 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical nitrogen reduction reaction over gallium – a computational and experimental study†
Vivek
Sinha
 *a,
Fateme
Rezai
b,
Nihat Ege
Sahin
b,
Jacopo
Catalano
b,
Espen Drath
Bøjesen
c,
Farnaz
Sotoodeh
*a and
Emil
Dražević
*b
*a,
Fateme
Rezai
b,
Nihat Ege
Sahin
b,
Jacopo
Catalano
b,
Espen Drath
Bøjesen
c,
Farnaz
Sotoodeh
*a and
Emil
Dražević
*b
aC2CAT B.V., Magnoliastraat 13, Lisserbroek 2165CK, The Netherlands. E-mail: vivek.sinha@c2cat.eu; farnaz.sotoodeh@c2cat.eu
bDepartment of Biological and Chemical Engineering – Process and Materials Engineering, Aarhus University, Åbogade 40, Aarhus N 8200, Denmark. E-mail: edrazevic@bce.au.dk
cInterdisciplinary Nanoscience Center & Aarhus University Centre for Integrated Materials Research, Aarhus University, Gustav Wieds Vej 14, Aarhus C 8000, Denmark
First published on 23rd December 2022
Abstract
Ga was identified earlier as one of the “overlooked” metals for catalyzing the electrochemical nitrogen reduction reaction (ENRR). We investigate here the electrocatalytic activity of Ga towards the nitrogen reduction reaction. We used a combination of molecular modelling and simulations using periodic density functional theory calculations (DFT), and experimental ENRR measurements. The ENRR was found to proceed via an associative mechanism where the first PCET to dinitrogen forming the surface adsorbed N2H* species is the overpotential limiting step. The bare Ga cathode has a high overpotential (>2 V (SHE)) for the ENRR. We also investigated the effect of a water-in-salt electrolyte (WISE) on the rate of ammonia formation. The addition of an Li salt lowers the overpotential to 1.88 V (SHE). DFT calculations revealed that the H-adatom was more favorably bound than the N-adatom, and the hydrogen evolution reaction (HER) is expected to dominate at high cathodic potentials. Experimental ENRR tests corroborate our results wherein no significant NH3 formation was detected. The low electrochemical activity of Ga is attributed to poor binding and activation of N2 which originates from an electropositive surface charge distribution.
Introduction
Ammonia (NH3) is a key chemical in human society. It finds widespread use across key sectors such as food, agriculture, and defence. In recent times ammonia has often been mentioned in the context of green transition and as a key chemical to decarbonize not only the agricultural sector but also the maritime industry.1 One of the main points for considering ammonia as a key “green chemical” is it can be made in unlimited quantities in any place in the world where water, nitrogen, and energy are found at the same time. It is also considered the cheapest e-chemical, as it is made from earth-abundant precursors. The downside of ammonia is its toxicity and burning products such as unreacted toxic ammonia, and nitrous oxide, which is a 280 times worse greenhouse gas than CO2 over a 20 year period.2Synthesis of ammonia is a chemically challenging reaction. It is produced with high energy efficiency in the well-known and established Haber–Bosch process. In the Haber–Bosch process, ammonia is formed by reacting two gaseous molecules: N2 and H2. N2 has a N–N triple bond which confers a thermodynamically inert nature to the molecule, making it inherently difficult to react at reasonable rates. In a Haber–Bosch reactor, commercially relevant ammonia production rates are reached at high temperatures (around 400 °C), while high pressures in the reactor (200 bar) ensure reasonable ammonia conversion (shifts the equilibria to the right) and efficient separation of liquid anhydrous ammonia from reactant gases by condensation. Nitrogen is obtained by removing oxygen from air, either by cryogenic distillation or pressure swing adsorption. H2 on the other hand is conventionally produced via fossil-based resources which increases the carbon-footprint of NH3 synthesis. However, there is now large political momentum that supports green hydrogen production, using renewable energy to power the water electrolyser, combined with a Haber–Bosch reactor. Powering the electrolyser with renewable energy would enable CO2-free production of NH3.3–6 The large heat losses at small scales constrain the suitability of the Haber–Bosch process to large scales typically exceeding 100 kg of NH3 per hour, limiting NH3 production as a centralized activity.3
Decentralized ammonia production can be made feasible by an electrochemical approach, often referred to in the literature as the Electrochemical Nitrogen Reduction Reaction (ENRR).4–7 Today, the ENRR can be performed at very high selectivity and production rates in non-aqueous media, using either ethanol or hydrogen as a proton/electron source using a Li-mediated approach. These Li-mediated approaches in non-aqueous media, although promising, suffer from high energy consumption inherently linked to the use of Li at the cathode. With respect to aqueous media, running the ENRR in aqueous media can potentially lead to better energy efficiencies and use of one electrolysis device that performs electrolysis of N2 with water as a proton and electron source. However, the ENRR in aqueous media suffers from a lack of suitable cathode materials that can produce ammonia at industrially viable rates.8–11 The cathode surface, which needs to be available for catalyzing the ENRR is instead poisoned by excessive H+ and H. Moreover, the high overpotentials at which cathode materials catalyze the ENRR often lead to dominant hydrogen production by the competing hydrogen evolution reaction (HER).8,9 The design and development of appropriate cathode materials for the ENRR is therefore an area of active research. Several studies have focused on the design and development of cathode materials with higher activity towards the ENRR.12–18 Using DFT calculations in conjunction with the computational hydrogen electrode model, Skúlason et al. constructed a volcano plot of transition metal catalysts for the ENRR. Their investigation revealed Mo, Fe, Rh and Ru metals to have the most active surfaces for the ENRR but with a strongly competitive HER at cathodic potentials. Early transition metals such as Sc, Ti, Zr and Y were found to have a higher preference for the ENRR compared to the HER. The N-adatom binding energy was shown to be a descriptor of the catalytic activity and was found to scale linearly with the Gibbs free energy to form other intermediates such as N2H*, NH*, etc. More recently, Dražević and Skúlason reported the largest volcano plot for the ENRR using a combination of experimental data on nitride formation with DFT computed limiting potentials for the ENRR.19 Their work identified Ga, In and Mn as “overlooked” catalysts for the ENRR. Ga has found widespread use in catalytic applications in the form of GaN, Ga2O3 and alloys such as NiGa or Pd–Ga. However, Ga metal has been particularly less explored for (electro)catalytic applications and computational/experimental studies into the mechanism of Ga catalyzed electrochemical conversions are rare. In the present work, we have explored the electrocatalytic potential of Ga as a cathode for the ENRR via DFT based computational modelling and electrochemical experiments.
