
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceα-Diazo-λ3-iodanes and α-diazo sulfonium salts: the umpolung of diazo compounds
Sven
Timmann
and
Manuel
Alcarazo
 *
*
Institut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen Tammannstr. 2, 37077 Göttingen, Germany. E-mail: manuel.alcarazo@chemie.uni-goettingen.de
First published on 6th June 2023
Abstract
This minireview aims to cover the developments of the last few years in the design of reagents able to accomplish the umpolung of the azomethine carbon in diazo compounds namely, α-diazo-λ3-iodanes and α-diazo sulfonium salts. Specifically, the routes available for their preparation and a classification of their unique reactivity patterns (either acting as equivalents of carbene-radical or carbene-carbocation synthons) are discussed. We also present a detailed overview of the synthetic utility of such species and, when possible, a critical comparation their reactivity and properties.
1. Introduction
Due to the broad variety of chemical transformations that they are able to promote, diazo compounds have well deserved their position as indispensable reagents in the toolbox of the synthetic practitioner. By irreversible elimination of N2, a process that can be induced thermally, photochemically or by treatment with transition metals, diazo compounds generate short lived carbenes or carbene–metal complexes,1 which further react either via addition to unsaturated substrates (e.g. cyclopropanation reactions)2 or via insertion into σ-bonds (e.g. C–H or X–H functionalizations).3,4 Diazo compounds also react with electrophiles at their carbon atom delivering aliphatic diazonium ions that subsequently lose N2 to form carbocations;5 and not less important, diazo compound additionally participate in a number of 1,3-dipolar cycloadditions in which the N2-fragment stays as a constitutional fragment of the newly-constructed heterocycle.6Interestingly, these three modes of reaction have a common origin, a nucleophilic character at the carbon atom of the CN2 moiety, which can be understood by observation of the frontier orbitals and charge distribution already in the parent diazomethane 3. Natural population analysis at the B3LYP7/def2SVP8,9 level indicates that in 3 the C-atom bears a substantial negative charge (−0.483 e), while both N-atoms remain nearly neutral (+0.020 e (N1); −0.033 e (N2)). Moreover, inspection of the HOMO orbital in 3 reveals that the electron pair is mainly localized in a p-orbital at C1 (Fig. 1a and b). This situation also explains why the stability of diazo compounds increases when electron withdrawing groups (EWG) are attached to the carbon bearing the diazo functionality. The decomposition enthalpies for model compounds 1–3 are also shown in Fig. 1c.10
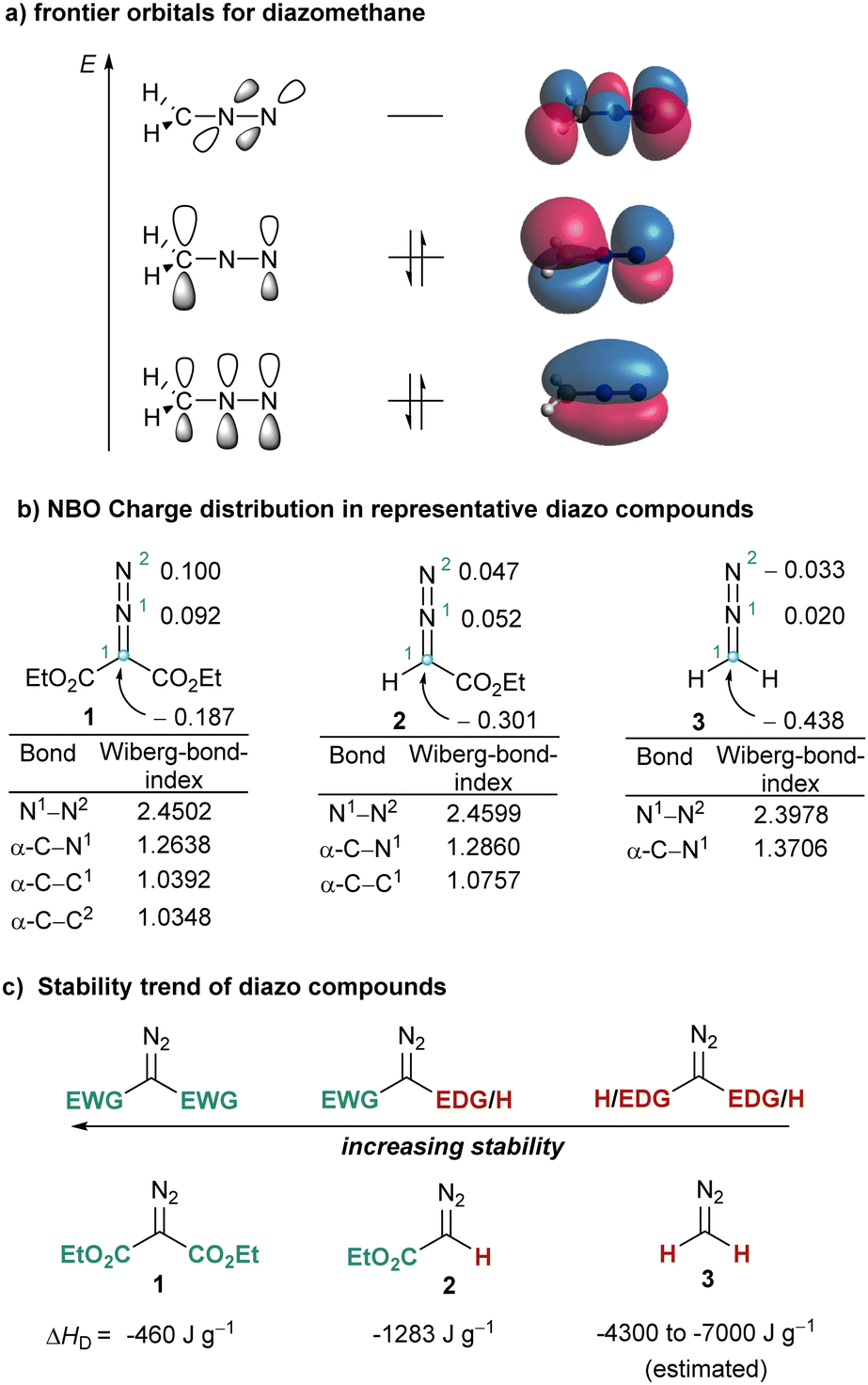 | ||
| Fig. 1 Structural features of diazo compounds. (A) Representation of the frontier orbitals of diazomethane calculated at B3LYP/def2SVP level; (b) partial NBO charges and Wiberg bond orders at the same level; (c) reactivity trend. EWG = electron-withdrawing group, EDG = electron-donating group. ΔHD = average enthalpy of decomposition. | ||
To better harvest the synthetic potential of diazo compounds, chemist have unsurprisingly tried to reverse their inherent polarity from nucleophilicity to electrophilicity by introducing a good leaving group at the C-atom of the diazo unit. That was probably what led Weiss to directly attach a positively charged and extremely nucleofuge phenyl iodonium moiety to the diazo unit in 4a.11 However, comparison of the NBO charge distribution in 2 and 4a counterintuitively reveals that the partial negative charge at the azomethine carbons remains basically unchanged in both compounds. The umpolung of C1 does not seem to be achieved by introduction of the phenyl iodonium group (Fig. 1 and 2). On the other hand, in 4a the iodine atom bears a complete positive charge (+1.020 e−), and the low lying LUMO, which is characterized by its considerable coefficient at the I-atom, corresponds to the σ*(I–C1) orbital. These two properties make possible the direct attack of nucleophiles (Nu−) to the iodine centre, a process that is often followed by reductive elimination of the Nu–R moiety from the iodine centre. It is actually that reaction pathway the one that explains the apparent umpolung of the azomethine carbon in 4a and structurally related compounds.
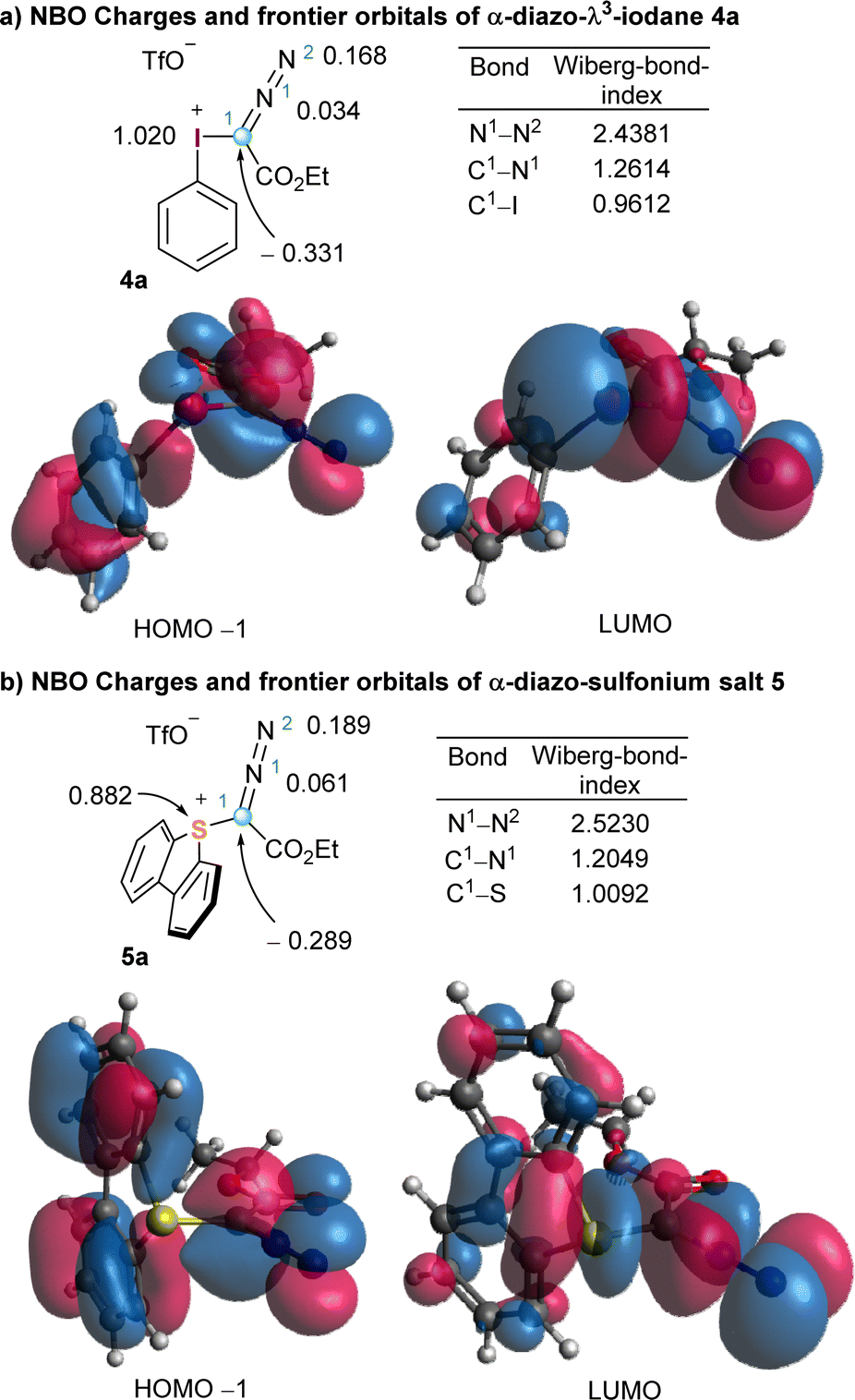 | ||
| Fig. 2 Representation of the HOMO−1 (left) and LUMO (right) of iodonium salt 4 (a) and sulfonium salt 5 (b). Partial NBO charges and Wiberg bond orders are calculated at B3LYP/def2SVP level. | ||
The parallelism in terms of reactivity often observed between the reactivity of hypervalent I(III) reagents and sulfonium salts is also illuminated when the same analysis is carried out for compound 5a (Fig. 2b). The sulfur atom is highly electron deficient and also bears an almost full positive charge (+0.882 e−). In addition, the LUMO corresponds to the σ*(S–C1) orbital, which is again significantly localized at the S-atom. A similar electronic structure generates analogue reactivity, and hence, sulfonium salts are often employed in synthesis when the necessary iodine-based reagent is thermally too unstable to be used in multigram scale, or simply when there is no protocol available for its preparation.10,12
One of the key features of reagents 4 and 5 is the presence of two orthogonal leaving groups attached to the same carbon-atom. Careful control of the reaction conditions allows each of the groups to be selectively addressed, and by this, different reaction pathways arise. The reactivity of a formal carbene–carbocation can be achieved when 4 or 5 react with a transition-metal catalyst delivering a metal–carbene species and subsequently the iodoarene or sulfide moieties are released. Alternatively, utilizing photoredox-catalytic conditions, the homolytic cleavage of the sulfur– or iodine–carbon bond takes place first, resulting in the formation a carbon-centered diazo radical. Once the radical has been trapped, the carbene reactivity can be accessed by release of dinitrogen, either thermally or through metal catalysis (Scheme 1).
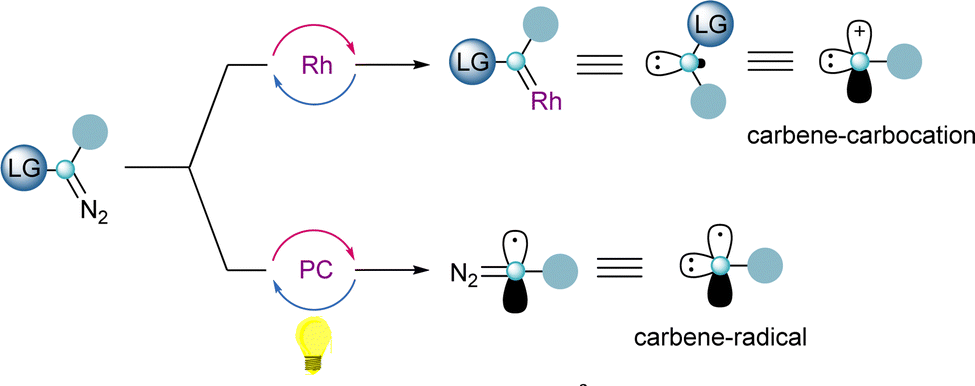 | ||
| Scheme 1 Dual reactivity character of α-diazo-λ3-iodanes and α-diazo-sulfonium salts. | ||
With this minireview we intend to provide the reader with an overview on the preparation and synthetic potential of these two families of compounds (α-diazo-λ3-iodanes and α-diazo sulfonium salts), together with a critical comparison between their reactivity and scope. Unsolved problems derived from the use of these reagents will be indicated, and possible developments in the area will also be anticipated.
2. Synthesis of α-diazo-λ3-iodanes and their sulfonium salts analogue
The prototype α-diazo-λ3-iodane 4a can be accessed in one-pot by initial activation of commercially available λ3-iodane precursors, such as the diacetoxy iodobenzene 6 (PIDA), with trimethylsilyl triflate and subsequent reaction with excess of the desired diazo compound.11 This route seems to be quite general and works equally well for several stabilized diazocompounds; substituents of different nature on the arene ring are tolerated as well. The use of reagent 4a presents, however, a practical limitation. It is not thermally stable and needs to be stored under refrigeration.13 Besides that, 4a is characterized by a highly exothermic decomposition upon moderate heating to 80 °C (472.4 J g−1),13 which limits its utility to small scale reactions. To mitigate, at least partially, this inconvenience and improve the safety profile of these structures, cyclic derivatives have been prepared using, for example, the benziodoxolone platform. This strategy has proven to be satisfactory for many other I(III)-based reagents.14α-Diazo-benziodoxolone reagent 9a is prepared in a similar fashion as its non-cyclic analogues; namely, initial activation of the acetoxy precursor 7 with TMS triflate followed by addition of the diazo compound under basic conditions. Pyridine seems to be the ideal base to trap the triflic acid generated and avoid decomposition of 9a. Alternatively, treatment of 8 with TMS triflate and excess of the desired diazo compound in the absence of base delivers the benzoate derivative 10a. The formation of this species can be explained by protonation of 2 followed by loss of molecular nitrogen and trapping of the corresponding carbocation by the carbonyl moiety of in situ generated 9a.15
The X-ray structures of 9a and 10a are depicted in Scheme 2; interestingly, the benzoyl oxygen atom in 10a also interacts with the neighboring iodine delivering an iodaxolane-type architecture, although not as tight as the one in 9a. Actually, the reactivity of 10a and analogue reagents can be tuned by addition of Lewis acids such as Zn(NTf)2; they coordinate the benzoyl moiety and, consequently, reduce the donation of this group towards the λ3-iodane unit.15
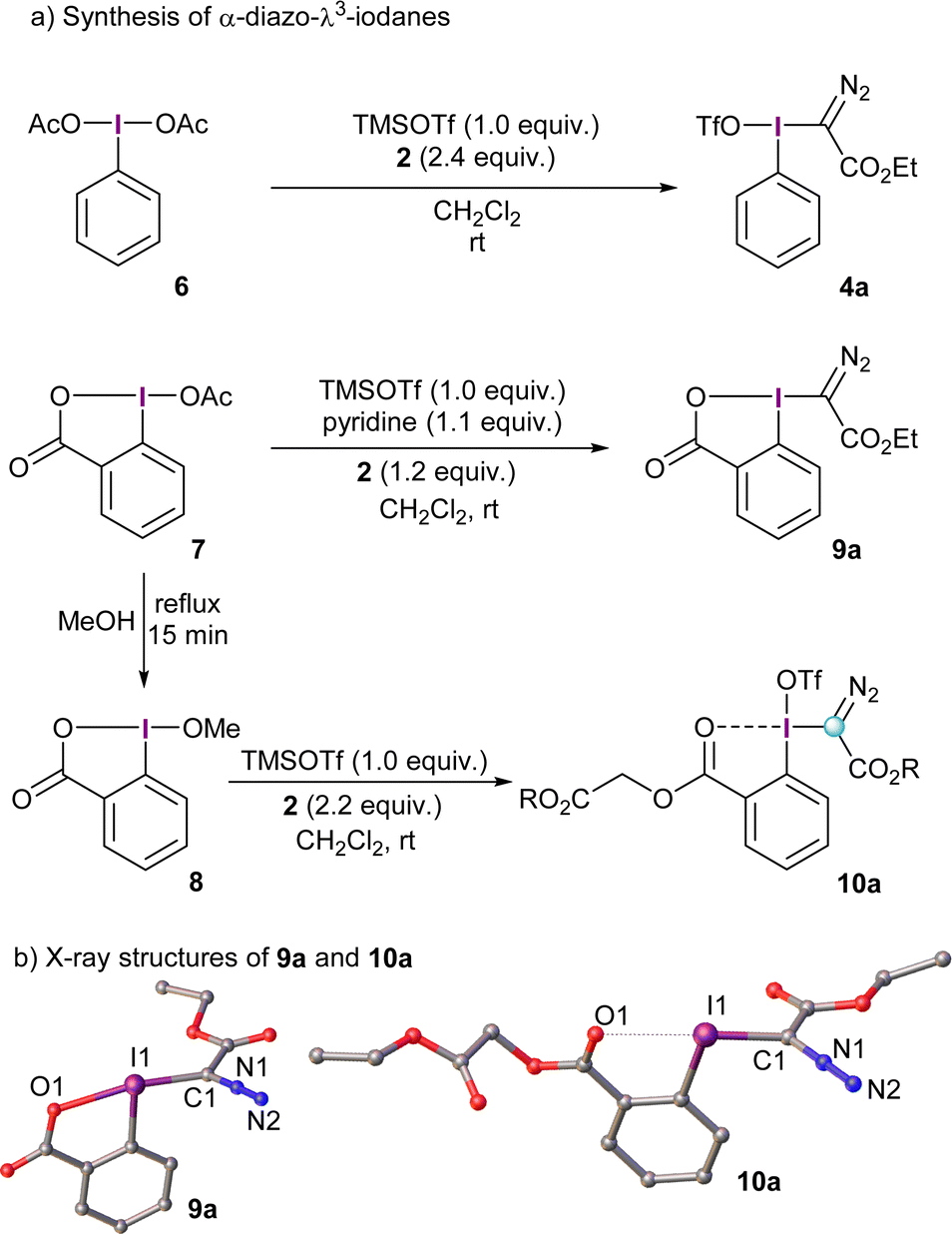 | ||
| Scheme 2 Synthetic pathways towards α-diazo reagents 4a, 9a and 10a and X-ray structures of 9a and 10a. Hydrogen atoms and triflate-anion (10a) omitted for clarity; ellipsoids shown at 50% probability level. | ||
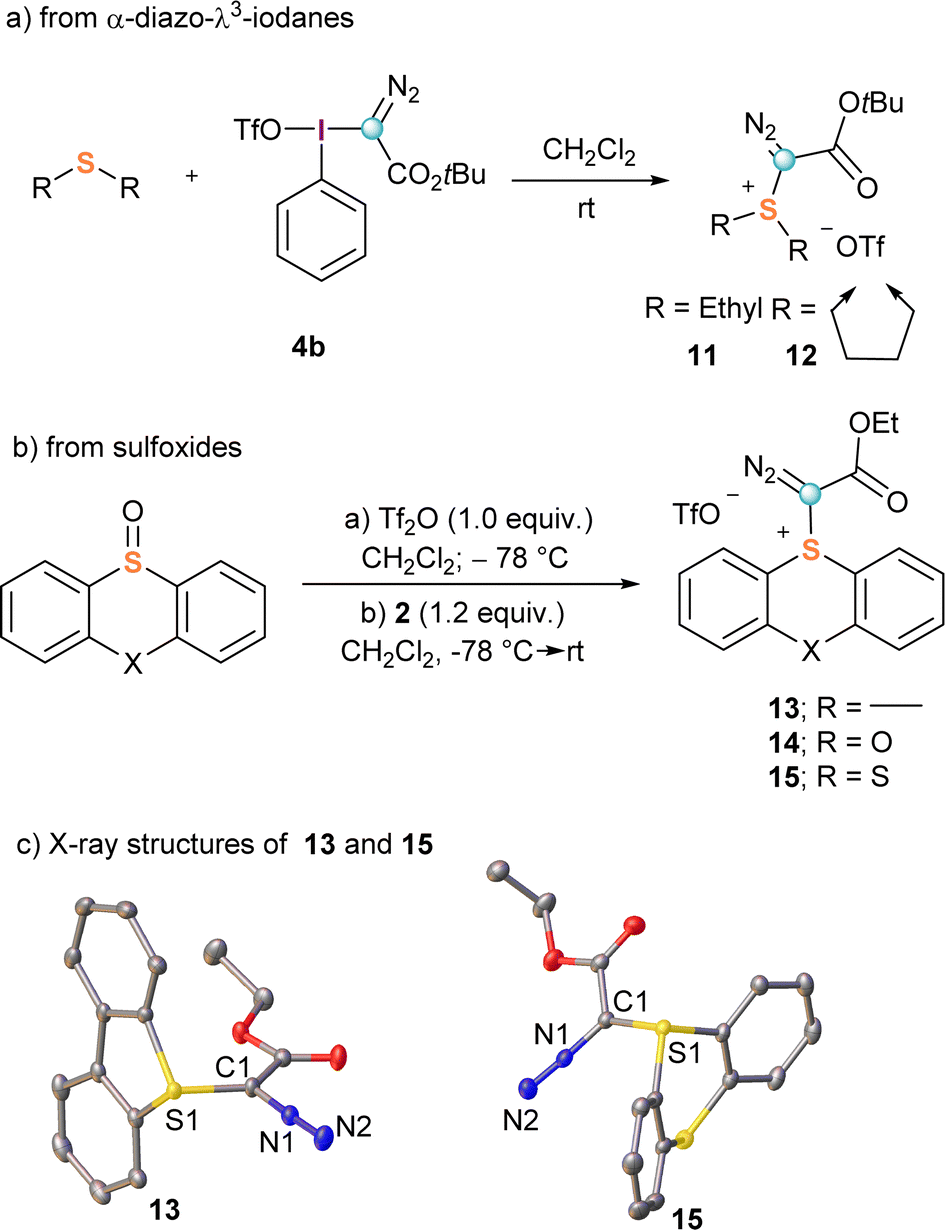
The first synthesis of α-diazo substituted sulfonium salts, compounds 11 and 12, was also reported by Weiss.11,16 These species were obtained with excellent yields by direct reaction of 4b with dimethyl sulphide or tetrahydrothiophene, respectively (Scheme 3a); however, this route has two drawbacks that severely limit its practical use: (i) it requires the previous preparation of the corresponding λ3-iodane precursors 4, and (ii) the diazo transfer process is not general. Typically, only electron rich dialkyl sulphides are nucleophilic enough for the reaction to proceed.
 | ||
| Scheme 3 Synthetic pathways towards α-diazo reagents 11–15 and X-ray structures of 13 and 15. Hydrogen atoms and triflate-anions omitted for clarity; ellipsoids shown at 50% probability level. | ||
To circumvent these shortcomings, we recently developed a general synthetic protocol towards α-diazo sulfonium salts starting from easily available sulfoxides. Activation of these starting materials with triflic or trifluoroacetic acid anhydride at low temperatures generates a highly electrophilic sulfurane intermediate, which is able to react with α-diazo compounds to afford the expected α-diazo-sulfonium salts.17 Following that route, salts derived from dibenzothiophene (13), phenoxatiine (14) and thianthrene (15) were prepared in a one-pot procedure (Scheme 3b). The X-ray structures of 13 and 15 are shown in Scheme 3c. Interestingly, differential scanning calorimetry of a sample of 13 shows decomposition above 130 °C leading to a significative energy release of 400 J g−1;17 for comparison, compound 9a starts decomposing at 120 °C with a larger release of energy >600 J g−1.15 On the basis of these results, it can be concluded that the use of 13 is relatively safer; in particular for large scale laboratory syntheses.
3. Reactivity of α-diazo-λ3-iodanes
The transformations in which α-diazo-λ3-iodanes are involved have been grouped in three different sections according to the operating mechanism proposed by the authors. Section 3.1 describes reactions in which the diazo group transfer occurs via typical nucleophilic attack at its carbon atom. Section 3.2 lumps together those processes in which the diazo transfer reaction takes place via formation of a diazoalkyl radical, and finally, Section 3.3 compiles the family of reactions that initially exploit the carbene reactivity of the diazo group in 4, 5, 9 or 10.3.1 Transfer of the diazoalkyl group by reaction with nucleophiles
Initial studies by Weiss and co-workers showed that reagent 4a is able to transfer the diazoalkyl moiety to a series of neutral nucleophiles such as tertiary amines, pyridines, arsines, stilbines and dialkyl sulfides furnishing the corresponding -onio products 17a–e (Scheme 4a). Interestingly, the reaction with triphenylphosphine (18) resulted in the formation of a phosphino phosphazine 19, regardless of the reaction conditions applied and the stoichiometry used (Scheme 4b).11 This particular reactivity has been further investigated by Podrugina et al. using phosphines with different substitution pattern.16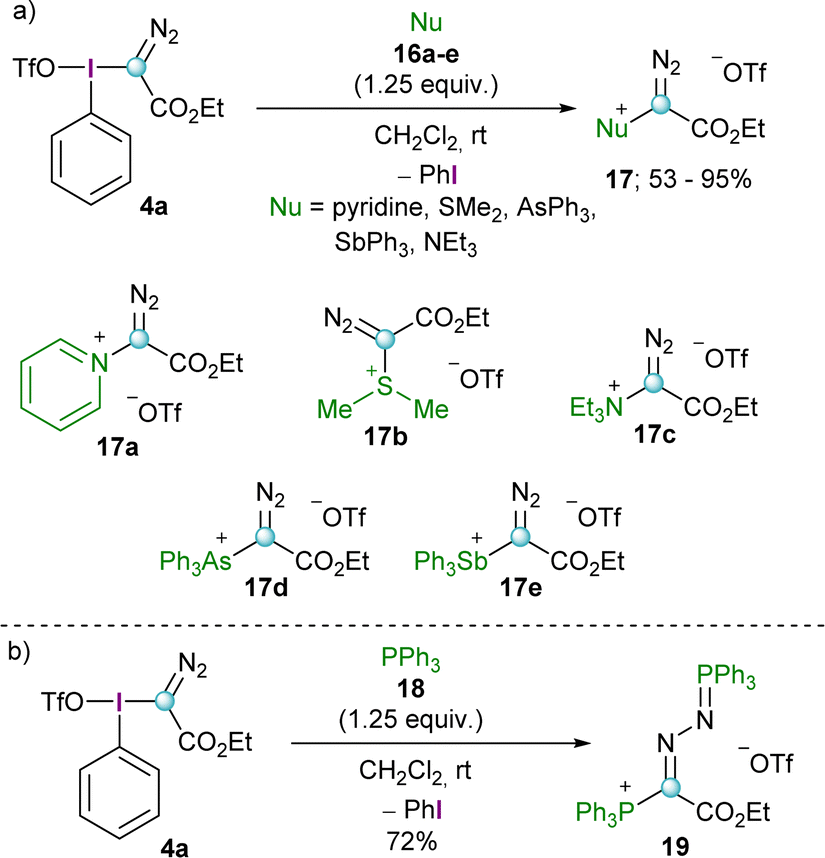 | ||
| Scheme 4 Nucleophilic substitution reaction of the α-diazo aryliodonio compounds with neutral nucleophiles by Weiss and co-workers. | ||
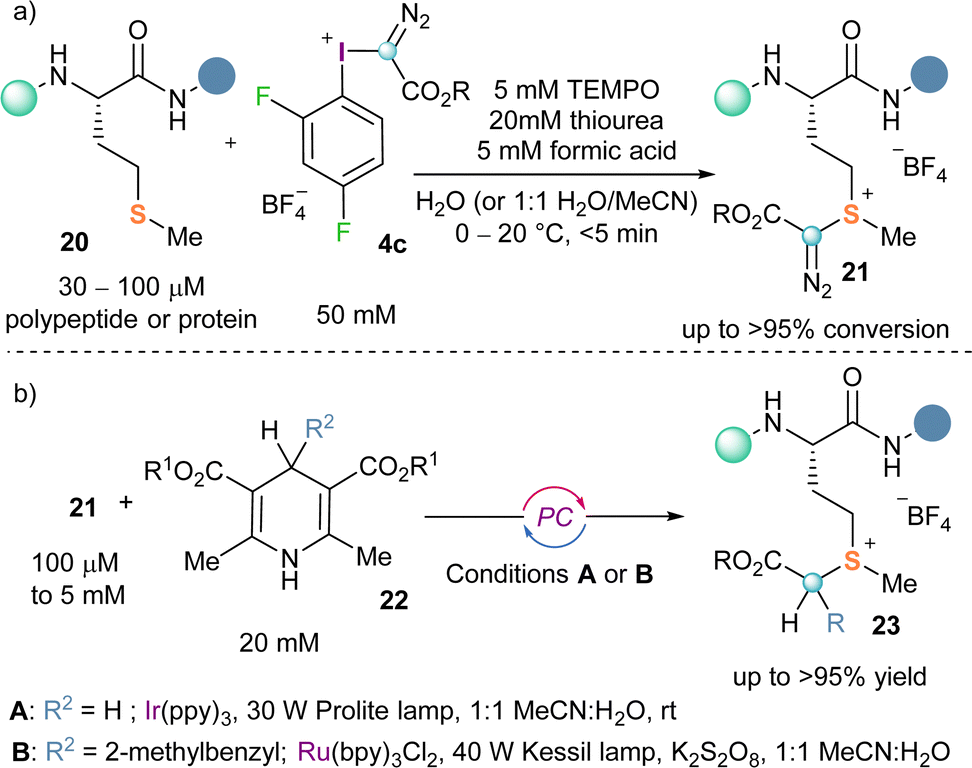
Making use of this reactivity mode Gaunt and co-workers recently conceived that α-diazo-λ3-iodanes could be applied to the selective functionalization of polypeptides and proteins at methionine residues. For this purpose, they designed an electron poorer derivative than the original Weiss reagent by introducing two fluorine atoms in the arene moiety (4c). The desired bioconjugation reaction using 4c, proceeds selectively delivering the expected sulfonium salt 21 (Scheme 5a).
 | ||
| Scheme 5 (a) Methionine-selective bioconjugation strategy; (b) protein diversification via photo-redox catalysis. | ||
The advantages associated with the targeting of the methionine residue are two-folded; its low abundance in proteins (around 2%) makes the reaction extremely site-selective and, additionally, the functionalization of methionine residues is not likely to interfere with the protein function.18,19 Also remarkable is the fact that the diazo unit once attached to the methionine residue could be used for further derivatization of the protein via photocatalytic approaches (Scheme 5b).20
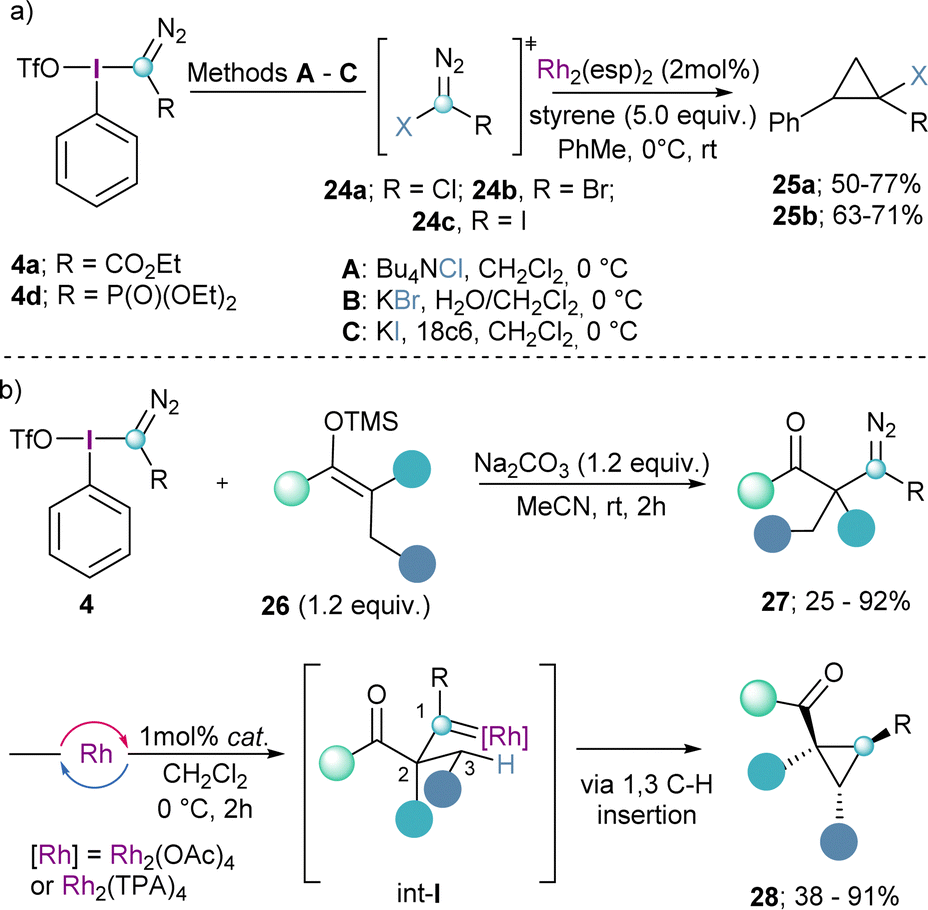
The same reaction mode has been employed by the group of Bonge-Hansen on the synthesized α-halo diazocompounds 24a–c by reaction of 4a and 4d with halide anions. These compounds were subsequently converted into the corresponding halo-substituted cyclopropane derivates 25a and 25bvia catalytic carbene addition to the double bond of styrenes employing Rh2(esp)2 as catalyst (Scheme 6a).21
 | ||
| Scheme 6 (a) Nucleophilic halogenation of α-aryliodonio diazo reagents with following catalytic cyclopropanation; (b) electrophilic diazomethylation of ketone silyl ethers. | ||
The synthesis of α-diazocarbonyl compounds is straightforward and can be achieved through diazo-transfer reactions such as the Regitz diazo-transfer, which makes react sulfonylazides with enolates.22 However, the synthesis of the β-homologes 27 is much more challenging and the diazo products lack the stabilization of an electron withdrawing group. Suero and co-workers realized that by reaction of the Weiss-reagent 4 with silylenol ethers (26), the desired β-diazocarbonyl compounds 27 could be obtained (Scheme 6b). Subsequently, compounds of general formula 27 were transformed into the corresponding cyclopropanes 28via initial Rh-promoted carbene formation (int-I) followed by the highly diastereoselective insertion of the carbene into the C(sp3)–H bond in position 3 of that species (Scheme 6b).13
A related transformation that also exploits the reactivity of electron rich olefins with 4 has been reported by the group of Wang; however, in this case the nucleophile was a Breslow intermediate catalytically generated by the umpolung of aldehydes with a bicyclic N-heterocyclic carbene 30. The products obtained from this transformation are 2,5-disubstituted-1,3,4-oxadiazoles 31, which originate from the attack of the electron rich olefin to the electrophilic terminal nitrogen of the diazo group, and not to the azomethine carbon.
Fundamental for the understanding of this unique mechanism and reaction outcome was the isolation of Int-II, which is formed by the initial reaction of TMEDA with 4 under the conditions applied (Scheme 7a).23
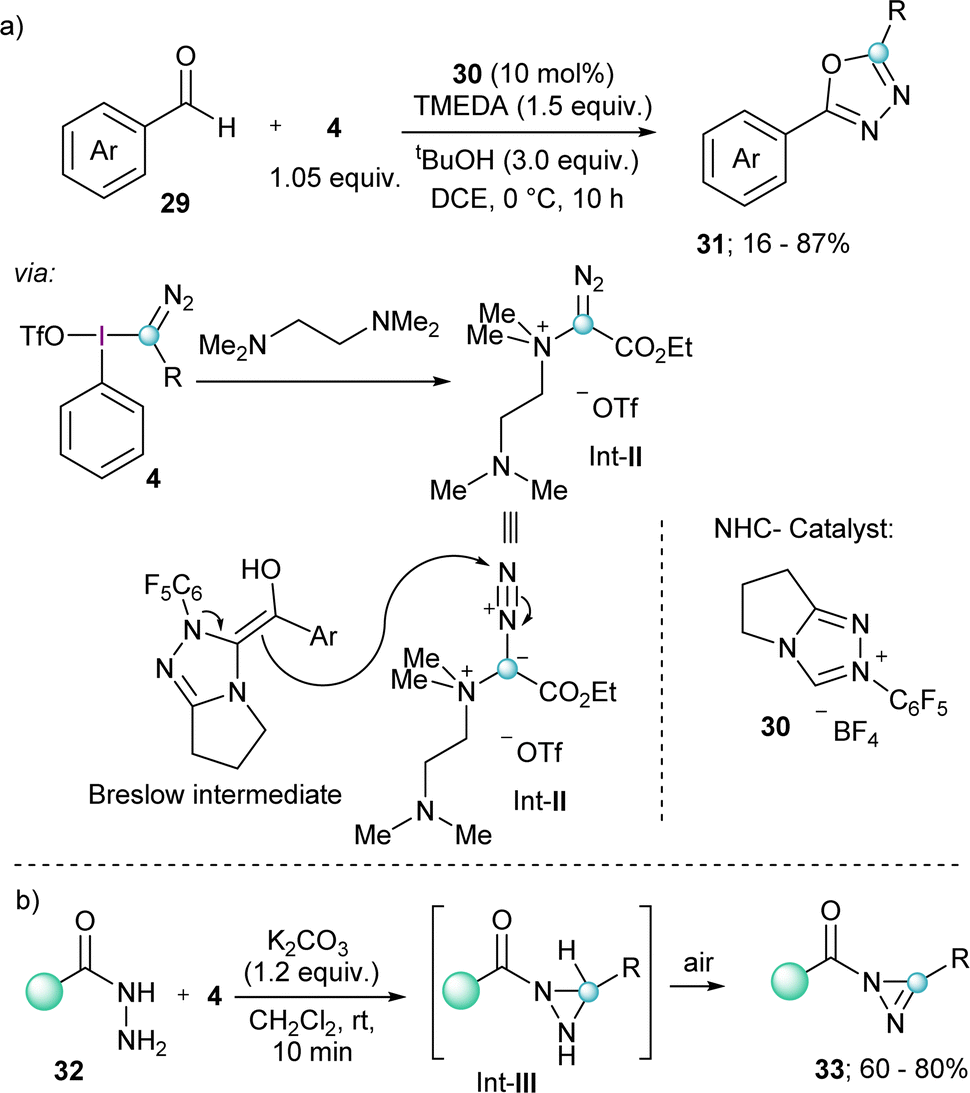 | ||
| Scheme 7 (a) NHC-catalyzed formation of 2,5-disubstituted 1,3,4-oxadiazoles; (b) formation of substituted diazirines. | ||
Finally, two additional transformations were reported in 2022 exploiting the electrophilicity of 4 and the excellent properties of iodobenzenes as leaving groups. The group of Singh described a novel route towards the synthesis of substituted diazirines 33 by reaction of semicarbazides 32 with 4, followed by oxidation of the diaziridine intermediate (Int-III) by air (Scheme 7b).24 Also in the same year Rastogi and coworkers published a base assisted synthesis of [1,2,4]triazolo[4,3-a]pyridines 35, by reaction of 4 with pyridines and isoquinolines (Scheme 8).25 Intermediates in this reaction are α-diazoalkyl pyridinium salts (Int-IV); compounds already described by Weiß in his seminal work.11 Treatment of these species with base result in the deprotonation of the iminium moiety and subsequent cyclization towards the desired heterocycles.25 A variant of this reaction, but using piperidines and tetrahydroisoquinolines as the nucleophilic reaction partner has been very recently reported by Singh and coworkers.26
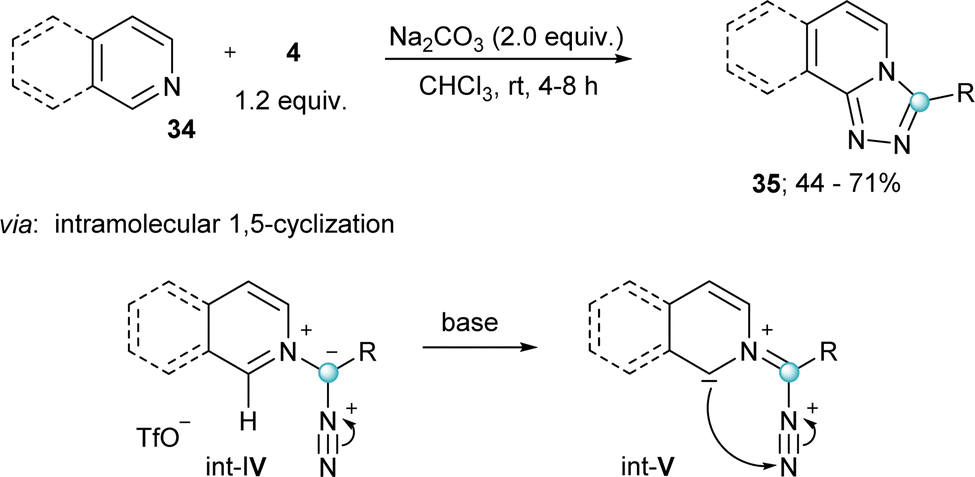 | ||
| Scheme 8 Synthesis of [1,2,4]triazolo[4,3-a]pyridines starting from pyridines. | ||
3.2 Transfer of the diazo group via radical processes
As previously indicated, α-diazo-λ3-iodanes of general formula 4 have been known for three decades; however, they were sporadically used as transfer reagents. It was only after the work of Suero in 2018 that their broad synthetic potential was first recognized, and their reactivity intensively explored.Probably inspired by the pioneering work of Strausz and Patrick towards the generation of carbynes by treatment of Hg{C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N2)CO2Et}2 with ultraviolet light,27 Suero and co-workers hypothesized that the generation of diazomethyl radicals (Int-VI) could be achieved more efficiently under the mild conditions offered by photocatalysis; specifically, by single electron transfer to 9 and 10 with concomitant elimination of the iodoarene moiety (Scheme 9b). After optimizing the experimental conditions, these authors succeeded in bringing to practise their hypothesis using white light as radiation source and [Ru(bpy)3](PF6)2 as the most adequate photocatalyst. Furthermore, they developed a C–H arene diazomethylation reaction by trapping the thus generated diazomethyl radicals with an array of (hetero)arenes. The transformation showed impressively broad functional group tolerance, and even a series of different pharmaceutically relevant molecules could be functionalized in acceptable yields. Subsequently, the initially obtained diazo-compounds were further elaborated into α-amino, α-thio, α-bora, α-haloesters and cyclopropanes 37 using the native carbene-type reactivity of the diazo moiety (Scheme 9c).15
N2)CO2Et}2 with ultraviolet light,27 Suero and co-workers hypothesized that the generation of diazomethyl radicals (Int-VI) could be achieved more efficiently under the mild conditions offered by photocatalysis; specifically, by single electron transfer to 9 and 10 with concomitant elimination of the iodoarene moiety (Scheme 9b). After optimizing the experimental conditions, these authors succeeded in bringing to practise their hypothesis using white light as radiation source and [Ru(bpy)3](PF6)2 as the most adequate photocatalyst. Furthermore, they developed a C–H arene diazomethylation reaction by trapping the thus generated diazomethyl radicals with an array of (hetero)arenes. The transformation showed impressively broad functional group tolerance, and even a series of different pharmaceutically relevant molecules could be functionalized in acceptable yields. Subsequently, the initially obtained diazo-compounds were further elaborated into α-amino, α-thio, α-bora, α-haloesters and cyclopropanes 37 using the native carbene-type reactivity of the diazo moiety (Scheme 9c).15
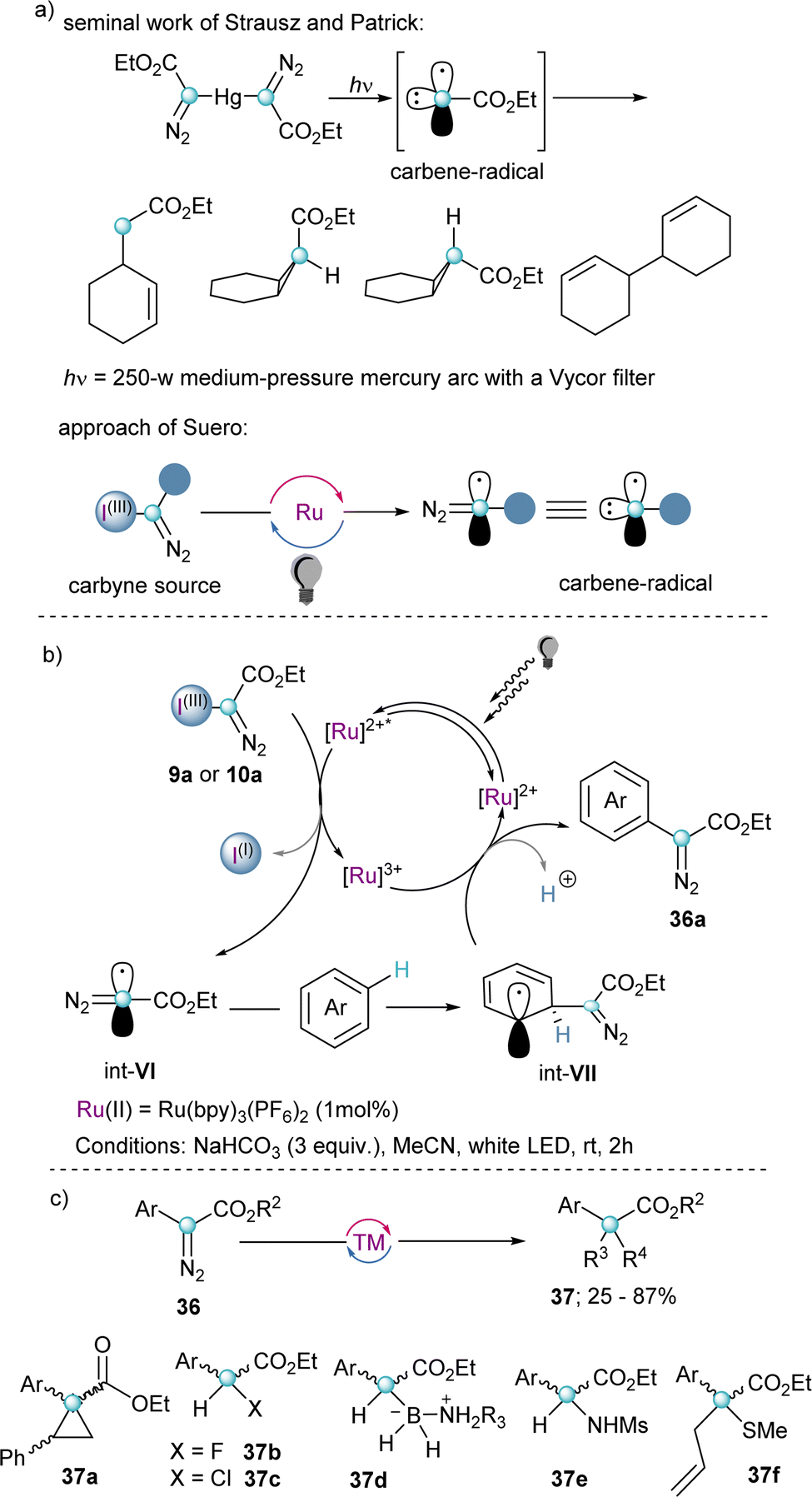 | ||
| Scheme 9 (a) Seminal work of Strausz and Patrick. (b) Photoredox catalysed C–H arene diazomethylation; proposed mechanism. (c) Diazo diversification. | ||
Inspired by this work, Wang and coworkers hypothesized that a good radical scavenger such as isonitrile 38 should also be able to react with electrophilic diazomethyl radicals (int-VI) to afford ethyl diazo-6-phenanthridinyl acetates. Interestingly, no trace of the expected product was formed; in fact, this reaction effectively delivers phenanthridine-6-carboxylates 39. Radical trapping experiments using 2,2,6,6-tetramethylpyperidine oxide (TEMPO) as scavenger allowed the isolation of the carboxylic ester adduct 40, which is a clear proof of the formation of the carboxylic radical int-VIII. Hence, Wang and co-workers proposed that under the reaction conditions employed, the initially formed diazomethyl radical thermally decomposed via extrusion of diazocarbene to form the corresponding carboxylate radical.28 Reaction of this open shell species with 38 followed by radical addition to the neighbouring phenyl ring, oxidation, and deprotonation, leads to the formation of 39 (Scheme 10).29
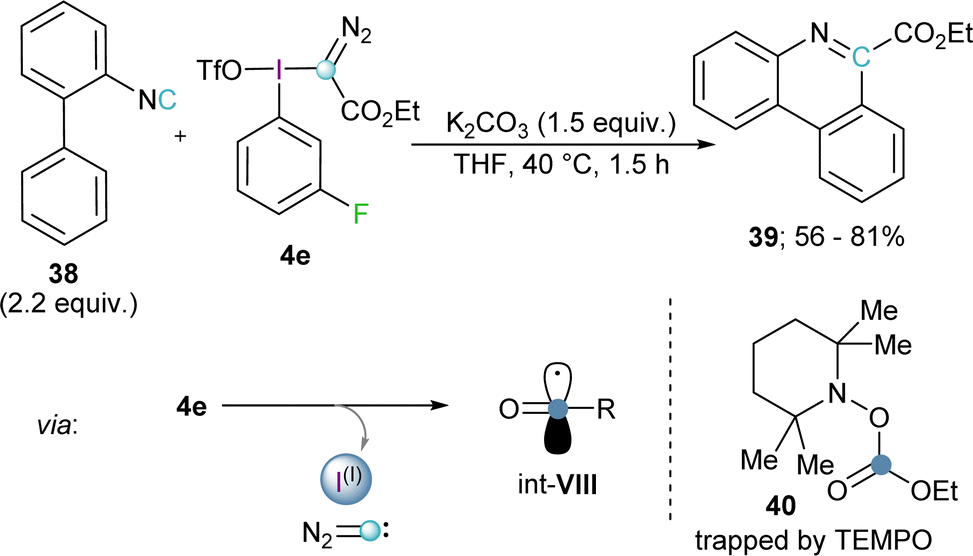 | ||
| Scheme 10 Phenanthridine synthesis via radical, diazo-methane elimination pathway. | ||
Also making use of the mesolitic C–I bond cleavage after one electron reduction, Li and co-workers recently reported a photoredox catalysed cascade transformation that delivers substituted acridines by reaction of 4 with diarylamine substrates 41. The authors propose the initial generation of the diazomethyl radical intermediate (int-VI) from 4 and subsequent interception of this species by 41 to generate the radical adduct (int-IX). Subsequent single-electron oxidation delivers int-X. Finally, photochemical decomposition of the diazo compound by irradiation with blue light delivers a free carbene (int-XI), which is intramolecularly trapped by the second aromatic ring of the diarylamine-substrate (int-XII). Oxidation of this intermediate produces the observed acridines 42 (Scheme 11).30 Other unsaturated organic nucleophiles can serve as diazomethyl radical acceptors as well. For example, a visible light-induced [3+2] cyclization reaction of hydrazones (43) with reagent 10, to form 1-amino-1,2,3-triazoles (44) was reported by the group of Li in 2021 (Scheme 12a). The reaction is initiated by single-electron reduction of reagent 10 under photochemical conditions to generate the diazomethyl radical (int-VI). Subsequent addition to the aldehyde-derived hydrazone 43 forms a N-centred radical intermediate (int-XIII), which is oxidized to int-XIV by the photocatalyst. In the presence of a base, deprotonation of int-XIV delivers an α-diazoalkylhydrazone, which cyclizes to the desired 1-(dialkylamino)-1,2,3-triazole product 44 (Scheme 12b).31 Interestingly, the same transformation was shortly earlier published by the Alcarazo group using α-diazo sulfonium triflates as the radical precursor;17 this variant of the reaction will be discussed in the next section of this minireview. A structurally related photoredox catalyzed [3+2] cyclization able to deliver 1,2,3-triazole fused quinoxalinones via an analogue mechanism has also been reported.32
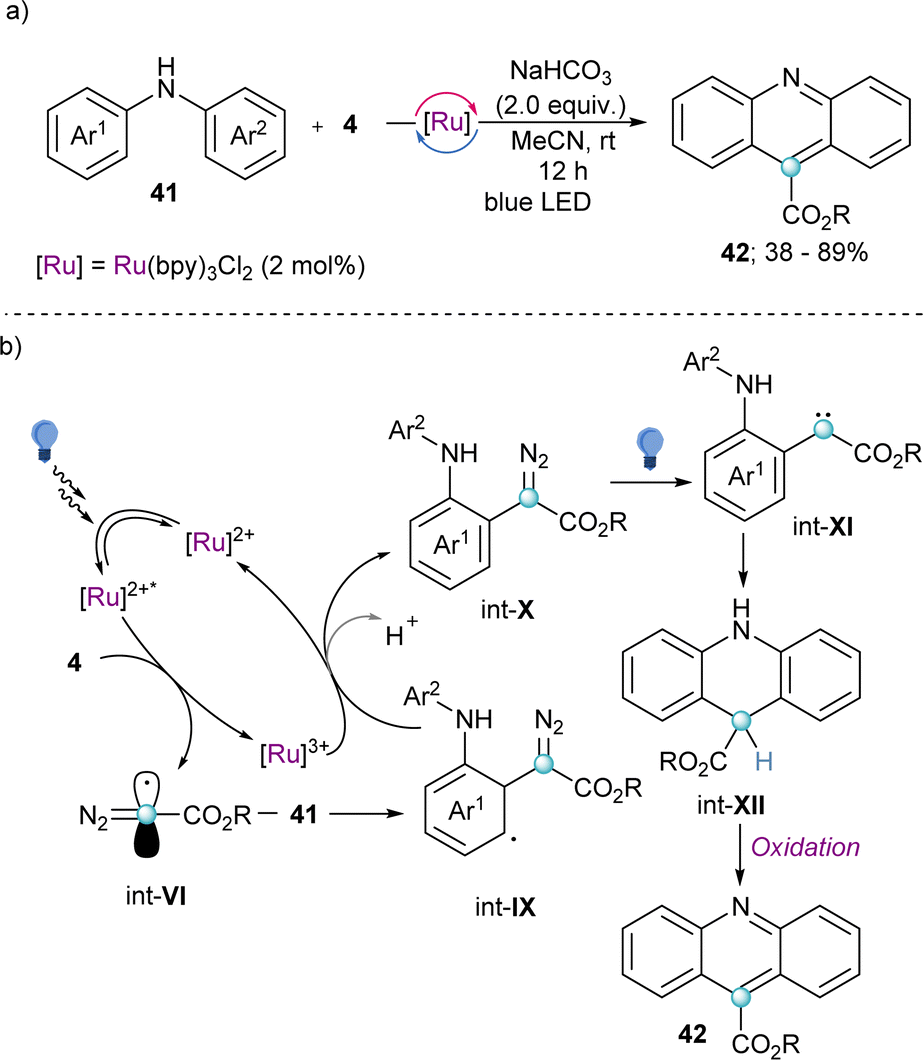 | ||
| Scheme 11 (a) Photoredox catalysed cascade reaction towards substituted acridines. (b) Proposed mechanistic pathway for the formation of acridines. | ||
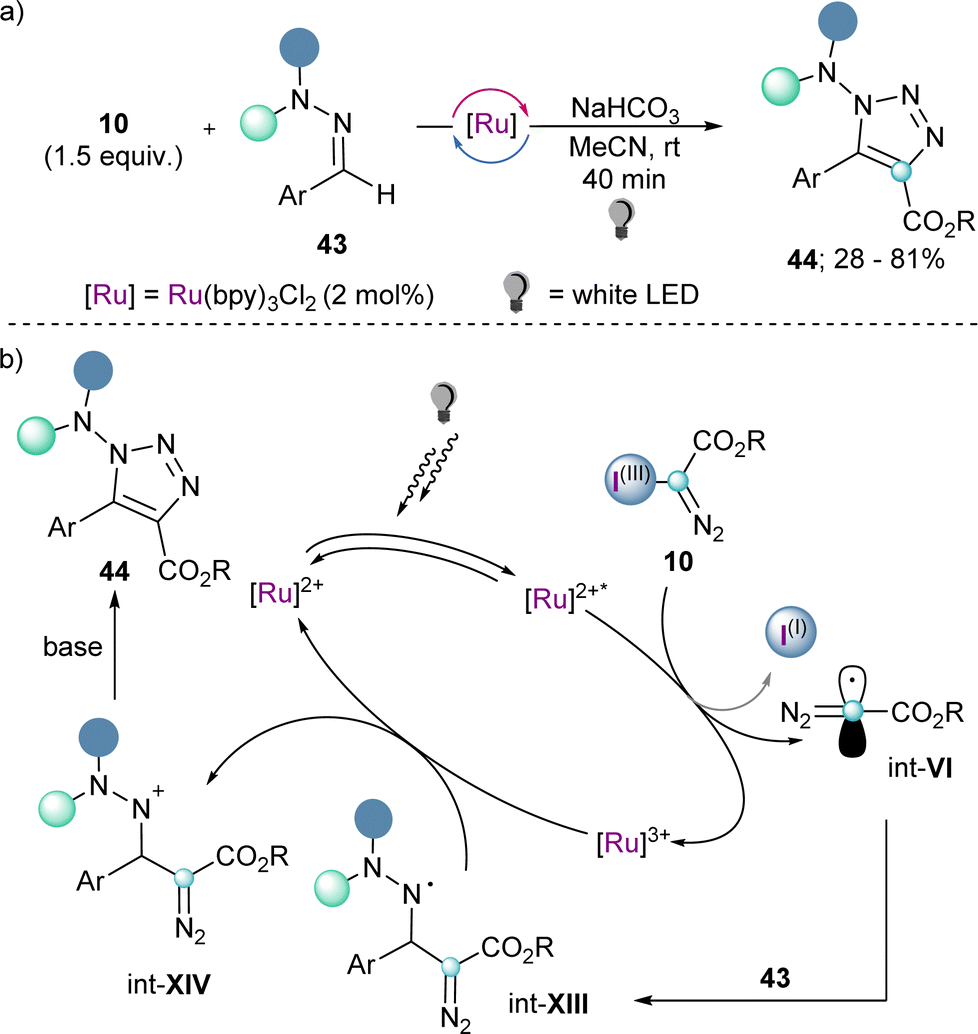 | ||
| Scheme 12 (a) Visible light-induced [3+2] cyclization of hydrazones to 1-amino-1,2,3-triazoles. (b) Proposed mechanism for the cyclization towards 1-amino-1,2,3-triazoles. | ||
The synthetic potential of int-VI might be even wider if this species could be trapped by another radical. Willing to explore this possibility and being aware of the fact that acyl radicals can be prepared from the decarboxylation of α-ketoacids by photoredox catalysis, Liu and co-workers developed a decarboxylative coupling towards 2,5-disubstituted 1,3,4-oxadiazoles from 10a and α-oxocarboxylic acids that relays on the simultaneous generation of both radicals (Scheme 13a). After excitation of the organic photocatalyst, single-electron oxidation of the α-oxocarboxylic acid delivers the corresponding radical cation, which undergoes decarboxylation to generate radical intermediate int-XV. Subsequently, the reduced photocatalyst is able to reduce the I(III)-reagent 10a liberating the diazomethyl radical int-VI. Radical combination leads to the formation of int-XVI, which finally delivers the desired 2,5-disubstituted 1,3,4-oxadiazoles 46 after intramolecular ring-closure.33 Unfortunately, the extension of this method to heterocyclic or aliphatic α-oxocarboxylic acids was unsuccessful; this remains as a clear synthetic limitation of this route.
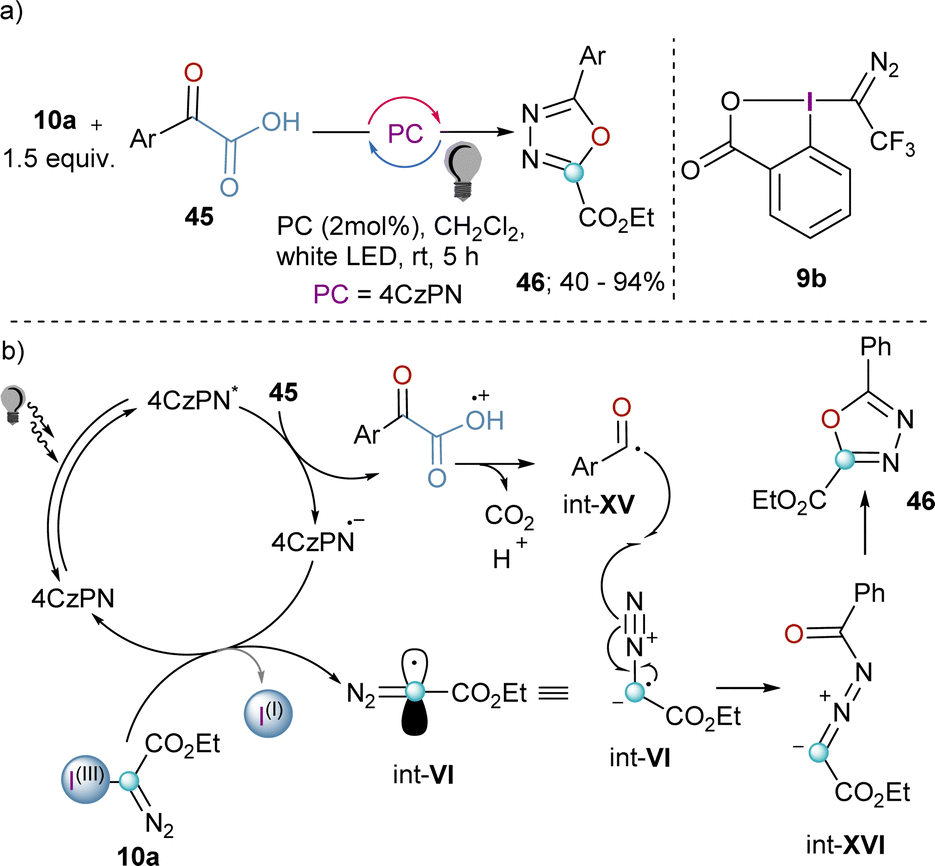 | ||
| Scheme 13 (a) Photoredox catalytic decarboxylative cyclization of α-oxocarboxylic acid and hypervalent iodine(III) reagents. (b) Proposed mechanism. | ||
In 2021 the group of Han also reported a variant of this reaction using the CF3-substituted reagent 9b. In this case no photocatalyst is needed and the diazotrifluoroethyl radical is directly generated from the hypervalent iodine reagent by visible-light irradiation. Moreover, the methodology developed allowed the introduction of trifluoromethyl-substituted 1,3,4-oxadiazole moieties in pharmaceuticals and other bioactive molecules.34
3.3 Initial reaction of the diazo group
In all the examples shown so far, from the two innate reactivities present in α-diazo-λ3-iodanes, the nucleofuge character of the iodonium group has always been exploited in the first instance. However, there is no reason to believe that in the presence of the appropriate catalyst, the carbene-type reactivity might be used primarily, followed by elimination of the aryl iodide moiety. This succession of steps turns α-diazo-λ3-iodanes into synthetic equivalents of carbene carbocations (Scheme 14).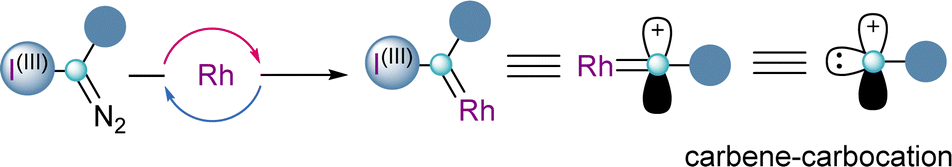 | ||
| Scheme 14 Dual reactivity character of hypervalent iodine(III) diazo reagents. | ||
The group of Suero pioneered the work in this direction and published in 2019 a strategy to insert a carbon atom into the double bond of olefins by using the well-established dirhodium-catalyst Rh2(esp)2. Specifically, they discovered that after initial generation of a highly electrophilic Rh-carbynoid int-XVII by reaction of the Rh-catalyst with 10a, insertion of the carbine into the C(sp2)–C(sp2) double bond generates the transient cyclopropyl-I(III) intermediate int-XVIII, which undergoes disrotatory electrocyclic ring-opening with concomitant elimination of the iodo arene fragment (Scheme 15a).35 The mechanism of that step was studied in detail in the 60's of the twentieth century using cyclopropyl tosylates or bromides as model substrates.36 Subsequent attack of nucleophiles to the allylic cation int-XIX delivers the corresponding building block 48. The scope of the reaction is broad, and halides, alcohols, azide anions and electron rich arenes serve as productive nucleophiles.35 By applying this methodology to terminal alkenes in combination with a fluoride source, the late-stage incorporation of fluorine-substituents to structurally complex olefins is also possible. Indeed, Suero has shown that this method provides access to the desired fluorinated targets with high selectivity towards the branched products (51) (Scheme 15c).37
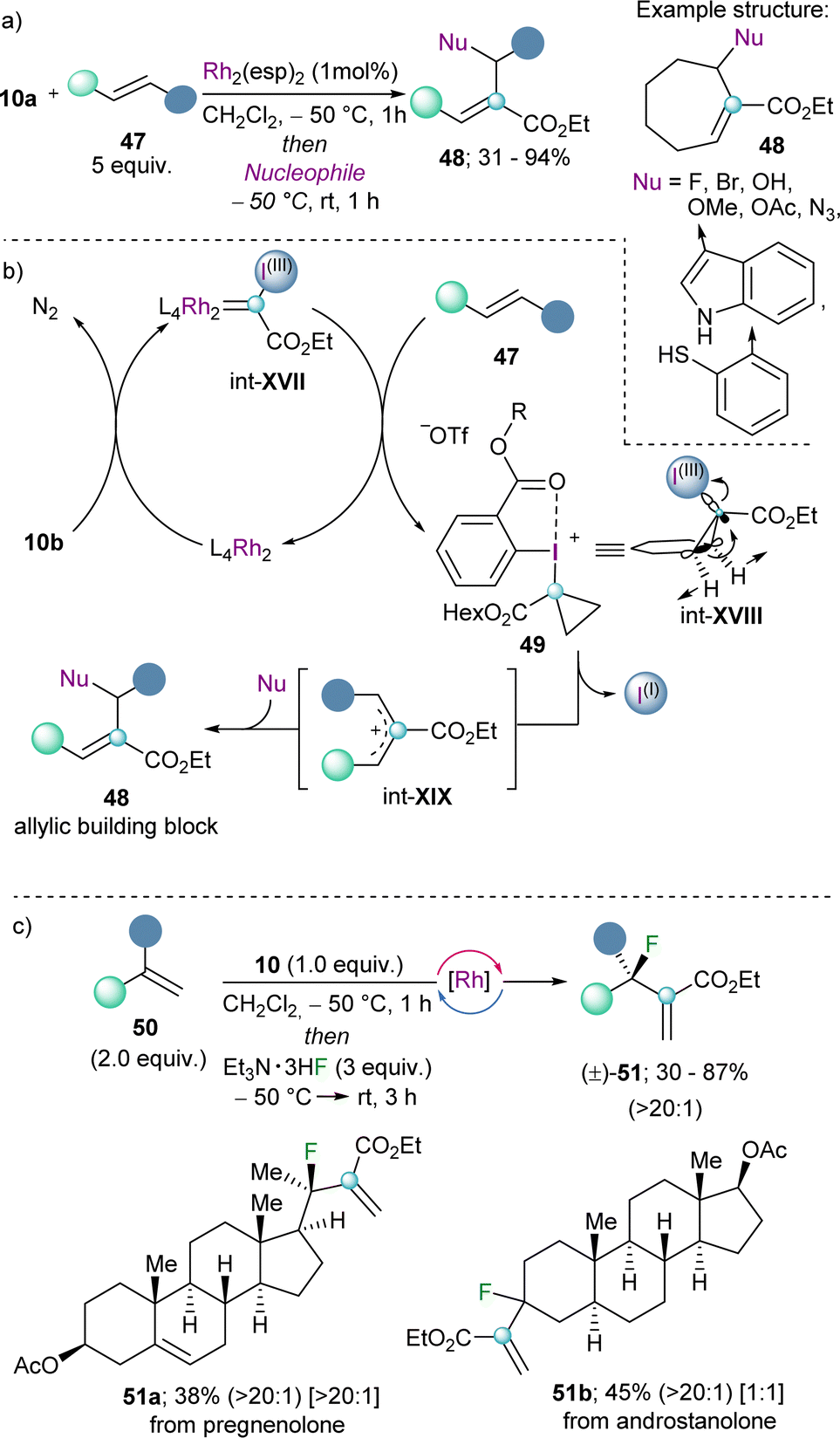 | ||
| Scheme 15 (a) Catalytic scission of C(sp2)–C(sp2) by Rh-carbynoids. (b) Proposed mechanism for the construction of allylic building blocks. (c) Generation of fluorinated cuaternary stereocenters; branched/linear ratio, indicated in parentheses, diastereoselectivity ratio indicated in brackets. | ||
Cyclopropenium cations 53 can also be synthesized using the same concept via the Rh-catalysed carbene addition to alkynes. The reaction seems to proceed by initial formation of cyclopropene intermediate int-XX, followed by elimination of the iodoarene moiety. This step is facilitated by the aromatization of the cyclopropenium cations, which were isolated with a variety of different substituents (53). Moreover, the addition of nucleophiles of different nature to 53 proceeds efficiently and is highly regioselective at the carbon atom that bears the ester functionality (Scheme 16).38
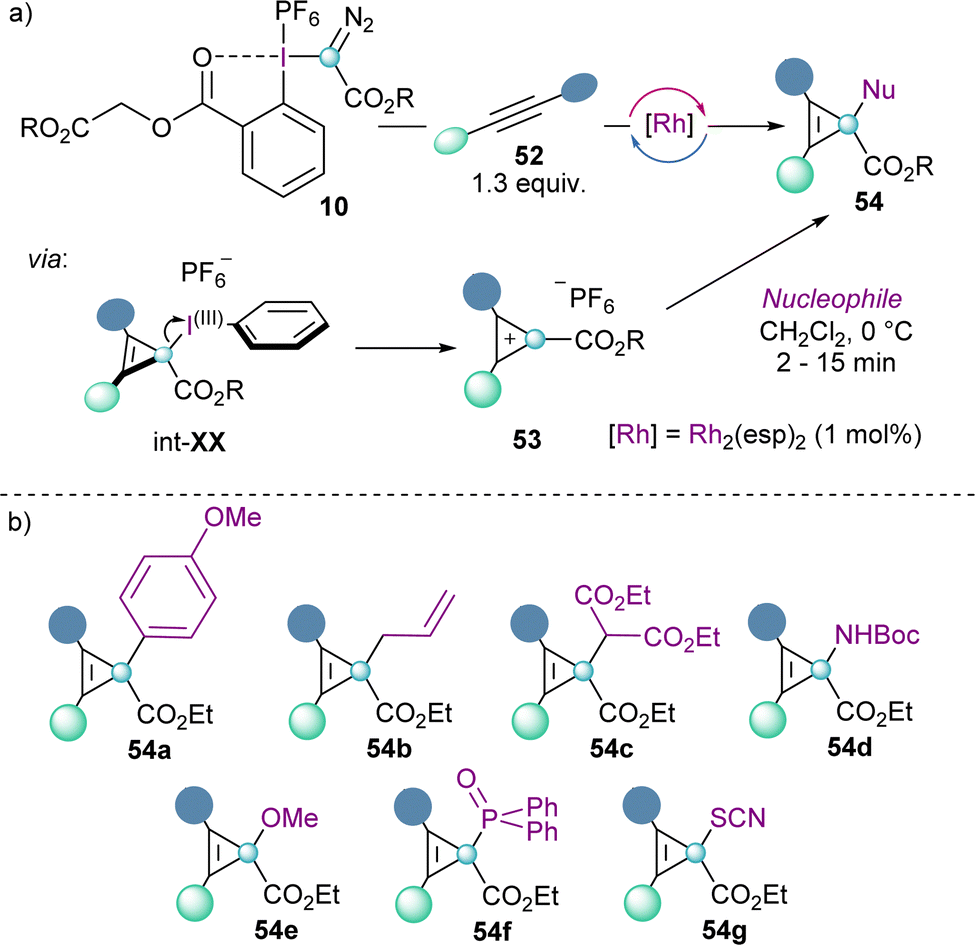 | ||
| Scheme 16 Rh-catalysed synthesis of cyclopropenium cations via carbene insertion. | ||
Finally, early in 2023 the group of Suero reported the reaction of unsaturated carboxylic acids with in situ generated int-XVII to form transient Fisher-type acyloxy Rh(II)-carbenes (int-XXI) in a catalytic fashion (Scheme 17a). The subsequent intramolecular alkene cyclopropanation of such intermediate leads to the formation of cyclopropyl-fused δ-lactones 56 with high diastereoselectivity (Scheme 17b). Moreover, complex tricyclic lactones (56b) could be obtained by using already cyclic alkenyl carboxylic acids as starting materials (Scheme 17c).39
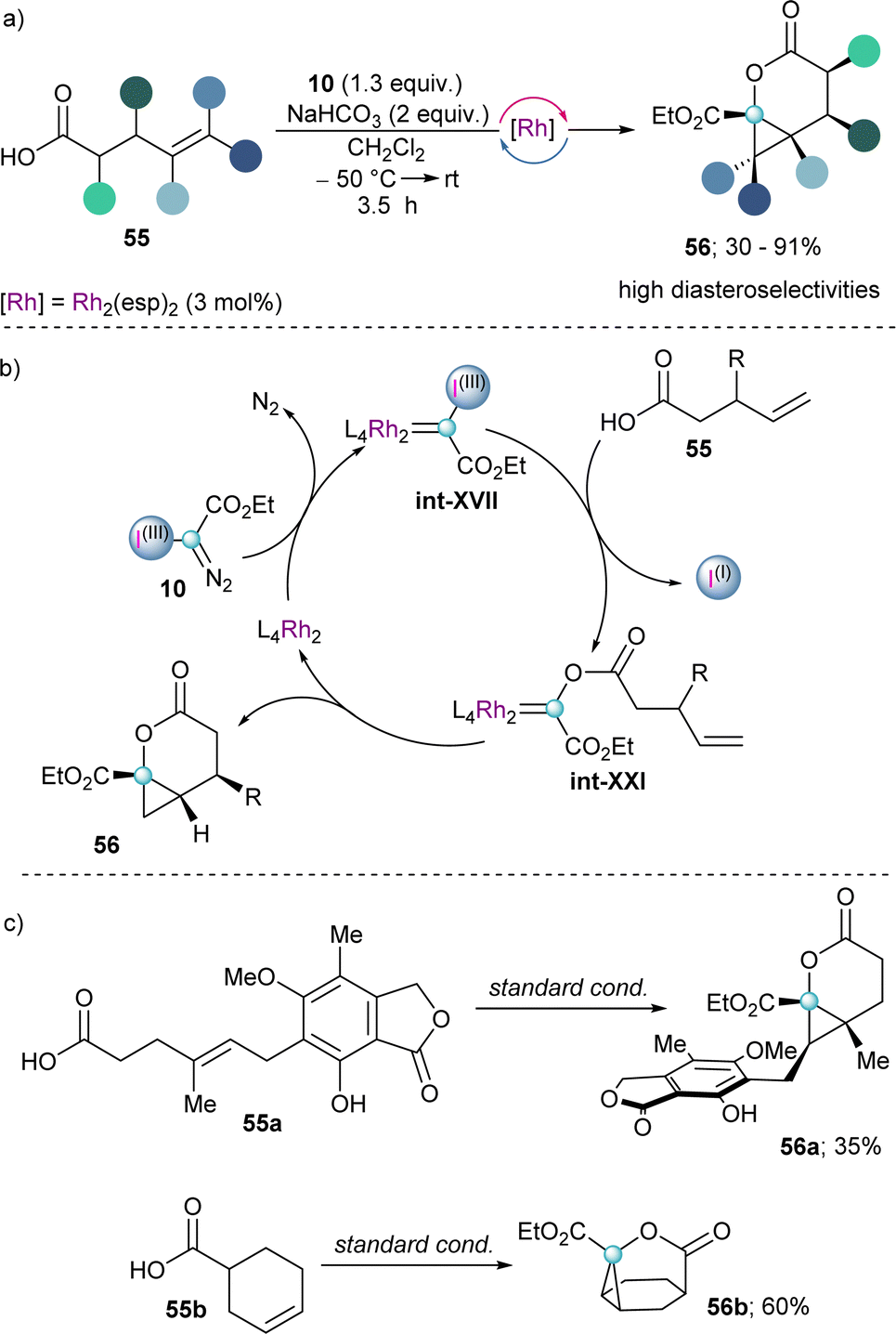 | ||
| Scheme 17 (a) Rh-catalysed carbyne transfer with alkenyl carboxylic acids. (b) Proposed mechanism for the formation of a Fisher-type carbene with follow up intramolecular alkene cyclopropanation. | ||
4. α-Diazo sulfonium salts
During the last years sulfonium salts have gained rising attention as versatile reagents in synthetic chemistry.40 As mentioned in the introduction, they share many aspects of their reactivity with hypervalent I(III) reagents of analogue structure and, consequently, they are often employed as their surrogates.Awared of the synthetic versatility of reagents 9 and 10, we asked ourselves about the possibility to prepare a sulfur(IV) based reagent bearing a diazoester substituent directly attached to the S-atom. Actually, the preparation of such structures was possible using the same synthetic strategy that we had previously developed for the synthesis of cyano-,41 alkyne-,42 and aryl-substituted43 dibenzothiophenium salts. As originally hypothesized, α-diazo sulfonium triflate 5a demonstrated to be a suitable precursor of diazomethyl radicals int-VI under photocatalytic conditions identical to those used for 9a or 10a. In addition, we were able to employ the electrophilic radical int-VI thus prepared on the synthesis of 1-amino-1,2,3-triazoles from hydrazones (Scheme 18a and b).17 Interestingly, after N-alkylation, decarboxylation and deprotonation of 44, we were also able to obtain the corresponding 1-dialkylamino-1,2,3-triazolium salts 57. These compounds were later used as mesoionic carbene precursors, and the corresponding Rh and Pd-complexes were prepared (Scheme 18c).17,44
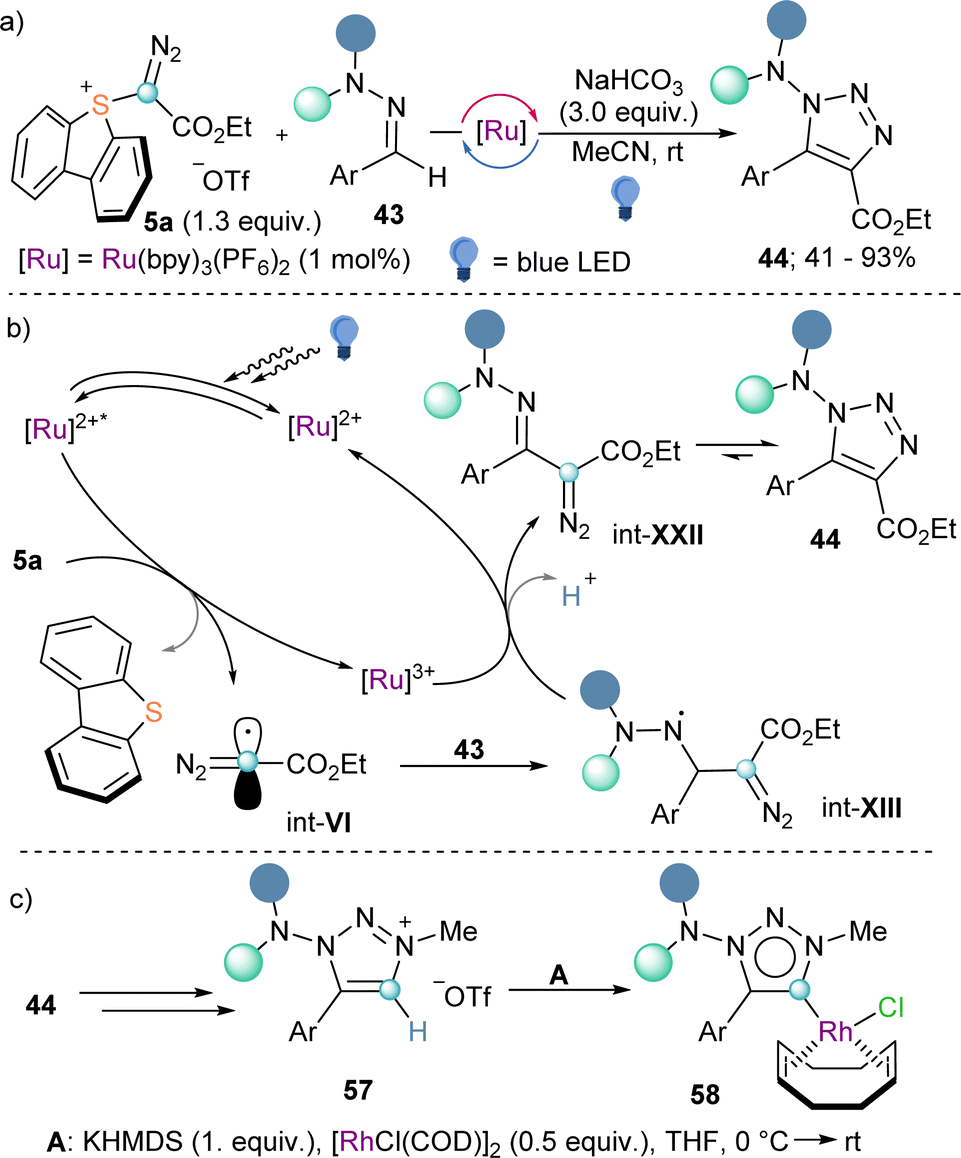 | ||
| Scheme 18 (a) Photoredox catalysed 1-amino-1,2,3-triazole formation by α-diazo sulfonium salts and hydrazones. (b) Proposed mechanism of the 1-amino-1,2,3-triazole formation. (c) Utility as mesoionic 1-amino-1,2,3-triazol-4-ylidene ligands. | ||
Finally, in 2022 the group of Wang published the coupling of internal alkynes 52 with α-diazo sulfonium triflate 17b in the presence of water to afford 1,4-dicarbonyl Z-alkenes (Scheme 19a).45 Reagent 17b had been first published by Weiss while studying the reactivity of compound 4a, but it was not employed for any synthetic porpoise since then.11 To understand how the reaction proceeds Wang and co-workers also conducted a variety of mechanistic investigations. The use of 18O-labelled and deuterated water as coupling partners in the reaction suggested that both the oxygen atom and the hydrogen one derived from water. Interestingly, when reagent 5a was employed no conversion was detected, whereas the addition of Me2S to that reaction promoted the conversion of 52 into 59, albeit in low yield. This observation together with cyclic voltammetry analyses and fluorescent quenching experiments indicate that Me2S, which is generated in situ during the progression of the reaction, is needed for an initial single-electron reduction of the photo catalyst that initiates the catalytic cycle. The reduced photocatalyst can be excited once more and undergoes single-electron transfer with reagent 17b, resulting in the formation of radical carbyne intermediate int-XXIII. By addition of this species into the C–C triple bond of the alkyne substrate, int-XXIV is formed, which is oxidized by the excited photocatalyst to the corresponding cyclopropenium cation (int-XXV). This step also delivers the reduced form of the photocatalyst, closing the catalytic cycle. Subsequent attack of water to int-XXV followed by KH2PO4 assisted ring-opening of the corresponding cyclopropanol intermediate furnishes the Z-configurated 1,4-dicarbonyl alkenes 59 (Scheme 19b).45
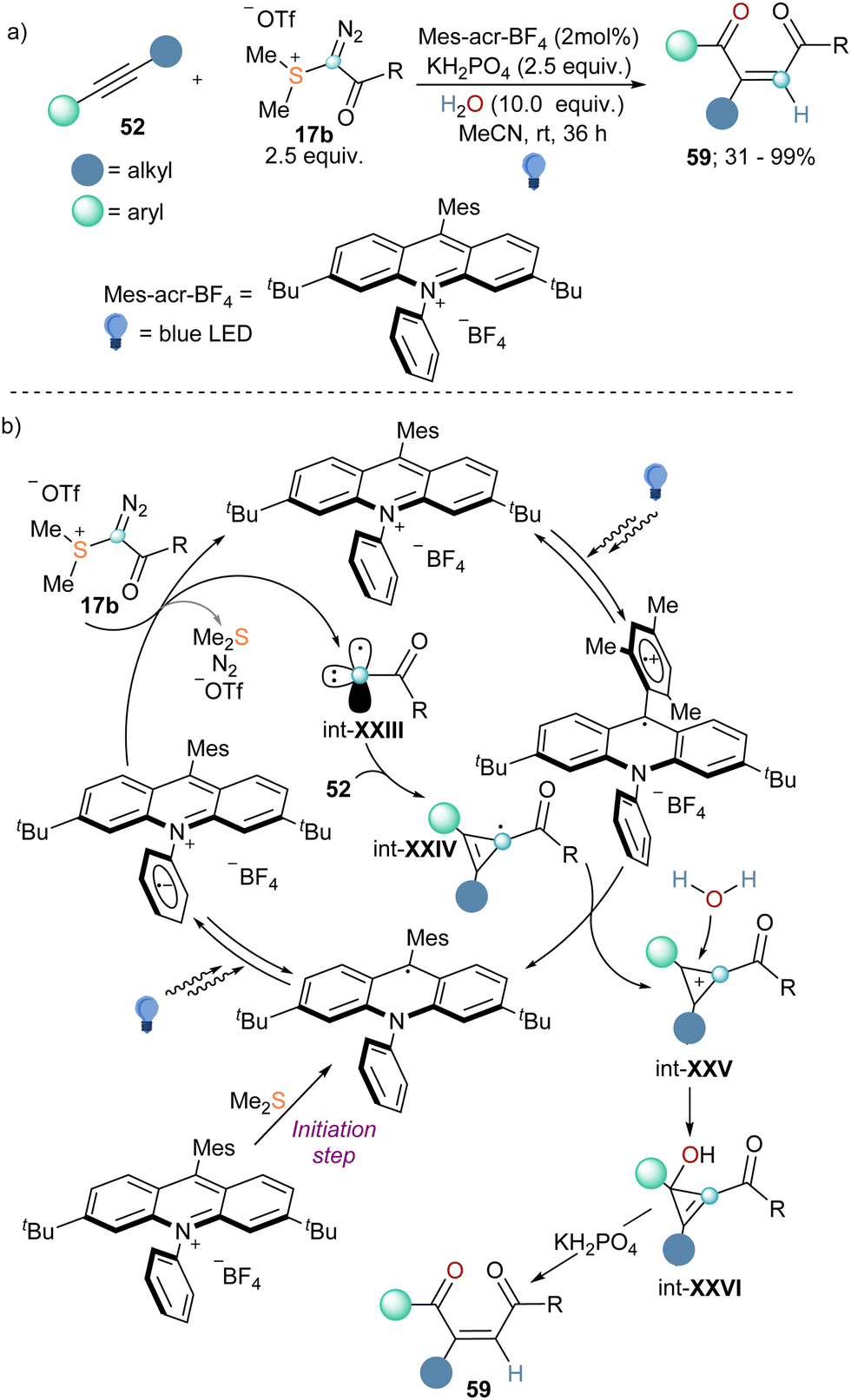 | ||
| Scheme 19 (a) Formation of 1,4-dicarbonyl Z-alkenes by coupling of alkynes, α-diazo sulfonium triflate, and water under photoredox catalysis. (b) Proposed mechanism for the three-compound reaction towards 1,4-dicarbonyl Z-alkenes. | ||
5. Summary and outlook
α-Diazo-λ3-iodanes combine on the same carbon atom two exceptionally versatile functionalities from a synthetic point of view; namely, an excellent leaving group and a masked carbene moiety. For this reason, it results difficult to understand why for the first 20 years after their introduction they were considered just curiosities, and practically ignored by the synthetic community. The answer to this question can be found in their reactive nature. Probably, the difficulty to control the reactivity of the first synthesized species of this type discouraged synthetic chemists from using them. It has not been until the change from the iodobenzene scaffold to the benziodoxolone one, and the subsequent development of the more stable α-diazo sulfonium salts that synthetic applications of these reagents have flourished. Nowadays, it is possible to selectively make react each of the two functionalities by controlling the reaction conditions. Especially useful in this regard has been the use of photocatalytic conditions to generate diazoalkyl radicals and the use of Rh catalysts able to transfer a carbene bearing an excellent leaving group as substituent. Just by gearing these two modes of reactivity we predict these compounds to find new applications in areas such as late-stage functionalization or (hetero)cycle editing. In addition, there are still several challenges ahead that deserve to be explored, such as the combination of (photochemically generated) diazoalkyl radicals with C–H activation strategies, the enantioselective transfer of the diazoalkyl or carbyne groups, or the design of stable and easy to handle “nitrene”-analogues of these reagents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the European Research Council (ERC CoG 771295) and the University of Göttingen is gratefully acknowledged.Notes and references
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS PubMed; (b) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed; (b) H. M. L. Davies and S. J. Hedley, Chem. Soc. Rev., 2007, 36, 1109–1119 RSC; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (d) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC.
- (a) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed; (b) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918–4931 RSC.
- J. Fink and M. Regitz, Synthesis, 1985, 569–585 CrossRef CAS.
- (a) T. V. Baiju and I. N. N. Namboothiri, Chem. Rec., 2017, 17, 939–955 CrossRef CAS PubMed; (b) M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) F. Weigend and R. Alrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- Calculations were performed using Gaussian16, Revision A.03.; M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scal-mani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16 Rev. A.03, Wallingford, CT, 2016 Search PubMed.
- S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67–84 CrossRef CAS PubMed.
- R. Weiss, J. Seubert and F. Hampel, Angew. Chem., Int. Ed. Engl., 1994, 33, 1952–1953 CrossRef.
- (a) V. V. Zhdankin and J. D. Protasiewicz, Coord. Chem. Rev., 2014, 275, 54–62 CrossRef CAS; (b) S. V. Kohlhepp and T. Guldner, Chem. Soc. Rev., 2016, 45, 6270–6288 RSC; (c) S. Alazet, J. Preindl, R. Simonet-Davin, S. Nicolai, A. Nanchen, T. Meyer and J. Waser, J. Org. Chem., 2018, 83, 12334–12356 CrossRef CAS PubMed.
- L. Jiang, Z. Wang, M. Armstrong and M. G. Suero, Angew. Chem., Int. Ed., 2021, 60, 6177–6184 CrossRef CAS PubMed.
- (a) V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, M. S. Formaneck and J. T. Bolz, Tetrahedron Lett., 1994, 35, 9677–9680 CrossRef CAS; (b) A. P. Krasutsky, C. J. Kuehl and V. V. Zhdankin, Synlett, 1995, 1081–1082 CrossRef CAS; (c) Y. Kita, S. Akai, T. Okuno, M. Egi, T. Takada and H. Tohma, Heterocycles, 1996, 42, 47 CrossRef; (d) V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192–5197 CrossRef CAS; (e) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed; (f) V. V. Zhdankin, Hypervalent Iodine Chemistry, John Wiley & Sons Ltd, Chichester, UK, 2013 CrossRef.
- Z. Wang, A. G. Herraiz, A. M. Del Hoyo and M. G. Suero, Nature, 2018, 554, 86–91 CrossRef CAS PubMed.
- T. A. Podrugina, A. S. Pavlova, D. S. Vinogradov, M. V. Shuvalov, I. D. Potapov, I. I. Levina, A. V. Mironov and R. Gleiter, Russ. Chem. Bull., 2019, 68, 284–292 CrossRef CAS.
- X. Li, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2021, 60, 6943–6948 CrossRef CAS PubMed.
- D. B. Cowie, G. N. Cohen, E. T. Bolton and H. de Robichin-Szulmajster, Biochim. Biophys. Acta, 1959, 34, 39–46 CrossRef CAS PubMed.
- R. L. Levine, J. Moskovitz and E. R. Stadtman, IUBMB Life, 2000, 50, 301–307 CrossRef CAS PubMed.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS PubMed.
- C. Schnaars, M. Hennum and T. Bonge-Hansen, J. Org. Chem., 2013, 78, 7488–7497 CrossRef CAS PubMed.
- M. Regitz, Ann. Chem. Pharm., 1964, 676, 101–109 CrossRef CAS.
- H. Huang, X. Zou, S. Cao, Z. Peng, Y. Peng and X. Wang, Org. Lett., 2021, 23, 4185–4190 CrossRef CAS PubMed.
- M. A. Ansari, G. Kumar and M. S. Singh, Org. Lett., 2022, 24, 2815–2820 CrossRef CAS PubMed.
- L. Devi, P. Kumar, R. Kant and N. Rastogi, Chem. Commun., 2022, 58, 7062–7065 RSC.
- M. A. Ansari, S. Khan, S. Ray, G. Shukla and M. S. Singh, Org. Lett., 2022, 24, 6078–6082 CrossRef CAS PubMed.
- (a) T. Do Minh, H. E. Gunning and O. P. Strausz, J. Am. Chem. Soc., 1967, 89, 6785–6787 CrossRef; (b) O. P. Strausz, D. M. Thap and J. Font, J. Am. Chem. Soc., 1968, 90, 1930–1931 CrossRef CAS; (c) O. P. Strausz, G. J. A. Kennepohl, F. X. Garneau, T. DoMinh, B. Kim, S. Valenty and P. S. Skell, J. Am. Chem. Soc., 1974, 96, 5723–5732 CrossRef CAS; (d) T. B. Patrick and G. H. Kovitch, J. Org. Chem., 1975, 40, 1527–1528 CrossRef CAS; (e) T. B. Patrick and T.-T. Wu, J. Org. Chem., 1978, 43, 1506–1509 CrossRef CAS.
- Y. Yamaguchi and H. Schaefer, J. Chem. Phys., 2004, 120, 9536–9546 CrossRef CAS PubMed.
- X. Ma, A. Sun and K.-K. Wang, New J. Chem., 2022, 46, 6856–6859 RSC.
- L. Liu, Y. Zhang, W. Zhao, J. Wen, C. Dong, C. Hu and J. Li, Org. Lett., 2023, 25, 592–596 CrossRef CAS PubMed.
- J.-Y. Dong, H. Wang, S. Mao, X. Wang, M.-D. Zhou and L. Li, Adv. Synth. Catal., 2021, 363, 2133–2139 CrossRef CAS.
- J. Wen, W. Zhao, X. Gao, X. Ren, C. Dong, C. Wang, L. Liu and J. Li, J. Org. Chem., 2022, 87, 4415–4423 CrossRef CAS PubMed.
- J. Li, X.-C. Lu, Y. Xu, J.-X. Wen, G.-Q. Hou and L. Liu, Org. Lett., 2020, 22, 9621–9626 CrossRef CAS PubMed.
- W.-W. Zhao, Y.-C. Shao, A.-N. Wang, J.-L. Huang, C.-Y. He, B.-D. Cui, N.-W. Wan, Y.-Z. Chen and W.-Y. Han, Org. Lett., 2021, 23, 9256–9261 CrossRef CAS PubMed.
- Z. Wang, L. Jiang, P. Sarró and M. G. Suero, J. Am. Chem. Soc., 2019, 141, 15509–15514 CrossRef CAS PubMed.
- (a) M. S. Baird, D. G. Lindsay and C. B. Reese, J. Chem. Soc. C, 1969, 1173 RSC; (b) P. von, R. Schleyer, G. W. van Dine, U. Schöllkopf and J. Paust, J. Am. Chem. Soc., 1966, 88, 2868–2869 CrossRef; (c) M. Fedoryński, Chem. Rev., 2003, 103, 1099–1132 CrossRef PubMed.
- L. Jiang, P. Sarró, W. J. Teo, J. Llop and M. G. Suero, Chem. Sci., 2022, 13, 4327–4333 RSC.
- H.-F. Tu, A. Jeandin and M. G. Suero, J. Am. Chem. Soc., 2022, 144, 16737–16743 CrossRef CAS PubMed.
- E. Palomo, A. K. Sharma, Z. Wang, L. Jiang, F. Maseras and M. G. Suero, J. Am. Chem. Soc., 2023, 145, 4975–4981 CrossRef CAS PubMed.
- (a) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (b) S. I. Kozhushkov and M. Alcarazo, Eur. J. Inorg. Chem., 2020, 2486–2500 CrossRef CAS PubMed; (c) A. Péter, G. J. P. Perry and D. J. Procter, Adv. Synth. Catal., 2020, 362, 2135–2142 CrossRef; (d) L. van Dalsen, R. E. Brown, J. A. Rossi-Ashton and D. J. Procter, Angew. Chem., Int. Ed., 2023, e202303104 Search PubMed.
- X. Li, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2019, 58, 9496–9500 CrossRef CAS PubMed.
- (a) B. Waldecker, F. Kraft, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2018, 130, 12718–12722 CrossRef; (b) K. Kafuta, C. J. Rugen, T. Heilmann, T. Liu, C. Golz and M. Alcarazo, Eur. J. Org. Chem., 2021, 4038–4048 CrossRef CAS PubMed.
- (a) K. Kafuta, A. Korzun, M. Böhm, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 1950–1955 CrossRef CAS PubMed; (b) M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163–169 CrossRef CAS; (c) P. Xu, D. Zhao, F. Berger, A. Hamad, J. Rickmeier, R. Petzold, M. Kondratiuk, K. Bohdan and T. Ritter, Angew. Chem., Int. Ed., 2020, 59, 1956–1960 CrossRef CAS PubMed; (d) S. Karreman, S. B. H. Karnbrock, S. Kolle, C. Golz and M. Alcarazo, Org. Lett., 2021, 23, 1991–1995 CrossRef CAS PubMed.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- X. Wang, W.-Y. Tong, B. Huang, S. Cao, Y. Li, J. Jiao, H. Huang, Q. Yi, S. Qu and X. Wang, J. Am. Chem. Soc., 2022, 144, 4952–4965 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |