
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew cationic coinage metal complexes featuring silyl group functionalized phosphine: syntheses, structures and catalytic studies in alkyne–azide cycloaddition reactions†
Amiya Kumar
Sahoo
a,
Ashish
Kumar Sahoo
a,
Bhagyashree
Das
a,
Subhra Jyoti
Panda
b,
Chandra Shekhar
Purohit
 b and
Adinarayana
Doddi
*a
b and
Adinarayana
Doddi
*a
aDepartment of Chemical Sciences; Indian Institute of Science Education and Research Berhampur; Transit Campus, Industrial Training Institute (ITI); Engineering School Road, Ganjam, Odisha 760010, India. E-mail: adoddi@iiserbpr.ac.in
bSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), Bhubaneswar, 752050, India
First published on 12th September 2023
Abstract
A series of coinage metal complexes bearing rarely explored ortho-silylated phosphine is reported. The treatment of diphenyl(2-(trimethylsilyl)phenyl)phosphine (1) with CuCl and [Cu(CH3CN)4]BF4 furnished the corresponding neutral [(1)CuCl]2 (2) and mono-cationic [(1)2Cu(CH3CN)]BF4 (3) complexes, respectively. The reactions of 1 with AgX (X = BF4−, NO3−) in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio furnished the corresponding mono cationic dicoordinate silver(I) complexes of the type [(1)2Ag]X (X = BF4− (4a), NO3− (4b)). The ortho-silylated phosphine ligand (1) was conveniently converted into the corresponding sulfide (5a) and selenide (5b) species, and their reactions with [Cu(CH3CN)4]BF4 yielded mono-cationic, homoleptic tris(silylphosphinochalcogenide)copper(I) complexes of the type [(5a/5b)3Cu]BF4 (6a/6b). The molecular structures of 2–4 and 6 were established by single-crystal X-ray diffraction analysis. The copper complexes 2, 3, and 6a were employed as catalysts in azide–alkyne cycloaddition reactions. Among these complexes, 3 was extensively used in the preparation of various mono- and bis-triazoles consisting of tolyl, benzyl, carbazolyl, and propargylic ether groups. Three sets of substituted triazole derivatives were achieved under mild conditions by employing copper(I) catalytic systems. The mechanistic studies indicated the formation of a heteroleptic copper(I) triazolide intermediate which was detected by high-resolution mass spectral analysis.
:1 ratio furnished the corresponding mono cationic dicoordinate silver(I) complexes of the type [(1)2Ag]X (X = BF4− (4a), NO3− (4b)). The ortho-silylated phosphine ligand (1) was conveniently converted into the corresponding sulfide (5a) and selenide (5b) species, and their reactions with [Cu(CH3CN)4]BF4 yielded mono-cationic, homoleptic tris(silylphosphinochalcogenide)copper(I) complexes of the type [(5a/5b)3Cu]BF4 (6a/6b). The molecular structures of 2–4 and 6 were established by single-crystal X-ray diffraction analysis. The copper complexes 2, 3, and 6a were employed as catalysts in azide–alkyne cycloaddition reactions. Among these complexes, 3 was extensively used in the preparation of various mono- and bis-triazoles consisting of tolyl, benzyl, carbazolyl, and propargylic ether groups. Three sets of substituted triazole derivatives were achieved under mild conditions by employing copper(I) catalytic systems. The mechanistic studies indicated the formation of a heteroleptic copper(I) triazolide intermediate which was detected by high-resolution mass spectral analysis.
Introduction
Over the last two decades, the organometallic chemistry community has witnessed tremendous developments in the design and isolation of stereoelectronically tuneable ligands featuring various phosphine groups and their applications in the stabilization and isolation of novel metal complexes.1 It is noteworthy that phosphine ligands functionalized with Lewis acidic moieties such as the group 13 and 14 elements in their +3 and +4 oxidation states, respectively, have been widely explored in coordination and organometallic chemistry as ambiphilic ligands (Chart 1).2,3 This type of ligand stabilizes metal centers in an intramolecular fashion in two different bonding modes, namely by donor atoms from the Lewis basic center and a simultaneous back donation from the metal atoms to the Lewis acidic centers of the ligand. Therefore, such a class of ligand systems has been found to serve as effective ligands in electronic and steric tuning around the metal centres of interest to achieve specific properties such as inducing more electrophilic nature in well-defined and soluble transition metal catalysts in homogeneous catalytic pathways.3,4 In this context, the notable examples of such type of reactive species can be considered from the pioneering reports by the group of Bourissou and co-workers.5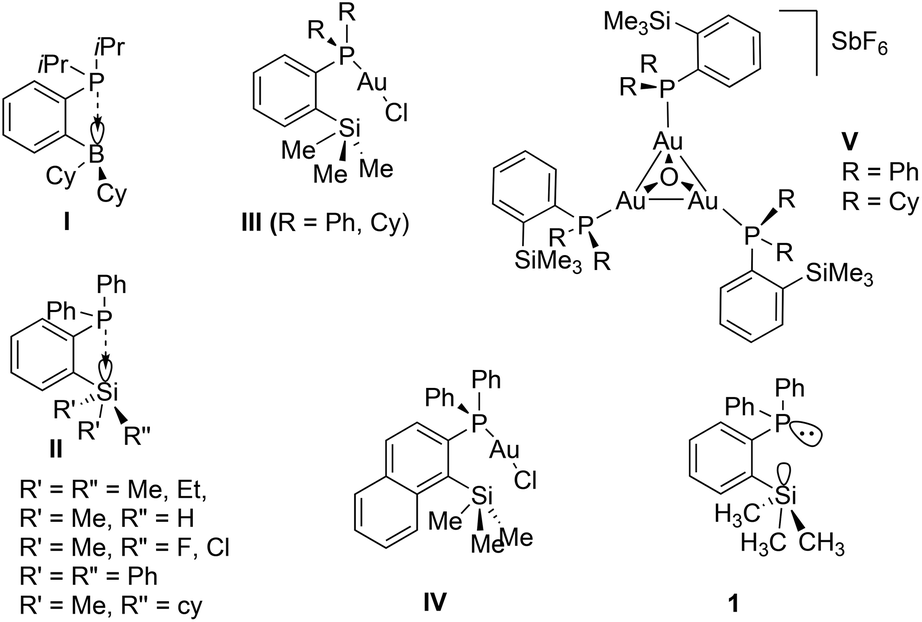 | ||
| Chart 1 Examples of ortho-position substituted electronically modified phosphorus(III) compounds and their gold complexes (R = alkyl or aryl group). | ||
As shown in Chart 1, group 13 and 14 element substituted phosphines of types I and II were isolated and used in both main group and transition metal chemistry. Among these two classes of phosphines, type I was introduced recently;6–8 however, species of type II (R = Me, Cl, H) were isolated several years ago.9–11 Type II phosphines consisting of ortho-silylated groups and their transition metal complexes have been shown to exhibit enhanced solubility in various non-polar solvents which is in contrast to many typical aryl phosphine ligands.4,12–14 Additionally, the type II species have also been employed as mono-dentate ligands in coinage metal chemistry,15 for instance gold(I) chloride complexes of type III [(IIR)AuCl] (R = Ph, Cy) and IV have been isolated.15 Furthermore, the corresponding cationic complexes featuring trimetallic gold complexes of the type [{(IIR)Au}3O]SbF6 (V, R = Ph, Cy) have been isolated and structurally characterized. In a similar fashion, Shi and co-workers demonstrated the isolation of gold(I) chloride complexes of type III.11
In view of the general interest, various phosphines functionalized with group 136,7,16 and 14 element-containing groups (as Lewis acidic moieties) as monodentate ligands17 have not been studied extensively in transition metal organometallic chemistry and homogeneous catalysis as in the case of their pincer counterparts (PSiP).2,3,18 Their transition metal chemistry is still in its infancy. These systems can be considered as more electron rich species; in addition, they can provide high solubility to their metal complexes compared to classical triphenylphosphines. In this contribution, we wish to report the synthesis and structural characterization of a series of coinage metal(I) complexes (Cu and Ag) bearing monodentate phosphine (1) and copper complexes of sulfides and selenides of 1. In addition, one of the bis(phosphine)copper(I) complexes was introduced as a catalyst in the synthesis of various substituted simple to complex triazole derivatives in good to high yields.
Results and discussion
The ortho-silylated phosphine 1 was prepared as a colourless oily substance by a slightly modified synthetic procedure.10 As shown in Scheme 1, the in situ generated lithium salt was treated with trimethylchlorosilane at 0 °C, and the crude product was purified by silica gel column chromatography. The spectroscopic (1H, 13C, and 31P NMR) and CHNS data were similar to the reported data.10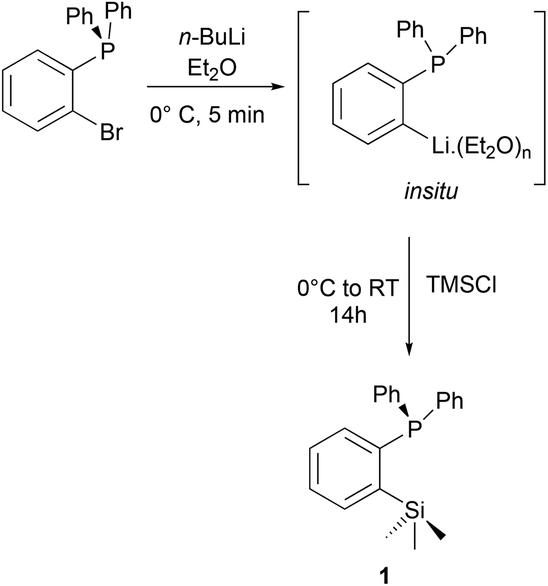 | ||
| Scheme 1 Preparation of compound 1 from bromophenyldiphenylphosphine. | ||
Single crystals suitable for X-ray diffraction analysis were grown by slow evaporation of n-hexane solution at RT. The molecular structure is similar to those that have been established for similar phosphine ligands (Fig. 1). For instance, the average P⋯Si separation in 1 is about 3.39 Å which is in the range observed for the reported P⋯Si separation (3.31 Å) in ortho-diisopropyl chlorosilyl-functionalized triarylphosphane.9
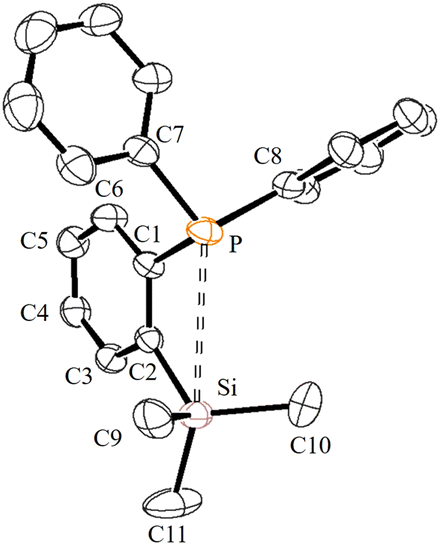 | ||
| Fig. 1 ORETP view of one of the two molecules of 1 in the crystal lattice showing 30% ellipsoid probability. Hydrogen atoms are omitted for clarity. Important bond lengths [Å] and angles [°]: P–C1 1.846(2), P⋯Si 3.381, C1–C2 1.411(3), C2–Si 1.894(2), C7–P–C8 102.57(11), and C9–Si–C10 110.65(16). | ||
Preparation and characterization of copper(I) and silver(I) cationic and neutral complexes bearing 1
The reactivity of ortho-silylated phosphine 1 was studied towards various copper and silver metal precursors (Scheme 2). The treatment of 1 with copper(I) chloride in THF at 60 °C afforded the neutral complex [(1)2CuCl]2 (2) as a pale-yellow solid in 87% yield, whereas the phosphine and cationic copper sources [Cu(CH3CN)4]BF4 or AgX (X = BF4−, or NO3−) in a 2:1 ratio in THF at 60 °C furnished the corresponding bis(phosphine) containing cationic copper(I) and silver(I) complexes 3 and 4a/4b in 89%, 88%, and 86% isolated yields, respectively, as off-white solids. It should be noted that 1:1 reactions of ligand and metal precursors also yielded the same complexes. The copper complexes 2 and 3 were found to be stable toward moisture and air; however, in the case of silver complexes, signs of decomposition were observed especially when exposed to light. 2–4 are highly soluble in polar solvents such as THF, CH2Cl2, and CHCl3 but are insoluble in non-polar solvents. These complexes were fully characterized by spectroscopic techniques (1H, 13C, 31P NMR, and MS) and by single crystal X-ray diffraction analysis.
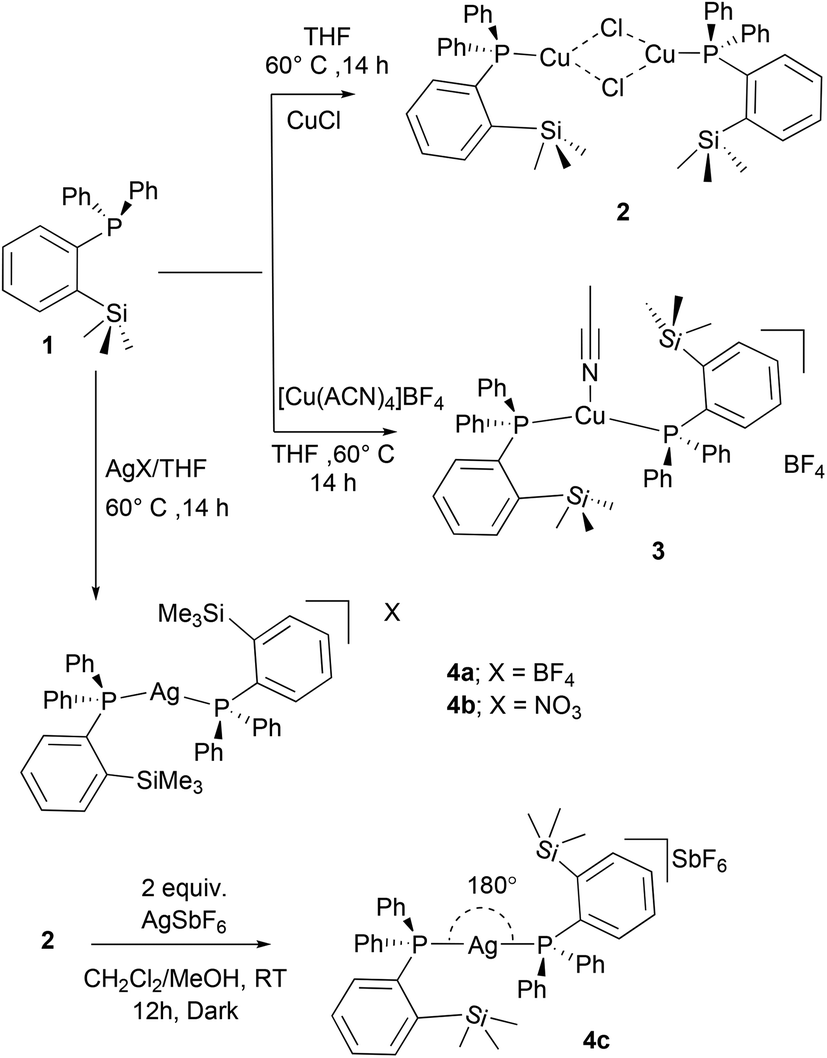 | ||
| Scheme 2 Preparation of ortho-silylated phosphine-supported copper and silver complexes. | ||
The 1H NMR spectra of 2–4 in CDCl3 show the presence of trimethyl silyl groups as sharp and intense signals in the range of 0.28–0.38 ppm. The corresponding 13C NMR spectrum of 3 displays a doublet at 2.05 (4JPC = 2.8 Hz) ppm, whereas in the case of cationic 4a (2.67 ppm) and 4b (2.72, 4JPC = 3.9 Hz), broad doublets were observed. These chemical shifts are downfield shifted when compared with those of the free ligand (doublet at 1.6 ppm, 4JPC = 9.0 Hz).10 Two sharp signals at 2.4 and 117.8 ppm in the 13C NMR spectrum of 3 indicate the presence of the CH3CN ligand attached to the copper center. The 31P{1H} NMR spectral data clearly indicate the coordination of the phosphine ligand to Cu and Ag metals. The copper complexes 2 (2.02 ppm) and 3 (0.29 ppm) show broad signals, which are significantly downfield shifted when compared to those of the free phosphine (−10.24 ppm) (1).10 The broadening of these signals can be ascribed to the coupling of the 31P nucleus with the quadrupole relaxation of 63Cu and 65Cu nuclei.19 The signals observed at m/z 438.0845 (ESI Fig. S11/21†) in the ESI-MS spectral analysis of 2 and 3 could be assigned to the cationic species [(1)Cu(CH3CN)]+.
In comparison, the 31P{1H} NMR spectra of the cationic silver(I) complexes 4a/4b in CDCl3 show the silver-phosphorus couplings at RT (ESI; Fig. S23/31†). Complex 4a exhibits a doublet resonance at δ = 13.3 ppm due to coupling with two silver isotopes 107Ag (d, 2JP,Ag(107) = 746 Hz) and 109Ag (d, 2JP,Ag(109) = 848 Hz), which are in the range reported for aminophosphine ligand-supported cationic silver complexes of the type [Ag(PR)3]BF4 (R = NMe2, NMeCH2CMe); (JAgP = 811 and 910 Hz).20,21 Similarly, 4b with the NO3− anion shows a doublet at 13.9 ppm with slightly lower coupling constants with 107Ag (d, 2JP,Ag(107) = 714) and 109Ag (d, 2JP,Ag(109) = 821) isotopes (Fig. S31†). It can be observed that these coupling constants are significantly higher than those reported for two-coordinated phosphine-silver complexes,20–22 and also considerably higher than those of the phosphorus(I) ligand-supported silver(I) complexes.23
In addition, we aimed to convert the dimeric copper(I) complex [(1)CuCl]2 (2) to the corresponding cationic complex by chloride abstraction reactions. However, the reaction of 2 with AgSbF6 in MeOH/CH2Cl2 mixture readily afforded another dicoordinate cationic silver(I) complex 4c in 81% isolated yield as an off-white solid (Scheme 2). Similar to 4a/4b, a doublet resonance in 31P NMR (in CDCl3) of 4c at 12.4 ppm indicates the silver metal coordination. Interestingly, compared to the other silver complexes, significantly lower Ag–P coupling constants (d, 2JP,Ag(107) = 510 Hz, and 2JP,Ag(109) = 586 Hz) were observed as in the case of bis(phosphine)silver complexes.24 This difference in coupling constants can be attributed to the presence of different weakly coordinating anions in 4a–4c. In contrast to 2 and 3, the ESI-MS spectral data clearly indicated the cationic nature of complexes 4a–4c, and the m/z values at 777.1696 can be attributed to the cationic moieties of the type [(1)2Ag]+ (ESI, Fig. S28/33/37/39†). Additionally, the anionic SbF6− of 4c was also detected at m/z 234.8944.