Materials and methods
The carbon material used herein was Ketjenblack EC-600JD (called KJ) purchased from Akzo Nobel Polymer Chemicals (Netherlands). 10 wt% KJ carbon supported gallium nanoparticles (Ga/KJ10) were supplied by C2CAT (Netherlands). The details of physicochemical characterization (thermogravimetric (TG) analysis in Fig. S3a,† X-ray diffraction (XRD) analysis in Fig. S3b,† scanning transmission electron microscopy (STEM) analysis in Fig. S4†) are presented in the ESI.† The details of the flow cell setup, membrane and electrode preparation, ENRR measurements, control experiments, and ammonia analysis are presented in the ESI.†Computational methods
The Materials Project database was used to create the input for the periodic surface slab model of gallium. We chose the 100 facet which was reported as being the most stable. The ASE20,21 package was used to create a supercell of 6 layers of gallium with 16 atoms in each layer. The surface slab was modelled as being periodic in all directions with a vacuum spacing of 8 Å on both sides of the surface in a cell of dimensions a = 15.4606 Å, b = 9.1217 Å, c = 27.4505 Å, and α = β = γ = 90°. Periodic density functional theory (DFT) calculations were carried out using the Projector Augmented Wave (PAW) method as implemented in the GPAW22,23 suite of software using a plane wave cut-off of 500 eV, 4 × 4 × 1 k-points and the PBE XC functional.24 In addition to the bare Ga 100 surface facet, the effect of a water in salt electrolyte (WISE) was also considered. The WISE was modelled as a hydrated Li cation cluster, Li(H2O)4, over the catalytically active side, and a Cl anion adsorbed on the other side of the slab (see Fig. 1). Ga was modelled as being a diamagnetic solid which corresponds to its magnetic state at room temperature in the solid form. The Computational Hydrogen Electrode (CHE) was used as a reference for all elementary electrochemical steps in the ENRR.25 The slab geometries for Mn and In 111 surface facets were taken from the Catalysis-Hub repository.26 For Mn, the supercell consisted of three layers with 9 atoms of Mn in each layer with cell dimensions a = b = 7.44217 Å, c = 18.051 Å, and angles α = β = 90°, γ = 60° with ∼7 Å of vacuum spacing on each side. The bottom two layers were frozen. For In, the supercell consisted of three layers with 4 atoms of In in each layer with cell dimensions a = b = 6.61761 Å, c = 25.4033 Å, and angles α = β = 90°, γ = 60°, and ∼10 Å of vacuum spacing on each side. The bottom layer was frozen.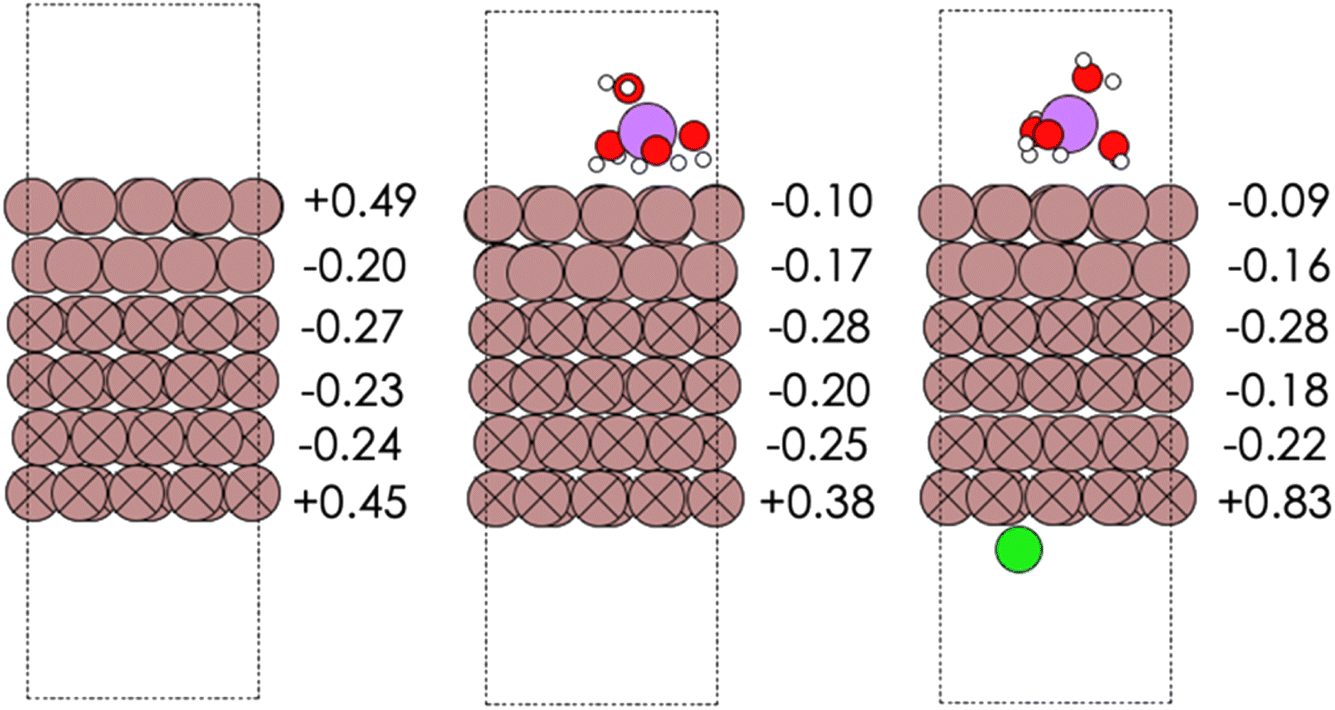 | ||
| Fig. 1 (Left to right) Optimized surface slabs for the Ga 100 surface facet for bare Ga (left), with the Li(H2O)4 cluster (middle), and with Li(H2O)4 and Cl ions (right). The computed Bader charges for each layer are also shown. | ||
Results and discussion
Mechanism of the ENRR
Two mechanisms have primarily been reported for the ENRR, namely associative and dissociative. In the associative mechanism the N2 molecule binds thermochemically on the catalyst surface and undergoes sequential proton-coupled electron transfer (PCET) steps to form 2 equivalents of ammonia. Possible surface adsorbed intermediates in the associative mechanism include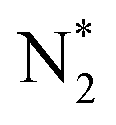 , N2H*,
, N2H*, 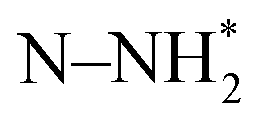 , N*, NH*,
, N*, NH*,  , NH–NH*, and
, NH–NH*, and 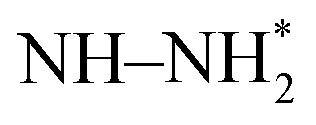 . In the dissociative mechanism the N2 molecule undergoes thermochemical splitting to form two surface adsorbed N* adatoms which undergo sequential PCET steps to form two equivalents of ammonia. The dissociative mechanism proceeds through N*, NH*, and
. In the dissociative mechanism the N2 molecule undergoes thermochemical splitting to form two surface adsorbed N* adatoms which undergo sequential PCET steps to form two equivalents of ammonia. The dissociative mechanism proceeds through N*, NH*, and  as possible surface adsorbed intermediates. Both mechanisms are shown in Scheme 1 below. The H+ transfer to surface adsorbed species is considered to proceed via the Heyrovsky mechanism (proton transfer by electrolyte), and the Tafel type mechanism for H+ transfer (i.e. protonation by recombination with H*) is excluded based on earlier suggestions.27
as possible surface adsorbed intermediates. Both mechanisms are shown in Scheme 1 below. The H+ transfer to surface adsorbed species is considered to proceed via the Heyrovsky mechanism (proton transfer by electrolyte), and the Tafel type mechanism for H+ transfer (i.e. protonation by recombination with H*) is excluded based on earlier suggestions.27
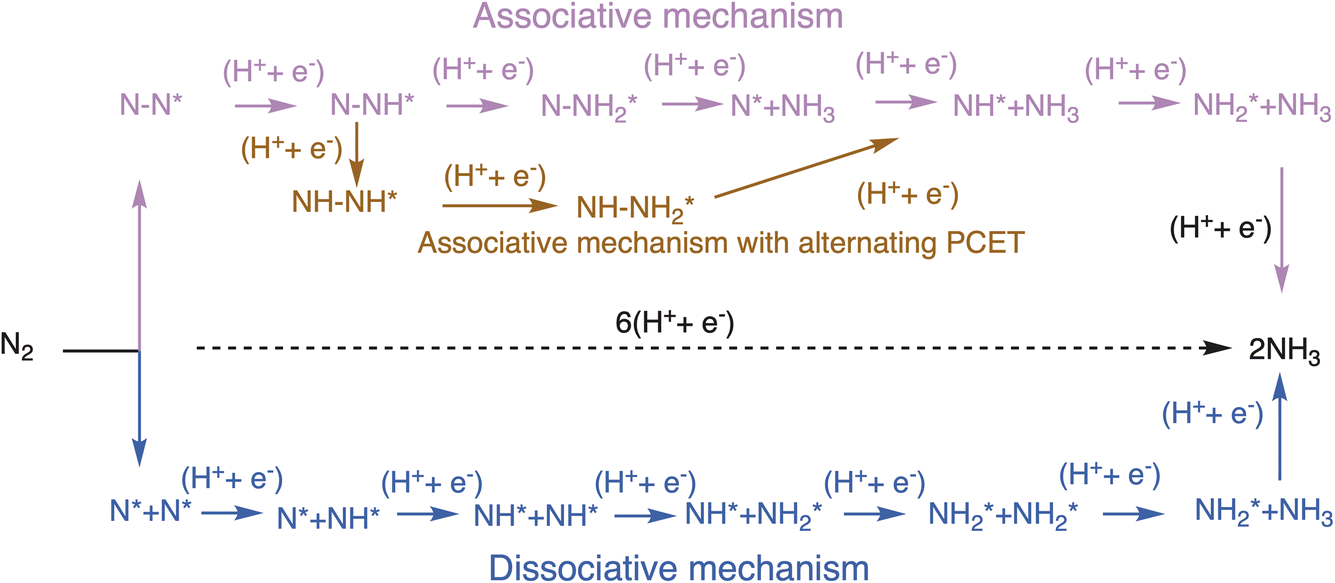 | ||
| Scheme 1 Schematic depiction of the mechanisms of the ENRR. The mechanistic pathway at the top shows the associative mechanisms while the pathway at the bottom shows the dissociative mechanism. | ||
Computational mechanistic modelling
The associative and dissociative mechanisms for the ENRR were investigated on bare Ga and in the presence of a model Li electrolyte over the Ga cathode surface. The Ga cathode was modelled as a periodic surface slab of the Ga 100 surface facet. The bare slab, the slab with hydrated Li on the reactive side, and the slab with hydrated Li and Cl ions are shown in Fig. 1. The Bader charges have also been shown for the respective layers of the surface slab. The bare Ga slab has positively charged surfaces. There is accumulation of a small negative surface charge in the presence of Li but the majority of the electronic charge is buried. For a comparison, the Ag 111 surface accumulates about −1.3 units of negative Bader charge density in response to 4 K+ cations in explicitly modelled layers of water.28 The water molecules coordinated to the Li point their protons towards the surface.We first investigated the interaction of N2 over the Ga 100 surface. N2 was found to interact weakly (ΔEN2 = −0.08 eV; ΔGN2 = +0.41 eV) with the bare Ga surface. This endergonic binding is consistent with the reported linear free energy scaling of N2 adsorption with the N-adatom adsorption on various metal surfaces which predicts ΔEN2 = −0.11 eV.24Fig. 2 shows the computed Gibbs free energy profiles for the ENRR for dissociative and associative mechanisms. In the dissociative mechanism, the first step is the thermally activated dissociation of dinitrogen. The dinitrogen dissociation was found to be endergonic by +2.4 eV and led to the formation of surface adsorbed N-adatoms (N* + N*). The N-adatom coordinated with three neighbouring Ga atoms on the surface. The computed N* binding energy was +0.91 eV which is consistent with the reported value of ∼1 eV for the Ga 111 surface facet.26 Subsequently the N* species underwent 6 exergonic PCET steps to release two equivalents of ammonia. The dissociative mechanism was modelled to occur on a single reactive site in the computed supercell of Ga and as such any lateral interactions between  (x = 0, 1, 2, 3) species were ignored. The associative mechanism commenced with an electrochemical PCET step involving the hydrogenation of a distal N species (ΔGN2H = 2.22 eV) of surface adsorbed dinitrogen. The subsequent PCET was slightly exergonic and formed a
(x = 0, 1, 2, 3) species were ignored. The associative mechanism commenced with an electrochemical PCET step involving the hydrogenation of a distal N species (ΔGN2H = 2.22 eV) of surface adsorbed dinitrogen. The subsequent PCET was slightly exergonic and formed a 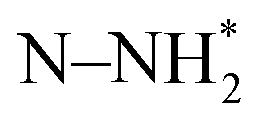 species (ΔGN2H = 2.16 eV). The
species (ΔGN2H = 2.16 eV). The 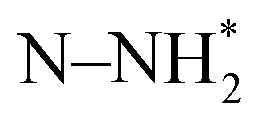 species underwent a further PCET step to evolve the first equivalent of NH3, resulting in an N-adatom on the surface. The subsequent steps were similar to the dissociative mechanism and led to the generation of the second equivalent of ammonia. Another possibility in the associative mechanism involves an alternating PCET step wherein instead of forming the
species underwent a further PCET step to evolve the first equivalent of NH3, resulting in an N-adatom on the surface. The subsequent steps were similar to the dissociative mechanism and led to the generation of the second equivalent of ammonia. Another possibility in the associative mechanism involves an alternating PCET step wherein instead of forming the 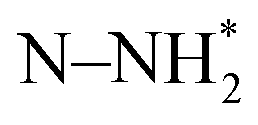 species, the N moiety adsorbed at the surface is protonated to form the NHNH* species which subsequently forms
species, the N moiety adsorbed at the surface is protonated to form the NHNH* species which subsequently forms 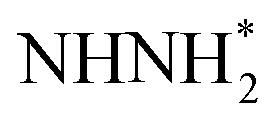 and
and 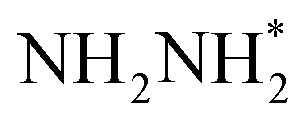 intermediates via successive PCETs. The alternating pathway for the associative mechanism was found to be less favorable. Among the dissociative and associative ENRR mechanisms, the associative mechanism is thermodynamically more favorable.
intermediates via successive PCETs. The alternating pathway for the associative mechanism was found to be less favorable. Among the dissociative and associative ENRR mechanisms, the associative mechanism is thermodynamically more favorable.
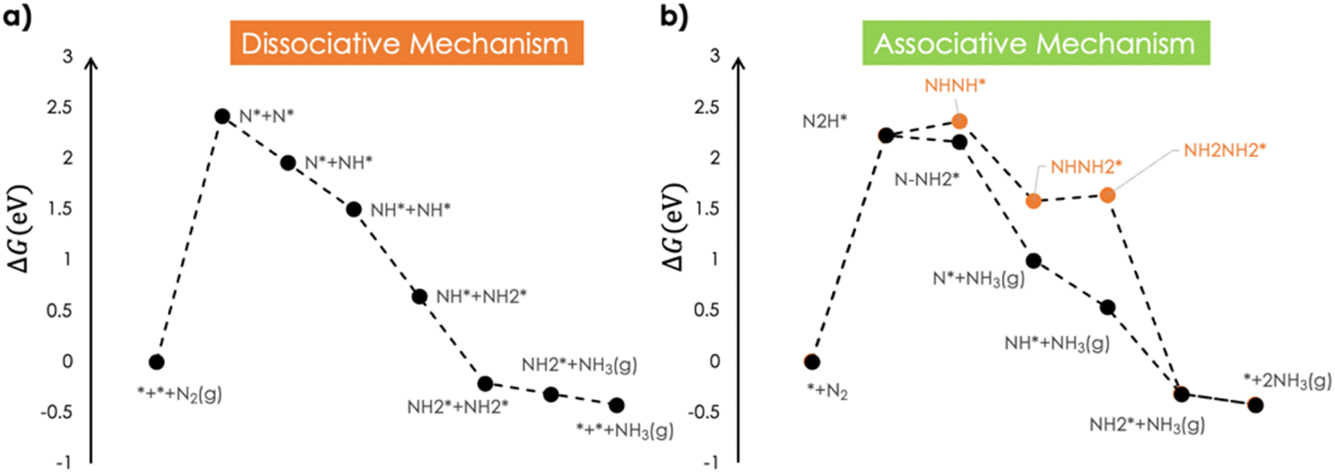 | ||
| Fig. 2 (a) Dissociative and (b) associative mechanisms for the ENRR over bare Ga. | ||
The thermochemical splitting of dinitrogen into two surface adsorbed N-adatoms was identified as the most endergonic elementary step in the mechanism and is expected to be more-or-less unaffected by an applied cathodic potential. Therefore, even at negative applied potentials one must thermally supply > 2 eV of free energy to convert N2 into ammonia via the dissociative pathway. It is noteworthy that the dissociative mechanism has been reported to be less favorable for late transition metals and only early transition metals which bind N2 and N-adatoms very strongly have been reported to proceed via the dissociative mechanism.27 In the associative mechanism, the first PCET to form the N2H* species is the potential determining step (PDS). Therefore, at potentials below −2.22 V (SHE) all steps in the associative mechanism will be downhill. The equilibrium potential for ammonia formation, computed at the same level of theory, is +0.07 V. The ENRR via the associative mechanism proceeds with an overpotential of 2.29 V (SHE).
Effect of Li
Both the associative and the dissociative mechanisms were computed in the presence of a solvated Li species over the catalyst surface and a Cl− ion adsorbed at the other side of the surface to maintain electroneutrality of the simulation cell. The presence of Li had minimum impact on the N2 binding and activation, with ΔGN2 = +0.23 eV being about 0.2 eV smaller than on the bare Ga surface. The computed Gibbs free energy profiles are shown in Fig. 3.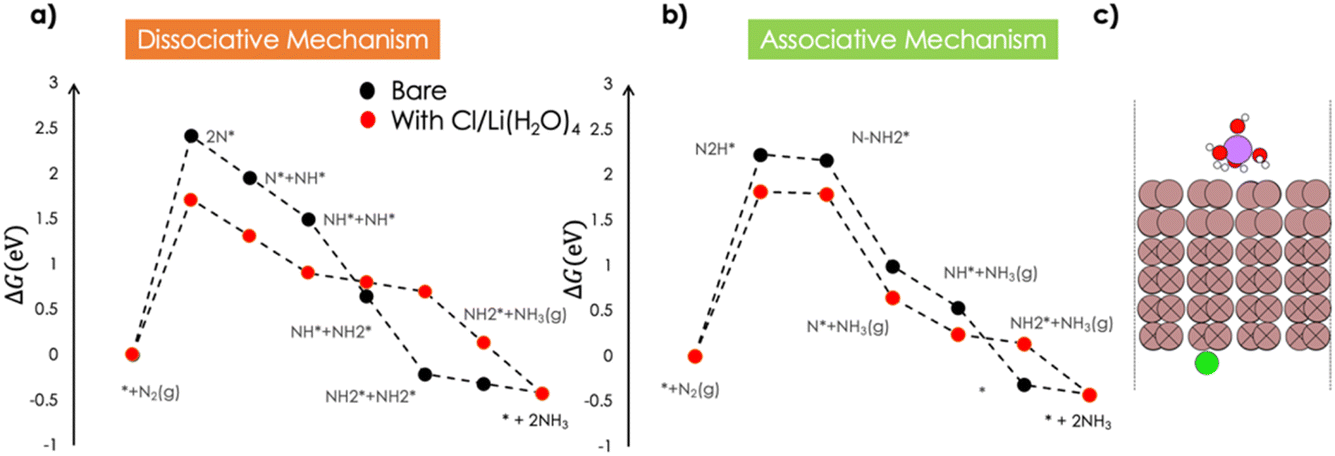 | ||
| Fig. 3 Gibbs free energy profile for the ENRR over the Ga 100 surface facet in the presence of hydrated Li and Cl: (a) dissociative mechanism, (b) associative mechanism. (c) Snapshot of the Ga 100 electrode with the electrolyte. Color code (brown: Ga, red: O, green: Cl, pink: Li, white: H). The Ga atoms with an ‘X’ were frozen during the geometry optimization steps. | ||
Li stabilizes the dinitrogen splitting in the dissociative mechanism by +0.7 eV. The presence of Li also influences the hydrogenation of NH* species. The formation of  from NH* was found to be less exergonic in the presence of the electrolyte. In the associative mechanism, the first PCET step forming the N2H* species is still the PDS and is stabilized by +0.41 eV. Therefore, the overpotential for NH3 formation is reduced to +1.88 V (SHE) when the effect of the electrolyte is considered.
from NH* was found to be less exergonic in the presence of the electrolyte. In the associative mechanism, the first PCET step forming the N2H* species is still the PDS and is stabilized by +0.41 eV. Therefore, the overpotential for NH3 formation is reduced to +1.88 V (SHE) when the effect of the electrolyte is considered.
Snapshots of the optimized geometries of the intermediates in the ENRR are shown in Fig. 4. The N2H*,  and
and  species coordinate in a bridging manner between two Ga atoms, in contrast to N* and NH* species which are triply coordinated. The N*, N2H* and
species coordinate in a bridging manner between two Ga atoms, in contrast to N* and NH* species which are triply coordinated. The N*, N2H* and  species engage in hydrogen bonding with the water molecules of the electrolyte (Li(H2O)4/Cl). The Ga–Ga–Ga–N dihedral angles are 29.5° for bare Ga, and 34.9° in the presence of Li(H2O)4/Cl. For a comparison the H–H–H–N dihedral angle is 35.3° in NH4+. Therefore, the electrolyte helps in achieving a more favorable tetrahedral configuration which is reflected in a more stabilized ΔEN* = +0.61 eV.
species engage in hydrogen bonding with the water molecules of the electrolyte (Li(H2O)4/Cl). The Ga–Ga–Ga–N dihedral angles are 29.5° for bare Ga, and 34.9° in the presence of Li(H2O)4/Cl. For a comparison the H–H–H–N dihedral angle is 35.3° in NH4+. Therefore, the electrolyte helps in achieving a more favorable tetrahedral configuration which is reflected in a more stabilized ΔEN* = +0.61 eV.
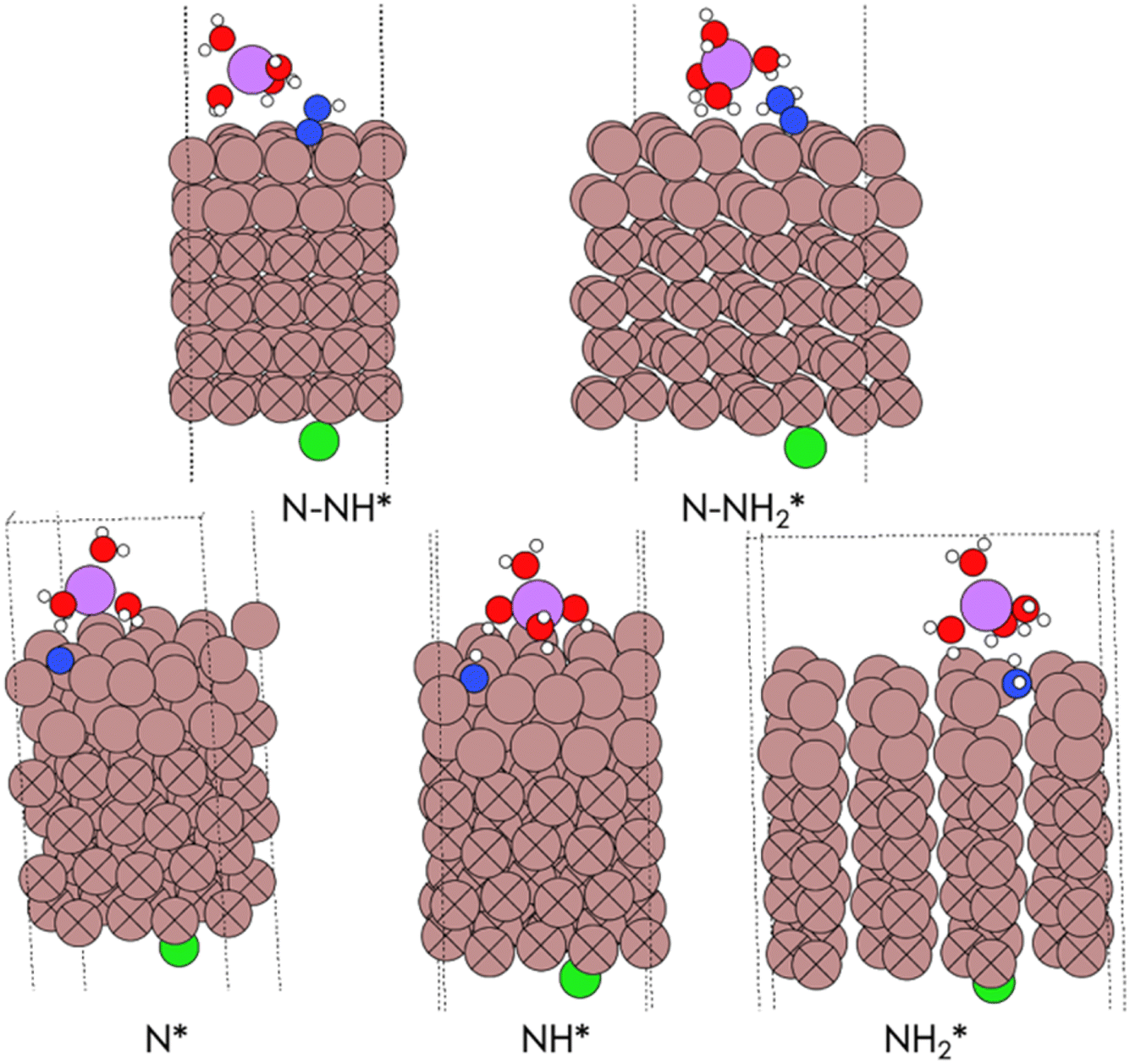 | ||
| Fig. 4 Snapshots of DFT optimized geometries of intermediates in the ENRR computed over a single reactive site on Ga 100. Color code (brown: Ga, red: O, green: Cl, pink: Li, white: H, blue: N). | ||
Competition between the ENRR and HER
The hydrogen evolution reaction (HER) competes with ammonia formation at cathodic potentials. The HER occurs via hydrogenation of surface adsorbed H adatoms. For a favorable ENRR, surface coverage by H* must be suppressed to make the catalytic surface available for ENRR intermediates. The N* species is a key intermediate in the mechanism of the ENRR and the adsorption energies of all other intermediates scale linearly with ΔEN*.27 Therefore, a comparison of the adsorption energies of N* and H* species is a good descriptor for the competition between the HER and ENRR. Moreover, the adsorption energies for H and N adatom adsorption are linearly related.27Fig. 5 shows a plot of H vs. N adsorption energies on flat surfaces of various transition metals. The data is taken from ref. 27. Entries for Ga, Mn and In are added from our present work. The adsorption of H adatoms is generally favorable for most metals. For Ga, we obtained ΔEH* = +0.2 eV whereas ΔEN* = +0.9 eV.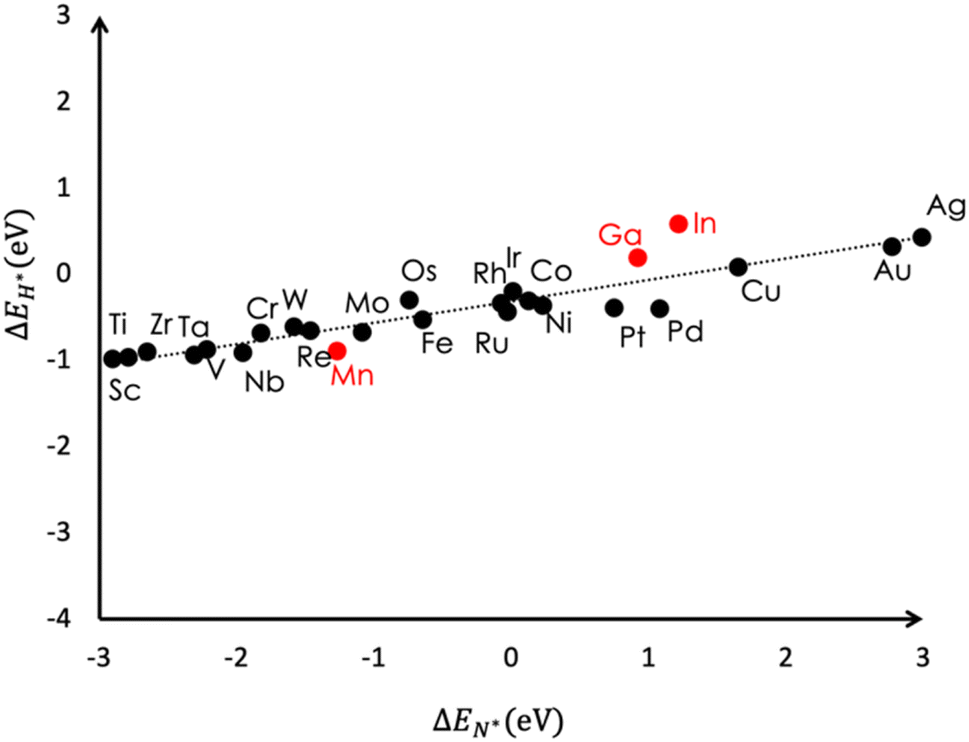 | ||
| Fig. 5 Plot showing the relative stabilities of surface adsorbed H species versus N species. This data is taken from the literature (black circles) and the data for Ga, In and Mn have been added from our calculations (red circles). | ||
