
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycopolymers against pathogen infection
Ulla I. M.
Gerling-Driessen
 a,
Miriam
Hoffmann
a,
Stephan
Schmidt
ab,
Nicole L.
Snyder
c and
Laura
Hartmann
*a
a,
Miriam
Hoffmann
a,
Stephan
Schmidt
ab,
Nicole L.
Snyder
c and
Laura
Hartmann
*a
aInstitute of Organic Chemistry and Macromolecular Chemistry, Heinrich-Heine-University Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany. E-mail: laura.hartmann@hhu.de
bInstitute for Macromolecular Chemistry, University of Freiburg, Stefan-Meier-Str. 31, 79104 Freiburg, Germany
cDepartment of Chemistry, Davidson College, Davidson, North Carolina 28035, USA
First published on 23rd February 2023
Abstract
Pathogens including viruses, bacteria, fungi, and parasites continue to shape our lives in profound ways every day. As we have learned to live in parallel with pathogens, we have gained a better understanding of the rules of engagement for how they bind, adhere, and invade host cells. One such mechanism involves the exploitation of host cell surface glycans for attachment/adhesion, one of the first steps of infection. This knowledge has led to the development of glycan-based diagnostics and therapeutics for the treatment and prevention of infection. One class of compounds that has become increasingly important are the glycopolymers. Glycopolymers are macromolecules composed of a synthetic scaffold presenting carbohydrates as side chain motifs. Glycopolymers are particularly attractive because their properties can be tuned by careful choice of the scaffold, carbohydrate/glycan, and overall presentation. In this review, we highlight studies over the past ten years that have examined the role of glycopolymers in pathogen adhesion and host cell infection, biofilm formation and removal, and drug delivery with the aim of examining the direct effects of these macromolecules on pathogen engagement. In addition, we also examine the role of glycopolymers as diagnostics for the detection and monitoring of pathogens.
 Ulla I. M. Gerling-Driessen | Ulla I. M. Gerling-Driessen studied Chemistry at Freie Universitaet Berlin, Germany. Working in the group of Prof. Beate Koksch at the Department of Organic Chemistry, she obtained her PhD in 2013. After a postdoc, working with Prof. Rainer Haag and Prof. Beate Koksch, she joined the group of Prof. Carolyn Bertozzi at Stanford University in 2016 as a DFG-funded postdoctoral fellow. In 2021, she joined the Chair of Macromolecular Chemistry at the Heinrich Heine University in Duesseldorf, headed by Prof. Laura Hartmann. Her research is centered around the development of novel chemical tools to understand the pathology of aberrant protein glycosylation. |
 Miriam Hoffmann | Miriam Hoffmann received her M.Sc. degree in Chemistry in 2018 from the Heinrich-Heine-University in Düsseldorf, Germany. Her master's thesis, completed under the supervision of Prof. Dr Laura Hartmann in the Institute of Organic and Macromolecular Chemistry, focused on the synthesis of glycomacromolecule-functionalized liposomes. Her current doctoral work, which is also being completed under the direction of Prof. Dr Laura Hartmann, focuses on the synthesis and membrane presentation of sulfonated and sulfated glycomacromolecules. |
 Stephan Schmidt | Stephan Schmidt studied chemistry and polymer science in Berlin and Potsdam. After doctoral studies in Bayreuth, he returned to Potsdam as a postdoc at the Max-Planck-Institute of Colloids and Interfaces and the Fraunhofer IZI. He then started his independent research career at the Universität Leipzig before moving to Düsseldorf as an assistant professor in 2016. In 2022 he was awarded with a Heisenberg professorship of the DFG and relocated to Freiburg. He is interested in interactive polymer materials, in providing insights for their biomedical applications and the underlying molecular mechanism mainly using atomic force microscopy and other microscopy techniques. |
 Nicole L. Snyder | Nicole L. Snyder received B.S. degrees in Chemistry and Biology in 2000 from Westminster College, New Wilmington, Pennsylvania, United States, and her PhD in Chemistry in 2005 from the University of Connecticut in Storrs, Connecticut. She is currently a Professor of Chemistry at Davidson College in Davidson, North Carolina. Her research focuses on the preparation and characterization of glycoconjugates as tools for studying the role of carbohydrates in microbial infectivity, tumor metastasis and transformation, and biomolecule stability. |
 Laura Hartmann | Laura Hartmann studied chemistry in Cologne and Freiburg. She obtained her PhD in 2007 at the Max-Planck-Institute of Colloids and Interfaces. After a postdoc at Stanford University, she returned to the MPI as an independent Emmy Noether fellow. In 2014 she became a full professor for Macromolecular Chemistry at Heinrich-Heine-University Duesseldorf. In 2022, she accepted a new position at the Institute for Macromolecular Chemistry, University of Freiburg and will be moving there in 2023. Her research interests focus on the synthesis of biomimetic polymers with controlled monomer sequences and their use in biomedical applications, especially the fight against pathogen infections. |
Introduction
Pathogen adhesion is mediated by glycans and glycan conjugates
It is part of our daily life to deal with infections caused by pathogens such as bacteria and viruses. The ongoing COVID-19 pandemic has served to reinforce this. Yet, even before SARS-CoV-2, viruses drastically shaped human history and society – for example polio, the 1918 Influenza Pandemic or HIV.1 Other pathogens continue to shape our world. As more and more bacteria develop resistance to current antibiotic regimens, new drugs for treating bacterial infections must be found.2 One of the most important tools in the fight against infections are vaccines. However, some pathogens continue to remain difficult to vaccinate against, including HIV and malaria, in part due to the complexity of their cell surfaces and, in the case of HIV, a high mutation rate that makes it difficult to target. In some cases, there are pathogens for which we have effective vaccines e.g., influenza A virus (IAV) and SARS-CoV-2, but the mutation rate of these pathogens is so rapid that new therapeutics must be designed and developed annually. As such, we find ourselves in an arms race; as therapeutics are developed, pathogens evolve to circumvent their function.In lieu of vaccines, drugs have been developed that can either help to prevent an infection as part of a prophylactic treatment or serve as therapeutics to treat the infection to support immunological targeting, neutralizing and pathogen clearance. This is often achieved by targeting crucial functions of the pathogen, including its entry mechanism, metabolism, reproduction cycle, or structural stability. Pathogenesis is a complex process that includes multiple steps from the initial contact with the host cell (attachment) to the cellular uptake (invasion), reproduction, and release. Viruses but also other pathogens, including bacteria such as Chlamydiae, can only reproduce inside a host cell. Other pathogens, for example bacteria such as Escherichia coli, do not critically rely on cellular uptake, but they can use it to their advantage, e.g., for protection against the immune system. Pseudomonas aeruginosa or Staphylococcus aureus, two examples of hospital acquired bacteria that have become resistant to many first line antibiotics, do not enter cells but still require tissue or epithelial cell adhesion and use this adhesive process to form biofilms comprised of aggregates of multiple bacteria. Thus, for most pathogens, cell attachment is a prerequisite for successful infection and intervention of the attachment step therefore represents another suitable target to inhibit infections.
Because the attachment step is critical in the infection process, pathogens have evolved a variety of mechanisms to bind to the host cells. Upon initial contact, pathogens often form rather weak non-specific interactions with the host cell surface. This non-receptor-specific and reversible interaction is mainly based on the overall physicochemical properties of the pathogen and host cell surfaces, such as charge and hydrophobicity. Initial contact can then be followed by a more specific adhesion, which is typically mediated by interactions of distinct residues on surface molecules such as proteins, lectins or glycans.
Today, it is well understood that glycans, glycoconjugates, and polysaccharides are key classes of molecules mediating pathogen–cell attachment processes. Host cell surfaces are decorated with a dense layer of carbohydrate structures known as the glycocalyx, thus offering a multitude of potential interaction partners for the pathogen's cell contact (Fig. 1). Pathogens also carry carbohydrate structures on their surface e.g., in the form of glycosylated proteins. These can mediate the interaction with carbohydrate recognizing receptors e.g., on cells of the immune system such as dendritic cell-specific intercellular adhesion molecule-grabbing nonintegrin (DC-SIGN) receptors on macrophages and thereby promote the infection process.3 Additionally, extracellular glycans can play a role in pathogen cell attachment as well as the protection against it, such as the human milk oligosaccharides (HMOs), a class of structurally diverse carbohydrates produced in breast milk that are known for their antiadhesive properties.4
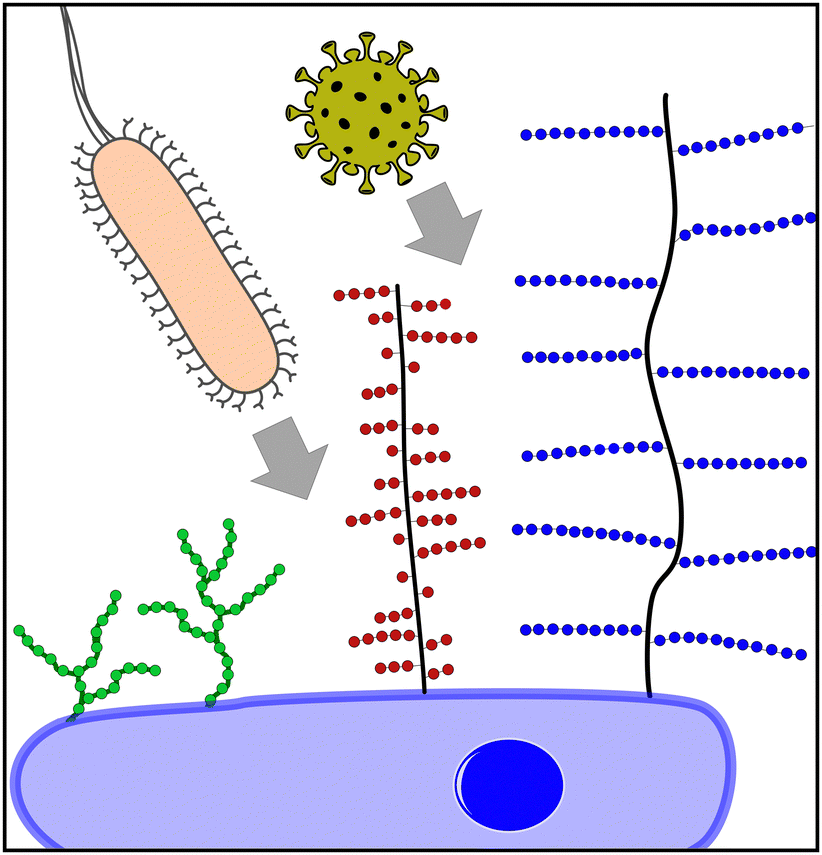 | ||
| Fig. 1 Pathogens using extracellular carbohydrate structures for host cell attachment as one of the first steps of the infection process. Three important classes are schematically depicted here: oligomannoside structures (green) e.g., binding bacteria such as E. coli, mucins (red) e.g., binding viruses such as IAV and proteoglycans (blue) e.g., binding viruses such as SARS-CoV-2. | ||
Focusing on pathogen interactions as mediated by the mammalian glycocalyx, larger glycoconjugates such as the proteoglycans5 or mucins6,7 are well known for their role in pathogen attachment. Mucins are high molecular weight, heavily glycosylated proteins presenting terminal sialic acids. It is well known that IAV can bind to sialic acids via hemagglutinin receptors. Interestingly, cell–surface mucins might serve as both attachment points to the host cell and thus promote infection, or as a steric barrier blocking virus–cell interactions due to their gel-like properties.8 Proteoglycans consist of a protein core with long polysaccharide brushes, consisting of glycosaminoglycan (GAG) structures such as heparan sulfate. A great variety of viruses, as well as bacteria, engage in proteoglycan binding through their interactions with GAGs. A recent example is the GAG-mediated cell attachment of SARS-CoV-2.9 Other glycan structures are also known to mediate pathogen interactions. For example, bacteria present pili or fimbriae,10 hair-like protein structures that they use for attachment to the host tissue and in biofilm formation. FimH, one part of the E. coli fimbriae, specifically recognizes mannose structures that are present on the host cell surface, typically in the form of oligomannosides. A simple inhibitor therefore is mannose itself; mannose binding to the FimH blocks receptor interactions, and thereby competes for the pathogen–host cell interaction. However, monosaccharide–protein interactions are usually very weak, in the μM to mM range. High affinity binding is only achieved through multivalent presentation and engagement, e.g., in more complex glycans (such as the oligomannosides on the cell surface), glycan-conjugates or polysaccharide structures. Additionally, secondary binding sites or the simultaneous interaction of different carbohydrate binding motifs can increase the binding affinity as well as the selectivity of the carbohydrate-mediated interactions.
Glycopolymers as glycan mimetics
Great efforts have been devoted to the isolation or synthesis of carbohydrates to be employed as inhibitors of pathogen adhesion. Carbohydrate and glycan mimetics offer an alternative to natural carbohydrate structures. Like other biomimetics, such as peptidomimetics, they retain enough structural similarity to enable biological activity but optimize on other parameters such as affinity, selectivity and stability, as well as ease in synthesis or cost of production.There exists a large variety of carbohydrate and glycan mimetics. One simple differentiation is between mono- and multivalent mimetics, where monovalent carbohydrate mimetics are, for example, C-glycosides or fluorinated mono- and oligosaccharides11–13 that are designed to engage a carbohydrate binding protein or enzyme in a specific way. In contrast, multivalent glycan mimetics are constructed using a scaffold presenting multiple carbohydrate side chains, and are often further classified according to the scaffold type, e.g., glycopolymers or glycodendrimers (Fig. 2). Multivalency is an important factor that contributes to the bioactivity of glycan mimetics as individual carbohydrate−protein interactions have weak binding affinities.14,15 It has been long recognized that the strength of binding, and also the specificity of the mimetic, can be improved by multivalent interactions. This, in turn, has been the basis for the development of multivalent glycan mimetics in the fight against pathogen infections.
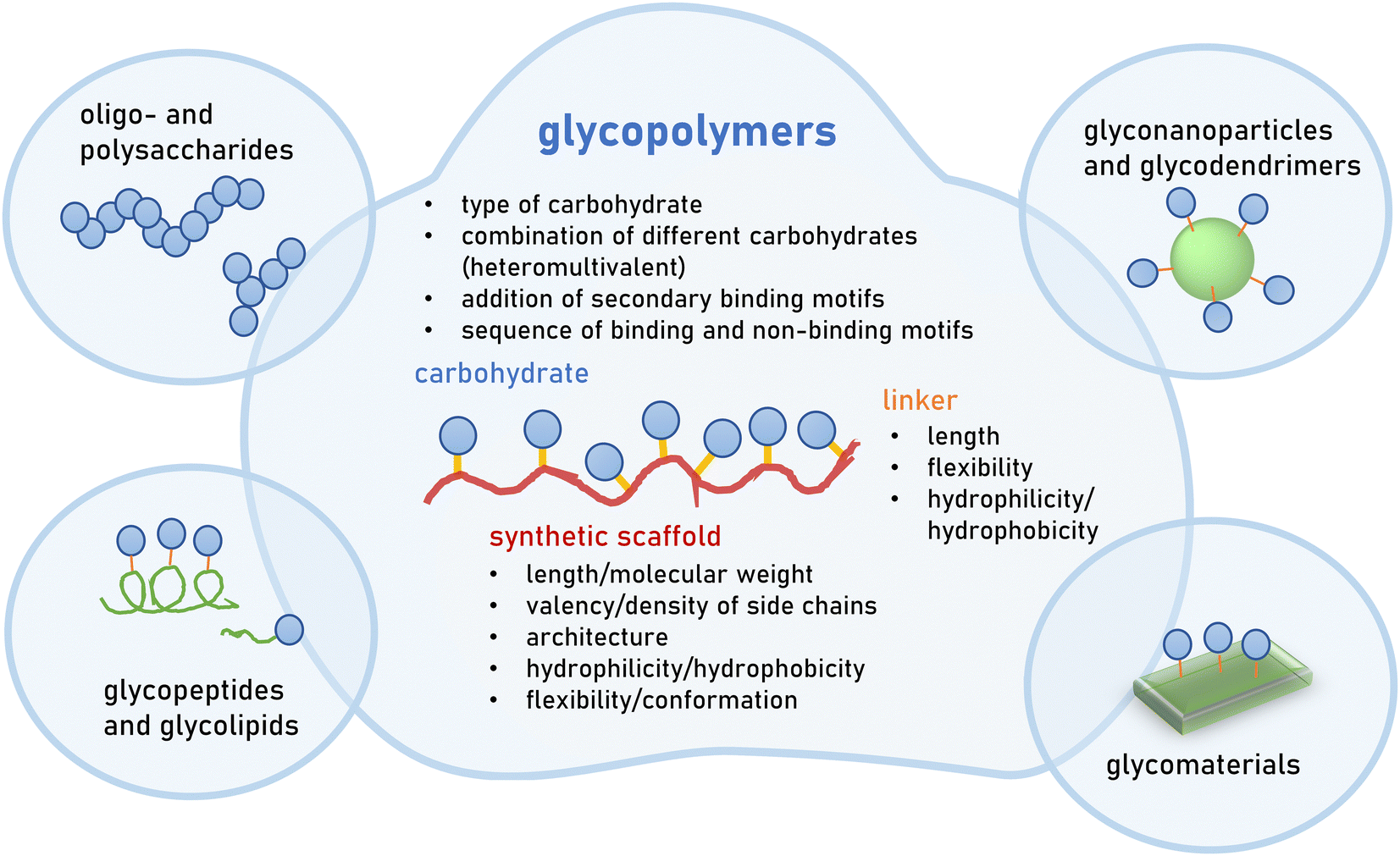 | ||
| Fig. 2 Multivalent glycan mimetics can be classified according to their scaffold e.g., polysaccharides, glycopeptides, glycoparticles, glycodendrimers and glycopolymers. Glycopolymers are multivalent glycan mimetics composed of a synthetic macromolecular or polymeric scaffold presenting carbohydrate motifs in the side chain. Different structural parameters are known to affect the overall avidity as well as selectivity of glycopolymers and thus their bioactivity. | ||
This review will exclusively focus on glycopolymers as multivalent glycan mimetics composed of a synthetic macromolecular or polymeric scaffold presenting carbohydrate motifs in the side chain. This is in contrast to similar macromolecular glycoconjugates such as glycopeptides,16–19 glycodendrimers,20,21 glycan-functionalized nanomaterials22–24 or combinations thereof25–27 (Fig. 2). For readers interested in the synthesis of glycomacromolecules and glycopolymers (in the following only referred to as glycopolymers) as well as their potential applications outside of targeting pathogens, we kindly refer to several recent reviews that are already available on the topic.28–36
In the design of glycopolymers, different structural parameters can affect and thus be used to tune biological activity e.g., their binding to a selected protein or pathogen target. The most obvious parameter is the choice of the carbohydrate motif that is attached to the polymeric scaffold. In the following subchapter, the most employed glycan motifs in the synthesis of glycopolymers to target pathogens will be discussed in more detail (Table 1). In principle, a glycopolymer can carry the same glycan motif multiple times, giving rise to so-called homomultivalent constructs, while combinations of different glycan motifs lead to heteromultivalent constructs.37,38 While the combination of two or more glycan motifs that are known to bind to the protein/pathogen target can increase the overall specificity, combinations of binding and weaker or non-binding glycan motifs have been shown to also affect binding as they alter the density of the binding epitope presentation along the polymeric scaffold. This, in turn, has been shown to affect the overall avidity.39–43 Heteromultivalent constructs have also been designed by combining glycan and secondary binding motifs, e.g., inducing aromatic or hydrophobic interactions with neighbouring sites of the carbohydrate recognition domain on the protein target.38,44,45 The use of controlled polymerization methods, as well as the development of sequence-controlled polymers, has also led to first insights into the effects of the carbohydrate sequence along the polymer scaffold.36,46,47 For homomultivalent glycopolymers, this has provided insights into the effects of glycan density as well as valency, and in recent studies also of the chain architecture – e.g., linear vs. branched/brushed or cyclic – on avidity and selectivity in protein binding.48,49 For heteromultivalent glycopolymers, sequence-control is less established; however, heteromultivalency has already proven a powerful tool to tune bioactivity. Thus, it can be expected that sequence-controlled heteromultivalent glycopolymers will become increasingly important in the future.
