
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembly of chemically modified protein nanocages into 3D materials for the adsorption of uremic toxins†
Hendrik
Böhler
a,
Setareh
Orth-Alampour
b,
Constance
Baaten
bc,
Maria
Riedner
d,
Joachim
Jankowski
b and
Tobias
Beck
 *ae
*ae
aUniversität Hamburg, Department of Chemistry, Institute of Physical Chemistry, Grindelallee 117, Hamburg 20146, Germany. E-mail: tobias.beck@chemie.uni-hamburg.de
bUniversitätsklinikum Aachen, Institute for Molecular Cardiovascular Research IMCAR, Pauwelsstraße, 30, Aachen 52074, Germany
cMaastricht University, Department of Biochemistry, Cardiovascular Research Institute Maastricht, Maastricht 6229 ER, The Netherlands
dUniversität Hamburg, Technology Platform Mass Spectrometry, Mittelweg 177, Hamburg 20148, Germany
eThe Hamburg Centre of Ultrafast Imaging, Hamburg, Germany
First published on 5th December 2022
Abstract
Hemodialysis fails to remove protein-bound uremic toxins that are attributed with high cardiovascular risk. Application of adsorption materials is a viable strategy, but suitable biocompatible adsorbents are still not available. Here, we demonstrate that adsorbents based on the bottom-up assembly of the intrinsically biocompatible protein cage ferritin are applicable for toxin adsorption. Due to the size-exclusion effect of its pores, only small molecules such as uremic toxins can enter the protein cage. Protein redesign techniques that target selectively the inner surface were used to introduce anchor sites for chemical modification. Porous crystalline adsorbents were fabricated by bottom-up assembly of the protein cage. Linkage of up to 96 phenylic or aliphatic molecules per container was verified by ESI-MS. Materials based on unmodified ferritin cages can already adsorb the uremic toxins. The adsorption capacity could be increased by about 50% through functionalization with hydrophobic molecules reaching 458 μg g−1 for indoxyl sulfate. The biohybrid materials show no contamination with endotoxins and do not activate blood platelets. These findings demonstrate the great potential of protein-based adsorbents for the clearance of uremic toxins: modifications enhance toxin adsorption without diminishing the biocompatibility of the final protein-based material.
About 50% of all fatalities in dialysis patients with end-stage CKD can be attributed to cardiovascular events linked to impaired kidney function.1 In recent years, many risk factors promoting cardiovascular diseases have been linked to protein-bound uremic toxins (PBUT).2,3 In clinical trials, it was shown that the PBUT p-cresyl sulfate (pCS) is associated with increased risk for cardiovascular and all-cause mortality in CKD patients.4–6 Additionally, pCS and other PBUTs such as indoxyl sulfate (IS) can induce kidney and renal fibrosis,3,7 inhibit endothelial proliferation,8 induce oxidative stress,9 and vascular calcification.6,10 Due to their hydrophobic character, a high fraction of the PBUTs is bound to plasma proteins, for example serum albumin, preventing an efficient diffusion through the pores of the dialysis membranes.11,12 Therefore, even with prolonged and more frequent sessions, common membrane-based dialysis techniques fail to sufficiently clear PBUTs from the blood.13 Several techniques for improving hemodialysis are under investigation, including the use of binding competitors,14 oral adsorbents,15 or synbiotika.16 A promising alternative method is the application of extracorporeal adsorbents.17 For easy and cost-effective broad-scale application, the adsorbent needs to possess a high bio- and hemocompatibility, allowing full-blood contact and simple integration in existing dialysis treatments.18 Some adsorption materials have been investigated for their ability of binding PBUTs such as zeolites,19,20 carbon-based materials,21 or zirconium-based metal–organic frameworks (MOF).22 However, only a few studies address the challenges in bio- and hemocompatibility during the application of these materials. Even if they do, the focus is on increasing the biocompatibility of conventional adsorption materials as recently demonstrated for activated charcoal.18,23 Instead of improving the biocompatibility of conventional adsorbents, starting with intrinsically biocompatible materials and increasing their binding affinity towards PBUTs is a promising alternative strategy. Here we report an unprecedented approach for the fabrication of a heterogeneous adsorbent for PBUTs. It is based on a bottom-up assembly strategy of protein cages with high intrinsic biocompatibility. We aim on answering two main questions. First: Is it possible to create a solid porous protein-based material, which can be applied to the adsorption of PBUTs? Secondly, can the binding capacity of such a material be improved by chemical modification, without diminishing its biocompatibility?
For the generation of a protein-based adsorption material, the protein cage ferritin was chosen. In this work, apoferritin variants, that is protein cages without the iron oxide core, are used for the formation of the materials. To make this publication more readable, these variants are referred to as ferritin. Ferritin is composed of 24 identical subunits, which self-assemble to form a nearly spherical quaternary structure with an outer diameter of 12–13 nm and an inner cavity of 6–8 nm.24 Its cage-like architecture makes it well-suited for the fabrication of functional adsorbents due to its inherent large surface area, in contrast to native globular proteins such as human serum albumin. Formation of macroscopic materials from the soluble protein cages allow the easy separation of the adsorbent and the patient blood in later applications. Furthermore, crystalline ferritin assemblies or other protein crystals have already reported to trap proteins in vitro25 and in vivo26 as well as inorganic materials27 or a combination of organic and inorganic components.28 In this study, a variant of the human heavy chain ferritin with negatively charged outer surface was utilized.29 It differs from the native variant by four additional glutamic acids, which can help to prevent the adsorption of negatively charged plasma proteins such as albumin. For ferritin, small-sized pores (3–5 Å)30 in the protein shell allow only small molecules such as PBUTs to enter the cage, while larger macromolecules or even cells are excluded. Due to this size-exclusion effect, the inner cavity can be selectively modified, for example, by conjugation of guest molecules without affecting the biocompatibility of the system. The covalent linkage of hydrophobic guest molecules to the inner surface can enhance the affinity towards toxins. Since these modifications are restricted to the inner surface, they should not affect the assembly or the biocompatibility of the protein cages. Strategies to decorate the inner surface with hydrophobic guest molecules are discussed in the following section.
We hypothesized that the introduction of hydrophobic ligands into the inner cavity could enhance the PBUT adsorption. Since it was not clear which type of ligand would benefit the adsorption, we designed a modular material to allow easy adaptation. This was achieved by the introduction of a generic site for modification, namely incorporating cysteine residues that are amendable through their thiol group as an anchor site for chemical derivatization. On the other hand, native cysteine residues were exchanged with alanine residues to prevent modification at undesired positions. For the introduction of cysteine residues on the cavity surface, important design criteria included the distance between the sites and the solvent-accessible surface area (SASA). A high value of SASA should correspond to a high reactivity. To this end, ferritin variants were designed with three and four introduced cysteines per subunit, named Ftn(neg)-3xCys and Ftn(neg)-4xCys (for respective SASA and position see Table S5 and Fig. S1, ESI†). The total number of anchor sites per assembled protein cage is 72 or 96. The mutations were introduced in the Ftn(neg) gene with mutagenesis techniques and the variants were overexpressed in E. coli bacteria. Purification followed closely the published protocol for Ftn(neg).27 No notable change in elution behavior during ion-exchange chromatography (IEC), (Fig. S2a, b and c, ESI†) and size-exclusion chromatography (SEC) (Fig. S2d, ESI†) compared to the parental protein is observed. The introduction of the desired mutations was verified by electrospray ionization mass spectrometry (ESI-MS), where the measured mass was found to fit the calculated mass (Table 1 and Fig. S6, ESI†).
For chemical modification of cysteine thiol-groups, the α-halocarbonyls 2-iodo-N-phenyl acetamide (Phe) and 2-bromo-N-decyl acetamide (C10) were used. The overall strategy for chemical modification of the inner surface and subsequent assembly yielding a heterogeneous material is summarized in Fig. 1. First, the cage is disassembled into its subunits under acidic conditions.31 In the disassembled state, the thiol groups are readily accessible for the halogen acetamide derivates. The agents themselves are not soluble in an aqueous solution. Therefore, the functionalization is done in a solution containing high fractions of ethanol to facilitate the solubility of the hydrophobic molecules. After incubation of the reaction partners, the mixture is diluted, and the cage is allowed to reassemble. Purification by SEC shows a similar elution volume as the unfunctionalized cage (Fig. 1b and Fig. S3, ESI†), indicating a complete reassembly of the cage. This finding is confirmed in the negative-stained TEM images, which show intact cage structures (Fig. S4, ESI†). The SEC of functionalized Ftn(neg)-4xCys shows a slight shift towards higher elution volume linked to a slightly increased size, which is in good agreement with the results from dynamic light scattering (DLS) measurements (Table 1). To verify the successful and complete functionalization of the cysteine anchor sites, ESI-MS measurements were conducted. For the first trials, multiple mass peaks per charged species could be detected (Fig. S5, ESI†). Each of them could be attributed to the ferritin subunit with one to four of the desired molecules attached, indicating that a mixture of different degrees of functionalization is present in the sample. Fine-tuning of the reaction conditions, especially the fraction of ethanol in the mixture, the ratio of reactive molecules to cysteines, and the addition of a reducing agent (TCEP) enabled reproducible and complete functionalization at all sites, proven by ESI-MS as seen in Table 1 and Fig. S7 (full spectrum Fig. S7 and S8, ESI†). Derivatization was also shown with X-ray crystallography of the functionalized ferritin (Fig. 1c and Fig. S9, ESI†). Strategies for the assembly of the protein cages to macroscopic materials are summarized in the next section.
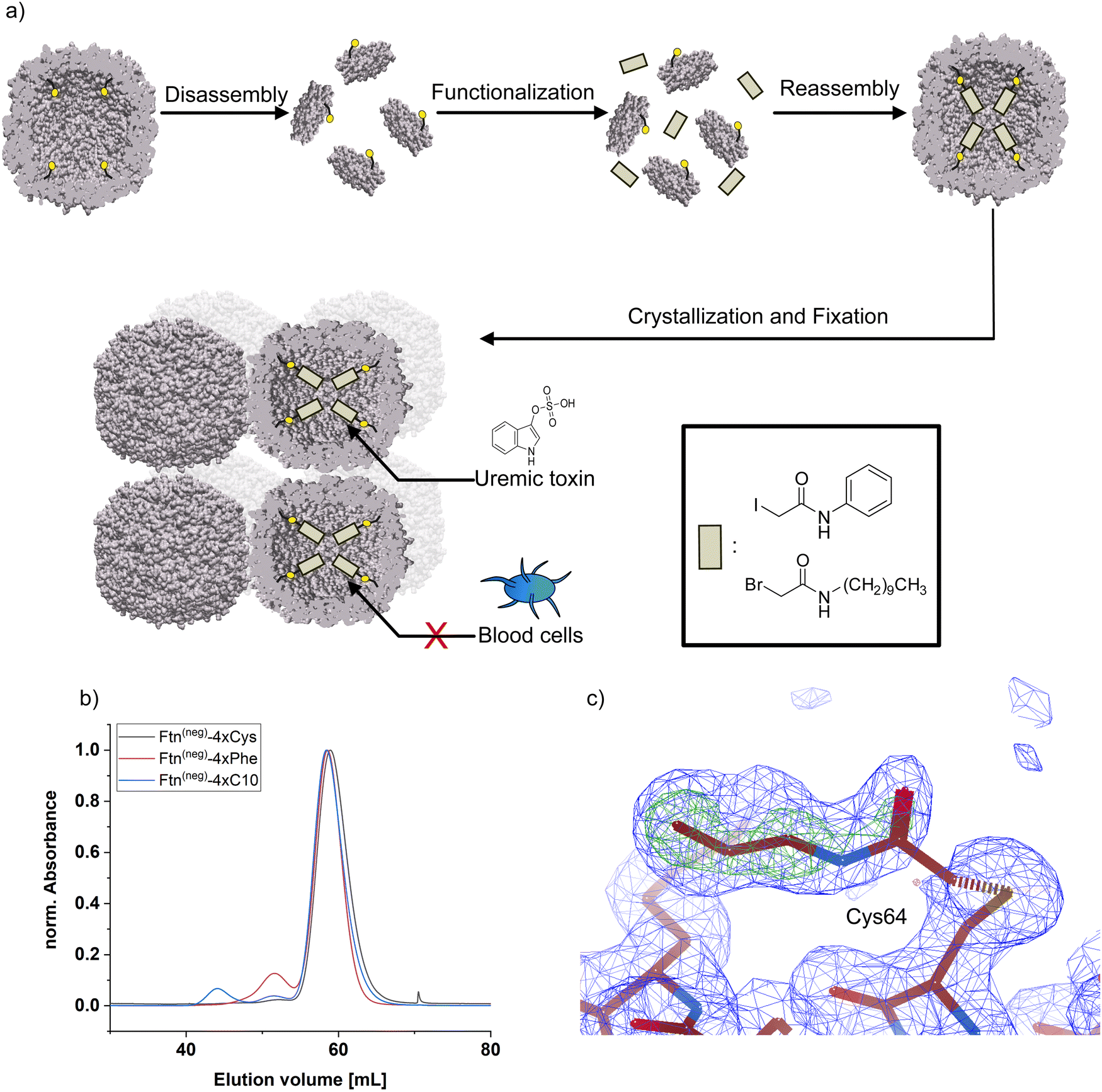 | ||
| Fig. 1 (a) Schematic overview of the strategy for protein functionalization. Protein cage is disassembled into subunits and incubated with respective ligand. After reassembly, cages self-assemble into 3D-material and are chemical fixated. (b) Results from size-exclusion chromatography of ferritin before and after functionalization. (c) Electron density (2FO–FC omit map, blue) and difference electron density (FO–FC, green) map for a functionalized cysteine residue with the (truncated) aliphatic molecule. 2FO–FC map (blue): 1 rmsd, FO–FC (green): 5 rmsd. Maps were calculated without ligand atoms. | ||
By self-assembly of the protein cages, a well-defined crystalline material can be created with uniformly distributed solvent channels. We envisioned that this material structure ensures high purity of the material, comparability of different modified derivates and accessibility of the cavities inside the material. Assembly into a macroscopic material with the protein cage as a building block is achieved by a batch crystallization technique. The general procedure is schematically shown in Fig. 2. The protein solution is cautiously mixed under constant shaking while adding the precipitant solution. After approximately 24 h, first crystals can be observed as shown in the left panel in Fig. 2. The size of the crystals can be tuned by altering protein and precipitant concentration (Fig. S11, ESI†). The protein forms a cubic lattice in the space group P23 and possesses uniformly distributed solvent channels with a diameter of around 3 nm at its narrowest part (Fig. S12, ESI†).29 To increase the stability, the crystals were fixated with a cross-linking agent. First experiments were conducted with glutaraldehyde. However, during stability tests in a 60 mg mL−1 BSA solution, chosen to simulate the high protein content of the blood, the crystals dissolve (Fig. S13, ESI†). After an additional cross-linking step, the crystals tolerate the condition but the respective adsorption capacity is greatly diminished, which is probably due to the formation of oligomeric glutaraldehyde blocking the pores. Therefore, we chose a sulfo-SMCC cross-linker, which is unable to undergo reactions with itself (Fig. 2), allowing stable fixation by maintaining adsorption capacity. The sulfo-SMCC linker bears a maleimide group and an activated ester group. At the pH value of the mother liquor of pH 8.5, both groups can undergo reactions with the primary amine group of the amino acid lysine. The resulting crosslinks between different protein cages stabilize the crystal and increase its tolerance against aqueous solutions with high concetrations of BSA. Macroscopic crystalline materials from the functionalized variants could be fabricated under the same conditions, indicating that the functionalization did not influence the outer surface.
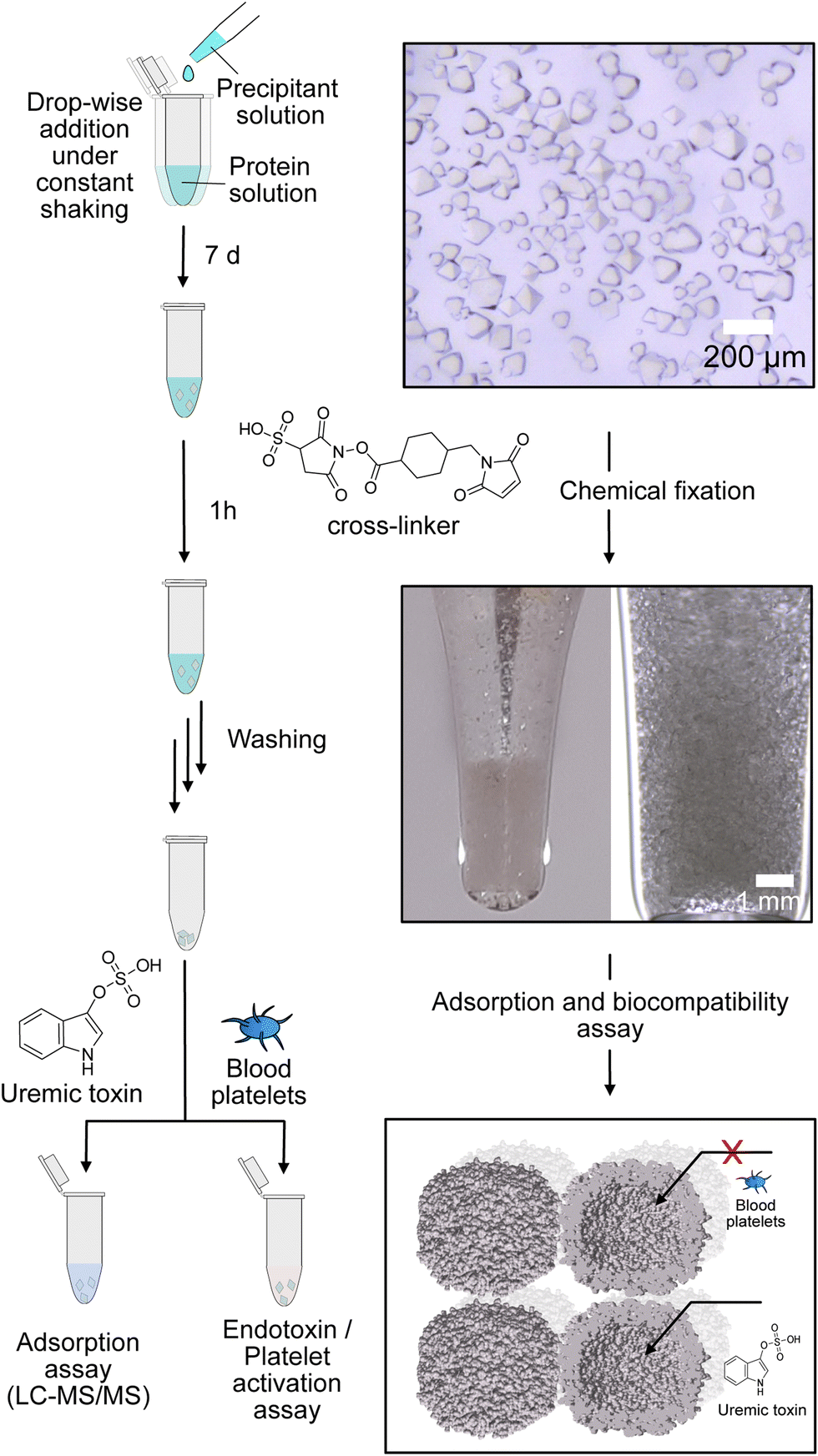 | ||
| Fig. 2 Schematic overview of batch crystallization technique for fabrication of protein-based adsorption materials, images of the material before and after fixation and schematic depiction of the size-exclusion effect for excluding blood platelets from the inner cavity. | ||
Subsequently, PBUT adsorption assays with the unfunctionalized protein-based material and the material functionalized with aliphatic or phenylic molecules were carried out. To this end, the respective sample was incubated in solutions of three different uremic toxins indoxyl sulfate (IS), p-cresyl sulfate (pCS), and phenylacetic acid (PheAc). Here, toxin concentrations expected in an end-stage CKD patient were used.10,32,33 After incubation for 3 h, the PBUT concentration in the supernatant and respective control samples were determined by HPLC-MS/MS techniques. Finally, the protein-based material is vacuum-dried and weighed to determine the mass. The adsorption capacity, which is the ratio of the adsorbed mass of PBUT to the total mass of the material, is calculated and shown in Fig. 3.
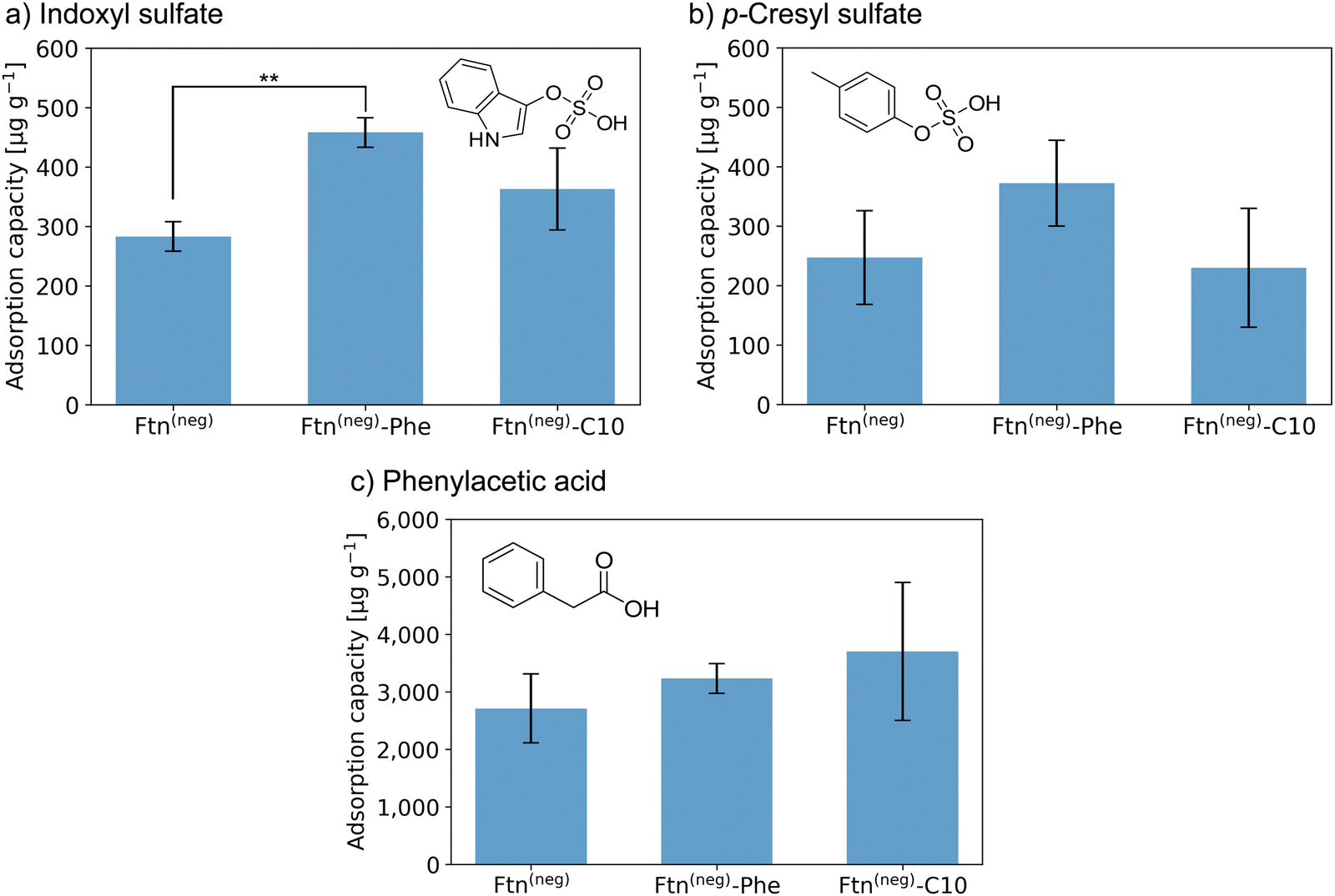 | ||
Fig. 3 Adsorption capacity of crystalline protein-based adsorber material composed of different ferritin variants towards (a) indoxyl sulfate (IS), (b) p-cresyl sulfate (pCS), and (c) phenylacetic acid (PheAc). All measurements were performed in a triplicate. The error bar represents the deviation in adsorption capacity between the three measurements. Confidence values for comparison to an untreated control sample can be found in Fig. S14 (ESI†). In addition, two-sample t-test were performed to determine if the improvement of adsorption capacity of the functionalized ferritin is significant in comparison to the unmodified sample Ftn(neg). Except the indicated sample (**P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ≤ 0.01), all changes were not significant (P ≥ 0.05). Starting concentrations were as follow: IS 44 mg L−1; pCS 41 mg L−1; PheAc 474 mg L−1. ≤ 0.01), all changes were not significant (P ≥ 0.05). Starting concentrations were as follow: IS 44 mg L−1; pCS 41 mg L−1; PheAc 474 mg L−1. | ||
Crystals of the unfunctionalized protein Ftn(neg) adsorb all three tested toxins as seen in Fig. 3a–c with a capacity of 283 and 247 μg g−1 for IS and pCS, respectively, and 2710 μg g−1 for PheAc. The reason for the much higher capacity of the PheAc adsorption is most likely the 10-fold higher concentration of the PBUT in the assay (see above). For IS and pCS, functionalization with phenylic molecules (Phe) leads to an increase in adsorption capacity to 458 and 372 μg g−1, respectively. However, based on statistical tests (see Fig. 3 and caption), only the capacity improvement for Ftn(neg)-Phe with IS is significant. For functionalization with the aliphatic molecules (C10), the improvement of adsorption capacity is rather small (Fig. 3a and b). For the toxin PheAc, no significant enhancement of the adsorption could be observed after the incorporation of the hydrophobic molecules (Fig. 3c). The capacities of the protein-based adsorbent are in a similar regime but smaller than the values of other published materials, for example the P87 zeolites with capacities of up to 1000 μg mL−134 or carbon-based adsorbents with capacities of up to 3200 μg mL−135 with respect to IS. To the best of our knowledge, the adsorbent with the highest capacity published so far is a zirconium-based MOF with a capacity of up to 156 mg g−1.22 However, efforts to install their biocompatibility need to be taken, which can further affect the adsorption capacity. On the other hand, our protein-based material approaches the problem from the other side: it has intrinsic biocompatibility, but its adsorption capacity can be further improved. Regarding this, capacities in the range of several hundred ug mL−1 are a promising starting point for future work in this direction.
First investigations in the direction of biocompatibility were promising by proving that there are no residual endotoxin contaminations left from the bacterial production strain. Furthermore, no activation of blood platelets upon incubation with the crystalline adsorbent could be detected (Fig. S15 and S16, ESI†). Importantly, more in-depth investigations to prove complete hemocompatibility need to be performed for future application of this new material class.
In conclusion, we demonstrated the synthesis of macroscopic materials based on the bottom-up assembly of protein cages for blood purification applications. The resulting material shows stability and adsorption of three PBUTs. Furthermore, it was demonstrated that through the introduction of anchor sites, up to 96 water-insoluble aliphatic and phenylic molecules were incorporated inside the cavity of the ferritin protein cage. No decrease in biocompatibility could be found after these modifications. An increase in adsorption capacity due to the chemical modifications is observable. However, exclusively hydrophobic ligands are probably not optimal, since PBUTs possess hydrophobic and hydrophilic characteristics. Because the aliphatic and phenolic agents used in this study were extreme cases with regard to hydrophobicity, this paves the road for further studies, with other ligands designed for binding uremic toxins. The modular character makes adaption for other applications possible, for example, treatment strategies for heavy metal intoxication by incorporation of chelating agents. Moreover, through genetic modifications, positively charged amino acids around the present anchor sites could be introduced to satisfy the negative charge of the PBUTs while the toxins’ hydrophobic part is bound to the inserted molecules.
Conflicts of interest
A patent application is currently being prepared.Acknowledgements
We thank Dr Made Budiarta for initial coupling assays, Dr Marcel Lach for adaption of the batch crystallization conditions, Gaby Graack for support with the HPLC-MS/MS system and Michael Rütten for general support. Furthermore, we thank Heidi Noels for help with the platelet isolation. This work was supported by a Liebig Scholarship to TB, the Deutsche Forschungsgemeinschaft (DFG) with project ID 401323995, by the Cluster of Excellence “CUI: Advanced Imaging of Matter“ of the Deutsche Forschungsgemeinschaft (DFG)–EXC 2056–project ID 390715994 and by a support within the major research instrumentation programme – project ID 413227073. We also thank the DFG research training group “Nanohybrid” (GRK 2536) for financial support.References
- G. Schlieper, K. Hess, J. Floege and N. Marx, Nephrol., Dial., Transplant., 2016, 31, 382–390 CrossRef PubMed.
- S. Ito and M. Yoshida, Toxins, 2014, 6, 665–678 CrossRef PubMed.
- S. Lekawanvijit, A. R. Kompa and H. Krum, Am. J. Physiol., 2016, 311, F52–F62 CAS.
- B. Bammens, P. Evenepoel, H. Keuleers, K. Verbeke and Y. Vanrenterghem, Kidney Int., 2006, 69, 1081–1087 CrossRef CAS PubMed.
- I. W. Wu, K. H. Hsu, H. J. Hsu, C. C. Lee, C. Y. Sun, C. J. Tsai and M. S. Wu, Nephrol., Dial., Transplant., 2012, 27, 1169–1175 CrossRef CAS.
- S. Liabeuf, et al. , Nephrol., Dial., Transplant., 2010, 25, 1183–1191 CrossRef CAS.
- C. Y. Sun, S. C. Chang and M. S. Wu, PLoS One, 2012, 7, e34026 CrossRef CAS PubMed.
- L. Dou, E. Bertrand, C. Cerini, V. Faure, J. Sampol, R. Vanholder, Y. Berland and P. Brunet, Kidney Int., 2004, 65, 442–451 CrossRef CAS.
- M. Yu, Y. J. Kim and D. H. Kang, Clin. J. Am. Soc. Nephrol., 2011, 6, 30–39 CrossRef CAS PubMed.
- F. C. Barreto, et al. , Clin. J. Am. Soc. Nephrol., 2009, 4, 1551–1558 CrossRef CAS.
- R. Vanholder, et al. , Kidney Int., 2003, 63, 1934–1943 CrossRef CAS.
- J. Jansen, J. Jankowski, P. R. Gajjala, J. F. M. Wetzels and R. Masereeuw, Clin. Sci., 2017, 131, 1631–1647 CrossRef CAS.
- T. L. Sirich, et al. , Kidney Int., 2017, 91, 1186–1192 CrossRef CAS.
- M. Madero, et al. , Clin. J. Am. Soc. Nephrol., 2019, 14, 394–402 CrossRef CAS PubMed.
- S. Yamamoto, et al. , Sci. Rep., 2015, 5, 14381 CrossRef CAS PubMed.
- C. Cosola, et al. , Toxins, 2021, 13, 334 CrossRef PubMed.
- V. Saar-Kovrov, W. Zidek, S. Orth-Alampour, D. Fliser, V. Jankowski, E. A. L. Biessen and J. Jankowski, J. Intern. Med., 2021, 290, 499–526 CrossRef CAS PubMed.
- M. Sternkopf, et al. , Toxins, 2019, 11, 389 CrossRef CAS PubMed.
- D. Bergé-Lefranc, C. Vagner, R. Calaf, H. Pizzala, R. Denoyel, P. Brunet, H. Ghobarkar and O. Schäf, Microporous Mesoporous Mater., 2012, 153, 288–293 CrossRef.
- V. Wernert, O. Schäf, H. Ghobarkar and R. Denoyel, Microporous Mesoporous Mater., 2005, 83, 101–113 CrossRef CAS.
- D. Pavlenko, D. Giasafaki, G. Charalambopoulou, E. van Geffen, K. G. F. Gerritsen, T. Steriotis and D. Stamatialis, Sci. Rep., 2017, 7, 14914 CrossRef CAS.
- S. Kato, K. I. Otake, H. Chen, I. Akpinar, C. T. Buru, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2568–2576 CrossRef CAS.
- S. R. Sandeman, et al. , Biomed. Mater., 2017, 12, 035001 CrossRef.
- P. D. Hempstead, S. J. Yewdall, A. R. Fernie, D. M. Lawson, P. J. Artymiuk, D. W. Rice, G. C. Ford and P. M. Harriso, J. Mol. Biol., 1997, 268, 424–448 CrossRef CAS PubMed.
- K. Han, Y. Na, L. Zhang and F. A. Tezcan, J. Am. Chem. Soc., 2022, 144, 10139–10144 CrossRef CAS PubMed.
- B. S. Heater, Z. Yang, M. M. Lee and M. K. Chan, J. Am. Chem. Soc., 2020, 142, 9879–9883 CrossRef CAS PubMed.
- B. Maity, S. Abe and T. Ueno, Nat. Commun., 2017, 8, 14820 CrossRef CAS PubMed.
- A. Shaukat, E. Anaya-Plaza, N. K. Beyeh and M. A. Kostiainen, Chem. – Eur. J., 2022, 28, e202104341 CAS.
- M. Künzle, T. Eckert and T. Beck, J. Am. Chem. Soc., 2016, 138, 12731–12734 CrossRef.
- T. Tosha, H. L. Ng, O. Bhattasali, T. Alber and E. C. Theil, J. Am. Chem. Soc., 2010, 132, 14562–14569 CrossRef CAS PubMed.
- M. Lach, C. Strelow, A. Meyer, A. Mews and T. Beck, ACS Appl. Mater. Interfaces, 2022, 14, 10656–10668 CrossRef CAS.
- J. Jankowski, et al. , J. Clin. Invest., 2003, 112, 256–264 CrossRef CAS.
- M. Hida, Y. Aiba, S. Sawamura, N. Suzuki, T. Satoh and Y. Koga, Nephron, 1996, 74, 349–355 CrossRef CAS PubMed.
- L. Lu and J. T. W. Yeow, Mater. Des., 2017, 120, 328–335 CrossRef CAS.
- D. Pavlenko, D. Giasafaki, G. Charalambopoulou, E. van Geffen, K. G. F. Gerritsen, T. Steriotis and D. Stamatialis, Sci. Rep., 2017, 7, 14914 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb02386e |
| This journal is © The Royal Society of Chemistry 2023 |