These binding energies correspond to ΔGH* = +0.4 eV and ΔGN* = +1.2 eV at pH = 0. Therefore, the Ga surface will be preferentially covered with H* species at all applied potentials. Only at pH values close to 14 would N* adsorption become favorable. Comparing the adsorption of H adatoms versus N2 (ΔGN2 = +0.4 eV (bare); ΔGN2 = +0.2 eV (with Li(H2O)4/Cl)), the adsorption of dinitrogen will be competitive at pH = 0, and more favorable at higher pH values. However, since N2 adsorption is independent of applied potential, H adatom adsorption will dominate at cathodic potentials. At pH = 6 where we conducted ENRR measurement, ΔGH* (pH = 6; U) = 0.8 + U eV where U is the applied potential. At U < −0.4 V (SHE) (or −0.6 V with Li(H2O)4/Cl), H adatom adsorption will outcompete dinitrogen adsorption.
In shows similar binding characteristics with ΔEH* = +0.6 eV and ΔEN* = +1.6 eV and is expected to perform worse than Ga. For the In 111 surface the N adatom was found to be more stable in a tetrahedral binding mode where it coordinates with one of the In species from the second layer. The trigonal coordination of the N adatom with three neighbouring In atoms (similar to Ga) was less favorable by ∼0.3 eV. We note that a tetrahedral coordination mode will likely be catalytically inactive. The tetrahedral N adatom binding mode was found to be less stable for Ga (ΔEN* = +2.2 eV). For the Mn 111 surface, the N adatom was found to adopt a trigonal coordination mode with three Mn atoms on the surface. The H adatom was found to bind in a trigonal coordination mode over Mn and In but a bridging coordination mode between two Ga species over the Ga 100 surface. Snapshots of N* and H* species over In, Mn and Ga are shown in Fig. S4 (ESI†). The binding energies for Mn are ΔEH* = −0.9 eV and ΔEN* = −1.3 eV. Using the linear free energy scaling relations (LFER) reported by Skúlason and co-workers we obtain ΔGN2→N2H* = +0.18 eV, and ΔGN2→2N* = −2.0 eV.27 Therefore, Mn is expected to catalyze the ENRR via the dissociative mechanism. Due to the more favourable adsorption of the N adatom, the surface coverage of N* species will be higher than that of H* which should suppress the HER relative to the ENRR. The estimated ΔGH* = −0.7 eV indicates that at cathodic potentials below −1.3 V (SHE), the formation of H* species via H+(aq) + e− → H* will be more favored over N2(g) + 2* → N* + N* and the HER is expected to occur. Based on the relative stabilities of H* and N* species, Mn seems to be an interesting catalyst for the ENRR, and a detailed investigation of Mn for the ENRR is an ongoing effort in our group.