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|
 : N-acetylglucosamine, : N-acetylglucosamine,  : glucuronic acid. : glucuronic acid. : N-acetylgalactosamine, : N-acetylgalactosamine,  : iduronic acid. : iduronic acid. |
||||||
| Escherichia coli | Pseudomonas aeruginosa | Norovirus | Influenza | Clostridium perfringens | Norovirus | Papillomavirus |
| Lactobacillus plantarum | Fusobacterium nucleatum | Pseudomonas aeruginosa | Streptococcus oralis | Escherichia coli | Rotavirus | Coronavirus |
| Klebsiella pneumoniae | Streptococcus suis Actinomyces | Helicobacter pylori | Helicobacter pylori | Pseudomonas aeruginosa | Coronavirus | Chlamydia pneumoniae |
| Salmonella typhimurium | Burkholderia cenocepacia | Mycoplasma genitalium | Helicobacter pylori | Helicobacter pylori | Pseudomonas aeruginosa | |
| Saccharomyces cerevisiae | Escherichia coli | |||||
| Vibrio cholerae | ||||||
One of the fundamental debates in the design of multivalent ligands is the advantage or disadvantage of the rigidification of the scaffold.50–53 In a simplified view, a rigid multivalent ligand will lose most of the entropy upon first engagement with the target. Additional binding to the same target will come at a much lower entropy cost but at similarly high enthalpy wins as the first binding, thus overall promoting the multivalent vs. monovalent binding with the same number of interactions. However, not only the rigidity of the ligand but also the protein target can tremendously affect the binding event. Overall, multivalent binding events are complex, and more insights are required, especially for larger multivalent constructs derived from low affinity ligands such as glycopolymers. Nevertheless, in contrast to natural scaffolds, such as DNA or highly ordered proteins, it is important to keep in mind that synthetic polymer scaffolds are mostly flexible structures. Rigidification can be introduced e.g., by choosing polymer scaffolds with higher chain stiffness. Similarly, the linker connecting the carbohydrate side chain and the polymer scaffold can be chosen with different levels of flexibility, often determined by the linker length and morphology.
In conclusion, there is a fine balance of structural parameters that affects the overall properties of a glycopolymer, especially its biological activity. In the following chapters of our review, we will focus on the interaction of glycopolymers and pathogens to prevent or treat infections. For readers interested in more details on the multivalent binding modes of glycopolymers, as well as other glycoconjugates and their structure–property–correlations, we kindly refer to other reviews on the topic29,54–60 including two very recent reviews by Bhattacharya et al. from the Singha group61 and Kim et al. from the Shin group62 as well as a review article by Stenzel63 on the use of glycopolymers in drug delivery.
Glycan motifs employed to derive glycopolymers in the fight against infections
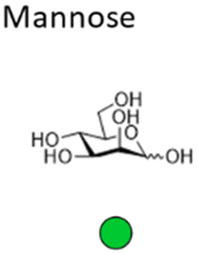
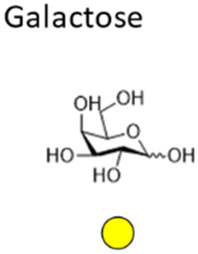
Mannose is a six-carbon monosaccharide and C-2 epimer of glucose. It plays an important role in the biosynthesis of N-glycosylated proteins as it is the main component of the highly conserved lipid-bound oligosaccharide consisting of 15 sugars (Glc3Man9GlcNAc2-P-P-Dol) that serves as a precursor for all N-glycosylated proteins. After en block transfer to proteins, this precursor is processed in the Golgi apparatus, where mannose moieties are hydrolyzed by different mannosidases. Thus, mature human glycoproteins typically only contain three mannose residues buried under sequential modification by N-acetylglucosamine, galactose, and sialic acid. Exposed mannose residues are recognized by the innate immune system in mammals. Mannose residues on the cell surface serve as important recognition motifs for pathogen adhesins. One example is Escherichia coli, a human pathogen that can cause a range of diseases from enteritis to urinary tract infections and meningitis. E. coli expresses different types of pili on its cell surface to mediate adhesion to host cell surfaces. Type 1 pili are filamentous protein complexes that are anchored to the outer membrane of uropathogenic E. coli that carry a mannose-specific lectin on its tip, the so-called type 1 fimbrin D-mannose specific adhesin (FimH). FimH can differ in its ligand binding specificity depending on the origin of the particular E. coli. FimH on E. coli in the gut bind to monomannosylated host glycans. In contrast, E. coli commonly causing urinary tract infections (UTIs), bind to oligomannose glycans.64 The binding specificity of the latter strain was successfully used to design C1-modified α-mannoside-based FimH antagonists (e.g. mannosides and biphenyl α-D-mannopyranosides) as therapeutics for UTI infections.65–67Galactose is the C-4 epimer of glucose and is an abundant component of N- and O-linked glycoproteins. D-Galactose does not occur in nature in an uncombined state. It is released when lactose, a disaccharide of glucose and galactose, is enzymatically hydrolysed by lactase. The galactose used in the biosynthesis of lactose is produced through metabolic epimerization of D-glucose to D-galactose. Galactose is also present in glycolipids of cells in the brain or the nervous system. Some pathogens, such as Pseudomonas aeruginosa, use galactose-binding lectins, such as LecA/PA-IL to adhere to host cells.68 The lectin is specific for α-galactose that is present on glycosphingolipids of the globoside family of epithelial cells. LecA binds galactose in a tetrameric complex and is involved in bacterial adhesion as well as biofilm formation.
Fucose is a six-carbon monosaccharide that is missing the C6 hydroxy group and is often found in the terminal positions of mucins and other glycoconjugates, such as N-linked glycans. Fucose is also an important motif of blood group antigens (BGAs) and Lewis epitopes,69 another BGA related oligosaccharide often found on cell surfaces. As such, fucose is also well known as a mediator of pathogen cell binding e.g., of Norovirus70 or P. aeruginosa71 that expresses, in addition to the above-mentioned mannose-recognizing LecA, a second lectin, LecB, on its surface. LecB is a homotetramer with a high affinity for L-fucose and its derivatives but can also bind to mannose and mannose-containing oligosaccharides. LecB plays a role in the adhesion of P. aeruginosa to endothelial cells but was also recently reported to be involved in biofilm formation.72 While LecA is primarily involved in host cell invasion and cytotoxicity, LecB was found to arrest the ciliary beating of human airway epithelia, an essential mechanism to transport foreign particles (including pathogens) trapped in the mucous layer out of the upper airway.
Sialic acids (SiA) are negatively charged nine-carbon neuraminic acid derivatives, that can be enzymatically modified e.g., at the C5 carbon as N-acetylneuraminic acid (Neu5Ac). SiA are often displayed as a terminal monosaccharide on various cell surface glycoconjugates such as glycolipids or glycoproteins e.g., of the mucin type. Mucins are a class of high-molecular-weight glycoproteins with a high density of short glycan side chains carrying terminal sialic acid residues. Many viruses, such as influenza, bind to SiA containing oligosaccharides and glycan conjugates and make use of this interaction in their infection process.73 However, SiAs, especially as part of the mucus in healthy individuals, are also used to trap viruses and prevent cellular uptake or even remove viruses from the system through regular mucus removal. Patients suffering from cystic fibrosis with increased mucus production and obstruction are thus more prone to viral and other pathogen infections.74,75
Human milk oligosaccharides (HMOs) are structurally diverse oligosaccharides produced by mothers as part of their breast milk.4 Their structure and composition can differ depending on the mother's genetic background, leading for example to the presence or absence of fucose in HMO oligosaccharides. HMOs identified so far contain five monosaccharides: glucose, galactose, N-acetylglucosamine, fucose, and sialic acid. Several in vitro and in vivo studies have shown that HMOs can act as inhibitors of pathogen attachment in bacterial, viral and parasitic host cell interactions.4,76
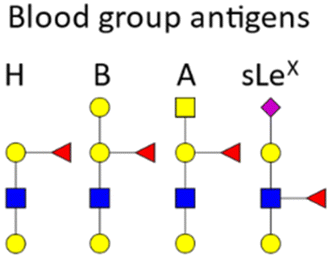
BGAs of the ABO type are oligosaccharide structures attached to the red blood cell membrane.77 BGAs are primarily composed of unique combinations of N-acetylglucosamine, glucosamine, N-acetylgalactosamine, galactose, and fucose. As cell surface receptors, these BGAs can mediate pathogen cell adhesion, and colonization as well as intracellular uptake. However, BGAs can also serve as decoys or false receptors to prevent binding to the actual target tissue.78 Some bacteria and other pathogens can even induce ABO antibody formation.77 Such antibodies can be considered part of the innate immune system, as they inhibit ABO BGA-mediated cell binding of pathogens.
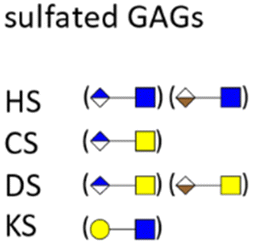
Glycosaminoglycans (GAGs) as part of the glycocalyx are typically displayed as proteoglycans (PGs).79 PGs are composed of different core proteins that carry side chains of anionic polysaccharides. GAGs are generated from repeating disaccharide units, the composition of which defines their classification, such as heparin/heparan sulfate (HP/HS), chondroitin/dermatan sulfate (CS/DS), keratan sulfate (KS), and hyaluronic acid (HA). Most GAGs are composed of distinct combinations of N-acetylglucosamine, glucosamine, N-acetylgalactosamine, galactose, and uronic acids. Viruses, bacteria and other pathogens are known to interact with PGs and sulfated GAGs not only to achieve adherence and/or colonization, but also in invasion, internalization, dissemination, and toxicity.80 Some pathogens are even able to induce shedding of cell surface PGs to promote their infection process.81
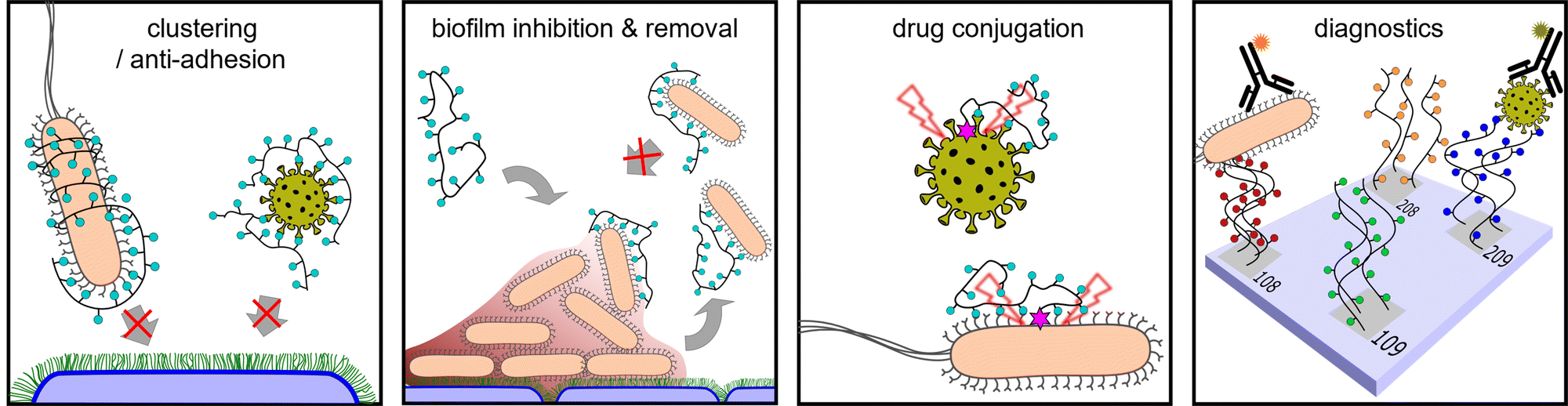 | ||
| Fig. 3 General modes of actions of glycopolymers in the fight against infections. | ||
The focus of this review is the development of glycopolymers for the fight against pathogen infections – viral, bacterial and other pathogens, including general concepts and recent progress in this field over the last 10 years (2012–2022). Here we exclusively highlight studies that have examined the effects of glycopolymers on pathogen adhesion, biofilm formation or host cell infection. In Chapter 1, glycopolymers as inhibitors of viral attachment are discussed. In the light of the recent COVID-19 pandemic, several reviews have been published on the development of polymers, including glycopolymers, for their use in fighting antiviral infections. Therefore, in this chapter we will only present work from the last two years (2020–2022). In Chapter 2, glycopolymers and their application in anti-adhesion therapy against bacterial infections, as well as their potential to disrupt biofilm formation and improve drug delivery to bacteria are presented. In Chapter 3, efforts towards the use of glycopolymers in the fight against other pathogens including fungi, helminths, and protozoa, are explored. We specifically exclude the use of glycopolymers and similar glycan-conjugates in the development of carbohydrate-based vaccines and kindly refer our readers to other articles on this specific topic.31,87–93 As the recent COVID-19 pandemic has taught us, the straightforward and unambiguous detection of specific pathogens and thereby infections can be an important tool in the fight against infections as well. Thus, in Chapter 4, we will highlight the use of glycopolymers also in isolating and/or detecting pathogens.
Chapter 1: Glycopolymers in the fight against viral infections
Viruses are obligate intracellular parasites, which means they require a host cell for replication. The process of engaging and infecting a host cell is a multistep process that begins with viral engagement of the host cell and typically ends with the release of viral genome inside the cell.94 There are two classes of viruses, enveloped and non-enveloped, and two different routes of cell entry, endocytic and non-endocytic. Enveloped viruses carrying a lipid membrane on the outside can enter the cell by membrane fusion, a non-endocytic pathway. Both enveloped and non-enveloped viruses can also enter cells through endocytic pathways such as clathrin-mediated uptake, but other receptor mediated entry mechanisms are known as well. In all cases, uptake is only achieved if the virus is first brought into close proximity or contact with the cell surface, usually through attachment. Virus cell attachment can be mediated by different cell surface molecules (proteins, lipids, glycans), but glycans and glycan-conjugates (glycoproteins, glycolipids) seem to play a particularly important role in the attachment of various viruses.95,96 One hypothesis for this is the great abundance of glycan motifs on the host cell surface and the increased affinity (avidity) of viruses through binding to these glycans in a multivalent fashion in comparison (or in competition) to other glycan-recognizing cell surface receptors.97 In principle, glycans can serve as both attachment and entry receptors and as such are potential targets to block virus attachment and entry for antiviral therapies. Glycans are also present on the viral surface, specifically as glycosylated proteins e.g., as part of the viral membrane in enveloped viruses such as influenza or the HIV protein gp120, the latter of which is one of the most heavily glycosylated proteins known.98 Here again, glycans mainly act as modulators and receptors of virus attachment and entry, and can serve as a blueprint for the development of molecules blocking these first steps of a viral infection.Developing inhibitors of glycan-mediated cell attachment and/or cell entry is typically based on the idea of competing for the natural glycan-recognition site. A molecule (the inhibitor) that has a higher affinity than the natural glycan motif will occupy the glycan-recognition sites, thereby blocking cell attachment or entry. Such high affinity ligands can be derived by different means e.g., from synthetic or biological resources, and can have different sizes and shapes, from small molecules to high molecular weight materials. However, in line with the topic of this review, we will exclusively focus on glycopolymers as so-called attachment or entry inhibitors. For glycopolymers, two main parameters are routinely examined in their development as efficient inhibitors. First, intrinsic multivalency typically leads to a significant increase in binding avidity in comparison to the natural counterparts they need to compete with. Second, their hydrodynamic size can enable so-called sterical shielding where non-binding parts of the glycopolymer sterically hinder the diffusion and binding of competing ligands, thereby increasing the overall blocking and thus inhibition efficiency.50
Notably, glycopolymers are not the only class of antiviral polymers. Polyanions, which can also include anionic glycopolymers, have long been recognized for their antiviral activity. In the light of the recent COVID-19 pandemic, antiviral polymers have regained increasing attention, and a number of recent reviews from the last two years have highlighted the different types of anionic antiviral polymers, including glycopolymers, from their synthesis to biomedical applications.99–101 Here, we highlight the review by Bianculli et al. from the Schulze group on antiviral polymers,102 a review by Huang et al. from the Ding group on antiviral biomaterials,103 a review by Jung et al. from the Boyer group on bioactive polymers including antiviral and antibacterial polymers104 and a review from our own group on polymers inspired by heparin and heparan sulfate and their use in antiviral therapy.9 The following section will therefore only highlight very recent studies from the years 2020–2022 to complement these previous review articles.
In a recent report, Stadtmueller and co-workers107 combined sialyllactose presenting glycopolymers with polyglycerol scaffolds, as developed by the Haag lab.108 Earlier studies for sialylated compounds had shown that it is challenging to elicit antiviral activity with such compounds against a broad spectrum of IAV strains. In comparison to directly SiA modified polyglycerol compounds, their sialyllactose conjugates with optimized linker chemistry now achieved efficient inhibition of different IAV strains for potential use as broad-spectrum antivirals revealing the importance of both the right choice of carbohydrate motif as well as presenting scaffold. They also described that, potentially in dependence on the overall avidity of the glycopolymer, glycopolymers can bind to the virus but not inhibit viral attachment.
