
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design and topochemical synthesis of polymorphs of a polymer†
Vignesh
Athiyarath
 a,
Liby Ann
Mathew
a,
Yakai
Zhao
b,
Ravichandran
Khazeber
a,
Upadrasta
Ramamurty
b and
Kana M.
Sureshan
*a
a,
Liby Ann
Mathew
a,
Yakai
Zhao
b,
Ravichandran
Khazeber
a,
Upadrasta
Ramamurty
b and
Kana M.
Sureshan
*a
aSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Kerala 695551, India. E-mail: kms@iisertvm.ac.in
bSchool of Mechanical and Aerospace Engineering, Nanyang Technological University, 639798, Singapore
First published on 19th April 2023
Abstract
Packing a polymer in different ways can give polymorphs of the polymer having different properties. β-Turn forming peptides such as 2-aminoisobutyric acid (Aib)-rich peptides adopt several conformations by varying the dihedral angles. Aiming at this, a β-turn-forming peptide monomer would give different polymorphs and these polymorphs upon topochemical polymerization would yield polymorphs of the polymer, we designed an Aib-rich monomer N3-(Aib)3-NHCH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH. This monomer crystallizes as two polymorphs and one hydrate. In all forms, the peptide adopts β-turn conformations and arranges in a head-to-tail manner with their azide and alkyne units proximally placed in a ready-to-react alignment. On heating, both the polymorphs undergo topochemical azide–alkyne cycloaddition polymerization. Polymorph I polymerized in a single-crystal-to-single-crystal (SCSC) fashion and the single-crystal X-ray diffraction analysis of the polymer revealed its screw-sense reversing helical structure. Polymorph II maintains its crystallinity during polymerization but gradually becomes amorphous upon storage. The hydrate III undergoes a dehydrative transition to polymorph II. Nanoindentation studies revealed that different polymorphs of the monomer and the corresponding polymers exhibited different mechanical properties, in accordance with their crystal packing. This work demonstrates the promising future of the marriage of polymorphism and topochemistry for obtaining polymorphs of polymers.
CH. This monomer crystallizes as two polymorphs and one hydrate. In all forms, the peptide adopts β-turn conformations and arranges in a head-to-tail manner with their azide and alkyne units proximally placed in a ready-to-react alignment. On heating, both the polymorphs undergo topochemical azide–alkyne cycloaddition polymerization. Polymorph I polymerized in a single-crystal-to-single-crystal (SCSC) fashion and the single-crystal X-ray diffraction analysis of the polymer revealed its screw-sense reversing helical structure. Polymorph II maintains its crystallinity during polymerization but gradually becomes amorphous upon storage. The hydrate III undergoes a dehydrative transition to polymorph II. Nanoindentation studies revealed that different polymorphs of the monomer and the corresponding polymers exhibited different mechanical properties, in accordance with their crystal packing. This work demonstrates the promising future of the marriage of polymorphism and topochemistry for obtaining polymorphs of polymers.
Introduction
Nature manifests variation in the properties of its components by tuning the conformation and packing of the comprising molecules. For instance, multifunctional proteins exhibit different functions via changing their conformation.1–4 In the abiotic domain also, variation in the structure imparts different properties to the same material.5 Polymorphism, the property of compounds to pack in different crystal forms,6–12 has been widely exploited to tune the properties of small molecules and has found immense use in the area of pharmaceuticals,13 optoelectronics,14 solar cells,15 functional materials,16etc. It is expected that different polymorphs of a particular polymer can have different properties. However, due to the difficulty in crystallizing polymers, accessing polymorphs of polymers is an unresolved challenge. However, topochemical polymerization17–37 is promising as different reactive polymorphs of a monomer can yield different polymorphs of a polymer. The main challenge lies in the design of monomers that can form different polymorphs. Here we report a rational design of a monomer that forms polymorphs and the topochemical polymerization of these different polymorphs to yield different polymer polymorphs.Results and discussion
Topochemical azide–alkyne cycloaddition (TAAC) polymerization has emerged as an ideal method to access crystalline polymers.38–46 We have adopted this methodology for the synthesis of several peptide-based polymers having various secondary structures such as a β-sheet,47 helix,48 helical sheet,49 and β-meander.50 The β-turn, characterized by an intra-chain NH⋯CO H-bond between the ith and i + 3rd amino acids, is the third most important secondary structure found in proteins and peptides after helices and β-strands. β-Turns are known to adopt at least nine different conformations (types I, II, VIII, I′, II′, Via1, Via2, Vib, and IV) having different dihedral angles of i + 1 and i + 2 positions of the turn.51–54 We hypothesized that an azide and an alkyne-modified peptide-based monomer having a sequence that can adopt a β-turn conformation might crystallize in several polymorphic forms due to the subtle variations in these dihedral angles. Such polymorphs upon topochemical polymerization would yield polymorphs of the polymer (Fig. 1a and b). Short peptides composed of 2-aminoisobutyric acid (Aib) residues are known to adopt β-turn structures.55–57 Also, Aib-containing tripeptides are known to adopt multiple conformations.58–61 In view of these, we designed a tripeptide-monomer N3-(Aib)3-CH2–CCH (1) with an aim to get multiple polymorphs and to polymerize them topochemically. We synthesized tripeptide 1 by following the standard peptide synthesis protocol (ESI, Scheme S1, Section S2†). We obtained two different polymorphs and a hydrate when crystallized from different solvents, and they were characterized by single-crystal X-ray diffraction (SCXRD) analysis (ESI, Section S3†).62
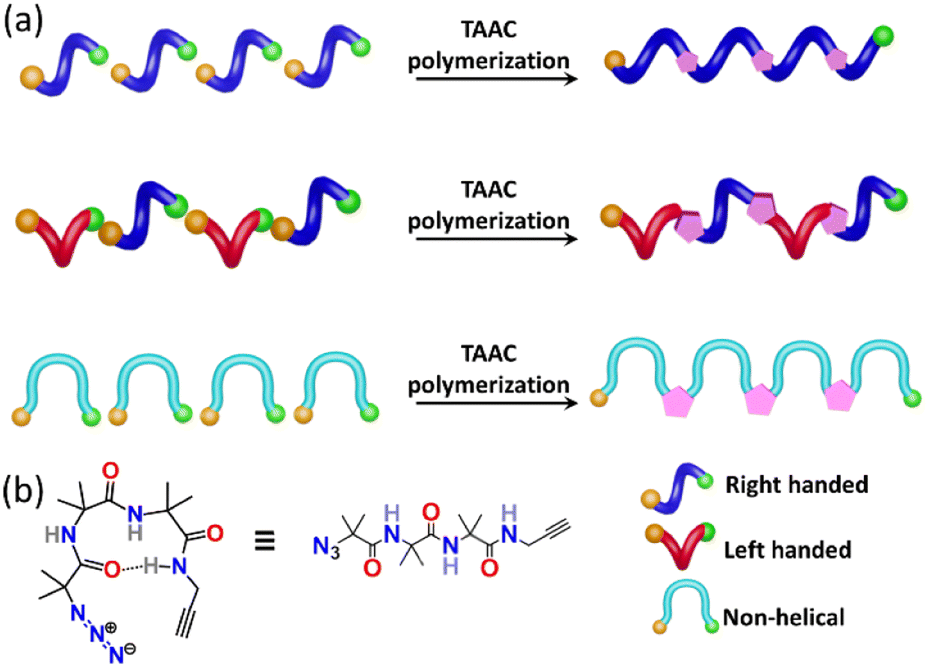 | ||
| Fig. 1 Design of a β-turn forming monomer that can form polymorphs via adopting various β-turn conformations. (a) Schematic showing plausible conformations of azide–alkyne modified β-turns in various polymorphic forms and their TAAC polymerization to yield polymorphs of a polymer. Azide and alkyne are represented as orange and green balls, respectively. (b) Chemical structure of the designed tripeptide 1. | ||
Polymorph I
We obtained polymorph I as rectangular flake-like crystals (mp 133 °C) when the tripeptide 1 (50 mg) was crystallized from a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (v/v, 7 mL) mixture of dichloromethane:petroleum ether (Fig. 2a). The molecule adopts a bent conformation and crystallizes in the P21/c space group (Table S1†) and the asymmetric unit has two conformers, a & b (Fig. 2b). The Ramachandran dihedral angles (Table 1) of this polymorph suggest that the tripeptide molecule adopts a type-I β-turn conformation.63 Intermolecular NH⋯O (N5–H5⋯O3) hydrogen bonding connects two conformers alternately in a head-to-tail fashion, forming a supramolecular chain along the crystallographic ‘b’ axis. The adjacent conformers are of opposite helical sense (Fig. 2c). The azide and alkyne units of adjacent conformers are arranged in an orientation (parallel; interplanar angle ω = 20.6°) and distance (dC⋯N = 3.4 Å & 3.6 Å) suitable for their topochemical cycloaddition reaction to yield a 1,5-linked triazolyl peptide polymer. Such chains are arranged parallelly along the ‘a’ axis and antiparallelly along the ‘c’ axis, such that each chain is surrounded by six other chains (Fig. 2d). Such an arrangement is stabilized by N4A–H4A⋯O2A hydrogen bonding, C–H⋯O, C–H⋯N, and weak dispersion interactions between adjacent chains (Table S2†).
:3 (v/v, 7 mL) mixture of dichloromethane:petroleum ether (Fig. 2a). The molecule adopts a bent conformation and crystallizes in the P21/c space group (Table S1†) and the asymmetric unit has two conformers, a & b (Fig. 2b). The Ramachandran dihedral angles (Table 1) of this polymorph suggest that the tripeptide molecule adopts a type-I β-turn conformation.63 Intermolecular NH⋯O (N5–H5⋯O3) hydrogen bonding connects two conformers alternately in a head-to-tail fashion, forming a supramolecular chain along the crystallographic ‘b’ axis. The adjacent conformers are of opposite helical sense (Fig. 2c). The azide and alkyne units of adjacent conformers are arranged in an orientation (parallel; interplanar angle ω = 20.6°) and distance (dC⋯N = 3.4 Å & 3.6 Å) suitable for their topochemical cycloaddition reaction to yield a 1,5-linked triazolyl peptide polymer. Such chains are arranged parallelly along the ‘a’ axis and antiparallelly along the ‘c’ axis, such that each chain is surrounded by six other chains (Fig. 2d). Such an arrangement is stabilized by N4A–H4A⋯O2A hydrogen bonding, C–H⋯O, C–H⋯N, and weak dispersion interactions between adjacent chains (Table S2†).
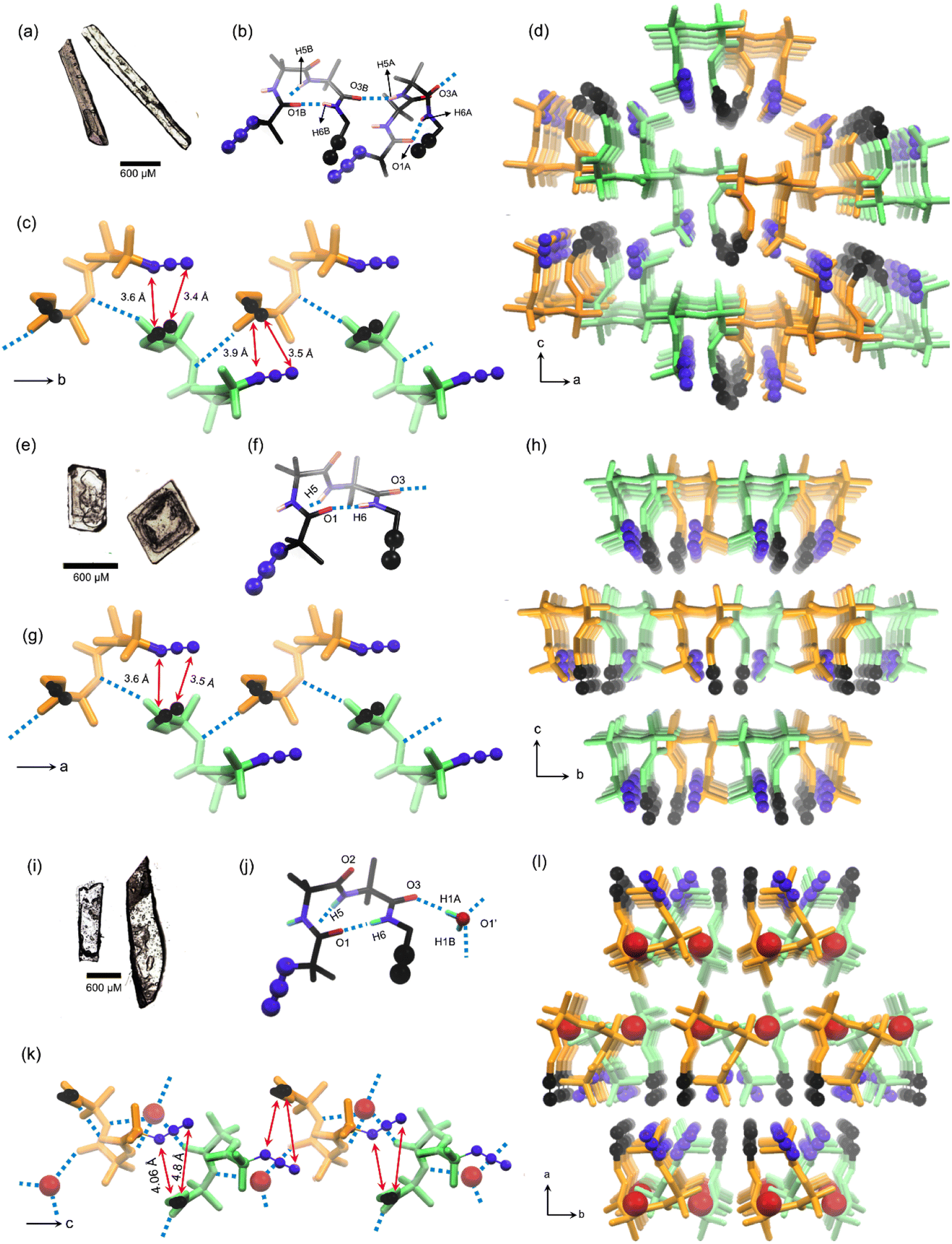 | ||
| Fig. 2 Crystal-structure analysis of monomers. The shape of single crystals of (a) polymorph I, (e) polymorph II, and (i) hydrate III. The conformation of tripeptide 1 in asymmetric units of (b) polymorph I, (f) polymorph II, and (j) hydrate III, adopting a β-turn conformation. A supramolecular chain comprising a head-to-tail arrangement of tripeptide 1 adopting β-turn conformations along (c) the b-axis in polymorph I, (g) the a-axis in polymorph II, and along (k) the c-axis in hydrate III. A supramolecular chain of tripeptide molecules surrounded by six chains, viewed along the (d) b-axis in polymorph I, (h) a-axis in polymorph II, and (l) c-axis in hydrate III. The backbone of left-handed and right-handed β-turns is colored in green and orange, respectively. Azide, alkyne, and water molecules in hydrate III are highlighted using a ball-and-stick model. H-atoms for alkyl groups are omitted for clarity. N5H5⋯O3 hydrogen bonding is shown by cyan-colored dotted lines. | ||
| Polymorph | Φ i+1 | Ψ i+1 | Φ i+2 | Ψ i+2 |
|---|---|---|---|---|
| I Conformer a | 52.6° | 49.2° | 78.6° | 3.1° |
| I Conformer b | −52.6° | −47.1° | −72.8° | −7.6° |
| II | 52° | 47.99° | 74.3° | 8.3° |
| III | 48.5° | 47.4° | 71.7 ° | 9.5° |
Polymorph II
Crystallization of the tripeptide 1 (50 mg) from a 2:3 (v/v, 4 mL) mixture of chloroform and hexane yielded polymorph II as cube-like crystals (Fig. 2e), which have a melting point of 135 °C. SCXRD analysis revealed that the polymorph II adopts the orthorhombic space group Pna21 (ESI, Table S3†) with one molecule in the asymmetric unit (Fig. 2f). This polymorph also shows a type-I β-turn conformation for the tripeptide as evidenced from its dihedral angles (Table 1 and Fig. 2f). Along the crystallographic ‘a’ axis, β-turns of opposite helicity arrange alternately in a head-to-tail manner and are connected by intermolecular NH⋯O hydrogen bonds, to form a supramolecular chain (Fig. 2g). Such chains are parallelly arranged along the crystallographic ‘b’ and ‘c’ axes. Each chain is surrounded by six similar chains coaxially oriented along the crystallographic ‘a’ axis (Fig. 2h), and this arrangement is stabilized by weak C–H⋯O, C–H⋯N, and dispersion interactions between chains (Table S4†). In this polymorph also, the azide and alkyne units of adjacent molecules in a supramolecular chain are parallelly arranged (ω = 25.5°) at a distance (dC⋯N = 3.5 Å & 3.6 Å) suitable for orbital overlap and thus, for topochemical polymerization.