Results from ENRR experiments
To investigate the electrocatalytic activity of the Ga/KJ10 catalyst, ENRR experiments were performed in a membrane electrode assembly cell, using dry and purified nitrogen at the cathode and water at the anode side. The cathode and anode sides were separated by a cation exchange membrane, Nafion 117, with a reference hydrogen electrode placed in proximity to the membrane surface in the cathode compartment. The cyclic voltammetry (CV) curves of Ga/KJ10 were collected with both pure N2 and He gas with MiliQ water as the anolyte in the same potential range (Fig. 6a). From the evaluation above, the ENRR activity of the catalyst will increase with a more negative applied potential, where potentials from −1.88 to −2.29 V (SHE) are needed to activate nitrogen in the associative mechanism with and without Li present. Therefore, the chronoamperometric tests of Ga/KJ10 were performed at different working electrode potentials of −1.60, −1.70, −1.85 and −2 V (SHE), which correspond to −1.25, −1.35, −1.5, and −1.65 V (RHE) at pH = 6. The tests were run for 1 h to determine the dominant electrocatalytic process (Fig. 6c). The results showed that the detected ammonia in none of these tests was higher than background ammonia concentrations detected under He gas chronoamperometry. Only one test at −1.85 V (SHE) showed detectable ammonia, with a faradaic efficiency of 6.6%. However this result could not be reproduced in two subsequent tests at −1.85 V (SHE) for 1 h (Fig. 6b). Although the coulombic charge passing through the cell during chronoamperometric tests was higher under N2, compared to under He, the analysis of ammonia using spectrophotometry methods revealed that the ammonia detected during N2 electrolysis was not higher than that detected under He which suggests that the Ga surface was not ENRR active in pure MiliQ water with no ions present.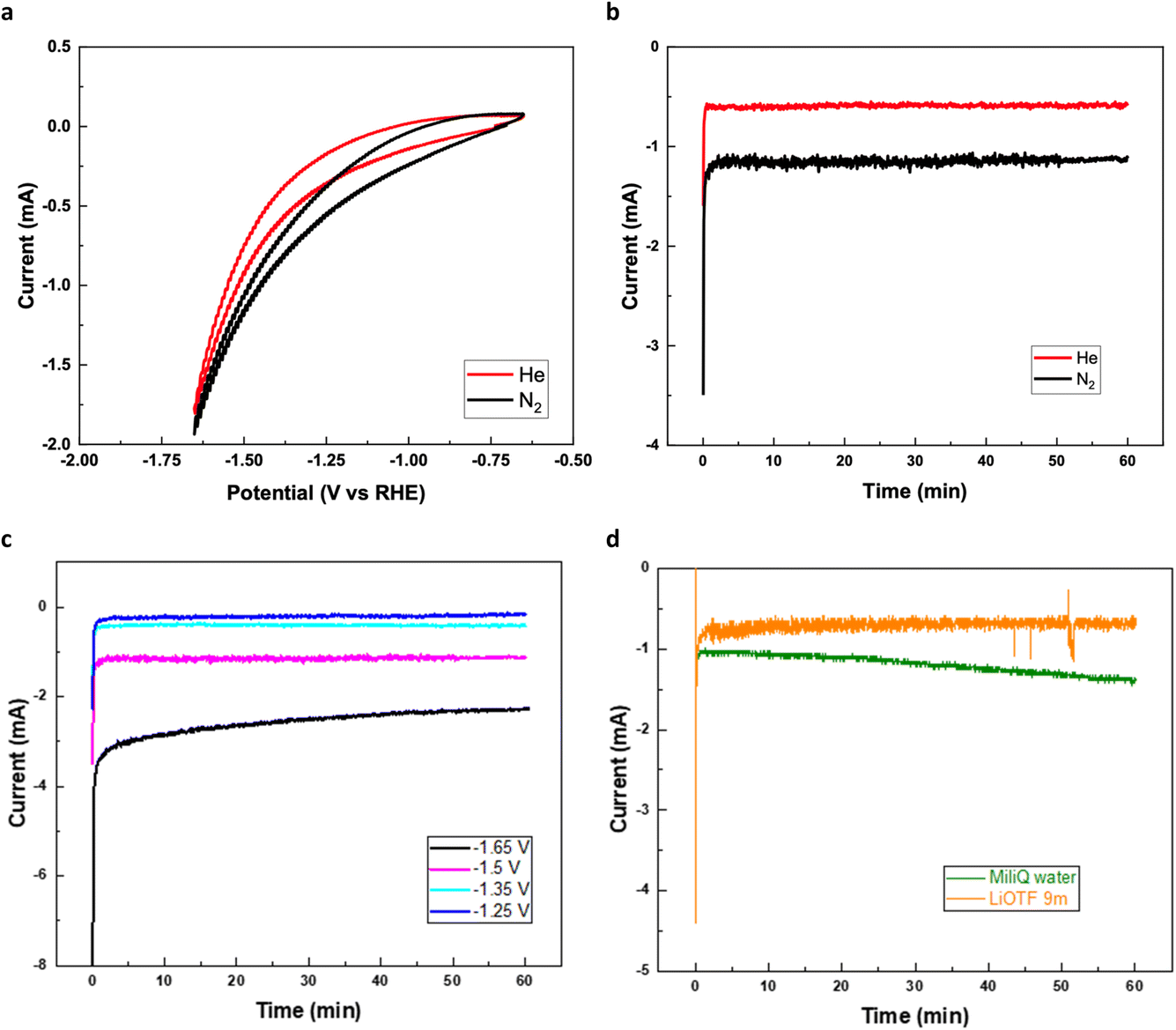 | ||
| Fig. 6 Electrochemical characterization. (a) The cyclic voltammetry curves of Ga/KJ10 in both N2 and He gas with MiliQ water as the anolyte, (b) chronoamperometric tests of Ga/KJ10 with N2 and He as background at −1.5 V (RHE) (−1.85 V (SHE)) for 1 h with MiliQ water as the anolyte, (c) chronoamperometric tests of Ga/KJ10 at different potentials of −1.60, −1.70, −1.85 and −2 V (SHE), which correspond to −1.25, −1.35, −1.5, and −1.65 V (RHE) at pH = 6. The tests were run for 1 h with MiliQ water as the anolyte. (d) The chronoamperometric tests of Ga/KJ10 with N2 at −1.85 V (SHE) for 1 h in MiliQ water and 9 m (mol kg−1) LiOTf as the electrolyte at the anode. | ||
Role of Li in Ga ENRR activity
DFT calculations hinted at the possibility of reducing the overpotential for NH3 formation in the presence of Li salts. On the other hand, applying a water in salt electrolyte (WISE) could suppress the HER by reducing proton accessibility and increasing the ENRR selectivity.29 Therefore, a concentrated Li salt (the molality of LiOTf was 9 m) was utilized as the electrolyte at the anode side, and chronoamperometric tests of Ga/KJ10 were carried out with N2 at −1.85 V (SHE) for 1 h (Fig. 6d). As shown in Fig. 6d, the absolute value of the current decreased when the concentrated salt was used at the anode side, and we ascribe this to a lower water activity at the anode side. This is a normal effect of concentrated salt solutions which have been shown earlier to extend the electrochemical window of stability of water. In the same manner, lower water activity and proton concentration can also improve the ENRR selectivity by suppressing the HER at the cathode. However, the detected ammonia was not higher than the He background, which might indicate that the Ga/KJ10 electrocatalyst is not an ENRR active catalyst.Ga as an overlooked catalyst
Earlier calculations used experimental enthalpies of metal nitride formation as descriptors of nitrogen binding energies and predicted Ga and In as strong nitrogen binders, where both could favourably activate dinitrogen and enable electrocatalysis of dinitrogen in water media.19 However, the DFT calculations and experiments in the present study show that dinitrogen is not very easily activated on Ga. It is not completely clear why a metal that has a very exothermic enthalpy of nitride formation is not able to activate N2, as our calculations and experiments show here. However, metal nitrides from different blocks in the periodic system of elements have quite different properties. For instance, s-group metals, alkali and alkaline earth metals, form stoichiometric metal nitrides that have a strong ionic character, which is less pronounced for bigger alkaline earth metals. These metal nitride forming reactions are very exothermic. Most of the d-block metals form interstitial and non-stoichiometric nitrides with moderately low enthalpies of nitridation. p-block metals, such as Ga and In, form well defined tetrahedrally coordinated nitrides, where the enthalpies of nitridation are quite negative. The enthalpy of formation of metal nitrides does not seem to be a good descriptor of activity for p-block elements, here in particular Ga and In. The activity of the transition metal Mn, on the other hand, seems very promising from Fig. 5 and this agrees with the prediction from the enthalpy of metal nitride formation.Conclusion
The ENRR requires a high overpotential over Ga. The lack of surface electronic charge results in weaker activation of dinitrogen which in turn raises the overpotential required for the first PCET. A Li salt has a stabilizing effect on the overpotential for both associative and dissociative mechanisms, the effect on the latter being more pronounced. Even with the stabilizing effect of Li, a rather high overpotential remains. Comparative binding of N and H adatoms revealed more favored adsorption of H. Therefore, being thermodynamically more favored, the competing HER is expected to dominate the ENRR at high cathodic potentials. These predictions are corroborated by ENRR experiments. Comparison of H and N adatom binding energies shows that among the previously proposed overlooked catalysts, Mn is a promising metal to be explored for the ENRR which is a topic of ongoing development in our groups.Author contributions