X-ray structural characterization of complexes 2–4
The solid-state molecular structures of 2–4 could be established by the single-crystal X-ray diffraction analysis. Single crystals suitable for the XRD measurements of copper complexes 2 (Fig. 2) and 3 (Fig. 3) were obtained by the vapor diffusion method by using the CH2Cl2/n-hexane combination. The complex 2 displays a dimeric structure in the solid state with an ortho-silylated phosphine-supported Cu2(μ2-Cl)2 core with a Cu⋯Cu separation of 3.00 Å.25 It can be observed that simple triphenyl phosphine–CuCl attains a cubane structure in the solid state having tetrameric [PPh3CuCl]4 nature with long Cu⋯Cu distances.26 In contrast to this structure, complex 2 attains a dimeric nature in the solid state presumably due to the presence of a sterically bulky silyl group in the phosphine moiety, whereas the cationic complex 3 shows the formation of a tri-coordinate copper complex with two phosphine ligands and one acetonitrile coordination. The copper atom in 3 exhibits a distorted trigonal planar geometry with an average bond angle of 119.9° around the copper centre. The copper–phosphorus distances in 2 (av. 2.181 Å) are considerably shorter than those in 3 (av. 2.268 Å) due to the cationic nature of the complex. The dihedral angles between the two Cl–Cu–Cl in 2 are 97.28° (Cl1–Cu1–Cl2) and 98.15° (Cl1–Cu2–Cl2), which are consistent with the values reported in similar chloro-bridged copper complexes.25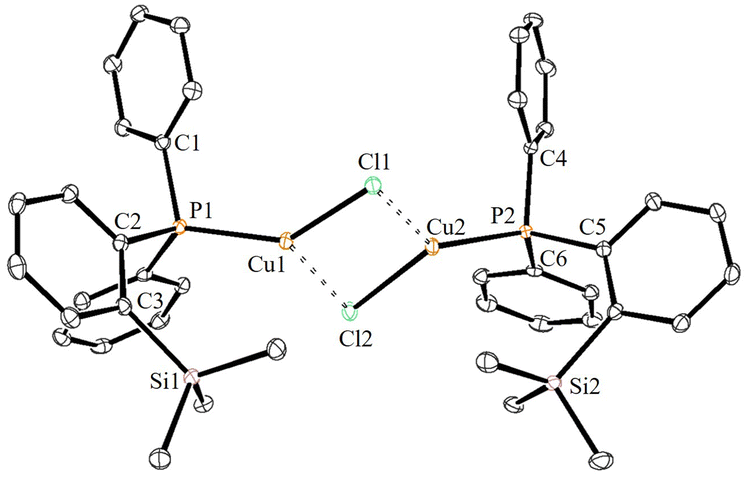 | ||
| Fig. 2 ORTEP view of complex 2 with thermal displacement parameters drawn at 30% probability. Hydrogen atoms are omitted for clarity. Important bond lengths [Å] and angles [°]: P1–Cu1 2.1867(4), P2–Cu2 2.1759(4), Cu1–Cl1 2.3089 (4), Cu1–Cl2 2.3135 (5), Cu2–Cl2 2.2867(4), Cu2–Cl1 2.3056 (4), Cu1–Cl1–Cu2 81.320(15), Cu1–Cl2–Cu2 81.625(15), Cl1–Cu1–Cl2 97.294(16), Cl1–Cu2–Cl2 98.151(16), P1–Cu1–Cl1135.184(18), P1–Cu1–Cl2 127.086(18), P2–Cu2–Cl1 131.683(18), and P2–Cu2–Cl2 128.684(19). | ||
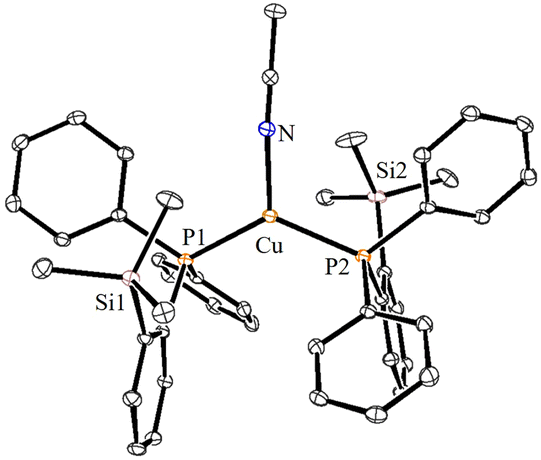 | ||
| Fig. 3 ORTEP view of the cationic part of complex 3 with 30% ellipsoid probability. The BF4− counter anionic moiety and hydrogen atoms are omitted for clarity. Important bond lengths [Å] and angles [°]: P1–Cu 2.2694(5), P2–Cu 2.2681(5), Cu–N 1.9910(17), P1–Cu–P2 127.66(2), N–Cu–P1 118.26(5), and N–Cu–P2 113.99(5). | ||
The silver complexes 4a–4c were also structurally characterized. Single crystals suitable for XRD analysis were grown by the vapor diffusion of dichloromethane solutions with n-hexane, and colorless crystals were used for the measurements. Their molecular structures are given in Fig. 4, Fig. S165† and Fig. 5, respectively. Although 4a–4c contain the same phosphine 1, the geometrical environment around the cationic silver(I) atoms is different. The average silver–phosphorus bond lengths in 4a (2.423 Å) and 4b (2.431 Å) are relatively longer than those observed in 4c (2.393 Å); however, they are slightly shorter than those of similar tertiary phosphines that feature cationic silver complexes, for instance, [(Mes3P)2Ag]BF4 (2.4409(9) Å),27 but are comparable to those found for several other bis(phosphine)silver complexes.28
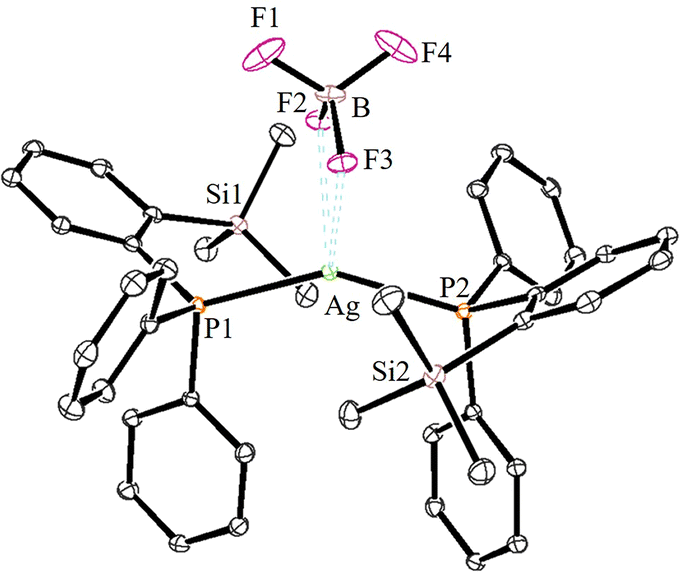 | ||
| Fig. 4 ORTEP view of 4a with thermal displacement parameters drawn at 30% probability. Hydrogen atoms are omitted for clarity. Important bond lengths [Å] and angles [°]: P1-Ag1 2.4241(5), P2–Ag1 2.4223(5), B1–F1 1.366(3), B1–F2 1.404(3), B1–F3 1.409(3), B1–F4 1.368(3), Ag1–F2 2 2.644, Ag1–F3 2.626, P1–Ag1–P2 147.34(2), and F3–Ag–F2 50.91. | ||
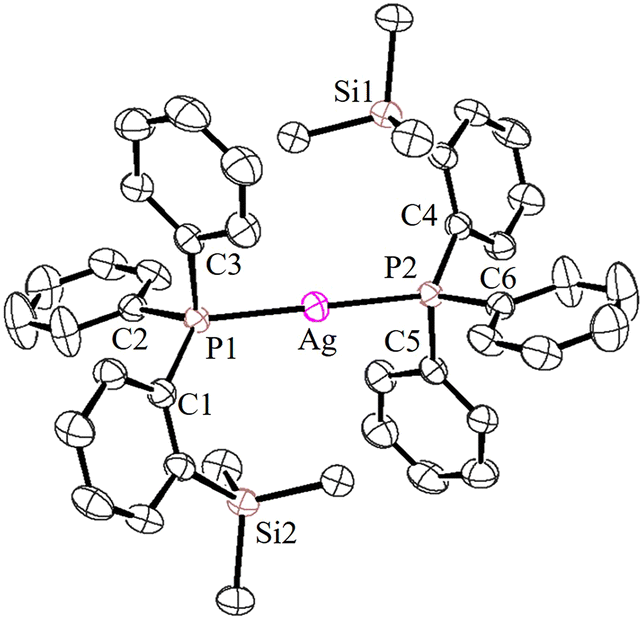 | ||
| Fig. 5 ORTEP view of the cationic part of 4c with thermal displacement parameters drawn at 30% probability. Counter anions and hydrogen atoms are omitted for clarity. Important bond lengths [Å] and angles [°]: P1–Ag 2.3936(6), P2–Ag 2.3936(6), and P1–Ag–P2 180.0 (exact linear unit). | ||
The P–Ag–P bond angles in 4a (147.34(2)°) and 4b (143.85(3)°) are far from linearity; however, complex 4c with the SbF6− counter anion clearly exhibits a perfectly linear geometry with the P–Ag–P bond angle of 180.0°. This can be attributed to the interaction of anions with the Ag(I) centre in the case of 4a/4b. It is noteworthy that, in general, bis(phosphine)silver complexes exhibit an almost linear geometry as reported in numerous previously reported examples.27–29 Therefore, complex 4c can be considered as one of the few bis(phosphine) supported dicoordinate silver complexes with a perfect linear P–Ag–P unit.
Furthermore, in contrast to 4c, complexes 4a and 4b show weak interactions between the counter anions (BF4− and NO3−) and the cationic Ag(I) center. All the B–F bond distances in the tetrahedral BF4 unit are different (free B–F bonds; 1.367 Å), and two of the B–F bonds show slightly elongated (B–F; av 1.406 Å) distances due to the weak ligating property toward the Ag(I) center. The observed average F⋯Ag distances (2.635 Å) in 4a are shorter than those in BF4-containing coordination polymers such as {[Ag(1,4-dithiane)]BF4}∞ (F⋯Ag; 2.886(2)).30 Similarly, complex 4b displays weak O, O-chelation of the NO3− anion with the Ag(I) center (Fig. S165†).
Reactivity of 1 with elemental sulfur, selenium, and their cationic homoleptic copper(I) complexes
In addition to the aforementioned class of ortho-silylated phosphine-containing cationic copper and silver complexes, we have also isolated ortho-silylated phosphorus(V) compounds and investigated their reactivity toward [Cu(CH3CN)4]BF4.The treatment of 1 with elemental sulfur (S8) and selenium (Se) powders in benzene at 50 °C readily afforded the corresponding phosphorus(V) compounds 5a and 5b in 88% and 93% isolated yields, respectively (Scheme 3). These compounds are highly soluble in both polar and nonpolar solvents. Their formation was further confirmed by 1H and 31P NMR spectroscopy and mass spectral analysis. As expected, the 31P NMR spectra (in CDCl3) of 5a (46.6 ppm) and 5b (37.7 ppm) showed sharp singlet resonances, which are significantly downfield shifted when compared with those of the free ligand 1 (δ = −10.24 ppm). These shifts are slightly downfield shifted compared to the corresponding triphenylphosphine sulphide and selenides.31 Furthermore, their formation was supported by ESI-HRMS data (Fig. S43/47†). The signal at 367.1111 of 5a can be assigned to [M + H]+ (calcd for C21H24PSSi 367.1106); similarly, for 5b, m/z at 415.0550 can be assigned to [M + H]+.
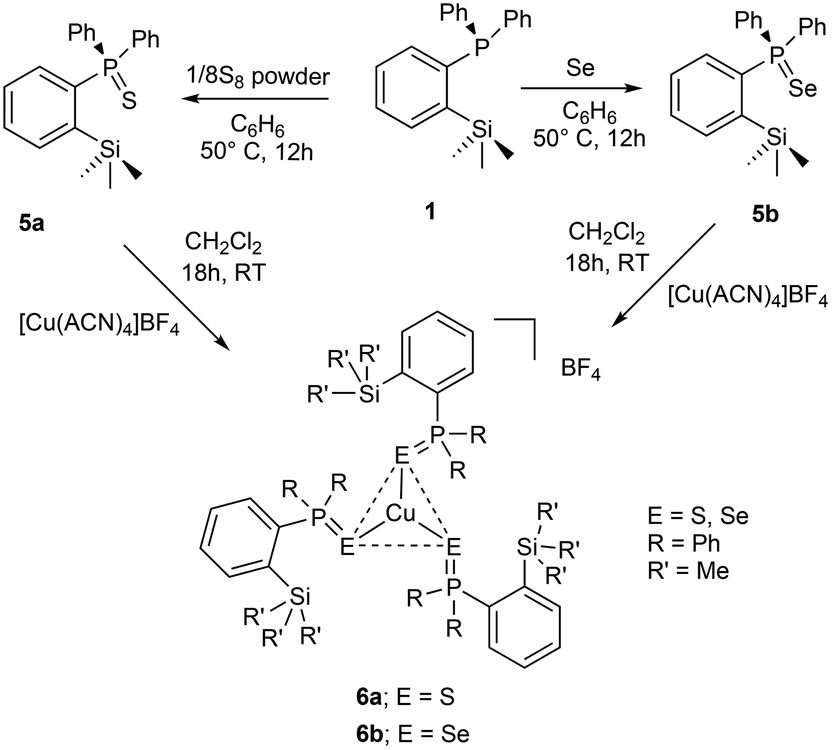 | ||
| Scheme 3 Preparation of ortho-silylated phosphinochalcogenides and their cationic copper(I) complexes (ACN = CH3CN). | ||
Furthermore, the reactions of 5a/5b with [Cu(CH3CN)4]BF4 in a 3:1 ratio in CH2Cl2 at RT cleanly furnished the corresponding homoleptic and mono-cationic tris(phosphinochalcogenide)copper complexes of the type [(5a/5b)3Cu]BF4 in 85% (6a) and 79% (6b) isolated yields as stable solids (Scheme 3). The complexes 6a/6b are soluble in polar solvents such as THF, CH2Cl2, and CHCl3 but are insoluble in nonpolar solvents. The 1H NMR spectra of 6a/6b in CDCl3 showed considerably up-field shifted (in comparison with the free ligands) singlet signals at δ = 0.09 and δ = 0.06 ppm, which can be assigned to the SiMe3 groups. The corresponding 13C NMR chemical shifts observed at δ = 2.74 (6a) and 2.97 (6b) can be assigned to the CH3 carbons. The 31P NMR spectra of 6a and 6b showed sharp singlets at 46.3 and 34.5 ppm, respectively. Furthermore, the CHNS data support the predicted composition of these complexes consisting of three phosphine chalcogenide ligands.32 Single crystals suitable for XRD measurements of 6a were grown by the vapor diffusion method from CH2Cl2 and n-hexane solution at RT. The molecular structure (Fig. 6) indicates the formation of a tri-coordinated and mono-cationic copper(I) complex, with C3 symmetry in the molecule. The sum of the bond angles 119.42° and 106.93° corresponding to S–Cu–S and Cu–S–P, respectively, indicates the presence of a trigonal planar geometry around the copper center. Furthermore, the average copper–sulfur (S–Cu 2.254 Å) and phosphorus–sulfur bond distances (P–S 1.990 Å) fall in the range reported for three-coordinate copper(I) complexes featuring tertiary phosphine sulfides.32,33
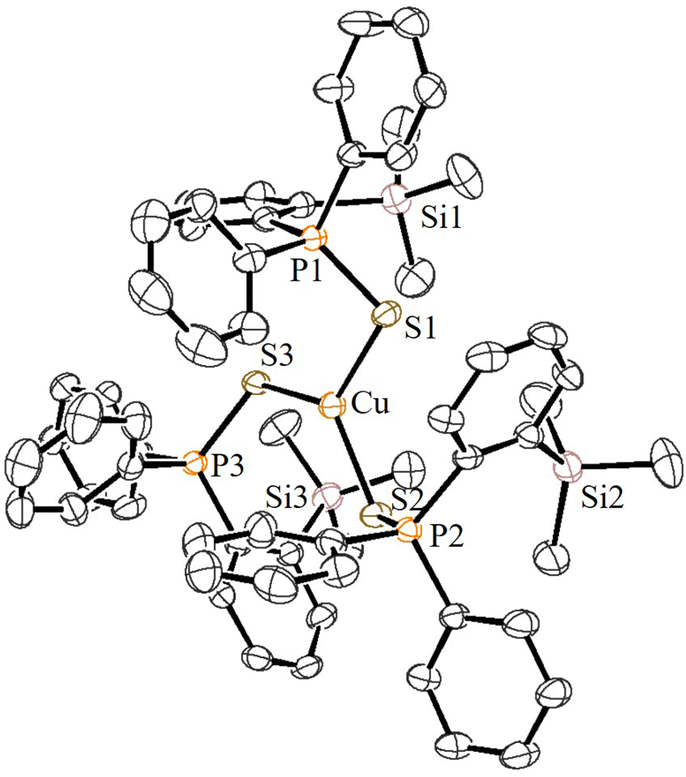 | ||
| Fig. 6 ORTEP view of the cationic part of 6a with thermal displacement parameters drawn at 30% probability. Hydrogen atoms are omitted for clarity. Important bond lengths [Å] and angles [°]: P1–S1 1.990(2), P2–S2 1.990(2), P3–S3 1.990(2), S1–Cu 2.2538(17), S2–Cu 2.2538(17), S3–Cu 2.2538(17), P1–Si1 3.690, P2–Si2 3.690, P3–Si3 3.690, S1–Si1 3.752, S2–Si2 3.752, S3–Si3 3.752, P1–S1–Cu 106.92(10), P2–S2–Cu 106.92(10), P3–S3–Cu 106.92(10), S1–Cu–S2 119.42(2), S2–Cu–S3 119.42(2), and S3–Cu–S1 119.42(2). | ||
Catalytic studies of cationic copper(I) complexes (2, 3 and 6a) in azide–alkyne cycloaddition reactions
To our knowledge, the transition metal complexes featuring ligands of type-II (Chart 1) in their mono-coordination fashion have rarely been explored in homogeneous catalysis as ancillary ligands. Recently, Si–H bond-containing type II (R′ = Me, R′′ = H) phosphine ligand-supported metal complexes such as phosphinobenzylsilane–ruthenium,34 iron,13,14 cobalt,4,35 rhodium,36 and iridium17 have been employed as molecular catalysts in various organic transformations. With this motivation and the newly synthesized copper(I) complexes in hand, we chose to study the catalytic performance of [(1)CuCl]2 (2), [(1)2Cu(CH3CN)]BF4 (3) and [(5a)3Cu]BF4 (6a) in azide–alkyne cycloaddition reactions.37,38 We first tested a series of reactions under various experimental conditions, such as by employing different solvents (CH3CN, and CH2Cl2), temperatures, and catalyst loadings together with various other commercially available copper(I) complexes (Table 1).|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions yield of productb (%) | |
| a Reaction conditions: azide (0.1 mmol), alkyne (0.12 mmol), cat. 3 (2 mol%), neat conditions, RT, 4 h. b isolated yields. | ||
| 1 | None | 99 |
| 2 | Dichloromethane as reaction solvent | 85 |
| 3 | MeCN as a reaction solvent | 79 |
| 4 | 1 mol% cat. 3 | 89 |
| 5 | 0.5 mol% cat. 3 | 81 |
| 6 | Alkyne 1 equiv. | 90 |
| 7 | Alkyne 1.5 equiv | 98 |
| 8 | 2 mol% complex 2 | 89 |
| 9 | 2 mol% (PPh3)CuCI as catalyst | 88 |
| 10 | 2 mol% CuCI | 70 |
| 11 | 2 mol% CuI | 79 |
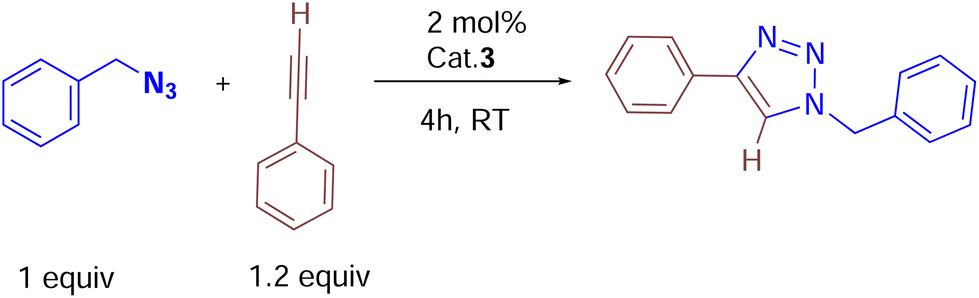
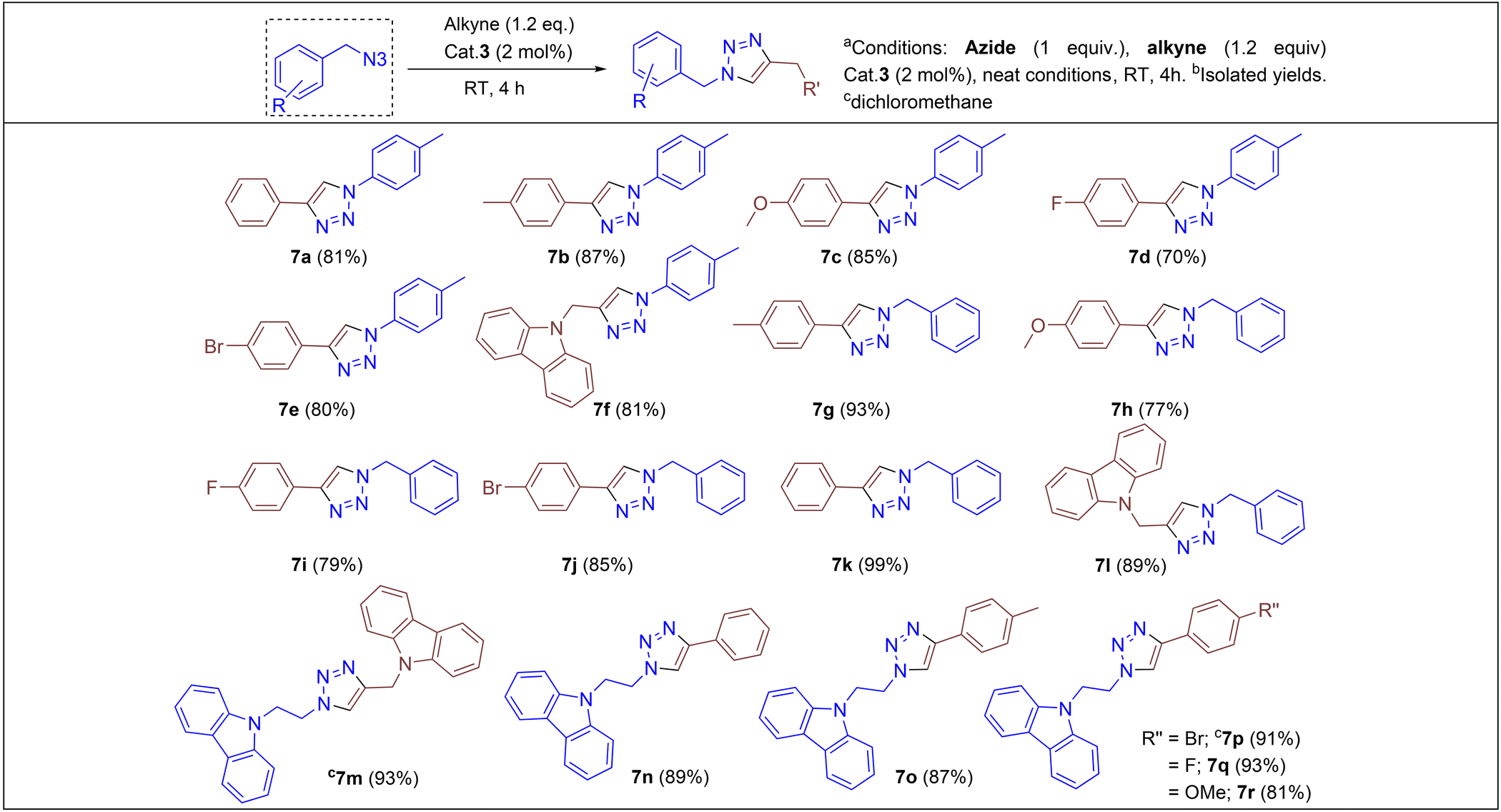
We began by screening 2 and 3 for their ability to synthesize various triazoles. Under neat solvent-free conditions (entry 1), the reaction of benzyl azide with phenyl acetylene and 2 mol% 3 at room temperature produced the corresponding triazole 7k in a 99% isolated yield (Table 2), while lower catalyst loadings (entry 5; 0.5 mol%) showed lower yields. Furthermore, catalyst 3 showed better conversion rates (99% entry 1) within 4 h than the neutral complex 2 (89%, entry 8), showing the superiority of cationic complex 3 over the neutral complex 2. In addition, under the same optimized experimental conditions, we screened other commercially available copper(I) halides by keeping complex 3 as the reference. For instance, as shown in entries 10 and 11, ligand-free copper halides furnished moderate yields; however, [Ph3PCuCl] gave relatively higher yields, indicating the requirement of ligand-supported cationic copper systems (Table 1). The polar solvents like CH3CN and CH2Cl2 provided good yields, but the majority of the reactions furnished excellent yields under solvent-free conditions. It should be noted that based on the physical state of substrates, in some cases, (carbazolyl triazole as well as bis-triazole derivatives) dichloromethane was employed as the reaction medium.
|
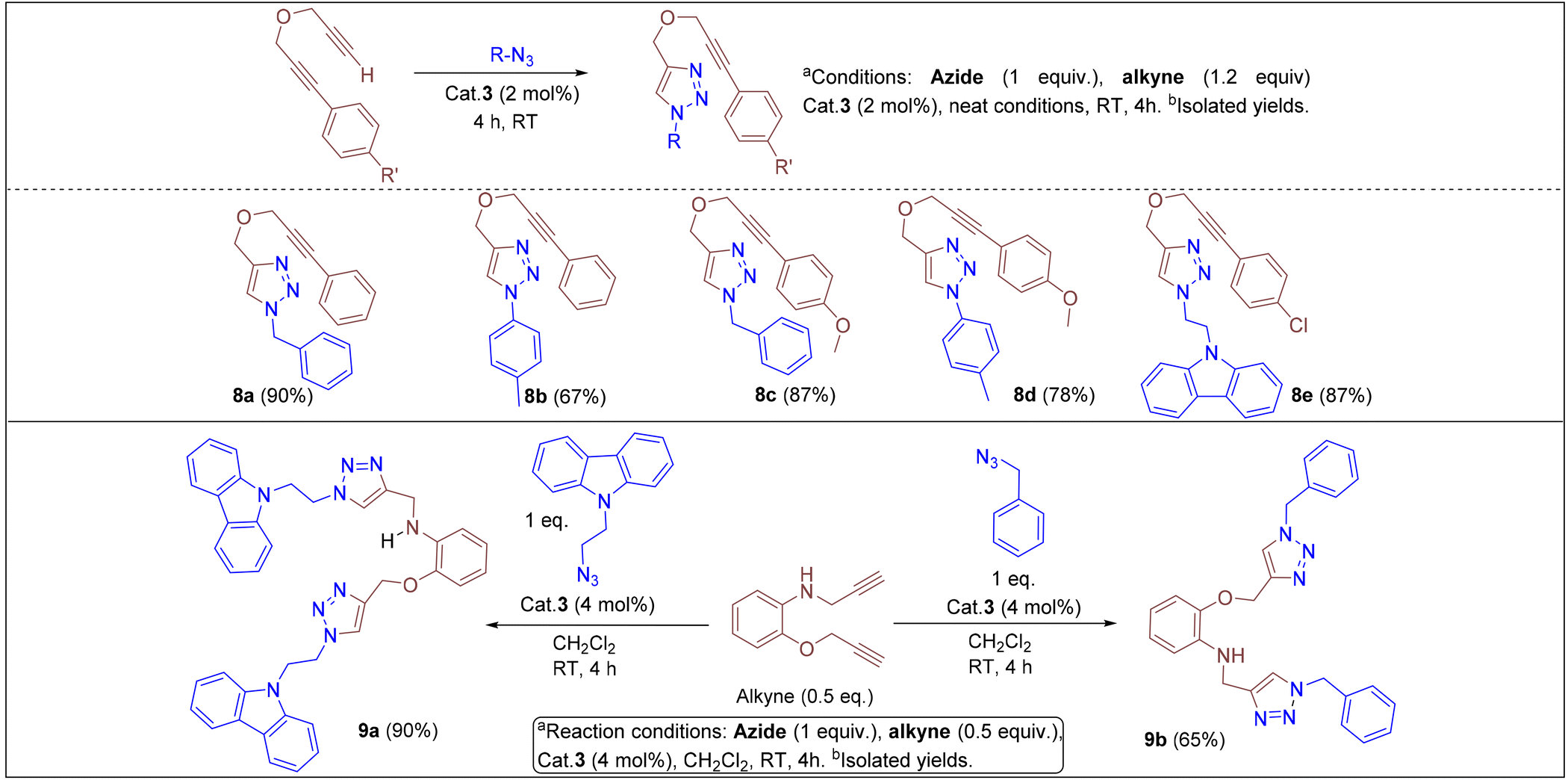
Under the optimized conditions, a series of three different classes of triazole derivatives (7, Table 2; 8 and 9, Table 3) were obtained. The first set of triazoles 7a–7r was achieved in 70–99% isolated yields. In the second set of five triazoles, 8a–8e39 bearing alkyl-phenyl propargylic ether groups were prepared by employing a series of azides (benzyl azide, tolylazide, and carbazolyl azides) and terminal-internal alkynes as coupling partners (Table 3). It should be noted that, triazoles 8a–8d are yellow viscous compounds, whereas 8e is solid in nature at room temperature. The molecular compositions of triazole derivatives 8 were assigned by 1H, 13C NMR, and HRMS analysis (Fig. S129–145†). The presence of propargylic ether groups in triazoles 8 could be observed in the range of chemical shifts δ = 83–89 ppm in their 13C NMR spectra (in CDCl3) in addition to their aromatic carbons. Additionally, the solid-state molecular structure of the triazole 8e was established. Single crystals suitable for XRD measurements were grown by slow evaporation of the dichloromethane solution at RT. Compound 8e crystallizes in the monoclinic system, and its structure clearly displays the presence of carbazolyl and phenyl propargylic ether groups at the 1 and 4-positions of the triazole (Fig. 7).