Hydrate III
Slow evaporation of a solution of tripeptide 1 (50 mg) in a mixture of ethyl acetate and petroleum ether (1:1 v/v, 7 mL), yielded the hydrate III, which crystallizes in the P21/c space group (Table S5†) with a rectangular plate-like morphology (mp 125 °C, Fig. 2i). This hydrate has one molecule of tripeptide and a molecule of water in the asymmetric unit. The source of water could be the adventitious residual water in the solvent or the moisture in the atmosphere. Some anhydrous crystals are known to absorb water from the atmosphere.42 The alkyne is disordered over two positions with an occupancy ratio of 0.73:0.27 (Fig. 2j). Analysis of dihedral angles (Table 1) suggested that this is yet another crystal form having a tripeptide in a type-I β-turn conformation. β-Turns of opposite helicity are alternately bridged via an intermittent water molecule through hydrogen bonding forming a head-to-tail arranged supramolecular chain along the crystallographic ‘c’ axis (Fig. 2k). Also, such supramolecular chains are stabilized by an intermolecular azide–oxygen interaction between adjacent molecules (Table S6†).64 These water molecules bridge similar chains along the crystallographic ‘b’ axis. Also, such chains are parallelly arranged along the crystallographic ‘a’ axis through weak van der Waals interaction (Fig. 2l). In each water-bridged chain, the azide and alkyne are arranged at an interplanar angle of 44.6°, and interterminal distances of 4.06 Å & 4.8 Å (Fig. 2m).
Thus, as designed, in both the polymorphs and the hydrate, the molecule adopts a β-turn conformation through an intramolecular H-bonding between ith and i + 3rd amino acids, and the monomer molecules are arranged in a head-to-tail fashion forming supramolecular chains. The azide and alkyne units of adjacent molecules in these chains are proximally placed in an orientation suitable for their TAAC polymerization to yield 1,5-disubstituted triazole-linked polypeptide. While it is ideal to have a parallel or anti-parallel arrangement of azide and alkyne with a distance of <4 Å between them for their topochemical cycloaddition reaction, upon heating, molecules can undergo various motions in crystals and reach a different orientation suitable or unsuitable for their reaction.45 Phase purities of the crystals obtained from each solvent were established by comparing the simulated powder X-ray diffraction (PXRD) patterns obtained from the SCXRD data with the experimental PXRD patterns of the powdered forms of the corresponding crystals (ESI, Fig. S4†). A perfect match of the PXRD patterns confirmed that all crystals obtained from a particular solvent are of only one type of polymorphic form.
Dehydrative transition of hydrate III to polymorph II
Differential-scanning calorimetry (DSC) profiles of crystals (heating rate 5 °C min−1) of all these crystal forms confirmed their melting points (Fig. 3a). The large exothermic peaks after melting (in the 120–190 °C range) found in the DSC profiles of all these forms correspond to the heat generated due to the azide–alkyne cycloaddition in their melt. While polymorphs I and II showed endothermic peaks only at temperatures corresponding to their melting points, hydrate III showed an additional endothermic peak at 85 °C. We analyzed thermo-gravimetric analysis (TGA) and the DSC profile of the hydrate III at a slower heating rate of 1 °C min−1. At this heating rate, the endothermic peak shifted to 80 °C. We also observed a weight loss of 4.7% in the range of 80–119 °C. This weight loss corresponds to the theoretical weight of the water present in the lattice, suggesting that the additional endothermic peak corresponds to the heat of dehydration (Fig. 3b). We heated the crystals of hydrate III at 50 °C, 60 °C, 70 °C and 80 °C; and recorded their PXRD profiles. We did not observe any changes in PXRD patterns when heated up to 70 °C for one week. But when hydrate III was heated at 80 °C for 15 min, its PXRD pattern matched with that of polymorph II (ESI, Fig. S5†), confirming that the endothermic peak at 80 °C observed in the DSC profile corresponds to the dehydrative conversion of hydrate III to polymorph II. The dehydration generates free space in the lattice which would permit molecules to reorganize to reach a stable polymorphic form.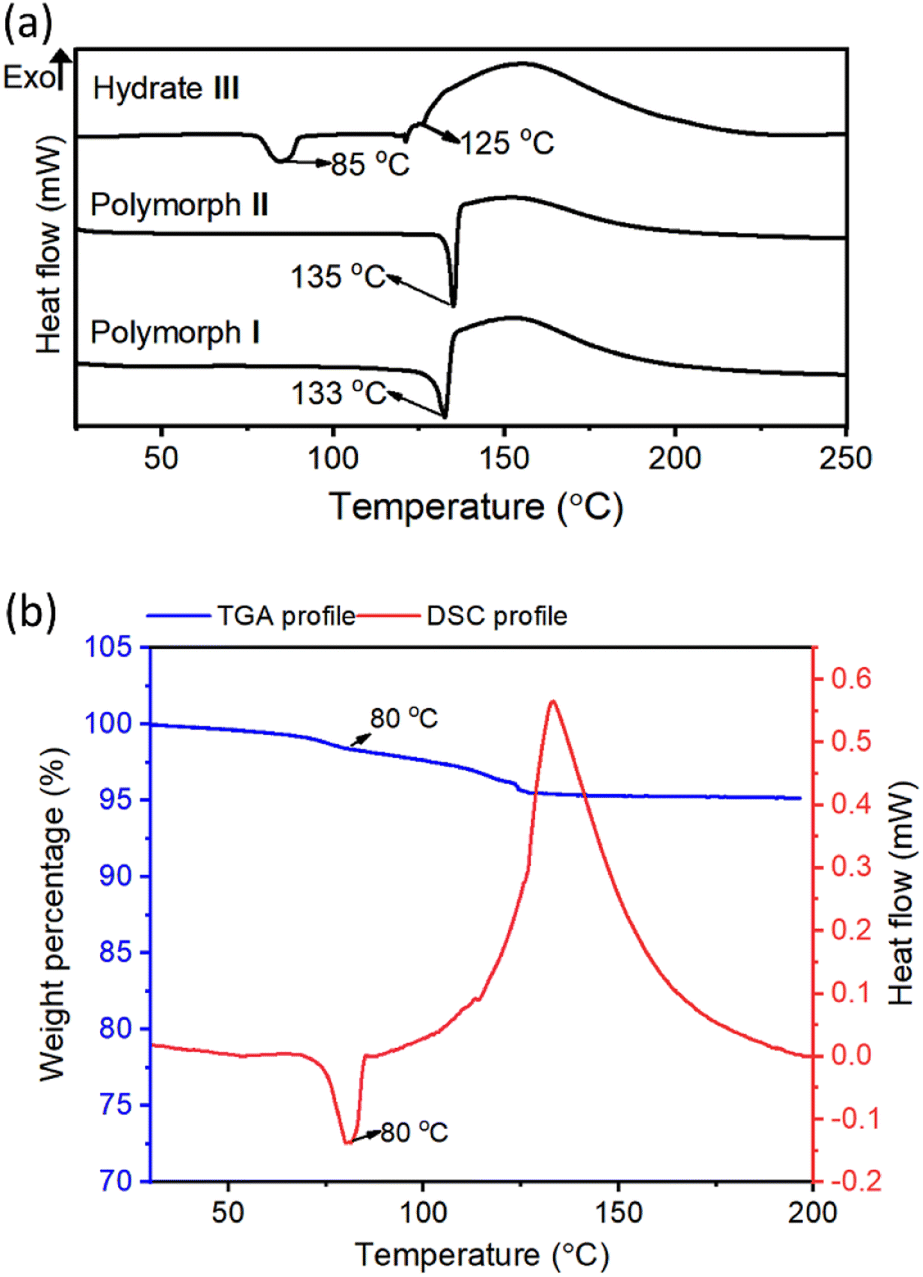 | ||