VS and FR equally contributed to the manuscript. Conceptualization: VS, FS, NS, ED; methodology: VS, FS, NS, EB, ED, JC; software: VS; validation: VS, NS, FR; formal analysis: VS, NS; investigation: VS, FR, NS; resources: VS, FS, ED; data curation: VS, NS, FR; writing – original draft: VS; writing – review & editing: all the authors; visualization: VS, FR, NS; supervision: VS, FS, ED; project administration: FS and ED; funding acquisition: FS and ED.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 101022738. This work was supported in part by Oracle Cloud credits and related resources provided by the Oracle for Research program. Calculations were partly performed on the LUMI-C supercomputer funded jointly by the EuroHPC joint undertaking, through the European Union's Connecting Europe Facility and the Horizon 2022 research and innovation program. We thank Aarhus University for granting us 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours on the LUMI-C.
000 hours on the LUMI-C.
Notes and references
- B. Wang, T. Li, F. Gong, M. H. D. Othman and R. Xiao, Ammonia as a green energy carrier: Electrochemical synthesis and direct ammonia fuel cell – a comprehensive review, Fuel Process. Technol., 2022, 235, 107380 CrossRef CAS.
- Intergovernmental Panel On Climate Change IPCC Second Assessment Climate Change 1995 A Report Of The Intergovernmental Panel On Climate Change Search PubMed.
- K. H. R. Rouwenhorst, A. G. J. van der Ham, G. Mul and S. R. A. Kersten, Islanded ammonia power systems: Technology review & conceptual process design, Renewable Sustainable Energy Rev., 2019, 114, 109339 CrossRef CAS.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- J. Deng, J. A. Iñiguez and C. Liu, Electrocatalytic Nitrogen Reduction at Low Temperature, Joule, 2018, 2, 846–856 CrossRef CAS.
- T. Wu, W. Fan, Y. Zhang and F. Zhang, Electrochemical synthesis of ammonia: Progress and challenges, Mater. Today Phys., 2021, 16, 100310 CrossRef CAS.
- B. M. Comer, P. Fuentes, C. O. Dimkpa, Y.-H. Liu, C. A. Fernandez, P. Arora, M. Realff, U. Singh, M. C. Hatzell and A. J. Medford, Prospects and Challenges for Solar Fertilizers, Joule, 2019, 3, 1578–1605 CrossRef CAS.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Rational design of electrocatalysts and photo(electro)catalysts for nitrogen reduction to ammonia (NH3) under ambient conditions, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Catalysts for nitrogen reduction to ammonia, Nat. Catal., 2018, 1, 490–500 CrossRef.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Review of electrochemical ammonia production technologies and materials, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Electrocatalytic Synthesis of Ammonia at Room Temperature and Atmospheric Pressure from Water and Nitrogen on a Carbon-Nanotube-Based Electrocatalyst, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- X. Zhao and G. Hu, in Nanomaterials for Electrocatalysis, ed T. Maiyalagan, M. Khandelwal, A. Kumar, T. A. Nguyen and G. Yasin, Elsevier, 2022, pp. 273–298 Search PubMed.
- H. He, H.-M. Wen, H.-K. Li and H.-W. Zhang, Recent advances in metal–organic frameworks and their derivatives for electrocatalytic nitrogen reduction to ammonia, Coord. Chem. Rev., 2022, 471, 214761 CrossRef CAS.
- X. Fu, J. Zhang and Y. Kang, Recent advances and challenges of electrochemical ammonia synthesis, Chem Catal., 2022, 2, 2590–2613 CrossRef CAS.
- S. Lin, X. Zhang, L. Chen, Q. Zhang, L. Ma and J. Liu, A review on catalysts for electrocatalytic and photocatalytic reduction of N2 to ammonia, Green Chem., 2022, 24, 9003–9026 RSC.
- V. C. D. Graça, F. J. A. Loureiro, L. I. v Holz, S. M. Mikhalev, A. J. M. Araújo and D. P. Fagg, in Heterogeneous Catalysis, ed. M. R. Cesario and D. A. de Macedo, Elsevier, 2022, pp. 497–514 Search PubMed.
- J. Hou, M. Yang and J. Zhang, Recent advances in catalysts, electrolytes and electrode engineering for the nitrogen reduction reaction under ambient conditions, Nanoscale, 2020, 12, 6900–6920 RSC.
- C. Lee and Q. Yan, Electrochemical reduction of nitrogen to ammonia: Progress, challenges and future outlook, Curr. Opin. Electrochem., 2021, 29, 100808 CrossRef CAS.
- E. Dražević and E. Skúlason, Are There Any Overlooked Catalysts for Electrochemical NH3 Synthesis—New Insights from Analysis of Thermochemical Data, iScience, 2020, 23(12), 101803 CrossRef PubMed.
- S. R. Bahn and K. W. Jacobsen, An object-oriented scripting interface to a legacy electronic structure code, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The atomic simulation environment—a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, Electronic structure calculations with GPAW: a real-space implementation of the projector augmented-wave method, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Real-space grid implementation of the projector augmented wave method, Phys. Rev. B, 2005, 71, 35109 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- O. Mamun, K. T. Winther, J. R. Boes and T. Bligaard, High-throughput calculations of catalytic properties of bimetallic alloy surfaces, Sci. Data, 2019, 6, 76 CrossRef PubMed.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- V. Sinha, E. Khramenkova and E. A. Pidko, Solvent-mediated outer-sphere CO2 electro-reduction mechanism over the Ag111 surface, Chem. Sci., 2022, 13, 3803–3808 RSC.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706–709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2fd00169a |
| This journal is © The Royal Society of Chemistry 2023 |