
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodissociation of permanganate (MnO4−) produces the manganese dioxide anion (MnO2−) in an excited triplet state†
Jemma A.
Gibbard
 *,
Jonathan
Reppel
and
Jan. R. R.
Verlet
*,
Jonathan
Reppel
and
Jan. R. R.
Verlet
Department of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: jemma.gibbard@durham.ac.uk
First published on 29th November 2023
Abstract
Photoelectron imaging, electron action spectroscopy and electronic structure calculations are used to probe the structure and dynamics of MnO4−. Following excitation to the first bright absorption band of MnO4− (11T2), photodetachment, via ground state electron loss, and photodissociation, to produce MnO2−, are both observed to occur simultaneously. MnO2− is produced in an excited electronic state, identified as a triplet state, which indicates that the dissociation proceeds on singlet potential energy surfaces via spin conservation. Furthermore, electronic structure calculations indicate that both photodetachment and photodissociation are multiple photon processes that are mediated by the same 11T2 excited state. Taken together this data indicates that photodissociation of MnO4− occurs via a statistical dissociation on the MnO4− ground state at visible wavelengths.
Introduction
Permanganate (MnO4−) is an archetypal transition metal complex, familiar to chemistry students worldwide for its deep purple colour. The well-known absorption spectrum of KMnO4(aq) shows a strong visible absorption band near hν = 2.25 eV (λ ∼ 550 nm), assigned to the first fully allowed transition from the tetrahedral ground state of MnO4−, 1A1, to the 11T2 excited state.1 The 1A1 → 11T2 absorption band displays vibronic structure predominantly associated with a vibrational progression of the Mn–O stretching mode (770 cm−1).1 Photoexcitation to the 11T2 state has also been known to lead to MnO2− + O2 in aqueous solution for many years, although the underlying photodissociation mechanism remains unclear.2,3 Early work postulated that dissociation occurred on the ground state via a unimolecular decomposition after the rapid formation of a vibrationally excited ground state anion, due to an increasing propensity for MnO2− formation with increasing photon energy.3,4 This mechanism has been challenged, as the quantum yield for photodissociation seemed linked to the specific electronically excited state accessed, leading to speculation that the red-tail of a higher-lying UV transition was responsible for dissociation in the visible band.5 It has also been postulated that electron loss may precede dissociation.5 More recent work has proposed that MnO2− production occurs following the formation of a long-lived intermediate, potentially a Mn(V) peroxo-complex, which undergoes reductive elimination.4,6 However, this long-lived intermediate has only been observed following excitation at UV wavelengths suggesting this process might not contribute in the visible region.4,6 Recent femtosecond transient absorption spectroscopy showed that excitation to the 11T2 state is followed by a rapid (∼16 ps) return to the ground state, via a cascade of radiationless transitions.4 Supported by quantum chemical calculations, the mechanism was determined to involve very fast internal conversion (IC) of the 11T2 state to a lower-lying 1T1 state, which is accompanied by a Jahn–Teller (JT) distortion. Subsequently, this 1JT1 state undergoes intersystem crossing (ISC) to the triplet-manifold, followed by IC to the lowest lying triplet state (3JT1), which is more stable than the lowest energy singlet in the JT-distorted geometry. Afterwards, the JT-distorted triplet decays by ISC back to the 1A1 electronic ground state in ∼16 ps with the accompanying structural relaxation to the tetrahedral geometry.4 These dynamics are summarised in Fig. 1.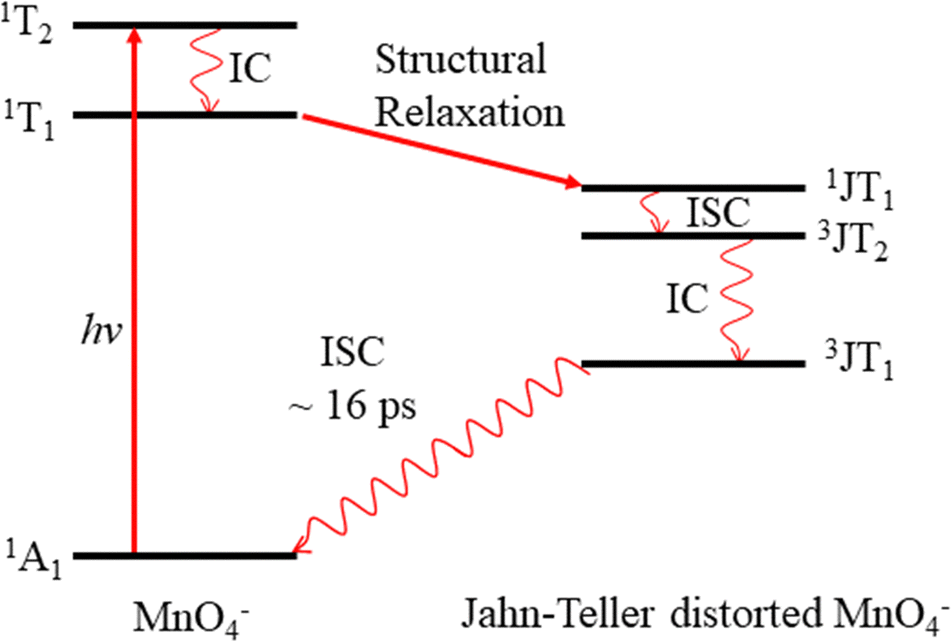 | ||
| Fig. 1 Excitation of MnO4− in the visible to 11T2, followed by rapid relaxation via the 3JT1 state of a Jahn–Teller distorted MnO4− back to 1A1, via structural relaxation, internal conversion (IC) and intersystem crossing (ISC). The transition from 1A1 to 1T1 is symmetry forbidden. | ||
In order to elucidate the intrinsic photodissociation mechanism of permanganate, a number of experimental and computational studies of isolated (gas phase) MnO4− have also been performed.7–9 Fragment ion action spectroscopy by Houmoller et al. showed that photodissociation of MnO4− produces both MnO3− and MnO2− following 1A1 → 11T2 excitation (2.48 ≤ hν ≤ 2.07 eV, 500 ≤ λ ≤ 600 nm).7,10 Fluence dependent measurements indicated that the photodissociation is a multiple photon process in this wavelength region.7,8 This was in agreement with theoretical studies which suggest that both the MnO4− dissociation asymptotes, MnO2− + O2 and MnO3− + O, lie approximately 5 eV above the MnO4− ground state.8 Photoelectron spectroscopy of MnO4− has also been performed by Gutsev et al., reporting the electron affinity of MnO4 to be 4.8 ± 0.1 eV, as well as calculations which suggested that photodissociation of MnO4− in the visible must involve a spin–flip and be a multiple photon process.8,11,12 Finally, Gutsev et al. also measured the photoelectron spectrum of MnO2−, where they were able to discern that MnO2− was produced in both the ground electronic state (5B2) and the first excited triplet state (3B1), in a ratio which depends upon the initial temperature of the ions.11
Despite the wealth of studies investigating the structure and dynamics of MnO4−, in both the solution and gas phase, fundamental questions remain about the photodissociation process. For example, what is the dissociation mechanism following excitation at visible wavelengths in the 1A1 → 11T2 band and what is the quantum state distribution of the products. Here, using a combination of photoelectron imaging, electronic structure calculations and electron action spectroscopy on MnO4−, we show that photodissociation forms MnO2− in an electronically excited state, which is consistent with a thermal unimolecular dissociation mechanism on the 1A1 ground state.
Experimental methods
The experimental apparatus has been described in detail elsewhere and will only be summarised here.13,14 Anions are produced via electrospray ionization of a 5 mM solution of KMnO4 in acetonitrile. Anions enter the apparatus via a capillary, before being guided and trapped using a series of RF guides and subsequent acceleration to 2.2 kV using a Wiley–McLaren time of flight spectrometer.15 Mass selected anions are intersected with the variable wavelength output of a nanosecond optical parametric oscillator (OPO) pumped via the third harmonic of a Nd:YAG, operating at 10 Hz. Ejected electrons are focused onto the position sensitive microchannel plate and phosphor screen detector using a velocity map imaging setup. From the resulting photoelectron images, the electron kinetic energy (eKE) and photoelectron angular distribution (PAD) are extracted using the polar onion peeling algorithm.16 From the eKE spectrum and the photon energy, hν, the electron binding energy (eBE) can be extracted (eKE + eBE = hν). The PADs are characterised by an anisotropy parameter (−1 ≤ β2 ≤ 2) and provide information on the molecular orbital from which the ejected photoelectron originated.17 Photoelectron imaging is performed over a range of photon energies, to determine how the spectral features shift with wavelength. As the features in the spectra presented here are consistent across the wavelengths, only a sample of the spectra will be reported.The electron action spectrum of MnO4− was recorded by monitoring the yield of photoelectrons, with the electron imaging detector, as a function of wavelength. The data was recorded whilst scanning the OPO at 0.1 nm s−1 and is smoothed by a 20-point moving average. The laser power from the OPO will vary somewhat across the wavelengths studied and is independently measured using a power meter.
Electronic structure calculations to determine the relative energetics of the possible electron loss and photodissociation channels of MnO4− were performed using DFT at the B3LYP level of theory with the aug-cc-pVTZ basis set via Gaussian 16.18–20 For each species the geometry was optimised and confirmed to be a true minima by vibrational analysis. All calculated energies are zero point energy corrected. This theoretical methodology has previously been used to study the electronic and nuclear structure of MnO3− and MnO3, where it was found to offer reliable results.21
Results
(a) Photoelectron imaging of MnO4−
Fig. 2 shows photoelectron spectra of MnO4− as well as an exemplar raw photoelectron image. Despite the large electron affinity of MnO4,12 photoelectrons are clearly ejected from MnO4− after irradiation with hν ∼ 2.5 eV (λ ∼ 500 nm) light. There are two dominant features in the photoelectron spectra shown in Fig. 2(a): a broad band of electrons peaking near eKE ∼ 0 eV; and a relatively narrow feature at fixed electron binding energy, eBE = 1.9 ± 0.1 eV (corresponding to an eKE of 0.6 eV at hν = 2.48 eV). From the image in Fig. 2(b), the feature near eKE ∼ 0 eV and labelled thermionic emission is isotropic (β2 = 0), while the outer ring at higher eKE (eBE = 1.9 eV), labelled direct detachment, is anisotropic and shows that photoelectrons are emitted predominantly parallel to the polarisation axis of the light (β2 ≈ +1.0).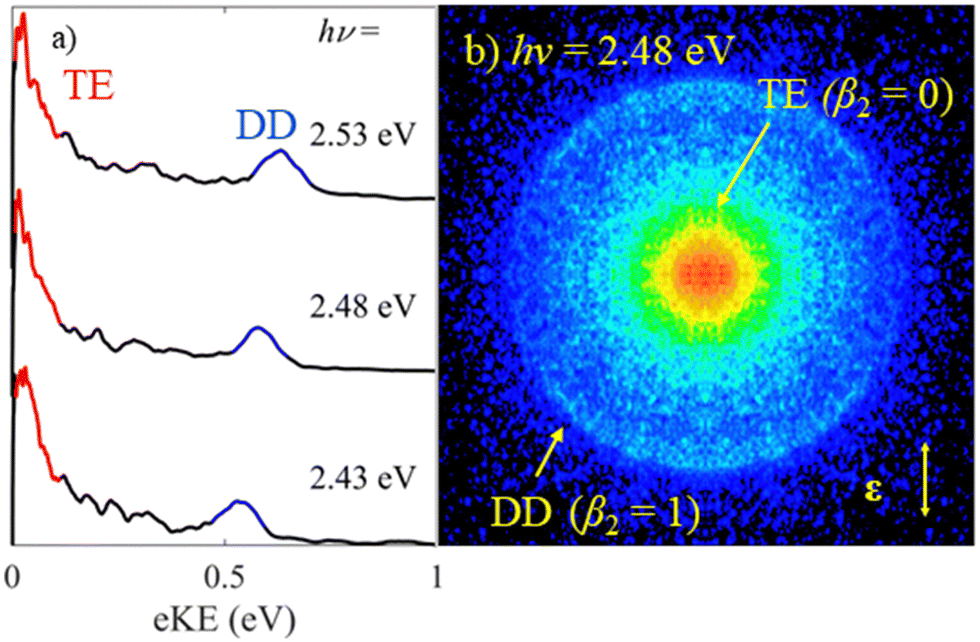 | ||
| Fig. 2 (a) Photoelectron spectrum of MnO4− recorded at hν = 2.53 eV (490 nm), 2.48 eV (500 nm) and 2.43 eV (510 nm). (b) Raw symmetrised photoelectron image of MnO4− recorded at hν = 2.48 eV, where the laser polarisation axis is indicated by the double arrow. The spectral features associated with thermionic emission (TE) and direct detachment (DD) are highlighted in red and blue respectively, in both the photoelectron spectra and image. | ||
The spectral feature peaking near eKE ∼ 0 eV has a characteristic distribution (both in terms of energy and PAD) associated with unimolecular loss of electrons from a vibrationally hot ground state (thermionic emission).22–25 The second narrow spectral feature increases in eKE with increasing hν, indicating a direct photodetachment process. Given that eBE = 1.9 eV is significantly lower than the reported electron affinity of MnO4,12 this feature is attributable to a multiple-photon process in which a final photon detaches an electron either from an excited state of MnO4− with eBE of 1.9 eV or from a photofragment with this eBE. Photodissociation of MnO4− produces MnO2−, making this a likely candidate for an anionic fragment.3,4,6,7 It is possible that some, or all, of the thermionic emission arises from this fragment too.
(b) Electron action spectroscopy
In order to gain insights into the electronic states which mediate the electron loss pathways observed in the photoelectron spectra of MnO4−, we recorded the electron action spectrum, i.e., the yield of photoelectrons as a function of wavelength, which is shown in Fig. 3. A gradual increase in electron yield is seen with increasing hν, followed by a large increase around hν = 2.4 eV (λ = 520 nm), beyond which vibrational structure becomes apparent, with a spacing of ∼770 cm−1. Both photo-processes observed in the photoelectron spectra in Fig. 2 (i.e., thermionic emission and multiple-photon photodetachment) contribute electrons to the electron action spectrum. The laser power variation across the spectrum is shown in red on the right-side axis, but the spectrum is not normalised to account for this, due to the contribution of multiple photon processes.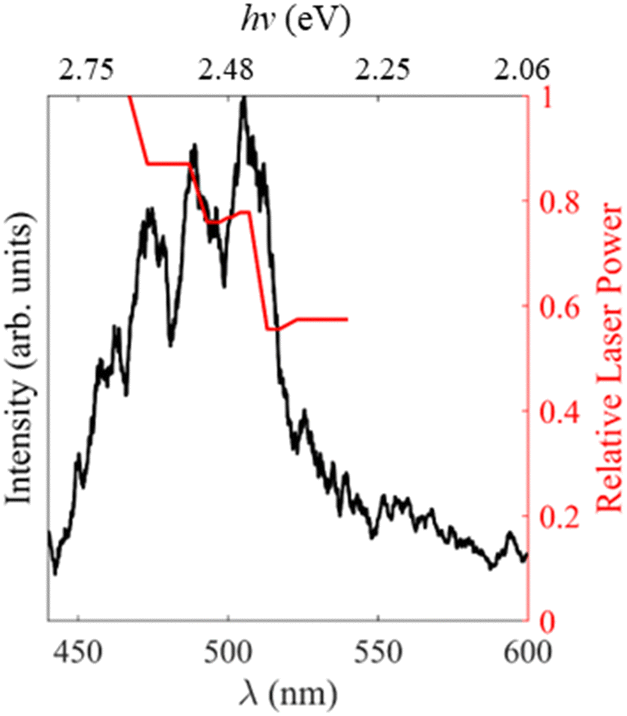 | ||
| Fig. 3 Electron action spectrum of MnO4− recorded between hν = 2.81 – 2.06 eV (λ = 440 and 600 nm) in black. The relative laser power is shown in red on the right. | ||
(c) Calculations
The calculated relative energetics for the photodetachment and photodissociation channels of MnO4− are shown in Table 1 and compared to experimental data where available.8,11,12 In order to test the validity of our methodology, we computed the bond dissociation energy of O2 and found it to be D0 = 5.2 eV, which is close to the experimental value of 5.12 eV.26 There is also good agreement between our calculated electron affinities and the experimentally observed detachment energies for MnO2−, MnO3− and MnO4−.11,12 Finally, we have previously shown that the computational methodology predicts electronic and geometric structures with good accuracy for MnO3 and its anion.21 The high electron affinity of MnO4 suggests that electron emission is likely to be a multiple photon process at visible wavelengths.In addition to the electron emission energetics, we also consider the dissociation energetics, which are summarised in Table 2 for a range of possible channels involving the MnO2− product. Our calculations indicate that photodissociation is a lower energy pathway than photodetachment of MnO4− and suggest that photodissociation is likely to be a two-photon process for 3.31 eV < hν < 1.65 eV (375 nm < λ < 750 nm). The lowest energy photodissociation channel is predicted to form MnO2− + O2 in their respective ground electronic states. The lowest energy MnO3− + O asymptote lies 0.4 eV higher in energy (3.7 eV). We do not include this channel in Table 2 because we have not observed MnO3− in our experiments, but we will return to this channel later. To the best of our knowledge, there are no reported experimental measurements of the dissociation asymptotes for comparison. Table 2 shows that the ground state of MnO2− is a quintet state (5B2), with the triplet state (3B1) lying 0.4 eV higher in energy. The lowest energy channels to form singlet states of either MnO2− or O2 are much higher in energy (note that the 1 eV gap between O2 singlet and triplet dissociation channels is consistent with the energy 3Σg− − 1Δg difference of O2).27
| Dissociation asymptote | Relative energy (eV) |
|---|---|
| MnO2− (5B2) + O2 (3Σg−) | 3.3 |
| MnO2− (3B1) + O2 (3Σg−) | 3.7 |
| MnO2− (5B2) + O2 (1Δg) | 4.3 |
| MnO2− (3B1) + O2 (1Δg) | 4.7 |
| MnO2− (1A1) + O2 (3Σg−) | 4.9 |
| MnO2− (1A1) + O2 (1Δg) | 5.9 |
Discussion
(a) Assignment of triplet MnO2− as photodissociation product
We first consider the assignment of the features in the photoelectron spectra in Fig. 2. Given the electron affinity of MnO4 is 4.8 ± 0.1 eV, all the photoelectrons in Fig. 2 arise via multiple photon processes.12 Light around hν ∼ 2.5 eV (λ ∼ 500 nm) is resonant with the bright 1A1 → 11T2 transition, and given the rapid non-radiative decay observed in aqueous solution from the 11T2 state back to the ground state, thermionic emission is likely to occur via photon cycling.7,10,28,29 In such a scenario, a second photon excites the hot-ground state via the same 1A1 → 11T2 transition to leave MnO4− in the 1A1 ground state with sufficient internal energy to statistically dislodge the electron (i.e. internal energy is 2 × hν). This thermionic emission feature was not seen in the previously reported photoelectron spectrum of MnO4−, because this spectrum was measured at a much higher photon energy (hν = 6.39 eV, λ = 194 nm) that is not resonant with the 1A1 → 11T2 transition, and because of the low detection efficiency for low eKE electrons in their electron spectrometer (based on a magnetic bottle).12The second narrow feature can be assigned to photodetachment from an excited state of MnO2−, as both the binding energy and the shape of the feature are consistent with the previously reported spectrum of MnO2−.11,30,31 In Fig. 4(a), we overlay the photoelectron spectrum of Gustev et al. taken at hν = 3.49 eV (λ = 355 nm) with our spectrum of MnO4− taken at hν = 2.48 eV (λ = 500 nm), which clearly shows that the peak at eBE = 1.9 eV arises from MnO2−. However, the peak at eBE = 1.9 eV (Fig. 4(a)) is not the ground electronic state of MnO2−; temperature dependent measurements by Gutsev et al. showed that this feature in fact arises from an excited state anion, which they assigned to the 3B1 state.11 Hence, we conclude from the photoelectron spectrum in Fig. 2 that photodissociation leaves at least some of the MnO2− product in the 3B1 excited state.11 It should also be noted that MnO2− may lose electrons via thermionic emission and contribute to the low eKE signal observed. None of the previously reported photoelectron spectra of MnO2− showed thermionic emission, although these were insensitive to low eKE electrons.11
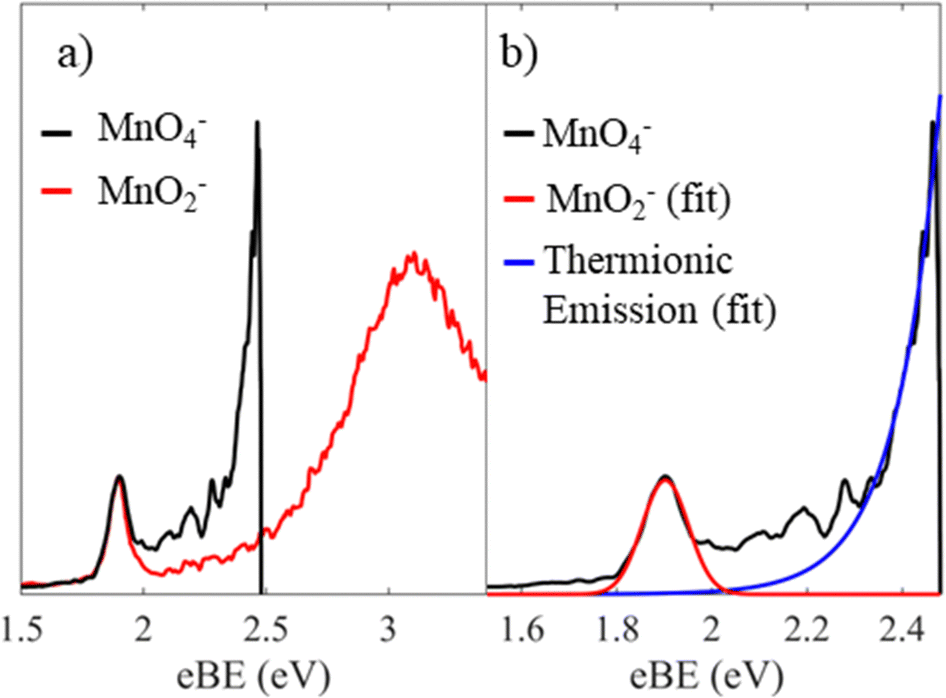 | ||
| Fig. 4 Photoelectron spectrum of MnO4− recorded at hν = 2.48 eV (black), where the energy axis is in electron binding energy, eBE = hν − eKE. In (a) this is shown with the previously reported spectrum of MnO2− taken at hν = 3.49 eV (red) reprinted from J. Chem. Phys., 2000, 113, 1473, with the permission of AIP publishing.11 In (b), a Gaussian fit to the spectral features associated with the MnO2− triplet state (red) is shown as well as an exponential fit to the thermionic emission (blue). | ||
Our measurements contain additional information in the form of PADs, which report on the nature of the molecular orbital that the photoelectron is ejected from. In this case, photodetachment from the excited state of MnO2− results in photoelectrons with β2 ≈ +1.0. Given that photodissociation and photodetachment of MnO2− is a multiple photon process of distinct sequential steps and occurs under irradiation with nanosecond light (∼5 ns pulse duration), it is probable that the timescale of the process is much longer than rotational period of MnO2− and therefore that any potential alignment effects from the photodissociation process itself, can be ignored when interpreting the PADs. By Koopmans’ theory, photodetachment results in ejection of an electron from the highest lying molecular orbital (HOMO), suggesting that the HOMO has s- or σ-orbital character (given the outgoing wave of p-character). We can use the simple d-block model of transition metal complexes to estimate the dominant contributions to the highest lying molecular orbitals, which are typically the non-bonding or antibonding metal-localised d-orbitals.32 By considering the interaction of Mn with the two O atoms in the bent geometry, the dz2 would be non-bonding, whereas the other d-orbitals would be higher in energy and antibonding in character. As a result of interactions between the O localised p-orbitals and the Mn localised dxz, dyz and dx2−y2 orbitals, there would be three π* orbitals, and a σ* orbital from the interaction of dxy with s- and p-orbitals orientated along the Mn–O bonds. These high-lying orbitals are then populated with the valence shell electrons of the metal, i.e. d4 for Mn(III).32 However for bent ML2 complexes, a metal localised p-orbital and a s–p hybrid are strictly non-bonding and higher in energy than the d-block, and as such these four electrons also populate the d-block.32 The energy level diagram which arise from the d-block model for the triplet MnO2− is shown in Fig. 5.
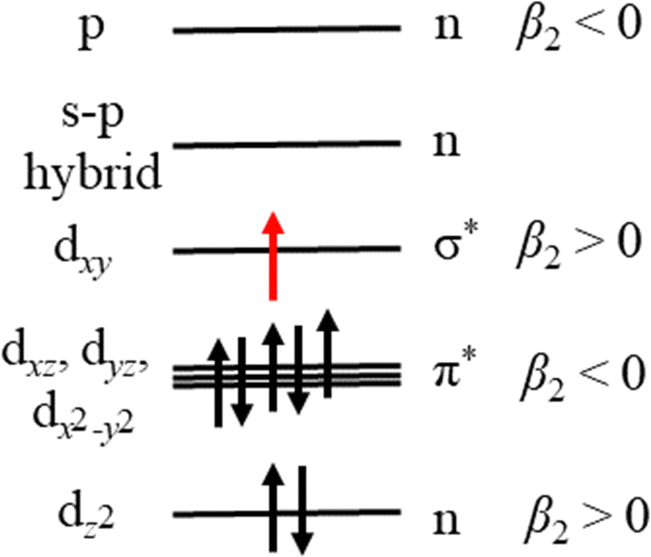 | ||
| Fig. 5 An energy level diagram of the d-block of triplet MnO2−, including the expected β2 values if the electron is lost. The molecular orbitals are populated with the d4 electrons of Mn(III) and 4 electrons from the high lying non-bonding orbitals of a bent ML2 complex, whilst the electron, which is lost in photodetachment, via Koopmans' correlations, is highlighted red. Expected anisotropy parameters are indicated. | ||
Our calculations, which agree with previous work by Gutsev at al., indicate that MnO2− preferentially adopts high spin electron configurations, i.e. the ground electronic state of MnO2− is 5B2.11 Effectively this suggests that the pairing energy to doubly occupy d-block orbitals is higher than the ligand field splitting in this case. Given the temperature dependent measurements of Gutsev et al., we know that photodetachment of MnO2− arises from an excited state i.e. a singlet or triplet.11 We have populated the d-block in Fig. 5 with the electron configuration of the 3B1 state of MnO2−, which calculations have consistently determined to be the first excited state (Table 2 and Gutsev et al.11). If photodetachment removed the red electron from the HOMO in Fig. 5, which is a σ* molecular orbital, it would be expected to result in a PAD characterised by β2 > 0, as observed experimentally and consistent with the assignment that MnO2− is produced in the 3B1 electronically excited state. If instead detachment took place from the lowest energy singlet electronic state, then we would expect the PAD to be characterised by β2 < 0 on account of its π-character, which is not observed.
It is more challenging to determine if ground state MnO2− is also produced. In the photoelectron spectrum of MnO2− by Gutsev et al. (Fig. 4(a)), photoelectrons with eBE > 2.1 eV were assigned to photodetachment from the ground 5B2 state of MnO2−.11 If ground state MnO2− was produced in our experiments, then the associated spectral features would overlap with the thermionic emission. In order to disentangle these potential contributions, we fit our spectrum with a Gaussian and exponential decay to account for the direct and thermionic detachment channels, respectively, as shown in Fig. 4(b). While the fit accounts for most signal, some signal is unaccounted for. However, the spectrum of the unaccounted for electrons does not match the ground state spectrum of MnO2−. Furthermore, the PAD in this region is isotropic, which would be very unusual for a transition metal complex.33,34 From Fig. 5 we may expect that the 5B2 state would lose an electron from the s–p hybrid molecular orbital, and would be expected to exhibit an anisotropic PAD. We attempted to verify this by recording the photoelectron spectrum of MnO2− directly but were unsuccessful as we could not produce MnO2−via electrospray ionization. Therefore, it is likely that the mismatch between the fits and the recorded data is either due to noise, which may be present because of the very low data acquisition rates for our photoelectron spectra, or due to additional thermionic emission leading to an emission profile that cannot be accounted for by a single exponential. Overall, we see no direct evidence for the formation of MnO2− in its electronic ground state. However, given that the vertical detachment energy of ground state MnO2− is near eBE ∼ 3.3 eV (see Fig. 4(a)), and the low signal levels in our experiments, it is not possible to conclusively rule it out. Ideally, we would like to have performed two-colour experiments, with a UV probe pulse, in order to search for ground state MnO2−, but the signal levels were too low for these measurements to be successful.