Matsuoka and co-workers recently synthesized another sialyllactose presenting glycopolymer and successfully tested its ability to inhibit mumps virus.109 Previous studies had identified sialyl Lewis X, one of the BGAs, as a strong inhibitor of mumps viral attachment. The multivalent presentation of the sialyllactose motif led to ten times increased inhibitory potency for their best binder; interestingly this glycopolymer exhibited a lower overall density of carbohydrate motifs per polymer construct than some of their other constructs.
Making use of their polyglycerol scaffolds, Wallert et al.110 introduced both SiA motifs as well as sulfates, thereby more closely mimicking the glycosylation of mucins. Furthermore, they applied their synthesis towards very high molecular weight (MDa) polyglycerols to match the size of natural mucins. They then tested their compounds as inhibitors of IAV attachment to model membranes and surprisingly found a two-phase behavior. Counterintuitively, at overall lower inhibitor concentrations, adding more glycopolymer increased virus attachment. Only at higher overall concentrations did the inhibitory efficiency increase with increasing glycopolymer concentration. The authors explained this by using a concentration dependent binding model with the glycopolymer binding first to the envelope protein neuraminidase, which enhances virus membrane interaction, followed by glycopolymer binding to a second envelope protein, hemagglutinin, now leading to inhibition of membrane attachment. Overall, the largest mucin mimetic glycopolymers presenting both SiA and sulfate groups showed the highest inhibitory potential in the pM range.
In a study from our own group,111 we have explored both, globally sulfated lower molecular weight glycomacromolecules derived by solid phase synthesis, as well as higher molecular weight glycopolymers from RAFT polymerization to derive simple GAG mimetics. Together with the Schelhaas team, we could show that higher molecular weight GAG mimetics can efficiently inhibit a variety of viruses in vitro and prevent human papillomavirus infection in vivo in a vaginal mice model (Fig. 4). Interestingly, cell studies with virus-like capsids of human papillomavirus indicated a different inhibition mode in dependence of the chain length, where shorter constructs exert an inhibition post attachment of the virus to the cell. This observation could be related to the biphasic behavior as observed by Wallert and coauthors110 as discussed in the previous example.
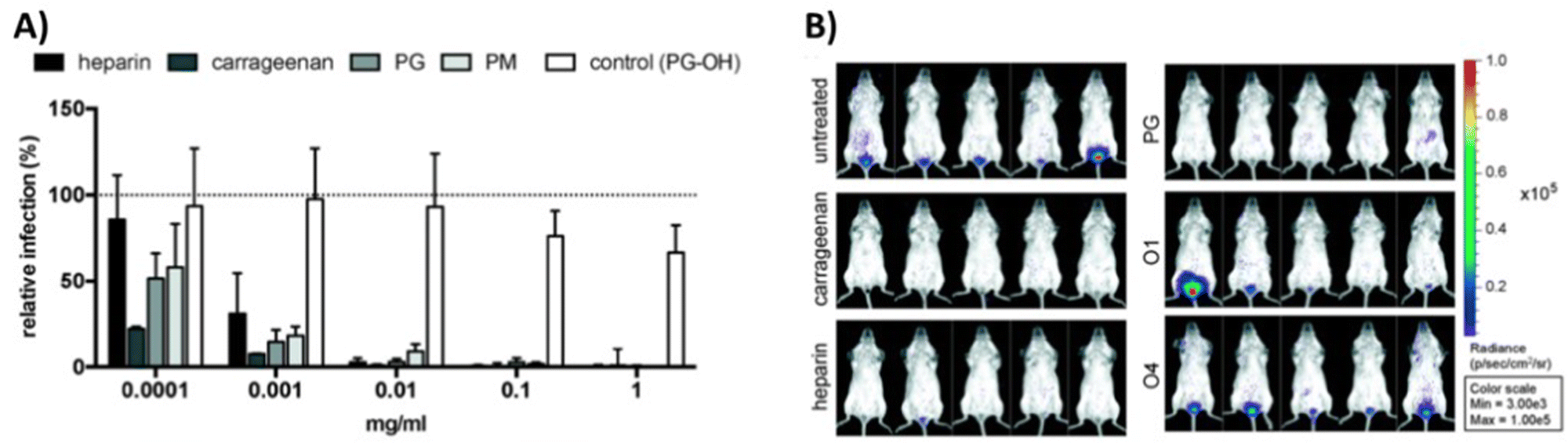 | ||
| Fig. 4 GAG mimetic polymers can inhibit virus attachment in vitro and in vivo. (A) Infection cell assay showing HPV-16 inhibition by polysaccharides and glycopolymers. (B) Vaginal mouse study visualizing the inhibition of HPV-16 (PG = polymers with sulfated glucose side chain, PM = sulfated mannose side chain, PG-OH = unsulfated control, glycomacromolecules presenting N-acetylglucosamine side chains – O1 (divalent) and O4 (decavalent)). Reprinted with permission from ref. 111 Copyright 2020 American Chemical Society. | ||
Viruses not only use glycans on the cell surface for attachment but also present glycan motifs on their own surface to engage in receptor binding on the host cell. An example of such interactions is the binding of the glycosylated spike proteins of SARS-CoV-2 to the DC-SIGN receptor of innate immune cells. DC-SIGN belongs to the class of C-type lectin receptors binding to oligomannose-based glycans and is involved in various pathogen infections. It serves as a receptor or co-receptor of cell attachment and entry to innate immune cells from which pathogens can further spread to other cells. Thus, blocking the DC-SIGN-pathogen interaction is another strategy to inhibit virus cell attachment and infection. In a recent study, Cramer et al. made use of this concept by exploring mannose-functionalized polylysine as a glycopolymer inhibitor of DC-SIGN mediated binding of viral envelope glycoproteins (Fig. 5).112 They then used what they learned from this study to prepare a series of monovalent mannose-based aglycons showing significantly higher affinity to DC-SIGN than the natural methyl-mannose ligand.113 Multivalent presentation of one of their aglycons on a polylysine scaffold provided a glycopolymer that was able to inhibit SARS-CoV-2 spike glycoprotein binding to DC-SIGN expressing cells in the nanomolar range, and importantly also blocked transfer of spike glycoprotein presenting virus-like particles to other cells.
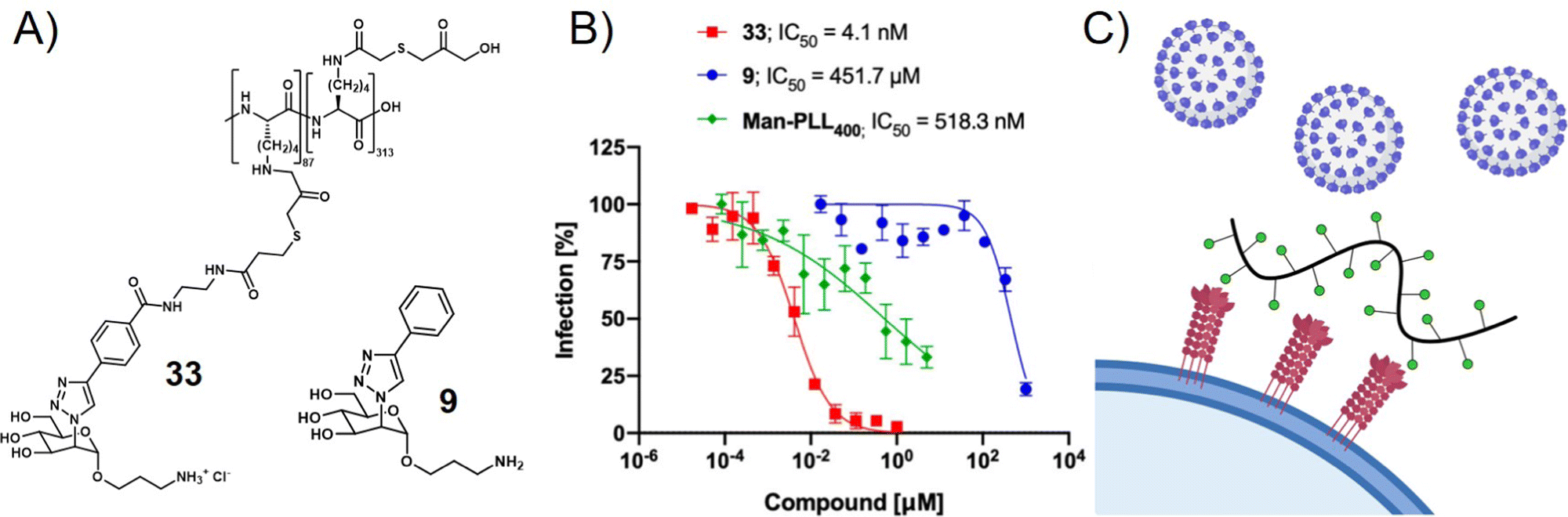 | ||
| Fig. 5 (A) Mannose-derived aglycon (9) and polymer thereof (33) and (B) trans-infection assay results showing the inhibition of DC-SIGN mediated trans-infection of VSV*ΔG-SARS-CoV-2-SΔ21 encoding the SARS-CoV-2 spike glycoprotein from DC-SIGN+ B-THP-1 cells to Vero E6 Cells. (C) Schematic presentation of virus inhibition by blocking DC-SIGN receptors on the cell surface. Parts A and B reprinted with permission from ref. 113 Copyright 2021 American Chemical Society. | ||
Interestingly, glycopolymers themselves have recently gained attention in more fundamental studies of pathogen cell interactions in the context of glycocalyx engineering.114,115 Here, glycopolymers carrying membrane anchors, such as cholesterol or lipids, are inserted into artificial membranes as well as cell membranes to mimic or alter the natural glycocalyx. The Bertozzi group, who had originally introduced the concept of glycocalyx engineering with glycopolymers, recently presented in a study by Delaveris et al.,116 the preparation of mucin-mimetic glycopolymers for incorporation into lipid bilayers and tested these for the effects of glycopolymer length, glycosylation, and surface density on the binding of influenza A virus to GD1, a SiA presenting glycolipid within the bilayer. They observed a change in polymer conformation in dependence of glycopolymer density on the membrane surface with higher densities leading to more stretched, brush-like conformations. Their results suggest that it is this stretched conformation that leads to more efficient virus inhibition for long chain glycopolymers at high surface densities. The Godula group, in a study by Honigfort et al.117 have recently used similar mucin-mimetic glycopolymers for reengineering the glycocalyx of red blood cells to study effects of crowding the membrane surface with non-binding glycopolymers on the attachment of IAV. Indeed, as would be expected, binding of the virus to the cell is reduced by higher molecular weight non-binding glycopolymers. However, the detachment of the virus is affected as well, with glycopolymers promoting receptor clustering and leading to higher cell surface retention times for viruses in the crowded environment (Fig. 6). In another study, the same group introduced mucin-mimetic glycopolymers with a photolabile linker between the glycopolymer and its membrane anchor.118 Through light-induced release of the glycopolymers, they explored the potential role of glycocalyx shedding in both the disruption of the glycocalyx to promote infection, or its protective function to defend against pathogen adhesion.
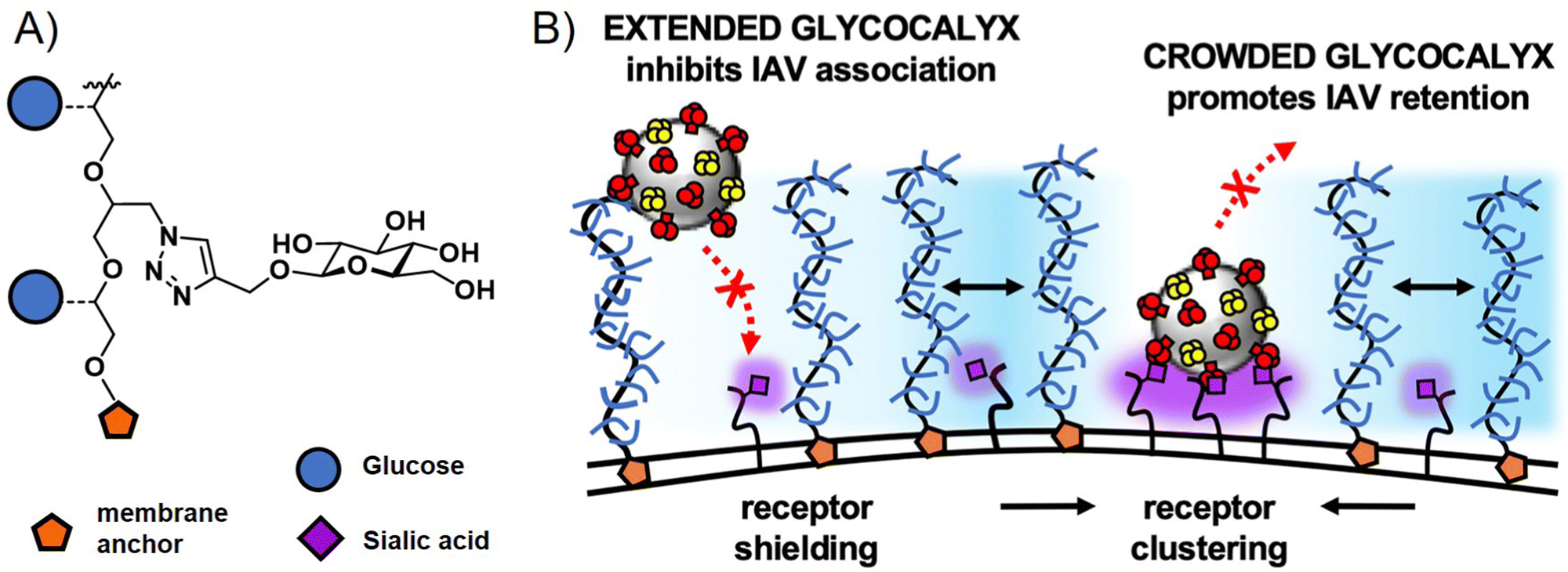 | ||
| Fig. 6 (A) Structure of non-binding glycopolymers and (B) proposed model for the influence of non-binding glycopolymers as glycocalyx modulators on cell–pathogen interactions. High molecular weight and dense presentation of glycopolymers shields cell surface receptors from viral adhesion. Glycopolymer crowding supports receptor clustering and thus enhances retention of bound viruses. Adapted with permission from ref. 117. Copyright 2021 National Academy of Sciences. | ||
Chapter 2: Glycopolymers in the fight against bacterial infections
Bacterial infection is a highly complex process that includes four steps to establish pathogenesis: exposure (contact), adhesion (colonization), invasion, and infection (Fig. 7). Exposure is the initial encounter with a potential pathogen. To cause a disease, the pathogen needs to be able to gain access into host tissue via a suitable anatomic site, a so-called portal of entry. These entry sites are often mucosal surfaces, including the mucous membranes of the respiratory tract, the gastrointestinal tract, and the genitourinary tract. The initial exposure is followed by adhesion of the pathogen to the entry portal of the host. | ||
| Fig. 7 Schematic representation of different steps of bacterial infection highlighting (1) the attachment where adhesion is mediated by non-covalent interactions of bacterial lectins with glycans on the host cell surface and (2) entry and invasion which is initiated upon secretion of bacterial virulence factors (e.g., either effector proteins inducing endocytosis or toxins damaging the host tissue). | ||
Pathogens use different mechanisms to adhere to host cells. Adhesins are proteins expressed on the surface of bacteria that bind to specific receptors (often glycoproteins or glycolipids) on host cells. Biofilms can also act as adhesion factors. A biofilm is a community of glycocalyx producing bacteria that provides protection against factors of the immune system and treatment with antibiotics. After successful adhesion, invasion can proceed. Invasion involves the dissemination of a pathogen throughout local tissues or systemically throughout the body and involves virulence factors to mediate the host cell entry. Virulence factors can be effector enzymes initiating host cell entry or toxins that cause damage to host tissues or compounds of the immune system defenses. The entry to a host cell usually occurs by endocytosis. Finally, following the successful invasion, multiplication of the pathogen and infection occurs.
Strategies to interfere at each stage of the bacterial infection process to inhibit infection have been subject to intense investigations.56,57,119 In this chapter, we focus on the use of glycopolymers preventing or reversing the adhesion process. This includes the design of glycopolymers that present antagonists of bacterial adhesins and glycopolymers that inhibit or disrupt biofilm formation. In addition, we will highlight examples where glycopolymers in combinations with inhibitors of virulence factors have been used to successfully fight bacterial infections.
E coli is a member of the family of Enterobacteriaceae, which are Gram-negative facultatively anaerobic single straight rods that have a typical size of 1.1–1.5 μm in width and 2–6 μm in length. E. coli normally live in the intestines of healthy people and animals but can cause urinary tract infections (UTI) if they enter the bladder. This is the leading cause of UTIs among women. The majority of E. coli stains are not considered harmful; in most cases infections cause only a mild case of diarrhea. In contrast, there are a few strains, such as E. coli O157:H7, that can cause more severe symptoms, such as vomiting or bloody diarrhea.123E. coli contains a D-mannose specific adhesin (FimH) at the tip of the type 1 fimbriae, which mediates adhesion to mannose containing receptors on the host cell.124
P. aeruginosa is a heterotrophic, motile, Gram-negative rod-shaped bacterium that is about 1–5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μm long and 0.5–1.0μm wide. Due to its capability of breaking down polycyclic aromatic hydrocarbons, P. aeruginosa is an important soil bacterium. The bacterium is commonly found in water sources that have been contaminated by animal or human waste, such as sewage and sinks in both hospital and non-hospital settings. P. aeruginosa is considered an ‘opportunistic’ pathogen because it rarely infects healthy individuals, but it can cause infections in the blood, lungs, or other parts of the body after surgery, particularly in patients who are immunocompromised due to a pre-existing condition, such as AIDS, cancer, cystic fibrosis, burn injuries, or non-healing wounds.125P. aeruginosa is often resistant to many classes of antibiotics and therapeutic agents, making it difficult to treat.126
μm long and 0.5–1.0μm wide. Due to its capability of breaking down polycyclic aromatic hydrocarbons, P. aeruginosa is an important soil bacterium. The bacterium is commonly found in water sources that have been contaminated by animal or human waste, such as sewage and sinks in both hospital and non-hospital settings. P. aeruginosa is considered an ‘opportunistic’ pathogen because it rarely infects healthy individuals, but it can cause infections in the blood, lungs, or other parts of the body after surgery, particularly in patients who are immunocompromised due to a pre-existing condition, such as AIDS, cancer, cystic fibrosis, burn injuries, or non-healing wounds.125P. aeruginosa is often resistant to many classes of antibiotics and therapeutic agents, making it difficult to treat.126
S. aureus is a Gram-positive, approximately 0.5–1.5μm in diameter, bacterium that causes a wide variety of clinical diseases. It commonly resides in skin and mucosa, but if it enters the body, it can cause skin infection, sometimes pneumonia, endocarditis, and osteomyelitis.127 This commonly leads to abscess formation and mild to life-threatening sepsis, especially in immunocompromised or immunosuppressed individuals. Routes of entry include broken skin or mucosa, and oral ingestion of infected food.