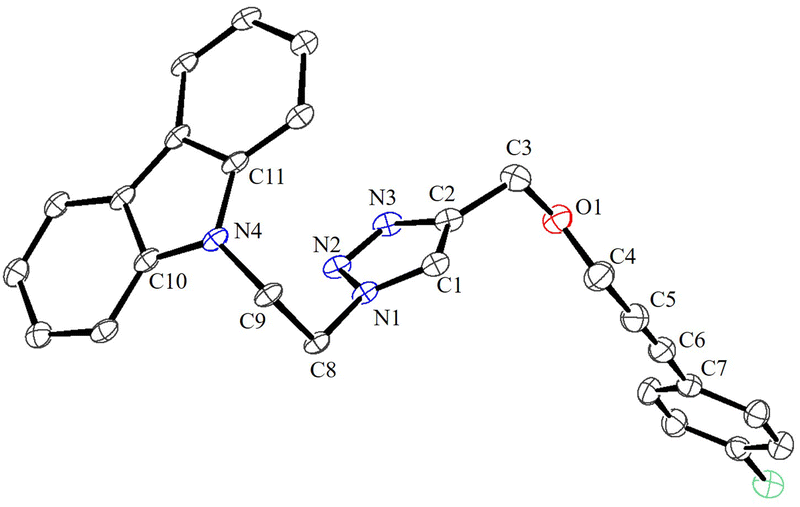 | ||
| Fig. 7 ORTEP view of 8e with thermal displacement parameters drawn at 30% probability. Hydrogen atoms are omitted for clarity. Important bond lengths [Å] and bond angles [°]: N1–C1 1.448 (4), C1–C2 1.528 (4), C2–N2 1.457 (4), O–C5 1.429 (4), O–C6 1.417 (5), and C6–C7 1.456 (6). | ||
|
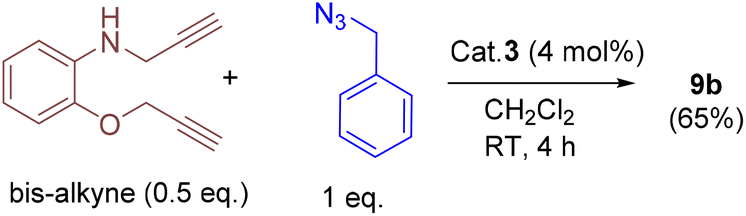
In addition, we tested the catalytic performance of 3 in the cyclization reactions of bis(terminal)alkyne with carbazolyl and benzyl azides (Table 3). Starting from the bis-alkyne, the new bis-triazoles 9a (90%) and 9b (65%) could be selectively achieved by employing 4 mol% catalyst loadings in dichloromethane at room temperature. The 1:2 reaction of bis(alkyne) and carbazolyl azide in the presence of 4 mol% catalyst showed a brown precipitate formation within 10 min; this precipitate further turned into a viscous yellow substance. Washing with CH2Cl2 yielded 9a as a pale-brown solid in 90% isolated yield without any further purification steps. Interestingly, 9a could also be obtained in 77% isolated yield within 10 min, indicating the efficient catalytic performance of 3 (Scheme S4†); however, for achieving further higher yields, it was stirred for 4 h. 9a was insoluble in CH2Cl2 and CHCl3, but had minimal solubility in ethanol and methanol. 9a was fully characterized by 1H and 13C NMR and by HMRS analytical data. A signal at m/z = 658.3066 in the HRMS spectrum can be assigned to [M + H]+ calcd for 658.3043 (C40H36N9O).
We have also examined the catalytic efficiency of silyl-phosphine sulfide ligand (5a) supported cationic complex 6a in the azide–alkyne cycloaddition reactions. Interestingly, 2 mol% catalyst loading of [(5a)3Cu]BF4 can also catalyze the cycloaddition of benzyl azide with phenylacetylene; by this reaction, we could achieve triazole 7k in 92% isolated yield as a clean product (Scheme S6, and Fig. S155†). This reaction shows the potential applications of silyl group-functionalized phosphine sulfide systems as ancillary ligands in homogeneous catalysis. For a detailed understanding of the reaction mechanism, a series of supporting experiments (Scheme S1–3†) were performed. As shown in Fig. S64,† the 1:1:1 reaction of benzyl azide, phenylacetylene, and cat. 3 in an NMR tube clearly showed the progression of the reaction toward the anticipated cyclization reaction to give the triazole derivative. In the presence of cat. 3, the a and b (Scheme S3†) protons of the corresponding azide and phenyl acetylenes vanish during the course of the reaction to selectively afford triazole 7k with complete conversion. Noteworthily, the catalytic efficiency of the reported complex is comparable to that of a series of other phosphine,40 phosphinite and phosphonite41 ligand-supported copper(I) systems; however, with cat. 3, triazoles consisting of internal alkyne groups could be achieved selectively.
For further understanding of the homogeneity of the catalytic reaction with cat. 3, we have performed a series of mercury drop experiments (Scheme S7†) in 2:150 mol% of cat. 3 to Hg metal. Four different substrates were screened, and no significant changes in the overall isolated yields were observed, which indicates the homogeneous pathway of the reported catalytic cycle.
Azide–alkyne cycloaddition reactions catalysed by copper(I) catalysts have been widely studied, and numerous studies extensively investigated their mechanistic pathways both by theoretical and experimental methods.38,42,43 Accordingly, a plausible mechanism could be derived as depicted in Fig. 8. The bis(phosphine) copper complex 3 would be transformed into acetylide A by releasing one phosphine ligand, which then coordinates with azide to form intermediate B. It could then further rearrange to a six-membered metallacycle intermediate C.44 Intermediate C can further rearrange to form ortho-silylated phosphine containing copper(I) triazolide intermediate D. Furthermore, proteolysis of D releases the corresponding triazole product by regenerating the catalyst. For further detection of intermediate C, two reactions with different substrates were performed, and they were analysed by the in situ ESI-MS technique at different intervals of the reaction course.
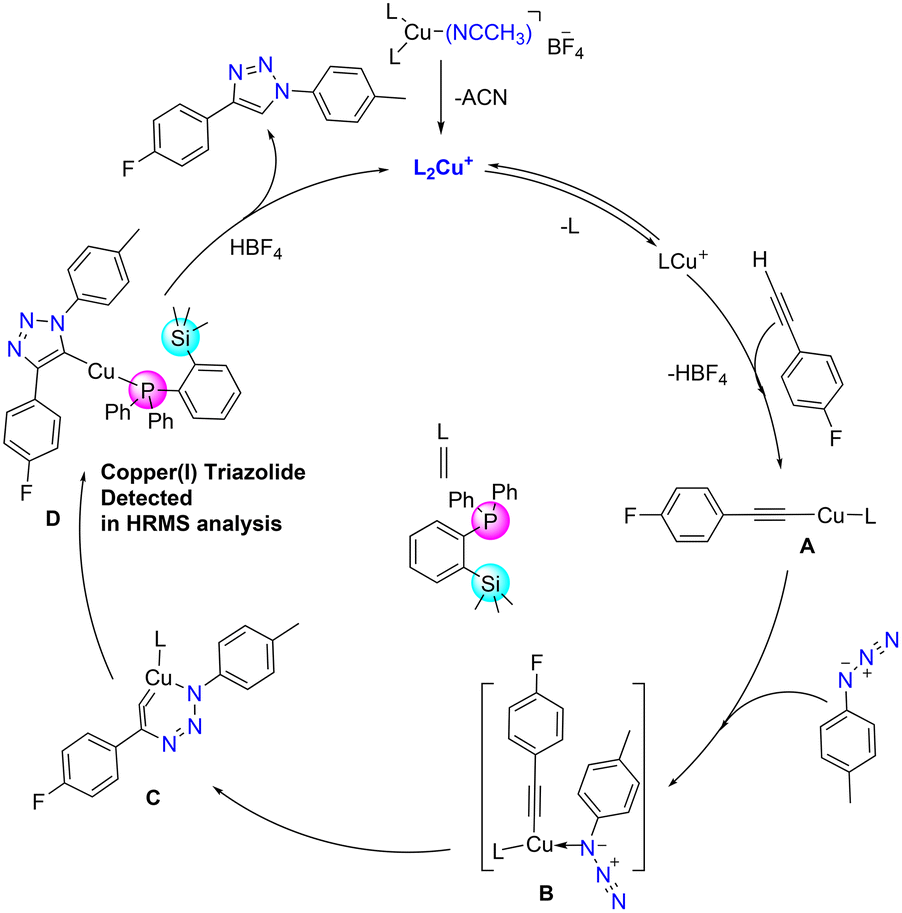 | ||
| Fig. 8 Proposed reaction mechanism for the formation of 1,4-substituted triazole derivatives catalyzed by cat. 3. | ||
The HRMS spectrum showed the presence of the copper(I) triazolide intermediate [(triazolide)Cu(1)] consisting of the meso-ionic carbene45,46 and the phosphine 1 as shown in Fig. S62† (exp. 650.1650 [M + H]+, calcd 650.1618 for C36H35CuFN3PSi) and Fig. S63† (exp. 632.1751 [M + H]+, calcd 632.1712 for C36H36CuN3PSi). Several attempts to isolate these intermediates resulted in the formation of the corresponding triazoles, indicating their highly reactive nature. It should be noted that Straub and co-workers isolated heteroleptic copper(I)triazolide (click intermediate) for the first time and also established its molecular structure.43,46 In addition to the above catalytic reactions by conventional methods, cat. 3 was also employed in the mechanical grinding method using 2 mol% catalyst loading (Scheme S5†). Under solvent-free conditions, azide/alkyne/catalysts were mixed and mortared for 5 min for the isolation of pure triazole 7k in 86% yield (Fig. S67†).
Conclusions
In summary, a series of neutral and mono-cationic complexes of copper(I) and silver(I) bearing a sterically bulky diphenyl(2-(trimethylsilyl)phenyl)phosphine ligand were synthesized in excellent yields and structurally characterized. In the case of silver complexes, the solid-state molecular structures show the presence of linear and nonlinear P–M–P (M = Ag) units depending on the counter anions and their interaction with the cationic central metal atoms. The ortho-silylated phosphine was conveniently converted to its sulfide and selenides, and their cationic homoleptic copper(I) complexes of the type [(5a/5b)3Cu]BF4 were isolated. The latter phosphine sulfide complex displayed a trigonal planar environment around the tricoordinate copper center.The catalytic performance of 2, 3 and 6a was tested in the azide–alkyne cycloaddition reactions (CuAAC); among them, complex 3 displayed high catalytic activity for the preparation of three sets of triazoles which were achieved in good to excellent yields under very mild conditions. Cat. 3 displayed high selectivity towards terminal alkynes over internal alkyne groups. The present work showed the formation of the click intermediate [(triazolide)Cu(phosphine)], indicating the presence of silylated phosphine-bound copper species during the catalytic reaction. Overall, the ortho-silylated phosphine and its sulfide derivatives were first time introduced as potential ancillary ligands in homogeneous catalysis.
Experimental section
Materials and methods
All the reactions were performed under a strictly dry argon/nitrogen atmosphere using the standard Schlenk line technique. Freshly distilled solvents (CH3CN, THF, CH2Cl2, n-hexane, diethyl ether, and ethylacetate) were used for the reactions reported in this study. All the chemical shifts are reported in ppm and spectra are referenced to the corresponding residual solvent peaks. Elemental analysis was measured on an Elimentar instrument. The NMR spectra (1H, 13C, 31P, 19F, and 11B NMR) were obtained using Bruker Avance 400 and 700 MHz NMR spectrometers. The ESI mass spectral data were measured using a Waters Xevo G2-XS QTof instrument in CH3OH or CH3CN solvents. The required compound [Cu(CH3CN)4]BF4 was purchased from commercial sources. The phosphine-copper complex ClCu(PPh3) was prepared according to a reported procedure.47Crystallography
Single crystals of 1–3, 4a, 4b, 4c, 6a and 8e were mounted on a nylon loop under a microscope using a paraffin bar, and X-ray data were collected on the Rigaku Super Nova fine-focused dual diffractometer, with CuKα radiation (λ = 1.54178 Å) and MoKα radiation (0.71073 Å) equipped with a PILATUS200K. Using Olex2, the structures were solved with the SHELXS structure solution program using direct methods and refined with the SHELXL refinement package. The CCDC numbers of the compounds reported in this study are: 2264786 (1), 2264791 (2), 2264787 (3), 2264784 (4a), 2264789 (4b), 2264788 (4c), 2264785 (6a), and 2264790 (8e).†Synthesis of the reported compounds 1–9
Catalytic reactions and general experimental procedures for substituted triazoles
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 85.5 (CC), 62.5 (OCH2), 57.5 (OCH2), 43.4 (NCH2CH2N), and 31.7 (NCH2CH2N). Mass spectral data (HRMS, CH3CN, m/z): found 441.1461; calcd for C26H22ClN4O; 441.1482 [M + H]+. IR (ATR mode, cm−1): 1599, 1485, 1451, 1330, 1222, 1073, 921, 823, 748, 718, and 524.
C), 85.5 (CC), 62.5 (OCH2), 57.5 (OCH2), 43.4 (NCH2CH2N), and 31.7 (NCH2CH2N). Mass spectral data (HRMS, CH3CN, m/z): found 441.1461; calcd for C26H22ClN4O; 441.1482 [M + H]+. IR (ATR mode, cm−1): 1599, 1485, 1451, 1330, 1222, 1073, 921, 823, 748, 718, and 524.
A 25 mL Schlenk tube was charged with carbazolyl azide (2 equiv.), bis-terminal alkyne (1 equiv.) and cat. 3 (4 mol%) under a nitrogen atmosphere. To this solid mixture, CH2Cl2 was added and stirred for 4 h at RT. A white precipitate formed was filtered and washed with CH2Cl2 (2 × 3 mL), followed by n-hexane (2 × 3 mL). The resulting solid was dried under vacuum to afford 9a as a pale brown solid. Yield: 75 mg (90%). 1H NMR (400 MHz, DMSO-d6): δ = 8.10 (d, J = 8.0 Hz, 5H), 7.77 (s, 1H), 7.34 (m, J = 18.5, 8H), 7.16 (m, 4H), 6.87 (d, J = 7.7 Hz, 1H), 6.73 (t, J = 7.4 Hz, 1H), 6.52 (t, J = 7.4 Hz, 1H), 6.43 (d, J = 7.5 Hz, 1H), 4.98 (s, 3H), 4.90–4.66 (m, 8H), and 4.19 (d, J = 4.5 Hz, 2H). 13C NMR (101 MHz, DMSO-d6): δ = 145.1, 143.1, 139.8, 137.6, 125.7, 125.61, 124.9, 122.2, 122.1, 121.5, 120.2, 120.16, 119.1, 119.0, 115.9, 111.3, 109.7, 108.8, 108.7, 61.5 (OCH2), 48.4 (NCH2CH2), 48.4 (NCH2CH2), 42.9 (NCH2CH2), 42.8 (NCH2CH2), and 38.5 (HNCH2−). Melting point: 211 °C (decomposed to black solid). HRMS (MeOH, m/z): 658.3066; calcd for C40H36N9O; 658.3043 [M + H]+. IR (ATR mode, cm−1): 3051, 1598, 1516, 1452, 1331, 1214, 1130, 1046, 1016, 847, 745, 666, 615, and 561.

To a Schlenk tube containing benzyl azide (2 equiv.), bis-terminal alkyne (1 equiv.), and cat. 3 (4 mol%) was added CH2Cl2 (1 mL) and the mixture was stirred for 4 h at RT. The solvent was removed under reduced pressure, and it was further purified by flash column (silica gel) chromatography using ethyl acetate (15 mL). After the removal of the solvent, the residue was washed again with n-hexane (4 mL), and dried under vacuum to afford 9b as a yellow oily substance. Yield: 44 mg (65%). 1H NMR (400 MHz, CDCl3): δ = 7.53 (s, 1H), 7.33 (m, 9H, ArH), 7.24–7.20 (m, 3H, ArH), 6.94–6.81 (m, 2H, ArH), 6.64 (dd, J = 7.9 Hz, 2H, ArH), 5.51 (s, 2H, BnCH2), 5.46 (s, 2H, BnCH2), 5.18 (s, 2H, OCH2), and 4.42 (s, 2H, HNCH2). 13C NMR (101 MHz, CDCl3): δ = 147.1, 145.8, 144.6, 138.1, 134.8, 134.6, 129.3, 128.8, 128.9, 128.8, 128.2, 128.1, 123.0, 122.3, 121.8, 117.3, 112.1, 110.96, 62.8 (OCH2), 54.4 (BnCH2), 54.3 (BnCH2), and 39.9 (HNCH2). HRMS (ESI, MeOH, m/z): 452.2208; calcd for C26H26N7O: 452.2199 [M + H]+. IR (ATR mode, cm−1): 3052, 1600, 1511, 1449, 1334, 1262, 1208, 1122, 1048, 900, 847, 805, and 732.
Author contributions
AD supervised the project work. AKS prepared the metal complexes and carried out all the experiments. BD helped in substrate synthesis. All authors contributed to writing the manuscript. SJP and CP performed the X-ray diffraction analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AD thanks IISER Berhampur for the CAIF facility and SERB for the Start-Up Research Grant (SRG/2020/000216), GOI for providing financial assistance. AKS, BD and AKS thank IISER Berhampur for PhD and DST Inspire fellowships respectively.References
- (a) H. Shet, U. Parmar, S. Bhilare and A. R. Kapdi, Org. Chem. Front., 2021, 8, 1599–1656 RSC; (b) K. Izod, Coord. Chem. Rev., 2013, 257, 924–945 CrossRef CAS; (c) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; (d) F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840–1843 CrossRef PubMed.
- G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC.
- T. Komuro, Y. Nakajima, J. Takaya and H. Hashimoto, Coord. Chem. Rev., 2022, 473, 214837 CrossRef CAS.
- L. Denker, D. Wullschläger, J. P. Martínez, S. Świerczewski, B. Trzaskowski, M. Tamm and R. Frank, ACS Catal., 2023, 13, 2586–2600 CrossRef CAS.
- P. Gualco, S. Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2011, 50, 8320–8324 CrossRef CAS PubMed.
- A. A. Omaña, R. K. Green, R. Kobayashi, Y. He, E. R. Antoniuk, M. J. Ferguson, Y. Zhou, J. G. C. Veinot, T. Iwamoto, A. Brown and E. Rivard, Angew. Chem., 2021, 133, 230–233 CrossRef.
- C. A. Theulier, Y. García-Rodeja, S. Mallet-Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Organometallics, 2021, 40, 2409–2414 CrossRef CAS.
- C. A. Theulier, Y. García-Rodeja, N. Saffon-Merceron, K. Miqueu, G. Bouhadir and D. Bourissou, Chem. Commun., 2021, 57, 347–350 RSC.
- D. Quintard, M. Keller and B. Breit, Synthesis, 2004, 905–908 CAS.
- A. Kawachi, T. Yoshioka and Y. Yamamoto, Organometallics, 2006, 25, 2390–2393 CrossRef CAS.
- J. Wen, B. Dong, J. Zhu, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2020, 59, 10909–10912 CrossRef CAS PubMed.
- (a) R. Imayoshi, K. Nakajima, J. Takaya, N. Iwasawa and Y. Nishibayashi, Eur. J. Inorg. Chem., 2017, 2017, 3769–3778 CrossRef CAS; (b) F. Montilla, A. Galindo, V. Rosa and T. Aviles, Dalton Trans., 2004, 2588–2592 RSC.
- G. Chang, P. Zhang, W. Yang, Y. Dong, S. Xie, H. Sun, X. Li, O. Fuhr and D. Fenske, Dalton Trans., 2021, 50, 17594–17602 RSC.
- S. Ren, S. Xie, T. Zheng, Y. Wang, S. Xu, B. Xue, X. Li, H. Sun, O. Fuhr and D. Fenske, Dalton Trans., 2018, 47, 4352–4359 RSC.
- X.-L. Pei, A. Pereira, E. S. Smirnova and A. M. Echavarren, Chem. – Eur. J., 2020, 26, 7309–7313 CrossRef CAS PubMed.
- (a) R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS; (b) M. Boudjelel, E. D. Sosa Carrizo, S. Mallet-Ladeira, S. Massou, K. Miqueu, G. Bouhadir and D. Bourissou, ACS Catal., 2018, 8, 4459–4464 CrossRef CAS; (c) M. Boudjelel, S. Mallet-Ladeira, G. Bouhadir and D. Bourissou, Chem.Comm., 2019, 55, 12837–12840 RSC.
- B. Ghaffari, S. M. Preshlock, D. L. Plattner, R. J. Staples, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2014, 136, 14345–14348 CrossRef CAS PubMed.
- (a) H. Kameo, J. Yamamoto, A. Asada, H. Nakazawa, H. Matsuzaka and D. Bourissou, Angew. Chem., Int. Ed., 2019, 58, 18783–18787 CrossRef CAS PubMed; (b) F. Ritter, L. John, T. Schindler, J. P. Schroers, S. Teeuwen and M. E. Tauchert, Chem. – Eur. J., 2020, 26, 13436–13444 CrossRef CAS PubMed.
- (a) A. Marker and M. J. Gunter, J. Magn. Reson., 1982, 47, 118–132 CAS; (b) R. J. Bowen, M. Navarro, A.-M. J. Shearwood, P. C. Healy, B. W. Skelton, A. Filipovska and S. J. Berners-Price, Dalton Trans., 2009, 10861–10870 RSC; (c) J. V. Hanna, S. E. Boyd, P. C. Healy, G. A. Bowmaker, B. W. Skelton and A. H. White, Dalton Trans., 2005, 2547–2556 RSC.
- S. M. Socol, R. A. Jacobson and J. G. Verkade, Inorg. Chem., 1984, 23, 88–94 CrossRef CAS.
- S. M. Socol and J. G. Verkade, Inorg. Chem., 1984, 23, 3487–3493 CrossRef CAS.
- R. E. Bachman and D. F. Andretta, Inorg. Chem., 1998, 37, 5657–5663 CrossRef CAS PubMed.
- A. Doddi, D. Bockfeld, A. Nasr, T. Bannenberg, P. G. Jones and M. Tamm, Chem. – Eur. J., 2015, 21, 16178–16189 CrossRef CAS PubMed.
- C. Zovko, S. Bestgen, C. Schoo, A. Görner, J. M. Goicoechea and P. W. Roesky, Chem. – Eur. J., 2020, 26, 13191–13202 CrossRef CAS PubMed.
- A. Doddi, M. Peters, D. Bockfeld and M. Tamm, Z. Anorg. Allg. Chem., 2023, 649, e202200364 CrossRef CAS.
- M. R. Churchill and K. L. Kalra, J. Am. Chem. Soc., 1973, 95, 5772–5773 CrossRef CAS.
- A. Bayler, A. Schier, G. A. Bowmaker and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 7006–7007 CrossRef CAS.
- M. Altaf and H. Stoeckli-Evans, Polyhedron, 2010, 29, 701–708 CrossRef CAS.
- M. Nazish, H. Bai, C. M. Legendre, R. Herbst-Irmer, L. Zhao, D. Stalke and H. W. Roesky, Chem. Commun., 2022, 58, 12704–12707 RSC.
- (a) A. J. Blake, N. R. Brooks, N. R. Champness, J. W. Cunningham, P. Hubberstey and M. Schroeder, CrystEngComm, 2000, 2, 41–45 RSC; (b) L. Poorters, D. Armspach, D. Matt, L. Toupet and P. G. Jones, Angew. Chem., Int. Ed., 2007, 46, 2663–2665 CrossRef CAS PubMed.
- R. Kumar, S. Kumar, M. K. Pandey, V. S. Kashid, L. Radhakrishna and M. S. Balakrishna, Eur. J. Inorg. Chem., 2018, 2018, 1028–1037 CrossRef CAS.
- H. Wu, Y. Qu, C. Wang, Y. Wu and K. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 195, 88–95 CrossRef CAS.
- W. E. Slinkard and D. W. Meek, Inorg. Chem., 1969, 8, 1811–1816 CrossRef CAS.
- V. Montiel-Palma, M. A. Muñoz-Hernández, T. Ayed, J.-C. Barthelat, M. Grellier, L. Vendier and S. Sabo-Etienne, Chem. Commun., 2007, 3963–3965 RSC.
- S. Xu, P. Zhang, X. Li, B. Xue, H. Sun, O. Fuhr and D. Fenske, Chem. – Asian J., 2017, 12, 1234–1239 CrossRef CAS PubMed.
- U. Prieto-Pascual, A. Rodríguez-Diéguez, Z. Freixa and M. A. Huertos, Inorg. Chem., 2023, 62, 3095–3105 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- B. Das, A. K. Sahoo, S. K. Banjare, S. J. Panda, C. S. Purohit and A. Doddi, Dalton Trans., 2023 10.1039/D3DT01989F.
- D. Wang, N. Li, M. Zhao, W. Shi, C. Ma and B. Chen, Green Chem., 2010, 12, 2120–2123 RSC.
- S. Lal, J. McNally, A. J. P. White and S. Díez-González, Organometallics, 2011, 30, 6225–6232 CrossRef CAS.
- (a) L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef PubMed; (b) Y.-C. Lin, Y.-J. Chen, T.-Y. Shih, Y.-H. Chen, Y.-C. Lai, M. Y. Chiang, G. C. Senadi, H.-Y. Chen and H.-Y. Chen, Organometallics, 2019, 38, 223–230 CrossRef CAS.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- C. Nolte, P. Mayer and B. F. Straub, Angew. Chem., Int. Ed., 2007, 46, 2101–2103 CrossRef CAS PubMed.
- E. Fossum, Z. Yu and L.-S. Tan, ARKIVOC, 2010, 2009, 255–265 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and spectral data. CCDC 2264786 (1), 2264791 (2), 2264787(3), 2264784 (4a), 2264789 (4b), 2264788 (4c), 2264785 (6a) and 2264790 (8e). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01692g |
| This journal is © The Royal Society of Chemistry 2023 |