
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved electrode reversibility of anionic redox with highly concentrated electrolyte solution and aramid-coated polyolefin separator†
Nanaka
Shimada
a,
Yosuke
Ugata
 ab,
Satoshi
Nishikawa
c,
Daisuke
Shibata
d,
Toshiaki
Ohta
d and
Naoaki
Yabuuchi
*ab
ab,
Satoshi
Nishikawa
c,
Daisuke
Shibata
d,
Toshiaki
Ohta
d and
Naoaki
Yabuuchi
*ab
aDepartment of Chemistry and Life Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama, Kanagawa 240-8501, Japan. E-mail: yabuuchi-naoaki-pw@ynu.ac.jp
bAdvanced Chemical Energy Research Center, Institute of Advanced Sciences, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama, Kanagawa 240-8501, Japan
cBattery Materials Business Department, Teijin Limited, 2-1 Hinode-cho, Iwakuni, Yamaguchi 740-8511, Japan
dSR Center, Ritsumeikan University, 1-1-1 Noji-Higashi, Kusatsu, Shiga 525-8577, Japan
First published on 3rd March 2023
Abstract
High-capacity electrode materials made of abundant elements are necessary to develop cost-effective energy storage applications. An emerging new chemistry has involved a series of electrode materials with a cation-disordered rock salt structure, and Li-excess and Mn-based oxides with disordered structures are attractive candidates for this purpose. Large reversible capacities for these electrode materials are obtained through both cationic/anionic redox. However, cyclability associated with insufficient reversibility of anionic redox in carbonate-based electrolyte solutions is not acceptable for practical applications. Herein, a significant improvement of electrode reversibility was achieved with highly concentrated electrolyte consisting of LiN(SO2F)2 and dimethyl carbonate. A practical problem when using high-viscosity concentrated electrolyte, i.e., non-wettability of concentrated electrolyte to polyolefin separators with small pores, was also effectively solved by using an aramid-coated polyolefin membrane. The improvement of reversibility for anionic redox associated with the suppression of oxygen loss, was clearly demonstrated by the results of X-ray absorption spectroscopy of nanosized Li1.14Ti0.29Mn0.57O2 with the concentrated electrolyte solution and aramid-coated separator. These findings have shown the future possibility of developing applications of high-energy batteries without ions of non-abundant nickel/cobalt.
To develop a sustainable society without depending on fossil fuels, it is necessary to effectively use renewable energy resources, a goal for which energy storage technology has a central role. Lithium-ion batteries are partly used for grid energy storage, but reducing the costs of these battery materials, especially for positive electrodes, is a key challenge. Currently, electric vehicles applications constitute the largest market for lithium-ion batteries, and Ni-based layered oxides are widely used in these applications.1–5 State-of-the-art lithium-ion batteries provide gravimetric/volumetric high-energy density, which in turn yield driving distances competitive with those achieved by internal combustion engine vehicles. However, electric vehicles with Ni-based layered materials are expensive,6 and therefore developing cost-effective electrode materials is necessary.
For this purpose, Mn-based layered materials containing excess Li, namely Li2MnO3 and its derivatives, have been extensively studied as potential cost-effective positive electrode materials.7–11 The importance of these electrode materials is also found in anionic redox, in addition to cationic redox used for conventional positive electrode materials. Large reversible capacities are also obtained by using anionic redox. For electrode materials made of Li2MnO3 and its derivatives, the Fermi level mainly consists of oxygen 2p orbitals, which have a non-bonding nature associated with a linear Li–O–Li local environment.12,13 After the oxidation, oxygen in the Li–O–Li environment becomes energetically destabilized, but then can become restabilized as a result of electron donation from Mn t2g orbitals and the formation of π-type bonding.14 However, oxygen dimerization also progresses when the concentration of oxidized oxygen is increased.15 Oxygen dimerization is a partially reversible process, but gradual oxygen loss cannot be avoided; hence, thus far, Mn-based layered oxides have not found practical use for battery applications.
Recently, the concept of anionic redox has been extended to non-layered oxides, i.e., oxides with a cation-disordered rock salt structure.16–19 Electrode materials with the disordered rock salt structure were thought to be electrochemically inactive due to the absence of any Li migration path. However, these oxides have shown formation of percolative Li migration paths and are hence also used as electrode materials.20 Because the percolation probability is increased with site concentration of Li ions in host structures, the electrode kinetics are significantly improved by the enrichment of Li in host structures. In addition, disordered structures provide rigid three-dimensional host structures without non-isotropic volume expansion as observed in layered oxides.21 A new functionality of disordered structures as electrode materials has been reported in recent publications.22
Anionic redox has been used to achieve electrode materials with a large reversible capacity, but inferior electrode reversibility for layered and non-layered oxides is an unsolved problem, which is partly due to the insufficient stability of electrolyte solvent against the highly reactive character for charged samples.18,23 Therefore, in this study, the impact of concentrated electrolyte solution was systematically examined. Having all the solvent molecules in the concentrated electrolyte solutions solvated to Li ions, and therefore electrons of the solvent molecules partly donated to positively charged Li ions,24 resulted in a HOMO energy level shift and better oxidative stability of electrolyte solvent. Indeed, much improved electrode reversibility in the concentrated electrolyte solution was achieved for a Mn-based oxide with excess Li, namely Li1.14Ti0.29Mn0.57O2, which delivered a large reversible capacity using anionic redox. Based on these results, the effectiveness of concentrated electrolyte for anionic redox reaction is discussed in detail.
Li1.14Ti0.29Mn0.57O2 was synthesized from a mixture of Mn2O3, TiO2 (anatase, 98.5%, Wako), and Li2CO3 (98.5%, Kanto Kagaku). Mn2O3 was synthesized by heating MnCO3 (Wako) at 850 °C in air. These powders were mixed by using a planetary ball mill (PULVERISETTE 7; FRITSCH) with a ZrO2 pot (45 mL) and balls at 300 rpm for 5 h with ethanol. The mixture was dried, and then pelletized and heated at 900 °C for 12 h in Ar. An XRD pattern and particle morphology of as-prepared Li1.14Ti0.29Mn0.57O2 are shown in Fig. 1a; the data were collected using an X-ray diffractometer (D2 PHASER, Bruker) equipped with a one-dimensional X-ray detector using Cu Kα radiation generated at 300 W (30 kV and 10 mA) with a Ni filter. Li1.14Ti0.29Mn0.57O2 formed a solid solution, with a makeup between those of Li2TiO3 and LiMnO2 (Li2TiO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LiMnO2 = 1:2 in a molar ratio). Using a scanning electron microscope (SEM, JCM-6000, JEOL) with an acceleration voltage of 15 keV, the as-prepared Li1.14Ti0.29Mn0.57O2 was observed to consist of several micrometer-sized particles with round shapes.
:LiMnO2 = 1:2 in a molar ratio). Using a scanning electron microscope (SEM, JCM-6000, JEOL) with an acceleration voltage of 15 keV, the as-prepared Li1.14Ti0.29Mn0.57O2 was observed to consist of several micrometer-sized particles with round shapes.
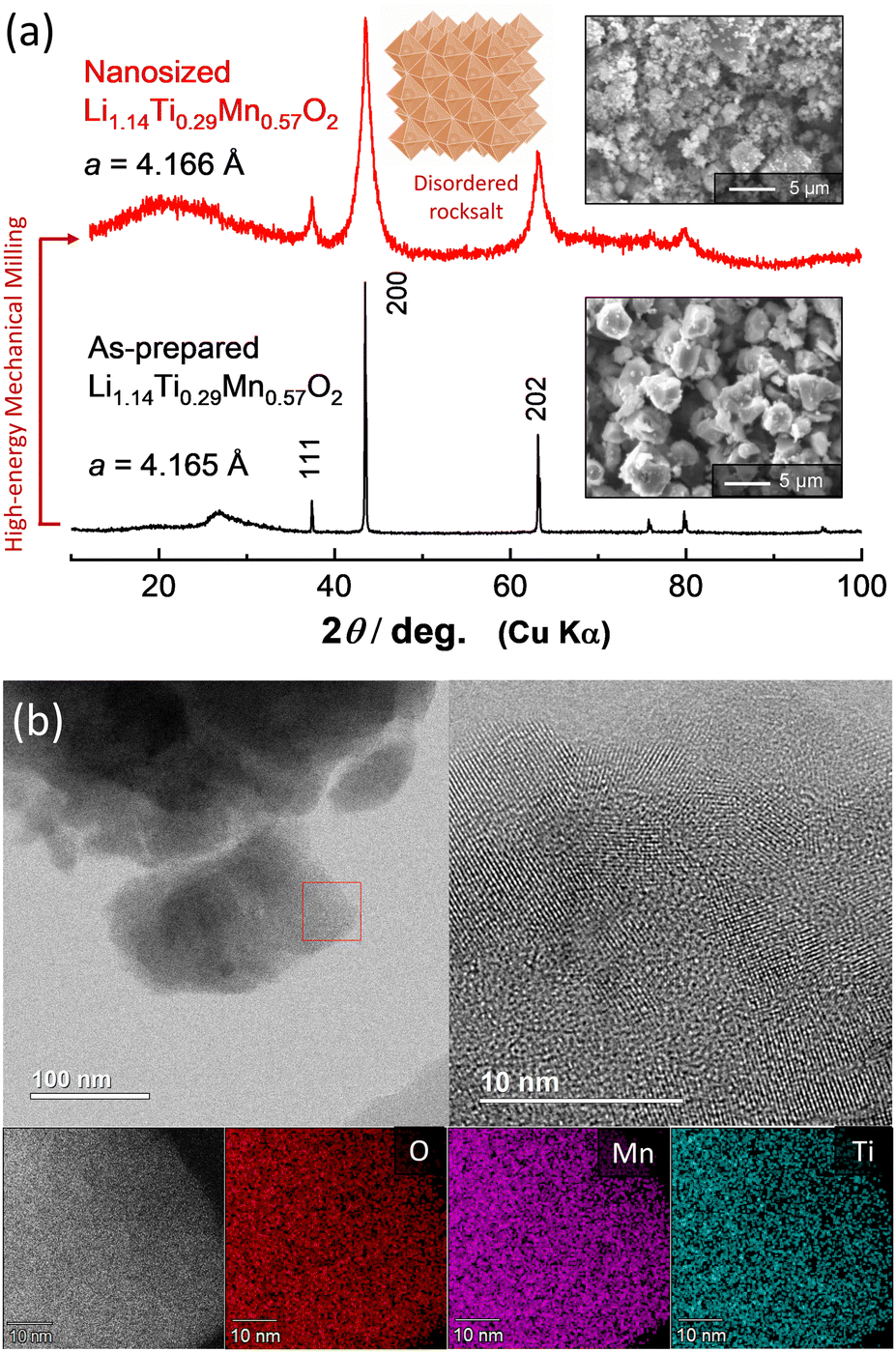 | ||
| Fig. 1 (a) XRD patterns and SEM images of as-prepared and nanosized Li1.14Ti0.29Mn0.57O2, and a schematic illustration of disordered rock salt structure drawn using the program VESTA.27 (b) STEM and EDX images of nanosized Li1.14Ti0.29Mn0.57O2. | ||
As-prepared Li1.14Ti0.29Mn0.57O2 was further subjected to high-energy mechanical milling using a planetary ball mill with a ZrO2 pot (45 mL) and balls at 600 rpm for 12 h. The ball-to-sample mass ratio was 20:1. After this milling, nanosized Li1.14Ti0.29Mn0.57O2 was successfully obtained, with the reduced particle size clearly shown in a comparison of SEM images for Li1.14Ti0.29Mn0.57O2 before and after milling. Moreover, peak widths of the corresponding XRD patterns were significantly broadened after milling, indicative of the domain size having been reduced to less than 10 nm. This hypothesis was directly supported by the scanning transmission electron microscopy (STEM) results shown in Fig. 1b and Figure S1 (ESI†). Nanosized Li1.14Ti0.29Mn0.57O2 was observed using a STEM apparatus (ARM200F, JEOL) with an acceleration voltage of 200 kV. Elemental maps were also collected by using a STEM apparatus equipped with an energy dispersive X-ray spectrometer (EDX, JED-2300T, JEOL) with a resolution of 256 × 256 pixels. Nanosized Li1.14Ti0.29Mn0.57O2 was observed to consist of nanocrystalline domains with dimensions of less than 10 nm, which is a common feature for samples produced using high-energy mechanical milling.25,26 STEM imaging with EDX spectroscopy also revealed that Mn and Ti ions were uniformly dispersed throughout each individual particle. Phase segregation was not evidenced for the nanosized Li1.14Ti0.29Mn0.57O2 subjected to the high-energy milling.
Electrochemical properties of as-prepared and nanosized Li1.14Ti0.29Mn0.57O2 were examined in Li cells with different electrolyte solutions. The electrode performances of the samples were assessed using two-electrode-type cells (TJ-AC, Tomcell Japan). Electrode performance of carbon composite samples was examined. A slurry consisting of 80 wt% active material, 10 wt% acetylene black (AB, HS-100, Denka), and 10 wt% poly(vinylidene fluoride) was made, mixed, and pasted on an aluminum foil. Metallic lithium (Honjo Metal) was used as a negative electrode. The electrolyte solution used was 1.0 mol dm−3 LiPF6 dissolved in ethylene carbonate:dimethyl carbonate (EC:DMC = 3:7 by volume) (battery grade, Kishida Chemical). The as-prepared sample was observed to be electrochemically less active, and a reversible capacity of only 25 mA h g−1 was obtained at a rate of 10 mA g−1 (Fig. S2a, ESI†). The obtained nanosized powder was further mixed with 10 wt% AB using the planetary ball mill at 300 rpm for 12 h. Compared to the material before this mixing, the Li1.14Ti0.29Mn0.57O2 afterwards showed a reduced particle size, in the sub-micrometer range, and showed improved electronic conductivity when serving as an electrode material, leading to a larger reversible capacity of ∼200 mA h g−1 at the same rate (Fig. S2b, ESI†). The reversible capacity was even higher for nanosized Li1.14Ti0.29Mn0.57O2, and this sample showed a value of ∼280 mA h g−1 at 10 mA g−1 at room temperature (Fig. S2c, ESI†). Nanosized Li1.14Ti0.29Mn0.57O2 also delivered a reversible capacity of ∼250 mA h g−1 at 50 mA g−1 as shown in Fig. 2a, but with a capacity retention not acceptable for practical applications. The discharge capacity decreased to 130 mA h g−1 over the course of 50 cycles (Fig. 2b), and 43% of the initial reversible capacity was lost. The coulombic efficiency for the second cycle was observed to be 96%; while it gradually increased with increasing number of electrochemical cycles, it still only reached 98.5% at the 50th cycle.
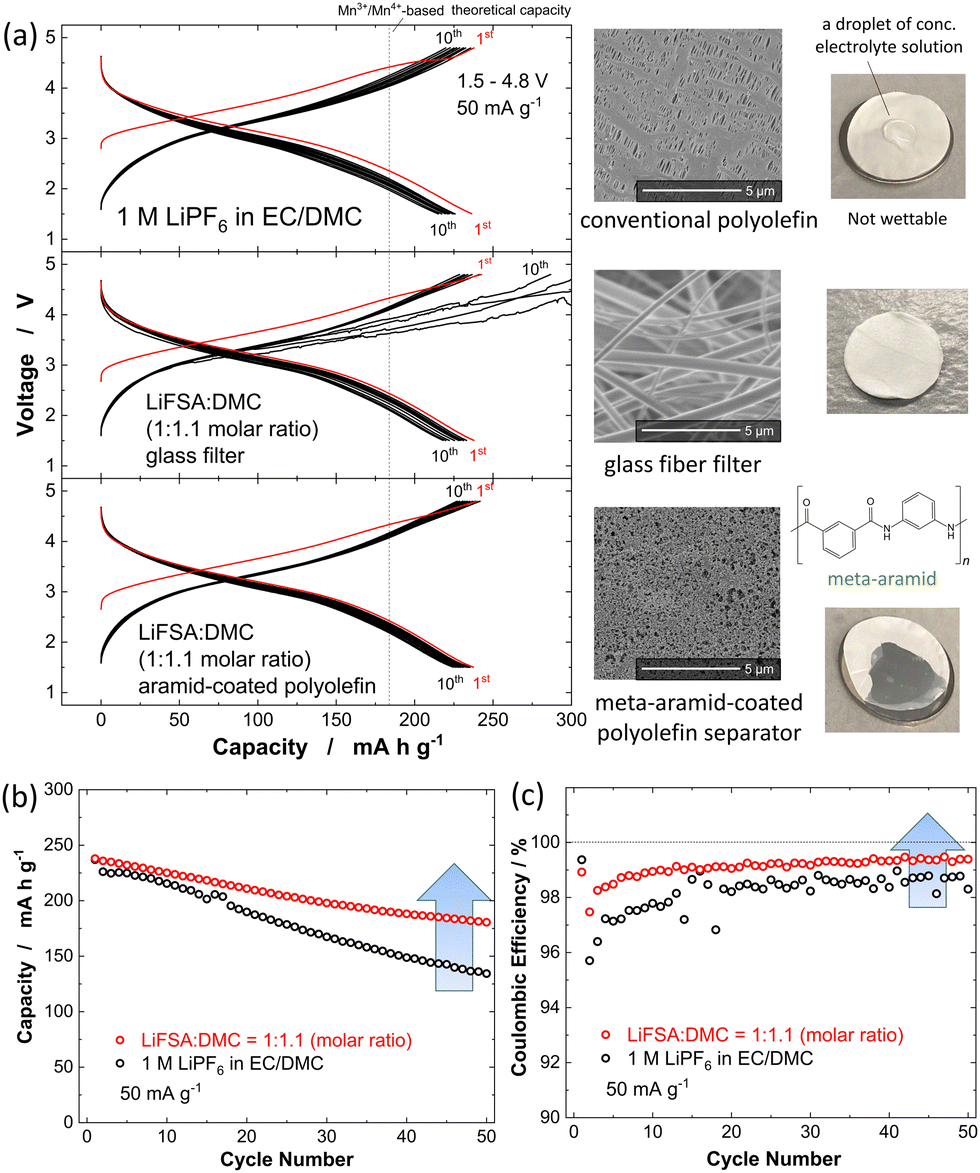 | ||
| Fig. 2 (a) Galvanostatic oxidation/reduction curves of nanosized Li1.14Ti0.29Mn0.57O2 at room temperature; with 1 M LiPF6 dissolved in EC/DMC at a rate of 50 mA g−1. (b) Capacity retention and (c) coulombic efficiency. The electrochemical data with HCE (LiFSA:DMC molar ratio of 1:1.1) are also shown. A glass fiber filter separator (GB-100R, Advantec, mass; 95 g m−2, porosity; 88.6%, and thickness; 0.38 mm), and aramid-coated polyolefin separator were used.29 Results with HCE and the aramid-coated separator are also shown. Electrode loadings were 2.53–4.64 mg cm−2, corresponding to the areal capacities of 0.63–1.16 mA h cm−2. | ||
To further improve electrode reversibility, highly concentrated electrolyte (HCE) with LiN(SO2F)2 (LiFSA) dissolved in DMC (LiFSA:DMC = 1:1.1 in a molar ratio) was used as the electrolyte solution.28 However, due to the overly high viscosity of HCE (238.9 mPa s at 30 °C),28 a conventional polyolefin separator with small pores was not wetted by HCE (Fig. 2a). A glass fiber filter with large pores is often used instead as a separator. But another problem was found: unstable voltage profiles were observed associated with the internal short circuit caused by growth of dendritic metallic lithium, which was used as a counter electrode for half-cell tests (Fig. 2a and Fig. S2d, ESI†). This growth appeared to be the critical problem when the areal capacity of the metallic lithium was increased to >1 mA h cm−2 with the glass fiber filter used in our study.29 Due to the ability of nanosized Li1.14Ti0.29Mn0.57O2 to deliver a reversible capacity of 280 mA h g−1, the maximum sample loading without the dendritic Li growth was expected to be only 3.57 mg cm−2, too low for practical applications (where >10 mg cm−2 is necessary).30 This issue was effectively solved by using HCE with an aramid-coated polyolefin separator. Polar functional groups in aramid effectively improved the wettability of HCE to the polyolefin membrane as shown in Fig. 2a. (The preparation of the aramid-coated polyolefin separator is described in the literature.29) Moreover, small pores of polyolefin separators effectively suppressed the dendritic Li growth, and a highly reversible cycling without internal shortage was achieved. A much improved capacity retention was achieved with HCE. A discharge capacity of 180 mA h g−1 after 50 cycles was observed, which corresponded to a 73% retention. Coulombic efficiency was also improved, and 99.2% efficiency was achieved at the 50th cycle. Rate capabilities of nanosized Li1.14Ti0.29Mn0.57O2 in two different electrolyte solutions were also compared (Fig. S3, ESI†). Despite the influence of the high viscosity for HCE on the ionic conductivity of electrolyte, similar rate capabilities were obtained for the two tested electrolyte solutions.
Conventional electrolyte and HCE with the aramid-coated separator yielded differences in electronic structures of nanosized Li1.14−yTi0.29Mn0.57O2. Soft X-ray absorption spectroscopy (XAS) data were collected at BL-11 (O K-edge and Mn LII, III-edges) in the synchrotron facility of Ritsumeikan University (Synchrotron Radiation Center). Absorption spectra were acquired using fluorescence yield mode. Samples for XAS measurements were handled in an Ar-filled glove box after disassembling electrochemical cells, and the electrode samples were transferred to the spectrometer without exposing them to air by using a laboratory-made transfer vessel. A study involving soft XAS at the Mn L-edge revealed, for both tested solutions, an initially trivalent oxidation state of Mn ions in nanosized Li1.14−yTi0.29Mn0.57O2, and a clear peak shift, attributed to oxidation of Mn ions into the tetravalent state. The different electrolytes yielded no clear difference for Mn ions after charge (Fig. 3a). In contrast, some differences were noted between their O K-edge XAS results, and a larger peak area was observed at 530 eV for the sample charged with HCE (Fig. 3b). A signal at 530 eV has been shown to be a specific feature for the activation of anionic redox and following oxygen dimerization.14,15 The observed differences between the O K-edge XAS data after charge were indicative of better stability of anionic redox for HCE. This trend was further supported by the Mn K-edge XAS results after discharge. Hard XAS was performed at beamline BL-12C of the Photon Factory Synchrotron Source in Japan. The composite electrodes were rinsed with DMC and sealed in a water-resistant polymer film in an Ar-filled glovebox. The Mn K-edge XAS signal was found in a slightly higher energy region for the sample charged in HCE. Moreover, the two electrolyte solutions yielded a clear difference between their discharge states (after charge to 4.8 V). The Mn K-edge XAS signal was observed to be at an energy position approximately 2.5 eV lower when using the conventional electrolyte than when using HCE (Fig. 3c). The thermal stability of electrode materials was further evaluated by performing differential scanning calorimetry (DSC; DSC-60 Plus, SHIMADZU) with a high-pressure-resistant container made from stainless steel. Fully charged (delithiated) samples, which were prepared by carrying out electrochemical oxidation in Li cells, were used. Electrolyte (0.75 μL) was added to 2.5 mg of samples. Containers were heated at 10 °C min−1 in a nitrogen atmosphere from room temperature to 400 °C. The DSC study revealed a significantly improved thermal stability of the fully charged sample when using HCE (Fig. 3d). Heat generation started from 150 °C with a sharp heat spike at 240 °C for conventional electrolyte. In contrast, a main heat spike was observed at 270 °C for HCE even though the two electrolyte solutions yielded similar total heat generation levels. Note that the onset temperature of electrolyte decomposition without the charged sample was also higher for HCE (Fig. S4, ESI†). These findings clearly provided evidence for the electrode reversibility of anionic redox being significantly improved and effectively stabilized by HCE.
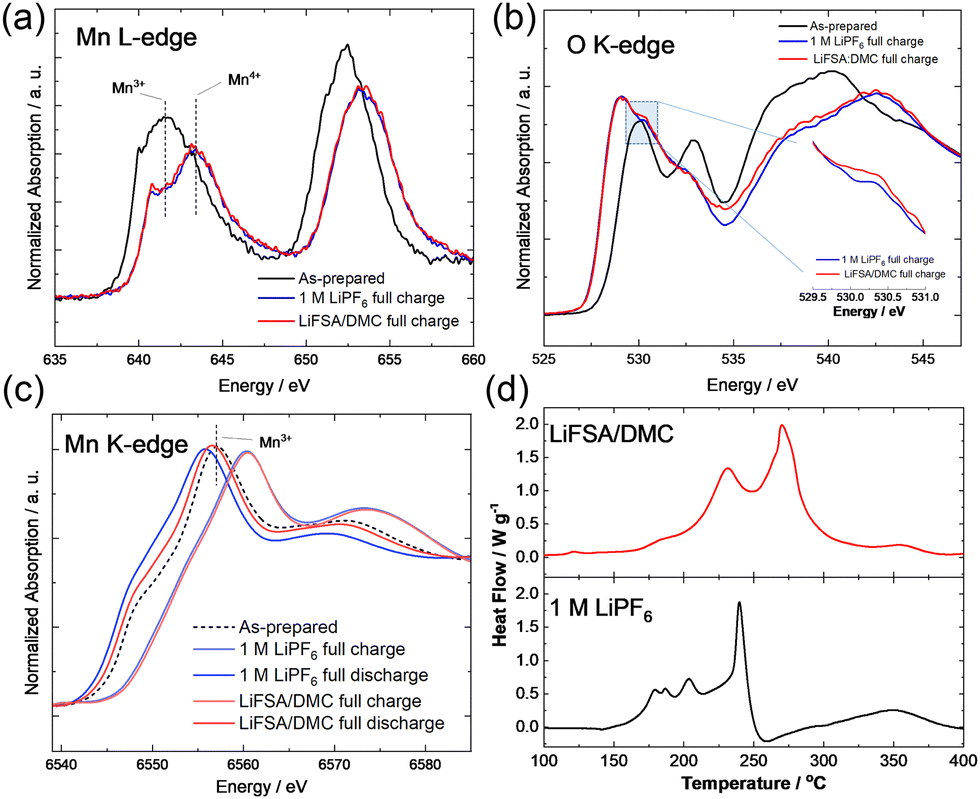 | ||
| Fig. 3 (a and b) Soft and (c) hard XAS spectra of nanosized Li1.14−yTi0.29Mn0.57O2 on charge/discharges. (d) DSC curves of charged nanosized Li1.14−yTi0.29Mn0.57O2 with different electrolyte solutions. | ||
In this study, nanosized Li1.14Ti0.29Mn0.57O2 has been studied as a Ni-/Co-free high-capacity and cost-effective electrode material for Li storage applications. Although a high energy density is obtained with Mn cationic redox coupled with O anionic redox, the reversibility of this electrode reaction with anionic redox is not high enough for a conventional electrolyte solution, which hinders its use for practical applications. Our study revealed a significantly improved electrode reversibility resulting from the use of HCE (LiFSA:DMC molar ratio of 1:1.1) coupled with the aramid-coated polyolefin separator, which effectively solved the problem of HCE with non-wettability to polyolefin separators. The superior tolerance to oxidative electrolyte decomposition for HCE without free solvent molecules was concluded to lead to the improved reversibility of electrode materials using anionic redox coupled with the partial suppression of oxygen loss. This finding has opened a new path to future designs of high-capacity electrode materials made from ions of abundant Mn and the further development of high-energy battery applications with improved reversibility for anionic redox through the optimized electrolyte solutions.
Author contributions
Nanaka Shimada: investigation, validation, writing – original draft. Yosuke Ugata: validation, writing – review & editing. Satoshi Nishikawa: validation. Daisuke Shibata: investigation. Toshiaki Ohta: investigation, validation. Naoaki Yabuuchi: conceptualization, validation, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NY acknowledges partial support from JSPS, Grant-in-Aid for Scientific Research (Grant Numbers 19H05816, 21H04698, and 21K18815). This work was partially supported by JST, CREST Grant Number JPMJCR21O6, Japan. The work involving hard X-ray absorption was performed under the approval of the Photon Factory Program Advisory Committee (proposal 2021G039). The experiment involving soft XAS was performed at the SR Center of Ritsumeikan University under the approval of the Program Review Committee (S22025). We thank Dr Yoshinobu Miyazaki and Dr Tomohiro Saito from Sumika Chemical Analysis Service, Ltd for the STEM observation.Notes and references
- S. H. Jung, U.-H. Kim, J.-H. Kim, S. Jun, C. S. Yoon, Y. S. Jung and Y.-K. Sun, Adv. Energy Mater., 2020, 10, 1903360 CrossRef CAS.
- C. Geng, D. Rathore, D. Heino, N. Zhang, I. Hamam, N. Zaker, G. A. Botton, R. Omessi, N. Phattharasupakun, T. Bond, C. Yang and J. R. Dahn, Adv. Energy Mater., 2022, 12, 2103067 CrossRef CAS.
- N. Ikeda, I. Konuma, H. B. Rajendra, T. Aida and N. Yabuuchi, J. Mater. Chem. A, 2021, 9, 15963–15967 RSC.
- C. Xu, S. Guan, L. Li, C. Sun, B. An and X. Geng, Coatings, 2021, 11, 932 CrossRef CAS.
- D. Weber, J. Lin, A. Pokle, K. Volz, J. Janek, T. Brezesinski and M. Bianchini, J. Electrochem. Soc., 2022, 169, 030540 CrossRef.
- L. Mauler, X. Lou, F. Duffner and J. Leker, Energy Adv., 2022, 1, 136–145 RSC.