(b) Assignment of electron action spectrum
Our electron action spectrum of MnO4− and the previously reported MnO2− action spectrum of MnO4− by Houmoller et al. are plotted together in Fig. 6.7 Both spectra show an identical vibrational progression, associated with the Mn–O stretch vibrational mode of MnO4− in the 11T2 state and observed in the absorption spectrum of aqueous MnO4−.1 The similarity indicates that both photodissociation to produce MnO2− and thermionic emission involve photoexcitation of the 11T2 state of MnO4−. However, there are some clear differences too. Specifically, the first three vibrational peaks appear to be missing in the electron action spectrum. These differences arise as there are different photoprocesses contributing to each of the action spectra. Photodissociation to produce MnO2− is the only contributor to the ion action spectrum, whereas both photodetachment from MnO2− as well as thermionic emission (from either MnO2− or MnO4−) contribute to the electron action of MnO4− (see Fig. 2).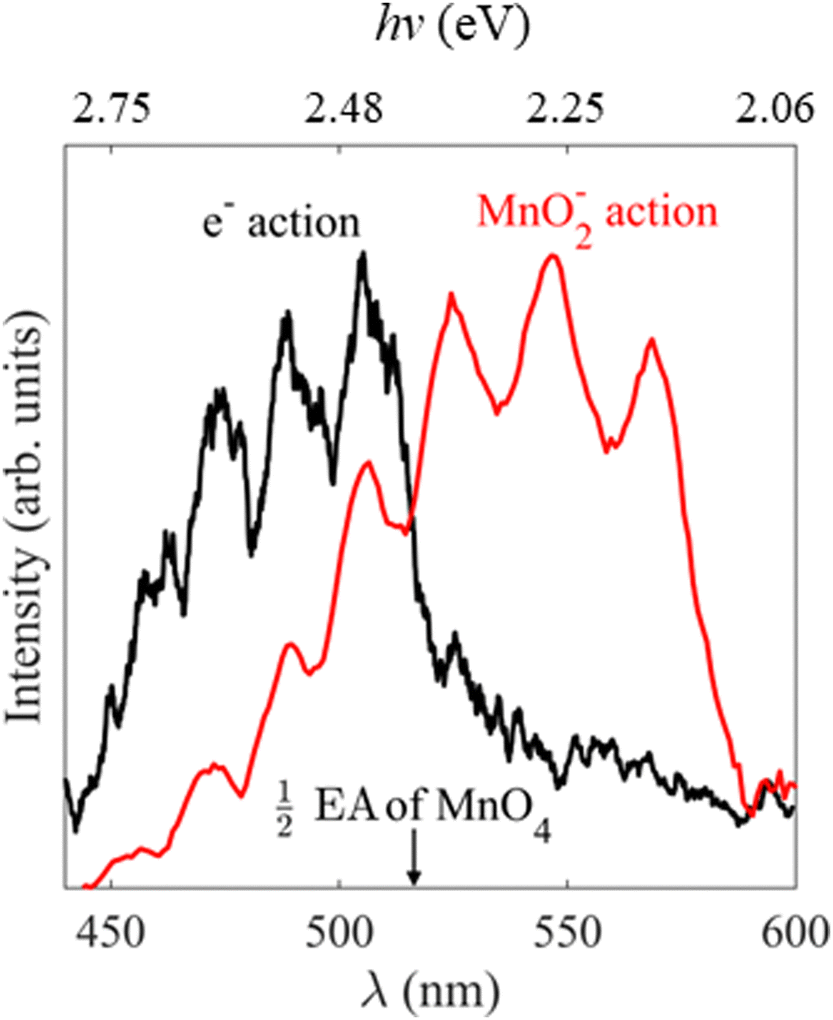 | ||
| Fig. 6 Electron action spectrum of MnO4− recorded between hν = 2.82 and 2.07 eV (λ = 440 and 600 nm) in black, with the previously reported MnO2− action spectrum of MnO4− in red, reprinted from Chem. Phys. Chem., 2013, 14, 1133 with the permission of John Wiley and Sons.7 The wavelength at which two-photon photodetachement of MnO4− becomes energetically accessible i.e., half the previously reported EA of MnO4, is indicated by the black arrow.12 | ||
Our calculations indicate that MnO2− (3B1) + O2 (3Σg−) lies 3.7 eV above MnO4−, requiring two visible photons with hν > 1.85 eV (λ < 670 nm) to photodissociate to our observed products. Houmoller et al. confirm that the photodissociation is a two-photon process at these wavelengths by performing fluence dependent measurements and suggest that the dissociation asymptote is <4 eV, in accordance with our calculations.7 In our experiments a further photon is required to photodetach an electron from MnO2−, which we subsequently detect in our electron action spectrum, making this channel a three-photon process overall. From our photoelectron spectra in Fig. 2, the triplet state of MnO2− has an eBE ∼ 1.9 eV (two-photons at λ = 650 nm), meaning that the three-photon process is energetically accessible at all the wavelengths studied here, as long as a dissociation takes place on a timescale sufficiently short that the third photon can be observed during the laser pulse (∼5 ns).
In addition, thermionic emission contributes to the MnO4− electron action spectrum, whether from MnO4− directly or the photodissociation product of MnO2−. The electron affinity of MnO4− is 4.8 eV, requiring a three-photon process at the longest wavelengths studied, but a two-photon electron loss process would become energetically accessible at hν ∼ 2.4 eV (λ ∼ 516 nm). If photodissociation is the dominant product channel following excitation of MnO4− at visible wavelengths, then the thermionic emission may also arise from MnO2−via a three-photon process.
A large increase in signal is seen in the electron action spectrum near hν ∼ 2.4 eV (λ ∼ 520 nm), but the origin of this increase is not clear cut. From Fig. 2 it is seen that this increase coincides somewhat with a ∼75% increase in laser power, which would be expected to rapidly increase the likelihood of multiple photon processes (the rate of an n-photon process is typically ∝ Pn, where P is the laser fluence). Alternatively, electron loss from MnO4− switches from a two-to-three photon transition for hν > 2.4 eV (λ < 516 nm), which is indicated in Fig. 6, and also coincides with the increase in overall electron signal. In order to test these different hypothesises, we performed additional measurements which were inconclusive. Fluence dependent measurements (Fig. S1, ESI†) at hν = 2.46 eV (λ = 505 nm) showed no difference in the ratio of direct detachment to thermionic emission with laser power, indicating that dynamics of both photodissociation and photodetachment are limited by the same step, likely to be the initial photoexcitation from the 1A1 → 11T2 transition. Furthermore, the photoelectron spectrum of MnO4− at hν = 2.34 eV (λ = 530 nm) (Fig. S1, ESI†), where only three-photon processes are energetically accessible, unexpectedly showed a higher proportion of thermionic emission compared to direct detachment from MnO2−, than spectra recorded at shorter wavelengths. Ultimately it is probable that a combination of increased laser power and electron loss from MnO4− switching from a three- to a two-photon process, contribute to the shape of the electron action spectrum. However, it is challenging to unravel comprehensively, as these factors are seen against a backdrop of competing multiple photon photoprocesses, with different cross-sections for each step at each wavelength.
(c) Photodissociation mechanism
The production of MnO2− in an excited state must result directly from the photodissociation process. This preference may be due to the requirement to conserve spin during bond breaking. Given that the ion action spectrum of Houmoller et al. indicated that MnO2− was produced via a two-photon process at hν = 4.27 eV (2 × λ = 580 nm), only the lowest two energy asymptotes from Table 2 are energetically accessible, with the third being within the error of the calculations.The requirement to conserve spin is applied to the final electronic state of MnO4− before dissociation, and MnO4− has singlet, triplet, quintet and septet electronic states.8 Removal of two electrons from the bonding orbitals of MnO4− with spin conservation, to account for the breaking of two Mn–O bonds during photodissociation, allows us to predict the spin of the products. This process is illustrated in Fig. 7 for the energetically accessible dissociation asymptotes and demonstrates that the spin of the parent anion dictates the spin state of the products (in the absence of spin–orbit coupling). Therefore, by conservation of spin angular momentum, the observed MnO2− (3B1) can only arise from dissociation of a singlet MnO4− state, whereas ground state MnO2− (5B2) may be produced via dissociation of a triplet state of MnO4−.
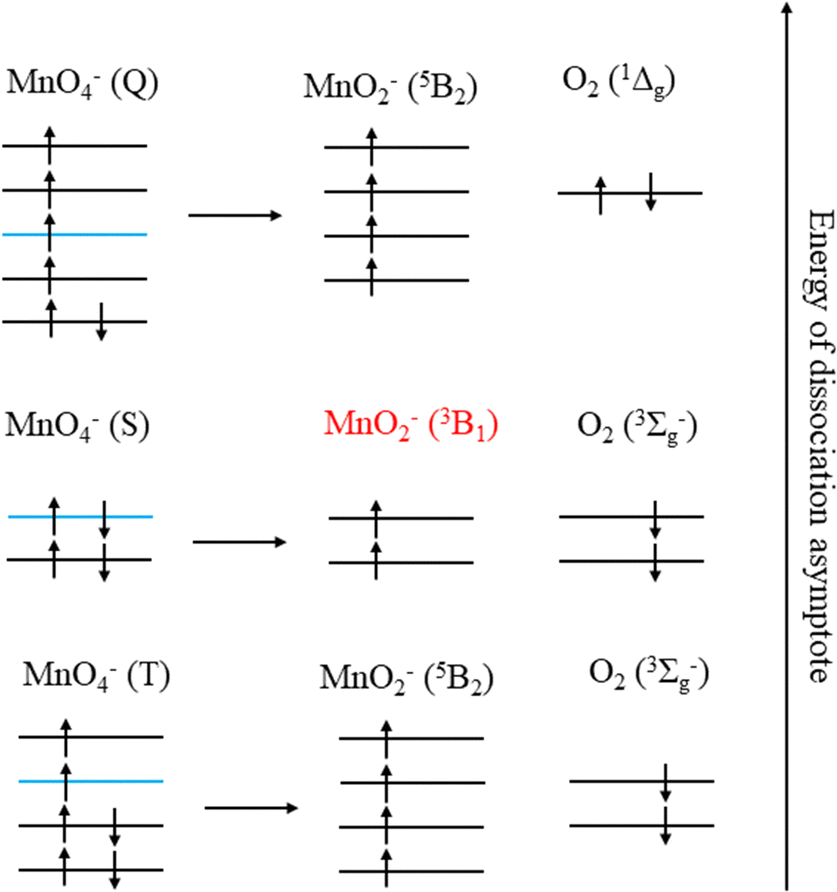 | ||
| Fig. 7 The singlet (S), triplet (T) and quintet (Q) spin states of MnO4− which can produce the three energetically accessible dissociation asymptotes of MnO2− + O2. The experimentally observed spin state of MnO2− is highlighted in red, with the MnO4− singlet HOMO being highlighted in blue for all the spin states. | ||
From spin conservation only, the singlet state of MnO4− upon which dissociation occurs may be either the ground state (1A1), the excited state accessed at visible wavelengths (11T2) or the 11T1 transient intermediate that may be populated in the decay mechanism of the 11T2 state (see Fig. 1). Evidence for a ground state dissociation of MnO4− is found in the presence of thermionic emission in the photoelectron spectra, the structure of our electron action spectrum, and the previously reported ion action spectrum of MnO4.7 Therefore, the overall mechanism of non-radiative (NR) decay from 11T2 to 1A1 is likely to be the dominant decay pathway, as observed in aqueous solution to take place on a 16 ps timescale.4 The vibrational structure in the both the electron and the ion action spectrum of MnO4− also indicates that both photodetachment and photodissociation are initiated by the same 1A1 → 11T2 transition, and again provides evidence for a ground state mechanism.7 A cartoon of the two-photon process which results in a vibrationally hot ground state MnO4−, and acts as the precursor to electron loss or dissociation, is shown in Fig. 8.
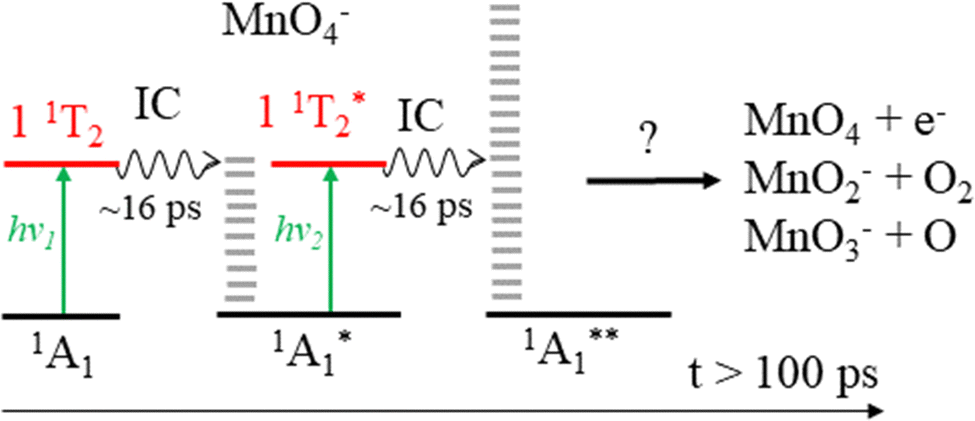 | ||
| Fig. 8 Sequential absorption of two photons near hν = 2.48 eV (λ = 500 nm) by MnO4− produces a highly internally excited MnO4− in its electronic ground state (where * indicates a photon's worth of internal excitation), which subsequently can lose either an electron, an O atom or O2via a statistical process. The IC process involves several nonradiative transitions as shown in Fig. 1. | ||
The overall two-photon process for thermionic emission can be described as follows, where the * represents a photon's worth of internal excitation (i.e. heat):

An analogous two-photon ground state dissociation mechanism for the production of MnO2− may also be proposed:
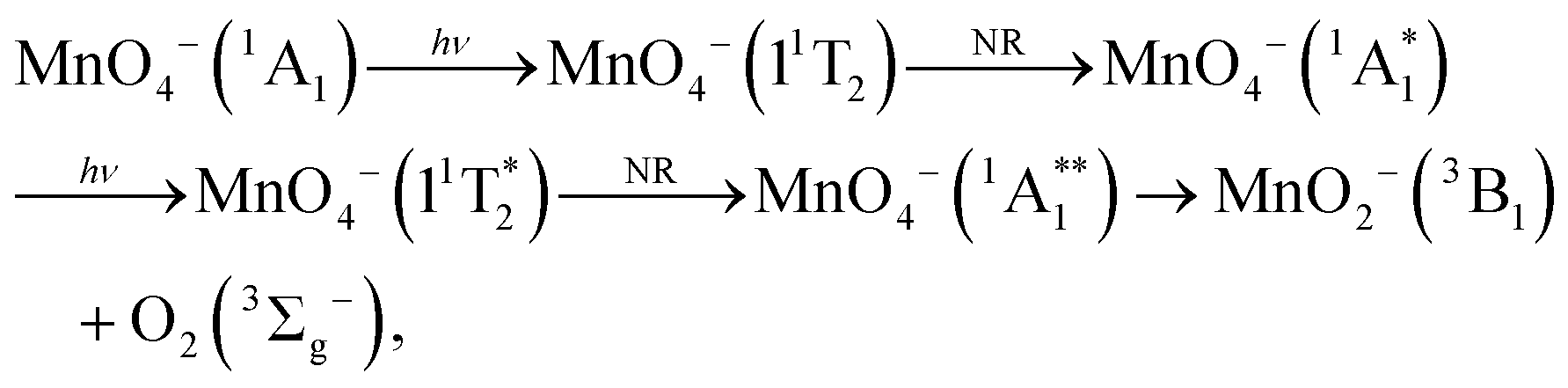
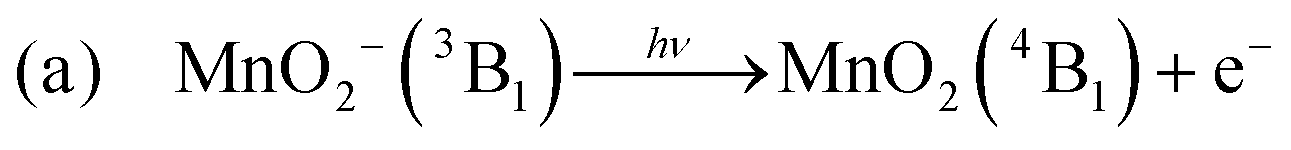

It is clear from our measurements that MnO2− (3B1) is a long-lived electronically excited state, with a lifetime on the order of ns, which does not readily return to the ground state. This must be the case as we record photodetachment from the excited state, indicating that the absorption of a third photon occurs on a faster, or at least similar, timescale to excitation (laser pulse is ∼5 ns in duration). Furthermore Gutsev et al. observed some MnO2− (3B1) directly in their anion beam, which suggests a lifetime on the order of their time of flight, which is many μs.11
In aqueous solution, excitation to the 1T2 band also shows the formation of MnO2−, although it is not known in what electronic state. It is also produced in a very low yield. These differences are likely associated with the fact that the hot ground state formed after non-radiative decay can dissipate its internal energy before absorbing a second photon, inhibiting dissociation.
Finally, we also consider the MnO3− + O channel. Previous gas phase ion action spectroscopy has reported the production of MnO3− from irradiation of MnO4− in the visible region. We were unable to observe any MnO3− produced via photodissociation of MnO4− in our experiments, as the electron affinity (∼3.1 eV) is larger than the photon energies used (2.0 < hν < 2.75 eV). However, it was observed as an almost equal yield channel as the MnO2− + O2 channel by Houmoller et al. and this result is consistent with our data. The onset for the MnO3− + O channel is 0.4 eV above that of the lowest MnO2− (5B2) + O2 (3Σg−) channel. Hence, statistically, the former should be much less likely. However, as the MnO2− (5B2) + O2 (3Σg−) channel is closed because of the required spin–flip, the relevant channel is the MnO2− (3B1) + O2(3Σg−) dissociation channel, which is 0.4 eV higher in energy and essentially degenerate with the MnO3− + O channel (according to our calculations). It is interesting to note that the lowest energy dissociation asymptote is predicted to be MnO3− (1A1′) + O (3P2), which would only be spin-allowed from a triplet state of MnO4− (Fig. 7). However, calculations have indicated that there is a low-lying triplet state of MnO3−, which using our theoretical methodology lies 0.1 eV above the ground state, and could be produced with conservation of spin from a singlet state of MnO4−i.e., via a ground state dissociation.11 Ideally two-colour pump–probe measurements to probe the production of MnO3− would be undertaken. However, the attempted time-resolved measurements to study the production of MnO2− were unsuccessful, due to the very low signal levels observed as a result of the two-photons required for photodissociation. Note also that MnO3− was not observed as a product in aqueous solution, which might indicate that dissociation in solution is preferred to proceed to the ground state product of MnO2−, where perhaps a spin–flip is mediated by collisions with solvent molecules.
Conclusions
It has been known for many decades that aqueous permanganate photodissociates to produce MnO2− following excitation with green light, but the mechanism has remained the subject of active research.3 In this work we demonstrate that the gas-phase photodissociation of MnO4− in the visible region is a multiple photon ground state process, which produces MnO2− in an electronically excited triplet state. Our proposed mechanism of ground state dissociation is consistent with recent femtosecond transient absorption spectroscopy in the solution phase which unravelled the rapid non-radiative decay dynamics of MnO4−.4 Furthermore, it may explain why MnO3− has been observed as a photodissociation product of MnO4− in the gas phase, but not the solution phase.7 Our conclusions are reached via photoelectron imaging, electron action spectroscopy and electronic structure calculations, where competition is observed between a statistical photodissociation and ground state electron loss from MnO4−, via thermionic emission.Data availability statement
The data that support the findings of this study are avaliable in Zenodo at https://zenodo.org/records/10204163.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Jemma Gibbard is grateful for the support of a Royal Society University Research Fellowship (URF\R1\221140).References
- S. L. Holt and C. J. Ballhausen, Low temperature absorption spectra of KMnO4 in KClO4, Theor. Chim. Acta, 1967, 7(4), 313–320 CrossRef CAS.
- J. H. Mathews and L. H. Dewey, A quantitative study of some photochemical effects produced by ultra-violet light, J. Phys. Chem., 1913, 17(3), 211–218 CrossRef.
- G. Zimmerman, Photochemical Decomposition of Aqueous Permanganate Ion, J. Chem. Phys., 1955, 23(5), 825–832 CrossRef CAS.
- O. S. Haggag, P. Malakar, P. Pokhilko, J. F. Stanton, A. I. Krylov and S. Ruhman, The elusive dynamics of aqueous permanganate photochemistry, Phys. Chem. Chem. Phys., 2020, 22(18), 10043–10055 RSC.
- A. W. Adamson, W. L. Waltz, E. Zinato, D. W. Watts, P. D. Fleischauer and R. D. Lindholm, Photochemistry of transition-metal coordination compounds, Chem. Rev., 1968, 68(5), 541–585 CrossRef CAS.
- D. G. Lee, C. R. Moylan, T. Hayashi and J. I. Brauman, Photochemistry of aqueous permanganate ion, J. Am. Chem. Soc., 1987, 109(10), 3003–3010 CrossRef CAS.
- J. Houmøller, S. H. Kaufman, K. Støchkel, L. C. Tribedi, S. Brøndsted Nielsen and J. M. Weber, On the Photoabsorption by Permanganate Ions in Vacuo and the Role of a Single Water Molecule. New Experimental Benchmarks for Electronic Structure Theory, ChemPhysChem, 2013, 14(6), 1133–1137 CrossRef PubMed.
- G. L. Gutsev, B. K. Rao and P. Jena, Photodecomposition of MnO4−: A Theoretical Study, J. Phys. Chem. A, 1999, 103(50), 10819–10824 CrossRef CAS.
- H. Nakai, Y. Ohmori and H. Nakatsuji, Theoretical Study on the Photochemical Decomposition Reaction of Permanganate Ion, MnO4−, J. Phys. Chem., 1995, 99(21), 8550–8555 CrossRef CAS.
- L. Jose, M. Seth and T. Ziegler, Molecular and Vibrational Structure of Tetroxo d0 Metal Complexes in their Excited States. A Study Based on Time-Dependent Density Functional Calculations and Franck–Condon Theory, J. Phys. Chem. A, 2012, 116(7), 1864–1876 CrossRef CAS PubMed.
- G. L. Gutsev, B. K. Rao, P. Jena, X. Li and L.-S. Wang, Experimental and theoretical study of the photoelectron spectra of MnOx−(x = 1–3) clusters, J. Chem. Phys., 2000, 113(4), 1473–1483 CrossRef CAS.
- G. L. Gutsev, B. K. Rao, P. Jena, X.-B. Wang and L.-S. Wang, Origin of the unusual stability of MnO4−, Chem. Phys. Lett., 1999, 312(5), 598–605 CrossRef CAS.
- J. Lecointre, G. M. Roberts, D. A. Horke and J. R. R. Verlet, Ultrafast Relaxation Dynamics Observed Through Time-Resolved Photoelectron Angular Distributions, J. Phys. Chem. A, 2010, 114(42), 11216–11224 CrossRef CAS PubMed.
- L. H. Stanley, C. S. Anstöter and J. R. R. Verlet, Resonances of the anthracenyl anion probed by frequency-resolved photoelectron imaging of collision-induced dissociated anthracene carboxylic acid, Chem. Sci., 2017, 8(4), 3054–3061 RSC.
- W. C. Wiley and I. H. Mclaren, Time-of-Flight Mass Spectrometer with Improved Resolution, Rev. Sci. Instrum., 1955, 26(12), 1150–1157 CrossRef CAS.
- G. M. Roberts, J. L. Nixon, J. Lecointre, E. Wrede and J. R. R. Verlet, Toward real-time charged-particle image reconstruction using polar onion-peeling, Rev. Sci. Instrum., 2009, 80(5), 053104 CrossRef CAS PubMed.
- A. Sanov, Laboratory-Frame Photoelectron Angular Distributions in Anion Photodetachment: Insight into Electronic Structure and Intermolecular Interactions, Annu. Rev. Phys. Chem., 2014, 65(1), 341–363 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96(9), 6796–6806 CrossRef CAS.
- J. A. Gibbard, J. Reppel and J. R. R. Verlet, Photoelectron imaging of MnO3− to probe its nuclear and electronic structure, J. Chem. Phys., 2023, 159(14), 146101 CrossRef CAS PubMed.
- B. Baguenard, J. C. Pinaré, C. Bordas and M. Broyer, Photoelectron imaging spectroscopy of small tungsten clusters: Direct observation of thermionic emission, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 63(2), 023204 CrossRef.
- B. Baguenard, J. C. Pinaré, F. Lépine, C. Bordas and M. Broyer, Thermionic emission in small carbon cluster anions, Chem. Phys. Lett., 2002, 352(3), 147–153 CrossRef CAS.
- K. Hansen, K. Hoffmann and E. E. B. Campbell, Thermal electron emission from the hot electronic subsystem of vibrationally cold C60, J. Chem. Phys., 2003, 119(5), 2513–2522 CrossRef CAS.
- M. Kjellberg, O. Johansson, F. Jonsson, A. V. Bulgakov, C. Bordas, E. E. B. Campbell and K. Hansen, Momentum-map-imaging photoelectron spectroscopy of fullerenes with femtosecond laser pulses, Phys. Rev. A: At., Mol., Opt. Phys., 2010, 81(2), 023202 CrossRef.
- K. P. Huber and G. Herzberg, Constants of diatomic molecules, in Molecular Spectra and Molecular Structure: IV. Constants of Diatomic Molecules, ed. K. P. Huber and G. Herzberg, Springer US, Boston, MA, 1979, pp. 8–689 Search PubMed.
- C. Schweitzer and R. Schmidt, Physical Mechanisms of Generation and Deactivation of Singlet Oxygen, Chem. Rev., 2003, 103(5), 1685–1758 CrossRef CAS PubMed.
- J. N. Bull, C. W. West and J. R. R. Verlet, On the formation of anions: frequency-, angle-, and time-resolved photoelectron imaging of the menadione radical anion, Chem. Sci., 2015, 6(2), 1578–1589 RSC.
- J. A. Gibbard, C. J. Clarke and J. R. R. Verlet, Photoelectron spectroscopy of the protoporphyrin IX dianion, Phys. Chem. Chem. Phys., 2021, 23(34), 18425–18431 RSC.
- K. M. Ervin, I. Anusiewicz, P. Skurski, J. Simons and W. C. Lineberger, The Only Stable State of O2− Is the X 2Πg Ground State and It (Still!) Has an Adiabatic Electron Detachment Energy of 0.45 eV, J. Phys. Chem. A, 2003, 107(41), 8521–8529 CrossRef CAS.
- S. J. Cavanagh, S. T. Gibson, M. N. Gale, C. J. Dedman, E. H. Roberts and B. R. Lewis, High-resolution velocity-map-imaging photoelectron spectroscopy of the O−photodetachment fine-structure transitions, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 76(5), 052708 CrossRef.
- Y. Jean, Molecular orbitals of transtion metal complexes, OUP, Oxford, UK, 2005 Search PubMed.
- J. A. Gibbard and J. R. R. Verlet, Photoelectron Imaging Study of the Diplatinum Iodide Dianions [Pt2I6]2− and [Pt2I8]2−, J. Phys. Chem. A, 2022, 126(22), 3495–3501 CrossRef CAS PubMed.
- J. A. Gibbard and J. R. R. Verlet, Photoelectron imaging of PtI2− and its PtI− photodissociation product, J. Chem. Phys., 2022, 156(13), 134303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04576e |
| This journal is © the Owner Societies 2023 |