It has been established for more than 50 years that bacteria make use of carbohydrate-mediated adhesion and that this adhesion can be inhibited using monosaccharides.64 Thus, it is not surprising that glycans, glycan conjugates, as well as glycan mimetics have been extensively investigated for their suitability as inhibitors of adhesins for so-called anti-adhesion therapy. In the last ten years, glycopolymers, especially mannose-derived glycopolymers, have been used to provide new insights into structure-property correlations, e.g., the effects of valency and introduction of secondary binding motifs, including the combination of polycations and glycopolymers, to improve the activity of glycopolymers as adhesion inhibitors and to further their development towards a potential clinical use.
A recent study investigating the structure-property relation of glycopolymers presenting mannose in various valencies was reported by Yan et al.128 Here, the authors designed a library of 27 mannosylated copolymers with varying compositions and microstructures by combining different RAFT polymerization procedures. Depending on the copolymerization method that was used, the glycopolymers differed in the neighbouring groups (number and type of the side group) flanking the mannose residues. Anti-adhesive properties were tested against E. coli but the authors could not reveal a clear correlation between the microstructure, the density of epitopes, the nature of the mannose neighbouring groups and the anti-adhesive properties. However, the glycopolymers that were able to efficiently inhibit bacterial adhesion were effective in pre- and post-incubation adhesion assays, demonstrating that they could not only prevent E. coli from adhering, but also cause bacteria that had already attached to T84 cells to detach (Fig. 8). In another study, Yan and co-workers showed the effect of a mannose-functionalized glycopolymer on bacterial binding in vivo. The authors used glycopolymers consisting of multivalent n-heptyl-α-D-mannose (HM) antagonist of FimH.129 Their glycopolymers were 102 and 106 times more potent than n-heptyl-α-D-mannose or D-mannose alone in the sequestration of free bacteria in the lumen of the gut and also in the disruption of already established E. coli cell interactions, highlighting their potential in both preventing and treating an infection in the animal model.
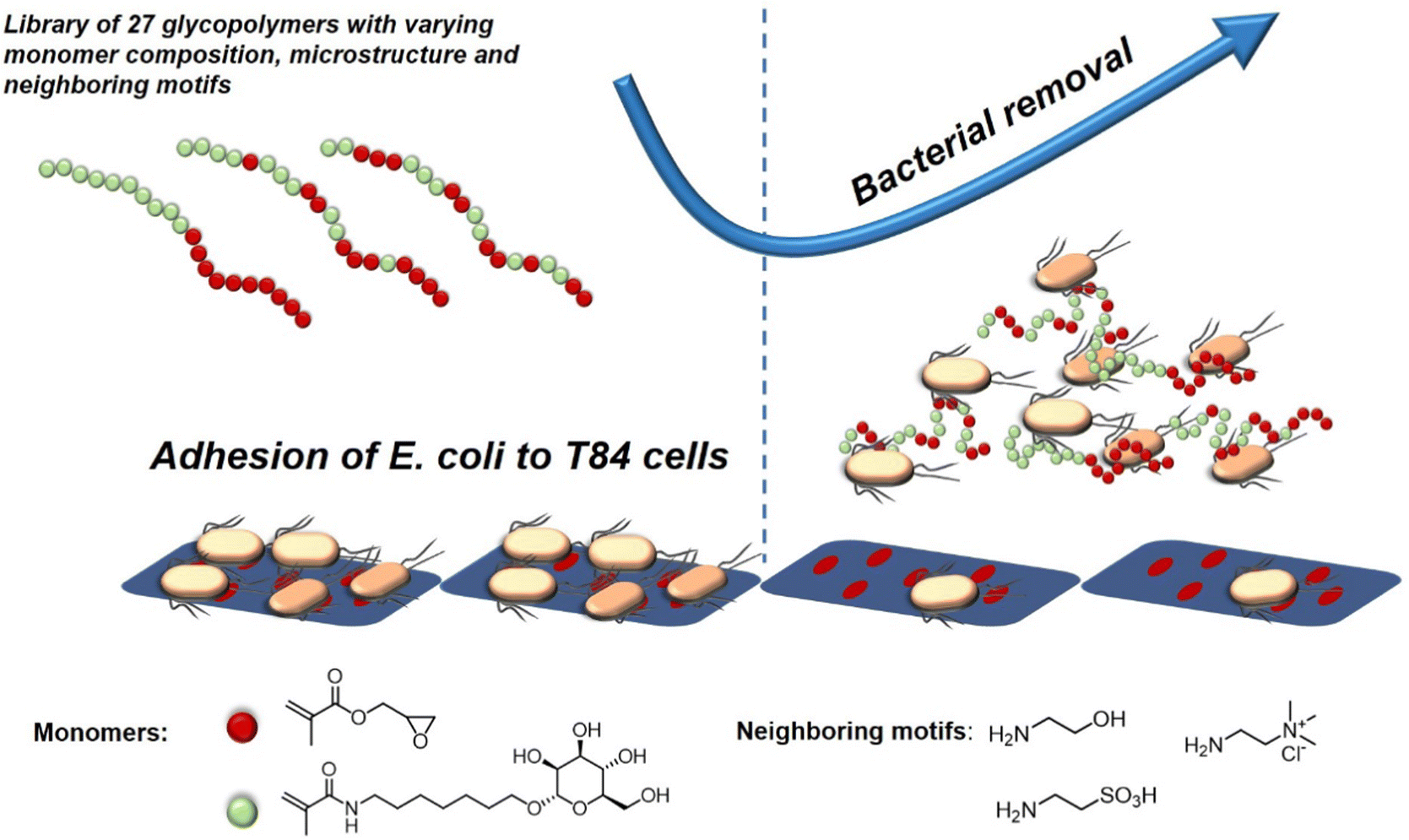 | ||
| Fig. 8 Schematic representation of glycopolymers binding to E. coli and thereby removing adherent bacteria and inhibiting new adhesion. Adapted from ref. 128 with permission from the Royal Society of Chemistry. | ||
The effect of carbohydrate valency in combination with hydrophobic motifs as potential secondary binding motifs was investigated in our lab in a study by Boden et al.130 The established concept of solid phase polymer synthesis131 using tailor-made buildings blocks was employed to generate a library of 16 sequence-defined glycomacromolecules presenting increasing numbers of either α-D-mannopyranoside (Man) or β-D-galactopyranoside (Gal) ligands. The backbone composition was varied by including either hydrophilic, aliphatic, or aromatic spacer groups between carbohydrate carrying building blocks. E. coli adhesion was shown to be more strongly inhibited by glycomacromolecules with hydrophobic backbones most likely due to additional secondary interactions with hydrophobic regions of the FimH protein receptor.
The knowledge of the binding of pathogens to certain carbohydrate motifs can be utilized to target specific pathogens and associated diseases. However, many of the carbohydrate motifs utilized by pathogens are also recognized by other cell receptors, e.g., mannose is a ligand for both the FimH receptors of E. coli and DC-SIGN receptors of immune cells. Luo et al.132 presented an elegant approach to achieve selectivity by using the targeted pathogen as a template to create glycopolymers that match the distance and density of mannose epitopes on the bacterial surface. E. coli from the MG1655 strain were used as living templates to synthesize glycopolymers in situ by adding a sugar-containing monomer and a non-binding monomer acting as a spacer in the formed polymer chain (Fig. 9). Conveniently, the reducing properties of the bacteria itself served to enable the polymerization via activator regenerated electron transfer atom transfer radical polymerization (ARGET ATRP). The resulting glycopolymers were then isolated from E. coli by washing with high concentrations of mannose. For comparison, the same monomers were polymerized in absence of the bacteria using the same polymerization method in solution. Indeed, the templated glycopolymers had a much higher affinity to the E. coli strain MG1655 than the control polymers generated in solution. Interestingly, in a mixture of two E. coli strains, MG1655 and E. coli DH5α, with slightly different genomes, the templated glycopolymers bound with higher affinity to the MG1655 indicating varying distances of surface receptors in different strains. The authors term this bacteria-templated polymerization method bacteria-sugar-monomer-aptation-polymerization (BS-MAP).
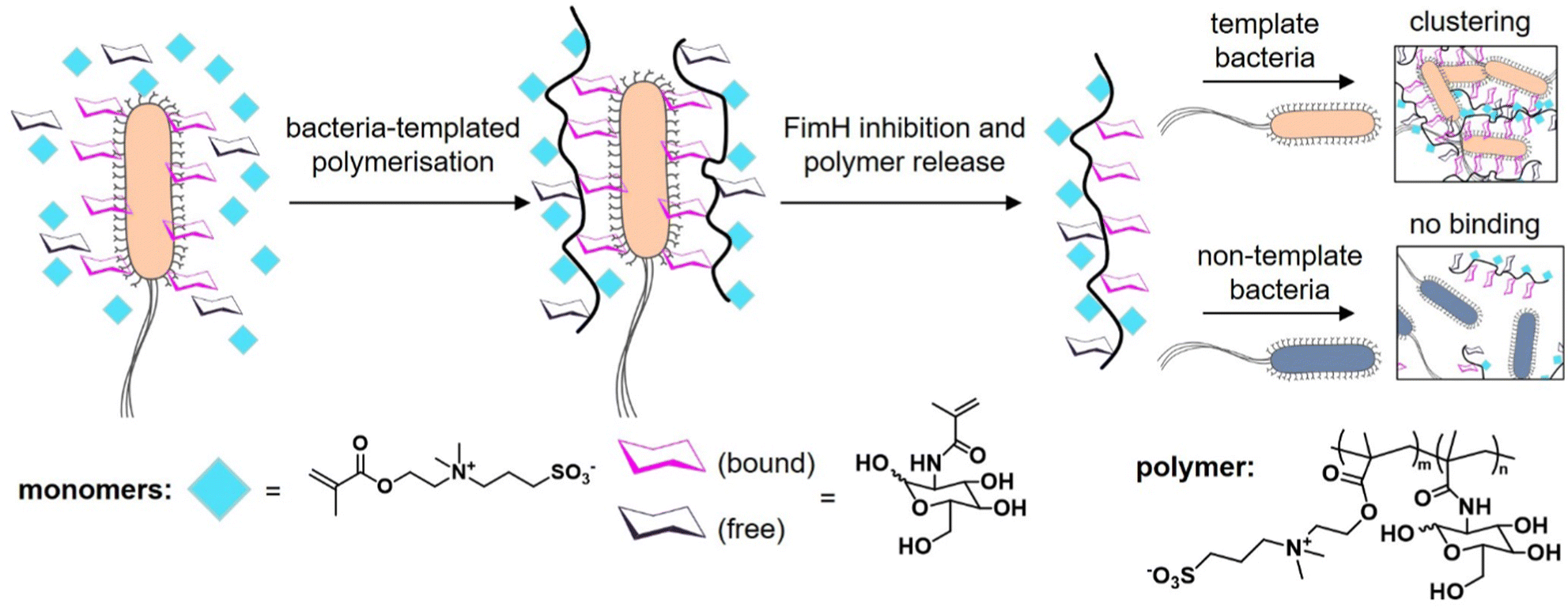 | ||
| Fig. 9 Schematic representation of the concept of bacteria-assisted polymerization yielding two different polymers: in solution (SP) and on the surface of bacteria (BP). The BP polymer shows only efficient binding to the template strain of E. coli but not to a different E. coli strain. Adapted from ref. 132 with permission from The Royal Society of Chemistry. | ||
In a study from our own lab, Banger et al.134 demonstrated that fixing spherical architectures of glycomacromolecules by cross-linking can benefit the binding of bacteria. Using solid-phase polymer synthesis with tailor-made building blocks, a small library of monodisperse amphiphilic glycomacromolecules (APGs) was generated. Carbohydrate side chains and cross-linker units were varied at defined positions within the macromolecules. Depending on their structure, the APG self-assembled into spherical or worm-like micelles, which could be fixed by a polymerizable unit within the APGs. Carbohydrate presenting micelles were then evaluated as inhibitors of E. coli adhesion, where the crosslinked worm-like micelles showed strong binding cooperativity and multivalent binding to bacteria (Fig. 10).
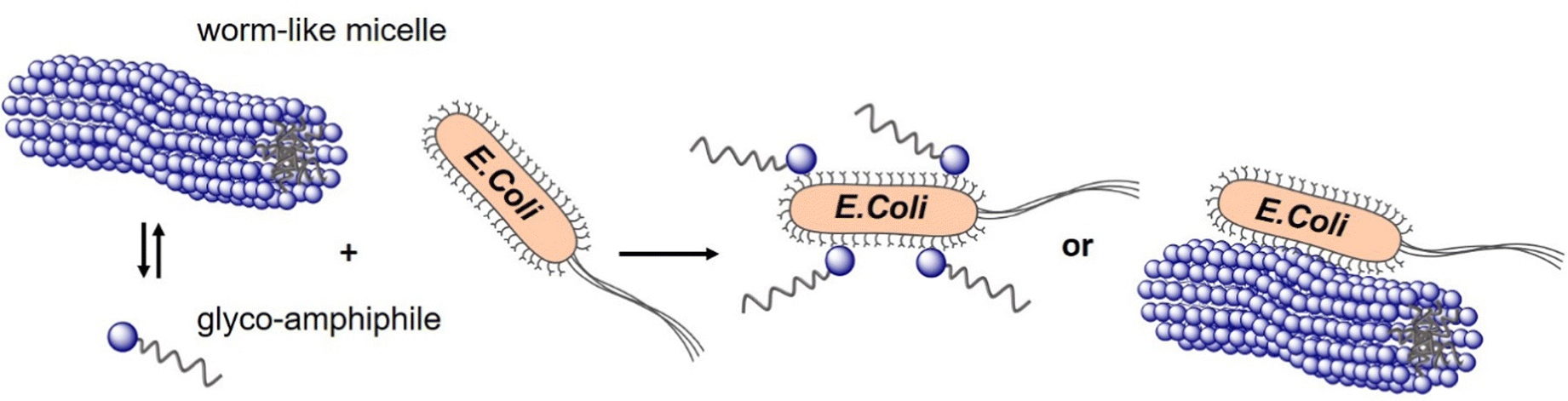 | ||
| Fig. 10 Crosslinked and non-crosslinked mannose functionalized micelles from glycomacromolecules inhibit E. coli adhesion either in a mono- or multivalent fashion. Reproduced from ref. 134 with permission from The Royal Society of Chemistry. | ||
Inspired by the design of CAMPs and synthetic antibacterial cationic polymers, cationic glycopolymers have attracted great attention as a new type of biocompatible antibacterial materials. On the one hand, glycan motifs establish the selectivity towards particular pathogens. On the other hand, the introduction of pathogen-targeting glycans allows limiting electrostatic interactions while maintaining the bactericidal activity.
Following this concept, Zheng et al.137 synthesized sugar-containing poly(ionic liquids) (PILs) with mannose or glucose groups incorporated into the polymers by quaternization of the dimethylamino unit. The corresponding PILs showed suitable killing of typical Gram-negative E. coli and Gram-positive S. aureus while exhibiting a lower hemolytic rate of blood cells compared to those quaternized by bromohydrin as a non-sugar-containing polycationic control. In another study from the same group,138 they performed a quaternization reaction of poly(4-vinyl pyridine) with halogen-functionalized D-mannose and tetraphenylethylene, a known aggregation-induced emission (AIE) fluorophore. Through this combination, their cationic glycopolymers retained their antibacterial activity against Gram-positive S. aureus and Gram-negative E. coli, and now also allowed for detection and imaging of the bacteria through fluorescence being induced only upon clustering of the bacteria with the polymer. In a follow-up study,139 the group also reported a new one-step reaction to produce mannose or glucose-containing N-alkyl imidazole-based block-copolymers, which were used to construct core or surface-functionalized glyconanoparticles from different solvents. By tuning selected structural parameters such as the molecular weight, they determined thresholds for maximization of antibacterial activity while reducing hemolytic activity.
Pranantyo et al.140 recently used ATRP to synthesize a series of four-arm star glycopolymer–polypeptide conjugates that contained either glucose, galactose, or mannose glycopolymer arms with well-defined chain length in combination with antimicrobial polylysine arms. The glycopolymer–polypeptide conjugates did not affect red blood cells and exhibited higher cytocompatibility than linear α-polylysine, which was used as a cationic control polymer. Anti-infective properties against Gram-positive bacteria (S. aureus and S. epidermidis) and Gram-negative bacteria (E. coli and P. aeruginosa) were tested. The ratio of glycopolymer-to-cationic polylysine domains was optimized for efficient bacteria killing. It was shown that the antimicrobial effect per lysine units is increased when including bacteria-binding glycopolymers.
In a recent study by our group,144 glycopolymers presenting fucose in varying number and density were studied as ligands of P. aeruginosa LecB showing increasing affinity with increasing valency. When adding fucosylated structures to the bacteria and examining their subsequent ability to form biofilms, a reduction of 20% in biofilm formation was observed compared to monovalent fucose with only a 7% reduction in biofilm formation. However, no correlation between the different glycopolymer structures and inhibitory efficiency was found.
One cationic glycopolymer, poly(acetyl, arginyl)glucosamine (PAAG) (Fig. 11A), has been extensively investigated for its antimicrobial activity against a broad range of bacterial species, in addition to its mucolytic activity, which is the ability to disrupt thick, adherent mucus commonly found in diseases like cystic fibrosis. Based on our categorization in the beginning of this article, this would rather belong to the group of polysaccharides and not glycopolymers. However, we include PAAG here, as it showcases the potential of polymeric compounds for the inhibition and removal of biofilm formation, also already in in vivo and preclinical studies, and thus demonstrates the potential for the development of glycopolymers in this field.
