
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral organophosphates as ligands in asymmetric metal catalysis
Nikolai
Brodt
and
Jochen
Niemeyer
 *
*
Faculty of Chemistry (Organic Chemistry) and Centre of Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstr. 7, 45141 Essen, Germany. E-mail: jochen.niemeyer@uni-due.de
First published on 29th March 2023
Abstract
Chiral organophosphoric acids have made a huge contribution in the context of asymmetric organocatalysis. Surprisingly, the application of chiral organophosphates as ligands in asymmetric metal-catalysis is less well-studied. However, based on the number of different readily available chiral phosphates (e.g. biphenyl phosphates, binaphthyl phosphates, spirocyclic phosphates or vaulted biphenanthrol phosphates) and the large number of available metal sources, this area of research offers many exciting possibilities. In this Minireview, the most recent examples of chiral metal-phosphate catalysis are discussed, including main-group catalysis using Li-, Mg-, Ca-, In- or Bi-phosphates, transition-metal catalysis using Ti-, Mn-, Rh-, Pd-, Ag- and Au-phosphates and catalysis by rare-earth metals based on Yb-phosphates.
 Jochen Niemeyer and Nikolai Brodt | Nikolai Brodt (right) studied Chemistry at the University of Duisburg-Essen (Germany). After his bachelor-thesis in the group of Mathias Ulbricht (Technical chemistry), he moved to the group of Jochen Niemeyer for his master-thesis, working on novel synthetic methods for interlocked molecules. After completing his master's degree in 2022, Nikolai is now working towards his PhD and continues to work on the synthesis and application of interlocked molecules. |
Jochen Niemeyer (left) studied chemistry at the University of Muenster (Germany) and obtained his PhD in 2009 in the group of Prof. Gerhard Erker. After postdoctoral work with Prof. Simon Aldridge in Oxford (UK) from 2010–2011, he worked in the R&D department of Evonik Industries AG. In 2014, Jochen started his independent academic career at the University of Duisburg-Essen (Germany) and was appointed as Heisenberg-Professor for Organic and Supramolecular Chemistry in 2020. The Niemeyer-group is working on the development of novel chemosensors and organocatalysts. Here, the group has developed functional interlocked molecules featuring chiral organophosphosphoric acids as a novel class of organocatalysts. |
1. Introduction
Chiral phosphoric acids (CPAs) are among the most widely used organocatalysts.1 Since their initial organocatalytic application in 2004,2 CPAs have enabled a plethora of enantioselective transformations. Their Brønsted acidic nature enables the activation of Lewis-basic substrates, most commonly imines, quinolines or other nitrogen-containing compounds.3 More recently, the use of highly acidic CPAs has also allowed for activation of less activated electrophiles, such as simple olefins.4 In terms of catalyst structure, 1,1′-binaphthyl-2,2′-diol (BINOL) based phosphoric acids have been used most commonly,5 but other chiral backbones such as 1,1′-biphenyl-2,2′-diols, SPINOL or VAPOL have also found application (see Fig. 1a for the structure of these backbones).6 In addition, the nature of the acidic center has been varied: next to OH-acidic compounds of the type (ArO)2P(O)OH, related CH acids, NH acids or combined acids have also been used.7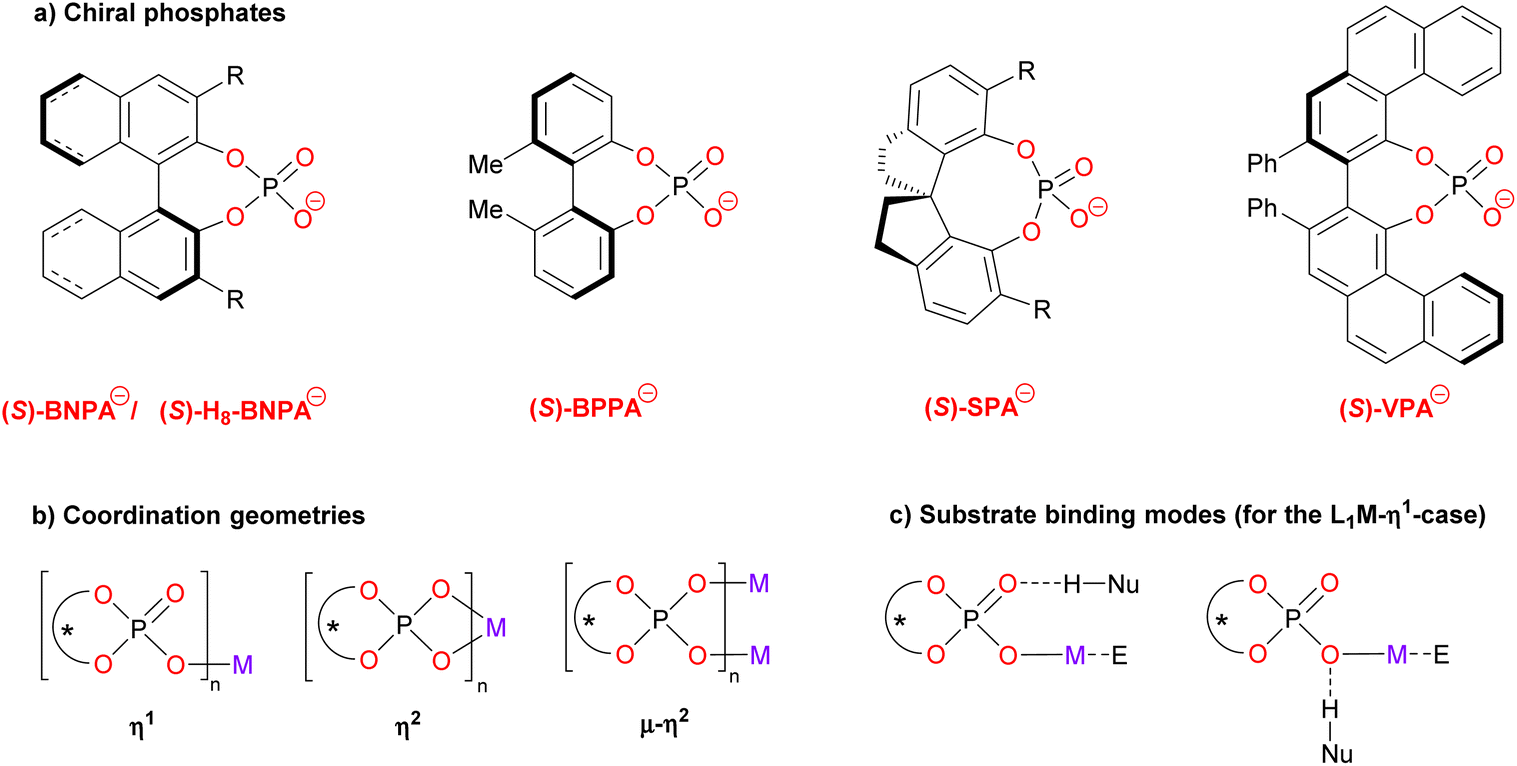 | ||
| Fig. 1 (a) Chiral phosphate ligands discussed in this review. BNPA−: binaphthylphosphoric acid anion, BPPA−: biphenylphosphoric acid anion, SPA−: spirocyclic phosphoric acid anion, VPA−: vaulted biphenanthrol phosphate anion. (b) Possible metal coordination geometries. (c) Possible substrate binding modes. | ||
Soon, it was realized that chiral phosphoric acids can also be used as the corresponding phosphate anions in different contexts: List and others developed asymmetric counteranion directed catalysis (ACDC), which is based on the ionic interaction of the phosphate anions with cationic electrophiles, such as iminium-ions.8 Toste and others discovered that chiral phosphates can act as anionic phase-transfer catalysts, e.g. by solubilizing cationic reagents such as Selectfluor© in organic solvents.9
Last but not least, chiral phosphates have also been used as chiral ligands in enantioselective metal catalysis, using main group metals, transition metals and rare-earth metals. However, when compared to the extensive application of chiral phosphoric acids in organocatalysis, the number of examples of enantioselective catalysis with phosphate-metal complexes is still small. In our opinion, this leaves a lot of space for further developments in this exciting research area, so that a timely review of the state-of-the-art in this area would be highly helpful.
Accordingly, this Minireview will discuss recent examples of chiral phosphates that have been used as ligands in enantioselective metal catalysis. Previous developments in this field have been reviewed by Rueping in 2014,1 followed by an update in 2017.10 To clearly set the scope for this Minireview, we will mention all examples (to the best of our knowledge) that are not included in Rueping's review. Thus, to give a full picture of recent developments we have included some examples that were mentioned in reviews on related topics, such as enantioselective Friedel–Crafts reactions,11 Mannich reactions,12 radical reactions,13 C–H functionalization reactions14 and multicomponent reactions,15 or in reviews about enantioselective catalysis with complexes of rhodium,16 silver,17 gold18 or lanthanides.19 In order to focus on phosphates as chiral ligands, we have excluded examples of tandem catalysis, in which a phosphoric acid and a metal-catalyst act in independent catalytic cycles.
The structure of this Minireview is based on the metals that are involved in the catalytic reactions, starting with main group metals (i.e. Li, Mg, Ca, In, Bi; chapters 2.1–2.5), followed by transition metals (i.e. Ti, Mn, Rh, Pd, Ag, Au; chapters 3.1–3.6) and rare-earth metals (Yb, chapter 4). The number of phosphate ligands that have been employed in these examples is limited (see Fig. 1a for the structures): in most cases, binaphtyl-phosphates (BNPA−) or the partially hydrogenated derivatives (H8-BNPA− ) were employed.5 However, some examples also feature biphenyl phosphates (BPPA−), spirocyclic phosphates (SPA−)6a and vaulted biphenanthrol phosphates (VAPOL-PA−).6b Some very recent examples that were published during the writing and peer-review process of this Minireview can be found in ref. 20.
Commonly, the chiral ligands are used as the monoanionic phosphates which can coordinate cationic metal centers in various coordination geometries (see Fig. 1b). Thus, they can act as monodentate ligands (η1), as bidentate ligands (η2) or as bridging bidentate ligands (μ–η2). As for the substrate binding, the metal will serve as the Lewis-acidic binding site for the electrophile in most cases. A nucleophile with an H-bond donor can bind to either of the phosphate oxygens (e.g. the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O fragment or the P–O–M fragment, see Fig. 1c for the η1-case). Wherever possible, we will depict and discuss the coordination geometries of these complexes (as suggested by the authors), because we believe that a deeper understanding of the coordination chemistry will give insights for further catalyst development.
O fragment or the P–O–M fragment, see Fig. 1c for the η1-case). Wherever possible, we will depict and discuss the coordination geometries of these complexes (as suggested by the authors), because we believe that a deeper understanding of the coordination chemistry will give insights for further catalyst development.
2. Main-group metals
Chiral phosphates have been used as ligands for different main-group metals, such as lithium, magnesium, calcium, indium and bismuth. Here, the phosphate-bound metal center commonly acts as Lewis acid that binds one of the substrates. This chapter will describe recent examples (since 2014) involving the metals mentioned above. Earlier examples based on the same metals, but also based on sodium- or aluminium-phosphates can be found in Rueping's review.12.1 Lithium-catalysts
In case of lithium-phosphate based catalysts, Niemeyer and coworkers used interlocked rotaxane-catalysts (1a/b, 2a/b, see Fig. 2), consisting of a chiral BNPA-based macrocycle and an ammonium thread.21 The rotaxanes were used as catalysts for the asymmetric Michael addition of diethyl malonate (4) to cinnamaldehyde and its derivatives (3). When used as the ammonium-phosphate zwitterion (ArO)2POO− H2N+R2, no conversion was observed due to the effective shielding of the ammonium station by the macrocycle.22 Thus, different metal hydroxides (KOH, NaOH, LiOH) were used to deprotonate the ammonium-thread, giving the rotaxanes with a metal-phosphate macrocycle and an amine-thread (ArO2POO-M HNR2). Lithium as a counterion gave the best results in terms of conversion and enantioselectivity. Four different rotaxanes were investigated as catalysts, trying to evaluate the influence of the length of the thread (rotaxanes 1/2) and the substitution pattern of the macrocycle (rotaxanes a/b) on the performance of the catalysts. In order to investigate the influence of the mechanical bond of the catalytic outcome, the non-interlocked mixtures of the corresponding macrocycle and thread were used as reference catalysts.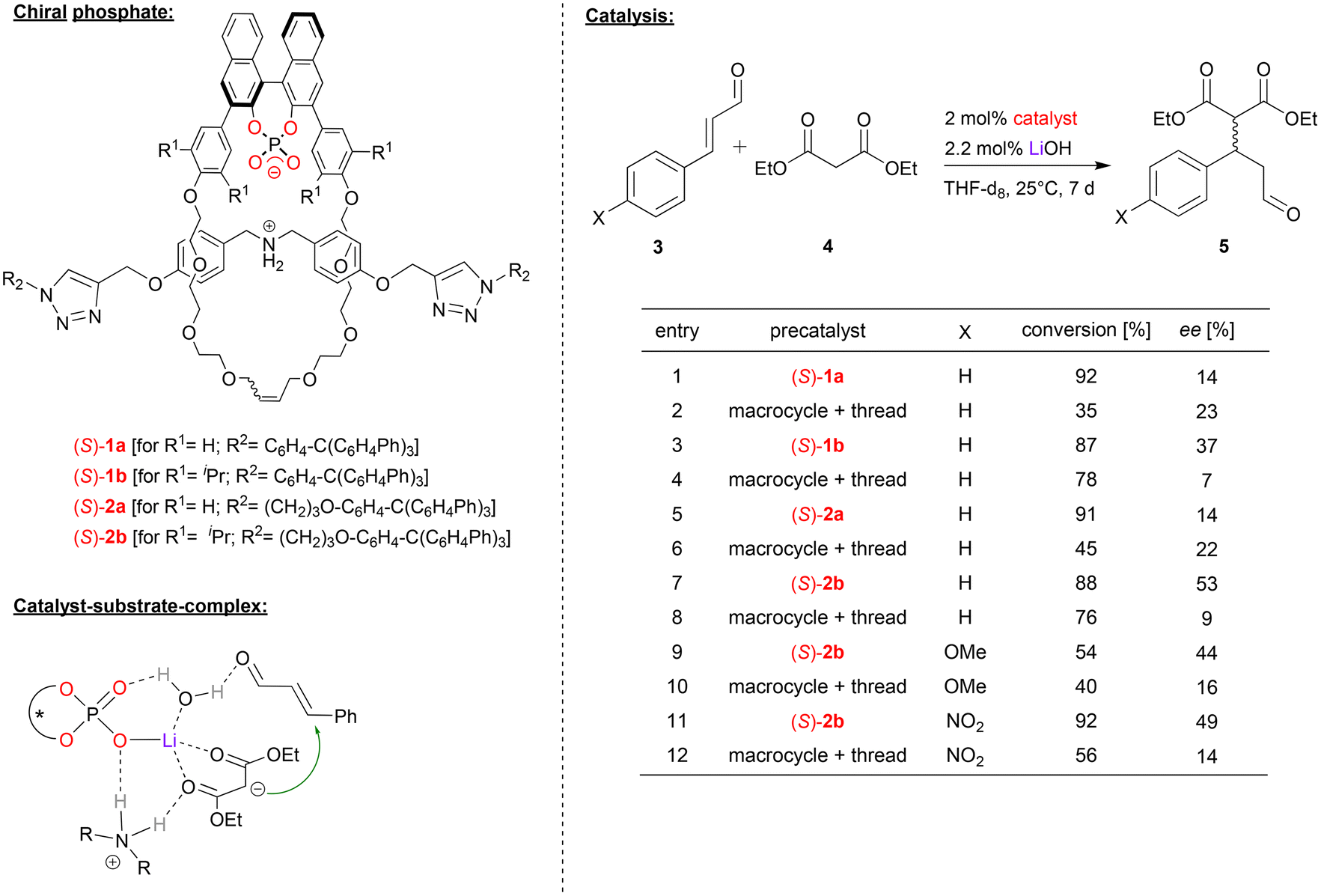 | ||
| Fig. 2 Enantioselective Michael-addition catalyzed by Li-phosphate rotaxanes.21 | ||
For the Michael-addition of diethylmalonate to cinnamaldehyde, all rotaxane-catalysts give better conversions than the control catalysts. The length of the thread (i.e. catalyst (S)-1avs.2a and (S)-1bvs.2b) has no significant influence. This is different for the macrocycle-substitution: for the unsubstituted macrocycles (R1 = H, i.e. catalysts (S)-1a/2a), enantioselectivities are low for both the control catalysts and the rotaxanes (14–23% ee). However, for the sterically more hindered catalysts (R1 = iPr, i.e. catalysts (S)-1b/2b), enantioselectivities are significantly higher for the rotaxanes (37/53% ee) than for the control catalysts (7/9% ee). This was also found for substituted cinnamaldehyde derivatives using catalyst (S)-2b.
DFT-calculations show that the η1-phosphate-lithium complex acts as a Lewis-acid towards the diethylmalonate, this facilitating its deprotonation by the amine-thread. The resulting malonate anion and ammonium cation remain coordinated to the Li-phosphate. In the Michael-addition step, the cinnaldehyde can also be bound via a Li-coordinated water-molecule, thus placing both substrates in the chiral environment of the Li-phosphate (see Fig. 2). According to the calculations, the interlocked nature of the catalyst not only facilitates the necessary deprotonation/reprotonation steps by the amine/ammonium-function (increasing the reaction rate), but also leads to a slightly increased stereoselectivity, as observed experimentally.
Wang and coworkers used the Li-phosphate Li[(R)-6] together with a photoredox catalyst in an asymmetric dicarbofunctionalization of enamides (see Fig. 3).23 This work is based on an earlier publication by the same group, where they described the Li-phosphate catalyzed addition of indoles to imines, giving the corresponding indolyl-1-alkylamines in good enantioselectivities (up to 97% ee). The corresponding imines were generated in situ from amino-acid derivatives (protected as the N-(acyloxy)phthalimides) that served as redox-active esters (RAEs). These undergo oxidation and decarboxylation to the corresponding imines when irradiated in the presence of a photocatalyst such as Ru(bpy)3(PF6)2.24 In this work, this was now extended towards a multicomponent reaction, where the imines are generated in situ from α-phenyl enamides 7 and phenylacetic acid derived RAEs 8 (see Fig. 3). Here, the Li-phosphate plays a double role: first, the catalyst, the enamide and the RAE form a ternary complex, which upon irradiation in presence of the photosensitizers leads to addition of a benzyl-radical (from the RAE) to the enamide, resulting in the corresponding imine, CO2 and phthalimide. Then, the η1-phosphate-lithium complex coordinates both the prochiral imine and the indole-nucleophile, catalyzing the enantioselective addition to give the tertiary amines 10.
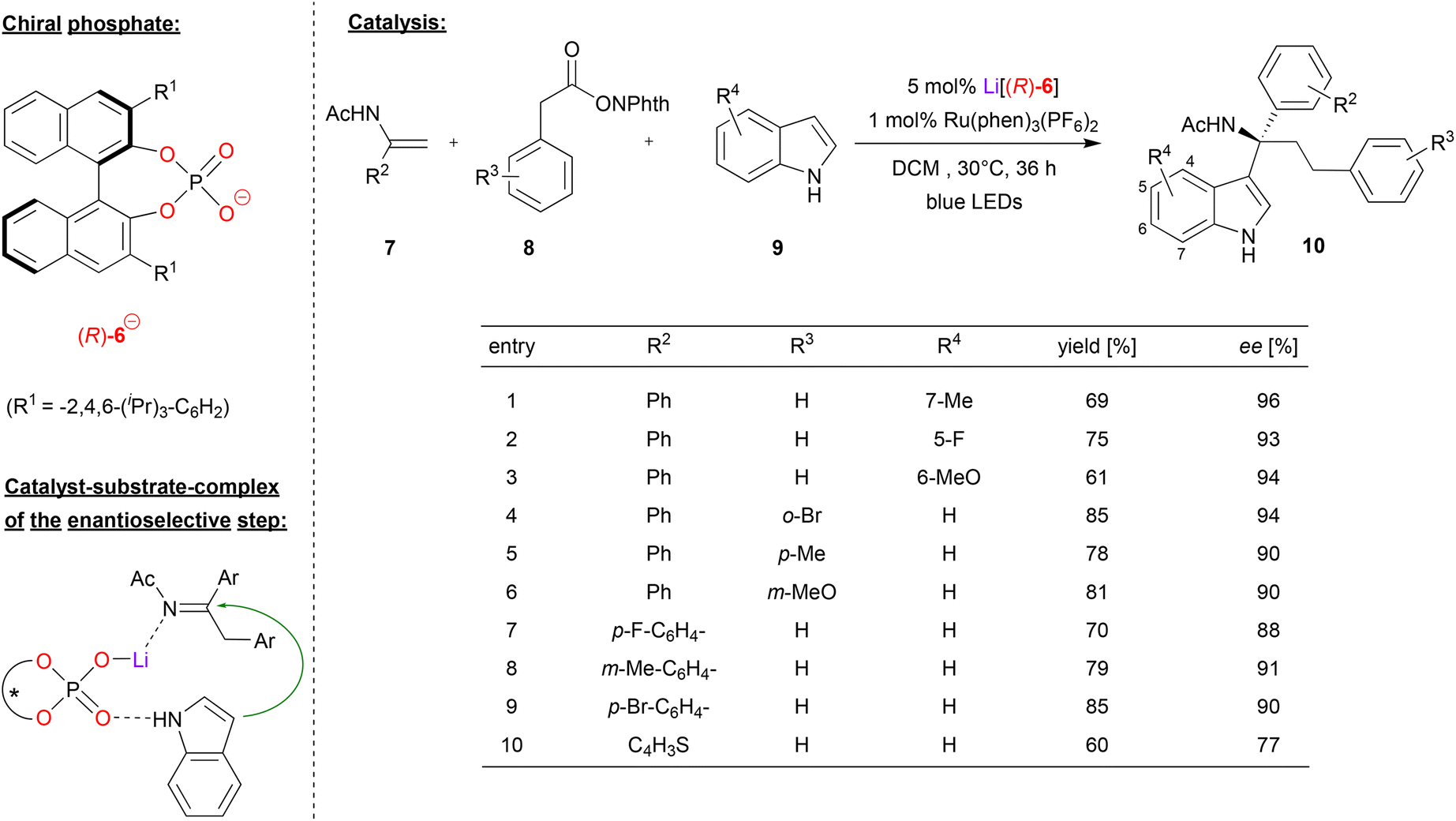 | ||
| Fig. 3 Light mediated enantioselective dicarbofunctionalization of enamides catalyzed by a Li-phosphate.23 | ||
Regarding the substrate scope (see Fig. 3 for selected examples), different substituents could be used on all three components. In terms of the indole-substrate, a wide range of electron-donating and electron-withdrawing groups in 5, 6 and 7 positions resulted in high levels of enantioselectivity (93–96% ee), as was the case for different aryl-substituents on the phenylacetic acid derivative (90–94% ee). Substitution on the enamide gave slightly lower enantioselectivities (88–91% ee), which was also observed for thiophene-analogue of the enamide (77% ee).
Antilla and coworkers used the H8-BINOL-phosphate (R)-11 as the corresponding Li-phosphate for the asymmetric ring opening of meso-epoxides with aromatic thiols (see Fig. 4).25 As exemplified in Fig. 4, cyclohexene epoxide, but also trans-cyclohexadiene-bis-epoxide and benzo-cyclohexene epoxide were opened by thiophenol to give the trans-2-hydroxy-1-thioethers 14 in good enantioselectivities (87–93% ee). For cyclopentane epoxide, the enantioselectivity was lower (75% ee), while cyclohexadiene monoepoxide gave the best selecitivity (94% ee). With regard to the nucleophile, differently substituted thiophenols were well-tolerated with consistently high stereoselectivities: thiophenols with electron withdrawing groups, (e.g. Cl, CF3), electron donating groups (e.g. Me, OMe), and even aminothiophenol were successfully used, giving the products with 88–96% ee. Aliphatic thiols were also tested as nucleophiles but gave no product. Mechanistically, the authors suggest a Lewis-acid activation of the epoxide by an η1-phosphate-lithium complex, together with coordination of the thiophenol by the Lewis-basic PO unit. This ternary complex allows facile and stereoselective opening the meso-epoxides with high enantioselectivities.
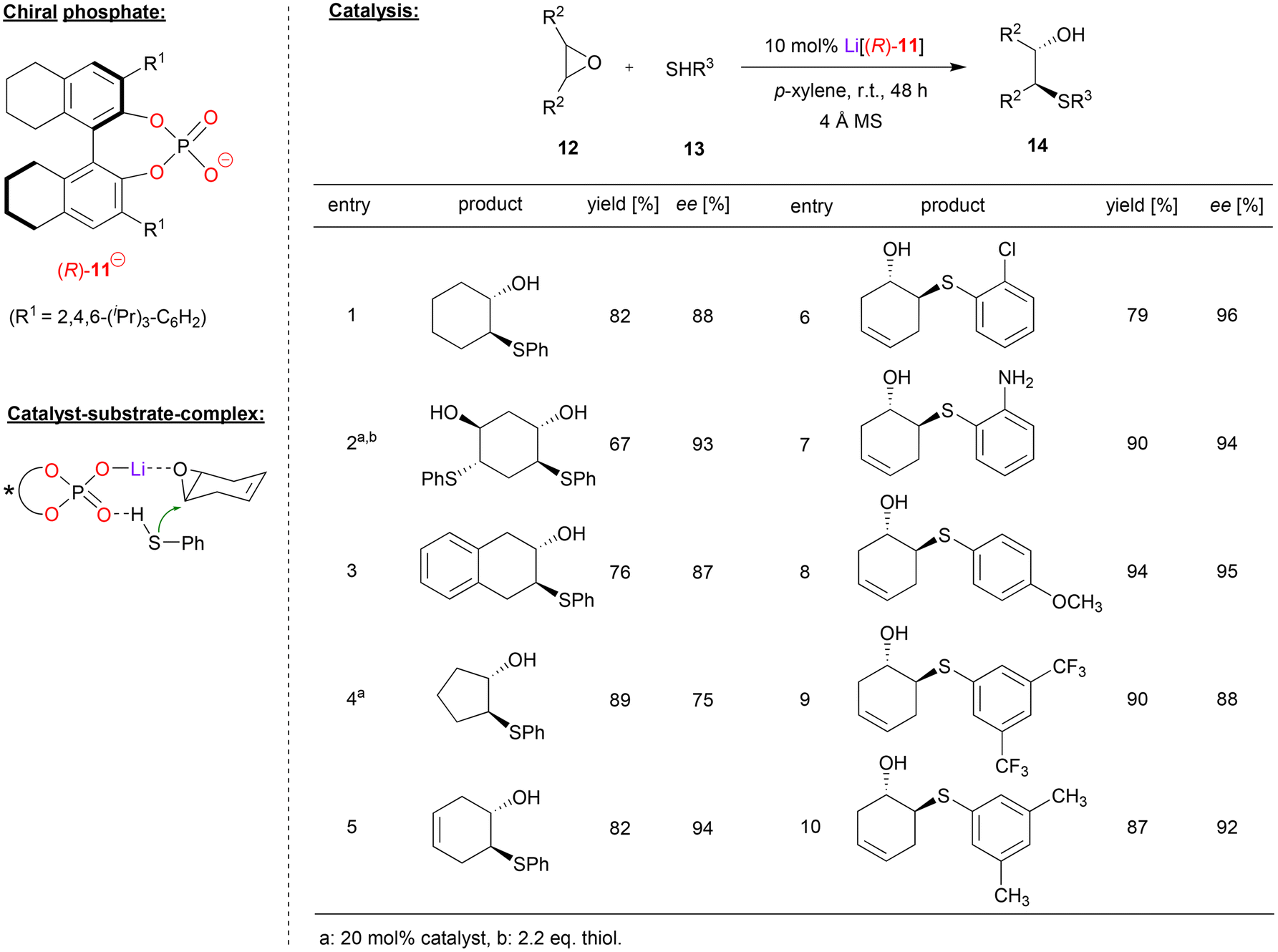 | ||
| Fig. 4 Enantioselective desymmetrization of meso-epoxides catalyzed by a Li-phosphate.25 | ||
Shao and coworkers accomplished the N-propargylation of indoles by using the SPINOL-phosphate (R)-15 as its lithium complex (see Fig. 5).26 They employed the alkynyl-substituted N,O-acetals 16, which upon liberation of ethanol deliver the corresponding α-alkynyl imines in situ (this first step is also catalyzed by the lithium phosphate). Subsequent addition of the indole then delivers the desired N-propargylated indoles, namely the chiral N,N-acetals 18. As can be seen from selected examples in Fig. 5, overall good yields (61–80%) and good to excellent enantioselectivities (90–99% ee) could be achieved in many cases. Different alkyne and indole substituents were well-tolerated and even high temperatures (110 °C) do not affect the enantioselectivity. Importantly, no competitive C3-reaction is observed and only the N1-position serves as the nucleophile.
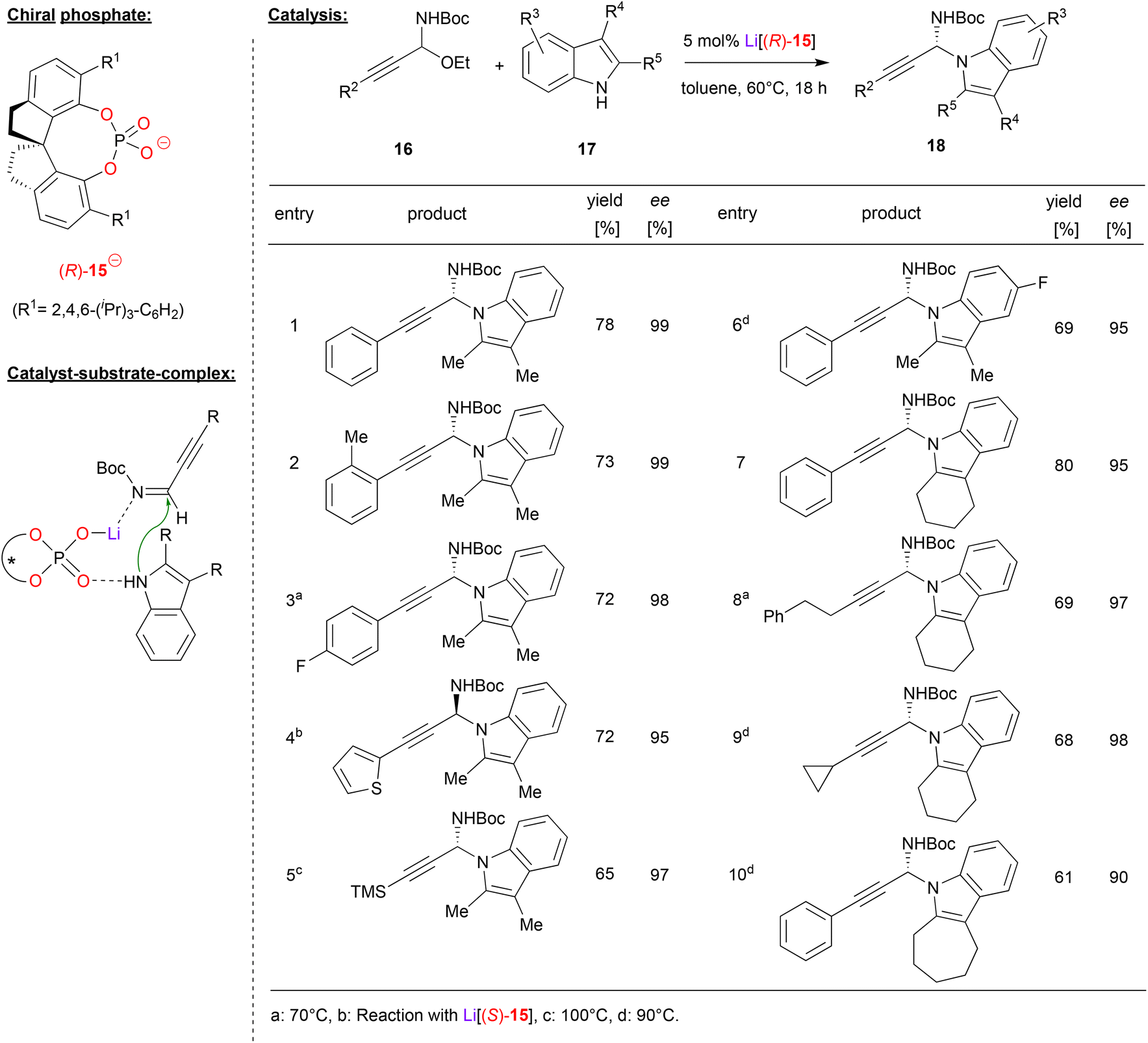 | ||
| Fig. 5 Enantioselective N-propargylation of indoles and carbazoles catalyzed by a Li-phosphate.26 | ||
Shao and coworkers suggest a dual activation mode of the catalyst. The indole binds to the Lewis-basic PO unit, while the η1-O–Li fragment serves as a Lewis acid to coordinate the imine.
2.2 Magnesium-catalysts
Chiral phosphates can also form efficient catalysts when combined with dicationic alkaline earth metals like magnesium or calcium.Antilla and coworkers used (R)-H8-BNPA 19 in combination with magnesium to generate the homoleptic complex Mg[(R)-19]2. This complex catalyzes the asymmetric Friedel–Crafts alkylation/N-hemiacetalization of 4-aminoindoles (see Fig. 6).27 The first reaction step involves the Michael addition of 4-aminoindoles 21 to the β,γ-unsaturated α-ketoesters 20, which occurs via the nucleophilic C7 of the aminoindole framework. Then, intramolecular attack of the indole N1 leads to formation of the stable hemiacetals. As can be seen for selected examples in Fig. 6, different substituents were used on the α-ketoesters, giving high enantioselectivities (86–99% ee). Different substituents on the 4-amino group of the indole were also tolerated with virtually no change in ee (94–99% ee), probably due to the fact that these groups are remotely placed from the reaction center.
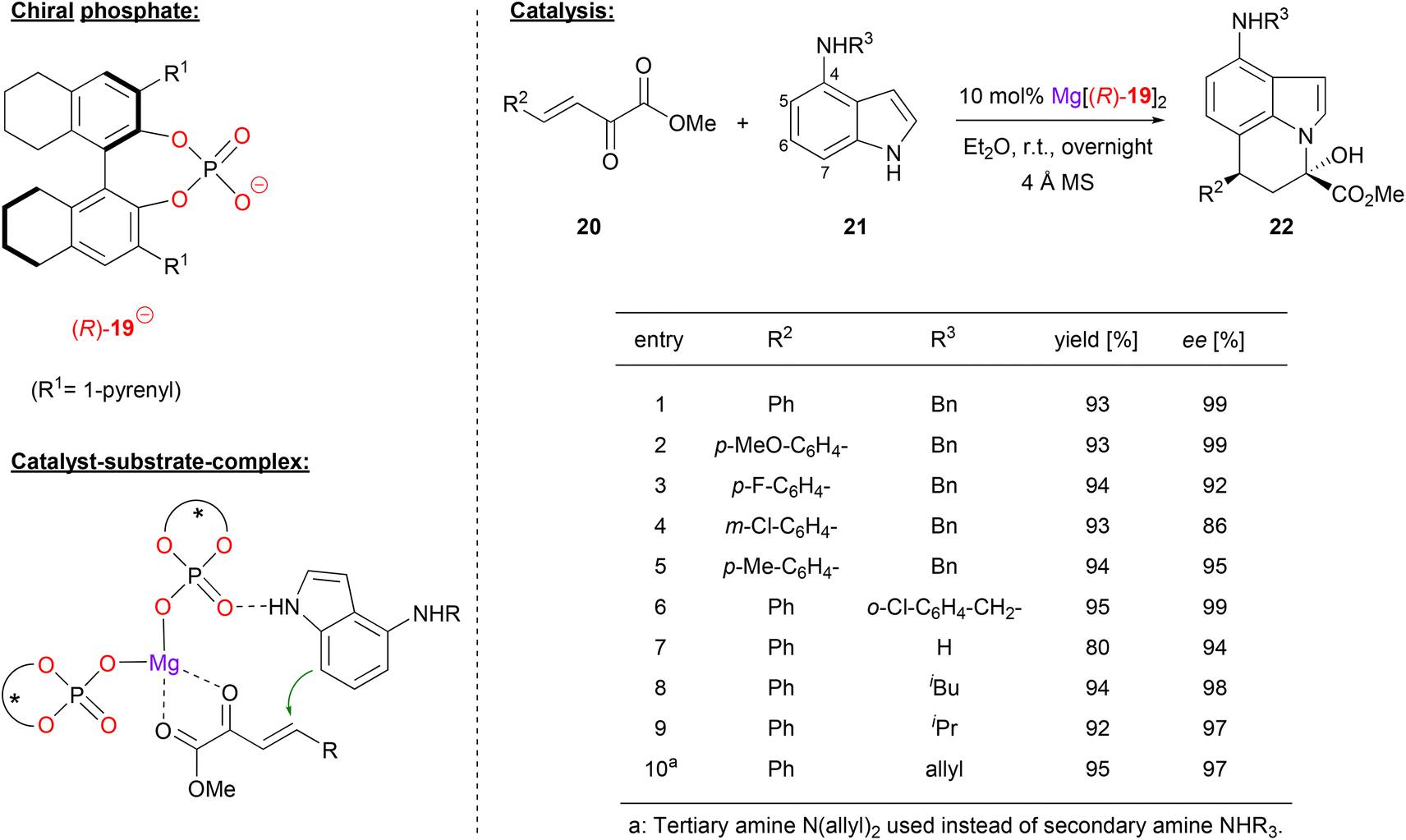 | ||
| Fig. 6 Enantioselective Friedel–Crafts-alkylation/N-hemiacetalization catalyzed by a Mg-bisphosphate catalyst.27 | ||
Mechanistically, the authors propose coordination of the ketoester to the Lewis-acidic magnesium center in the bis-(η1-phosphate)-magnesium complex. Once again, coordination of the nucleophile is also possible, in this case most likely via the indole NH-group.
Lin and coworkers used the chiral BINOL derivative (R)-23 together with MgSO4 to promote the [3 + 3] annulation of aminopyrazoles with trifluoromethyl-methylidene oxindoles 25 for construction of trifluoromethylated pyrazolo[3,4-b]pyridine-6-ones (see Fig. 7).28 In contrast to the other examples in this review, Lin used the catalyst as a combination of phosphoric acid (not phosphate) and magnesium sulfate, so this protocol might be termed as Lewis acid assisted phosphoric acid catalysis (not Lewis acid catalysis with a magnesium phosphate, as in Antilla's work).
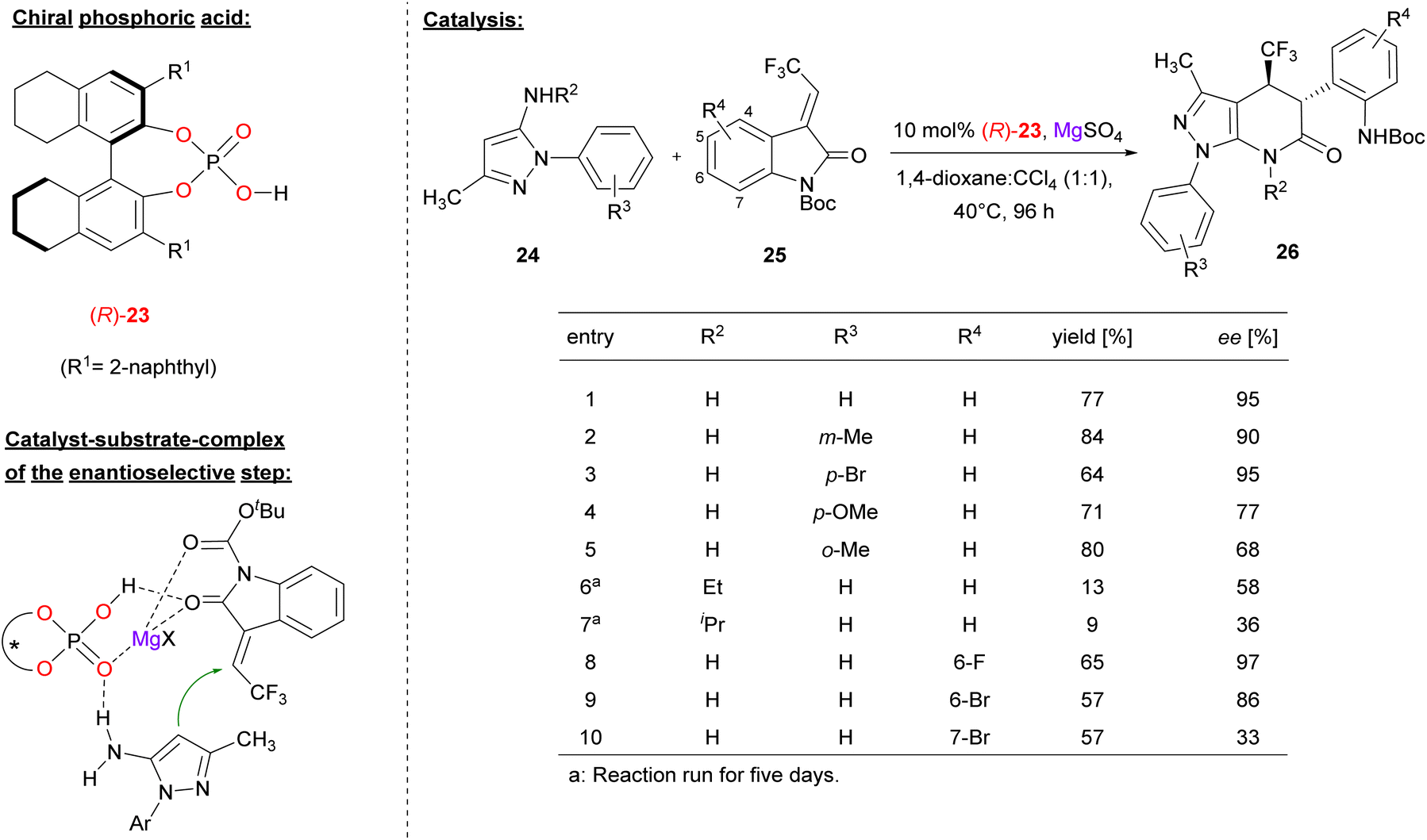 | ||
| Fig. 7 Enantioselective [3 + 3] annulation catalyzed by synergistic action of a phosphoric acid and Mg2+.28 | ||
In the reaction, the nucleophilic C4-position of the aminopyrazole 24 performs a Michael addition to the methylidene oxindole 25. Subsequent transamidation via intramolecular attack of the amino-group onto the oxoindole gives the pyridone products 26 with two adjacent stereocenters.
Regarding the substrate scope (see Fig. 7 for selected examples), the pyrazole could be equipped with different aryl groups bearing electron withdrawing groups (e.g. Br) or electron-donating groups (e.g. Me or OMe) to give to products in good yields (64–84%) and good to excellent enantioselectivities (68–95% ee). Variation of the primary amine by using the N-ethyl/isopropyl derivatives led to diminished enantioselectivities (58%/36% ee). With regard to the indoles, substituents in the 6-positions were tolerated (e.g. 6-F/Br, giving 97%/86% ee), while substitutents in the 7-position were not (e.g. 7-Br, giving 33% ee).
Mechanistically, the authors proposed a synergistic action of the phosphoric acid (R)-23 and Mg2+: binding of both oxygen donors of the Boc-protected oxindole (to the Mg-center and the P(O)OH-group, respectively) enhances its electrophilicity, while simultaneous binding of the aminopyrazol is possible via a PO⋯HN hydrogen bond.
Mg-bisphosphates were also used for the synthesis of 1,3-oxazolidines and 1,3-oxazinanes,29 for the Diels–Alder reaction of benzoquinones with vinylindoles30 as well as for the synthesis of spirocyclic dihydroquinolones31 (not shown in detail).
2.3 Calcium-catalysts
Akiyama and coworkers used (R)-BNPA 27 in combination with calcium for the enantioselective Friedel–Crafts alkylation of indoles 29 with α-substituted-β-nitrostyrenes 28 (see Fig. 8).32 This reaction gives rise to alkylated indole-derivates 30 with an all-carbon quaternary stereocenter.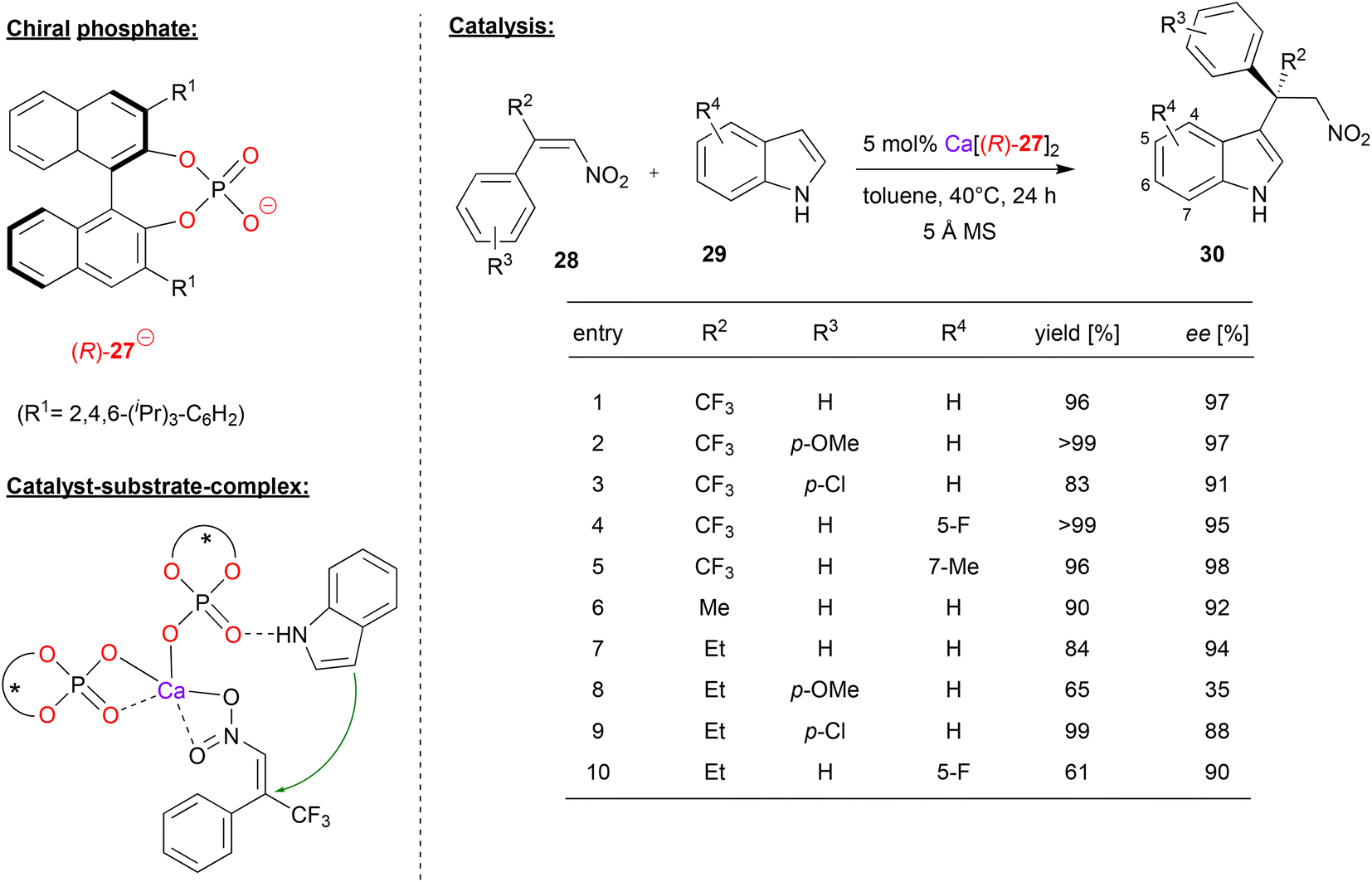 | ||
| Fig. 8 Enantioselective Friedel–Crafts alkylation of indoles catalyzed by a Ca-bisphosphate.32 | ||
First, the authors investigated the use of α-trifluoromethyl-β-nitrostyrene as the electrophile. As exemplified in Fig. 8, differently substituted nitrostyrenes and indoles could be used to give the corresponding products in excellent stereoselectivities (95–98% ee). When using α-alkyl-β-nitrostyrenes, stereoselectivites are generally lower: for the parent α-methyl- or α-ethyl-β-nitrostyrene together with indole, stereoselectivities are still high (92/94% ee), but substitution on the styrene (especially for the p-OMe case) or the indole leads to decreased stereoselectivities (35–90% ee).
According to the proposed mechanism, which was based on by DFT-calculations, the preferred transition state (leading to the experimentally observed product enantiomer) features a calcium-bisphosphate complex with one phosphate acting as a bidentate η2-ligand and one phosphate acting as a monodentate η1-ligand (see Fig. 8). The latter phosphate can coordinate the indole by a PO⋯HN hydrogen-bond, while the nitrostyrene can coordinate to the calcium via both oxygen-donors. Interestingly, the disfavoured diastereomeric transition state (2.8 kcal mol−1 above the favoured transition state) features a distorted geometry, which only allows for monodentate η1-binding of both phosphates (not shown).
Masson and coworkers also used Ca-bisphosphate catalysts, in their case for the asymmetric synthesis of 1,2-hydrazinoamines.33 To this end, the enamides 32 were reacted with azodicarboxylates 33 to give the corresponding hydrazinoamines 34 after reductive workup (see Fig. 9). The enamides were used as the (E)-isomers, with aryl-substituents in the α-position and alkyl-groups in the β-position, respectively. As can be seen from selected examples in Fig. 9, different aryls as well as different n-alkyl chains were well tolerated (83–95% ee). Only for the thiophene-derivative, the stereoselectivity dropped significantly (22% ee), possibly due to competitive binding of the sulfur to the catalyst.
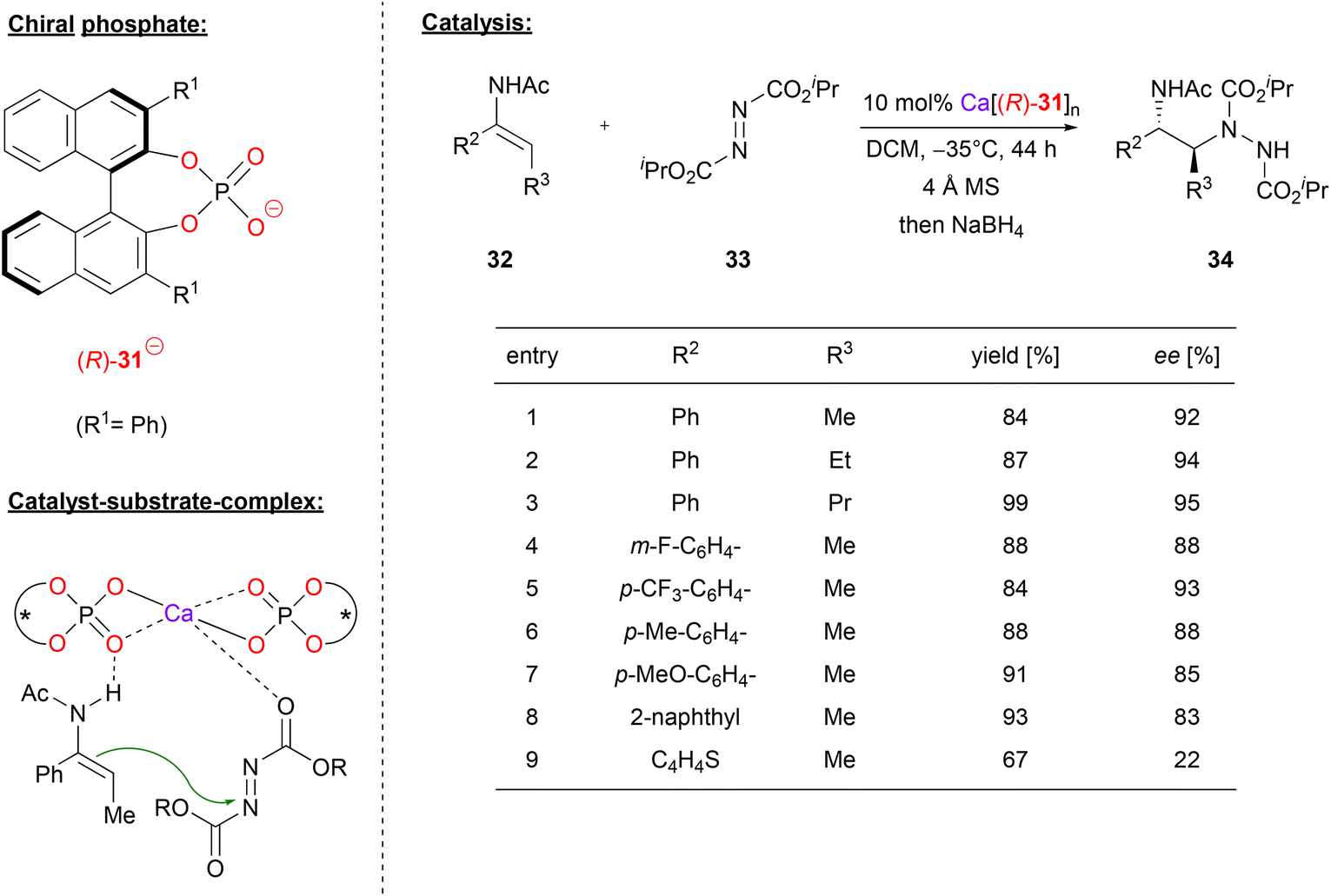 | ||
| Fig. 9 Enantioselective synthesis of 1,2-hydrazinoamines catalyzed by a Ca-bisphosphate.33 | ||
The authors point out that the exact stoichiometry of the complex Ca[(R)-31]n might be variable, even if both components are used in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. This is supported by NMR and MALDI-TOF measurements (although the species distribution in the MALDI-TOF might differ from that in solution). A strong negative nonlinear effect was observed between the enantiopurity of the phosphate and the enantiopurity of the product, clearly showing the involvement of more than one phosphate in the stereodetermining transition state. The nonlinear effect is especially strong in this case, because the heterochiral meso-complex seems to be significantly more active than its homochiral counterpart. Based on DFT-calculations, the authors suggest that the main catalytically active species is a bis-(η2-phosphate)-calcium complex. This acts as a Lewis-acid to coordinate the azodicarboxylate via one of its oxygen-atoms, while the enamide-nucleophile is complexed via a PO⋯HN hydrogen bond (see Fig. 9). In this arrangement, the major transition state is favoured by 1.1 kcal mol−1, giving the (2S)-product, as observed experimentally.
:2 ratio. This is supported by NMR and MALDI-TOF measurements (although the species distribution in the MALDI-TOF might differ from that in solution). A strong negative nonlinear effect was observed between the enantiopurity of the phosphate and the enantiopurity of the product, clearly showing the involvement of more than one phosphate in the stereodetermining transition state. The nonlinear effect is especially strong in this case, because the heterochiral meso-complex seems to be significantly more active than its homochiral counterpart. Based on DFT-calculations, the authors suggest that the main catalytically active species is a bis-(η2-phosphate)-calcium complex. This acts as a Lewis-acid to coordinate the azodicarboxylate via one of its oxygen-atoms, while the enamide-nucleophile is complexed via a PO⋯HN hydrogen bond (see Fig. 9). In this arrangement, the major transition state is favoured by 1.1 kcal mol−1, giving the (2S)-product, as observed experimentally.
Antilla's group successfully applied Ca-bisphosphate catalysts for various reactions, e.g. for the synthesis of chlorinated oxindoles and geminal diamines,34 for the amidation of imines,35 as well as for the amination of benzofuranones36 and β-keto esters37 (not shown in detail).
In a recent example, Antilla's group also used an azodicarboxylate electrophile (cf. Masson's work in Fig. 9), in this case for an enantioselective amination of 3-aryl-2-oxindoles, effected by a calcium bisphosphate catalyst.38 As exemplified in Fig. 10, differently substituted oxindoles 36 could be used in combination with dibenzylazodicarboxylate 37 to give the 3-aryl-3-amino-oxindoles 38. With regard to substituents in the 3-position of the indole (the position of the newly formed stereocenter), aryl-groups give high stereoselectivity (92–94% ee). Interestingly, a simple methyl group is also tolerated (87% ee), but a benzyl-group gives low stereoselectivity (20% ee). Substitution at the 5- or 7-position has no significant influence on stereoselectivity, both for electron-withdrawing and electron-donating groups (93–97% ee).
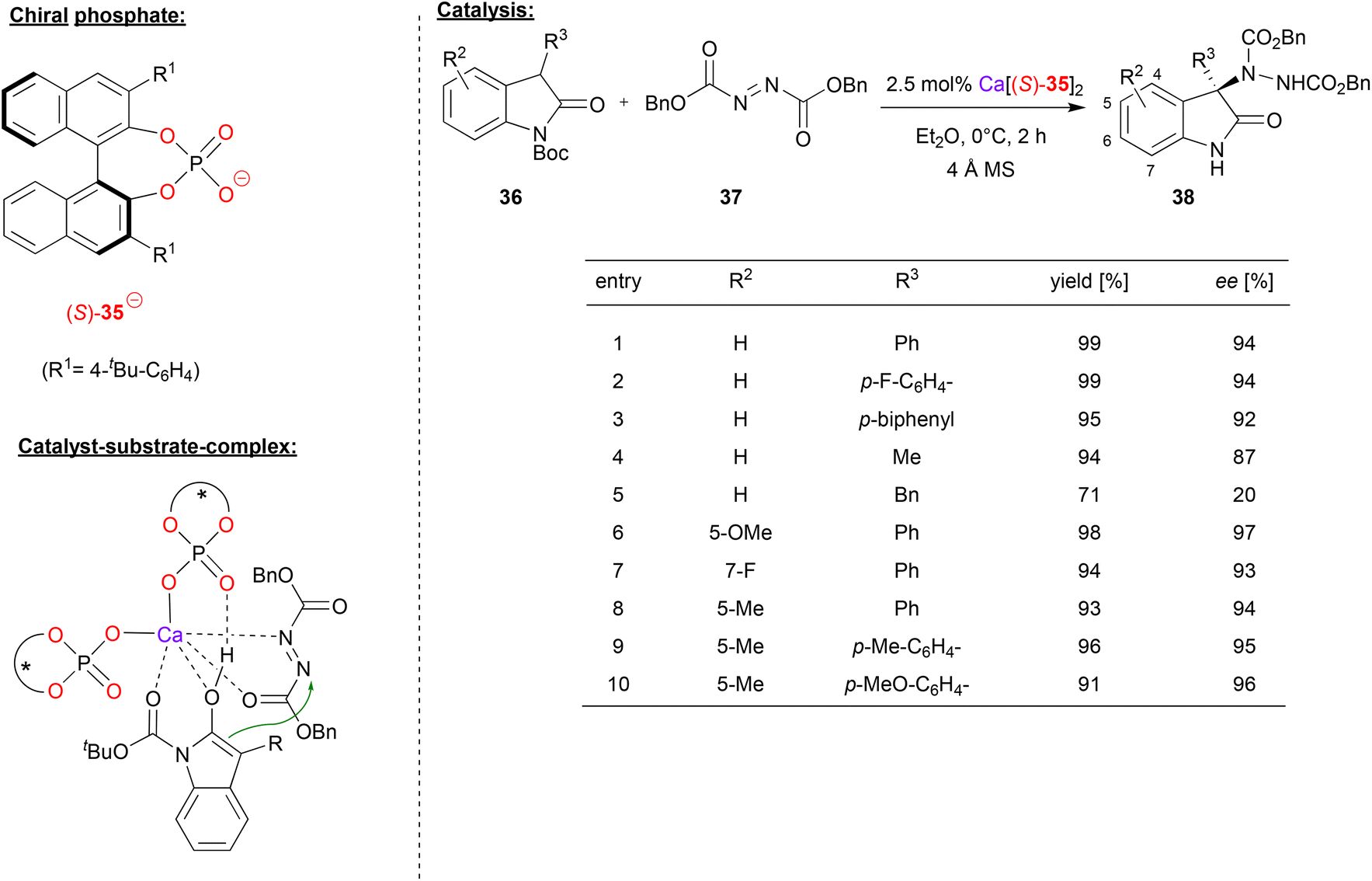 | ||
| Fig. 10 Enantioselective amination of 3-aryl-2-oxindoles catalyzed by a Ca-bisphosphate.38 | ||
Mechanistically, the authors propose a monodentate η1-binding of both phosphates to calcium, enabling bidentate N,O-coordination of the azodicarboxylate and bidentate O,O-coordination of the Boc-protected indole (in its enol-form). Additionally, the enol OH can form a hydrogen-bond to the PO fragment of one phosphate (see Fig. 10).
Calcium bisphosphate catalysts were also used to promote asymmetric hetero-Diels–Alder reactions. Antilla and coworkers showed that different 1,2-dicarbonyl compounds, such as the ketoesters 40 or isatin 41 can react with Danishefsky's diene 42 to give the corresponding 6,6-disubstituted dihydropyranones 43 (see Fig. 11 for selected examples).39
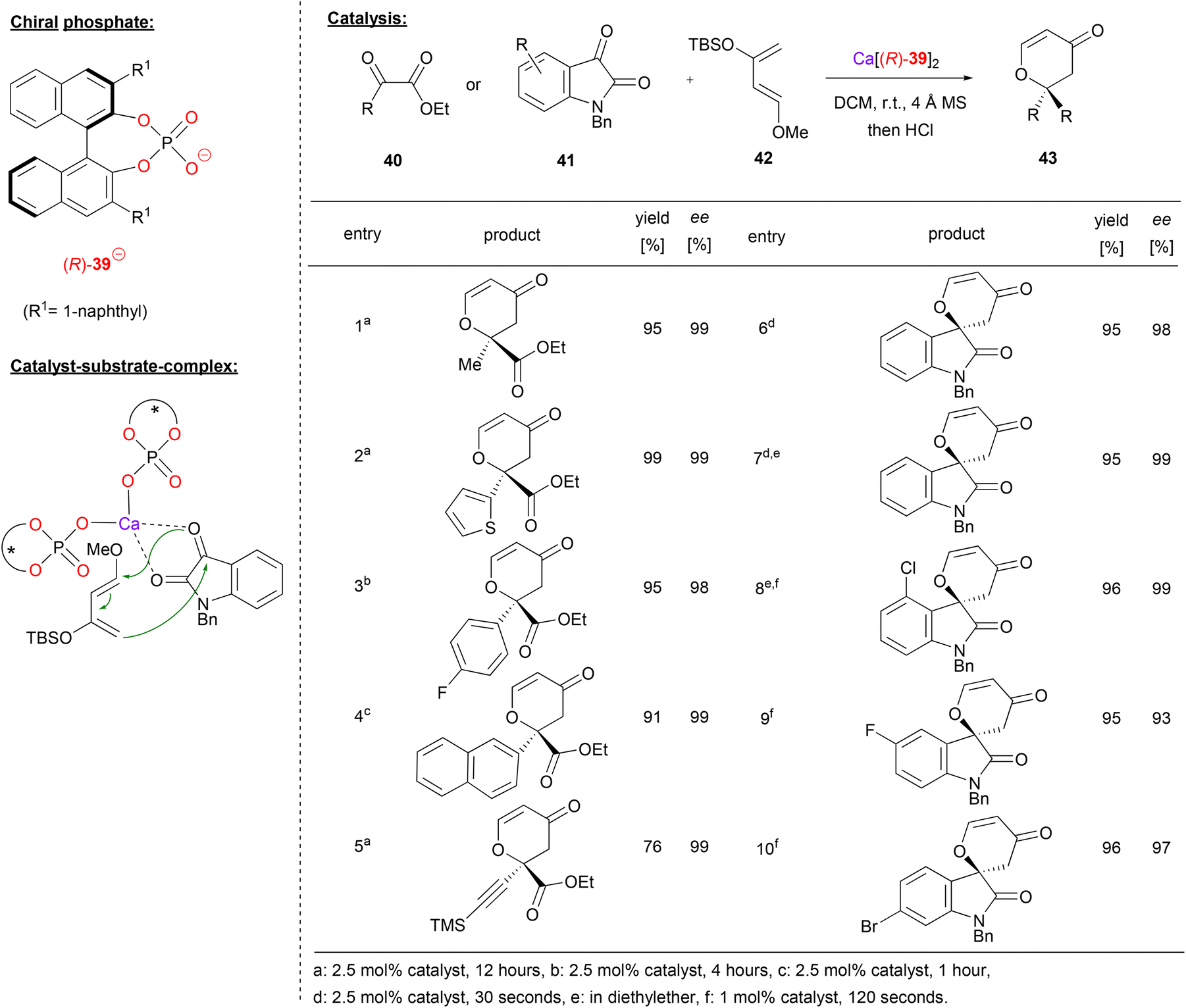 | ||
| Fig. 11 Enantioselective hetero-Diels–Alder reaction catalyzed by a Ca-bisphosphate.39 | ||
Using different ketoesters 40 (featuring alkyl-, heteroaryl-, aryl- or even alkynylketones) as the dienophile, the products were obtained in excellent stereoselectivities (98–99% ee).
When using isatin as the electrophile, the corresponding spirocyclic products were obtained. The more reactive isatin gave full conversion in only 30–120 seconds with 1–2.5 mol% catalyst loading (cf. 1–12 hours with 2.5 mol% catalyst for the ketoesters). Different electron withdrawing substituent (e.g. F, Cl, Br) were tolerated on the isatin-backbone to give the products 43 in excellent enantioselectivities (93–99% ee).
The authors investigated the substrate–catalyst interaction by Raman-spectroscopy. Upon mixing of the catalyst with isatin, both carbonyl-bands shift to lower wavenumbers, indicating a bidentate coordination. Based on this, a bis-(η1-phosphate)-calcium complex was suggested, which binds the isation via O,O-coordination. In this chiral environment, the re-face of isatin is shielded, so that the Diels–Alder reaction leads to the experimentally observed (R)-enantiomer.
Sun and coworkers used the calcium-complex of (S)-BNPA 44 to perform an enantioselective peroxidation of C-alkynyl imines (see Fig. 12).40 Here, the N,O-acetals 45 were used as precursors, liberating EtOH in situ to deliver the corresponding imines. These were reacted with alkyl- or arylhydroperoxides 46 to give the corresponding peroxo-N,O-acetals 47.
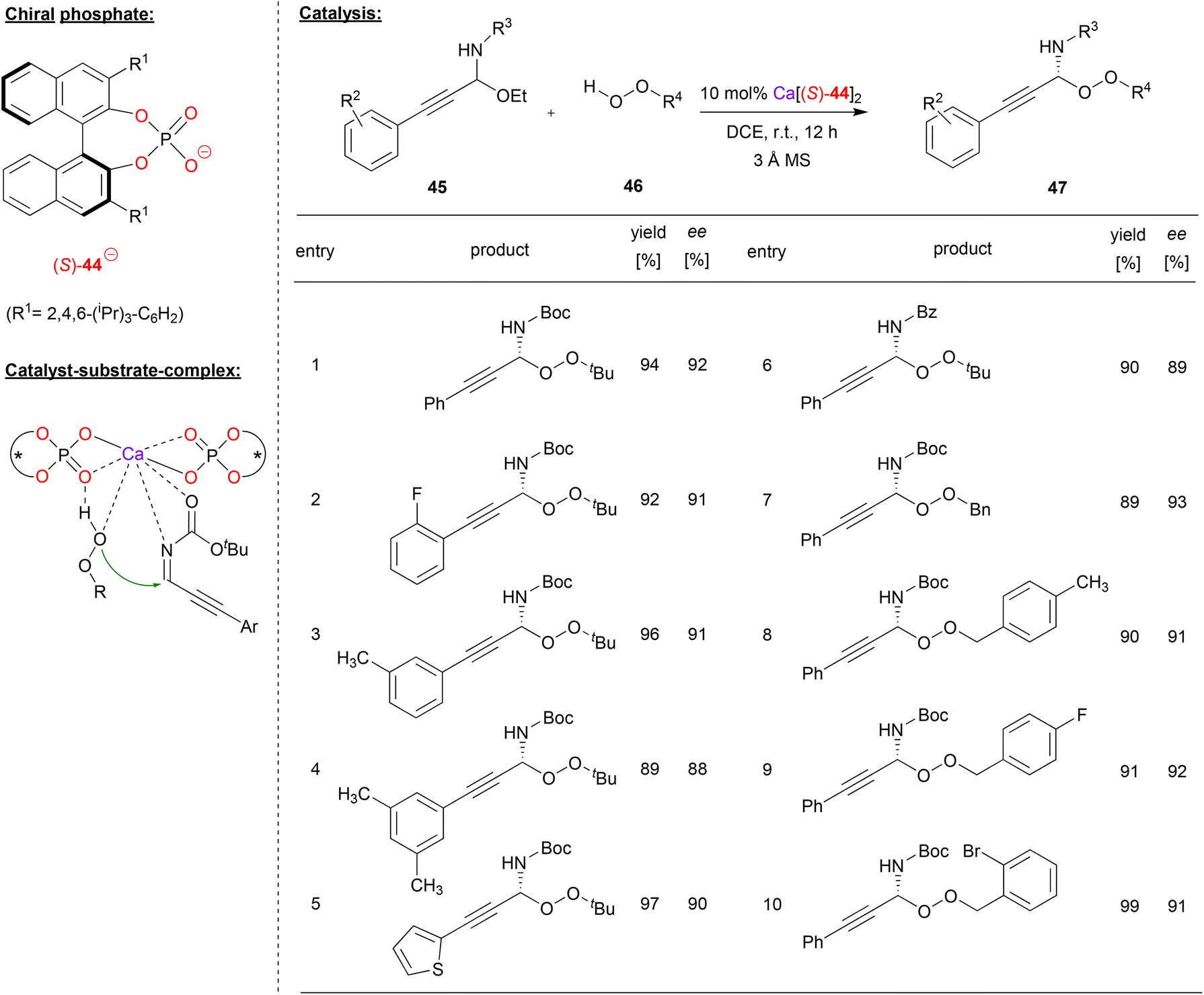 | ||
| Fig. 12 Enantioselective peroxidation of C-alkynyl imines catalyzed by a Ca-bisphosphate.40 | ||
As can be seen from the selected examples in Fig. 12, different aryl-groups were well tolerated on the arylethinyl-N,O-acetals, such as substituted phenyl-groups, but also a heteroaromatic thiophenyl-group. When reacted with tert-butylhydroperoxide, the products were obtained in good stereoselectivities (88–93% ee). Also, the N-protecting group could be changed without effecting the stereoselectivity (93%/89% ee for N-Boc/N-Cbz). Furthermore, differently substituted benzylhydroperoxides could be used as oxidants, delivering the products in 91–93% ee.
The authors found a negative nonlinear effect between the enantiopurity of the catalyst and the enantiopurity of the product, indicating the involvement of more than one phosphate ligand in the stereodetermining step. A bis-(η1-phosphate)-calcium complex was postulated as the active species, which can coordinate both the alkynylimine and the hydroperoxide via the Ca-center, aided by additional hydrogen bonding of the hydroperoxide OH-group by one of the Lewis-basic PO fragments.
2.4 Indium-catalysts
Luo and coworkers used a cationic indium-complex of (S)-BNPA (48) to catalyze the [4 + 2] annulation of non-activated allenes (see Fig. 13).41 Previous work by the same group had shown that β,γ-unsaturated α-keto esters react with diazoacetate to give the corresponding chiral cyclopropanes in high stereoselectivites, effected by chiral Ca-phosphates in the presence of additional InBr3.42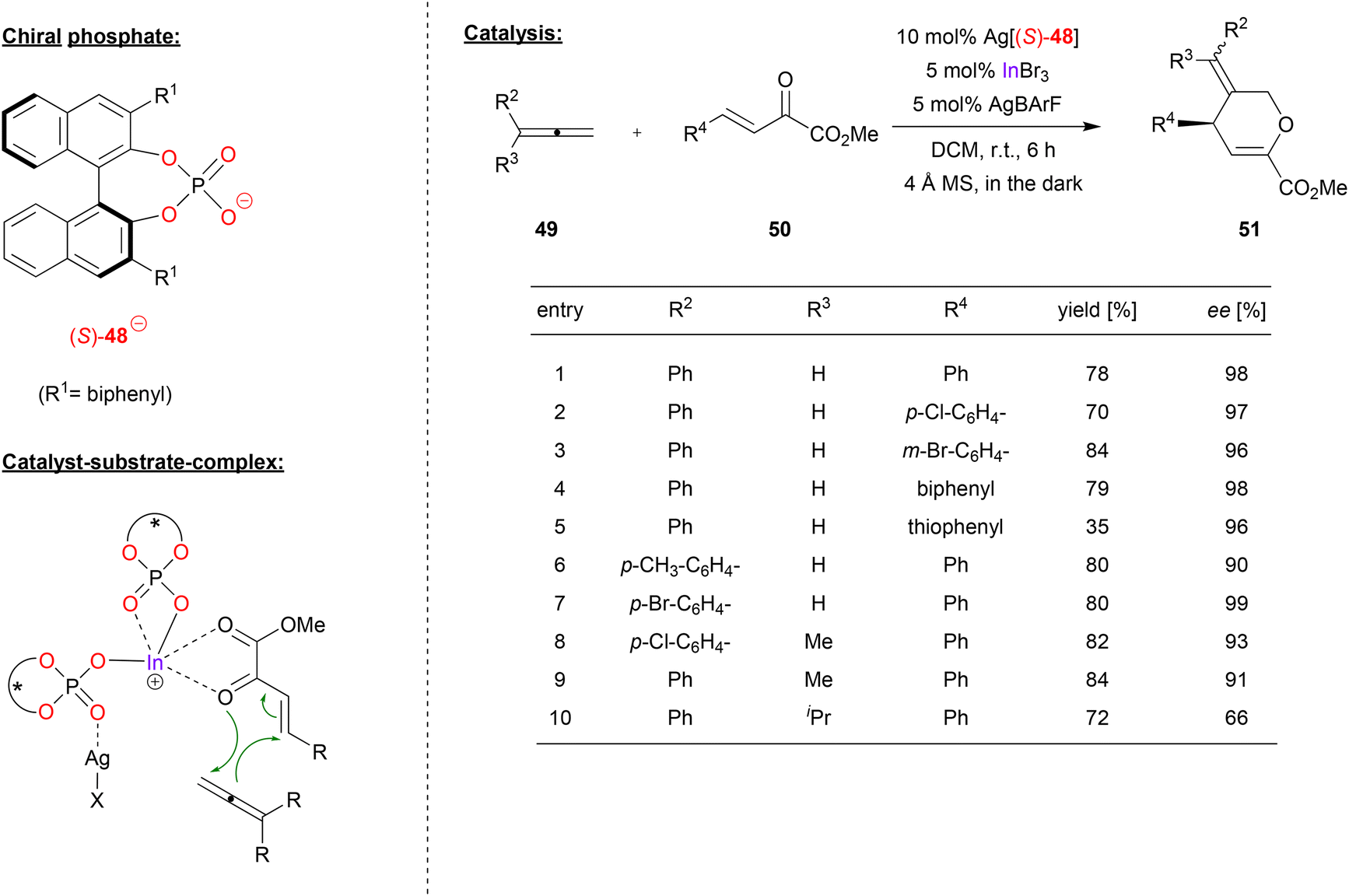 | ||
| Fig. 13 [4 + 2] annulation of non-activated allenes catalyzed by a cationic In-bisphosphate.41 | ||
Now, the β,γ-unsaturated α-keto esters 50 were reacted with allenes in a [4 + 2] annulation reaction, resulting in the formation of the corresponding dihydropyrans 51 (see Fig. 13). Here, the catalyst was formed in situ from InBr3, two equivalents of Ag-phosphate Ag[48] and one equivalent of AgBArF, resulting in formation of the cationic complex In(48)2+ BArF− with release of three equivalents of AgBr.
As shown in Fig. 13 for selected examples, different α-ketoesters can be reacted with phenylallene to give the products in good yields (70–84%) and excellent enantioselectivities (96–98% ee), regardless of electron-withdrawing or electron donating on substituents. Even the thiophene-analogue was tolerated, albeit resulting in lower yield (35% yield, 96% ee). Different 1-arylallenes and 1-aryl-1-methyl allenes were also successfully employed (90–99% ee), only for the 1-phenyl-1-isopropylallene a drop in enantioselectivity was observed (66% ee).
Upon investigation of the reaction mechanism, the authors found a positive nonlinear effect between enantiopurity of phosphate and enantiopurity of product, suggesting a higher-order ligand–metal complex. In addition, it was found that removal of the AgBr (formed upon in situ catalyst preparation, vide supra) by filtration leads to an inactive catalyst, while readdition of AgBr reactivates the catalyst. Based on these findings, the authors suggest a bimetallic complex composed of In, Ag and two phosphates. One phosphate acts as an η2-ligand to the indium, the other phosphate as a bridging μ-η1-η1 ligand to indium and silver. The In-center then binds the diene in a bidentate fashion, leading to its activation for the cycloaddition reaction (see Fig. 13).
2.5 Bismuth-catalysts
A bismuth-phosphate catalyst was used by Cheng and coworkers in the enantioselective allylation of oxocarbenium ions (see Fig. 14).43 This was based on previous work by the same group on the asymmetric allylation of isatin44 and cyclic imines.45 Now the Cheng-group reports the reaction of 3-hydroxyisobenzofuran-1(3H)-ones 53 and allylboranes 54 to give the corresponding 3-allylisobenzofuraone-1(3H)-ones 55 in the presence of a bismuth-monophosphate-diacetate complex.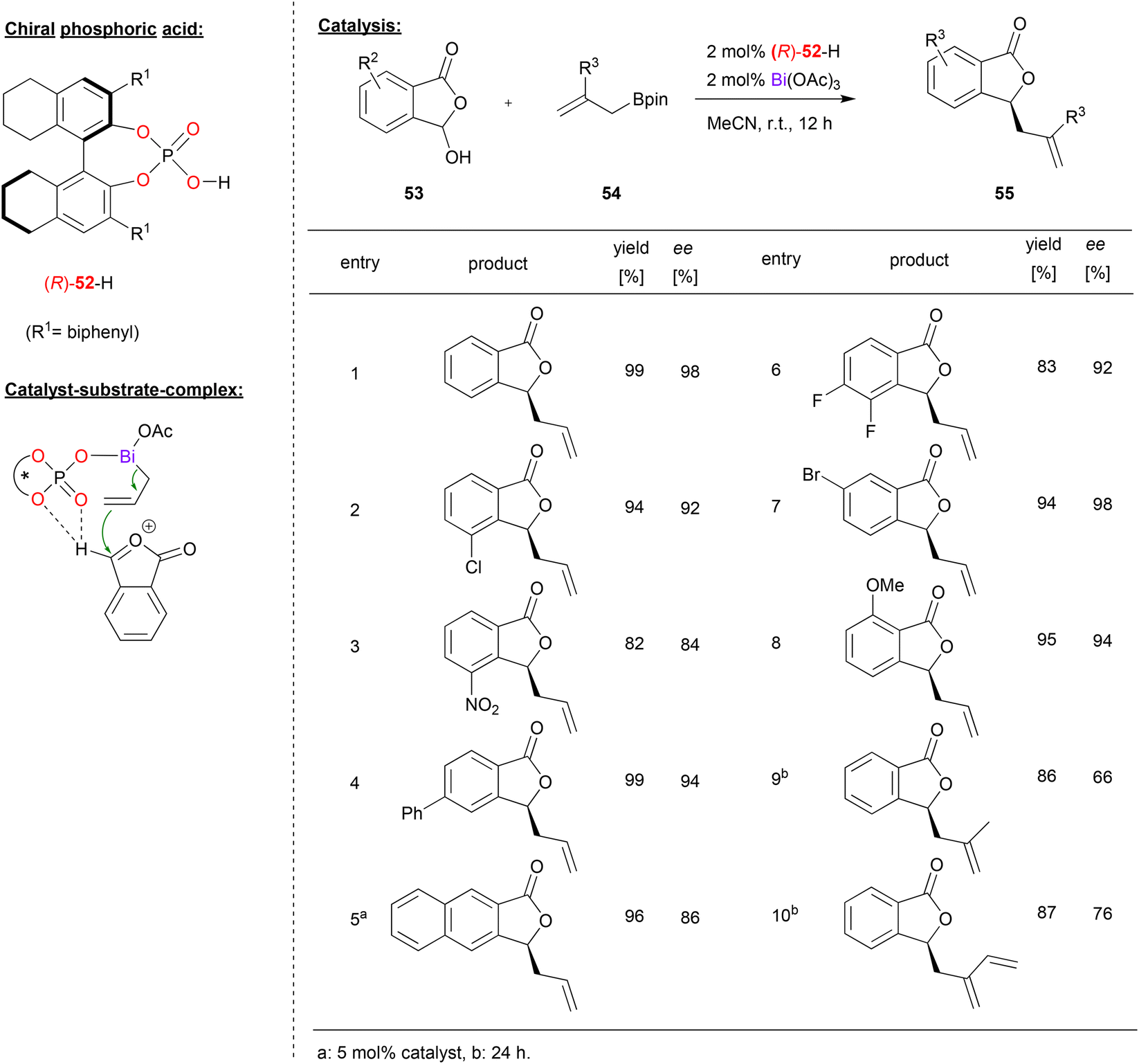 | ||
| Fig. 14 Enantioselective allylation of oxocarbenium ions catalyzed by a Bi-phosphate.43 | ||
As shown in Fig. 14 for selected examples, different isobenzofuranones with electron withdrawing (e.g. Cl, F, Br or NO2) or electron donating substituents (e.g. OMe) could be used to give the products in good enantioselectivities (84–98% ee). Using branched allylboronates was found to be more challenging and the products were obtained in slightly lower enantioselectivity (66–76% ee).
Mechanistically, the reaction proceeds via multiple steps: the hydroxyisobenzofuranone undergoes acid-catalyzed dehydratization to give the oxocarbenium ion. Simultaneously, the allylborane reacts with the catalyst to form an allyl bismuth complex of the type BiIII(allyl)(acetoxy)(phosphate) under liberation of acetoxyborane. Coordination of the oxocarbenium-ion to this complex is enabled by a bifurcated hydrogen-bond of the oxocarbenium CH group to the PO and one P-OAr oxygen (see Fig. 14). DFT-calculations of these transition states show that the experimentally observed (S)-product is favoured by 2.6 kcal mol−1.
3. Transition metals
In the main-group metal phosphates described above, the metal center commonly acts as a Lewis-acid to interact with one or both substrates. Transition-metals offer additional patterns of reactivity (e.g. oxidative addition/reductive elimination processes), so that these have also been actively investigated in combination with chiral phosphate ligands.This chapter will describe recent examples (since 2014) of metal-phosphate complexes of titanium, manganese, rhodium, palladium, silver and gold and their use in enantioselective catalysis. Earlier examples with these metals, but also examples for the use of yttrium, scandium, iron, copper, ruthenium, iridium and zinc can be found in Rueping's review.1
3.1 Titanium-catalysts
Leibfarth and coworkers reported a pioneering work in a polymerization reaction of vinyl ethers using phosphate (S)-56 in combination with titanium (see Fig. 15).46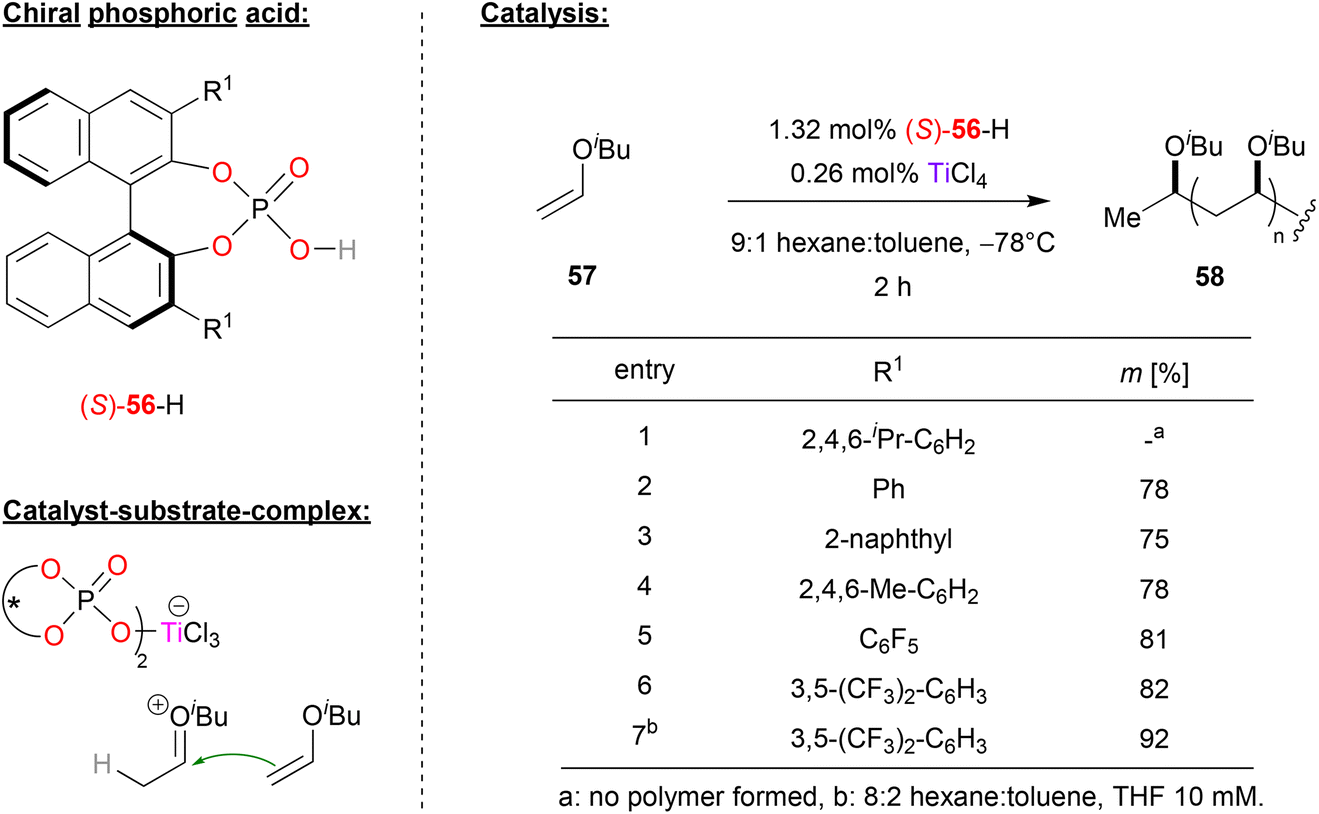 | ||
| Fig. 15 Stereoselective polymerization of vinyl ethers catalyzed by a Ti-phosphate.46 | ||
Here, phosphoric acid (S)-56 and titanium tetrachloride were used to polymerize isobutyl-vinyl ether 57 to polyisobutylvinyl ether 58. Phosphoric acids with different aryl-substitutents in the 3,3′-positions were investigated, with the aim of generating a polyvinylether with high isotacticity. Using sterically hindered TRIP-phosphate resulted in no polymer at all, while other electron-neutral aryl- or alkylaryl-substituents such as phenyl, naphthyl or mesityl gave isotacticities between 75–78%. Next, electron deficient substituents were investigated: pentafluorophenyl-groups already gave 81% isotacticity, and the best results were obtained with 3,5-bis(trifluoromethyl)phenyl groups (82% isotacticity). Further optimization of the reaction conditions by adding THF to the hexane/toluene solvent mixture led an even higher isotacticity of 92%.
In terms of mechanism, the authors suggest the initial formation of a Markovnikow-adduct that is formed by protonation of the vinyl ether by the phosphoric acid, followed by addition of the phosphate to the oxocarbenium-ion. TiCl4 then enables liberation of the protonated vinyl ether by binding the phosphate in a titanium-bisphosphate-trichloride complex featuring two η1-phosphate ligands (see Fig. 15). The titan-phosphate acts as the chiral counteranion to the cationic oxocarbenium-ion, which then undergoes stereoselective polymerization.
3.2 Manganese-catalysts
Schneider and coworkers used the chiral phosphate (R)-59 together with manganese(III) to catalyze the enantioselective addition of β-dicarbonyls to o-qiunone methides (see Fig. 16).47 Here, acetylacetone 61 (or its ketoester derivatives) react with o-quinone methides, which are generated in situ from the ortho-arylmethyl or ortho-alkylmethyl-phenols 60 by oxidation. As a result, the chiral 4H-chromenes 62 are formed.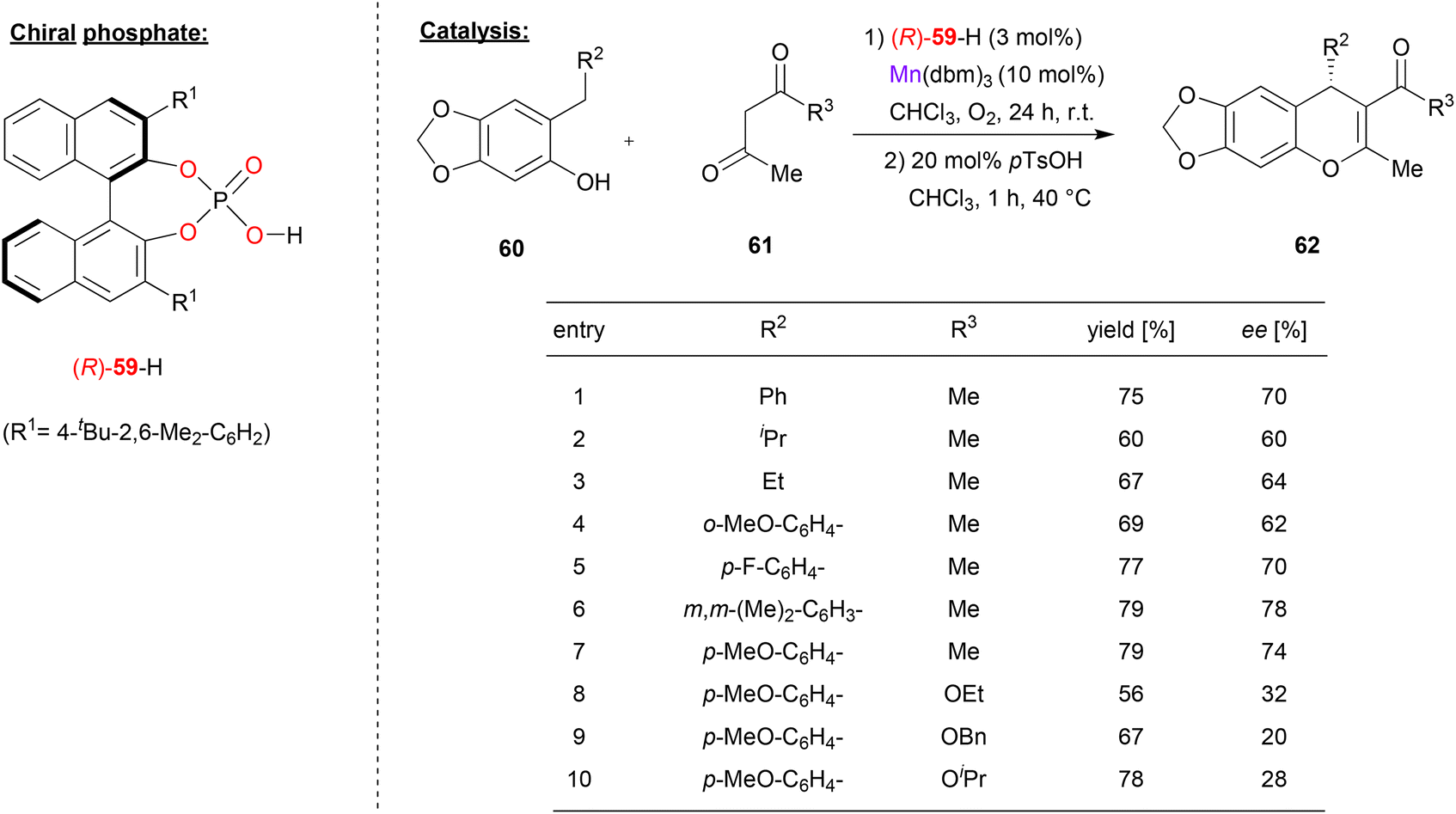 | ||
| Fig. 16 Enantioselective addition of dicarbonyls to ortho-quinone methides catalyzed by a Mn-phosphate.47 | ||
As can be seen from the selected examples in Fig. 16, different phenols 60 with alkyl- or aryl-substituents on the ortho-methylene group can be used to give the products in moderate stereoselectivities (62–78% ee). However, when using ketoesters instead of diketones (61), stereoselectivity drops significantly to 20–32% ee.
Mechanistically, the authors suggest that the manganese precatalyst Mn(dbm)3 (used with 10 mol%) reacts with excess acetylacetone 61 to form the Mn(acac)3 complex. This complex then reacts with one equivalent of the phosphoric acid (R)-59 (used with 3 mol%) to generate the heteroleptic catalyst Mn(acac)2(59), leaving 7 mol% of the Mn(acac)3 untouched. Excess of Mn(acac)3 is needed for oxidation of the phenol to the ortho-quinone methide, with the catalyst being reoxidized by molecular oxygen. The addition reaction is then catalyzed by the chiral Mn(acac)2(59)-complex to yield the products in an enantioselective fashion.
3.3 Rhodium-catalysts
Rhodium-phosphate catalysts have also been applied in asymmetric catalysis. Especially the so called “paddle-wheel”-complexes (dirhodium complexes with four bridging ligands, e.g. of the type Rh2(μ–η2-phosphate)4) have found widespread application.16Davies and coworkers used a rhodium paddlewheel-complex featuring (R)-BNPA 63 to catalyze a cyclopropanation reaction (see Fig. 17).48 To this end, styrene 65 was reacted with aryldiazoacetates 64 to give the 1,1,2-trisubstituted cyclopropanes 66.
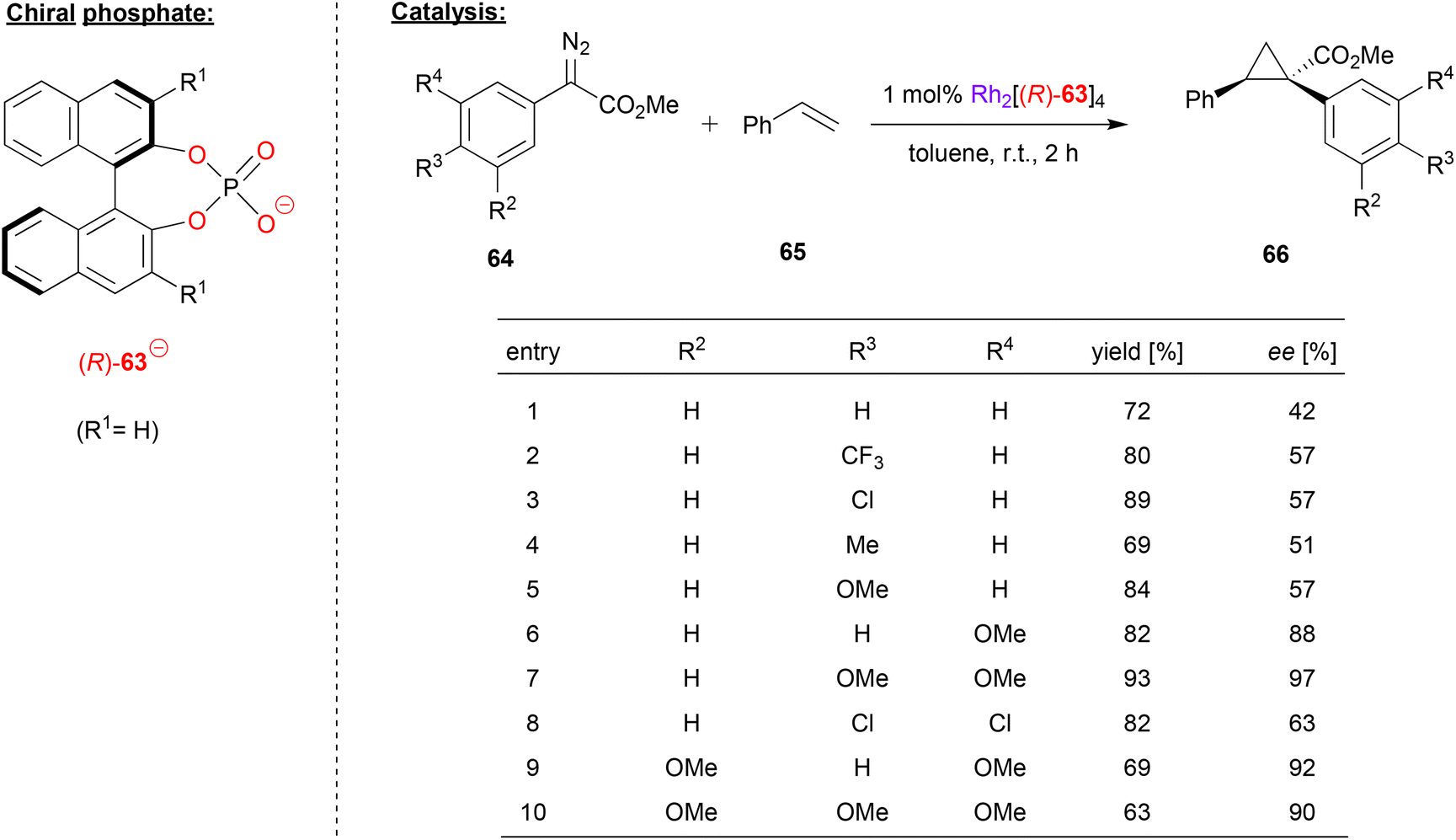 | ||
| Fig. 17 Enantioselective cyclopropanation catalyzed by a Rh-phosphate paddlewheel complex.48 | ||
As shown in Fig. 17 for selected examples, the reaction turned out to be rather sensitive to the substitution pattern of the aryldiazoacetates. Compounds with no aryl-substituent or a single para-substituent (independent of electronic nature), gave only moderate enantioselectivity (42–57% ee). Using the meta-OMe derivative increases the enantioselectivity (88% ee), as does the use of multiple substituents, especially when using multiple OMe groups (63–97% ee).
Recently, Zhou and coworkers applied a rhodium-paddlewheel complex which features the spirocyclic phosphate ligand (S)-67, in order to promote the enantioselective diarylcarbene insertion into Si–H bonds (see Fig. 18).49 To this end, diaryldiazomethanes 68 were reacted with dimethylphenylsilane 69 to yield the chiral diarylsilylmethanes 70.
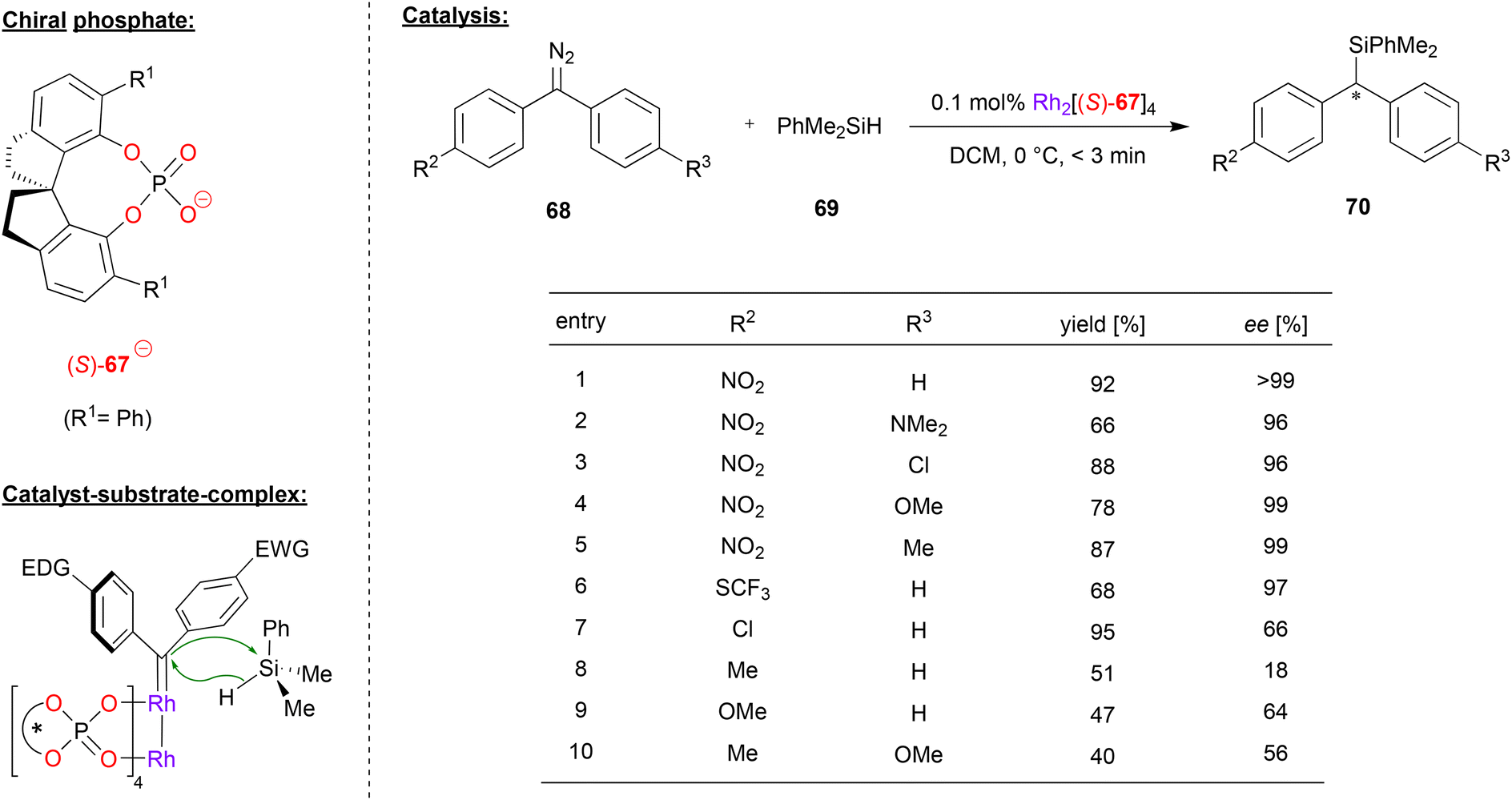 | ||
| Fig. 18 Enantioselective diarylcarbene insertion into Si–H bonds catalyzed by a Rh-phosphate paddlewheel-complex.49 | ||
As shown for selected examples in Fig. 18, differently para-substituted diaryldiazomethanes were tested, showing that at least one electron-withdrawing substituent (e.g. NO2, SCF3) is key to good enantioselectivities (96–99% ee). However, if substituents with a weak electronic effect are used (e.g. Cl, Me, OMe) the enantioselectivity drops significantly (18–66% ee). The authors were able to find that as long as the difference between the Hammett substituent constants between both para-substituents is over 0.5, the ee of the product is generally over 90%.
Mechanistically, the rhodium-complex enables the decomposition of the diazomethane reagent, leading to the corresponding Rh–carbene complex. The transition state of the reaction was calculated by DFT, showing that the Si–H insertion proceeds in a concerted manner from the Rh–carbene. Interestingly, the diarlycarbene adopts a conformation where the more electron-rich aryl group is coplanar with the carbene-plane, allowing for efficient electronic interaction of the aryl-ring with the empty carbene p-orbital, while the electron-poor aryl ring is perpendicular to the carbene plane. Because of this orientation, the silane approaches with the phenyl-ring pointing towards the electron-poor aryl-ring of the diarylcarbene. In the chiral environment of the catalyst, this leads to a 2.2 kcal preference for the experimentally observed product isomer.
This pioneering work on Si–H activation by diaryldiazomethanes was extended to the use of arylalkynyldiazomethanes50 and diazoacetates51 (not shown in detail).
3.4 Palladium-catalysts
In many cases, chiral phosphates have been using in combination with Pd and additional chiral co-ligands featuring nitrogen-donors. For example, Beller and coworkers combined (S)-BNPA 71 with Pd(dba)2 and the chiral phosphoramidite (S,S,S)-72 to generate a catalyst system for the enantioselective amination of allylic alcohols (see Fig. 19).52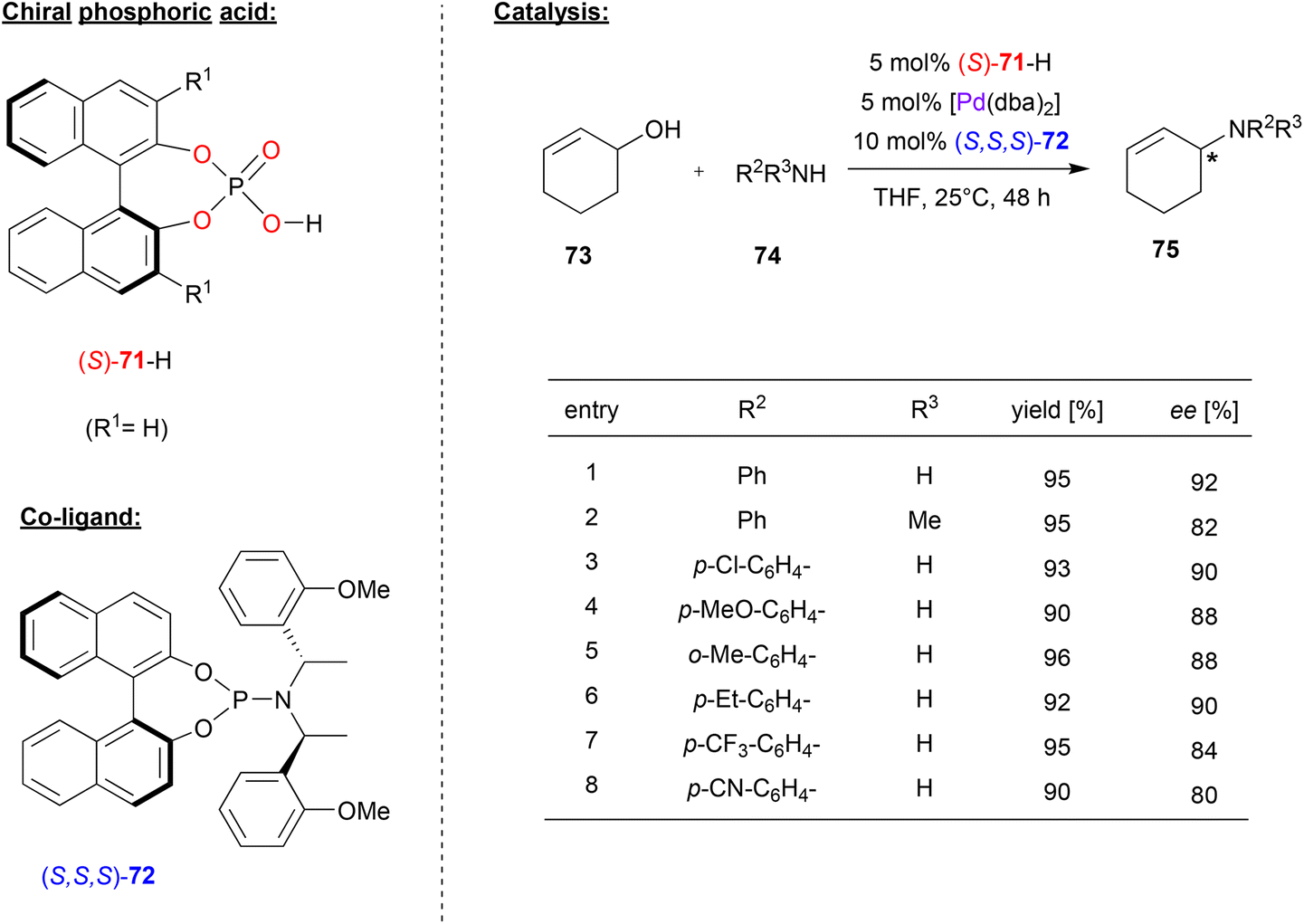 | ||
| Fig. 19 Enantioselective amination of racemic allylic alcohols catalyzed by Pd in the presence of a chiral phosphate and a chiral phosphoramidite.52 | ||
Here, cyclohexenol 73 was used as a precursor for a Pd(cyclohexenyl)-complex, which undergoes nucleophilic attack from anilines 74 to give the N-cyclohexenyl-N-arylamines 75 as the reaction products. Different N-donor ligands and different phosphoric acids were tested and it was found that the combination of (S)-71 and (S,S,S)-72 acts in a highly synergistic way, giving the products in high enantioselectivities.
As shown in Fig. 19 for selected examples, differently substituted primary aryl amines, but also secondary arylalkylamines were well-tolerated and the products were obtained in 80–92% ee. Other cyclic and acyclic allyl alcohols could also be used as reagents (not shown). Surprisingly, no side-products from further reaction of the products 75, which would result in bis-allylic amines, were formed.
Although the exact nature of the active complex is unknown, the synergistic action of the phosphate and the phosphoramidite indicates that both ligands are involved and presumably bound to the Pd-center. It was also found that the predefined palladium-cyclohexenyl-phosphate complex PdII(71)(η3-cyclohexenyl) can be used in combination with phosphoramidite (S,S,S)-72 and aniline to obtain the reaction product (N-cyclohexenyl-N-phenylamine) with high stereoselectivity of 80% ee (cf. 92% ee for the same product when forming the catalyst in situ). This also hints at a direct binding of the phosphate to the Pd-center, presumably with additional coordination of the phosphoramidite ligand.
In a similar catalyst system, Gong and coworkers used the phosphoric acid (R)-76 together with Pd(dba)2 and the phosphoramidite ligand (R)-77 to achieve the asymmetric allylic C–H alkylation of terminal olefins (see Fig. 20).53 Here, pyrazol-5-ones 78 were reacted with allylarenes 79 (or 1,4-pentadienes, not shown) in the presence of 2,5-dimethoxybenzoquinone to give the 4,4-disubstiuted pyrazol-5-ones 80 featuring all-carbon quaternary stereocenters. As shown in Fig. 20 for selected examples, different pyrazol-5-ones 78 and different allylarenes could be used to give the products in consistently high enantioselectivities (87–96% ee).
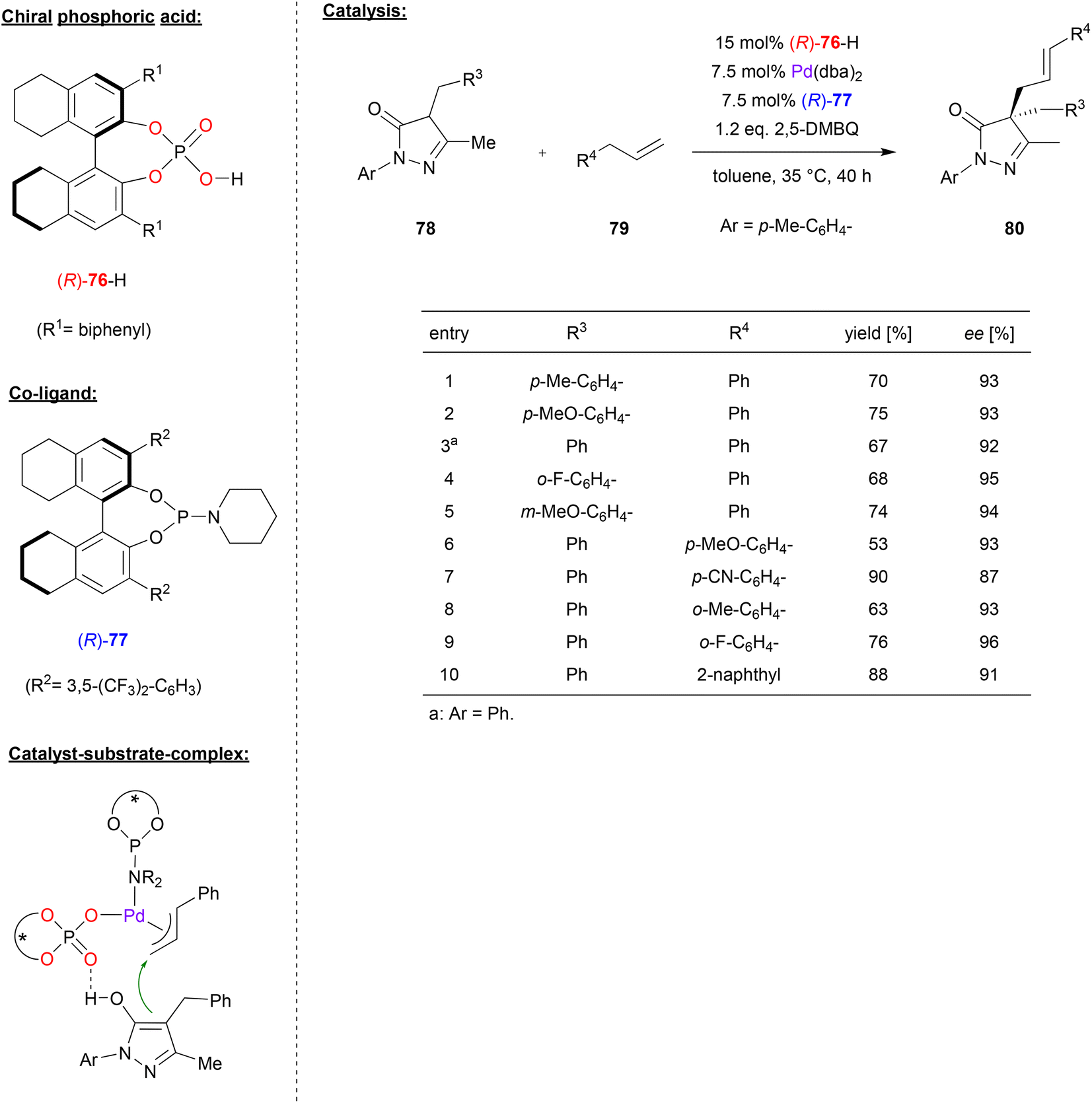 | ||
| Fig. 20 Enantioselective allylic C–H alkylation of olefins catalyzed by Pd in the presence of a chiral phosphate and a chiral phosphoramidite.53 | ||
The authors proposed a mechanism where the phosphoramidite-Pd(0) complex can undergo an oxidative addition of the allylarene (79) in the presence of (R)-76 and 2,5-dimethoxybenzoquinone. This results in formation of the PdII(allyl)(phosphate)(phosporamidite) complex, which can bind the pyrazol-5-one (in its enol-form) via hydrogen-bonding of the OH group to the PO fragment of the phosphate (see Fig. 20). Allylation then gives the product and regenerates the Pd(0) species.
A multi-catalyst relay catalysis was performed by Gong and coworkers, who employed the chiral phosphoric acid (R)-81, a rhodium-bisposphine complex, Pd(Ph3P)4 and an achiral amine. Here the α-quaternary chiral aldehydes 85 were generated from styrenes 83, allylic alcohols 84 and syngas (see Fig. 21).54
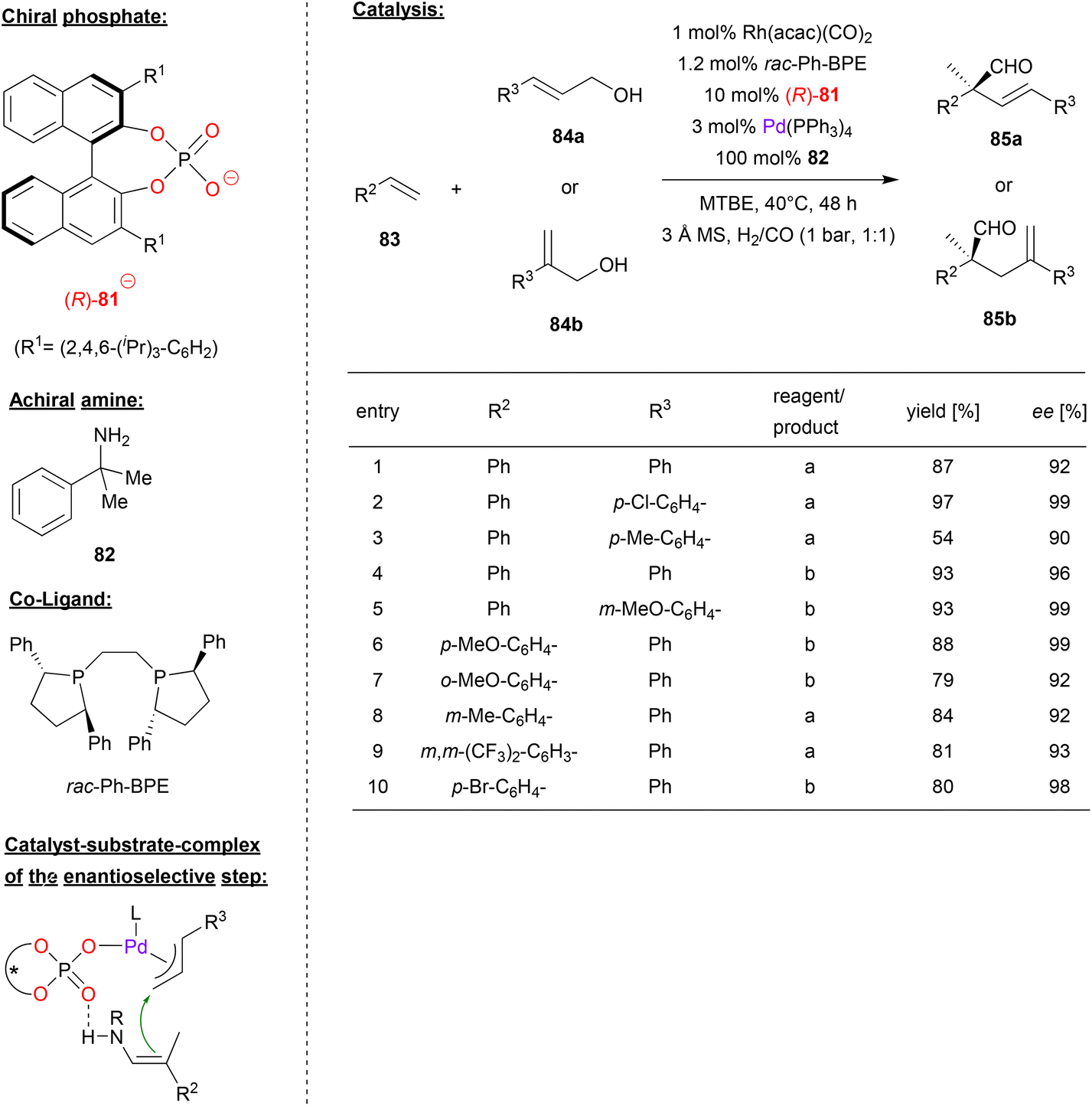 | ||
| Fig. 21 Enantioselective synthesis of α-quaternary chiral aldehydes by a Pd-phosphate, a Rh-bisphosphine and an achiral amine.54 | ||
As can be seen from Fig. 21 for selected examples, different cinnamyl alcohols 84a and 2-aryl allylic alcohols 84b gave the products in moderate to excellent yields (54–97%) and good to excellent enantioselectivities (90–99% ee). As for the styrene substrates 83, the reaction proceeds in good yields (79–88%) and good to excellent enantioselectivity (92–99% ee) for differently substituted styrenes.
Mechanistically the authors suggest a multi-catalyst relay catalysis: the rhodium-bisphosphine-complex reacts with the styrene 83 and syngas (CO/H2) to give the hydroformylation product, namely the 2-arylpropionaldehyde. This reacts with the primary amine 82 to give the corresponding enamine. At the same time, the allylic alcohol 84 is transformed into the π-allyl Pd complex which features a chiral phosphate ligand. The enamine-nucleophile can coordinate by PO⋯HN hydrogen-bonding, setting both substrates up for the enantioselective α-allylation (see Fig. 21). Subsequent hydrolysis liberates the product 85 and the achiral amine.
Further examples of enantioselective reactions (not shown in detail) involving chiral allyl–Pd-phosphate complexes were published by Gong, who reported the α-allylation of aldehydes with alkynes,55 and by Hu, who reported the three-component reaction α-diazoketones with alcohols and allyl carbonates to give the corresponding α,α-disubstituted ketones,56 the three-component reaction of diazoamides, enamines and aldehydes for the synthesis of 3-substituted oxindoles57 and a three component reaction of pyrrole, diazoesters and imines to give α-pyrrolyl-β-amino acids with two adjacent stereocenters.58
Zhu and coworkers investigated a desymmetrizing aza-Wacker-reaction, using the biphenylphosphate 86 as chiral ligand for Pd, together with the pyrox ligand (4R,5S)-87 as a co-ligand (see Fig. 22).59 Here, 3-aminoethyl-cyclohexa-1,4-dienes 88 were cyclized via an enantioselective aza-Wacker reaction in the presence of oxygen as the terminal oxidant to give the corresponding pyrrolidine-products 89.
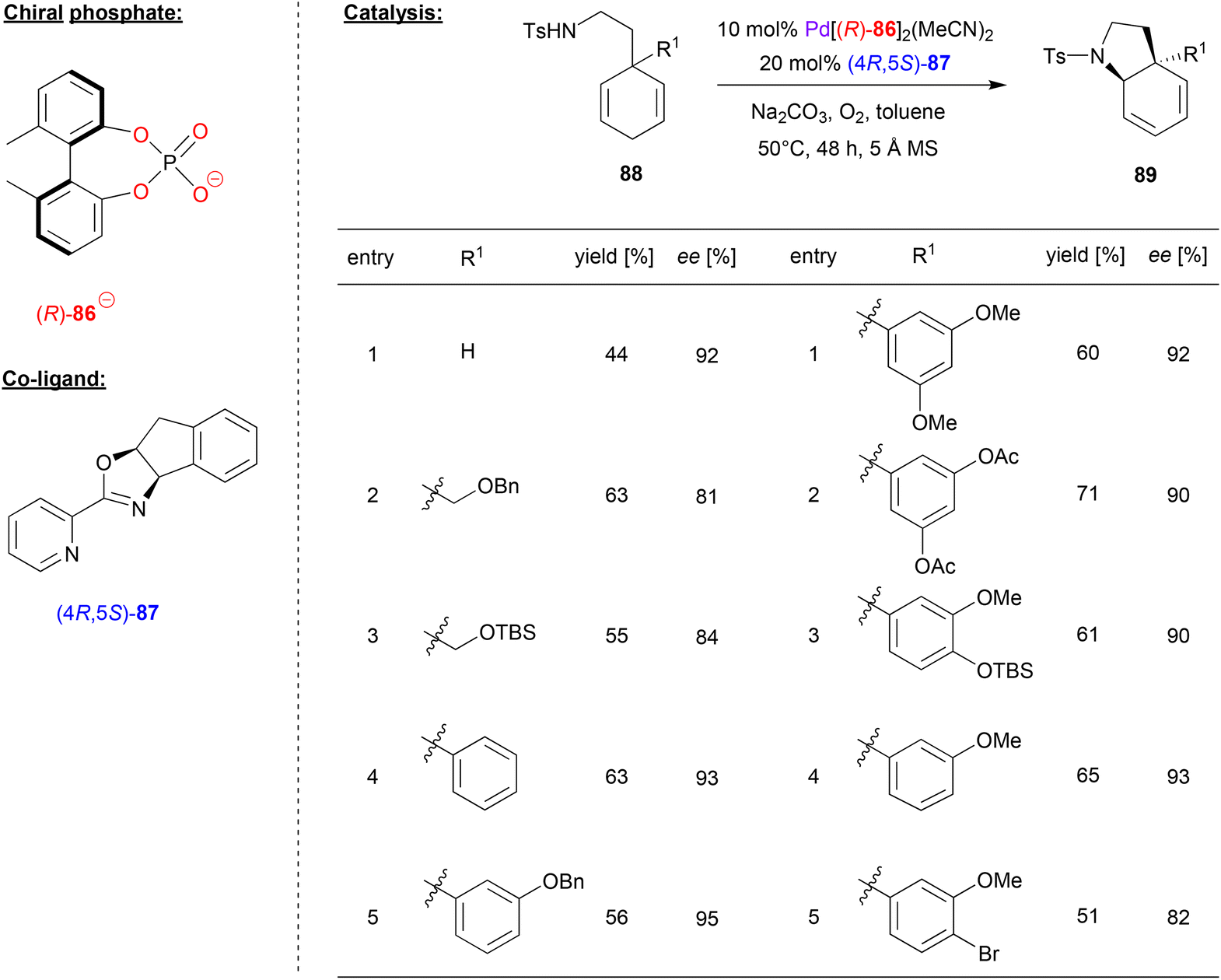 | ||
| Fig. 22 Desymmetrizing aza-Wacker reaction catalyzed by Pd in the presence of a chiral phosphate and a chiral pyrox-ligand.59 | ||
With regard to the substrate scope (see selected examples in Fig. 22), the parent system featuring no second substituent in the 3-position gave the cyclization product in 92% ee. 3-Alkyl-substituted systems gave lightly lower stereoselectivities (81–84% ee), while a range of different aryl-groups in the 3-position were well-tolerated to give the products in 90–95% ee. Even a para-substituted aryl bromide was stable despite the presence of a Pd-catalyst, to give the product with a moderate yield of 51% and an enantiomeric excess of 82% ee. Based on this protocol, the authors also achieved the synthesis of natural products like (−)-mesembrane and (+)-crinane in good yields and excellent enantioselectivity.
With regard to the synergistic action of both chiral ligands, it was found that the pyrox ligand determines the sense of chirality of the product, while the chiral phosphoric acid is crucial to obtain high yields and the enantioselectivities. The authors did not comment on the mechanism of the reaction, but most likely the reaction takes place via an aminopalladation involving one of the two enantiotopic double-bonds of the cyclohexadiene-substrate, followed by β-hydride elimination and reoxidation of the catalyst.
Shi and coworkers achieved the enantioselective synthesis of biaryl atropoisomers by a palladium-phosphate catalyzed C–H olefination (see Fig. 23).60 Here, the palladium-complex of phosphate (R)-90 catalyzes the reaction between quinoline-derived biaryls 91 and acrylic esters 92 in the presence of silver acetate.
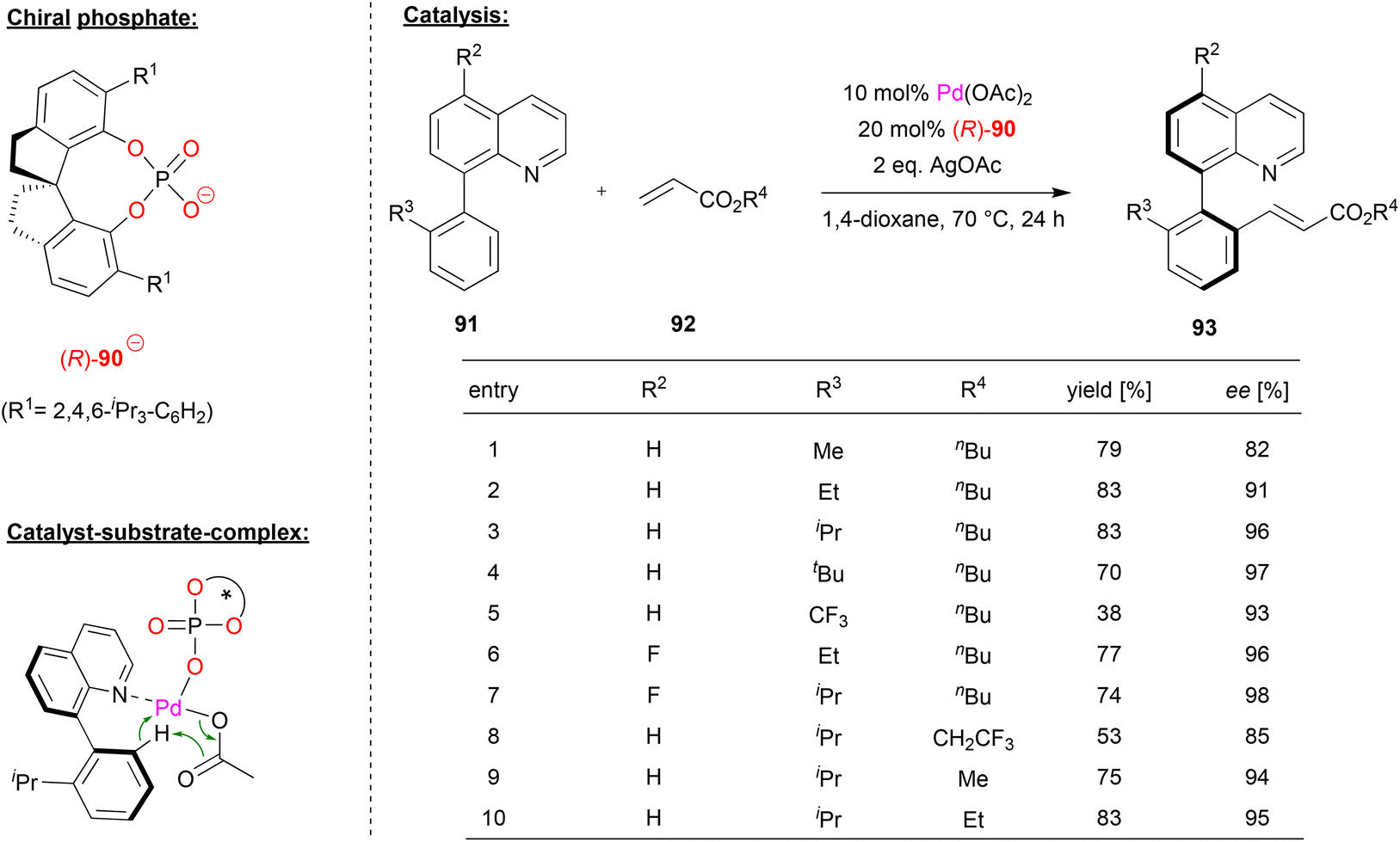 | ||
| Fig. 23 Enantioselective C–H olefination for the formation of atropisomers catalyzed by a Pd-phosphate.60 | ||
First, the authors investigated the reaction scope with regard to the phenyl-quinolines 91. The ortho-substituent on the phenyl-group, which is necessary for the formation of stable atropisomers, had a strong influence on the enantioselectivity, with bulky groups giving the best stereoselectivities (96/97% ee for R3 = iPr/tBu). Using an electron deficient CF3-group drastically lowers the yield of the reaction to 38%. With regard to substitution on the quinoline, it was found that 5-fluoro-substituents led to even better stereoselectivities (up to 98% ee). Subsequent investigation of different acrylic esters showed that variation of the n-butyl group led to decreased stereoselectivities.
Mechanistically, the authors suggest that a heteroleptic Pd(acetate)(phosphate) complex is involved. This can be coordinated by the quinoline-nitrogen, which then allows a stereoselective C–H activation in a cyclometallation–deprotonation (CMD) type mechanism (see Fig. 23).
Based on this pioneering work, Shi and coworkers published a further example of an atroposelective C–H allylation of 2-amino-biphenyls (not shown in detail).61
Baudoin and coworkers used a chiral palladium-phosphate complex for the enantioselective intramolecular C(sp3)–H arylation (see Fig. 24).62 Here, 2-bromo-aniline-carbamates 95 that feature an N-alkyl substituent (e.g. isopropyl) with two enantiotopic methyl groups were cyclized to the corresponding indoline-derivatives 96.
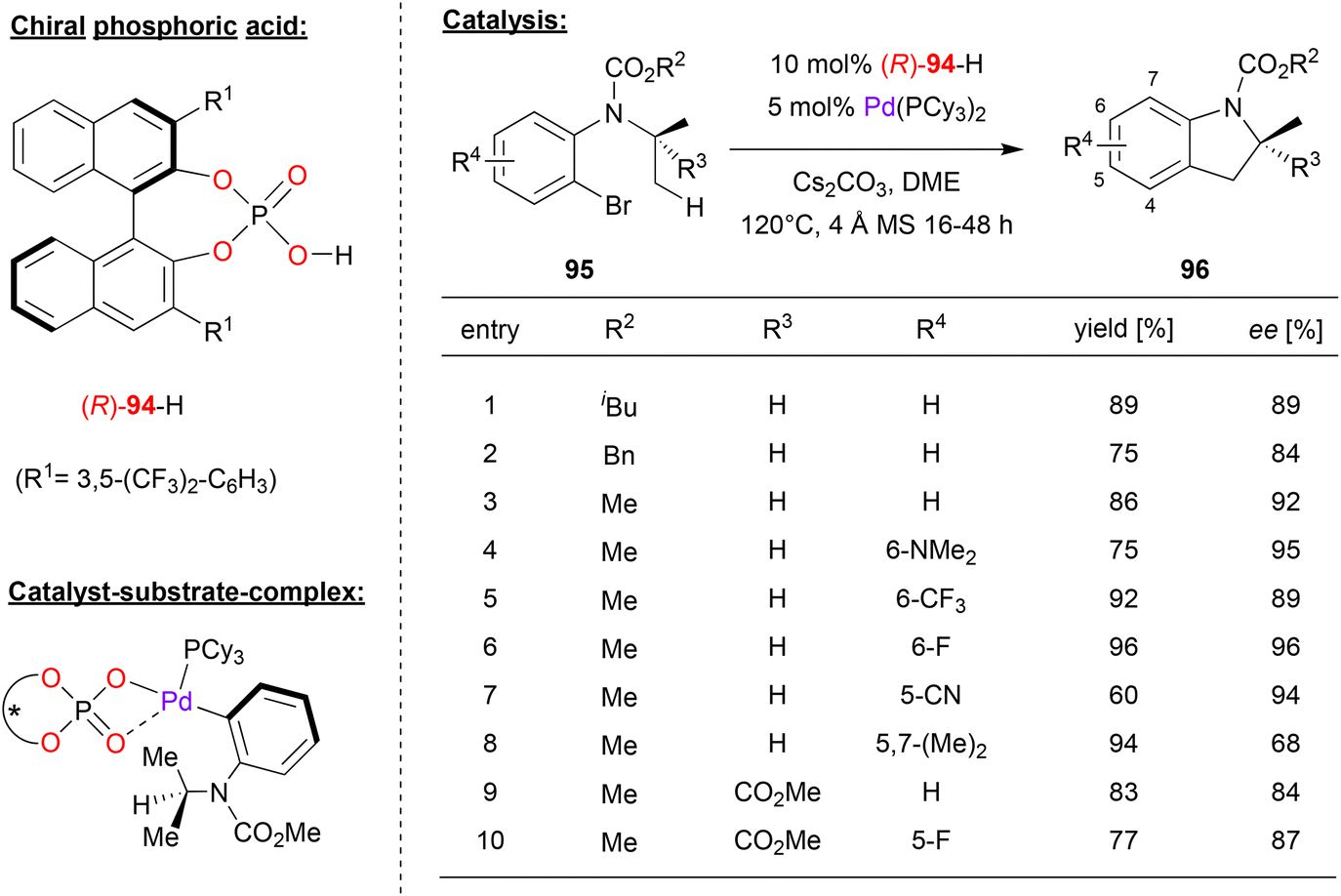 | ||
| Fig. 24 Enantioselective C(sp3)–H arylation catalyzed by chiral Pd-phosphates.62 | ||
A wide range of chiral phosphoric acid precursors was tested, identifying the 3,5-bis(trifluoromethyl)phenyl-derivative 94 as the optimal choice. Using Pd(PCy3)2 and the phosphoric acid in presence of cesium carbonate, a wide range of anilines were successfully converted to the cyclized products in high enantioselectivities. As shown in Fig. 24 for selected examples, different carbamates (N-COOiBu/Bn/Me) could be used. Furthermore, different substitution patterns on the aromatic core of the aniline-substrate could be employed: introduction of dimethylamino-, fluoro-, trifluoromethyl- or cyano-groups in the 5- or 6-positions was well-tolerated (87–95% ee), only a substituent the 7-position (i.e. in the 5,7-dimethyl case) led to decreased stereoselectivity (68% ee). Finally, it was shown that even the introduction of a further-substituent (here: a COOMe group) on the N-isopropyl group was possible, giving the products featuring a quaternary stereocenter in high enantioselectivities (84–87% ee).
Mechanistically, the authors suggest that oxidative addition in the aryl–Br bond leads to formation of a Pd(II)-complex with the aryl-ligand, one chiral η2-phosphate ligand an one ancillary phosphine ligand (see Fig. 24). This would place the N-isopropyl-substituent in close contact to the phosphate and one of its 3,3′-substituents. This complex could then undergo a phosphate-assisted cyclometallation–deprotonation involving one of the enantiotopic methyl-groups, which upon reductive elimination of Pd delivers the reaction product.
This pioneering work by Baudoin on intramolecular asymmetric C–H arylation of methyl-groups was followed by work by Shi on intramolecular asymmetric CH-arylation of methylene-groups63 and on intermolecular asymmetric CH functionalization of 2-amino-biaryls to give the axially chiral 2-amino-2′-alkenyl-biaryls64 (not shown in detail).
Bäckvall and coworkers used a palladium complex of (R)-VPA (97) for the carbonylative cyclization of enallenes (see Fig. 25).65 Specifically, the allyl-substituted allenes 98 were reacted with terminal alkynes 99 in the presence of CO and benzoquinone as oxidant, resulting in formation of the chiral cyclopentenones 100.
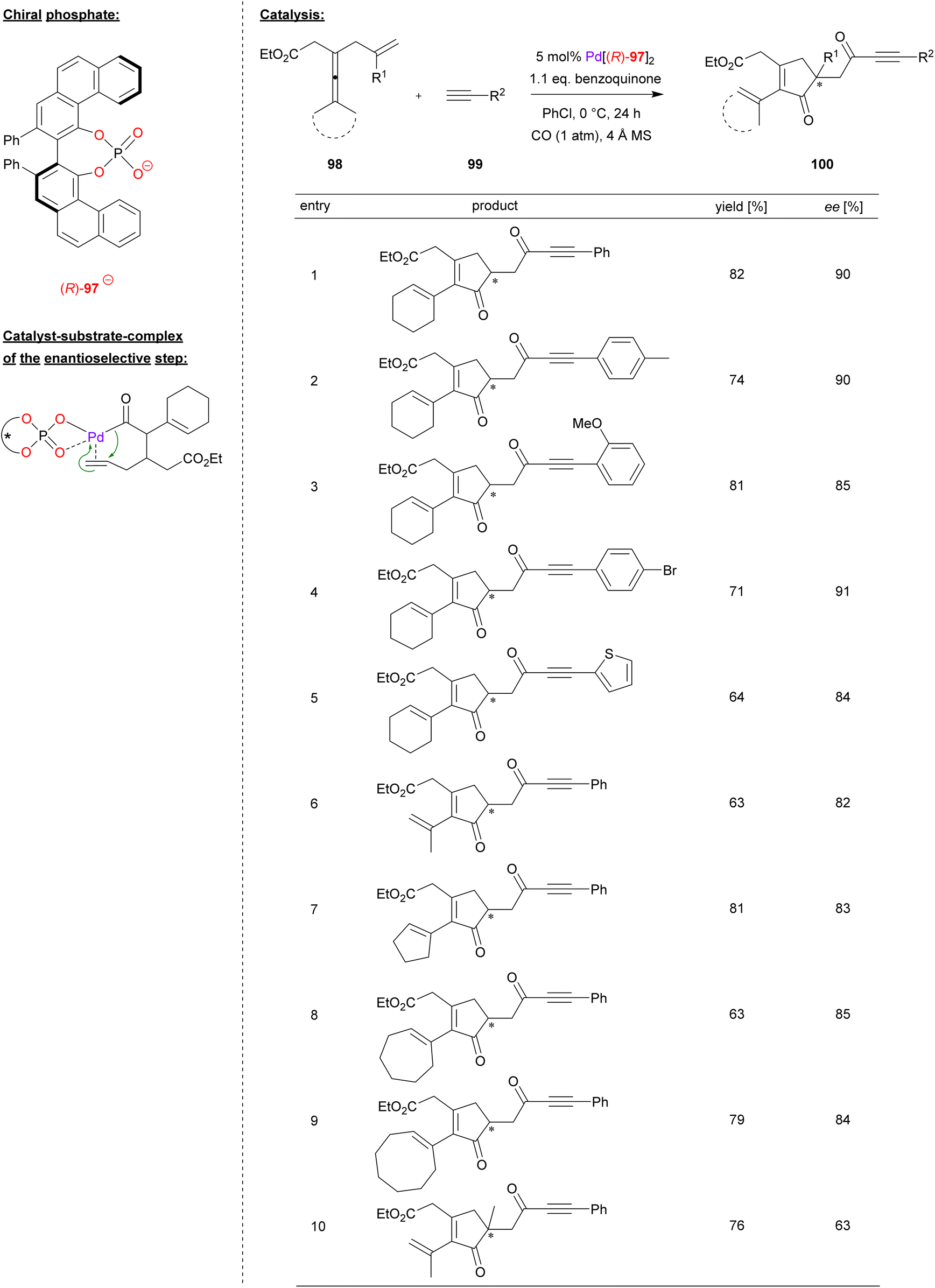 | ||
| Fig. 25 Enantioselective carbonylative carbocyclization of enallenes catalyzed by a Pd-phosphate.65 | ||
To determine the substrate scope, a screening of different alkynes and enallenes was carried out (see Fig. 25 for selected examples). In case of terminal alkynes, electron withdrawing and electron donating substituents, as well as heteroaryl variants like ethinylthiophene, are well tolerated to give the products in high enantioselectivities (84–91% ee). Different enallenes featuring a terminal dimethyl-substitution or cyclooligomethylene-substitution (e.g. cyclo-C5/6/8) could also be used (82–85% ee). Only when changing the terminal olefin from a simple allyl to a branched 2-methylallyl-group, the enantiomeric excess dropped significantly to 63% ee.
The reaction proceeds via π-coordination of the enallene, followed by allene-attack and CO-insertion to give the corresponding acyl-palladium complex (see Fig. 25 for a putative structure of this complex). Enabled by the chiral phosphate, an enantioselective migratory insertion of the terminal double-bond (from the allyl-fragment) leads to the chiral cyclopentenone, which reacts further with CO and the terminal alkyne to form the ynone-substituent after reductive elimination. In summary, two equivalents of CO are inserted and one equivalent of hydrogen is generated, so that this represents a dehydrogenative cross-coupling. To close the catalytic cycle, Pd(0) is reoxidized by quinone (which forms hydroquinone by formal uptake of the hydrogen-equivalent).
3.5 Silver-catalysts
Wang and coworkers published an enantioselective tandem cyclization reaction of alkyne-tethered indoles, catalyzed by a combination of silver tetrafluoroborate and (S)-BNPA 101 (see Fig. 26).66 As a substrate, they used an indole that features a pentynyl-substituent in the 2-position and a carbamoylethyl-substituent in the 3-position. This substrate undergoes an enantioselective 6-exo-dig cyclization between the indole-enamine and the alkyne, followed by attack of the carbamate onto the resulting iminium-ion. As a result, the products 103 are formed, which feature a tricyclic skeleton with C2 and C3 as bridgehead atoms.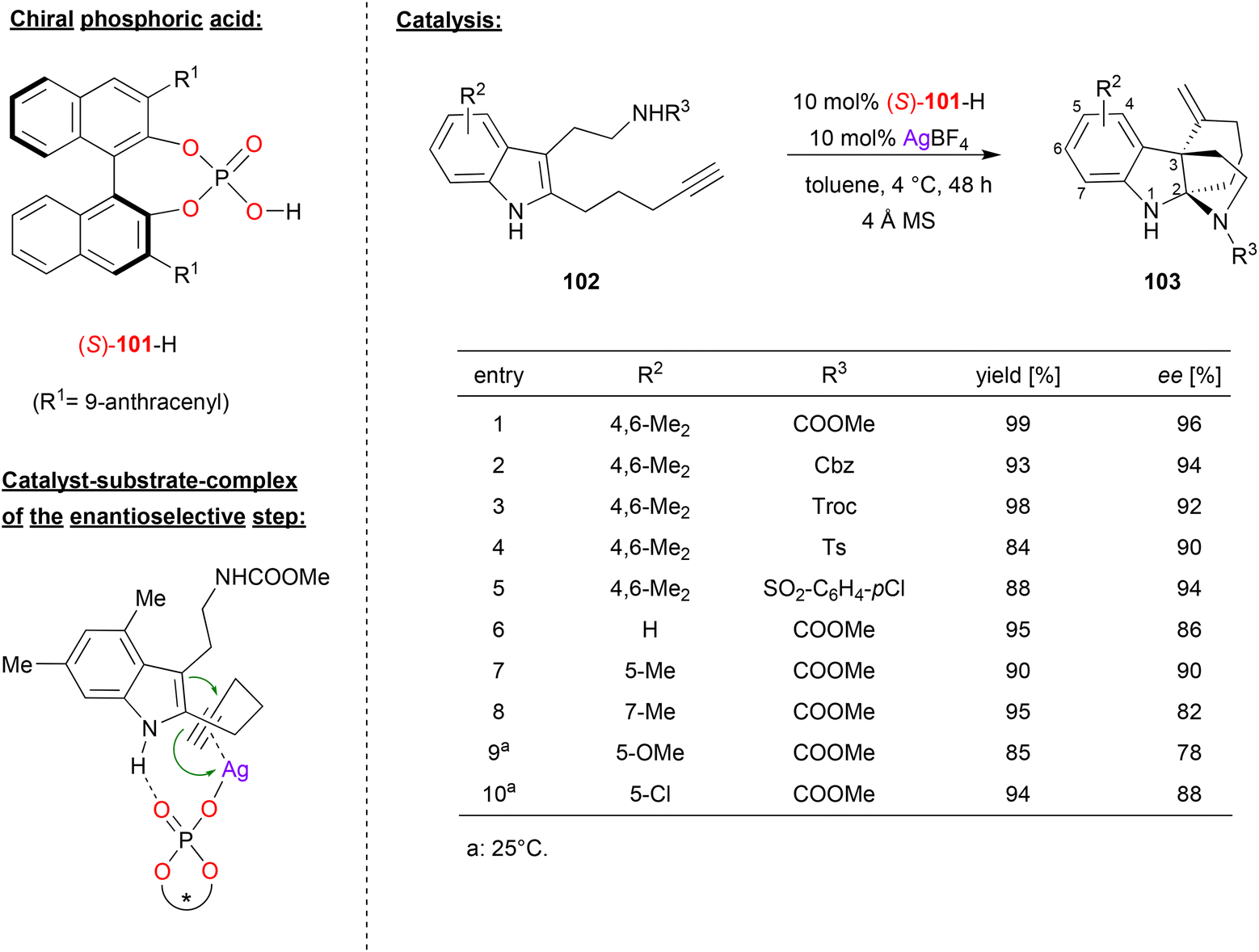 | ||
| Fig. 26 Enantioselective cyclization of alkyne-tethered indoles catalyzed by a Ag-phosphate.66 | ||
As exemplified in Fig. 26, the authors tested different variants of the carbamate side-chain and different substituents on the indole-backbone. Changing the carbamate from NH-COOMe to other carbamates (NH-Cbz, NH-Troc), but also to sulfonamides (NH-Ts, NH-SO2C6H4Cl) gave consistently high stereoselectivities (90–96% ee). Variation of the substituents on the indole-backbone had a stronger impact: changing from the original 4,6-Me2-system (96% ee) to other substitution patterns (e.g. unsubstituted, 5-Me/OMe/Cl or 7-Me) gave slightly diminished enantioselectivities (78–90% ee). No products could be formed from internal alkynes, and substitution on the indole NH led to decreased stereoselectivities.
As for the mechanism, the authors suggest a dual activation in the enantioselective 6-exo-dig cyclization: the phosphate is coordinated to the Ag+ in an η1-fashion, allowing for π-coordination of the alkyne, thus activating this for a nucleophilic attack of the enamine. At the same time, hydrogen bonding between the N–H of the indole and the Brønsted basic PO fragment leads to a well-defined transition state. According to DFT-calculations, the energy difference between the diastereomeric transition states amounts to 2.4 kcal mol−1, preferentially resulting in formation of the experimentally observed enantiomer.
Unsworth and coworkers used a chiral silver phosphate complex as catalyst in a dearomatization reaction of ynone-substituted indoles (see Fig. 27).67 The substrates 105 underwent a 5-endo-dig cyclization to give the corresponding spirocyclic products 106.
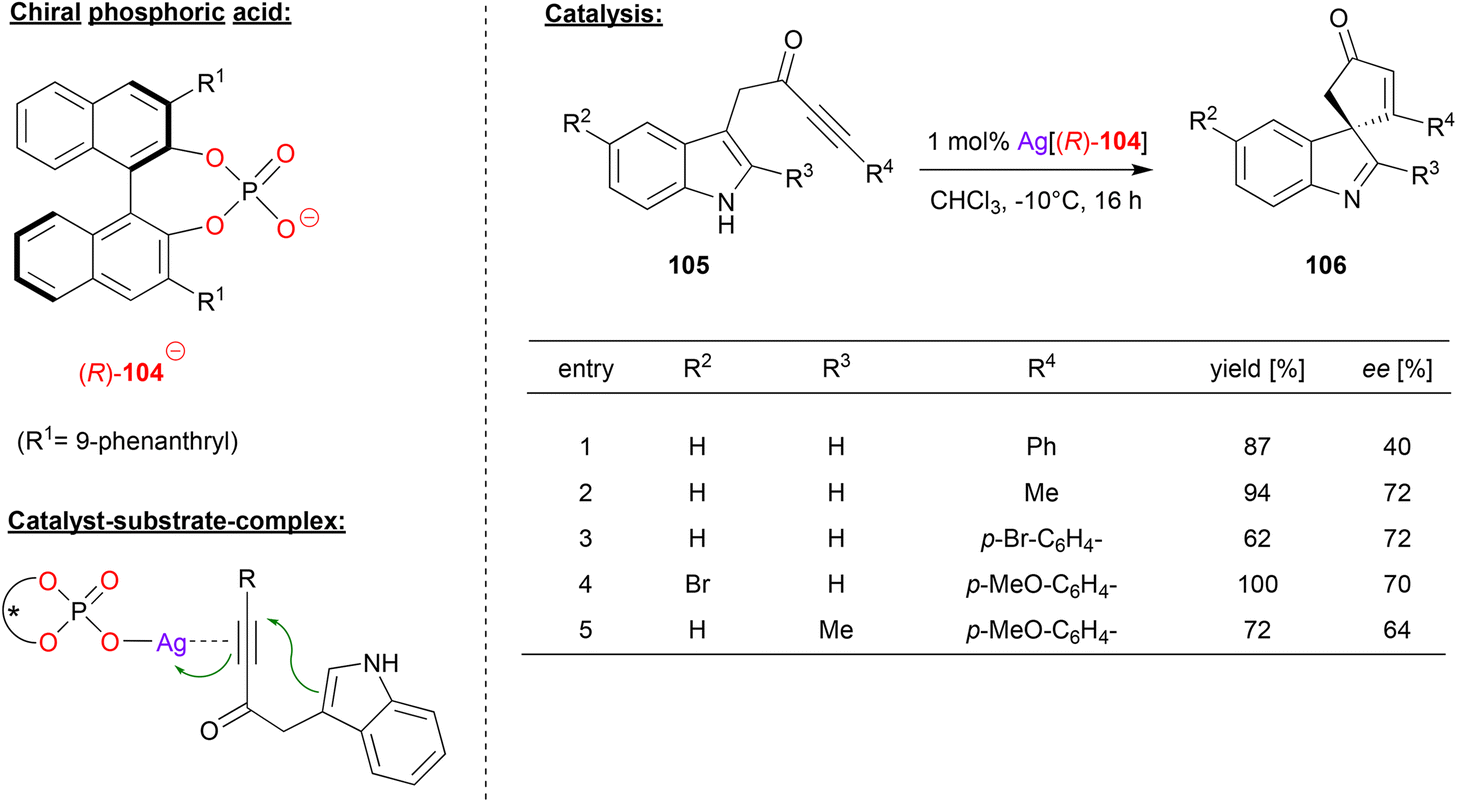 | ||
| Fig. 27 Cyclization of ynone-substituted indoles catalyzed by a Ag-phosphate.67 | ||
For the non-stereoselective case, the authors found that silver triflate enables excellent to quantitative yields in the spirocyclization. To render this transformation enantioselective, the authors used the Ag-complex of the phosphate-ligand 104. As shown in Fig. 27 for selected examples, different substituents were tolerated in the 2- and 5-position of the indole-backbone to give the products in moderate to good stereoselectivities (40–72% ee).
Mechanistically, the authors suggest an η1-silver phosphate complex, which coordinates to the ynone via the triple-bond. Although not suggested explicitly by the authors, additional PO⋯HN hydrogen bonding of the indole seems likely, which would lead to a divalent binding of the substrate.
Cossío and coworkers reported the cooperative catalysis by a silver phosphate and a chiral Chincona alkaloid base for 1,3-dipolar cycloaddition reactions (see Fig. 28).68 They used glycine ester imines 110 as precursors for the corresponding azomethine ylides, which were reacted with olefins 109 to the corresponding bicyclic proline-derivatives 111.
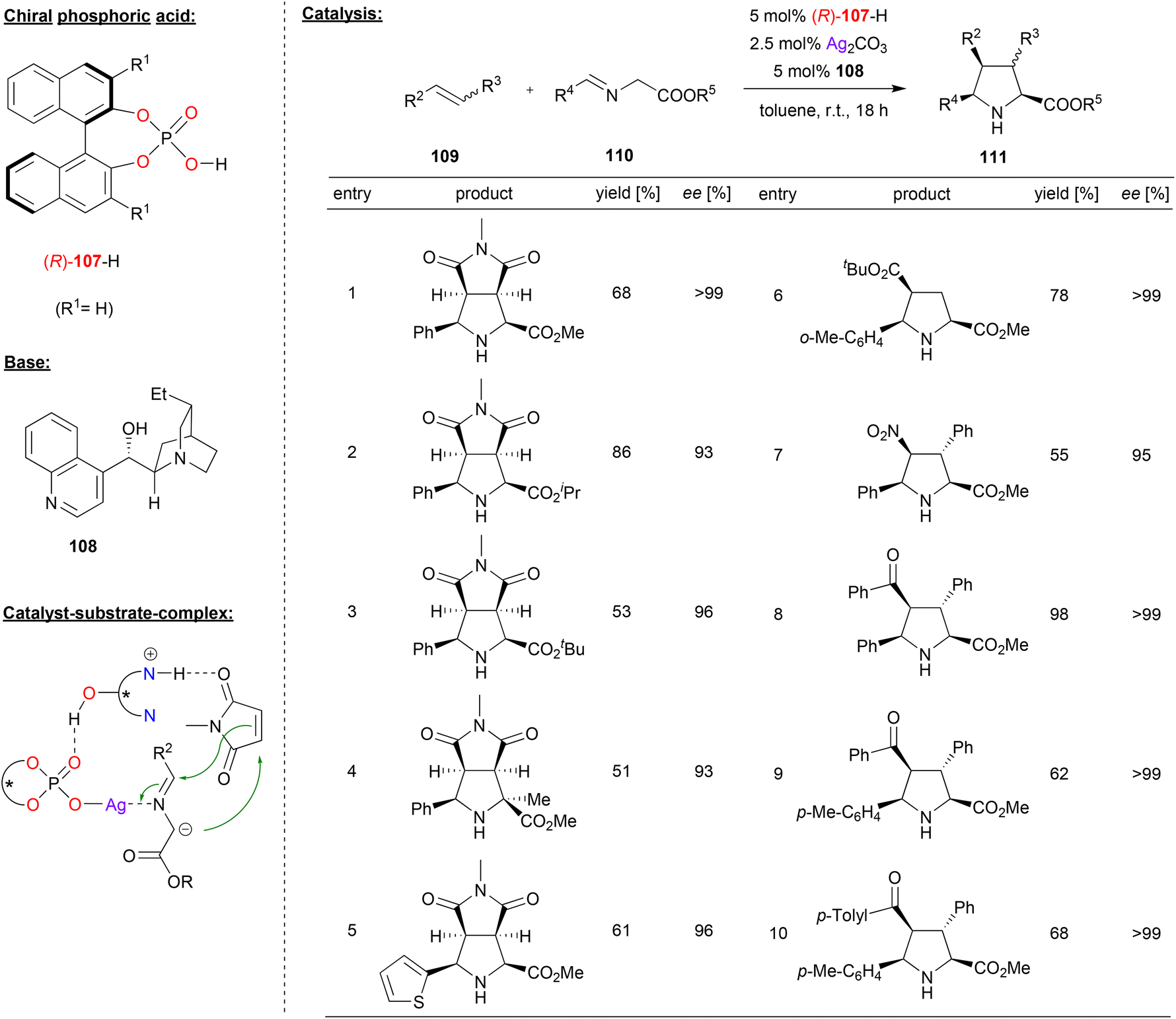 | ||
| Fig. 28 Enantioselective 1,3-dipolar cycloadditions of azomethine ylides catalyzed by a Ag-phosphate.68 | ||
It was found that the order of addition was crucial for high enantioselectivities, and the best results were obtained when Ag2CO3 and the chiral phosphoric acid (R)-107 were premixed in one flask, to which a mixture of the Chincona alkaloid 108 and the substrates 109 and 110 was added. As shown in Fig. 28 for selected examples, this gave excellent results for a variety of glycine ester imines in the reaction with N-methylmaleimide as the olefin (93–99% ee). Also, different acyclic acceptor-substituted olefins were successfully employed to give the products in excellent enantioselectivities (95–99% ee).
Mechanistically, the authors suggest the formation of an N-metalated azomethine ylide from the silver phosphate and the imine 110 on one hand and a complex between the substituted olefin and the protonated Chincona alkaloid on the other hand. Both complexes can then interact via a PO⋯HO hydrogen-bond to form a quaternary complex, which leads to product formation (see Fig. 28).
Gong and coworkers also used chiral silver phosphates for cycloaddition-reactions, in this case for a hetero-Diels–Alder (see Fig. 29).69 Here, the diazene 114 was reacted with the pentadiene-derivatives 113 to give the corresponding chiral 1,2-piperazines 115a/b.
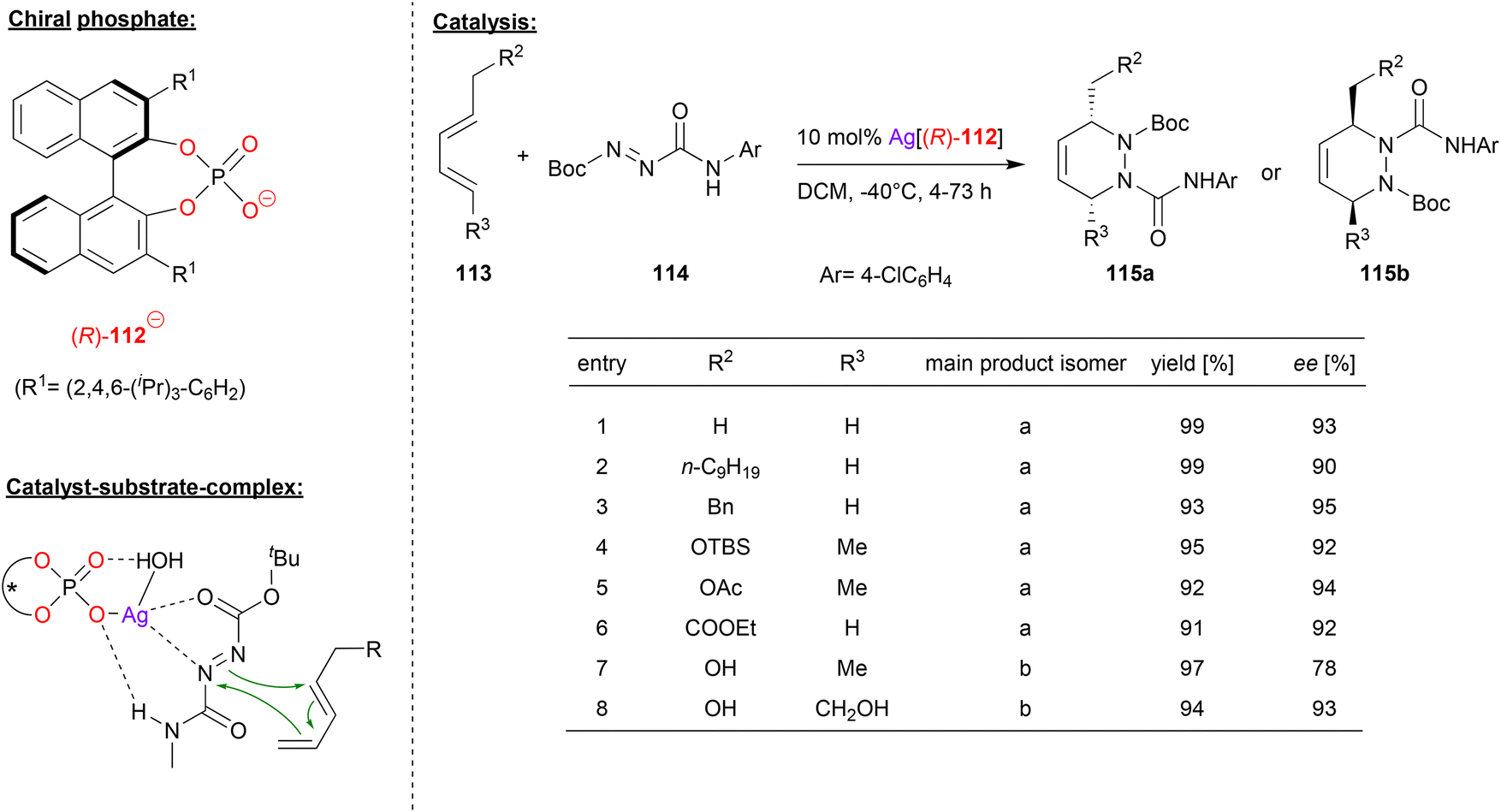 | ||
| Fig. 29 Enantioselective hetero-Diels–Alder reaction catalyzed by a Ag-phosphate.69 | ||
As can be seen from the selected examples in Fig. 29, different substituents on both ends of the pentadiene-substrate are tolerated to furnish the products 115a in high yields and enantioselectivities (90–95% ee). Interestingly, when a pentadiene featuring one or two unprotected hydroxy-groups is employed, the other regioisomer of the product (115b) is formed, also in good enantioselectivity (78–93% ee).
To investigate the structure of the transition-state, DFT-calculations were carried out. These suggest that an η1-phosphate-Ag complex coordinates the diazene via one nitrogen of the diazo-group and via the CO group of the Boc protecting group. In addition, the NH-function of the NN-CONHR protecting group forms a hydrogen-bond with the silver-bound P–O unit. In their calculations, the authors also take into account an additional water molecule that is bound the silver atom and forms an additional PO⋯HO hydrogen bond, thus stabilizing the entire complex.
For the pentadiene-substrates without free OH-groups, the substituent R2 (see Fig. 29) points away from the silver-phosphate, resulting in formation of the observed regioisomer 115a. For the OH-functionalized dienes, an additional hydrogen-bond to the PO unit would induce a 180° rotation of the diene, thus leading to the other regioisomer 115b. In both cases, the DFT calculations show a stereoselectivity of 4.6–5.2 kcal mol−1, giving the product enantiomers that were also favoured experimentally.
Next to the works describe above, organic silver phosphates have also been used for other transformations, such as the alkynylation of cyclic imines,70 the annulation of alkynylacrylaldehydes with styrenes71 and the intramolecular dearomatization of α-diazoacetamide-functionalized indoles72 (not shown in detail).
3.6 Gold-catalysts
Gong and coworkers used a chiral gold-phosphate complex, formed in situ from phosphoric acid 116 and the gold–carbene IMesAuMe, for a tandem hydroamination/asymmetric transfer hydrogenation reaction (see Fig. 30).73 Here, 2-propargylated anilines were cyclized in a 6-endo-dig fashion to give the corresponding 1,4-dihydroquinolines. After tautomerization to the 3,4-dihydroquinolines, these are enantioselectively reduced by Hantzsch-ester 118 to give the chiral tetrahydroquinolines 119.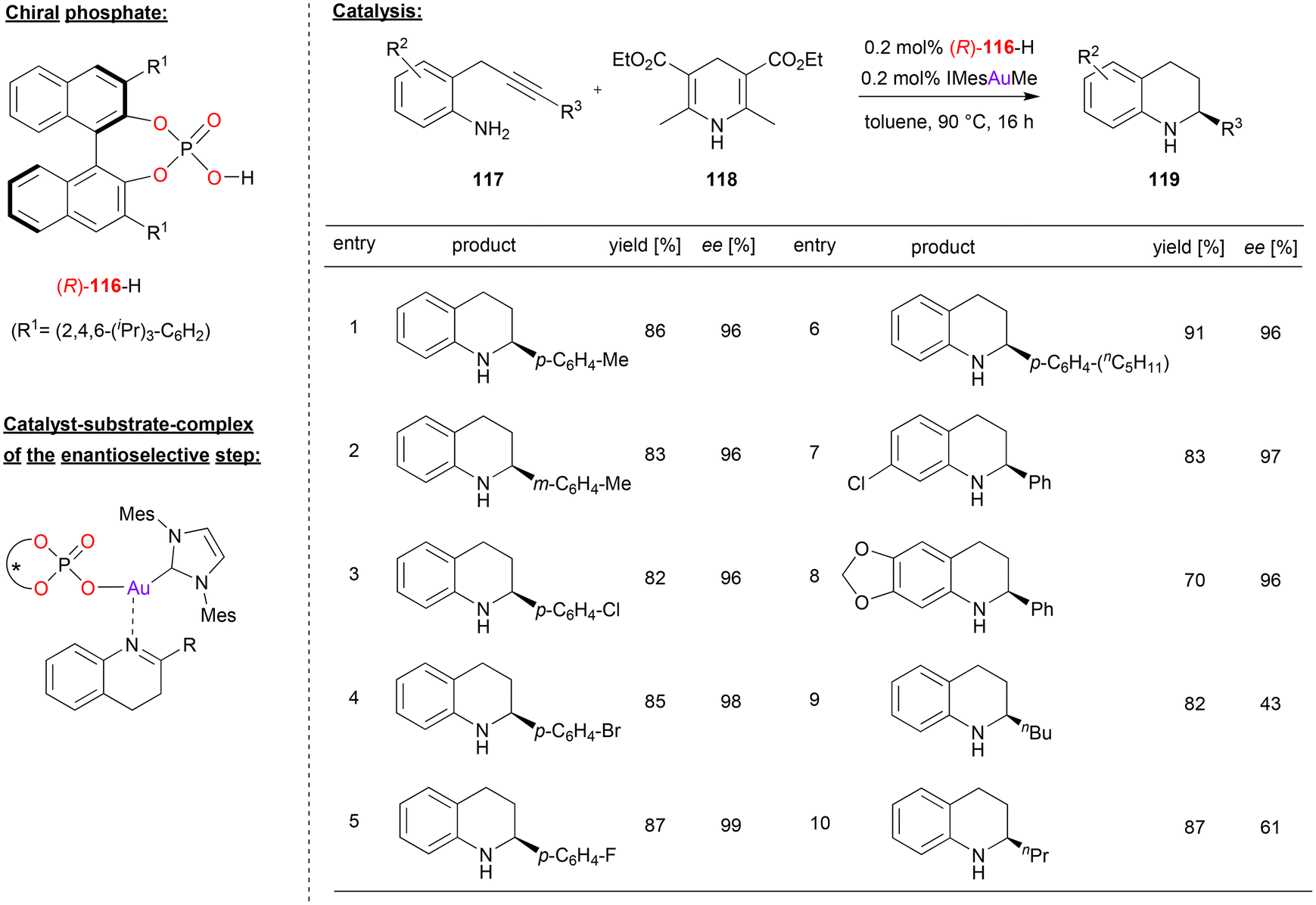 | ||
| Fig. 30 Hydroamination/enantioselective transfer hydrogenation catalyzed by an Au-phosphate.73 | ||
A variety of substrates underwent the tandem cyclization/transfer-hydrogenation in good yields (70–91%, see Fig. 30 for selected examples). Various aryl-substituents on the alkyne (i.e. in the 2-position of the dihydroquinoline-intermediate) were tolerated, as were different substituents on the aniline backbone, so that the products were obtained in high stereoselectivities (96–99% ee). However, when changing to alkyl-substituents on the alkyne, stereoselectivities dropped significantly (43/61% ee for n-butyl/n-propyl).
The authors suggest that the active catalyst is composed of the carbene-ligand and an η1-phosphate ligand to give an (IMes)AuI(phosphate) complex. This first acts as π-Lewis acid to enable the cyclization to the 4-dihydroquinolines. After tautomerization, the catalyst then acts as a chiral Lewis-acid to activate the 3,4-dihydroquinoline for the transfer-hydrogenation (see Fig. 30). Although not suggested explicitly by the authors, it seems likely that the phosphate ligand allows for simultaneous binding of the Hantzsch-ester via a PO⋯HN hydrogen-bond, which would lead to a compact transition state involving both substrates.
Togni, Hii and coworkers used the chiral phosphoric acid 120 in combination with different chiral ferrocenyl phosphanes (121a/b/c) in order to achieve the gold-catalyzed cyclization of γ-allenols (see Fig. 31).74 Here, the 2,2-diphenyl allenol 122 was subjected to cyclization to give the corresponding chiral tetrahydrofuranes 123.
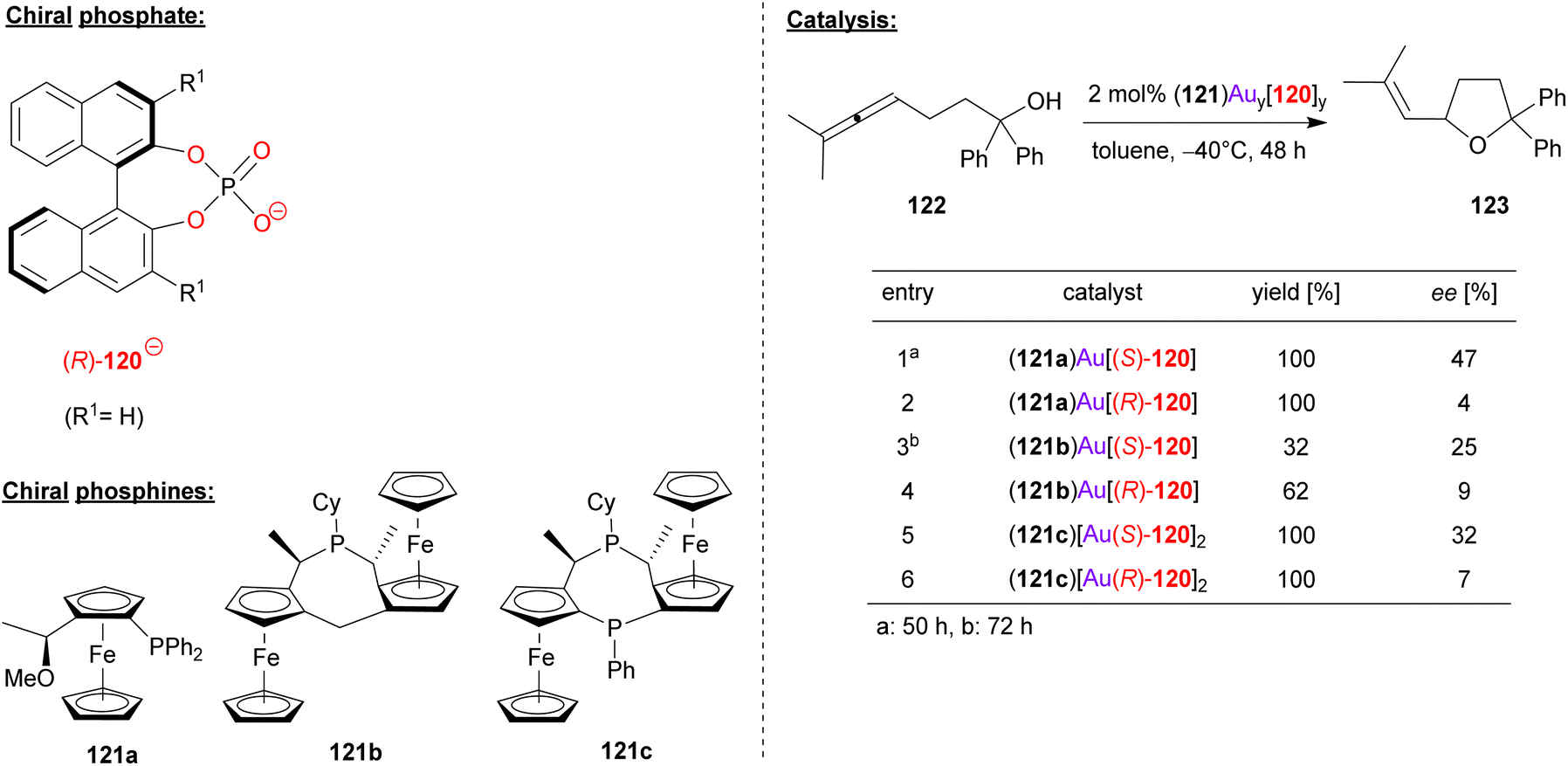 | ||
| Fig. 31 Enantioselective cyclization of a γ-allenol catalyzed by Au-phosphates.74 | ||
Three different ferrocene-based mono- and bisphosphines (121a/b/c) were transformed into the corresponding R3P-AuCl complexes [i.e. (121a)AuCl, (121b)AuCl and (121c)(AuCl)2], all of which were characterized by X-ray diffraction. By reaction with the appropriate amount of silver phosphate Ag(120), the heteroleptic complexes (121a)Au(120), (121b)Au(120) and (121c)[Au(120)]2 were formed. Both the (R)-BINOL- and (S)-BINOL-phosphates were used to generate all three pairs of diastereomeric complexes. In the application in catalysis, a matched/mismatched effect can be observed, but unfortunately the overall enantioselectivities were moderate (7–47% ee).
4. Rare-earth metals
Besides main-group-metal- and transition-metal-phosphates, rare-earth metal-phosphates have also been used as chiral catalysts. Next to examples based on ytterbium and cerium, which were already described in Ruepings review,1 two more recent examples have been reported. In 2015, Wang reported the use of homobimetallic yttrium-phosphates as catalysts for the hetero-Diels–Alder reaction between enamines and α,β-unsaturated imines to give the corresponding dihydropyridines (not shown in detail).75In 2019, the same group reported an extension of this work, based on the use of a heterobimetallic catalyst obtained from mixing a chiral ytterbium-trisphosphate and yttrium-tristriflate. Based on this catalyst system, they achieved the enantioselective ring-opening of the racemic donor–acceptor substituted cyclopropanes 125 with anilines 126 to give the chiral γ-amino acid derivatives 127 (see Fig. 32).76 This reaction is based on kinetic resolution of the racemic cyclopropanes, which are used in excess (2.4 eq.) with regard to the aniline nucleophile (1 eq.).
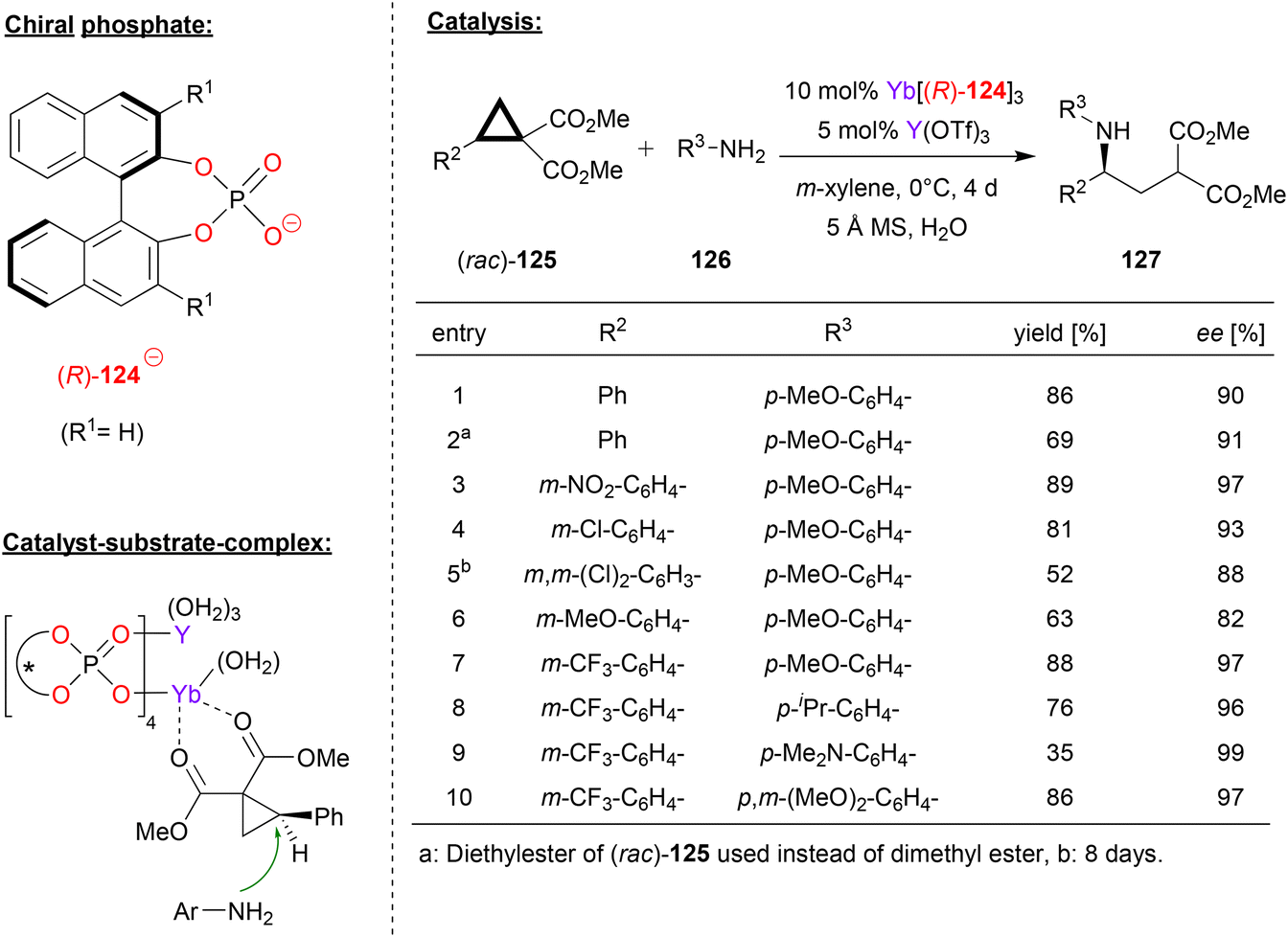 | ||
| Fig. 32 Enantioselective ring opening of cyclopropanes catalyzed by an Yb–Y-phosphate.76 | ||
As exemplified in Fig. 32, the substitution pattern on the aryl group of the cyclopropane and on the aniline were systematically varied. With regard to the aryl-group on the cyclopropane, electron-neutral and electron-withdrawing groups are well-tolerated to give the products in high stereoselectivities (88–97% ee), only a strongly electron-donating group led to slightly diminished enantiomeric excess (82% ee for OMe). When using the m-trifluoromethylphenyl-version of the cyclopropane, substitution on the aniline had only little effect, independent of the electronic nature of the substituents, resulting in formation of the products in high stereoselectivities (96–99% ee).
As for the catalyst structure, the authors found that mixing Yb(124)3 with Yb(OTf)3 leads to formation of the homobimetallic complex [Yb2(124)4]2+, which has a paddle-wheel geometry with four bridging μ–η2-phosphate ligands. From this, it was concluded that upon mixing Yb(124)3 with Y(OTf)3, as done for the catalyst preparation in the catalytic reaction, the isostructural heterobimetallic complex [(Yb)(Y)(124)4]2+ will be formed. Based on this complex, DFT-calculations suggest binding of the cyclopropane to the ytterbium–metal via both carbonyl-groups, placing the substrate in the chiral environment of the catalyst and activating it for intermolecular attack of the aniline (see Fig. 32).
5. Conclusion
In summary, the use of metal-phosphates has made possible a variety of different enantioselective transformations, enabled by the excellent enantioinduction delivered by many chiral phosphates and by the different modes of reactivity offered by the different metal centers.Here, the role of the metal in phosphate-complexes of main-group metals, early transition metals or rare-earth metals is mainly to serve as a Lewis-acid, e.g. for activation of the electrophile of the reaction. Here, special nature of the phosphate ligands, which offer Lewis-basic coordination sites on the P–O or PO fragments even when already coordinated to the metal, is highly beneficial. These sites allow additional coordination of electrophiles if they possess suitable hydrogen-bond donors. This leads to a tight precoordination of both substrates, allowing for high enantioselectivities.
In phosphate-metal complexes featuring late transition-metals, the role of the metal-center is naturally more diverse: rhodium-phosphates have been used as the corresponding carbene-complexes, enabling cyclopropanation and Si–H insertion chemistry. Phosphate-palladium complexes oftentimes perform their catalytic reactions involving oxidative addition, reductive elimination or migratory insertion steps. This enables transformations such as C–H activation or carbonylative couplings. The Pd-phosphates are mostly used in combination with additional co-ligands featuring N-donors, which offers additional potential for catalyst tuning.
Silver and gold metal centers serve as Lewis-acids for coordination of O- or N-donor atoms, but also enable π-activation of carbon–carbon multiple bonds.
In summary, there is certainly a lot of room for further development in this exciting area. For example, the use of multidentate ligands, either involving two phosphate units or involving one phosphate and a different second donor, has not been reported. Also, exciting new chiral frameworks have emerged in the area of Brønsted-acid catalysis, such as the imidodiphosphates (IDP), imino-imidodiphosphates (iIDP) and imido-diphosphorimidates (IDPi).4 These are highly interesting candidates to be used as ligands for metal-centers, which will certainly be evaluated by the community in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the DFG (Research Grant NI1273/2-2 and Heisenberg-Professorship NI1273/4-1 to J.N.) and the University of Duisburg-Essen is gratefully acknowledged.References
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2014, 114, 9047–9153 CAS.
- (a) D. Uraguchi and M. Terada, Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation, J. Am. Chem. Soc., 2004, 126, 5356–5357 CAS; (b) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CAS.
- (a) M. Rueping, A. Kuenkel and I. Atodiresei, Chiral Brønsted acids in enantioselective carbonyl activations - activation modes and applications, Chem. Soc. Rev., 2011, 40, 4539–4549 CAS; (b) M. Terada, Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations, Synthesis, 2010, 1929–1982 CAS.
- L. Schreyer, R. Properzi and B. List, IDPi Catalysis, Angew. Chem., Int. Ed., 2019, 58, 12761–12777 CAS.
- A. Zamfir, S. Schenker, M. Freund and S. B. Tsogoeva, Chiral BINOL-derived phosphoric acids: privileged Brønsted acid organocatalysts for C–C bond formation reactions, Org. Biomol. Chem., 2010, 8, 5262–5276 CAS.
- (a) X. Lin, L. Wang, Z. Han and Z. Chen, Chiral Spirocyclic Phosphoric Acids and Their Growing Applications, Chin. J. Chem., 2021, 39, 802–824 CAS; (b) Y. Zhang, Z. Lu and W. D. Wulff, Catalytic Asymmetric Aziridination with Catalysts Derived from VAPOL and VANOL, Synlett, 2009, 2715–2739 CAS.
- (a) M. C. Benda and S. France, Chiral disulfonimides: a versatile template for asymmetric catalysis, Org. Biomol. Chem., 2020, 18, 7485–7513 CAS; (b) L. Liu, P. S. J. Kaib, A. Tap and B. List, A General Catalytic Asymmetric Prins Cyclization, J. Am. Chem. Soc., 2016, 138, 10822–10825 CAS; (c) T. James, M. van Gemmeren and B. List, Development and Applications of Disulfonimides in Enantioselective Organocatalysis, Chem. Rev., 2015, 115, 9388–9409 CAS; (d) I. Čorić and B. List, Asymmetric spiroacetalization catalysed by confined Brønsted acids, Nature, 2012, 483, 315–319 Search PubMed; (e) R. Mitra and J. Niemeyer, Dual Brønsted-acid Organocatalysis: Cooperative Asymmetric Catalysis with Combined Phosphoric and Carboxylic Acids, ChemCatChem, 2018, 10, 1221–1234 CAS.
- M. Mahlau and B. List, Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications, Angew. Chem., Int. Ed., 2013, 52, 518–533 CAS.
- (a) S. Kaneko, Y. Kumatabara and S. Shirakawa, A new generation of chiral phase-transfer catalysts, Org. Biomol. Chem., 2016, 14, 5367–5376 CAS; (b) R. J. Phipps, G. L. Hamilton and F. D. Toste, The progression of chiral anions from concepts to applications in asymmetric catalysis, Nat. Chem., 2012, 4, 603–614 CAS.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Addition and Correction to Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2017, 117, 10608–10620 CAS.
- T. Ahmad, S. Khan and N. Ullah, Recent Advances in the Catalytic Asymmetric Friedel–Crafts Reactions of Indoles, ACS Omega, 2022, 7, 35446–35485 CAS.
- M. Hatano and K. Ishihara, Highly Practical BINOL-Derived Acid-Base Combined Salt Catalysts for the Asymmetric Direct Mannich-Type Reaction, Synthesis, 2010, 3785–3801 CAS.
- S. Mondal, F. Dumur, D. Gigmes, M. P. Sibi, M. P. Bertrand and M. Nechab, Enantioselective Radical Reactions Using Chiral Catalysts, Chem. Rev., 2022, 122, 5842–5976 CAS.
- P.-S. Wang and L.-Z. Gong, Asymmetric C–H Functionalization Enabled by Pd/Chiral Phosphoric Acid Combined Catalysis, Synthesis, 2021, 4795–4801 Search PubMed.
- D. Zhang and W. Hu, Asymmetric Multicomponent Reactions Based on Trapping of Active Intermediates, Chem. Rec., 2017, 17, 739–753 CAS.
- J. Hansen and H. M. L. Davies, High symmetry dirhodium(II) paddlewheel complexes as chiral catalysts, Coord. Chem. Rev., 2008, 252, 545–555 CAS.
- H. Pellissier, Enantioselective Silver-Catalyzed Transformations, Chem. Rev., 2016, 116, 14868–14917 CAS.
- (a) A. Pradal, P. Y. Toullec and V. Michelet, Recent Developments in Asymmetric Catalysis in the Presence of Chiral Gold Complexes, Synthesis, 2011, 1501–1514 CAS; (b) A. S. K. Hashmi and C. Hubbert, Gold and Organocatalysis Combined, Angew. Chem., Int. Ed., 2010, 49, 1010–1012 CAS.
- J. Inanaga, H. Furuno and T. Hayano, Asymmetric Catalysis and Amplification with Chiral Lanthanide Complexes, Chem. Rev., 2002, 102, 2211–2226 CAS.
- (a) Z. Wu, H. He, M. Chen, L. Zhu, W. Zheng, Y. Cao and J. C. Antilla, Asymmetric Reductive Amination with Pinacolborane Catalyzed by Chiral SPINOL Borophosphates, Org. Lett., 2022, 24, 9436–9441 CAS; (b) H. He, X. Shen, X. Ding and J. C. Antilla, Enantioselective Mukaiyama–Michael Reaction of β,γ-Unsaturated α-Keto Esters with Silyl Ketene Acetals Catalyzed by a Chiral Magnesium Phosphate, Org. Lett., 2023, 25, 782–787 CAS; (c) H. He, K. S. S. Tummalapalli, L. Zhu, M. Chen, S. Krishnamurthy and J. C. Antilla, Asymmetric Rubottom-Type Oxidation Catalyzed by Chiral Calcium Phosphates, Chem. – Eur. J., 2023, 29, e202203720 CAS.
- N. Pairault, H. Zhu, D. Jansen, A. Huber, C. G. Daniliuc, S. Grimme and J. Niemeyer, Heterobifunctional Rotaxanes for Asymmetric Catalysis, Angew. Chem., Int. Ed., 2020, 59, 5102–5107 CAS.
- (a) C. Kwamen and J. Niemeyer, Functional Rotaxanes in Catalysis, Chem. – Eur. J., 2021, 27, 175–186 CAS; (b) V. Blanco, D. A. Leigh and V. Marcos, Artificial switchable catalysts, Chem. Soc. Rev., 2015, 44, 5341–5370 CAS.
- Y. Shen, M.-L. Shen and P.-S. Wang, Light-Mediated Chiral Phosphate Catalysis for Asymmetric Dicarbofunctionalization of Enamides, ACS Catal., 2020, 10, 8247–8253 CAS.
- M.-L. Shen, Y. Shen and P.-S. Wang, Merging Visible-Light Photoredox and Chiral Phosphate Catalysis for Asymmetric Friedel–Crafts Reaction with in Situ Generation of N-Acyl Imines, Org. Lett., 2019, 21, 2993–2997 CAS.
- G. Ingle, M. G. Mormino and J. C. Antilla, Lithium BINOL phosphate catalyzed desymmetrization of meso-epoxides with aromatic thiols, Org. Lett., 2014, 16, 5548–5551 CAS.
- Y. Wang, S. Wang, W. Shan and Z. Shao, Direct asymmetric N-propargylation of indoles and carbazoles catalyzed by lithium SPINOL phosphate, Nat. Commun., 2020, 11, 226 CAS.
- H. He, Y. Cao, J. Xu and J. C. Antilla, Catalytic Asymmetric C-7 Friedel–Crafts Alkylation/N-Hemiacetalization of 4-Aminoindoles, Org. Lett., 2021, 23, 3010–3014 CAS.
- A. G. Woldegiorgis, Z. Han and X. Lin, Asymmetric [3 + 3] Annulation to Construct Trifluoromethylated Pyrazolo[3,4-b]pyridin-6-ones via Chiral Phosphoric Acid and MgSO4 Synergistic Catalysis, Org. Lett., 2022, 24, 4058–4063 CAS.
- S. K. Nimmagadda, Z. Zhang and J. C. Antilla, Asymmetric One-Pot Synthesis of 1,3-Oxazolidines and 1,3-Oxazinanes via Hemiaminal Intermediates, Org. Lett., 2014, 16, 4098–4101 CAS.
- Y. Bai, J. Yuan, X. Hu and J. C. Antilla, Catalytic Enantioselective Diels–Alder Reactions of Benzoquinones and Vinylindoles with Chiral Magnesium Phosphate Complexes, Org. Lett., 2019, 21, 4549–4553 CAS.
- T. Hodík and C. Schneider, A Highly Enantio– and Diastereoselective Synthesis of Spirocyclic Dihydroquinolones via Domino Michael Addition–Lactamization of ortho –Quinone Methide Imines, Chem. – Eur. J., 2018, 24, 18082–18088 Search PubMed.
- I. Ibáñez, M. Kaneko, Y. Kamei, R. Tsutsumi, M. Yamanaka and T. Akiyama, Enantioselective Friedel–Crafts Alkylation Reaction of Indoles with α-Trifluoromethylated β-Nitrostyrenes Catalyzed by Chiral BINOL Metal Phosphate, ACS Catal., 2019, 9, 6903–6909 Search PubMed.
- C. Lalli, A. Dumoulin, C. Lebée, F. Drouet, V. Guérineau, D. Touboul, V. Gandon, J. Zhu and G. Masson, Chiral calcium-BINOL phosphate catalyzed diastereo- and enantioselective synthesis of syn-1,2-disubstituted 1,2-diamines: scope and mechanistic studies, Chem. – Eur. J., 2015, 21, 1704–1712 CAS.
- X. Fang, Z. Deng, W. Zheng and J. C. Antilla, Catalytic One-Pot Double Asymmetric Cascade Reaction: Synthesis of Chlorinated Oxindoles and Geminal Diamines, ACS Catal., 2019, 9, 1748–1752 CAS.
- R. Cao and J. C. Antilla, Imine Amidation Catalyzed by a Chiral VAPOL Calcium Phosphate, Org. Lett., 2020, 22, 5958–5962 CAS.
- R. Liu, S. Krishnamurthy, Z. Wu, K. S. S. Tummalapalli and J. C. Antilla, Chiral Calcium Phosphate Catalyzed Enantioselective Amination of 3-Aryl-2-benzofuranones, Org. Lett., 2020, 22, 8101–8105 CAS.
- Z. Wu, S. Krishnamurthy, K. S. Satyanarayana Tummalapalli, J. Xu, C. Yue and J. C. Antilla, Enantioselective Amination of β -Keto Esters Catalyzed by Chiral Calcium Phosphates, Chem. – Eur. J., 2022, 28, e202200907 CAS.
- Z. Wu, S. Krishnamurthy, K. S. S. Tummalapalli and J. C. Antilla, Chiral Calcium Phosphate-Catalyzed Enantioselective Amination of 3-Aryl-2-oxindoles with Dibenzyl Azodicarboxylate, J. Org. Chem., 2022, 87, 8203–8212 CAS.
- T. Liang, G. Li, L. Wojtas and J. C. Antilla, Chiral metal phosphate catalysis: highly asymmetric hetero-Diels-Alder reactions, Chem. Commun., 2014, 50, 14187–14190 CAS.
- Z. Sun, L. Chen, K. Qiu, B. Liu, H. Li and F. Yu, Enantioselective Peroxidation of C-alkynyl imines enabled by chiral BINOL calcium phosphate, Chem. Commun., 2022, 58, 3035–3038 CAS.
- L. Wang, J. Lv, L. Zhang and S. Luo, Catalytic Regio- and Enantioselective [4 + 2] Annulation Reactions of Non-activated Allenes by a Chiral Cationic Indium Complex, Angew. Chem., Int. Ed., 2017, 56, 10867–10871 CAS.
- X. Zhong, J. Lv and S. Luo, Enantio- and Diastereoselective Cyclopropanation of β,γ-Unsaturated α-Ketoester by a Chiral Phosphate/Indium(III) Complex, Org. Lett., 2017, 19, 3331–3334 CAS.
- Y.-L. Pan, H.-L. Zheng, J. Wang, C. Yang, X. Li and J.-P. Cheng, Enantioselective Allylation of Oxocarbenium Ions Catalyzed by Bi(OAc)3/C hiral Phosphoric Acid, ACS Catal., 2020, 10, 8069–8076 CAS.
- J. Wang, Q. Zhang, Y. Li, X. Liu, X. Li and J.-P. Cheng, Bi(OAc)3/C hiral phosphoric acid catalyzed enantioselective allylation of isatins, Chem. Commun., 2020, 56, 261–264 CAS.
- L. Cai, Y.-L. Pan, L. Chen, J.-P. Cheng and X. Li, Bi(OAc)3/c hiral phosphoric acid catalyzed enantioselective allylation of seven-membered cyclic imines, dibenzo[b,f][1,4]oxazepines, Chem. Commun., 2020, 56, 12383–12386 CAS.
- A. J. Teator and F. A. Leibfarth, Catalyst-controlled stereoselective cationic polymerization of vinyl ethers, Science, 2019, 363, 1439–1443 CAS.
- K. Gebauer, F. Reuß, M. Spanka and C. Schneider, Relay Catalysis: Manganese(III) Phosphate Catalyzed Asymmetric Addition of β-Dicarbonyls to ortho-Quinone Methides Generated by Catalytic Aerobic Oxidation, Org. Lett., 2017, 19, 4588–4591 CAS.
- K. M. Chepiga, C. Qin, J. S. Alford, S. Chennamadhavuni, T. M. Gregg, J. P. Olson and H. M. L. Davies, Guide to Enantioselective Dirhodium(II)-Catalyzed Cyclopropanation with Aryldiazoacetates, Tetrahedron, 2013, 69, 5765–5771 CAS.
- L.-L. Yang, D. Evans, B. Xu, W.-T. Li, M.-L. Li, S.-F. Zhu, K. N. Houk and Q.-L. Zhou, Enantioselective Diarylcarbene Insertion into Si-H Bonds Induced by Electronic Properties of the Carbenes, J. Am. Chem. Soc., 2020, 142, 12394–12399 CAS.
- L.-L. Yang, J. Ouyang, H.-N. Zou, S.-F. Zhu and Q.-L. Zhou, Enantioselective Insertion of Alkynyl Carbenes into Si–H Bonds: An Efficient Access to Chiral Propargylsilanes and Allenylsilanes, J. Am. Chem. Soc., 2021, 143, 6401–6406 CAS.
- L.-L. Yang, J. Cao, T.-Y. Zhao, S.-F. Zhu and Q.-L. Zhou, Chiral Dirhodium Tetraphosphate-Catalyzed Enantioselective Si–H Bond Insertion of α-Aryldiazoacetates, J. Org. Chem., 2021, 86, 9692–9698 CAS.
- D. Banerjee, K. Junge and M. Beller, Cooperative catalysis by palladium and a chiral phosphoric acid: enantioselective amination of racemic allylic alcohols, Angew. Chem., Int. Ed., 2014, 53, 13049–13053 CAS.
- H.-C. Lin, P.-S. Wang, Z.-L. Tao, Y.-G. Chen, Z.-Y. Han and L.-Z. Gong, Highly Enantioselective Allylic C–H Alkylation of Terminal Olefins with Pyrazol-5-ones Enabled by Cooperative Catalysis of Palladium Complex and Brønsted Acid, J. Am. Chem. Soc., 2016, 138, 14354–14361 CAS.
- J. Meng, L.-F. Fan, Z.-Y. Han and L.-Z. Gong, α-Quaternary Chiral Aldehydes from Styrenes, Allylic Alcohols, and Syngas via Multi-catalyst Relay Catalysis, Chem, 2018, 4, 1047–1058 CAS.
- Y.-L. Su, L.-L. Li, X.-L. Zhou, Z.-Y. Dai, P.-S. Wang and L.-Z. Gong, Asymmetric α-Allylation of Aldehydes with Alkynes by Integrating Chiral Hydridopalladium and Enamine Catalysis, Org. Lett., 2018, 20, 2403–2406 CAS.
- Z. Kang, W. Chang, X. Tian, X. Fu, W. Zhao, X. Xu, Y. Liang and W. Hu, Ternary Catalysis Enabled Three-Component Asymmetric Allylic Alkylation as a Concise Track to Chiral α,α-Disubstituted Ketones, J. Am. Chem. Soc., 2021, 143, 20818–20827 CAS.
- R. Y. Hua, S. F. Yu, X. T. Jie, H. Qiu and W. H. Hu, Multicomponent Assembly of Complex Oxindoles by Enantioselective Cooperative Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202213407 CAS.
- D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Enantioselective Palladium(II) Phosphate Catalyzed Three-Component Reactions of Pyrrole, Diazoesters, and Imines, Angew. Chem., Int. Ed., 2013, 52, 13356–13360 CAS.
- X. Bao, Q. Wang and J. Zhu, Palladium-Catalyzed Enantioselective Desymmetrizing Aza-Wacker Reaction: Development and Application to the Total Synthesis of (-)-Mesembrane and (+)-Crinane, Angew. Chem., Int. Ed., 2018, 57, 1995–1999 CAS.
- J. Luo, T. Zhang, L. Wang, G. Liao, Q. J. Yao, Y. J. Wu, B. B. Zhan, Y. Lan, X. F. Lin and B. F. Shi, Enantioselective Synthesis of Biaryl Atropisomers by Pd–Catalyzed C–H Olefination using Chiral Spiro Phosphoric Acid Ligands, Angew. Chem., Int. Ed., 2019, 58, 6708–6712 CAS.
- B.-B. Zhan, Z.-S. Jia, J. Luo, L. Jin, X.-F. Lin and B.-F. Shi, Palladium-Catalyzed Directed Atroposelective C–H Allylation via β-H Elimination: 1,1-Disubstituted Alkenes as Allyl Surrogates, Org. Lett., 2020, 22, 9693–9698 CAS.
- L. Yang, R. Melot, M. Neuburger and O. Baudoin, Palladium(0)-catalyzed asymmetric C(sp3)-H arylation using a chiral binol-derived phosphate and an achiral ligand, Chem. Sci., 2017, 8, 1344–1349 CAS.
- S.-Y. Yan, Y.-Q. Han, Q.-J. Yao, X.-L. Nie, L. Liu and B.-F. Shi, Palladium(II)-Catalyzed Enantioselective Arylation of Unbiased Methylene C(sp3)–H Bonds Enabled by a 2-Pyridinylisopropyl Auxiliary and Chiral Phosphoric Acids, Angew. Chem., Int. Ed., 2018, 57, 9093–9097 CAS.
- B. B. Zhan, L. Wang, J. Luo, X. F. Lin and B. F. Shi, Synthesis of Axially Chiral Biaryl-2-amines by PdII -Catalyzed Free–Amine–Directed Atroposelective C–H Olefination, Angew. Chem., Int. Ed., 2020, 59, 3568–3572 CAS.
- B. Yang, Y. Qiu, T. Jiang, W. D. Wulff, X. Yin, C. Zhu and J.-E. Bäckvall, Enantioselective Palladium-Catalyzed Carbonylative Carbocyclization of Enallenes via Cross-Dehydrogenative Coupling with Terminal Alkynes: Efficient Construction of α-Chirality of Ketones, Angew. Chem., Int. Ed., 2017, 56, 4535–4539 CAS.
- Y. Zhu, W. He, W. Wang, C. E. Pitsch, X. Wang and X. Wang, Enantioselective Tandem Cyclization of Alkyne-Tethered Indoles Using Cooperative Silver(I)/Chiral Phosphoric Acid Catalysis, Angew. Chem., Int. Ed., 2017, 56, 12206–12209 CAS.
- M. J. James, J. D. Cuthbertson, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Silver(I)- or copper(II)-mediated dearomatization of aromatic ynones: direct access to spirocyclic scaffolds, Angew. Chem., Int. Ed., 2015, 54, 7640–7643 CAS.
- A. Cayuelas, O. Larrañaga, V. Selva, C. Nájera, T. Akiyama, J. M. Sansano, A. De Cózar, J. I. Miranda and F. P. Cossío, Cooperative Catalysis with Coupled Chiral Induction in 1,3-Dipolar Cycloadditions of Azomethine Ylides, Chem. – Eur. J., 2018, 24, 8092–8097 CAS.
- B. Liu, T.-Y. Liu, S.-W. Luo and L.-Z. Gong, Asymmetric hetero-Diels-Alder reaction of diazenes catalyzed by chiral silver phosphate: water participates in the catalysis and stereocontrol, Org. Lett., 2014, 16, 6164–6167 CAS.
- Y.-Y. Ren, Y.-Q. Wang and S. Liu, Asymmetric Alkynylation of Seven-Membered Cyclic Imines by Combining Chiral Phosphoric Acids and Ag(I) Catalysts: Synthesis of 11-Substituted-10,11-dihydrodibenzo[b, f][1,4]oxazepine Derivatives, J. Org. Chem., 2014, 79, 11759–11767 CAS.
- H. Zhang, L. Zhu, S. Wang and Z.-J. Yao, Asymmetric Annulation of 3-Alkynylacrylaldehydes with Styrene-Type Olefins by Synergetic Relay Catalysis from AgOAc and Chiral Phosphoric Acid, J. Org. Chem., 2014, 79, 7063–7074 CAS.
- J. Ueda, S. Harada, A. Kanda, H. Nakayama and T. Nemoto, Silver-Catalyzed, Chemo- and Enantioselective Intramolecular Dearomatization of Indoles to Access Sterically Congested Azaspiro Frameworks, J. Org. Chem., 2020, 85, 10934–10950 CAS.
- Y.-L. Du, Y. Hu, Y.-F. Zhu, X.-F. Tu, Z.-Y. Han and L.-Z. Gong, Chiral Gold Phosphate Catalyzed Tandem Hydroamination/Asymmetric Transfer Hydrogenation Enables Access to Chiral Tetrahydroquinolines, J. Org. Chem., 2015, 80, 4754–4759 CAS.
- E. M. Barreiro, D. F. D. Broggini, L. A. Adrio, A. J. P. White, R. Schwenk, A. Togni and K. K. Hii, Gold(I) Complexes of Conformationally Constricted Chiral Ferrocenyl Phosphines, Organometallics, 2012, 31, 3745–3754 CAS.
- Y. Deng, C. V. Karunaratne, E. Csatary, D. L. Tierney, K. Wheeler and H. Wang, Chiral Bimetallic Catalysts Derived from Chiral Metal Phosphates: Enantioselective Three-Component Asymmetric Aza-Diels–Alder Reactions of Cyclic Ketones, J. Org. Chem., 2015, 80, 7984–7993 CAS.
- W. Luo, Z. Sun, E. H. N. Fernando, V. N. Nesterov, T. R. Cundari and H. Wang, Asymmetric Ring-Opening of Donor–Acceptor Cyclopropanes with Primary Arylamines Catalyzed by a Chiral Heterobimetallic Catalyst, ACS Catal., 2019, 9, 8285–8293 CAS.
| This journal is © the Partner Organisations 2023 |