| Fig. 3 Thermal analysis of monomers. (a) DSC profiles for crystals of the polymorphs I and II, and the hydrate III at a heating rate of 5 °C min−1. (b) Comparison of DSC and TGA profiles of hydrate III crystals recorded at a heating rate of 1 °C min−1. | ||
Topochemical polymerization
Initially, we heated the crystals of polymorphs I and II at various temperatures in the range of 40–100 °C and monitored the progress of the TAAC reaction by 1H NMR spectroscopy. We have withdrawn small fractions from each sample at different intervals of time and recorded the 1H NMR spectra after dissolving them in DMSO-d6. None of the polymorphs reacted when heated at any temperature below 70 °C for two weeks.When we heated at 80 °C the peak corresponding to the alkyne proton at 2.97 ppm started reducing its intensity with time, and the peak due to the methylene protons gradually shifted from 3.8 to 4.31 ppm in all the forms, suggesting a gradual TAAC reaction (ESI, Fig. S6†). The peak corresponding to the triazolyl proton merges with the amide proton signals of the oligomer/polymer. Polymerization of polymorph I was completed in 30 days (ESI, Fig. S6a†), but polymorph II reacted slowly and it took 45 days for completion (ESI, Fig. S6b†). The rate of the reaction of both the polymorphs increased drastically when heated at 90 °C and 100 °C, but resulted in the decrease of diffraction quality of single crystals with time. Additionally, the exothermic nature of azide–alkyne cycloaddition might increase the local temperature further, resulting in the melting of the crystals before complete polymerization. Hence, we performed the TAAC reaction at 80 °C.
1H NMR spectra of polymers of both forms were identical and the peaks corresponding to the triazoles merged with that of amide protons (ESI, Fig. S6a and b†). Thus, the regioselectivity of cycloaddition could not be assigned by 1H NMR spectroscopy. The 13C NMR spectrum of polymers of both forms showed the presence of peaks corresponding to both 1,4-linked and 1,5-linked triazoles (Fig. 4a). NOESY spectral analysis of the polymer of both forms also showed evidence for the presence of both the regioisomers (ESI, Section S7†). Both polymorphs I and II exhibited sigmoidal kinetics for the polymerization reaction (Fig. 4b) and the extents of the reaction were similar in both cases. Gel permeation chromatography (GPC) analysis of the polymers obtained from polymorphs I and II showed the average molecular weights to be 14 kDa and 11 kDa, respectively (ESI, Section S8†).
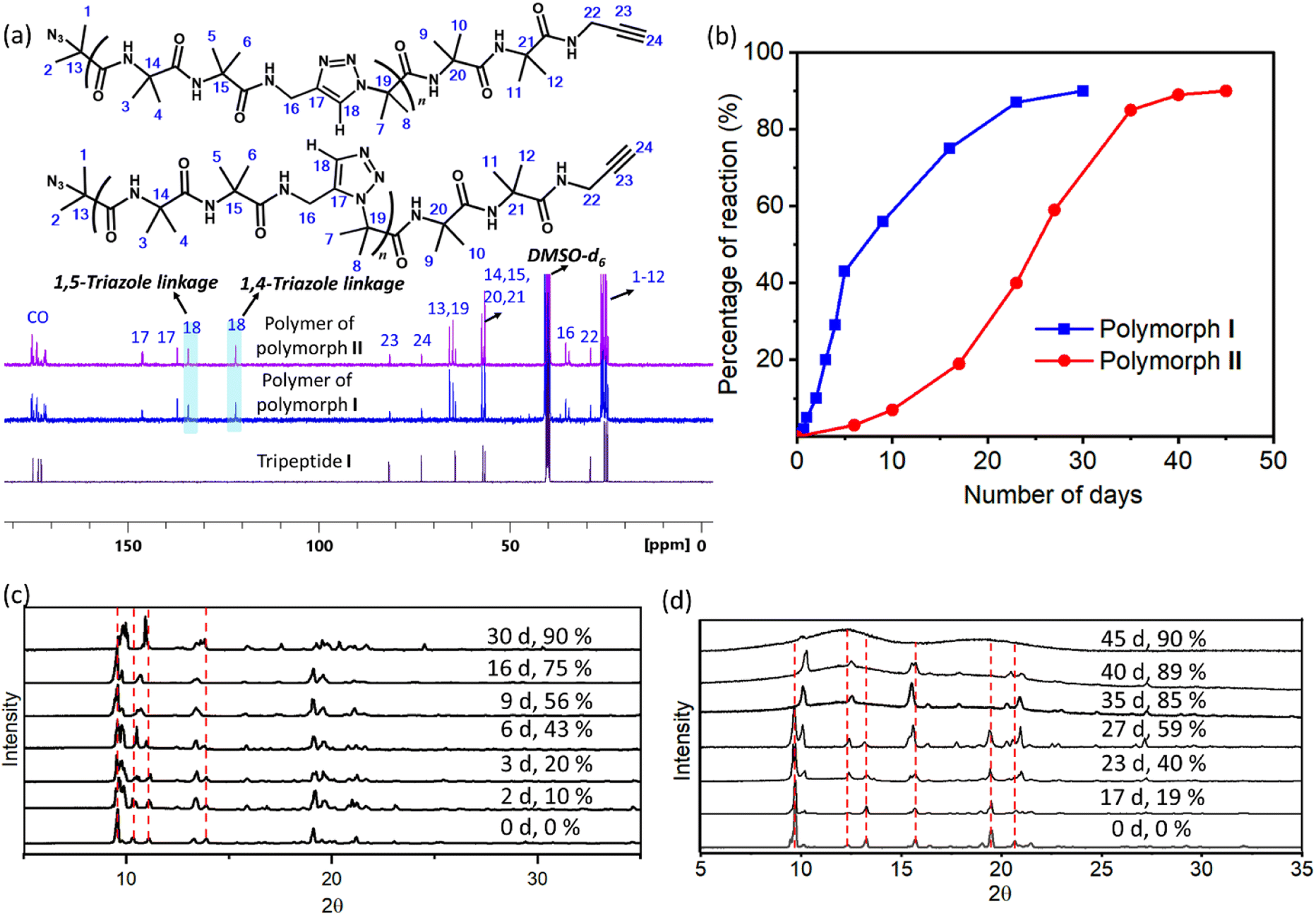 | ||
| Fig. 4 Characterization of TAAC polymerization. (a) 13C NMR spectra of polymers of polymorphs I and II (recorded in DMSO-d6) showing the presence of peaks corresponding to both 1,4- and 1,5-triazole linkages. (b) Comparison of kinetic plots for the TAAC reaction of polymorphs I and II. (c and d) Time-dependent PXRD patterns recorded during the polymerization of polymorphs (c) I and (d) II. The peaks which undergo changes are highlighted using a red dashed line. | ||
In order to assess the crystallinity of the sample during polymerization, we have utilized optical polarizing microscopy imaging of monomer polymorphs and heated samples (polymers). Both the polymorphs retained their shapes and dimensions even after complete polymerization. While the crystals of polymorph I were translucent and showed birefringence even after complete polymerization, the crystals of polymorph II have become opaque and lost their birefringence after polymerization (ESI, Fig. S9†). This suggests that polymorph I retains single-crystallinity while polymorph II loses its single-crystalline nature after complete polymerization.
We have also monitored the crystallinity of the sample during the polymerization reaction using time-dependent PXRD measurements. In the case of polymorph I, at all stages of the reaction, sharp peaks were observed in the PXRD diffractogram (Fig. 4c). The gradual disappearance or shifting of peaks due to the monomer and the appearance and growth of new peaks were very smooth. In the case of polymorph II, the peaks corresponding to the polymer phase appeared prominently after 40% reaction (23 days). After 35 days (85% reaction) the monomer phase completely disappeared and only the polymer phase was present. It is to be noted that the percentage of the reaction was calculated using end group analysis, using 1H NMR spectroscopy, and the remaining 15% is not the monomer but the reactable end groups. The reaction was stagnant at 89% (40 days) of conversion and even at this stage, the polymer retained crystallinity, as evident from the sharp peaks in the PXRD diffractogram. However, upon further aging, the polymer of polymorph II became amorphous, as indicated by the broad peaks in the PXRD profile of a 45 day old sample (Fig. 4d). The FTIR spectra of the aged polymer of polymorph II showed an additional broad peak due to OH stretching compared to those of the fresh polymer (ESI, Fig. S10a†). Also, the TGA profile of this aged polymer showed a weight loss of 6.7% in comparison to that of the fresh polymer during heating (Fig. S10b†). These observations suggest that the amorphization of the polymer of polymorph II could be due to the absorption of moisture by the polymer. Thus, both the polymorphs retained the crystallinity throughout the reaction, but polymorph II gradually lost its crystallinity upon storage.
Crystal structure and structural analysis of polymers
Next, we performed SCXRD analysis of the polymer crystals obtained from both polymorphs. In agreement with the PXRD analysis and microscopy, only the crystal structure of the polymer of polymorph I could be determined by SCXRD after complete polymerization (ESI, Section S11†). Polymorph I underwent single-crystal-to-single-crystal (SCSC)65–69 polymerization and the space group changed from P21/c to Pbca after polymerization. As expected from the NMR analysis, both 1,4-linked and 1,5-linked triazolyl linkages in a 1:1 occupancy ratio were seen (Fig. 5a). The latter was disordered with relative occupancies of 0.43:0.07 (Fig. 5b). These triazole linkages exist between tripeptide molecules with opposite helical sense, thus forming an achiral helical polymer chain. Similar to the packing of a monomer, each polymer chain of polymorph I is co-axially surrounded by six other chains (Fig. 5c). Although one can predict the formation of only 1,5-disubstituted triazole linkages from the monomer structure, the actual formation of both the isomers suggests significant molecular motion in the crystals. The PXRD pattern simulated from the polymer crystal structure matched with that of the bulk polymer sample obtained by heating the powdered polymorph I (ESI, Fig. S12†).
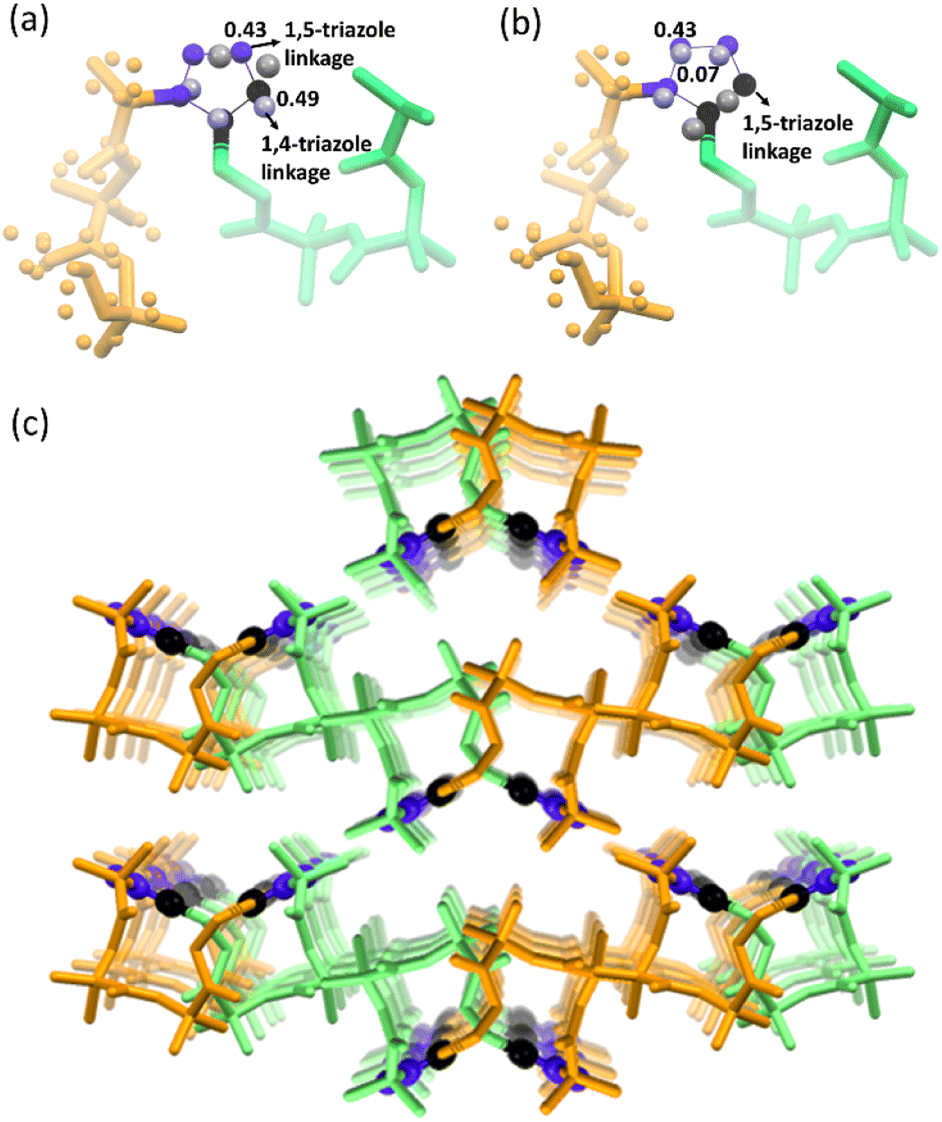 | ||
| Fig. 5 Crystal-structure analysis of the polymer of polymorph I. (a) Crystal structure of the polymer of polymorph I showing the presence of 1,5-triazole linkage and 1,4-triazole linkages. (b) Polymer strand with a 1,5-triazole linkage disordered with relative occupancies of 0.43:0.07. (c) A covalently linked polymer strand of the polymer of polymorph I with alternative left- and right-handed helical units surrounded by six coaxially arranged similar polymer strands. N atoms and C atoms of triazole are colored in blue and black, respectively, and are shown in a ball-and-stick model. Right- and left-handed helical turns present in the polymer of polymorph I are colored in orange and green, respectively. H-atoms are avoided for clarity. | ||
The formation of 1,4-linked and 1,5-linked triazolyl linkages in a 1:1 ratio was surprising given the preorganization of azide and alkyne for the exclusive formation of only the 1,5-linked triazolyl linkage. This prompted us to investigate the regiochemical outcome of the TAAC reaction in the other polymorph too. As 1H NMR spectral analysis was cumbersome due to the merging of the triazolyl proton signal(s) with amide proton signals, we recorded the 13C spectra of samples taken at various intervals of time for both the polymorphs. Surprisingly, we observed carbon signals corresponding to both 1,4-linked and 1,5-linked triazolyl linkages at every stage of polymerization in both the polymorphs, even though both the polymorphs suggested the formation of only the 1,5-linked triazolyl linkage (ESI, Fig. S13†). We analyzed the possibilities for the rotation of the bonds that connect azide and alkyne to the peptide backbone. We observed that voids, which facilitate motion, are present in the vicinity of azide and alkyne groups in both the polymorphs (ESI, Fig. S14a and b†). We observed that the azide and the alkyne can reach an arrangement suitable for the formation of 1,4-linked triazole by rotating the bonds connecting them to the peptide in both the polymorphs (ESI, Fig. S14c†).
Peptides having β-turns show specific signatures in their FTIR spectra. In order to gain an idea about the conformation of the polymer chain, we have compared the FT-IR spectra of polymorphs I and II with that of their corresponding polymers. Both the polymorphs showed amide I (1661 cm−1), amide II (1529 cm−1), and amide III (1274–1290 cm−1) bands in accordance with their β-turn conformation (ESI, Fig. S15†).70 These values did not vary even after polymerization. For both the polymorphs, the peaks corresponding to asymmetric stretching of azide at 2108–2114 cm−1 and bending of alkynyl C–H at 636 cm−1 reduced considerably after polymerization. The residual peak at 2114–2118 cm−1 after polymerization is due to the end group azide present in the polymer chains. A comparison of PXRD patterns of fully reacted polymers of polymorphs I and II unambiguously establishes that they are polymorphs of the polymer (ESI, Fig. S16†).The solid-state 13C CP/MAS NMR spectra of polymers of polymorphs I and II exhibited only subtle differences in the δ values (ESI, Fig. S17†).
Thermal and mechanical properties of polymorphs
TGA analysis of the polymers obtained from polymorphs I and II exhibited different thermal stabilities. While the polymer of polymorph I decomposed at 280 °C, that of polymorph II decomposed at 260 °C (ESI, Fig. S18†). This difference in thermal stability could be due to the difference in their packing (established by PXRD) and non-covalent interactions. However, the difference in molecular weight of the polymers as a reason for their differences in thermal stability cannot be ruled out. When we attempted to find the melting point using a melting point apparatus, both the polymers did not melt but underwent decomposition when heated above 280 °C. The DSC profile of the polymer of polymorph II showed endothermic peaks with negligible intensities around 110–132 °C presumably due to the melting of partially reacted oligomer domains. We have removed such short oligomers by washing the polymer samples with DCM and the insoluble residues were again analyzed by DSC. No endothermic transition ascribable to the melting or glass transition was observed for these polymers (ESI, Fig. S19†). There are several polymers that undergo decomposition before melting or glass transition.71,72It is expected that different polymorphs of the monomer and polymer will have different mechanical properties, in view of the difference in their packing and intermolecular interactions. We studied the mechanical properties of these crystals by nanoindentation.73,74 For this, we have face-indexed the crystals of all the polymorphs; some are then heat-polymerized and all the crystals (monomer and polymer) were fixed on glass slides by exposing the faces to be indented. Indentations were performed along the direction in which the tripeptide molecules undergo polymerization (the crystallographic b-axis for polymorph I, a-axis for polymorph II, and crystallographic c-axis for hydrate III; ESI, Section S19†).
As anticipated, monomers and polymers of different polymorphs show different mechanical properties (Fig. 6). Apart from compressing the molecules along the direction of indenting, the indenter has to overcome the attractive forces that hold each molecule in the plane perpendicular to the indenting direction. Although polymorphs I and II have similar hydrogen-bonded supramolecular chains, Young's modulus E (4.2 GPa) and hardness H (231 MPa) of polymorph I are higher than those of polymorph II (E = 3.2 GPa & H = 182 MPa). This can be rationalized based on the presence of N–H⋯O hydrogen bonding interaction along two crystallographic axes: the b-axis (along the direction of indentation) and a-axis (perpendicular to the direction of indentation) in polymorph I (ESI, Fig. S20 and Table S8†). Since these interactions stabilize the molecules in the ‘ab’-plane, the molecules could deform with relative ease only along the crystallographic c-axis during the indentation. But in polymorph II, N–H⋯O hydrogen bonding interaction exists only along the a-axis (direction of indentation). In the ‘bc’ plane (plane perpendicular to the direction of indentation), the molecules interact only through weaker interactions such as C–H⋯O and van der Waal's interactions (ESI, Fig. S21 and Table S9†). Thus, the molecules are susceptible to slipping along the b- and c-axes, and this, in turn, would reduce the restoring force along the direction of indentation. The hydrate III exhibited higher E (7.8 GPa) than both polymorphs I and II because of the O–H⋯O hydrogen bonding interactions with water molecules along both b- (perpendicular to the direction of indentation) and c-axes (parallel to the direction of indentation, ESI, Fig. S22 and Table S10†).
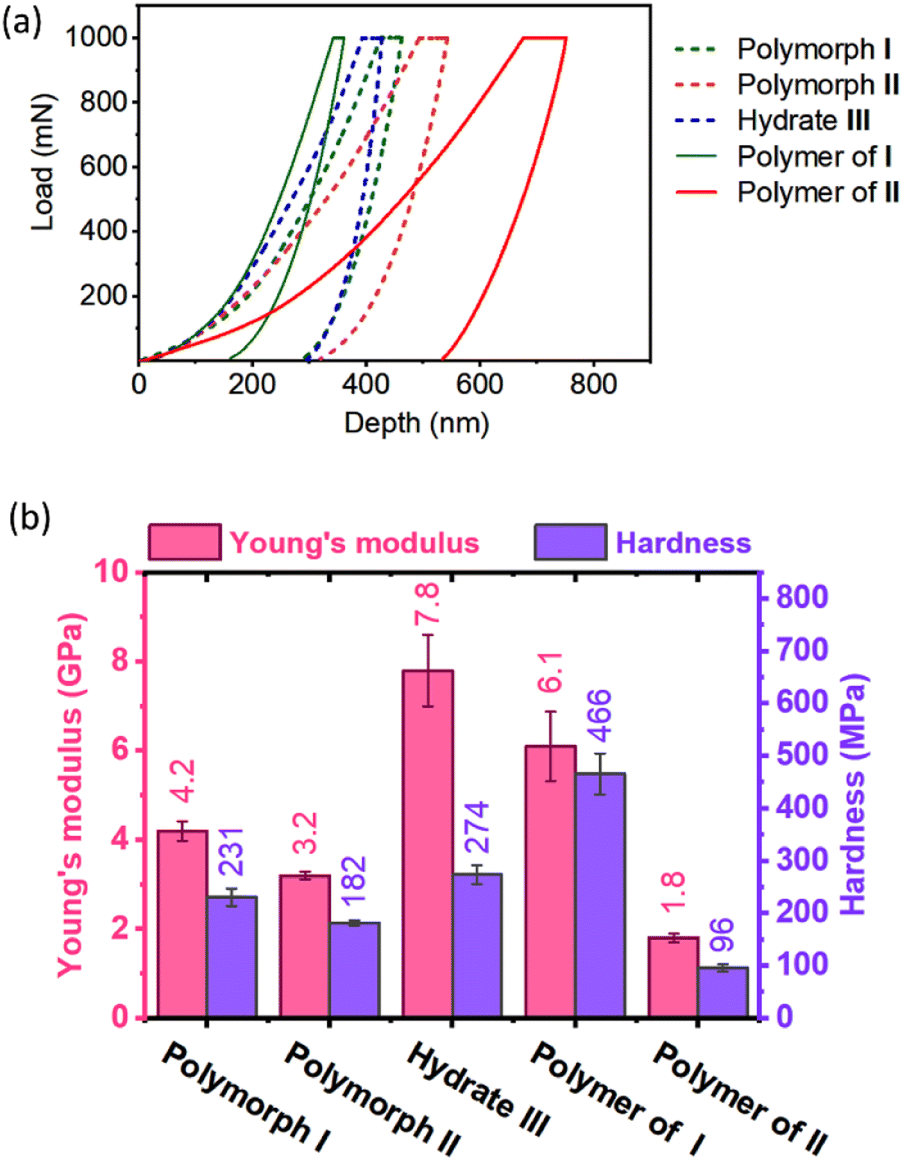 | ||
| Fig. 6 Analysis of mechanical properties of polymorphic monomers and polymers. (a) Representative load–displacement responses obtained from the nanoindentation experiments on the monomer crystals of polymorphs I and II, and hydrate III, and polymer crystals of polymorphs I and II. (b) Comparison of Young's modulus and hardness of monomer and polymer crystals of various polymorphic forms. Error bars represent the standard deviations obtained for different measurements taken at various positions of the corresponding face of the several crystals. | ||
Upon polymerization, as the monomers are connected by covalent linkages at the expense of non-covalent linkages, it is expected that E and H values would increase along the direction of polymerization. As anticipated the polymer of polymorph I showed higher E and H values compared to the corresponding monomer. But the polymer formed from polymorph II showed lower values of E and H compared to its monomer. This may be due to disruption in the non-covalent interactions between polymer chains due to the drastic molecular motion during polymerization. However, the role of amorphization on storage in decreasing the mechanical strength for the polymer of polymorph II cannot be ruled out.
Conclusions
In summary, inspired by the various conformations exhibited by β-turn-forming peptides by varying their dihedral angles, we anticipated that a short peptide which can adopt a β-turn conformation in a crystalline state would form various conformational polymorphs with varying dihedral angles. We have established that this property can be exploited to obtain polymers with different packings via topochemical polymerization of such polymorphic monomers. A designed β-turn forming peptide monomer decorated with an azide and an alkyne at its termini, yielded two different polymorphs and a hydrate, each having a ready-to-react, head-to-tail arrangement of molecules in their crystal lattice. Two polymorphs, upon heating, underwent TAAC polymerization yielding the respective polymers. Polymorph I polymerized completely in an SCSC fashion to result in an achiral helical polymer. Polymorph II retained its crystallinity during TAAC polymerization but became amorphous upon storage. The hydrate III underwent dehydration upon heating and transformed into polymorph II before polymerization. The polymers obtained from these different polymorphs exhibited different mechanical strengths and thermal stabilities. This work demonstrates that the rational design of monomers to obtain different topochemically polymerizable polymorphs can be successful and has the potential to achieve access to various polymorphs of a polymer via topochemical polymerization.Data availability
The authors confirm that all the data supporting this study are available within the article and its ESI.† Raw data will be available upon reasonable request.Author contributions
K. M. S. conceived the idea, V. A. carried out the experiments, and K. M. S. and V. A. wrote the manuscript. L. A. M. recorded the solid-state CP-MAS 13C NMR spectra of polymers. R. K. performed the control experiments during revision. Y. Z. performed nanoindentation under the guidance of U. R. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Babu Varghese, consultant to the single crystal X-ray diffraction laboratory in SAIF at IIT Madras, for solving the single crystal structures of the polymer of polymorph I. K. M. S. thanks the Department of Science and Technology, Ministry of Science and Technology, Government of India, for a SwarnaJayanti fellowship (DST/SJF/CSA02/2012-13), and the Science and Engineering Research Board, India, for the research grant (SERB/CRG/000577/2018).References
- N. Leloup, et al. , Nat. Commun., 2017, 8, 1708 CrossRef PubMed.
- S. Crennell, T. Takimoto, A. Portner and G. Taylor, Nat. Struct. Biol., 2000, 7, 1068–1074 CrossRef CAS PubMed.
- E. Campbell, et al. , Nat. Chem. Biol., 2016, 12, 944–950 CrossRef CAS PubMed.
- K. Frontzek, et al. , Nat. Struct. Mol. Biol., 2022, 29, 831–840 CrossRef CAS PubMed.
- H. O. Pierson, in Handbook of Carbon, Graphite, Diamonds and Fullerenes, ed. William Andrew Publishing, Oxford, 1993, pp. 244–277 Search PubMed.
- E. D. Whitney, Nature, 1963, 199, 278–280 CrossRef CAS.
- S. Bram and P. Tougard, Nat. New Biol., 1972, 239, 128–131 CrossRef CAS PubMed.
- J. Bernstein, in Strength from Weakness: Structural Consequences of Weak Interactions in Molecules, Supermolecules, and Crystals, ed. A. Domenicano and I. Hargittai, Springer Netherlands, Dordrecht, 2002, pp. 247–260 Search PubMed.
- M. Wehner, et al. , J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- C.-A. Shen, et al. , Angew. Chem., Int. Ed., 2021, 60, 11949–11958 CrossRef CAS PubMed.
- Z. Li, et al. , J. Am. Chem. Soc., 2019, 141, 19448–19457 CrossRef CAS PubMed.
- R. Liu, et al. , J. Am. Chem. Soc., 2021, 143, 6622–6633 CrossRef CAS PubMed.
- M. A. Neumann, J. van de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef CAS PubMed.
- M. Li, et al. , Nat. Commun., 2019, 10, 2867 CrossRef PubMed.
- S. Marina, et al. , Adv. Funct. Mater., 2021, 31, 2103784 CrossRef CAS.
- D. Gentili, M. Gazzano, M. Melucci, D. Jones and M. Cavallini, Chem. Soc. Rev., 2019, 48, 2502–2517 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- K. Hema, et al. , Chem. Soc. Rev., 2021, 50, 4062–4099 RSC.
- M. Hasegawa, Pure Appl. Chem., 1986, 58, 1179 CrossRef CAS.
- K. Hema, A. Ravi, C. Raju and K. M. Sureshan, Chem. Sci., 2021, 12, 5361–5380 RSC.
- G. Wegner, Makromol. Chem., 1972, 154, 35–48 CrossRef CAS.
- P. R. Pokkuluri, J. R. Scheffer, J. Trotter and M. Yap, J. Org. Chem., 1992, 57, 1486–1494 CrossRef CAS.
- V. V. Boldyrev, React. Solids, 1990, 8, 231–246 CrossRef CAS.
- S. Nagahama, T. Tanaka and A. Matsumoto, Angew. Chem., Int. Ed., 2004, 43, 3811–3814 CrossRef CAS PubMed.
- G. Kaupp, CrystEngComm, 2003, 5, 117–133 RSC.
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- Q. Chu, et al. , J. Am. Chem. Soc., 2018, 140, 4940–4944 CrossRef CAS PubMed.
- D. de Loera, A. Stopin and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2013, 135, 6626–6632 CrossRef CAS PubMed.
- S. Kusaka, et al. , J. Am. Chem. Soc., 2019, 141, 15742–15746 CrossRef CAS PubMed.
- L. Dou, et al. , Science, 2014, 343, 272 CrossRef CAS PubMed.
- M. J. Kory, et al. , Nat. Chem., 2014, 6, 779–784 CrossRef CAS PubMed.
- G. Campillo-Alvarado, et al. , Angew. Chem., Int. Ed., 2019, 58, 5413–5416 CrossRef CAS PubMed.
- C. L. Anderson, et al. , Nat. Commun., 2021, 12, 6818 CrossRef CAS PubMed.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed.
- X. Jiang, et al. , J. Am. Chem. Soc., 2019, 141, 10915–10923 CrossRef CAS PubMed.
- T. Jadhav, et al. , J. Am. Chem. Soc., 2020, 142, 8862–8870 CrossRef CAS PubMed.
- T. N. Hoheisel, et al. , Nat. Chem., 2013, 5, 327–334 CrossRef CAS PubMed.
- R. Rai and K. M. Sureshan, Angew. Chem., Int. Ed., 2022, 61, e202111623 CrossRef CAS PubMed.
- B. P. Krishnan and K. M. Sureshan, J. Am. Chem. Soc., 2017, 139, 1584–1589 CrossRef CAS PubMed.
- R. Rai, B. P. Krishnan and K. M. Sureshan, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 2896–2901 CrossRef CAS PubMed.
- K. Hema and K. M. Sureshan, Acc. Chem. Res., 2019, 52, 3149–3163 CrossRef CAS PubMed.
- R. Mohanrao, K. Hema and K. M. Sureshan, Nat. Commun., 2020, 11, 865 CrossRef CAS PubMed.
- V. Athiyarath and K. M. Sureshan, Angew. Chem., Int. Ed., 2020, 59, 15580–15585 CrossRef CAS PubMed.
- R. Mohanrao, K. Hema and K. M. Sureshan, ACS Appl. Polym. Mater., 2020, 2, 4985–4992 CrossRef CAS.
- A. Ravi, S. Z. Hassan, S. Bhandary and K. M. Sureshan, Angew. Chem., Int. Ed., 2022, 61, e202200954 CAS.
- C. Raju, S. Kunnikuruvan and K. M. Sureshan, Angew. Chem., Int. Ed., 2022, 61, e202210453 CrossRef CAS PubMed.
- B. P. Krishnan, R. Rai, A. Asokan and K. M. Sureshan, J. Am. Chem. Soc., 2016, 138, 14824–14827 CrossRef CAS PubMed.
- R. Khazeber and K. M. Sureshan, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2205320119 CrossRef CAS PubMed.
- K. Hema and K. M. Sureshan, Angew. Chem., Int. Ed., 2020, 59, 8854–8859 CrossRef CAS PubMed.
- V. Athiyarath, M. C. Madhusudhanan, S. Kunnikuruvan and K. M. Sureshan, Angew. Chem., Int. Ed., 2022, 61, e202113129 CAS.
- J. S. Richardson, in Advances in Protein Chemistry, ed. C. B. Anfinsen, J. T. Edsall and F. M. Richards, Academic Press, 1981, vol. 34, pp. 167–339 Search PubMed.
- A. W. Chan, E. G. Hutchinson, D. Harris and J. M. Thornton, Protein Sci., 1993, 2, 1574–1590 CrossRef CAS PubMed.
- A. G. de Brevern, Sci. Rep., 2016, 6, 33191 CrossRef CAS PubMed.
- S. Mondal, et al. , J. Am. Chem. Soc., 2019, 141, 363–369 CrossRef CAS PubMed.
- R. Gessmann, H. Brückner and K. Petratos, J. Pept. Sci., 2021, 27, e3307 CrossRef CAS PubMed.
- H. Hikawa, et al. , CrystEngComm, 2020, 22, 8353–8361 RSC.
- M. De Poli, et al. , J. Org. Chem., 2013, 78, 2248–2255 CrossRef CAS PubMed.
- R. Schweitzer-Stenner, W. Gonzales, G. T. Bourne, J. A. Feng and G. R. Marshall, J. Am. Chem. Soc., 2007, 129, 13095–13109 CrossRef CAS PubMed.
- A. Dutt, R. Fröhlich and A. Pramanik, Org. Biomol. Chem., 2005, 3, 661–665 RSC.
- R. Gessmann, H. Brueckner and M. Kokkinidis, Biochem. Biophys. Res. Commun., 1991, 174, 878–884 CrossRef CAS PubMed.
- N. Jayashree, C. G. Suresh, M. Vijayan, C. Toniolo and G. M. Bonora, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 1618–1621 CrossRef.
- CCDC Deposition numbers 2208396–9 and 2247295 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge crystallographic data centre access structures service.
- C. M. Venkatachalam, Biopolymers, 1968, 6, 1425–1436 CrossRef CAS PubMed.
- M. C. Madhusudhanan, H. Balan, D. B. Werz and K. M. Sureshan, Angew. Chem., Int. Ed., 2021, 60, 22797–22803 CrossRef CAS PubMed.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed.
- Q.-H. Guo, et al. , J. Am. Chem. Soc., 2020, 142, 6180–6187 CrossRef PubMed.
- Z. Li, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2009, 131, 634–643 CrossRef CAS PubMed.
- M. Garai, R. Santra and K. Biradha, Angew. Chem., Int. Ed., 2013, 52, 5548–5551 CrossRef CAS PubMed.
- T.-J. Hsu, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2012, 134, 142–145 CrossRef CAS PubMed.
- A. Adochitei and G. Drochioiu, Rev. Roum. Chim., 2011, 56, 783–791 CAS.
- R. M. Gohil, C. K. Patel, K. C. Patel and R. D. Patel, Starch, 1976, 28, 95–100 CrossRef CAS.
- H. R Kricheldorf and T. Krawinkel, J. Macromol. Sci., Part A: Pure Appl.Chem., 1998, A35, 1853–1873 CrossRef.
- M. K. Mishra, G. R. Desiraju, U. Ramamurty and A. D. Bond, Angew. Chem., Int. Ed., 2014, 53, 13102–13105 CrossRef CAS PubMed.
- M. K. Mishra, U. Ramamurty and G. R. Desiraju, Curr. Opin. Solid State Mater. Sci., 2016, 20, 361–370 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2208396–2208399 and 2247295. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00053b |
| This journal is © The Royal Society of Chemistry 2023 |