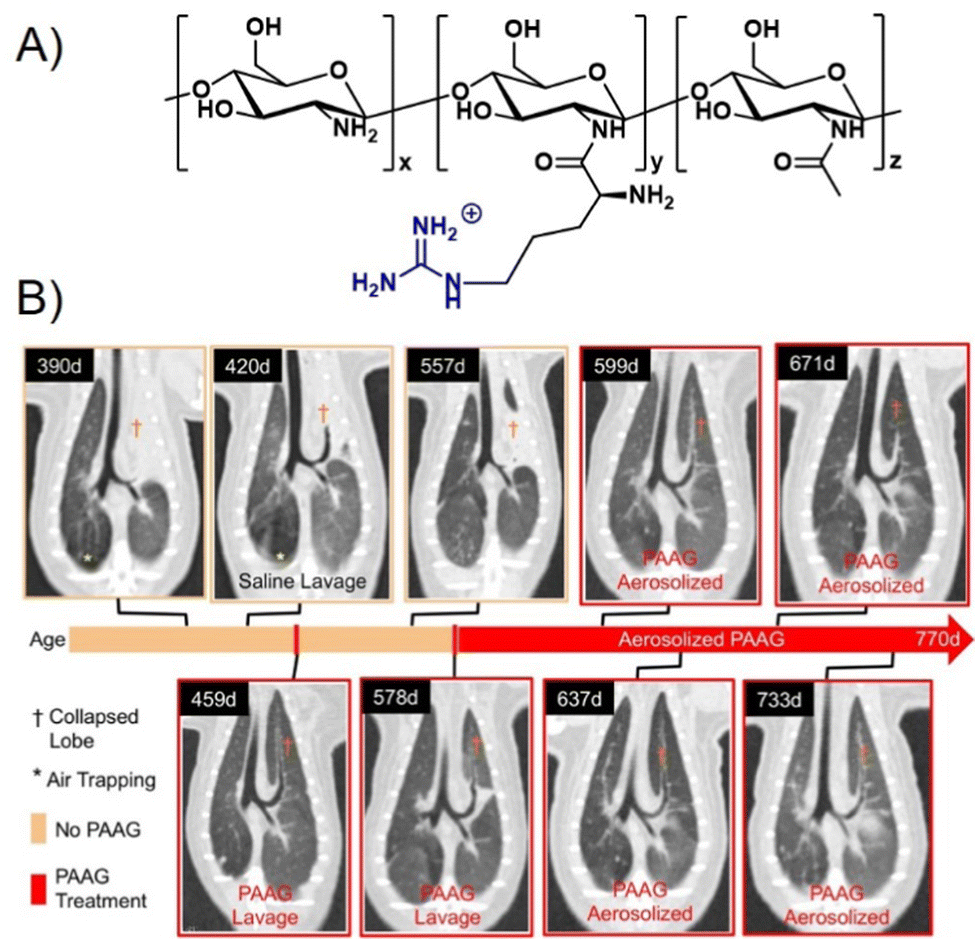 | ||
| Fig. 11 (A) Chemical structure of PAAG. (B) The effect of PAAG in an animal model (ferret) for cystic fibrosis. CT images show mucus obstructing the left cranial lobe marked by (†). At 420 days, local washing with saline was tested to remove mucus without success. Next, PAAG was applied at the same location, resulting in mucus removal (459 days). Three months after the single PAAG application, a mucus obstruction was present again (557 days), which could also be removed by PAAG washing (578 days). After that, the animal was treated with aerosolized PAAG twice a week suppressing further mucus growth. Part B taken with permission from ref. 151. | ||
A major recent finding was that PAAG acts against biofilm formation of different bacteria such as P. aeruginosa,145Burkholderia cepacian complex (BCC),146,147 methicillin-resistant S. aureus (MRSA),148 and non-tuberculosis mycobacteria (NTM),149 all of which play a pronounced role in pulmonary infections including cystic fibrosis. Narayanaswamy et al.150 also showed that PAAG is particularly effective against, so called persister cells of P. aeruginosa present in biofilms that are otherwise very difficult to treat with antibiotics. PAAG eliminated persister cells at concentrations low enough to prevent human lung epithelial cell cytotoxicity. PAAG also showed rapid bactericidal activity against two different forms of induced P. aeruginosa persister cells and demonstrated greater efficacy against persisters in vitro than the currently used antibiotics to treat persistent chronic infections.
Recently, using animal models, Fernandez-Petty and co-workers demonstrated that PAAG may be indeed beneficial for cystic fibrosis patients.151 Direct engagement of mucin by PAAG was shown to both; reduce mucous viscosity and enhance mucous transport in a calcium-dependent manner. PAAG reduced the density of purified mucin MUC5B and favourably altered the viscoelastic properties indicating that the desired mucin expansion was achieved. Using PAAG nebulization in vivo, mucus plugging was eliminated in cystic fibrosis ferrets and rats with successful mucus clearance and enhanced mucociliary transport (Fig. 11B). Preclinical toxicology studies were performed in two species, and phase I studies in humans were successfully completed.152 PAAG was found to be non-toxic, safe, and well tolerated, underlining the high potential for this polymer to be used in the treatment of cystic fibrosis.
This concept is nicely demonstrated by Boffoli and co-workers153 who developed an alternative class of anti-biofilm agents that use polyanionic glycopolymers for the targeted delivery of cationic antibacterial drugs. The anionic glycopolymers are targeting the adhesion lectins LecA and LecB of P. aeruginosa via mannose and galactose residues in combination with the antibiotic reagent (tobramycin) to prevent and disrupt biofilm formation. The tobramycin-loaded block copolymer complexes were shown to suppress the in vitro biofilm formation.
Li and co-workers154 combined RAFT-derived trehalose-based polymers with a synthetic antibiotic on a cellulose nanofibers (CNFs) scaffold. The anti-adhesion effect was evaluated using the S. aureus-HUVECs infection system where the glyco-functionalized CNFs showed a significant reduction of infection (80%) while non-glyco-functionalized CNF decreased infections only by 54%, most likely due to steric shielding of the bacteria by the CNF. In the next step, the commercially available antibiotic Ciprofloxacin was loaded onto glyco-functionalized CNFs by ionic interaction with the cellulose nanofibers. The resulting drug-loaded conjugates showed an antibacterial activity against S. aureus and P. aeruginosa at the same level of free ciprofloxacin, indicating the successful release of loaded ciprofloxacin. Based on the more selective anti-adhesive properties through the glyco-functionalization, a targeted release of the drug in proximity to the bacteria is envisioned. However, it was also noted that the release was very fast, suggesting that tighter binding of the drug to the scaffold might be beneficial.
A critical challenge for the use of glycopolymers in anti-bacterial therapy is their administration, which typically would be oral or intravenous. However, effective treatment of lung-residing bacteria would also benefit from pulmonary administration e.g., delivering antibiotics or other antibacterial agents directly to the lung. Chen and co-workers155 synthesized mannosylated Ciprofloxacin polymeric prodrugs for this purpose. Their conjugates were designed for efficient, targeted, pulmonary delivery and subsequent internalization by alveolar macrophages through mannose-recognizing cell surface receptors. The authors demonstrate the use of their prodrug in a murine model and were able to show significant improvement in efficacy against intracellular infections of airborne Francisella novicida. When administered to the lungs of mice in a prophylactic regimen, the mannosylated Ciprofloxacin polymeric prodrugs led to 50% survival rate for the otherwise lethal infection. In a treatment regimen that was concurrent with infection, the survival of mice increased to 87.5%. Free Ciprofloxacin antibiotic was ineffective in both cases.
Addressing another target and thus delivery site for antibiotics or antibacterials, Zhang et al.156 developed epithelium-penetrable polymer micelles with enhanced antibiotic internalization for treating bacterial keratitis (Fig. 12). Bacterial keratitis is an infection of the cornea with bacteria such as P. aeruginosa or S. aureus that, if left untreated, can cause blindness. Poor epithelial penetration and a short corneal retention time is a common challenge when administering drugs to treat such an eye infection. The authors proposed a new strategy for transporting antibiotics to bacteria-infected corneas via topical administration of epithelium-penetrable polymer micelles. The amphiphilic glycopolymers examined in their study contained boron dipyrromethene and boronic acid moieties to target bacterial cell wall residues. Glycopolymers were chosen due to their bioadhesive properties, enabling prolonged contact and improved drug accumulation at the target site. Efficient cellular internalization of the micelles was demonstrated, together with enhanced drug penetration and retention inside the pathogenic bacteria. Compared with the drug alone, the delivery system achieved enhanced bacterial mortality and attenuated inflammation associated with S. aureus-induced keratitis in rats, demonstrating the potential and benefit of targeted ocular drug delivery.
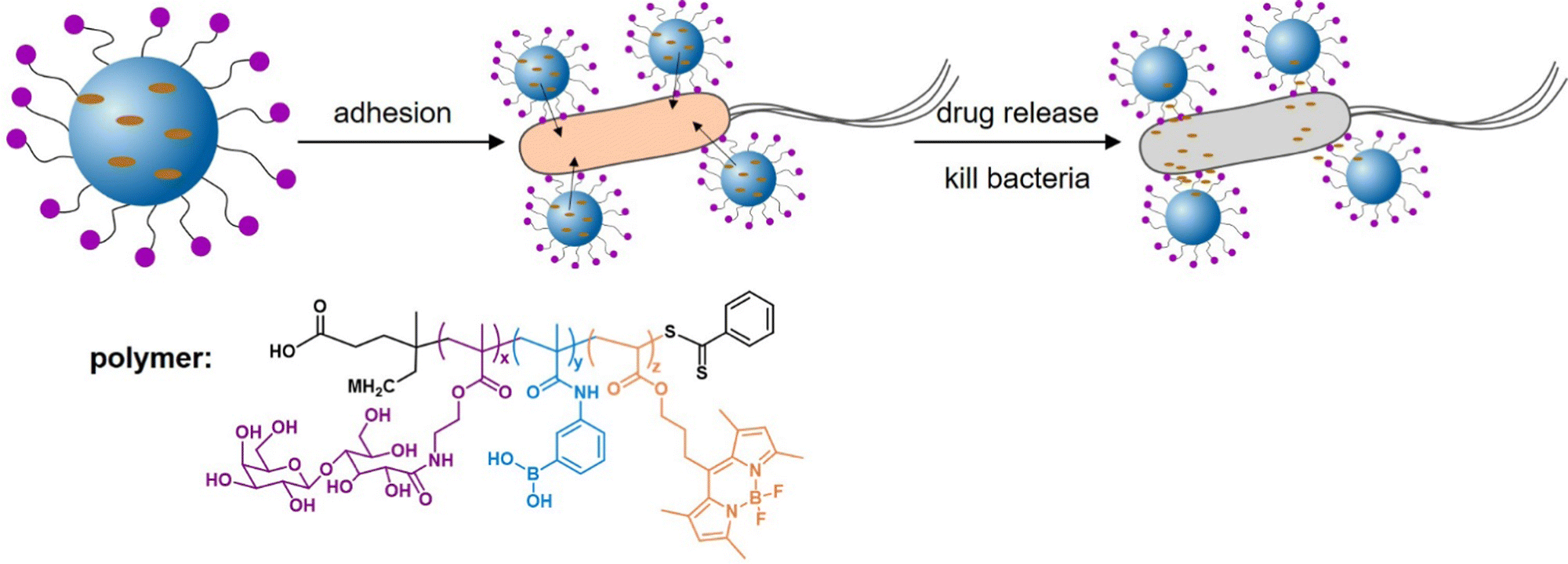 | ||
| Fig. 12 Glycopolymer micelles for the delivery of antibacterial compounds to treat infections of the cornea. (A) Self-assembly of the glycopolymer into nanotherapeutics and a schematic illustration of drug release and bacterial mortality. (B) Proposed mode of action of the glycopolymer-derived micelles. Adapted with permission from ref. 156 Copyright 2021 American Chemical Society. | ||
Kitov and Bundle are well known for their groundbreaking work on the STARFISH molecule as a multivalent nanomolar inhibitor of the Shiga-like toxin as produced by E. coli.161 Kitov et al. recently presented the design, synthesis and evaluation of a library of glycopolymers as antagonists of CTx.162 Polymer design was based on knowledge from crystal structures and thermodynamic analyses which led to the identification of the galactose residue of the GM1 ligand as the most important motif for affinity and selectivity; overall affinity is still low, in the mM range. To further increase affinity and selectivity, galactose-presenting glycopolymers were synthesized carrying different secondary binding motifs adjacent to the carbohydrate side chain. Interestingly, homomultivalent galactose polymers showed no inhibition, while heteromultivalent glycopolymers, including those incorporating SiA motifs as another carbohydrate building block of the GM1 unit, showed modest inhibition. Combinations of galactose and non-carbohydrate secondary binding motifs provided a further increase in toxin inhibition, which the authors attributed to the high levels of multivalency of binding components rather than the high binding constants of the individual binding motifs.
With the aim to more systematically study the effects of individual structural parameters of galactose glycopolymers in the inhibition of CTx, Richards et al., a study from the Gibson group,163 applied their advanced synthetic methodology of tandem post-polymerization modifications to derive a series of galactose-functionalized glycopolymers with selectively varied chain length, carbohydrate density and linker lengths. Their study showed that glycopolymers with longer linkers had a higher inhibition potency against the B subunit of CTx, which was attributed to the depth of the binding pocket of CTx. Interestingly, a nonlinear relationship was observed for the correlation between carbohydrate density and inhibitory potency where both, the highest and the lowest density structures tested, showed the highest activity when normalized to the number of galactose residues. In a follow-up report,44 the authors extended their approach to include secondary binding motifs adjacent to the galactose motif within the same side chain of the polymer. Using this approach, they were able to further increase the overall affinity of the glycopolymers in addition to the selectivity, as demonstrated by comparison in binding to peanut agglutinin. Combining this approach with thiolactone building blocks, Wilkings et al.164 introduced a third generation of glycopolymers targeting CTx by placing the carbohydrate and non-carbohydrate motif in a branched side chain on the polymer scaffold. Here, a lower carbohydrate density proved beneficial for toxin binding, but, surprisingly, introduction of benzyl motifs next to the carbohydrate completely diminished toxin affinity while another galactose recognizing lectin, Ricinus communis agglutinin, could still bind to the same glycopolymer.
Making use of hyperbranched polyglycerol scaffolds by Pouyan et al.108 as well as polyacrylamide and dextran-based linear scaffolds, Haskar et al., in a study from the Pieters lab,165 synthesized a series of multivalent constructs presenting meta-nitrophenyl α-galactoside (MNPG), a monovalent inhibitor of CTx with 100-fold increased activity compared to galactose. Their study revealed that the best results were obtained for the polyglycerol–MNPG conjugates with an almost 600 fold increase in inhibition potential when normalized to the number of ligands (MNPG motifs). The authors attributed this to the similar size and globular shape of these constructs, which is beneficial for maximum CTx binding and thus inhibition. In a follow-up report, Haskar and co-authors166 derived a series of mono- and multivalent galabiose-conjugates, including hyperbranched polyglycerol scaffolds, and demonstrated their ability to also bind Shiga toxin, another bacterial toxin of the AB5 type produced by Shigella Gram-negative bacteria.
Kimoto et al., in a study from the Miura group, systematically investigated a series of galactose-presenting glycopolymers and the incorporation of different hydrophobic units – tert-butyl, hexyl or phenyl side chains – as secondary binding elements in targeting cholera toxin B subunits (CTB).167 They demonstrated an increase by a factor of 8 in CTB affinity upon the addition of phenyl side chains as the hydrophobic unit. No additional contribution of the hydrophobic units to the glycopolymer binding was observed for other galactose-recognizing lectins. Thus, this study showcases the potential of such heteromultivalent glycopolymers to increase both affinity and selectivity.
Mahon et al. in a study from the Turnbull group168 prepared glycopolymers with switchable affinity for CTB by attaching GM1 oligosaccharides as a known binding motif of CTB to a thermoresponsive polymer scaffold, namely poly-N-ispropylacrylamide (PNIPAAm). The derived copolymers showed very high affinity towards CTB with an inhibitory concentration in the nanomolar range. Upon attaching the polymer to agarose beads, the authors demonstrated isolation of CTB from complex cell culture mixtures and showed the ability of the temperature transition of the polymer scaffold to release the toxin again. This is most likely due to a highly reduced accessibility of the GM1 units upon coil to globule transition of the polymer above its lower critical solution temperature.
Chapter 3: Glycopolymers in the fight against other pathogens
While there are several applications of synthetic glycopolymers for targeting viruses and bacteria, only a handful of examples exist for targeting other microbes such as fungi and parasites, including helminths and protozoa. One of the major reasons for this is the limited information regarding pathogen host interactions for these microorganisms. We are only now developing a better understanding of the unique roles that carbohydrates play in fungal and parasite invasion. As a result, most reports that have been published over the last 10–15 years have focused more on elucidating and understanding the unique roles of fungal/parasite-specific glycans in host–pathogen immunity,169 than the role of host glycans in engaging fungal/parasite lectins. This work has led to advancements in vaccines addressing infection,90–93 as well as the use of lectins as potential therapeutics.170 However, much work remains in developing the understanding of glycans that could lead to alternative therapeutic approaches, such as fungal/parasite inhibition, through the decoy methods described above for viruses and bacteria. Nevertheless, a few examples that show the promise of this area are highlighted here.An early study by Tøndervik et al. revealed that an oligosaccharide known as oligosaccharide G, an alginate derived from seaweed, could be used as an antifungal agent against multiple strains of Candida and Aspergillus.86 The authors used SEM and AFM to show that OligoG at ≥2% concentration significantly disrupted fungal biofilm formation even without the addition of fluconazole, a known antifungal. Further results revealed that using varying concentrations of OligoG (2, 6, 10%) in conjunction with antifungals nystatin, amphotericin B, fluconazole, miconazole, voriconazole or terbinafine could reduce the amount of these antimycotic/fungal medications required for treatment up to 16-fold for Aspergillus strains. OligoG used in conjunction with nystatin or fluconazole resulted in a 16- to 8-fold reduction in the MIC of these antimycotic/fungal medications for targeting Candida spp. Together, these results show the ability of alginate oligosaccharides such as OligoG to both serve as direct antifungal agents and to support the management of fungal treatment in conjunction with other known antifungal agents by reducing the amount of antifungal required for treatment.
More recently, Dong et al. prepared a series of microporous zwitterionic composite cryogens comprised of chitosan oligosaccharides and poly(N-methacryl arginine) (PMarg).173 Chitosan oligosaccharides, which maintain an overall positive charge under physiological conditions due to the free amine of the monomeric glucosamine subunit, have been shown to play an important role as antifungals due to their ability to bind to the negatively charged antifungal cell wall.174,175 The goal of this work was to overcome the challenges associated with using chitosan-based oligosaccharides as antifungals. Their work revealed that the zwitterionic composites could increase the duration of action while effectively inhibiting C. albicans growth through a sequential “sterilization-release” mechanism, thus revealing the importance of using copolymers to improve the antifungal properties of native oligosaccharides.
HMOs were more recently identified for their antifungal properties against C. albicans. Gonia et al.171 revealed that treatment with a solution of pooled HMOs derived from breast milk could inhibit C. albicans invasion of premature intestinal epithelial cells in a dose dependent manner by 14–67%. Furthermore, it was found that a physiologically relevant concentration of HMOs (15 mg mL−1) could prevent invasion by up to 52%, suggesting that HMO serve a crucial role in protecting the premature infant intestine from C. albicans infection. The HMOs were shown to play a specific role in reducing fungal filamentation, though the authors did not provide a mechanism for how HMOs serve in this role or which HMOs might be responsible for the observed activity.
More recently, Takagi et al.172 revealed that mucin O-glycans from three different major mucosal surfaces could also serve to attenuate C. albicans’ invasion. Mucins are a complex series of glycopolymers that form a mucous barrier that plays an important role in limiting pathogen invasion as part of the innate immune system. In their study, mucins were shown to inhibit the yeast-to-hyphal transition that is responsible for the pathogenesis of C. albicans. In addition, the mucins examined were shown to inhibit surface adhesion, biofilm formation, and cross kingdom competition between C. albicans and P. aeruginosa. Finally, evaluation of a series of synthetic O-glycans revealed that three specific structures (core 1, core 1 + fucose and core 2 + galactose) could inhibit the filamentation at the same rate as a complex mucin pool, demonstrating the therapeutic potential of these mucins.
Neqal et al.85 recently demonstrated that glycopolymers could be used to form biocides for preventing the fungal contamination of aircraft fuel tanks which are particularly susceptible to contamination by Hormoconis resinae, a filamentous fungus. Current methods to eliminate this fungus from fuel tanks often involve mixing biocides directly with the fuel. However, these additives are often toxic to the environment. To address this issue, the authors synthesized a galactosylamine polymer, which was subsequently functionalized with dodecylamine to form biocidal polymers C12Gal (NPC12Gal). The corresponding polymers were shown to inhibit the growth of H. resinae at concentrations as low as 2 × 10−5 mol mL−1 (Fig. 13), although they were not able to achieve total inhibition of mycelial growth. Nevertheless, this work demonstrated the potential for glycopolymers to serve as additives to prevent the biofouling of fuel.
 | ||
| Fig. 13 (A) NPC12Gal. (B) Inhibition of H. resinae mycelium after ten days of incubation at 25 °C and 75% RH (left, H. resinae control; middle, DMSO control; right NPC12Gal doped media). Reprinted with permission from ref. 85 Copyright 2018 Elsevier. | ||
Brissonnet et al.176 recently prepared a series of multivalent thiosialosides to inhibit the sialidases of Trypanosoma cruzi, a protozoan parasite which is responsible for Chagas Disease in humans (Fig. 14). T. cruzi sialidases are responsible for hydrolyzing host sialic acid residues leading to pathogenesis. The authors prepared a series of non-hydrolyzable thialosialoside-based glycopolymers through grafting and revealed that the corresponding polymers could significantly reduce the binding of sialidases NanA and NanA-L (3.5 and 2.3 μM) in comparison to a monomer thiosialoside reference (890 and 2160 μM). This corresponds to high RIP values of 245 and 939 for the polymer with NanA and NanA-L, respectively. While this work has not yet been applied to the pathogen itself, it demonstrates the potential for glycopolymers to serve an indirect role in parasite engagement with host cells.
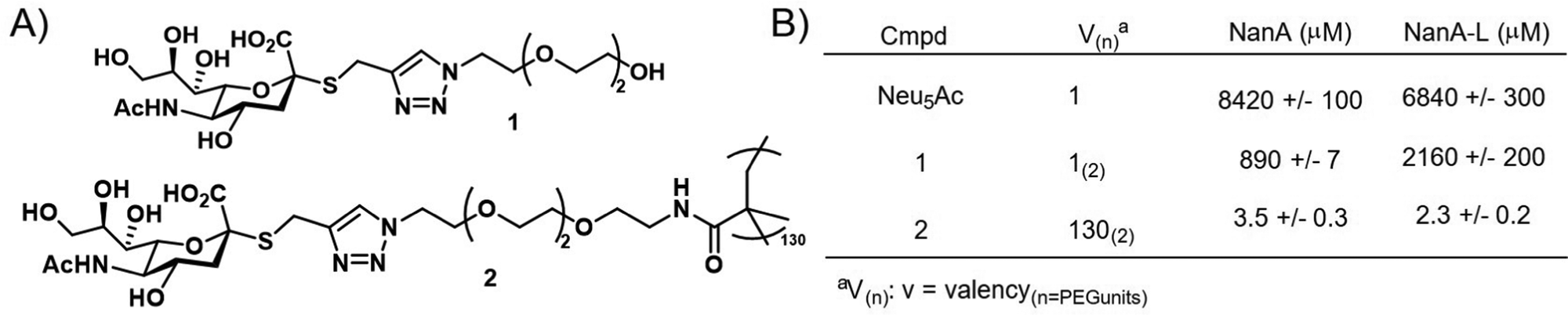 | ||
| Fig. 14 (A) Thialosialoside-based glycopolymer (2) and monomer thialosialoside reference (1). (B) Binding inhibition of thialosialoside-based glycopolymer (2) and monomer thialosialoside reference (1) against WGA, NanA and NanA-L determined using a chip (structure and data taken from ref. 176). | ||
One recent report by Liu et al.177 examining glycol-functionalized polystyrene nanospheres as Cryptosporidium surrogates shows the potential application of glycopolymers for developing new detection techniques for parasites. Cryptosporidium is a protozoan pathogen that is often found in surface waters used for drinking. The pathogen often leads to gastrointestinal illness and can be difficult to filter from surface water.178 Owing to the pathogenic nature of this parasite, this process has been difficult to study under regular laboratory conditions. To overcome these challenges, Liu and co-workers developed two polystyrene microspheres loaded with either lactose and N-(3-aminopropyl) pendant groups (poly(LAEMA-co-APMA)) or zwitterionic sulfobetaine and N-(3-aminopropyl) pendant groups (poly(SBMA-Co-APMA)) that resembled Cryptosporidium in size, density and shape. They then used these structures in filtration studies to reveal the crucial nature of surface charge and hydrophobicity on filtration. Glycopolymer microspheres showed superior particle deposition rates and were comparable to Cryptosporidium oocysts.
Chapter 4: Glycopolymers for the detection and isolation of pathogens
There is increasing interest in glycopolymers for isolating and detecting pathogens to develop treatments at early stages of infection or to prevent them entirely. For these purposes, glycopolymers are particularly well-suited given their multivalency and labelling ability. Thus, they can serve as molecular labels or sensor surfaces while retaining specific carbohydrate-pathogen interactions. Polymeric structures can also be readily designed with responsive properties to enable more controlled isolation and new sensing principles. In the spirit of the present article, this section focuses on the detection and isolation of pathogens. A more general review on glycopolymer materials for the detection of not only pathogens but also carbohydrate binding proteins, was recently published by Thalji and co-workers.27A few studies have employed glycopolymer functionalized 2D surfaces for the capture and label-free detection of pathogens. For example, Seto and co-workers187 used a copolymer with α-D-mannose and silane units to coat nickel-based micro-mashes that were able to bind and detect E. coli by IR-readout. A fast, point-of-care compatible method for detecting the influenza viruses using sialyl-functionalized glycopolymers was established by Erofeev et al.188 Here the virus-binding anionic polymers were physisorbed on cationic amine-functionalized gold coated piezo disks. The piezo resonance frequency shifts upon virus binding serving as a fast readout signal. Conductive polymers with a conjugated π-system are also often used to construct sensing surfaces with label-free optoelectronic readout capabilities. A recent review presents sensors with sugar-functionalized conductive polymers and their application for detecting lectins and pathogens.189 Also antifouling and antiviral properties can be achieved with surfaces decorated by natural or synthetic glycopolymers.190
 | ||
| Fig. 15 A fast point-of-care compatible SARS-CoV-2 paper strip test detects the presence of sialic acid binding sites on the spike protein of the virus. (A) The virus binds to gold nanoparticles functionalized with sialic acid glycopolymers followed by flow to the test line also presenting sialic acid motifs that capture the virus-carrying particles to signify a positive result. (B) Various sialic acid motifs for polymer analog reactions to prepare the glycopolymers. Reprinted with permission from ref. 192 Copyright 2020 American Chemical Society. https://pubs.acs.org/doi/10.1021/acscentsci.0c00855. | ||
Gold nanoparticles can also be used directly in colorimetric assays for pathogen detection due to their plasmon resonance, which leads to high extinction in the visible to near-infrared range depending on the size, shape and aggregation state of the particles.193 Zhang and co-workers194 used this approach to detect influenza virus upon binding to sialylated gold nanoparticles. To prepare the particles, first a copolymer with primary amine groups was synthesized via RAFT followed by post functionalization of the amine groups with α-2,6-sialyllactose and further immobilization on gold nanoparticles via a thiol end group. Using polyethylene glycol with thiol and mannose units as end groups, Richards and co-workers195 prepared gold nanoparticles that were capable of distinguishing E. coli strains with and without the mannose binding lectin FimH. Using the PEGylated gold nanoparticles, the detection of FimH expressing E. coli via UV was rapid and reliable also in media of increased ionic strength.
By applying a strong magnet, superparamagnetic Fe2O3 particles can be used to isolate bound pathogens from complex media, which could be useful for more reliable diagnostics. Furthermore, suitable libraries of functional Fe2O3 particles are commercially available. This approach was used by Li and co-workers196 who aimed at isolating and detecting influenza viruses. Commercial streptavidin-coated magnetic beads were functionalized with biotinylated glycopolymers that were modified by different sialoside ligands via click chemistry. The authors showed that, via the capture and isolation of virus with optimized magnetic beads, the detection limit of a clinically relevant rapid immuno-chromatography kit was increased by a factor of 30. Petch and co-wokers197 prepared magnetic mannosylated nanoparticles by directly binding the polymer via catechol end groups to the Fe2O3 surface. Using this approach, they could selectively isolate E. coli strains expressing FimH by means of magnetic separation.
Recently, Liu and co-workers198 grafted polymannose glycopolymers on silica nanoparticles that were further modified by europium as a fluorescent marker. The particles showed distinct fluorescence, but in presence of black phosphorus nanosheets, the fluorescence was attenuated due to the coordination between europium and phosphorous. When mannose binding E. coli were added, this fluorescence was again increased due to ablation of the nanoparticles from the nanosheets via FimH-mannose binding. This fluorescence increase was markedly lower for non-mannose binding P. aeruginosa, indicating potential use for detecting specific bacteria.
Ensuring selective binding is also a current key challenge in a polymicrobial environment. Preferably, the glycopolymers should discriminate between “bad” pathogenic bacteria and “good” commensal symbiotic bacteria that are part of the mycobiome. Hussain and co-workers202 explored this issue with a Förster resonance energy transfer (FRET)-based approach with a cationic conjugated glycopolymer. They synthesized a water-soluble polyfluorene derivative bearing mannose and quaternary ammonium groups showing only weak FRET in water. Only upon incubation with E. coli a strong FRET signal was observed. In contrast, negative controls not having mannose specificity (fungus C. albicans) and lower net negative surface charge (Gram-positive bacteria (S. aureus)), showed much lower readout. Thus, different types of bacteria could be separated as well as discriminated from fungi via fluorescence imaging even in a mixed environment.
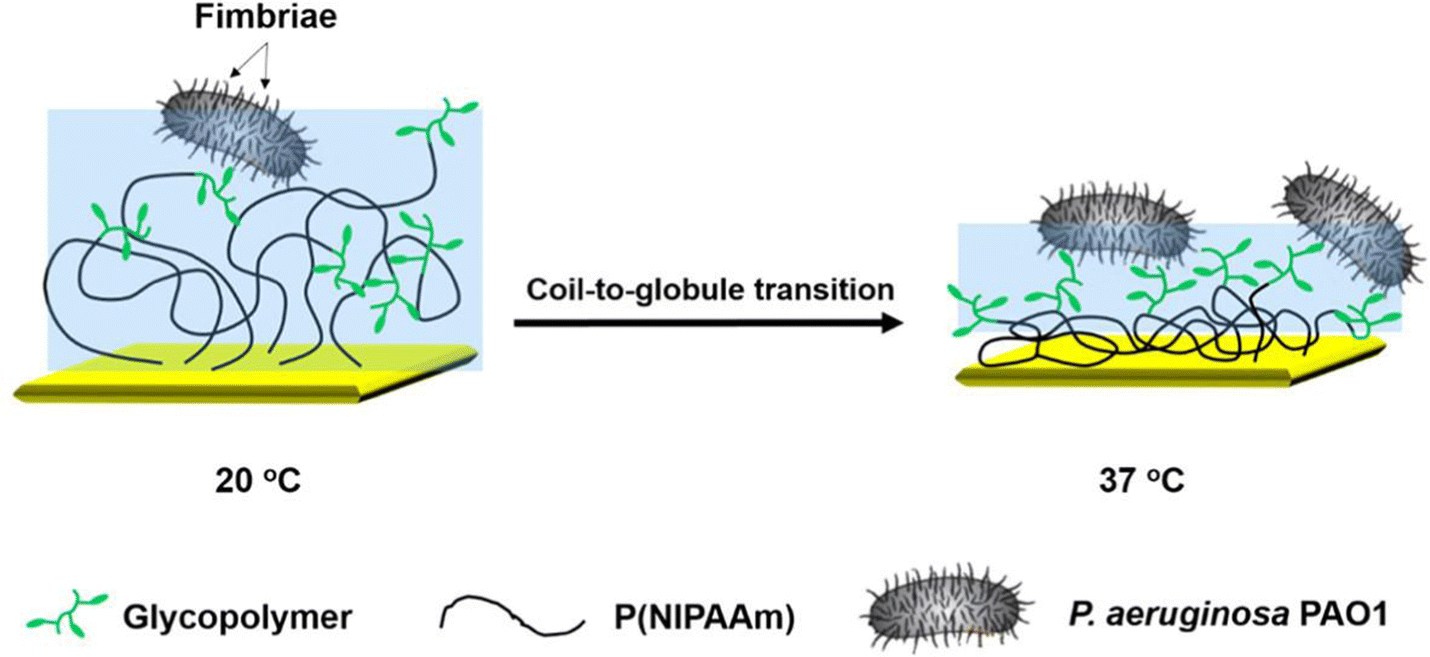 | ||
| Fig. 16 The adhesion of P. aeruginosa to a thermoresponsive polymer coating results in stronger adhesion in the collapsed state of the polymers above the LCST due to an increase in carbohydrate density and hydrophobic interactions. Reprinted with permission from ref. 203 Copyright 2015 American Chemical Society. | ||
Conclusions
In summary, it can be concluded that many of the above described glycopolymers show a high potential for their applicability as anti-microbial materials. While the inhibitory potential can be easily applied in sterilizing applications, the development of glycopolymers as potent anti-pathogenic therapeutics is accompanied by several challenges. One challenge is the selectively towards certain pathogens in a complex biological environment. The examples discussed, demonstrate the progress that has been made by careful glycopolymer design to address this issue. However, most studies still investigate pathogen engagement in in vitro settings and in a narrow parameter range; glycopolymer/pathogen selectivity has yet to be examined in more complex in vivo settings. Once undertaken, these studies will reveal how much material would be lost due to unspecific binding to other cells or microbiota components, which will be crucial information to determine realistic inhibitory potentials for in vivo applications. Currently, the realization of systemic administrations seems still pretty far in the future and will also require toxicity studies. However, many of the examples herein hold a high potential for local administration, such as anti-bacterial creams, nasal sprays, inhalants, etc. Some studies have even shown successful transfer of the materials to applications in animal models. PAAG is one example with high clinical relevance due to its biofilm-disrupting properties, even for different antibiotic-resistant bacterial strains. Successful applications in animal model and preclinical studies have been demonstrated, representing an important milestone on the way towards clinical studies in humans.Overall, the selected examples highlight the significant potential these glycopolymer conjugates have for targeted pathogen inhibition, killing, and detection. This field is still in an exploratory phase offering room for the development of new materials. It can be predicted that more sophisticated glycopolymers are currently under development, such as heteromultivalent, glycan-mimicking structures or systems with defined three-dimensional structures will enable more precise functional properties. This synthetic progress will give new opportunities to better understand infections and to develop new therapeutics. To put these concepts into practice, improved molecular understanding of the glycopolymers biological activity, biodegradability, and safety assessments should be the prime foci in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
LH thanks Jonathan Cramer for support in generating Fig. 5. LH thanks the DFG for financial support through the CRC 1208. MH and LH thank the DFG for financial support through the FOR 2327. UGD and LH thank the SFF – Strategic Research Fund of the Heinrich Heine University Düsseldorf for financial support.Notes and references
- J. Piret and G. Boivin, Front. Microbiol., 2020, 11, 631736 CrossRef PubMed.
- Y. Ben, C. Fu, M. Hu, L. Liu, M. H. Wong and C. Zheng, Environ. Res., 2019, 169, 483–493 CrossRef CAS PubMed.
- L. Marongiu, M. Valache, F. A. Facchini and F. Granucci, Clin. Sci., 2021, 135, 2217–2242 CrossRef CAS PubMed.
- M. Wicinski, E. Sawicka, J. Gebalski, K. Kubiak and B. Malinowski, Nutrients, 2020, 12, 266–279 CrossRef CAS PubMed.
- N. K. Karamanos, Z. Piperigkou, A. D. Theocharis, H. Watanabe, M. Franchi, S. Baud, S. Brezillon, M. Gotte, A. Passi, D. Vigetti, S. Ricard-Blum, R. D. Sanderson, T. Neill and R. V. Iozzo, Chem. Rev., 2018, 118, 9152–9232 CrossRef CAS PubMed.
- S. K. Linden, P. Sutton, N. G. Karlsson, V. Korolik and M. A. McGuckin, Mucosal Immunol., 2008, 1, 183–197 CrossRef CAS PubMed.
- B. O. Schroeder, Gastroenterol. Rep., 2019, 7, 3–12 CrossRef PubMed.
- J. L. McAuley, L. Corcilius, H. X. Tan, R. J. Payne, M. A. McGuckin and L. E. Brown, Mucosal. Immunol., 2017, 10, 1581–1593 CrossRef CAS PubMed.
- M. Hoffmann, N. L. Snyder and L. Hartmann, Macromolecules, 2022, 55, 7957–7973 CrossRef CAS PubMed.
- T. K. Lindhorst, Glycopolymer Code, The Royal Society of Chemistry, 2015, pp. 1–16 10.1039/9781782622666-00001.
- D. C. Koester, A. Holkenbrink and D. B. Werz, Synthesis, 2010, 3217–3242, DOI:10.1055/s-0030-1258228.
- R. Hevey, Eur. J. Chem., 2021, 27, 2240–2253 CrossRef CAS PubMed.
- A. Sadurni and R. Gilmour, Eur. J. Org. Chem., 2018, 3684–3687 CrossRef CAS PubMed.
- M. Delbianco, P. Bharate, S. Varela-Aramburu and P. H. Seeberger, Chem. Rev., 2016, 116, 1693–1752 CrossRef CAS PubMed.
- S. H. Liyanage and M. Yan, Chem. Commun., 2020, 56, 13491–13505 RSC.
- G. C. Daskhan, N. Berthet, B. Thomas, M. Fiore and O. Renaudet, Carbohydr. Res., 2015, 405, 13–22 CrossRef CAS PubMed.
- M. C. Galan, P. Dumy and O. Renaudet, Chem. Soc. Rev., 2013, 42, 4599–4612 RSC.
- W. Doelman and S. I. van Kasteren, Org. Biomol. Chem., 2022, 20, 6487–6507 RSC.
- C. S. Bennett, R. J. Payne, K. M. Koeller and C.-H. Wong, in Glycoscience, ed. B. O. Fraser-Reid, K. Tatsuta and J. Thiem, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, ch. 45, pp. 1795–1857 DOI:10.1007/978-3-540-30429-6_45.
- L. Mousavifar and R. Roy, Molecules, 2021, 26, 2428 CrossRef CAS PubMed.
- Y. M. Chabre and R. Roy, Curr. Top. Med. Chem., 2008, 8, 1237–1285 CrossRef CAS PubMed.
- G. Rivero-Barbarroja, J. M. Benito, C. Ortiz Mellet and J. M. Garcia Fernandez, Nanomaterials, 2020, 10, 2517 CrossRef CAS PubMed.
- F. Seidi, R. Jenjob, T. Phakkeeree and D. Crespy, J. Controlled Release, 2018, 284, 188–212 CrossRef CAS PubMed.
- P. Bojarova and V. Kren, Biomater. Sci., 2016, 4, 1142–1160 RSC.
- Z. S. Clauss and J. R. Kramer, Adv. Drug Delivery Rev., 2021, 169, 152–167 CrossRef CAS PubMed.
- C. Bonduelle and S. Lecommandoux, Biomacromolecules, 2013, 14, 2973–2983 CrossRef CAS PubMed.
- M. R. Thalji, A. A. Ibrahim, K. F. Chong, A. V. Soldatov and G. A. M. Ali, Top. Curr. Chem., 2022, 380, 45 CrossRef CAS PubMed.
- I. Pramudya and H. Chung, Biomater. Sci., 2019, 7, 4848–4872 RSC.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS PubMed.
- Q. Zhang and D. M. Haddleton, in Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II, ed. V. Percec, Springer International Publishing, Cham, 2013, pp. 39–59 DOI:10.1007/12_2013_254.
- Q. Qin, S. Lang and X. Huang, J. Carbohydr. Chem., 2021, 40, 1–44 CrossRef CAS PubMed.
- S. R. S. Ting, G. J. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- J. Wang, D. Wang, Y. Zhang and J. Dong, ACS Biomater. Sci. Eng., 2021, 7, 963–982 CrossRef CAS PubMed.
- C. S. Kwan, A. R. Cerullo and A. B. Braunschweig, ChemPlusChem, 2020, 85, 2704–2721 CrossRef CAS PubMed.
- C. R. Becer, Macromol. Rapid Commun., 2012, 33, 742–752 CrossRef CAS PubMed.
- J. L. Jimenez Blanco, C. Ortiz Mellet and J. M. Garcia Fernandez, Chem. Soc. Rev., 2013, 42, 4518–4531 RSC.
- C. Muller, G. Despras and T. K. Lindhorst, Chem. Soc. Rev., 2016, 45, 3275–3302 RSC.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615–1619 CrossRef CAS PubMed.
- B. Martyn, C. I. Biggs and M. I. Gibson, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 40–47 CrossRef CAS.
- Z. F. Liu, Y. Zhu, W. L. Ye, T. Wu, D. Y. Miao, W. Deng and M. N. Liu, Polym. Chem., 2019, 10, 4006–4016 RSC.
- D. Ponader, P. Maffre, J. Aretz, D. Pussak, N. M. Ninnemann, S. Schmidt, P. H. Seeberger, C. Rademacher, G. U. Nienhaus and L. Hartmann, J. Am. Chem. Soc., 2014, 136, 2008–2016 CrossRef CAS PubMed.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130–15132 CrossRef CAS PubMed.
- M. W. Jones, L. Otten, S. J. Richards, R. Lowery, D. J. Phillips, D. M. Haddleton and M. I. Gibson, Chem. Sci., 2014, 5, 1611–1616 RSC.
- T. Freichel, V. Heine, D. Laaf, E. E. Mackintosh, S. Sarafova, L. Elling, N. L. Snyder and L. Hartmann, Macromol. Biosci., 2020, 20, e2000163 CrossRef PubMed.
- S. G. Spain, M. I. Gibson and N. R. Cameron, J, Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059–2072 CrossRef CAS.
- T. Pelras and K. Loos, Prog. Polym. Sci., 2021, 117, 101393 CrossRef CAS.
- W. Choi, H. Sun, C. Battistella, O. Berger, M. A. Vratsanos, M. M. Wang and N. C. Gianneschi, Angew. Chem., Int. Ed., 2020, 59, 19762–19772 CrossRef CAS PubMed.
- Y. Abdouni, G. Yilmaz and C. R. Becer, Macromol. Rapid Commun., 2017, 38, 1700212 CrossRef PubMed.
- S. Bhatia, L. C. Camacho and R. Haag, J. Am. Chem. Soc., 2016, 138, 8654–8666 CrossRef CAS PubMed.
- S. Liese and R. R. Netz, Beilstein J. Org. Chem., 2015, 11, 804–816 CrossRef CAS PubMed.
- H. Q. Wang, F. Jacobi, J. Waschke, L. Hartmann, H. Lowen and S. Schmidt, Adv. Funct. Mater., 2017, 27, 1702040 CrossRef.
- R. S. Kane, Langmuir, 2010, 26, 8636–8640 CrossRef CAS PubMed.
- L. Su, Y. Feng, K. Wei, X. Xu, R. Liu and G. Chen, Chem. Rev., 2021, 121, 10950–11029 CrossRef CAS PubMed.
- G. Yilmaz and C. R. Becer, Front. Bioeng. Biotechnol., 2014, 2, 39 Search PubMed.
- S. Behren and U. Westerlind, Eur. J. Org. Chem., 2022, e202200795 Search PubMed.
- A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penades, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schaffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- X. Wei and R. J. Pieters, ARKIVOC, 2021, 2021, 297–312 Search PubMed.
- V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492–4503 RSC.
- R. J. Pieters, Adv. Exp. Med. Biol., 2011, 715, 227–240 CrossRef CAS PubMed.
- K. Bhattacharya, U. Kalita and N. K. Singha, Polym. Chem., 2022, 13, 1458–1483 RSC.
- Y. Kim, J. Y. Hyun and I. Shin, Chem. Soc. Rev., 2021, 50, 10567–10593 RSC.
- M. H. Stenzel, Macromolecules, 2022, 55, 4867–4890 CrossRef CAS.
- J. Poole, C. J. Day, M. von Itzstein, J. C. Paton and M. P. Jennings, Nat. Rev. Microbiol., 2018, 16, 440–452 CrossRef CAS PubMed.
- T. Klein, D. Abgottspon, M. Wittwer, S. Rabbani, J. Herold, X. Jiang, S. Kleeb, C. Luthi, M. Scharenberg, J. Bezencon, E. Gubler, L. Pang, M. Smiesko, B. Cutting, O. Schwardt and B. Ernst, J. Med. Chem., 2010, 53, 8627–8641 CrossRef CAS PubMed.
- P. S. Guiton, C. K. Cusumano, K. A. Kline, K. W. Dodson, Z. Han, J. W. Janetka, J. P. Henderson, M. G. Caparon and S. J. Hultgren, Antimicrob. Agents Chemother., 2012, 56, 4738–4745 CrossRef CAS PubMed.
- C. K. Cusumano, J. S. Pinkner, Z. Han, S. E. Greene, B. A. Ford, J. R. Crowley, J. P. Henderson, J. W. Janetka and S. J. Hultgren, Sci. Transl. Med., 2011, 3, 109ra115 Search PubMed.
- S. Kuhaudomlarp, E. Gillon, A. Varrot and A. Imberty, Methods Mol. Biol., 2020, 2132, 257–266 CrossRef CAS PubMed.
- H.-L. Chen, Adv. Exp. Med. Biol., 2011, 705, 53–80 CrossRef CAS PubMed.
- T. Peters, R. Creutznacher, T. Maass, A. Mallagaray, P. Ogrissek, S. Taube, L. Thiede and C. Uetrecht, Biochem. Soc. Trans., 2022, 50, 347–359 CrossRef CAS PubMed.
- E. P. Mitchell, C. Sabin, L. Snajdrova, M. Pokorna, S. Perret, C. Gautier, C. Hofr, N. Gilboa-Garber, J. Koca, M. Wimmerova and A. Imberty, Proteins, 2005, 58, 735–746 CrossRef CAS PubMed.
- D. Passos da Silva, M. L. Matwichuk, D. O. Townsend, C. Reichhardt, D. Lamba, D. J. Wozniak and M. R. Parsek, Nat. Commun., 2019, 10, 2183 CrossRef PubMed.
- J. E. Stencel-Baerenwald, K. Reiss, D. M. Reiter, T. Stehle and T. S. Dermody, Nat. Rev. Microbiol., 2014, 12, 739–749 CrossRef CAS PubMed.
- L. M. Filkins, T. H. Hampton, A. H. Gifford, M. J. Gross, D. A. Hogan, M. L. Sogin, H. G. Morrison, B. J. Paster and G. A. O'Toole, J. Bacteriol., 2012, 194, 4709–4717 CrossRef CAS PubMed.
- J. J. Wine, G. C. Hansson, P. Konig, N. S. Joo, A. Ermund and M. Pieper, J. Cystic Fibrosis, 2018, 17, S35–S39 CrossRef PubMed.
- A. Rousseaux, C. Brosseau, S. Le Gall, H. Piloquet, S. Barbarot and M. Bodinier, Front. Immunol., 2021, 12, 680911 CrossRef CAS PubMed.
- L. Cooling, Clin. Microbiol. Rev., 2015, 28, 801–870 CrossRef CAS PubMed.
- J. E. Heggelund, A. Varrot, A. Imberty and U. Krengel, Curr. Opin. Struct. Biol., 2017, 44, 190–200 CrossRef CAS PubMed.
- B. Garcia, J. Merayo-Lloves, C. Martin, I. Alcalde, L. M. Quiros and F. Vazquez, Front. Microbiol., 2016, 7, 220 Search PubMed.
- E. Kamhi, E. J. Joo, J. S. Dordick and R. J. Linhardt, Biol. Rev. Cambridge Philos. Soc., 2013, 88, 928–943 CrossRef PubMed.
- T. Manon-Jensen, Y. Itoh and J. R. Couchman, FEBS J., 2010, 277, 3876–3889 CrossRef CAS PubMed.
- M. Chakravarty and A. Vora, Drug Delivery Transl. Res., 2021, 11, 748–787 CrossRef CAS PubMed.
- M. B. Calvert, V. R. Jumde and A. Titz, Beilstein J. Org. Chem., 2018, 14, 2607–2617 CrossRef CAS PubMed.
- Z. Si, W. Zheng, D. Prananty, J. Li, C. H. Koh, E. T. Kang, K. Pethe and M. B. Chan-Park, Chem. Sci., 2022, 13, 345–364 RSC.
- M. Neqal, A. Voisin, V. Neto, V. Coma and V. Heroguez, Eur. Polym. J., 2018, 105, 304–312 CrossRef CAS.
- A. Tondervik, H. Sletta, G. Klinkenberg, C. Emanuel, L. C. Powell, M. F. Pritchard, S. Khan, K. M. Craine, E. Onsoyen, P. D. Rye, C. Wright, D. W. Thomas and K. E. Hill, PLoS One, 2014, 9, e112518 CrossRef PubMed.
- R. Sunasee and R. Narain, Macromol. Biosci., 2013, 13, 9–27 CrossRef CAS PubMed.
- D. Shae, A. Postma and J. T. Wilson, Ther Delivery, 2016, 7, 193–196 CrossRef CAS PubMed.
- M. Anderluh, F. Berti, A. Bzducha-Wrobel, F. Chiodo, C. Colombo, F. Compostella, K. Durlik, X. Ferhati, R. Holmdahl, D. Jovanovic, W. Kaca, L. Lay, M. Marinovic-Cincovic, M. Marradi, M. Ozil, L. Polito, J. J. Reina, C. A. Reis, R. Sackstein, A. Silipo, U. Svajger, O. Vanek, F. Yamamoto, B. Richichi and S. J. van Vliet, FEBS J., 2022, 289, 4251–4303 CrossRef CAS PubMed.
- L. Del Bino and M. R. Romano, Drug Discovery Today: Technol., 2020, 38, 45–55 CrossRef PubMed.
- L. C. Garcia-Carnero, L. A. Perez-Garcia, J. A. Martinez-Alvarez, J. E. Reyes-Martinez and H. M. Mora-Montes, Infect Drug Resist., 2018, 11, 903–913 CrossRef CAS PubMed.
- J. A. Jaurigue and P. H. Seeberger, Front. Cell. Infect. Microbiol., 2017, 7, 248 CrossRef PubMed.
- Y. L. Huang and C. Y. Wu, Expert Rev. Vaccines, 2010, 9, 1257–1274 CrossRef CAS PubMed.
- D. S. Dimitrov, Nat. Rev. Microbiol., 2004, 2, 109–122 CrossRef PubMed.
- L. J. Stroh and T. Stehle, Annu. Rev. Virol., 2014, 1, 285–306 CrossRef PubMed.
- T. Suenaga and H. Arase, Viral Interactions with Glycans, Glycoscience: Biol. Med, 2014, 785–794, DOI:10.1007/978-4-431-54841-6_152.
- J. Wang, Trends Biochem. Sci., 2002, 27, 122–126 CrossRef CAS PubMed.
- D. J. Vigerust and V. L. Shepherd, Trends Microbiol., 2007, 15, 211–218 CrossRef CAS PubMed.
- A. Kuroki, J. Tay, G. H. Lee and Y. Y. Yang, Adv. Healthcare Mater., 2021, 10, e2101113 CrossRef PubMed.
- Y. Umar, S. Al-Batty, H. Rahman, O. Ashwaq, A. Sarief, Z. Sadique, P. A. Sreekumar and S. K. M. Haque, J. Polym. Environ., 2022, 30, 1244–1263 CrossRef CAS PubMed.
- A. Akbari, A. Bigham, V. Rahimkhoei, S. Sharifi and E. Jabbari, Polymers, 2022, 14, 1634 CrossRef CAS PubMed.
- R. H. Bianculli, J. D. Mase and M. D. Schulz, Macromolecules, 2020, 53, 9158–9186 CrossRef CAS.
- X. Huang, W. G. Xu, M. Q. Li, P. Zhang, Y. S. Zhang, J. X. Ding and X. S. Chen, Matter, 2021, 4, 1892–1918 CrossRef CAS.
- K. Jung, N. Corrigan, E. H. H. Wong and C. Boyer, Adv. Mater., 2022, 34, e2105063 CrossRef PubMed.
- A. Spaltenstein and G. M. Whitesides, J. Am. Chem. Soc., 1991, 113, 686–687 CrossRef CAS.
- M. N. Matrosovich, L. V. Mochalova, V. P. Marinina, N. E. Byramova and N. V. Bovin, FEBS Lett., 1990, 272, 209–212 CrossRef CAS PubMed.
- M. N. Stadtmueller, S. Bhatia, P. Kiran, M. Hilsch, V. Reiter-Scherer, L. Adam, B. Parshad, M. Budt, S. Klenk, K. Sellrie, D. Lauster, P. H. Seeberger, C. P. R. Hackenberger, A. Herrmann, R. Haag and T. Wolff, J. Med. Chem., 2021, 64, 12774–12789 CrossRef CAS PubMed.
- P. Pouyan, M. Cherri and R. Haag, Polymers, 2022, 14, 2684 CrossRef CAS PubMed.
- K. Matsuoka, T. Kaneshima, R. Adachi, J. Sasaki, T. Hashiguchi, T. Koyama, T. Matsushita and K. Hatano, Bioorg. Med. Chem. Lett., 2021, 52, 128389 CrossRef CAS.
- M. Wallert, C. Nie, P. Anilkumar, S. Abbina, S. Bhatia, K. Ludwig, J. N. Kizhakkedathu, R. Haag and S. Block, Small, 2020, 16, e2004635 CrossRef PubMed.
- L. Soria-Martinez, S. Bauer, M. Giesler, S. Schelhaas, J. Materlik, K. Janus, P. Pierzyna, M. Becker, N. L. Snyder, L. Hartmann and M. Schelhaas, J. Am. Chem. Soc., 2020, 142, 5252–5265 CrossRef CAS PubMed.
- J. Cramer, B. Aliu, X. Jiang, T. Sharpe, L. Pang, A. Hadorn, S. Rabbani and B. Ernst, ChemMedChem, 2021, 16, 2345–2353 CrossRef CAS PubMed.
- J. Cramer, A. Lakkaichi, B. Aliu, R. P. Jakob, S. Klein, I. Cattaneo, X. Jiang, S. Rabbani, O. Schwardt, G. Zimmer, M. Ciancaglini, T. Abreu Mota, T. Maier and B. Ernst, J. Am. Chem. Soc., 2021, 143, 17465–17478 CrossRef CAS PubMed.
- S. C. Purcell and K. Godula, Interface Focus, 2019, 9, 20180080 CrossRef PubMed.
- M. Critcher, T. O'Leary and M. L. Huang, Biochem. J., 2021, 478, 703–719 CrossRef CAS PubMed.
- C. S. Delaveris, E. R. Webster, S. M. Banik, S. G. Boxer and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12643–12650 CrossRef CAS PubMed.
- D. J. Honigfort, M. O. Altman, P. Gagneux and K. Godula, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2107896118 CrossRef CAS PubMed.
- S. C. Purcell, M. H. Zhang, D. J. Honigfort, H. J. C. Ng, A. L. Michalak and K. Godula, Chem. Sci., 2022, 13, 6626–6635 RSC.
- N. P. Pera and R. J. Peters, MedChemComm, 2014, 5, 1027–1035 RSC.
- C. S. Hayes, S. K. Aoki and D. A. Low, Annu. Rev. Genet., 2010, 44, 71–90 CrossRef CAS PubMed.
- E. V. Sokurenko, V. Chesnokova, D. E. Dykhuizen, I. Ofek, X. R. Wu, K. A. Krogfelt, C. Struve, M. A. Schembri and D. L. Hasty, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8922–8926 CrossRef CAS PubMed.
- A. Hendriks, R. van Dalen, S. Ali, D. Gerlach, G. A. van der Marel, F. F. Fuchsberger, P. C. Aerts, C. J. C. de Haas, A. Peschel, C. Rademacher, J. A. G. van Strijp, J. D. C. Codee and N. M. van Sorge, ACS Infect. Dis., 2021, 7, 624–635 CrossRef CAS PubMed.
- J. Vila, E. Saez-Lopez, J. R. Johnson, U. Romling, U. Dobrindt, R. Canton, C. G. Giske, T. Naas, A. Carattoli, M. Martinez-Medina, J. Bosch, P. Retamar, J. Rodriguez-Bano, F. Baquero and S. M. Soto, FEMS Microbiol. Rev., 2016, 40, 437–463 CrossRef CAS PubMed.
