
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient photoelectrochemical Kolbe C–C coupling at BiVO4 electrodes under visible light irradiation†
William A.
Swansborough-Aston
 a,
Ayman
Soltan
a,
Ben
Coulson
b,
Andrew
Pratt
b,
Victor
Chechik
*a and
Richard E.
Douthwaite
*a
a,
Ayman
Soltan
a,
Ben
Coulson
b,
Andrew
Pratt
b,
Victor
Chechik
*a and
Richard E.
Douthwaite
*a
aDepartment of Chemistry, University of York, York, YO10 5DD, UK. E-mail: richard.douthwaite@york.ac.uk
bDepartment of Physics, University of York, Heslington, York YO10 5DD, UK
First published on 16th January 2023
Abstract
Electrochemical Kolbe C–C coupling of carboxylic acids at Pt electrodes has been studied for over 150 years and remains relevant today because renewable electricity is envisaged to make an increasing contribution to clean chemical processes and carboxylic acids are readily available precursors for chemical synthesis. Traditional electrochemical Kolbe occurs typically at very high potential (>10 V) which is required to achieve high selectivity for C–C coupling. Here we describe porous BiVO4 photoelectrodes that mediate C–C Kolbe coupling with near quantitative faradaic efficiency under visible light irradiation at <2 V. High substrate concentrations are also found to stabilise the double layer avoiding the need for additional supporting electrolyte. Comparison with related literature describing photocatalytic Kolbe C–C coupling shows that the apparent quantum yield can be raised from <1% to 12% demonstrating the distinct advantage of using photoelectrochemistry in this system.
Introduction
There is an increasing drive to develop technologies for chemical synthesis that mitigate the use of fossil fuels and possibly open new reaction pathways. Approaches to more renewable chemical synthesis have seen significant advances in recent years, including heterogeneous, electro- and photocatalytic transformations which exploit renewable electricity and solar energy.1–3 In general, heterogeneous catalysis offers the key advantage of simple product separation, which has motivated the use of solid electro- and colloidal photocatalysts.4 Photoelectrochemistry (PEC) combines aspects of electro- and photocatalysis, comprising a photoactive semiconductor for light absorption, application of a bias to promote electron–hole separation and surmount overpotential, and offers the spatial separation of redox catalytic sites.5 Efficient PEC requires optimization of light absorption, the electrode–electrolyte interface, and catalysis, which is technically very demanding. However, in comparison to traditional dark electrochemistry, an advantage of PEC is the generation of photoexcited states which reduce the required bias potential to induce a reaction, and mitigates substrate or solvent degradation observed at large applied potentials. Furthermore, compared to colloidal photocatalysis, applying a bias to a photoelectrode allows manipulation of charge carrier energies enhancing electron–hole separation, and supports the spatial separation of reduction and oxidation products where necessary.6,7Photoelectrochemistry has been studied extensively for the water splitting reaction but there is much less work targeting organic transformations.5,8 Related processes, also sometimes described as photoelectrochemistry, have been developed more recently where one or both of photon absorption and redox catalysis can occur in solution and not at an electrode. To minimise confusion, these have been separately classified as interfacial photoelectrochemistry and decoupled photoelectrochemistry, respectively.5 Interfacial photoelectrochemistry includes direct oxidative transformations postulated to occur at the surface of tungsten oxide (WO3) and bismuth vanadate (BiVO4) photoelectrodes for selective C–H oxidation of alkanes and alkenes to corresponding ketones and alkenones in both aqueous and organic media.9,10 Additionally, a number of studies have been carried out on the simple oxidation of alcohols to aldehydes including for applications in biomass valorization.9,11–15 Unbiased oxidation of N-heterocycles to cyano derivatives have also been reported.16 Examples of late-stage functionalisation and cross-coupling have also begun to emerge, including the coupling of pyrazole to substituted arenes at hematite (α-Fe2O3) photoelectrodes and amination of pharmaceutical compounds, demonstrating the potential of synthetic interfacial photoelectrochemistry.17 It should be noted that in all cases high concentrations of supporting electrolytes are required to reduce solution resistance which complicates product isolation.
This work is focused on oxidative decarboxylation of carboxylic acids which is an attractive route to form carbon radicals for C–C bond formation due to the ready availability of carboxylic acid derivatives.18–23 Seminal work by Bard first described TiO2 (anatase) photoanodes under UV illumination with a 450 W Xe lamp for a Kolbe type C–C coupling reaction using acetate-acetic acid mixtures in a supporting electrolyte to generate methyl radicals to form ethane.24 More recently TiO2 photoanodes have been reported for trifluoroacetic acid decarboxylation under similar conditions.25 Given the prevalence of carboxylic acids, photoelectrochemical oxidative decarboxylation could offer a useful route to C–C bonds via renewable electricity and solar energy if the semiconductor could operate under visible light. Carboxylic acids and associated carboxylates also offer the opportunity to reduce cell resistance and the need for additional supporting electrolyte. The complementary reductive byproduct is hydrogen which simplifies product isolation and can allow single compartment electrolysis to be performed avoiding the need for more complex cell designs that require separation of anodic and cathodic compartments. Furthermore, as synthetic applications of PEC continue to expand there is a need to improve our understanding of the processes that occur at the electrode surface that control reaction rate and selectivity.
Here, we report a study of a simple photoelectrochemical oxidative decarboxylation of phenylacetic acid derivatives at porous bismuth vanadate (BiVO4) photoanodes for Kolbe C–C coupling under visible light irradiation. BiVO4 was chosen because it is a known visible light-active material used in photocatalysis and photoelectrochemistry and has a valence band potential sufficiently positive to oxidize carboxylate anions. In addition, Kolbe C–C coupling is in principle a simple one electron oxidation reaction that has been studied extensively using dark electrochemistry since its discovery over 150 years ago,26 supporting comparison between electrochemical and photoelectrochemical processes. In addition, a wide range of aromatic and aliphatic carboxylic acids are available providing insight into substrate scope and mechanistic information occurring at the electrode surface. We find that near quantitative faradaic efficiencies can be achieved at low voltage and current densities with an apparent quantum yield of 12% for benzylic substrates, and product selectivity is maximized in the absence of supporting electrolyte. Data are indicative of a step-wise intramolecular dissociative electron transfer mechanism, proceeding via initial oxidation of an aryl moiety which is in contrast to electrochemical Kolbe coupling at Pt electrodes.
Experimental
Electrode fabrication
Electrodes were fabricated using a home-made electrospray apparatus which deposits BiVO4 films onto a heated fluorine-doped tin oxide (FTO) electrode. Film thickness is controlled by deposition time. Specially, FTO glass (30 × 10 mm) was cleaned by soaking in Piranha solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2SO4:H2O2) for 1 h then rinsed sequentially with deionised water and ethanol and dried under a N2 stream. Bismuth 2-ethylhexanoate (0.5 mmol, 319.3 mg) and vanadyl acetylacetonate (0.5 mmol, 132.6 mg) were separately dissolved in 1:1 ethyl acetate:dimethyl sulfoxide (50 mL). Solutions were separately pumped through 1/16th in. i.d. PTFE tubing to a PEEK mixing chamber and subsequently to a flat metal nozzle at a flow rate of 0.03 mL min−1. Solutions were sprayed onto FTO substrates on a heated baseplate held at 350 °C at an applied potential of 9.5 kV between the nozzle and base plate for 30 min. Deposited films were subsequently transferred to a muffle furnace and annealed in air at 450 °C for 2 h to give a bright yellow film of BiVO4.
:1 H2SO4:H2O2) for 1 h then rinsed sequentially with deionised water and ethanol and dried under a N2 stream. Bismuth 2-ethylhexanoate (0.5 mmol, 319.3 mg) and vanadyl acetylacetonate (0.5 mmol, 132.6 mg) were separately dissolved in 1:1 ethyl acetate:dimethyl sulfoxide (50 mL). Solutions were separately pumped through 1/16th in. i.d. PTFE tubing to a PEEK mixing chamber and subsequently to a flat metal nozzle at a flow rate of 0.03 mL min−1. Solutions were sprayed onto FTO substrates on a heated baseplate held at 350 °C at an applied potential of 9.5 kV between the nozzle and base plate for 30 min. Deposited films were subsequently transferred to a muffle furnace and annealed in air at 450 °C for 2 h to give a bright yellow film of BiVO4.
Electrochemistry and photoelectrochemistry
All (photo)electrochemical measurements were recorded using a Biologic SP-150 potentiostat and illumination used a commercial blue, 450 nm, LED array (32 mW cm−2 irradiance at the electrode). A BiVO4 working electrode (15 × 10 mm), Pt counter electrode and Ag wire reference electrode isolated in a Luggin capillary were used for electrochemical measurements and referenced internally to ferrocene by replacing BiVO4 for a Pt working electrode. The voltage between the working and counter electrode was measured using a multimeter. All controlled potential electrolyses were undertaken in dry degassed acetonitrile in a single compartment cell made from a boiling tube with an attached Luggin capillary housing for the pseudo-reference electrode. The cell was sealed with a rubber septum through which electrical connections could be made. An over pressure of Ar was provided using an inflated balloon to exclude atmospheric oxygen. Electrolyte solutions comprised MeCN (8 mL) containing phenylacetic acid derivative, and triethylamine in concentrations as described in the manuscript. Reaction using inert electrolyte was undertaken in the same cell with 8 mL MeCN contained phenylacetic acid derivative (0.4 mmol), triethylamine (0.02 mmol) and tetrabutylammonium hexafluorophosphate (TBAPF6) (0.8 mmol). Reactions were stirred using a magnetic stir bar and purged with N2 for 5 minutes prior to applying a voltage. The electrolytic cell was submerged in a water bath to maintain a constant temperature of 25 °C.Electrochemical impedance spectroscopy (EIS)
Were recorded using a BioLogic SP-150 potentiostat in a 3-electrode, 1-compartment electrolytic cell containing a BiVO4 working electrode, a Pt wire counter electrode and an Ag wire reference electrode isolated in a Luggin capillary. Data were collected using an a.c. potential amplitude of 10 mV between 500 kHz–50 mHz, with 10 points per frequency decade. Applied voltage was increased in 200 mV steps between −0.66 to 0.54 V vs. Fc/Fc+ in the dark and under illumination by a 450 nm LED array (32 mW cm−2). Mott–Schottky analysis was recorded between −0.76 to 0.74 V vs. Fc/Fc+ in 75 mV steps. Measurements were recorded between 100 kHz–100 mHz with a sinus amplitude of 10 mV.Characterisation methods
Grazing incidence X-ray diffraction was measured using a Rigaku SmartLab X-Ray diffractometer with a Cu Kα source. The incident X-ray beam was held at 0.5° whilst 2θ was scanned between 5–75° with 0.05° step size. SEM samples (approx. 1 × 1 cm) were coated with 10 nm of Pt/Pd alloy using a JEOL JFC High Resolution Fine Coater and Agar scientific silver paint to reduce sample charging. Surface and cross-section images were taken using a JSM 7800 F Prime Field Emission Scanning Electron Microscope at an accelerating voltage of 10 kV. XPS experiments were performed in an ultrahigh vacuum system with a base pressure of <3 × 10−10 mbar using a monochromated Al Kα source at 1486.6 eV (Omicron XM 1000) and a power of 220 W. An aperture diameter of 6 mm was used with the sample normal at 45° to both the X-ray source and the entrance optics of the hemispherical energy analyser (Omicron EA 125). XPS spectra were referenced to adventitious carbon peak at 284.8 eV. Peak deconvolution and fitting was carried out manually using XPSPEAK4.1 software. Diffuse Reflectance measurements were recorded using an Ocean Optics HR2000+ spectrometer with Mikropak DH-2000-BAL UV-VIS-NIR light source. Spectra were recorded and averaged over 10 scans with 5 seconds integration time. Reference spectra were collected using a flat polystyrene standard in the dark and under illumination. NMR spectra were recorded on a JEOL ECS 400 NMR spectrometer. The volatiles from a reaction solution were removed under vacuum, and the resulting solid dissolved in a saturated NaHCO3 solution (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The CH2Cl2 extracts were dried over MgSO4, filtered and volatiles removed under vacuum. The resulting solid was then redissolved in CDCl3 containing dimethylterephthalate (10 mg mL−1) as an internal standard and peaks referenced to the residual CHCl3 solvent peak. GC analysis was performed on a ThermoTrace 1300 GC. For GC analysis reaction aliquots were initially diluted 1 in 50 using 1:1 MeOH:CH2Cl2. GC analysis was improved by methylating phenylacetic acid derivatives, which was achieved by adding trimethylsilyl diazomethane (30 μL, 2.0 M in hexanes) to 1 mL of diluted solution and allowing the solution to stand for 30 min followed by removal of volatiles under vacuum and redissolving in 1:1 MeOH:CH2Cl2 (1 mL) Product mixtures were analysed with the GC inlet held at 280 °C. Separation of products was achieved using a fused silica capillary column (Rxi-17, 30 m × 0.25 mm I.D. × 0.25 μm film thickness), with an oven temperature program as follows: 50 °C (0.5 min) to 90 °C at 100 °C min−1, then 300 °C (1 min) at 15 °C min−1. A hydrogen carrier gas was used at a flow of 1.5 mL min−1. Products were detected using a flame ionisation detector held at 330 °C.
Results
Photoelectrode synthesis and reaction optimization
BiVO4 is a well-known visible light responsive n-type semiconductor used extensively in photocatalysis and interfacial photoelectrochemistry primarily for the oxygen evolution reaction of water splitting.27,28 In this work photoelectrodes were prepared by an electrostatic spray pyrolysis methodology that allows reproducible control of film thickness and morphology to optimize light absorption and electrode–electrolyte interface area. Solutions of bismuth tris (2-ethylhexanoate) and vanadyl bis(acetylacetonate) were simultaneously deposited onto a heated FTO substrate and subsequently sintered as 450 °C for 2 h. Photoelectrode synthesis was initially optimized with respect to maximizing the photocurrent for phenylacetic acid oxidation under illumination with a blue LED (450 nm) as described below. Modifying the precursor solution concentration, deposition time, and substrate temperature showed the photocurrent reduced if films became too thick or deposition occurred above 400 °C. Visually the BiVO4 photoelectrodes are bright yellow (Fig. 1) and powder X-ray diffraction (PXRD) is consistent with a monoclinic polymorph (Fig. S1†). Scanning electron microscopy (SEM) (Fig. 1a and b) shows a porous morphology with a film thickness of approximately 500 nm and macroporous features >100 nm which can support penetration by the electrolyte. Energy dispersive X-ray spectroscopy (EDS) shows a slight excess of vanadium, with a Bi:V ratio of 1:1.08 (Fig. S2a†). X-ray photoelectron spectroscopy (XPS) shows signals attributable to Bi(III) and V(V) (Fig. S3–S5†) with a surface composition deficient in bismuth (Table S3†) which is consistent with other reports for BiVO4 materials.29 Optical properties were measured using diffuse reflectance spectroscopy which showed a broad visible light response <500 nm (Fig. S6†) and a band gap of 2.5 eV was determined from Tauc plot analysis (Fig. S7†) comparable to reported values 2.3–2.5 eV.30,31
 | ||
| Fig. 1 SEM micrographs of BiVO4 photoelectrodes. (a) Plane view with photograph of a typical electrode, inset; and (b) cross section view. | ||
Optimization of photoelectrochemical C–C coupling of phenyl acetic acid
Linear sweep voltammetry (LSV) was used initially to identify the most promising electrodes, followed by controlled potential electrolysis (CPE) and electrolyte analysis to determine the identity and faradaic efficiency (FE) of reaction products. Homo C–C coupling of phenyl acetic acid (PAA) (Scheme 1) was initially targeted because the product of C–C coupling, bibenzyl, and likely oxidation byproducts, benzaldehyde and benzyl alcohol, are not very volatile but are amenable to GC analysis allowing accurate FE determination. Furthermore, many PAA derivatives are readily available allowing study of substrate effects. Photoelectrochemistry was performed in acetonitrile and was supported by the addition of typically 0.05 equivalents of triethylamine which reduces the solution resistance by in situ formation of triethylammonium carboxylate ions. In addition, carboxylate anions typically undergo electrochemical oxidation at applied potentials 100's of mV lower than the corresponding acid32 and the ammonium ion also supports complementary proton reduction to dihydrogen at the cathode. Control experiments showed in the absence of phenyl acetic acid, no significant photocurrent was observed on illumination with a 450 nm blue LED, however above 0.1 M photocurrent is detected from −0.3 V vs. an internal reference of ferrocene (Fc/Fc+) (Fig. 2). Photocurrent is also commensurate with evolution of CO2 at the cathode and H2 at the anode, respectively which was confirmed using gas chromatography analysis (Fig. S8†). Analysis of reaction electrolytes (Table 1) showed the only products detected in significant concentration were the product of C–C Kolbe coupling, bibenzyl, and the oxidation products, benzaldehyde and benzyl alcohol. These data are consistent with photocurrent arising from oxidative decarboxylation of PAA to give CO2 and a benzyl radical which either undergoes subsequent C–C Kolbe coupling to bibenzyl or is further oxidised to benzaldehyde and benzyl alcohol with complementary dark electrochemical proton reduction to H2 at the cathode (Scheme 1).33 Further control experiments over 2 h also showed that below 0.25 V vs. Fc/Fc+, bibenzyl does not exhibit further reactivity from reductive or oxidative processes allowing a single compartment electrochemical cell to be used for CPE experiments. In addition, throughout this study LSV of multiple electrodes were tested showing good reproducibility (Fig. S9†).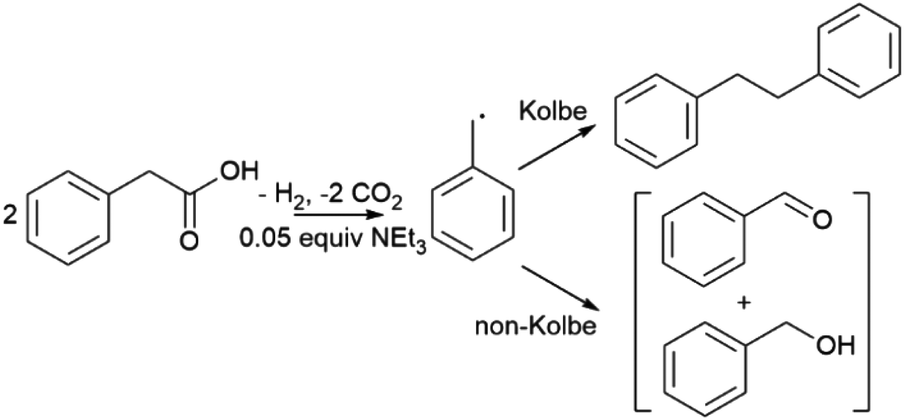 | ||
| Scheme 1 Kolbe and non-Kolbe reaction pathways of phenylacetic acid at BiVO4 photoelectrodes. | ||
 | ||
| Fig. 2 Linear sweep voltammograms of BiVO4 photoelectrodes in electrolytes containing 2 M (black line), 1 M (red), 0.5 M (blue), 0.25 M (green), and 0.01 M (purple) PAA with 0.05 equiv. NEt3. Voltammograms were recorded in the dark (dashed) and under 450 nm LED illumination (solid). Scan rate 10 mV s−1. | ||
| [PAA] (M) | Et3N equiv. | j (mA cm−2) | Chargeb (C) | FEc (%) |
|---|---|---|---|---|
|
a Average current density over 3 h at +0.09 V vs. Fc/Fc+ under 450 nm LED illumination for a 1.5 cm2 electrode.
