | −52 | 573.3 | +4 | |
| 462.2 | 436.2 | −26 | 464.2 | +2 | |
The MS analysis revealed interesting details concerning the relationship between the 613.3, 569.3 and 462.2 Da fragments. However, the precise structure of the 613.3 HBP (and therefore of the other significant structures) remained undetermined. We know that hydride transfer was occurring, and surmised that the HBP must be cleaved at one of the meso-carbon positions, but it was unclear how this process would give a m/z value of 613.3, from the m/z value of 616.5 (614.5 when the propionates are deprotonated) of heme.
Nuclear magnetic resonance (NMR) spectroscopy presented many challenges. Firstly, as heme limits its own breakdown at high concentrations, only small quantities of the HBP could be produced. There were also difficulties in extracting the HBP to an appropriate solvent. Some conditions led to removal of the iron ion, and hence further fragmentation of the HBP. Paradoxically, other conditions allowed for the iron ion (and hence HBP stability) to be retained but, because this ion was in a paramagnetic state, peak-broadening made the NMR spectra uninterpretable.
Crystals were grown with the HBP retained in the HemS pocket. Crystal growth was good, and a purple color indicated that the iron ion remained in the HBP. However, when it came to adding cryoprotectants, the purple color was lost, suggesting that the HBP was further degraded. These crystals diffracted and were analyzed. Electron density from the protein was good but that of the remaining HBP was low resolution; the density observed in the pocket did confirm that fragmentation of the HBP within the HemS pocket had indeed taken place (an example is given in Fig. S29, ESI†). To date we have not managed to keep the breakdown product stable in the protein crystals.
3.5 NADH-binding pocket
The breakdown of heme via hydride transfer from NADH constitutes new chemistry. Details of NADH-binding within the HemS pocket were therefore sought, but standard biophysical investigations were limited by the relatively high KM for NADH (900 μM at pH 7.0).Bioinformatics was therefore used to determine whether any possible NADH-binding regions within HemS have any well-studied analogues in the literature. The package Relibase+104 (now retired by the Cambridge Crystallographic Data Centre, with most of its functions being embedded in its CSD Python API) was used to scan the protein cavity and surface to compare structurally against known NADH-binding pockets. This package was selected due to its inclusion of CavBase,105,106 a program explicitly designed to be independent of sequence and fold homology, whilst proficient in the identification of unexpected similarities between protein cavities.97,98
Relibase+ identified a portion of the large heme-binding pocket as a possible NADH-binding site, and inspection revealed that it was actually a fusion of two smaller cavities, separated by a double phenylalanine (phe-)gate (see Fig. 7).
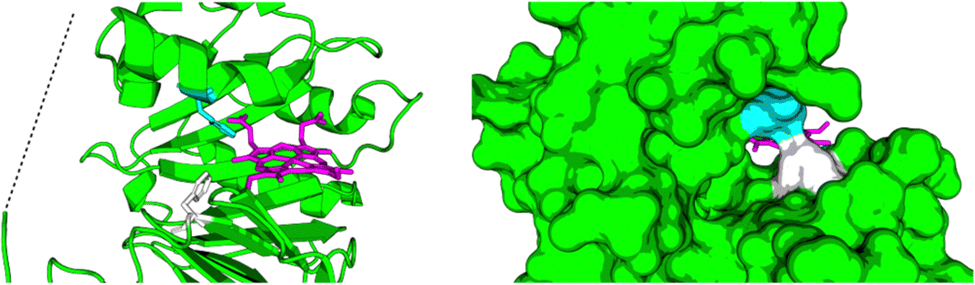 | ||
| Fig. 7 Large cavity of HemS. It is split into two smaller cavities by a central pair of phenylalanine residues, F104 (white) and F199 (cyan). In one cavity, heme binds. In the other, Relibase+ identified a possible NADH-binding site. A dashed line has been included to represent the missing loop region from the crystal structure (2J0P). | ||
The first of these smaller pockets contained the well-defined heme-binding site, whereas the function of the other pocket was unknown. This cavity was tested against every cavity in the PDB, giving one weak hit to a known NADH-binding site. This hit was with a mutant of the 2-trans enoyl–acyl carrier protein reductase enzyme InhA, from Mycobacterium tuberculosis (PDB: 2AQI).107 Despite this weak hit, superimposing the coordinates of NADH from this structure onto holo-HemS (PDB: 2J0P) produced a remarkable overlap; not only did NADH fit favourably in the HemS pocket, but its conformation was stretched out in such a way that the hydride-bearing nicotinamide group was pointing directly at the β-meso carbon of heme.
Although this analysis provided a promising indication that NADH can indeed bind close to the heme-binding site, the superimposition did not provide the actual binding mode of NADH in atomic detail, nor did it provide a mechanism for close approach of NADH to the heme. To answer these questions, we employed computations based on the potential energy landscape approach. Details of the algorithms and force fields employed are given in the ESI.†
GMIN,108 a program which features basin-hopping global optimization algorithms,66,109,110 was used to sample the potential energy landscape surrounding the putative NADH-binding site. Using the NADH structure from 2AQI superimposed on the 2J0P structure of holo-HemS as a starting point, basin-hopping allowed us to determine the most important residue-ligand interactions required for NADH-binding. These residues are shown in Fig. 8. Whilst Q132, S171 and R250 all interact with the adenine base (the latter with the adenosine ribose oxygen and phosphate groups as well), K203 was found to always interact with the amide group of nicotinamide, and T312 with the hydroxyl groups of the adenosine ribose and the phosphate groups.
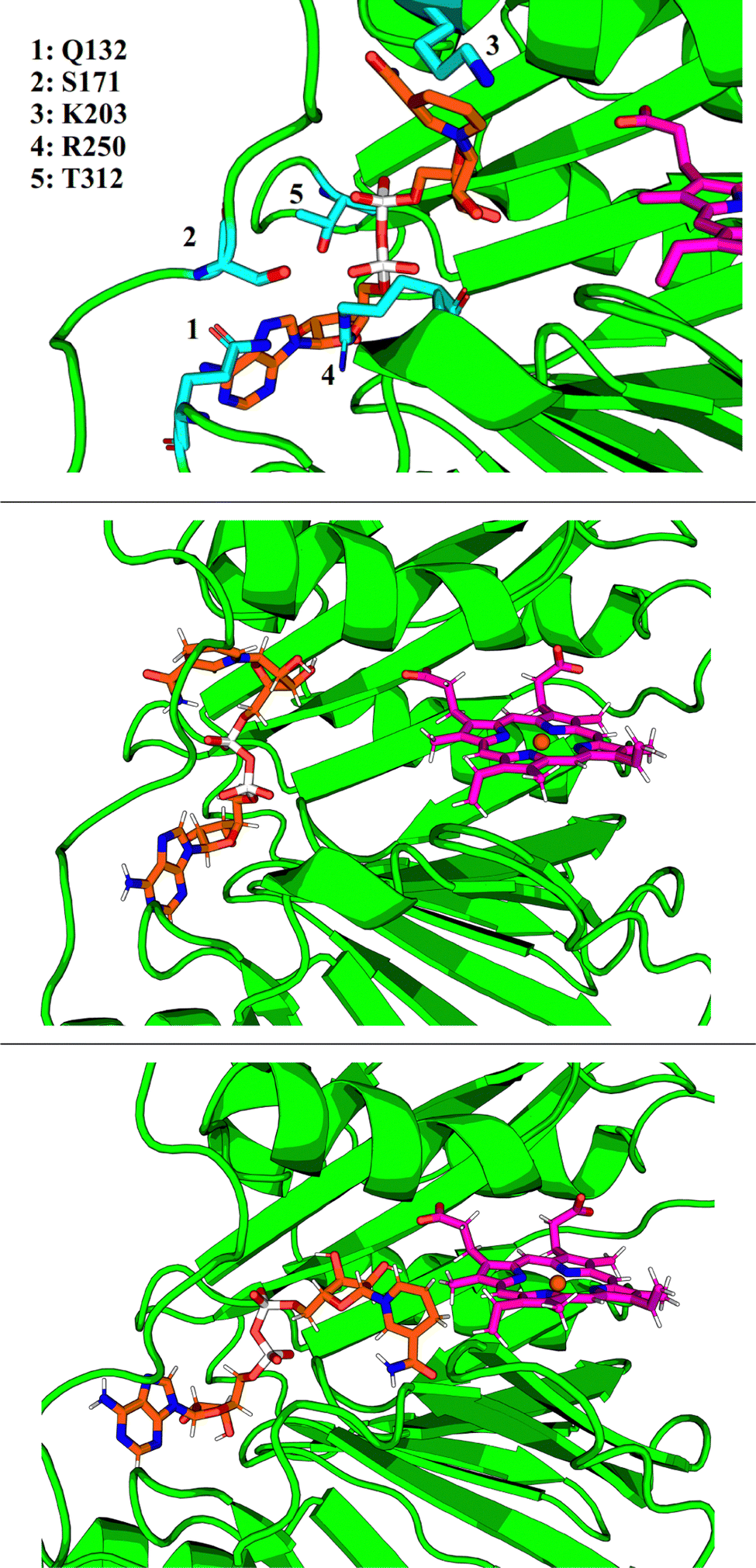 | ||
| Fig. 8 Top: Close-up representation of the holo-HemS main pocket with NADH (computational reconstruction). NADH is shown in orange. The most important residues implicated in NADH-binding are shown in cyan. Middle: Representative structure of folded NADH docking to the heme-binding cavity. Bottom: Representative structure of unfolded NADH, where it can access heme. | ||
3.6 Docking of NADH to HemS
Whilst providing details on residue-ligand interactions, this strategy focused only on the small region of the PES surrounding the Relibase+ superimposition, and therefore yielded little information concerning the movement of NADH within the pocket, from the cavity entrance towards the heme molecule. The basin-hopping step size was therefore increased. Although this approach increases the chances of the structural perturbations initially producing high energy structures, as well as the likelihood of cis-/trans-isomerism and chirality flipping, GMIN has strategies to reject these configurations, and the larger steps enable a greater area of the PES to be explored. This strategy allowed for structures to be identified where NADH was in a dramatically different position (and conformation) compared to its Relibase+-identified starting point. Interestingly, NADH occupies a folded conformation towards the edge of the cavity, and unfolds as it enters further into the pocket to access the heme (see Fig. 8). Such behaviour is consistent with the finding that NADH preferentially occupies a folded conformation when free in solution, as demonstrated experimentally,111,112 because it is more energetically stable than its unfolded analogue. A folded conformation often precludes NADH from easy access to protein cavities, including the center of the holo-HemS pocket. Therefore, after docking to HemS at the edge of the cavity, NADH must be induced to unfold to access the heme molecule. Computations and further experimental studies demonstrated in detail which residues are important to bring about this unfolding of NADH, as shall be described in a separate report.3.7 NADH unfolding pathway
The extended GMIN calculations served to provide a series of minima representing NADH at various positions within the holo-HemS pocket. However, they do not provide detailed pathways for NADH approaching heme within the HemS pocket. To generate such pathways, selected minima were connected using OPTIM113 (a program that uses the discrete path sampling strategy)55,114,115 and PATHSAMPLE116 (a program which acts as a driver for OPTIM). Once connected pathways were identified that showed NADH approaching heme within the HemS pocket, refinement strategies were used (see Section A of the ESI,† and Joseph et al.62 for details). Due to the size of the system (5501 atoms), GPUs were employed,117 and various strategies were implemented to facilitate efficient sampling, as outlined in the ESI.†This approach generated a large database of interconnected minima and transition states describing the behaviour of NADH and heme within the HemS pocket. Fig. 9 visualizes the landscape with a disconnectivity graph. From this database, multiple competing pathways (including the fastest pathway, also shown in Fig. 9), can be extracted for further analysis.
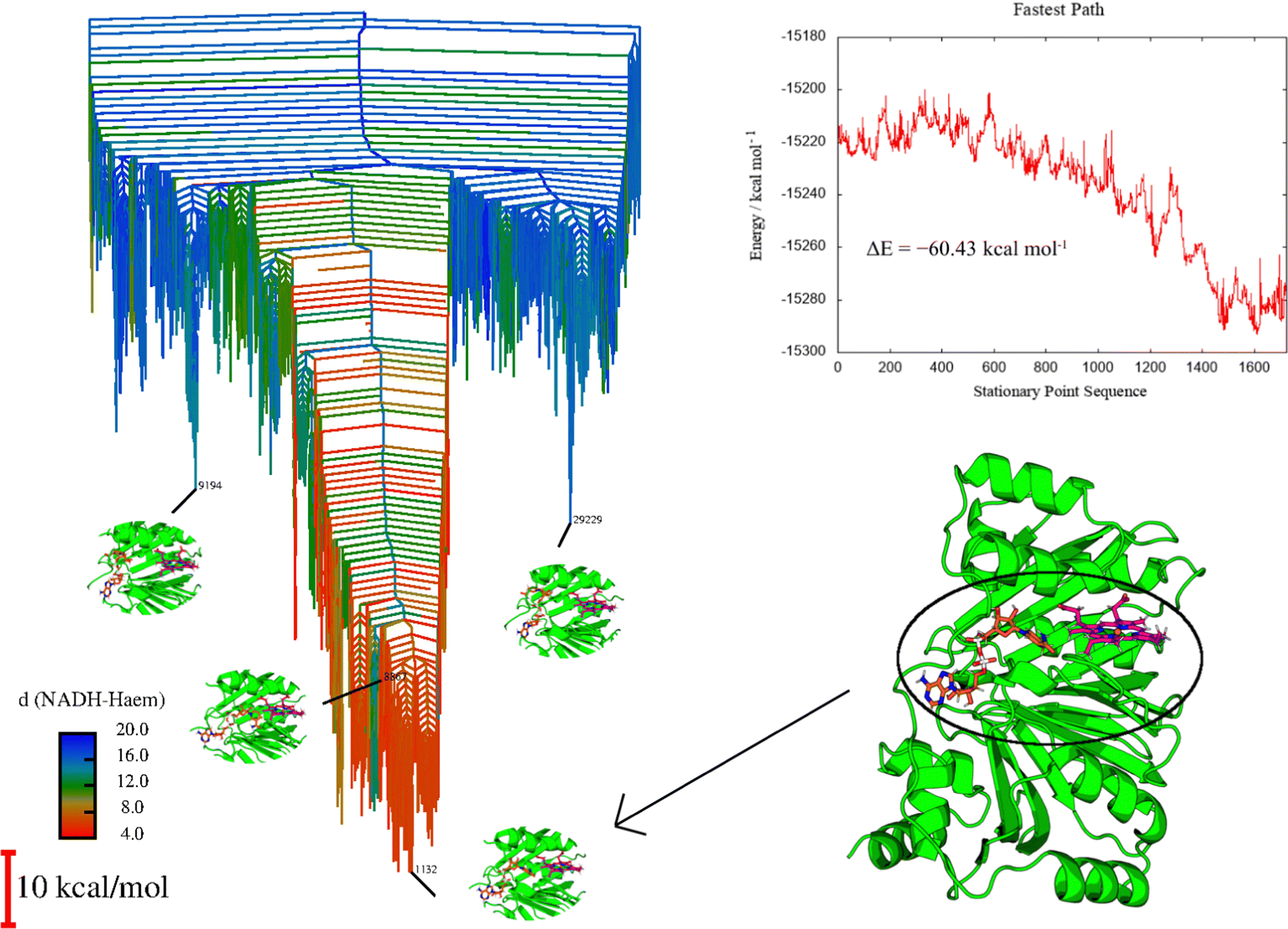 | ||
| Fig. 9 Left: Disconnectivity graph for HemS with its heme and NADH ligands, color-coded according to NADH-heme distance, as shown by the color-bar, which is labeled in Angstroms. Relevant magnified structures are highlighted. Minima 9194 and 24229 correspond to structures outside of the main funnel, and show NADH folded at the edge of the cavity, far from heme. Minimum 1132 is the global minimum, and shows NADH in close proximity to heme, with a hydride pointing towards heme, ready to transfer. Minimum 8867 is the minimum where NADH and heme are in closest proximity, but the hydride is not necessarily in a good location to transfer. Top right: Fastest pathway between representative minima showing NADH unfolding and approaching heme. The stationary point sequence at index 1 corresponds to a structure with NADH folded and at the edge of the cavity, whereas index 1723 corresponds to NADH fully stretched, with a hydride in close proximity to heme. All odd indices on the stationary point sequence correspond to minima, and even indices to transition states. The process of NADH approach to heme is clearly stabilizing, with few significant barriers, despite NADH being distorted from its natural conformation. Bottom right: Full representation of minimum 1132, giving a clear representation of the protein conformation in its entirety. HemS is represented in green, heme in magenta and NADH in orange. | ||
Fig. 9 displays clear funnelling properties, with little frustration in terms of barriers separating either low- or high-energy states from the global basin. Such PESs are typical for evolved biomolecules that perform a single function, and have been illustrated for various protein folding84–87 and DNA base-pairing problems.92,93 Whereas the funnel for a single protein provides information on the nature of protein folding, in the protein–ligand case the protein already occupies a conformation close to its native state, and so the funnel instead represents the energetics of interactions with the ligand(s). A well-defined funnel for a protein–ligand system therefore indicates that the ligand(s) is directed to a specific location, where binding is strong and there is significant energetic stabilization. The disconnectivity graph in Fig. 9 is further color-coded according to NADH-heme distance, where red represents minima with these two ligands in close proximity. The fact that these minima are clustered at the bottom of the funnel suggests that there is a strong energetic driving force to bring NADH and heme into close proximity within the HemS pocket. Visualization of these lowest energy minima shows that these structures do indeed have NADH and heme close together and primed to react. Representative minima, including the global minimum, are shown superimposed on the disconnectivity graph in Fig. 9. Remarkably, these minima are still the lowest-energy ones despite NADH being in a destabilized unfolded conformation, demonstrating the significant stabilization effects of residue-phosphate interactions within the HemS pocket. A deep, uninhibited funnel is heavily suggestive of precise functionality (in this case bringing heme and NADH together to react), which typically only arise from evolved or designed systems. Such a PES therefore strongly implies that HemS has specifically evolved to catalyze the reaction of these two ligands. Furthermore, the fastest pathway, attained using Dijkstra analysis,118,119 shows that the barriers to be overcome in order for NADH to unfold and approach heme successfully are relatively small. The energy landscape therefore provides strong evidence that NADH is indeed a natural ligand of HemS in addition to heme, and that the function of this biomolecule is to bring the two ligands together and prime them for reaction.
3.8 Homologues
Despite this evidence, there was the possibility that the reaction is merely an evolutionary artefact of HemS with no real biological significance. To resolve this issue, relevant homologues of HemS were also studied. Should they also catalyze anaerobic heme breakdown, there would be further evidence to suggest that the reaction is indeed biologically relevant.Phylogenetic analysis revealed that HemS has at least 5000 known homologues, from 218 different genera. A maximum-likelihood phylogenetic tree is given in Fig. S4 (ESI†). Homologues from Yersinia pestis (HmuS), Escherichia coli O157:H7 (ChuS) and Shigella dysenteriae (ShuS) were selected for further studies, mainly due to the fact that they are relatively well-characterized in the literature. As such, they have already been discussed in the introduction to this Perspectives article. These homologues are derived from interesting strains of bacteria: Y. pestis was responsible for the Black Death, S. dysenteriae is responsible for dysentery, and Escherichia coli is a major foodborne pathogen.
3.9 Sequence analysis and conservation
The 218 sequences selected to construct the phylogenetic tree were aligned, and the consensus sequence taken to be 345 residues long. As shown in Table S4 (ESI†), the residues involved in heme-binding showed remarkable conservation. R102, H196 and R209 all showed 100% conservation across the 218 homologues, and K294 and R321 98.1% and 99.1%, respectively. The residue at 318 showed 69.7% conservation for phenylalanine, although HemS has a tyrosine at this position. Tyrosine and phenylalanine are both aromatic residues, and so presumably this change does not significantly alter the heme-binding properties. Together, they account for 97.2% of residues at this position. The residues implicated in NADH-binding, meanwhile, were not as faithfully conserved, with approximately one-third of homologues retaining each of these residues. These data suggest that virtually all of the homologues are hemoproteins, but not all of them are heme breakdown enzymes, or at least they do not necessarily use NADH as the ligand required to effect heme breakdown.Given that neither of them are directly involved in heme- or NADH-binding, the conservation of the two residues comprising the double phenylalanine gate (F104 and F199) is significant. F104 is conserved across 98.6% of the homologues, and F199 across 82.6% (rising to 90.8% when the other possible aromatic residues, histidine and tyrosine, are included). This result is consistent with the hypothesis (studied in detail in a separate paper which is in preparation) that this double phe-gate is performing an important regulatory function, controlling access to heme. The fact that this gate is better conserved than the residues for NADH-binding suggests that it regulates access not just for NADH, but for other ligands too.
3.10 Computational investigation of selected homologues
To expedite calculations, an alternative ‘templating’ strategy (described in Section A of the ESI†) was used to generate the databases for HmuS, ChuS and ShuS. Fig. 10 shows the disconnectivity graphs arising from these databases. The HmuS graph shows a clear, deep funnel very similar to that seen for HemS. This feature is unsurprising given that HemS and HmuS are close (89.6% identity) homologues, and strongly suggests that HmuS (like HemS) has specifically evolved to bring NADH and heme into close proximity in order to break down the latter molecule. However, the graphs for ChuS and ShuS show no clear funnelling properties and certainly do not exhibit a funnel with a cluster of low-lying minima that feature NADH and heme in close proximity. This result does not necessarily mean that these homologues will not be able to engage in heme breakdown using NADH (as will be demonstrated using biophysical techniques), but that it is most likely not these proteins' primary function. Possible alternative functions are discussed later in the results.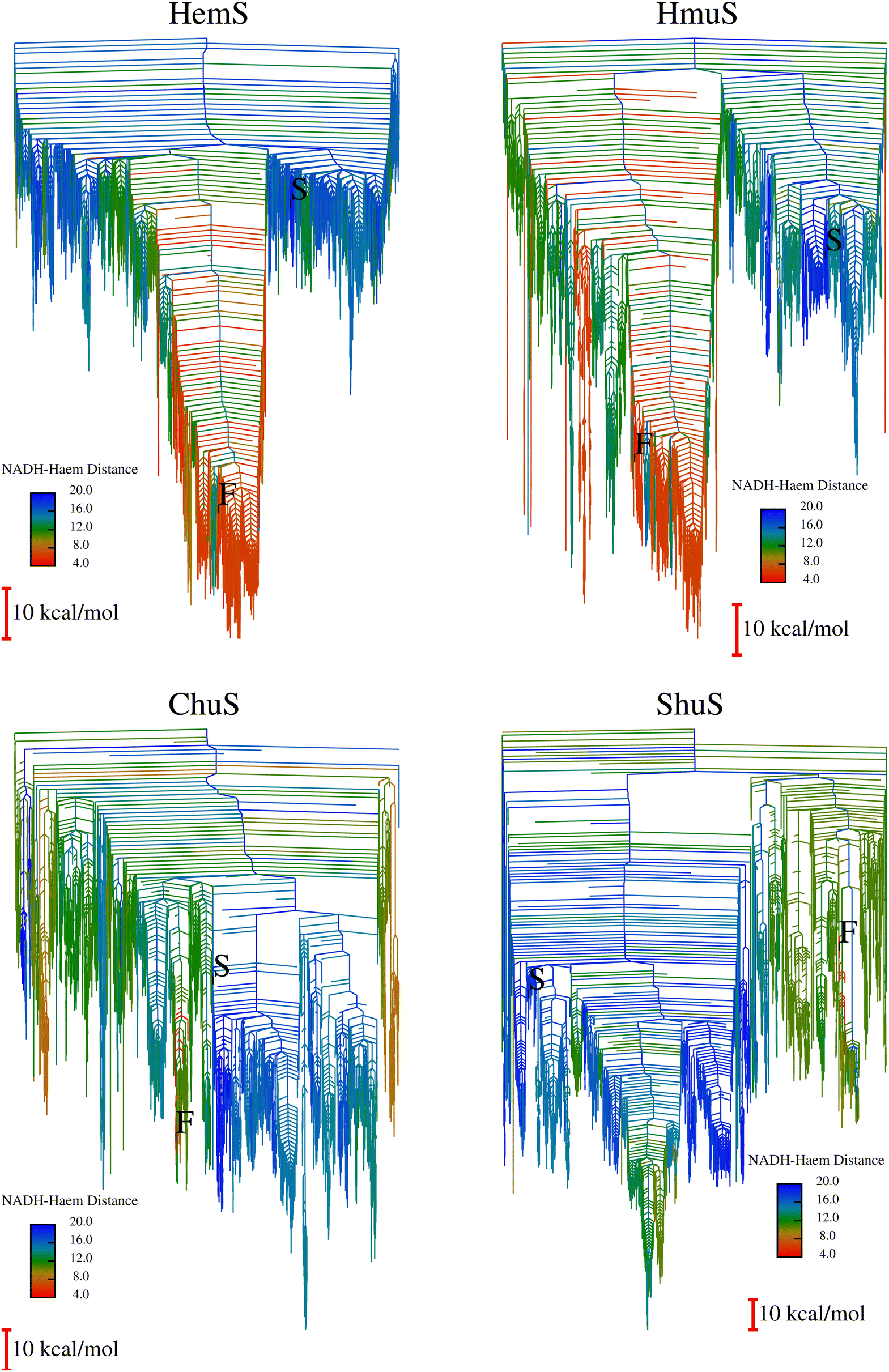 | ||
| Fig. 10 HemS, HmuS, ChuS and ShuS Disconnectivity Graphs. The HemS graph is repeated for comparison. In each graph, S represents a selected minimum featuring NADH in a folded state. It represents the same minimum in all four systems, having been reoptimized from the original HemS template minimum for the other three systems. F, meanwhile, represents the minimum with closest NADH-heme proximity found in each system. HmuS is most like HemS, displaying clear funnelling properties, which is not surprising given that these proteins are closest in homology. This disconnectivity graph has some dangling red minima that are not part of the main pocket, most likely due to artificially large barriers separating them from the main basin. Usually, these artefacts would be corrected, but due to the large system size, such calculations would have been time- and resource-consuming. Nonetheless, the main feature of the system is clear: as for HemS, a clear and deep funnel bringing NADH and heme into close proximity is apparent. For the ChuS and ShuS systems there are no clear funnelling properties, and very few minima with short NADH-heme distances. In the case of ShuS, the selected F minimum (where NADH is unfolded) is higher in energy than the S minimum (where it is folded, and far from heme). | ||
3.11 Experimental investigation of selected homologues
HmuS, ChuS and ShuS were all incubated with heme and NADH to determine whether they could indeed catalyze heme breakdown to give the HBP, with characteristic peak at 591 nm. A series of UV-Visible experiments, displayed in Fig. 11, showed that they all could, with varying degrees of efficacy. This result strongly suggests that these proteins have evolved specifically to carry out this reaction, and that it was not merely an incidental artefact unique to HemS that it could catalyze heme breakdown via the reductive route. Both ChuS and especially ShuS displayed slower rates of heme breakdown compared to HmuS or HemS. This observation is consistent with the computational data, which imply that ChuS and ShuS may have alternative functions, working orthogonally, or even in competition, with heme breakdown.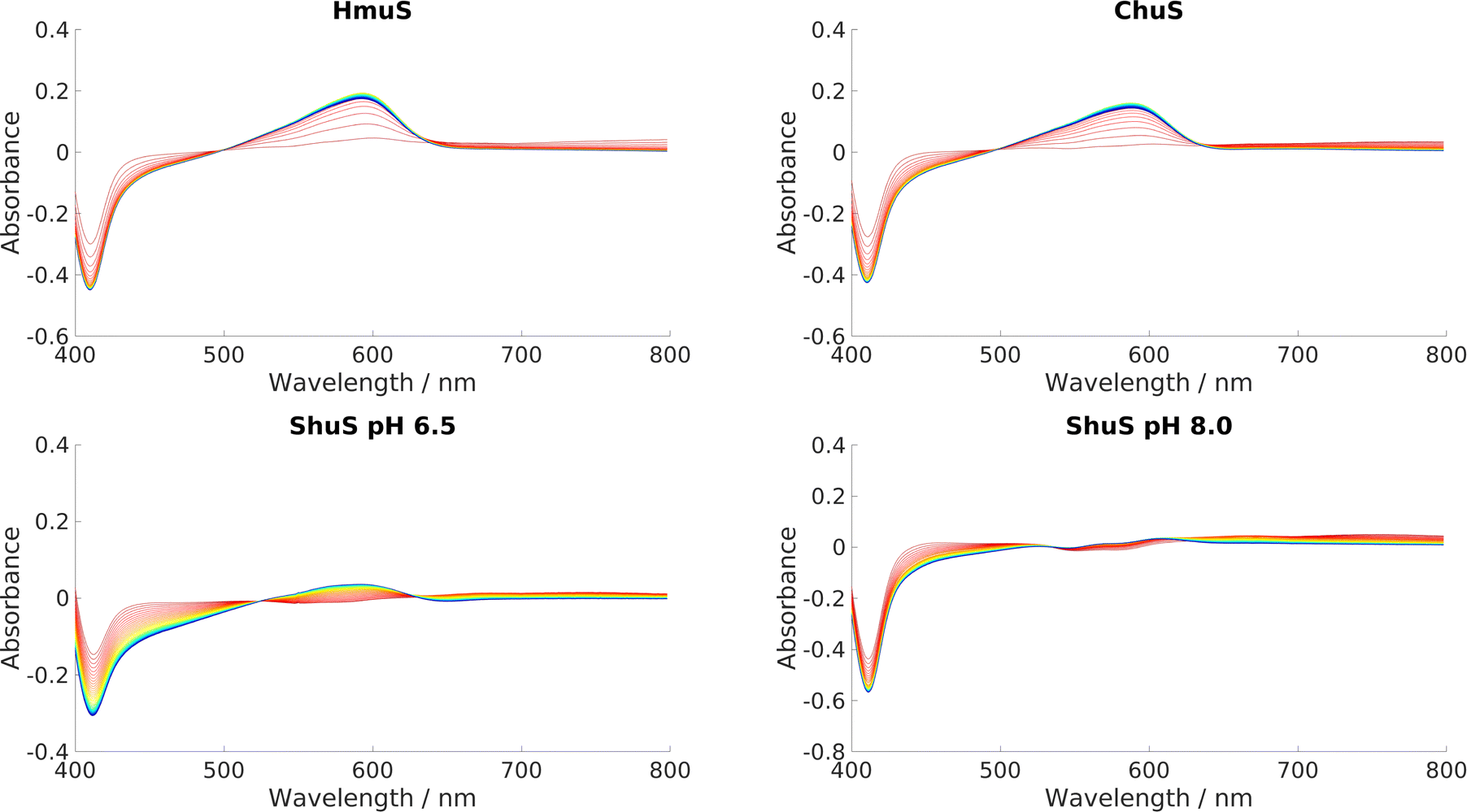 | ||
| Fig. 11 UV-visible spectra of HmuS, ChuS and ShuS. Reaction stoichiometry was 8 μM protein: 8 μM heme: 2000 μM NADH. Scans were run every minute for 50 minutes, indicated by the color scheme change from red (1 minute) to blue/purple (50 minutes). ShuS was tested at pH 8.0 as well as 6.5. | ||
3.12 Possible alternative functions to heme breakdown
As mentioned in the introduction, some of the homologues have been demonstrated to be capable of binding with DNA. ShuS is one such homologue, and this behaviour was observed during cell lysis in our investigations. Lysing BL21-Gold (DE3) E. coli cells containing overexpressed ShuS resulted in the protein aggregating and precipitating out of solution. Addition of benzonase nuclease immediately prior to cell lysis prevented this aggregation, suggesting that ShuS otherwise binds to the bacterial DNA. Furthermore, unlike the other homologues studied, ShuS proved to be unstable if left at pH 6.5 for extended periods of time. This problem was resolved by increasing the pH to 8.0.Given that ChuS and ShuS differ by only five residues, it was noteworthy that one homologue so readily bound to DNA, and yet the other one did not appear to display such properties. The bioinformatics package, DNAbind,120 was used to investigate this observation further, by predicting the number of DNA-binding residues within each homologue. HmuS was excluded since no crystal structure has been solved for it; though this is also true for ShuS, it was reconstructed using SwissModel.121 Eight DNA-binding residues were identified in HemS, although these did not form a contiguous surface, suggesting limited DNA-binding capabilities for this homologue. Seventeen were identified for PhuS, and these were clustered around the large cavity. Ten residues apiece were identified for ChuS and ShuS, although none corresponded to the five residues that differed between the two homologues. However, these predicted DNA-binding residues were clustered around the small cavity, within which were two of the five divergent residues, thus suggesting an indirect impact. One of these divergent residues is S123 in ChuS, but R123 in ShuS. As serine is a polar, neutral residue but arginine is cationic, this mutation may explain why the ChuS pocket does not bind to anionic DNA but ShuS does. The other different residue is A19 in ChuS but V19 in ShuS. Both of these residues are hydrophobic, but valine has significantly more steric bulk, allowing it to interact with R123 and thus hold it in place, which A19 is unable to do with S123 (see Fig. S31, ESI†). Such anchoring of R123 to the center of the cavity may further assist ShuS to bind to DNA.
Calculations also suggest that this small cavity is important in ShuS, but not the other homologues. Instead of a clear funnel bringing heme and NADH into close proximity, the primary (very shallow) funnel instead showed significant movement of the N-terminal α-helix, which opens up the small cavity, as illustrated in Fig. S31 (ESI†). The stabilization brought about by this movement indicates that ShuS may be priming itself to bind to a ligand (such as DNA) in this small cavity.
4 Conclusions
The cytosolic family of hemoproteins to which HemS, HmuS, ChuS and ShuS belong has been extensively studied, prompting much debate as to their function. An integral product of heme-uptake operons, it has been unclear whether such proteins are merely heme transporters, or are heme degrading enzymes. The observation that HemS only breaks down heme when the heme concentration is low suggests that this protein, at least, has a dual role. It appears to be a heme breakdown enzyme at low heme concentration to release iron, and a heme-storage protein at higher heme concentration, protecting against toxicity and retaining the excess heme for future use. This functionality may correlate with the observation that HemS can bind more than one heme, whether as a dimer, or in a second, low-affinity pocket. Evidence of transcriptional regulation performed by some members within this hemoprotein family further complicates the analysis. Heme breakdown behavior catalyzed by ChuS has been demonstrated previously,39,52 although (like all HOs) this process required oxygen to proceed. HemS has challenged us for some time, in that its heme-binding (and subsequently NADH-binding) properties studied by traditional biophysical methods suggested a subtly complex hemoprotein with unusual characteristics.Our results demonstrate anaerobic enzymatic behaviour within this family of hemoproteins. All four members, HemS, HmuS, ChuS and ShuS, demonstrated this capability to varying extents. Our results suggest that these hemoproteins are the end-users of heme for the proteins produced from their respective operons, which may themselves either adapt to oxygen-limiting conditions, or may be backup systems that only switch on when oxygen levels are low. These operons themselves differ markedly from the typical heme-uptake and utilization operons, centered around HOs, found in bacteria. Pathogenic bacteria typically require exogenous heme to survive and replicate, and so effective strategies to acquire and degrade heme are essential. In the absence of oxygen, typical pathways reliant on HOs become obsolete. And yet, many species of bacteria are known to specifically target oxygen-depleted regions of their host organism. Of the four homologues studied in this work, three belong to bacteria that target the terminal ileum of the human small intestine: HemS from Yersinia enterocolitica; ChuS from Escherichia coli O157:H7 and ShuS from Shigella dysenteriae. Anaerobic heme-uptake and degradation strategies are therefore required for pathogenic survival under these conditions, and this research presents one such feasible strategy. This insight could prove important when formulating disease-prevention strategies in the future, especially considering the global effect of dysentery and other gastrointestinal conditions.
This heme breakdown reaction has many unusual features. It demonstrates, for the first time, hydride transfer from NAD(P)H to a heme molecule. Deuterium-labeled experiments demonstrated that the reaction is not stereospecific, although there is a kinetic preference for one face of the nicotinamide head to transfer hydride over the other. Both NADH and NADPH can be used as a reagent for heme breakdown, and it is unclear which one is, or whether both are, the natural ligand. NAD(P)H was demonstrated to unfold as it progressed through the cavity in its approach to heme, a requirement due to steric considerations.
LCMS and X-ray crystallographic studies revealed that the HBP was cleaved consistently at its β-meso-carbon or δ-meso-carbon positions; the β-meso-carbon would seem more likely given the proximity with which the nicotinamide head of NADH reaches that atom. The cleavage would facilitate extraction of heme. The exact structure of this 613.3 Da HBP is unknown, although further properties and details of formation are to be discussed in a further paper currently in preparation.
Due to the weak, transient nature of NADH-binding, bioinformatics was used to identify possible docking sites for this ligand within the cavity of HemS. This search only gave very weak ‘hits’, of which the strongest was with a mutated version of InhA, thus emphasizing the originality of this class of reaction.
From this hit, the energy landscape approach was used to calculate possible binding modes of NADH and heme within the HemS pocket, plus pathways demonstrating the unfolding and approach of NADH to heme. Further landscapes were generated for HmuS, ChuS and ShuS.
The potential energy surfaces arising from these studies revealed that the HemS and HmuS systems both possess funnelled, unfrustrated landscapes, where the lowest energy conformations correspond to the shortest distances between the NADH and heme ligands. Strongly funnelled landscapes suggest clear functionality associated with a particular structural configuration, and so these results suggest that these proteins have specifically evolved to bring these ligands into close proximity to facilitate reaction. These landscapes therefore provide compelling additional evidence to support the experimental data that these proteins are indeed heme-breakdown enzymes, and that heme and NADH, or perhaps NADPH, are the associated ligands.
The potential energy surfaces for ChuS and ShuS were not so clearly funnelled. These results suggest that these proteins may not be as efficient at heme breakdown as their homologues, and that it may not be their primary function. The experimental data corroborate this conclusion by showing that ChuS and ShuS are capable of heme breakdown, but are less effective than HemS or HmuS.
DNA-binding was indirectly demonstrated with ShuS, in agreement with the literature. This behaviour, as already discussed elsewhere,47 would suggest that at least some members of this hemoprotein family can act as transcriptional modulators. It is perhaps the case that these proteins alternate between controlling heme flux via promoter suppression and reductively degrading heme according to heme availability and demand.
Though only four homologues were studied in detail, a phylogenetic analysis revealed that there were over 5000 related sequences from 218 different genera, including both pathogenic and non-pathogenic types. There is therefore broad scope for further study of possible anaerobic breakdown of heme across a wide variety of species, some of which could be of potential medical importance.
The mutual benefit from conducting both experiments and energy landscape theory calculations side-by-side has been invaluable to understand the system. Further mechanistic details of the reaction and the possible role of the ‘phe-gate’, resulting from experiment and further ELT calculations, are to be described in a separate report.
Author contributions
ADK wrote the original draft of the manuscript. ADK, EBS, DCYC, DJW and PDBa all contributed to manuscript review and editing. Project conceptualization was by ADK, EBS, DCYC, DJW and PDBa. Unless otherwise stated, laboratory-based experiments were performed by EBS, DCYC and ADK. Computational experiments were by DCYC and ADK. The corresponding programs were written by DJW and his group. YX ran the phylogenetics analysis under the supervision of ADK and PDBa. GSB ran mass spectrometry experiments and was consulted on project conceptualization. OJK ran NMR experiments and was consulted on project conceptualization. PDBr ran X-ray crystallography experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ADK thanks the Cambridge Commonwealth, European and International Trust for a Cambridge Vice-Chancellor's PhD scholarship. EBS, GSB and OJK thank the EPSRC for studentships. DJW gratefully acknowledges funding from the EPSRC. The authors thank Jonathan Gregory for experiments with Co-PPIX and Zn-PPIX, Joanna Ashton for producing some of the mutant HemS proteins and Chris Whittleston for assistance on initial computational setup. We thank Max Paoli and Sabine Schneider for the original HemS-containing plasmid and for piquing our interest in HemS.Notes and references
- N. D. Hammer and D. Weakland, Bacterial iron acquisition strategies, Elsevier Inc., 4th edn, 2019, pp. 410–417 Search PubMed.
- H. A. Dailey, S. Gerdes, T. A. Dailey, J. S. Burch and J. D. Phillips, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2210–2215 CrossRef CAS.
- J. E. Choby and E. P. Skaar, J. Mol. Biol., 2016, 428, 3408–3428 CrossRef CAS PubMed.
- A. Wilks and G. Heinzl, Arch. Biochem. Biophys., 2014, 544, 87–95 CrossRef CAS PubMed.
- A. Wilks and M. Ikeda-Saito, Acc. Chem. Res., 2014, 47, 2291–2298 CrossRef CAS PubMed.
- K. V. Lyles and Z. Eichenbaum, Front. Cell. Infect. Microbiol., 2018, 8, 1–13 CrossRef PubMed.
- M. D. Maines and A. Kappas, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4293–4297 CrossRef CAS PubMed.
- M. Unno, T. Matsui and M. Ikeda-Saito, Nat. Prod. Rep., 2007, 24, 553–570 RSC.
- B. Wegiel and L. E. Otterbein, Front. Pharmacol., 2012, 3, 1–8 Search PubMed.
- T. Ingi, J. Cheng and G. V. Ronnett, Neuron, 1996, 16, 835–842 CrossRef CAS PubMed.
- K. A. Hanafy, J. Oh and L. E. Otterbein, Curr. Pharm. Des., 2013, 19, 2771–2775 CrossRef CAS PubMed.
- K. Ueno, J. Morstein, K. Ofusa, S. Naganos, E. Suzuki-Sawano, S. Minegishi, S. P. Rezgui, H. Kitagishi, B. W. Michel, C. J. Chang, J. Horiuchi and M. Saitoe, J. Neurosci., 2020, 40, 3533–3548 CrossRef CAS PubMed.
- H. Parfenova, C. W. Leffler, S. Basuroy, J. Liu and A. L. Fedinec, J. Cereb. Blood Flow Metab., 2012, 32, 1024–1034 CrossRef CAS PubMed.
- J. P. Berne, B. Lauzier, L. Rochette and C. Vergely, Cell. Physiol. Biochem., 2012, 29, 475–484 CrossRef CAS PubMed.
- E. Lehmann, W. H. El-Tantawy, M. Ocker, R. Bartenschlager, V. Lohmann, S. Hashemolhosseini, G. Tiegs and G. Sass, Hepatology, 2010, 51, 398–404 CrossRef CAS PubMed.
- Z. Zhu, A. T. Wilson, B. A. Luxon, K. E. Brown, M. M. Mathahs, S. Bandyopadhyay, A. P. McCaffrey and W. N. Schmidt, Hepatology, 2010, 52, 1897–1905 CrossRef CAS PubMed.
- R. Stocker, A. N. Glazer and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5918–5922 CrossRef CAS PubMed.
- R. Stocker, Y. Yamamoto, A. F. McDonagh, A. N. Glazer and B. N. Ames, Science, 1987, 235, 1043–1046 CrossRef CAS PubMed.
- T. Ohrui, H. Yasuda, M. Yamaya, T. Matsui and H. Sasaki, J. Exp. Med., 2003, 199, 193–196 Search PubMed.
- D. E. Barañano, M. Rao, C. D. Ferris and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16093–16098 CrossRef PubMed.
- E. P. Skaar, A. H. Gaspar and O. Schneewind, J. Biol. Chem., 2004, 279, 436–443 CrossRef CAS PubMed.
- E. P. Skaar, A. H. Gaspar and O. Schneewind, J. Bacteriol., 2006, 188, 1071–1080 CrossRef CAS PubMed.
- N. Chim, A. Iniguez, T. Q. Nguyen and C. W. Goulding, J. Mol. Biol., 2010, 395, 595–608 CrossRef CAS PubMed.
- I. Stojiljkovic and K. Hantke, EMBO J., 1992, 11, 4359–4367 CrossRef CAS PubMed.
- J. W. LaMattina, D. B. Nix and W. N. Lanzilotta, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12138–12143 CrossRef CAS PubMed.
- Q. Zhang, W. A. van der Donk and W. Liu, Acc. Chem. Res., 2012, 45, 555–564 CrossRef CAS PubMed.
- W. Huang, H. Xu, Y. Li, F. Zhang, X. Y. Chen, Q.-L. He, Y. Igarashi and G.-L. Tang, J. Am. Chem. Soc., 2012, 134, 8831–8840 CrossRef CAS PubMed.
- M. D. Suits, J. Lang, G. P. Pal, M. Couture and Z. Jia, Protein Sci., 2009, 18, 825–838 CrossRef CAS PubMed.
- V. Braun and K. Hantke, Genetics of bacterial iron transport in Handbook of Microbial Iron Chelates, CRC Press, Boca Raton, Florida, 1991 Search PubMed.
- J. Shine and L. Dalgarno, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1342–1346 CrossRef CAS PubMed.
- I. Stojiljkovic and K. Hantke, Mol. Microbiol., 1994, 13, 719–732 CrossRef CAS PubMed.
- D. Mattle, A. Zeltina, J. S. Woo, B. A. Goetz and K. P. Locher, J. Mol. Biol., 2010, 404, 220–231 CrossRef CAS PubMed.
- J. S. Woo, A. Zeltina, B. A. Goetz and K. P. Locher, Nat. Struct. Mol. Biol., 2012, 19, 1310–1315 CrossRef CAS.
- J. E. Walker, M. Saraste, M. J. Runswick and N. J. Gay, EMBO J., 1982, 1, 945–951 CrossRef CAS.
- P. I. Hanson and S. W. Whiteheart, Nat. Rev. Mol. Cell Biol., 2005, 6, 519–529 CrossRef CAS PubMed.
- The UniProt Consortium, Nucleic Acids Res., 2021, 49, 480–489 CrossRef PubMed.
- S. Schneider, K. H. Sharp, P. D. Barker and M. Paoli, J. Biol. Chem., 2006, 281, 32606–32610 CrossRef CAS PubMed.
- S. Schneider and M. Paoli, Acta Crystallogr., Sect. A: Found. Crystallogr., 2005, 61, 343 Search PubMed.
- M. D. Suits, G. P. Pal, K. Nakatsu, A. Matte, M. Cygler and Z. Jia, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16955–16960 CrossRef CAS.
- M. D. Suits, N. Jaffer and Z. Jia, J. Biol. Chem., 2006, 281, 36776–36782 CrossRef CAS PubMed.
- L. G. Mathew, N. R. Beattie, C. Pritchett and W. N. Lanzilotta, Biochemistry, 2019, 58, 4641–4654 CrossRef CAS PubMed.
- S. Tripathi, M. J. O'Neill, A. Wilks and T. L. Poulos, J. Inorg. Biochem., 2013, 128, 131–136 CrossRef CAS.
- M. J. Lee, D. Schep, B. McLaughlin, M. Kaufmann and Z. Jia, J. Mol. Biol., 2014, 426, 1936–1946 CrossRef CAS PubMed.
- I. B. Lansky, G. S. Lukat-Rodgers, D. Block, K. R. Rodgers, M. Ratliff and A. Wilks, J. Biol. Chem., 2006, 281, 13652–13662 CrossRef CAS PubMed.
- L. Avila, H.-w Huang, C. O. Damaso, S. Lu, P. Moënne-Loccoz and M. Rivera, J. Am. Chem. Soc., 2003, 125, 4103–4110 CrossRef CAS PubMed.
- M. J. O'Neill, M. N. Bhakta, K. G. Fleming and A. Wilks, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5639–5644 CrossRef PubMed.
- T. Wilson, S. Mouriño and A. Wilks, J. Biol. Chem., 2021, 296, 100275–100285 CrossRef CAS PubMed.
- P. J. Wilderman, N. A. Sowa, D. J. FitzGerald, P. C. FitzGerald, S. Gottesman, U. A. Ochsner and M. L. Vasil, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9792–9797 CrossRef CAS PubMed.
- A. G. Oglesby, J. M. Farrow, J. H. Lee, A. P. Tomaras, E. P. Greenberg, E. C. Pesci and M. L. Vasil, J. Biol. Chem., 2008, 283, 15558–15567 CrossRef CAS PubMed.
- A. A. Reinhart, D. A. Powell, A. T. Nguyen, M. O'Neill, L. Djapgne, A. Wilks, R. K. Ernst and A. G. Oglesby-Sherrouse, Infect. Immun., 2015, 83, 863–875 CrossRef PubMed.
- A. Wilks, Arch. Biochem. Biophys., 2001, 387, 137–142 CrossRef CAS.
- Y. H. Ouellet, C. T. Ndiaye, S. M. Gagné, A. Sebilo, M. D. Suits, É. Jubinville, Z. Jia, A. Ivancich and M. Couture, J. Inorg. Biochem., 2016, 154, 103–113 CrossRef CAS PubMed.
- A. P. Kaur and A. Wilks, Biochemistry, 2007, 46, 2994–3000 CrossRef CAS.
- D. J. Wales, J. P. K. Doye, M. A. Miller, P. N. Mortenson and T. R. Walsh, Adv. Chem. Phys., 2000, 115, 1–111 CrossRef.
- D. J. Wales, Energy Landscapes, Cambridge University Press, Cambridge, 2003 Search PubMed.
- D. J. Wales and T. V. Bogdan, J. Phys. Chem. B, 2006, 110, 20765–20776 CrossRef CAS PubMed.
- D. J. Wales, Phil. Trans. Roy. Soc. A, 2012, 370, 2877–2899 CrossRef CAS.
- D. J. Wales, Annu. Rev. Phys. Chem., 2018, 69, 401–425 CrossRef CAS PubMed.
- J. N. Onuchic, Z. Luthey-Schulten and P. G. Wolynes, Annu. Rev. Phys. Chem., 1997, 48, 545–600 CrossRef CAS PubMed.
- P. G. Wolynes, Q. Rev. Biophys., 2005, 38, 405–410 CrossRef CAS PubMed.
- K. Klenin, B. Strodel, D. J. Wales and W. Wenzel, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 977–1000 CrossRef CAS PubMed.
- J. A. Joseph, K. Röder, D. Chakraborty, R. G. Mantell and D. J. Wales, Chem. Commun., 2017, 53, 6974–6988 RSC.
- O. M. Becker and M. Karplus, J. Chem. Phys., 1997, 106, 1495–1517 CrossRef CAS.
- D. J. Wales, M. A. Miller and T. R. Walsh, Nature, 1998, 394, 758–760 CrossRef CAS.
- V. A. Sharapov, D. Meluzzi and V. A. Mandelshtam, Phys. Rev. Lett., 2007, 98, 105701 CrossRef PubMed.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS.
- J. P. K. Doye, D. J. Wales and M. A. Miller, J. Chem. Phys., 1998, 109, 8143–8153 CrossRef CAS.
- J. P. K. Doye, M. A. Miller and D. J. Wales, J. Chem. Phys., 1999, 110, 6896–6906 CrossRef CAS.
- J. P. Neirotti, F. Calvo, D. L. Freeman and J. D. Doll, J. Chem. Phys., 2000, 112, 10340–10349 CrossRef CAS.
- F. Calvo, J. P. Neirotti, D. L. Freeman and J. D. Doll, J. Chem. Phys., 2000, 112, 10350–10357 CrossRef CAS.
- P. A. Frantsuzov and V. A. Mandelshtam, Phys. Rev. E, 2005, 72, 037102 CrossRef PubMed.
- C. Predescu, P. A. Frantsuzov and V. A. Mandelshtam, J. Chem. Phys., 2005, 122, 154305 CrossRef.
- H. Liu and K. D. Jordan, J. Phys. Chem. A, 2005, 109, 5203–5207 CrossRef CAS PubMed.
- V. A. Sharapov and V. A. Mandelshtam, J. Phys. Chem. A, 2007, 111, 10284–10291 CrossRef CAS PubMed.
- F. Rao and A. Caflisch, J. Mol. Biol., 2004, 342, 299–306 CrossRef CAS PubMed.
- F. Noé and S. Fischer, Curr. Opin. Struct. Biol., 2008, 18, 154–162 CrossRef PubMed.
- D. Prada-Gracia, J. Gómez-Gardenes, P. Echenique and F. Falo, PLoS Comput. Biol., 2009, 5, e1000415 CrossRef PubMed.
- D. J. Wales, Curr. Opin. Struct. Biol., 2010, 20, 3–10 CrossRef CAS PubMed.
- B. Strodel and D. J. Wales, Chem. Phys. Lett., 2008, 466, 105–115 CrossRef CAS.
- M. R. Hoare and J. McInnes, Faraday Discuss. Chem. Soc., 1976, 61, 12–24 RSC.
- F. H. Stillinger and T. A. Weber, Science, 1984, 225, 983 CrossRef CAS PubMed.
- D. J. Wales, Mol. Phys., 1993, 78, 151–171 CrossRef CAS.
- F. H. Stillinger, Science, 1995, 267, 1935–1939 CrossRef CAS PubMed.
- J. M. Carr and D. J. Wales, J. Phys. Chem. B, 2008, 112, 8760–8769 CrossRef CAS PubMed.
- M. C. Prentiss, D. J. Wales and P. G. Wolynes, PLoS Comput. Biol., 2010, 6, 1–12 CrossRef PubMed.
- J. A. Joseph, D. Chakraborty and D. J. Wales, J. Chem. Theory Comput., 2019, 15, 731–742 CrossRef CAS PubMed.
- S. Neelamraju, S. Gosavi and D. J. Wales, J. Phys. Chem. B, 2018, 122, 12282–12291 CrossRef CAS.
- D. J. Wales, Philos. Trans. R. Soc., A, 2005, 363, 357–377 CrossRef CAS.
- D. J. Wales, J. Theor. Med., 2005, 6, 107–110 CrossRef.
- S. N. Fejer, T. R. James, J. Hernández-Rojas and D. J. Wales, Phys. Chem. Chem. Phys., 2009, 11, 2098–2104 RSC.
- M. Martín-Bravo, J. M. Llorente, J. Hernández-Rojas and D. J. Wales, ACS Nano, 2021, 15, 14873–14884 CrossRef PubMed.
- D. Chakraborty and D. J. Wales, J. Phys. Chem. Lett., 2018, 9, 229–241 CrossRef CAS PubMed.
- S. Xiao, D. J. Sharpe, D. Chakraborty and D. J. Wales, J. Phys. Chem. Lett., 2019, 10, 6771–6779 CrossRef CAS PubMed.
- K. Röder and D. J. Wales, J. Phys. Chem. Lett., 2018, 9, 6169–6173 CrossRef PubMed.
- K. Röder and D. J. Wales, J. Phys. Chem. B, 2018, 122, 10989–10995 CrossRef PubMed.
- E. B. Sawyer, PhD thesis, University of Cambridge, 2009.
- D. C. Y. Choy, PhD thesis, University of Cambridge, 2015.
- A. D. Keith, PhD thesis, University of Cambridge, 2021.
- O. Warburg and E. Negelein, Chem. Ber., 1930, 63, 1816–1819 CrossRef.
- R. Lemberg, Biochem. J., 1935, 29, 1322–1336 CrossRef CAS.
- A. I. Celis and J. L. DuBois, Curr. Opin. Struct. Biol., 2019, 59, 19–28 CrossRef CAS PubMed.
- O. P. Charkin, N. M. Klimenko, P. T. Nguyen, D. O. Charkin, A. M. Mebel, S. H. Lin, Y. S. Wang, S. C. Wei and H. C. Chang, Chem. Phys. Lett., 2005, 415, 362–369 CrossRef CAS.
- E. R. Sekera and T. D. Wood, Mass Spectrom. Lett., 2018, 9, 91–94 CAS.
- M. Hendlich, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 1178–1182 CrossRef CAS PubMed.
- S. Schmitt, M. Hendlich and G. Klebe, Angew. Chem., Int. Ed., 2001, 40, 3141–3144 CrossRef CAS.
- S. Schmitt, D. Kuhn and G. Klebe, J. Mol. Biol., 2002, 323, 387–406 CrossRef CAS PubMed.
- J. S. Oliveira, J. H. Pereira, F. Canduri, N. C. Rodrigues, O. N. de Souza, W. F. de Azevedo, L. A. Basso and D. S. Santos, J. Mol. Biol., 2006, 359, 646–666 CrossRef CAS PubMed.
- D. J. Wales, GMIN: A program for basin-hopping global optimisation, 2021, https://www-wales.ch.cam.ac.uk/GMIN.
- Z. Li and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 6611–6615 CrossRef CAS PubMed.
- Z. Li and H. A. Scheraga, J. Mol. Struct., 1988, 179, 333–352 CrossRef.
- G. Weber, Nature, 1957, 180, 1409 CrossRef CAS.
- D. J. Patel, Nature, 1969, 221, 825–828 CrossRef CAS PubMed.
- D. J. Wales, OPTIM: A program for optimising geometries and calculating pathways, 2021, https://www-wales.ch.cam.ac.uk/OPTIM.
- D. J. Wales, Mol. Phys., 2002, 100, 3285–3305 CrossRef CAS.
- D. J. Wales, Mol. Phys., 2004, 102, 891–908 CrossRef CAS.
- D. J. Wales, PATHSAMPLE: A driver for optim to create stationary point databases using discrete path sampling and perform kinetic analysis, 2021, https://www-wales.ch.cam.ac.uk/PATHSAMPLE.
- R. G. Mantell, C. E. Pitt and D. J. Wales, J. Chem. Theory Comput., 2016, 12, 6182–6191 CrossRef CAS.
- E. Dijkstra, Numer. Math., 1959, 1, 269–271 CrossRef.
- D. A. Evans and D. J. Wales, J. Chem. Phys., 2004, 121, 1080–1090 CrossRef CAS PubMed.
- R. Liu and J. Hu, Proteins, 2013, 81, 1885–1899 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. De Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, 296–303 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03897a |
| ‡ Present address: Department of Biochemistry, Emory University, Atlanta, GA 30329, USA. Email: adkeith@emory.edu |
| This journal is © the Owner Societies 2024 |