- M. M. Sauer, R. P. Jakob, T. Luber, F. Canonica, G. Navarra, B. Ernst, C. Unverzagt, T. Maier and R. Glockshuber, J. Am. Chem. Soc., 2019, 141, 936–944 CrossRef CAS PubMed.
- M. F. Moradali, S. Ghods and B. H. Rehm, Front. Cell. Infect. Microbiol., 2017, 7, 39 Search PubMed.
- Z. Pang, R. Raudonis, B. R. Glick, T. J. Lin and Z. Cheng, Biotechnol. Adv., 2019, 37, 177–192 CrossRef CAS.
- D. Balasubramanian, L. Harper, B. Shopsin and V. J. Torres, Pathog. Dis., 2017, 75, ftx005 CrossRef PubMed.
- X. B. Yan, A. Sivignon, N. Barnich, S. G. Gouin, J. Bouckaert, E. Fleurya and J. Bernard, Polym. Chem., 2016, 7, 2674–2683 RSC.
- X. Yan, A. Sivignon, N. Yamakawa, A. Crepet, C. Travelet, R. Borsali, T. Dumych, Z. Li, R. Bilyy, D. Deniaud, E. Fleury, N. Barnich, A. Darfeuille-Michaud, S. G. Gouin, J. Bouckaert and J. Bernard, Biomacromolecules, 2015, 16, 1827–1836 CrossRef CAS.
- S. Boden, F. Reise, J. Kania, T. K. Lindhorst and L. Hartmann, Macromol. Biosci., 2019, 19, e1800425 CrossRef.
- S. A. Hill, C. Gerke and L. Hartmann, Chem. – Asian J., 2018, 13, 3611–3622 CrossRef CAS PubMed.
- Y. Luo, Y. Gu, R. Feng, J. Brash, A. M. Eissa, D. M. Haddleton, G. Chen and H. Chen, Chem. Sci., 2019, 10, 5251–5257 RSC.
- L. Zheng, Y. Luo, K. Chen, Z. Zhang and G. Chen, Biomacromolecules, 2020, 21, 5233–5240 CrossRef CAS PubMed.
- A. Banger, J. Sindram, M. Otten, J. Kania, D. Wilms, A. Strzelczyk, S. Miletic, T. C. Marlovits, M. Karg and L. Hartmann, Polym. Chem., 2021, 12, 4795–4802 RSC.
- S. Omardien, S. Brul and S. A. Zaat, Front. Cell Dev. Biol., 2016, 4, 111 Search PubMed.
- R. A. Dorschner, V. K. Pestonjamasp, S. Tamakuwala, T. Ohtake, J. Rudisill, V. Nizet, B. Agerberth, G. H. Gudmundsson and R. L. Gallo, J. Invest. Dermatol., 2001, 117, 91–97 CrossRef CAS PubMed.
- Z. Q. Zheng, B. Y. Wang, J. Chen, Y. Wang, Z. Y. Miao, C. Y. Shang and Q. Zhang, Eur. Polym. J., 2021, 158, 110702 CrossRef CAS.
- D. Li, J. Chen, M. Hong, Y. Wang, D. M. Haddleton, G. Z. Li and Q. Zhang, Biomacromolecules, 2021, 22, 2224–2232 CrossRef CAS PubMed.
- J. Chen, C. Y. Bao, R. Han, G. Z. Li, Z. Q. Zheng, Y. Wang and Q. Zhang, Polym. Chem., 2022, 13, 2285–2294 RSC.
- D. Pranantyo, L. Q. Xu, Z. Hou, E.-T. Kang and M. B. Chan-Park, Polym. Chem., 2017, 8, 3364–3373 RSC.
- L. K. Vestby, T. Gronseth, R. Simm and L. L. Nesse, Antibiotics, 2020, 9, 59–87 CrossRef CAS PubMed.
- C. Moser, H. T. Pedersen, C. J. Lerche, M. Kolpen, L. Line, K. Thomsen, N. Hoiby and P. O. Jensen, APMIS, 2017, 125, 320–338 CrossRef PubMed.
- R. Roy, M. Tiwari, G. Donelli and V. Tiwari, Virulence, 2018, 9, 522–554 CrossRef CAS PubMed.
- K. S. Bucher, N. Babic, T. Freichel, F. Kovacic and L. Hartmann, Macromol. Biosci., 2018, 18, e1800337 CrossRef PubMed.
- B. A. Garcia, M. S. McDaniel, A. J. Loughran, J. D. Johns, V. Narayanaswamy, C. Fernandez Petty, S. E. Birket, S. M. Baker, R. Barnaby, B. A. Stanton, J. B. Foote, S. M. Rowe and W. E. Swords, Microbiology, 2022, 168, 001121 CrossRef CAS PubMed.
- V. P. Narayanaswamy, A. P. Duncan, J. J. LiPuma, W. P. Wiesmann, S. M. Baker and S. M. Townsend, Antimicrob. Agents Chemother., 2019, 63, e00498–19 CrossRef CAS PubMed.
- V. P. Narayanaswamy, S. Giatpaiboon, S. M. Baker, W. P. Wiesmann, J. J. LiPuma and S. M. Townsend, PLoS One, 2017, 12, e0179776 CrossRef PubMed.
- V. P. Narayanaswamy, S. A. Giatpaiboon, J. Uhrig, P. Orwin, W. Wiesmann, S. M. Baker and S. M. Townsend, PLoS One, 2018, 13, e0191522 CrossRef PubMed.
- V. P. Narayanaswamy, S. M. Townsend, A. J. Loughran, W. Wiesmann and S. Baker, Front. Microbiol., 2022, 13, 821820 CrossRef PubMed.
- V. P. Narayanaswamy, L. L. Keagy, K. Duris, W. Wiesmann, A. J. Loughran, S. M. Townsend and S. Baker, Front. Microbiol., 2018, 9, 1724 CrossRef PubMed.
- C. M. Fernandez-Petty, G. W. Hughes, H. L. Bowers, J. D. Watson, B. H. Rosen, S. M. Townsend, C. Santos, C. E. Ridley, K. K. Chu, S. E. Birket, Y. Li, H. M. Leung, M. Mazur, B. A. Garcia, T. I. A. Evans, E. F. Libby, H. Hathorne, J. Hanes, G. J. Tearney, J. P. Clancy, J. F. Engelhardt, W. E. Swords, D. J. Thornton, W. P. Wiesmann, S. M. Baker and S. M. Rowe, JCI Insight, 2019, 4, 125954 CrossRef PubMed.
- A Study of the Safety and Tolerability of Inhaled SNSP113 in Healthy Subjects and Subjects With Stable Cystic Fibrosis, (https://ClinicalTrials.gov/show/NCT03309358).
- D. Boffoli, F. Bellato, G. Avancini, P. Gurnani, G. Yilmaz, M. Romero, S. Robertson, F. Moret, F. Sandrelli, P. Caliceti, S. Salmaso, M. Camara, G. Mantovani and F. Mastrotto, Drug Delivery Transl. Res., 2022, 12, 1788–1810 CrossRef CAS PubMed.
- Y. M. Li, M. Milewska, Y. Y. Khine, N. Ariotti and M. H. Stenzel, Polym. Chem., 2022, 13, 1502–1509 RSC.
- J. Chen, F. Y. Su, D. Das, S. Srinivasan, H. N. Son, B. Lee, F. Radella, 2nd, D. Whittington, T. Monroe-Jones, T. E. West, A. J. Convertine, S. J. Skerrett, P. S. Stayton and D. M. Ratner, Biomaterials, 2019, 195, 38–50 CrossRef CAS PubMed.
- Y. Zhang, Y. Yu, G. Li, X. Zhang, Z. Wu and L. Lin, Biomacromolecules, 2021, 22, 2020–2032 CrossRef CAS PubMed.
- A. M. Wands, A. Fujita, J. E. McCombs, J. Cervin, B. Dedic, A. C. Rodriguez, N. Nischan, M. R. Bond, M. Mettlen, D. C. Trudgian, A. Lemoff, M. Quiding-Jarbrink, B. Gustavsson, C. Steentoft, H. Clausen, H. Mirzaei, S. Teneberg, U. Yrlid and J. J. Kohler, eLife, 2015, 4, e09545 CrossRef PubMed.
- A. M. Wands, J. Cervin, H. Huang, Y. Zhang, G. Youn, C. A. Brautigam, M. Matson Dzebo, P. Bjorklund, V. Wallenius, D. K. Bright, C. S. Bennett, P. Wittung-Stafshede, N. S. Sampson, U. Yrlid and J. J. Kohler, ACS Infect. Dis., 2018, 4, 758–770 CrossRef CAS PubMed.
- J. E. Heggelund, D. Burschowsky, V. A. Bjornestad, V. Hodnik, G. Anderluh and U. Krengel, PLoS Pathog., 2016, 12, e1005567 CrossRef PubMed.
- A. Holmner, M. Lebens, S. Teneberg, J. Angstrom, M. Okvist and U. Krengel, Structure, 2004, 12, 1655–1667 CrossRef CAS PubMed.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS PubMed.
- J. M. Jacobson, P. I. Kitov and D. R. Bundle, Carbohydr. Res., 2013, 378, 4–14 CrossRef CAS PubMed.
- S. J. Richards, M. W. Jones, M. Hunaban, D. M. Haddleton and M. I. Gibson, Angew. Chem., Int. Ed., 2012, 51, 7812–7816 CrossRef CAS PubMed.
- L. E. Wilkins, N. Badi, F. Du Prez and M. I. Gibson, ACS Macro Lett., 2018, 7, 1498–1502 CrossRef CAS PubMed.
- D. Haksar, E. de Poel, L. Q. van Ufford, S. Bhatia, R. Haag, J. Beekman and R. J. Pieters, Bioconjugate Chem., 2019, 30, 785–792 CrossRef CAS PubMed.
- D. Haksar, M. Asadpoor, T. Heise, J. Shi, S. Braber, G. Folkerts, L. Ballell, J. Rodrigues and R. J. Pieters, J. Med. Chem., 2021, 64, 6059–6069 CrossRef CAS PubMed.
- Y. Kimoto, Y. Terada, Y. Hoshino and Y. Miura, ACS Omega, 2019, 4, 20690–20696 CrossRef CAS PubMed.
- C. S. Mahon, G. C. Wildsmith, D. Haksar, E. de Poel, J. M. Beekman, R. J. Pieters, M. E. Webb and W. B. Turnbull, Faraday Discuss., 2019, 219, 112–127 RSC.
- C. M. Verissimo, C. Graeff-Teixeira, M. K. Jones and A. L. Morassutti, Parasitology, 2019, 146, 1217–1232 CrossRef PubMed.
- V. M. Castillo-Acosta, J. Balzarini and D. Gonzalez-Pacanowska, Trends Parasitol., 2017, 33, 775–787 CrossRef CAS PubMed.
- S. Gonia, M. Tuepker, T. Heisel, C. Autran, L. Bode and C. A. Gale, J. Nutr., 2015, 145, 1992–1998 CrossRef CAS PubMed.
- J. Takagi, K. Aoki, B. S. Turner, S. Lamont, S. Lehoux, N. Kavanaugh, M. Gulati, A. Valle Arevalo, T. J. Lawrence, C. Y. Kim, B. Bakshi, M. Ishihara, C. J. Nobile, R. D. Cummings, D. J. Wozniak, M. Tiemeyer, R. Hevey and K. Ribbeck, Nat. Chem. Biol., 2022, 18, 762–773 CrossRef CAS PubMed.
- P. Dong, X. Shu, R. Peng, S. Lu, X. Xie and Q. Shi, Mater. Sci. Eng., C, 2021, 128, 112327 CrossRef CAS PubMed.
- F. Seyfarth, S. Schliemann, P. Elsner and U. C. Hipler, Int. J. Pharm., 2008, 353, 139–148 CAS.
- A. Saeed, A. Haider, S. Zahid, S. A. Khan, R. Faryal and M. Kaleem, Int. J. Biol. Macromol., 2019, 125, 761–766 CrossRef CAS PubMed.
- Y. Brissonnet, C. Assailly, A. Saumonneau, J. Bouckaert, M. Maillasson, C. Petitot, B. Roubinet, B. Didak, L. Landemarre, C. Bridot, R. Blossey, D. Deniaud, X. Yan, J. Bernard, C. Tellier, C. Grandjean, F. Daligault and S. G. Gouin, Eur. J. Chem., 2019, 25, 2358–2365 CrossRef CAS PubMed.
- L. Liu, Y. Wang, R. Narain and Y. Liu, Colloids Surf., B, 2019, 175, 680–687 CrossRef CAS PubMed.
- N. Tufenkji, D. R. Dixon, R. Considine and C. J. Drummond, Water Res., 2006, 40, 3315–3331 CrossRef CAS PubMed.
- S. Fukui, T. Feizi, C. Galustian, A. M. Lawson and W. Chai, Nat. Biotechnol., 2002, 20, 1011–1017 CrossRef CAS PubMed.
- C. Gao, M. Wei, T. R. McKitrick, A. M. McQuillan, J. Heimburg-Molinaro and R. D. Cummings, Front. Chem., 2019, 7, 833 CrossRef CAS PubMed.
- J. Y. Hyun, J. Pai and I. Shin, Acc. Chem. Res., 2017, 50, 1069–1078 CrossRef CAS PubMed.
- Z. Li and T. Feizi, FEBS Lett., 2018, 592, 3976–3991 CrossRef CAS PubMed.
- O. Blixt and U. Westerlind, Curr. Opin. Chem. Biol., 2014, 18, 62–69 CrossRef CAS PubMed.
- S. Park, J. C. Gildersleeve, O. Blixt and I. Shin, Chem. Soc. Rev., 2013, 42, 4310–4326 RSC.
- C. von der Ehe, C. Weber, M. Gottschaldt and U. S. Schubert, Prog. Polym. Sci., 2016, 57, 64–102 CrossRef CAS.
- D. J. Valles, Y. S. Zholdassov, J. Korpanty, S. Uddin, Y. Naeem, D. R. Mootoo, N. C. Gianneschi and A. B. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 20350–20357 CrossRef CAS PubMed.
- H. Seto, S. Kamba, T. Kondo, M. Hasegawa, S. Nashima, Y. Ehara, Y. Ogawa, Y. Hoshino and Y. Miura, ACS Appl. Mater. Interfaces, 2014, 6, 13234–13241 CrossRef CAS PubMed.
- A. S. Erofeev, P. V. Gorelkin, D. V. Kolesov, G. A. Kiselev, E. V. Dubrovin and I. V. Yaminsky, R. Soc. Open Sci., 2019, 6, 190255 CrossRef CAS PubMed.
- X. Zeng, K. Qu and A. Rehman, Acc. Chem. Res., 2016, 49, 1624–1633 CrossRef CAS PubMed.
- O. Lishchynskyi, Y. Shymborska, Y. Stetsyshyn, J. Raczkowska, A. G. Skirtach, T. Peretiatko and A. Budkowski, Chem. Eng. J., 2022, 446, 137048 CrossRef PubMed.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y. M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed.
- A. N. Baker, S. J. Richards, C. S. Guy, T. R. Congdon, M. Hasan, A. J. Zwetsloot, A. Gallo, J. R. Lewandowski, P. J. Stansfeld, A. Straube, M. Walker, S. Chessa, G. Pergolizzi, S. Dedola, R. A. Field and M. I. Gibson, ACS Cent. Sci., 2020, 6, 2046–2052 CrossRef CAS PubMed.
- H. Aldewachi, T. Chalati, M. N. Woodroofe, N. Bricklebank, B. Sharrack and P. Gardiner, Nanoscale, 2017, 10, 18–33 RSC.
- Z. Zhang, B. Schepens, L. Nuhn, X. Saelens, M. Schotsaert, N. Callewaert, R. De Rycke, Q. Zhang, S. Moins, S. Benali, L. Mespouille, R. Hoogenboom and B. G. De Geest, Chem. Commun., 2016, 52, 3352–3355 RSC.
- S. J. Richards, E. Fullam, G. S. Besra and M. I. Gibson, J. Mater. Chem. B, 2014, 2, 1490–1498 RSC.
- G. Li, W. Ma, J. Mo, B. Cheng, S. I. Shoda, D. Zhou and X. S. Ye, ACS Appl. Mater. Interfaces, 2021, 13, 46260–46269 CrossRef CAS PubMed.
- J. E. Petch, P. Gurnani, G. Yilmaz, F. Mastrotto, C. Alexander, S. Heeb, M. Camara and G. Mantovani, ACS Appl. Mater. Interfaces, 2021, 13, 19230–19243 CrossRef CAS PubMed.
- W. X. Liu, D. X. Xiao, Z. F. Tao and A. Dong, ACS Appl. Nano Mater., 2022, 5, 11948–11955 CrossRef CAS.
- T. Zhao, R. Terracciano, J. Becker, A. Monaco, G. Yilmaz and C. R. Becer, Biomacromolecules, 2022, 23, 543–575 CrossRef CAS PubMed.
- L. Q. Xu, C. Huang, R. Wang, K. G. Neoh, E. T. Kang and G. D. Fu, Polymer, 2011, 52, 5764–5771 CrossRef CAS.
- W. Wang, D. L. Chance, V. V. Mossine and T. P. Mawhinney, Glycoconjugate J., 2014, 31, 133–143 CrossRef CAS PubMed.
- S. Hussain, F. Lv, R. Qi, T. Senthilkumar, H. Zhao, Y. Chen, L. Liu and S. Wang, ACS Appl. Bio Mater., 2020, 3, 20–28 CrossRef CAS PubMed.
- Y. Wang, Y. Kotsuchibashi, Y. Liu and R. Narain, ACS Appl. Mater. Interfaces, 2015, 7, 1652–1661 CrossRef CAS PubMed.
- J. S. J. Tang, S. Rosencrantz, L. Tepper, S. Chea, S. Klopzig, A. Kruger-Genge, J. Storsberg and R. R. Rosencrantz, Molecules, 2019, 24, 1865 CrossRef CAS PubMed.
- C. von der Ehe, T. Bus, C. Weber, S. Stumpf, P. Bellstedt, M. Hartlieb, U. S. Schubert and M. Gottschaldt, ACS Macro Lett., 2016, 5, 326–331 CrossRef CAS PubMed.
- S. Schmidt, T. J. Paul and A. K. Strzelczyk, Macromol. Chem. Phys., 2019, 220, 1900323 CrossRef CAS.
- J. Siirila, S. Hietala, F. S. Ekholm and H. Tenhu, Biomacromolecules, 2020, 21, 955–965 CrossRef CAS PubMed.
- T. J. Paul, A. K. Strzelczyk, M. I. Feldhof and S. Schmidt, Biomacromolecules, 2020, 21, 2913–2921 CrossRef CAS PubMed.
- D. Wilms, J. Muller, A. Urach, F. Schroer and S. Schmidt, Biomacromolecules, 2022, 23, 3899–3908 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |