
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe heat-promoted metal–support interaction of a PtCu/SiO2 carbon-free catalyst for the methanol oxidation and oxygen reduction reactions†
Quanqing
Zhao
a,
Han
Zhi
b,
Liu
Yang
a and
Feng
Xu
 *ab
*ab
aCollege of Materials Science and Engineering, Fuzhou University, Fuzhou, 350108, China. E-mail: xufeng@fzu.edu.cn
bSchool of Advanced Manufacturing, Fuzhou University, Jinjiang, 362200, China
First published on 28th August 2023
Abstract
Pt shows excellent catalytic activity and acid stability towards the methanol oxidation reaction (MOR) and oxygen reduction reaction (ORR), but suffers from deactivation due to the weak interaction with the carbon support and the oxidation of carbon under operation. To solve the problem, herein, silica nanospheres were used to load PtCu nanoparticles as the carbon-free catalyst for the MOR and ORR. It is found that the heat treatment is crucial to regulate the metal–support interaction and enhance the catalytic performance. The PtCu/SiO2 catalyst after heat treatment exhibits a specific activity (SA) nearly 7 times the value of Pt/C toward the MOR, and much better durability. The MOR mass activity of heat-treated PtCu/SiO2 is over twice the value of the untreated one. The electron transfer promoted by heat treatment leads to an upshift of the d-band center of Pt, resulting in the increase of the absorption rate of methanol and the intermediates.
Sustainability spotlightDirect methanol fuel cells (DMFCs) are widely accepted as one of the promising green energy resources to replace fossil fuels and are now on their way to commercialization. Pt/C is the commonly used catalyst in DMFCs. The carbon support leads to deactivation during long-term operation. Moreover, carbon is a product of the petrochemical industry. In this work, SiO2 was studied as the non-carbon support to load PtCu alloy. The results showed better performance towards the methanol oxidation reaction and comparable activity towards the oxygen reduction reaction of the PtCu/SiO2. Hence, the activity and durability of the Pt-based catalyst were enhanced and the carbon consumption of DMFCs was further cut, promoting the sustainability of our world. |
Introduction
Platinum (Pt) shows excellent catalytic activity and acid stability towards the methanol oxidation reaction (MOR) and oxygen reduction reaction (ORR), and hence, is regarded as the state-of-the-art catalyst of direct methanol fuel cells (DMFCs).1 Carbon, widely used as the support for the Pt catalyst, possesses high electronic conductivity and large specific surface area,2 resulting in high catalytic activity of the Pt/C catalyst. But the catalytic activity of Pt/C decreases owing to the oxidation of the carbon support under the operating conditions such as high potential, frequent start–stop cycles, and strongly acidic environment, and the interaction between Pt and carbon will be undermined which causes Pt migration and aggregation.3 To address these issues, many researchers have focused on developing alternative support materials which interact strongly with metal nanoparticles and remain stable under operation.4Non-carbon support materials, such as titania, alumina, ceria, silica, and tungsten oxide, have received wide attentions recently.5 Most of the oxide materials are inert in an acidic environment and exhibit fascinating advantages over carbon. For example, the surficial groups (such as –OH) on silica promote the MOR through reacting with intermediate species (such as CO*), and reduce the impact of Pt poisoning.6 However, most non-carbon materials exhibit low conductivity which decreases the catalytic activity compared to the carbon support, due to the inherent wide bandgap. In the last decade, it has been revealed that tuning the metal–support interaction (MSI) is effective in tailoring the electronic structure and hence the catalytic performance. MSI engineering includes downsizing the metals, tuning the defects/interface and morphology, and so on.7–9 Fundamentally, the electron transfer involved in the MSI is crucial to the catalytic performance. During electron transfer, excess electrons tend to accumulate at the interface of the support and metal,10 which significantly enhances the adsorption and reactions of adsorbates,11 leading to improved activity and higher conductivity.12 The electron transfer from the support to Pt results in a downshift of the Pt d-band center and consequently weakens the adsorption of methanol and intermediate species,13,14 which promotes the catalytic activity and durability.15
Another urgent issue is the rarity and high cost of Pt, making the DMFCs expensive and hindering their commercialization.16 Using non-noble metals (such as Co, Ni, Cu) to form Pt based alloys is an effective way to solve the issue, and improve electrocatalytic activity.17 Lu18 and co-workers found that Cu could change the binding energy of Pt due to the strong electronic interaction between Pt and Cu. Cu donates electrons and the electron density increases around Pt sites, resulting in weaker chemisorption of intermediates.
Herein, we present a PtCu alloy catalyst supported on silica nanospheres. SiO2, an oxide widely used in industries, was herein studied as a non-carbon support to promote the MSI and hence the activities toward the methanol oxidation and oxygen reduction reactions. Through thermal treatments, the MSI was engineered and the d-band center of Pt was tuned, leading to enhanced MOR performance.
Results and discussion
Fig. 1A shows the X-ray diffraction (XRD) patterns of the synthesized Cu/SiO2 before and after heat treatment. The Cu/SiO2 before heat treatment exhibited diffraction peaks centered at 43.2°, 50.3°, and 73.8°, which were assigned to copper (JCPDS 04-0836). The Cu/SiO2 after heat treatment (HC300) showed not only copper diffraction signals, but also peaks at 36.5°, 42.4°, and 61.5°, relating to Cu2O (JCPDS 05-0667). The appearance of Cu2O was because the copper became activated and was slowly oxidized in the air after heat treatment.19 The peaks are steep with a narrow half-peak width and strong intensity, indicating a large average grain size, which is calculated to be about 50 nm using the Debye–Scherrer formula below:20,21D = Kλ/(β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) cosθ) | (1) |
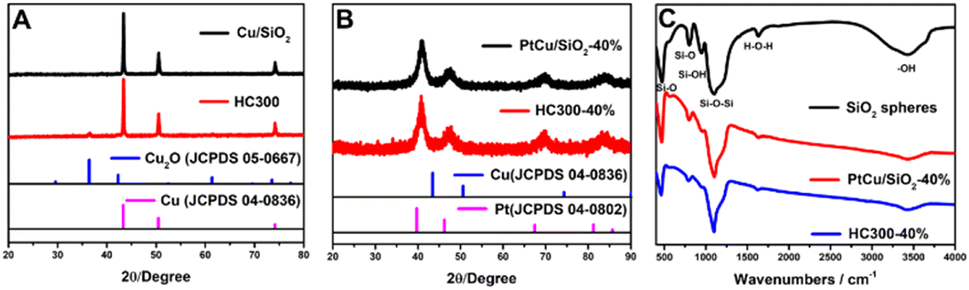 | ||
| Fig. 1 (A) XRD patterns of Cu/SiO2 and HC300; (B) XRD patterns of PtCu/SiO2-40% and HC300-40%; (C) FTIR spectra of SiO2 spheres, PtCu/SiO2-40% and HC300-40%. | ||
The Fourier transform infrared spectroscopy (FTIR) results of SiO2 nanospheres, PtCu/SiO2-40%, and HC300-40% are exhibited in Fig. 1C. The peaks centered at 800 and 471 cm−1 represent the symmetrical stretching vibration and bending vibration of Si–O. The peaks centered at 949 cm−1 represent the Si–O stretching vibrations of Si–O–H, revealing the rich silanol groups (Si–OH) on the SiO2 surface. The peaks at 1100 cm−1 represent the anti-symmetric stretching vibration of Si–O–Si.23 The peaks at 1633 cm−1 represent the bending or deformation mode of molecularly coordinated water and the peaks at 3424 cm−1 represent the stretching vibration of O–H that were part of the water molecule and Si–OH.24 These peaks were also observed on the other as-prepared catalysts (Fig. S2†). The Pt, Cu, and SiO2 contents were calculated based on the ICP analysis (Table S1†). The Pt, Cu, and SiO2 contents of PtCu/SiO2-40% were 58, 13, and 29 wt% respectively. After heat treatment, HC300-40% showed higher SiO2 content (43 wt%) and lower Pt and Cu contents (46.6 and 10.4 wt% respectively) than PtCu/SiO2-40%, but the Pt:Cu atomic ratio remained almost unchanged (about 1.46). PtCu/SiO2-30% showed higher Pt content but lower SiO2 and Cu contents after heat treatment, and the Pt:Cu atomic ratio became larger (increasing from 0.81 to 1.36). The differences in the content changes on PtCu/SiO2-30% and PtCu/SiO2-40% before and after heat treatment demonstrated that the PtCu alloy of PtCu/SiO2-40% was more stable than that of PtCu/SiO2-30%.
The microstructures of the SiO2 nanospheres, PtCu/SiO2-40% and HC300-40% were investigated using a scanning electron microscope (SEM) and transmission electron microscope (TEM). The solid SiO2 nanospheres were mainly around 100 nm in diameter (Fig. 2A and B). The TEM and EDS images of PtCu/SiO2-40% showed the metal nanoparticles intimately attached on SiO2 nanospheres, thus the PtCu alloy interacted strongly with SiO2 nanospheres (Fig. 2C–G). The metal nanoparticles were mainly 5 nm in diameter (Fig. 2H) and a metal lattice spacing of 0.22 nm was observed, relating to the PtCu (111) facet (Fig. 2I). After heat treatment, most of the PtCu alloy was still loaded on the SiO2 surface and the morphology slightly changed probably because of the heat treatment (Fig. 2J). The measurements of the PtCu (111) crystal plane showed that the spacing of the HC300-40% crystal plane turned into 0.225 and 0.247 nm respectively and the average spacing showed a value of 0.23 nm (Fig. 2K and L), showing that after heat treatment the lattice spacing increased. The elemental mapping (Fig. S3†) of HC300-40% demonstrated the homogeneous dispersion of Pt and Cu elements, and the intimate contact of PtCu and SiO2.
 | ||
| Fig. 2 (A) SEM image of SiO2 nanospheres; (B) TEM image of SiO2 spheres; (C–G) TEM and EDS images of PtCu/SiO2-40%; (H) magnification of a specific region of (C); (I) High resolution image of PtCu/SiO2-40%, showing the magnification of a specific region of (H); (J) TEM image of HC300-40%; (K and L) high resolution images of HC300-40%. | ||
X-ray photoelectron spectroscopy (XPS) was carried out to investigate the electronic structures of Si, Cu, and Pt. The Si 2p orbital of PtCu/SiO2-40% exhibited a peak at around 103.68 eV, and that of HC300-40% exhibited a peak at 103.48 eV (Fig. 3A). There was a negative B.E. shift of the Si 2p after heat treatment, suggesting an increased electron density of HC300-40%. The Cu 2p orbital of PtCu/SiO2-40% showed peaks at 931.88 and 951.68 eV, and that of HC300-40% showed peaks at 932.48 and 952.18 eV (Fig. 3B). The Cu 2p orbital of HC300-40% showed a noticeably positive B.E. shift, suggesting that heat treatment leads to a decreased electron density. The Pt 4f orbital of PtCu/SiO2-40% showed peaks at 71.38 and 74.58 eV, and that of HC300-40% showed peaks at 71.68 and 74.88 eV (Fig. 3C). There was also a positive shift in Pt 4f binding energy after heat treatment, suggesting a decreased electron density. The HC300-30% showed similar shifts of Pt 4f, Cu 2p and Si 2p orbitals after heat treatment compared to HC300-40% (Table S2†). The positive shifts of Pt 4f and Cu 2p and the negative shift of Si 2p revealed that even though Cu donated electrons to Pt, the electron density of Pt still decreased and the electrons transferred from PtCu to SiO2, which demonstrated a strong MSI.7
 | ||
| Fig. 3 XPS spectra of the Si 2p (A), Cu 2p (B), and Pt 4f (C) orbitals of PtCu/SiO2-30%, HC300-30%, PtCu/SiO2-40% and HC300-40%. | ||
The MOR activity of PtCu/SiO2-10%, PtCu/SiO2-20%, PtCu/SiO2-30%, and PtCu/SiO2-40% is compared in Fig. S3.† As the Pt loading increases, the activity rises and the PtCu/SiO2-40% shows much higher activity than the others; therefore, the discussion hereafter focuses on PtCu/SiO2-40% and HC300-40%. The ECSA was calculated from CV curves (Fig. 4A) using a hydrogen adsorption theory by assuming a monolayer adsorption of hydrogen atoms on the surface of metallic catalysts (0.21 mC cm−2).25 The ECSA of Pt/C (30.8 m2 gPt−1) was higher than that of PtCu/SiO2-40% (3.53 m2 gPt−1) and HC300-40% (4.4 m2 gPt−1) because the Pt nanoparticles of Pt/C exhibited smaller size (<5 nm) and better dispersion, and the electronic conductivity of the carbon support is higher.26 The Nyquist plot illustrated that although the electronic resistance of SiO2 nanospheres was higher than that of carbon, it was low enough as a support (Fig. S4†). The PtCu/SiO2-40% demonstrated even lower resistance than SiO2, and the resistance was further reduced after heat treatment, which evidenced the strong MSI.
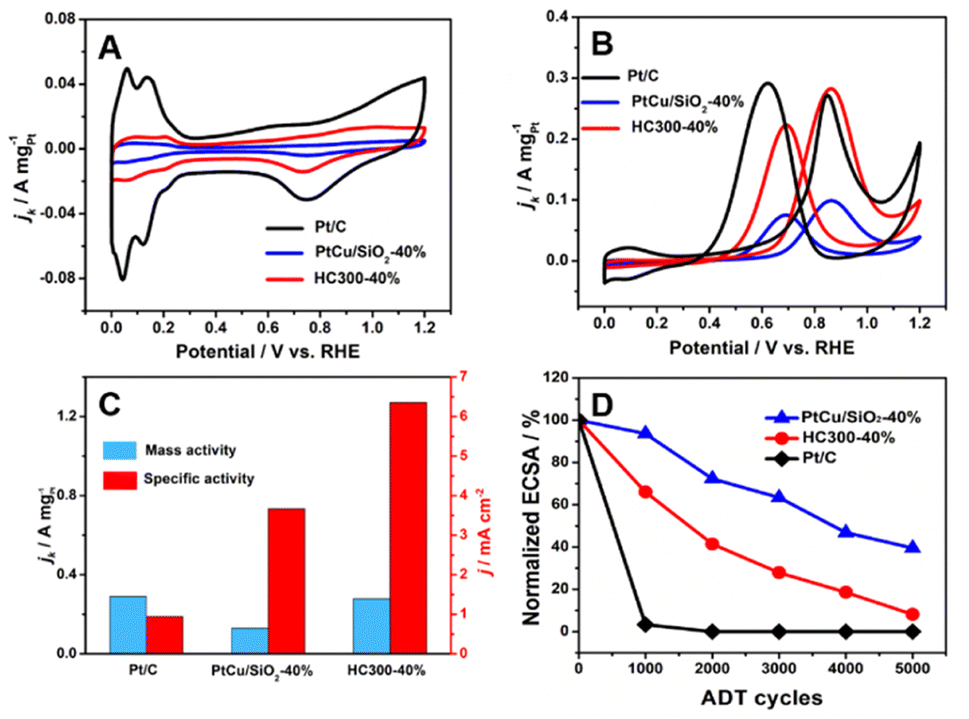 | ||
| Fig. 4 (A) CVs of commercial Pt/C (20 wt% Pt), PtCu/SiO2-40%, and HC300-40%; (B) MOR activity; (C) mass and specific activities obtained from MOR results; (D) accelerating durability tests (ADT) of commercial Pt/C (20 wt% Pt), PtCu/SiO2-40%, and HC300-40%. | ||
Relative to Pt/C, PtCu/SiO2-40% showed poor MOR activity, but HC300-40% exhibited comparable activity (Fig. 4B). The mass activities (MAs) of HC300-40% (0.28 A mgPt−1) and Pt/C (0.29 A mgPt−1) were very close (Fig. 4C); the MA of PtCu/SiO2-40% was 0.13 mgPt−1, less than half that of HC300-40%. The HC300-30% also showed an increase of MOR activity compared to PtCu/SiO2-30% (Table S3†).
The ratio of peak current densities of the forward scan (If) to backward scan (Ib) is commonly used to identify the poison resistance of the catalysts.27 In the forward scan, methanol was oxidized into intermediates which strongly adsorb on the active sites, then in the backward scan the intermediates resume being oxidized.28 Higher If:Ib ratios indicate that more intermediates are oxidized in the forward scan, leaving less to the backward scan. Compared to the If:Ib ratio of 0.93 (Pt/C), PtCu/SiO2-40% and HC300-40% demonstrate higher values of 1.30 and 1.27. Obviously, the poisoning resistance of the PtCu/SiO2 catalysts is improved significantly. The specific activities (SAs) of PtCu/SiO2-40% and HC300-40% are 3.66 and 6.35 mA cm−2 respectively, and the SA of HC300-40% is nearly 7 times the value of Pt/C (0.95 mA cm−2).
The durability of the catalyst is a highly important issue and was also tested (Fig. 4D). The ECSA of Pt/C decreased significantly in the first 1000 cycles, which meant poor durability due to the weak metal–support interaction and the oxidation of the carbon support.29 The PtCu/SiO2-40% showed the best durability with 39.49% ECSA remaining after 5000 cycles, and an ECSA of 8.14% for HC300-40% remained after 5000 cycles. PtCu/SiO2-40% and HC300-40% retained 32.7% and 38% of their MOR activity and all catalysts retained more than 30% after 5000 cycles, and the If:Ib ratios of PtCu/SiO2-40% and HC300-40% were 1.26 and 1.24 respectively (Fig. S5†). Such slight decreases after ADT demonstrated the minor decline of poisoning resistance.
The analysis of electrochemical performance showed that the heat treatment was crucial to regulate the MSI between the SiO2 and PtCu alloy and improve the MOR activity. The If:Ib ratios of PtCu/SiO2-40% and HC300-40% were 1.26 and 1.24 respectively (Fig. S4†). Such slight decreases after ADT demonstrated the minor decline of poisoning resistance. The electron transfer promoted by heat treatment led to an upshift of the d-band center of Pt,7 which meant easier charge donation from Pt to the adsorbates and the chemisorption on Pt became stronger, leading to the increase of the absorption rate of methanol and the intermediates.30–32 In the durability test, the ECSA of HC300-40% drops more than that of PtCu/SiO2-40%, owing to the increase of the absorption rate of the intermediates such as CO*. Meanwhile, the HC300-40% showed less MOR activity decrease than PtCu/SiO2-40% after ADT, due to the MSI which promoted charge accumulation at the interface during electron transfer and increased the specific activity of Pt active sites, and the existing –OH at the interface protecting the Pt active sites from poisoning. Thus, the electron transfer caused by heat treatment led to much better MOR activity and the durability also improved.
Many studies have shown that the shift of the d-band center of Pt can change the ORR activity.21,33,34 The ORR results (Fig. 5A) illustrated that PtCu/SiO2-40% and HC300-40% exhibited close onset potentials to Pt/C, but much better durability. After 10000 cycles, the ORR activity of PtCu/SiO2-40% became better and that of HC300-40% remained unchanged, while that of Pt/C showed a significant decline (Fig. 5B–D). The PtCu nanoparticles strongly bonded to SiO2 nanospheres due to the MSI, leading to improved ORR durability. The structural evolution of PtCu during the ORR was the reason for the better activity after ADT. Besides, the ascorbates were wiped out to form a clean surface during operation, leading to better activity.35
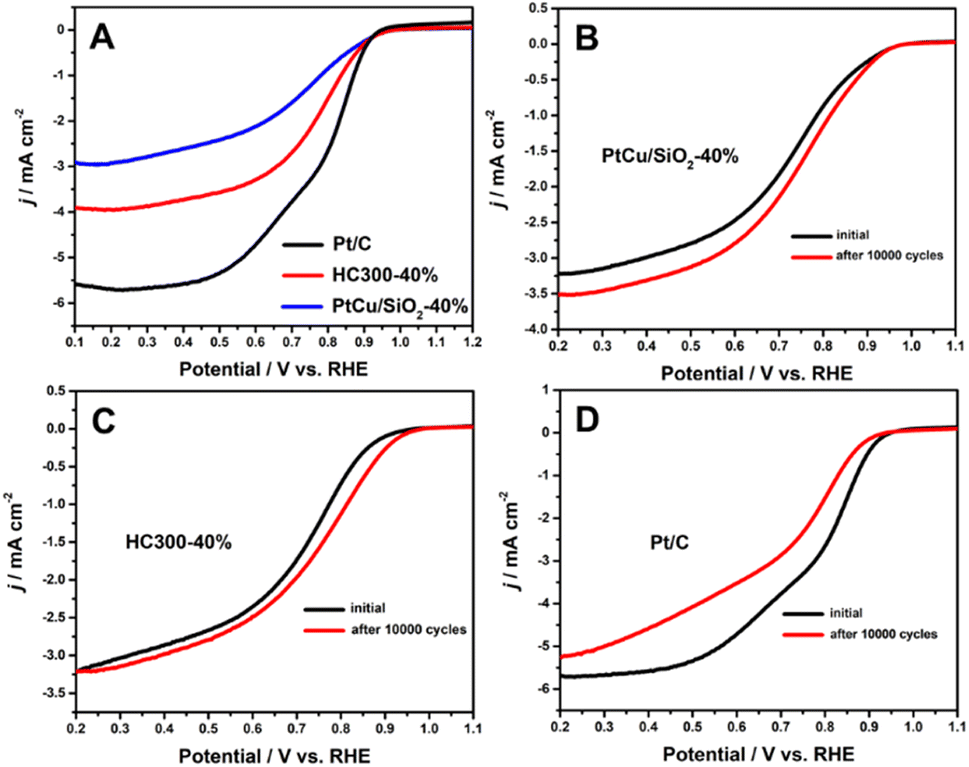 | ||
| Fig. 5 (A) The ORR curves of Pt/C, HC300-40%, and PtCu/SiO2-40%; the changes in ORR polarization curves under potential cycling (scan rate 10 mV s−1, O2 saturated) of PtCu/SiO2-40% (B), HC300-40% (C) and Pt/C (D). | ||
Conclusions
SiO2 nanosphere supported PtCu nanoparticles were successfully synthesized as carbon-free catalysts and exhibited much better activity and durability than Pt/C towards the methanol oxidation reaction. The as-prepared catalysts also showed comparable activity and better durability towards the oxygen reduction reaction. The heat treatment, which regulates the metal–support interaction, is crucial to the catalytic performance. The heat treatment promotes the electron transfer, which can adjust the d-band center of Pt. This work provides a strategy to synthesize a carbon-free catalyst with high MOR activity and durability, and reveals the importance of heat treatment for regulating the interaction between metal and non-carbon supports.Experimental
The preparation of SiO2 spheres
100 mL of ethanol and 10 mL of deionized water were mixed, then 3 mL of ammonium hydroxide was added to the ethanol stock and stirred for 10 minutes. Next, 1 mL of ethylsilicate was added drop by drop to the solution and stirred continuously for 24 h. Lastly, the solution was centrifuged at 12000 rpm for 10 minutes to obtain the silica spheres. Then the SiO2 spheres were washed three times with deionized water until pH = 7 and dried at 60 °C for 12 hours in the air.
The preparation of Cu/SiO2
With magnetic stirring, 100 mL ethylene glycol, 200 mg of the as-prepared silica spheres and 1.5 g of copper nitrate trihydrate were added to a 250 mL three-neck flask. The copper nitrate was dissolved completely by stirring for 10 minutes, and then the silica spheres were dispersed by ultrasonication for 10 minutes. With constant bubbling of N2 into the flask and stirring, the flask was refluxed at 210 °C in an oil bath for 30 minutes. Then it was kept stirring and cooled to room temperature. The solution was filtered and the obtained sample was washed three times with deionized water and lyophilized.The preparation of PtCu/SiO2
50 mg of the as-prepared Cu/SiO2 and 35 mL of deionized water were dispersed by ultrasonication in a 50 mL flask. With magnetic stirring, 1.3 mL of chloroplatinate aqueous solution (4 mg mL−1) was added to the flask drop by drop. The solution was stirred at 60 °C for 6 h, and the obtained solution was filtered and washed three times. Then, the sample was mixed with 30 mL of 1 M nitric acid solution and stirred slowly for 24 h to obtain the samples with 10 wt% Pt loading, which was labelled PtCu/SiO2-10%. The difference in the synthesis of samples with Pt loading of 20 wt%, 30 wt%, and 40 wt% was that the additive amount of chloroplatinate aqueous solution was changed to 2.7 mL, 4.6 mL and 7.3 mL and the obtained samples were labelled as PtCu/SiO2-20%, PtCu/SiO2-30%, and PtCu/SiO2-40%, respectively.50 mg of Cu/SiO2 was placed in a tube furnace and calcined at 300 °C for 2 hours in an atmosphere of 10 vol% H2/N2. The obtained sample was named HC300, loaded with 30 wt% and 40 wt% Pt and treated with nitric acid solution. Then the solution was filtered and the obtained samples were washed three times with water and lyophilized, and named HC300-30% and HC300-40% respectively.
The preparation of Pt/C
50 mg Vulcan XC-72, 42 mL ethylene glycol and 8 mL of chloroplatinate aqueous solution (4 mg mL−1) were mixed in a 250 mL flask and dispersed by ultrasonication for 10 min. The solution was stirred at 130 °C for 3 h in an oil bath. After cooling, the Pt/C catalyst was filtered and washed in deionized water 3 times and lyophilized.Electrochemical measurements
The electrochemical activity of the MOR and accelerated durability test (ADT) were tested on a CHI 660E. The tests were carried out in a three-electrode cell at room temperature. A glassy carbon electrode (GCE) (ϕ = 0.196 cm2) was used as the working electrode, and Ag/AgCl and platinum wire were used as the reference and counter electrodes, respectively. Electrochemical activity and durability tests of the ORR were performed in a three-electrode cell on a PGSTAT302N (Autolab). The ORR test was carried out using a rotating ring disk electrode (RRDE, ϕ = 0.196 cm2) as the working electrode.The GCE was finely polished to a mirror-like surface with an Al2O3 slurry (<50 nm) before use. Catalyst ink was prepared by mixing 2 mg of catalyst, 0.5 mL of isopropanol, 0.5 mL of deionized water and 10 μL of Nafion with ultrasonic dispersion. 10 μL of ink was dropped on the working electrode and dried in the air. Cyclic voltammetry (CV) was performed in a nitrogen saturated 0.5 M H2SO4 electrolyte in the potential range of 0–1.2 V vs. the reversible hydrogen electrode (RHE) at the scanning rate of 100 mV s−1. The MOR activity test was performed in a nitrogen saturated solution containing 0.5 M H2SO4 and 1 M methanol, and the CV program was selected for the MOR activity test. The MOR ADT was performed in 0.5 M H2SO4 electrolyte in the potential range of 0.6–1.2 V vs. RHE at the scanning rate of 50 mV s−1. Before the ADT and after every 1000 cycles, the MOR activity test was performed and recorded.
For the ORR tests, CV was performed in O2 saturated 0.1 M HClO4 aqueous solution in the potential range of 0–1.2 V vs. RHE at the scanning rate of 50 mV s−1. Linear scanning voltammetry (LSV) was performed in O2 saturated 0.1 M HClO4 aqueous solution in the potential range of 0–1.2 V at the scanning rate of 10 mV s−1 with the rotating speed of 1600 rpm. The ADT was completed by performing CV in N2 saturated 0.1 M HClO4 aqueous solution in the potential range of 0.6–1.2 V at the scanning rate of 100 mV s−1 for 5000 cycles.
Author contributions
Quanqing Zhao: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft. Liu Yang: methodology, formal analysis. Feng Xu: formal analysis, resources, validation, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the financial support from the Natural Science Foundation of Fujian Province (no. 2022J01089).Notes and references
- J. Zhao, H. Zeng and Z.-X. Lu, ACS Appl. Nano Mater., 2022, 5, 13594–13600 CrossRef CAS.
- R. Qu, S. Tang, J. Zhang and L. Wu, ACS Sustainable Chem. Eng., 2018, 6, 15560–15569 CrossRef CAS.
- S. Takao, O. Sekizawa, G. Samjeské, T. Kaneko, K. Higashi, Y. Yoshida, X. Zhao, T. Sakata, T. Yamamoto, T. Gunji, T. Uruga and Y. Iwasawa, ACS Appl. Mater. Interfaces, 2018, 10, 27734–27744 CrossRef CAS PubMed.
- S. G. Dali, J. Q. Bermejo, M. T. Izquierdo, J. C. Gutierrez, A. Celzard and V. Fierro, RSC Sustain., 2023, 1, 1270–1277 RSC.
- J. J. Ogada, A. K. Ipadeola, P. V. Mwonga, A. B. Haruna, F. Nichols, S. Chen, H. A. Miller, M. V. Pagliaro, F. Vizza, J. R. Varcoe, D. M. Meira, D. M. Wamwangi and K. I. Ozoemena, ACS Catal., 2022, 12, 7014–7029 CrossRef CAS.
- A. A. Melvin, V. S. Joshi, D. C. Poudyal, D. Khushalani and S. K. Haram, ACS Appl. Mater. Interfaces, 2015, 7, 6590–6595 CrossRef CAS PubMed.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- C.-C. Ting, C.-H. Chao, C. Y. Tsai, I. K. Cheng and F.-M. Pan, Appl. Surf. Sci., 2017, 416, 365–370 CrossRef CAS.
- Z. Wang, S.-M. Xu, L. Tan, G. Liu, T. Shen, C. Yu, H. Wang, Y. Tao, X. Cao, Y. Zhao and Y.-F. Song, Appl. Catal., B, 2020, 270 CAS.
- B. Zhang and Y. Qin, ACS Catal., 2018, 8, 10064–10081 CrossRef CAS.
- M. Farnesi Camellone, F. Negreiros Ribeiro, L. Szabová, Y. Tateyama and S. Fabris, J. Am. Chem. Soc., 2016, 138, 11560–11567 CrossRef CAS PubMed.
- N. K. Oh, J. Seo, S. Lee, H.-J. Kim, U. Kim, J. Lee, Y.-K. Han and H. Park, Nat. Commun., 2021, 12, 4606 CrossRef CAS PubMed.
- Z. Hou, L. Dai, J. Deng, G. Zhao, L. Jing, Y. Wang, X. Yu, R. Gao, X. Tian, H. Dai, D. Wang and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202201655 CrossRef CAS PubMed.
- Z. Hou, L. Dai, J. Deng, G. Zhao, L. Jing, Y. Wang, X. Yu, R. Gao, X. Tian, H. Dai, D. Wang and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202201655 CrossRef CAS PubMed.
- Z. X. Xia, X. L. Xu, X. M. Zhang, H. Q. Li, S. L. Wang and G. Q. Sun, J. Mater. Chem. A, 2020, 8, 1113–1119 RSC.
- X. Du, W. Cai, Q. Zhang, L. Yang and H. He, ACS Appl. Nano Mater., 2021, 4, 9729–9737 CrossRef CAS.
- L. Lu, S. Chen, S. Thota, X. Wang, Y. Wang, S. Zou, J. Fan and J. Zhao, J. Phys. Chem. C, 2017, 121, 19796–19806 CrossRef CAS.
- L. Lu, S. Chen, S. Thota, X. Wang, Y. Wang, S. Zou, J. Fan and J. Zhao, J. Phys. Chem. C, 2017, 121, 19796–19806 CrossRef CAS.
- S. J. Kim, Y. I. Kim, B. Lamichhane, Y.-H. Kim, Y. Lee, C. R. Cho, M. Cheon, J. C. Kim, H. Y. Jeong, T. Ha, J. Kim, Y. H. Lee, S.-G. Kim, Y.-M. Kim and S.-Y. Jeong, Nature, 2022, 603, 434–438 CrossRef CAS.
- L. Zhuo, Z. Zhao, Z. Qin, Q. Chen, S. Liang, X. Yang and F. Wang, Composites, Part B, 2019, 161, 336–343 CrossRef CAS.
- B. Liu, Q. Zhang, Y. Li, Y. Hao, U. Ali, L. Li, L. Zhang, C. Wang and Z. Su, CCS Chem., 2023, 5, 209–220 CrossRef CAS.
- F. R. Zhang, K. P. Jiang, G. S. Zhu, H. R. Xu, X. Y. Zhang, Y. Y. Zhao, Y. J. Zhang, Q. B. Wang, P. F. Pang and A. B. Yu, Sustainable Energy Fuels, 2023, 7, 1245–1255 RSC.
- G. Chen, Q. Hu, F. Schulz, W. J. Parak, L. Wang, X. Cui, K. Yang, Z. Luo, A. Zeng and Q. Fu, ACS Appl. Nano Mater., 2021, 4, 9060–9067 CrossRef CAS.
- Y. Takeuchi, S. Obata, K. Ohkura and Y. Nishina, ACS Mater. Lett., 2022, 4, 2590–2596 CrossRef CAS.
- R. Sharma, S. Gyergyek and S. M. Andersen, Appl. Catal., B, 2022, 311, 121351 CrossRef CAS.
- Z. Xu, H. Zhang, H. Zhong, Q. Lu, Y. Wang and D. Su, Appl. Catal., B, 2012, 111–112, 264–270 CrossRef CAS.
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- D. Paital, V. Thambi, M. S. Kutwal and S. Khatua, ACS Appl. Nano Mater., 2022, 5, 13286–13294 CrossRef CAS.
- L. Fan, J. Zhao, X. Luo and Z. Tu, Int. J. Hydrogen Energy, 2022, 47, 5418–5428 CrossRef CAS.
- S. Joo, K. Kim, O. Kwon, J. Oh, H. J. Kim, L. Zhang, J. Zhou, J.-Q. Wang, H. Y. Jeong, J. W. Han and G. Kim, Angew. Chem., Int. Ed., 2021, 60, 15912–15919 CrossRef CAS PubMed.
- D. Wang, Z. Chen, Y. Wu, Y. C. Huang, L. Tao, J. Chen, C. L. Dong, C. V. Singh and S. Wang, SmartMat, 2022, 4 Search PubMed.
- Y. Zhou, L. Yu, J. Chang, L. Feng and j. Zhang, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.08.007.
- H. Ma, Z. Zheng, H. Zhao, C. Shen, H. Chen, H. Li, Z. Cao, Q. Kuang, H. Lin and Z. Xie, J. Mater. Chem. A, 2021, 9, 23444–23450 RSC.
- Y. Yang, B. He, H. Ma, S. Yang, Z. Ren, T. Qin, F. Lu, L. Ren, Y. Zhang, T. Wang, X. Liu and L. Chen, Acta Phys.-Chim. Sin., 2022, 0, 2201050 Search PubMed.
- L.-M. Luo, W. Zhan, R.-H. Zhang, D. Chen, Q.-Y. Hu, Y.-F. Guo and X.-W. Zhou, J. Power Sources, 2019, 412, 142–152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00193h |
| This journal is © The Royal Society of Chemistry 2023 |