- A. D. Robertson and P. G. Bruce, Chem. Commun., 2002, 2790–2791, 10.1039/B207945C.
- M. Oishi, K. Yamanaka, I. Watanabe, K. Shimoda, T. Matsunaga, H. Arai, Y. Ukyo, Y. Uchimoto, Z. Ogumi and T. Ohta, J. Mater. Chem. A, 2016, 4, 9293–9302 RSC.
- N. Yabuuchi, K. Kubota, Y. Aoki and S. Komaba, J. Phys. Chem. C, 2016, 120, 875–885 CrossRef CAS.
- M. Freire, O. I. Lebedev, A. Maignan, C. Jordy and V. Pralong, J. Mater. Chem. A, 2017, 5, 21898–21902 RSC.
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. McIntire, J. Hong, J. H. Song, Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J.-H. Park, S.-K. Doo, M. F. Toney, W. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 2091 CrossRef PubMed.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef CAS.
- T. Sudayama, K. Uehara, T. Mukai, D. Asakura, X.-M. Shi, A. Tsuchimoto, B. Mortemard de Boisse, T. Shimada, E. Watanabe, Y. Harada, M. Nakayama, M. Okubo and A. Yamada, Energy Environ. Sci., 2020, 13, 1492–1500 RSC.
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS PubMed.
- R. Fukuma, M. Harada, W. Zhao, M. Sawamura, Y. Noda, M. Nakayama, M. Goto, D. Kan, Y. Shimakawa, M. Yonemura, N. Ikeda, R. Watanuki, H. L. Andersen, A. M. D’Angelo, N. Sharma, J. Park, H. R. Byon, S. Fukuyama, Z. Han, H. Fukumitsu, M. Schulz-Dobrick, K. Yamanaka, H. Yamagishi, T. Ohta and N. Yabuuchi, ACS Cent. Sci., 2022, 8, 775–794 CrossRef CAS PubMed.
- Y. Kobayashi, M. Sawamura, S. Kondo, M. Harada, Y. Noda, M. Nakayama, S. Kobayakawa, W. Zhao, A. Nakao, A. Yasui, H. B. Rajendra, K. Yamanaka, T. Ohta and N. Yabuuchi, Mater. Today, 2020, 37, 43–55 CrossRef CAS.
- M. Sawamura, S. Kobayakawa, J. Kikkawa, N. Sharma, D. Goonetilleke, A. Rawal, N. Shimada, K. Yamamoto, R. Yamamoto, Y. Zhou, Y. Uchimoto, K. Nakanishi, K. Mitsuhara, K. Ohara, J. Park, H. R. Byon, H. Koga, M. Okoshi, T. Ohta and N. Yabuuchi, ACS Cent. Sci., 2020, 6, 2326–2338 CrossRef CAS.
- R. J. Clément, Z. Lun and G. Ceder, Energy Environ. Sci., 2020, 13, 345–373 RSC.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed.
- R. Qi, I. Konuma, B. D. L. Campéon, Y. Kaneda, M. Kondo and N. Yabuuchi, Chem. Mater., 2022, 34, 1946–1955 CrossRef CAS.
- N. Yabuuchi, Curr. Opin. Electrochem., 2022, 34, 100978 CrossRef CAS.
- J. Lee, J. K. Papp, R. J. Clément, S. Sallis, D.-H. Kwon, T. Shi, W. Yang, B. D. McCloskey and G. Ceder, Nat. Commun., 2017, 8, 981 CrossRef.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS.
- K. Motohashi, A. Nasu, T. Kimura, C. Hotehama, A. Sakuda, M. Tatsumisago and A. Hayashi, Electrochemistry, 2022, 90, 067009 CrossRef CAS.
- R. Qi, B. D. L. CampeOn, I. Konuma, Y. Sato, Y. Kaneda, M. Kondo and N. Yabuuchi, Electrochemistry, 2022, 90, 037005 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- Y. Ugata, C. Motoki, S. Nishikawa and N. Yabuuchi, Energy Adv., 2023 10.1039/D2YA00359G.
- S. Chen, C. Niu, H. Lee, Q. Li, L. Yu, W. Xu, J.-G. Zhang, E. J. Dufek, M. S. Whittingham, S. Meng, J. Xiao and J. Liu, Joule, 2019, 3, 1094–1105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00066d |
| This journal is © The Royal Society of Chemistry 2023 |