b Integrated charge passed over 3 h.
c Faradaic efficiency for bibenzyl:benzyl alcohol:benzaldehyde respectively based on gas chromatography analysis.
|
||||
| 0.25 | 0.05 | 0.3 | 4.4 | 36:22:23 |
| 0.5 | 0.05 | 0.4 | 5.7 | 39:24:21 |
| 1 | 0.05 | 1.0 | 16.4 | 89:8:4 |
| 2 | 0.05 | 0.8 | 13.5 | 59:17:9 |
| 1 | 0.1 | 1.1 | 17.6 | 99:2:1 |
| 1 | 0.025 | 0.4 | 6.7 | 90:4:3 |
The photocurrent and product distributions were measured as a function of PAA and NEt3 concentration (Table 1). Increasing the concentration of PAA from 0.25 M to 1 M using a 0.05 equiv. of NEt3 (Fig. 2) increases the photocurrent which is stable, however at 2 M PAA, whilst initially the photocurrent is greatest, photocorrosion occurs causing a steady decrease in performance (Fig. S10†), commensurate with the known instability of BiVO4 in strongly acidic media. Increasing the relative concentration of NEt3 also increases the photocurrent which plateaus at ca. 0.05 equiv. (Fig. S11†) with no loss in photocurrent over 3 h (Fig. S12†). The FE for C–C coupling generally increases with increasing photocurrent to 1 M PAA from 36 to 99%, but significantly reduces at 2 M PAA due to photocorrosion. In a further control experiment, using 1 M PAA in the absence of light no significant current was observed below 1.5 V vs. Fc/Fc+ but at more positive potentials dark processes became significant (Fig S13†). Dark electrolysis at 2.5 V vs. Fc/Fc+ for 3 h (Fig. S14†) gave bibenzyl with a variable current of ca. 0.3 mA cm−2 and much reduced FE of 16%. The lower rate of radical production will disfavour radical dimerization and large positive bias will also promote secondary oxidation of benzyl radical to the carbocation and subsequent byproducts. Nonetheless, the comparison of dark and photoactivated processes clearly demonstrates the advantage of photoelectrochemistry in this system for C–C coupling.
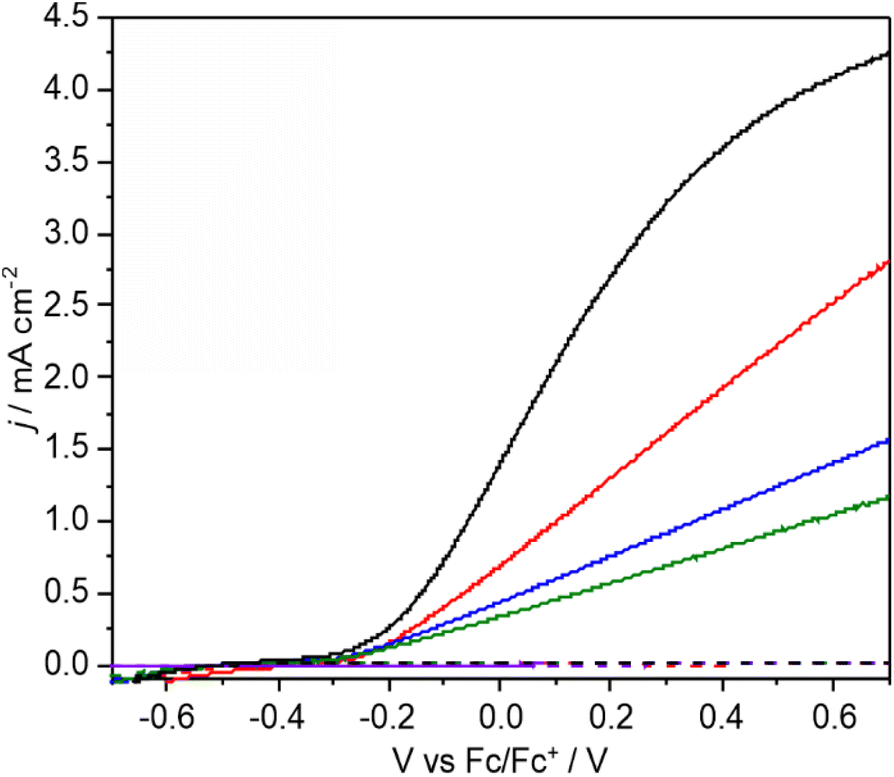
Over a typical CPE reaction time of 3 h the photocurrent tends to increase by ca. 5–10% (Fig. S10 and S12†). XPS after reaction (Table S4†) shows a lower V:Bi ratio indicating surface loss of V possibly indicating removal of an insulating amorphous VOx layer. The observation that lower photocurrents are correlated with lower FE for C–C coupling is also reflected in the product distribution found for CPE performed as a function of applied potential (Fig. 3a and b). As the applied bias is increased from −0.26 to +0.26 V vs. Fc/Fc+ the photocurrent increases from ca. 0.2 to 1.8 mA cm−2 (Fig. 2) which is reflected in the CPE (Fig. 3a). However, at +0.26 V a continual decrease in the photocurrent is observed, indicative of photocorrosion and in contrast to the stable photocurrents at lower potentials. The rate of bibenzyl production is reflective of the increase in photocurrent at more positive potentials again commensurate with a dimerization process, with the FE maximised at 0.09 V, in the absence of photocorrosion (Fig. 3b). Photostability was also examined by measuring the FE and bibenzyl production rate by removal of electrolyte aliquots over 6 h at 0.09 V vs. Fc/Fc+ (Fig. 3c) indicating that the rate of bibenzyl production and FE do not vary significantly confirming photostability under these conditions. The voltage across the working and counter electrode was also measured during CPE at 0.09 V vs. Fc/Fc+ which slowly increased from 1.5 to 1.7 V over 3 h (Fig. S15†).
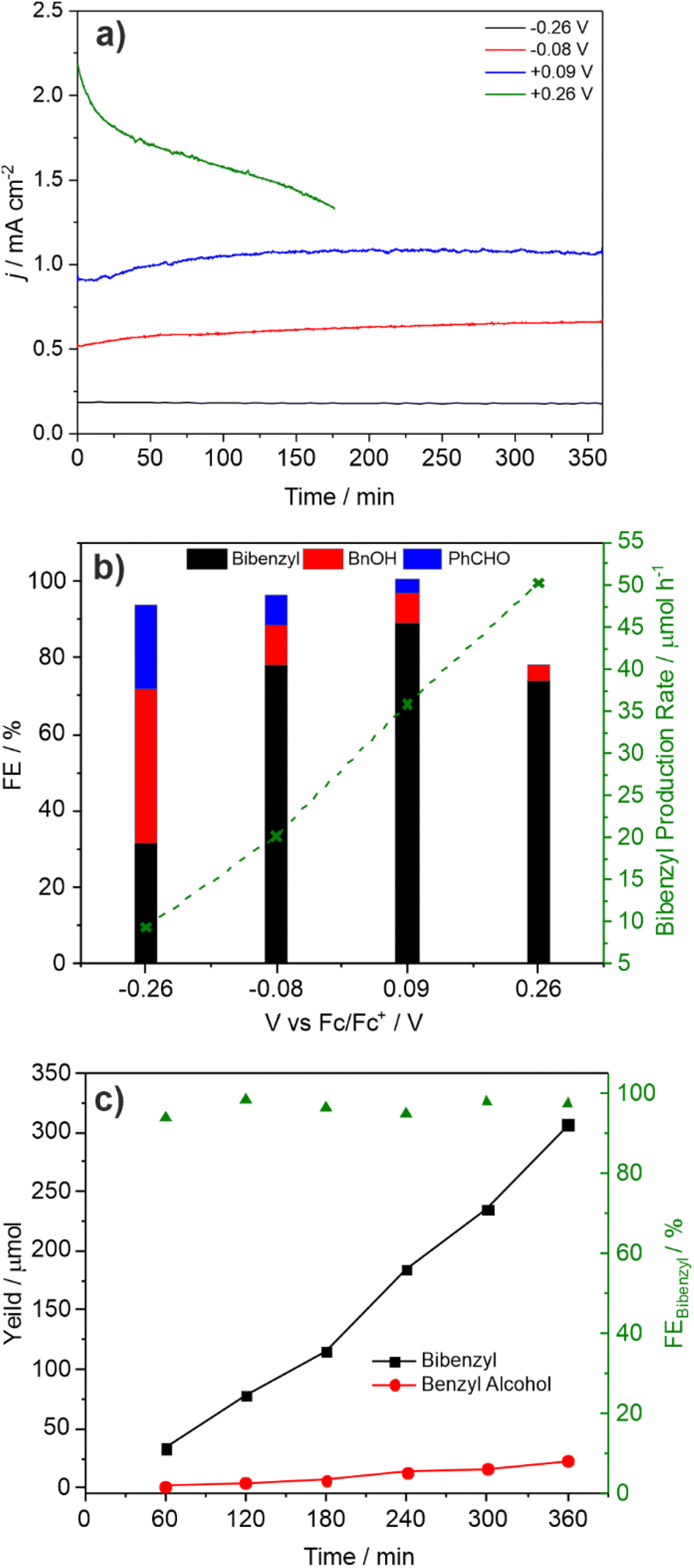 | ||
| Fig. 3 (a) CPE of 1 M PAA with 0.05 equiv. Et3N at −0.26 (black), −0.08 (red), +0.09 (blue) and +0.26 V (green) vs. Fc/Fc+; (b) faradaic efficiencies for bibenzyl (black), benzyl alcohol (red) and benzaldehyde (blue) as a function of electrolysis potential; (c) time-dependent bibenzyl (black) and benzyl alcohol (red) formation at +0.09 V vs. Fc/Fc+ of 1 M PAA with 0.05 equiv. Et3N. Overall faradaic efficiency for bibenzyl is given in green. | ||
Photoelectrochemical characterisation
Electrochemical impedance spectroscopy (EIS) and Mott–Schottky analysis were used to provide further insight into acid photooxidation using BiVO4 photoelectrodes as a function of applied potential. The equivalent circuit used to interpret EIS data is complicated by the electrode porosity and a constant phase element was used (Fig. 4) in place of a traditional capacitor to account for frequency dispersions caused by the heterogeneous surface. Impedance measurements were performed using 1 M 4-Cl PAA rather than PAA because the almost quantitative FE for this substrate (see below) simplified interpretation of processes occurring at the electrode surface. Photooxidation was recorded at 200 mV intervals (Fig. 4), which showed a potential independent high frequency feature attributed to the bulk of the BiVO4/FTO electrode and a potential dependent low frequency feature attributed to the space-charge region of the semiconductor-electrolyte interface.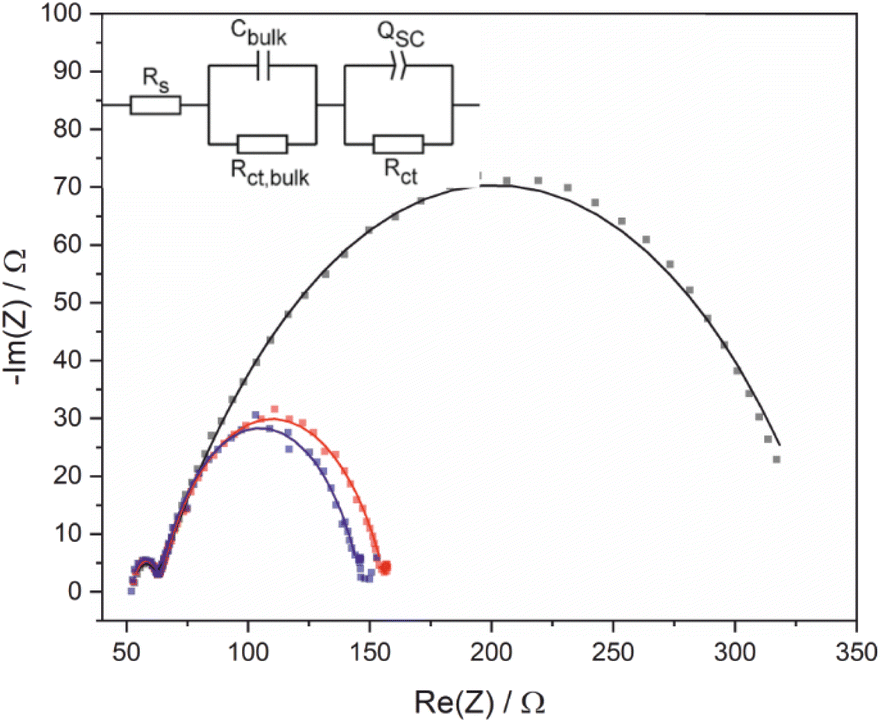 | ||
| Fig. 4 Nyquist plot and equivalent circuit for of an illuminated (450 nm, 32 mW cm−2) BiVO4 photoanode in 1 M 4-Cl PAA with 0.05 equiv. Et3N at −0.26 (grey circles), −0.06 (red) and 0.14 V (blue). Data were acquired between 500 kHz–50 mHz, using an a.c. potential amplitude of 10 mV. Simulated impedance spectra are shown by the solid lines. | ||
Fitting of the data (Table 2) shows that as the applied bias increases, the circuit elements representing the surface charge transfer resistance (RCT) and capacitance (CSC) decrease, whilst other elements remain largely unchanged. Decreasing RCT is consistent with the LSV data which shows an increasing current with increasing applied potential. The validity of data fitting is also supported by comparing the total resistance measured experimentally by LSV and the total resistance calculated from the EIS fitting which shows a strong correlation (Fig. S16†). The term CSC encompasses all phenomena within the space-charge region, including contributions from the Helmholtz capacitance and the capacitance of mid-bandgap surface states, and a decreasing value is consistent with depopulation of occupied Mott–Schottky analysis was used to determine the approximate flat-band potential, EFB, and overpotential under these conditions and to determine the donor density, ND, (Fig. S17a†). From −0.8 to −0.2 V vs. Fc/Fc+, the Mott–Schottky (M–S) analysis of BiVO4 electrodes shows typical behaviour, with a decreasing surface capacitance with applied potential and EFB estimated as −0.65 V vs. Fc/Fc+. However, at potentials greater than −0.2 V vs. Fc/Fc+ an inversion of CSC occurs which has also been observed for other nanostructured materials such as Fe2O3 nanorod electrodes.36 It should be noted that M–S analysis relies upon several assumptions, including a planar surface and that no other interfacial capacitances, e.g. Helmholtz, are significant.34 In addition high donor densities (>1023 cm−3) can affect estimations of flat-band potential by decreasing the slope of the linear region.35 Here, the donor density, ND, is estimated as ca. 1019 cm−3 (Table S7†) increasing the confidence in the measurement of flat-band potential although the nanostructuring will lead to a greater Helmholtz capacitance than is assumed. An overpotential of approximately 350 mV can be estimated for C–C coupling under these conditions, taken as the difference between photocurrent onset and flat-band potentials.17 Furthermore, ND, correlates well at all frequencies (Table S5†), and is similar to reported values of ca. 1019 cm−3 for other BiVO4 electrodes.37,38
| V/V | R s/Ω | C bulk/μF | R ct,bulk/Ω | C SC/μF | R ct/Ω |
|---|---|---|---|---|---|
| −0.26 | 53.12 | 0.972 | 8.226 | 380.8 | 280.8 |
| −0.06 | 52.84 | 0.912 | 9.971 | 150.5 | 94.52 |
| 0.14 | 52.33 | 0.844 | 10.52 | 117.8 | 84.26 |
Photoelectrode efficiency
Under the photoelectrochemical conditions studied (e.g.Fig. 2), photocurrent saturation is not observed below at least 0.7 V vs. Fc/Fc+ indicating that the generation of holes at the electrode surface is limited by electron–hole recombination at these potentials. Variation of light irradiance between 10 and 32 mW cm−2 (Table S5†) gave an increase of photocurrent from 1.3 to 2.1 mA cm−2 again showing that the photocurrent is not saturated at the potentials and light intensities investigated. The overall photon-product efficiency was investigated by measuring the apparent quantum yield (AQY) at 450 nm giving AQY = 12.0% for C–C coupling of 4-Cl PAA at 0.1 V vs. Fc/Fc+ under 32 mw cm−2 irradiance (Table S7†). Given the almost quantitative FE for this reaction under these conditions, the AQY is limited by, and approximates to, the incident photon-to-electron conversion efficiency (IPCE). As stated above, the LSV polarisation curves do not saturate at the applied potentials investigated, and the IPCE and AQY are primarily limited by electron–hole recombination. Based on an irradiance of 32 mw cm−2 and a theoretical IPCE = 100%, a maximum possible partial photocurrent density for C–C coupling of ca. 12 mA cm−2 is estimated (ESI†) indicating there is considerable scope for further optimization in this system.Photoelectrochemical C–C coupling of other carboxylic acids
Photoelectrochemical oxidation of substituted PAA was similarly undertaken at a bias of 0.09 V (Table 3 and Fig. S18, 19†). Aromatic ring substituents including Cl, F and Me are tolerated giving FE of 73–97% (entries 2–6) whereas MeO led to mainly oxidation products and no C–C coupled Kolbe product could be detected (entries 7 and 8). For electrochemical Kolbe of 4-MeO-PAA reported at a Pt electrode in methanol, two-electron oxidation pathways are dominant, suggesting that over-oxidation also occurs photoelectrochemically preventing C–C coupling.39,40 The FE and photocurrents do not show a clear trend with respect to the steric or electronic effects of the aryl substituents, which is similar to observations for electrochemical oxidations in methanol.39,41 As observed for PAA (Table 1) lower photocurrents are also generally associated with lower FE for C–C coupling.| Entry | Acid | j (mA cm−2) | FEb (%) |
|---|---|---|---|
|
a Average current density over 3 h at +0.09 V vs. Fc/Fc+ under illumination (450 nm, 32 mW cm−2).
b Faradaic efficiency for C–C coupled product: alcohol:aldehyde respectively based on gas chromatography analysis.
c Two diastereomers observed in 50:50 ratio.42
|
|||
| 1 |
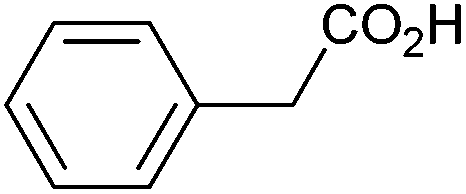
|
1.02 | 89:6:4 |
| 2 |
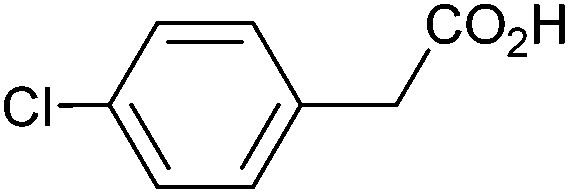
|
1.14 | 97:0:0 |
| 3 |
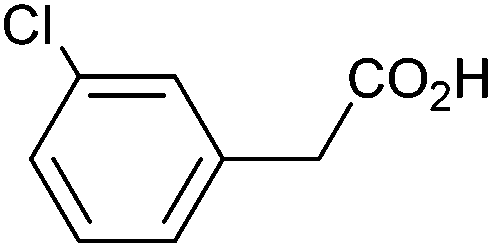
|
0.69 | 73:13:11 |
| 4 |
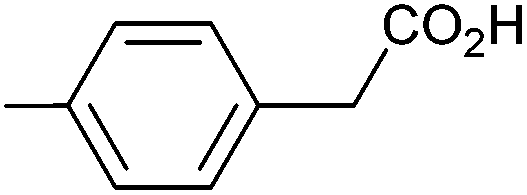
|
1.09 | 92:7:6 |
| 5 |
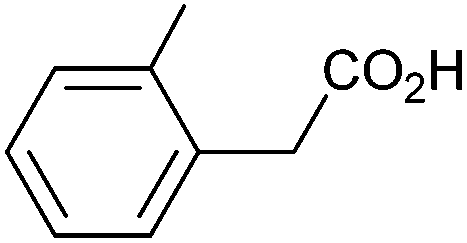
|
0.74 | 84:9:7 |
| 6 |
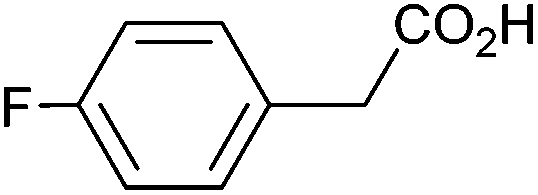
|
0.86 | 88:7:5 |
| 7 |
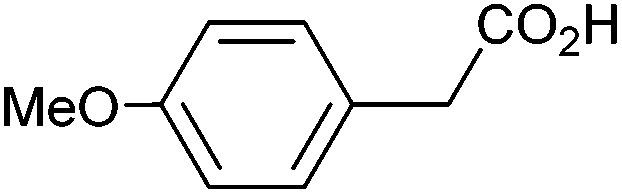
|
0.92 | 0:18:43 |
| 8 |
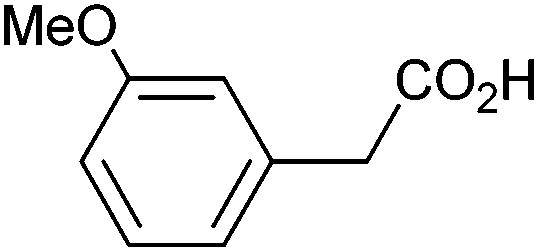
|
0.65 | 0:10:24 |
| 9 |
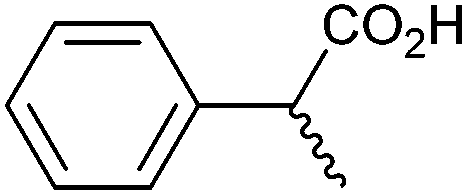
|
1.80 | 99c:0:0 |
| 10 |
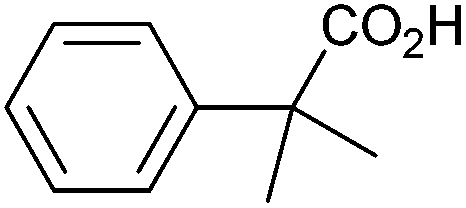
|
1.19 | 99:0:0 |
| 11 |
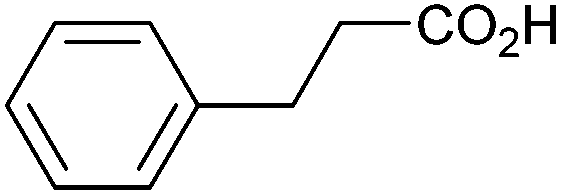
|
0.33 | 0:8:31 |
| 12 |
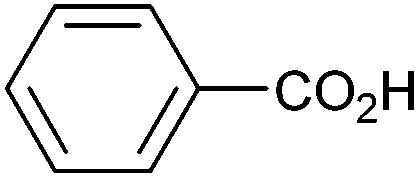
|
0.13 | 0:14:0 |
Methyl substitution of the benzylic methylene moiety (entries 9 and 10) was investigated because of the greater stability of the putative radical intermediates reflected in the bond dissociation energies of the analogous alkyl benzenes 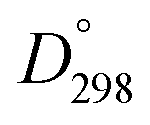 (R–H) toluene = 88.9, ethylbenzene = 86.2, and isopropylbenzene = 86.1 kcal mol−1, respectively.43 Photoelectrolysis of 2-phenylpropionic acid (PPA) and 2-phenylisobutyric acid (PIBA) (Fig. S20 and S21†) generate secondary and tertiary intermediate radicals respectively, and indeed there is an increase in the FE of C–C coupling for both to >99%, consistent with greater stabilisation of the substituted radicals. Both also exhibit greater photocurrent densities of ca. 1.8 and 1.2 mA cm−2 respectively, thus generating radicals at an increased rate compared to ca. 1 mA cm−2 for the other PAA derivatives studied (Fig. S21†). The oxidation potentials of the carboxylates derived from PAA and PIBA have been reported at a Pt electrode as +1.27 and +1.07 V vs. SCE respectively,32 indicating methyl substitution likely leads to faster oxidation as observed here. However, it is not clear why the mono-methyl derivative, PPA gives the greatest photocurrent, although the additional methyl group of PIBA is likely to result in greater steric effects and weaker adsorption to the electrode causing a reduction in the rate of photooxidation.
(R–H) toluene = 88.9, ethylbenzene = 86.2, and isopropylbenzene = 86.1 kcal mol−1, respectively.43 Photoelectrolysis of 2-phenylpropionic acid (PPA) and 2-phenylisobutyric acid (PIBA) (Fig. S20 and S21†) generate secondary and tertiary intermediate radicals respectively, and indeed there is an increase in the FE of C–C coupling for both to >99%, consistent with greater stabilisation of the substituted radicals. Both also exhibit greater photocurrent densities of ca. 1.8 and 1.2 mA cm−2 respectively, thus generating radicals at an increased rate compared to ca. 1 mA cm−2 for the other PAA derivatives studied (Fig. S21†). The oxidation potentials of the carboxylates derived from PAA and PIBA have been reported at a Pt electrode as +1.27 and +1.07 V vs. SCE respectively,32 indicating methyl substitution likely leads to faster oxidation as observed here. However, it is not clear why the mono-methyl derivative, PPA gives the greatest photocurrent, although the additional methyl group of PIBA is likely to result in greater steric effects and weaker adsorption to the electrode causing a reduction in the rate of photooxidation.
Other carboxylic acids investigated were 3-phenyl propanoic acid (entry 11) and benzoic acid (entry 12) to further probe the scope of radical formation and C–C coupling. Both exhibit much lower photocurrents (Fig. S22 and 23†) and no products attributable to C–C coupling were detected. For benzoic acid, the putative intermediate phenyl radical is less stable than benzylic radical,44 In addition the oxidation potential of phenyl carboxylate in acetonitrile at Pt has been reported as ca. 1.9 V vs. SCE45 leading to slow radical production and less probable C–C coupling. Furthermore, oxidation of benzoic acid at platinum electrodes in methanol is known to occur without decarboxylation, resulting in esterification reactions.46 In contrast aliphatic acids such as 3-phenyl propanoic acid typically undergo decarboxylation and exhibit C–C coupling Kolbe electrolysis with high faradaic efficiencies at sufficient current densities but can be prone to isomerisation.47 Several aliphatic carboxylates have reported oxidation potentials of ca. 1.2 V vs. SCE in acetonitrile at a carbon electrode very similar to benzylic carboxylates,48,49 demonstrating a reactivity difference between photoelectrochemical and traditional electrochemical Kolbe coupling.
Photoelectrochemical coupling in the presence of electrolyte
Comparison was also made with conditions used in preparative organic synthetic electrochemistry which is typically performed in the laboratory at <15 mA cm−2 for hours on the mM scale using a supporting inert electrolyte and various additives. LSV of an acetonitrile solution containing 0.1 M TBAF supporting electrolyte in the absence of substrate showed no significant oxidative photocurrent below 0.4 V vs. Fc/Fc+. On addition of 0.05 M 4-Cl PAA photocurrent onset occurs at 0.2 V vs. Fc/Fc+ (Fig. 5a) and on subsequent addition of 0.05 equiv. NEt3 the onset shifts 0.4 V to −0.2 V, approaching a plateau between 0 and 0.2 V before following a similar photocurrent profile to the polarisation curve without Et3N. These data are consistent with oxidation of 4-Cl PAA at more positive potentials than 0.2 V, and on addition of Et3N, deprotonation supports oxidation of the complementary carboxylate anion above −0.2 V. In the plateau region, photooxidation of the carboxylate anion is limited by its concentration within the double layer at the electrode surface. CPE at 0.25 V was undertaken until sufficient charge was passed to theoretically consume all the 4-Cl PAA substrate (Fig. S24†). The photocurrent density declined from ca. 0.5 to 0.15 mA cm−2 over 24 h and periodic analysis of the electrolyte showed that the FE for C–C coupling decreased throughout the reaction from 20 to 6%, whilst oxidation products 4-Cl-benzyl alcohol and particularly 4-Cl-benzaldehyde increased (Fig. 5b).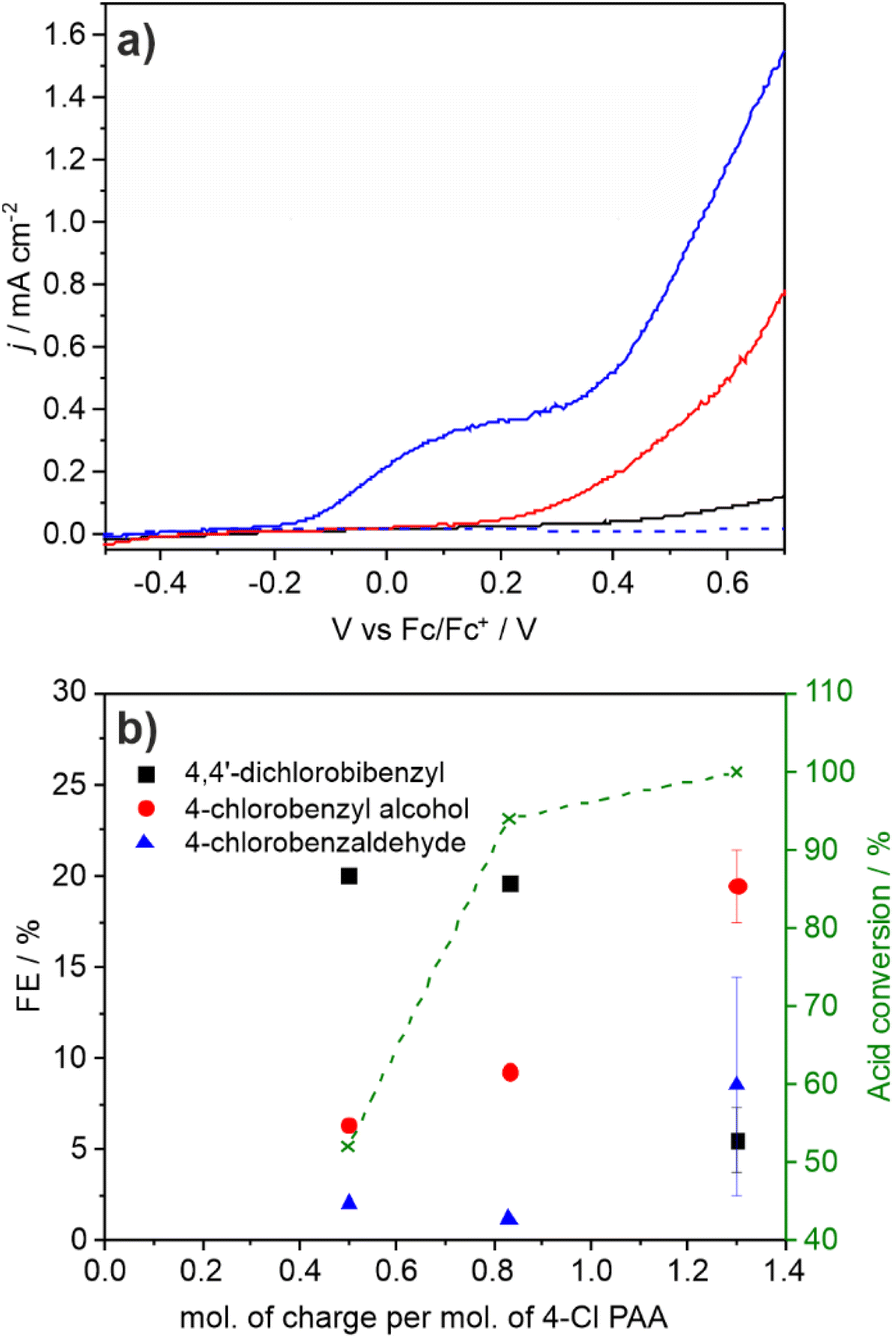 | ||
| Fig. 5 (a) Linear sweep voltammograms of a BiVO4 photoelectrode in 0.1 M TBAPF6 (black), 0.1 M TBAPF6 + 0.05 M 4-Cl PAA (red) and 0.1 M TBAPF6 + 0.05 M 4-Cl PAA + 0.05 equiv. Et3N (blue) solutions under illumination (450 nm, 32 mW cm−2). A scan of the full electrolyte solution in the absence of illumination is also given (blue, dashed). Scan rate 10 mV s−1; (b) FE for Kolbe and non-Kolbe products after passage of different equivalents of charge. Electrolyses at +0.26 V vs. Fc/Fc+ in 0.05 M 4-Cl-PAA with 0.05 equiv. Et3N and 0.1 M TBAPF6. | ||
EIS and M–S analysis was also performed in 0.05 M 4-Cl PAA and 0.1 M TBAPF6 at −0.06 V vs. Fc/Fc+ where LSV indicates a rising current and absence of dark electrochemistry (Fig. 5a). A Nyquist plot and fitting using a modified equivalent circuit (Fig. 6) shows a diffusional component at lower frequencies, which is also indicated by the plateau region of the LSV most clearly between 0 and 0.2 V (Fig. 5a). Again, two other features in the Nyquist plot are observed at higher frequencies similar to data acquired using 1 M 4-Cl PAA, attributed to the same bulk and interfacial processes. However, under these conditions, a considerable overlap of the two features was observed indicating more comparable timescales for bulk and surface processes. Comparison of the fitted parameters (Table S6†) shows significant differences compared to 1 M 4-Cl PAA particularly for Cbulk (39.5 vs. 0.912 μF), and to a lesser extent QSC (422.4 vs. 150.5 μF), and RCT (430.9 vs. 94.52 Ω). These data are consistent with lower band-bending due to the change in electrolyte which is also reflected in the more anodic flat-band potential estimated at −1.1 V vs. Fc/Fc+, as measured by Mott–Schottky analysis (Fig. S17b†). When compared to the photocurrent onset of the LSV at ∼−0.2 V vs. Fc/Fc+, (Fig. 5a) an approximate overpotential of 0.9 V is estimated for the oxidation of 4-Cl PAA under these conditions. The significant 0.45 V cathodic shift of Efb on reducing 4-Cl-PAA concentration may have arisen from a non-negligible Helmholtz capacitance in these electrolytes due to the interference of PF6− anions on double layer formation.35 Notwithstanding the significant assumptions used in the EIS and particularly the M–S analysis, the difference between 1.0 M and 0.05 M 4-Cl PPA clearly suggest a greater overpotential and more sluggish electron transfer at low acid concentrations and in the presence of supporting electrolyte.
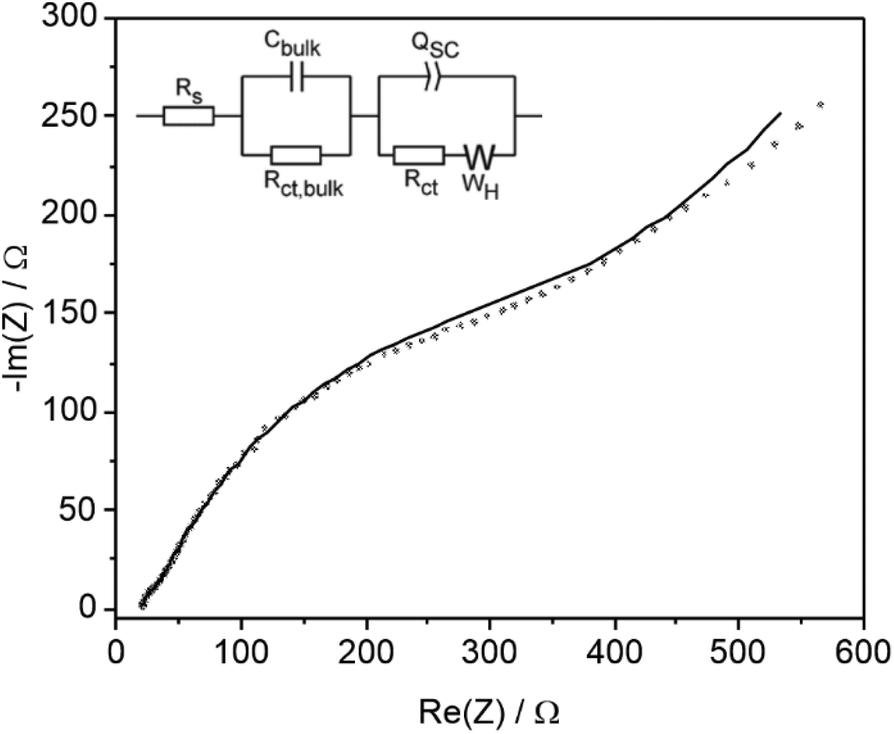 | ||
| Fig. 6 Nyquist plot and equivalent circuit for of an illuminated (450 nm, 32 mW cm−2) BiVO4 photoanode in 0.05 M 4-Cl PAA solution with 0.05 equiv. Et3N containing 0.1 M TBAPF6 supporting electrolyte at −0.06 V vs. Fc/Fc+. Data were acquired between 500 kHz–50 mHz, using an a.c. potential amplitude of 10 mV. Simulated impedance spectra are shown by the solid lines. | ||
Discussion
Here we first summarise aspects of traditional electrochemical oxidation of carboxylic acids for Kolbe C–C coupling which has been of interest since its discovery over 150 years ago with mechanistic studies reported from at least the 1890′s.33,50 As may be expected, Kolbe coupling has been shown to be dependent on the carboxylic acid substrate, electrode composition and morphology, electrolyte and the operating conditions. Most Kolbe studies have been performed on simple aliphatic carboxylic acids, their halogenated derivatives, and benzylic acids as described here. Radical formation is supported by electron-withdrawing groups at the α-position to the acid, whereas electron-donating groups support carbocation formation.49 Thermodynamically, oxidation of the intermediate hydrocarbyl radical is much more favourable than the parent carboxylic acid leading to competitive two electron oxidation of the carboxylate substrate to give carbocation intermediates which participate in non-Kolbe chemistry such as that described by Hofer–Moest reactions to give alcohols or carbonaceous deposits on an electrode.40 Mechanistic studies support a step-wise mechanism comprising initial rate-limiting oxidation of carboxylate to form an acyloxy radical intermediate followed by rapid elimination of CO2 to give a hydrocarbyl radical prior to dimerisation.51,52 In addition competitive solvent oxidation occurs particularly in water and alcohol electrolytes. Therefore Kolbe C–C coupling is promoted by dissociation of the initial acyloxy and/or hydrocarbyl radical from the electrode surface to avoid secondary oxidation, and large voltage to increase the electrode surface coverage by the anionic carboxylate ion.53 Most mechanistic studies have been undertaken in water and methanol solutions, and less frequently in non-aqueous solvents including acetonitrile.47,52,54,55 Platinum is by far the most common electrode material followed by carbonaceous electrodes. In general, in water and methanol solvents, alkaline salts of carboxylic acids undergo Kolbe C–C coupling reactions at planar electrodes at large voltages and current density (>100 mA cm−2) where one-electron oxidation is favoured when the electrode is sufficiently polarised to rapidly adsorb a high surface concentration of carboxylate anions.56–58 At lower current densities and voltages the thermodynamically preferred water and methanol oxidation occurs and competes with unselective secondary oxidative C–H activation of carboxylic acid substituents.56 In acetonitrile, solvent oxidation is much less competitive but it is found that secondary oxidation to the carbocation is often observed leading to non-Kolbe products52,59 including N-acylamides which are formed from addition of the carbocation to the nucleophilic acetonitrile solvent.60 Carbocation formation is also found to be further enhanced at porous carbon electrodes presumably due to slower diffusion of radicals within the pores.61 In contrast, oxidations at smooth metal electrodes exhibit greater desorption of radicals also reflecting the relative adsorption energies of radicals at metal and carbon surfaces.41,62,63 Electrochemical studies to determine the nature of adsorption of Kolbe intermediates indicate chemisorption of radicals to the electrode surface.33 The presence of surface bound intermediates is evidenced by using a platinum rotating disk electrode which shows the rate of dimer formation is independent of rotation speed.54,64 Conversely, EPR evidence for solution-phase radicals generated by oxidation of carboxylic acids on irradiation of TiO2 powders,19,65,66 showed narrow linewidths, associated with isotropic tumbling of radicals in solution, although surface bound radicals could not be excluded. Notwithstanding the preceding observations, for significant FE of C–C coupling to occur, clearly dimerization is required to be faster than secondary oxidation, emphasising the importance of relative rates of electron transfer and electrode adsorption kinetics. In this regard some carboxylate substrates support initial oxidation at a substituent followed by rearrangement to obtain the intermediate acyloxy radical that can subsequently eliminate CO2.52 The time required for rearrangement during this intramolecular dissociative electron transfer (IDET) pathway has been proposed to support radical dissociation from the electrode surface therefore reducing carbocation formation.52Considering this context of dark electrochemical Kolbe chemistry there are several notable features of the photoelectrochemical system described here. Remarkably high FE's for Kolbe C–C coupling are observed for a range of benzylic carboxylic acids at low current density ca. >1 mA cm−2. In part, this is supported by use of acetonitrile avoiding competitive solvent oxidation but clearly the relative adsorption of carboxylate anions and radical intermediates is critical to the observed selectivity to avoid secondary oxidation to carbocations. Overoxidation to the carbocation is not observed for the PAA substrates investigated here even within the porous environment of the BiVO4 photoelectrode showing that under the studied conditions C–C coupling occurs faster than a second oxidation to carbocation. This could be explained by either relatively slow radical oxidation or fast desorption of radicals from the BiVO4 surface. Given that radical oxidation is thermodynamically easier than for the parent carboxylate, fast radical desorption promoted by carboxylate substrate is more likely. This is supported experimentally (Table 1) where photocurrent correlates with acid concentration and a greater bulk acid concentration can drive a high carboxylate concentration within the double layer supporting more rapid desorption of the intermediate radical. At low acid and carboxylate concentration, the radical remains adsorbed for greater time on the photoelectrode surface supporting additional oxidation to the carbocation, and subsequent reaction with an oxygen source such as lattice oxide, residual oxygen or water to give alcohol and aldehyde by-products.67,68 Quantitative XPS analysis of the BiVO4 photoelectrode surface after electrolysis (Tables S3 and S4†) shows relative loss of V and O, indicating lattice oxygen may contribute to the formation of oxygenated products.18,40
With respect to substrate selectivity the photoelectrochemical behaviour of PAA derivatives, benzoic and 3-phenyl propanoic acid (Table 3) as described above are not fully explained by oxidation potentials or carbocation formation. A rapid secondary oxidation to a carbocation would result in a greater photocurrent density if initial carboxylate oxidation occurred at similar overpotential and rates for all substrates. Whilst benzoic acid does have an oxidation potential ca. 700 mV greater than the other substrates the lower photocurrent of 3-phenyl propanoic acid which has similar oxidation potential to PAA derivatives suggests that initial electron transfer from PAA derivatives to BiVO4 is much faster. Collectively, the data suggest that PAA and its derivatives studied here are oxidized via an IDET pathway. This mechanism is not available to other classes of substrate such as 3-phenyl propanoic acid which did not give either identifiable C–C or oxidation products and is reflective of the low current regime of electrochemical Kolbe that results in unselective substituent activation.56
We note that the few previous studies of photoelectrochemical Kolbe chemistry use acetic acids and TiO2 photoanodes which is known to bind carboxylic acids strongly via the carboxylate group. BiVO4 contains far fewer surface OH groups and carboxylate binding is much weaker promoting alternative binding modes. An IDET pathway gives additional time for dissociation of the acyloxy radical prior to rapid CO2 elimination reducing the probability of secondary oxidation to the carbocation resulting in the high FE observed. Furthermore, comparison of the FE and photocurrent of PAA and the α-methyl-substituted analogues PPA and PIBA show that whilst both PPA and PIBA show near quantitative C–C coupling, mono-α-methylation gives the greatest photocurrent (Table 2). The data suggest that the increased steric bulk of di-α-methylation reduces adsorption to the photoelectrode surface.
The critical role of surface adsorption is also evident in using carboxylic acids in lower concentration in the presence of supporting electrolytes. The photocurrent and FE of C–C coupling decreases as the acid concentration decreases reflecting the changes in the double layer where electrolyte anions are also present. EIS and M–S analysis indicate a greater interfacial charge transfer resistance resulting in greater overpotential. Consequently, a lower current density is observed at comparable applied bias resulting in slower electron transfer and radical formation and C–C coupling, promoting byproduct formation by direct oxygenation from the surface or carbocation formation via a second oxidation. The appearance of a diffusional element in the Nyquist plot at low acid concentration also indicates lower electrode coverage by carboxylate substrate reducing the rate of radical desorption.
Finally, our results can also be compared to closely related photocatalytic Kolbe processes that use powdered semiconductor photocatalyst. Kolbe coupling has predominantly been investigated using TiO2 and simple aliphatic acids but most commonly degradation of carboxylic acids occurs to a mixture of H2, CO2 and alkanes depending on the reaction conditions.69–73 Reported quantum efficiencies for Kolbe coupling are typically <1% due to electron hole recombination and byproduct formation.71 Clearly PEC supports electron–hole separation and the fabrication of a porous semiconductor electrode allows more straightforward optimisation of light absorption and semiconductor-electrolyte interface and the double layer where photooxidation and radical dissociation occur.
Conclusions
BiVO4 photoelectrodes can catalyse the oxidation of benzylic carboxylic acids to give C–C coupled Kolbe products with near quantitative faradaic efficiency and a quantum efficiency over 10%. This occurs at photocurrent densities of ca. 1 mA cm−2 in acetonitrile without the need for additional electrolyte contrasting traditional electrochemical Kolbe reactions at meal and carbon electrodes. Furthermore, reaction proceeds at low voltages with an estimated overpotential in acetonitrile of 350 mV which contrasts with traditional Kolbe electrolysis in alcohols operating at 10's V needed to overcome competitive solvent oxidation. Compared to Kolbe electrolysis in acetonitrile, significant carbocation formation and nitrile addition products are not observed for photooxidation of benzylic substrates. The relative adsorption energies of carboxylic acid, acyloxy and radical intermediates at the illuminated semiconductor-electrolyte interface are clearly key to the observed selectivity. Substrate scope suggests that for the photoelectrodes used here, adsorption and initial electron transfer may proceed via the arene group of benzylic substrates and intramolecular dissociative electron transfer pathway. Furthermore, additional electrolyte is detrimental to the rate and selectivity of reaction suggesting that a stable double layer is a critical feature. Collectively, interfacial photoelectrochemistry presents an advantage in this system compared to electrolysis and access to alternative electrode–substrate interactions compared to a strong carboxylate–Pt interaction at high voltage. There is clearly considerable scope to control reaction rate and selectivity by modification of the semiconductor surface to differentiate substrate and intermediate binding modes, and to increase product diversity by introducing radical coupling partners. Further nanostructuring can also improve light collection and electron–hole separation which currently limit the quantum efficiency. Ultimately, even with improved efficiencies, the need for scale-up will require overcoming additional engineering challenges including, but not limited to, reactor design product separation in a batch or flow photoelectrochemical system.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. R. E. D. and V. C. designed the experiment; W. A. S. performed the preparation, characterization, and (photo)electrochemical experiments; A. S. performed additional photoelectrochemical experiments; B. A. C. and A. P. performed XPS analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the EPSRC (EP/N509802/1), the British Council Newton-Mosharafa Fund (A. S.) and University of York for financial support.References
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2022, 122, 3180–3218 CrossRef CAS PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- L. M. Peter, in Photocatalysis: Fundamentals and Perspectives, The Royal Society of Chemistry, 2016, pp. 1–28 Search PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. C. Wu, R. J. Song and J. H. Li, Org. Chem. Front., 2020, 7, 1895–1902 RSC.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 390 CrossRef PubMed.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed.
- L. Samiolo, M. Valigi, D. Gazzoli and R. Amadelli, Electrochim. Acta, 2010, 55, 7788–7795 CrossRef CAS.
- H. Tateno, Y. Miseki and K. Sayama, ChemElectroChem, 2017, 4, 3283–3287 CrossRef CAS.
- R. Zhang, M. Shao, Z. Li, F. Ning, M. Wei, D. G. Evans and X. Duan, Chem. – Eur. J., 2017, 23, 8142–8147 CrossRef CAS PubMed.
- Y. J. Zhang, G. H. Zhao, Y. N. Zhang and X. F. Huang, Green Chem., 2014, 16, 3860–3869 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- M. Gong, M. M. Huang, Y. B. Li, J. Y. Zhang, J. K. Kim, J. S. Kim and Y. J. Wu, Green Chem., 2022, 24, 837–845 RSC.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed.
- M. C. Leech and K. Lam, Acc. Chem. Res., 2020, 53, 121–134 CrossRef CAS PubMed.
- D. W. Manley, R. T. McBurney, P. Miller, R. F. Howe, S. Rhydderch and J. C. Walton, J. Am. Chem. Soc., 2012, 134, 13580–13583 CrossRef CAS PubMed.
- M. Quertenmont, I. Goodall, K. Lam, I. Markó and O. Riant, Org. Lett., 2020, 22, 1771–1775 CrossRef CAS PubMed.
- Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913–17918 CrossRef CAS PubMed.
- K. Neubert, M. Hell, M. Chávez Morejón and F. Harnisch, ChemSusChem, 2022, 15, e202201426 CrossRef CAS PubMed.
- H. A. Hintz and C. S. Sevov, Nat. Commun., 2022, 13, 1319 CrossRef CAS PubMed.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1977, 99, 7729–7731 CrossRef CAS.
- V. A. Grinberg, V. V. Emets, A. D. Modestov, N. A. Maiorova, E. V. Ovsyannikova, O. V. Bukhtenko and D. A. Maslov, Russ. J. Electrochem., 2017, 53, 217–222 CrossRef CAS.
- H. Kolbe, Ann. Chem., 1849, 69, 257–294 CrossRef.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- D. Lee, W. Wang, C. Zhou, X. Tong, M. Liu, G. Galli and K.-S. Choi, Nat. Energy, 2021, 6, 287–294 CrossRef CAS.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781–1787 RSC.
- B.-C. Xiao, L.-Y. Lin, J.-Y. Hong, H.-S. Lin and Y.-T. Song, RSC Adv., 2017, 7, 7547–7554 RSC.
- L. Capaldo, L. Buzzetti, D. Merli, M. Fagnoni and D. Ravelli, J. Org. Chem., 2016, 81, 7102–7109 CrossRef CAS PubMed.
- A. Vijh and B. Conway, Chem. Rev., 1967, 67, 623–664 CrossRef CAS.
- R. De Gryse, W. P. Gomes, F. Cardon and J. Vennik, J. Electrochem. Soc., 1975, 122, 711–712 CrossRef CAS.
- A. Hankin, F. E. Bedoya-Lora, J. C. Alexander, A. Regoutz and G. H. Kelsall, J. Mater. Chem. A, 2019, 7, 26162–26176 RSC.
- L. M. Peter, Gurudayal, L. H. Wong and F. F. Abdi, J. Electroanal. Chem., 2018, 819, 447–458 CrossRef CAS.
- D.-D. Qin, T. Wang, Y.-M. Song and C.-L. Tao, Dalton Trans., 2014, 43, 7691–7694 RSC.
- S. Xie, T. Zhai, Y. Zhu, W. Li, R. Qiu, Y. Tong and X. Lu, Int. J. Hydrogen Energy, 2014, 39, 4820–4827 CrossRef CAS.
- J. P. Coleman, R. Lines, J. H. P. Utley and B. C. L. Weedon, J. Chem. Soc., Perkin Trans. 2, 1974, 1064–1069 RSC.
- H. Hofer and M. Moest, Ann. Chem., 1902, 323, 284–323 CrossRef CAS.
- W. S. Trahanovsky, J. Cramer and D. W. Brixius, J. Am. Chem. Soc., 1974, 96, 1077–1081 CrossRef CAS.
- R. Bigler, K. A. Mack, J. Shen, P. Tosatti, C. Han, S. Bachmann, H. Zhang, M. Scalone, A. Pfaltz, S. E. Denmark, S. Hildbrand and F. Gosselin, Angew. Chem., Int. Ed., 2020, 59, 2844–2849 CrossRef CAS PubMed.
- M. Mautner, J. Am. Chem. Soc., 1982, 104, 5–10 CrossRef CAS.
- J. Hioe and H. Zipse, Org. Biomol. Chem., 2010, 8, 3609–3617 RSC.
- Y. Matsuda, K. Kimura, C. Iwakura and H. Tamura, Bull. Chem. Soc. Jpn., 1973, 46, 430–434 CrossRef CAS.
- D. Hayrapetyan, V. Shkepu, O. T. Seilkhanov, Z. Zhanabil and K. Lam, Chem. Commun., 2017, 53, 8451–8454 RSC.
- G. S. Pande and S. N. Shukla, Electrochim. Acta, 1961, 4, 215–231 CrossRef CAS.
- M. Galicia and F. J. González, J. Electrochem. Soc., 2002, 149, D46 CrossRef CAS.
- M. Galicia, M. A. González-Fuentes, D. P. Valencia and F. J. González, J. Electroanal. Chem., 2012, 672, 28–33 CrossRef CAS.
- A. Brown and J. Walker, Liebigs Ann. Chem., 1891, 261, 107–128 CrossRef.
- A. A. Isse, A. Gennaro and F. Maran, Acta Chem. Scand., 1999, 53, 1013–1022 CrossRef CAS.
- C. Andrieux, F. Gonzalez and J.-M. Savéant, J. Electroanal. Chem., 2001, 498, 171–180 CrossRef CAS.
- J. W. Hilborn and J. A. Pincock, J. Am. Chem. Soc., 1991, 113, 2683–2686 CrossRef CAS.
- M. Fleischmann, J. Mansfield and L. Wynne-Jones, J. Electroanal. Chem., 1965, 10, 522–537 CAS.
- J. Wilshire, Aust. J. Chem., 1963, 16, 432–439 CrossRef.
- T. Dickinson and W. Wynne-Jones, Trans. Faraday Soc., 1962, 58, 382–387 RSC.
- Y. B. Vassiliev and V. Grinberg, J. Electroanal. Chem., 1990, 283, 359–378 CrossRef.
- Y. B. Vassiliev and V. Grinberg, J. Electroanal. Chem., 1991, 308, 1–16 CrossRef CAS.
- B. Sim, P. Milne, D. Griller and D. Wayner, J. Am. Chem. Soc., 1990, 112, 6635–6638 CrossRef CAS.
- J. Kornprobst, A. Laurent and D. Laurent, Bull. Soc. Chim. Fr., 1968, 3657 CAS.
- D. L. Muck and E. R. Wilson, J. Electrochem. Soc., 1970, 117, 1358–1362 CrossRef CAS.
- S. D. Ross and M. Finkelstein, J. Org. Chem., 1969, 34, 2923–2927 CrossRef CAS.
- D. D. Tanner and S. A. Osman, J. Am. Chem. Soc., 1968, 90, 6572–6574 CrossRef CAS.
- M. Fleischmann, J. Mansfield, H. Thirsk, H. Wilson and L. Wynne-Jones, Electrochim. Acta, 1967, 12, 967–982 CrossRef CAS.
- B. Kraeutler, C. D. Jaeger and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 4903–4905 CrossRef CAS.
- M. Kaise, H. Kondoh, C. Nishihara, H. Nozoye, H. Shindo, S. Nimura and O. Kikuchi, J. Chem. Soc., Chem. Commun., 1993, 395–396 RSC.
- S. Civiš, M. Ferus, M. Zukalová, A. Zukal, L. Kavan, K. D. Jordan and D. C. Sorescu, J. Phys. Chem. C, 2015, 119, 3605–3612 CrossRef.
- J. F. Montoya, J. Peral and P. Salvador, ChemPhysChem, 2011, 12, 901–907 CrossRef CAS PubMed.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 2239–2240 CrossRef CAS.
- S. Hamid, I. Ivanova, T. H. Jeon, R. Dillert, W. Choi and D. W. Bahnemann, J. Catal., 2017, 349, 128–135 CrossRef CAS.
- S. Sato, J. Chem. Soc., Chem. Commun., 1982, 26–27 RSC.
- S. Ngo, L. M. Betts, F. Dappozze and C. Guillard, ChemistrySelect, 2018, 3, 12773–12781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04423d |
| This journal is © The Royal Society of Chemistry 2023 |